Кристаллическая форма эртапенема натрия и способ ее получения - RU2583052C2
Код документа: RU2583052C2
Чертежи

Описание
Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к исследованию кристаллической формы эртапенема натрия, особенно кристаллической формы E эртапенема натрия и способу ее получения.
Уровень техники настоящего изобретения
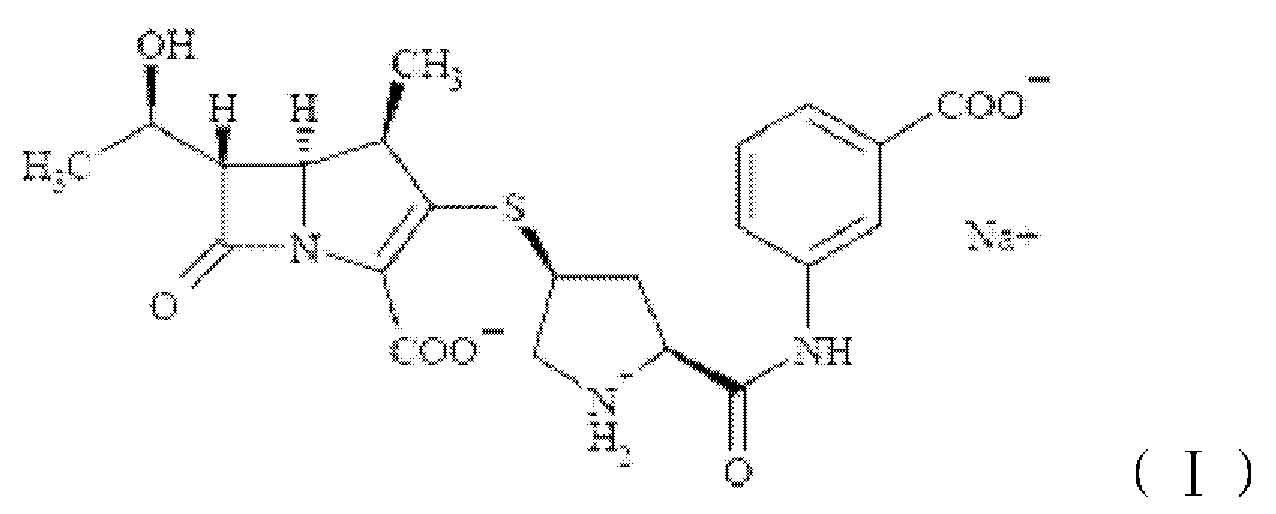
Эртапенем натрия имеет структуру формулы I, химическим названием которой является мононатриевая соль (1R,5S,6S,8R,2S*,4S*)-2-[2-[(3-карбоксифенил)карбамоил]пирролидинил-4-тио]-6-(1-гидроксиэтил)-1-метилкарбапенем-3-карбоновой кислоты. Эртапенем натрия представляет собой новый карбапенемовый антибиотик широкого спектра действия, совместно разрабатываемый Merck & Co. (U.S.) и AstraZeneca, который обладает хорошей антибактериальной активностью относительно грамположительных и грамотрицательных аэробных и анаэробных бактерий.
Полиморфизм является важным свойством соединения, и он обычно присутствует у большинства химических лекарственных средств. Вещество в специфической кристаллической форме оказывает важное влияние на стабильность, однородность, биодоступность и формулирование и т.д. Эртапенем натрия обладает низкой стабильностью и является крайне чувствительным к нагреванию, кислоте и т.д. Для того чтобы снизить разложение продукта и улучшить качество формулируемого продукта, исследователи провели обширные исследования кристаллических форм эртапенема натрия, и различные кристаллические формы эртапенема натрия и способы их получения описаны на предшествующем уровне техники.
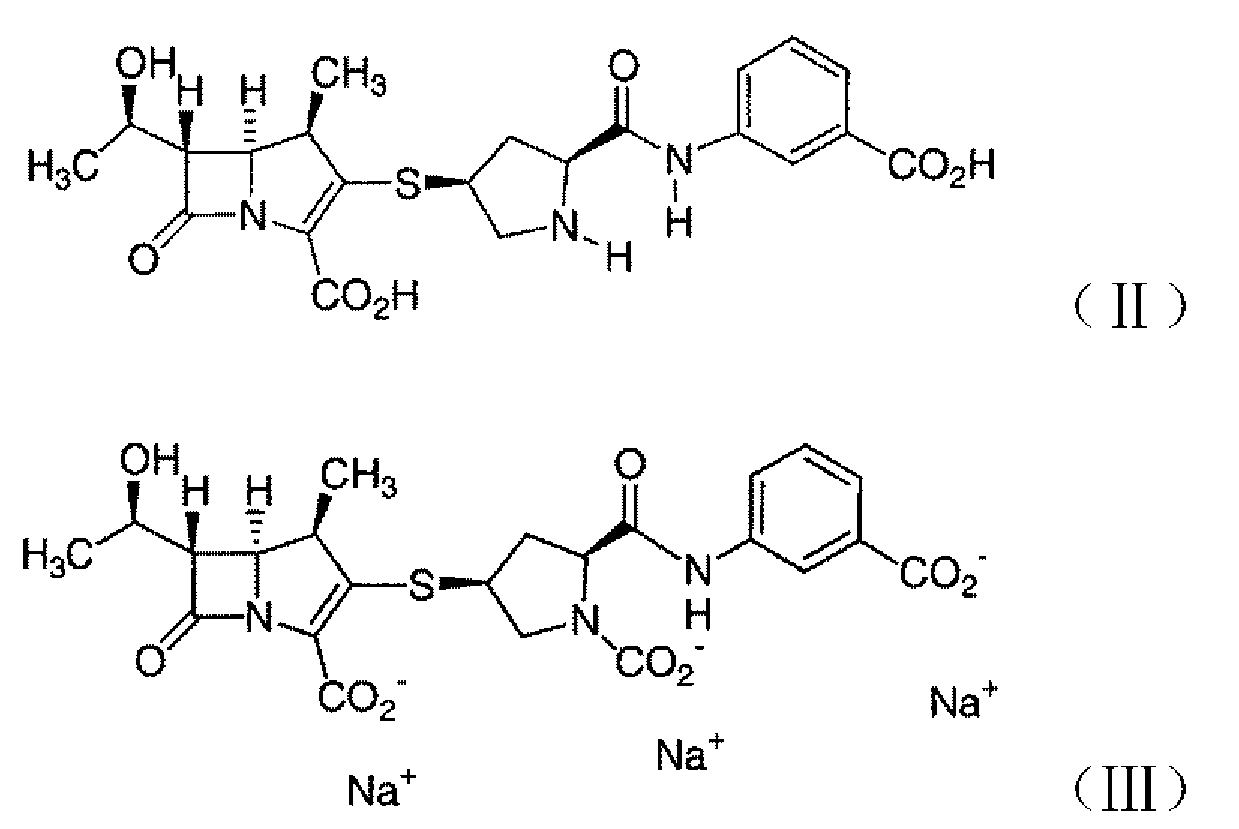
Например, WO03026572 описывает кристаллическую форму A и кристаллическую форму B эртапенема натрия и способ их получения. Кристаллическая форма A имеет основные дифракционные пики при приблизительно 4,8º, 6,7º, 10,5º, 11,7º, 13,6º, 14,4º, 16,0º, 17,2º, 18,4º, 19,7º, 20,8º, 21,6º, 22,1º, 23,1º, 24,1º, 26,1º, 26,6º, 27,0º, 27,4º, 28,6º и 31,1º в спектре дифракции рентгеновских лучей, представленные 2θ углом. Кристаллическая форма B имеет основные дифракционные пики при приблизительно 4,8º, 6,8º, 7,8º, 10,4º, 11,8º, 13,6º, 14,4º, 15,2º, 17,3º, 18,5º, 19,0º, 19,7º, 20,9º, 21,9º, 23,1º, 24,1º, 24,5º, 26,1º, 26,5º, 26,9º, 27,7º, 28,7º, 30,0º, 31,1º и 32,2º в спектре дифракции рентгеновских лучей, представленные 2θ углом.
Способ получения кристаллической формы A включает: a) добавление 1-пропанола к водному раствору, содержащему эртапенем формулы II и III и/или его солевые формы; b) охлаждение раствора ниже -5ºC; c) доведение pH до приблизительно 6 - приблизительно 5, применяя кислоту; d) кристаллизацию добавлением к раствору от приблизительно 0,5 до приблизительно 3 объемов метанола относительно объема водного раствора, и от приблизительно 0,5 до приблизительно 3 объемов 1-пропанола относительно объема водного раствора; и e) выделение с получением кристаллической формы A эртапенема натрия.
Способ получения кристаллической формы B включает стадии: промывки кристаллической формы A эртапенема натрия смесью воды и 2-пропанола с получением кристаллической формы B, где указанная смесь содержит от приблизительно 5% до приблизительно 25% воды (об./об.).
Кроме того, WO03027067 описывает кристаллическую форму C эртапенема натрия. В способе, описанном в данном патенте, кристаллическую форму C эртапенема натрия выделяют и получают после промывки приведенной выше кристаллической формы A или кристаллической формы B эртапенема натрия водным раствором этилацетата, ацетона или их смеси. Кристаллическая форма C соединения имеет основные дифракционные пики при приблизительно 4,8º, 6,8º, 7,8º, 10,7º, 11,8º, 13,7º, 14,6º, 17,3º, 18,6º, 19,14º, 19,9º, 21,0º, 22,1º, 24,2º, 26,1º, 27,9º, 28,7º, 31,3º и 32,5º в спектре дифракции рентгеновских лучей, представленные 2θ углом.
Все способы получения кристаллической формы A, кристаллической формы B и кристаллической формы C эртапенема натрия имеют следующие недостатки: в приведенных выше способах при кристаллизации эртапенема натрия требуется, чтобы все концентрации для растворов кристаллизации эртапенема натрия были выше 100 мг/мл. Ввиду того факта, что эртапенем натрия легко разлагается и полимеризуется, общепринятое концентрирование и нанофильтрация будут вызывать значительное разложение продукта. Следовательно, из-за чрезмерного концентрирования раствора эртапенема натрия, трудно удовлетворить требования, касающиеся и чистоты и цветности кристалла. Кроме того, большое количество растворителя также включено в приведенный выше способ, который не приспособлен к защите окружающей среды.
WO2009150630 описывает кристаллическую форму D эртапенема натрия. Данная кристаллическая форма D имеет основные дифракционные пики при приблизительно 4,44º, 5,26º, 7,44º, 8,12º, 10,98º, 12,74º, 19,28º, 22,93º, 23,51º, 25,07º и 30,15º в спектре дифракции рентгеновских лучей, представленные 2θ углом. Способ получения кристаллической формы D включает следующие стадии: a) обработка эртапенема натрия водой и метанолом; b) обработка раствора, полученного на стадии a), 1-пропанолом; c) перемешивание смеси, полученной на стадии b), при температуре приблизительно 0ºC или ниже 0ºC, для осаждения твердых веществ; d) обработка твердого остатка, полученного на стадии c), ацетоном с получением кристаллической формы D эртапенема натрия. Недостатки кристаллической формы D эртапенема натрия заключаются в том, что кристаллизационные свойства являются плохими, частицы небольшими и фильтрация с отсасывание является сложной.
В добавление к приведенным выше различным продуктам в виде кристаллических форм эртапенема натрия, описанным на предшествующем уровне техники, несколько способов получения аморфных продуктов эртапетанема натрия также описаны на предшествующем уровне техники, например:
В CN1752090A, ацетон и пропанол добавляют в реакционную систему эртапенема натрия после экстракции, и нерастворившиеся вещества удаляют, и продукт осаждают посредством кристаллизации выпариванием, который затем промывают 95% этанолом и метилацетатом и очищают, получая аморфный продукт эртапенема натрия.
В качестве другого примера, способ получения твердого эртапенема натрия описан в “Synthesis of carbapenem antibiotic ertapenem” ZHANG Yi-feng (Journal of China Pharmaceutical University, 2007, 38(4): 305-310), который включает следующие стадии: реакционную жидкость эртапенема натрия фильтруют, затем фильтрат экстрагируют дихлорметаном, и водный слой концентрируют при пониженном давлении, удаляя органический растворитель, затем очищают CHP-20P смолой, и затем лиофилизуют, получая белый порошок эртапенема натрия, который оказался аморфным продуктом после исследования рентгеновским анализом.
Основными недостатками приведенного выше аморфного продукта эртапенема натрия являются: низкая стабильность, низкая чистота, и что трудно удовлетворить требования к степени окраски.
Сущность настоящего изобретения
Техническая проблема, которую решает настоящее изобретение, заключается в обеспечении кристаллической формы E эртапенема натрия. По сравнению с предшествующим уровнем техники, в способе получения кристаллической формы E эртапенема натрия, относящемся к настоящему изобретению, требуемая концентрация раствора эртапенема натрия является низкой, и полученная кристаллическая форма E является очень чистой и обладает высокой стабильностью.
Для того чтобы решить приведенные выше технические проблемы, настоящее изобретение обеспечивает кристаллическую форму E эртапенема натрия, показанную формулой (I)
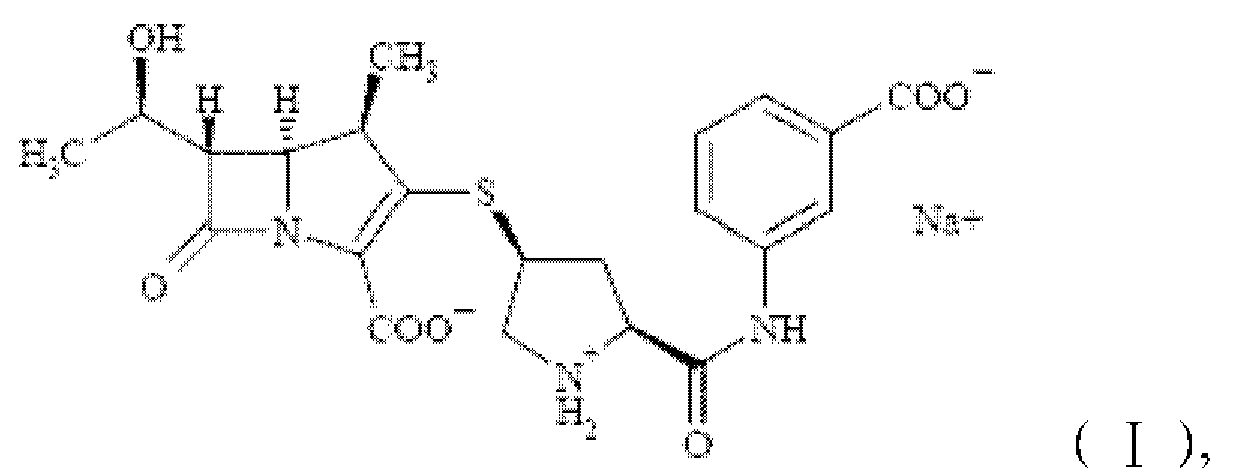
При применении CuKα излучения спектр дифракции рентгеновских лучей кристаллической формы имеет основные дифракционные пики при брэгговских углах 2θ (º) 4,220, 4,900, 6,980, 8,000, 10,720, 11,960, 13,958, 14,740, 17,319, 18,641, 19,200, 22,060, 24,780, 26,299 и 27,920±0,2º.
Кроме того, настоящее изобретение относится к способу получения кристаллической формы E эртапенема натрия, включающему стадии:
a) получения водного раствора эртапенема натрия при концентрации 40~100 мг/мл;
b) доведения величины pH водного раствора эртапенема натрия до значений 5,3~5,6 путем добавления кислоты, в условиях 0~20ºC;
c) добавления по каплям метанола и 1-пропанола к раствору, полученному на стадии b), до того как объемное отношение вода:метанол:1-пропанол будет составлять 1:0,5~2:0,25~1,5, с последующим охлаждением до -10~-5ºC и выдерживанием;
d) добавления по каплям метанола и 1-пропанола к раствору, полученному на стадии c), до того как объемное отношение вода:метанол:1-пропанол будет составлять 1:0,8~3:0,8~3,5, с последующим охлаждением до -30~-10ºC и осаждением кристаллов с получением кристаллической формы E эртапенема натрия.
Предпочтительно, чтобы концентрация водного раствора эртапенема натрия на стадии a) составляла 50~90 мг/мл.
Предпочтительно, чтобы кислота на стадии b) представляла собой одну или более кислот, выбранных из группы, состоящей из муравьиной кислоты, уксусной кислоты, пропановой кислоты и хлористоводородной кислоты, предпочтительно уксусную кислоту.
Предпочтительно, чтобы величину pH водного раствора эртапенема натрия на стадии b) доводили до значений 5,4~5,5 кислотой.
Предпочтительно, чтобы температура для осаждения кристаллов на стадии d) составляла -25~-15ºC.
Предпочтительно, чтобы на стадии c) метанол и 1-пропанол добавляли по каплям к раствору, полученному на стадии b), до того как объемное отношение вода:метанол:1-пропанол будет составлять 1:0,8~1,5:0,4~1.
Предпочтительно, чтобы на стадии d) метанол и 1-пропанол добавляли по каплям к раствору, полученному на стадии c), до того как объемное отношение вода:метанол:1-пропанол будет составлять 1:1~2,5:1~3.
Предпочтительно, способ дополнительно включает стадию добавления по каплям зародыша кристалла к раствору, полученному после добавления метанола и 1-пропанола на стадии c).
Кроме того, настоящее изобретение относится к применению кристаллической формы E эртапенема натрия для получения лекарственного средства для лечения инфекции.
Кроме того, настоящее изобретение относится к фармацевтической композиции, содержащей кристаллическую форму E эртапенема натрия согласно техническим решениям, приведенным выше. Фармацевтическая композиция предпочтительно представляет собой лиофилизованный порошок для инъекции.
Настоящее изобретение относится к кристаллической форме E эртапенема натрия. Кристаллическую форму E, относящуюся к настоящему изобретению, легко фильтровать, сушить, и она является стабильной по свойствам в процессе способа сушки, и чистота кристаллов является большой и может достигать более чем 98,5%. Более того, в способе получения кристаллической формы E эртапенема натрия, относящемся к настоящему изобретению, благодаря меньшим концентрациям применяемого раствора эртапенема натрия, трудности и потребление энергии для концентрирования снижаются, что создает условия для промышленного получения.
Описание чертежей
Фигура 1 представляет собой спектр дифракции рентгеновских лучей кристаллической формы E эртапенема натрия, полученной в примере 1 настоящего изобретения.
Подробное описание вариантов осуществления настоящего изобретения
Настоящее изобретение описывает кристаллическую форму E эртапенема натрия. При применении CuKα излучения спектр дифракции рентгеновских лучей кристаллической формы имеет основные дифракционные пики при брэгговских углах 2θ (º) 4,220, 4,900, 6,980, 8,000, 10,720, 11,960, 13,958, 14,740, 17,319, 18,641, 19,200, 22,060, 24,780, 26,299 и 27,920. Специалисту в данной области техники ясно, что дифракционные пики кристаллической формы согласно настоящему изобретению не ограничиваются пиками, возникающими при приведенных выше 2θ углах, и дополнительно включают дифракционные пики, возникающие в пределах приведенных выше 2θ углов ±0,2º из-за экспериментальной ошибки и других факторов, которые также включены в объем притязаний настоящего изобретения. Концентрация водного раствора эртапенема натрия, приведенная в настоящем изобретении, представляет собой отношение растворенной массы к объему растворителя.
Настоящее изобретение описывает способ получения кристаллической формы E эртапенема натрия, включающий:
a) получение водного раствора эртапенема натрия при концентрации 40~100 мг/мл;
b) доведение величины pH водного раствора эртапенема натрия до значений 5,3~5,6 путем добавления кислоты в условиях 0~20ºC;
c) добавление по каплям метанола и 1-пропанола к раствору, полученному на стадии b), до того как объемное отношение вода:метанол:1-пропанол будет составлять 1:0,5~2:0,25~1,5 с последующим охлаждением до -10~-5ºC и выдерживанием;
d) добавление по каплям метанола и 1-пропанола к раствору, полученному на стадии c), до того как объемное отношение вода:метанол:1-пропанол будет составлять 1:0,8~3:0,8~3,5, с последующим охлаждением до -30~-10ºC и осаждением кристаллов с получением кристаллической формы E эртапенема натрия.
Согласно настоящему изобретению на стадии a) неочищенный эртапенем натрия можно растворять в воде, получая водный раствор при концентрации 40~100 мг/мл. Что касается неочищенного эртапенема натрия, его получают согласно способам, описанным на предшествующем уровне техники, конкретными примерами являются способы получения, описанные в CN93101472.7, CN02803742.1, CN9880609.1, CN200510030660.5, патенте США US6504027, WO03026572, WO2008062279, “Synthesis of carbapenem antibiotic ertapenem” ZHANG Yi-feng (Journal of China Pharmaceutical University, 2007, 38(4): 305-310) и т.д., но не ограничиваются ими. Содержание приведенных выше документов включено в настоящее изобретение с помощью ссылки. Концентрация водного раствора эртапенема натрия на стадии a) предпочтительно составляет 50~90 мг/мл.
Согласно настоящему изобретению на стадии b) величину pH водного раствора эртапенема натрия доводят путем добавления кислоты до значений 5,3~5,6, предпочтительно до 5,4~5,5, в условиях предпочтительно 2~18ºC, более предпочтительно 5~15ºC. Указанная кислота предпочтительно представляет собой одну или более кислот, выбранных из группы, состоящей из муравьиной кислоты, уксусной кислоты, пропановой кислоты и хлористоводородной кислоты, более предпочтительно уксусную кислоту.
Согласно настоящему изобретению на стадии c) предпочительно добавлять по каплям метанол и 1-пропанол к раствору, полученному на стадии b), до того как объемное отношение вода:метанол:1-пропанол будет составлять 1:0,8~1,5:0,4~1, с последующим охлаждением до -10~-5ºC и выдерживанием. Перед выдерживанием, предпочтительно добавлять зародыш кристалла, затем выдерживать, и раствор перемешивают до того, как он становится мутным. Что касается времени выдерживания, оно предпочтительно составляет 5~30 минут.
На стадии d) предпочтительно поддерживать температуру на стадии c) неизменной, затем добавляют по каплям метанол и 1-пропанол, до того как объемное отношение вода:метанол:1-пропанол будет составлять 1:0,8~3:0,8~3,5, предпочтительно 1:1~2,5:1~3, с последующим охлаждением до -30~-10ºC, более предпочтительно -25~-15ºC, затем перемешиванием в течение 1~10 часов, и кристаллы осаждаются с получением кристаллической формы E эртапенема натрия.
Согласно настоящему изобретению после стадии d) оно может дополнительно включать стадии выделения кристаллической формы E эртапенема натрия. Что касается способа выделения, он может представлять собой фильтрование и другие способы, хорошо известные специалисту в данной области техники, такие как фильтрование и затем сушка, но не ограничиваются ими.
Кроме того, настоящее изобретение относится к применению кристаллической формы E эртапенема натрия для получения лекарственного средства для лечения инфекции.
Кроме того, настоящее изобретение относится к фармацевтической композиции, содержащей кристаллическую форму E эртапенема натрия, относящуюся к настоящему изобретению, в качестве активного ингредиента. Фармацевтическая композиция предпочтительно представляет собой лиофилизованный порошок для инъекции.
По сравнению с предшествующем уровнем техники, кристаллическую форму E эртапенема натрия, относящуюся к настоящему изобретению, легко фильтровать, сушить, и она является стабильной по свойствам в процессе сушки, и чистота кристаллов является высокой и может достигать более чем 98,5%. Более того, в способе получения кристаллической формы E эртапенема натрия, относящемся к настоящему изобретению, благодаря меньшим концентрациям применяемого раствора эртапенема натрия, трудности и потребление энергии для концентрирования снижаются, что создает условия для промышленного получения.
Для того чтобы лучше понять настоящее изобретение, новую кристаллическую форму эртапенема натрия, относящуюся к настоящему изобретению, описывают вместе с примерами, однако, объем притязаний настоящего изобретения не ограничивается следующими примерами.
Спектры дифракции ренгеновских лучей образцов в следующих примерах получали в следующих условиях:
Прибор: Rigaku D/max-2550 порошковый рентгеновский дифрактомер, Япония;
Условия: CuKα излучение, графитовый монохроматор, напряжение на лампе 40 кВ, сила тока лампы 40 мА, 2θ сканирующий диапазон 2~40º, скорость сканирования 8º/минуту, ширина шага 0,02º.
Пример 1: Получение кристаллической формы E эртапенема натрия, согласно следующим стадиям:
a) 10 г неочищенного эртапенема натрия растворяли в воде для получения раствора 80 мг/мл;
b) величину pH раствора, полученного на стадии a), доводили до значения 5,5 добавлением хлористоводородной кислоты при 0ºC;
c) метанол и 1-пропанол добавляли по каплям к раствору, полученному на стадии b), до того как объемное отношение вода:метанол:1-пропанол составляло 1:1:0,6, охлаждали до -8ºC, и добавляли зародыш кристалла и выдерживали в течение 30 минут, и перемешивали до того как раствор становился мутным;
d) температуру раствора, полученного на стадии c), поддерживали неизменной, и метанол и 1-пропанол дополнительно добавляли к раствору, до того как объемное отношение вода:метанол:1-пропанол составляло 1:1,2:1,5, и охлаждали до -18ºC, перемешивали в течение 5 часов с образованием кристаллов; и
e) 8,5 г твердого остатка получали фильтрацией и сушкой.
Полученный твердый остаток испытывали ВЭЖХ, и чистота эртапенема натрия составляла 99,3%.
Полученный твердый остаток дополнительно исследовали рентгеновским анализом, и данные спектра дифракции рентгеновских лучей показаны в таблице 1:
Примеры 2~6
Кристаллическую форму E эртапенема натрия получали в различных технологических условиях, и подробные технологические параметры показаны в таблице 2:
Сравнительные примеры 1~2
Кристаллическую форму E эртапенема натрия получали в различных технологических условиях, и подробные технологические параметры показаны в таблице 3:
Вес, выход, ВЭЖХ чистоту и кристаллическую форму продукта сравнительных примеров 1~2 определяли соответственно, и результаты показаны в таблице 4:
Результаты в таблице 4 показывают, что, когда концентрация раствора эртапенема натрия находится в пределах диапазона 40~100 мг/мл и в условиях настоящего изобретения, можно получить кристаллическую форму E эртапенема натрия. Полнота извлечения образца увеличивалась с увеличением кристаллизационной концентрации эртапенема натрия; кристаллизация была затруднена, когда концентрация раствора эртапенема натрия была ниже чем 40 мг/мл, и когда она была большей чем 100 мг/мл, полученная кристаллическая форма эртапенема натрия была отличной от кристаллической формы E.
Пример 7:
Сравнительные испытания на стабильность кристаллической формы E и других кристаллических форм
Кристаллическую форму A и кристаллическую форму B эртапенема натрия получали согласно способам, описанным в WO03026572, кристаллическую форму C эртапенема натрия получали согласно способу, описанному в WO03027067, кристаллическую форму D эртапенема натрия получали согласно способу, описанному в WO2009150630, и аморфный продукт эртапенема натрия получали согласно способу, описанному в CN1752090A.
Приведенные выше кристаллические формы A, B, C, D, аморфный твердый эртапенем натрия и кристаллическую форму E, полученную согласно примеру 1, хранили в течение 1 года в холодильнике при 6°C и замораживали до -20ºC, и отбирали образцы через 1, 3, 6, 9 и 12 месяцев, соответственно, и исследовали суммарное количество примесей и содержание хранимых образцов, и конкретные данные показаны в таблице 5:
Можно видеть из результатов в таблице 5, что кристаллическая форма E и кристаллические формы A, B, C эртапенема натрия обладают сравнимой стабильностью, которые являются более стабильными, чем аморфный продукт и кристаллическая форма D.
Пример 8: Лиофилизованный порошок эртапенема натрия
Формула:
Способ получения для формулы является следующим:
Во-первых, взвешивали бикарбонат натрия и гидроксид натрия в количествах формулы и растворяли в воде для инъекции, и охлаждали ниже 5ºC в бане со льдом, и образец примера 1 в количестве формулы добавляли и растворяли, и величину pH доводили до значения 7,5, применяя раствор гидроксида натрия, затем продукт получали фильтрацией и лиофилизацией.
Описанные выше примеры приведены только с целью лучшего понимания способа настоящего изобретения и его основной идеи. Следует указать, что настоящее изобретение может быть также улучшено и изменено специалистом в данной области техники, не выходя за пределы принципа настоящего изобретения, и данные улучшения и модификации также включены в объем притязаний формулы изобретения настоящего изобретения.
Описание вышеописанных примеров позволяет специалисту в данной области техники реализовать или применять настоящее изобретение. Различные модификации данных примеров являются очевидными специалисту в данной области техники, и общие принципы, определенные в настоящем изобретении, можно реализовать в других примерах, не выходя за пределы сущности и объема настоящего изобретения. Следовательно, настоящее изобретение не будет ограничено данными примерами, показанными в настоящем изобретении, но будет согласовываться с самым широким объемом, который соответствует принципу и новым характеристикам, описанным в настоящем изобретении.
Реферат
Изобретение относится к кристаллической форме E эртапенема натрия и к способу получения кристаллической формы E эртапенема натрия, характеризующемуся применением водного раствора эртапенема натрия при низкой концентрации в качестве исходного вещества. Кристаллическую форму E можно легко фильтровать и сушить, свойства при сушке являются неизменными, и чистота кристаллов является высокой и может достигать вплоть до 98,5% или более. 4 н. и 8 з.п. ф-лы, 1 ил., 5 табл., 8 пр.
Формула
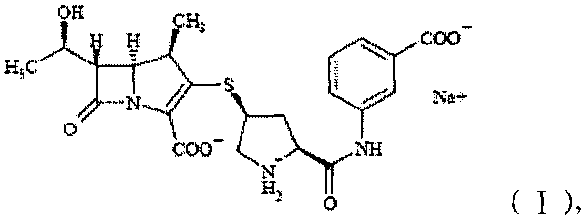
отличающаяся тем, что при применении CuKα излучения спектр дифракции рентгеновских лучей кристаллической формы имеет основные дифракционные пики при брэгговских углах 2θ (°) 4,220, 4,900, 6,980, 8,000, 10,720, 11,960, 13,958, 14,740, 17,319, 18,641, 19,200, 22,060, 24,780, 26,299 и 27,920±0,2°.
a) получения водного раствора эртапенема натрия при концентрации 40~100 мг/мл;
b) доведения величины рН водного раствора эртапенема натрия до значения 5,3~5,6 путем добавления кислоты в условиях 0~20°С;
c) добавления метанола и 1-пропанола по каплям к раствору, полученному на стадии b), до того как объемное отношение вода:метанол:1-пропанол будет составлять 1:0,5~2:0,25~1,5, с последующим охлаждением до -10 ~ -5°С и выдерживанием;
d) добавления метанола и 1-пропанола по каплям к раствору, полученному на стадии с), до того как объемное отношение вода:метанол:1-пропанол будет составлять 1:0,8~3:0,8~3,5, с последующим охлаждением до -30 ~ -10°С и осаждением кристаллов с получением кристаллической формы Е эртапенема натрия.
Комментарии