Композиции без приона на основе наночастиц и способы их получения - RU2559570C2
Код документа: RU2559570C2
Чертежи





























































Описание
Родственные заявки
Данная заявка притязает на преимущество приоритета предварительной заявки на патент США № 61/169665, поданной 15 апреля 2009 года, и предварительной заявки на патент США № 61/238052, поданной 28 августа 2009 года, которые полностью включены в настоящее описание изобретения.
Область техники, к которой относится изобретение
Настоящее изобретение относится к композициям без приона, содержащим наночастицы, включающие альбумин и по существу не растворимые в воде лекарственные средства, к способам получения указанных композиций и способам их применения.
Уровень техники
Прионные заболевания, известные также как трансмиссивные губчатые энцефалопатии (TSE), составляют группу смертельных трансмиссивных нейродегенеративных заболеваний. Конкретные примеры TSE включают болезнь скрепи, поражающую овец и коз, губчатую энцефалопатию крупного рогатого скота (BSE), трансмиссивную энцефалопатию норок, губчатую энцефалопатию кошек и хроническую болезнь, вызывающую истощение (CWD). Заболевания TSE, поражающие человека, могут представлять собой куру, болезнь Крейтцфельда-Якоба (CJD), синдром Герстманна-Страуслера-Шенкера (GSS), смертельную инсомнию и разновидность болезни Крейтцфельда-Якоба (vCJD). vCJD возникла у людей в результате эпидемии BSE в Великобритании и, вероятнее всего, была вызвана потреблением пищевых продуктов, изготовленных из мяса крупного рогатого скота, инфицированного BSE или “коровьим бешенством” (Will et al., (1006) Lancet 347:921-925). Поскольку инкубационный период заболевания при пероральном контактировании может составлять у человека более 20 лет, истинное заболевание vCJD может не проявляться в течение многих лет.
Помимо потребления инфицированных продуктов, изготовленных из мяса крупного рогатого скота, другим способом переноса vCJD от одного человека к другому может быть переливание крови или трансплантация органов (Brown et al. (1998) Transfusion 38:810-816; Diringer et al. (1984) Archives of Virology 82:105-109; Manuelidis et al. (1978) Nature 271:778-779). В середине 1990-х годов возникло опасение, что vCJD может передаваться в результате переливания крови или других фракций крови, полученных у людей, инфицированных TSE. У таких субъектов могут отсутствовать симптомы заболевания в течение длительного времени до проявления клинических признаков и инкубационного периода vCJD, и кровь, полученная у таких доноров, может быть причиной заражения данным заболеванием лиц, которым была перелита кровь или фракции крови донора.
В настоящее время в Соединенном Королевстве Великобритании известны по меньшей мере четыре случая заболевания vCJD вследствие переливания крови. Из 64 человек, которым была перелита кровь 22 доноров, у 4 человек возникла vCJD. В первом случае реципиент заболел через 7 лет после переливания эритроцитов донора, у которого отсутствовали симптомы заболевания, и признаки vCJD появились через 3 года после сдачи крови (Llewely et al. (2004) Lancet 363: 417-421). Во втором случае донор умер от vCJD через два года после сдачи крови, и реципиент умер от аневризма (а не от vCJD) через 5 лет после переливания крови (Peden et al. (2004) Lancet 364: 527-529). При вскрытии реципиента, PrPsc был обнаружен в лимфатическом узле и селезенке, а не в головном мозге. В третьем случае реципиент умер от vCJD через семь с половиной лет после переливания крови донора, у которого vCJD возникла через 20 месяцев после сдачи крови (Wroe et al. (2006) Lancet 368: 2061-2067). Четвертый случай заражения реципиента произошел через восемь с половиной лет после переливания крови того же донора, что и в третьем случае (Health Protection Agency - Health Protection Report, (2007) Vol. 1, No. 3, 26. Информация доступна на сайте http://www.hpa.org.uk/hpr/archives/2007/news2007/news0307.htm).
Общим признаком всех прионных заболеваний является превращение нормального клеточного прионного белка (PrPc) в аномальную изоформу (PrPsc). Считается, что различие между PrPc и PrPsc является чисто структурным, при этом PrPc имеет в основном альфа-спиральную структуру и PrPsc имеет в основном бета-складки, которые часто образуют агрегаты. PrPsc действует в качестве матрицы, вызывающей превращение нормальных белковых молекул в аномальную изоформу, которая затем в свою очередь превращает новый PrPc в PrPsc (Prusiner et al. (1998) Proc. Natl. Acad. Sci. USA 95: 13363-13383). Такой автокаталитический процесс ведет к экспоненциальному образованию нейротоксичных агрегатов PrPsc (Aguzzi et al., (2007) Nat. Rev. Mol. Cell Biol. 8: 552-561). Лиганды прионного белка и их применение описаны в публикациях WO04/050851, WO06/010915, WO04/090102 и WO06/044459.
Исследования показали, что инфективность приона в крови может проявляться на ранней стадии инкубационного периода данного заболевания (Brown et al. (2006) Blood infectivity in the transmissible spongiform encephalopathies. Chapter 4 In: Turner ML, ed. 95-118). Поскольку может пройти много времени до появления симптомов заболевания, бессимптомно инфицированные субъекты могут все это время считаться здоровыми активными донорами крови. Кроме того, некоторые субъекты могут быть постоянно или временно инфицированы без возникновения заболевания. Поэтому трудно, если вообще возможно, гарантировать, что источники крови для получения фракций крови не содержат приона.
Композиции на основе наночастиц, содержащих альбумин, были созданы в качестве систем доставки по существу не растворимых в воде лекарственных средств. Смотрите, например, патенты США №№ 5916596, 6506405, 6749868 и 6537579, а также публикации патентов США №№ 2005/0004002 и 2007/0082838. В технологии получения наночастиц на основе альбумина использованы уникальные природные свойства белка альбумина, позволяющие переносить и доставлять по существу не растворимые в воде лекарственные средства к очагу заболевания. Указанные наночастицы легко встраиваются в собственные процессы переноса организма и могут использовать свойство опухолей притягивать альбумин, что позволяет доставлять к очагу-мишени активное лекарственное средство в более высоких концентрациях. Кроме того, методы создания наночастиц на основе альбумина позволяют повысить растворимость лекарственного средства без использования токсичных химических веществ, таких как растворители, что потенциально повышает безопасность благодаря устранению побочных эффектов, связанных с использованием растворителей.
Все публикации, патенты, заявки на патент и опубликованные заявки на патент полностью включены в настоящее описание изобретения в качестве ссылки.
Сущность изобретения
Одним объектом настоящего изобретения являются композиции без приона на основе наночастиц (в частности, фармацевтические композиции). Некоторые варианты осуществления изобретения относятся к композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, при этом в указанной композиции по существу отсутствует прионный белок. В некоторых вариантах осуществления изобретения композиция является стерильной. В некоторых вариантах осуществления изобретения композиция стерилизована фильтрованием. В некоторых вариантах осуществления изобретения композиция дополнительно содержит фармацевтически приемлемый носитель.
В некоторых вариантах осуществления изобретения композиция содержит менее примерно 100 фг/мл прионного белка. В некоторых вариантах осуществления изобретения композиция характеризуется инфективностью приона менее примерно 10 ин.ед.-иц/мл. В некоторых вариантах осуществления изобретения композиция характеризуется инфективностью приона менее примерно 1 LD50/мл.
В некоторых вариантах осуществления изобретения в композиции отсутствует прионный белок по результатам анализа методом циклической амплификации с ошибочной укладкой цепи белка (РМСА). В некоторых вариантах осуществления изобретения в композиции отсутствует прионный белок по результатам анализа методом ИПЦР. В некоторых вариантах осуществления изобретения композиция характеризуется инфективностью приона менее примерно 10 ин.ед.-иц/мл и отсутствием прионного белка по результатам анализа РМСА. В некоторых вариантах осуществления изобретения композиция характеризуется инфективностью приона менее примерно 10 ин.ед.-иц/мл и отсутствием прионного белка по результатам анализа методом ИПЦР.
Композиции по настоящему изобретению обычно по существу не содержат PrPsc. В некоторых вариантах осуществления изобретения композиция также не содержит PrPc. В некоторых вариантах осуществления изобретения молярное соотношение PrPsc и PrPc в композиции не превышает примерно 1:1, в частности, не превышает примерно 1:10, 1:100, 1:1000, 1:10000 или 1:100000.
В некоторых вариантах осуществления изобретения композиция по настоящему изобретению содержат некоторое количество (например, следовое количество) веществ, введенных в процессе удаления приона. Например, некоторые варианты осуществления изобретения относятся к композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, при этом в указанной композиции по существу отсутствует прионный белок и присутствует некоторое количество (например, следовое количество) лиганда, способного связываться с прионным белком. Некоторые варианты осуществления изобретения относятся к композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, при этом в указанной композиции по существу отсутствует прионный белок и присутствует некоторое количество (например, следовое количество) носителя (в частности, носителя по настоящему изобретению, включающего смолу). Некоторые варианты осуществления изобретения относятся к композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, при этом в указанной композиции по существу отсутствует прионный белок и присутствует некоторое количество смолы PRDT (например, следовое количество смолы PRDT). Некоторые варианты осуществления изобретения относятся к композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, при этом в указанной композиции по существу отсутствует прионный белок и присутствует некоторое количество смолы DRV (например, следовое количество смолы DRV).
В некоторых вариантах осуществления изобретения содержание стабилизатора альбумина в композиции меньше, чем в композиции, в которой альбумин не был очищен в соответствии с процессом удаления приона. Указанные стабилизаторы альбумина включают, например, N-ацетилтриптофанат и каприлат натрия.
В некоторых вариантах осуществления изобретения указанная композиция биологически эквивалента композиции, в которой альбумин не был очищен в соответствии с процессом удаления приона.
Некоторые варианты осуществления изобретения относятся к композиции, содержащей наночастицы, включающие альбумин и по существу не растворимаый в воде фармакологически активный агент, при этом альбумин в композиции был получен в соответствии с процессом удаления приона, который включает приведение исходной композиции альбумина в контакт с лигандом, способным связываться с прионным белком. В некоторых вариантах осуществления изобретения процесс удаления приона дополнительно включает удаление указанного лиганда и связанных с ним белков из композиции альбумина.
Некоторые варианты осуществления изобретения относятся к композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, при этом альбумин в композиции был получен способом, включающим: а) приведение исходной композиции альбумина в контакт с лигандом, способным связываться с прионным белком с образованием комплекса лиганда с прионным белком, и b) удаление полученного комплекса из исходной композиции.
В некоторых вариантах осуществления изобретения исходная композиция альбумина является продуктом, выделенным из крови. В некоторых вариантах осуществления изобретения исходная композиция альбумина является композицией, полученной из биологической жидкости (такой как кровь). В некоторых вариантах осуществления изобретения альбумин является сывороточным альбумином человека.
В некоторых вариантах осуществления изобретения лиганд является пептидом (в частности, любым пептидом, указанным в таблице 1). В некоторых вариантах осуществления изобретения лиганд является антителом, узнающим прионный белок. В некоторых вариантах осуществления изобретения лиганд является химическим соединением (в частности, соединением на основе триазина). В некоторых вариантах осуществления изобретения лиганд включает аминогруппу, например, аминогруппу в полиамине.
Лиганд может быть присоединен к носителю, включающему, например, колонку, гранулу, матрицу, фильтр и мембрану.
Другим объектом изобретения являются способы получения композиций без приона на основе наночастиц. Например, некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который состоит в том, что смесь, включающую раствор альбумина и органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе, подвергают воздействию высокого сдвигающего усилия, при этом альбумин был получен способом, включающим удаление прионного белка из исходной композиции альбумина. Некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который состоит в том, что смесь, включающую раствор альбумина и органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе, подвергают воздействию высокого сдвигающего усилия, при этом альбумин был получен способом, включающим приведение исходной композиции альбумина в контакт с лигандом, способным связываться с прионным белком. Некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который состоит в том, что смесь, включающую раствор альбумина и органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе, подвергают воздействию высокого сдвигающего усилия, при этом альбумин был получен способом, включающим: а) приведение исходной композиции альбумина в контакт с лигандом, способным связываться с прионным белком, и b) удаление лиганда и связанного с ним белка из исходной композиции.
Некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает: а) удаление прионного белка из исходной композиции альбумина; b) воздействие высоким сдвигающим усилием на смесь, включающую раствор альбумина без приона и органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе. Некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастиицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает: а) приведение исходной композиции альбумина в контакт с лигандом, способным связываться с прионным белком с образованием комплекса лиганда с прионным белком, b) удаление полученного комплекса из исходной композиции альбумина и с) воздействие высоким сдвигающим усилием на смесь, включающую раствор альбумина без приона и органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе.
Некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает: а) приведение раствора альбумина в контакт с лигандом, способным связываться с прионным белком, b) удаление лиганда и связанных с ним белков из раствора альбумина и c) воздействие высоким сдвигающим усилием на смесь, включающую указанный раствор альбумина и органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе. В некоторых вариантах осуществления изобретения смесь по существу не содержит поверхностно-активных веществ.
Прионы могут быть удалены во время образования наночастиц. Например, некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает: а) приведение т смеси, включающей раствор альбумина и органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе, в контак с лигандом, способным связываться с прионным белком. В некоторых вариантах осуществления изобретения указанный способ дополнительно включает: b) удаление из смеси лиганда и связанных с ним белков. В некоторых вариантах осуществления изобретения указанный способ дополнительно включает с) воздействие на смесь высоким сдвигающим усилием. В некоторых вариантах осуществления изобретения смесь по существу не содержит поверхностно-активных веществ.
Прионные белки могут быть также удалены после образования композиции на основе наночастиц. Например, некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает приведение смеси, включающей органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе, и раствор альбумина, в контакт с лигандом, способным связываться с прионным белком, при этом смесь подвергают воздействию высокого сдвигающего усилия до приведения в контакт с лигандом. Некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает: а) воздействие высоким сдвигающим усилием на смесь, включающую органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органической фазе, и раствор альбумина, и b) приведение смеси в контакт с лигандом, способным связываться с прионным белком. В некоторых вариантах осуществления изобретения указанный способ дополнительно включает: с) удаление из смеси лиганда и связанных с ним белков. В некоторых вариантах осуществления изобретения смесь по существу не содержит поверхностно-активных веществ.
Некоторые варианты осуществления изобретения относятся к способу удаления прионного белка из композиции с подозрением на наличие прионного белка, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает а) приведение композиции на основе наночастиц в контакт с лигандом, способным связываться с прионным белком, b) удаление лиганда и связанных с ним белков из композиции на основе наночастиц. Некоторые варианты осуществления изобретения относятся к способу удаления прионного белка из композиции альбумина с подозрением на наличие аномального прионного белка, который включает: а) приведение композиции альбумина в контакт с лигандом, способным связываться с прионным белком, b) удаление лиганда и связанных с ним белков из композиции альбумина, при этом композицию альбумина используют для получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент.
Некоторые варианты осуществления изобретения относятся к способу удаления прионного белка из композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает: а) определение наличия или отсутствия в композиции прионного белка, b) приведение композиции в контакт с лигандом, способным связываться с прионным белком, и с) удаление из композиции лиганда и связанных с ним белков.
Настоящее изобретение также относится к композициям, полученным при выполнении процесса удаления приона. Например, некоторые варианты осуществления изобретения относятся к композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, а также лиганд, способный связываться с прионным белком. Некоторые варианты осуществления изобретения относятся к смеси, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, а также лиганд, способный связываться с прионным белком, присоединенным к носителю, в частности, к одному или нескольким носителям, рассмотренным в настоящем описании изобретения. Некоторые варианты осуществления изобретения относятся к колонке, заполненной композицией, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, при этом указанная колонка содержит лиганд, способный связываться с прионным белком.
Настоящее изобретение относится также к композициям, полученным способами по настоящему изобретению. Настоящее изобретение относится также к способам применения композиций без приона по настоящему изобретению. Например, некоторые варианты осуществления изобретения относятся к способу введения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, в которой по существу отсутствует прионный белок. Некоторые варианты осуществления изобретения относятся к способу лечения болезни (такой как рак), который включает введение композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, в которой по существу отсутствует прионный белок.
Некоторые варианты осуществления изобретения относятся к способу введения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, при этом альбумин в композиции был получен в соответствии с процессом удаления приона, который включает приведение исходной композиции альбумина в контакт с лигандом, способным связываться с прионным белком. В некоторых вариантах осуществления изобретения процесс удаления приона дополнительно включает удаление указанного лиганда и связанных с ним белков из композиции альбумина. Некоторые варианты осуществления изобретения относятся к способу лечения болезни (такой как рак), который включает введение композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, при этом альбумин в композиции был получен в соответствии с процессом удаления приона, который включает приведение исходной композици альбумиина в контакт с лигандом, способным связываться с прионным белком.
Настоящее изобретение относится также к лекарственным формам (таким как ампулы, например, запаянные ампулы), включающим композиции без приона на основе наночастиц по настоящему изобретению, и наборам, используемым при осуществлении способов по настоящему изобретению. Настоящее изобретение относится также к системам (включая устройства), предназначенным для осуществления одного или нескольких способов по настоящему изобретению.
Вышеуказанные и другие объекты и преимущества настоящего изобретению будут более понятны из последующего подробного описания изобретения и прилагаемой формулы изобретения. Следует отметить, что один, некоторые или все признаки разных вариантов осуществления изобретения могут быть объединены с созданием других вариантов осуществления настоящего изобретения.
Краткое описание чертежей
На фиг.1 показаны вестерн-блоты и гели смолы DVR для SDS-PAGE, стимулированные 0,01% и 0,005% гомогената головного мозга хомячка, страдающего болезнью скрепи, в 20% или 25% растворе альбумина. AMN31 является положительной контрольной смолой. Наблюдаемый сигнал относится к связанной фракции. Символы “-PK” и “+РК” означают отсутствие или наличие расщепления протеиназой К.
На фиг.2 показаны вестерн-блоты и гели смол YVHEA и SYA для SDS-PAGE, стимулированные 0,01% и 0,005% гомогената головного мозга хомячка, страдающего болезнью скрепи, в 20% или 25% растворе альбумина. AMN31 является положительной контрольной смолой. Наблюдаемый сигнал относится к связанной фракции. Символы “-PK” и “+РК” означают отсутствие или наличие расщепления протеиназой К.
На фиг.3 показаны вестерн-блоты и гели смолы D4 для SDS-PAGE, стимулированные 0,01% и 0,005% гомогената головного мозга хомячка, страдающего болезнью скрепи, в 20% или 25% растворе альбумина. AMN31 является положительной контрольной смолой. Наблюдаемый сигнал относится к связанной фракции. Символы “-PK” и “+РК” означают отсутствие или наличие расщепления протеиназой К.
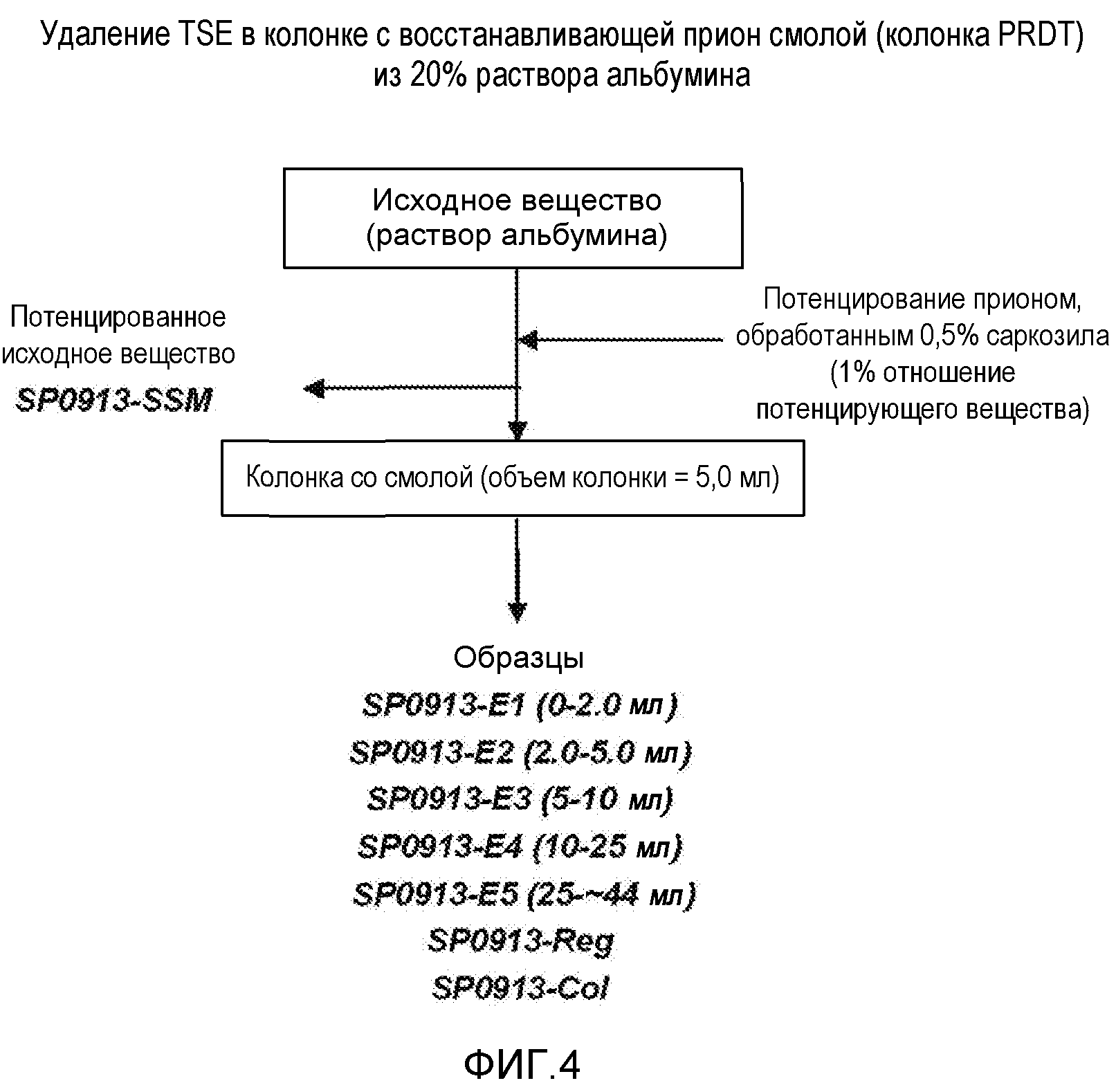
На фиг.4 показана блок-схема процесса удаления TSE в колонке с восстанавливающей прион смолой (колонка PRDT (для диагностики и удаления патогенных микроорганизмов) для 20% раствора альбумина.
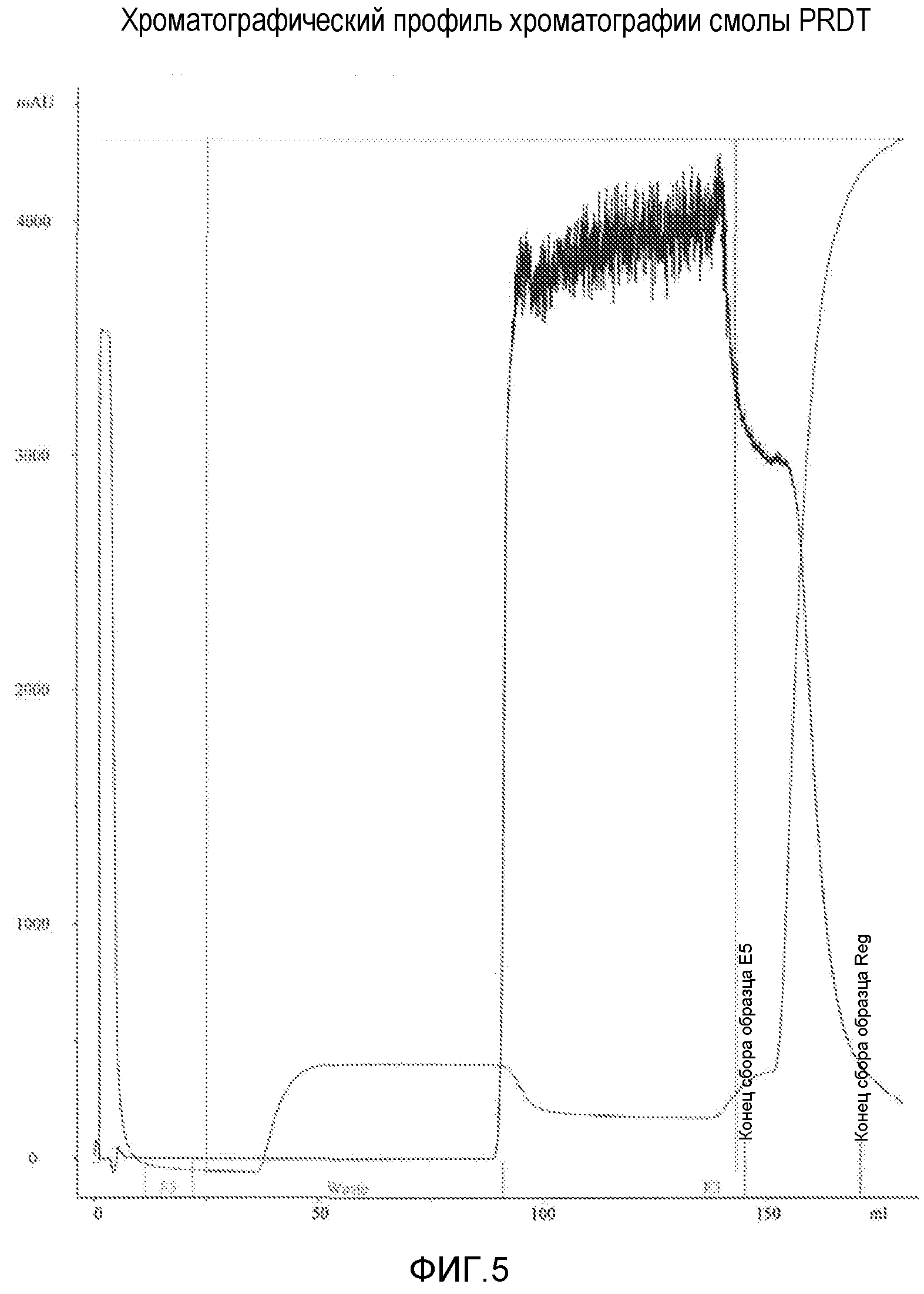
На фиг.5 показан хроматографический профиль потенцированного исследования удаления TSE в колонке PRDT для 20% раствора альбумина.
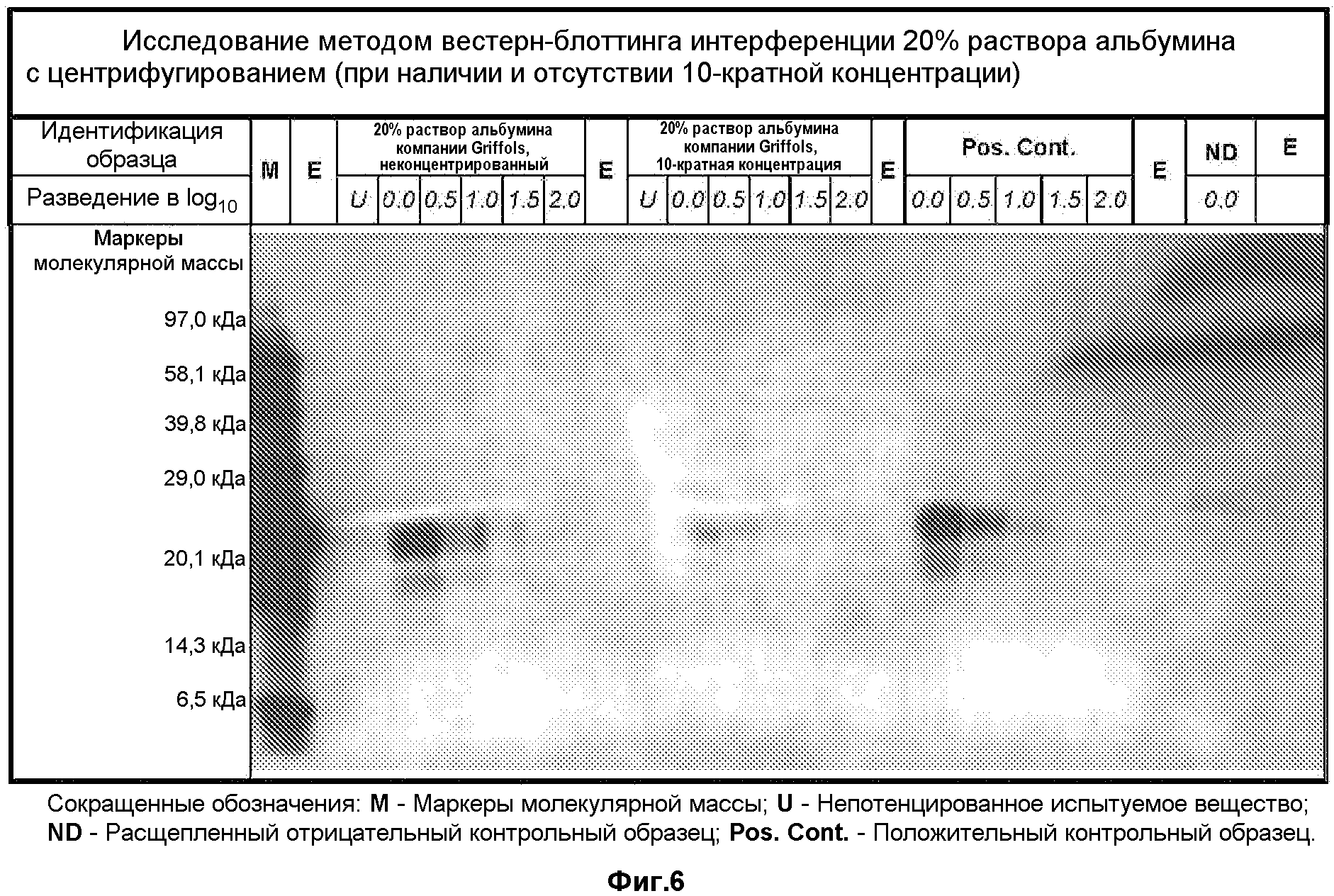
На фиг.6 показано исследование интерференции 20% раствора альбумина методом вестерн-блоттинга с центрифугированием (при наличии или отсутствии 10-кратной концентрации).
На фиг.7 показана блок-схема процесса удаления TSE в колонке с восстанавливающей прион смолой (колонка PRDT) для 25% раствора альбумина.
На фиг.8 показан хроматогорафический профиль потенцированного исследования удаления TSE в колонке PRDT для 25% раствора альбумина.
На фиг.9 показано исследование интерференции 25% раствора альбумина методом вестерн-блоттинга с центрифугированием (при наличии или отсутствии 10-кратной концентрации).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к композициям без приона (таким как фармацевтические композиции), содержащим наночастицы, включающие альбумин и по существу не растворимые в воде фармакологические активные агенты, и к способам получения композиций без приона.
Одним объектом настоящего изобретения являются композиции (в частности, фармацевтические композиции), содержащие наночастицы, включающие альбумин и по существу не растворимые в воде фармакологически активные агенты, при этом в указанной композиции по существу отсутствует прионный белок.
Другим объектом настоящего изобретения является способ получения композиций без приона (таких как фармацевтические композиции), содержащих наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент. Настоящее изобретение также относится к композициям, приготовленным соответствующим способом получения.
Другим объектом настоящего изобретения является способ применения композиций без приона (таких как фармацевтические композиции), содержащих наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент.
Настоящее изобретение также относится к лекарственным формам (таким как ампулы, например, запаянные ампулы), включающим композиции без приона на основе наночастиц по настоящему изобретению, к наборам и системам, используемым при осуществлении способов по настоящему изобретению.
Термин “без приона” использован в настоящем описании изобретения для удобства и обычно служит для описания новых композиций; в определение вышеуказанного термина входят все варианты осуществления изобретения, представленные в настоящем описании изобретения.
Термин “примерно” применительно к использованному значению или параметру служит для описания вариантов, относящихся непосредственно к данному значению или параметру. Например, указание “примерно Х” включает само значение “Х”.
Следует отметить, что объекты и варианты осуществления изобретения, рассмотренные в настоящем описании изобретения, включают определения “состоящий из” и/или “в основном состоящий из”, относящиеся к объектам и вариантам осуществления изобретения.
Композиции без приона на основе наночастиц
Настоящее изобретение относится к композиции (такой как фармацевтическая композиция), содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, при этом в указанной композиции по существу отсутствует прионный белок. В некоторых вариантах осуществления изобретения композиция является стерильной. В некоторых вариантах осуществления изобретения композиция стерилизована фильтрованием. В некоторых вариантах осуществления изобретения композиция дополнительно содержит фармацевтически приемлемый носитель.
В некоторых вариантах осуществления изобретения прионный белок нельзя обнаружить в композиции при помощи метода обнаружения с чувствительностью примерно 100 фг/мл или выше, то есть результат исследования является отрицательным при выполнении анализа методом обнаружения с чувствительностью примерно 100 фг/мл или выше (например, с чувствительностью обнаружения примерно 50 фг/мл, примерно 10 фг/мл, примерно 1 фг/мл, примерно 0,1 фг/мл). Содержание приона в композиции можно обнаружить непосредственно в композиции. Альтернативно композицию можно обработать перед определением, например, концентрировать или обогатить, чтобы облегчить обнаружение и количественное определение прионного белка в композиции.
Одним способом определения наличия или отсутствия в композиции прионного белка является выполнение анализа инфективности in vivo. Напрнимер, инфективность in vivo можно определить, инкубируя испытуемую композицию в таких животных моделях как мышь, норка, хомячок или коза. Инфективность можно определить в виде смертельной дозы (LD50), то есть дозы, которая при введении данным способом (таким как интрацеребральный способ введения) вызывает заболевание у 50% зараженных животных. Альтернативно инфективность можно определить в инфекционных единицах, то есть в виде минимальной инфекционной дозы, способной переносить заболевание от одного экспериментального животного другому животному при данном способе введения. Как правило, 100 ин.ед.-иц/мл (инфекционная единица, определенная при интрацеребральном способе введения) соответствует примерно 10 LD50/мл и 1 пг/мл прионного белка.
Другим способом определения прионного белка в композиции является метод иммунополимеразной цепной реакции (ИПЦР), в котором способность экспоненциальной амплификации ПЦР объединена с обнаружением белков антителами в формате ELISA и использована в модифицированном методе ИПЦР в реальном времени для обнаружения прионного белка на сверхнизких уровнях. Смотрите публикацию Barletta et al., J. Virology Methods, 127 (2005): 154-104. При помощи ИПЦР рекомбинантный PrPc хомячка был обнаружен на уровне 1 фг/мл в расщепленных протеиназой К (РК) гомогенатах головного мозга хомячка, инфицированного болезнью скрепи, разведенных до 10-8 (приблизительно 10-100 инфекционных единиц), с использованием полуколичественного метода определения зависимости между дозой и эффектом.
В некоторых вариантах осуществления изобретения прионный белок в композиции определяют методом циклической амплификации с ошибочной укладной цепи белка (РМСА). Указанный метод был использован для обнаружения PrPsc в крови. Другие методы, пригодные для определения прионного белка в композиции, включают, не ограничиваясь ими: количественный сэндвич-метод ELISA с использованием усиливающей диссоциацию флуоресценции с временным разрешением; двухцветное флуоресцентное конфокальное сканирование; конформационный иммуноанализ (CDI). Для обнаружения прионных белков в композиции также могут быть использованы такие методы как вестерн-блоттинг, гибридизация с гранулами, анализы сдвига подвижности в геле, флуоресцентные анализы гибридизации in situ (FISH), обнаружение радиоактивных или биолюминесцентных маркеров, ядерный магнитный резонанс, электронный парамагнитный резонанс, спектроскопия в остановленном потоке, хроматография на колонке, капиллярный электрофорез или другие методы.
В некоторых вариантах осуществления изобретения композиция по существу не содержит прионного белка по результатам одного из анализов обнаружения прионных белков (в частности, одного из вышеописанных анализов). В некоторых вариантах осуществления изобретения для исследования композиции могут быть использованы два или большее число анализов, при этом для определения наличия или отсутствия прионного белка в композиции используют корреляцию результатов разных анализов.
Таким образом, некоторые варианты осуществления изобретения относятся к композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, при этом в композиции по существу отсутствует прионный белок. В некоторых вариантах осуществления изобретения композиция характеризуется инфективностью приона менее примерно 100 фг/мл. В некоторых вариантах осуществления изобретения композиция характеризуется инфективностью приона менее примерно 10 ин.ед.-иц/мл. В некоторых вариантах осуществления изобретения композиция характеризуется инфективностью приона менее примерно 1 LD50/мл. В некоторых вариантах осуществления изобретения в композиции отсутствует прионный белок по результатам анализа методом циклической амплифимкации с ошибочной укладкой цепи белка (РМСА). В некоторых вариантах осуществления изобретения в композиции отсутствует прионный белок по результатам анализа ИПЦР. В некоторых вариантах осуществления изобретения композиция характеризуется инфективностью приона менее примерно 10 ин.ед.-иц/мл и не содержит прионного белка по результатам анализа РМСА. В некоторых вариантах осуществления изобретения композиция характеризуется инфективностью приона менее примерно 10 ин.ед.-иц/мл и не содержит прионного белка по результатам анализа методом ИПЦР. В некоторых вариантах осуществления изобретения композиция не содержит прионного белка на основании стандарта, представленного в Руководстве по исследованию процессов приготовления выделенных из плазмы медицинских продуктов в отношении риска заражения vCJD (CPMP5136/03), которое доступно на сайте http://www.emea.europa.eu/pdfs/human/bwp/513603en.pdf и полностью включено в настоящее описание изобретения.
Композиции по настоящему изобретению обычно по существу не содержат PrPsc. В некоторых вариантах осуществления изобретения в композиции также по существу отсутствует PrPc. В некоторых вариантах осуществления изобретения молярное соотношение PrPsc и PrPc в композиции не превышает примерно 1:1, в частности, не превышает примерно 1:10, 1:100, 1:1000, 1:10000 или 1:100000.
В некоторых вариантах осуществления изобретения в композиции по существу отсутствует прионный белок человека, коровы, овцы и грызуна (такого как хомячок, мышь и норка). В некоторых вариантах осуществления изобретения в композиции по существу отсутствует прионный белок Н-типа, прионный белок L-типа или обоих типов. В некоторых вариантах осуществления изобретения в композиции по существу отсутствует прионный белок, связанный с клеткой, свободный прионный белок или оба прионных белка.
Термин “PrPc” в использованном здесь значении означает молекулу нативного прионного белка, которая обычно экспрессирована в организме млекопитающего. Термин “PrPsc” в использованном здесь значении означает структурно измененную форму молекулы PrPc, которая, как известно, является инфекционной.
Термин "альбумин" в использованном здесь значении означает природный альбумин, и в определение данного термина не входит альбумин, полученный методами рекомбинантных ДНК. Природный альбумин обладает преимуществами по сравнению с рекомбинантным альбумином, так как является природным лигандом для рецептором альбумина in vivo. Альбумин, используемый в способах по настоящему изобретению, обычно сохраняет посттрансляционные модификации альбумина и, таким образом, характеризуется меньшим риском иммуногенности. В некоторых вариантах осуществления изобретения альбумин получают из композиции, выделенной из крови. Термин “композиция, выделенная из крови” в использованном здесь значении означает цельную кровь, концентрат эритроцитов, плазму, сыворотку, фракцию с высоким и низким содержанием тромбоцитов, концентрат тромбоцитов, лейкоциты, преципитат плазмы крови, фракционированный преципитат и супернатант плазмы крови, промежуточные фракции крови, разные другие вещества, выделяемые из крови, и тому подобные. В некоторых вариантах осуществления изобретения альбумин получают у человека. В некоторых вариантах осуществления изобретения альбумин получают у животного, такого как корова, овца и грызун (такой как мышь, хомячок и норка). В некоторых вариантах осуществления изобретения альбумин получают у группы субъектов (таких как человек), из которых по меньшей мере некоторые субъекты инфицированы прионами. В некоторых вариантах осуществления изобретения альбумин получают у группы субъектов (таких как человек), в которой по меньшей мере некоторые субъекты являются субъектами с подозрением на инфицирование прионами. В некоторых вариантах осуществления изобретения альбумин получают у большой группы субъектов.
В некоторых вариантах осуществления изобретения композиция по настоящему изобретению содержит следовое количество веществ, введенных в процессе удаления приона. Например, некоторые варианты осуществления изобретения относятся к композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, при этом в композиции по существу отсутствует прионный белок и присутствует следовое количество лиганда, способного связываться с прионным белком. Некоторые варианты осуществления изобретения относятся к композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, при этом в композиции по существу отсутствует прионный белок и присутствует следовое количество носителя (в частности, носителя, рассмотренного в настоящем описании изобретения, включая смолу). Некоторые варианты осуществления изобретения относятся к композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, при этом в композиции по существу отсутствует прионный белок и присутствует следовое количество лиганда, способного связываться с прионным белком, и следовое количество носителя (в частности, носителя, рассмотренного в настоящем описании изобретения, включая смолу).
Термин “следовое количество” означает обнаруживаемое количество, которое не влияет на свойство композиции, например, с точки зрения биологической доступности и/или билологической эквивалентности.
В некоторых вариантах осуществления изобретения указанная композиция биологически эквивалентна композиции, в которой альбумин не был очищен в соответствии с процессом удаления приона. Биологическая эквивалентность может быть выражена, например, в виде 90% доверительного интервала в пределах от 0,80 до 1,25 для значений Cmax и AUC или 90% доверительного интервала в пределах от 0,80 до 1,25 для значений AUC и 90% доверительного интервала в пределах от 0,70 до 1,43 для значений Cmax.
В некоторых вариантах осуществления изобретения содержание стабилизатора альбумина в композиции меньше аналогичного показателя в композиции, в которой альбумин не был очищен в соответствии с процессом удаления приона. Указанные стабилизаторы альбумина включают, например, N-ацетилтриптофанат и каприлат натрия.
Композиции по настоящему изобретению обычно состоит из наночастиц, включающих по существу не растворимый в воде фармацевтически активный агент и альбумин. В некоторых вариантах осуществления изобретения композиция на основе наночастиц содержит наночастицы, включающие по существу не растворимый в воде фармокологически активный агент и альбумин. В некоторых вариантах осуществления изобретения наночастицы в композиции по настоящему изобретению имеют средний диаметр не более примерно 1000 нм, в том числе, например, не более примерно 900, 800, 700, 600, 500, 400, 300, 200, 190, 180, 170, 160, 150, 140, 130, 120, 110, 100, 90, 80, 70 или 60 нм. В некоторых вариантах осуществления изобретения по меньшей мере примерно 50% (например, по меньшей мере примерно 60%, 70%, 80%, 90%, 95% или 99%) всех наночастиц в композиции имеют диаметр не более примерно 1000 нм, в том числе, например, не более примерно 900, 800, 700, 600, 500, 400, 300, 200, 190, 180, 170, 160, 150, 140, 130, 120, 110, 100, 90, 80, 70 или 60 нм. В некоторых вариантах осуществления изобретения по меньшей мере примерно 50% (например, по меньшей мере 60%, 70%, 80%, 90%, 95% или 99%) всех наночастиц в композиции имеют диаметр в пределах от примерно 20 до примерно 200 нм, в том числе, например, от примерно 30 до примерно 180 нм, от примерно 40 до примерно 150 нм, от примерно 50 до примерно 120 нм и от примерно 60 до примерно 100 нм.
В некоторых вариантах осуществления изобретения по меньшей мере примерно 5% (в том числе, например, по меньшей мере примерно 10%, 15%, 20%, 25%, 30%, 40%, 50%, 60%, 70%, 80% или 90%) альбумина в наночастицах композиции перекрестно связаны (например, перекрестно связаны одной или несколькими дисульфидными связями).
В некоторых вариантах осуществления изобретения наночастицы включают по существу не растворимый в воде фармакологически активный агент (такой как паклитаксел), покрытый альбумином (например, сывороточным альбумином человека). В некоторых вариантах осуществления изобретения композиция содержит по существу не растворимый в воде фармакологически активный агент как в форме наночастиц, так и в формах, отличных от наночастиц, при этом по меньшей мере примерно 50%, 60%, 70%, 80%, 90%, 95% или 99% по существу не растворимого в воде фармакологически активного агента находится в композиции в форме наночастиц. В некоторых вариантах осуществления изобретения по существу не растворимый в воде фармакологически активный агент в наночастицах составляет более примерно 50%, 60%, 70%, 80%, 90%, 95% или 99% наночастиц в расчете на массу. В некоторых вариантах осуществления изобретения наночастицы имеют неполимерную матрицу. В некоторых вариантах осуществления изобретения наночастицы имеют сердцевину из по существу не растворимого в воде фармакологически активного агента, которая по существу не содержит полимерных материалов (таких как полимерная матрица).
В некоторых вариантах осуществления изобретения композиция на основе наночастиц по существу не содержит поверхностно-активных веществ или органического растворителя (таких как кремофор®, твин-80 или любых других органических растворителей, используемых для введения по существу не растворимых в воде фармакологически активных агентов). В некоторых вариантах осуществления изобретения композиция на основе наночастиц содержит менее примерно 20%, 15%, 10%, 7,5%, 5%, 2,5%, 1% или меньше органического растворителя.
Удаление приона из композиции, содержащей альбумин, позволяет вводить большие количества альбумина без риска попадания прионов. Таким образом, настоящее изобретение относится также к композициям (таким как фармацевтические композиции), содержащим наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, при этом массовое отношение альбумина к по существу не растворимому в воде фармацевтическому агенту составляет примерно 20:1 или больше, в частности, примерно 30:1 или больше, примерно 40:1 или больше или примерно 50:1 или больше. Типичные отношения включают, например, от примерно 20:1 до примерно 40:1, от примерно 40:1 до примерно 60:1, от примерно 60:1 до примерно 80:1 или от примерно 90:1 до примерно 100:1. В некоторых вариантах осуществления изобретения массовое соотношение альбумина и по существу не растворимого в воде фармакологически активного агента в композиции на основе наночастиц составляет примерно 18:1 или меньше, в частности, примерно 15:1 или меньше, например, примерно 10:1 или меньше. В некоторых вариантах осуществления изобретения массовое соотношение альбумина и по существу не растворимого в воде фармакологически активного агента в композиции находится в пределах от примерно 1:1 до примерно 18:1, от примерно 2:1 до примерно 15:1, от примерно 3:1 до примерно 13:1, от примерно 4:1 до примерно 12:1, от примерно 5:1 до примерно 10:1. В некоторых вариантах осуществления изобретения массовое соотношение альбумина и по существу не растворимого в воде фармакологически активного агента в наночастицах композиции составляет примерно 1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8, 1:9, 1:10, 1:11, 1:12, 1:13, 1:14, 1:15 или меньше.
В некоторых вариантах осуществления изобретения композиция в виде частиц характеризуется наличием одного или нескольких вышеуказанных признаков.
В некоторых вариантах осуществления изобретения композиция на основе наночастиц является абраксаном™. Композиции на основе наночастиц, содержащие другие по существу не растворимые в воде фармакологически активные агенты (такие как доцетаксел и ортатаксел), также могут обладать одним или несколькими вышеуказанными признаками.
Некоторые варианты осуществления изобретения относятся к композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, в которой альбумин получен методом удаления приона, включающим приведение исходной композиции альбумина в контакт с лигандом, способным связываться с прионным белком. В некоторых вариантах осуществления изобретения процесс удаления приона дополнительно включает удаление указанного лиганда и связанных с ним белков из композиции альбумина.
Некоторые варианты осуществления изобретения относятся к композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, при этом альбумин в композиции получают способом, включающим: а) приведение исходной композиции альбумина в контакт с лигандом, способным связываться с прионным белком с образованием комплекса лиганда с прионным белком, и b) удаление полученного комплекса из исходной композиции.
Наночастицы, включающие альбумин и по существу не растворимые в воде лекарственные средства, более подробно описаны ниже. Способ удаления прионов из композиции (такой как исходная композиция альбумина, композиция на основе наночастиц, включающих альбумин и по существу не растворимый в воде фармакологически активный агент, или промежуточная композиция, образованная в процессе получения наночастиц) более подробно описаны ниже. В объем настоящего изобретения входят композиции, полученные любыми способами по настоящему изобретению.
Композиции по настоящему изобретению обычно характеризуются более низким уровнем прионного белка по сравнению с композициями, в которых альбумин не очищен в соответствии с процессом удаления приона. Например, в некоторых вариантах осуществления изобретения композиция содержит примерно на 50%, 40%, 30%, 20%, 10%, 5%, 4%, 3%, 2%, 1% или меньше прионного белка, чем композиция, в которой альбумин не был очищен в соответствии с процессом удаления приона. В некоторых вариантах осуществления изобретения композиция содержит примерно на 1, 2, 2,5, 3, 3,5, 4, 4,5, 5, 6, 7 или 8 log меньше прионного белка, чем композиция, в которой альбумин не был очищен в соответствии с процессом удаления приона. В некоторых вариантах осуществления изобретения инфективность композиции примерно на 1, 2, 2,5, 3, 3,5, 4, 4,5, 5, 6, 7 или 8 log меньше по сравнению с композицией, в которой альбумин не был очищен в соответствии с процессом удаления приона. В некоторых вариантах осуществления изобретения композиция по настоящему изобретению биологически эквивалентна колмпозиции, в которой альбумин не был очищен в соответствии с процессом удаления приона.
Хотя целью настоящего изобретения является альбумин, должно быть понятно, что в объем изобретения также входят другие белки, обычно обнаруживаемые в крови или плазме, которые включают, не ограничиваясь ими, иммуноглобулин (в том числе IgA и IgG), липопротеины, аполипопротеин В, гликопротеин альфа-кислоты, бета-2-макроглобулин, тироглобулин, трансферин, фибронектин, фактор VII, фактор VIII, фактор IX, фактор Х и тому подобные. Все описания, относящиеся к альбумину, в равной степени относятся к другим белкам, используемым при образовании наночастиц.
Способы получения композиций без приона на основе наночастиц
Другим объектом настоящего изобретения являются способы получения композиций без приона на основе наночастиц. Например, некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, в соответствии с которым смесь, включающую раствор альбумина и органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе, подвергают воздействию высокого сдвигающего усилия, при этом альбумин получают способом, включающим удаление прионного белка из исходной композиции альбумина. Некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, в соответствии с которым смесь, включающую раствор альбумина и органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе, подвергают воздействию высокого сдвигающего усилия, при этом альбумин получают способом, включающим приведение исходной композиции альбумина в контакт с лигандом, способным связываться с прионным белком. Некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, в соответствии с которым смесь, включающую раствор альбумина и органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе, подвергают воздействию высокого сдвигающего усилия, при этом альбумин получают способом, включающим: а) приведение исходной композиции альбумина в контакт с лигандом, способным связываться с прионным белком, и b) удаление лиганда и связанного с ним белка из исходной композиции.
Некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает: а) удаление прионного белка из исходной композиции альбумина; b) воздействие высоким сдвигающим усилием на смесь, включающую раствор альбумина без приона и органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе. Некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает: а) приведение исходной композиции альбумина в контакт с лигандом, способным связываться с прионным белком с образованием комплекса лиганда с прионным белком, b) удаление полученного комплекса из исходной композиции альбумина и с) воздействие высоким сдвигающим усилием на смесь, включающую раствор альбумина без приона и органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе.
Некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает: а) приведение раствора альбумина в контакт с лигандом, способным связываться с прионным белком, b) удаление лиганда и связанных с ним белков из раствора альбумина, с) воздействие высоким сдвигающим усилием на смесь, включающую раствор альбумина и органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе. В некоторых вариантах осуществления изобретения смесь по существу не содержит поверхностно-активных веществ.
Прионы могут быть удалены во время образования наночастиц. Например, некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает: а) приведение смеси, включающей раствор альбумина и органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе, в контакт с лигандом, способным связываться с прионным белком. В некоторых вариантах осуществления изобретения указанный способ дополнительно включает: b) удаление из смеси лиганда и связанных с ним белков. В некоторых вариантах осуществления изобретения указанный способ дополнительно включает: с) воздействие на смесь высоким сдвигающим усилием. В некоторых вариантах осуществления изобретения указанный способ дополнительно включает удаление органического растворителя из смеси. В некоторых вариантах осуществления изобретения смесь по существу не содержит поверхностно-активных веществ.
Некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает: а) воздействие высоким сдвигающим усилием на смесь, включающую раствор альбумина и органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе, и b) приведение смеси в контакт с лигандом, способным связываться с прионным белком. В некоторых вариантах осуществления изобретения указанный способ дополнительно включает: с) удаление лиганда и связанных с ним белков из смеси. В некоторых вариантах осуществления изобретения указанный способ дополнительно включает удаление из смеси органического растворителя. В некоторых вариантах осуществления изобретения смесь по существу не содержит поверхностно-активных веществ.
Некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает приведение смеси, включающей органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе, и раствор альбумина, в контакт с лигандом, способным связываться с прионным белком, в соответствии с которым смесь подвергают воздействию высокого сдвигающего усилия до приведения в контакт с лигандом. Некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает: а) воздействие высоким сдвигающим усилием на смесь, включающую органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе, и раствор альбумина, и b) приведение смеси в контакт с лигандом, способным связываться с прионным белком. В некоторых вариантах осуществления изобретения указанный способ дополнительно включает: с) удаление из смеси лиганда и связанных с ним белков. В некоторых вариантах осуществления изобретения указанный способ дополнительно включает: d) удаление из смеси водной фазы. В некоторых вариантах осуществления изобретения смесь по существу не содержит поверхностно-активных веществ.
Некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает: а) воздействие высоким сдвигающим усилием на смесь, включающую органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе, и раствор альбумина, b) удаление указанного органического растворителя и с) приведение смеси, не содержащей органического растворителя, в контакт с лигандом, способным связываться с прионным белком. В некоторых вариантах осуществления изобретения указанный способ дополнительно включает: d) удаление из смеси лиганда и связанных с ним белков. В некоторых вариантах осуществления изобретения указанный способ дополнительно включает: d) удаление из смеси водной фазы. В некоторых вариантах осуществления изобретения смесь по существу не содержит поверхностно-активных веществ.
Некоторые варианты осуществления изобретения относятся к способу получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает: а) воздействие высоким сдвигающим усилием на смесь, включающую органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе, и раствор альбумина b) удаление органического растворителя, с) добавление альбумина к смеси и d) приведение смеси в контакт с лигандом, способным связываться с прионным белком. В некоторых вариантах осуществления изобретения указанный способ дополнительно включает: е) удаление из смеси лиганда и связанных с ним белком. В некоторых вариантах осуществления изобретения указанный способ дополнительно включает: f) удаление из смеси водной фазы. В некоторых вариантах осуществления изобретения смесь по существу не содержит поверхностно-активных веществ.
Способы по настоящему изобретению обычно включают стадию воздействия высоким сдвигающим усилием на смесь, включающую органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе, и раствор альбумина. В некоторых вариантах осуществления изобретения высокое сдвигающее усилие представляет собой гомогенизацию под высоким давлением, например, под давлением в диапазоне от примерно 210 атм. (3000 фунтов/кв.дюйм) до примерно 2100 атм. (30000 фунтов/кв.дюйм), в том числе, например, от примерно 420 атм. (6000 фунтов/кв.дюйм) до примерно 1750 атм. (25000 футов/кв.дюйм), от примерно 630 атм. (9000 фунтов/кв.дюйм) до примерно 1260 атм. (18000 фунтов/кв.дюйм), от примерно 700 атм. (10000 фунтов/кв.дюйм) до примерно 1750 атм. (25000 фунтов/кв.дюйм), от примерно 1050 атм. (15000 фунтов/кв.дюйм) до примерно 1750 атм. (25000 фунтов/кв.дюйм). В некоторых вариантах осуществления изобретения органический растворитель является смесью по существу не смешивающегося с водой органического растворителя (такого как хлороформ или метиленхлорид) и водорастворимого органического растворителя (такого как водорастворимый спирт, в том числе этанол и трет-бутанол). В некоторых вариантах осуществления изобретения соотношение (об./об.) по существу не смешивающегося с водой органического растворителя и водорастворимого органического растворителя (например, соотношение хлороформа/этанола или хлороформа/бутанола) составляет примерно 1:9, 1:8, 1:7, 1:6, 1:5, 1:4, 1:3, 1:2, 1:1, 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1 или 9:1 или примерно 3:7, 5:7, 4:6, 6:4, 5:5, 6:5, 8:5, 9:5, 9,5:5, 5:3, 7:3, 6:4 или 9,5:0,5.
В некоторых вариантах осуществления изобретения указанный способ дополнительно включает удаление из смеси органической фазы (например, удаление выпариванием при пониженном давлении). В некоторых вариантах осуществления изобретения указанный способ дополнительно включает удаление из смеси водной фазы. В некоторых вариантах осуществления изобретения указанный способ дополнительно включает стерилизацию фильтрованием наночастиц, полученных вышеописанным способом.
Некоторые варианты осуществления изобретения относятся к способу удаления прионного белка из композиции с подозрением на наличие прионного белка, содержащей альбумин и по существу не растворимый в воде фармакологически активный агент, который включает: а) приведение композиции на основе наночастиц в контакт с лигандом, способным связываться с прионным белком, b) удаление лиганда и связанных с ним белков из композиции на основе наночастиц. Некоторые варианты осуществления изобретения относятся к способу удаления прионного белка из композиции, содержащей альбумин, с подозрением на наличие аномального прионного белка, который включает: а) приведение композиции альбумина в контакт с лигандом, способным связываться с прионным белком, b) удаление лиганда и связанных с ним белков из композиции альбумина, при этом композицию альбумина используют для получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент.
Некоторые варианты осуществления изобретения относятся к способу удаления прионного белка из композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает: а) определение наличия или отсутствия прионного белка в композиции, b) приведение композиции в контакт с лигандом, способным связываться с прионным белком, и с) удаление из композиции лиганда и связанных с ним белков.
В некоторых вариантах осуществления изобретения одну или несколько стадий способов по настоящему изобретению выполняют в периодическеом режиме. В некоторых вариантах осуществления изобретения одну или несколько стадий способов по настоящему изобретению выполняют в непрерывном режиме.
Удаление приона до образования наночастиц
Прионный белок может быть удален из исходной композиции альбумина до получения композиций на основе наночастиц, содержащих альбумин. Указанный способ обычно включает приведение исходной композиции альбумина в контакт с лигандом, способным связываться с прионным белком, и удаление лиганда и связанного с ним белка из композиции альбумина. Указанный процесс может быть повторен один или несколько раз при использовании одного и того же или другого лиганда. В процессе удаления приона могут быть также одновременно использованы два или более лигандов.
В некоторых вариантах осуществления изобретения исходная композиция альбумина является композицией, выделенной из крови. Например, в некоторых вариантах осуществления изобретения исходная композиция альбумина представляет собой цельную кровь, концентрат эритроцитов, плазму, сыворотку, фракцию с высоким и низким содержанием тромбоцитов, концентрат тромбоцитов, лейкоциты, преципитат плазмы крови, фракционированный преципитат и супернатант плазмы крови или промежуточную фракцию плазмы. В некоторых вариантах осуществления изобретения исходную композицию альбумина получают у человека. В некоторых вариантах осуществления изобретения исходную композицию альбумина получают у животного, такого как корова, овца и грызун (такой как мышь, хомячок и норка). В некоторых вариантах осуществления изобретения исходную композицию альбумина получают у группы субъектов (таких как человек), в которой по меньшей мере некоторые субъекты инфицированы прионами. В некоторых вариантах осуществления изобретения исходную композицию альбумина получают у группы субъектов (таких как человек), в которой по меньшей мере некоторые субъекты являются субъектами с подозрением на инфицирование прионом.
В некоторых вариантах осуществления изобретения исходная композиция альбумина является композицией, полученной из биологической жидкости (такой как кровь) любыми способами, известными в данной области, которые включают ионообменную хроматографию, аффинную хроматографию, гель-хроматографию, гидрофобную хроматографию и/или дифференцированную преципитацию. В некоторых вариантах осуществления изобретения исходная композиция альбумина очищена от крови (такой как кровь человека). В некоторых вариантах осуществления изобретения исходная композиция альбумина очищена от сыворотки (такой как сыворотка человека). В некоторых вариантах осуществления изобретения исходная композиция альбумина характеризуется инфективностью приона примерно 100 ин.ед.-иц/мл, 90 ин.ед.-иц/мл, 50 ин.ед-иц./мл или 10 ин.ед.-иц/мл. В некоторых вариантах осуществления изобретения концентрация альбумина в исходной композиции альбумина равна примерно 1% (масс./об.), в том числе, например, примерно 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 15%, 20%, 25% или 30%.
В процессе удаления приона лиганд вводят в соприкосновение с исходной композицией альбумина и оставляют для связывания с прионными белками в исходной композиции альбумина. Условия, пригодные для связывания, можно определить и оптимизировать для облегчения связывания лиганда с приорнным белком в зависимости от природы лиганда и его специфичности связывания с прионным белком. В некоторых вариантах осуществления изобретения связывание выполняют при температуре от примерно 0°С до примерно 39°С, в том числе, например, от примерно 20°С до примерно 25°С. Связывание можно выполнять при значении рН от примерно 4 до примерно 10, в том числе, например, от примерно 5 до примерно 9, от примерно 6 до примерно 8, от примерно 6,8 до примерно 7,5, от примерно 6,9 до примерно 7,4 или примерно 7. Для уменьшения неспецифического связывания с лигандом могут быть необязательно использованы блокирующие агенты.
После выполнения стадии приведения в контакт лиганд и связанные с ним белки удаляют из остальной части композиции. Термин “удаление” в использованном здесь значении означает отделение лиганда и связанного с ним белка от композиции альбумина. Отделение может быть выполнено разными способами в зависимости от природы лиганда и носителя (при его наличии), используемого для облегчения отделения. Например, лиганд и связанные с ним белки можно отделить при помощи хроматографии, которая включает, не ограничиваясь ими, тонкослойную хроматографию, хроматографию на колонке и периодическую хроматографию; путем разделения на мембране и твердом носителе; разделения в реакторе; разделения в магнитном поле; иммуноразделения; коллоидного разделения; осаждения; преципитации или центрифугирования.
В некоторых вариантах осуществления изобретения лиганд может быть присоединен к носителю, такому как гранулы или мембрана, который в свою очередь может приводить в контакт с исходной композицией альбумина. Носитель с иммобилизованным лигандом вводят в соприкосновение с исходной композицией альбумина в условиях, пригодных для образования комплекса прион-лиганд. Затем твердую фазу отделяют от композиции, удаляя таким образом прионный белок, связанный с лигандом. Например, в одном типичном варианте осуществления изобретения лиганды иммобилизуют на колонке, такой как хроматографическая колонка, образец (такой как исходная композиция альбумина) пропускают через колонку под действием силы тяжести или под давлением, например, как в колонке для жидкостной хроматографии высокого давления. Прионные белки в образце связываются с лигандом, иммобилизованным на колонке, и образец, проходящий через колонку, собирают. Указанный процесс можно повторять несколько раз для достижения требуемого результата, используя один и том же или разные лиганды.
Скорость потока образца (такого как исходная композиция альбумина) в колонке можно отрегулировать так, чтобы максимально увеличить связывание лиганда с прионными белками в образце. В некоторых вариантах осуществления изобретения связывание выполняют при скорости потока от примерно 0,1 мл/мин до примерно 5,0 мл/мин, от примерно 0,1 мл/мин до примерно 2,5 мл/мин, от примерно 0,1 мл/мин до примерно 0,25 мл/мин, от примерно 0,25 мл/мин до примерно 0,5 мл/мин, от примерно 0,5 мл/мин до примерно 1,0 мл/мин, от примерно 1,0 мл/мин до примерно 1,5 мл/мин, от примерно 1,5 мл/мин до примерно 2,0 мл/мин, от примерно 2,0 мл/мин до примерно 2,5 мл/мин, от примерно 2,5 мл/мин до примерно 3,0 мл/мин, от примерно 3,0 мл/мин до примерно 3,5 мл/мин, от примерно 3,5 мл/мин до примерно 4,0 мл/мин, от примерно 4,0 мл/мин до примерно 4,5 мл/мин или от примерно 4,5 мл/мин до примерно 5,0 мл/мин, в том числе, например, при скорости потока, равной примерно 0,1 мл/мин, 0,25 мл/мин, 0,5 мл/мин, 1,0 мл/мин, 1,5 мл/мин, 1,7 мл/мин, 1,8 мл/мин, 1,9 мл/мин, 2,0 мл/мин, 2,1 мл/мин, 2,3 мл/мин, 2,5 мл/мин, 2,7 мл/мин, 3,0 мл/мин, 3,5 мл/мин, 4,0 мл/мин, 4,5 мл/мин или 5,0 мл/мин. В некоторых вариантах осуществления изобретения скорость потока равна по меньшей мере примерно 10 мл/мин, в частности, по меньшей мере примерно 20 мл/мин, 30 мл/мин, 40 мл/мин, 50 мл/мин.
Общий объем проходящего потока или общее время прохождения потока в процессе связывания также можно отрегулировать так, чтобы максимально увеличить связывание лиганда с прионными белками в образце (таком как исходная композиция альбумина). В некоторых вариантах осуществления изобретения общий объем проходящего потока составляет от примерно 1-кратного до примерно 1000-кратного объема колонки, в том числе, например, от примерно 2-кратного до примерно 10-кратного объема колонки, от примерно 10-кратного до примерно 20-кратного объема колонки, от примерно 20-кратного до примерно 30-кратного объема колонки, от примерно 30-кратного до примерно 40-кратного объема колонки, от примерно 40-кратного до примерно 50-кратного объема колонки, от примерно 50-кратного до примерно 1000-кратного объема колонки, от примерно 50-кратного до примерно 500-кратного объема колонки, от примерно 100-кратного до примерно 600-кратного объема колонки или от примерно 200-кратного до примерно 800-кратного объема колонки. В некоторых вариантах осуществления изобретения общий объем проходящего потока равен примерно 100-кратному объему колонки. В других вариантах осуществления изобретения общий объем проходящего потока равен примерно 500-кратному объему колонки. В некоторых вариантах осуществления изобретения общее время прохождения потока составляет от примерно 1 часа до примерно 30 часов, в том числе, например, от примерно 2 часов до примерно 25 часов, от примерно 3 часов до примерно 20 часов или от примерно 3 часов до примерно 17 часов. В некоторых вариантах осуществления изобретения общее время прохождения потока составляет примерно 3 часа, 4 часа, 6 часов, 8 часов, 10 часов, 12 часов, 14 часов, 16 часов, 17 часов, 18 часов, 19 часов или 20 часов. В некоторых вариантах осуществления изобретения общее время прохождения потока составляет более примерно 24 часов. В некоторых вариантах осуществления изобретения общее время прохождения потока составляет менее примерно 8 часов, в том числе, например, 7, 6, 5, 4, 3, 2, 1 или 0,5 часа.
Альтернативно лиганд сначала может быть введен в соприкосновение с исходной композицией альбумина в условиях, обеспечивающих образование комплекса прион-лиганд. Комплекс прион-лиганд затем удаляют, используя колонку для аффинной хроматографии. Для облегчения отделения лиганд может быть конъюгирован с партнером связывания, благодаря чему комплекс лиганд-прион может быть удален при помощи указанного партнера связывания, например, на колонке для аффинной хроматографии, содержащей молекулу, узнающую данный партнер связывания.
Помимо периодической хроматографии или хроматографии на колонке, существует целый ряд других конфигураций, модификаций и вариантов использования лигандов для связывания прионных белков. Такие варианты и модификации включают, не ограничиваясь ими: периодические процессы, непрерывные процессы, хроматографию в подвижном слое; процессы, выполняемые при низком, среднем или высоком давлении; лабораторные, среднемасштабные или крупномасштабные процессы. В некоторых вариантах осуществления изобретения лиганды наносят на мембрану, гранулы, погружают в нетканое сито или наносят на покрывающие волокна, находящиеся в корпусе фильтра.
В некоторых вариантах осуществления изобретения стадия удаления не вызывает значительных потерь продукта и/или изменений свойств и/или устойчивости альбумина. В некоторых вариантах осуществления изобретения восстановление альбумина в первоначальном биологическом состоянии превышает 50% и составляет, например, 80%, 90% или больше. В некоторых вариантах осуществления изобретения степень восстановления альбумина в результате выполнения процесса удаления приона составляет более примерно 80%, 90%, 95% или 99%. В некоторых вариантах осуществления изобретения концентрация альбумина в исходной композиции альбумина регулируют или контролируют до стадии удаления приона с тем, чтобы свести к минимуму неспецифическое связывание и потерю альбумина при выполнении данного процесса. Например, концентрация альбумина может находиться в пределах от примерно 1% до примерно 50%, от примерно 5% до примерно 25%, от примерно 5% до примерно 30%, от примерно 5% до примерно 40%, от примерно 5% до примерно 10%, от примерно 10% до примерно 15%, от примерно 15% до примерно 20%, от примерно 20% до примерно 25%, от примерно 25% до примерно 30% и т.д. и, например, может быть равна примерно 5%, 10%, 15%, 20%, 25% или 30% альбумина.
Полученная композиция альбумина может быть подвергнута анализу для определения степени очистки в процессе удаления приона. Лиганд со связанными прионными белками также может быть подвергнут анализу (сразу же или после элюирования) для определения степени очистки.
Удаление прионных белков можно оценить с учетом уменьшения количества прионного белка или уменьшения инфективности. В некоторых вариантах осуществления изобретения из исходной композиции альбумина может быть удалено по меньшей мере примерно 50%, в том числе, например, по меньшей мере примерно 60%, 70%, 80%, 90%, 95%, 99% или 100% прионных белков. В некоторых вариантах осуществления изобретения инфективность композиции альбумина после удаления приона по меньшей мере примерно в 10 раз, 20 раз, 30 раз, 40 раз, 50 раз, 80 раз, 100 раз, 200 раз, 500 раз, 1000 раз, 104 раз, 105 раз, 106 раз, 107 раз, 108 раз, 109 раз меньше, чем в исходной композиции альбумина.
В некоторых вариантах осуществления изобретения для определения степени очистки композиции в процессе удаления приона используют инфективность серийных разведений. Серийные разведения образцов исследуют в отношении инфекционной активности, например, с использованием экспериментальных животных. Разведение, при котором инфицируется половина животных, является инфекционным титром. Например, при 5-кратном разведении инфективность образца может быть равна 5 log. Сравнивая число log инфективности исходной композиции альбумина и композиции альбумина после удаления приона, можно определить степень очистки в процессе удаления приона. В некоторых вариантах осуществления изобретения указанный способ удаления приона позволяет уменьшить инфективность на 1, 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9, 2, 3 или 4, 5, 6, 7, 8, 9 или 10 log.
В некоторых вариантах осуществления изобретения степень очистки в процессе удаления приона определяют при помощи экспериментов по потенцированию инфекционными веществами, выполняя стадии, рассмотренные в настоящем описании изобретения для данного способа удаления приона. Приемлемые потенцирующие агенты включают, не ограничиваясь ими, гомогенаты головного мозга, микросомы, ямкоподобные домены, очищенный PrPsc и фибриллы приона. В некоторых вариантах осуществления изобретения потенцирующий агент растворяют в детергенте (например, в саркозиле). В некоторых вариантах осуществления изобретения содержание потенцирующего агента в композиции находится в пределах от примерно 0,001% до примерно 5%, от примерно 0,001% до примерно 0,25%, от примерно 0,001% до примерно 0,1%, от примерно 0,001% до примерно 0,005%, от примерно 0,005% до примерно 0,075%, от примерно 0,075% до примерно 0,01%, от примерно 0,01% до примерно 0,1%, от примерно 0,1% до примерно 0,5%, от примерно 0,5% до примерно 0,75%, от примерно 0,75% до примерно 1%, от примерно 1% до примерно 2%, от примерно 2% до примерно 3% или от примерно 3% до примерно 5% и, например, составляет примерно 0,001%, 0,005%, 0,075%, 0,01%, 0,1%, 0,5%, 0,75%, 1%, 2%, 3% или 5%.
В некоторых вариантах осуществления изобретения для определения степени очистки в процессе удаления приона используют коэффициенты восстановления (RF). RF можно вычислить по формуле:
RF=(V1×T1)/(V2×T2)
или
Log10[RF]=[Log10(V1)+Log10(T1)]-[Log10(V2)+Log10(T2)],
где V1 и Т1 означают соответственно объем и титр исходной композиции альбумина, V2 и Т2 означают объем и титр композиции альбумина после удаления приона. Коэффициенты восстановления могут быть округлены до одной десятой после окончательного вычисления. В некоторых вариантах осуществления изобретения инфективность прионных белков может быть уменьшена в исходной композиции альбумина на коэффициент восстановления, равный по меньшей мере примерно 1,0, 1,5, 2,0, 2,5, 3,0, 3,5, 4,0, 4,5, 5,0, 5,5, 6,0, 6,5, 7,0, 7,5 или 8,0 log10. Например, содержание прионного белка может быть уменьшено на коэффициент восстановления, который больше или равен 2,5 log10 во фракции, растворенной в 0,5% саркозиле, которой была потенцирована композиция, содержащая 20% альбумина. В качестве другого примера можно отметить, что содержание прионного белка может быть уменьшено на коэффициент восстановления, который больше или равен 2,0 log10 во фракции, растворенной в 0,5% саркозиле, которой была потенцирована композиция, содержащая 25% альбумина.
Удаление прионов можно определить при помощи стандартного анализа методом вестерн-блоттинга. Например, лиганды после связывания с прионом сначала обрабатывают протеиназой К, которая расщепляет весь PrPc и не расщепляет PrPsc. Гидролизат затем исследуют на геле с додецилсульфатом натрия (SDS) и переносят на лист нитроцеллюлозы или мембрану из PVDF. Выделенные полосы PrPsc затем визуализируют, используя 3F4 или 6Н4. 3F4 взаимодействует с аминокислотными остатками 109-112 PrP человека, хомячков и кошек. В одном типичном варианте осуществления изобретения инкубацию выполняют при концентрации 0,6 мкг/мл в течение минимум одного часа, после чего избыток антитела вымывают и мембраны инкубируют с конъюгатом пероксидазы из хрена с антителом кролика против мыши (степень разведения 1:1000) в течение минимум одного часа. После обильной промывки TTBS мембраны проявляют при повышенной хемилюминесценции. В некоторых вариантах осуществления изобретения удаление прионных белков оценивают в соответствии с указаниями, приведенными в публикации Guideline for the Investigation of Manufacturing Processes for Plasma-Derived Medicinal Products with Regard to vCJD Risk (CPMP5136/03).
Лиганды, способные связываться с прионным белком и носителем
Термин “лиганд” в использованном здесь значении означает молекулу, с которой связывается прионный белок или пептид. Термин “лиганд, способный связываться с прионным белком” означает лиганд, который специфически связывается с прионным белком в приемлемым условиях. В некоторых вариантах осуществления изобретения лиганд специфически связывается с прионным белком человека. В некоторых вариантах осуществления изобретения лиганд специфически связывается с прионным белком хомячка. В некоторых вариантах осуществления изобретения лиганд специфически связывается с прионным белком мыши. В некоторых вариантах осуществления изобретения лиганд связывается с прионными белками нескольких видов животных. Например, в некоторых вариантах осуществления изобретения лиганд связывается с прионным белком человека, прионным белком хомячка и прионным белком мыши.
В некоторых вариантах осуществления изобретения лиганд связывается с прионным белком (таким как прионный белок человека, прионный белок хомячка и/или прионный белок мыши) с высоким сродством связывания. Например, в некоторых вариантах осуществления изобретения лиганд характеризуется сродством связывания более примерно 107, 108, 109, 1010 или 1011.
Идентифицирован целый ряд лигандов, связывающихся с прионным белком, которые могут быть использованы в способах по настоящему изобретению. Смотрите, например, публикации WO04/090102, WO04/050851, WO06/010915 и WO06/044459. Такие лиганды включают, например, пептиды, химические соединения и антитела, которые специфически узнают белок прион. Указанный лиганд может быть использован для удаления всех форм прионных белков из композиции или может быть избирательно выбран для обнаружения или удаления одной формы прионного белка.
В некоторых вариантах осуществления изобретения лиганд, используемый для удаления приона, является пептидом, в частности, пептидами, описанными в опубликованной заявке РСТ № WO04/05051. Например, в некоторых вариантах осуществления изобретения лиганд является пептидом, содержащим аминокислоту, имеющую любую SEQ ID NO: 1-232, указанную в таблице 1. В некоторых вариантах осуществления изобретения лиганд является трипептидом, таким как пептиды, содержащие аминокислотную последовательность, включающую любую SEQ ID NO: 48, 102, 105, 108, 109, 110, 111, 143, 148, 193, 194, 195, 202, 203, 204 или 210. В некоторых вариантах осуществления изобретения лиганд является пептидом, содержащим шесть аминокислот, таким как шестимер, содержащий аминокислотную последовательность, включающую любую SEQ ID NO: 152, 153, 180, 181, 182, 183, 185, 186, 187, 188, 189 или 190. В некоторых вариантах осуществления изобретерия лиганд имеет аминокислотную последовательность SEQ ID NO: 150 или 151.
В некоторых вариантах осуществления изобретения лиганд является пептидом аминокислотной последовательности DVR, SYA, AMN31, D4 или YVHEA. В некоторых вариантах осуществления изобретения лиганд является пептидом, связывающимся с прионным белком со сродством, которое подобно или выше аналогичного показателя DVR, SYA, AMN31, D4 или YVHEA. В некоторых вариантах осуществления изобретения лиганд является пептидом DVR. В других вариантах осуществления изобретения лиганд является пептидом AMN31. В некоторых вариантах осуществления изобретения лиганды, являющиеся пептидами, использованы в форме смол (в частности, связанными со смолами).
В некоторых вариантах осуществления изобретения лиганд является антителом, узнающим прионный белок. В данной области известны антитела, узнающие прионные белки. Такие антитела включают, не ограничиваясь ими, моноклональные антитела 3F4, 6H4 или 16А18. В некоторых вариантах осуществления изобретения антитело является антителом, специфичным к гликоформе, таким как ICSM-4 и ICSM-10. Антитела, пригодные для использования в способах по настоящему изобретению, включают поликлональные и моноклональные антитела, одноцепочечные антитела, Fab-фрагменгты и Fv-фрагменты.
В некоторых вариантах осуществления изобретения лиганд связывается со специфической последовательностью прионного белка. Например, в некоторых вариантах осуществления изобретения лиганд (такой как пептид или антитело) связывается с одной из последовательностей прионного белка SEQ ID NO: 133-146, приведенных в таблице 2.
В некоторых вариантах осуществления изобретения лиганд является химическим соединением. Соединения, способные связываться с прионным белком, представлены, например, в публикации заявки РСТ № WO06/010915, которая полностью включена в настоящее описание изобретения. В некоторых вариантах осуществления изобретения лиганд является замещенным триазином. В некоторых вариантах осуществления изобретения лиганд является соединением формулы (I):

где R1 и R2 имеют одинаковые или разные значения и означают необязательно замещенный алкил, необязательно замещенный циклоалкил, необязательно замещенный арил или необязательно замещенный гетероарил; R3 означает водород или замещающую арильную группу либо R3 является твердым носителем, необязательно присоединенным с помощью спейсера; Z означает атом кислорода, атом серы или NR4; Y означает атом кислорода, атом серы или NR5, где R4 и R5, которые могут иметь одинаковые или разные значения, означают водород, необязательно замещенный алкил, содержащий 1-6 атомов углерода, необязательно замещенный фенил, необязательно замещенный бензил или необязательно замещенный β-фенилэтил; и один из элементов Х1 и Х2 означает атом азота и другой из элементов Х1 и Х2 означает атом азота или CR6, где R6 означает водород или арильную группу, и предназначен для аффинного связывания с прионным белком.
В некоторых вариантах осуществления изобретения лиганд является соединением формулы (II):
где R1 означает группу -(СН2)m-Q1, где m означает целое число от 0 до 7 и Q1 означает -CR11R12R13 или -NR11R12, где R11, R12 и R13 независимо означают водород, алкил, циклоалкил или гетероциклоалкил, либо два элемента из R11, R12 и R13 вместе с атомом углерода или азота, к которому они присоединены, образуют необязательно замещенную циклоалкильную группу или необязательно замещенную гетероциклоалкильную группу; R2 означает группу -(СН2)n-Q2, где n означает целое число от 0 до 7, и Q2 означает -CR21R22R23 или -NR21R22, где R21, R22 и R23 независимо означают водород, алкил, циклоалкил или гетероциклоалкил, либо два элемента из R11, R12 и R13 вместе с атомом углерода или азота, к которому они присоединены, образуют необязательно замещенную циклоалкильную группу или необязательно замещенную гетероциклоалкильную группу, и R3 означает водород или замещающую арильную группу либо R3 означает твердый носитель, необязательно присоединенный с помощью спейсера; Z означает атом кислорода, атом серы или NR4; Y означает атом кислорода, атом серы или NR5, где R4 и R5, которые могут иметь одинаковые или разные значения, означают водород, необязательно замещенный алкил, содержащий 1-6 атомов углерода, необязательно замещенный фенил, необязательно замещенный бензил или необязательно замещенный β-фенилэтил; и один из элементов Х1 и Х2 означает атом азота и другой из элементов Х1 и Х2 означает атом азота или CR6, где R6 означает водород или арильную группу, и предназначен для аффинного связывания с прионным белком.
В некоторых вариантах осуществления изобретения а) Х1 и Х2, оба означают атом азота; b) Z и Y, оба означают NR4, в частности, когда R4 означает водород; с) m означает целое число от 0 до 4, более предпочтительно от 0 до 3 и наиболее предпочтительно от 1 до 3; d) Q1 означает -NR11R12, где R11 и R12 вместе с атомом азота, к которому они присоединены, предпочтительно образуют гетероциклоалкильную группу; е) n означает целое число от 0 до 4, более предпочтительно от 0 до 2 и наиболее предпочтительно 0; и f) R21 и R22 вместе с атомом углерода или атомом азота, к которому они присоединены, образуют циклоалкильную или гетероциклоалкильную группу.
В некоторых вариантах осуществления изобретения Q2 означает гидрофобную циклоалкильную или гетероциклоалкильную группу, особенно в кольцевых системах, включающих по меньшей мере шесть атомов.
В некоторых вариантах осуществления изобретения Q1 означает гетероциклоалкильную группу, предпочтительно пиперидил, в частности, 1-пиперидил, или пиперазинил, в частности, 1-пиперазинил.
В некоторых вариантах осуществления изобретения лиганд является соединением формулы (III):
где R1 означает алкиленовую цепь -(СН2)р-СН3, где р означает целое число от 0 до 6, замещенную одной или несколькими карбоксильными группами и необязательно замещенную одной или несколькими другими алкильными группами; R2 означает группу -(СН2)q-Ar, где q означает целое число от 0 до 7 и Ar означает необязательно замещенную арильную группу; и R3 означает водород или замещающую арильную группу либо R3 является твердым носителем, необязательно присоединенным с помощью спейсера; Z означает атом кислорода, атом серы или NR4; Y означает атом кислорода, атом серы или NR5, где R4 и R5, которые могут иметь одинаковые или разные значения, означают водород, необязательно замещенный алкил, содержащий 1-6 атомов углерода, необязательно замещенный фенил, необязательно замещенный бензил или необязательно замещенный β-фенилэтил; и один из элементов Х1 и Х2 означает атом азота и другой из элементов Х1 и Х2 означает атом азота или CR6, где R6 означает водород или арильную группу, и предназначен для аффинного связывания с прионным белком.
В некоторых вариантах осуществления изобретения а) Х1 и Х2 оба означают атом азота; b) Z и Y, оба означают NR4, в частности, когда R4 означает водород; с) р означает целое число от 0 до 4, более предпочтительно от 0 до 3; d) R1 замещен одной или двумя карбоксильными группами, при этом по меньшей мере одна из указанных карбоксильных групп присоединена к концевому атому углерода алкиленовой цепи -(СН2)p-СН3; е) q означает целое число от 0 до 4, более предпочтительно от 0 до 3 и наиболее предпочтительно 1 или 2; и f) Ar означает моноциклическую карбоциклическую или гетероциклическую ароматическую группу, необязательно замещенную одним или несколькими заместителями, выбираемыми из группы, состоящей из фенила, фенокси, толила, хлорбензила, метоксибензила, фторбензила, пиридила и индоила.
В некоторых вариантах осуществления изобретения R1 означает карбоксиметил, 4-карбоксибутил или 1-(1,3-дикарбокси)пропил.
В некоторых вариантах осуществления изобретения Ar означает фенил, 4-гидроксифенил или пиридил, в частности, 2-пиридил.
В некоторых вариантах осуществления изобретения R3 означает водород или замещающую арильную группу либо R3 означает твердый носитель, необязательно присоединенный с помощью спейсера; Х1 и Х2, оба означают N; Y и Z, оба означают NH; m означает 2; Q1 означает пиперидил или пиперазинил; n означает 0 или 2; и Q2 означает 1-пиперидил или адамантил.
В некоторых вариантах осуществления изобретения R3 означает водород или замещающую арильную группу либо R3 означает твердый носитель, необязательно присоединенный с помощью спейсера; Х1 и Х2, оба означают N; Y и Z, оба означают NH; R1 означает карбоксиметил, 4-карбоксибутил и 1-(1,3-дикарбокси)пропил; q означает 2 и Ar означает фенил, 2-пиридил или 4-гидроксифенил.
В некоторых вариантах осуществления изобретения лиганд является неорганическим соединением или компонентом, который включает, не ограничиваясь ими, алюминий (в частности, оксид алюминия) или диоксид кремния (в частности, кварцевое стекло). В некоторых вариантах осуществления изобретения неорганическим соединением является Al2O3 или SiO2.
В некоторых вариантах осуществления изобретения лиганд включает одну или несколько функциональных групп. Термин “функциональная группа” в использованном здесь значении означает химические группы, подгруппы или подструктуры, которые сообщают отличительные химические, физические или физико-химические признаки молекуле или веществу. Функциональные группы по настоящему изобретению включают, не ограничиваясь ими, гидрофильные группы, такие как положительно заряженные, отрицательно заряженные, незаряженные, нейтральные или гидрофобные группы. В объем настоящего изобретения также входят амфифильные или многофункционные функциональные группы. Функциональные группы включают органические или неорганические функциональные группы. Предпочтительные функциональные группы содержат аминогруппы, фенильные или сульфитные группы. Предпочтительной аминогруппой является ион первичного, вторичного, третичного или четвертичного аммония, такой как диметиламиноэтил (DMAE) или триметиламиноэтил (ТМАЕ).
Другие типичные функциональные группы включают, не ограничиваясь ими: -СН2-СНОН-CH2NH2, -C6H5, -(CH2)3-CH3, CH2-CH2-NH(C2H5)2, -SO2-CH2-CF3'-CH2-CH2-H(CH3)2, -CH2-CH2-(CH3)3, -SO2. Приемлемые функциональные группы дополнительно включают сульфонильные группы и трезильные группы. Следует отметить, что функциональные группы могут изначально присутствовать в лиганде или могут быть добавлены к лиганду в результате химической модификации.
В некоторых вариантах осуществления изобретения лиганд содержит аминогруппу. Такие лиганды включают, например, полиамин, такой как Toyopearl™ амино-650M, Toyopearl™ амино-AMN31, TSK-GEL™ амино-750С или их функциональные эквиваленты. В некоторых вариантах осуществления изобретения лиганд включает фенильную группу. Такие лиганды включают, например, TSK-GEL™ фенил-5PW или его функциональный эквивалент.
В некоторых вариантах осуществления изобретения лиганд является полимерным материалом, таким как хрооматографические смолы (например, полиамины), которые избирательно и специфически связываются с прионными белками. Примером хроматографических смол являются восстанавливающие прион смолы PRDT (ProMetic Biosciences, Ltd, 211 Cambridge Science Park, Milton Road, Cambridge, CB4 0WA, UK). В некоторых вариантах осуществления изобретения полимерный материал содержит одну или несколько функциональных групп, подобных вышеописанным функциональным группам. В некоторых вариантах осуществления изобретения лиганд является полимерным материалом с метакрилатным остовом, который включает, не ограничиваясь ими, коммерчески доступную смолу TSK, TOYOPEARL или FACTOGEL (Tosoh Bioscience, Montgomeryville, PA).
Другие лиганды, которые могут быть использованы в способах по настоящему изобретению, включают, например, лиганды, взаимодействующие с амилоидной бляшкой, например, Congo:E;Led (Ingrosso, L., et al., Congo Red: Prolongs the Incubation Period in Scrapie-infected Hamsters, Virology 69: 506-508 (1995)); 1,4-иод,4-дезоксидоксорубицин (Tagliavini, F., et al., Effectiveness of Anthracycline Against Experimental Prion Diseases in Syrian Hamsters. Science 276: 1119-1122 (1997)); амфотерицин 13, порфирины и фталоцианины (Priola, S.A., et al., Porphyrin and Phthalocyanine Antiscrapie Compounds, Science 287: 1503-1506 (2000)); металлы (Stocker et al., Biochemistry, 37, 7185-7193 (1998)); пептиды, взаимодействующие с PrP с образованием комплексов (смотрите патент США № 5750361, выданный Prusiner et al. and Solo, C. et al., Reversion of Prion Protein Conformational Changes in Synthetic p-sheet Breaker Peptides, Lances, 355: 192-197 (2000)); гепарин и другие полисульфатированные полианионы (Caughey, B., et al., Binding of the Protease-sensitive Form of Prion Protein PrP to Sulphated Glycosaminoglycan and Congo Red, J. Virology 68: 2135-2141 (1994)); антитела (Kascsak, R.J., et al., Immunodiagnosis of Prion Disease, Immunological Invest., 26: 259-268 (1997)); и другие белки, например, плазминоген (Fischer, M.B. et al., Binding of Disease-associated Prion Protein to: Plasminogen., Nature 408: 479-483 (2000)).
Лиганды могут быть присоединены к любым носителям. Термин “носитель” в использованном здесь значении означает любое соединение или вещество, которое является физическим или химическим средством отделения лиганда и связанных с ним белков от остальной композиции. Носитель может быть в форме частиц или не быть частицами, может быть растворимым или нерастворимым, пористым или непористым.
Примеры носителей включают, не ограничиваясь ими, природные полимеры, например, полисахарид, такой как агароза, альгинат, каррагенан, хитин, целлюлоза, декстран или крахмал; синтетические полимеры, такие как полиакриламид, полистирол, полиакролеин, поливиниловый спирт, полиметилакрилат, перфторуглерод; неорганические соединения, такие как кремнезем, стекло, кизельгур, глинозем, оксид железа и оксиды других металлов либо сополимеры, состоящие из любой комбинации двух или более природных полимеров, синтетических полимеров или неорганических соединений. В объем настоящего изобретения также входят растворимые носители, включающие такие полимеры как декстран, полиэтиленгликоль, поливиниловый спирт или гидролизованный крахмал, которые представляют собой конъюгаты аффинного лиганда с матрицей, предназначенные для разделения жидкостей.
В некоторых вариантах осуществления изобретения носитель является твердым носителем, таким как колонка, гранула, мембрана, картридж, фильтр, погружаемый стержень, титрационный микропланшет, пробирка, твердый порошок, литейный или экструзионный модуль, сито, композиционный материал из магнитных частиц или любые другие твердые материалы. Указанные твердые материалы могут быть покрыты таким веществом, как полиэтилен, полипропилен, поли(4-метилбутен), полистирол, полиакрилат, полиэтилентерефталат, гидратцеллюлозное волокно, найлон, поливинилбутират, поливинилидендифторид (PVDF), силиконы, полиформальдегид, целлюлоза, ацетат целлюлозы, нитроцеллюлоза и тому подобные. Альтернативно могут быть использованы вещества, образующие гель, такие как белки (например, желатины), липополисахариды, силикаты, агароза и полиакриламиды. Могут быть также использованы такие полимеры как декстраны, полиалкиленгликоли или поверхностно-активные вещества, такие как фосфолипиды, длинноцепные соли алкиламмония (12-24 атома углерода) и тому подобные. Лиганды присоединяют к носителям или диспергируют в носителях.
В некоторых вариантах осуществления изобретения носителем является активированная агароза, кремнезем, целлюлоза, стекло, метакрилат, гидроксиэтилметакрилат, полиакриламид, стиролдивинилбензол, Hyper D или перфторуглероды. В некоторых вариантах осуществления изобретения носитель является метакрилатом, продаваемым под торговым названием Toyopearl (компании Tosoh Bioscience LLC, 156 Keystone Drive, Montgomeryville, PA I 18936, USA). В публикации WO 97/10887 описаны методы присоединения аффинных лигандов к несущим матрицам, например, методы активации и присоединения аффинного лиганда к матрице с помощью спейсера, например, путем конденсации, с образованием конъюгатов аффинного лиганда с матрицей.
В некоторых вариантах осуществления изобретения лиганды и/или носители могут быть повторно использованы. В некоторых вариантах осуществления изобретения лиганды и/или носители предназначены только для однократного использования.
Способность лиганда связываться с прионом можно оценить, определяя инфекционный титр в лиганде. Например, полученную серию разведений эталонного исходного раствора исследуют, выполняя стандартный анализ методом вестерн-блоттинга. Титры, полученные при исследования эталонного исходного раствора, сравнивают с соответствующими титрами, полученными при выполнении биоанализа. Титры, полученные методом вестерн-блоттинга, затем можно преобразовать в инфекционные титры по формуле: Титр(биоанализ)=Титр(вестерн-блоттинг)+(отсечение значений линейной регрессии)/(наклон значений линейной регрессии). После вычисления инфекционного титра на мл можно определить общее содержание прионного белка, связанного в колонке. Способность приона связываться с лигандом определяют по количеству прионного белка, непосредственно связанного с лигандом. В некоторых вариантах осуществления изобретения способность лиганда связываться с прионом равна по меньшей мере примерно 2,0, 2,5, 3,0, 3,5, 4,0, 4,5, 5,0, 5,5, 6,0, 6,5, 7,0, 7,1, 7,2, 7,3, 7,4, 7,5, 7,6, 7,7, 7,8, 7,9, 8,0, 8,1, 8,2, 8,3, 8,4, 8,5, 8,6, 8,7, 8,8, 8,9, 9,0, 9,5 или 10,0 log10 ID50 на мл лиганда. В некоторых вариантах осуществления изобретения способность лиганда связываться с прионом равна по меньшей мере примерно 102, 103, 104, 105, 106, 107 или 108 ID50 на мл лиганда.
Способ получения наночастиц
В данной области известны методы получения композиций, содержащих альбумины и по существу не растворимые в воде фармакологически активные агенты. Например, наночастицы, включающие по существу не растворимые в воде фармакологически активные агенты и альбумины, могут быть получены в условиях высоких сдвигающих усилий (таких как обработка ультразвуком, гомогенизация под высоким давлением или тому подобные). Указанные методы описаны, например, в патентах США №№ 5916596, 6506405, 6749868 и 6537579, а также в публикациях патентов США №№ 2005/0004002 и 2007/0082838 и публикации РСТ WO00/00113, которые полностью включены в настоящее описание изобретения в качестве ссылки.
В одном типичном варианте осуществления изобретения по существу не растворимый в воде фармакологически активный агент (например, паклитаксел) растворяют в органическом растворителе. Приемлемые органические растворители включают, например, кетоны, сложные эфиры, простые эфиры, хлорсодержащие растворители и другие растворители, известные в данной области. Например, органическим растворителем может быть метиленхлорид. В некоторых вариантах осуществления изобретения органическим растворителем может быть смесь не смешивающегося с водой растворителя (такого как хлороформ) и смешивающегося с водой растворителя (такого как смешивающийся с водой спирт, в частности, хлороформ/метанол, хлороформ/этанол, хлороформ/пропанол или хлороформ/бутанол (например, в соотношении (об./об.) примерно 1:9, 1:8, 1:7, 1:6, 1:5, 1:4, 1:3, 1:2, 1:1, 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1 или 9:1 или в соотношении (об./об.) примерно 3:7, 5:7, 4:6, 5:5, 6:5, 8:5, 9:5, 9,5:5, 5:3, 7:3, 6:4 или 9,5:0,5). Полученный раствор добавляют к альбумину (например, сывороточному альбумину человека). Смесь гомогенизируют под высоким давлением (например, в стандартных устройствах для гомогенизации). Эмульсия может быть пропущена через гомогенизатор высокого давления на протяжении от примерно 2 до примерно 100 циклов, например, от примерно 5 до примерно 50 циклов или от примерно 8 до примерно 20 циклов (например, на протяжении примерно 8, 10, 12, 14, 16, 18 или 20 циклов). Органический растворитель затем может быть удален выпариванием в известном оборудовании, предназначенном для данной цели, которое включает, не ограничиваясь им, роторные испарители, пленочные испарители, испарители с падающей пленкой, испарители с очищаемой пленкой, распылительные сушилки и тому подобные. Растворитель может быть удален, например, при пониженном давлении (например, примерно при 5 мм Hg, 10 мм Hg, 15 мм Hg, 20 мм Hg, 25 мм Hg, 30 мм Hg, 40 мм Hg, 50 мм Hg, 100 мм Hg, 200 мм Hg или 300 мм Hg). Продолжительность удаления растворителя при пониженном давлении можно отрегулировать на основании объема композиции. Например, для композиции объемом 300 мл растворитель может быть удален под давлением от примерно 1 до примерно 300 мм Hg (например, примерно 5-100 мм Hg, 10-50 мм Hg, 20-40 мм Hg или 25 мм Hg) в течение от примерно 1 минуты до примерно 120 минут, в том числе от примерно 5 минут до примерно 60 минут (например, в течение примерно 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 18, 20, 25 или 30 минут).
Раствор альбумина (например, раствор альбумина после удаления приона) при желании может быть добавлен к дисперсии для регулирования соотношения альбумина и лекарственного средства (например, паклитаксела) или для регулирования концентрации таксана (например, паклитаксела) в дисперсии. Например, раствор альбумина (например, 25% масс./об.) может быть добавлен для доведения соотношения альбумина и по существу не растворимого в воде фармакологически активного агента (например, паклитаксела) примерно до 18:1, 15:1, 14:1, 13:1, 12:1, 11:1, 10:1, 9:1, 8:1, 7,5:1, 7:1, 6:1, 5:1, 4:1, 3:1, 2:1, 1:1, 1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8, 1:9, 1:10, 1:11, 1:12, 1:13, 1:14, 1:15, 1:16, 1:17 или 1:18. Например, раствор альбумина (например, 25% масс./об.) или другой раствор добавляют для доведения концентрации по существу не растворимого в воде фармакологически активного агента (например, паклитаксела) в дисперсии до примерно 0,5 мг/мл, 1,3 мг/мл, 1,5 мг/мл, 2 мг/мл, 3 мг/мл, 4 мг/мл, 5 мг/мл, 6 мг/мл, 7 мг/мл, 8 мг/мл, 9 мг/мл, 10 мг/мл, 15 мг/мл, 20 мг/мл, 25 мг/мл, 30 мг/мл, 40 мг/мл или 50 мг/мл. Дисперсия может быть отфильтрована через один или несколько фильтров, таких как 1,2 мкм, 0,8 мкм, 0,45 мкм и 0,22 мкм фильтры; через комбинацию двух или более фильтров или комбинацию любых других фильтров, известных в данной области.
В композицию при желании может быть также введено средство для вторичной терапии (например, одно или несколько соединений, используемых для лечения рака), антимикробный агент (такой как цитрат или эдетат), сахар (такой как сахароза) и/или стабилизирующий агент. Указанный дополнительный агент может быть смешан с по существу не растворимым в воде фармакологически активным агентом (например, паклитакселом) и/или альбумином в процессе получения композиции либо добавлен после получения композиции на основе наночастиц. В некоторых вариантах осуществления изобретения указанный агент смешивают с композицией на основе наночастиц до лиофилизации. В некоторых вариантах осуществления изобретения указанный агент добавляют к лиофилизованной композиции. В некоторых вариантах осуществления изобретения в том случае, если добавление указанного агента изменяет значение рН композиции, показатель рН в композиции обычно (но не обязательно) доводят до желаемого значения рН. Типичные значения рН композиции, например, находятся в пределах от примерно 5 до примерно 8,5. В некоторых вариантах осуществления изобретения значение рН композиции доводят не менее, чем до примерно 6, в том числе, например, не менее, чем до примерно 6,5, 7 или 8 (например, примерно 8).
Как подробно описано ниже, процесс удаления приона может быть выполнен одновременно в процессом приготовления композиции. Например, процесс удаления приона может быть выполнен после получения смеси альбумина и по существу не растворимого в воде фармакологически активного агента и до воздействия на смесь высокого сдвигающего усилия. В некоторых вариантах осуществления изобретения процесс удаления приона может быть выполнен после воздействия на смесь высокого сдвигающего усилия и до удаления органического растворителя. В некоторых вариантах осуществления изобретения процесс удаления приона выполняют после удаления органического растворителя. В некоторых вариантах осуществления изобретения дополнительный альбумин к суспензии после выпаривания до выполнения процесса удаления приона. В некоторых вариантах осуществления изобретения процесс удаления приона выполняют в стерильных условиях. В некоторых вариантах осуществления изобретения процесс удаления приона выполняют, используя картридж, который одновременно стерилизует композицию фильтрованием.
Способ удаления прионов из композиций на основе наночастиц или промежуточных композиций
Одним объектом настоящего изобретения являются способы удаления прионных белков из композиций на основе наночастиц, полученных вышеописанными способами. Другим объектом изобретения являются способы удаления прионных белков из промежуточных композиций, образованных в процессе получения наночастиц (дополнительно именуемых “промежуточная композиция”).
Указанные способы обычно включают приведение композиции, включающей альбумин и по существу не растворимый в воде фармакологически активный агент, в контакт с лигандом, способным связываться с прионным белком, и удаление лиганда и связанного с ним белка из композиции на основе наночастиц. Указанный процесс может быть повторен один или несколько раз при использовании одного и того же или другого лиганда. В процессе удаления приона могут быть одновременно использованы два или более лигандов.
В некоторых вариантах осуществления изобретения композиция на основе наночастиц или промежуточная композиции характеризуется инфективностью приона примерно 100 ин.ед.-иц/мл, 90 ин.ед.-иц/мл, 50 ин.ед.-иц/мл или 10 ин.ед.-иц/мл.
В процессе удаления приона лиганд вводят в соприкосновение с композицией на основе наночастиц или промежуточной композицией и оставляют для связывания с прионными белками в композиции на основе наночастиц или промежуточной композиции. Условия, приемлемые для связывания, можно определить и оптимизировать для облегчения связывания лиганда с прионным белком в зависимости от природы лиганда и его специфичности связывания с прионным белком. В некоторых вариантах осуществления изобретения связывание выполняют при температуре от примерно 0°С до примерно 39°С, в том числе, например, от примерно 20°С до примерно 25°С. Связывание можно выполнять при значении рН от примерно 4 до примерно 10, в том числе, например, от примерно 5 до примерно 9, от примерно 6 до примерно 8, от примерно 6,8 до примерно 7,5, от примерно 6,9 до примерно 7,4 или примерно 7. Для уменьшения неспецифического связывания с лигандом могут быть необязательно использованы блокирующие агенты.
После стадии приведения в контакт лиганд и связанные с ним белки удаляют из остальной композиции. Удаление может быть выполнено разными способами в зависимости от природы лиганда и носителя (при его наличии). Например, лиганд и связанные с ним белки могут быть отделены при помощи хроматографии, которая включает, не ограничиваясь ими, тонкослойную хроматографию, хроматографию на колонке и периодическую хроматографию; путем разделения на мембране и твердом носителе; разделения в реакторе; разделения в магнитном поле; иммуноразделения и коллоидного разделения.
В некоторых вариантах осуществления изобретения лиганд может быть иммобилизован на носителе, таком как гранула или мембрана, который в свою очередь приводит в контакт с композицией на основе наночастиц или промежуточной композицией. Носитель с иммобилизованным лигандом оставляют приводить в контакт с композицией на основе наночачстиц или промежуточной композицией в условиях, необходимых для образования комплекса прион-лиганд. Твердую фазу затем отделяют от композиции, удаляя из образца прионный белок, связанный с лигандом. Например, в одном типичном варианте осуществления изобретения лиганды иммобилизуют на колонке, такой как хроматографическая колонка, образец (такой как композиция на основе наночастиц) пропускают через колонку под воздействием силы тяжести или под давлением, как это имеет место в колонке для жидкостной хроматографии высокого давления. Прионные белки в образце связываются с лигандом, иммобилизованным на колонке, и образец, проходящий через колонку, собирают. Указанный процесс можно повторить несколько раз до достижения требуемого результата.
Скорость потока образца (такого как композиция на основе наночастиц или промежуточная композиция) в колонке можно отрегулировать так, чтобы максимально увеличить связывание лиганда с прионными белками в образце. В некоторых вариантах осуществления изобретения связывание выполняют при скорости потока от примерно 0,1 мл/мин до примерно 5,0 мл/мин, от примерно 0,1 мл/мин до примерно 2,5 мл/мин, от примерно 0,1 мл/мин до примерно 0,25 мл/мин, от примерно 0,25 мл/мин до примерно 0,5 мл/мин, от примерно 0,5 мл/мин до примерно 1,0 мл/мин, от примерно 1,0 мл/мин до примерно 1,5 мл/мин, от примерно 1,5 мл/мин до примерно 2,0 мл/мин, от примерно 2,0 мл/мин до примерно 2,5 мл/мин, от примерно 2,5 мл/мин до примерно 3,0 мл/мин, от примерно 3,0 мл/мин до примерно 3,5 мл/мин, от примерно 3,5 мл/мин до примерно 4,0 мл/мин, от примерно 4,0 мл/мин до примерно 4,5 мл/мин или от примерно 4,5 мл/мин до примерно 5,0 мл/мин, в том числе, например, при скорости потока, равной примерно 0,1 мл/мин, 0,25 мл/мин, 0,5 мл/мин, 1,0 мл/мин, 1,5 мл/мин, 1,7 мл/мин, 1,8 мл/мин, 1,9 мл/мин, 2,0 мл/мин, 2,1 мл/мин, 2,3 мл/мин, 2,5 мл/мин, 2,7 мл/мин, 3,0 мл/мин, 3,5 мл/мин, 4,0 мл/мин, 4,5 мл/мин или 5,0 мл/мин. В некоторых вариантах осуществления изобретения скорость потока равна по меньшей мере примерно 10 мл/мин, в частности, по меньшей мере примерно 20 мл/мин, 30 мл/мин, 40 мл/мин, 50 мл/мин.
Общий объем проходящего потока или общее время прохождения потока в процессе связывания также можно отрегулировать так, чтобы максимально увеличить связывание лиганда с прионными белками в образце (таком как композиция на основе наночастиц или промежуточная композиция). В некоторых вариантах осуществления изобретения общий объем проходящего потока составляет от примерно 50-кратного до примерно 1000-кратного объема колонки, в том числе, например, от примерно 50-кратного до примерно 500-кратного объема колонки, от примерно 100-кратного до примерно 600-кратного объема колонки или от примерно 200-кратного до примерно 800-кратного объема колонки. В некоторых вариантах осуществления изобретения общий объем проходящего потока равен примерно 100-кратному объему колонки. В других вариантах осуществления изобретения общий объем проходящего потока равен примерно 500-кратному объему колонки. В некоторых вариантах осуществления изобретения общее время прохождения потока составляет от примерно 1 часа до примерно 30 часов, в том числе, например, от примерно 2 часов до примерно 25 часов, от примерно 3 часов до примерно 20 часов или от примерно 3 часов до примерно 17 часов. В некоторых вариантах осуществления изобретения общее время прохождения потока составляет примерно 3 часа, 4 часа, 6 часов, 8 часов, 10 часов, 12 часов, 14 часов, 16 часов, 17 часов, 18 часов, 19 часов или 20 часов. В некоторых вариантах осуществления изобретения общее время прохождения потока составляет более примерно 24 часов. В некоторых вариантах осуществления изобретения общее время прохождения потока составляет менее примерно 8 часов, в том числе, например, 7, 6, 5, 4, 3, 2, 1 или 0,5 часа.
Альтернативно лиганд сначала может быть введен в соприкосновение с композицией на основе наночастиц или промежуточной композицией в условиях, обеспечивающих образование комплекса прион-лиганд. Комплекс прион-лиганд затем удаляют, используя колонку, такую как колонка для аффинной хроматографии. Для облегчения отделения лиганд может быть конъюгирован с партнером связывания, благодаря чему комплекс прион-лиганд может быть удален при использовании колонки для аффинной хроматографии, содержащей молекулу, узнающую данный партнер связывания.
Помимо периодической хроматографии или хроматографии на колонке, существует целый ряд других конфигураций, модификаций и вариантов использования лигандов для связывания прионных белков. Такие варианты и модификации включают, не ограничиваясь ими: периодические процессы, непрерывные процессы, хроматографию в подвижном слое; процессы, выполняемые при низком, среднем или высоком давлении; лабораторные, среднемасштабные или крупномасштабные процессы. В некоторых вариантах осуществления изобретения лиганды наносят на мембрану, гранулы, погружают в нетканое сито или наносят на покрывающие волокна, находящиеся в корпусе фильтра.
В некоторых вариантах осуществления изобретения стадия удаления не вызывает значительных потерь продукта и/или изменений свойства и/или устойчивости альбумина. В некоторых вариантах осуществления изобретения восстановление альбумина в первоначальном биологическом состоянии превышает по меньшей мере 50% и составляет, например, 80%, 90% или больше. В некоторых вариантах осуществления изобретения степень восстановления альбумина в результате выполнения процесса удаления приона составляет более примерно 80%, 90%, 95% или 99%. В некоторых вариантах осуществления изобретения концентрация альбумина в композиции на основе наночастиц регулируют или контролируют до стадии удаления приона с тем, чтобы свести к минимуму неспецифическое связывание и потерю альбумина при выполнении данного процесса. Например, концентрация альбумина может находиться в пределах от примерно 1% до примерно 50%, от примерно 5% до примерно 25%, от примерно 5% до примерно 30%, от примерно 5% до примерно 40%, от примерно 5% до примерно 10%, от примерно 10% до примерно 15%, от примерно 15% до примерно 20%, от примерно 20% до примерно 25%, от примерно 25% до примерно 30% и т.д. и, например, может быть равна примерно 5%, 10%, 15%, 20%, 25% или 30% альбумина.
В некоторых вариантах осуществления изобретения стадия удаления не вызывает значительной потери продукта, изменения свойства или устойчивости наночастиц в композиции.
В некоторых вариантах осуществления изобретения стадия удаления не вызывает значительной потери продукта и/или изменения свойств или содержания по существу не растворимого в воде фармакологически активного агента в композиции.
В некоторых вариантах осуществления изобретения стадия удаления не вызывает значительного изменения соотношения альбумина и по существу не растворимого в воде фармакологического агента в композиции.
После удаления приона композиция может быть подвергнута анализу для определения степени очистки в процессе удаления приона. Лиганд со связанными прионными белками также может быть подвергнут анализу (сразу же или после элюирования) для определения степени очистки.
Удаление прионного белка можно определить с учетом уменьшения содержания прионного белка и/или уменьшения инфективности. В некоторых вариантах осуществления изобретения из композиции на основе наночастиц может быть удалено по меньшей мере примерно 50%, в том числе, например, 60%, 70%, 80%, 90%, 95%, 99% или 100% прионных белков. В некоторых вариантах осуществления изобретения инфективность композиции альбумина после удаления приона по меньшей мере примерно в 10 раз, 20 раз, 30 раз, 40 раз, 50 раз, 80 раз, 100 раз, 200 раз, 500 раз, 1000 раз, 104 раз, 105 раз, 106 раз меньше, чем в исходной композиции на основе наночастиц.
В некоторых вариантах осуществления изобретения для определения степени очистки композиции в процессе удаления приона используют инфективность серийных разведений. Серийные разведения образцов исследуют в отношении инфекционной активности, например, с использованием экспериментальных животных. Разведение, при котором инфицируется половина животных, является инфекционным титром. Например, при 5-кратном разведении инфективность образца может быть равна 5 log. Сравнивая число log инфективности исходной композиции на основе наночастиц и композиции на основе наночастиц после удаления приона, можно определить степень очистки в процессе удаления приона. В некоторых вариантах осуществления изобретения указанный способ удаления приона позволяет уменьшить инфективность на 1, 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9, 2, 3 или 4, 5, 6, 7, 8, 9 или 10 log.
В некоторых вариантах осуществления изобретения степень очистки в процессе удаления приона определяют при помощи экспериментов по потенцированию с использованием инфекционных материалов, выполняя стадии, рассмотренные в настоящем описании изобретения для данного способа удаления приона. Приемлемые потенцирующие агенты включают, не ограничиваясь ими, гомогенаты головного мозга, микросомы, ямкоподобные домены, очищенный PrPsc и фибриллы приона. В некоторых вариантах осуществления изобретения потенцирующий агент растворяют в детергенте (например, в саркозиле). В некоторых вариантах осуществления изобретения содержание потенцирующего агента в композиции находится в пределах от примерно 0,001% до примерно 5%, от примерно 0,001% до примерно 0,25%, от примерно 0,001% до примерно 0,1%, от примерно 0,001% до примерно 0,005%, от примерно 0,005% до примерно 0,075%, от примерно 0,075% до примерно 0,01%, от примерно 0,01% до примерно 0,1%, от примерно 0,1% до примерно 0,5%, от примерно 0,5% до примерно 0,75%, от примерно 0,75% до примерно 1%, от примерно 1% до примерно 2%, от примерно 2% до примерно 3% или от примерно 3% до примерно 5% и, например, составляет примерно 0,001%, 0,005%, 0,075%, 0,01%, 0,1%, 0,5%, 0,75%, 1%, 2%, 3% или 5%.
В некоторых вариантах осуществления изобретения для определения степени очистки в процессе удаления приона используют коэффициенты восстановления (RF). RF можно вычислить по формуле:
RF=(V1×T1)/(V2×T2)
или
Log10[RF]=[Log10(V1)+Log10(T1)]-[Log10(V2)+Log10(T2)],
где V1 и Т1 означают соответственно объем и титр исходной композиции альбумина, V2 и Т2 означают объем и титр композиции альбумина после удаления приона. Коэффициенты восстановления могут быть округлены до одной десятой после окончательного вычисления. В некоторых вариантах осуществления изобретения инфективность прионных белков может быть уменьшена в исходной композиции альбумина на коэффициент восстановления, равный по меньшей мере примерно 1,0, 1,5, 2,0, 2,5, 3,0, 3,5, 4,0, 4,5, 5,0, 5,5, 6,0, 6,5, 7,0, 7,5 или 8,0 log10. Например, содержание прионного белка может быть уменьшено на коэффициент восстановления, который больше или равен 2,5 log10 во фракции, растворенной в 0,5% саркозиле, которой была потенцирована композиция, содержащая 20% альбумина. В качестве другого примера можно отметить, что содержание прионного белка может быть уменьшено на коэффициент восстановления, который больше или равен 2,0 log10 во фракции, растворенной в 0,5% саркозиле, которой была потенцирована композиция, содержащая 25% альбумина.
Удаление прионов можно определить при помощи стандартного анализа методом вестерн-блоттинга. Например, лиганды после связывания с прионом сначала обрабатывают протеиназой К, которая расщепляет весь PrPc и не расщепляет PrPsc. Гидролизат затем исследуют на геле с додецилсульфатом натрия (SDS) и переносят на лист нитроцеллюлозы или мембрану из PVDF. Выделенные полосы PrPsc затем визуализируют, используя 3F4 или 6Н4. 3F4 взаимодействует с аминокислотными остатками 109-112 PrP человека, хомячков и кошек. В одном типичном варианте осуществления изобретения инкубацию выполняют при концентрации 0,6 мкг/мл в течение минимум одного часа, после чего избыток антитела вымывают и мембраны инкубируют с конъюгатом пероксидазы из хрена с антителом кролика против мыши (степень разведения 1:1000) в течение минимум одного часа. После обильной промывки TTBS мембраны проявляют при повышенной хемилюминесценции. В некоторых вариантах осуществления изобретения удаление прионных белков оценивают в соответствии с указаниями, приведенными в публикации Guideline for the Investigation of Manufacturing Processes for Plasma-Derived Medicinal Products with Regard to vCJD Risk (CPMP5136/03).
В объем настоящего изобретения также входят композиции, полученные в процессе удаления приона. Так, некоторые варианты осуществления изобретения относятся к композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, которая дополнительно включает лиганд, способный связываться с прионным белком. Некоторые варианты осуществления изобретения относятся к смеси, содержащей наночастицы, включающие альбумин, по существу не растворимый в воде фармакологически активный агент и лиганд, способный связываться с прионным белком, присоединенным к носителю, такому как один или несколько носителей, рассмотренных в настоящем описании изобретения. Некоторые варианты осуществления изобретения относятся к колонке, в которую вводят композицию, содержащую наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, при этом в указанной колонке находится лиганд, способный связываться с прионным белком. Некоторые варианты осуществления изобретения относятся к композиции, содержащей наночастицы, включающие альбумин, по существу не растворимый в воде фармакологически активный агент и смолу, содержащую лиганд, способный связываться с прионным белком. Например, некоторые варианты осуществления изобретения относятся к композиции, содержащей наночастицы, включающие альбумин, по существу не растворимый в воде фармакологически активный агент и смолу DVR. Некоторые варианты осуществления изобретения относятся к композиции, содержащей наночастицы, включающие альбумин, по существу не растворимый в воде фармакологически активный агент и смолу PRDT.
Некоторые варианты осуществления изобретения относятся к композиции, содержащей смесь водного раствора альбумина и по существу не растворимого в воде фармакологически активного агента, диспергированного в органическом растворителе, которая дополнительно включает лиганд, способный связываться с прионным белком. Некоторые варианты осуществления изобретения относятся к композиции, содержащей смесь водного раствора альбумина, по существу не растворимого в воде фармакологически активного агента, диспергированного в органическом растворителе, и лиганд, способный связываться с прионным белком, который присоединен к носителю, такому как один или несколько носителей, рассмотренных в настоящем описании изобретения. Некоторые варианты осуществления изобретения относятся к колонке, в которую вводят композицию, содержащую смесь водного раствора альбумина и по существу не растворимого в воде фармакологически активного агента, диспергированного в органическом растворителе, при этом в указанной колонке находится лиганд, способный связываться с прионным белком.
Настоящее изобретение относится также к композициям, полученным способами по настоящему изобретению. В некоторых вариантах осуществления изобретения такая композиция биологически эквивалента композиции, которую не подвергали процессу удаления приона.
Композиции на основе наночастиц
Композиции на основе наночастиц по настоящему изобретению содержат наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент (такой как паклитаксел) (которые в разных вариантах осуществления изобретения с основном состоят из указанных компонентов). Наночастицы плохо растворимых в воде лекарственных средств (таких как таксан) описаны, например, в патентах США №№ 5916596, 6506405, 6749868 и 6537579, а также в публикациях патентов США №№ 2005/0004002 и 2007/0082838 и заявке на патент РСТ WO08/137148, которые полностью включены в настоящее описание изобретения в качестве ссылки.
В некоторых вариантах осуществления изобретения композиция содержит наночастицы со средним диаметром не более примерно 1000 нанометров (нм), в частности, не более примерно 900, 800, 700, 600, 500, 400, 300, 200 и 100 нм. В некоторых вариантах осуществления изобретения средний диаметр наночастиц не превышает примерно 200 нм. В некоторых вариантах осуществления изобретения средний диаметр наночастиц не превышает примерно 150 нм. В некоторых вариантах осуществления изобретения средний диаметр наночастиц не превышает примерно 100 нм. В некоторых вариантах осуществления изобретения средний диаметр наночастиц равен от примерно 20 до примерно 400 нм. В некоторых вариантах осуществления изобретения средний диаметр наночастиц равен от примерно 40 до примерно 200 нм. В некоторых вариантах осуществления изобретения наночастицы стерилизуют фильтрованием.
В некоторых вариантах осуществления изобретения средний диаметр наночастиц в композиции по настоящему изобретению не превышает примерно 200 нм, в том числе, например, не превышает примерно 190, 180, 170, 160, 150, 140, 130, 120, 110, 100, 90, 80, 70 или 60 нм. В некоторых вариантах осуществления изобретения по меньшей мере примерно 50% (например, по меньшей мере примерно 60%, 70%, 80%, 90%, 95% или 99%) всех наночастиц в композиции имеет диаметр не более примерно 200 нм, в том числе, например, не более примерно 190, 180, 170, 160, 150, 140, 130, 120, 110, 100, 90, 80, 70 или 60 нм. В некоторых вариантах осуществления изобретения по меньшей мере примерно 50% (например, по меньшей мере 60%, 70%, 80%, 90%, 95% или 99%) всех наночастиц в композиции имеют диаметр в пределах от примерно 20 до примерно 200 днм, в том числе, например, от примерно 30 до примерно 180, от примерно 40 до примерно 150, от примерно 50 до примерно 120 и от примерно 60 до примерно 100 нм.
В некоторых вариантах осуществления изобретения по меньшей мере примерно 5% (в том числе, например, по меньшей мере примерно 10%, 15%, 20%, 25%, 30%, 40%, 50%, 60%, 70%, 80% или 90%) альбумина в наночастицах композиции перекрестно связаны (например, перекрестно связаны одной или несколькими дисульфидными связями).
В некоторых вариантах осуществления изобретения наночастицы включают по существу не растворимый в воде фармакологически активный агент (такой как паклитаксел), покрытый альбумином (например, сывороточным альбумином человека). В некоторых вариантах осуществления изобретения композиция содержит по существу не растворимый в воде фармакологически активный агент в форме, отличной от наночастиц, при этом по меньшей мере примерно 50%, 60%, 70%, 80%, 90%, 95% или 99% по существу не растворимого в воде фармакологически активного агента в композиции имеет форму наночастиц. В некоторых вариантах осуществления изобретения по существу не растворимый в воде фармакологически активный агент в наночастицах составляет более примерно 50%, 60%, 70%, 80%, 90%, 95% или 99% наночастиц в расчете на массу. В некоторых вариантах осуществления изобретения наночастицы имеют неполимерную матрицу. В некоторых вариантах осуществления изобретения наночастицы включают сердцевину из по существу не растворимого в воде фармакологически активного агента, в которой по существу отсутствуют полимерные материалы (такие как полимерная матрица).
В некоторых вариантах осуществления изобретения композиция содержит альбумин как в форме наночастиц, так и форме, отличной от наночастиц, при этом по меньшей мере примерно 50%, 60%, 70%, 80%, 90%, 95% или 99% альбумина в композиции имеет форму, отличную от наночастиц.
В некоторых вариантах осуществления изобретения композиция на основе наночастиц по существу не содержит поверхностно-активных веществ (таких как кремофор®, твин-80 или другие органические растворители, используемые для введения по существу не растворимых в воде фармакологически активных агентов). В некоторых вариантах осуществления изобретения композиция на основе наночастиц содержит менее примерно 20%, 15%, 10%, 7,5%, 5%, 2,5%, 1% или меньше органического растворителя. В некоторых вариантах осуществления изобретения массовое соотношение альбумина и по существу не растворимого в воде фармакологически активного агента в композиции на основе наночастиц составляет примерно 18:1 или меньше, в частности, примерно 15:1 или меньше, например, примерно 10:1 или меньше. В некоторых вариантах осуществления изобретения массовое соотношение альбумина и по существу не растворимого в воде фармакологически активного агента в композиции находится в пределах от примерно 1:1 до примерно 18:1, от примерно 2:1 до примерно 15:1, от примерно 3:1 до примерно 13:1, от примерно 4:1 до примерно 12:1, от примерно 5:1 до примерно 10:1. В некоторых вариантах осуществления изобретения массовое соотношение альбумина и по существу не растворимого в воде фармакологически активного агента в наночастицах композиции составляет примерно 1:2, 1:3, 1:4, 1:5, 1:10, 1:15 или меньше. В некоторых вариантах осуществления изобретения массовое соотношение альбумина (такого как сывороточный альбумин человека) и по существу не растворимого в воде фармакологически активного агента в композиции находится в следующих пределах: от примерно 1:1 до примерно 18:1, от примерно 1:1 до примерно 15:1, от примерно 1:1 до примерно 12:1, от примерно 1:1 до примерно 10:1, от примерно 1:1 до примерно 9:1, от примерно 1:1 до примерно 8:1, от примерно 1:1 до примерно ?:1, от примерно 1:1 до примерно 6:1, от примерно 1:1 до примерно 5:1, от примерно 1:1 до примерно 4:1, от примерно 1:1 до примерно 3:1, от примерно 1:1 до примерно 2:1 или равно примерно 1:1.
В некоторых вариантах осуществления изобретения композиция на основе наночастиц имеет один или несколько вышеуказанных отличительных признаков.
Наночастицы по настоящему изобретению могут находиться в сухом препарате (таком как лиофилизованная композиция) или могут быть суспендированы в биологически совместимой среде. Приемлемые биологически совместимые среды включают, не ограничиваясь ими, воду, забуференную водную среду, физиологический раствор, забуференный физиологический раствор, необязательно забуференные растворы аминокислот, необязательно забуференные растворы белков, необязательно забуференные растворы сахаров, необязательно забуференные растворы витаминов, необязательно забуференные растворы синтетических полимеров, эмульсии, содержащие липиды, и тому подобные.
В некоторых вариантах осуществления изобретения фармацевтически приемлемый носитель включает сывороточный альбумин человека. Сывороточный альбумин человека (HSA) является хорошо растворимым глобулярным белком Mr 65K и состоит из 585 аминокислот. HSA присутствует в плазме в наибольшем количестве и определяет 70-80% коллоидного осмотического давления плазмы человека. Аминокислотная последовательность HSA содержит в общей сложности 17 дисульфидных мостиков, один свободный тиол (Cys 34) и один триптофан (Trp 214). Было установлено, что внутривенное введение раствора HSA позволяет предотвратить и лечить гиповолемический шок и может быть использовано в сочетании с обменным переливанием крови при лечении неонатальной гипербилирубинемии. В объем настоящего изобретения входят также другие альбумины, такие как сывороточный альбумин коровы. Альбумины, отличные от человеческих, могут быть использованы в композициях, предназначенных для лечения млекопитающих, отличных от человека, например, в ветеринарии (в том числе при лечении домашних и сельскохозяйственных животных).
Сывороточный альбумин человека (HSA) имеет множество гидрофобных сайтов связывания (в общей сложности восемь для жирных кислот, эндогенного лиганда HSA) и связывает разные по существу не растворимые в воде фармакологически активные агенты, в частности, нейтральные и отрицательно заряженные гидрофобные соединения. В субдоменах IIA и IIIA HSA было предположено наличие двух высокоаффинных сайтов связывания, представляющих собой сильно удлиненные гидрофобные карманы с заряженными остатками лизина и аргинина около поверхности, которые действует в качестве точек присоединения для полярных лигандов.
Альбумин в композиции обычно служит в качестве носителя для по существу не растворимого в воде фармакологически активного агента, то есть альбумин в композиции делает по существу не растворимый в воде фармакологически активный агент более суспендируемым к водной среде или помогает сохранять суспензию по сравнению с композициями, не содержащими альбумин. Таким образом можно избежать использования токсичных растворителей (или поверхностно-активных веществ) для растворения по существу не растворимого в воде фармакологически активного агента и уменьшить число побочных эффектов, возникающих при введении по существу не растворимого в воде фармакологически активного агента субъекту (такому как человек). Так, в некоторых вариантах осуществления изобретения композиция по настоящему изобретению по существу не содержит поверхностно-активных веществ, таких как кремофор (в том числе кремофор EL® (BASF)). В некоторых вариантах осуществления изобретения композиция на основе наночастиц по существу не содержит поверхностно-активных веществ. Композиция “по существу не содержит кремофор” или “по существу не содержит поверхностно-активное вещество” в том случае, если количество кремофора или поверхностно-активного вещества в композиции не вызывает одного или нескольких побочных эффектов при введении субъекту композиции на основе наночастиц.
Количество альбумина в композиции по настоящему изобретению является различным в зависимости от наличия других компонентов в композиции. В некоторых вариантах осуществления изобретения композиция содержит альбумин в количестве, достаточном для стабилизации по существу не растворимого в воде фармакологически активного агента в водной суспензии, например, в форме устойчивой коллоидной суспензии (такой как устойчивая суспензия наночастиц). В некоторых вариантах осуществления изобретения альбумин использован в количестве, уменьшающем скорость осаждения по существу не растворимого фармакологически активного агента в водной среде. Для композиций, содержащих частицы, количество альбумина также зависит от размера и плотности наночастиц по существу не растворимого в воде фармакологически активного агента.
По существу не растворимый в воде фармакологически активный агент “стабилизирован” в водной суспензии, если он остается суспендированным в водной среде (без видимой преципитации или седиментации) в течение длительного периода времени, например, в течение по меньшей мере, 0,1, 0,2, 0,25, 0,5, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 24, 36, 48, 60 или 72 часов. Указанная суспензия обычно, но не обязательно, предназначена для введения субъекту (такому как человек). Устойчивость суспензии обычно (но не обязательно) определяют при температуре хранения (такой как комнатная температура (например, 20-25°С) или в условиях охлаждения (например 4°С). Например, суспензия является устойчивой при комнатной температуре при отсутствии флокуляции или агломерации частиц, видимой невооруженным глазом или наблюдаемой при помощи оптического микроскопа с 1000-кратным увеличением, в течение примерно пятнадцати минут после получения суспензии. Устойчивость также определить в условиях ускоренного испытания при температуре выше примерно 40°С.
В некоторых вариантах осуществления изобретения альбумин присутствует в количестве, достаточном для стабилизации по существу не растворимого в воде фармакологически активного вещества в водной суспензии в определенной концентрации. Например, концентрация по существу не растворимого в воде фармакологически активного агента в композиции составляет от примерно 0,1 до примерно 100 мг/мл, в том числе, например, от примерно 0,1 до примерно 50 мг/мл, от примерно 0,1 до примерно 20 мг/мл, от примерно 1 до примерно 10 мг/мл, от примерно 2 мг/мл до примерно 8 мг/мл, от примерно 4 до примерно 6 мг/мл, примерно 5 мг/мл. В некоторых вариантах осуществления изобретения концентрация по существу не растворимого в воде фармакологически активного агента равна по меньшей мере примерно 1,3 мг/мл, 1,5 мг/мл, 2 мг/мл, 3 мг/мл, 4 мг/мл, 5 мг/мл, 6 мг/мл, 7 мг/мл, 8 мг/мл, 9 мг/мл, 10 мг/мл, 15 мг/мл, 20 мг/мл, 25 мг/мл, 30 мг/мл, 40 мг/мл и 50 мг/мл. В некоторых вариантах осуществления изобретения альбумин присутствует в количестве, позволяющем избежать использования поверхностно-активных веществ (таких как кремофор), поэтому композиция не содержит или по существу не содержит поверхностно-активного вещества (такого как кремофор).
В некоторых вариантах осуществления изобретения композиция в жидкой форме содержит от примерно 0,1% до примерно 50% (масс./об.) (например, примерно 0,5% (масс./об.), примерно 5% (масс./об.), примерно 10% (масс./об.), примерно 15% (масс./об.), примерно 20% (масс./об.), примерно 25% (масс./об.), примерно 30% (масс./об.), примерно 40% (масс./об.) или примерно 50% (масс./об.) альбумина. В некоторых вариантах осуществления изобретения композиция в жидкой форме содержит от примерно 0,5% до примерно 5% (масс./об.) альбумина.
В некоторых вариантах осуществления изобретения массовое соотношение альбумина и по существу не растворимого в воде фармакологически активного агента в композиции на основе наночастиц делает возможным связывание или перенос клеткой достаточного количества по существу не растворимого в воде фармакологически активного агента. Несмотря на то, что массовое соотношение альбумина и по существу не растворимого в воде фармакологически активного агента может быть оптимизировано для разных комбинаций альбумина и по существу не растворимого в воде фармацевтически активного агента, массовое соотношение альбумина и по существу не растворимого в воде фармакологически активного агента (масс./масс.) обычно составляет от примерно 0,01:1 до примерно 100:1, от примерно 0,02:1 до примерно 50:1, от примерно 0,05:1 до примерно 20:1, от примерно 0,1:1 до примерно 20:1, от примерно 1:1 до примерно 18:1, от примерно 2:1 до примерно 15:1, от примерно 3:1 до примерно 12:1, от примерно 4:1 до примерно 10:1, от примерно 5:1 до примерно 9:1 или примерно 9:1. В некоторых вариантах осуществления изобретения массовое соотношение альбумина и по существу не растворимого в воде фармакологически активного агента равно примерно 18:1 или меньше, 15:1 или меньше, 14:1 или меньше, 13:1 или меньше, 12:1 или меньше, 11:1 или меньше, 10:1 или меньше, 9:1 или меньше, 8:1 или меньше, 7:1 или меньше, 6:1 или меньше, 5:1 или меньше, 4:1 или меньше и 3:1 или меньше.
В некоторых вариантах осуществления изобретения очищенный альбумин делает возможным введение композиции субъекту (такому как человек) без возникновения значительных побочных эффектов. В некоторых вариантах осуществления изобретения альбумин используют в количестве, позволяющем эффективно уменьшать один или несколько побочных эффектов, возникающих при введении человеку по существу не растворимого в воде фармакологически активного агента. Термин “уменьшение одного или нескольких побочных эффектов, возникающих при введении по существу не растворимого в воде фармакологически активного агента” означает уменьшение, ослабление, устранение или избежание возникновения одного или нескольких нежелательных эффектов, вызываемых по существу не растворимым в воде фармакологически активным агентом, а также побочных эффектов, вызываемых средствами доставки (такими как растворители, которые делают по существу не растворимые в воде фармакологически активные агенты пригодными для инъекций), используемыми для доставки по существу не растворимого в воде фармакологически активного агента. Такие побочные эффекты включают, например, подавление функции костного мозга, нейротоксичность, аллергию, воспаление, заболевание вен, флебит, боль, раздражение кожи, периферическую невропатию, лихорадку нейрогенного происхождения, анафилактическую реакцию, тромбоз вен, транссудацию и их комбинации. Однако указанные побочные эффекты приведены только в качестве примера, и благодаря настоящему изобретению могут быть ослаблены другие побочные эффекты или комбинации побочных эффектов, ассоциированных с по существу не растворимыми в воде фармакологически активными агентами.
В некоторых вариантах осуществления изобретения композиция включает абраксан® (или Nab-паклитаксел). В некоторых вариантах осуществления изобретения композиция представляет собой абраксан® (или Nab-паклитаксел). Абраксан® является препаратом паклитаксела, стабилизированным альбумином человека, соответствующим требованиям фармакопеи США (USP), который может быть диспергирован непосредственно в инъецируемом физиологическом растворе. При диспергировании в приемлемой водной среде, такой как инъекционный раствор, содержащий 0,9% хлорида натрия или 5% декстрозы, абраксан™ образует устойчивую коллоидную суспензию паклитаксела. Средний размер наночастиц в коллоидной суспензии составляет примерно 130 нанометров. Так как сывороточный альбумин человека (HSA) легко растворяется в воде, абраксан™ может быть восстановлен в широком диапазоне концентраций от разбавленного (0,1 мг/мл паклитаксела) до концентрированного (20 мг/мл паклитаксела), в том числе, например, от примерно 2 мг/мл до примерно 8 мг/мл, примерно 5 мг/мл.
По существу не растворимый в воде фармакологически активный агент
Композиции по настоящему изобретению содержат по существу не растворимые в воде фармакологически активные агенты. Например, растворимость в воде плохо растворимого в воде агента при температуре примерно 20-25°С может быть менее примерно 10 мг/мл, в том числе, например, менее примерно 5, 2, 1, 0,5, 0,2, 0,1, 0,05, 0,2 или 0,01 мг/мл. В некоторых вариантах осуществления изобретения по существу не растворимый в воде фармакологически активный агент является твердым веществом. В некоторых вариантах осуществления изобретения по существу не растворимый в воде фармакологически активный агент является жидкостью. По существу не растворимый в воде фармакологически активный агент по настоящему изобретению может быть, например, фармацевтическим агентом, диагностическим агентом или пищевым агентом.
Приемлемые фармацевтические агенты включают, не ограничиваясь ими, противораковые или противоопухолевые средства, антимикробные средства, иммуносупрессоры, анестетики, гормоны, средства для лечения сердечно-сосудистых нарушений, средства от аритмии, антибиотики, противогрибковые средства, гипотензивные средства, антиастматические средства, противовоспалительные средства, средства от артрита, вазоактивные средства, аналгетики/жаропонижающие средства, антидепрессанты, средства от диабета, средства от состояния тревоги, средства от головной боли, седативные средства, средства от стенокардии, антипсихотические средства, антиманиакальные средства, средства от артрита, средства от подагры, антикоагулянты, тромболитические средства, антифибринолитические средства, антигеморрагические средства, антитромбоцитарные средства, противосудорожные средства, антипаркинсонические средства, антигистамины/противозудные средства, средства для регулирования кальция, антивирусные средства, антимикробные средства, антибактериальные средства, бронходилататоры, гормоны, гипогликемические средства, гиполипидемические средства, средства от язвы/средства от рефлюкса, противорвотные средства и растворимые в масле витамины (например, витамины А, D, E, K и тому подобные).
В некоторых вариантах осуществления изобретения по существу не растворимый в воде фармакологически активный агент является одним из нижеследующих веществ, включающих ингибитор тирозинкиназы, ингибитор серин/треонинкиназы, ингибитор гена hedgehog, ингибитор топоизомеразы, ингибитор образования микротрубочек, ингибитор сигнального пути AKT-киназы, ингибитор протеасом, антиметаболит и средство на основе платины.
В некоторых вариантах осуществления изобретения по существу не растворимый в воде фармакологически активный агент является противоопухолевым средством. В некоторых вариантах осуществления изобретения по существу не растворимый в воде фармакологически активный агент является химиотерапевтическим средством.
Приемлемые по существу не растворимые в воде фармакологически активные агенты включают, не ограничиваясь ими, таксаны (такие как паклитаксел, доцетаксел, ортатаксел и другие таксаны), эпофилоны, камптотецины, колцицины, геладамицины, амиодароны, тиреоидные гормоны, амфотерицин, кортикостероиды, пропофол, мелатонин, циклоспорин, рапамицин (сиролимус) и его производные, такролимус, микофенольные кислоты, ифосфамид, винорелбин, ванкомицин, гемцитабин, SU5416, тиотепа, блеомицин, диагностические радиоактивные агенты и их производные. Другие по существу не растворимые в воде фармакологически активные агенты, пригодные для использования в композициях по настоящему изобретению, описаны, например, в патентах США №№ 5916596, 6096331, 6749868 и 6537539. Дополнительные примеры по существу не растворимых в воде фармакологически активных агентов включают соединения, которые плохо растворяются в воде и приведены в публикации “Therapeutic Category and Biological Activity Index” of The Merck Index (12th Edition, 1996).
В некоторых вариантах осуществления изобретения по существу не растворимый в воде фармакологически активный агент является любым веществом, выбираемым из группы, включающей паклитаксел, доцетаксел, CY196, октатаксел и другие таксаны или аналоги таксанов, 17-аллиламиногелданамицин (17-AAG), производный 18-гелданамицин, камптотецин, пропофол, амиодарон, циклоспорин, эпотилон, радицикол, комбретастатин, рапамицин, амфотерицин, лиотиронин, эпотилон, колцицин, тиоколцицин и его димеры, тиреоидный гормон, вазоактивный кишечный пептид, кортикостероиды, мелатонин, такролимус, микофенольные кислоты, эпотилоны, радициколы, комбретастатины и их аналоги или производные. В некоторых вариантах осуществления изобретения по существу не растворимый в воде фармакологически активный агент является любым веществом, выбираемым из группы, включающей паклитаксел, доцетаксел, CY196, ортатаксел или другие таксаны, гелданамицин, 17-аллиламиногелданамицин, тиоколцицин и его димеры, рапамицин, циклоспорин, эпотилон, радицикол и комбретастатин. В некоторых вариантах осуществления изобретения по существу не растворимый в воде фармакологически активный агент является рапамицином. В некоторых вариантах осуществления изобретения по существу не растворимый в воде фармакологически активный агент является 17-AAG. В некоторых вариантах осуществления изобретения по существу не растворимый в воде фармакологически активный агент является димером тиоколцицина (таким как IDN5404). В некоторых вариантах осуществления изобретения по существу не растворимый в воде фармакологически активный агент является таксаном. В некоторых вариантах осуществления изобретения по существу не растворимый в воде фармакологически активный агент является паклитакселом. В некоторых вариантах осуществления изобретения по существу не растворимый в воде фармакологически активный агент является доцетакселом. В некоторых вариантах осуществления изобретения по существу не растворимый в воде фармакологически активный агент является CY196.
В некоторых вариантах осуществления изобретения по существу не растворимый в воде фармакологически активный агент является таксаном или его производным, который включает, не ограничиваясь ими, паклитаксел, доцетаксел и IDN5109 (ортатаксел) или их производное. В некоторых вариантах осуществления изобретения композиция содержит некристаллический и/или аморфный таксан (такой как паклитаксел или его производное). В некоторых вариантах осуществления изобретения композицию получают, используя безводный таксан (такой как безводный доцетаксел или его производное). Установлено, что безводный доцетаксел позволяет получить более устойчивый препарат по сравнению с гидратированным доцетакселом, таким как тригидрат или полугидрат доцетаксела.
Другие компоненты, входящие в состав композиций на основе наночастиц
Наночастицы по настоящему изобретению могут присутствовать в композиции, содержащей также другие агенты, наполнители или стабилизаторы. Например, для повышения устойчивости путем увеличения отрицательного дзета-потенциала наночастиц в композицию могут быть введены определенные отрицательно заряженные компоненты. Такие отрицательно заряженные компоненты включают, не ограничиваясь ими, соли желчных кислот, состоящие из гликохолевой кислоты, холевой кислоты, хенодезоксихолевой кислоты, таурохолевой кислоты, гликохенодезоксихолевой кислоты, таурохенодезоксихолевой кислоты, литохолевой кислоты, урсодезоксихолевой кислоты, дегидрохолевой кислоты и других; фосфолипиды, включающие фосфолипиды на основе лецитина (яичный желток), которые включают следующие фосфатидилхолины: пальмитоилолеоилфосфатидилхолин, пальмитоиллинолеоилфосфатидилхолин, стеароиллинолеоилфосфатидилхолин, стеароилолеоилфосфатидилхолин, стеароиларахидоилфосфатидилхолин и дипальмитоилфосфатидилхолин. Другие фосфолипиды включают L-a-димиристоилфосфатидилхолин (DMPC), диолеоилфосфатидилхолин (DOPC), дистеариолфосфатидилхолин (DSPC), гидрированный соевый фосфатидилхолин (HSPC) и другие родственные соединения. В качестве добавок также могут быть использованы отрицательно заряженные поверхностно-активные вещества или эмульгаторы, например, холестерилсульфат натрия и тому подобные.
В некоторых вариантах осуществления изобретения указанная композиция предназначена для введения человеку. В некоторых вариантах осуществления изобретения композиция предназначена для использования в ветеринарии для введения млекопитающим, таким как домашние животные и сельскохозяйственные животные. Существует целый ряд приемлемых препаратов, включающих композиции на основе наночастиц (смотрите, например, патенты США №№ 5916596 и 6096331). Рассмотренные ниже препараты и способы их получения являются иллюстративными и не ограничивают объем изобретения. Препараты, предназначенные для перорального введения, могут включать (а) жидкие растворы, получаемые в результате растворения эффективного количества соединения в таких разбавителях как вода, физиологический раствор или апельсиновый сок, (b) капсулы, саше или таблетки, содержащие заранее определенное количество активного ингредиента в виде твердых веществ или гранул, (с) суспензии в соответствующей жидкости и (d) приемлемые эмульсии. Лекарственные формы в виде таблетки могут включать один или несколько следующих компонентов, таких как лактоза, маннит, кукурузный крахмал, картофельный крахмал, микрокристаллическая целлюлоза, аравийская камедь, желатин, коллоидный кремнезем, натрий-кроскармеллоза, тальк, стеарат магния, стеариновая кислота и другие наполнители, красители, разбавители, буферы, смачивающие вещества, консерванты, ароматизаторы и фармакологически совместимые наполнители. Лекарственные формы в виде лепешек могут включать активный ингредиент в ароматизаторе, таком как сахароза, аравийская камедь или трагант; лекарственные формы в виде пастилок включают активный ингредиент в инертном основании, таком как желатин и глицерин или сахароза и аравийская камедь; эмульсии, гели и тому подобные формы, помимо активного ингредиента, содержат наполнители, известные в данной области.
Примеры приемлемых носителей, наполнителей и разбавителей включают, не ограничиваясь ими, лактозу, декстрозу, сахарозу, сорбит, маннит, крахмалы, аравийскую камедь, фосфат кальция, альгинаты, трагант, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду, физиологический раствор, сироп, метилцеллюлозу, метил- и пропилгидроксибензоаты, тальк, стеарат магния и минеральное масло. Указанные препараты могут дополнительно включать смазывающие вещества, смачивающие вещества, эмульгирующие и суспендирующие вещества, консерванты, подсластители или ароматизаторы.
Препараты, предназначенные для парентерального введения, включают водные и неводные, изотонические стерильные инъекционные растворы, которые могут содержать антиоксиданты, буферы, бактериостаты и растворенные вещества, которые делают препарат совместимым с кровью предполагаемого реципиента, и водные и неводные стерильные суспензии, которые могут включать суспендирующие вещества, растворители, загустители, стабилизаторы и консерванты. Препараты могут находиться в герметичных упаковках для однократного или многократного введения, таких как ампулы и флаконы, и могут храниться в лиофилизованном состоянии, требующем только добавления стерильного жидкого наполнителя, например, воды для инъекций, непосредственно перед использованием. Инъекционные растворы и суспензии могут быть получены по индивидуальному рецепту из стерильных порошков, гранул и таблеток вышеописанного типа. Инъецируемые препараты являются более предпочтительными.
В некоторых вариантах осуществления изобретения композицию получают со значением рН в интервале от примерно 4,5 до примерно 9,0, в том числе, например, со значением рН в интервале от примерно 5,0 до примерно 8,0, от примерно 6,5 до примерно 7,5 и от примерно 6,5 до примерно 7,0. В некоторых вариантах осуществления изобретения значение рН композиции равно не менее примерно 6, в том числе, например, не менее примерно 6,5, 7 или 8 (например, примерно 8). Композиция может быть также получена в виде соответствующего крови изотонического раствора путем добавления приемлемого модификатора тоничности, такого как глицерин.
Способ применения композиций без приона на основе наночастиц
Настоящее изобретение также относится к способам применения композиций без приона по настоящему изобретению. Например, некоторые варианты осуществления изобретения относятся к способу введения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, в которой по существу отсутствует прионный белок. Некоторые варианты осуществления изобретения относятся к способу лечения болезни (такой как рак), который включает введение эффективного количества композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, в которой по существу отсутствует прионный белок.
Некоторые варианты осуществления изобретения относятся к способу введения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, в которой альбумин получен способом, включающим процесс удаления приона, который включает приведение исходной композиции альбумина в контакт с лигандом, способным связываться с прионным белком. В некоторых вариантах осуществления изобретения процесс удаления приона дополнительно включает удаление указанного лиганда и связанных с ним белков из композиции альбумина. Некоторые варианты осуществления изобретения относятся к способу лечения у субъекта болезни (такой как рак), который включает введение указанному субъекту эффективного количества композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, в которой альбумин получен способом, включающим процесс удаления приона, который включает приведение исходной композиции альбумина в контакт с лигандом, способным связываться с прионным белком.
В некоторых вариантах осуществления изобретения субъект страдает разновидностью болезни Крейтцфельда-Якоба (vCJD). В некоторых вариантах осуществления изобретения субъект уже инфицирован прионным белком. В некоторых вариантах осуществления изобретения существует подозрение, что субъект болен vCJD или инфицирован прионным белком. В некоторых вариантах осуществления изобретения субъект является бессимптомным носителем прионного белка. В некоторых вариантах осуществления изобретения субъекту переливали кровь по меньшей мере один раз. В некоторых вариантах осуществления изобретения субъекту по меньшей мере 60 лет, например, по меньшей мере 65, 70 или 75 лет. В некоторых вариантах осуществления изобретения у субъекта ослаблена иммунная система. В некоторых вариантах осуществления изобретения субъект болен раком.
Термин “эффективное количество” в использованном здесь значении означает количество соединения или композиции, достаточное для лечения определенного нарушения, состояния или болезни с целью ослабления, облегчения и/или задержки возникновения одного или нескольких симптомов. Применительно к раку или другой нежелательной пролиферации клеток эффективное количество означает количество, достаточное для уменьшения объема опухоли и/или замедления роста опухоли (например, подавления роста опухоли). В некоторых вариантах осуществления изобретения эффективное количество представляет собой количество, достаточное для замедления прогрессирования болезни. В некоторых вариантах осуществления изобретения эффективное количество является количеством, достаточным для предотвращения возникновения и/или рецидива заболевания. Эффективное количество препарата можно вводить один или несколько раз.
Разные типы рака, предназначенные для лечения композициями по настоящему изобретению (например, композицией, включающей противоопухолевое средство, такое как таксан, рапамицин и 17-AAG), включают, не ограничиваясь ими, карциному, лимфому, бластому, саркому и лейкоз. Примеры разных типов рака, которые можно лечить композициями по настоящему изобретению включают, не ограничиваясь ими, плоскоклеточнывй рак, рак легкого (в том числе мелкоклеточный рак легкого, немелкоклеточный рак легкого, аденокарциному легкого и плоскоклеточную карциному легкого, включая плоскоклеточный NSCLC), рак брюшины, печеночно-клеточный рак, рак желудка (в том числе желудочно-кишечный рак), рак поджелудочной железы (такой как запущенный рак поджелудочной железы), глиобластому, рак шейки матки, рак яичника, рак печени (такой как печеночно-клеточная карцинома), рак мочевого пузыря, гепатому, рак молочной железы, рак ободочной кишки, меланому, карциному эндометрия или матки, карциному слюнной железы, рак почки, рак печени, рак предстательной железы (такой как запущенный рак предстательной железы), рак женских наружных половых органов, рак щитовидной железы, карциному печени, рак головы и шеи, колоректальный рак, рак прямой кишки, саркому мягких тканей, саркому Капоши, В-клеточную лимфому (в том числе низкодифференцированную/фолликулярную неходжкинскую лимфому (NHL), В-клеточную (SL) NHL, среднедифференцированную/фолликулярную NHL, среднедифференцированную генерализованную NHL, высокодифференцированную иммунобластную NHL, высокодифференцированную лимфобластную NHL, высокодифференцированную мелкоклеточную NHL с нерассеченными ядрами, множественную NHL, лимфому клеток коры головного мозга, лимфому, обусловленную СПИД, и макроглобулинемию Вальденстрема), хронический лимфолейкоз (CLL), острый лимфобластный лейкоз (ALL), миелому, волосатоклеточный лейкоз, хронический миелобластный лейкоз и посттрансплантационное лимфопролиферативное нарушение (PTLD), а также аномальную пролиферацию сосудов, ассоциированную с фагоматозом, водянку (например, ассоциированную с опухолями головного мозга) и синдром Мейгса. Некоторые варианты осуществления изобретения относятся к способу лечения метастазирующего рака (то есть рака, метастазирующего из первичной опухоли). Некоторые варианты осуществления изобретения относятся к способу уменьшения пролиферации и/или миграции клеток. Некоторые варианты осуществления изобретения относятся к способу лечения гиперплазии.
Некоторые варианты осуществления изобретения относятся к способам лечения рака в запущенной стадии. Некоторые варианты осуществления изобретения относятся к способам лечения рака молочной железы (который может быть HER2-положительным или HER2-отрицательным), включающего, например, запущенный рак молочной железы, рак молочной железы IV стадии, локальный запущенный рак молочной железы и метастазирующий рак молочной железы. В некоторых вариантах осуществления изобретения рак является раком легкого, в том числе, например, немелкоклеточным раком легкого (NSCLC, таким как запущенный NSCLC), мелкоклеточным раком легкого (SCLC, таким как запущенный SCLC) и запущенной солидной злокачественной опухолью в легком. В некоторых вариантах осуществления изобретения рак является раком яичника, раком головы и шеи, злокачественной опухолью желудка, меланомой (включая метастазирующую меланому), колоректальным раком, раком поджелудочной железы и солидными опухолями (такими как запущенные солидные опухоли). В некоторых вариантах осуществления изобретения рак выбирают из группы, включающей рак молочной железы, колоректальный рак, рак прямой кишки, немелкоклеточный рак легкого, неходжкинскую лимфому (NHL), рак почки, рак предстательной железы, рак печени, рак поджелудочной железы, саркому мягких тканей, саркому Капоши, карциноидную карциному, рак головы и шеи, меланому, рак яичника, мезотелиому, глиомы, глиобластомы, нейробластомы и множественную миелому. В некоторых вариантах осуществления изобретения рак является солидной опухолью.
Выбор субъекта, которому могут быть введены указанные композиции, зависит от природы плохо растворимого в воде фармацевтического средства, а также от подлежащего лечению и/или предотвращению заболевания/состояния/нарушения. Таким образом, термин “субъект” означает любых позвоночных, млекопитающих и человека. В некоторых вариантах осуществления изобретения субъект является млекопитающим и включает, не ограничиваясь ими, человека, корову, лошадь, кошку, собаку, грызуна или примата. В некоторых вариантах осуществления изобретения субъект является человеком.
Вводимая субъекту (такому как человек) доза композиции по настоящему изобретению может быть различной в зависимости от конкретной композиции, способа введения и подлежащей лечению болезни. Доза должна быть достаточной для достижения требуемой реакции, такой как терапевтическая или профилактическая реакция против конкретной болезни. Например, доза паклитаксела в композиции может составлять 100-400 мг/м2 при введении один раз в 3 недели или 50-250 мг/м2 при введении один раз в неделю. Кроме того, при ежедневном введении или введении несколько раз в неделю доза может составлять примерно 5-75 мг/м2.
Композиции по настоящему изобретению можно вводить субъекту (такому как человек) разными способами, включающими, например, внутривенное, внутриартериальное, внутрилегочное, внутрипортальное, внутрипеченочное, пероральное, ингаляционное, внутрипузырное, внутримышечное, внутритрахеальное, подкожное, внутриглазное, внутриоболочечное, трансдермальное введение и введение через слизистую оболочку. Например, композиция по настоящему изобретению может быть введена путем ингаляции для лечения заболеваний дыхательных путей. Композиция может быть использована для лечения респираторных заболеваний, таких как фиброз легких, облитерирующий бронхиолит, рак легкого, бронхоальвеолярная карцинома и тому подобных.
В некоторых вариантах осуществления изобретения композицию вводят вместе с фильтром для удаления приона.
Настоящее изобретение также относится к способа уменьшения побочных эффектов, ассоциированных с введением композиции на основе наночастиц. Например, настоящее изобретение относится к способам уменьшения разных побочных эффектов, ассоциированных с введением плохо растворимого в воде фармацевтического средства, которые включают, не ограничиваясь ими, подавление функции костного мозга, нейротоксичность, аллергию, воспаление, заболевание вен, флебит, боль, раздражение кожи, периферическую невропатию, лихорадку нейрогенного происхождения, анафилактическая реакцию, токсическое воздействие на кроветворную систему, токсическое воздействие на головной мозг и нервную систему и их комбинации. Некоторые варианты осуществления изобретения относятся к способу уменьшения аллергических реакций, ассоциированных с введением плохо растворимого в воде фармацевтического средства, включающих, например, сильную сыпь на коже, крапивницу, покраснение, одышку, тахикардию и другие побочные эффекты.
Наборы и системы
Настоящее изобретение также относится к наборам, используемым при осуществлении способов по настоящему изобретению. Наборы по настоящему изобретению включают одну или несколько емкостей, в которых находятся композиции без приона на основе наночастиц, и в некоторых вариантах осуществления изобретения наборы дополнительно включают инструкции по использованию в соответствии с любыми описанными способами. Набор может дополнительно включать описание выбора субъекта, отвечающего требованиям лечения. Инструкции, предоставляемые в наборах по настоящему изобретению, обычно напечатаны на этикетке или вложены в упаковку (например, лист бумаги, вложенный в набор), но также могут представлять собой инструкции, считываемые компьютером (например, инструкции на магнитном или оптическом запоминающем диске).
Наборы по настоящему изобретению находятся в соответствующей упаковке. Такая упаковка включает, не ограничиваясь ими, флаконы, бутылки, гибкую упаковку (например, герметичные майларовые или пластиковые пакеты) и тому подобные. В комплект наборов могут необязательно входить дополнительные компоненты, такие как буферы и разъясняющая информация.
Инструкции по использованию композиций на основе наночастиц обычно включают информацию о дозах, схеме приема и способе введения, соответствующих предполагаемому лечению. Емкости могут содержать унифицированные дозы, упаковки лекарственных средств для многократного приема или разделенные дозы. Например, наборы могут содержать дозы по существу не растворимого в воде фармакологически активного агента по настоящему изобретению, обеспечивающие эффективное лечение субъекта в течение длительного периода времени, например, в течение одной недели, 2 недель, 3 недель, 4 недель, 6 недель, 8 недель, 3 месяцев, 4 месяцев, 5 месяцев, 7 месяцев, 8 месяцев, 9 месяцев или больше. Наборы могут также включать большие количества унифицированных доз по существу не растворимого в воде фармакологически активного агента, фармацевтические композиции и инструкции по применению, в упаковках, предназначенных для хранения и использования в аптеках, например, в больничных аптеках и аптеках, занимающихся приготовлением лекарственных средств.
Некоторые варианты осуществления изобретения относятся к набору для удаления прионного белка из композиции на основе наночастиц, включающих альбумин и по существу не растворимый в воде фармакологически активный агент, в комплект которого входит лиганд, способный связываться с прионным белком. В некоторых вариантах осуществления изобретения набор дополнительно включает носитель. В некоторых вариантах осуществления изобретения набор дополнительно включает инструкцию по использованию лиганда для удаления приона из композиции на основе наночастиц.
Настоящее изобретение также относится к системам для осуществления способов по настоящему изобретению. Например, некоторые варианты осуществления изобретения относятся к системе получения композиции без приона на основе наночастиц, включающих альбумин и по существу не растворимый в воде фармакологически активный агент, которая включает 1) устройство для получения композиции на основе наночастиц и 2) устройство для удаления прионных белков из альбумина, используемого для получения композиции на основе наночастиц. В некоторых вариантах осуществления изобретения устройство для удаления прионных белков из альбумина, используемого для получения композиции на основе наночастиц, встроено в устройство для получения композиции на основе наночастиц. В некоторых вариантах осуществления изобретения устройство для получения композиции на основе наночастиц отделено от устройства для удаления прионных белков из альбумина, используемого для получения композиции на основе наночастиц.
Некоторые варианты осуществления изобретения относятся к системе получения композиции без приона на основе наночастиц, включающих альбумин и по существу не растворимый в воде фармакологически активный агент, которая включает 1) устройство для получения композиции на основе наночастиц и 2) устройство для удаления прионных белков (например, из промежуточной композиции, полученной в процессе создания наночастиц, или из созданной композиции на основе наночастиц). В некоторых вариантах осуществления изобретения устройство для удаления прионных белков встроено в устройство для получения композиции на основе наночастиц. В некоторых вариантах осуществления изобретения устройство для получения композиции на основе наночастиц отделено от устройства для удаления прионных белков.
Специалистам в данной области должно быть понятно, что в объем настоящего изобретения могут входить несколько вариантов осуществления изобретения.
Все материалы, приведенные в настоящем изобретении, в том числе публикации, заявки на патент и патенты, включены в настоящее описание изобретения в качестве ссылки так, как если бы каждая публикация была приведена отдельно в полном объеме.
В настоящем описании изобретения приведены предпочтительные варианты осуществления изобретения, включая лучшие способы осуществления изобретения, известные авторам настоящего изобретения. Предпочтительные варианты осуществления изобретения станут очевидны специалистам в данной области при ознакомлении с нижеследующим описанием изобретения. Авторы изобретения считают, что квалифицированные специалисты смогут использовать такие варианты осуществления изобретения должным образом, при этом авторы изобретения предполагают наличие других вариантов осуществления изобретения, помимо рассмотренных в настоящем описании изобретения. Поэтому в объем настоящего изобретения входят все модификации и эквиваленты объекта изобретения, представленного в прилагаемой формуле изобретения, как это допускается соответствующим законом. Кроме того, в объем изобретения входит любая комбинация вышеописанных элементов во всех возможных вариантах осуществления изобретения за исключением случаев, специально оговоренных в настоящем описании изобретения или каким-либо другим образом противоречащих контексту изобретения.
Типичные варианты осуществления изобретения по настоящей заявке
Одним объектом настоящего изобретения является композиция, содержащая наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, в которой по существу отсутствует прионный белок. В некоторых вариантах осуществления изобретения композиция характеризуется инфективностью приона менее примерно 100 фг/мл. В некоторых вариантах осуществления изобретения указанная композиция является любой из вышеописанных композиций, в которой инфективность приона составляет менее примерно 10 ин.ед.-иц/мл. В некоторых вариантах осуществления изобретения указанная композиция является любой из вышеописанных композиций, в которой отсутствует прионный белок по результатам анализа методом циклической амплификации с ошибочной укладкой цепи белка (РМСА) или анализа методом ИПЦР. В некоторых вариантах осуществления изобретения указанная композиция является любой из вышеописанных композиций, которая дополнительно содержит следовое количество лиганда, способного связываться с прионным белком. В некоторых вариантах осуществления изобретения указанная композиция является любой из вышеописанных композиций, которая дополнительно содержит следовое количество носителя.
Другим объектом настоящего изобретения является композиция, содержащая наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, в которой альбумин получен способом, включающим процесс удаления приона, который включает приведение исходной композиции альбумина в контакт с лигандом, способным связываться с прионным белком. В некоторых вариантах осуществления изобретения процесс удаления приона дополнительно включает удаление указанного лиганда и связанных с ним белков из альбумина и композиции. В некоторых вариантах осуществления изобретения лиганд является пептидом. В некоторых вариантах осуществления изобретения лиганд является соединением на основе триазина.
Другим объектом настоящего изобретения является способ получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает: а) удаление прионного белка из исходной композиции альбумина; b) воздействие высоким сдвигающим усилием на смесь, включающую раствор альбумина без приона и органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, диспергированный в органическом растворителе. В некоторых вариантах осуществления изобретения стадия а) включает: 1) приведение исходного раствора альбумина в контакт с лигандом, способным связываться с прионным белком. В некоторых вариантах осуществления изобретения стадия а) дополнительно включает: 2) удаление лиганда и связанных с ним белков из раствора альбумина. В некоторых вариантах осуществления изобретения указанный способ является любым из вышеописанных способов, который дополнительно включает удаление указанного органического растворителя из смеси. В некоторых вариантах осуществления изобретения органический растворитель удаляют выпариванием. В некоторых вариантах осуществления изобретения указанный способ является любым из вышеописанных способов, в котором указанный лиганд является пептидом. В некоторых вариантах осуществления изобретения указанный способ является любым из вышеописанных способов, в котором лиганд является соединением на основе триазина. В некоторых вариантах осуществления изобретения указанный способ является любым из вышеописанных способов, в котором исходная композиция альбумина является продуктом, выделенным из крови.
Другим объектом настоящего изобретения является способ получения композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает: а) воздействие высоким сдвигающим усилием на смесь, включающую органическую фазу, содержащую по существу не растворимый в воде фармакологически активный агент, и раствор альбумина, и b) удаление прионного белка из указанной смеси. В некоторых вариантах осуществления изобретения стадия b) включает: 1) приведение смеси в контакт с лигандом, способным связываться с прионным белком. В некоторых вариантах осуществления изобретения стадия b) дополнительно включает: 2) удаление лиганда и связанных с ним белков из указанной смеси. В некоторых вариантах осуществления изобретения лиганд является пептидом. В некоторых вариантах осуществления изобретения лиганд является соединением на основе триазина.
Настоящее изобретение также относится к композиции, полученной способом по любому из пп.11-23. Настоящее изобретение также относится к применению любой из вышеописанных композиций для лечения такой болезни, как рак.
Другим объектом настоящего изобретения является способ удаления прионного белка из композиции с подозрением на наличие прионного белка, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, который включает: а) приведение композиции в контакт с лигандом, способным связываться с прионным белком, b) удаление лиганда и связанных с ним белков из композиции. В некоторых вариантах осуществления изобретения лиганд является пептидом. В некоторых вариантах осуществления изобретения лиганд является соединением на основе триазина. Настоящее изобретение также относится к композициям, полученным указанным способом.
Другим объектом настоящего изобретения является композиция, содержащая наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент, которая дополнительно включает лиганд, способный связываться с прионным белком. В некоторых вариантах осуществления изобретения лиганд является пептидом. В некоторых вариантах осуществления изобретения лиганд является соединением на основе триазина.
Приведенные ниже примеры являются иллюстративными и не ограничивают объем изобретения. Следует отметить, что примеры, приведенные в настоящем описании изобретения, предназначены только для иллюстративных целей, и в объем данной заявки на патент входят разные модификации или изменения, очевидные специалистам в данной области.
ПРИМЕРЫ
Пример 1. Создание аффинного сорбента для удаления прионного белка из препаратов, содержащих альбумин
В результате выполнения данного эксперимента была исследована состоящая из четырех смол панель лигандов, связывающихся с прионом, на которые воздействовали двумя разными коммерчески доступными препаратами, содержащими альбумин (20% масс./об. или 25% масс./об. альбумина), которые были потенцированы гомогенатом головного мозга хомячка, страдающего болезнью скрепи, в двух разных концентрациях (0,01% или 0,005%). Методом PRDT (Pathogen Removal and Diagnostic Technologies Inc.; ProMetic Biosciences Ltd., Cambridge, UK) предварительно было установлено, что все четыре смолы панели хорошо связываются с прионом в присутствии 25% альбумина. Выбирая оптимальную смолу, можно оптимизировать стадию удаления приона при получении наночастиц, включающих альбумин.
Методика выполнения исследования
В комплект набора для выделения шести белков, предназначенного для идентификации сорбента (PIKSI™, ProMetic Biosciences Ltd), входили двенадцать колонок (примерно 0,5 мл) для каждой из четырех смол PRDT и контрольной смолы (Toyopearl амино-AMN31). Каждую смолу, помещаемую в три колонки, обрабатывали растворами, содержащими 20% и 25% альбумина, потенцированного 0,01% или 0,005% гомогената головного мозга хомячка, страдающего болезнью скрепи (SBH), для оценки способности каждой смолы связываться с прионными белками. Рабочие характеристики смол сравнивали на основании связывания с прионным белком, определяемого при помощи вестерн-блоттинга и денситометрии. Общий профиль связывания белка определяли при помощи гелей для SDS-PAGE. Связанные белки удаляли из смол для обнаружения связывания прионного белка и связывания общего белка. Связывание с альбумином определяли при помощи спектрофотометра NanoDrop® ND-1000, предназначенного для измерения оптической плотности при 280 нм. Сигналы, полученные в вестерн-блоттах и гелях для SDS-PAGE, соответствуют связанной фракции прионного белка и общего белка.
В настоящем изобретении были использованы следующие коммерческие препараты, содержащие альбумин: альбумин (человека), соответствующий требованиям фармакопеи США (USP), грифолз®, 20%, партия № IBAB8MJ001, и альбумин (человека), соответствующий требованиям фармакопеи США (USP), 25% раствор бакстера, партия № LA06D04AA.
РЕЗУЛЬТАТЫ
Вестерн-блоттинг и SDS-PAGE
Полученные результаты показывают, что все четыре смолы связывали PrPsc, которым были потенцированы 20% или 25% растворы альбумина человека. Интенсивность сигнала PrPsc при выполнении вестерн-блоттинга (фиг.1-3) позволяет предположить, что наибольшим связыванием приона характеризовалась смола DVR и затем смолы YVHEA, SYA и D4. Контрольная смола AMN31 характеризовалась ожидаемым сильным сигналом. Уровень сигнала, полученного при использовании смолы DVR, позволяет предположить, что высокие концентрации альбумина не препятствовали связыванию приона.
Детектируемый сигнал не был обнаружен во второй и третьей колонках при использовании DVR (фиг.1) в разных условиях испытания, даже при более продолжительном времени воздействия, из чего следует, что весь детектируемый PrPsc был связан в первой колонке с DVR.
Интенсивность сигнала была слабее для других смол в первой колонке, при этом прионный белок был обнаружен во второй колонке серии (фиг.2 и 3), что указывает на возможную интерференцию альбумина в отношении связывания прионного белка. В случае всех трех лигандов не был обнаружен сигнал приона в третьей колонке, из чего следует, что указанные смолы могут избирательно удалять прионы из SBH-потенцированных растворов альбумина. Концентрация PrPsc в головном мозге хомячка была равна примерно 50 мкг/г, что эквивалентно примерно 5 нг/мл PrPsc в 0,01% SBH, введенного в 250 мг/мл раствора альбумина, с образованием 50000000-кратного избытка альбумина.
Картина общего белка в окрашенных кумасси гелях для SDS-PAGE (фиг.1-3) показывает, что смола DVR характеризуется гораздо более низким уровнем связывания общего белка по сравнению с остальными смолами, связывающими прион, что считается преимуществом несмотря на то, что большая часть полос в геле с общим белком была получена из потенцирующего гомогената головного мозга. Как ожидалось, одна видимая полоса белка в геле DVR имеет среднюю молекулярную массу, аналогичную альбумину (66,5 кДа).
Денситометрия
Удаление приона также определяли, вычисляя соотношение денситометрического сигнала PrPsc, связанного со смолой, и сигнала в растворах альбумина, потенцированных SBH. Способность смолы DVR связывать прион была сравнима с положительной контрольной смолой, адсорбирующей практически весь имеющийся PrPsc в первой колонке. При использовании инфекционных доз в качестве меры связывания приона в колонке, содержащей 0,5 мл DVR, прион был удален в 10 мл SBH-потенцированных растворах альбумина, что эквивалентно примерно 106 ID50, из чего следует, что 0,1% SBH содержит примерно 106 ID50/мл. Аналогично результатам вестерн-блоттинга смола DVR характеризовалась лучшим связыванием приона, чем смолы YVHEA, D4 и SYA. Для всех трех смол была необходима вторая колонка в каждой серии для дальнейшего связывания любого обнаруживаемого прионного белка в SBH-потенцированных растворах альбумина.
Связывание альбумина
Концентрацию альбумина определяли, используя спектрофотометр NanoDrop® ND-1000, предназначенный для количественного измерения белка при оптической плотности, равной 280 нм. Концентрацию альбумина измеряли после прохождения SBH-потенцированных растворов альбумина через каждую из четырех смол (DVR, YVHEA, SYA и D4) и контрольную смолу (AMN31). Концентрацию белка в коммерческих растворах альбумина с концентрацией 20% и 25% измеряли при отсутствии или наличии 0,01% или 0,005% SBH до прохождения через колонки со смолами. Полученные результаты приведены в таблице 3.
Концентрации коммерческих растворов альбумина были выше маркированного значения, особенно для препарата, содержащего 20% масс./об. раствора альбумина. Однако, поскольку исследователи имели дело со сравнительными величинами, такое расхождение не имело значения. Было получено среднее значение для трех величин, при этом количество SBH считалось пренебрежимым по сравнению с количеством альбумина, присутствующего в растворах, при определении концетрации белка. Концентрацию альбумина определяли после прохождения SBH-потенцированных растворов альбумина через смолы, как показано в таблицах 4 и 5.
Ни в одной из испытанных смол не было обнаружено значительной потери альбумина. Потеря альбумина фактически не была обнаружена для большинства условий. Полученные значения характеризуются разбросом величин в пределах примерно ±5%. Полученные данные позволяют предположить, что потеря альбумина, по-видимому, не является фактором, определяющим выбор одной из смолы в испытанных условиях.
Заключение
На основании полученных результатов можно сделать вывод о том, что DVR является лучшей смолой из четырех испытанных смол, предназначенных для удаления приона из коммерческих растворов альбумина. Указанная смола позволила удалить примерно 106 ID50 в 0,5-мл колонке. Данное значение аналогично значениям, полученным в предыдущих испытаниях при стимулировании другими веществами. Была обнаружена небольшая потеря альбумина при испытанном соотношении стимуляции 20% или 25% альбумина/смола, равном 20. Связывание альбумина должно быть ниже в технологических условиях, так как приведенное выше соотношение, вероятно, увеличится.
Пример 2. Исследование возможности удаления приона из суспензий, содержащих паклитаксел и раствор альбумина человека
Возможность применения технологии удаления приона в композиции на основе наночастиц исследовали с использованием суспензий, содержащих паклитаксел (например, абраксан™) и раствор альбумина человека с разным процентным содержанием альбумина (% масс./об.).
ЭКСПЕРИМЕНТАЛЬНЫЕ УСЛОВИЯ
Системы, используемые в исследовании
Были исследованы суспензии для абраксана™ и препарата, содержащего сахар и паклитаксел. Суспензия (FS) для абраксана™ была создана при использовании упаренной суспензии (РЕ), полученной в результе приготовления раствора альбумина человека (25%, Baxter, Deerfield, IL), содержащего примерно 7 мг/мл паклитаксела и 56 мг/мл альбумина человека. Суспензия (FS) для препарата, содержащего сахар и паклитаксел, была создана при использовании упаренной суспензии (РЕ), полученной в результате приготовления раствора альбумина человека (25%, Baxter), сахарозы, хлорида натрия и динатрийдигидрата эдетата, содержащего примерно 7 мг/мл паклитаксела, 56 мг/мл альбумина человека, 32 мг/мл сахарозы, 8,4 мг/мл NaCl и 0,07 мг/мл ЭДТА (этилендиаминтетрауксусаная кислота).
Были также исследованы растворы альбумина человека с концентрацией 25%, 20% и 5%. Раствор альбумина человека с концентрацией 25% или 250 мг/мл был предоставлен компанией Baxter; раствор альбумина человека с концентрацией 20% или 200 мг/мл (например, Grifols®) был предоставлен компанией Grifols Biologicals, Inc. (Los Angeles, CA). Раствор альбумина человека с концентрацией 5% был получен путем разведения 25% раствора альбумина человека (Baxter) стериальной водой для инъекций.
Колонки для удаления приона
В качестве колонок для удаления приона (1 мл) в данном исследовании были использованы колонки из коммерчески доступного набора PIKSI® (набор для выделения белка с целью идентификации сорбента), содержащие смолу Toyopearl амино-650CU (образец AMN31), предоставленную компанией ProMetic Biosciences, Ltd (Cambridge, UK).
Условия прохождения потока
При исследовании препарата в виде суспензии общий объем проходящего потока был равен примерно 500 мл, что было в 500 раз больше объема колонки (1 мл). Скорость потока была равна примерно 0,5 мл/мин. Общее время прохождения потока составило более 16 часов. Образцы отбирали через каждые два часа. Не было отмечено закупоривания колонки.
При исследовании раствора альбумина общий объем проходящего потока был равен по меньшей мере 100 мл, что в 100 раз больше объема колонки (1 мл). Скорость потока была равна примерно 0,5 мл/мин. Общее время прохождения потока составило более 3 часов. Образцы отбирали через каждый час. Не было отмечено закупоривания колонки.
Результаты и их рассмотрение
Результаты исследования возможности удаления приона в суспензии суммированы в таблицах 6 и 7. Результаты физического и химического исследования суспензии для абраксана™ свидетельствуют об отсутствии существенных различий в суспензии до и после прохождения потока через колонку, при этом исследуемые параметры включали размер частиц, рН, анализ паклитаксела и загрязняющих примесей, анализ альбумина человека и композиции альбумина человека. Аналогичным образом результаты физического и химического исследования суспензии для препарата, содержащего сахар и паклитаксел, свидетельствуют об отсутствии существленных различий в суспензии до и после прохождения через колонку, при этом исследуемые параметры включали размер частиц, рН, анализ паклитаксела и загрязняющих примесей, анализ альбумина человека, композиции альбумина человека, сахарозы и ЭДТА.
Результаты, полученные при исследовании возможности удаления приона из растворов альбумина, суммированы в таблице 8. Не было обнаружено существенных различий в растворе до и после прохождения через колонку по результатам анализа альбумина человека и композиции альбумина человека.
Данное исследование показывает, что прохождение через колонку для удаления приона не оказывает вредного влияния на физические и химические свойства раствора альбумина человека и суспензии для абраксана™ и препарата, содержащего сахар и паклитаксел.
Пример 3. Удаление TSE восстанавливающими прион смолами в 20% растворе альбумина
При выполнении данного исследования оценивали возможное удаление TSE восстанавливающими прион смолами (колонка PRDT; ProMetic Biosciences, Ltd) в 20% (масс./об.) растворе альбумина (Grifols®). Исходное вещество для стадии удаления TSE потенцировали агентом, моделирующим TSE. Указанную стадию выполняли в лаборатории VirusSure (Virusure Forschung und Entwicklung GmbH, Vienna, Austria). Во время выполнения указанной стадии собирали разные фракции и эффективность удаления TSE вычисляли, определяя уровни TSE при помощи вестерн-блоттинга для обнаружения PrPsc.
Данное исследование было выполнено в соответствии со следующими указаниями: 1) CPMP/BWP/268/95 (revised in 1996), Note for Guidance on Virus Validation Studies: The Design, Contribution, and Interpretation of Studies Validating the Inactivation and Removal of Viruses; 2) CPMP/BWP/5136/03 Guideline on the Investigation of Manufacturing Processes for Plasma-Derived Medicinal Products with Regards to vCJD Risk; 3) OECD Principles of Good Laboratory Practice as outlined in ENV/MC/CHEM(98)17, revised in 1997; 4) 21 CFR part 58, Good Laboratory Practice, US FDA; 5) EU directive 2004/9/EG, Inspection und Uberpriifung der Guten Laborpraxis (GLP); 6) EU Directive 2004/10/EG, Anwendung der Grundsatze der guten Laborpraxis und zur Kontrolle ihrer Anwendung bei Versunchen mit chemischen Stoffen; 7) Austrian BGBI. II, 211. Verordnung, Chemikalien-GLP-Inspektionsverordnung, Jahrgang 2000; and 8) Austrian BGBI. II, 450. Verordnung, gute Laborpraxis 2006, Jahrgang 2006.
Материалы и методы
При исследовании возможности удаления TSE восстанавливающими прион смолами (колонка PRDT) в 20% растворе альбумина (Grifols®) были использованы следующие изделия, реагенты и материалы.
Для потенцированного исследования и исследования интерференции был использован альбумин (человека) Grifols® (20% раствор, соответствующий требованиям фармакопеи США (партия: IBAB7GX001/TA09/0122)).
Для выполнения процесса была использована одноразовая колонка SepFast™ (ProMetic Biosciences Ltd.), содержащая 5 мл смолы для удаления приона в 9% физиологическом растворе.
Были приготовлены разные буферы. Указанные буферы представляли собой следующие буферы:
1) буфер, содержащший 0,9% NaCl (9 г/л), был получен в качестве уравновешивающего буфера для восстанавливающих прион смол;
2) физиологический раствор с трис-буфером (TBS) был получен в качестве буфера для повторного суспендирования смол после хроматографии;
3) 2 М раствор NaCl (116,88 г/л) был получен в качестве буфера для регенерации восстанавливающих прион смол;
4) буферы, содержащие NaOH (0,1 М, 0,5 М и 1,0 М) были получены для регулирования значений рН в образцах, используемых в потенцированном исследовании, и инактивации инфекционного вещества в промежуточных продуктах; и
5) буферы, содержащие HCl (0,1 М и 1,0 М) были получены для регулирования значения рН в образцах, используемых в потенцированном исследовании, и промежуточных продуктах.
Штамм клеток 263 болезни скрепи
В данном исследовании был использован штамм клеток 263К болезни скрепи, адаптированной для хомячка (обработанный 0,5% саркозила). Штамм клеток 263К болезни скрепи, адаптированной для хомячка, позволяет получить высокие титры в головном мозге хомячков. Типичные титры для 10% гомогената головного мозга находятся в пределах 108-1010 единиц ID50/мл. Белок PrPsc, осажденный указанным агентом, является относительно устойчивым к расщеплению протеиназой К, что позволяет отличить форму белка PrPc, не ассоциированную с болезнью. Предназначенный для посева штамм клеток 263К болезни скрепи, адаптированной для хомячка, был предоставлен в виде 10% гомогената лабораторией доктора Роберта Рохвера (Baltimore Research and Education Foundation, Mail Stop 151-A, 10 North Greene Street, Baltimore, MD 21201, USA). Для данного эксперимента была выбрана фракция, обработанная 0,5% саркозила. Указанная фракция была получены из неочищенного гомогената головного мозга (из которого предварительно была удалена микросомная/цитозольная фракция клеток 263К) путем обработки 0,5% саркозила с последующим дифференциальным центрифугированием для удаления болоее крупных агрегатов, в результате чего в супернатанте оставались только фракции, раствориемые детергентом. Фракция, обработанная 0,5% саркозила, была получена для имитации загрязнения при растворении в детергенте (то есть, как это имеет место в процессах с использованием детергента в качестве растворителя), причем фракция такого типа широко используется в исследованиях по удалению приона из плазмы человека и рекомбинантных продуктов.
Анализ методом вестерн-блоттинга для обнаружения PrPsc
Анализ методом вестерн-блоттинга для обнаружения PrPsc был использован для полуколичественного определения уровней TSE (PrPsc) в разных образцах. Динамический диапазон анализа методом вестерн-блоттинга обычно находится в области 4-5 log10 разведений до потери сигнала, поэтому указанный анализ является менее чувствительным, чем биоанализ хомячка. Однако анализ методом вестерн-блоттинга является полезным средством оценки удаления приона при выполнении биофармацевтических процессов приготовления лекарственных средств.
При выполнении анализа методом вестерн-блоттинга образец сначала расщепляли протеиназой К для удаления нормальной формы белка, PrPc (все образцы расщепляли протеиназой К с концентрацией 83 пг/мл). Протеолитическую реакцию блокировали, образец смешивали с буфером, содержащим SDS, и кипятили для денатурации PrPsc из агрегированной формы. Затем образец разводили до 0,5 log10 и помещали на гель для SDS-PAGE вместе с маркером молекулярной массы. После электрофореза гель при помощи вестерн-блоттинга переносили на мембрану из PVDF, блокировали и зондировали антителами, позволяющими обнаружить белок PrPsc, связанный с антителом 3F4. В штамме клеток 263К болезни скрепи была обнаружена характерная картина полос в области 25-33 кДа, которая подтверждает наличие белка PrPsc в образцах. Конечная точка титра образца была определена в виде первого разведения с отсутствием сигнала при выполнении вестерн-блоттинга.
Вычисление коэффициентов восстановления
Коэффициенты восстановления (RF) вычисляли следующим образом:
RF=(V1×T1)/(V2×T2)
где:
V1 и Т1 означают соответственно объем и титр исходного вещества;
V2 и Т2 означают соответственно объем и титра фракции продукта.
В логарифмическом выражении указанное уравнение можно представить следующим образом:
Log10[RF]=[Log10(V1)+Log10(T1)]-[Log10(V2)+Log10(T2)]
Коэффициенты восстановления были округлены до одной десятой только после окончательного вычисления.
Исследование интерференции
Сначала исследовали неразведенное исходное вещество и затем вещество, предварительно разведенное до 1,0 log10 в TBSA. После потенцирования штаммом клеток 263К до конечной концентрации, эквивалентной титру в пределах примерно 2 log10 конечной точки исходного раствора клеток болезни скрепи, использованного для потенцирования, неразведенный образец и предварительно разведенный образец исходного вещества центрифугировали с ускорением 15,558×g в течение 60 минут при комнатной температуре. После центрифугирования супернатант осторожно сливали и осадок ресуспендировали с использованием TBSA в 1/10 первоначального объема центрифугированного образца (не эквивалентного эффективной концентрации для 1,0 log10 предварительно разведенного образца и эквивалентного 10-кратной концентрации для неразведенного образца). Затем образец расщепляли протеазой К и исследовали стандартным методом вестерн-блоттинга. Регенерированный образец и образец из колонки разводили до 0,5 log10 или исследовали в неразведенном виде до выполнения анализа методом вестерн-блоттинга (то есть без центрифугирования).
Регулирование значения рН
В образцах проверяли значение рН, которое должно быть равно 6-8 (регулирование значения рН не требовалось для некоторых образцов), затем брали аликвотную пробу и хранили образцы при температуре ≤-60°С.
Оборудование
Для выполнения данного исследования были использованы следующие основные компоненты оборудования. Стерильная камера для защиты от биологической опасности класса II, камеры замораживания до ≤-60°С Sanyo & Angelantoni, холодильники с температурой 2-8°С Angelantoni и камеры замораживания до ≤-15°С, аналитические весы и принтер Sartorius или Kern, электронный термометр Hanna, измеритель рН Mettler Toledo, водяные бани Grant или Selecta, лабораторный таймер Oregon, микроцентрифуга Hettich, ванна для электрофореза BioRad Criterion, устройство для анализа методом вестерн-блоттинга BioRad Criterion, хроматографическая система АКТА, источник питания Wealtec, проявитель пленки Agfa и колонка Biotoolomics, заполненная смолой PRDT.
Выполнение процесса
Блок-схема процесса вместе с полученными образцами показана на фиг.4. Объемы соответствующих образцов указаны в таблице 9 раздела “Результаты”.
При подготовке к выполнению процесса очищали камеру защиты от ламинарнорго потока (LF), которую включали по меньше мере на 10-15 минут для уравновешивания. Водяную баню уравновешивали до 30±2°С и исходное вещество уравновешивали до достижения температуры, равной 29,5°С. SBH, растворенный в 0,5% саркозила, оттаивали на той же водяной бане и после оттаивания помещали на лед до использования. Колонку PRDT уравновешивали до комнатной температуры (23,0±5,0°С) в течение ночи.
Входной трубопровод колонки (сверху) присоединяли к выходному отверстию хроматографической системы AKTA. Трубопровод выходного отверстия колонки (снизу) вводили в соответствующий сборник (химический стакан или 50 мл одноразовая центрифужная пробирка). Все трубопроводы были предварительно заполнены водой для инъекций (WFI) для предупреждения попадания пузырьков воздуха в систему.
Колонку сначала уравновешивали 5 объемами (CV) воды для инъекций (WFI) и затем 10 объемами уравновешивающего буфера. Скорость потока была равна 2,0±0,1 мл/минуту.
Получение образцов и хроматография восстанавливающей прион смолы
50,7 мл уравновешенного исходного вещества потенцировали 0,51 мл гомогената клеток 263К, растворенного в 0,5% саркозила. Затем проверяли значение рН, которое находилось в пределах 6,9-7,4. Отбирали 0,5 мл аликвоту (образец SSM) и хранили при ≤-60°С.
Потенцированный образец затем вводили в вышеуказанную уравновешенную колонку PRDT со скоростью потока 1,8±0,1 мл/мин и собирали выходящий из колонки поток в виде следующих фракций:
Образец Е1 начинали собирать сразу же после достижения оптической плотности, равной 80% полной шкалы. Фракцию выходящего потока собирали для каждого исследования (объем каждого собранного элюированного образца определяли взвешиванием). После введения потенцированного раствора альбумина колонку проомывали 10,0 мл уравновешивающего буфера. Образец Е5 прекращали собирать сразу же после того, как оптическая плотность падала ниже 80% полной шкалы. Колонку промывали уравновешивающим буфером и регенерировали, используя >20,0 мл 2 М раствора NaCl (образец REG), после регенерации смолу удаляли и ресуспендировали в 5 мл TBS (образец COL).
РЕЗУЛЬТАТЫ
Образцы, полученные в результате потенцированных исследований
В таблице 9 представлены образцы, собранные в результате выполнения потенцированного исследования, с указанием объема каждого образца. В тех случаях, когда размер образца определяли взвешиванием, предполагалось, что объем каждого образца может быть вычислен на основании плотности, равной 1,0 г/мл. Все образцы хранили в виде аликвот при -60°С до выполнения анализа. Сканированное воспроизведение хроматографического профиля, полученного при выполнении потенцированного исследования, показано на фиг.5.
Результаты исследования интерференции
Для преодоления интерференции все образцы, за исключением регенерированного образца и образца смолы, разводили до 1,0 log10 в TBS, содержащем 0,1% BSA, с последующим центрифугированием и концентрированием до 1/10 объема. Неразведенный образец исследовали в отношении интерференции при 10-кратной концентрации, вызывающей сильную интерференцию. Регенерированные образцы разводили до 0,5 log10, чтобы уменьшить концентрацию NaCl. Образцы смолы исследовали без предварительного разведения, так как смола была ресуспендирована в буфере TBS.
Разведение образца, необходимое для устранения интерференции альбумина, производили путем предварительного разведения до 1,0 Log10 с последующим центрифугированием и ресуспендированием в 1/10 части первоначального объема. Смотрите фиг.6.
Данные титрования приона и вычисления коэффициентов восстановления
Вычисление коэффициентов восстановления приона при выполнении данного процесса показано в таблице 10. В таблице 10 также показано разведение образца с целью устранения интерференции.
Коэффициент восстановления (RF), равный 2,5 log10, относительно потенцированного исходного вещества вычисляли для образца Е1, который соответствовал первым 2,0 мл, прошедшим через колонку PRDT. В образцах Е2-Е5 было отмечено проникновение PrPsc в выходящую из колонки фракцию (коэффициент восстановления 1,5 log10 для образца Е5). PrPsc не был обнаружен в регенерированном образце, и эквиваленты PrPsc, связанные со смолой PRDT, более подробно рассмотрены в нижеследующем разделе, посвященном вычислению способности смолы PRDT связывать прион.
Вычисление способности восстанавливающих прион смол связывать прион
Для определения способности восстанавливающих прион смол (смола PRDT) связывать инфекционный прионный белок титры, полученные при выполнении вестерн-блоттинга, сравнивали с инфекционными титрами. При выполнении анализа методом вестерн-блоттинга использовали исходный раствор клеток 263К с известным титром. Серию произведенных разведений эталонного исходного раствора исследовали методом вестерн-блоттинга. Строили график полученных титров относительно соответствующих титров, полученных в результате биоанализа хомячка, и выполняли линейный регрессионный анализ для оценки взаимосвязи между двумя испытуемыми системами. Вычисленные значения наклона и пересечения кривой регрессии были соответственно равны 1,0667 и -4,5867. Параметры регрессии использовали для преобразования титров вестерн-блоттинга в инфекционные титры по следующей формуле:
Титр[биоанализ]=(Титр[вестерн-блоттинг]+4,5867)/(1,0667).
После вычисления инфекционного титра на мл можно определить общий прион, связанный в колонке. Вычисленная способность восстанавливающей прион смолы связывать прионный белок показана в приведенной ниже таблице 11. Указанную способность определяли, используя количество PrPsc, непосредственно связанное с восстанавливающей прион смолой (образцы Reg и Col).
Общее связывание PrPsc со смолой PRDT после промывки 2 М солевым раствором было равно 7,4 log10 ID50/мл. По результатам анализа методом вестерн-блоттинга PrPsc не был обнаружен во фракциях, промытых солевым раствором. Также было отмечено значительное удаление PrPsc (≥2,5 log10).
Для исследования интерференции использовали неразведенное исходное вещество, выполняя центрифугирование и 10-кратное концентрирование. Таким образом можно определить возможность концентрирования образцов и достижения более высоких коэффициентов восстановления.
Пример 4. Удаление TSE восстанавливающими прион смолами в 25% растворе альбумина
В данном исследовании определяли потенциальное удаление TSE восстанавливающими прион смолами (колонка PRDT) в 25% растворе альбумина (Baxter, Deerfield, IL). Были использованы такие же материалы и методы, что и в примере 3, за исключением использования в качестве исходного вещества для потенцированного исследования и исследования интерференции 25% раствора альбумина (человека) компании Baxter (партия: LA07D051AB/ТА09/0123), соответствующего требованиям фармакопеи США.
Выполнение процесса
Процесс был выполнен в лабораторном масштабе в компании ViruSure. Блок-схема процесса наряду с полученными образцами показана на фиг.7. Объемы соответствующих образцов указаны в таблице 12, приведенной в разделе “Результаты”. Параметры и способы, использованные при подготовке процесса, включая присоединение хрорматографической системы AKTA к колонке и уравновешение колонки, аналогичны описанным в примере 3.
Получение образца и хроматогорафия восстанавливающей прион смолы
50,1 мл уравновешенного исходного вещества потенцировали 0,51 мл гомогената клеток 263К, растворенного в 0,5% саркозила. Затем регулировали значение рН, которое находилось в пределах 6,9-7,4, добавляя 0,1 М раствор HCl или 0,5 М раствор NaOH. Значение рН потенцированного исходного вещества (6,85) выходило за пределы целевого диапазона в пределах 6,9-7,4 (смотрите раздел “Отклонения”). После добавления 65 мкл 0,1 М раствора NaOH и 50 мкл 1 М раствора NaOH значение рН не изменилось, поэтому было принято решение загрузить исходное вещество в колонку при данном значении рН. Затем отбирали 0,5 мл аликвоты (образец SSM) и хранили при ≤-60°С.
Потенцированный образец вводили в вышеуказанную уравновешенную колонку PRDT со скоростью потока 1,8±0,1 мл/мин и собирали выходящий из колонки поток в виде следующих фракций:
Образец Е1 начинали собирать сразу же после достижения оптической плотности, равной 80% полной шкалы. Фракцию выходящего потока собирали для каждого исследования (объем каждого собранного элюированного образца определяли взвешиванием). После введения потенцированного раствора альбумина колонку проомывали 10,0 мл уравновешивающего буфера. Образец Е5 прекращали собирать сразу же после того, как оптическая плотность падала ниже 80% полной шкалы. Колонку промывали уравновешивающим буфером и регенерировали, используя >20,0 мл 2 М раствора NaCl (образец REG), после регенерации смолу удаляли и ресуспендировали в 5 мл TBS (образец COL).
РЕЗУЛЬТАТЫ
Образцы, полученные в результате потенциированных исследований
В нижеследующей таблице 12 представлены образцы, собранные в результате выполнения потенцированного исследования, с указанием объема каждого образца. В тех случаях, когда размер образца определяли взвешиванием, предполагалось, что объем каждого образца может быть вычислен на основании плотности, равной 1,0 г/мл. Все образцы хранили в виде аликвот при ≤-60°С до выполнения анализа. Сканированное воспроизведение хроматографического профиля, полученного при выполнении потенцированного исследования, показано на фиг.8.
Результаты исследования интерференции
Для преодоления интерференции альбумина все образцы, за исключением регенерированного образца и образца смолы, разводили до 1,0 log10 в TBS, содержащем 0,1% BSA, с последующим центрифугированием и концентрированием до 1/10 объема. Неразведенный образец исследовали в отношении интерференции при 10-кратной концентрации, вызывающей сильную интерференцию. Регенерированные образцы разводили перед исследованием до 0,5 log10, чтобы уменьшить концентрацию NaCl. Образцы смолы исследовали без предварительного разведения, так как смола была ресуспендирована в буфере TBS.
Разведение образца, необходимое для устранения интерференции альбумина, производили путем предварительного разведения до 1,0 Log10 с последующим центрифугированием и ресуспендированием в 1/10 части первоначального объема. Смотрите фиг.9.
Данные титрования приона и вычисления коэффициентов восстановления
Вычисление коэффициентов восстановления приона при выполнении данного процесса показано в таблице 13. В таблице 13 также показано разведение образца с целью устранения интерференции.
Коэффициент восстановления (RF) ≥2,5 log10 относительно потенцированного исходного вещества вычисляли для образца Е1, который соответствовал первым 2,0 мл, прошедшим через колонку PRDT. В образцах Е2-Е5 было отмечено проникновение PrPsc в выходящую из колонки фракцию (коэффициент восстановления 1,5 log10 для образца Е5). PrPsc не был обнаружен в регенерированном образце, и эквиваленты PrPsc, связанные со смолой PRDT, более подробно рассмотрены в нижеследующем разделе, посвященном вычислению способности смолы PRDT связывать прион.
Вычисление способности восстанавливающих прион смол связывать прион
Для определения способности восстанавливающих прион смол (смола PRDT) связывать инфекционный прионный белок титры, полученные при выполнении вестерн-блоттинга, сравнивали с инфекционными титрами. При выполнении анализа методом вестерн-блоттинга использовали исходный раствор клеток 263К с известным титром. Серию произведенных разведений эталонного исходного раствора исследовали методом вестерн-блоттинга. Строили график полученных титров относительно соответствующих титров, полученных в результате биоанализа хомячка, и выполняли линейный регрессионный анализ для оценки взаимосвязи между двумя испытуемыми системами. Вычисленные значения наклона и пересечения кривой регрессии были соответственно равны 1,0667 и -4,5867. Параметры регрессии использовали для преобразования титров вестерн-блоттинга в инфекционные титры по следующей формуле:
Титр[биоанализ]=(Титр[вестерн-блоттинг]+4,5867)/(1,0667).
После вычисления инфекционного титра на мл можно определить общий прион, связанный в колонке. Вычисленная способность восстанавливающей прион смолы связывать прионный белок показана в приведенной ниже таблице 14. Указанную способность определяли, используя количество PrPsc, непосредственно связанное со смолой PRDT (образцы Reg и Col).
Общее связывание PrPsc с матрицей PRDT после промывки 2 М солевым раствором было равно 7,4 log10 ID50/мл. PrPsc не был обнаружен во фракциях, промытых солевым раствором, по результатам анализа методом вестерн-блоттинга. Также было отмечено значительное удаление PrPsc (≥2,0 log10).
Значение рН потенцированного исходного вещества было равно 6,85. После добавления 65 мкл 0,1 М раствора NaOH и 50 мкл 1 М раствора NaOH значение рН не изменилось. Такой эффект, по-видимому, связан со значительной буферной способностью 25% раствора альбумина.
Для исследования интерференции использовали неразведенное исходное вещество, выполняя центрифугирование и 10-кратное концентрирование. Такое исследование выполняли дополнительно к исследованию интерференции, описанному в плане исследования, для оценки возможности концентрирования образцов и, следовательно, для достижения более высоких коэффициентов восстановления.
Пример 5. Анализ устойчивости альбумина человека после удаления приона в ходе выполнения процесса
В данном эксперименте определяли влияние колонки для удаления приона на концентрацию и композицию альбумина человека. В двух отдельных экспериментах один литр альбумина обрабатывали в колонке PRIOCLEAR™ В (50 мл) (ProMetic Biosciences). Необработанный образец, образец, полученный в начале исследования, и образец, полученный в конце исследования, затем концентрировали и анализировали композицию. Результаты приведены в таблицах 15 и 16.
Не было обнаружено существенных различий в альбумине до и после выполнения процесса удаления приона.
Далее была определена технологическая устойчивость альбумина человека, обработанного в соответствии с процессом удаления приона, и суспензий абраксана®, полученных с использованием альбумина человека, не содержащего приона. Полученные результаты приведены в таблицах 17 и 18.
Повышенную устойчивость альбумина человека в готовых продуктах абраксана®, приготовленных с использованием альбумина человека, обработанного в соответствии с процессом удаления приона (партия, изготовленная в опытной установке с использованием альбумина человека, обработанного в соответствии с процессом удаления приона), сравнивали с готовыми продуктами абраксана®, приготовленными с использованием альбумина человека, не обработанного в соответстви с процессом удаления приона (демонстрационная партия абраксана, сертифицированная партия абраксана и партия абраксана, полученная в опытной установке). Полученные результаты приведены в таблице 19.
Сравнение данных устойчивости показывает, что хранение в течение 2 недель при 55°С эквивалентно хранению в течение 3 месяцев при 40°С. В процессе изготовления и во время технологического тестирования не было обнаружено существенных различий между партиями, полученными в опытных установках с использованием альбумина человека, обработанного в соответствии с процессом удаления приона, и партиями, полученными в опытных установках с использованием необработанного альбумина человека. Отсутствуют существенные различия в свойствах готовых продуктов, полученных с использованием альбумина человека, обработанного в соответствии с процессом удаления приона, и готовых продуктов, полученных с использованием необработанного альбумина человека.
Пример 6. Оценка влияния колонки для удаления приона на технологическую суспензию абраксана®
Целью данного эксперимента является оценка удаления приона из технологической суспензии абраксана®, а именно из суспензии абраксана® до лиофилизации. В данном эксперименте были использованы колонки (1 см3) из коммерчески доступного набора PIKSI, содержащие смолу Toyopearl амино-650CU, ProMetic Biosciences.
Технологическую суспензию абраксана® объемом 0,5 л, содержащую альбумин человека, пропускали через колонку в двух отдельных экспериментах. Результаты анализа суммированы в таблице 20.
Образец 1: технологическая суспензия абраксана, необработанная
Образец 2: технологическая суспензия абраксана после прохождения через колонку для удаления приона; образец, полученный в начале эксперимента
Образец 3: технологическая суспензия абраксана после проохождения через колонку для удаления приона; образец, полученный в конце эксперимента (0,5 л).
Как показано в таблице 20, физическое и химическое исследование технологической суспензии абраксана®, обработанной в соответствии с процессом удаления приона, не выявило осуществленных различий между необработанной и обработанной суспензиями в отношении размера частиц, рН, анализа паклитаксела и загрязняющих примесей, анализа альбумина человека (НА) и композиции альбумина человека.
Реферат
Группа изобретений относится к медицине и касается фармацевтической композиции для лечения рака, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный противораковый агент, приготовленные из смеси, содержащей органическую фазу, включающую по существу не растворимый в воде фармакологически активный противораковый агент и раствор альбумина, которая была подвергнута воздействию высокого сдвигающего усилия, где в указанной композиции по существу отсутствует прионный белок. Группа изобретений также касается способа получения фармацевтической композиции, содержащей наночастицы, включающие альбумин и по существу не растворимый в воде фармакологически активный агент; способа удаления прионного белка из фармацевтической композиции с подозрением на наличие прионного белка. Группа изобретений обеспечивает отсутствие пагубного воздействия на физические и/или химические свойства наночастиц при удалении прионного белка воздействием высокого сдвигающего усилия/лиганда. 4 н. и 15 з.п. ф-лы, 6 пр., 9 ил., 20 табл.
Формула
где в указанной композиции по существу отсутствует прионный белок.
Комментарии