Фармацевтическая наносуспензия для терапии вич-инфекции - RU2665383C1
Код документа: RU2665383C1
Чертежи





Описание
Данное изобретение относится к фармацевтической композиции (наносуспензии) для длительно действующего инъекционного (LAI - long-acting injectable) препарата для долгосрочно поддерживающей терапии ВИЧ/СПИД.
Вирус иммунодефицита человека - ретровирус из рода лентивирусов, вызывающий медленно прогрессирующее заболевание - ВИЧ-инфекцию [Weiss R.A. How does HIV cause AIDS. Science 1993, 260(5112), 1273-1279. Douek D.C., Roederer M, Koup R.A. Emerging Concepts in the Immunopathogenesis of AID». Annu. Rev. Med. 2009, 60, 471-84]. Вирус иммунодефицита человека независимо открыли в 1983 году в двух лабораториях: в Институте Пастера во Франции под руководством Люка Монтанье и в Национальном институте рака в США под руководством Роберта Галло. Результаты исследований, в которых из тканей пациентов с симптомами СПИДа впервые удалось выделить новый ретровирус, были опубликованы 20 мая 1983 года в журнале Science [

Вирус поражает клетки иммунной системы, имеющие на своей поверхности рецепторы CD4: Т-хелперы, моноциты, макрофаги, клетки Лангерганса, дендритные клетки, клетки микроглии. В результате работа иммунной системы угнетается и развивается синдром приобретенного иммунного дефицита (СПИД), организм больного теряет возможность защищаться от инфекций и опухолей, возникают вторичные оппортунистические заболевания, которые не характерны для людей с нормальным иммунным статусом. Без врачебного вмешательства ВИЧ вызывает смерть пациента в среднем через 9-11 лет после заражения (в зависимости от подтипа вируса) [https://ru.wikipedia.org/wiki/Вирус_иммунодефицита_человека].
Согласно глобальной статистике в 2015 г. во всем мире: жили с ВИЧ 36,7 миллиона человек, 2,1 миллиона человек были инфицированы ВИЧ, 1,1 миллиона человек умерли от болезней, обусловленных СПИДом, 78 миллионов человек были инфицированы ВИЧ с момента начала эпидемии, из них 35 миллионов человек умерли от болезней, обусловленных СПИДом, с момента начала эпидемии.
По состоянию на декабрь 2015 года 17 миллионов человек, живущих с ВИЧ, имели доступ к антиретровирусной терапии, в то время как в июне 2015 года это число составляло 15,8 миллиона, а в 2010 году - 7,5 миллиона человек.
В 2015 году 46% всех взрослых, живущих с ВИЧ, имели доступ к лечению, в то время как в 2010 году этот показатель составлял 23%.
С 2010 года число новых случаев инфицирования ВИЧ снизилось на 6%. Во всем мире число людей, инфицированных ВИЧ в 2015 году, составило 2,1 миллиона человек, а в 2010 году это число составляло 2,2 миллиона.
По сравнению с пиковым показателем в 2005 году число смертей, связанных со СПИДом, снизилось на 45%, и в 2015 году число людей, умерших в связи со СПИДом, во всем мире составило 1,1 миллиона человек. Для сравнения в 2005 году это число составляло 2 миллиона.
ВИЧ можно ослаблять с помощью комбинированной антиретровирусной терапии (APT), состоящей из трех или более антиретровирусных препаратов. APT не излечивает ВИЧ-инфекцию, но контролирует репликацию вируса в организме человека и содействует укреплению иммунной системы и восстановлению ее способностей бороться с инфекциями. При проведении APT продолжительность жизни пациента может быть продлена до 70-80 лет.
В 2016 году ВОЗ выпустила второе издание «Руководства в отношении начала антиретровирусной терапии и предэкспозиционной профилактики ВИЧ».
К середине 2016 года 18,2 миллиона человек с ВИЧ в мире получали APT, что означает 46% [43-50%] всех людей с ВИЧ получали APT.
С учетом новых рекомендаций ВОЗ в отношении лечения всех людей с ВИЧ и предложения антиретровирусных препаратов в качестве дополнительного варианта профилактики людям, подвергающимся «значительному» риску, число людей, отвечающих критериям антиретровирусной терапии, возрастает с 28 миллионов до 36,7 миллиона человек. Расширение доступа к лечению является одной из центральных задач, выдвинутых на 2020 год с целью ликвидации эпидемии СПИДа к 2030 году [http://www.who.int/mediacentre/factsheets/fs360/ru/].
В настоящее время достигнуты значительные успехи в терапии ВИЧ/СПИД с помощью APT - эффективных и безопасных схем профилактики и лечения ВИЧ. Наиболее продвинутым препаратом, который в настоящий момент находится на регистрации в Минздраве России, является Элсульфавирин (Elsulfavirine, Elpida®, VM1500) для APT ВИЧ/СПИД [(a) A. Kravchenko et al. Safety and antiviral effect of Elpida (VM-1500), a novel NNRT1 (+Truvada) in

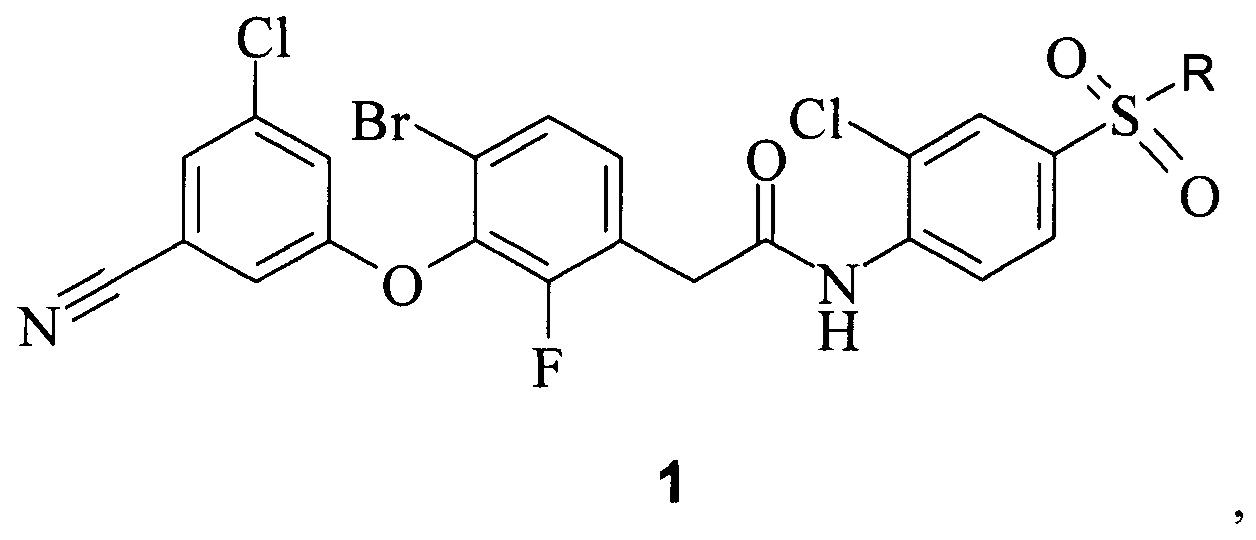
Элсульфавирин/Elpida/VM-1500 (1а) является пролекарством активного соединения VM-1500A (1b), которое является мощным ингибитором репликации ВИЧ-1 штамма НХВ2 в клетках линии МТ-4 и относится к классу ненуклеозидных ингибиторов обратной транскриптазы (Non-Nucleoside Reverse Transcriptase Inhibitors - NNRTI). Среднее значение IC50, полученное для VM-1500A (1b) на штамме НХВ2 дикого типа и для ингибирования репликации мутантных вирусов ВИЧ-1, содержащих мутации V106A, G190A, L100I/K103N и K103N/Y181C имеют значение для: НХВ2 1,3±0,4 нМ, V106A 1,2±0,2 нМ, G190A 0,6±0,6 нМ, L100I/K103N 1,3±0,3 нМ, K103N/Y181C 1,3±0,4 нМ.

где R представляет собой C2H5CON-Na+, NH2;

Несмотря на успехи, достигнутые в терапии ВИЧ/СПИД с помощью APT, актуальной остается разработка более эффективных и безопасных схем профилактики и лечения ВИЧ.
В частности, к настоящему моменту в мире не зарегистрировано длительно действующих (Long-Acting - LA) препаратов для долгосрочно поддерживающей терапии ВИЧ/СПИД.
Данное изобретение относится к фармацевтической композиции (наносуспензии) для длительно действующего инъекционного (LAI - long-acting injectable) препарата для долгосрочно поддерживающей терапии ВИЧ/СПИД.
Ниже приведены определения различных терминов, используемых для описания данного изобретения. Эти определения применимы к терминам, как они использованы в данном описании и формуле изобретения, если иным не ограничены в конкретных случаях либо по отдельности, либо как часть большей группы.
Термин «активный компонент» (лекарственное вещество) относится к физиологически активному веществу синтетического или иного (биотехнологического, растительного, животного, бактерицидного и так далее) происхождения, обладающему фармакологической активностью, которое является активным ингредиентом фармацевтической композиции.
Термин «кристаллическая форма» означает структуру вещества, характеризующуюся упаковкой образующих ее молекул в один из видов кристаллической решетки.
Термин «поликристаллическая форма» означает структуру вещества, имеющую поликристаллическое строение, т.е. состоящую из множества мелких монокристаллов, т.е. кристаллитов определенной кристаллической формы.
Термин «лекарственный препарат» означает вещество (или смесь веществ в виде фармацевтической композиции) в виде таблеток, капсул, инъекций, мазей и др. готовых форм, предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
Термин «фармацевтическая композиция» обозначает композицию, включающую в себя соединение общей формулы 1 и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустите ли, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например, сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например, моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, альгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного начала, одного или в комбинации с другим активным началом, может быть введена животным и людям в стандартной форме введения, в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения.
Термин «инертный наполнитель», используемый в данном описании, относится к соединению, которое используют для получения фармацевтической композиции, и, как правило, безопасному, нетоксичному, ни биологически, ни иным образом нежелательному, и включает в себя вспомогательные вещества, которые являются приемлемыми для применения в ветеринарии, а также фармакологически приемлемыми для использования человеком. Соединения по данному изобретению могут быть введены отдельно, но обычно их будут вводить в смеси с одним или более фармацевтически приемлемыми эксципиентами, разбавителями или носителями, выбранными с учетом предполагаемого пути введения и стандартной фармацевтической практики.
Термин «терапевтически эффективное количество», используемый здесь, означает количество субстанции, пролекарства или лекарства, необходимое для уменьшения симптомов заболевания у субъекта. Доза субстанции, пролекарства или лекарства будет соответствовать индивидуальным требованиям в каждом конкретном случае. Эта доза может варьироваться в широких пределах в зависимости от многочисленных факторов, таких как тяжесть заболевания, подлежащего лечению, возраста и общего состояния здоровья пациента, других лекарственных средств, с помощью которых пациент проходит лечение, способа и формы введения и опыта лечащего врача. Для перорального введения суточная доза составляет приблизительно от 0,01 до 10 г, включая все значения между ними, в день в монотерапии и/или в комбинированной терапии. Предпочтительная суточная доза составляет примерно от 0,1 до 7 г в день. Как правило, лечение начинают с большой начальной «нагрузочной дозы», чтобы быстро уменьшить или устранить вирус, сопровождающей убывающую дозу до уровня, достаточного для предотвращения всплеска инфекции.
Термин «субъект» означает млекопитающее, предпочтительно субъектом является человек. Предполагается, что в способе лечения субъекта может быть любое из пролекарств общей формулы 1, его изотопно-обогащенный аналоги, его фармацевтически приемлемые соли, гидраты, сольваты, кристаллические и полиморфные формы, либо в сочетании их с другими соединениями и препаратами для комбинированной антиретровирусной терапии ВИЧ/СПИД.
Неожиданно изобретатели обнаружили, что фармацевтическая суспензия, включающая в качестве активного вещества соединение общей формулы 1, эффективна в качестве инъекционного препарата для долгосрочно поддерживающей терапии ВИЧ-инфекции.
Предметом данного изобретения является композиция, включающая в качестве активного вещества соединение общей формулы 1 в терапевтически эффективном количестве необязательно в комбинации с фармацевтически приемлемым наполнителем, добавкой или разбавителем для производства фармацевтической суспензии.
Более предпочтительной является лиофилизированная композиция, включающая в качестве активного вещества соединение формулы lb, в комбинации с солюбилизатором (например, с полоксамером Р338) и детергентом (например, с маннитолом или сахарозой).
Более предпочтительной является лиофилизированная композиция с размером частиц от 200 нм до 900 нм.
Предметом данного изобретения является также фармацевтическая суспензия, включающая описанную выше композицию, фосфатно-буферный солевой раствор (PBS) и воду для инъекций для долгосрочно поддерживающей терапии субъектов инфицированных ВИЧ/СПИД.
Предметом данного изобретения является также способ получения лиофилизированной композиции, заключающийся во вращательном перетирании с помощью циркониевого песка в водном растворе полоксамера соединения общей формулы 1 в кристаллической или поликристаллической форме, солюбилизатора и детергента, последующем отделении циркониевого песка и лиофилизации полученной суспензии.
Предметом данного изобретения является также способ получения фармацевтической суспензии путем смешивания описанной выше лиофилизированной композиции и воды для инъекций.
Данное изобретение поясняется чертежами.
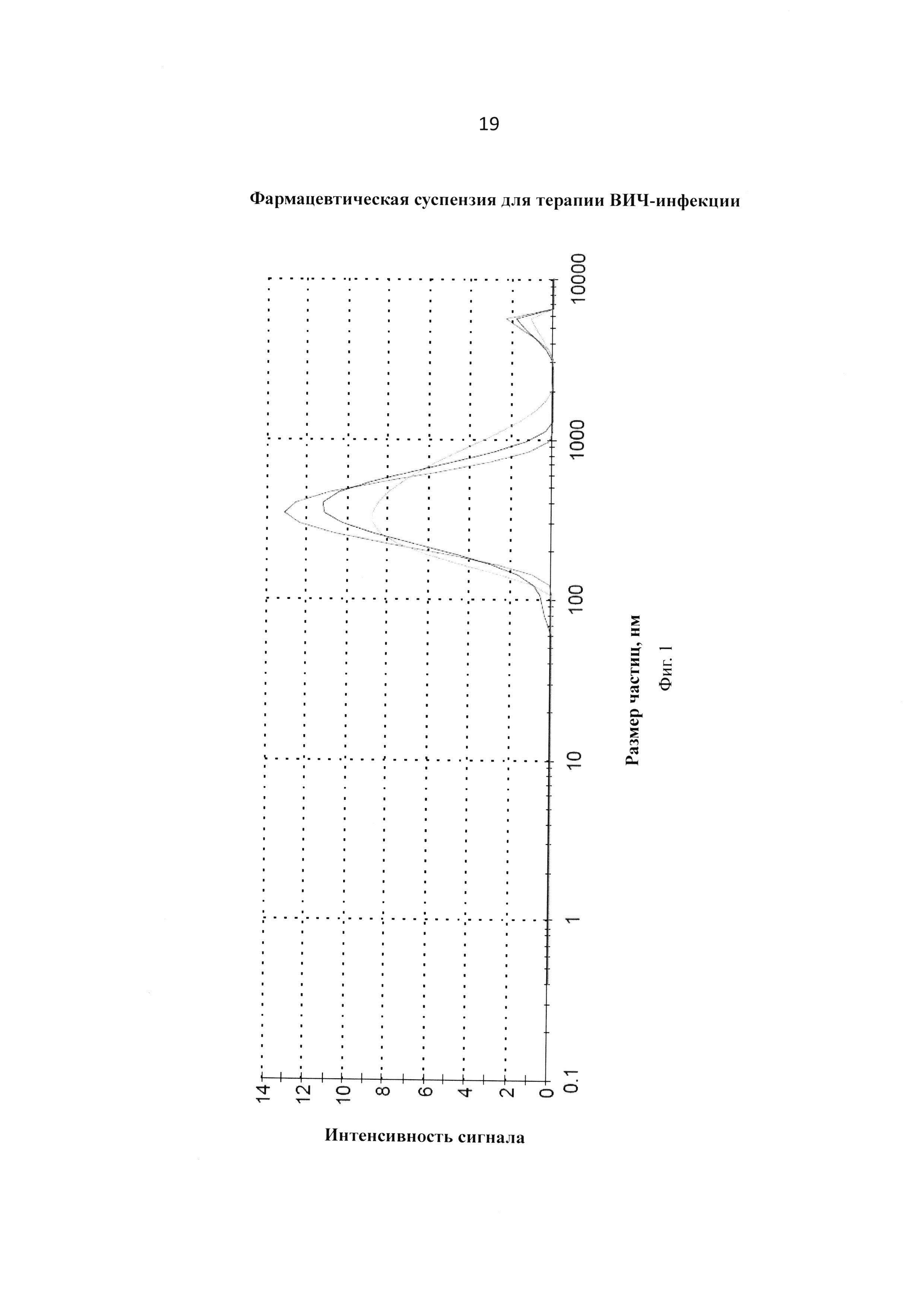
Фиг. 1. График измерения размера частиц трех образцов после 24-х часов перемалывания с сахарозой.
Фиг. 2. График измерения размера частиц трех образцов после 24-х часов перемалывания с маннитолом.
Фиг. 3. График измерения размера частиц трех образцов после 24-х часов перемалывания и 20-ти часов отстаивания (сахароза).
Фиг. 4. График измерения размера частиц трех образцов после 24-х часов перемалывания и 20-ти часов отстаивания (маннитол).
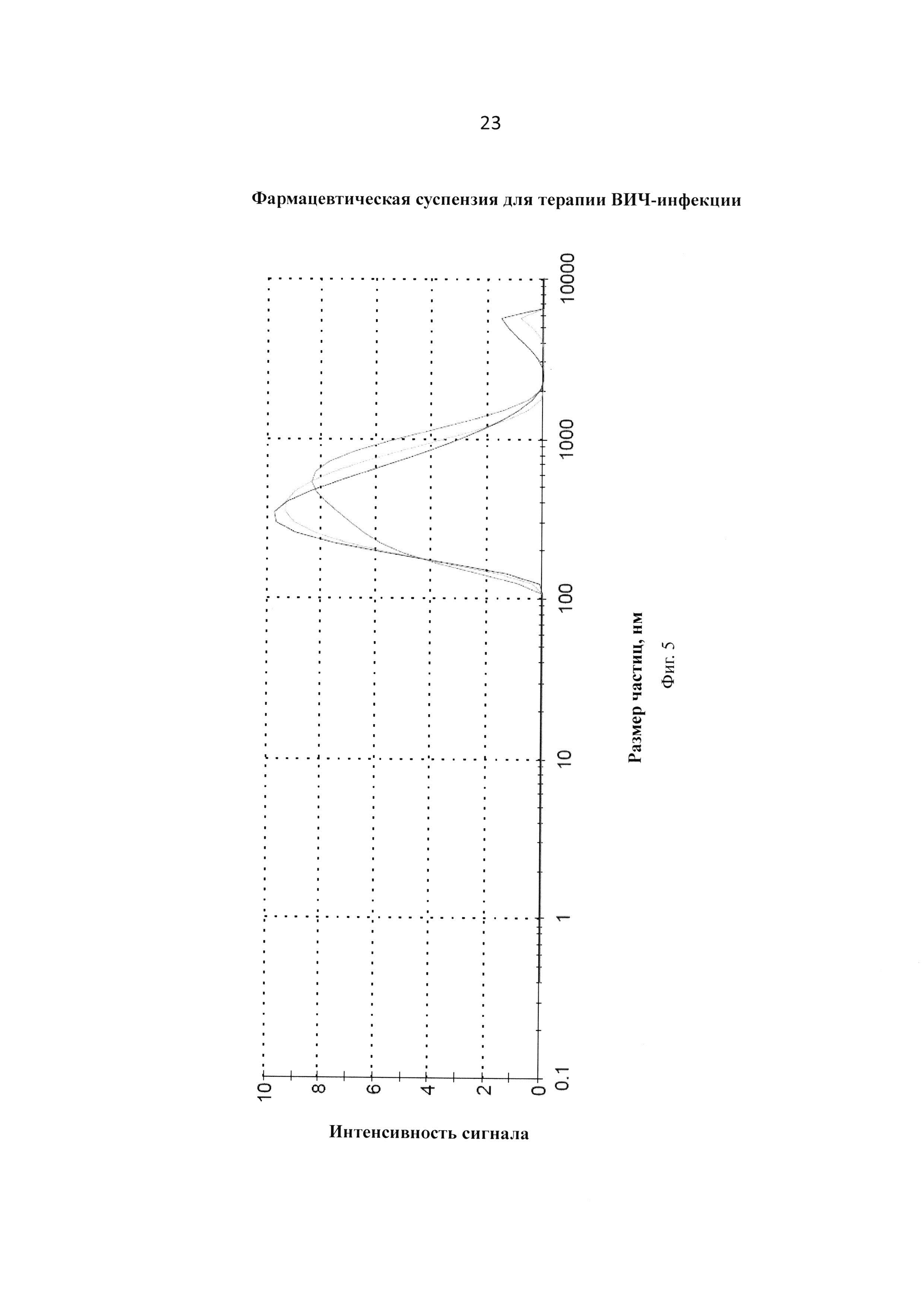
Фиг. 5. График измерения распределения частиц трех образцов Лота №740-1-101.1 (сахароза) после лиофилизации и ресуспендирования.
Фиг. 6. График измерения распределения частиц трех образцов Лота №740-1-101.2 (маннитол) после лиофилизации и ресуспендирования.
Фиг. 7. Фармакокинетические кривые VM-1500A в плазме крови собак при однократном SC и IM введении фармацевтических суспензий VM-1500-LAI и VM-1500A-LAI в дозе 10 мг/кг.
Настоящее изобретение далее будет описано в связи с определенными вариантами осуществления, которые не предназначены для ограничения его объема. Напротив, настоящее изобретение охватывает все альтернативы, модификации и эквиваленты, которые могут быть включены в объем формулы изобретения. Таким образом, следующие примеры, которые включают в себя конкретные варианты, иллюстрируют, но не ограничивают настоящее изобретение.
Пример 1. Способ получения лиофилизированной композиции.
В сосуд (Jar) загружают циркониевый песок (150 мл) (grinding media: 0.5 mm YTZ® Zirconia Grinding and Dispersion Media; Tosoh USA, Inc.) и раствор полоксамера P338 (10,0 г) и сахарозы или маннитола (11,6 г) в 400 мл фосфатного буферного раствора (PBS, рН=7.4). 150 мл полученного раствора помещают в сосуд (Jar), прибавляют циркониевый песок (150 мл) (grinding media: 0.5 mm YTZ® Zirconia Grinding and Dispersion Media; Tosoh USA, Inc.) и соединение VM1500A (15,07 г). Сосуд (Jar) помещают на мельничный аппарат (U.S. Stoneware 700 Series jar mill) и устанавливают вращение 104 об/мин и перетирают в течение 24 часов. Окончание процесса контролируется анализами распределения частиц на приборе Malvern Zetasizer Nano ZS. По окончании процесса перемалывания вращение останавливают и оставляют на 16-20 часов при температуре 2-8°С для осаждения циркониевого песка. Суспензию фильтруют через стеклянный фильтр пористостью 5-15 микрон и анализируют на установление содержания (концентрации) соединения VM1500A (92-97% от номинальной). Получают суспензию, которую в стерильных условиях разливают по 2 мл в стеклянные стерильные флаконы емкостью 5 мл, замораживают и лиофилизируют. Получают лиофилизированную композицию, характеристики которой представлены в Таблицах 1 и 2 и на Фигурах 1, 2, 3 и 4.


*PDI - Здесь и далее - индекс дисперсности, характеризующий однородность частиц в смеси.

Пример 2. Способ получения фармацевтической суспензии.
К полученной в Примере 1 суспензии прибавляют предварительно приготовленный стерильный раствор PBS с рН=6.8 из расчета 2.2 мл на флакон емкостью 5 мл. Получают фармацевтическую суспензию, готовую к использованию, характеристики которой представлены в Таблице 3 и на Фигурах 5 и 6.


Пример 3. Фармакокинетика фармацевтических суспензий VM1500LAI и VM1500A-LAI в собаках.
суспензии VM1500LAI и VM1500A-LAI, полученные в Примере 2, вводили собакам породы Бигль однократно подкожно и внутримышечно в дозе 10 мг/кг и анализировали содержание VM1500 в плазме крови собак на протяжении 120-ти дней после инъекций. Определение концентрации исследуемых соединений проводили с помощью ВЭЖХ-МС/МС метода.
Для введения дозы приготовленные суспензии вводят собакам подкожно или внутримышечно в объеме 0.1 мл/кг в одно и то же место. Кровь собирали через 0, 0.5, 1, 2, 4, 8, 24 ч. и 2, 3, 6, 8, 12, 15, 18, 22, 29, 36, 43, 50, 57, 64, 71, 78, 85, 92, 107, 114 и 121 день после инъекции. Цельную кровь для получения плазмы собирали в пробирки, содержащие натрий-гепарин. Немедленно после взятия крови пробирку осторожно переворачивали 8 раз для лучшего перемешивания крови и натрий-гепарина, после чего инкубировали при комнатной температуре 15 минут. Затем плазму отделяли центрифугированием в течение 10 минут при 3000 об/мин. После центрифугирования аликвоту плазмы крови объемом 1 мл аккуратно переносили в криопробирку при помощи автоматической пипетки, избегая касания носиком клеточной массы/разделительного геля. Из одной пробирки Vacuette образцы плазмы переносили в две криопробирки. Полученные образцы замораживали. До анализа образцы плазмы крови и ФЭК хранятся при температуре -80°С.
Для определения концентрации VM-1500 и VM-1500A использовали ВЭЖХ-МС/МС анализ. В качестве внутреннего стандарта (IS) использовали близкое по структуре соединение VM-6819. При сканировании в режиме полного ионного тока (MS1) определяли молекулярный ион исследуемых соединений и IS, основные ионы-продукты фиксировали в режиме MS2. Для количественного анализа была проведена оптимизация МС/МС метода в режиме MRM для достижения максимальной чувствительности. При анализе регистрировали как минимум два MRM-перехода для каждого аналита, при количественной обработке хроматограмм использовали один из них.
Хроматографические условия были подобраны при вводе раствора исследуемого соединения и IS в смеси MeCN:Н2О (1:1) в МС/МС детектор через ВЭЖХ систему для достижения оптимальных хроматографических параметров. Стандартные калибровочные образцы VM1500 и VM1500A в плазме крови готовили в диапазоне концентраций 1,0-2000,0 нг/мл. Помимо калибровочных стандартов готовили нулевую пробу k0, k0 с IS и по две серии контрольных образцов (QC) с низкой, средней и высокой концентрациями, которые служили показателем качества количественных расчетов. Из сток-растворов VM1500 (2,0 мг/мл) и VM1500A (2,0 мг/мл) в ДМСО готовили 10-кратные рабочие растворы в MeCN:Н2О (1:1). Растворы для калибровочных стандартов (k) и QC образцов готовились из отдельных аликвот сток-растворов VM1500 и VM1500A.
Экспериментальные образцы плазмы, используемые для приготовления стандартных образцов, были разморожены при комнатной температуре, перемешаны и центрифугированы в течение 5 мин при 14000 об/мин. 90 мкл интактной плазмы переносили в пробирку на 1,5 мл, добавляли 10 мкл 10-кратного стандартного раствора смеси исследуемых веществ в смесь ацетонитрил : вода 1:1. К 90 мкл экспериментального образца добавляли 10 мкл чистой смеси ацетонитрил : вода 1:1. Пробы тщательно перемешивали на вортексе в течение 10 сек. Добавляли 10 мкл IS (VM-6819 5 мг/мл в ацетонитрил : вода 1:1), перемешивали на вортексе в течение 10 сек. К образцам добавляли 1 мл смеси этилацетат : гексан (60:40) и перемешивали на вортексе в течение 20 сек, а затем на шейкере при 1000 об/мин в течение 5 мин. Затем образцы центрифугировали при 14000 об/мин 10 мин.
0,8 мл органической фазы отбирали в пробирки на 1,5 мл и упаривали под током азота в эвапораторе при температуре 40°С. Сухие экстракты разводили в 150 мкл смеси ацетонитрил : вода (1:1), перемешивали на вортексе в течение 10 сек и переносили в чистую плашку для ВЭЖХ/МС/МС анализа.
Концентрации VM1500 и VM1500A в пробах рассчитывали по калибровочной кривой, построенной по площадям хроматографических пиков стандартных образцов VM1500 и VM1500A в матрице (плазма крови или ФЭК), нормированных на площадь IS. Построение калибровочной кривой проводилось путем подбора простейшей модели, адекватно описывающей зависимость аналитического сигнала от концентрации, и используя подходящее нормирование. Основные фармакокинетические параметры рассчитывали внемодельным методом с помощью компьютерной программы WinNonlin Professional 6.3 (Pharsight Corporation) на основании экспериментально полученных данных «концентрация - время» для каждого животного:
- Cmax - максимальная концентрация вещества в плазме крови;
- Tmax - время достижения максимальной концентрации в плазме крови;
- AUC0-t - площадь под фармакокинетической кривой от момента введения препарата до последней точки наблюдения с измеряемой концентрацией;
- AUC0-inf - площадь под фармакокинетической кривой от момента введения препарата до бесконечности;
- Т1/2 - период полувыведения из плазмы крови;
- kel - константа скорости элиминации - параметр, характеризующий скорость выведения вещества из плазмы крови;
- MRT - среднее время удержания препарата в организме от момента приема препарата.
Статистическую обработку результатов проводили с помощью описательной статистики. Рассчитывали средние арифметические значения параметров (М), стандартное отклонение (SD), стандартную ошибку среднего (SE), коэффициент вариации (CV), медиану. Статистический анализ был выполнен с помощью программы WinNonlin Professional 6.3 (Pharsight Corporation).
В Таблице 4 суммированы фармакокинетические параметры, характеризующие поведение VM-1500 и VM-1500A после введения собакам наносуспензий VM-1500-LAI и VM-1500A-LAI подкожно (SC) и внутримышечно IM.
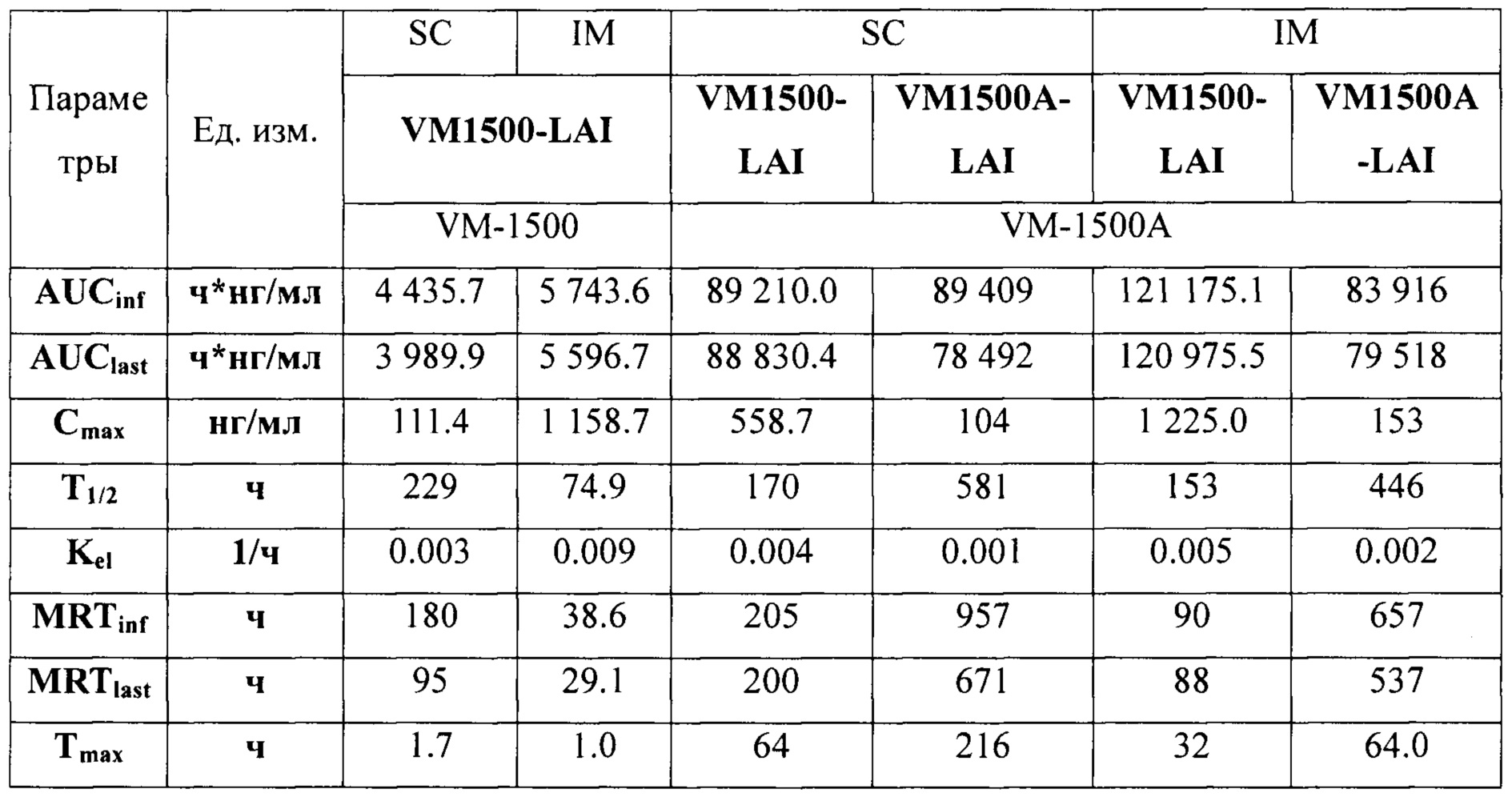
При внутримышечной инъекции достигаются более высокие значения Cmax как самого VM-1500, так и его метаболита VM-1500A, чем при подкожном введении. Однако при использовании формуляции VM-1500A-LAI эта разница не так существенна. При этом общая экспозиция VM-1500 и VM-1500A при внутримышечном введении в виде VM-1500-LAI выше, чем при подкожном, тогда как экспозиция VM-1500A не зависит от пути введения формуляции VM-1500A-LAI. Также при внутримышечном введении VM-1500A раньше достигает максимальной концентрации, чем при подкожном введении. Время полувыведения не зависит существенно от пути введения.
Средние концентрации (С) VM-1500A в плазме крови собак после однократного SC и IM введения собакам в виде суспензий VM1500-LAI и VM1500A-LAI приведены в Таблице 5 и на Фигуре 7.


BLQ - ниже предела количественного определения
Как видно из Таблицы 5 и Фигуры 7, при IM введении фармацевтической наносуспензии VM-1500-LAI в дозе 10 мг/кг собакам эффективная концентрация препарата VM1500A (IC50=1,3 нМ или 0,74 нг/мл) в плазме поддерживается в течение месяца (через 29 дней CVM-1500А=2,61 нг/мл), а при SC введении - в течение полутора месяцев (через 43 дня CVM-1500A=4,83 нг/мл).
Еще более убедительный результат наблюдается при IM и SC введении наносуспензии VM-1500-LAI в дозе 10 мг/кг собакам. В этом случае эффективная концентрация VM1500A поддерживается в течение четырех месяцев (через 121 день CVM-1500A=5,70 нг/мл при IM введении и CVM-1500A=11,47 нг/мл при SC введении). Фармацевтическая суспензия для терапии ВИЧ-инфекции
Данное изобретение относится к фармацевтической композиции (суспензии) для длительно действующего инъекционного (LAI - long-acting injectable) препарата для долгосрочно поддерживающей терапии ВИЧ/СПИД.
Вирус иммунодефицита человека - ретровирус из рода лентивирусов, вызывающий медленно прогрессирующее заболевание - ВИЧ-инфекцию [Weiss R.A. How does HIV cause AIDS. Science 1993, 260(5112), 1273-1279. Douek D.C., Roederer M., Koup R.A. Emerging Concepts in the Immunopathogenesis of AID». Annu. Rev. Med. 2009, 60, 471-84]. Вирус иммунодефицита человека независимо открыли в 1983 году в двух лабораториях: в Институте Пастера во Франции под руководством Люка Монтанье и в Национальном институте рака в США под руководством Роберта Галло. Результаты исследований, в которых из тканей пациентов с симптомами СПИДа впервые удалось выделить новый ретровирус, были опубликованы 20 мая 1983 года в журнале Science [Barre-Sinoussi F. at al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220 (4599), 868-871. Gallo R.C. at al. Isolation of human T-cell leukemia virus in acquired immune deficiency syndrome (AIDS). Science 1983, 220 (4599), 865-867.]. В 2008 году Люк Монтанье и Франсуаза Барр-Синусси были удостоены Нобелевской премии в области физиологии или медицины «за открытие вируса иммунодефицита человека».
Вирус поражает клетки иммунной системы, имеющие на своей поверхности рецепторы CD4: Т-хелперы, моноциты, макрофаги, клетки Лангерганса, дендритные клетки, клетки микроглии. В результате работа иммунной системы угнетается и развивается синдром приобретенного иммунного дефицита (СПИД), организм больного теряет возможность защищаться от инфекций и опухолей, возникают вторичные оппортунистические заболевания, которые не характерны для людей с нормальным иммунным статусом. Без врачебного вмешательства ВИЧ вызывает смерть пациента в среднем через 9-11 лет после заражения (в зависимости от подтипа вируса) [https://ru.wikipedia.org/wiki/Вирус_иммунодефицита_человека].
где R представляет собой C2H5CON-Na+, NH2;
Несмотря на успехи, достигнутые в терапии ВИЧ/СПИД с помощью APT, актуальной остается разработка более эффективных и безопасных схем профилактики и лечения ВИЧ.
В частности, к настоящему моменту в мире не зарегистрировано длительно действующих (Long-Acting - LA) препаратов для долгосрочно поддерживающей терапии ВИЧ/СПИД.
Реферат
Группа изобретений относится к химико-фармацевтической промышленности и представляет собой фармацевтические суспензии в качестве инъекционных препаратов для долгосрочно поддерживающей терапии ВИЧ-инфекции, включающие композиции, содержащие в качестве активного вещества соединение общей формулыгде R представляет собой CHCONNa, NH, или соединение формулы 1bДанную композицию можно получать в форме лиофилизата с размером частиц от 200 нм до 900 нм. Также изобретения представлены способом получения композиций, описанных ранее, который заключается во вращательном перетирании с помощью циркониевого песка в водном растворе полоксамера активных соединений, солюбилизатора и детергента, последующем отделении циркониевого песка и лиофилизации полученной суспензии, а также способом получения фармацевтической суспензии путем смешивания заявленной композиции, фосфатно-буферного солевого раствора PBS с рН=6.8 и воды для инъекций. 5 н. и 5 з.п. ф-лы, 7 ил., 5 табл., 3 пр.
Формула

Комментарии