Способ получения субмикронных частиц паклитаксела - RU2402313C2
Код документа: RU2402313C2
Чертежи




















Описание
Данная заявка является частичным продолжением заявки № 10/390333, поданной 17 марта 2003 г., которая является частичным продолжением заявки № 10/246802, поданной 17 сентября 2002 г., которая является частичным продолжением заявки № 10/035821, поданной 19 октября 2001 г., которая является частичным продолжением заявки № 09/953979, поданной 17 сентября 2001 г., которая является частичным продолжением заявки № 09/874637, поданной 5 июня 2001 г., в которой испрашивается приоритет предварительной заявки № 60/258160, поданной 22 декабря 2000 г. Все указанные выше заявки включены в данное описание в качестве ссылки и составляют его часть.
Спонсируемое федерально исследование или разработка:
Не заявляются.
Настоящее изобретение касается создания субмикронных частиц антинеопластического агента, в частности паклитаксела, или соединений, являющихся его производными, с помощью осаждения антинеопластического агента в водной среде для образования предварительной суспензии с последующей гомогенизацией. Поверхностно-активные вещества с фосфолипидами, конъюгированные с водорастворимым или гидрофильным полимером, таким как полиэтиленгликоль (ПЭГ), используют в качестве покрытия для частиц. Полученные частицы обычно имеют средний размер менее приблизительно 1000 нм и не являются быстро растворимыми.
Существует постоянно увеличивающееся количество органических соединений, которые включают в состав для терапевтических или диагностических целей, которые являются плохо растворимыми или нерастворимыми в водных растворах. Такие лекарственные средства сталкиваются с проблемой их доставки путем введений, подробно указанных ниже. Соединения, которые нерастворимы в воде, могут иметь существенные преимущества при составлении их в виде стабильной суспензии субмикронных частиц. Тщательный контроль размера частиц осуществляют для безопасного и эффективного применения данных составов. Частицы должны быть менее семи микрон в диаметре для безопасного прохождения через капилляры без индукции эмболии (Allen et al., 1987; Davis and Taube, 1978; Schroeder et al., 1978; Yokel et al., 1981). Одним решением данной проблемы является получение малых частиц нерастворимого кандидатного лекарственного средства и создание суспензии микрочастиц или наночастиц. Таким способом лекарственные средства, которые ранее невозможно было составить в системе на основе воды, могут быть сделаны в форме, подходящей для внутривенного введения. Пригодность для внутривенного введения включает малый размер частиц (<7 мкм), низкую токсичность (как в отношении компонентов состава, так и остаточных растворителей) и биодоступность частиц лекарственного средства после введения.
Препараты малых частиц нерастворимых в воде лекарственных средств могут также подходить для перорального, легочного, местного офтальмологического, интраназального, защечного, ректального, вагинального, чрескожного или других путей введения. Малый размер частиц улучшает скорость растворения лекарственного средства и поэтому улучшает его биодоступность и потенциально профили его токсичности. При введении данными путями может быть желательно иметь размер частиц в диапазоне от 5 до 100 мкм в зависимости от пути введения, состава, растворимости и биодоступности лекарственного средства. Например, для перорального введения желательно иметь размер частиц менее приблизительно 7 мкм. Для легочного введения размер частиц предпочтительно составляет менее приблизительно 10 мкм.
В настоящем изобретении предлагаются способы получения и композиции субмикронных частиц антинеопластического агента, в частности паклитаксела или соединений, являющихся его производными. Растворимость антинеопластического агента является более высокой в смешиваемом с водой первом растворителе, чем во втором растворителе, который является водным. Способы включают (i) перемешивание в смешиваемом с водой первом растворителе, или во втором растворителе, или в обоих смешиваемом с водой первом растворителе и во втором растворителе первого модификатора поверхности, включающего фосфолипид, конъюгированный с водорастворимым или гидрофильным полимером; (ii) растворение антинеопластического агента в смешиваемом с водой первом растворителе с образованием раствора; (iii) смешивание раствора со вторым растворителем для предопределения предварительной суспензии частиц; и (iv) гомогенизацию предварительной суспензии с образованием суспензии частиц, имеющих средний эффективный размер частиц менее приблизительно 1 мкм. Предпочтительно, чтобы частицы имели средний эффективный размер частиц менее приблизительно 400 нм, более предпочтительно менее 200 нм и наиболее предпочтительно менее приблизительно 150 нм.
В предпочтительном варианте осуществления водорастворимый или гидрофильный полимер, конъюгированный с фосфолипидом, представляет собой полиэтиленгликоль (ПЭГ). Необязательно второй модификатор поверхности может быть смешан со смешиваемым с водой первым растворителем, или вторым растворителем, или с обоими смешиваемым с водой первым растворителем и со вторым растворителем. Предпочтительный второй модификатор поверхности представляет собой полоксамер.
В варианте осуществления гомогенизацию осуществляют при приблизительно 30°С или выше.
Способы могут дополнительно включать удаление смешиваемого с водой первого растворителя или полной жидкой фазы из суспензии. В предпочтительном варианте осуществления смешиваемый с водой первый растворитель удаляют одновременно с гомогенизацией.
Способ может также дополнительно включать стерилизацию композиции.
В предпочтительном варианте осуществления частицы являются нерастворимыми.
В другом предпочтительном варианте осуществления частицы не агрегируют при напряженных состояниях или при хранении.
Данные и другие аспекты и свойства настоящего изобретения будут обсуждаться со ссылкой на последующие фигуры и сопровождающее описание.
На фиг.1 показано диаграммное представление одного способа настоящего изобретения.
На фиг.2 - диаграммное представление другого способа настоящего изобретения.
На фиг.3 представлены аморфные частицы до гомогенизации.
На фиг.4 - частицы после отжига гомогенизацией.
Фиг.5 представляет собой рентгенограмму дифракции рентгеновских лучей микроосажденного итраконазола с 12-гидроксистеаратом полиэтиленгликоля-660 до и после гомогенизации.
На фиг.6 представлены кристаллы карбамазепина до гомогенизации.
На фиг.7 - микрочастицы карбамазепина после гомогенизации (Avestin C-50).
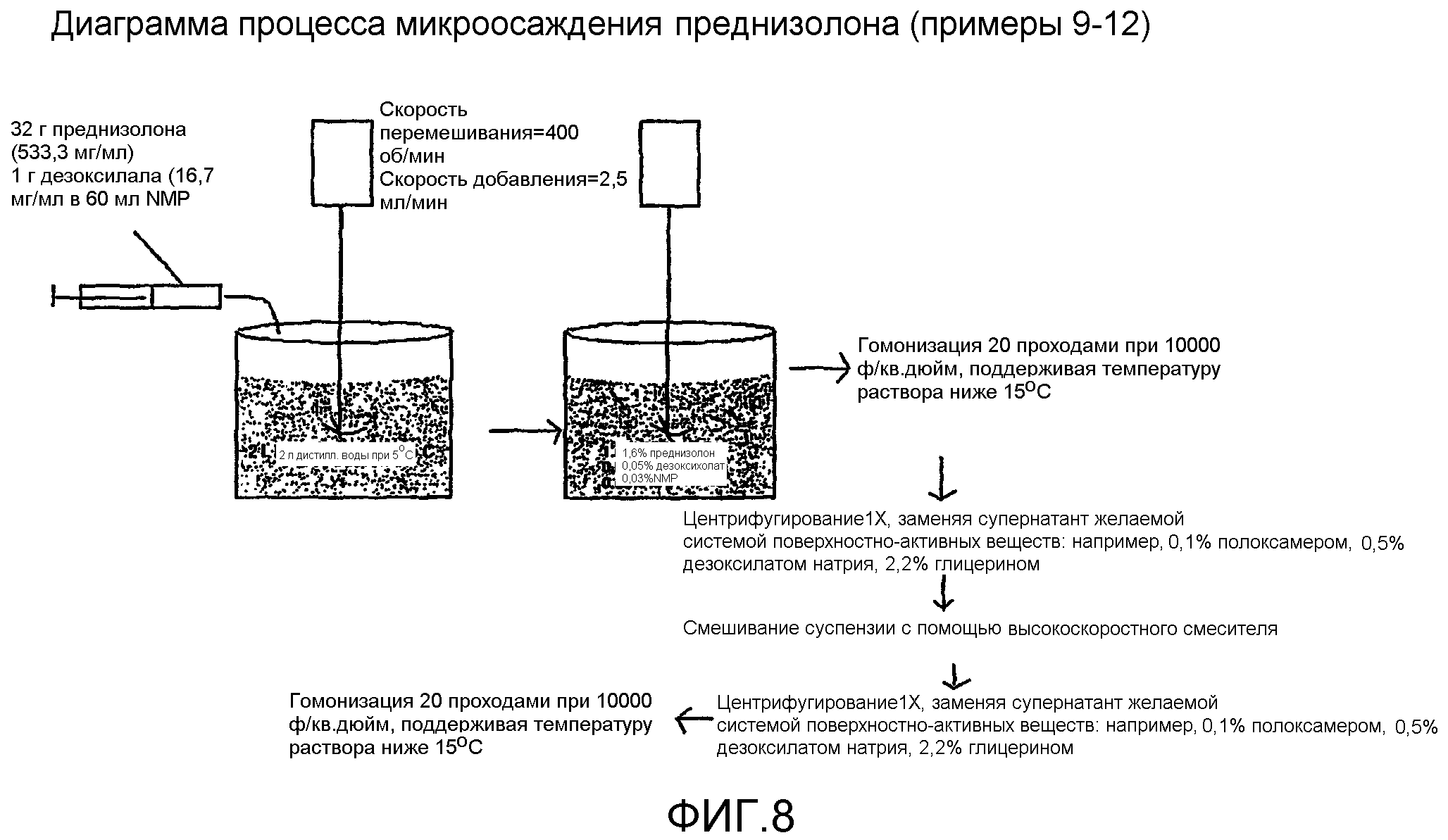
Фиг.8 представляет собой диаграмму, иллюстрирующую способ микроосаждения для преднизолона.
Фиг.9 - микрофотографию суспензии преднизолона до гомогенизации.
Фиг.10 - микрофотографию суспензии преднизолона после гомогенизации.
На фиг.11 иллюстрируется сравнение распределений по размеру наносуспензий (данного изобретения) и имеющейся в продаже эмульсии жиров.
На фиг.12 представлена порошковая рентгенограмма для исходного материала итраконазола (верх) и SMP-2-PRE (низ). Рентгенограмма исходного материала смещена вверх для ясности.
На фиг.13a - запись DSC для исходного материала итраконазола.
На фиг.13b - запись DSC для SMP-2-PRE.
На фиг.14 иллюстрируется запись DSC для SMP-2-PRE, показывающая плавление менее стабильного полиморфа при нагревании до 160°С, событие повторной кристаллизации при охлаждении и последующее плавление более стабильного полиморфа при повторном нагревании до 180°С.
На фиг.15 - сравнение образцов SMP-2-PRE после гомогенизации. Сплошная линия=образец с затравкой исходным материалом итраконазола. Прерывистая линия=образец без затравки. Сплошная линия смещена на 1 Вт/г для ясности.
На фиг.16 - эффект внесения затравки в процессе осаждения. Прерывистая линия = образец без затравки, сплошная линия=образец с затравкой исходным материалом итраконазола. Прерывистая линия смещена выше на 1,5 Вт/г для ясности.
На фиг.17 - эффект затравки концентрата лекарства в результате старения. Верхняя рентгенограмма представлена для кристаллов, полученных из свежего концентрата лекарства, и совместима со стабильным полиморфом (см. фиг.12, верх). Нижняя рентгенограмма представлена для кристаллов, полученных из состаренного (содержащего затравку) концентрата лекарства, и совместима с метастабильным полиморфом (см. фиг.12, низ). Верхний график смещен вверх для ясности.
На фиг.18 показано растворение двух составов субмикронных частиц паклитаксела.
На фиг.19 - влияние различных напряженных состояний на размер частиц субмикронных частиц паклитаксела.
На фиг.20 - влияние хранения на размер частиц субмикронных частиц паклитаксела.
Настоящее изобретение допускает варианты осуществления во многих различных формах. Предпочтительные варианты осуществления изобретения раскрыты с пониманием того, что настоящее раскрытие предназначено для рассмотрения в качестве примеров принципов изобретения и не предназначено для ограничения широких аспектов изобретения проиллюстрированными вариантами осуществления.
В настоящем изобретении предлагаются композиции и способы создания малых частиц органического соединения. Органическое соединение для применения в способе данного изобретения представляет собой любое органическое химическое вещество, чья растворимость снижается от одного растворителя к другому. Данное органическое соединение может представлять собой фармацевтически активное соединение, которое может быть выбрано из терапевтических агентов, диагностических агентов, косметических агентов, пищевых добавок и пестицидов.
Терапевтические агенты могут быть выбраны из множества известных фармацевтических агентов, таких как, но не ограничиваясь этим, анальгетики, анестетики, аналептики, адренергические агенты, адренергические блокирующие агенты, адренолитики, адренокортикоиды, адреномиметики, антихолинергические агенты, антихолинэстеразы, антиконвульсанты, алкилирующие агенты, алкалоиды, аллостерические ингибиторы, анаболические стероиды, анорексигенные агенты, антациды, антидиаррейные агенты, антидоты, антифолаты, антипиретики, антиревматоидные агенты, психотерапевтические агенты, блокирующие нервную систему агенты, противовоспалительные агенты, антигельминтные агенты, антиаритмические агенты, антибиотики, антикоагулянты, антидепрессанты, противодиабетические агенты, антиэпилептические агенты, противогрибковые агенты, антигистамины, антигипертензивные агенты, антимускариновые агенты, антимикобактериальные агенты, противомалярийные агенты, антисептики, антинеопластические агенты, антипротозойные агенты, иммуносуппрессорные агенты, иммуностимуляторные агенты, антитиреоидные агенты, противовирусные агенты, анксиолитические седативные агенты, вяжущие агенты, блокаторы бета-адренорецепторов, контрастирующие среды, кортикостероиды, подавляющие кашель агенты, диагностические агенты, диагностические визуализирующие агенты, диуретики, дофаминергические агенты, гемостатики, гематологические агенты, модификаторы гемоглобина, гормоны, гипнотические агенты, замуровывающие агенты, антигиперлипидемические и другие регулирующие липиды агенты, мускариновые агенты, мышечные релаксанты, парасимпатомиметики, паратиреоидные агенты, кальцитонин, простагландины, радиоактивные фармацевтические агенты, седативные агенты, половые гормоны, противоаллергические агенты, стимуляторы, симпатомиметики, тиреоидные агенты, вазодилататоры, вакцины, витамины и ксантины. Антинеопластические, или противораковые, агенты включают, но не ограничиваются ими, паклитаксел и соединения, являющиеся его производными, и другие антинеопластические агенты, выбранные из группы, состоящей из алкалоидов, антиметаболитов, ингибиторов ферментов, алкилирующих агентов и антибиотиков. Терапевтический агент может также быть биологическим, который включает, но не ограничивается этим, белки, полипептиды, углеводы, полинуклеотиды и нуклеиновые кислоты. Белок может быть антителом, которое может быть поликлональным или моноклональным.
Диагностические агенты включают рентгеновские визуализирующие агенты и контрастирующие среды. Примеры рентгеновских визуализирующих агентов включают WIN-8883 (этил-3,5-диацетамидо-2,4,6-трийодбензоат), также известный как этиловый эфир диатразоевой кислоты (EEDA), WIN 67722, т.е. (6-этокси-6-оксогексил-3,5-бис(ацетамид)-2,4,6-трийодобензоат; этил-2-(3,5-бис(ацетамид)-2,4,6-трийодбензоилокси)бутират (WIN 16318); этилдиатризоксиацетат (WIN 12901); этил-2-(3,5-бис(ацетамид)-2,4,6-трийодбензоилокси)пропионат (WIN 16923); N-этил-2-(3,5-бис(ацетамид)-2,4,6-трийодбензоилоксиацетамид (WIN 65312); изопропил-2-(3,5-бис(ацетамид)-2,4,6-трийодбензоилокси)ацетамид (WIN 12855); диэтил-2-(3,5-бис(ацетамид)-2,4,6-трийодбензоилоксималонат (WIN 67721); этил-2-(3,5-бис(ацетамид)-2,4,6-трийодбензоилокси)фенилацетат (WIN 67585); [[3,5-бис(ацетиламино)-2,4,5-трийиодбензоил]окси]бис(l-метил)овый эфир малоновой кислоты (WIN 68165) и 3,5-бис(ацетиламино)-2,4,6-трийод-4-(этил-3-этокси-2-бутеноат)эфир бензойной кислоты, (WIN 68209). Предпочтительные контрастирующие агенты включают такие, от которых требуется относительно быстрый распад в физиологических условиях, сводя тем самым к минимуму любую часть, связанную с воспалительным ответом. Распад может быть результатом ферментативного гидролиза, солюбилизации карбоновых кислот при физиологическом рН или других механизмов. Таким образом, предпочтительными могут быть плохо растворимые йодированные карбоновые кислоты, такие как йодипамид, диатризоевая кислота и метризоевая кислота вместе с гидролитически лабильными йодированными типами, такими как WIN 67721, WIN 12901, WIN 68165 и WIN 68209 или другие.
Другие контрастирующие среды включают, но не ограничиваются ими, препараты частиц для визуализации магнитного резонанса, такие как хелаты гадолиния или другие парамагнитные контрастирующие агенты. Примерами таких соединений являются гадопентетат димеглумина (Magnevist (E)) и гадотеридол (Prohance®).
Описание таких классов терапевтических агентов и диагностических агентов и перечисление их видов в пределах каждого класса можно найти в Martindale, The Extra Pharmacopoeia, Twenty-ninth Edition, The Pharmaceutical Press, London, 1989, который включен в данное описание в качестве ссылки и составляет его часть. Терапевтические агенты и диагностические агенты коммерчески доступны и/или могут быть получены способами, известными в данной области.
Косметический агент представляет собой любой активный ингредиент, способный обладать косметической активностью. Примерами таких активных ингредиентов могут быть, среди прочего, смягчающие агенты, увлажняющие агенты, ингибиторы свободных радикалов, противовоспалительные агенты, витамины, депигментирующие агенты, противоугревые агенты, противосеборейные агенты, кератолитики, агенты для похудения, окрашивающие кожу агенты и солнцезащитные агенты и, в частности, линолевая кислота, ретинол, ретиноевая кислота, алкиловые эфиры аскорбиновой кислоты, полиненасыщенные жирные кислоты, сложные эфиры никотиновой кислоты, никотинат токоферола, неомыляемые агенты риса, сои или масляного дерева, керамиды, оксикислоты, такие как гликолевая кислота, производные селена, антиоксиданты, бета-каротин, гамма-оризанол и стеарилглицерат. Косметические агенты коммерчески доступны и/или могут быть получены способами, известными в данной области.
Примеры пищевых добавок, рассматриваемых для применения в практике настоящего изобретения, включают, но не ограничиваются ими, белки, углеводы, водорастворимые витамины (например, витамин C, комплекс витаминов B и тому подобное), жирорастворимые витамины (например, витамины A, D, E, K и тому подобное) и экстракты растений. Пищевые добавки коммерчески доступны и/или могут быть получены способами, известными в данной области.
Термин “пестицид” понимается как охватывающий гербициды, инсектициды, акарициды, нематоциды, эктопаразитоциды и фунгициды. Примеры классов соединений, к которым в настоящем изобретении может принадлежать пестицид, включают производные мочевины, триазины, триазолы, карбаматы, сложные эфиры фосфорной кислоты, динитроанилины, морфолины, ацилаланины, пиретроиды, сложные эфиры бензиловой кислоты, дифенилэфиры и полициклические галогенизированные углеводороды. Конкретные примеры пестицидов в каждом из указанных классов перечислены в Pesticide Manual, 9th Edition, British Crop Protection Council. Пестициды коммерчески доступны и/или могут быть получены способами, известными в данной области.
Предпочтительно, чтобы органическое соединение или фармацевтически активное соединение плохо растворялось в воде. Под «плохой растворимостью в воде» подразумевается растворимость соединения в воде менее приблизительно 10 мг/мл и предпочтительно менее 1 мг/мл. Такие плохо растворимые в воде агенты больше всего подходят для препаратов в виде водной суспензии, так как существуют ограниченные альтернативы для создания составов данных агентов в водной среде.
Настоящее изобретение может быть также осуществлено с растворимыми в воде фармацевтически активными соединениями с помощью включения данных соединений в матрикс из твердого носителя (например, сополимер полиактаида-полигликолида, альбумин, крахмал) или с помощью инкапсулирования данных соединений в окружающий их пузырек, который является непроницаемым для фармацевтического соединения. Инкапсулирующий пузырек может представлять собой полимерное покрытие, такое как полиакрилат. Далее, малые частицы, полученные из данных водорастворимых фармацевтических агентов, могут быть модифицированы для улучшения химической стабильности и контроля фармакокинетических свойств агентов с помощью контролирования высвобождения агентов из частиц. Примеры водорастворимых фармацевтических агентов включают, но не ограничиваются ими, простые органические соединения, белки, пептиды, нуклеотиды, олигонуклеотиды и углеводы.
Частицы настоящего изобретения имеют средний эффективный размер частиц обычно менее приблизительно 100 мкм при измерении с помощью способов динамического рассеивания света, например фотокорреляционной спектроскопии, лазерной дифракции, лазерного рассеивания света под малым углом (LALLS), лазерного рассеивания света под средним углом (MALLS), способов затемнения света (например, способ Коултера), реологии или микроскопии (световой или электронной). Однако частицы могут быть получены в широком диапазоне размеров, таких как от приблизительно 20 мкм до приблизительно 10 нм, от приблизительно 10 мкм до приблизительно 10 нм, от приблизительно 2 мкм до приблизительно 10 нм, от приблизительно 1 мкм до приблизительно 10 нм, от приблизительно 400 нм до приблизительно 50 нм, от приблизительно 200 нм до приблизительно 50 нм или в любом представленном в данном описании диапазоне или сочетании диапазонов. Предпочтительный средний эффективный размер частиц зависит от таких факторов, как определенный путь введения, состав, растворимость, токсичность и биодоступность соединения.
Чтобы подходить для парентерального введения, частицы предпочтительно имеют средний эффективный размер менее приблизительно 7 мкм, более предпочтительно менее приблизительно 2 мкм, или любой представленный в данном описании диапазон или сочетание диапазонов. Парентеральное введение включает внутривенное, внутриартериальное, подоболочечное, внутрибрюшинное, внутриглазное, внутрисуставное, интрадуральное, внутрижелудочковое, внутриперикардиальное, внутримышечное, внутрикожное или подкожное введение.
Размеры частиц для пероральных единичных дозированных форм могут превышать 2 мкм. Частицы могут находиться в диапазоне размеров до приблизительно 100 мкм, предлагаемые так, чтобы частицы имели достаточную биодоступность и другие характеристики пероральной единичной дозированной формы. Пероральные единичные дозированные формы включают таблетки, капсулы, капсулообразные таблетки, капсулы из мягкого и твердого геля или другой доставляющий носитель для доставки лекарственного средства с помощью перорального введения.
Настоящее изобретение дополнительно подходит для обеспечения частицами органического соединения в форме, подходящей для легочного введения. Размеры частиц для легочных единичных дозированных форм могут превышать 500 нм и обычно менее приблизительно 10 мкм. Частицы в суспензии могут быть превращены в аэрозоль и введены с помощью небулайзера для легочного введения. Альтернативно частицы могут быть введены в виде сухого порошка с помощью ингалятора сухого порошка после удаления жидкой фазы из суспензии или сухой порошок может быть введен в неводном пропелленте для введения с помощью ингалятора с контролируемой дозировкой. Примером подходящего пропеллента является гидрофторуглерод (HFC), такой как HFC-134a (1,1,1,2-тетрафторэтан) и HFC-227ea (1,1,1,2,3,3,3-гептафторпропан). В отличие от хлорфторуглеродов (CFC), HFC характеризуются отсутствием или низким потенциалом снижения озона.
Дозированные формы для других путей доставки, таких как интраназальная, местная, офтальмологическая, назальная, защечная, ректальная, вагинальная, трансдермальная и тому подобное, также могут быть составлены из частиц, полученных по настоящему изобретению.
Способ получения частиц может быть разделен на четыре общих категории. Каждая из категорий способов охватывает стадии: (1) растворения органического соединения в смешиваемом с водой первом растворителе с образованием первого раствора, (2) смешивания первого раствора со вторым растворителем воды для осаждения органического соединения и создания предварительной суспензии и (3) придания энергии предварительной суспензии в форме смешивания с высоким сдвигом или нагревания, или сочетания обоих для обеспечения стабильной формы органического соединения, имеющего желаемые диапазоны размеров, определенные выше. Стадии смешивания и стадия придания энергии могут быть выполнены в виде последовательных стадий или одновременно.
Категории способов подразделяются на основе физических свойств органического соединения, что определяется исследованиями дифракции рентгеновских лучей, исследованиями дифференциальной сканирующей калориметрии (DSC) или другим подходящим исследованием, проведенными перед стадией придания энергии или после стадии придания энергии. В первой категории способа перед стадией придания энергии органическое соединение в предварительной суспензии имеет аморфную форму, полукристаллическую форму или переохлажденную жидкую форму и имеет средний эффективный размер частиц. После стадии придания энергии органическое соединение находится в кристаллической форме, имеющей средний эффективный размер частиц по существу тот же самый или менее, чем в предварительной суспензии.
Во второй категории способа перед стадией придания энергии органическое соединение находится в кристаллической форме и имеет средний эффективный размер частиц. После стадии придания энергии органическое соединение находится в кристаллической форме, имеющей по существу тот же самый средний эффективный размер частиц, как перед стадией придания энергии, но кристаллы после стадии придания энергии агрегируют с меньшей вероятностью.
Более низкая тенденция органического соединения к агрегации наблюдается при лазерном динамическом рассеивании света и при световой микроскопии.
В третьей категории способа перед стадией придания энергии органическое соединение находится в кристаллической форме, которая является хрупкой, и имеет средний эффективный размер частиц. Под термином «хрупкий» подразумевается, что частицы являются ломкими и с большей легкостью разбиваются на более мелкие частицы. После стадии придания энергии органическое соединение находится в кристаллической форме, имеющей средний эффективный размер частиц, меньший, чем кристаллы в предварительной суспензии. С помощью использования стадий, необходимых для перевода органического соединения в кристаллическую форму, которая является хрупкой, последующая стадия придания энергии может быть осуществлена более быстро и эффективно по сравнению с органическим соединением с менее хрупкой морфологией кристаллов.
В четвертой категории способа первый раствор и второй растворитель одновременно подвергают стадии придания энергии. Таким образом, физические свойства органического соединения перед и после стадии придания энергии не измеряются.
Стадия придания энергии может быть осуществлена любым способом, в котором предварительная суспензия или первый раствор и второй растворитель подвергаются кавитации, сдвигу или ударной силе. В одной предпочтительной форме изобретения стадия придания энергии представляет собой стадию отжига. Отжиг определяется в данном изобретении как процесс превращения вещества, которое является термодинамически не стабильным, в более стабильную форму с помощью единственного или повторяющегося приложения энергии (прямого нагревания или механического напряжения), с последующей тепловой релаксацией. Данное снижение энергии может быть достигнуто с помощью превращения твердой формы из менее упорядоченной в более упорядоченную решетчатую структуру. Альтернативно данная стабилизация может осуществляться с помощью реорганизации молекул поверхностно-активного вещества на поверхности раздела твердая-жидкая фазы.
Данные четыре категории способа будут обсуждаться ниже по отдельности. Однако должно быть понятно, что условия способа, такие как выбор поверхностно-активных веществ или сочетания поверхностно-активных веществ, количество используемого поверхностно-активного вещества, температура реакции, скорость смешивания растворов, скорость осаждения и тому подобное, могут быть выбраны так, чтобы дать возможность получения любого лекарственного средства с помощью любой из категорий, обсуждаемых ниже.
Первая категория способа, так же как и вторая, третья и четвертая категории способа, могут быть дополнительно подразделены на две субкатегории, способ A и способ B, представленные в виде диаграмм на фиг.1 и 2.
Первый растворитель в соответствии с настоящим изобретением представляет собой растворитель или смесь растворителей, в которых представляющее интерес органическое соединение является относительно растворимым и которое смешивается со вторым растворителем. Такие растворители включают, но не ограничиваются ими, смешиваемые с водой протонные соединения, в которых атом водорода в молекуле связан с электроотрицательным атомом, таким как кислород, азот или другая группа VA, VIA и VII A в периодической таблице элементов. Примеры таких растворителей включают, но не ограничиваются ими, спирты, амины (первичные или вторичные), оксимы, гидроксамовые кислоты, карбоновые кислоты, сульфоновые кислоты, фосфоновые кислоты, фосфорные кислоты, амиды и производные мочевины.
Другие примеры первого растворителя включают апротонные органические растворители. Некоторые из данных апротонных растворителей могут образовывать водородные связи с водой, но могут действовать только в качестве акцепторов протонов, потому что они не имеют эффективных донорских групп протонов. Один класс апротонных растворителей представляет собой диполярный апротонный растворитель, определенный Международным объединением чистой и прикладной химии (IUPAC Compendium of Chemical Terminology, 2nd Ed., 1997) как:
“Растворитель со сравнительно высокой относительной диэлектрической проницаемостью (или диэлектрической константой), большей, чем приблизительно 15, и с существенным перманентным дипольным моментом, который не может быть донором пригодных лабильных атомов водорода для образования сильных водородных связей, например диметилсульфоксид.
Диполярные апротонные растворители могут быть выбраны из группы, состоящей из: амидов (полностью замещенных, с азотом без присоединенных атомов водорода), мочевин (полностью замещенных, с отсутствием присоединения атомов водорода к азоту), сложных эфиров, циклических эфиров, нитрилов, кетонов, сульфонов, сульфоксидов, полностью замещенных фосфатов, сложных эфиров фосфоновой кислоты, фосфорамидов, нитросоединений и тому подобное. Среди других членами данного класса являются диметилсульфоксид (ДМСО), N-метил-2-пирролидинон (NMP), 2-пирролидинон, 1,3-диметилимидазолидинон (DMI), диметилацетамид (DMA), диметилформамид (ДМФА), диоксан, ацетон, тетрагидрофуран (ТГФ), тетраметиленсульфон (сульфолан), ацетонитрил и гексаметилфосфорамид (HMPA), нитрометан.
Могут быть также выбраны растворители, которые обычно не смешиваются с водой, но имеют достаточную растворимость в воде при низких объемах (менее 10%), действуя в качестве смешиваемого с водой первого растворителя при данных пониженных объемах. Примеры включают ароматические углеводороды, алкены, алканы и галогенизированные ароматические соединения и галогенизированные алканы. Ароматические соединения включают, но не ограничиваются ими, бензол (замещенный или незамещенный) и моноциклические или полициклические арены. Примеры замещенных бензолов включают, но не ограничиваются ими, ксилолы (орто, мета или пара) и толуол. Примеры алканов включают, но не ограничиваются ими, гексан, неопентан, гептан, изооктан и циклогексан. Примеры галогенизированных ароматических соединений включают, но не ограничиваются ими, хлорбензол, бромбензол и хлортолуол. Примеры галогенизированных алканов и алкенов включают, но не ограничиваются ими, трихлорэтан, метиленхлорид, этилендихлорид (EDC) и тому подобное.
Примеры всех указанных выше классов растворителей включают, но не ограничиваются ими: N-метил-2-пирролидинон (также называемый N-метил-2-пирролидон), 2-пирролидинон (также называемый 2-пирролидон), 1,3-диметил-2-имидазолидиннон (DMI), диметилсульфоксид, диметилацетамид, уксусную кислоту, молочную кислоту, метанол, этанол, изопропанол, 3-пентанол, н-пропанол, бензиловый спирт, глицерин, бутиленгликоль (бутандиол), этиленгликоль, пропиленгликоль, моно- и диацилированные моноглицериды (такие как глицерилкаприлат), диметилизосорбид, ацетон, диметилсульфон, диметилформамид, 1,4-диоксан, тетраметиленсульфон (сульфолан), ацетонитрил, нитрометан, тетраметилмочевину, гексаметилфосфорамид (HMPA), тетрагидрофуран (ТГФ), диоксан, диэтиловый эфир, трет-бутилметиловый эфир (TBME), ароматические углеводороды, алкены, алканы, галогенизированные ароматические соединения, галогенизированные алкены, галогенизированные алканы, ксилол, толуол, бензол, замещенный бензол, этилацетат, метилацетат, бутилацетат, хлорбензол, бромбензол, хлортолуол, трихлорэтан, метиленхлорид, этилендихлорид (EDC), гексан, неопентан, гептан, изооктан, циклогексан, полиэтиленгликоль (ПЭГ, например, ПЭГ-4, ПЭГ-8, ПЭГ-9, ПЭГ-12, ПЭГ-14, ПЭГ-16, ПЭГ-120, ПЭГ-75, ПЭГ-150), эфиры полиэтиленгликоля (примеры, такие как ПЭГ-4 дилаурат, ПЭГ-20 дилаурат, ПЭГ-6 изостеарат, ПЭГ-8 пальмитостеарат, ПЭГ-150 пальмитостеарат), сорбиты полиэтиленгликоля (такие как ПЭГ-20 сорбит изостеарат), моноалкиловые эфиры полиэтиленгликоля (примеры, такие как ПЭГ-3 диметиловый эфир), ПЭГ-4 диметиловый эфир), полипропиленгликоль (PPG), полипропиленальгинат, PPG-10 бутандиол, PPG-10 метилглюкозный эфир, PPG-20 метилглюкозный эфир, PPG-15 стеариловый эфир, пропиленгликоль дикаприлат/дикапрат, пропиленгликоль лаурат и гликофурол (эфир тетрагидрофурфурилового спирта и полиэтиленгликоля). Предпочтительным первым растворителем является N-метил-2-пирролидинон. Другим предпочтительным первым растворителем является молочная кислота.
Второй растворитель представляет собой водный растворитель. Данный водный растворитель может быть водой самой по себе. Данный растворитель может также содержать буферы, соли, поверхностно-активное(ные) вещество(а), водорастворимые полимеры и сочетания данных наполнителей.
Способ А
В способе A (см. фиг.1) органическое соединение («лекарство») сначала растворяют в первом растворителе для создания первого раствора. Органическое соединение может быть добавлено от приблизительно 0,1% (мас./об.) до приблизительно 50% (мас./об.) в зависимости от растворимости органического соединения в первом растворителе. Нагревание концентрата от приблизительно 30°С до приблизительно 100°С может быть необходимым для обеспечения полного растворения соединения в первом растворителе.
Второй водный растворитель предлагается с добавлением в него одного или более необязательных модификаторов поверхности, таких как анионное поверхностно-активное вещество, катионное поверхностно-активное вещество, неионное поверхностно-активное вещество или поверхностно-активная биологическая молекула. Подходящие анионные поверхностно-активные вещества включают, но не ограничиваются ими, алкилсульфонаты, алкилфосфаты, алкилфосфонаты, лаурат калия, стеарат триэтаноламина, лаурилсульфат натрия, додецилсульфат натрия, сульфаты алкилполиоксиэтилена, альгинат натрия, диоктилсульфосукцинат натрия, фосфатидилхолин, фосфатидилглицерин, фосфатидилинозин, фосфатидилсерин, фосфатидная кислота и ее соли, эфиры глицерина, натриевая соль карбоксиметилцеллюлозы, холевая кислота и другие желчные кислоты (например, холевая кислота, дезоксихолевая кислота, гликохолевая кислота, таурохолевая кислота, гликодезоксихолевая кислота) и их соли (например, дезоксихолат натрия и т.д.). Подходящие катионные поверхностно-активные вещества включают, но не ограничиваются ими, четвертичные аммониевые соединения, такие как хлорид бензалкония, бромид цетилтриметиламмония, хитозаны, хлорид лаурилдиметилбензиламмония, гидрохлориды ацилкарнитина или галогениды алкилпиридиния. В качестве анионных поверхностно-активных веществ могут быть использованы фосфолипиды. Подходящие фосфолипиды включают, например, фосфатидилхолин, фосфатидилэтаноламин, диацилглицерофосфоэтаноламин (такой как димиристоилглицерофосфоэтаноламин (DMPE), дипальмитоилглицерофосфоэтаноламин (DPPE), дистеароилглицерофосфоэтаноламин (DSPE) и диолеолилглицерофосфоэтаноламин (DOPE)), фосфатидилсерин, фосфатидилинозитол, фосфатидилглицерин, фосфатидную кислоту, лизофосфолипиды, фосфолипид яйца или сои или их сочетание. Фосфолипид может быть в солевой или несолевой форме, гидрогенизированным или частично гидрогенизированным или природным полусинтетическим или синтетическим. Фосфолипид может также быть конъюгированным с водорастворимым или гидрофильным полимером. Предпочтительный полимер представляет собой полиэтиленгликоль (ПЭГ), который также известен как монометоксиполиэтиленгликоль (мПЭГ). Молекулярные массы ПЭГ могут варьироваться, например, от 200 до 50000. Некоторые обычно применяемые ПЭГ, которые коммерчески доступны, включают ПЭГ 350, ПЭГ 550, ПЭГ 750, ПЭГ 1000, ПЭГ 2000, ПЭГ 3000 и ПЭГ 5000. Фосфолипид или конъюгат ПЭГ-фосфолипид может также включать функциональную группу, которая может ковалентно присоединяться к лиганду, включая, но не ограничиваясь этим, белки, пептиды, углеводы, гликопротеиды, антитела или фармацевтически активные агенты. Данные функциональные группы могут конъюгироваться с лигандами через, например, образование амидной связи, образование дисульфида или тиоэфира или связывание биотин/стрептавидин. Примеры связывающих лиганд функциональных групп включают, но не ограничиваются ими, гексаноиламин, додеканиламин, 1,12-додекандикарбоксилат, тиоэтанол,
4-(п-малеимидфенил)бутирамид (MBP),
4-(п-малеимидметил)циклогексанкарбоксамид (MCC),
3-(2-пиридилдитио)пропионат (PDP), сукцинат, глютарат,
додеканоат и биотин.
Подходящие неионные поверхностно-активные вещества включают эфиры полиоксиэтилена с жирными спиртами (Macrogol и Brij), эфиры полиоксиэтиленсорбитана и жирных кислот (полисорбаты), эфиры полиоксиэтилена и жирных кислот (Myrj), эфиры сорбитана (Span), моностеарат глицерина, полиэтиленгликоли, полипропиленгликоли, цетиловый спирт, цетостеариловый спирт, стеариловый спирт, арилалкилполиэфирные спирты, полиоксиэтилен-полиоксипропилен сополимеры (полоксамеры), полоксамины, метилцеллюлозу, гидроксиметилцеллюлозу, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, некристаллическую целлюлозу, полисахариды, включая крахмал и производные крахмала, такие как гидроксиэтилкрахмал (HES), поливиниловый спирт и поливинилпирролидон. В предпочтительной форме изобретения неионное поверхностно-активное вещество представляет собой сополимер полиоксиэтилена и полиоксипропилена и предпочтительно блок сополимер пропиленгликоля и этиленгликоля. Такие полимеры продают под торговой маркой POLOXAMER и иногда также обозначают как PLURONIC®, и они продаются несколькими поставщиками, включая Spectrum Chemical и Ruger. Из эфиров полиоксиэтилена и жирных кислот включаются те, которые имеют короткие алкильные цепи. Одним примером такого поверхностно-активного вещества является SOLUTOL® HS 15, полиэтилен-660-гидроксистеарат, производимый BASF Aktiengesellschaft.
Поверхностно-активные биологические молекулы включают такие молекулы, как альбумин, казеин, гирудин или другие подходящие белки. Включаются также биологические полисахариды и они состоят из, но не ограничиваются ими, крахмалов, гепарина и хитозанов.
Может быть также желательным добавление ко второму растворителю агента, доводящего рН, такого как гидроксид натрия, хлористоводородная кислота, трис буфер или цитрат, ацетат, лактат, меглумин или тому подобное. Второй растворитель должен иметь рН в диапазоне от приблизительно 3 до приблизительно 11.
Для пероральных лекарственных форм могут быть применены один или более следующих эксципиентов: желатин, казеин, лецитин (фосфатиды), аравийская камедь, холестерин, трагакант, стеариновая кислота, хлорид бензалкония, стеарат кальция, глицерилмоностеарат, цетостеариловый спирт, цетомакроголевый эмульсифицирующий парафин, сложные эфиры сорбитана, полиоксиэтиленалкиловые эфиры, например, эфиры макроголя, такие как цетомакроголь 1000, производные полиоксиэтиленового касторового масла, эфиры полиоксиэтиленсорбитана с жирными кислотами, например коммерчески доступные Твины™, полиэтиленгликоли, полиоксиэтиленстеараты, коллоидный диоксид кремния, фосфаты, додецилсульфат натрия, кальциевая соль карбоксиметилцеллюлозы, натриевая соль карбоксиметилцеллюлозы, метилцеллюлоза, гидроксиэтилцеллюлоза, гидроксипропилцеллюлоза, фталат гидроксипропилметилцеллюлозы, некристаллическая целлюлоза, силикат алюмомагния, триэтаноламин, поливиниловый спирт (PVA) и поливинилпирролидон (PVP). Большинство из таких эксципиентов подробно описаны в Handbook of Pharmaceutical Excipients, опубликованной Американской фармацевтической ассоциацией совместно с Фармацевтическим обществом Великобритании, Pharmaceutical Press, 1986. Модификаторы поверхности коммерчески доступны и/или могут быть получены с помощью способов, известных в данной области. Два или более модификаторов поверхности могут быть использованы в сочетании.
В предпочтительной форме изобретения способ получения малых частиц органического соединения включает стадии добавления первого раствора ко второму растворителю. Скорость добавления зависит от размера партии и кинетики осаждения органического соединения. Обычно для мало масштабного лабораторного способа (получение 1 литра) скорость добавления составляет от приблизительно 0,05 см3 в минуту до приблизительно 10 см3 в минуту. В процессе добавления растворы должны постоянно перемешиваться. С использованием световой микроскопии обнаружено, что при создании предварительной суспензии образуются аморфные частицы, полукристаллические твердые или переохлажденные жидкие частицы. Способ дополнительно включает стадию подвергания предварительной суспензии стадии придания энергии для превращения аморфных частиц, переохлажденных жидких или полукристаллических твердых частиц в более стабильное кристаллическое твердое состояние. Полученные частицы должны иметь средний эффективный размер частиц при измерении с помощью способов динамического рассеивания света, (например, фотокорреляционной спектроскопии, лазерной дифракции, лазерного рассеивания света под малым углом (LALLS), лазерного рассеивания света под средним углом (MALLS), способов затемнения света (например, способом Коултера), реологии или микроскопии (световой или электронной) в диапазонах, указанных выше). В четвертой категории способа первый раствор и второй растворитель объединяют при одновременном проведении стадии придания энергии.
Стадия придания энергии включает придание энергии путем сонификации, гомогенизации, противоточной гомогенизации, микрофлуидизации или других способов придания ударной силы, сил сдвига или кавитации. В течение данной стадии образец может быть охлажден или нагрет. В одной предпочтительной форме изобретения стадия придания энергии совершается с помощью плунжерно-зазорного гомогенизатора, такого как аппарат, продаваемый Avestin Inc. под названием EmulsiFlex-C160. В другой предпочтительной форме изобретения стадия придания энергии может быть осуществлена с помощью ультрасонификации с использованием процессора для ультрасонификации, такого как Vibra-Cell Ultrasonic Processor (600W), выпускаемого Sonics and Materials, Inc. В еще одной предпочтительной форме изобретения стадия придания энергии может быть осуществлена с помощью использования прибора для эмульсификации, как описано в патенте США No. 5720551, который включен в данное описание в качестве ссылки и составляет его часть.
В зависимости от скорости придания энергии может быть желательно довести температуру обрабатываемого образца до диапазона от приблизительно -30°С до 30°С. Альтернативно, для того чтобы воздействовать на желаемое изменение фазы обрабатываемого твердого агента, может быть необходимым нагревание предварительной суспензии до температуры в диапазоне от приблизительно 30°С до приблизительно 100°С в течение стадии придания энергии.
Способ B
Способ B отличается от способа A в следующем отношении. Первое различие заключается в поверхностно-активном веществе или сочетании поверхностно-активных веществ, добавляемых к первому раствору. Поверхностно-активные вещества могут быть выбраны из групп анионных, неионных, катионных поверхностно-активных веществ и поверхностно-активных биологических модификаторов, указанных выше.
Сравнительный пример способа A и способа B и патента США USPN 5780062
В патенте Соединенных Штатов No. 5780062 раскрыт способ получения малых частиц органического соединения с помощью первоначального растворения соединения в подходящем смешиваемом с водой первом растворителе. Второй раствор получают с помощью растворения полимера и амфифильного агента в водном растворителе. Первый раствор затем добавляют ко второму раствору с образованием осадка, который состоит из органического соединения и комплекса полимер-амфифильный агент. В патенте 5780062 не раскрывается применение стадии придания энергии данного изобретения в способах A и B. Отсутствие стабильности обычно подтверждается быстрой агрегацией и ростом частиц. В некоторых примерах аморфные частицы повторно кристаллизуются в виде больших кристаллов. Придание энергии предварительной суспензии раскрытым выше способом обычно дает частицы, которые проявляют пониженные скорости агрегации и роста частиц, а также отсутствие повторной кристаллизации при хранении продукта.
Способы A и B дополнительно отличаются от способа патента 5780062 отсутствием стадии образования комплекса полимер-амфифильный агент перед осаждением. В способе A такой комплекс не может быть образован, так как полимер не добавляется к фазе разбавителя (водной). В способе B поверхностно-активное вещество, которое может также действовать в качестве амфифильного агента, или полимер растворяют с органическим соединением в первом растворителе. Это предотвращает образование любых комплексов амфифильный агент-полимер перед осаждением. В патенте 5780062 успешное осаждение малых частиц зависит от образования комплекса амфифильный агент-полимер перед осаждением. В патенте 5780062 раскрывается комплекс амфифильный агент-полимер, образующий агрегаты в водном втором растворе. В патенте 5780062 объясняется, что гидрофобное органическое соединение взаимодействует с комплексом амфифильный агент-полимер, тем самым снижая растворимость данных агрегатов и вызывая осаждение. В настоящем изобретении показано, что включение поверхностно-активного вещества или полимера в первый растворитель (способ B) ведет при последующем добавлении второго растворителя к образованию более однородных, более мелких частиц, чем достигается при способе, описанном в патенте 5780062.
С данной целью были получены и проанализированы два состава. Каждый из составов имеет два раствора, концентрат и водный разбавитель, которые смешиваются вместе и затем сонифицируются. Концентрат в каждом составе содержит органическое соединение (итраконазол), смешиваемый с водой растворитель (N-метил-2-пирролидинон, или NMP) и, возможно, полимер (полоксамер 188). Водный разбавитель содержит воду, трис буфер и, возможно, полимер (полоксамер 188) и/или поверхностно-активное вещество (дезоксихолат натрия). Средний диаметр частиц органической частицы измеряют перед сонификацией и после сонификации.
Первый состав A содержит концентрат итраконазола и NMP. Водный разбавитель включает воду, полоксамер 188, трис буфер и дезоксихолат натрия. Таким образом, водный разбавитель включает полимер (полоксамер 188) и амфифильный агент (дезоксихолат натрия), который может образовывать комплекс полимер/амфифильный агент и, следовательно, находится в соответствии с раскрытием патента 5780062. (Однако опять патент 5780062 не раскрывает стадию придания энергии).
Второй состав B содержит концентрат итраконазола, NMP и полоксамер 188. Водный разбавитель включает воду, трис буфер и дезоксихолат натрия. Данный состав образован в соответствии с настоящим изобретением. Так как водный разбавитель не содержит сочетания полимера (полоксамера) и амфифильного агента (дезоксихолата натрия), комплекс полимер/амфифильный агент не может образоваться перед стадией смешивания.
В таблице 1 представлены средние диаметры частиц, измеренные с помощью лазерной дифракции в трех параллельных препаратах суспензий. Определяли исходный размер, затем образец сонифицировали в течение 1 минуты. Определение размера затем повторяли. Большое снижение размера при сонификации способом A было показателем агрегации частиц.
Суспензия лекарственного средства, полученная при применении способов, описанных в данном изобретении, может быть введена прямо в виде инъецируемого раствора, в составе используют предлагаемую воду для инъекций и подходящие средства для стерилизации раствора. Стерилизация может быть осуществлена с помощью способов, хорошо известных в данной области, таких как паровая или тепловая стерилизация, гамма-облучение и тому подобное. Другие способы стерилизации, особенно для частиц, у которых более 99% частиц составляют менее 200 нм, должны также включать предварительную фильтрацию, сначала через 3,0 микронный фильтр с последующей фильтрацией через 0,45-микронный фильтр для частиц с последующей паровой или тепловой стерилизацией или стерильной фильтрацией через два дублирующих 0,2-микронных мембранных фильтра. Еще одним средством стерилизации является стерильная фильтрация концентрата, полученного из первого растворителя, содержащего лекарство и необязательное поверхностно-активное вещество или поверхностно-активные вещества, и стерильная фильтрация водного разбавителя. Их затем объединяют в стерильном смешивающем контейнере предпочтительно в изолированной стерильной среде. Смешивание, гомогенизацию и дополнительную обработку суспензии затем осуществляют в асептических условиях.
Еще одна процедура стерилизации должна состоять из стерилизации нагреванием или в автоклаве в самом гомогенизаторе перед, в течение или после стадии гомогенизации. Процесс после данной тепловой обработки должен осуществляться в асептических условиях.
Необязательно свободная от растворителя суспензия может быть получена с помощью удаления растворителя после осаждения. Это может быть осуществлено с помощью центрифугирования, диализа, диафильтрации, фракционирования силовым полем, фильтрации высокого давления, обратного осмоса или других способов разделения, хорошо известных в данной области. Полного удаления N-метил-2-пирролидинона обычно достигали с помощью одного-трех последовательных циклов центрифугирования; после каждого центрифугирования (18000 об/мин в течение 30 минут) супернатант сливали и отбрасывали. Новый объем носителя суспензии без органического растворителя добавляли к оставшемуся твердому веществу и смесь диспергировали гомогенизацией. Специалистам в данной области будет очевидно, что другие способы смешивания с высоким сдвигом могут быть использованы на данной стадии восстановления. Альтернативно свободные от растворителя частицы могут быть составлены в различных единичных дозированных формах, как это желательно для разных способов введения, таких как пероральный, легочный, интраназальный, местный, внутримышечный и тому подобное.
Более того, нежелательные эксципиенты, такие как поверхностно-активные вещества, могут быть заменены более желаемыми эксципиентами, используя способы разделения, описанные в представленном выше абзаце. Растворитель и первый эксципиент могут быть слиты с супернатантом после центрифугирования или фильтрации. Потом может быть добавлен новый объем суспензии носителя без растворителя и без первого эксципиента. Альтернативно может быть добавлено новое поверхностно-активное вещество. Например, суспензия, состоящая из лекарства, N-метил-2-пирролидинона (растворителя), полоксамера 188 (первого эксципиента), дезоксихолата натрия, глицерина и воды, может быть замещена фосфолипидами (новое поверхностно-активное вещество), глицерином и водой после центрифугирования и удаления супернатанта.
I. Первая категория способа
Способы первой категории способа обычно включают стадию растворения органического соединения в смешиваемом с водой первом растворителе с последующей стадией смешивания данного раствора с водным растворителем с образованием предварительной суспензии, где органическое соединение находится в аморфной форме, полукристаллической форме или в переохлажденной жидкой форме при определении с помощью исследований рентгеноструктурного анализа, DSC, световой микроскопии или другими аналитическими способами и имеет средний эффективный размер частиц в пределах диапазонов эффективных размеров частиц, указанных выше. За стадией смешивания следует стадия придания энергии.
II. Вторая категория способа
Способы второй категории способов включают по существу те же стадии, что и стадии первой категории способов, но отличаются в следующем отношении. Рентгеноструктурный анализ, DSC, световая микроскопия или другие подходящие аналитические способы обнаруживают органическое соединение в кристаллической форме, которое имеет средний эффективный размер частиц. Органическое соединение после стадии придания энергии имеет по существу тот же средний эффективный размер частиц, что и до стадии придания энергии, но характеризуется меньшей тенденцией к агрегации в более крупные частицы при сравнении с данным свойством частиц в предварительной суспензии. Не ограничиваясь рамками теории, считается, что различия в стабильности частиц могут быть обусловлены реорганизацией молекул поверхностно-активного вещества на поверхности раздела твердая-жидкая фазы.
III. Третья категория способа
В способах третьей категории модифицируются первые две стадии таковых первой и второй категории способов для гарантии того, что органическое соединение в предварительной суспензии находится в хрупкой форме, имеющей средний эффективный размер частиц (например, таких как тонкие иглы и тонкие пластинки). Хрупкие частицы могут быть образованы с помощью выбора подходящих растворителей, поверхностно-активных веществ или сочетания поверхностно-активных веществ, температуры индивидуальных растворов, скорости смешивания и скорости осаждения и тому подобное. Хрупкость может также быть увеличена с помощью введения дефектов решетки (например, плоскостей спайности) в течение стадий смешивания первого раствора с водным растворителем. Это должно происходить с помощью быстрой кристаллизации, которая достигается на стадии осаждения. На стадии придания энергии данные хрупкие кристаллы превращаются в кристаллы, которые кинетически стабилизированы и имеют средний эффективный размер частиц, более мелкий, чем в предварительной суспензии. Кинетически стабилизированными считаются частицы, которые имеют пониженную тенденцию к агрегации при сравнении с частицами, которые кинетически не стабилизированы. В таком примере стадия придания энергии ведет к разбиванию хрупких частиц. При гарантии того, что частицы в предварительной суспензии находятся в хрупком состоянии, органическое соединение может быть легко и более быстро получено в виде частиц в пределах желаемых диапазонов размера при сравнении с получением органического соединения, когда не использовались стадии придания ему хрупкой формы.
IV. Четвертая категория способа
Способы четвертой категории способа включают стадии первой категории способа за исключением того, что стадию смешивания осуществляют одновременно со стадией придания энергии.
Контролирование полиморфов
В настоящем изобретении дополнительно предлагаются добавочные стадии для контролирования кристаллической структуры органического соединения с получением в конечном итоге суспензии соединения в желаемом диапазоне размеров и желаемой кристаллической структуры. Под термином «кристаллическая структура» подразумевается расположение атомов в элементарной ячейке кристалла. Соединения, которые могут кристаллизоваться в различные кристаллические структуры, как говорят, являются полиморфными. Идентификация полиморфов является важной стадией в составлении лекарственного средства, так как различные полиморфы одного и того же лекарственного средства могут проявлять различия в растворимости, терапевтической активности, биодоступности и стабильности суспензии. Соответственно, важно контролировать полиморфную форму соединения для уверенности в чистоте продукта и воспроизводимости от партии к партии.
Стадии контроля полиморфной формы соединения включают внесение затравки в первый раствор, второй растворитель или предварительную суспензию для гарантии образования желаемого полиморфа. Затравка включает использование соединения затравки или придание энергии. В предпочтительной форме изобретения соединение затравки представляет собой фармацевтически активное соединение в желаемой полиморфной форме. Альтернативно соединение затравки может также быть инертной примесью, соединением, не родственным по структуре с желаемым полиморфом, но с чертами, которые могут вести к моделированию ядра кристалла, или соединением со структурой, сходной со структурой желаемого полиморфа.
Соединение затравки можно осадить из первого раствора. Данный способ включает стадии добавления органического соединения в достаточном количестве для превышения растворимости органического соединения в первом растворителе, создавая перенасыщенный раствор. Перенасыщенный раствор обрабатывают для осаждения органического соединения в желаемой полиморфной форме. Обработка перенасыщенного раствора включает старение раствора в течение периода времени до выявления образования кристалла или кристаллов с созданием затравочной смеси. Возможно также придать энергию перенасыщенному раствору для индукции осаждения органического соединения из раствора в желаемой полиморфной форме. Энергия может быть придана различными способами, включая стадии придания энергии, описанные выше. Дополнительно энергия может быть придана нагреванием или действием на предварительную суспензию электромагнитной энергии, источником лучевой энергии или пучком электронов. Электромагнитная энергия включает световую энергию (ультрафиолетовую, видимого света, инфракрасную) или когерентное излучение, такое как обеспечиваемое лазером, микроволновую энергию, такую как обеспечиваемая мазером (усиление в СВЧ-диапазоне с помощью стимуляции индукции излучения), динамическую электромагнитную энергию или другие источники излучения. В качестве источника придания энергии рассматривается также применение ультразвука, статического электрического поля или статического магнитного поля или их сочетаний.
В предпочтительной форме изобретения способ получения затравочных кристаллов из зрелого перенасыщенного раствора включает стадии: (i) добавления количества органического соединения к первому органическому растворителю для создания перенасыщенного раствора, (ii) старения перенасыщенного раствора с образованием определяемых кристаллов для создания затравочной смеси; и (iii) смешивания затравочной смеси со вторым растворителем для осаждения органического соединения с созданием предварительной суспензии. Предварительную суспензию можно затем дополнительно обрабатывать, как подробно описано выше, с обеспечением водной суспензии органического соединения в желаемой полиморфной форме и в желаемом диапазоне размеров.
Затравка может осуществляться с помощью придания энергии первому раствору, второму раствору или предварительной суспензии, предлагаемых так, чтобы подвергаемые воздействию жидкость или жидкости содержали органическое соединение или материал затравки. Энергия может быть придана таким же способом, как описано выше для перенасыщенного раствора.
Соответственно, в настоящем изобретении предлагается композиция материала органического соединения в желаемой полиморфной форме, по существу свободной от неуточненных полиморфа или полиморфов. В предпочтительной форме настоящего изобретения органическое соединение представляет собой фармацевтически активное вещество. Один такой пример представлен в примере 16 ниже, где затравка в течение микроосаждения дает полиморф итраконазола по существу свободный от полиморфа исходного материала. Ожидается, что способы данного изобретения могут быть применены для избирательного получения желаемого полиморфа многочисленных фармацевтически активных соединений.
Субмикронные суспензии антинеопластических агентов
Способ, описанный выше, может быть применен для получения составов, содержащих суспензии субмикронных частиц нерастворимых в воде антинеопластических агентов, в частности паклитаксела или соединений, являющихся его производными, включая, но не ограничиваясь этим, доцетаксел и другие аналоги паклитаксела. В данных составах обычно допускается большое наполнение лекарством с содержанием 1-20% мас./об. лекарства. В данных составах может быть также достигнуто наполнение лекарством более 20% мас./об. Один и тот же состав может быть введен различными путями, например пероральным, парентеральным и легочным.
Частицы антинеопластического агента могут быть составлены как для удаления кремофора в качестве эксципиента, а также для достижения лекарственной формы, характеризующейся длительным временем циркуляции. Частицы, составленные с модификаторами поверхности с функциональной частью полиэтиленгликоля (ПЭГ), могут быть использованы для избежания опсонизации частиц и последующего захвата ретикулоэндотелиальной системой. Кроме того, частицы, имеющие размер частиц менее 200 нм, и особенно менее 150 нм, могут быть использованы для достижения длительного времени циркуляции, а также направленной доставки в опухоль с помощью проникновения через окончатую сосудистую сеть опухоли.
Предпочтительный способ получения субмикронных частиц данных неопластических агентов состоит из: (i) смешивания со смешиваемым с водой первым растворителем, или со вторым растворителем, или с обоими смешиваемым с водой первым растворителем и вторым растворителем первого модификатора поверхности, включающего фосфолипид, конъюгированный с водорастворимым или гидрофильным полимером; (ii) растворения антинеопластического агента в смешиваемом с водой первом растворителе с образованием раствора; (iii) смешивания раствора со вторым растворителем для предопределения предварительной суспензии частиц; и (iv) гомогенизации предварительной суспензии с образованием суспензии частиц, имеющих средний эффективный размер частиц менее приблизительно 1 мкм. Предпочтительный смешиваемый с водой первый растворитель представляет собой N-метил-2-пирролидинон. Предпочтительно, чтобы частицы имели средний эффективный размер частиц менее приблизительно 400 нм, более предпочтительно менее 200 нм и наиболее предпочтительно менее приблизительно 150 нм.
Используемый фосфолипид может быть природным или синтетическим. Примеры подходящих фосфолипидов включают, но не ограничиваются ими, фосфатидилхолин, фосфатидилэтаноламин, диацилглицерофосфоэтаноламин, фосфатидилсерин, фосфатидилинозитол, фосфатидилглицерин, фосфатидную кислоту, лизофосфолипиды, фосфолипид яйца или сои или их сочетание. Диацилглицерофосфоэтаноламин может быть выбран из:
димиристоилглицерофосфоэтаноламина (DMPE),
дипальмитоилглицерофосфоэтаноламина (DPPE),
дистеароилглицерофосфоэтаноламина (DSPE),
диолеолилглицерофосфоэтаноламина (DOPE) или тому подобное.
В предпочтительном варианте осуществления растворимый в воде или гидрофильный полимер, конъюгированный с фосфолипидом, представляет собой полиэтиленгликоль (ПЭГ), такой как, но не ограничиваясь этим, ПЭГ 350, ПЭГ 550, ПЭГ 750, ПЭГ 1000, ПЭГ 2000, ПЭГ 3000 и ПЭГ 5000. Могут быть также использованы другие конъюгированные гидрофильные полимеры, например декстран, гидроксипропилметакрилат (HPMA), полиглутамат и тому подобное.
Необязательно второй модификатор поверхности может быть смешан со смешиваемым с водой первым растворителем, или со вторым растворителем, или с обоими смешиваемым с водой первым растворителем и вторым растворителем. Второй модификатор поверхности может быть необходим для дальнейшей стабилизации частиц. Второй модификатор поверхности может быть выбран из анионных поверхностно-активных веществ, катионных поверхностно-активных веществ, неионных поверхностно-активных веществ и поверхностно-активных биологических модификаторов, как подробно описано в данной заявке выше. Предпочтительный второй модификатор поверхности представляет собой полоксамер, такой как полоксамер 188.
Размер полученных частиц можно контролировать с помощью температуры, при которой осуществляется гомогенизация, как показано в примерах, в примере 19. В варианте осуществления гомогенизацию выполняют при приблизительно 30°С или выше, такой как приблизительно 40°С или приблизительно 70°С.
Способы могут дополнительно включать удаление из суспензии смешиваемого с водой первого растворителя с образованием водной суспензии частиц, которая по существу свободна от растворителя. В предпочтительном варианте осуществления смешиваемый с водой первый растворитель удаляют одновременно гомогенизацией, как подробно описано в находящейся одновременно на рассмотрении и обычным образом переуступленной патентной заявке США с номером дела патентного поверенного 113957-375.
Способ может дополнительно включать полное удаление жидкой фазы из суспензии с образованием сухого порошка частиц. Сухой порошок может быть введен субъекту легочным путем, или он может быть ресуспендирован в подходящем разбавителе, таком как разбавитель, который подходит для парентерального или перорального введения. Частицы могут быть также составлены для перорального введения. Составы для парентерального и перорального введения хорошо известны специалистам в данной области. Один и тот же состав может быть применен для введения субъекту различными путями, такими как, но не ограничиваясь этим, парентеральный, пероральный, легочный, местный, офтальмологический, интраназальный, защечный, ректальный, вагинальный и чрескожный.
Способ может также дополнительно включать стерилизацию композиции, как описано выше. Способы стерилизации фармацевтических композиций включают, но не ограничиваются ими, фильтрацию, стерилизацию нагреванием и гамма-облучение. Стерилизация нагреванием может быть осуществлена с помощью нагревания в гомогенизаторе, где гомогенизатор служит в качестве источника нагревания и создания давления для стерилизации.
В предпочтительном варианте осуществления частицы являются нерастворимыми. Частицы могут быть протестированы на их растворимость с помощью анализа кинетики растворения с использованием % пропускания при 400 нм в качестве показателя растворения. Частицы являются нерастворимыми, если % пропускания не возвращается к 95% или более от исходной величины.
В другом предпочтительном варианте осуществления частицы не агрегируют в условиях напряжения или при хранении. Примеры условий напряжения включают, но не ограничиваются ими, термоциклы, циклы повторного замораживания-оттаивания, перемешивание и центрифугирование. Способы тестирования напряжения частиц хорошо известны в данной области. Типичные способы тестирования напряжения частиц подробно описаны в Novel Injectable Formulations of Insoluble Drugs, Pace et al., Pharm Tech, March 1999, pg 116-134. Агрегация может быть определена с помощью измерения размера частиц перед и после сонификации в течение 1 минуты и сравнения различия с помощью следующего уравнения:
% агрегации=


где P99 представляет собой 99-й процентиль распределения размеров частиц перед сонификацией частиц и P99S представляет собой 99-й процентиль распределения размеров частиц после сонификации частиц.
Примеры
A. Примеры категории 1 способа
Пример 1: Получение суспензии итраконазола с помощью категории 1 способа, способ A с гомогенизацией
В 3-л сосуд добавляли 1680 мл воды для инъекций. Нагревали жидкость до 60-65°С и затем медленно добавляли 44 грамм Pluronic F-68 (полоксамера 188) и 12 грамм дезоксихолата натрия, тщательно перемешивая после каждого добавления для растворения твердых веществ. После завершения добавления твердых веществ перемешивали еще в течение 15 минут при 60-65°С для обеспечения полного растворения. Приготавливали 50 мМ трис (трометаминовый) буфер растворением 6,06 грамм трис в 800 мл воды для инъекций. Титровали данный раствор до pH 8,0 0,1М хлористоводородной кислотой. Разбавляли полученный раствор до 1 л дополнительной порцией воды для инъекций. Добавляли 200 мл трис-буфера к раствору полоксамер/дезоксихолат. Тщательно перемешивали для смешивания растворов.
В 150-мл лабораторный стакан добавляли 20 грамм итраконазола и 120 мл N-метил-2-пирролидинона. Нагревали смесь до 50-60°С и перемешивали для растворения твердых веществ. После визуально определяемого общего растворения перемешивали еще в течение 15 минут для обеспечения полного растворения. Охлаждали раствор итраконазол-NMP до комнатной температуры. Заполняли шприцевой насос (два 60-мл стеклянных шприца) 120 мл заранее полученного раствора итраконазола. Тем временем выливали весь раствор поверхностно-активного вещества в контейнер для гомогенизации, который был охлажден до 0-5°С (это может быть осуществлено путем использования контейнера с рубашкой, через которую проходит охлаждающий агент, или помещением контейнера на лед). Помещали механическую мешалку в раствор поверхностно-активного вещества так, чтобы лопасти были полностью погружены в жидкость. С помощью шприцевого насоса медленно (1-3 мл/мин) добавляли весь раствор итраконазола к перемешиваемому, охлаждаемому раствору поверхностно-активного вещества. Рекомендуется скорость перемешивания, по меньшей мере, 700 об/мин. Аликвоту полученной суспензии (суспензия A) анализировали с помощью световой микроскопии (Hoffman Modulation Contrast) и лазерной дифракции (Horiba). Наблюдаемая с помощью световой микроскопии суспензия A состояла из примерно сферических аморфных частиц (менее 1 микрон), либо связанных между собой в агрегаты, либо свободно перемещающихся в результате броуновского движения. См. фиг.3. Измерения динамического рассеяния света обычно дают бимодальный тип распределения, означающий присутствие агрегатов (размером 10-100 микрон) и присутствие отдельных аморфных частиц со средним диаметром частиц в диапазоне 200-700 нм.
Суспензию немедленно гомогенизировали (при 10000-30000 фунт/кв. дюйм) в течение 10-30 минут. В конце гомогенизации температура суспензии в контейнере не превышала 75°С. Гомогенизированную суспензию собирали в 500-мл флаконы, которые немедленно охлаждали в холодильнике (2-8°С). Данную суспензию (суспензия B) анализировали с помощью световой микроскопии и обнаружили, что она состояла из маленьких удлиненных пластинок длиной в диапазоне от 0,5 до 2 микрон и шириной 0,2-1 микрон. См. фиг.4. Измерения динамического рассеяния света обычно показывают средний диаметр 200-700 нм.
Стабильность суспензии A («Предварительная суспензия») (Пример 1)
При микроскопическом исследовании аликвоты суспензии A прямо наблюдалась кристаллизация аморфного твердого вещества. Суспензию A хранили при 2-8°С в течение 12 часов и исследовали с помощью световой микроскопии. Грубая визуальная оценка выявила сильную флоккуляцию с осаждением части содержимого на дно контейнера. Микроскопическое исследование показало наличие больших, удлиненных, пластинчатых кристаллов длиной более 10 микрон.
Стабильность суспензии B
В противоположность нестабильности суспензии A, суспензия B была стабильна при 2-8°С на протяжении предварительного исследования стабильности (1 месяц). Микроскопия состарившегося образца четко показала, что значительного изменения морфологии или размера частиц не происходит. Это было подтверждено измерением рассеяния света.
Пример 2:Получение суспензии итраконазола с помощью категории 1 способа, способ A с применением ультразвука
В 500-мл сосуд из нержавеющей стали добавляли 252 мл воды для инъекций. Нагревали жидкость до 60-65°С и затем медленно добавляли 6,6 грамм Pluronic F-68 (полоксамера 188) и 0,9 грамм дезоксихолата натрия, перемешивая после каждого добавления для растворения твердых веществ. После завершения добавления твердых веществ перемешивали еще в течение 15 минут при 60-65°С для обеспечения полного растворения. Приготавливали 50 мМ трис (трометаминовый) буфер растворением 6,06 грамм трис в 800 мл воды для инъекций. Титровали данный раствор до pH 8,0 0,1М хлористоводородной кислотой. Разбавляли полученный раствор до 1 л дополнительной порцией воды для инъекций. Добавляли 30 мл трис-буфера к раствору полоксамер/дезоксихолат. Тщательно перемешивали для смешивания растворов.
В 30-мл контейнер добавляли 3 грамм итраконазола и 18 мл N-метил-2-пирролидинона. Нагревали смесь до 50-60°С и перемешивали для растворения твердых веществ. После визуально определяемого общего растворения перемешивали еще в течение 15 минут для обеспечения полного растворения. Охлаждали раствор итраконазол-NMP до комнатной температуры.
Заполняли шприцевой насос 18 мл раствора итраконазола, полученного на предыдущей стадии. Помещали механическую мешалку в раствор поверхностно-активного вещества так, чтобы лопасти были полностью погружены в жидкость. Охлаждали контейнер до 0-5°С погружением на ледяную баню. С помощью шприцевого насоса медленно (1-3 мл/мин) добавляли весь раствор итраконазола к перемешиваемому, охлаждаемому раствору поверхностно-активного вещества. Рекомендуется скорость перемешивания, по меньшей мере, 700 об/мин. Погружали ультразвуковой рожок в полученную суспензию так, чтобы зонд находился на расстоянии приблизительно в 1 см от дна сосуда из нержавеющей стали. Обрабатывали ультразвуком (10000-25000 Гц, по меньшей мере, 400 Вт) в течение от 15 до 20 минут с 5-минутными интервалами. После первой 5-минутной обработки ультразвуком удаляли ледяную баню и проводили дополнительную обработку ультразвуком. В конце обработки ультразвуком температура суспензии в сосуде не превышала 75°С.
Суспензию собирали в 500-мл стеклянный сосуд типа I, который немедленно охлаждали в холодильнике (2-8°С). Показатели морфологии частиц суспензии до и после обработки ультразвуком были очень похожи на наблюдавшиеся в способе A до и после гомогенизации (см. пример 1).
Пример 3: Получение суспензии итраконазола с помощью категории 1 способа, способ B с гомогенизацией
Приготавливали 50 мМ трис (трометаминовый) буфер растворением 6,06 грамм трис в 800 мл воды для инъекций. Титровали данный раствор до pH 8,0 0,1М хлористоводородной кислотой. Разбавляли полученный раствор до 1 л дополнительной порцией воды для инъекций. В 3-л сосуд добавляли 1680 мл воды для инъекций. Добавляли 200 мл трис-буфера к 1680 мл воды. Тщательно перемешивали для смешивания растворов.
В 150-мл контейнер добавляли 44 грамм Pluronic F-68 (полоксамера 188) и 12 грамм дезоксихолата натрия к 120 мл N-метил-2-пирролидинона. Нагревали смесь до 50-60°С и перемешивали для растворения твердых веществ. После визуально определяемого общего растворения перемешивали еще в течение 15 минут для обеспечения полного растворения. К данному раствору добавляли 20 грамм итраконазола и перемешивали до полного растворения. Охлаждали раствор итраконазол/поверхностно-активное вещество/NMP до комнатной температуры.
Заполняли шприцевой насос (два 60-мл стеклянных шприца) 120 мл концентрированного раствора итраконазола, полученного на предыдущей стадии. Тем временем выливали полученный ранее разбавленный раствор трис-буфера в контейнер для гомогенизации, который был охлажден до 0-5°С (это может быть осуществлено путем использования контейнера с рубашкой, через которую проходит охлаждающий агент, или помещением контейнера на лед). Помещали механическую мешалку в буферный раствор так, чтобы лопасти были полностью погружены в жидкость. С помощью шприцевого насоса медленно (1-3 мл/мин) добавляли весь концентрат итраконазол/поверхностно-активное вещество к перемешиваемому, охлаждаемому буферному раствору. Рекомендуется скорость перемешивания, по меньшей мере, 700 об/мин. Полученную охлажденную суспензию немедленно гомогенизировали (при 10000-30000 фунт/кв. дюйм) в течение 10-30 минут. В конце гомогенизации температура суспензии в контейнере не превышала 75°С.
Гомогенизированную суспензию собирали в 500-мл флаконы, которые немедленно охлаждали в холодильнике (2-8°С). Показатели морфологии суспензии до и после гомогенизации были очень похожими на наблюдавшиеся в примере 1, за исключением того, что в категории 1 способа B материал перед гомогенизацией обладал склонностью к образованию меньшего количества и меньших агрегатов, что приводило к значительно меньшему общему размеру частиц по данным измерения с помощью лазерной дифракции. После гомогенизации результаты динамического рассеяния света обычно были идентичны представленным в примере 1.
Пример 4:Получение суспензии итраконазола с помощью категории 1 способа, способ B с применением ультразвука
В 500-мл сосуд добавляли 252 мл воды для инъекций. Приготавливали 50 мМ трис (трометаминовый) буфер растворением 6,06 грамм трис в 800 мл воды для инъекций. Титровали данный раствор до pH 8,0 0,1М хлористоводородной кислотой. Разбавляли полученный раствор до 1 л дополнительной порцией воды для инъекций. Добавляли 30 мл трис-буфера к воде. Тщательно перемешивали для смешивания растворов.
В 30-мл контейнер добавляли 6,6 грамм Pluronic F-68 (полоксамера 188) и 0,9 грамм дезоксихолата натрия к 18 мл N-метил-2-пирролидинона. Нагревали смесь до 50-60°С и перемешивали для растворения твердых веществ. После визуально определяемого общего растворения перемешивали еще в течение 15 минут для обеспечения полного растворения. К полученному раствору добавляли 3,0 грамм итраконазола и перемешивали до полного растворения. Охлаждали раствор итраконазол/поверхностно-активное вещество/NMP до комнатной температуры.
Заполняли шприцевой насос (один 30-мл стеклянный шприц) 18 мл концентрированного раствора итраконазола, полученного на предыдущей стадии. Помещали механическую мешалку в буферный раствор так, чтобы лопасти были полностью погружены в жидкость. Охлаждали контейнер до 0-5°С погружением в ледяную баню. С помощью шприцевого насоса медленно (1-3 мл/мин) добавляли весь концентрат итраконазол/поверхностно-активное вещество к перемешиваемому, охлаждаемому буферному раствору. Рекомендуется скорость перемешивания, по меньшей мере, 700 об/мин. Полученную охлажденную суспензию немедленно обрабатывали ультразвуком (10000-25000 Гц, по меньшей мере, 400 Вт) в течение 15-20 минут с 5-минутными интервалами. После первой 5-минутной обработки ультразвуком удаляли ледяную баню и проводили дополнительную обработку ультразвуком. В конце обработки ультразвуком температура суспензии в контейнере не превышала 75°С.
Полученную суспензию собирали в 500-мл флакон, который немедленно охлаждали в холодильнике (2-8°С). Показатели морфологии суспензии до и после обработки ультразвуком были очень похожими на наблюдавшиеся в примере 1, за исключением того, что в категории 1 способа, способ B, материал перед обработкой ультразвуком обладал склонностью к образованию меньшего количества и меньших агрегатов, что приводило к значительно меньшему общему размеру частиц по данным измерения с помощью лазерной дифракции. После обработки ультразвуком результаты динамического рассеяния света обычно были идентичны представленным в примере 1.
B. Примеры категории 2 способа
Пример 5: Получение суспензии итраконазола (1%) с 0,75% Solutol® HR (PEG-660 12-гидроксистеарат) в категории 2 способа, способ B
В лабораторный стакан отвешивали солутол (2,25 г) и итраконазол (3,0 г) и добавляли 36 мл отфильтрованного N-метил-2-пирролидинона (NMP). Данную смесь перемешивали при слабом нагревании (до 40°С) в течение приблизительно 15 минут до растворения ингредиентов раствора. Раствор охлаждали до комнатной температуры и фильтровали через 0,2-микронный фильтр в вакууме. Два 60-мл шприца наполняли отфильтрованным концентратом лекарства и помещали в шприцевой насос. Насос устанавливали для доставки приблизительно 1 мл/мин концентрата к быстро перемешиваемому (400 об/мин) водному буферному раствору. Буферный раствор состоял из 22 г/л глицерина в 5 мМ трис-буфере. Во время добавления концентрата буферный раствор оставляли на ледяной бане при 2-3°С. В конце осаждения, после завершения добавления концентрата к буферному раствору, приблизительно 100 мл суспензии центрифугировали в течение 1 часа и супернатант отбрасывали. Осадок ресуспендировали в 20% растворе NMP в воде и вновь центрифугировали в течение 1 часа. Продукт сушили в течение ночи в вакуумной печи при 25°С. Высушенный продукт переносили во флакон и анализировали с помощью рентгеноструктурного анализа с применением излучения хрома.
Другую 100-мл аликвоту микроосажденной суспензии подвергали обработке ультразвуком в течение 30 минут при 20000 Гц, 80% от полной мощности (полная мощность=600 Вт). Обработанный ультразвуком образец гомогенизировали в 3 равных аликвотах, каждую в течение 45 минут (Avestin C5, 2-5°C, 15000-20000 фунт/кв. дюйм). Объединенные фракции центрифугировали в течение приблизительно 3 часов, супернатант удаляли и осадок ресуспендировали в 20% NMP. Ресуспендированную смесь вновь центрифугировали (15000 об/мин при 5°С). Супернатант сливали и осадок сушили в вакууме в течение ночи при 25°С. Осадок подвергали рентгеноструктурному анализу (см. фиг.5). Как видно на фиг.5, характер рентгеновской дифракции обработанных образцов до и после гомогенизации был практически одинаковым и в то же время существенно отличается от неочищенного исходного материала. Негомогенизированная суспензия нестабильна и агломерирует при хранении при комнатной температуре. Полагают, что стабилизация, которая происходит в результате гомогенизации, возникает из-за перестройки поверхностно-активного вещества на поверхности частицы. Данная перестройка должна приводить к меньшей склонности к агрегации частиц.
C. Примеры категории способа
Пример 6: Получение суспензии карбамазепина с помощью категории 3 способа, способ A с гомогенизацией
2,08 г карбамазепина растворяли в 10 мл NMP. 1 мл данного концентрата затем добавляли по каплям со скоростью 0,1 мл/мин в 20 мл перемешиваемого раствора 1,2% лецитина и 2,25% глицерина. Температуру лецитиновой системы поддерживали на уровне 2-5°С на протяжении всего добавления. Предварительную дисперсию затем гомогенизировали на холоде (5-15°С) в течение 35 минут при 15000 фунт/кв. дюйм. Давление увеличивали до 23000 фунт/кв. дюйм и гомогенизацию продолжали еще в течение 20 минут. Полученные с помощью данного способа частицы имели средний диаметр 0,881 мкм, причем 99% частиц были меньше 2,44 мкм.
Пример 7: Получение 1% суспензии карбамазепина с 0,125% Solutol® с помощью категории 3 способа, способ B с гомогенизацией
Получали концентрат лекарства из 20% карбамазепина и 5% гликодезоксихолевой кислоты (Sigma chemical Co.) в N-метил-2-пирролидиноне. Стадия микроосаждения включала добавление концентрата лекарства к принимающему раствору (дистиллированная вода) со скоростью 0,1 мл/мин. Принимающий раствор перемешивали и поддерживали при температуре приблизительно 5°С во время осаждения. После осаждения конечная концентрация ингредиентов составляла 1% карбамазепина и 0,125% Solutol®. Кристаллы лекарства исследовали под световым микроскопом с применением позитивного фазового контраста (400X). Осадок состоял из тонких игл диаметром приблизительно 2 микрона и длиной, варьирующей от 50 до 150 микрон.
Гомогенизация (Avestin C-50 плунжерно-зазорный гомогенизатор) при приблизительно 20000 фунт/кв. дюйм в течение приблизительно 15 минут давала мелкие частицы, размером менее 1 микрона и преимущественно неагрегированные. Лазерный дифракционный анализ (Horiba) гомогенизированного материала показал, что частицы имеют средний размер 0,4 микрон, притом что 99% частиц имеет размер менее 0,8 микрон. Обработка ультразвуком образца при низкой энергии, пригодная для разрушения агломерированных частиц, но не при высокой энергии для вызова дробления индивидуальных частиц перед анализом Horiba не влияла на результаты (величины были одинаковыми при обработке и без нее). Данный результат соответствовал отсутствию агломерации частиц.
Образцы, полученные с помощью описанного выше способа, центрифугировали и надосадочные растворы заменяли раствором, состоящим из 0,125% Solutol®. После центрифугирования и замены супернатанта концентрация ингредиентов суспензии составляла 1% карбамазепина и 0,125% Solutol®. Образцы повторно гомогенизировали с помощью плунжерно-зазорного гомогенизатора и хранили при 5°С. После 4 недель хранения средний размер частиц суспензии равнялся 0,751, притом что 99% были менее 1,729. Представленные величины получены с помощью анализа Horiba необработанных ультразвуком образцов.
Пример 8: Получение 1% суспензии карбамазепина с 0,06% гликодезоксихолатом натрия и 0,06% полоксамером 188 с помощью категории 3 способа, способ B с гомогенизацией
Получали концентрат лекарства, включающий 20% карбамазепина и 5% гликодезоксихолата в N-метил-2-пирролидиноне. Стадия микроосаждения включала добавление концентрата лекарства к принимающему раствору (дистиллированная вода) со скоростью 0,1 мл/мин. Таким образом, следующие примеры показывают, что добавление поверхностно-активного вещества или другого эксципиента к водному осаждающему раствору в описанных выше способах A и B является необязательным. Принимающий раствор перемешивали и поддерживали при температуре приблизительно 5°С во время осаждения. После осаждения конечная концентрация ингредиентов составляла 1% карбамазепина и 0,125% Solutol®. Кристаллы лекарства исследовали под световым микроскопом с использованием позитивного фазового контраста (400X). Осадок состоял из тонких игл диаметром приблизительно 2 микрона и длиной, варьирующей от 50 до 150 микрон. Сравнение осадка с исходным материалом до осаждения показало, что стадия осаждения в присутствии модификатора поверхности (гликодезоксихолевая кислота) приводит к очень тонким кристаллам, которые гораздо тоньше исходного материала (см. фиг.6).
Гомогенизация (Avestin C-50 плунжерно-зазорный гомогенизатор) при приблизительно 20000 фунт/кв. дюйм в течение приблизительно 15 минут дает мелкие частицы, размером менее 1 микрон и преимущественно неагрегированные. См. фиг.7. Лазерный дифракционный анализ (Horiba) гомогенизированного материала показал, что частицы имеют средний размер 0,4 микрон, притом что 99% частиц имеет размер менее 0,8 микрон. Обработка ультразвуком образца перед анализом Horiba не влияла на результаты (величины были одинаковыми при обработке ультразвуком и без нее). Данный результат соответствовал отсутствию агломерации частиц.
Образцы, полученные с помощью описанного выше способа, центрифугировали и надосадочные растворы заменяли раствором, состоящим из 0,06% гликодезоксихолевой кислоты (Sigma Chemical Co.) и 0,06% полоксамера 188. Образцы повторно гомогенизировали с помощью плунжерно-зазорного гомогенизатора и хранили при 5°С. После 2 недель хранения средний размер частиц суспензии равнялся 0,531 микрон, притом что 99% были менее 1,14. Представленные величины получены с помощью анализа Horiba не подвергнутых ультразвуковой обработке образцов.
Математический анализ (пример 8) силы, необходимой для переламывания осажденных частиц, при сравнении с силой, необходимой для переламывания исходного материала (карбамазепина)
Ширина наибольших кристаллов, наблюдаемых в исходном материале карбамазепина (фиг.6, изображение слева), примерно в 10 раз больше ширины кристаллов в микроосажденном материале (фиг.6, изображение справа). При допущении, что отношение толщины кристаллов (1:10) пропорционально отношению ширины кристаллов (1:10), момент силы, необходимой для перелома большего кристалла в исходном материале, должен быть приблизительно в 1000 раз больше силы, необходимой для перелома микроосажденного материала, поскольку:
eL=6PL/(Ewx2)
Равенство I
где
eL означает продольную деформацию, необходимую для перелома кристалла («величина выхода»),
P - нагрузку на стержень,
L - расстояние от нагрузки до точки опоры,
E - модуль упругости,
w - ширину кристалла,
x - толщину кристалла.
Если принять, что L и E одинаковы для исходного материала и осажденного материала; дополнительно принять, что w/w0=x/x0=10, тогда
(eL)0=6P0L/(Ew0x02), где индекс '0' относится к исходному материалу,
eL=6PL/(Ewx2) для микроосадка.
Приравнивая (eL)0 и eL:
6PL/(Ewx2)=6P0L/(Ew0x02).
После упрощения
P=P0(w/w0)(x/x0)2=P0(0,1)(0,1)2=0,001 P0.
Таким образом, результирующая сила P, необходимая для перелома микроосажденного твердого вещества, составляет одну тысячную от силы, необходимой для перелома исходного кристаллического твердого вещества. Если из-за быстрого осаждения возникают дефекты кристаллической решетки или свойства аморфности, то модуль (E) должен снижаться, что делает микроосадок даже еще более легко раскалываемым.
Пример 9: Получение 1,6% (мас./об.) суспензии преднизолона с 0,05% дезоксихолатом натрия и 3% N-метил-2-пирролидиноном с помощью категории 3 способа, способ B
Схема всего способа получения представлена на фиг.8. Готовили концентрированный раствор преднизолона и дезоксихолата натрия. Преднизолон (32 г) и дезоксихолат натрия (1 г) добавляли к достаточному объему 1-метил-2-пирролидинона (NMP) с получением конечного объема 60 мл. Полученная концентрация преднизолона составляла приблизительно 533,3 мг/мл, и концентрация дезоксихолата натрия равнялась приблизительно 16,67 мг/мл. 60 мл концентрата NMP добавляли к 2 л воды, охлажденной до 5°С, при скорости добавления 2,5 мл/мин при перемешивании при приблизительно 400 об/мин. Полученная суспензия содержала тонкие иглоподобные кристаллы шириной менее 2 мкм (фиг.9). В осажденной суспензии концентрация составляла для преднизолона 1,6% (мас./об.), для дезоксихолата натрия 0,05%, для NMP 3%.
pH осажденной суспензии доводили до 7,5-8,5 с помощью гидроксида натрия и хлористоводородной кислоты и затем гомогенизировали (Avestin C-50 плунжерно-зазорный гомогенизатор) 10 проходами при 10000 фунт/кв. дюйм. NMP удаляли проведением 2 последовательных стадий центрифугирования с заменой супернатанта каждый раз свежим раствором поверхностно-активного вещества, который содержал желаемые концентрации поверхностно-активных веществ, необходимые для стабилизации суспензии (см. таблицу 2). Суспензию гомогенизировали еще 10 проходами при 10000 фунт/кв. дюйм. Конечная суспензия содержала частицы со средним размером частиц менее 1 мкм, и 99% частиц были менее 2 мкм. Фиг.10 представляет собой микрофотографию конечной суспензии преднизолона после гомогенизации.
На стадии центрифугирования/замены поверхностно-активного вещества использовали множество различных поверхностно-активных веществ (см. таблицу 2). В таблице 2 перечислены сочетания поверхностно-активных веществ, которые были стабильны в отношении размера частиц (средний <1 мкм, 99% <2 мкм, pH 96-8), концентрации лекарства (потеря менее 2%) и ресуспендируемости (ресуспендирование за 60 секунд или менее).
Примечательно, что данный способ позволяет добавлять активное соединение к водному разбавителю в отсутствие поверхностно-активного вещества или другой добавки. Это является модификацией процесса способа B на фиг.2.
Пример 10: Получение суспензии преднизолона с помощью категории 3 способа, способ A с гомогенизацией
32 г преднизолона растворяли в 40 мл NMP. Для осуществления растворения требовалось легкое нагревание при 40-50°С. Концентрат лекарства в NMP затем добавляли по каплям со скоростью 2,5 мл/мин к 2 литрам перемешиваемого раствора, состоящего из 0,12% лецитина и 2,2% глицерина. Других модификаторов поверхности не добавляли. Систему поверхностно-активного вещества буферировали при pH 8,0 5 мМ трис-буфером, и температуру поддерживали на уровне от 0° до 5°С на протяжении всего процесса осаждения. Дисперсию после осаждения затем гомогенизировали на холоде (5-15°С) 20 проходами при 10000 фунт/кв. дюйм. После гомогенизации NMP удаляли центрифугированием суспензии, удалением супернатанта и заменой супернатанта свежим раствором поверхностно-активного вещества. Данную суспензию после центрифугирования затем повторно гомогенизировали на холоде (5-15°С) еще 20 проходами при 10000 фунт/кв. дюйм. Полученные с помощью данного способа частицы имели средний диаметр 0,927 мкм, и 99% частиц были менее 2,36 мкм.
Пример 11: Получение суспензии набуметона с помощью категории 3 способа, способ B с гомогенизацией
Поверхностно-активное вещество (2,2 г полоксамера 188) растворяли в 6 мл N-метил-2-пирролидинона. Данный раствор перемешивали при 45°С в течение 15 минут, после чего добавляли 1,0 г набуметона. Лекарство растворялось быстро. Получали разбавитель, который состоял из 5 мМ трис-буфера с 2,2% глицерина, и pH доводили до 8. 100-мл порцию разбавителя охлаждали на ледяной бане. Концентрат лекарства медленно (приблизительно 0,8 мл/мин) добавляли к разбавителю при интенсивном перемешивании. Данную неочищенную суспензию гомогенизировали при 15000 фунт/кв. дюйм в течение 30 минут и затем при 20000 фунт/кв. дюйм в течение 30 минут (температура=5°С). Было обнаружено, что конечная наносуспензия имеет эффективный средний диаметр 930 нм (по данным анализа лазерной дифракцией). 99% частиц были меньше приблизительно 2,6 микрон.
Пример 12: Получение суспензии набуметона с помощью категории 3 способа, способ B с гомогенизацией и с применением Solutol® HS 15 в качестве поверхностно-активного вещества. Замена надосадочной жидкости фосфолипидной средой.
Набуметон (0,987 грамм) растворяли в 8 мл N-метил-2-пирролидинона. К данному раствору добавляли 2,2 грамм Solutol® HS 15. Данную смесь перемешивали до полного растворения поверхностно-активного вещества в концентрате лекарства. Получали разбавитель, который состоял из 5 мМ трис-буфера с 2,2% глицерина и pH которого доводили до 8. Разбавитель охлаждали на ледяной бане и концентрат лекарства медленно (приблизительно 0,5 мл/мин) добавляли к разбавителю при интенсивном перемешивании. Полученную неочищенную суспензию гомогенизировали в течение 20 минут при 15000 фунт/кв. дюйм и в течение 30 минут при 20000 фунт/кв. дюйм.
Суспензию центрифугировали при 15000 об/мин в течение 15 минут, супернатант удаляли и отбрасывали. Оставшийся твердый осадок ресуспендировали в разбавителе, состоящем из 1,2% фосфолипидов. Данная среда имела тот же объем, что и супернатант, удаленный на предыдущей стадии. Полученную суспензию гомогенизировали при приблизительно 21000 фунт/кв. дюйм в течение 30 минут. Конечную суспензию анализировали с помощью лазерной дифракции, и было обнаружено, что она содержит частицы со средним диаметром 542 нм, и 99% функции распределения частиц имеет размер менее 1 микрон.
Пример 13: Получение 1% суспензии итраконазола с полоксамером с частицами со средним диаметром приблизительно 220 нм
Концентрат итраконазола получали растворением 10,02 грамм итраконазола в 60 мл N-метил-2-пирролидинона. Для растворения лекарства требовалось нагревание до 70°С. Затем раствор охлаждали до комнатной температуры. Получали часть 50 мМ трис(гидроксиметил)аминометанового буфера (трис-буфера) и pH доводили до 8,0 с помощью 5М хлористоводородной кислоты. Получали водный раствор поверхностно-активного вещества объединением 22 г/л полоксамера 407, 3,0 г/л яичных фосфатидов, 22 г/л глицерина и 3,0 г/л дигидрата холата натрия. 900 мл раствора поверхностно-активного вещества смешивали со 100 мл трис-буфера с получением 1000 мл водного разбавителя.
Водный разбавитель добавляли в бункер гомогенизатора (APV Gaulin Model 15MR-8TA), который охлаждали ледяной рубашкой. Раствор быстро перемешивали (4700 об/мин) и отслеживали температуру. Медленно добавляли концентрат итраконазола с помощью шприцевого насоса со скоростью приблизительно 2 мл/мин. Добавление заканчивали через приблизительно 30 минут. Полученную суспензию перемешивали еще в течение 30 минут, причем бункер все еще охлаждали с помощью ледяной рубашки и отбирали аликвоту для анализа световой микроскопией любого динамичного рассеяния света. Остальную суспензию затем гомогенизировали в течение 15 минут при 10000 фунт/кв. дюйм. К концу гомогенизации температура возрастала до 74°С. Гомогенизированную суспензию собирали в 1-л стеклянную бутыль типа I и герметично закрывали резиновой пробкой. Бутыль, содержащую суспензию, хранили в холодильнике при 5°С.
Было обнаружено, что образец суспензии перед гомогенизацией состоял как из свободных частиц, так и агрегатов частиц и из многослойных липидных телец. Свободные частицы нельзя было четко визуализировать из-за броуновского движения; но многие из агрегатов, по-видимому, состоят из аморфного, некристаллического материала.
Гомогенизированный образец содержал свободные субмикронные частицы, обладающие прекрасной однородностью по размеру, без видимых липидных везикул. Динамичное рассеяние света показало монодисперсное логарифмическое распределение по размеру со средним диаметром приблизительно 220 нм. Интегральная 99% верхняя граница по размеру равнялась приблизительно 500 нм. На фиг.11 показано сравнение распределения по размеру полученной наносуспензии с таковым типичного парентерального жирового эмульсионного продукта (10% Intralipid®, Pharmacia).
Пример 14: Получение 1% наносуспензии итраконазола с гидроксиэтилкрахмалом
Получение раствора A: Гидроксиэтилкрахмал (1 г, Ajinomoto) растворяли в 3 мл N-метил-2-пирролидинона (NMP). Полученный раствор нагревали на водяной бане до 70-80°С в течение 1 часа. В другой контейнер добавляли 1 г итраконазола (Wyckoff). Добавляли три мл NMP и смесь нагревали до 70-80°С для достижения растворения (приблизительно 30 минут). К горячему раствору добавляли фосфолипид (Lipoid S-100). Нагревание продолжали при 70-90°С в течение 30 минут до растворения всего фосфолипида. Раствор гидроксиэтилкрахмала соединяли с раствором итраконазол/фосфолипид. Смесь нагревали еще в течение 30 минут при 80-95°С для растворения смеси.
Добавление раствора A к трис-буферу: Девяносто четыре (94) мл 50 мМ трис(гидроксиметил)аминометанового буфера охлаждали на ледяной бане. При быстром перемешивании раствора трис по каплям медленно (менее 2 мл/мин) добавляли горячий раствор A (см. выше).
После завершения добавления полученную суспензию подвергали обработке ультразвуком (Cole-Palmer Ultrasonic Processor - 20000 Гц, установка 80% от максимальной мощности) при продолжении охлаждения на ледяной бане. Использовали однодюймовый твердый зонд. Обработку ультразвуком продолжали в течение 5 минут. Ледяную баню удаляли, зонд вынимали, перенастраивали и новь погружали в суспензию. Суспензию вновь подвергали обработке ультразвуком в течение еще 5 минут без ледяной бани. Ультразвуковой зонд снова вынимали, перенастраивали и после погружения зонда образец подвергали обработке ультразвуком еще в течение 5 минут. В этот момент температура суспензии возрастала до 82°С. Суспензию вновь быстро охлаждали на ледяной бане, и когда ее температура оказывалась ниже комнатной, выливали в стеклянную бутыль типа I и герметично закрывали. Микроскопическая визуализация частиц показала размер отдельных частиц порядка одного микрона или менее.
После одного года хранения при комнатной температуре суспензию вновь оценивали на предмет размера частиц, и было обнаружено, что они имеют средний диаметр приблизительно 300 нм.
Пример 15: Предсказывающий пример способа A с применением HES
В настоящем изобретении предусматривается получение 1% наносуспензии итраконазола с гидроксиэтилкрахмалом с применением способа A, следуя стадиям примера 14, за исключением того, что к трис-буферному раствору добавляли HES вместо раствора NMP. Может потребоваться нагревание водного раствора для растворения HES.
Пример 16: Использование затравки во время гомогенизации для превращения смеси полиморфов в более стабильный полиморф.
Получение образца. Наносуспензию итраконазола получали способом микроосаждения-гомогенизации следующим образом. Итраконазол (3 г) и Solutol HR (2,25 г) растворяли в 36 мл N-метил-2-пирролидинона (NMP) при легком нагревании и перемешивании с получением раствора концентрата лекарства. Раствор охлаждали до комнатной температуры и фильтровали через 0,2 мкм нейлоновый фильтр в вакууме для удаления нерастворившегося лекарства или частиц материала. Раствор рассматривали в поляризованном свете для подтверждения отсутствия кристаллического материала после фильтрования. Раствор концентрата лекарства затем добавляли со скоростью 1,0 мл/минуту к приблизительно 264 мл водного буферного раствора (22 г/л глицерина в 5 мМ трис-буфере). Водный раствор хранили при 2-3°С и непрерывно перемешивали при приблизительно 400 об/мин во время добавления концентрата лекарства. Приблизительно 100 мл полученной суспензии центрифугировали и твердое вещество ресуспендировали в предварительно отфильтрованном растворе 20% NMP в воде. Полученную суспензию повторно центрифугировали и твердое вещество переносили в вакуумную печь для сушки в течение ночи при 25°С. Полученный твердый образец обозначали как SMP 2 PRE.
Характеристика образца. Образец SMP 2 PRE и образец исходного материала итраконазола анализировали с помощью порошкового рентгеноструктурного анализа. Измерения производили с использованием аппарата Rigaku MiniFlex+ с медным излучателем с шагом 0,02° 22 и скоростью сканирования 0,25° 22/минуту. Полученные порошковые рентгенограммы показаны на фиг.12. Графики показывают, что SMP 2 PRE значительно отличается от исходного материала, что позволяет предполагать присутствие другого полиморфа или псевдополиморфа.
Записи дифференциальной сканирующей калориметрии (DSC) образцов показаны на фиг.13a и b. Оба образца нагревали при 2°/мин до 180°С в герметично закрытых алюминиевых емкостях.
Запись для исходного материала итраконазола (фиг.13a) показывает выраженную эндотермическую реакцию при приблизительно 165°С.
Запись для SMP 2 PRE (фиг.13b) демонстрирует две эндотермы при приблизительно 159°С и 153°С. Данный результат в сочетании с порошковой рентгенограммой позволяет предполагать, что SMP 2 PRE состоит из смеси полиморфов и что преобладающей формой является полиморф, который менее стабилен по сравнению с полиморфом, присутствующим в исходном материале.
Дополнительное доказательство в пользу данного заключения получено с помощью записи DSC на фиг.14, которая показывает, что после нагревания SMP 2 PRE до первого перехода, последующего охлаждения и повторного нагревания менее стабильный полиморф плавится и повторно кристаллизуется с образованием более стабильного полиморфа.
Использование затравки. Получали суспензию соединением 0,2 г твердого SMP 2 PRE и 0,2 г исходного материала итраконазола с дистиллированной водой до конечного объема 20 мл (образец с затравкой). Суспензию перемешивали до смачивания всего твердого вещества. Получали вторую суспензию таким же образом, но без добавления исходного материала итраконазола (образец без затравки). Обе суспензии гомогенизировали при приблизительно 18000 фунт/кв. дюйм в течение 30 минут. Конечная температура суспензий после гомогенизации равнялась приблизительно 30°С. Суспензии затем центрифугировали и твердое вещество сушили в течение приблизительно 16 часов при 30°С.
На фиг.15 показаны записи DSC образцов с затравкой и без затравки. Скорость нагревания обоих образцов составляла 2°/мин до 180°С в герметично закрытых алюминиевых емкостях. Запись для образца без затравки выявляет две эндотермы, что указывает на наличие после гомогенизации смеси полиморфов. Запись для образца с затравкой показывает, что внесение затравки и гомогенизация вызывают превращение твердого вещества в стабильный полиморф. Таким образом, затравка, по-видимому, влияет на кинетику перехода от менее стабильной к более стабильной полиморфной форме.
Пример 17: Использование затравки во время осаждения для преимущественного образования стабильного полиморфа
Получение образца. Концентрат лекарства итраконазол-NMP получали растворением 1,67 г итраконазола в 10 мл NMP при перемешивании и слабом нагревании. Раствор дважды фильтровали с помощью 0,2-мкм шприцевых фильтров. Затем получали наносуспензии итраконазола добавлением 1,2 мл концентрата лекарства к 20 мл водного принимающего раствора при приблизительно 3°С и перемешиванием при приблизительно 500 об/мин. Наносуспензию с затравкой получали с использованием смеси приблизительно 0,02 г исходного материала итраконазола в дистиллированной воде в качестве принимающего раствора. Наносуспензию без затравки получали с использованием в качестве принимающего раствора только дистиллированной воды. Обе суспензии центрифугировали, супернатанты декантировали и твердые вещества сушили в вакуумной печи при 30°С в течение приблизительно 16 часов.
Характеристика образца. На фиг.16 показано сравнение записей DSC для твердых веществ из суспензий с затравкой и без затравки. Образцы нагревали при 2°/мин до 180°С в герметично закрытых алюминиевых емкостях. Пунктирная линия, соответствующая образцу без затравки, показывает две эндотермы, что указывает на наличие полиморфной смеси.
Сплошная линия, соответствующая образцу с затравкой, показывает только одну эндотерму рядом с ожидаемой температурой плавления исходного материала, что указывает на то, что материал затравки вызвал образование исключительно более стабильного полиморфа.
Пример 18: Контроль полиморфов с помощью затравки концентрата лекарства
Получение образца. Экспериментально измеренная растворимость итраконазола в NMP при комнатной температуре (приблизительно 22°С) равнялась 0,16 г/мл. Получали 0,20 г/мл раствор концентрата лекарства растворением 2,0 г итраконазола и 0,2 г полоксамера 188 в 10 мл NMP при нагревании и перемешивании. Данному раствору затем давали остыть до комнатной температуры с получением перенасыщенного раствора. Немедленно производили эксперимент по микроосаждению, в котором 1,5 мл концентрата лекарства добавляли к 30 мл водного раствора, содержащего 0,1% дезоксихолата, 2,2% глицерина. Во время стадии добавления водный раствор поддерживали при ~2°С и скорости перемешивания 350 об/мин. Полученную предварительную суспензию гомогенизировали при ~13000 фунт/кв. дюйм в течение приблизительно 10 минут при 50°С. Суспензию затем центрифугировали, супернатант декантировали и твердые кристаллы сушили в вакуумной печи при 30°С в течение 135 часов.
Перенасыщенный концентрат лекарства затем подвергали старению путем хранения при комнатной температуре для индукции кристаллизации. Через 12 дней концентрат лекарства был мутным, что указывало на образование кристаллов. Из концентрата лекарства получали суспензию итраконазола тем же способом, что и в первом эксперименте, путем добавления 1,5 мл к 30 мл водного раствора, содержащего 0,1% дезоксихолата, 2,2% глицерина. Во время стадии добавления водный раствор поддерживали при ~5°С и скорости перемешивания 350 об/мин. Полученную предварительную суспензию гомогенизировали при ~13000 фунт/кв. дюйм в течение приблизительно 10 минут при 50°С. Суспензию затем центрифугировали, супернатант декантировали и твердые кристаллы сушили в вакуумной печи при 30°С в течение 135 часов.
Характеристика образца. Для определения морфологии высушенных кристаллов использовали порошковый рентгеноструктурный анализ. Полученные графики показаны на фиг.17. Было определено, что кристаллы из первого эксперимента (с использованием свежего концентрата лекарства) состоят из более стабильного полиморфа. Напротив, кристаллы из второго эксперимента (состарившийся концентрат лекарства) преимущественно состояли из менее стабильного полиморфа с присутствием также небольшого количества более стабильного полиморфа. Таким образом, полагают, что старение вызывало образование кристаллов из менее стабильного полиморфа в концентрате лекарства, которые затем действовали в качестве затравочного материала во время стадий микроосаждения и гомогенизации, в результате чего предпочтительно образовывался менее стабильный полиморф.
Пример 19: Способы микроосаждения и гомогенизации для получения частиц паклитаксела
Пример A:
Раствор паклитаксела в NMP осаждали в растворе поверхностно-активного вещества, содержащем 0,5% полоксамера 188 и 0,05% mPEG-DSPE (с 2% глицерина в качестве тоничного агента) при низкой температуре (<10°С). Общий объем суспензии составлял 10 мл при концентрации лекарства 1% (мас./об.). Сразу после осаждения проводили гомогенизацию при высоком давлении, ~25000 фунт/кв. дюйм, при температуре 40°С. После гомогенизации (20 минут) исследовали размер частиц суспензии с помощью рассеяния света. Средний размер частиц составлял 186 нм.
Пример B:
Раствор паклитаксела в NMP осаждали в растворе поверхностно-активного вещества, содержащем 0,5% полоксамера 188 и 0,05% mPEG-DSPE (с 2% глицерина в качестве тоничного агента) при низкой температуре (<10°С). Общий объем суспензии составлял 20 мл при концентрации лекарства 1% (мас./об.). Сразу после осаждения проводили гомогенизацию при высоком давлении, ~25000 фунт/кв. дюйм, при температуре 40°С. После 30 минут гомогенизации исследовали размер частиц суспензии с помощью рассеяния света. Средний размер частиц составлял 204 нм.
Пример C:
Раствор паклитаксела в NMP осаждали в растворе поверхностно-активного вещества, содержащем 0,5% полоксамера 188 и 0,05% mPEG-DSPE (с 2% глицерина в качестве тоничного агента) при низкой температуре (<10°С). Общий объем суспензии составлял 10 мл при концентрации лекарства 1% (мас./об.). Сразу после осаждения проводили гомогенизацию при высоком давлении, ~25000 фунт/кв. дюйм, при температуре 70°С. После гомогенизации исследовали размер частиц суспензии с помощью рассеяния света. Средний размер частиц составлял 158 нм. Приблизительно 45% частиц были менее 150 нм.
Пример D:
Раствор паклитаксела в NMP осаждали в растворе поверхностно-активного вещества, содержащем 0,05% mPEG-DSPE (с 2% глицерина в качестве тоничного агента) при низкой температуре (<10°С). Общий объем суспензии составлял 10 мл при концентрации лекарства 1% (мас./об.). Сразу после осаждения проводили гомогенизацию при высоком давлении, ~25000 фунт/кв. дюйм, при температуре 40°С. После гомогенизации исследовали размер частиц суспензии с помощью рассеяния света. Средний размер частиц составлял 244 нм.
Пример 20: Показатели растворения субмикронных частиц паклитаксела
Одним из желаемых свойств субмикронных составов антинеопластических лекарств является то, чтобы они не растворялись, обеспечивая длительную циркуляцию, при введении субъекту. Два состава частиц паклитаксела, полученные способами, описанными в примере 19, тестировали на предмет их растворимости по кинетике растворения с применением % пропускания при 400 нм в качестве показателя растворения. Частицы нерастворимы, если % пропускания не возвращается к 100% после добавления суспензии. Один состав содержит модификаторы поверхности полоксамер 188 (P188) и mPEG-DSPE. Другой состав содержит модификатор поверхности только mPEG-DSPE. Результаты показаны на фиг.18. В обоих случаях % пропускания не рос после исходного падения до приблизительно 60%, что указывает на то, что частицы не растворяются.
Пример 21: Стабильность субмикронных частиц паклитаксела в условиях напряжения и после хранения
Стабильность субмикронных частиц паклитаксела, полученных в примере A примера 19, тестировали с помощью теста ускоренного напряжения, а также после хранения при 5°С в течение одного месяца. Как показано на фиг.19 и 20, средний размер частиц и 99-й процентиль в обоих случаях оставались практически без изменений. Для любого состава, даже после всех тестов напряжения, агрегация не наблюдалась. Агрегацию определяли измерением размера частиц до и после 1-минутной обработки ультразвуком и сравнением разницы с помощью следующего равенства:

где P99 представляет собой 99-й процентиль распределения частиц по размеру до обработки ультразвуком, и P99S представляет собой 99-й процентиль распределения частиц по размеру после обработки ультразвуком.
Хотя были проиллюстрированы и описаны конкретные варианты осуществления, могут быть произведены многочисленные модификации, не отходя от существа и объема защиты, ограничиваясь лишь объемом прилагаемой формулы изобретения.
Реферат
Изобретение относится к лекарственным средствам и касается способа получения фармацевтической композиции субмикронных осажденных частиц паклитаксела или его производных, растворимость которых выше в смешивающемся с водой первом растворителе, чем во втором растворителе, который является водным, причем способ включает стадии: (i) смешивания со смешивающимся с водой первым или вторым растворителем или как со смешивающимся с водой первым растворителем, так и со вторым растворителем первого модификатора поверхности, включающего фосфолипид, конъюгированный с водорастворимым или гидрофильным полимером; (ii) смешивания со смешивающимся с водой первым или вторым растворителем или как со смешивающимся с водой первым растворителем, так и со вторым растворителем второго модификатора поверхности, выбранного из группы, состоящей из анионных, катионных и неионных поверхностно-активных веществ и поверхностно-активных биологических модификаторов; (iii) растворения паклитаксела или его производных в смешивающемся с водой первом растворителе с образованием раствора; (iv) смешивания раствора со вторым растворителем с получением предварительной суспензии частиц; и (v) гомогенизации предварительной суспензии с образованием суспензии осажденных мелких частиц, имеющих средний эффективный размер частиц менее приблизительно 1000 нм. 2 н. и 32 з.п. ф-лы, 21 ил., 2 табл.
Формула
(i) смешивания со смешивающимся с водой первым растворителем или со вторым растворителем или как со смешивающимся с водой первым растворителем, так и со вторым растворителем первого модификатора поверхности, включающего фосфолипид, конъюгированный с водорастворимым или гидрофильным полимером;
(ii) смешивания со смешивающимся с водой первым растворителем или со вторым растворителем или как со смешивающимся с водой первым растворителем, так и со вторым растворителем второго модификатора поверхности, выбранного из группы, состоящей из анионных поверхностно-активных веществ, катионных поверхностно-активных веществ, неионных поверхностно-активных веществ и поверхностно-активных биологических модификаторов;
(iii) растворения паклитаксела или его производных в смешивающемся с водой первом растворителе с образованием раствора;
(iv) смешивания раствора со вторым растворителем с получением предварительной суспензии частиц; и
(v) гомогенизации предварительной суспензии с образованием суспензии осажденных мелких частиц, имеющих средний эффективный размер частиц менее приблизительно 1000 нм.
димиристоилглицерофосфоэтаноламина (DMPE),
дипальмитоилглицерофосфоэтаноламина(DРРЕ),
дистеароилглицерофосфоэтаноламина (DSPE) и
диолеолилглицерофосфоэтаноламина(DОРЕ).
Комментарии