Микрочастица и ее фармацевтическая композиция - RU2490009C2
Код документа: RU2490009C2
Чертежи












Описание
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к микрочастице, включающей агломерат частиц, содержащих гидрофильные активные вещества, и к ее фармацевтической композиции. В частности, изобретение относится к микрочастице и ее фармацевтической композиции в качестве так называемой системы доставки лекарств. Более конкретно, например, изобретение относится к микрочастице, эффективно содержащей белок, пептидные лекарственные средства, лекарственные средства на основе нуклеиновых кислот и аналогичные, обладающие гидрофильным свойством и большой молекулярной массой, и к ее фармацевтической композиции.
УРОВЕНЬ ТЕХНИКИ
В настоящее время разрабатываются препараты в виде частиц, содержащие лекарственные средства, заключенные в мелкодисперсных частицах, называемых наночастицей, микрочастицей, наносферой, микросферой или микрокапсулой, и осуществляются попытки их использования в качестве агентов замедленного высвобождения для лекарственных средств.
Дисперсные препараты, использующие полимерные соединения в качестве основы, включают мелкодисперсные частицы, состоящие из биоразлагаемой полимолочной кислоты или сополимера молочной и гликолевой кислот. В данные дисперсные препараты трудно заключить белковое или пептидное лекарственное средство, обладающее гидрофильным свойством и большой молекулярной массой, в то же время сохраняя биологическую активность. Кроме того, при введении в организм человека известно, что лекарственное средство массированно высвобождается за короткое время, и данное явление называют начальным выбросом.
В качестве мелкодисперсных частиц, состоящих из полимера, представляющего ковалентно связанные сахарид и полигидроксикислоту, патентный документ 1 описывает микрокапсулу для доставки фармакологически активного вещества, состоящую из продукта реакции полиола и полимолочной кислоты. В данном способе полисахариды не используются, и ничего не указывается по поводу включения пептида или белка. Микрокапсула, изготовленная методом распылительной сушкой, высвобождала 62% инкапсулированного лекарственного средства в течение 24 часов. Данная скорость высвобождения является слишком быстрой, и микрокапсулу едва ли можно использовать в качестве агента замедленного высвобождения для лекарственных средств.
Патентный документ 2 и непатентный документ 1 описывает наночастицу или наночастицу, состоящую из материала, имеющего биоразлагаемый полимер, привитый к полисахаридам, но в данном документе ничего не указывается по поводу микрочастицы, состоящей из наночастиц. Патентный документ 2 описывает, например, метод двойной эмульсии, уже цитированный в других источниках, в качестве способа изготовления микрочастицы для инкапсулирования гидрофильного активного вещества, но в ней нет конкретного описания, и включение лекарственного средства в частицу или высвобождение лекарственного средства из частицы не осуществлено. Непатентный документ 1 описывает микрочастицу, инкапсулирующую альбумин, изготовленную методом двойной эмульсии, но эффективность инкапсулирования по отношению к включенному количеству альбумина составляет 53% или менее, и низкая эффективность инкапсулирования гидрофильного активного вещества имеет проблему стоимости изготовления.
Патентный документ 3 описывает мелкодисперсную частицу, содержащую амфифильный полимер, состоящий из полисахаридов и алифатического полиэфира, более конкретно, мелкодисперсную частицу, состоящую из внутреннего ядра из полисахаридов, гидрофобного внешнего слоя из алифатического полиэфира и модификатора поверхности, связанного с гидрофобным внешним слоем. Данная мелкодисперсная частица не имеет агломерированную структуру мелкодисперсных частиц, и конкретные примеры не демонстрируют, что диаметр частиц находится в микрометровом диапазоне. Эффективность инкапсулирования гидрофильного вещества составляет 50% или менее, и данная низкая эффективность инкапсулирования является проблемой, аналогичной указанному выше случаю.
Патентный документ 4 описывает наночастицу со средним диаметром частицы менее 300 нм, состоящую из полученного из природного источника полимера декстрана, но конкретные примеры не показаны. Она не является агломерированной структурой мелкодисперсных частиц, средний диаметр частиц составляет сотни нанометров, лекарственное средство, вероятно, диффундирует от места введения, и она не является предпочтительной в качестве средства замедленного высвобождения.
В качестве полимера для формирования частиц, патентный документ 5 и патентный документ 6 описывают и предлагают использование амфифильного блок-полимера, имеющего гидрофильную часть, такую как полиэтиленгликоль, и гидрофобную часть, такую как сополимер молочной и гликолевой кислот. Частицы мицеллы, использующие такой амфифильный блок-полимер, обычно являются гидрофобными изнутри и гидрофильными во внешнем слое, и они подходят для удержания гидрофобных низкомолекулярных лекарственных средств, но не подходят для удержания гидрофильных активных веществ, таких как белок или пептид.
Патентный документ 7 и непатентный документ 2 описывают попытки включить белок в частицу, используя амфифильный блок-полимер, но количество содержащегося лекарственного средства является незначительным, или начальный пик выделения является существенным, и до настоящего времени технология изготовления частиц, имеющих свойства, подходящие для введения гидрофильного лекарственного средства с замедленным высвобождением, еще не налажена.
Патентный документ 1: Публикация заявки на патент Японии № 8-19226
Патентный документ 2: Японский перевод публикации международной заявки РСТ № 2004-521152
Патентный документ 3: WO 2006/095668
Патентный документ 4: Японский перевод публикации международной заявки РСТ № 10-511957
Патентный документ 5: Японский перевод публикации международной заявки РСТ № 2004-513154
Патентный документ 6: Японский перевод публикации международной заявки РСТ № 2004-514734
Патентный документ 7: Японский перевод публикации международной заявки РСТ № 2000-501084
Непатентный документ 1: Yuichi Oya и 3 соавтора, “Encapsulation and/or Release Behavior of Bovine Serum Albumin within and from Polylactide-Grafted Dextran Microspheres” (Macromolecular Bioscience, 2004, vol. 4, pp. 458-463).
Непатентный документ 2: Anshu Yang и 5 соавторов, “Tumor necrosis factor alpha blocking peptide loaded PEG-PLGA nanopeptides: Preparation and in vitro evaluation” (International Journal of Pharmaceutics, 2007, vol. 331, pp. 123-132).
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ПРОБЛЕМЫ, КОТОРЫЕ НЕОБХОДИМО РЕШИТЬ ИЗОБРЕТЕНИЕМ
Как указано выше, были разработаны микрочастицы, использующие полимер, и, следовательно, основная цель изобретения состоит в предоставлении микрочастицы, способной эффективно инкапсулировать гидрофобное активное вещество, и, более конкретно, микрочастицы, способной высвобождать инкапсулированное лекарственное средство с соответствующей скоростью, не вызывая значительный начальный выброс.
СРЕДСТВА РЕШЕНИЯ ПРОБЛЕМ
Авторы настоящего изобретения выполнили интенсивные исследования для решения данных проблем и в конечном итоге осуществили данное изобретение.
Изобретение относится к микрочастице, включающей агломерат частиц, содержащих гидрофильное активное вещество, причем данная частица включает амфифильный полимер, состоящий из гидрофобного сегмента полигидроксикислоты и гидрофильного сегмента полисахаридов или полиэтиленгликоля, и гидрофильное активное вещество, или, более конкретно, к микрочастице, включающей агломерат частиц, содержащих гидрофильное активное вещество, причем данная частица имеет гидрофильный сегмент из амфифильного полимера во внутренней части и имеет внешний слой гидрофильного сегмента из амфифильного полимера, к способу ее получения, и к ее фармацевтической композиции.
ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
Микрочастица по изобретению способна эффективно инкапсулировать гидрофильное активное вещество и высвобождать гидрофильное активное вещество с соответствующей скоростью в теле человека и, следовательно, является применимой в качестве нового DDS препарата.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
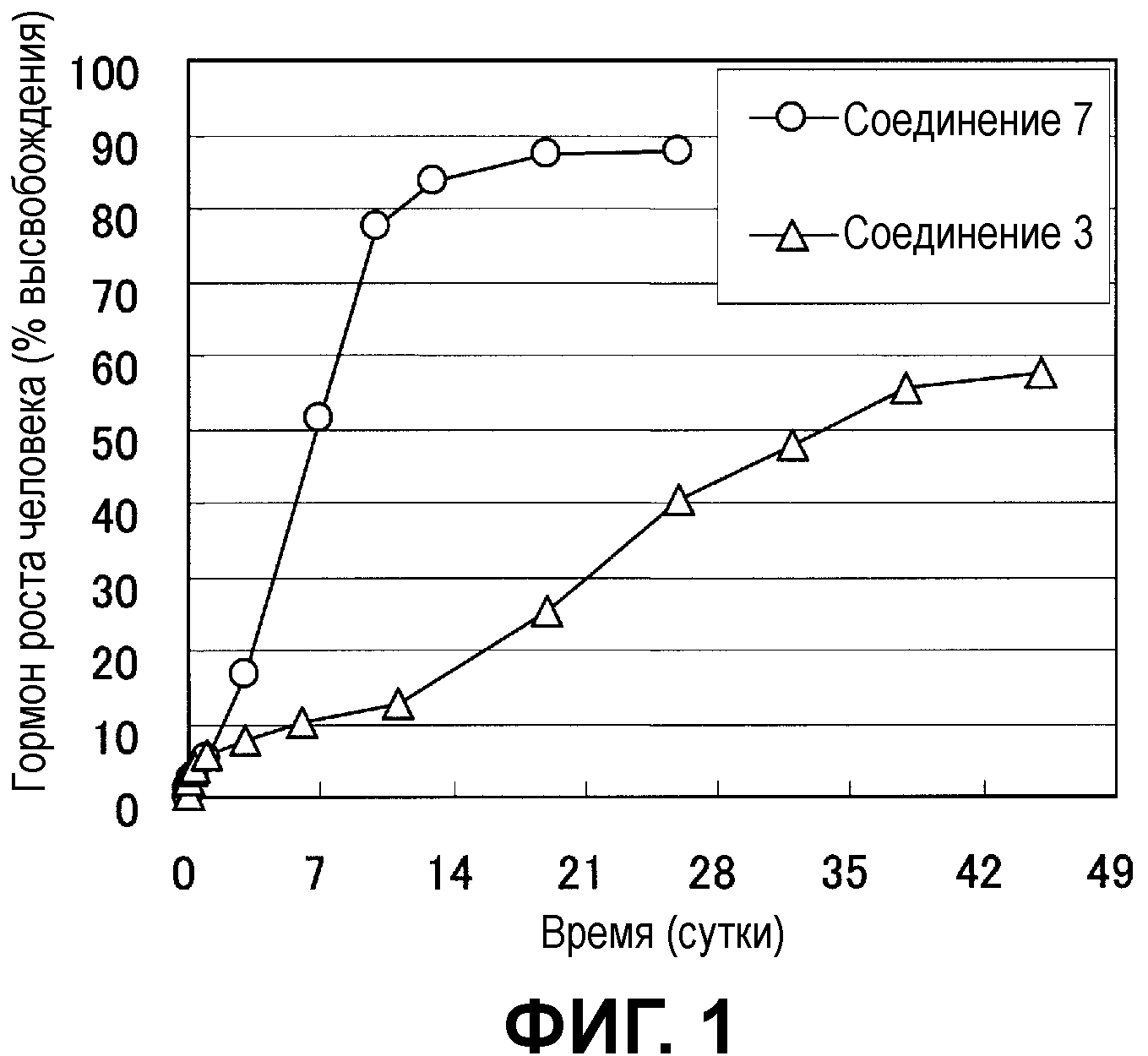
Фиг.1 показывает высвобождение лекарственного средства из микрочастиц с инкапсулированным гормоном роста человека.
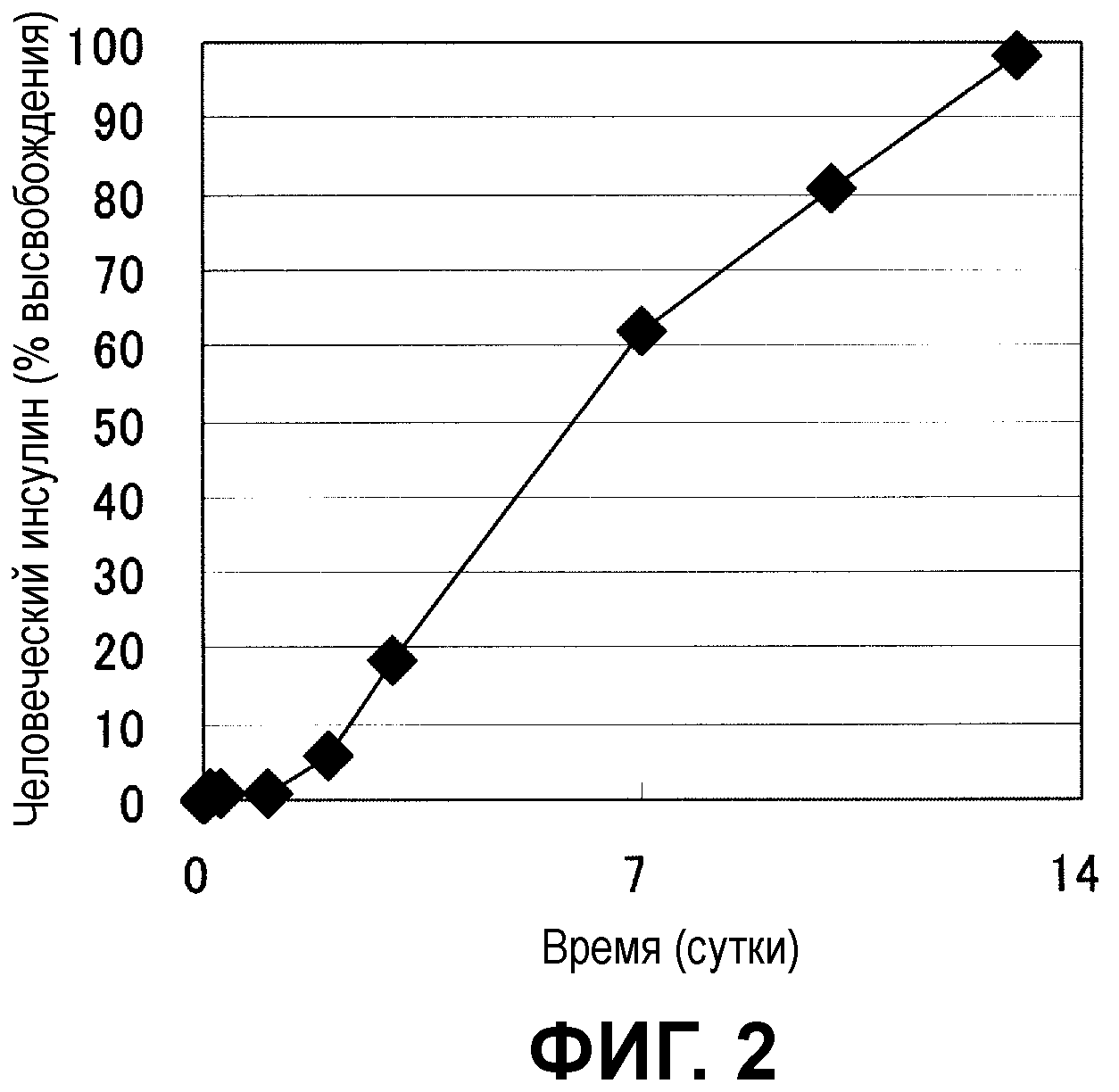
Фиг.2 показывает высвобождение лекарственного средства из микрочастиц декстран-PLGA с инкапсулированным человеческим инсулином.
Фиг.3 показывает изображение СЭМ микрочастиц декстран-PLGA.
Фиг.4 показывает изображение СЭМ микрочастицы полиэтиленгликоль-поли(эпсилон-капролактон).
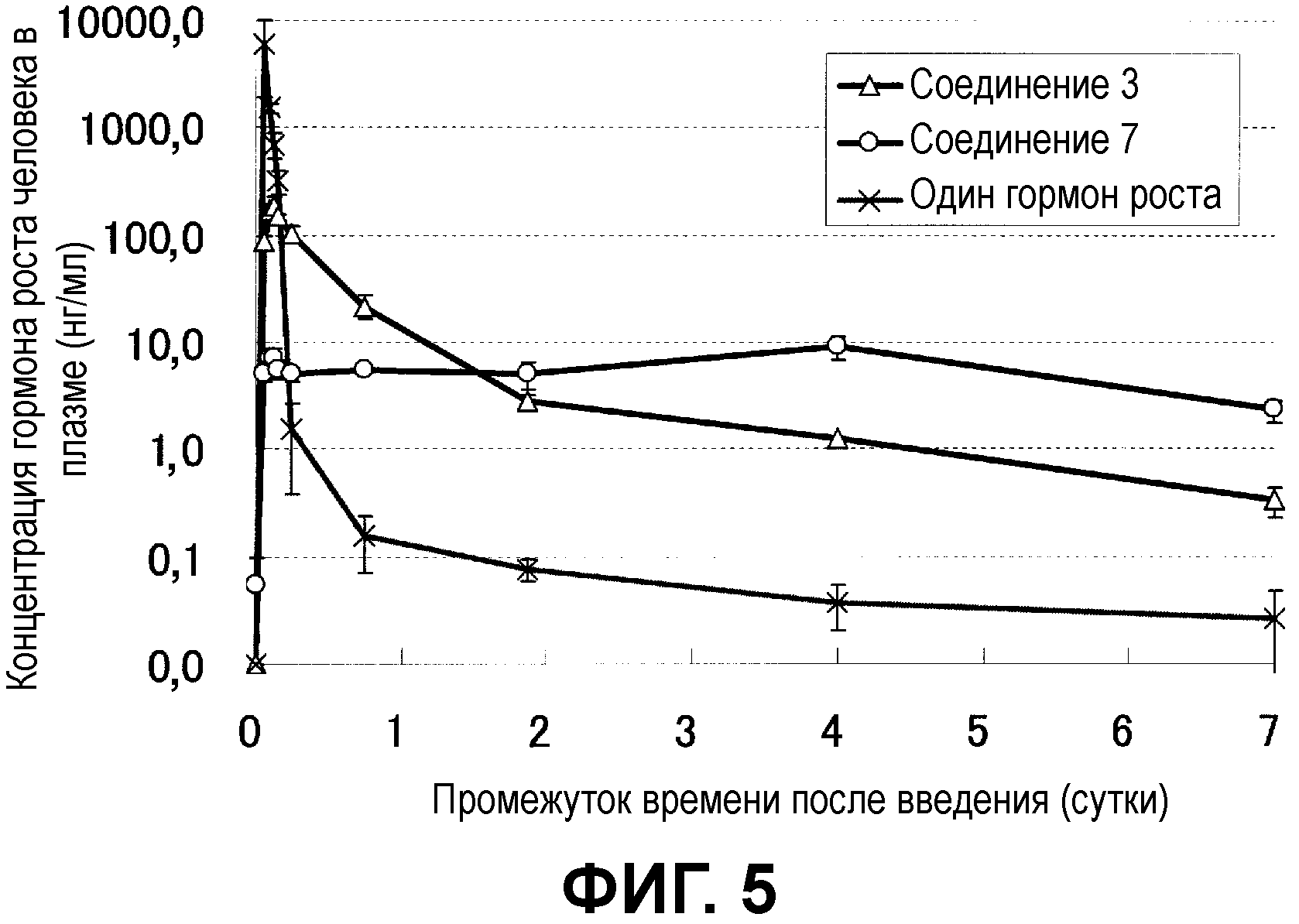
Фиг.5 показывает динамику изменений концентрации лекарственного средства в крови мыши, которой подкожно ввели частицы с инкапсулированным гормоном роста человека.
Фиг.6 показывает динамику изменений концентрации лекарственного средства в крови мыши, которой подкожно ввели микрочастицы с инкапсулированным гормоном роста человека.
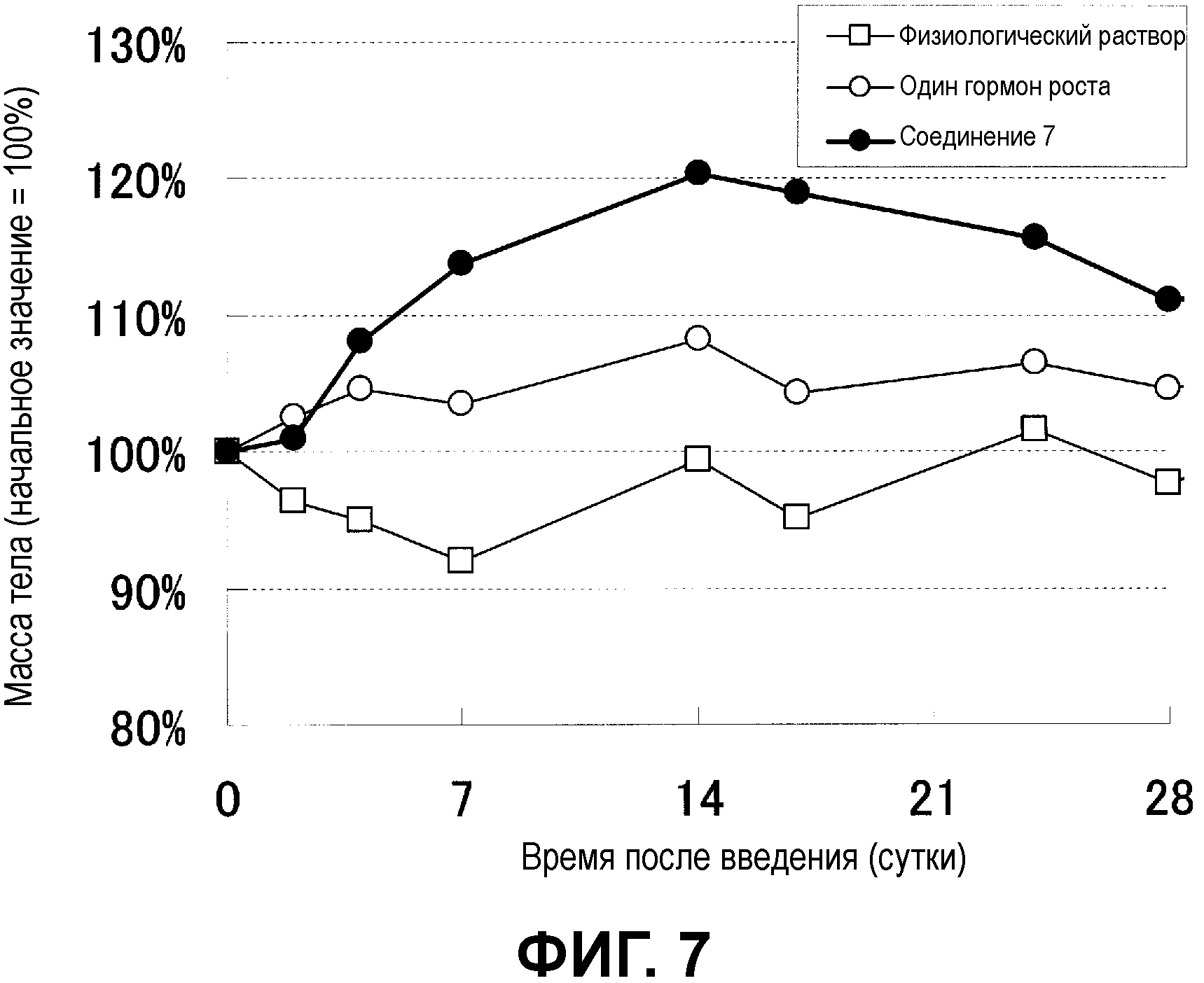
Фиг.7 показывает изменения массы тела мыши, которой подкожно ввели микрочастицы с инкапсулированным гормоном роста человека.
Фиг.8 показывает динамику изменений концентрации IGF-1 в крови мыши, которой подкожно ввели микрочастицы с инкапсулированным гормоном роста человека.
Фиг.9 показывает высвобождение лекарственного средства в буферный раствор из микрочастиц с инкапсулированным Эксендином-4.
Фиг.10 показывает динамику изменений концентрации лекарственного средства в крови мыши, которой подкожно ввели микрочастицы с инкапсулированным Эксендином-4.
Фиг.11 показывает высвобождение лекарственного средства из микрочастиц, состоящих из ассоциированных частиц, с инкапсулированным гормоном роста человека.
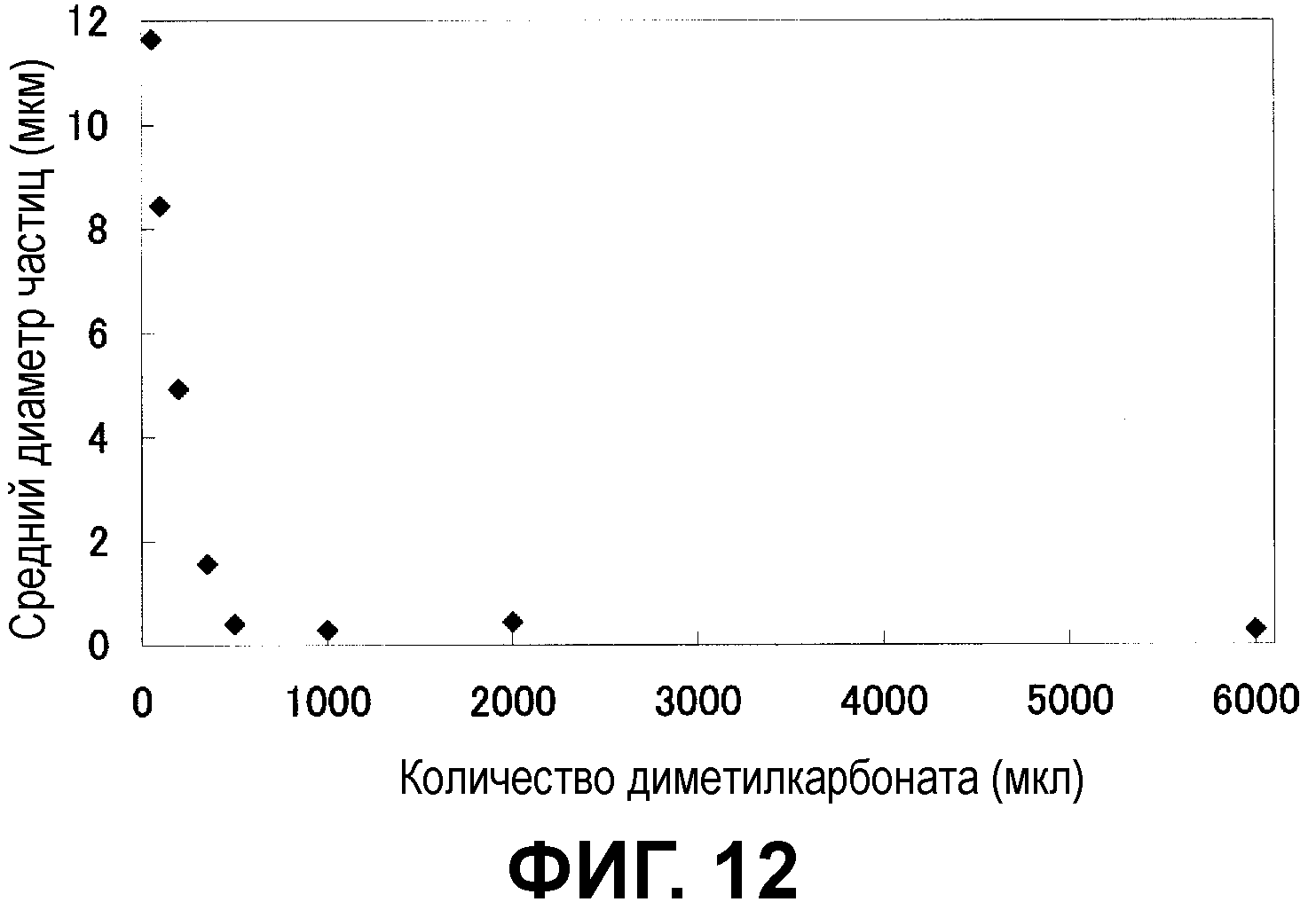
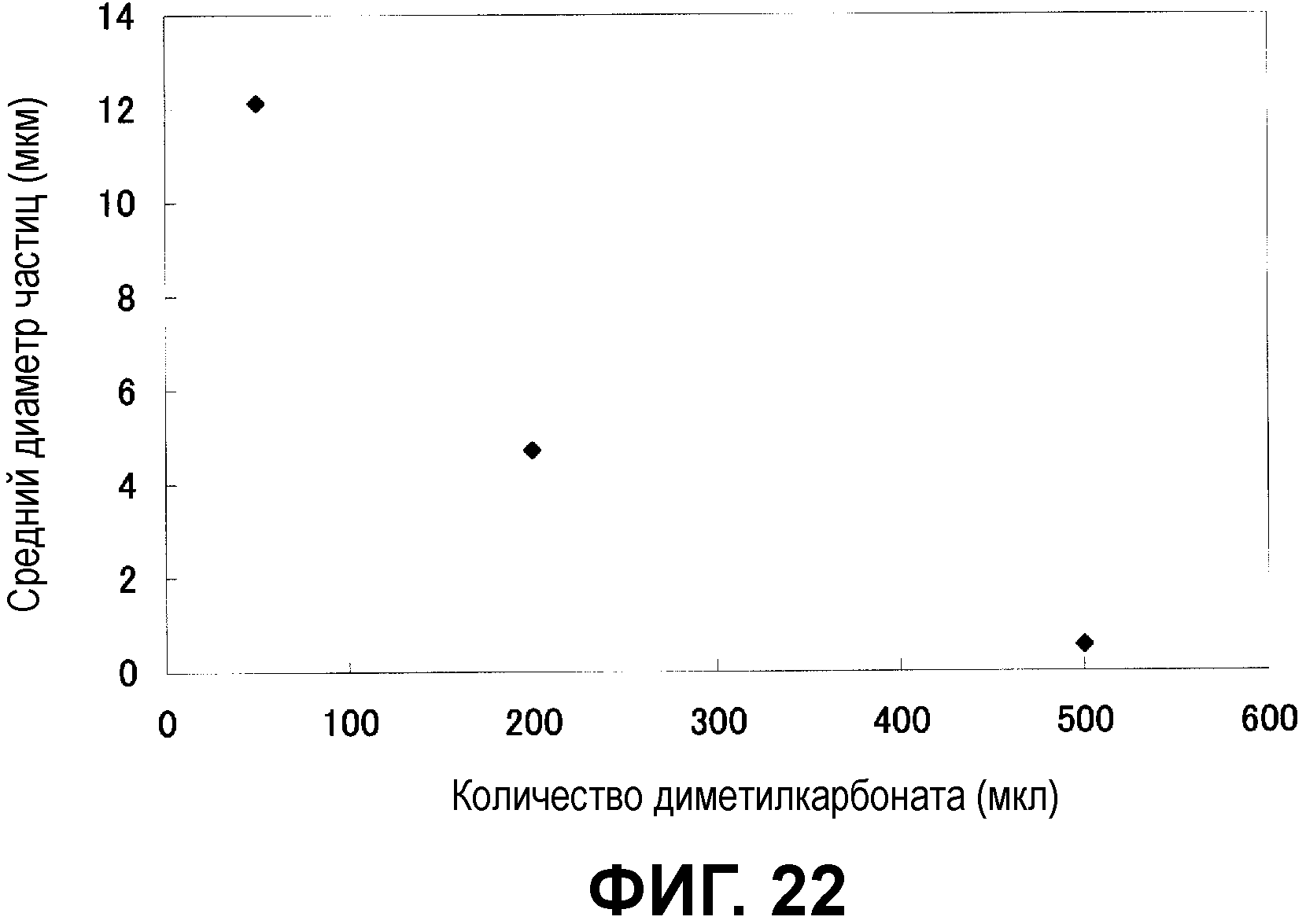
Фиг.12 показывает взаимосвязь между диаметром частицы и количеством диметилкарбоната, добавленного во время приготовления эмульсии типа S/O/W.
Фиг.13 показывает результаты по эффективности захватывания для микрочастиц с инкапсулированным FD40.
Фиг.14 показывает изображение СЭМ порошка микрочастиц, приготовленного из полимера PEG-PLGA (5k-10k).
Фиг.15 показывает изображение СЭМ порошка микрочастиц, приготовленного из полимера PEG-PLGA (5k-61k).
Фиг.16 показывает характер высвобождения FD40 из микрочастиц с инкапсулированным FD40.
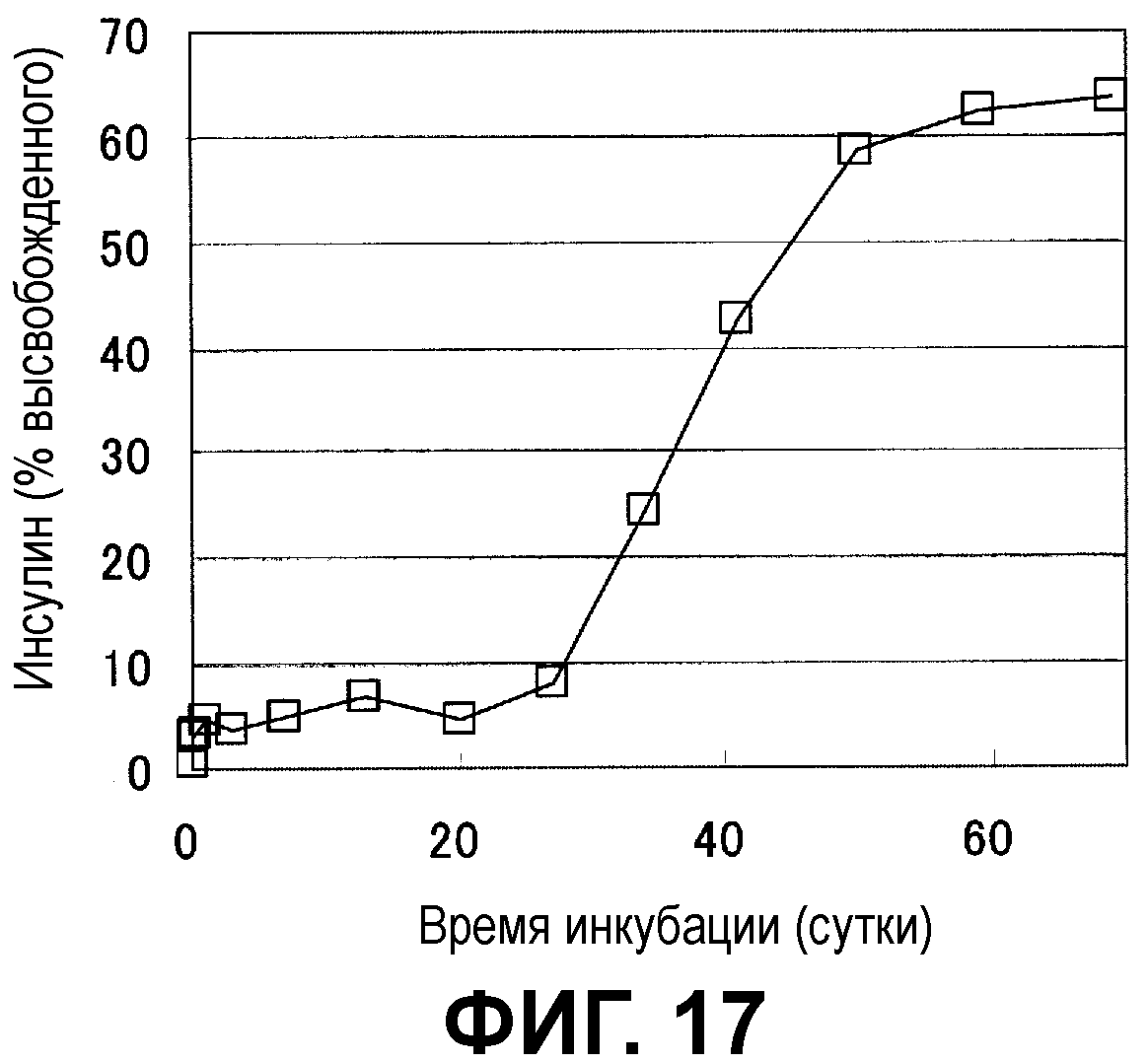
Фиг.17 показывает характер высвобождения лекарственного средства из микрочастиц с инкапсулированным человеческим инсулином.
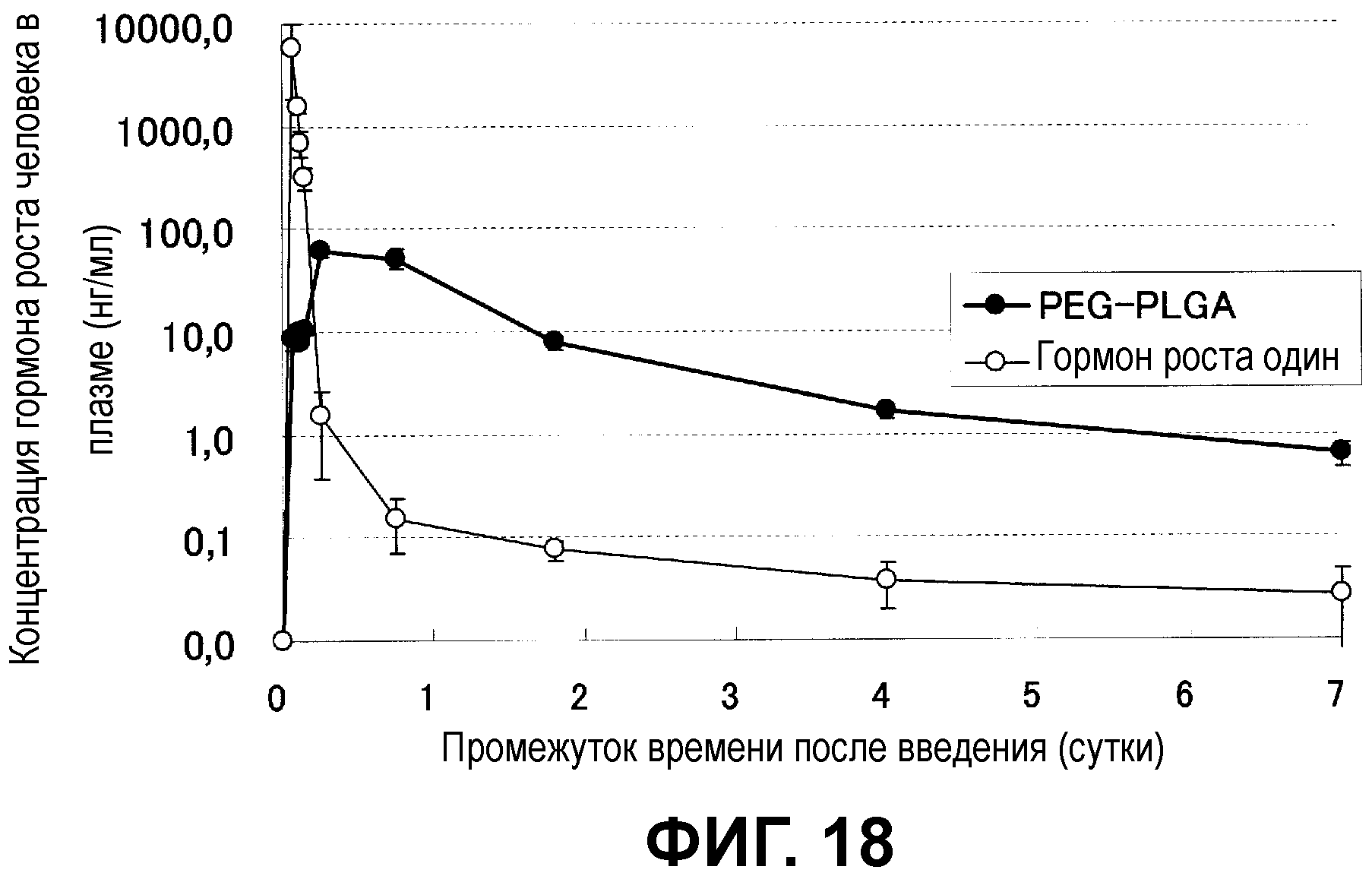
Фиг.18 показывает динамику изменений концентрации лекарственного средства в крови мыши, которой подкожно ввели микрочастицы с инкапсулированным гормоном роста человека.
Фиг.19 показывает динамику изменений концентрации лекарственного средства в крови мыши, которой подкожно ввели микрочастицы с инкапсулированным гормоном роста человека.
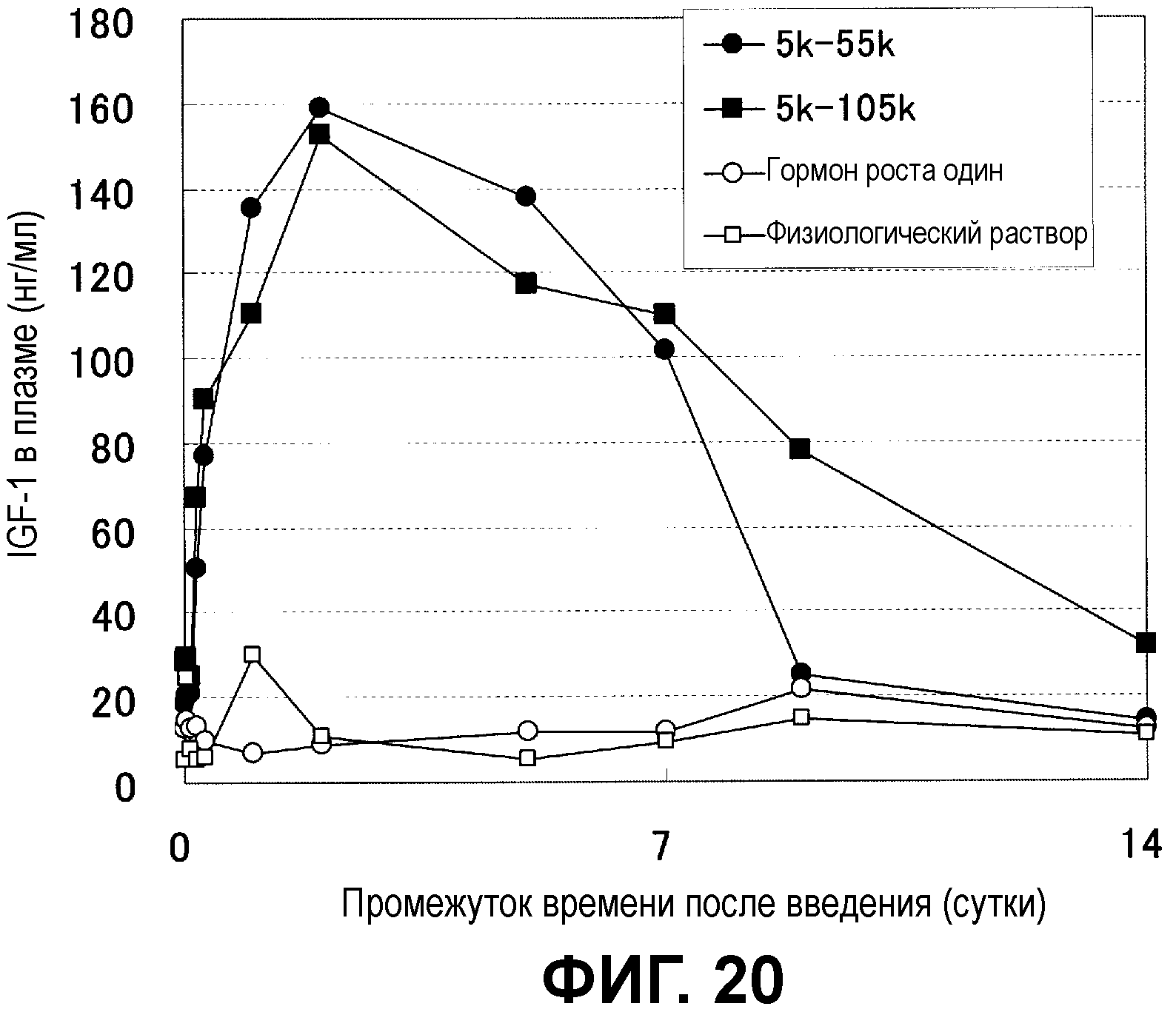
Фиг.20 показывает динамику изменений концентрации IGF-1 в крови мыши, которой подкожно ввели микрочастицы с инкапсулированным гормоном роста человека.
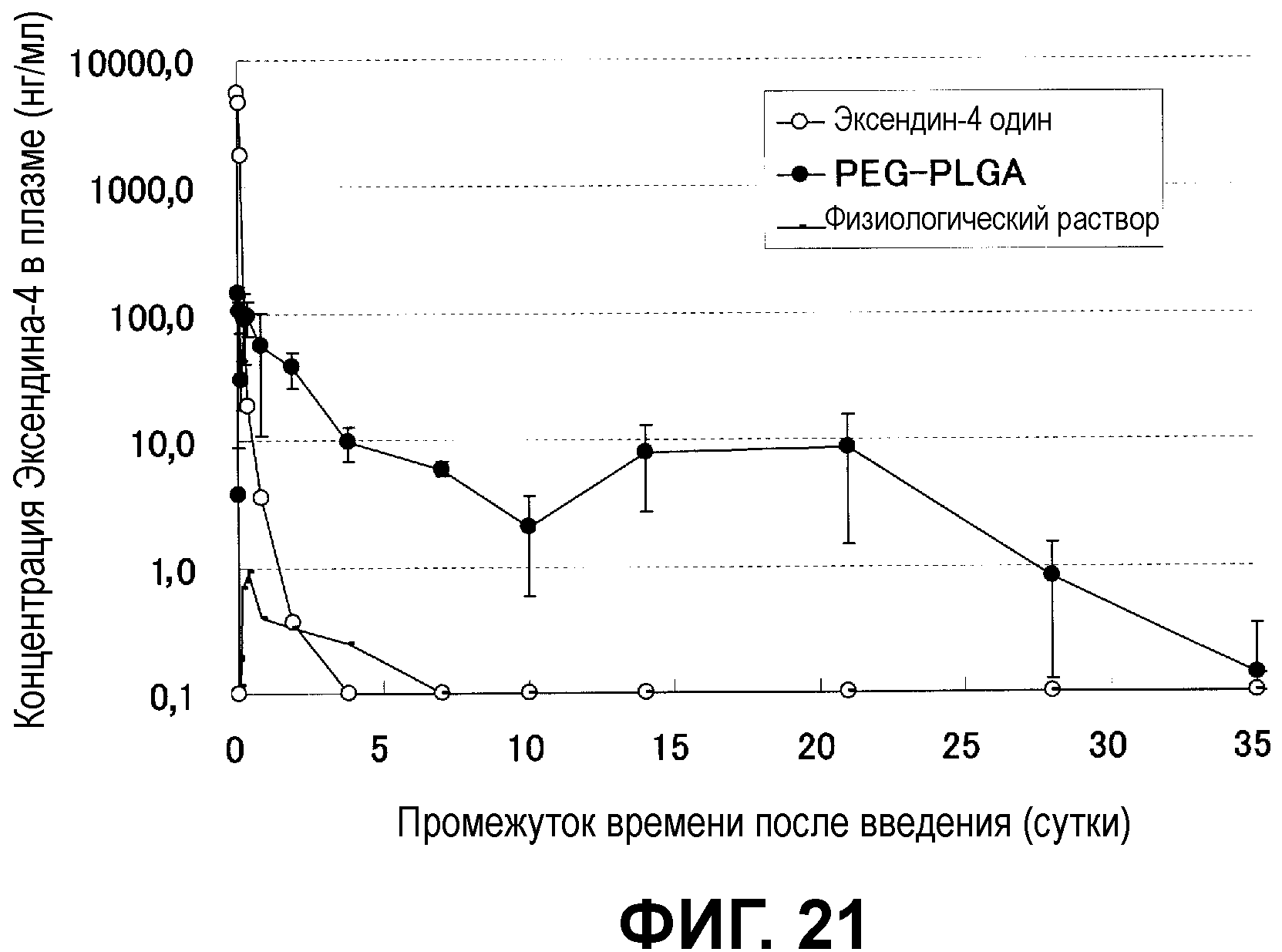
Фиг.21 показывает динамику изменений фармакокинетики в крови мыши, которой подкожно ввели микрочастицы с инкапсулированным Эксендином-4.
Фиг.22 показывает взаимосвязь между диаметром частицы и количеством диметилкарбоната, добавленного во время приготовления эмульсии типа S/O/W.
ЛУЧШИЙ СПОСОБ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Изобретение характеризуется формированием микрочастицы посредством агрегации частиц, содержащих гидрофильное активное вещество, причем частица включает амфифильный полимер и гидрофильное активное вещество. В настоящем описании, агрегация представляет собой связывание двух или более частиц посредством силы взаимодействия между частицами или другого вещества, и формирование группы. Сила взаимодействия между частицами конкретно не указывается, но приемлемые примеры включают гидрофобное взаимодействие, водородную связь и силу Ван-дер-Ваальса. Агрегация не ограничивается состоянием совместного контакта частиц, но между частицами могут присутствовать вещества, имеющие сродство к частицам, или частицы могут быть распределены в матрице. В качестве веществ, имеющих сродство к частицам, или матрицы, предпочтительным является полимер. В изобретении, агрегацией частиц, содержащих гидрофильное активное вещество, по сравнению с одиночной частицей, достигается эффект, состоящий в том, что эффективность инкапсулирования гидрофильного активного вещества выше. Диаметр частиц, содержащих гидрофильное активное вещество, которые должны ассоциироваться, является переменным параметром.
Микрочастицы представляют собой частицы, имеющие диаметр в диапазоне от субмикронного до субмиллиметрового. В изобретении, средний диаметр микрочастиц конкретно не ограничивается, но, в случае введения микрочастиц инъекцией в организм человека, чем больше средний диаметр частиц, тем больше игла шприца, и увеличивается нагрузка на пациента и, следовательно, с точки зрения уменьшения нагрузки на пациента, предпочтительно, чтобы он был в диапазоне от 1 мкм до 50 мкм. Средний диаметр микрочастиц можно определить анализом изображений, используя сканирующий электронный микроскоп.
Количество агломератов частиц, содержащих гидрофильное активное вещество, для составления микрочастицы, предпочтительно, находится в диапазоне от 10 до 10 в седьмой степени, более предпочтительно, в диапазоне от 10 в пятой степени до 10 в седьмой степени. Количество агломератов рассчитывают из среднего диаметра частиц, содержащих гидрофильное активное вещество, и среднего диаметра микрочастиц.
В изобретении, амфифильный полимер состоит из гидрофобного сегмента полигидроксикислоты и гидрофильного сегмента полисахаридов или полиэтиленгликоля. В настоящем описании амфифильное свойство является состоянием, имеющим как гидрофильные, так и гидрофобные свойства, и что касается гидрофильного свойства, когда растворимость в воде выше в определенном сегменте, чем в других сегментах, говорят, что такой сегмент является гидрофильным. Предпочтительно, гидрофильный сегмент растворим в воде, но если он трудно растворим, он является гидрофильным, если его растворимость в воде выше, чем у других сегментов. Определенный сегмент называют гидрофобным, если его растворимость в воде ниже, чем у других частей. Гидрофобный сегмент, предпочтительно, нерастворим в воде, но если он растворим, он может быть гидрофобным, если его растворимость в воде ниже, чем у других сегментов.
Конкретные примеры полигидроксикислоты амфифильного полимера включают полигликолевую кислоту, полимолочную кислоту, поли(2-гидроксимасляную кислоту), поли(2-гидроксивалериановую кислоту), поли(2-гидроксикапроновую кислоту), поли(2-гидроксикаприновую кислоту), полияблочную кислоту и производные и сополимеры данных высокомолекулярных соединений. Однако, поскольку желательно, чтобы микрочастицы по изобретению не оказывали существенного действия во время введения на организм человека, полигидроксикислота амфифильного полимера также предпочтительно является биосовместимым высокомолекулярным полимером. Биосовместимый высокомолекулярный полимер представляет собой вещество, не оказывающее значительных эффектов на организм человека при введении и, более конкретно, полулетальная доза LD50 предпочтительно составляет 2000 мг/кг или более при оральном введении высокомолекулярного полимера крысе.
В качестве полигидроксикислоты биосовместимого высокомолекулярного полимера предпочтительным является сополимер полимолочной кислоты и полигликолевой кислоты, или поли(молочная кислота-гликолевая кислота). Когда полигидроксикислота представляет собой сополимер полимолочной кислоты и полигликолевой кислоты, отношение компонентов сополимера полимолочной кислоты и полигликолевой кислоты (моль/моль%) конкретно не ограничивается, при условии, что достигаются цели изобретения, но данное отношение, предпочтительно, составляет от 10/0 до 30/70 или, более предпочтительно, от 60/40 до 40/60.
Когда гидрофильный сегмент амфифильного полимера представляет собой полисахариды, примеры полисахаридов могут включать целлюлозу, хитин, хитозан, геллановую камедь, альгиновую кислоту, гиалуроновую кислоту, пуллулан или декстран, и декстран является наиболее предпочтительным.
Амфифильный полимер, предпочтительно, получают привитой полимеризацией привитой(ых) цепи(ей) полигидроксикислоты на главную цепь полисахарида. В настоящем описании средняя молекулярная масса основной цепи полисахарида, предпочтительно, составляет от 1000 до 100000 или, более предпочтительно, от 2000 до 50000, и средняя молекулярная масса полигидроксикислоты, предпочтительно, составляет от 500 до 100000 или, более предпочтительно, от 1000 до 10000. Отношение средней молекулярной массы полигидроксикислоты к средней молекулярной массы полисахаридов, предпочтительно, составляет от 0,01 до 100, предпочтительно от 0,02 до 10 или, наиболее предпочтительно, от 0,02 до 1.
Количество привитых цепей полигидроксикислоты, связанных с основной цепью полисахаридов, предпочтительно, составляет от 2 до 50. Количество привитых цепей можно определить из средней молекулярной массы амфифильного полимера привитого типа, основной цепи полисахаридов и привитой цепи полигидроксикислоты.
Когда гидрофильный сегмент амфифильного полимера представляет собой полиэтиленгликоль, амфифильный полимер, предпочтительно, является блочным полимером полиэтиленгликоля и полигидроксикислоты. В изобретении термин ″блок″ относится к части сегмента полимерной молекулы, состоящей из, по меньшей мере, пяти или более мономерных звеньев, и являющейся отличной по химической структуре или конфигурации между данной частью сегмента и другой соседней частью сегмента, и полимер, образованный двумя или более блоками, связанными непосредственно, называют блок-полимером. Каждый блок, формирующий блок-полимер, может включать два или более мономерных звена, то есть может быть образован статистический, чередующийся или градиентный полимер. Когда гидрофильный сегмент амфифильного полимера представляет собой полиэтиленгликоль, амфифильный полимер, предпочтительно, представляет собой блок-полимер, связывающий блоки полиэтиленгликоля и полигидроксикислоты.
Когда гидрофильный сегмент амфифильного полимера представляет собой полиэтиленгликоль, конкретные примеры полиэтиленгликоля, который следует использовать, включают полиэтиленгликоль с прямой или разветвленной цепью или его производные, и предпочтительный пример производного полиэтиленгликоля представляет собой моноалкиловый эфир полиэтиленгликоля. Алкильная группа моноалкилового эфира полиэтиленгликоля представляет собой алкильную группу с прямой или разветвленной цепью, содержащую от 1 до 10 атомов углерода, и более предпочтительной является разветвленная алкильная группа, содержащая от 1 до 4 атомов углерода, и особенно предпочтительным являются метильные, этильные, пропильные и изопропильные группы.
Средняя молекулярная масса полиэтиленгликоля конкретно не ограничивается, но, предпочтительно, она составляет от 2000 до 15000, более предпочтительно, от 2000 до 12000, еще более предпочтительно, от 4000 до 12000 и, особенно предпочтительно, от 5000 до 12000.
Когда гидрофильный сегмент амфифильного полимера представляет собой полиэтиленгликоль, средняя молекулярная масса полигидроксикислоты конкретно не ограничивается, но, предпочтительно, она составляет от 5000 до 200000, более предпочтительно, от 15000 до 150000 или, еще более предпочтительно, от 20000 до 100000. Отношение средней молекулярной массы полигидроксикислоты к средней молекулярной массе полиэтиленгликоля, предпочтительно, составляет 1,0 или более, более предпочтительно, 2 или более, наиболее предпочтительно, 4 или более и, особенно предпочтительно, от 4 или более до 25 или менее.
В данном описании средняя молекулярная масса относится к среднечисленной молекулярной массе, если не указано иным образом, и среднечисленная молекулярная масса представляет собой среднюю молекулярную массу, вычисляемую методом, не учитывающим взвешивание абсолютной величины молекулы, и среднюю молекулярную массу амфифильного полимера, полисахаридов и полиэтиленгликоля можно получить в виде молекулярной массы, конвертированной в полистирол или пуллулан, измеренной гель-проникающей хроматографией (ГПХ). Среднюю молекулярную массу полигидроксикислоты можно определить из отношения пикового интегрального значения концевого остатка и пикового интегрального значения, отличного от концевого остатка, измеренного методом ядерного магнитного резонанса (1H-ЯМР).
Амфифильный полимер, состоящий из полисахаридов и полигидроксикислоты, используемых в изобретении, можно синтезировать любым известным методом, и при условии, что может сформироваться эмульсия с обращенной фазой, метод синтеза точно не указывается, и его можно изготовить, например, любым из следующих ниже методов (1), (2) и (3).
(1) В присутствии оловянного катализатора мономер, активирующий гидроксикислоту, добавляют к полисахаридам для осуществления реакции полимеризации, далее добавляют полигидроксикислоту, и получают амфифильный полимер привитого типа [Macromolecules, 31, 1032-1039 (1998)].
(2) Гидроксильную группу полисахаридов с частично снятой защитой, большинство гидроксильных групп которого защищено заместителем, активируют основанием, добавляют мономер, активирующий гидроксикислоту, чтобы образовать привитую(ые) цепь(и), состоящую из полигидроксикислоты и, наконец, удаляют защитную группу, и получают амфифильный полимер привитого типа [Polymer, 44, 3927-3933 (2003)].
(3) В полисахариды добавляют сополимер полигидроксикислоты, чтобы осуществить реакцию конденсации, используя дегидрирующий агент и/или функциональный активирующий агент, и получают амфифильный полимер привитого типа [Macromolecules, 33, 3680-3685 (2000)].
Амфифильный полимер, состоящий из полиэтиленгликоля и полигидроксикислоты, используемый в изобретении, можно синтезировать любым известным методом и, при условии, что можно получить эмульсию с обращенной фазой, метод синтеза не конкретизируется и, например, в присутствии оловянного катализатора мономер, активирующий гидроксикислоту, добавляют к полиэтиленгликолю для осуществления реакции полимеризации, чтобы получить полигидроксикислоту, и изготавливают амфифильный блок-полимер [Journal of Controlled Release, 71, 203-211 (2001)].
Структура содержащей гидрофильное активное вещество частицы, включающей амфифильный полимер и гидрофильное биологически активное вещество, конкретно не ограничивается, но поскольку частица, содержащая гидрофильное активное вещество, имеет гидрофильный сегмент амфифильного полимера во внутренней части и имеет внешний слой гидрофобного сегмента амфифильного полимера, это является предпочтительным, так как содержащееся гидрофильное активное вещество можно удерживать более стабильно.
Когда частица, содержащая гидрофильное активное вещество, представляет собой частицу, имеющую гидрофильный сегмент амфифильного полимера во внутренней части и имеющую внешний слой гидрофобного сегмента амфифильного полимера, одним из предпочтительных вариантов осуществления является, если модификатор поверхности связан с внешним слоем полигидроксикислоты. В настоящем изобретении связывание может представлять собой либо нековалентное связывание, либо ковалентное связывание. Нековалентное связывание, предпочтительно, представляет собой гидрофобное взаимодействие, но может включать электростатическое взаимодействие, водородную связь или силы Ван-дер-Ваальса, или их комбинации. При нековалентном связывании гидрофобный внешний слой мелкодисперсных частиц, содержащих амфифильный полимер, и гидрофобная часть модификатора поверхности, описанного ниже, предпочтительно, могут быть связаны друг с другом гидрофобным взаимодействием. В данном случае, диспергатор мелкодисперсных частиц, особенно предпочтительно, представляет собой воду, буферный раствор, физиологический солевой раствор, водный раствор модификатора поверхности или диспергатор мелкодисперсных частиц на основе гидрофильного растворителя.
Модификатор поверхности, предпочтительно, представляет собой соединение, способное стабилизировать поверхность раздела вода-масло эмульсии типа S/O/W, или поверхность раздела эмульсии масло-масло эмульсии типа S/O1/O2 и, более предпочтительно, соединение, обладающее свойствами усиливать коллоидную стабильность микрочастиц. Модификатор поверхности может быть одного типа или смесью множества типов. В настоящем описании свойство увеличивать коллоидную стабильность означает предотвращение или замедление агрегации микрочастиц в растворителе.
В настоящем изобретении модификатор поверхности, предпочтительно, представляет собой амфифильное соединение или гидрофильный полимер.
Гидрофильный полимер модификатора поверхности по изобретению, предпочтительно, выбран из группы, состоящей из полиэтиленгликоля, поливинилпирролидона, поливинилового спирта, полиэтиленимина, полиакриловой кислоты, полиметакриловой кислоты, поли-1,3-диоксолана, полимера 2-метакрилоилоксиэтилфосфорилхолина, поли-1,3,6-триоксана, полиаминокислоты, пептида, белка, сахаридов и их аналогов.
Аналоги гидрофильного полимера могут включать поверхностно-активное вещество, содержащее гидрофильный полимер, частично модифицированный гидрофобной группой, такой как алкил с длинной цепью, но они конкретно не ограничиваются этим.
В качестве полиэтиленгликолевого аналога модификатора поверхности по изобретению предпочтительно использовать Pluronic (зарегистрированный товарный знак), продаваемый BASF, или его эквиваленты.
В качестве полиаминокислоты модификатора поверхности по изобретению можно предпочтительно использовать полиаспаргиновую кислоту, полиглутаминовую кислоту или их аналоги. Аналог, водящий алкильную группу с длинной цепью в часть полиаспаргиновой кислоты или полиглутаминовой кислоты, является особенно предпочтительным.
В качестве пептида модификатора поверхности по изобретению можно использовать основной пептид.
В качестве белка модификатора поверхности по изобретению, предпочтительным для улучшения характеристик дисперсии частиц является желатин, казеин или альбумин. В качестве белка, одним из предпочтительных примеров является антитело.
В качестве сахаридов модификатора поверхности по изобретению предпочтительными являются моносахариды, олигосахариды и полисахариды. В качестве полисахаридов предпочтительными являются целлюлоза, хитин, хитозан, геллановая камедь, альгиновая кислота, гиалуроновая кислота, пуллулан и декстран. В частности, холестерил пуллулан является предпочтительным, принимая с точки зрения лучшей диспергируемости частиц. Предпочтительным являются аналоги любого вещества, выбранного из группы, состоящей из целлюлозы, хитина, хитозана, геллановой камеди, альгиновой кислоты, гиалуроновой кислоты, пуллулана и декстрана.
В качестве модификатора поверхности данные примеры пептида, белка и сахаридов, особенно предпочтительно, являются аналогами, частично модифицирующими гидрофобную группу алкила с длинной цепью, или аналогами, модифицирующими гидрофильный полимер или амфифильное соединение.
В модификаторе поверхности по изобретению амфифильное соединение включает липид в качестве одного из предпочтительных примеров.
В модификаторе поверхности по изобретению амфифильное соединение включает поверхностно-активное вещество в качестве одного из предпочтительных примеров. Предпочтительные примеры поверхностно-активного вещества включают: неионные активные вещества, такие как сополимер полиоксиэтилена и полипропиленгликоля, сложный эфир сахарозы и жирной кислоты, сложный эфир полиэтиленгликоля и жирной кислоты, сложный эфир полиоксиэтиленсорбитана и одноосновной жирной кислоты, сложный эфир полиоксиэтиленсорбитана и двухосновной жирной кислоты, сложный эфир полиоксиэтиленглицерина и одноосновной жирной кислоты, сложный эфир полиоксиэтиленглицерина и двухосновной жирной кислоты, полиглицерин жирную кислоту, полиоксиэтилен касторовое масло, полиоксиэтилен отвержденное касторовое масло; алкилсульфаты, такие как натрий лаурилсульфат, аммоний лаурилсульфат, натрий стеарилсульфат; или лецитин.
В изобретении гидрофильное активное вещество иллюстрируется низкомолекулярным соединением, белком, пептидом, ДНК, РНК или модифицирующей нуклеиновой кислотой. В микрочастицу по изобретению можно заключить даже гидрофобные лекарственные средства, если сделать их гидрофильными, используя солюбилизирующий агент. Солюбилизирующий агент в настоящем изобретении, предпочтительно, включает циклодекстрин и его аналоги.
Белок или пептид, используемый в изобретении в качестве гидрофильного активного вещества, конкретно не ограничивается, но предпочтительным является биоактивный белок или биоактивный пептид. Биоактивный белок или биоактивный пептид включает пептидный гормон, цитокин, ферментный белок или антитело. И конкретные примеры включают: пептид антагонист ГПП-1 рецептора, такой как эксендин-4, паратироидный гормон (ПТГ), кальцитонин, инсулин, инсулиноподобный фактор роста, ангиотензин, глюкагон, ГПП-1; бомбезин, мотилин, гастрин, гормон роста, пролактин (лютеотропный гормон), гонадотропин (гонадотропный гормон), тиротропный гормон, адренокортикотропный гормон (ACTH), производное ACTH (эбиратид), меланоцит-стимулирующий гормон, фолликуло-стимулирующий гормон (ФСГ), серморелин, вазопрессин, окситоцин, протирелин, лютеинизирующий гормон (ЛГ), кортикотропин, секретин, соматропин, тиротропин (тироидный стимулирующий гормон), стоматостатин, гонадотропин-высвобождающий гормон (GnRH), G-CSF, эритропоэтин (EPO), тромбопоэтин (ТРО), потенциатор мегакариоцитов, фактор роста гепатоцитов, ЭФР, фактор роста эндотелия сосудов, интерферон-α, интерферон-β, интерферон-γ, интерлейкины, ФРФ (фактор роста фибропластов), BMP (морфогенетический белок кости), гуморальный фактор тимуса (THF), сывороточный тимический фактор (FTS), супероксид димустазу (СОД), урокиназу, лизоцим, тканевой активатор плазминогена, аспарагиназу, калликреин, грелин, адипонектин, лептин, предсердный натрийуретический пептид, предсердный натрийуретический фактор, мозговой натрийуретический пептид (BNP), конантокин G, динорфин, эндорфин, киоторфин, энкефалин, нейротензин, ангиостин, брадикинин, вещество Р, калидин, гемоглобин, протеин C, фактор VIIa, гликоцеребросидазу, стрептокиназу, стафилокиназу, тимозин, панкреозимин, холецистокинин, плацентарный лактоген человека, фактор некроза опухолей (ФНО), полимиксин В, холистин, грамицидин, бацитрацин, тимопоэтин, бомбецин, церулеин, тимостимулин, секретин, резистин, гепцидин, нейропептид Y, нейропептид S, холецистокинин-панкреозимин (CCK-PZ), нейротрофический фактор головного мозга (BDNF), вакцину и аналогичное. Данные биоактивные белки или биоактивные пептиды могут являться природными белками или пептидами или производными, модифицированными в части их последовательности, или соединениями, модифицированными цепью полиэтиленгликоля или сахара.
Когда гидрофильное активное вещество представляет собой ДНК, РНК или модифицирующую нуклеиновую кислоту, оно может быть любым катионным поверхностно-активным веществом, катионным липидом, катионным полимером, или другими соединениями, образующими комплексы с их аналогами.
В изобретении сахариды, используемые в качестве гидрофильного активного вещества, включают гиалуроновую кислоту, гепарин, декстран сульфат, декстран или меченный ФИТЦ декстран (например, FD40 и т.д.).
Изобретение также относится к способу изготовления микрочастицы, сформированной агрегацией частиц, содержащих гидрофильное активное вещество, причем способ включает:
(a) стадию формирования эмульсии с обращенной фазой смешиванием водного растворителя, включающего гидрофильное активное вещество, и несмешивающегося с водой органического растворителя, растворяющего амфифильный полимер,
(b) стадию получения сухого остатка, содержащего гидрофильное активное вещество, посредством удаления растворителя из эмульсии с обращенной фазой, и
(c) стадию введения сухого остатка или дисперсии жидкости, содержащей сухой остаток, в жидкую фазу, содержащую модификатор поверхности.
В способе изготовления микрочастицы, сформированной агрегацией частиц, содержащих гидрофильное активное вещество, по изобретению, эмульсию с обращенной фазой формируют, добавляя водный растворитель, содержащий гидрофильное активное вещество к несмешивающемуся с водой органическому растворителю, растворяющему амфифильный полимер, и перемешивая их. Если необходимо, можно использовать, например, перемешивающее устройство, такое как, магнитная мешалка, турбинное перемешивающее устройство, гомогенизатор или мембранное эмульгирующее устройство, снабженное пористой пленкой. Несмешивающийся с водой органический растворитель в изобретении представляет собой органический растворитель, растворимость которого в воде составляет 30 г (несмешивающийся с водой органический растворитель)/100 мл (воды) или менее, а другие органические растворители, растворимость которых в воде выше, чем указанное значение, характеризуются как смешивающиеся с водой органические растворители.
В качестве водного раствора в стадии (a) используют воду или водный раствор, содержащий водорастворимое вещество. Водорастворимое вещество может представлять собой любое вещество из группы, состоящей из неорганических солей, сахаридов, органических солей, аминокислоты и аналогичного.
Свойство несмешивающегося с водой органического растворителя в стадии (a) конкретно не ограничивается, но, предпочтительно, он представляет собой растворитель, способный растворять полигидроксикислоту в качестве гидрофобного сегмента амфифильного полимера, и с трудом растворять или не растворять гидрофильный сегмент. Несмешивающийся с водой органический растворитель, предпочтительно, рассеивается и удаляется сушкой вымораживанием или аналогичным и, предпочтительно, составляет 0,1 г (несмешивающегося с водой органического растворителя)/100 мл (воды) или менее. Конкретные примеры несмешивающегося с водой органического растворителя включают этилацетат, изопропилацетат, бутилацетат, диметилкарбонат, диэтилкарбонат, метиленхлорид и хлороформ. Отношение несмешивающегося с водой органического растворителя к водному растворителю составляет, предпочтительно, от 1000:1 до 1:1, более предпочтительно, от 100:3 до 3:1. Концентрация амфифильного полимера в несмешивающемся с водой органическом растворителе варьируется от типа несмешивающегося с водой органического растворителя или амфифильного полимера, но, предпочтительно, составляет от 0,01 до 90% (масс./масс.), более предпочтительно от 0,1 до 50% (масс./масс.) или, еще более предпочтительно, от 1 до 20% (масс./масс.).
На стадии (a) в процессе формирования эмульсии с обращенной фазой водным растворителем, содержащим гидрофильное активное вещество, и несмешивающимся с водой органическим растворителем, растворяющим амфифильный полимер, в зависимости от фармакологической цели эмульсию с обращенной фазой можно получить, используя несмешивающийся с водой органический растворитель, растворяющий два или более типов амфифильного полимера.
На стадии (a) в процессе формирования эмульсии с обращенной фазой водным растворителем, содержащим гидрофильное активное вещество, и несмешивающимся с водой органическим растворителем, растворяющим амфифильный полимер, чтобы содействовать формирования эмульсии с обращенной фазой и чтобы сформировать однородную и тонкую эмульсию с обращенной фазой, можно добавить вспомогательный агент. Таким вспомогательным агентом, предпочтительно, может быть соединение, выбранное из группы, состоящей из алкилового спирта, содержащего от 3 до 6 атомов углерода, алкиламина, содержащего от 3 до 6 атомов углерода, и алкиловой карбоновой кислоты, содержащей от 3 до 6 атомов углерода. Структура алкильной цепи данных вспомогательных агентов конкретно не уточняется, и можно применить либо структуру с прямой цепью, либо разветвленную структуру, или можно использовать насыщенный алкил или ненасыщенный алкил. В частности, в изобретении предпочтительными в качестве вспомогательного агента являются трет-бутанол, изобутанол и пентанол.
Средний диаметр частиц эмульсии с обращенной фазой на стадии (a) варьируется с диаметром частицы желаемой микрочастицы по изобретению и конкретно не ограничивается, но для изготовления микрочастицы для фармацевтического препарата, что является одной из областей использования микрочастицы по изобретению, верхний предел среднего диаметра частиц, предпочтительно, составляет 50 мкм, более предпочтительно, 5 мкм, еще более предпочтительно, 500 нм, особенно предпочтительно, 150 нм и, наиболее предпочтительно, 100 нм. Нижний предел среднего диаметра частиц эмульсии с обращенной фазой, предпочтительно, составляет 10 нм или, более предпочтительно, 50 нм.
Далее, в способе изготовления микрочастицы важно включить стадию (b) для получения сухого остатка, содержащего гидрофильное активное вещество, удалением растворителя из эмульсии с обращенной фазой, полученной в стадии (a).
На стадии (b) метод удаления растворителя из эмульсии с обращенной фазой конкретно не ограничивается, но может включать, например, нагревание, высушивание в вакууме, диализ, сушку вымораживанием, операцию центрифугирования, фильтрование, повторное осаждение и их комбинации.
Среди данных методов удаления растворителя из эмульсии с обращенной фазой особенно предпочтительной является сушка вымораживанием, поскольку она приводит к незначительным структурным изменениям вследствие объединения частиц в эмульсии с обращенной фазой, и способна избежать деградации гидрофильного активного вещества из-за высокой температуры. Условие и устройство для сушки вымораживанием включают процесс замораживания и процесс сушки при пониженном давлении, и особенно предпочтительно, способ состоит из предварительной стадии замораживания в качестве обычного метода сушки вымораживанием, стадии первичной сушки при пониженном давлении и низкой температуре и стадии вторичной сушки при пониженном давлении. Например, охлаждая и замораживая ниже температуры плавления водного раствора и несмешивающегося с водой органического растворителя, для составления эмульсии с обращенной фазой, и затем высушивая при пониженном давлении, получают подвергнутый сушке вымораживанием сухой остаток, содержащий гидрофильное активное вещество. Температуру предварительного замораживания можно надлежащим образом определить экспериментом, принимая во внимание состав растворителя и, как правило, предпочтительно, она составляет -20°C или менее. Степень пониженного давления в процессе сушки можно надлежащим образом определить экспериментом, принимая во внимание состав растворителя и, как правило, предпочтительно, оно составляет 3000 Па или менее или, более предпочтительно, 500 Па или менее, для сокращения времени сушки. Для сушки вымораживанием, предпочтительно, использовать лабораторное устройство для сушки вымораживанием, имеющее холодную ловушку и присоединяемое к вакуумному насосу, или вакуумную установку для сушки вымораживанием стеллажного типа, используемую при изготовлении фармацевтических препаратов, и после предварительного замораживания посредством жидкого азота или хладагента, выполняют сушку при пониженном давлении при пониженной температуре или при комнатной температуре, используя вакуумный насос или другое устройство для уменьшения давления.
Сухой остаток, содержащий гидрофильное активное вещество, полученный в стадии (b), получают в виде агрегата частиц, содержащих гидрофильное активное вещество, включающих амфифильный полимер, причем данные агрегаты соответствуют структуре эмульсии с обращенной фазой. В настоящем описании агрегат представляет собой неупорядоченную массу, накапливающую мелкодисперсные частицы силой взаимодействия между частицами, и она четко отличается по форме от микрочастицы по изобретению. Средний диаметр мелкодисперсных частиц, содержащих гидрофильное активное вещество, для формирования данного агрегата меняется в зависимости от диаметра желаемой микрочастицы по изобретению, и конкретно не ограничивается, но для изготовления микрочастицы для фармацевтического препарата, что является одной из областей использования микрочастицы по изобретению, верхний предел среднего диаметра частиц, предпочтительно, составляет 50 мкм, более предпочтительно, 5 мкм, наиболее предпочтительно, 500 нм, особенно 150 нм, в частности, 100 нм. Нижний предел среднего диаметра мелкодисперсных частиц, содержащих гидрофильное активное вещество, составляет, предпочтительно, 10 нм или, более предпочтительно, 50 нм.
В способе изготовления микрочастицы по изобретению важно включить стадию (c) введения сухого остатка, содержащего гидрофильное активное вещество, или жидкой дисперсии, содержащей сухой остаток, в жидкую фазу, содержащую модификатор поверхности.
На стадии (c) метод введения сухого остатка или жидкой дисперсии, содержащей сухой остаток, в жидкую фазу, содержащую модификатор поверхности, включает, например, метод добавления сухого остатка в жидкую фазу, содержащую модификатор поверхности, и метод диспергирования сухого остатка сразу в дисперсионной среде, и добавления полученной жидкой дисперсии (суспензии твердое вещество в масле (S/O)) в жидкую фазу, содержащую модификатор поверхности.
При диспергировании сухого остатка, содержащего гидрофильное активное вещество, сразу в дисперсионной среде, дисперсионная среда конкретно не ограничивается, но, предпочтительно, представляет собой растворитель, способный растворять полигидроксикислоту, но по существу не способный растворять гидрофильный сегмент, составляющий амфифильный полимер, для цели сохранения структуры частицы, содержащей гидрофильное активное вещество, состоящей из амфифильного полимера, имеющего структуру эмульсии с обращенной фазой, для составления сухого остатка, содержащего гидрофильное активное вещество. Растворитель, способный растворять полигидроксикислоту, но по существу не способный растворять гидрофильный сегмент, является растворителем, в котором растворимость гидрофильного сегмента составляет 50 мг/мл или менее, предпочтительно, 10 мг/мл или менее.
Дисперсионная среда может представлять собой любой несмешивающийся с водой органический растворитель или смешивающийся с водой органический растворитель при условии, что он обладает указанными выше чертами, и более предпочтительным является несмешивающийся с водой органический растворитель. Конкретные примеры несмешивающегося с водой органического растворителя, способного растворять полигидроксикислоту амфифильного полимера, но по существу не растворяющегося в гидрофильном сегменте, включают этилацетат, изопропилацетат, бутилацетат, диметилкарбонат, диэтилкарбонат, метиленхлорид, хлороформ, диоксан, толуол и ксилол.
Дисперсионная среда для диспергирования сухого остатка, содержащего гидрофильное активное вещество, может содержать различные добавки, растворимые в дисперсионной среде, для цели контроля скорости высвобождения гидрофильного активного вещества вследствие разложения или дезинтеграции частиц, содержащих гидрофильное активное вещество, например, кислотное соединение, основное соединение, амфифильный полимер или биоразложимый полимер.
Жидкая фаза в стадии (с), предпочтительно, способна растворять модификатор поверхности, и имеет более высокую температуру кипения, чем дисперсионная среда сухого остатка, содержащего гидрофильное активное вещество, и может включать водный растворитель, несмешивающийся с водой органический растворитель и смешивающийся с водой органический растворитель. В настоящем изобретении водный растворитель представляет собой воду, или водный раствор, содержащий водорастворимый компонент, и водорастворимый компонент включает, например, неорганические соли, сахариды, органические соли и аминокислоты; несмешивающийся с водой органический растворитель включает, например, силиконовое масло, кунжутное масло, соевое масло, кукурузное масло, хлопковое масло, кокосовое масло, льняное масло, минеральное масло, касторовое масло, отвержденное касторовое масло, жидкий парафин, н-гексан, н-гептан, глицерин и олеиновое масло; и смешивающийся с водой органический растворитель включает, например, глицерин, ацетон, этанол, уксусную кислоту, дипропиленгликоль, триэтаноламин и триэтиленгликоль. В изобретении жидкая фаза в стадии (c), предпочтительно, представляет собой водный растворитель или смешивающийся с водой органический растворитель. Когда жидкая фаза представляет собой водный растворитель, и дисперсионная среда представляет собой несмешивающийся с водой органический растворитель, суспензия, получаемая в стадии (c) представляет собой так называемую эмульсию типа твердое вещество-масло-вода (S/O/W), а когда жидкая фаза представляет собой несмешивающийся с водой органический растворитель или смешивающийся с водой органический растворитель, и является несмешивающейся в дисперсионной среде, она представляет собой эмульсию типа твердое вещество-масло-масло (S/O1/O2).
Объемное отношение жидкой фазы к дисперсионной среде для диспергирования сухого остатка, содержащего гидрофильное активное вещество, как правило, составляет от 1000:1 до 1:1000 или, предпочтительно, от 100:1 до 1:100.
Концентрация модификатора поверхности в жидкой фазе по изобретению различается в зависимости от типа модификатора поверхности и, предпочтительно, составляет от 0,01 до 90% (масс./об.), более предпочтительно, от 0,1 до 50% (масс./об.) и, еще долее предпочтительно, от 5 до 10% (масс./об.).
Модификатор поверхности может быть связан с полигидроксикислотным внешним слоем амфифильного полимера микрочастицы по изобретению, и связывающее количество в данном случае, предпочтительно, составляет от 0,0001% до 1% от массы микрочастицы.
В жидкую фазу на стадии (c), кроме модификатора поверхности, могут быть добавлены различные добавки в зависимости от фармакологической цели, такие как буферный агент, антиоксидант, соль, полимер или сахар.
На стадии (c) также предпочтительно в жидкую фазу добавить неорганические соли. Предпочтительно, неорганические соли представляют собой соль щелочного металла или соль щелочноземельного металла, и особенно предпочтительным является хлорид натрия. Концентрация неорганических солей в жидкой фазе, предпочтительно, составляет от 0 до 1 М, более предпочтительно, от 10 мМ до 1 М или, еще более предпочтительно, от 10 мМ до 100 мМ.
На стадии (c), чтобы изготовить микрочастицу меньшего размера, образованную эмульсию типа твердое вещество-масло-вода (S/O/W) или эмульсию типа твердое вещество-масло-масло (S/O1/O2) можно обработать операцией эмульгирования. Метод эмульгирования конкретно не ограничивается при условии, что можно получить стабильную эмульсию. Например, метод включает метод перемешивания или метод с использованием гомогенизатора высокого давления, или высокоскоростного смесителя-гомогенизатора.
На стадии (c), когда жидкая дисперсия получена диспергированием сухого остатка, содержащего гидрофильное активное вещество, как только в дисперсионную среду добавляют в жидкую фазу, содержащую модификатор поверхности, удаляя дисперсионную среду, получают желаемую суспензию сформированных микрочастиц посредством агрегации частиц, содержащих гидрофильное активное вещество. Метод удаления дисперсионной среды конкретно не ограничивается, но он может включать способы сушки в жидкости, диализа, сушки вымораживанием, операцию центрифугирования, фильтрование, повторное осаждение, и особенно предпочтительными могут быть сушка в жидкости и сушка вымораживанием. На стадии (c), когда в качестве жидкой фазы используют водный растворитель, в данном процессе получают водный диспергатор микрочастиц.
Удаляя жидкую фазу из диспергатора микрочастиц, полученного в данном способе, можно получить микрочастицу по изобретению. Метод удаления жидкой фазы конкретно не ограничивается, но, предпочтительно, может включать методы отгонки выпариванием, диализа, сушки вымораживанием, операцию центрифугирования и фильтрование.
Области применения микрочастицы, полученной в изобретении, являются обширными и разносторонними, и в частности она используется в качестве фармацевтического препарата. Когда микрочастицу по изобретению используют в качестве фармацевтического препарата, кроме микрочастиц могут содержаться различные фармакологические полезные добавки, и применимые добавки включают буферный агент, антиоксидант, соль, полимер или сахар.
Когда микрочастицу по изобретению используют в качестве фармацевтического препарата, способ введения включает, например, оральное введение и парентеральное введение, и парентеральное введение является предпочтительным. Парентеральное введение включает подкожное введение, внутримышечное введение, энтеральное введение, ингаляционное введение, местное введение (нос, кожа, глаз) и введение в брюшную полость и, в частности, подкожное и внутримышечное введение являются предпочтительными. Доза и количество раз введения фармацевтического препарата по изобретению в тело пациента может быть надлежащим образом выбрано в зависимости от гидрофильного активного вещества, маршрута введения, возраста и массы тела пациента, или серьезности симптомов, но обычно взрослому пациенту в сутки вводят дозу от 0,1 мкг до 100 мг, предпочтительно, от 1 мкг до 10 мг.
ПРИМЕРЫ
Ниже показаны примеры, но изобретение не ограничивается описанными здесь примерами.
Пример 1
Синтез декстран-полимолочной кислоты (PLA)
1.1 Синтез TMS-декстрана (соединение 1)
Декстран (NACALAI TESQUE, INC. NAKARAI продукт, соответствующий стандартному особому сорту, среднечисленная молекулярная масса: 13000, 5,0 г) добавляли к формамиду (100 мл) и нагревали до 80°C. В данный раствор по каплям в течение 20 минут добавляли 1,1,1,3,3,3-гексаметилдисилазан (100 мл). После добавления раствор перемешивали в течение 2 часов при 80°C. После завершения реакции реакционный раствор охлаждали до комнатной температуры, и раствор делили на два слоя с помощью дозирующей воронки. Верхний слой концентрировали при пониженном давлении, добавляли метанол (300 мл), и полученный сухой остаток фильтровали и сушили, получая TMS-декстран (11,4 г) в виде белого сухого остатка.
1.2 Синтез TMS-декстран-PLA (соединение 2)
Соединение 1 (0,5 г) и трет-бутоксикалий (35 мг) сушили в течение 1 часа при пониженном давлении, добавляли тетрагидрофуран (20 мл), и смесь перемешивали в течение 1 часа при комнатной температуре. В данный раствор по каплям добавляли раствор (L)-лактида (4,49 г) в тетрагидрофуране (20 мл), и смесь перемешивали в течение 5 минут. После завершения реакции растворитель концентрировали при пониженном давлении и очищали повторным осаждением с системой хлороформ-метанол, получая TMS-декстран-PLA (1,9 г) в виде белого сухого остатка.
1.3 Синтез декстран-PLA (соединение 3)
В раствор соединения 2 (1,9 г) в хлороформе (24 мл) добавляли метанол (10,8 мл) и 12 н. хлористоводородную кислоту (1,2 мл) и перемешивали в течение 30 минут при комнатной температуре. Растворитель отгоняли при пониженном давлении, остаток растворяли в хлороформе (10 мл) и по каплям добавляли в диэтиловый эфир, охлажденный до 0°C, и осаждался продукт. Осажденное вещество отфильтровывали и концентрировали при пониженном давлении, получая декстран-PLA (1,6 г). Среднемассовая молекулярная масса данного полимера составляла 48720, а среднечисленная молекулярная масса составляла 43530. (Измерения ГПХ: колонка Toso TSK-gel α-5000×2, система растворителя ДМФА, детектор RI, стандартный продукт, пуллулан). Средняя молекулярная масса привитой цепи данного полимера, определенная измерением1Н-ЯМР, составляла 2300. Количество привитых цепей составляло от 10 до 12.
Пример 2
Синтез декстран-сополимера молочной и гликолевой кислот (PLGA)
2.1 Синтез TMS-декстран-PLGA (соединение 4, соединение 5, соединение 6)
Соединение 1 (0,5 г) и трет-бутоксикалий (35 мг) сушили в течение 1 часа при пониженном давлении, добавляли тетрагидрофуран (10 мл), и смесь перемешивали в течение 1 часа при комнатной температуре. В данный раствор по каплям добавляли раствор (DL)-лактида (1,12 г) и гликолида (0,9 г) в тетрагидрофуране (15 мл), и смесь перемешивали в течение 5 минут. После завершения реакции растворитель концентрировали при пониженном давлении и очищали повторным осаждением системой хлороформ-метанол, получая TMS-декстран-PLGA (1,96 г) в виде белого сухого остатка (соединение 4). Таким же способом, загружая количество (DL)-лактида (0,784 г) и гликолида (0,63 г) синтезировали соединение 5, и, загружая количество (DL)-лактида (1,12 г) и гликолида (0,9 г), синтезировали соединение 6.
2.2 Синтез декстран-PLGA (соединение 7, соединение 8, соединение 9)
В раствор соединения 4 (1,96 г) в хлороформе (14 мл) добавляли метанол (6,3 мл) и 12 н. хлористоводородную кислоту (0,7 мл) и перемешивали в течение 30 минут при комнатной температуре. Растворитель отгоняли при пониженном давлении, остаток растворяли в хлороформе (10 мл) и по каплям добавляли в диэтиловый эфир, охлажденный до 0°C, и осаждался продукт. Осажденное вещество отфильтровывали и концентрировали при пониженном давлении, получая декстран-PLGA (1,25 г) (соединение 7). Из соединений 5 и 6 таким же образом были получены декстран-PLGA продукты за исключением того, что использовали трифторуксусную кислоту (соединение 8, соединение 9). Среднемассовую молекулярную массу и среднечисленную молекулярную массу полимера соединений 7-9 определяли с помощью измерений ГПХ (колонка Toso TSK-gel α-5000×2, система растворителя ДМФА, детектор RI, стандартный продукт, пуллулан). Среднюю молекулярную массу привитой цепи и количество привитых цепей определяли измерением1Н-ЯМР.
Что касается соединения 7, среднемассовая молекулярная масса составляла 43820, среднечисленная молекулярная масса составляла 33422, молекулярная масса привитой цепи составляла 1900, и количество привитых цепей составляло от 7 до 10.
Что касается соединения 8, среднемассовая молекулярная масса составляла 94088, среднечисленная молекулярная масса составляла 81250, молекулярная масса привитой цепи составляла 3250, и количество привитых цепей составляло 21.
Что касается соединения 9, среднемассовая молекулярная масса составляла 137695, среднечисленная молекулярная масса составляла 109630, молекулярная масса привитой цепи составляла 6442, и количество привитых цепей составляло 15.
Пример 3
Способ получения микрочастиц с инкапсулированным гормоном роста человека (hGH)
5 мг декстрана-полимолочной кислоты (PLA) примера 1 (средняя молекулярная масса декстрана равна 13000, средняя молекулярная масса PLA равна 2300, количество привитых цепей PLA составляет от 10 до 12, соединение 3) или декстрана-сополимера молочной и гликолевой кислот (PLGA) примера 2 (средняя молекулярная масса декстрана равна 13000, средняя молекулярная масса PLGA равна 19000, количество привитых цепей PLGA равно от 7 до 10, соединение 7) растворяли в 100 мкл диметилкарбоната, получая раствор полимера с концентрацией 50 мг/мл. В данный раствор полимера добавляли 20 мкл трет-бутанола, по каплям добавляли 20 мкл 2 мг/мл водного раствора hGH и перемешивали вихревым перемешивающим устройством, получая эмульсию с обращенной фазой. Данную эмульсию с обращенной фазой предварительно замораживали и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов. Полученный сухой остаток диспергировали в 200 мкл диметилкарбоната, получая суспензию S/O. Данную суспензию S/O по каплям добавляли к 2 мл водного раствора, содержащего 10% Pluronic F-68 (зарегистрированная торговая марка BASF), и перемешивали и эмульгировали в вихревом перемешивающем устройстве, получая эмульсию типа S/O/W. Из данной эмульсии типа S/O/W сушкой в жидкости удаляли несмешивающийся с водой органический растворитель и получали жидкую дисперсию микрочастиц. Жидкую дисперсию микрочастиц предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов, и получали порошок микрочастиц с инкапсулированным hGH. Полученные частицы наблюдали сканирующим электронным микроскопом (СЭМ: HITACHI, S-4800), вычисляли средний диаметр частиц, и средний диаметр микрочастиц оказался равен 4,0 мкм.
Пример 4
Измерение эффективности инкапсулирования лекарственного средства микрочастицами с инкапсулированным гормоном роста человека (hGH)
20 мг микрочастиц с инкапсулированным гормоном роста человека, приготовленных способом примера 3 посредством использования полимера декстрана-PLA (соединение 3) или декстрана-PLGA (соединение 7), взвешивали, используя 1,5-мл пробирку Эппендорфа, и растворяли в 1 мл буферного раствора A (физиологический раствор с фосфатным буфером, содержащий 0,1% альбумина бычьей сыворотки, 0,1% Pluronic F-68 (зарегистрированная торговая марка BASF) и 0,02% азида натрия), центрифугировали в течение 10 минут при 18000×g и разделяли на частицы (осаждение) и надосадочную жидкость. Надосадочную жидкость собирали в другую пробирку, частицы снова суспендировали в 1 мл буферного раствора, и операцию центрифугирования и разделение на частицы и надосадочную жидкость снова проводили в тех же условиях. Данную операцию очистки повторяли еще один раз (всего три операции центрифугирования) и концентрацию гормона роста человека в каждой надосадочной жидкости, собранной с помощью операции центрифугирования измеряли, используя комплект для твердофазного иммуноферментного анализа (ELISA) (изготовленный R&D Systems). Из загруженного количества hGH во время приготовления частиц (на массу частиц равную 20 мг) вычитали общее количество hGH в трех надосадочных жидкостях, полученных операциями центрифугирования, и эффективность инкапсулирования рассчитывали согласно формуле ниже.
В микрочастицах декстран-PLA или микрочастицах декстран-PLGA эффективность инкапсулирования hGH составляла 92,6% в микрочастицах декстран-PLA и 85,7% в микрочастицах декстран-PLGA, и было доказано, что белковое лекарственное средство можно инкапсулировать с высокой эффективностью в частицах того и другого типа.
Пример 5
Анализ скорости высвобождения in vitro лекарственного средства из микрочастиц с инкапсулированным гормоном роста человека (hGH)
Микрочастицы, центрифугированные три раза в примере 4, суспендировали и диспергировали в 1,2 мл буферного раствора A. Из данного раствора часть (40 мкл) переносили в другую пробирку и центрифугировали в течение 10 минут при 18000×g для осаждения частиц, и 30 мкл надосадочной жидкости собирали в другую пробирку (0-часовой образец). Остающуюся суспензию частиц помещали в 1,5-мл пробирку Эппендорфа и медленно вращали и перемешивали в инкубаторе при 37°C, используя вращающее устройство, со скоростью 6 об./мин. Из данного раствора небольшую часть (40 мкл) дозировали при особых интервалах времени, и надосадочную жидкость отделяли аналогичным образом посредством операции центрифугирования. В образце надосадочной жидкости, собранном в каждый момент времени, измеряли концентрацию hGH, используя комплект для твердофазного иммуноферментного анализа, и высвобожденное количество (%) вычисляли по формуле, приведенной ниже.
Фиг.1 показывает динамику изменения высвобождения лекарственного средства из микрочастиц, изготовленных при использовании полимера декстран-PLA или декстран-PLGA. В частицах обоего типа начальный пик почти не наблюдался, лекарственное средство высвобождалось линейно пропорционально течению времени, и наблюдался благоприятный профиль. Время, требующееся для 50% высвобождения лекарственного средства, составляло примерно 1 месяц в микрочастице декстран-PLA и примерно 1 неделю в микрочастице декстран-PLGA, и было предположено, что скорость высвобождения можно контролировать выбором типа полигидроксикислоты.
Пример 6
Способ получения микрочастиц с инкапсулированным человеческим инсулином
5 мг декстрана-PLA (средняя молекулярная масса декстрана равна 13000, средняя молекулярная масса PLA равна 2300, количество привитых цепей PLA составляет от 10 до 12, соединение 3) или декстрана-PLGA (средняя молекулярная масса декстрана равна 13000, средняя молекулярная масса PLGA равна 19000, количество привитых цепей PLGA равно от 7 до 10, соединение 7) растворяли в 100 мкл диметилкарбоната, получая раствор полимера с концентрацией 50 мг/мл. В данный раствор полимера добавляли 20 мкл трет-бутанола, по каплям добавляли 20 мкл 2 мг/мл водного раствора человеческого инсулина и перемешивали вихревым перемешивающим устройством, получая эмульсию с обращенной фазой. Данную эмульсию с обращенной фазой предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов. Полученный сухой остаток диспергировали в 200 мкл диметилкарбоната, получая суспензию S/O. Данную суспензию S/O по каплям добавляли к 2 мл водного раствора, содержащего 10% Pluronic F-68 (зарегистрированная торговая марка BASF), и перемешивали и эмульгировали в вихревом перемешивающем устройстве, получая эмульсию типа S/O/W. Из данной эмульсии типа S/O/W сушкой в жидкости удаляли несмешивающийся с водой органический растворитель и получали жидкую дисперсию микрочастиц. Жидкую дисперсию микрочастиц предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов, и получали порошок микрочастиц с инкапсулированным человеческим инсулином. Полученные частицы наблюдали сканирующим электронным микроскопом (СЭМ: HITACHI, S-4800), вычисляли средний диаметр частиц, и средний диаметр микрочастиц оказался равен 6,4 мкм в микрочастицах, полученных из соединения 3, и 5,3 мкм в микрочастицах, полученных из соединения 7.
Пример 7
Измерение эффективности инкапсулирования лекарственного средства микрочастицами с инкапсулированным человеческим инсулином
20 мг микрочастиц с инкапсулированным человеческим инсулином, приготовленных способом примера 6 посредством использования полимера декстран-PLGA (соединение 7), взвешивали, используя 1,5-мл пробирку Эппендорфа, и растворяли в 1 мл буферного раствора A (физиологический раствор с фосфатным буфером, содержащий 0,1% альбумина бычьей сыворотки, 0,1% Pluronic F-68 (зарегистрированная торговая марка BASF) и 0,02% азида натрия), центрифугировали в течение 10 минут при 18800×g и разделяли на частицы (осаждение) и надосадочную жидкость. Надосадочную жидкость собирали в другую пробирку и частицы снова суспендировали в 1 мл буферного раствора, и операцию центрифугирования и разделение на частицы и надосадочную жидкость снова проводили в тех же условиях. Данную операцию очистки повторяли еще один раз (всего три операции центрифугирования) и концентрацию человеческого инсулина каждой надосадочной жидкости, собранной с помощью операции центрифугирования, измеряли, используя многослойным методом твердофазного иммуноферментного анализа. Из загруженного количества человеческого инсулина во время приготовления частиц (на массу частиц равную 20 мг) вычитали общее количество человеческого инсулина в трех надосадочных жидкостях, полученных операциями центрифугирования, и эффективность инкапсулирования рассчитывали согласно формуле ниже.
В микрочастицах декстран-PLA или микрочастицах декстран-PLGA эффективность инкапсулирования человеческого инсулина составляла 75,7%, и было доказано, что лекарственное средство можно инкапсулировать с высокой эффективностью.
Пример 8
Анализ скорости высвобождения in vitro лекарственного средства из микрочастиц с инкапсулированным человеческим инсулином
Микрочастицы, центрифугированные три раза в примере 7, суспендировали и диспергировали в 1,2 мл буферного раствора A. Из данного раствора часть (40 мкл) переносили в другую пробирку и центрифугировали в течение 10 минут при 18800×g для осаждения частиц, и 30 мкл надосадочной жидкости собирали в другую пробирку (0-часовой образец). Остающуюся суспензию частиц помещали в 1,5-мл пробирку Эппендорфа и медленно вращали и перемешивали в инкубаторе при 37°C, используя вращающее устройство, со скоростью 6 об/мин. Из данного раствора небольшую часть (40 мкл) дозировали при особых интервалах времени, и надосадочную жидкость отделяли аналогичным образом посредством операции центрифугирования. В образце надосадочной жидкости, собранном в каждый момент времени, концентрацию человеческого инсулина измеряли многослойным методом твердофазного иммуноферментного анализа, и высвобожденное количество (%) вычисляли по формуле, приведенной ниже.
Фиг.2 показывает динамику изменения высвобождения человеческого инсулина. Начальный пик почти не наблюдался, лекарственное средство высвобождалось линейно пропорционально течению времени, и наблюдался благоприятный профиль. Время, требующееся для 50% высвобождения лекарственного средства, составляло примерно 6 дней.
Пример 9
Динамика изменений морфологии микрочастиц
5 мг микрочастиц с инкапсулированным hGH, приготовленных в примере 3, взвешивали в пробирке Эппендорфа, диспергировали в 1 мл Milli-Q и разделяли центрифугированием в течение 30 минут при 13000 об/мин, отделяли от надосадочной жидкости, снова диспергировали в 1 мл Milli-Q и разделяли центрифугированием, и микрочастицы очищали. В раствор суспензии микрочастиц, инкубированных в течение определенного времени, добавляли 1 мл Milli-Q, раствор разделяли центрифугированием в течение 30 минут при 13000 об/мин, отделяли от надосадочной жидкости и снова диспергировали в 1 мл Milli-Q, разделяли центрифугированием, и микрочастицы очищали. Микрочастицы, полученные после очистки, диспергировали в 100 мкл Milli-Q и 3 мкл жидкой дисперсии микрочастиц капали на кремниевую подложку, оставляли стоять при комнатной температуре в течение 10 минут и сушили в течение 3 часов в десикаторе. Затем, используя прибор ионного распыления (HITACHI, E-1030), на поверхность образца осаждали платину (время осаждения 15 секунд), и форму и состояние поверхности микрочастиц наблюдали сканирующим электронным микроскопом (СЭМ: HITACHI, S-4800) при напряжении ускорения 1 кВ и высоком зондовом токе.
Как показано на фиг.3, непосредственно после изготовления поверхность была ровной и сферической, и частицы были явно деформированы после выдерживания в течение 13 дней при 37°C, образовалось множество пор, и было доказано, что частицы постепенно разлагались вместе с ходом высвобождения лекарственного средства.
Пример сравнения 1
5 мг полиэтиленгликоль-поли(эпсилон-капролактона) (средняя молекулярная масса полиэтиленгликоля 5000, средняя молекулярная масса поли(эпсилон-капролактона) 37000) растворяли в 100 мкл диметилкарбоната, получая раствор полимера с концентрацией 50 мг/мл. В данный раствор полимера добавляли 20 мкл трет-бутанола, по каплям добавляли 20 мкл 2 мг/мл водного раствора hGH и перемешивали вихревым перемешивающим устройством, получая эмульсию с обращенной фазой. Данную эмульсию с обращенной фазой предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов. Полученный сухой остаток диспергировали в 200 мкл диметилкарбоната, получая суспензию S/O. Данную суспензию S/O по каплям добавляли к 2 мл водного раствора, содержащего 10% Pluronic F-68 (зарегистрированная торговая марка BASF), и перемешивали и эмульгировали в вихревом перемешивающем устройстве, получая эмульсию типа S/O/W. Из данной эмульсии типа S/O/W сушкой в жидкости удаляли несмешивающийся с водой органический растворитель и получали жидкую дисперсию микрочастиц. Жидкую дисперсию микрочастиц предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов, и получали порошок микрочастиц с инкапсулированным hGH. Полученные микрочастицы наблюдали сканирующим электронным микроскопом (СЭМ: HITACHI, S-4800), вычисляли средний диаметр частиц, и средний диаметр микрочастиц оказался равен 8,0 мкм.
5 мг приготовленного порошка микрочастиц с инкапсулированным hGH взвешивали, используя пробирку Эппендорфа, диспергировали в 1 мл Milli-Q и центрифугировали в течение 30 минут при 13000, отделяли от надосадочной жидкости и снова диспергировали в 1 мл Milli-Q и разделяли центрифугированием аналогичным образом, и микрочастицы очищали. Микрочастицы, полученные после очистки, диспергировали в 100 мкл Milli-Q и 5 мкл жидкой дисперсии микрочастиц капали на кремниевую подложку, оставляли стоять при комнатной температуре в течение 10 минут и сушили в течение 3 часов в десикаторе. Затем, используя прибор ионного распыления (HITACHI, E-1030), на поверхность образца осаждали платину (время осаждения 15 секунд), и форму и состояние поверхности микрочастиц наблюдали сканирующим электронным микроскопом (СЭМ: HITACHI, S-4800) при напряжении ускорения 1 кВ и высоком зондовом токе.
Как показано на фиг.4, в отличие от микрочастиц декстран-PLGA в примере 9, после выдерживания в течение 21 дня при 37°C частицы почти не изменились морфологически, и существовала проблема в характеристике высвобождения гидрофильного активного вещества.
Пример 10
Подкожное введение мыши микрочастиц с инкапсулированным гормоном роста человека (hGH)
25 мг декстрана-полимолочной кислоты (PLA) (средняя молекулярная масса декстрана равна 13000, средняя молекулярная масса PLA равна 2300, количество привитых цепей PLA составляет от 10 до 12, соединение 3) или декстрана-сополимера молочной и гликолевой кислот (PLGA) (средняя молекулярная масса декстрана равна 13000, средняя молекулярная масса PLGA равна 19000, количество привитых цепей PLGA равно от 7 до 10, соединение 7) растворяли в 500 мкл диметилкарбоната, получая раствор полимера с концентрацией 50 мг/мл. В данный раствор полимера добавляли 100 мкл трет-бутанола, по каплям добавляли 250 мкл 10 мг/мл водного раствора hGH и перемешивали вихревым перемешивающим устройством, получая эмульсию с обращенной фазой. Данную эмульсию с обращенной фазой предварительно замораживали и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов. Полученный сухой остаток диспергировали в 1 мл диметилкарбоната, получая суспензию S/O. Данную суспензию S/O по каплям добавляли к 10 мл водного раствора, содержащего 10% Pluronic F-68 (зарегистрированная торговая марка BASF), и перемешивали и эмульгировали в вихревом перемешивающем устройстве, получая эмульсию типа S/O/W. Из данной эмульсии типа S/O/W сушкой в жидкости удаляли несмешивающийся с водой органический растворитель и получали жидкую дисперсию микрочастиц. Жидкую дисперсию микрочастиц предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов, и получали порошок микрочастиц с инкапсулированным hGH. Полученные микрочастицы наблюдали сканирующим электронным микроскопом (СЭМ: HITACHI, S-4800), вычисляли средний диаметр частиц, и средний диаметр микрочастиц оказался равен 4,9 мкм в микрочастицах, полученных из соединения 3, и 4,2 мкм в микрочастицах, полученных из соединения 7.
300 мг приготовленных микрочастиц суспендировали и диспергировали в 3 мл фосфатного физиологического буферного раствора (PBS) и центрифугировали в течение 5 минут при 80×g для осаждения микрочастиц, и надосадочную жидкость переносили в другую пробирку. Надосадочную жидкость снова центрифугировали в течение 5 минут при 80×g для осаждения остающихся частиц, и надосадочную жидкость удаляли. Повторным диспергированием в 1 мл физиологического раствора с фосфатным буфером после первого раза осаждения центрифугированием и второго раза осаждения центрифугированием, такую же операцию очистки центрифугированием повторяли три раза, и гормон роста, не инкапсулированный в микрочастицах, удаляли. Наконец, осадок вновь диспергировали в 200 мкл физиологического раствора с фосфатным буфером и получали раствор для введения. Количество гормона роста, инкапсулированного в декстран-PLA микрочастице и декстран-PLGA микрокапсуле измеряли комплектом для твердофазного иммуноферментного анализа, определяли концентрацию в очищающем растворе и вычитали из загруженного количества, и определяли количество, инкапсулированное в 300 мг частиц, вводимых одной мыши, и для микрочастицы декстран-PLA это количество составляло 590 мкг, а для микрочастицы декстран-PLGA - 536 мкг.
Данный раствор вводили подкожно в двух положениях в спину 10-недельного самца Balb/C мыши, и образцы крови отбирали в определенные промежутки времени из хвостовой вены. В образцы крови добавляли гепарин до конечной концентрации 3,3 межд. ед./мл, плазму собирали центробежным разделением в течение 5 минут при 5000 об./мин., и концентрацию гормона роста в плазме измеряли, используя комплект для твердофазного иммуноферментного анализа.
С целью сравнения раствор негранулированного белка гормона роста человека (700 мкг/0,2 мл) подкожно вводили мыши, и аналогичным образом отбирали образцы крови.
Для того чтобы подавить продукцию антител, образующихся в результате введения гормона роста человека, является чужеродным белком для мыши, за три дня до введения частиц подкожно вводили иммуносупрессор Такролимус гидрат (Astellas) 26 мкг/мышь и после этого 13 мкг/мышь во время введения лекарственного средства, и 3 днями и 7 днями позднее.
Фиг.5 показывает динамику изменения концентрации гормона роста человека в плазме. У мыши, которой ввели негранулированное лекарственное средство, уровень в крови через один час после введения был очень высоким, более 5000 нг/мл, и затем резко падал до уровня до введения в течение дня. С другой стороны, у мыши, которой вводили лекарственное средство в виде микрочастиц, приготовленный с использованием полимера декстран-PLA, кратковременное повышение уровня в крови непосредственно после введения подавлялось до 200 нг/мл или менее, и в течение семи последующих дней уровень в крови поддерживался при высоких уровнях. В микрочастицах декстран-PLA временное повышение концентрации после введения вовсе не наблюдалось, и приблизительно точная концентрация в крови поддерживалась в течение семи дней и наблюдалась превосходное замедленное высвобождение.
Пример 11
Подкожное введение мыши микрочастиц с инкапсулированным гормоном роста человека (hGH) (оценка фармакологической активности)
2 мг декстрана-сополимера молочной и гликолевой кислот (PLGA) (средняя молекулярная масса декстрана равна 13000, средняя молекулярная масса PLGA равна 1900, количество привитых цепей PLGA равно от 7 до 10, соединение 7) растворяли в 500 мкл диметилкарбоната, получая раствор полимера с концентрацией 50 мг/мл. В данный раствор полимера добавляли 100 мкл трет-бутанола, по каплям добавляли 250 мкл 10 мг/мл водного раствора hGH и перемешивали вихревым перемешивающим устройством, получая эмульсию с обращенной фазой. Данную эмульсию с обращенной фазой предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов. Полученный сухой остаток диспергировали в 1 мл диметилкарбоната, получая суспензию S/O. Данную суспензию S/O по каплям добавляли к 10 мл водного раствора, содержащего 10% Pluronic F-68 (зарегистрированная торговая марка BASF), и перемешивали и эмульгировали в вихревом перемешивающем устройстве, получая эмульсию типа S/O/W. Из данной эмульсии типа S/O/W сушкой в жидкости удаляли несмешивающийся с водой органический растворитель и получали жидкую дисперсию микрочастиц. Жидкую дисперсию микрочастиц предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов, и получали порошок микрочастиц с инкапсулированным hGH. Полученные микрочастицы наблюдали сканирующим электронным микроскопом (СЭМ: HITACHI, S-4800), вычисляли средний диаметр частиц, и средний диаметр микрочастиц оказался равен 4,1 мкм.
300 мг приготовленных микрочастиц суспендировали и диспергировали в 3 мл фосфатного физиологического буферного раствора (PBS) и частицы осаждали разделением с помощью центрифугирования в течение 5 минут при 80×g, и надосадочную жидкость переносили в другую пробирку. Надосадочную жидкость снова разделяли центрифугированием в течение 5 минут при 80×g, остающиеся частицы осаждали и надосадочную жидкость удаляли. Первый осадок после центрифугирования и второй осадок после центрифугирования объединяли и снова диспергировали в 1 мл PBS, и аналогичным образом проводили третью операцию центрифугирования, и гормон роста, не инкапсулированный в частицах, удаляли. Наконец, осадок вновь диспергировали в 200 мкл PBS, получая раствор для введения.
Данный раствор вводили подкожно в двух положениях в спину 8-недельной мыши с удаленным гипофизом (от японской SLC), и образцы крови отбирали в определенные промежутки времени из хвостовой вены. В образцы крови добавляли гепарин до конечной концентрации 3,3 межд. ед./мл и центрифугировали в течение 5 минут при 5000 об./мин., плазму собирали и концентрацию гормона роста в плазме и концентрацию инсулиноподобного фактора роста (IGF-1) мыши измеряли, используя метод твердофазного иммуноферментного анализа.
С целью сравнения раствор негранулированного белка гормона роста человека (700 мкг/0,2 мл) подкожно вводили мыши, и аналогичным образом отбирали образцы крови.
Для того чтобы подавить продукцию антител, образующихся в результате введения гормона роста человека, является чужеродным белком для мыши, за три дня до введения частиц подкожно вводили иммуносупрессор Такролимус гидрат (Astellas) 26 мкг/мышь и после этого 13 мкг/мышь во время введения лекарственного средства, и 3 днями и 7 днями позднее.
Фиг.6 показывает динамику изменения концентрации гормона роста человека в плазме. У мыши, которой ввели негранулированное лекарственное средство, уровень в крови через один час после введения был очень высоким, и затем резко падал до уровня до введения в течение двух дней. С другой стороны, в микрочастицах декстран-PLGA, кратковременное повышение уровня в крови непосредственно после введения подавлялось до низкого значения, и в течение десяти последующих дней после введения концентрация в плазме поддерживалась на высоких уровнях. В данный момент изменения массы тела мыши показаны на фиг.7. У мыши, которой вводили только гормон роста, увеличение массы тела подавлялось на уровне примерно 5%, но у мыши, которой вводили микрочастицы декстран-PLGA, масса тела увеличивалась примерно на 20%.
Фиг.8 показывает концентрацию IGF-1 в плазме. Концентрация IGF-1 в плазме соотносится с концентрацией гормона роста человека в крови, и у мыши, которой вводили микрочастицы декстран-PLGA, высокие уровни поддерживались в течение десяти дней после введения.
Пример 12
Анализ скорости высвобождения лекарственного средства в буферный раствор из микрочастиц с инкапсулированным Эксендином-4 (агонистом ГПП-1 рецептора)
25 мг декстрана-сополимера молочной и гликолевой кислот (PLGA) (средняя молекулярная масса декстрана равна 13000, средняя молекулярная масса PLGA равна 3250 (соединение 8) или 6442 (соединение 9), количество привитых цепей PLGA равно 21 (соединение 8) или 15 (соединение 9)) растворяли в 500 мкл диметилкарбоната, получая раствор полимера с концентрацией 50 мг/мл. В данный раствор полимера добавляли 100 мкл трет-бутанола, по каплям добавляли 250 мкл 10 мг/мл Эксендина-4 (синтезированного по поручению компанией Sigma Genosys), и перемешивали вихревым перемешивающим устройством, получая эмульсию с обращенной фазой. Данную эмульсию с обращенной фазой предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов. Полученный сухой остаток диспергировали в 1 мл диметилкарбоната, получая суспензию S/O. Данную суспензию S/O по каплям добавляли к 10 мл водного раствора, содержащего 10% Pluronic F-68 (зарегистрированная торговая марка BASF), перемешивали и эмульгировали в вихревом перемешивающем устройстве, получая эмульсию типа S/O/W. Из данной эмульсии типа S/O/W сушкой в жидкости удаляли несмешивающийся с водой органический растворитель и получали жидкую дисперсию микрочастиц. Жидкую дисперсию микрочастиц предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов, и получали порошок микрочастиц с инкапсулированным Эксендином-4. Полученные частицы наблюдали сканирующим электронным микроскопом (СЭМ: HITACHI, S-4800), вычисляли средний диаметр частиц, и средний диаметр микрочастиц составлял 4,3 мкм в соединении 8 и 4,5 мкм в соединении 9.
Данные микрочастицы три раза очищали согласно методу примера 4, суспендировали и диспергировали в 1,2 мл буферного раствора А. Из данного раствора часть (40 мкл) переносили в другую пробирку и центрифугировали в течение 10 минут при 18000×g, осаждая частицы, и 30 мкл надосадочной жидкости собирали в другую пробирку (0-часовой образец). Остающуюся суспензию частиц помещали в 1,5-мл пробирку Эппендорфа, медленно вращали и перемешивали в инкубаторе при 37°C, используя вращающее устройство, со скоростью 6 об./мин. Из данного раствора небольшую часть (40 мкл) дозировали при особых интервалах времени, и надосадочную жидкость отделяли аналогичным образом посредством операции центрифугирования. В образце надосадочной жидкости, собранном в каждый момент времени, методом твердофазного иммуноферментного анализа измеряли концентрацию Эксендина-4, и высвобожденное количество (%) вычисляли по формуле, приведенной ниже.
Фиг.9 показывает динамику изменений высвобождения лекарственного средства из микрочастиц, изготовленных с использованием каждого полимера декстран-PLGA. В тех и других микрочастицах начальный пик почти не наблюдался, лекарственное средство высвобождалось линейно пропорционально течению времени, и наблюдался благоприятный профиль.
Пример 13
Подкожное введение мыши микрочастиц с инкапсулированным Эксендином-4 (агонистом ГПП-1 рецептора)
300 мг микрочастиц, приготовленных в примере 12, суспендировали и диспергировали в 3 мл фосфатного физиологического буферного раствора (PBS) и частицы осаждали разделением с помощью центрифугирования в течение 5 минут при 80×g, и надосадочную жидкость переносили в другую пробирку. Надосадочную жидкость снова разделяли центрифугированием в течение 5 минут при 80×g, остающиеся частицы осаждали и надосадочную жидкость удаляли. Первый осадок после центрифугирования и второй осадок после центрифугирования объединяли и снова диспергировали в 1 мл PBS, и аналогичным образом проводили третью операцию центрифугирования, и Эксендин-4, не инкапсулированный в частицах, удаляли. Наконец, осадок вновь диспергировали в 200 мкл PBS, получая раствор для введения.
Данный раствор вводили подкожно в спину 8-недельной SCID мыши (CB17/lcr-Prkdcscid/CrlCrlk) (от Crea Japan Inc.), и образцы крови отбирали в определенные промежутки времени из хвостовой вены. В образцы крови добавляли гепарин до конечной концентрации 3,3 межд. ед./мл и центрифугировали в течение 5 минут при 5000 об/мин, плазму собирали и концентрацию Эксендина-4 в плазме измеряли методом твердофазного иммуноферментного анализа. С целью сравнения раствор негранулированного Эксендина-4 (700 мкг/0,2 мл) подкожно вводили мыши, и аналогичным образом отбирали образцы крови.
Фиг.10 показывает динамику изменений концентрации Эксендина-4 в плазме. У мыши, которой ввели негранулированное лекарственное средство, уровень в крови через 1 час после введения был очень высоким, и затем резко падал до уровня до введения. С другой стороны, в микрочастицах декстран-PLGA, кратковременное повышение концентрации после введения подавлялось до низкого значения, и в течение пяти последующих дней после введения, концентрация в плазме поддерживалась на высоких уровнях.
Пример 14
Синтез декстрана-сополимера молочной и гликолевой кислот (PLGA)
14.1 Синтез TMS-декстран-PLGA (соединение 10, соединение 11, соединение 12, соединение 13)
Соединение 1 (0,5 г) и трет-бутоксикалий (35 мг) сушили в течение 1 часа при пониженном давлении, добавляли тетрагидрофуран (10 мл), и смесь перемешивали в течение 1 часа при комнатной температуре. В данный раствор по каплям добавляли раствор (DL)-лактида (0,558 г) и гликолида (0,45 г) в тетрагидрофуране (15 мл), и смесь перемешивали в течение 5 минут. После завершения реакции растворитель концентрировали при пониженном давлении и очищали повторным осаждением системой хлороформ-метанол, получая TMS-декстран-PLGA (1,96 г) в виде белого сухого остатка (соединение 10).
Таким же способом, загружая количество (DL)-лактида (0,67 г) и гликолида (0,54 г) синтезировали соединение 11.
Таким же способом, загружая количество (DL)-лактида (0,781 г) и гликолида (0,629 г) синтезировали соединение 12.
Таким же способом, загружая количество (DL)-лактида (1,123 г) и гликолида (0,9 г) синтезировали соединение 13.
14.2 Синтез декстрана-PLGA (соединение 14, соединение 15, соединение 16, соединение 17)
В раствор соединения 10 в хлороформе (10 мл) добавляли трифторуксусную кислоту (1 мл) и перемешивали в течение 30 минут при комнатной температуре. Растворитель отгоняли при пониженном давлении, остаток растворяли в хлороформе (10 мл), по каплям добавляли в диэтиловый эфир, охлажденный до 0°C, и осаждался продукт. Осажденное вещество отфильтровывали и концентрировали при пониженном давлении, получая декстран-PLGA (0,44 г) (соединение 14).
Из соединений 11, 12 и 13 таким же образом были получены продукты декстран-PLGA (соединение 15, соединение 16, соединение 17). Среднемассовую молекулярную массу и среднечисленную молекулярную массу полимера соединений 14-17 определяли с помощью ГПХ (колонка Toso TSK-gel α-5000×2, система растворителя ДМФА, детектор RI, стандартный продукт, пуллулан). Средняя молекулярная масса привитой цепи и количество привитых цепей определяли измерением1Н-ЯМР.
Что касается соединения 14, среднемассовая молекулярная масса составляла 99462, среднечисленная молекулярная масса составляла 85101, среднечисленная молекулярная масса привитой цепи составляла 2167, и количество привитых цепей составляло 33.
Что касается соединения 15, среднемассовая молекулярная масса составляла 107779, среднечисленная молекулярная масса составляла 92134, среднечисленная молекулярная масса привитой цепи составляла 3127, и количество привитых цепей составляло 25.
Что касается соединения 16, среднемассовая молекулярная масса составляла 121281, среднечисленная молекулярная масса составляла 101873, среднечисленная молекулярная масса привитой цепи составляла 3000, и количество привитых цепей составляло 30.
Что касается соединения 17, среднемассовая молекулярная масса составляла 144838, среднечисленная молекулярная масса составляла 122151, среднечисленная молекулярная масса привитой цепи составляла 4864, и количество привитых цепей составляло 22.
Пример 15
Способ получения микрочастиц с инкапсулированным гормоном роста человека (hGH)
5 мг каждого полимера декстрана-сополимера молочной и гликолевой кислот (декстран-PLGA полимер, соединения 14-17) примера 14 растворяли в 100 мкл диметилкарбоната, получая раствор полимера с концентрацией 50 мг/мл. В данный раствор полимера добавляли 20 мкл трет-бутанола, по каплям добавляли 50 мкл 1 мг/мл водного раствора hGH и перемешивали вихревым перемешивающим устройством, получая эмульсию с обращенной фазой. Данную эмульсию с обращенной фазой предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов. Полученный сухой остаток диспергировали в 200 мкл диметилкарбоната, получая суспензию S/O. Данную суспензию S/O по каплям добавляли к 2 мл водного раствора, содержащего 10% Pluronic F-68 (зарегистрированная торговая марка BASF), и перемешивали и эмульгировали в вихревом перемешивающем устройстве, получая эмульсию типа S/O/W. Из данной эмульсии типа S/O/W сушкой в жидкости удаляли несмешивающийся с водой органический растворитель и получали жидкую дисперсию микрочастиц. Жидкую дисперсию микрочастиц предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов, и получали порошок микрочастиц с инкапсулированным hGH. Полученные частицы наблюдали сканирующим электронным микроскопом (СЭМ: HITACHI, S-4800), вычисляли средний диаметр частиц, и средний диаметр микрочастиц оказался в диапазоне от 1,0 до 10 мкм.
Пример 16
Измерение эффективности инкапсулирования лекарственного средства микрочастицами с инкапсулированным гормоном роста человека (hGH)
20 мг микрочастиц с инкапсулированным гормоном роста человека, приготовленных способом примера 15 посредством использования каждого полимера декстран-PLGA (соединения 14-17), взвешивали, используя 1,5-мл пробирку Эппендорфа, и растворяли в 1 мл буферного раствора A (физиологический раствор с фосфатным буфером, содержащий 0,1% альбумина бычьей сыворотки, 0,1% Pluronic F-68 (зарегистрированная торговая марка BASF) и 0,02% азида натрия), центрифугировали в течение 10 минут при 18000×g и разделяли на частицы (осаждение) и надосадочную жидкость. Надосадочную жидкость собирали в другую пробирку, частицы снова суспендировали в 1 мл буферного раствора, и операцию центрифугирования и разделения на частицы и надосадочную жидкость снова проводили в тех же условиях. Данную операцию очистки повторяли еще один раз (всего три операции центрифугирования) и концентрацию гормона роста человека каждой надосадочной жидкости, собранной с помощью операций центрифугирования, измеряли, используя комплект для твердофазного иммуноферментного анализа (изготовленный R&D Systems). Из загруженного количества hGH во время приготовления частиц (на массу частиц равную 20 мг) вычитали общее количество hGH в трех надосадочных жидкостях, полученных операциями центрифугирования, и эффективность инкапсулирования рассчитывали согласно формуле ниже.
В микрочастицах декстран-PLGA эффективность инкапсулирования hGH составляла 87,5% в микрочастицах соединения 14, 94,2% в микрочастицах соединения 15, 95,7% в микрочастицах соединения 16 и 97,5% в микрочастицах соединения 17, и было доказано, что белковое лекарственное средство можно инкапсулировать с высокой эффективностью во всех микрочастицах.
Пример сравнения 2
Изготовление частиц с инкапсулированным гормон роста и измерение эффективности инкапсулирования лекарственного средства
10 мг декстрана-сополимера молочной и гликолевой кислот (PLGA) (соединение 14 или соединение 17) растворяли в 2 мл этилацетата, получая раствор полимера. В данный раствор полимера по каплям добавляли 100 мкл водного раствора, содержащего 0,5 мг/мл hGH, и перемешивали. После операции перемешивания раствор добавляли к 20 мл диоксана. Растворитель выпаривали, раствор концентрировали примерно до 2 мл, и жидкую дисперсию частиц добавляли в воду, содержащую 500 мг Pluronic F-68 (зарегистрированная торговая марка BASF). Образец подвергали сушке вымораживанием и к 50 мг образца добавляли 1 мл воды, частицы вновь диспергировали и получали частицы, содержащие неассоциированное гидрофильное активное вещество. Средний диаметр частиц измеряли методом динамического светорассеивания, используя прибор ELS-Z (изготовленный Otsuka Denshi), и эффективность инкапсулирования лекарственного средства определяли также, как в примере 16.
В результате, в частицах соединения 14 средний диаметр частиц составлял 190,5 нм, и эффективность инкапсулирования составляла 73%, а в частицах соединения 17 средний диаметр частиц составлял 197,5 нм, и эффективность инкапсулирования составляла 70%, и эффективность инкапсулирования была ниже, чем в микрочастицах примера 16.
Пример 17
Анализ скорости высвобождения in vitro лекарственного средства из микрочастиц с инкапсулированным гормоном роста человека (hGH)
Микрочастицы, очищенные три раза в примере 16, суспендировали и диспергировали в 1,2 мл буферного раствора A. Из данного раствора часть (40 мкл) переносили в другую пробирку и центрифугировали в течение 10 минут при 18000×g для осаждения частиц, и 30 мкл надосадочной жидкости собирали в другую пробирку (0-часовой образец). Остающуюся суспензию частиц помещали в 1,5-мл пробирку Эппендорфа и медленно вращали и перемешивали в инкубаторе при 37°C, используя вращающее устройство, со скоростью 6 об/мин. Из данного раствора небольшую часть (40 мкл) дозировали при особых интервалах времени, и надосадочную жидкость отделяли аналогичным образом посредством операции центрифугирования. В образце надосадочной жидкости, собранном в каждый момент времени, концентрацию hGH измеряли с помощью комплекта для твердофазного иммуноферментного анализа, и высвобожденное количество (%) вычисляли по формуле, приведенной ниже.
Фиг.11 показывает динамику изменений высвобождения лекарственного средства из микрочастиц, изготовленных в примере 15. В данных частицах начальный пик почти не наблюдался, лекарственное средство высвобождалось линейно пропорционально течению времени, и наблюдался благоприятный профиль. Время, требующееся для 50% высвобождения лекарственного средства, составляло примерно 6 дней в микрочастицах соединения 14, примерно 9 дней в микрочастицах соединения 15, примерно 16 дней в микрочастицах соединения 16 и примерно 1 месяц в микрочастицах соединения 17, и было предположено, что скорость высвобождения можно контролировать, изменяя загружаемое количество лактида и гликолида во время синтеза TMS-декстран-PLGA.
Пример 18
Получение микрочастиц с инкапсулированным декстраном, меченым флуоресцеином (FD40), отличающихся диаметром частиц
5 мг декстрана-сополимера молочной и гликолевой кислот (PLGA) (соединение 7) примера 2 растворяли в 100 мкл диметилкарбоната, получая раствор полимера с концентрацией 50 мг/мл. В данный раствор полимера добавляли 20 мкл трет-бутанола, по каплям добавляли 20 мкл 1 мг/мл водного раствора FD40 и перемешивали вихревым перемешивающим устройством, получая эмульсию с обращенной фазой. Данную эмульсию с обращенной фазой предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов. Полученный сухой остаток диспергировали в 50 мкл, 100 мкл, 200 мкл, 350 мкл, 500 мкл, 1 мл, 2 мл и 6 мл диметилкарбоната, получая суспензию S/O. Данную суспензию S/O по каплям добавляли к 2 мл водного раствора, содержащего 10% Pluronic F-68 (зарегистрированная торговая марка BASF), и перемешивали и эмульгировали в вихревом перемешивающем устройстве, получая эмульсию типа S/O/W. Из данной эмульсии типа S/O/W сушкой в жидкости удаляли несмешивающийся с водой органический растворитель и получали жидкую дисперсию микрочастиц. Жидкую дисперсию микрочастиц предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов, и получали порошок микрочастиц с инкапсулированным FD40. Полученные микрочастицы наблюдали сканирующим электронным микроскопом (СЭМ: HITACHI, S-4800) и вычисляли средний диаметр частиц.
Фиг.12 показывает корреляцию между средним диаметром частиц и количеством диметилкарбоната, добавленного во время приготовления эмульсии типа S/O/W. В диапазоне от 50 мкл до 500 мкл, вместе с увеличением количества диметилкарбоната, наблюдали снижение среднего диаметра частиц. От 500 мкл до 6 мл никакой разницы в среднем диаметре частиц не наблюдалось.
Пример 19
Синтез полимера PEG-PLGA (серии PEG2k)
Монометиловый эфир полиэтиленгликоля (произведенный NOF Corp., SUNBRIGHT MEH-20H, среднечисленная молекулярная масса: 1862, Mw/Mn=1,03), (DL)-лактид и гликолид смешивали в заданную композицию, показанную в таблице 1, и нагревали при 140°C. После перемешивания в течение 20 минут, добавляли октилат олова (II) (0,05 масс.% относительно монометилового эфира полиэтиленгликоля) и перемешивали в течение 3 часов при 180°C. Реакционный раствор охлаждали до комнатной температуры, растворяли в хлороформе (до концентрации примерно 100 мг/мл), снова осаждали и очищали в диэтиловом эфире, охлажденном при 0°C, полученный сухой остаток фильтровали, разуплотняли и сушили, и полимер PEG-PLGA получали в виде белого или светло-коричневого сухого остатка. Среднечисленную молекулярную массу данного полимера определяли1Н-ЯМР (таблица 1).
Пример 20
Синтез полимера PEG-PLGA (серии PEG5k)
Монометиловый эфир полиэтиленгликоля (произведенный NOF Corp., SUNBRIGHT MEH-20H, среднечисленная молекулярная масса: 5128, Mw/Mn=1,02), (DL)-лактид и гликолид смешивали в заданную композицию, показанную в таблице 2, и нагревали при 140°C. После перемешивания в течение 20 минут, добавляли октилат олова (II) (0,05 масс.% относительно монометилового эфира полиэтиленгликоля) и перемешивали в течение 3 часов при 180°C. Реакционный раствор охлаждали до комнатной температуры, растворяли в хлороформе (до концентрации примерно 100 мг/мл), снова осаждали и очищали в диэтиловом эфире, охлажденном при 0°C, полученный сухой остаток фильтровали, разуплотняли и сушили, и полимер PEG-PLGA получали в виде белого или светло-коричневого сухого остатка. Среднечисленную молекулярную массу данного полимера определяли1Н-ЯМР (таблица 2).
Пример 21
Синтез полимера PEG-PLGA (серии PEG10k)
Монометиловый эфир полиэтиленгликоля (произведенный NOF Corp., SUNBRIGHT MEH-10H, среднечисленная молекулярная масса: 9975, Mw/Mn=1,02), (DL)-лактид и гликолид смешивали в заданную композицию, показанную в таблице 3, и нагревали при 140°C. После перемешивания в течение 20 минут, добавляли октилат олова (II) (0,05 масс.% относительно монометилового эфира полиэтиленгликоля) и перемешивали в течение 3 часов при 180°C. Реакционный раствор охлаждали до комнатной температуры, растворяли в хлороформе (до концентрации примерно 100 мг/мл), снова осаждали и очищали в диэтиловом эфире, охлажденном при 0°C, полученный сухой остаток фильтровали, разуплотняли и сушили, и полимер PEG-PLGA получали в виде белого или светло-коричневого сухого остатка. Среднечисленную молекулярную массу данного полимера определяли1Н-ЯМР (таблица 3).
Пример 22
Способ получения микрочастиц с инкапсулированным FD40
5 мг полимера PEG-PLGA, приготовленного в примерах 19-21, растворяли в 100 мкл диметилкарбоната, получая раствор полимера с концентрацией 50 мг/мл. В данный раствор полимера добавляли 20 мкл трет-бутанола, добавляли заданное количество 10 мг/мл водного раствора FD40, как показано в таблице 4, и перемешивали, получая эмульсию с обращенной фазой. Данную эмульсию с обращенной фазой предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов. Полученный сухой остаток диспергировали в 200 мкл диметилкарбоната, получая суспензию S/O. Данную суспензию S/O по каплям добавляли к 2 мл водного раствора, содержащего 10% Pluronic F-68 (зарегистрированная торговая марка BASF), и перемешивали и эмульгировали в вихревом перемешивающем устройстве, получая эмульсию типа S/O/W. Из данной эмульсии типа S/O/W сушкой в жидкости удаляли несмешивающийся с водой органический растворитель и получали жидкую дисперсию микрочастиц. Жидкую дисперсию микрочастиц предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов, получали порошок микрочастиц с инкапсулированным FD40, часть его наблюдали сканирующим электронным микроскопом (СЭМ: HITACHI, S-4800) и вычисляли средний диаметр частиц (таблица 4). Изображения СЭМ порошка полимера PEG-PLGA от 5k до 10k показаны на фиг.14, и изображения СЭМ порошка полимера PEG-PLGA от 5k до 61k показаны на фиг.15.
Пример 23
Измерение эффективности инкапсулирования микрочастиц с инкапсулированным FD40
Микрочастицы (5 мг) с инкапсулированным FD40, приготовленные методом примера 22 с использованием полимера PEG-PLGA, взвешивали, используя 1,5-мл пробирку Эппендорфа, диспергировали в Milli-Q (1 мл), центрифугировали в течение 30 минут и разделяли на надосадочную жидкость, содержащую неинкапсулированный FD40, и частицы с инкапсулированным FD40, и собирали. Собранные частицы с инкапсулированным FD40 растворяли в N,N-диметилформамиде (250 мкл) и частицы дезинтегрировались. Надосадочную жидкость, содержащую неинкапсулированный FD40, и раствор N,N-диметилформамида (50 мкл), содержащий инкапсулированный FD40, добавляли в Milli-Q (3 мл) по отдельности и интенсивно перемешивали, и FD40 количественно определяли, используя флуоресцентный спектрофотометр (HORIBA, Fluoro MAX-3, длина волны возбуждения 495 нм, длина волны флуоресценции 520 нм), и рассчитывали эффективность инкапсулирования во всем собранном объеме.
Фиг.13 показывает эффективность инкапсулирования FD40 в микрочастицах, приготовленных из полимера PEG-PLGA. Во всех сериях 2k, 5k, 10k молекулярной массы PEG, когда молекулярная масса PLG была высокой, эффективность инкапсулирования имела тенденцию быть высокой. В частности, в PEG5k сериях, при 5k-65k, эффективность инкапсулирования была очень высокой, составляя примерно 90%. Эффективность инкапсулирования составляла примерно 55% в 10k-95k (PLGA/PEG=9,5) примерно при том же отношении молекулярных масс как в 5k-47k *OKGA/PEG=9,4) с высокой эффективностью инкапсулирования (примерно 80%).
Пример сравнения 3
Изготовление частиц с инкапсулированным FD40
10 мг полимера PEG-PLGA (5k-61k) растворяли в 2 мл этилацетата, получая раствор полимера. В данный раствор полимера по каплям добавляли 100 мкл 2 мг/мл раствора гормона роста и перемешивали. После операции перемешивания раствор добавляли к 20 мл диоксана. Растворитель выпаривали и концентрировали примерно до 2 мл, и жидкую дисперсию частиц добавляли в воду, содержащую 500 мг Pluronic F-68 (зарегистрированная торговая марка BASF). Образец подвергали сушке вымораживанием, к 50 мг образца добавляли 1 мл воды, частицы вновь диспергировали и получали частицы, содержащие неассоциированное гидрофильное активное вещество. Средний диаметр частиц измеряли методом динамического светорассеяния, используя прибор ELS-Z (изготовленный Otsuka Denshi), и эффективность инкапсулирования лекарственного средства определяли так же как в примере 23.
В результате, эффективность инкапсулирования FD40 составляла 48%, средний диаметр частиц составлял 203,8 нм, и эффективность инкапсулирования была ниже, чем в микрочастицах примера 23.
Пример 24
Анализ скорости высвобождения in vitro FD40 из микрочастиц с инкапсулированным FD40
Для того чтобы оценить соотношение между поведением замедленного высвобождения и длиной PLGA цепи для составления полимерной частицы PEG-PLGA, оценивали поведение высвобождения в частицах 5k-23k, 5k-32,5k, 5k-47k и 5k-61k из микрочастиц с инкапсулированным FD40, приготовленных в примере 22.
Микрочастицы, сразу после приготовления, хранили в высушенном вымораживанием состоянии при -30°C и нагревали до нормальной температуры перед использованием. Точно 20 мг порошка частиц взвешивали и помещали в 1,5-мл пробирку (пробирку Эппендорфа) и добавляли 1 мл буфера для анализа (0,02% азида натрия, 0,1% Pluronic F-68 (зарегистрированная торговая марка BASF) и 0,1% физиологического раствора с фосфатным буфером, содержащего 0,1% альбумина бычьей сыворотки), интенсивно перемешивали в перемешивающем устройстве и суспендировали. Затем, используя высокоскоростную центрифугу Hitachi (CF16RX) раствор центрифугировали в течение 10 минут при 18900×g и удаляли 950 мкл фракции надосадочной жидкости, содержащей неинкапсулированный FD40, снова добавляли 950 мкл буфера для анализа, частицы суспендировали и центрифугировали и операцию очистки частиц повторяли в общем три раза.
В частицы, очищенные три раза, еще раз добавляли 950 мкл буфера для анализа, частицы суспендировали и 100 мкл каждой пробы распределяли в 1,5-мл пробирку. В каждую пробирку добавляли 900 мкл буфера для анализа, получая общий объем раствора 1 мл, который выдерживали в инкубаторе при 37°C, в то же время вращая со скоростью 10 об./мин. посредством вращающего устройства. Затем каждую инкубированную пробирку центрифугировали в течение 10 минут при 18900×g и 950 мкл надосадочной жидкости дозировали и хранили при 4°C до времени измерения интенсивности флуоресценции.
Интенсивность флуоресценции анализируемого раствора измеряли, используя 3 мл удаляемую кювету (KARTELL) и HORIBA, Fluoro MAX-3 при длине волны возбуждения 494 нм и длине волны флуоресценции 512 нм), и степень замедленного высвобождения определяли из отношения количества FD40, использованного при приготовлении частиц.
Фиг.16 показывает высвобожденное количество FD40 из различных микрочастиц, определенное оценкой высвобождения. Ось абсцисс обозначает время инкубации, а ось ординат представляет отношение высвобожденного количество к загруженному количеству. В частицах 5k-23k, имеющих короткую PLGA цепь, примерно 40% от загруженного количества высвобождались в течение 1 суток в начальный период инкубации, и через один месяц высвобождалось почти все количество за исключением части начального пика выделения. Противоположным образом, по мере того, как длина PLGA цепи становится больше, начальное высвобожденное количество снижалось, и в микрочастицах 5k-61k высвобожденное количество в первые сутки начального периода составляло 10% или менее.
Пример 25
Измерение эффективности инкапсулирования лекарственного средства микрочастицами с инкапсулированным человеческим инсулином
Используя полимер PEG-PLGA (5k-61k), приготовленный в примере 20, таким же методом, как в примере 22, приготовили микрочастицы с инкапсулированным человеческим инсулином. Полученные микрочастицы (20 мг) взвешивали, используя 1,5-мл пробирку Эппендорфа, и растворяли в 1 мл буферного раствора A (физиологический раствор с фосфатным буфером, содержащий 0,1% альбумина бычьей сыворотки, 0,1% Pluronic F-68 (зарегистрированная торговая марка BASF) и 0,02% азида натрия), центрифугировали в течение 10 минут при 18800×g и разделяли на частицы (осаждение) и надосадочную жидкость. Надосадочную жидкость собирали в другую пробирку и частицы снова суспендировали в 1 мл буферного раствора A, и операцию центрифугирования и разделения на частицы и надосадочную жидкость снова проводили в тех же условиях. Данную операцию очистки повторяли еще один раз (всего три операции центрифугирования) и концентрацию человеческого инсулина в каждой надосадочной жидкости, собранной с помощью операции центрифугирования, измеряли многослойным методом твердофазного иммуноферментного анализа.
Многослойный метод твердофазного иммуноферментного анализа проводили следующим образом. Античеловеческое моноклональное антитело к инсулину (изготовленное Fitzgerald, клон № Е6Е5) иммобилизовали на пластине ELISA (Maxisorp от Nunc Corp.) при концентрации 5 мкг/мл, добавляли 50 мкл буферного раствора ELISA (0,1 М Трис хлоратный буферный раствор, содержащий 0,25% альбумина бычьей сыворотки и 0,05% Tween 20, pH 8,0) и 50 мкл измеряемого образца или стандартного образца, разбавленного в разбавляющем растворе ELISA (PBS, содержащий 0,25% альбумина бычьей сыворотки и 0,05% Tween 20), и раствор реагировал при комнатной температуре при встряхивании в течение 1 часа. Пластину три раза очищали в очищающем растворе (PBS, содержащий 0,05% Tween 20), непрореагировавший реагент удаляли, и добавляли 0,5 мкг/мл меченного биотином античеловеческого моноклонального антитела (изготовленного Fitzgerald, клон № D4B8) и стрепто-авидин-HRP коньюгат (изготовленный Zymed), и растворы реагировали при комнатной температуре при встряхивании в течение 1 часа и 15 минут. После каждой реакции, пластину очищали три раза в очищающем растворе (PBS, содержащий 0,05% Tween 20) и непрореагировавший реагент удаляли. Наконец, добавляли субстрат HRP и HRP ферментную активность объединенного коньюгата определяли колориметрией, и на основании рабочей кривой, приготовленной проявлением цвета стандартного инсулина, определяли концентрацию инсулина в образце.
Из загруженного количества человеческого инсулина во время приготовления частиц (на массу частиц 20 мг) вычитали общее количество человеческого инсулина в трех надосадочных жидкостях, полученных операциями центрифугирования, и эффективность инкапсулирования рассчитывали согласно формуле ниже.
Средний диаметр полученных микрочастиц составлял 4,7 мкм. Эффективность инкапсулирования человеческого инсулина в микрочастицах составляла 86,75%, и было доказано, что белковое лекарственное средство можно содержать с высокой эффективностью.
Пример 26
Анализ скорости высвобождения in vitro лекарственного средства из микрочастиц с инкапсулированным человеческим инсулином
Микрочастицы, центрифугированные три раза в примере 25, суспендировали и диспергировали в 1,0 мл буферного раствора A. Из данного раствора пробы, равные 0,1 мл, распределяли в десять пробирок Эппендорфа (емкость 1,5 мл), и в каждую пробирку добавляли 0,9 мл буферного раствора A, и разбавляли в 10 раз. Сразу после разбавления, одну пробирку центрифугировали в течение 10 минут при 18800×g для осаждения частиц, и надосадочную жидкость собирали в другую пробирку (0-часовой образец). Оставшиеся девять пробирок вращали и медленно перемешивали в инкубаторе при 37°C, используя вращающее устройство со скоростью 6 об./мин. В определенные интервалы времени каждую пробирку центрифугировали аналогичным образом и надосадочную жидкость отделяли. В образце надосадочной жидкости, собранном в каждый момент времени, измеряли концентрацию инсулина, многослойным методом твердофазного иммуноферментного анализа, и высвобожденное количество инсулина (%) вычисляли по формуле, приведенной ниже.
Фиг.17 показывает динамику изменения высвобождения инсулина. С течением времени лекарственное средство высвобождалось постепенно, и скорость высвобождения увеличилась после 30 дней, и основная часть лекарственного средства высвобождалась в течение примерно 60 дней.
Пример 27
Подкожное введение мыши микрочастиц с инкапсулированным гормоном роста человека (hGH)
25 мг полимера PEG-PLGA растворяли в 500 мкл диметилкарбоната, получая раствор полимера с концентрацией 50 мг/мл. В данный раствор полимера добавляли 100 мкл трет-бутанола и по каплям добавляли 250 мкл 10 мг/мл водного раствора hGH и перемешивали вихревым перемешивающим устройством, получая эмульсию с обращенной фазой. Данную эмульсию с обращенной фазой предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов. Полученный сухой остаток диспергировали в 1 мл диметилкарбоната, получая суспензию S/O. Данную суспензию S/O по каплям добавляли к 10 мл водного раствора, содержащего 10% Pluronic F-68 (зарегистрированная торговая марка BASF), и перемешивали и эмульгировали в вихревом перемешивающем устройстве, получая эмульсию типа S/O/W. Из данной эмульсии типа S/O/W сушкой в жидкости удаляли несмешивающийся с водой органический растворитель и получали жидкую дисперсию микрочастиц. Жидкую дисперсию микрочастиц предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов, и получали порошок микрочастиц с инкапсулированным hGH. Средний диаметр полученных микрочастиц составлял 6,0 мкм.
300 мг приготовленных микрочастиц суспендировали и диспергировали в 3 мл фосфатного физиологического буферного раствора (PBS) и центрифугировали в течение 5 минут при 80×g для осаждения микрочастиц, и надосадочную жидкость переносили в другую пробирку. Надосадочную жидкость снова центрифугировали в течение 5 минут при 80×g для осаждения остающихся частиц, и надосадочную жидкость удаляли. Первый осадок от центрифугирования и второй осадок от центрифугирования объединяли и снова диспергировали в 1 мл физиологического раствора с фосфатным буфером, такую же операцию очистки центрифугированием повторяли в общем три раза, и гормон роста, не инкапсулированный в микрочастицах, удаляли. Наконец, осадок снова диспергировали в 200 мкл физиологического раствора с фосфатным буфером и получали раствор для введения. Количество гормона роста, инкапсулированного в PEG-PLGA частицах измеряли комплектом для твердофазного иммуноферментного анализа и вычитали из загруженного количества, определяли количество, инкапсулированное в 300 мг частиц, введенных мыши, и получали 700 мкг микрочастиц PEG-PLGA.
Данный раствор вводили подкожно в двух положениях в спину 10-недельного самца Balb/C мыши, и образцы крови отбирали в определенные промежутки времени из хвостовой вены. В образцы крови добавляли гепарин до конечной концентрации 3,3 межд. ед./мл, плазму собирали центробежным разделением в течение 5 минут при 5000 об./мин. и концентрацию гормона роста в плазме измеряли, используя комплект для твердофазного иммуноферментного анализа.
С целью сравнения раствор негранулированного белка гормона роста человека (700 мкг/0,2 мл) подкожно вводили мыши, и аналогичным образом отбирали образцы крови.
Для того чтобы подавить продукцию антител из-за введения гормона роста человека, который является чужеродным белком для мыши, за три дня до введения частиц подкожно вводили иммуносупрессор Такролимус гидрат (Astellas) 26 мкг/мышь и после этого 13 мкг/мышь во время введения лекарственного средства, и 3 днями и 7 днями позднее.
Фиг.18 показывает динамику изменения концентрации гормона роста человека в плазме. У мыши, которой ввели негранулированное лекарственное средство, уровень в крови через один час после введения был очень высоким, более 5000 нг/мл, и затем резко падал до уровня до введения в течение дня. С другой стороны, у мыши, которой вводили лекарственное средство в виде микрочастиц, приготовленный с использованием полимера PEG-PLGA, кратковременное повышение уровня в крови непосредственно после введения подавлялось до 100 нг/мл или менее, и в течение семи последующих дней уровень в крови поддерживался при высоких уровнях.
Пример 28
Изготовление микрочастиц с добавлением соли в жидкую фазу в стадии (с)
В 100 мкл 50 мг/мл раствора полимер PEG-PLGA (5k-61k)/диметилкарбонат добавляли 20 мкл трет-бутанола, добавляли 20 мкл водного раствора, содержащего 10 мг/мл FD40, и смесь перемешивали, получая раствор мицелл с обращенной фазой (эмульсию W/O). Полученный раствор предварительно замораживали жидким азотом и подвергали сушке вымораживанием в течение ночи, используя прибор для сушки вымораживанием, и получали сухой остаток, содержащий FD40. В полученный сухой остаток, содержащий FD40, добавляли 200 мкл диметилкарбоната и перемешивали в течение 10 секунд вихревым перемешивающим устройством, получая суспензию S/O, и ее по каплям добавляли в 2 мл водного раствора, содержащего 10% Pluronic F-68 (зарегистрированная торговая марка BASF) вместе с хлоридом натрия при заданных концентрациях (0 М, 10 мМ, 50 мМ, 1 М), перемешивали и эмульгировали в вихревом перемешивающем устройстве, получая эмульсию типа S/O/W. Из полученного раствора эмульсии типа S/O/W несмешивающийся с водой органический растворитель удаляли, используя испаритель (откаченный до 30 гПа (30·102 Па), и откачивали и отгоняли в течение 5 минут), получая диспергированное в воде вещество микрочастиц, содержащих FD40. Дисперсный водный раствор микрочастиц, содержащих FD40, предварительно замораживали жидким азотом и подвергали сушке вымораживанием в течение ночи, используя прибор для сушки вымораживанием, и получали порошок микрочастиц, содержащих FD40. Полученные частицы наблюдали сканирующим электронным микроскопом (СЭМ: HITACHI, S-4800), вычисляли средний диаметр частиц, и при всех концентрациях хлорида натрия средний диаметр микрочастиц составлял 6,5 мкм.
20 мг полученного порошка микрочастиц, содержащих FD40, взвешивали и диспергировали в 1 мл фосфатного физиологического буферного раствора (содержащего 0,1% Pluronic F-68 (зарегистрированная торговая марка BASF), 0,1% альбумина бычьей сыворотки и 0,01% азида натрия), и центрифугировали (14000 об./мин., 10 минут). После сбора надосадочной жидкости микрочастицы снова суспендировали в 1 мл фосфатного физиологического буферного раствора и центрифугировали, и микрочастицы очищали еще два раза. Очищенные микрочастицы снова суспендировали в 1 мл фосфатного физиологического буферного раствора, распределяли по 900 мкл в 1,5-мл пробирки Эппендорфа и добавляли 900 мкл фосфатного физиологического буферного раствора, раствор инкубировали при 37°C и образцы собирали после 24 часов. Собранные образцы центрифугировали в течение 10 минут при 14000 об./мин. и FD40, содержащийся в надосадочной жидкости, измеряли, используя флуоресцентный спектрофотометр (HORIBA, Fluoro MAX-3, длина волны возбуждения 495 нм, длина волны флуоресценции 520 нм), и рассчитывали высвобожденное количество. Количество FD40 в надосадочной жидкости, собранной во время очистки, измеряли аналогичным образом, и эффективность инкапсулирования рассчитывали из загруженного количества.
Эффективность инкапсулирования составляла 73%, 97%, 84% и 82% при концентрациях хлорида натрия 0 М, 10 мМ, 50 мМ и 1 М. Высвобожденное количество в 1 сутки составляло 14%, 7%, 15% и 11% при концентрациях хлорида натрия 0 М, 10 мМ, 50 мМ и 1 М, и при концентрации хлорида натрия 10 мМ эффективность инкапсулирования была наиболее высокой и высвобожденное количество в 1 сутки (начальный пик) было наименьшим.
Пример 29
Подкожное введение мыши микрочастиц с инкапсулированным гормоном роста человека (hGH) (оценка фармакологической активности)
25 мг каждого из полимеров PEG-PLGA (5k-55k) и PEG-PLGA (5k-105k) примера 20 растворяли в 500 мкл диметилкарбоната, получая раствор полимера с концентрацией 50 мг/мл. В данный раствор полимера добавляли 100 мкл трет-бутанола и по каплям добавляли 250 мкл 10 мг/мл водного раствора hGH и перемешивали вихревым перемешивающим устройством, получая эмульсию с обращенной фазой. Данную эмульсию с обращенной фазой предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов. Полученный сухой остаток диспергировали в 1 мл диметилкарбоната, получая суспензию S/O. Данную суспензию S/O по каплям добавляли к 10 мл водного раствора, содержащего 10% Pluronic F-68 (зарегистрированная торговая марка BASF), и перемешивали и эмульгировали в вихревом перемешивающем устройстве, получая эмульсию типа S/O/W. Из данной эмульсии типа S/O/W сушкой в жидкости удаляли несмешивающийся с водой органический растворитель и получали жидкую дисперсию микрочастиц. Жидкую дисперсию микрочастиц предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов, и получали порошок микрочастиц с инкапсулированным hGH. Полученные микрочастицы наблюдали сканирующим электронным микроскопом (СЭМ: HITACHI, S-4800), вычисляли средний диаметр частиц, и средний диаметр полученных микрочастиц составлял 4,2 мкм в микрочастицах из полимера PEG-PLGA (5k-55k) (микрочастицы 5k-55k) и 7,5 мкм в микрочастицах из PEG-PLGA (5k-105k) (микрочастицы 5k-105k).
300 мг микрочастиц каждого типа, приготовленных выше, суспендировали и диспергировали в 3 мл фосфатного физиологического буферного раствора (PBS) и частицы осаждали центробежным разделением в течение 5 минут при 80×g, и надосадочную жидкость переносили в другую пробирку. Надосадочную жидкость снова разделяли центрифугированием в течение 5 минут при 80×g, осаждали остающиеся частицы и надосадочную жидкость удаляли. Первый осадок от осаждения центрифугированием и второй осадок от осаждения центрифугированием объединяли и снова диспергировали в 1 мл физиологического раствора с фосфатным буфером, аналогичным образом проводили третью операцию очистки центрифугированием, и гормон роста, не инкапсулированный в микрочастицах, удаляли. Наконец, осадок вновь диспергировали в 200 мкл физиологического раствора с фосфатным буфером, чтобы получить раствор для введения.
Данный раствор вводили подкожно в спину 8-недельной ICR мыши с удаленным гипофизом (от японской SLC), и образцы крови отбирали в определенные промежутки времени из хвостовой вены. В образцы крови добавляли гепарин до конечной концентрации 3,3 межд. ед./мл, центрифугировали в течение 5 минут при 5000 об./мин., плазму собирали и концентрацию гормона роста в плазме концентрацию мышиного IGF-1 измеряли методом твердофазного иммуноферментного анализа.
С целью сравнения раствор негранулированного белка гормона роста человека (700 мкг/0,2 мл) подкожно вводили мыши, и аналогичным образом отбирали образцы крови.
Для того чтобы подавить продукцию антител из-за введения гормона роста человека, который является чужеродным белком для мыши, за три дня до введения частиц подкожно вводили иммуносупрессор Такролимус гидрат (Astellas) 26 мкг/мышь и после этого 13 мкг/мышь во время введения лекарственного средства, и дважды неделей позже.
Фиг.19 показывает динамику изменения концентрации гормона роста человека в плазме. У мыши, которой ввели негранулированное лекарственное средство, уровень в крови через 1 час после введения был очень высоким, и затем резко падал до уровня до введения в течение одного дня. С другой стороны, у мыши, которой вводили лекарственный препарат в виде микрочастиц, приготовленный с использованием полимера PEG-PLGA, кратковременное повышение концентрации в крови непосредственно после введения подавлялось до низкого уровня, примерно 1/100 от уровня у мыши, которой вводили негранулированное лекарственное средство, и в течение более девяти последующих дней после введения уровень в крови поддерживался при высоких уровнях.
Фиг.20 показывает концентрацию IGF-1 в плазме в течение данного периода времени. Концентрация IGF-1 в плазме повысилась после введения как в случае 5k-55k микрочастиц, так и 5k-105k микрочастиц, и высокие уровни сохранялись в течение 7 дней для 5k-55k микрочастиц и в течение более 14 дней для 5k-105k микрочастицах.
Пример 30
Подкожное введение мыши микрочастиц с инкапсулированным Эксендином-4 (агонистом ГПП-1 рецептора)
25 мг полимера PEG-PLGA (5k-61k) в примере 20 растворяли в 500 мкл диметилкарбоната, получая раствор полимера с концентрацией 50 мг/мл. В данный раствор полимера добавляли 100 мкл трет-бутанола и по каплям добавляли 250 мкл 10 мг/мл водного раствора Эксендина-4 (синтезированного по поручению компанией Sigma Genosys) и перемешивали вихревым перемешивающим устройством, получая эмульсию с обращенной фазой. Данную эмульсию с обращенной фазой предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов. Полученный сухой остаток диспергировали в 1 мл диметилкарбоната, получая суспензию S/O. Данную суспензию S/O по каплям добавляли к 10 мл водного раствора, содержащего 10% Pluronic F-68 (зарегистрированная торговая марка BASF), и перемешивали и эмульгировали в вихревом перемешивающем устройстве, получая эмульсию типа S/O/W. Из данной эмульсии типа S/O/W сушкой в жидкости удаляли несмешивающийся с водой органический растворитель и получали жидкую дисперсию микрочастиц. Жидкую дисперсию микрочастиц предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов, и получали порошок микрочастиц с инкапсулированным Эксендином-4. Полученные микрочастицы наблюдали сканирующим электронным микроскопом (СЭМ: HITACHI, S-4800), вычисляли средний диаметр частиц, и средний диаметр микрочастиц составлял 6,0 мкм.
300 мг приготовленных микрочастиц суспендировали и диспергировали в 3 мл фосфатного физиологического буферного раствора (PBS), частицы осаждали центрифугированием в течение 5 минут при 80×g и надосадочную жидкость переносили в другую пробирку. Надосадочную жидкость снова центрифугировали в течение 5 минут при 80×g и осаждали остающиеся частицы, и надосадочную жидкость удаляли. Первый осадок от центрифугирования и второй осадок от центрифугирования объединяли и снова диспергировали в 1 мл физиологического раствора с фосфатным буфером, и аналогичным образом проводили третью операцию центрифугирования, и Эксендин-4, не инкапсулированный в частицах, удаляли. Наконец, осадок вновь диспергировали в 200 мкл физиологического раствора с фосфатным буфером, получая раствор для введения.
Данный раствор вводили подкожно в двух положениях в спину 10-недельного самца Balb/C мыши (от японской SLC), и образцы крови отбирали в определенные промежутки времени из хвостовой вены. В образцы крови добавляли гепарин до конечной концентрации 3,3 межд. ед./мл и плазму собирали центробежным разделением в течение 5 минут при 5000 об./мин., и концентрацию гормона роста в плазме измеряли методом твердофазного иммуноферментного анализа.
С целью сравнения раствор негранулированного Эксендина-4 (700 мкг/0,2 мл) подкожно вводили мыши, и аналогичным образом отбирали образцы крови.
Для того чтобы подавить продукцию антител из-за введения Эксендина-4, который является чужеродным белком для мыши, за три дня до введения частиц подкожно вводили иммуносупрессор Такролимус гидрат (Astellas) 26 мкг/мышь и после этого 13 мкг/мышь во время введения лекарственного средства, и дважды неделей позже.
Фиг.21 показывает динамику изменений концентрации Эксендина-4 в плазме. У мыши, которой ввели негранулированное лекарственное средство, уровень в крови через 1 час после введения был очень высоким, и затем резко падал до уровня до введения в течение дня. С другой стороны, у мыши, которой вводили лекарственное средство в виде микрочастиц, приготовленный с использованием полимера PEG-PLGA, кратковременное повышение уровня в крови непосредственно после введения подавлялось до уровня менее примерно 1/100, и уровень в крови поддерживался высоким в течение месяца.
Пример 31
Получение микрочастиц с инкапсулированным декстраном, меченым флуоресцеином (FD40), отличающихся диаметром частиц
5 мг полимера PEG-PLGA (5k-55k) в примере 20 растворяли в 100 мкл диметилкарбоната, получая раствор полимера с концентрацией 50 мг/мл. В данный раствор полимера добавляли 20 мкл трет-бутанола и по каплям добавляли 20 мкл 1 мг/мл водного раствора FD40 и перемешивали вихревым перемешивающим устройством, получая эмульсию с обращенной фазой. Данную эмульсию с обращенной фазой предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов. Полученный сухой остаток диспергировали в 50 мкл, 200 мкл и 500 мкл диметилкарбоната, получая суспензию S/O. Данную суспензию S/O по каплям добавляли к 2 мл водного раствора, содержащего 10% Pluronic F-68 (зарегистрированная торговая марка BASF), и перемешивали и эмульгировали в вихревом перемешивающем устройстве, получая эмульсию типа S/O/W. Из данной эмульсии типа S/O/W сушкой в жидкости удаляли несмешивающийся с водой органический растворитель и получали жидкую дисперсию микрочастиц. Жидкую дисперсию микрочастиц предварительно замораживали жидким азотом и подвергали сушке вымораживанием, используя прибор для сушки вымораживанием (EYELA, FREEZE DRYER FD-1000) при температуре охлаждающей ловушки -45°C и вакууме 20 Па в течение 24 часов, и получали порошок микрочастиц с инкапсулированным FD40. Полученные микрочастицы наблюдали сканирующим электронным микроскопом (СЭМ: HITACHI, S-4800) и вычисляли средний диаметр частиц.
Фиг.22 показывает корреляцию между средним диаметром частиц и количеством диметилкарбоната, добавленного во время приготовления эмульсии типа S/O/W. В диапазоне от 50 мкл до 500 мкл, с увеличением количества диметилкарбоната наблюдали снижение среднего диаметра частиц.
ПРИМЕНИМОСТЬ В ПРОМЫШЛЕННОСТИ
Микрочастица по изобретению высвобождает гидрофильное активное вещество с подходящей скоростью в теле человека и применима в качестве DDS фармацевтического препарата.
Реферат
Микрочастица включает агломерат частиц, содержащих гидрофильное активное вещество, причем частица включает амфифильный полимер, состоящий из гидрофобного сегмента полигидроксикислоты и гидрофильного сегмента полисахарида или полиэтиленгликоля, и гидрофильное активное вещество. Также описан способ изготовления агломерированной микрочастицы, который включает (a) стадию получения эмульсии с обращенной фазой, (b) стадию получения твердого остатка, содержащего гидрофильное активное вещество, и (c) стадию введения твердого остатка в жидкую фазу, содержащую модификатор поверхности. Агломерированные микрочастицы обеспечивают эффективное инкапсулирование гидрофильного активного вещества и высвобождение гидрофильного активного вещества с соответствующей скоростью. 2 н. и 12 з.п. ф-лы, 22 ил., 4 табл., 31 пр.
Формула
(a) стадию получения эмульсии с обращенной фазой смешиванием водного растворителя, содержащего гидрофильное активное вещество, и не смешивающегося с водой органического растворителя, растворяющего амфифильный полимер, состоящий из гидрофобного сегмента полигидроксикислоты и гидрофильного сегмента полисахарида или полиэтиленгликоля,
(b) стадию получения твердого остатка, содержащего гидрофильное активное вещество, посредством удаления растворителя из эмульсии с обращенной фазой и
(c) стадию введения твердого остатка или жидкой дисперсии, содержащей твердый остаток, в жидкую фазу, содержащую модификатор поверхности.
Комментарии