Папилломавирусные вакцины - RU2206608C2
Код документа: RU2206608C2
Чертежи












Описание
Настоящая заявка является частичным продолжением заявки на Патент США Peг. 08/242794, поданной 16 мая 1994, находящейся на рассмотрении.
Область техники, к которой относится изобретение
Разработаны рекомбинантные векторы экспрессии, кодирующие белки
L1 и L2 папилломавирусов, способы создания рекомбинантных белков и способы применения рекомбинантных белков.
Известный уровень техники
Папилломавирусные инфекции встречаются у
самых разных животных, включая человека, овец, собак, кошек, кроликов, обезьян, змей и коров. Папилломавирусы поражают эпителиальные клетки, обычно вызывая на инфицированном участке образование
доброкачественных эпителиальных или фиброэпителиальных опухолей. Папилломавирусы являются видоспецифичными инфекционными факторами, папилломавирусы человека не могут поражать животных, не
принадлежащих к человеческому роду.
Папилломавирусы можно классифицировать в отдельные группы по хозяину, которого они инфицируют. Папилломавирусы человека (НPV), в частности, классифицированы более чем в 60 типов на основании данных о гомологии последовательности ДНК (см. обзор "Папилломавирусы и рак у человека", Н. Pfister (еd), СRС Рrеss, Inc., 1990). Типы папилломавирусов ведут себя как тип-специфические иммуногены, которые, создавая иммунитет, нейтрализующий инфекцию папилломавирусов одного типа, не создают иммунитет против папилломавирусов другого типа.
У человека отличающиеся типы НРV вызывают разные заболевания. HPV типов 1, 2, 3, 4, 7, 10 и 26-29 вызывают у людей с нормальным или неблагополучным иммунитетом образование доброкачественных бородавок. HPV типов 5, 8, 9, 12, 14, 15, 17, 19-25, 36 и 46-50 вызывают у людей с неблагополучным иммунитетом патологию эпителия. HPV типов 6, 11, 34, 39, 41-44 и 51-55 вызывают образование незлокачественных кондилом на слизистой половых органов или дыхательных путей.
HPV типов 16 и 18 вызывают эпителиальную дисплазию слизистой половых органов и ассоциированы с большинством преинвазивных и инвазивных карцином шейки, влагалища, вульвы и анального канала. HPV 6 и HPV 11 являются факторами, вызывающими более 90% всех кондилом (генитальные бородавки) и папиллом гортани. Наиболее часто встречаемым субтипом HPV типа 6 является HPV 6а.
Иммунологическими исследованиями на животных показано, что образование нейтрализующих антител к папилломавирусным антигенам предотвращает инфекцию гомологичным вирусом. Созданию эффективных папилломавирусных вакцин препятствовали затруднения, связанные с проведением культивирования папилломавирусов in vitro. Созданию эффективной HPV - вакцины особенно препятствовало отсутствие подходящей животной модели.
Нейтрализация папилломавирусов антителами осуществляется тип-специфически и зависит от конформации эпитопов на поверхности вируса.
Папилломавирусы - это маленькие (50-60 нм) вирусы, содержащие безоболочечную икосаэдрическую ДНК, которая кодирует до восьми ранних и два поздних гена. Открытые рамки считывания (ORFs) вирусных геномов обозначены Е1-Е7 и L1 и L2, где Е означает ранний, а L - поздний ген. Гены L1 и L2 кодируют белки капсида вируса. Ранние (Е) гены ассоциированы с такими функциями, как репликация вируса и клеточная трансформация.
Белок L1 является главным капсидным белком с молекулярным весом 55-60 кД. Белок L2 является минорным капсидным белком с предсказанным молекулярным весом 55-60 кД и уточненным, с помощью электрофореза в полиакриламидном геле, молекулярным весом, равным 75-100 кД. Иммунологические данные свидетельствуют, что белок L2 является внутренним по отношению к белку L1. У разных папилломавирусов L2-белки высококонсервативны, особенно 10 основных аминокислот на С-конце. У разных папилломавирусов ORF L1 высококонсервативна.
Гены Ll и L2 использовали для производства вакцин, предотвращающих и лечащих папилломавирусную инфекцию у животных. Zhou et al. (1991; 1992) клонировали гены L1 и L2 HPV типа 16 в векторе вируса коровьей оспы и заражали клетки СV-1 млекопитающих рекомбинантным вектором для получения вирусоподобных частиц (VLP).
С помощью рекомбинантных бактериальных папилломавирусов получили белки L1 и L2. Нейтрализующая сыворотка к рекомбинантным бактериальным белкам, перекрестно реагировала с нативным вирусом на низком уровне, что преимущественно связано с различиями в конформации белков, нативного и полученного с помощью бактерий.
Рекомбинантные бакуловирусы, экспрессирующие ОRFs белка L1 НРV 6, HPV 11, HPV 1, HPV 18, HPV 31 или белка L2 НРV 16, использовали для инфицирования SF9-клеток насекомых и получения белков L1 и L2. С помощью вестерн-блот-анализа показали, что белки L1 и L2, продуцируемые бакуловирусом, реагируют с антителами к НРV типа 16. Бакуловирус, продуцирующий белок L1, образует VLPS.
Carter et al. (1991) показали, что рекомбинантные штаммы Saccharomyces cerevisiae продуцируют белок L1 HPV типа 16 и белок L2 HPV типа 16.
Carter et al. продемонстрировали также получение белков L1 и L2 HPV типа 6b. Белок Ll HPV 6b не является полноразмерным белком L1. Рекомбинантные белки получали в виде секретируемых и в виде внутриклеточных продуктов. Рекомбинантные белки L1 и L2 были сходны с нативными белками по молекулярным весам. Было обнаружено, что когда белки экспрессировались внутриклеточно, главный белок оказывался нерастворим при лизисе клеток, в отсутствие денатурирующих агентов. Хотя нерастворимость может содействовать очистке белка, она может препятствовать анализу нативных эпитопов белка.
Было показано, что рекомбинантные белки, секретируемые дрожжами, содержат углеводы, синтезируемые дрожжами. Присутствие этих N-связанных олигосахаридов может маскировать нативные эпитопы. Кроме того, рекомбинантные белки могут иметь другие модификации, такие как удерживание секреторной лидерной последовательности.
Было бы полезно разработать способы получения больших количеств папилломавирусных белков любого вида и типа в результате культивирования рекомбинантных дрожжей. Было бы полезно получать большие количества папилломавирусных белков, обладающих свойствами природных белков, придающих иммунитет, таких как конформация природного белка.
Настоящее изобретение касается получения рекомбинантных папилломавирусных белков, обладающих свойствами природных папилломавирусных белков и иммунитетом, также как и способов их получения и использования. Настоящее изобретение относится прежде всего к получению профилактической и, возможно, терапевтической вакцины от папилломавирусной инфекции. Рекомбинантные белки настоящего изобретения способны производить вирусоподобные частицы. Эти VLP иммуногенны и предотвращают образование бородавок на модельных животных. В настоящем изобретении использовали папилломавирусы (CRPV) американского жесткошерстного кролика и HPV типа 6 (субтип 6а) в качестве модельных систем.
Краткое изложение существа изобретения
Разработаны рекомбинантные экспрессионные векторы,
кодирующие папилломавирусные белки L1 и L2, способы создания рекомбинантных белков и способы использования рекомбинантных белков.
Краткое описание чертежей
Фигура 1
представляет двунаправленный экспрессионный вектор дрожжей, использованный для экспрессии капсидных белков L1 и/или L2 папилломавируса.
Фигура 2 (A и В) представляет экспрессию L1 HPV типа 6а у дрожжей (иммуноблот).
Фигура 3 представляет экспрессию L2 HPV типа 6b у дрожжей. Иммуноблот экспрессированного в дрожжах белка L2 HPV типа 6а: дорожка 1, маркеры молекулярного веса; дорожка 2, слитой белок trpE-L2, экспрессированный в Е.coli в качестве положительного контроля; дорожка 4, белок L1 HPV типа 6а, экспрессированный в дрожжах в качестве негативного контроля; дорожка 5, белок L2 HPV типа 6а, экспрессированный в дрожжах (детали эксперимента смотрите в тексте).
Фигура 4 представляет электронную микрофотографию VLPs белка L2 HPV типа 6а, экспрессированного в дрожжах.
Фигура 5 представляет электронную микрофотографию VLPs белков Ll/L2 HPV 6а, экспрессированных в дрожжах.
Фигура 6 представляет иммуноблот белков L1 и L2 CRPV и белков L1+L2, экспрессированных в дрожжах. Панель (А): экспрессия белка L1 CRPV измерена по реакции с анти-СRРV L1-антисывороткой. Панель (В): экспрессия белка L2 CRPV измерена по реакции с анти-СRРV L2-анти-сывороткой. Дорожка 1, маркеры молекулярных весов в кД (Маркеры Amersham Rainbow, 14300-200000 дальтон), дорожка 2, штамм ВJ5462, содержащий экспрессионный вектор белка L2 CRPV, дорожка 3, штамм 1569, содержащий экспрессионный вектор белка L2 CRPV, дорожки 4 и 5, два изолята штамма 1569, котрансформированного двумя плазмидами для экспрессии белка L1 CRPV и белка L2 СRРV, дорожки 6-8, три изолята штамма 1569, содержащего единственный вектор для коэкспрессии белков L1 и L2 CRPV; дорожки 9 и 10, два изолята штамма BJ 5462, содержащего единственный вектор для коэкспрессии белков L1 и L2 СRРV. Местоположение миграции белков L1 и L2 отмечено на правой оси соответствующей панели.
Фигура 7 дает схематическое изображение конструирования штамма 1558 S. cerevisiae.
Фигура 8 дает схематическое изображение конструирования штамма 1569 S. cerevisiae.
Фигура 9 представляет основные этапы методики выделения очисткой.
Фигура 10 представляет перечень штаммов.
Фигура 11 отражает объем проанализированных в SDS/ПАГ-электрофорезе, выделенных очисткой из дрожжей белков L1+L2 HPV типа 16. Кроме конечного очищенного продукта VLP, показаны результаты выделения на промежуточных этапах. Таблица позволяет идентифицировать образец на каждой дорожке. Представлены результаты окрашивания геля коллоидальным Кумасси и Вестерн-блот с антисывороткой к белкам L1 и L2.
Фигура 12 представляет альтернативную схему процесса выделения очисткой белка L1 папилломавируса американского жесткошерстного кролика.
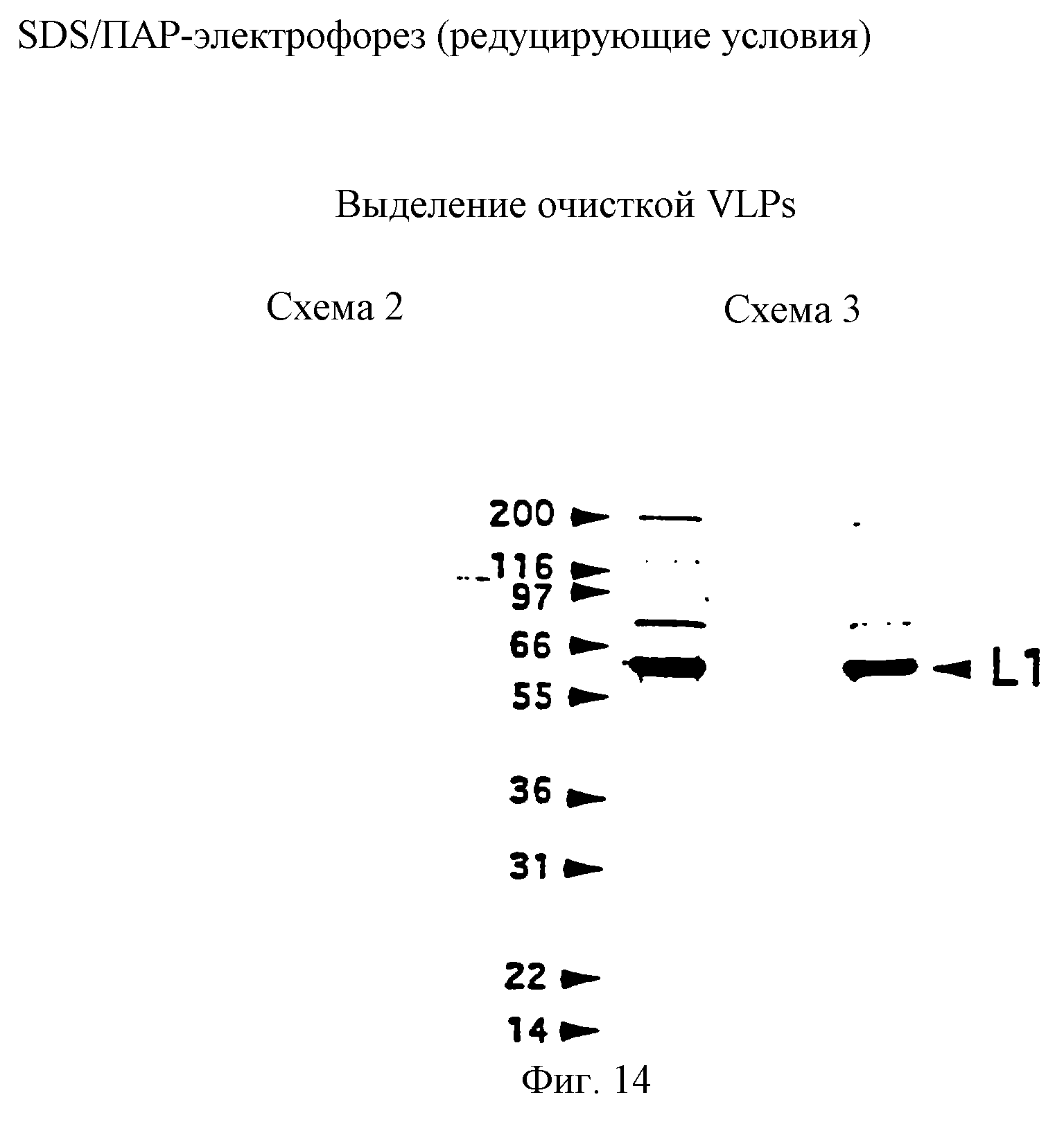
Фигуры 13 и 14 представляют результаты анализа очищенного препарата VLP в SDS/ПАГ-электрофорезе.
Подробное
описание изобретения
Разработаны способы, препараты и процедуры по предотвращению, характеристике, обнаружению и лечению папилломавирусной (PV) инфекции. Способы основаны на получении в
дрожжах рекомбинантного белка L1 или рекомбинантного белка L2 или рекомбинантных белков L1 и L2. Рекомбинантные белки обладают способностью имитировать конформационную нейтрализацию эпитопов
природного PV. Рекомбинантные белки L1 или L1 и L2 способны также образовывать вирусоподобные частицы (VLP). Препараты настоящего изобретения включают, но не ограничены ими, рекомбинантные молекулы
ДНК, кодирующие белки L1 или L2 или L1 и L2, рекомбинантные белки либо отдельно, либо в комбинации с другими рекомбинантными белками, VLP, включающие в себя не менее одного рекомбинантного белка,
фрагменты рекомбинантных белков, фармацевтические препараты, включающие в себя рекомбинантные белки, препараты вакцин, включающие в себя рекомбинантные белки, антитела к рекомбинантным белкам или VLP,
иммуногенные препараты, включающие в себя не менее одного рекомбинантного белка, и диагностические комплекты, включающие в себя рекомбинантные молекулы ДНК или рекомбинантные белки. Способы настоящего
изобретения включают, но не ограничены ими, способ получения рекомбинантного белка, включающего в себя трансформацию соответствующей дрожжевой клетки-хозяина с рекомбинантной молекулой ДНК,
культивирование трансформированных дрожжей в условиях, которые позволяют экспрессировать ДНК, кодирующую рекомбинантный белок и выделять рекомбинантный белок. Способы настоящего изобретения включают
также введение рекомбинантного белка, препаратов рекомбинантного белка или VLP животным, включая человека, но не ограничены им. Соответствующие клетки-хозяина включают, но не ограничены ими, штаммы
дрожжей из родов Saccharomyces, Pichia, Kluvvermyces, Schizosaccharomyces и Hansenula.
Папилломавирусные инфекции наблюдаются у разных животных, включая человека, овец, собак, кошек, кроликов, обезьян, змей и коров. Папилломавирусы поражают эпителиальные клетки, обычно индуцируя на инфицированном участке образование доброкачественных эпителиальных и фиброэпителиальных опухолей.
Папилломавирусы могут быть классифицированы в отдельные группы по хозяину, которого они инфицируют. Папилломавирусы человека (HPV), в частности, классифицированы на основании данных о гомологии последовательности ДНК более чем в 60 типов (см. обзор "Папилломавирусы и рак у человека", Н. Pfister (ed), CRC Press, Inc., 1990). Типы папилломавирусов ведут себя как иммуногены, которые создавая иммунитет, нейтрализующий инфекцию папилломавирусов одного типа, не создают иммунитета против папилломавирусов другого типа.
У человека отличающиеся типы HPV вызывают разные заболевания. HPV типов 1, 2, 3, 4, 7, 10 и 26-29 вызывают у людей с нормальным или неблагополучным иммунитетом образование доброкачественных бородавок. HPV типов 5, 8, 9, 12, 14, 15, 17, 19-25, 36 и 46-50 вызывают у индивидуумов с неблагополучным иммунитетом патологию эпителия. HPV типов 6, 11, 34, 39, 41-44 и 51-55 вызывают образование незлокачественных кондилом на слизистых половых органов и дыхательных путей. HPV типов 16 и 18 вызывают эпителиальную дисплазию половых путей и ассоциированы с большинством преинвазивных и инвазивных карцином шейки, влагалища, вульвы и анального канала. HPV типа 6 и HPV типа 11 вызывают в большинстве случаев образование генитальных бородавок и папиллом гортани.
Иммунологическими исследованиями на животных показано, что образование нейтрализующих антител к белкам капсида папилломавирусов защищает от инфицирования гомологичным вирусом. Созданию эффективных папилломавирусных вакцин препятствовали затруднения, связанные с осуществлением культивирования папилломавирусов in vitro. Созданию эффективной HPV-вакцины особенно препятствовало отсутствие подходящей модели на животных.
Нейтрализация папилломавирусов антителами осуществляется тип-специфически и зависит от конформации эпитопов на поверхности вируса.
Папилломавирусы - это маленькие (50-60 нм) вирусы, содержащие безоболочечную икосаэдрическую ДНК, которая кодирует до восьми ранних и два поздних гена. Открытые рамки считывания(ORFs ) получили обозначения Е1-Е7 и L1 и L2, где Е означает ранний, а L - поздний ген. Гены L1 и L2 кодируют белки капсида вируса. Ранние (Е) гены ассоциированы с такими функциями, как репликация вирусов и трансформация. Белок L1 является главным белком капсида и имеет молекулярный вес 55-60 кД. Белок L2 является минорным белком капсида с предсказанным ранее молекулярным весом 55-60 кД и уточненным, с помощью электрофореза в полиакриламидном геле, молекулярным весом 75-100 кД.
Было сообщено о получении рекомбинантных белков L1 НРV типа 16, L2 HPV типа 16b, L1 HPV типа 6 с помощью рекомбинантных штаммов Saccharomyces cerevisiae. Было бы полезно разработать способы получения больших количеств папилломавирусных белков любых видов и типа путем культивирования рекомбинантных дрожжей. Было бы полезно получать большие количества папилломавирусных белков, обладающих свойствами природных белков, придающими иммунитет, такими как конформация природного белка.
Настоящее изобретение касается получения рекомбинантных папилломавирусных белков, обладающих иммунитетом, придающим им свойства природных папилломавирусных белков, также как и способов по их получению и использованию. Настоящее изобретение относится прежде всего к получению профилактической вакцины от папилломавирусной инфекции. Настоящее изобретение иллюстрируется папилломавирусом американского жесткошерстного кролика (CRPV) и человеческим папилломавирусом типа 6 (HPV типа 6, или HPV 6) как модельными системами. Иллюстрация не ограничивается объемом изобретения, которое включает другие типы и субтипы папилломавирусов (PV), включая, но не ограничиваясь ими, тип 11 HPV, тип 16 HPV и тип 18 HPV, также как и субтип 6а HPV и субтип 6b HPV.
Фармацевтически полезный препарат, включающий в себя белки или VLP, может быть получен согласно известным способам, таким как примешивание фармацевтически переносимого носителя. Примеры таких носителей и способы их использования можно найти в Фармацевтических науках Ремингтона. Для образования фармацевтически переносимого препарата, пригодного для эффективного применения, такие препараты должны содержать достаточное количество белка или VLP. Такие препараты могут содержать белки или VLP, выделенные из более чем одного типа HPV.
Терапевтические или диагностические препараты настоящего изобретения вводили индивидуумам, инфицированным PV, в количествах, достаточных для лечения или диагностики. Эффективное количество может изменяться в соответствии с разнообразными факторами, такими как состояние индивидуума, его вес, пол и возраст. Другие факторы охватывают режим введения. Обычно препараты должны вводиться в дозах, колеблющихся от 1 до 250 мкг.
Фармацевтические препараты можно разрабатывать для индивидуального применения с использованием разнообразных способов введения, таких как подкожный, местный, пероральный, через слизистую и внутримышечный.
Вакцины настоящего изобретения включают в себя рекомбинантные белки или VLP, которые содержат антигенные детерминанты, необходимые для стимуляции образования нейтрализующих антител у хозяина. Такие вакцины достаточно безопасны для введения, без риска клинического инфицирования, не проявляют побочных токсических эффектов, могут быть введены эффективным способом, стабильны и обладают совместимостью с носителями для вакцин.
Вакцины можно вводить с помощью разнообразных способов, например перорально, парэнтерально, подкожно, через слизистую или внутримышечно. Вводимую дозу можно варьировать в зависимости от состояния индивидуума, его веса, пола и возраста; способа применения и типа PV-вакцины. Вакцину можно использовать в таких лекарственных формах, как капсулы, суспензии, эликсиры или жидкости. Вакцину можно приготовить с иммунологически приемлемым носителем.
Вакцины вводят в терапевтически эффективных количествах, т.е. в количествах, достаточных для получения защитного иммунного ответа. Терапевтически эффективное количество может изменяться в зависимости от типа PV. Вакцину можно вводить в виде однократной или многократной доз.
Способы, изложенные в настоящем изобретении, делают возможным готовить субвирусные вакцины для защиты от PV -инфекции. Согласно указанным способам можно создавать моновалентные или поливалентные PV-вакцины. Например, моновалентную вакцину HPV типа 16 можно создать с помощью рекомбинатных белков L1 или L2 или L1 и L2. Изготовить же поливалентную вакцину можно путем смешивания белков L1 или L2, или L1 и L2, или VLР от различных типов HPV.
Рекомбинантные белки и VLP настоящего изобретения можно использовать для приготовления иммуногенных препаратов. Такими препаратами, которые вводят в соответствующего хозяина, можно индуцировать у него иммунный ответ.
Рекомбинантные белки и VLP можно использовать для образования антител. Использованный здесь термин "антитело" охватывает поликлональные и моноклональные антитела, также как и их фрагменты, такие как Fv, Fab и F(ab)2-фрагменты, способные связывать антиген или гаптен.
Рекомбинантные белки, VLР и антитела настоящего изобретения можно использовать для серотипирования инфекции HPV и скринирования HPV. Рекомбинантные белки, VLP и антитела сами по себе пригодны для создания наборов по обнаружению и серотипированию HPV. Такой набор должен представлять собой изолированный несущий элемент, пригодный для удержания в ограниченном пространстве, как минимум, одного контейнера. Далее, несущий элемент должен содержать такие реагенты, как рекомбинантный белок HPV или VLP, или анти-HPV антитела, необходимые для обнаружения разных типов HPV. Несущий элемент должен также содержать средства для обнаружения таких веществ, как меченый антиген или ферментные субстраты и т.п.
Рекомбинантные белки и VLP настоящего изобретения пригодны также для использования в качестве маркеров молекулярного веса и размера молекул.
Нижеследующие примеры приводятся для дополнительного раскрытия существа изобретения, не ограничивая вместе с тем рамок изобретения спецификой этих примеров.
Пример 1
Получение
штамма U9 дрожжей
Штамм 2150-2-3 Saccharomyces cerevisiae (MATa, 1eu 2-04, ade1, cir) получен от Dr. Leland Hartwell (University of Washington, Seattle, WA). Клетки штамма 2150-2-3 размножили
в течение ночи при 30oС в 5 мл YEHD-среды (Carty et аl. , J. Ing. Micro, 2 (1987) 117-121). Клетки отмывали 3 раза стерильной дистиллированной водой, ресуспендировали в 2 мл стерильной
дистиллированной воды и 0,1 мл суспензии клеток внесли в каждую из шести чашек, содержащих 5-фтороротовую кислоту (FOA), с целью отбора ura3-мутантов (Cold Spring Harbor Laboratory, Руководство по
генетике дрожжей). Чашки инкубировали при 30oС. Среда содержала на 250 мл дистиллированной воды: 3,5 г дрожжевого азотного основания Difco без аминокислот и сульфата аммония, 0,5 г
5-фтороротовой кислоты, 25 мг урацила и 10 г декстрозы.
Среду стерилизовали фильтрацией через 0,2 мкм мембранный фильтр и затем смешивали с 250 мл 4% бактоагара (Difco), выдержанного при 50oС, 10 мл раствора аденина (1,2 мг/мл) и 5 мл раствора L - лейцина (180 мг/мл). Полученную таким образом среду разливали в чашки Петри по 20 мл.
После 5 дней инкубации появились многочисленные колонии. Одиночные колонии выделяли в результате перештрихования колоний с исходных FOA-чашек на свежие FОА-чашки, которые затем инкубировали при 30oС. Ряд колоний из второго комплекта FОА-чашек тестировали на наличие ura3-мутации путем реплицирования колоний на YFHD-чашку и на чашку без урацила. Получили один изолят (U9), который обладал указанными свойствами. Его хранили замороженным про запас в глицерине (штамм 325) при -70oС для последующего использования.
Пример 2
Приготовление вектора для разрыва гена MNN9 дрожжей
С целью изготовления вектора для разрыва гена МNN9 необходимо было прежде всего клонировать ген MNN9 из геномной ДНК S. cerevisiae. Это достигали с помощью стандартной технологии полимеразной
цепной реакции (ПЦР). 5'-смысловой праймер и 3'-антисмысловой праймер для ПЦР кодирующего последовательность полноразмерного гена МNN9, был разработан на основе опубликованной последовательности для
гена МNN9 дрожжей (Zymogenetics: EPO Patent Application 88117834.7, Publication 0-314-096-A2). Использовали нижеследующие олигодезоксинуклеотидные праймеры, содержащие фланкирующие HindIII-сайты:

Инициирующий метионин кодон гена МNN-9 светился на темном фоне. Проводили ПЦР, используя в качестве матрицы геномную ДНК штамма JRY 188 S. сerevisiae Таq-полимеразу ДНК (Perkin Elmer) и 25 циклов амплификации (94oС 1 мин, 37oС 2 мин, 72oС 3 мин). Полученный ПЦР-фрагмент размером 1,2 т.п.н. разрушали рестриктазой HindIII, извлекали из геля и лигировали с плазмидой pUC13 (Pharmacia), расщепленной с помощью рестриктазы НindIII и обработанной щелочной фосфатазой. Полученную плазмиду обозначили р1183.
С целью отделить ген МNN9 от дрожжевого гена URA3, плазмиду pBR322-URA3 (которая содержала фрагмент Hind III, размером 1,1 т.п.н., кодирующий ген URA3 S. cerevisiae, субклонированный в Нind III - сайте рВR322) расщепляли рестриктазой Hind III и фрагмент ДНК размером 1,1 т.п.н., переносящий функциональный ген URA3, извлекали из геля, тупили концы с помощью Т4 ДНК-полимеразы и затем лигировали с плазмидой, расщепленной с помощью рестриктазы Pm 11 (Pm 11 разрезала последовательность, кодирующую ген MNN9). Полученная плазмида р1199 содержала ген MNN9, разорванный функциональным геном URA3.
Пример 3
Конструирование U9-производного штамма 1372, содержащего разорванный ген MNN9
Для разрыва
гена MNN9 в штамме U9 ( 325) 30 мкг плазмиды р1199 расщепляли рестриктазой Hind III для создания линеаризированной разрывающей кассеты mnn 9:: URA3. Клетки штамма 325 трансформировали рестриктазой
Hind III, расщепляющей ДНК плазмиды р1199 методом сферопластов (Hinnen et al., 1978, Proc. Natl. Acad. Sci. USA & 5:1929-1933) и трансформанты отбирали на синтетической агаровой среде, не
содержащей урацила и содержащей 1,0 М сорбитола. Синтетическая среда содержала в расчете на 1 литр дистиллированной воды: агара 20 г, дрожжевых азотноосновных (w/о) аминокислот 6,7 г, аденина 0,04 г,
L-тирозина 0,05 г, сорбитола 182 г, глюкозы 20 г и безлейционового раствора # 2 10 мл. Не содержащий лейцина раствор # 2 содержал в расчете на 1 л дистиллированной воды: L-аргинина 2 г, L-гистидина 1
г; L-лейцина 6 г, L-изолейцина 6 г, L-лизина 4 г, L-метионина 1 г, L-фенилаланина 6 г, L-треонина 6 г, L-триптофана 4 г.
Чашки инкубировали при температуре 30oС в течение пяти дней и на них появлялись многочисленные колонии. Из 10 колоний готовили препараты хромосомной ДНК и затем обрабатывали ее рестриктазами ЕсоR1 плюс Hind III. Затем обработанную ДНК оценивали Саузерн-блотом (J. Sambrook et аl., Молекулярное клонирование: Руководство для лабораторных работ, 2-е издание, Соlg Spring Harbor Laboratory Press, 1989), используя в качестве зонда фрагмент Hind III размером 1,2 т. п.н., несущий ген MNN 9 (изолированный из плазмиды р1199). Изолят идентифицировали (штамм # 1372) и показали ожидаемый сдвиг полосы ДНК в Саузерн-блоте, также как и чрезвычайную скученность, обычно проявляемую мутантами MNN5.
Пример 4
Конструирование гена для разрыва гена HIS5 дрожжей
Чтобы сконструировать разорванную кассету, в которой ген
HIS3 S. cerevisiae разорван геном URA3, плазмиду YЕрб (К. Struhl et al., 1979, Proc. Natl. Acad. Sci., USA, 76:1035) расщепляли рестриктазой BаmHI. Фрагмент BamHI размером 1,7 т.п.н., несущий ген HIS3
извлекали из геля, тупили концы с помощью Т4 ДНК-полимеразы и лигировали с плазмидой pUC 18, которую предварительно расщепляли рестриктазой BamHI и обрабатывали Т4 ДНК-полимеразой. Результирующую
плазмиду (обозначенную р1501 или pUC18-HIS3) расщепляли рестриктазой NheI (которая разрезала последовательность, кодирующую HIS3) и фрагмент вектора извлекали из геля, тупили концы ДНК-полимеразой
фага Т4 и затем обрабатывали щелочной фосфатазой из телячьего кишечника. Ген URAS выделяли из плазмиды pBR322-URA3 в результате расщепления рестриктазой Hind III и фрагмент размером 1,1 т.п.н.,
несущий ген URA3, извлекали из геля, тупили концы ДНК-полимеразой и лигировали с вышеуказанным NheI-фрагментом pUC18-HIS3. Полученная плазмида (обозначенная pUC18-his3:: URA3 или р1505) содержала
разорванную кассету, в которой ген HIS3 был разорван функциональным геном URA3.
Пример 5
Конструирование вектора для разрыва гена PRB1 дрожжей геном HIS3
Плазмида FP8
Н, несущая ген PRBl S. cerevisiae создана Dr. E. Jones из университета Карнеги-Меллон (С.М. Моеhle et аl., 1987, Genetics, 115:255-263). Ее расщепляли рестриктазами Нind III плюс XhoI и фрагмент ДНК
размером 3,2 т.п.н., несущий ген PRBl, извлекали из геля, тупили концы обработкой Т4 ДНК-полимеразой. Плазмиду pUC18 расщепляли рестриктазой ВаmH1, извлекали из геля и тупили концы обработкой Т4
ДНК-полимеразой. Полученный фрагмент вектора лигировали с вышеуказанным фрагментом гена PRB1, чтобы получить плазмиду pUCl8-PRBl. Плазмиду YEр6, содержащую ген HIS3, расщепляли рестриктазой BamHI.
Полученный фрагмент BamHI размером 1,7 т.п.н., несущий функциональный ген HIS3, извлекали из геля и затем тупили концы обработкой Т4 ДНК-полимеразой. Плазмиду pU C18-PRB1 расщепляли смесью рестриктаз
EcoRV и NcoI, которые разрезали последовательность, кодирующую ген РRВ1, а протеазой В удаляли активный сайт и фланкирующую последовательность. EccRV-NcoI-фрагмент, несущий остатки 5' и 3'-частей гена
PRBl, кодирующего последовательность в pUC18, извлекали из геля, тупили концы путем обработки Т4 ДНК-полимеразой, дефосфорилировали щелочной фосфатазой из кишечника теленка и лигировали с
представленным выше затупленным фрагментом HIS3. Результирующая плазмида (обозначенная pUC18-prbl: : HIS3, штамм 1245) содержала функциональный ген HIS3, в месте расположения части гена РRВ1, который
ранее был делетирован.
Пример 6
Конструирование U9-связанного штамма дрожжей, содержащего разрывы генов MNN9 и РRВ1
U9 - связанный штамм 1372, который содержит
разорванный ген MNN9, представлен в примере 3. Выделенные клоны штамма 1372 пассировали на FОА-чашки для отбора ura3-мутантов. Получили ряд ura3-изолятов штамма 1372 и один из них (штамм
12930-190-S1-1) для последующего разрыва геном HIS3. Вектор (р1505) pUC18-his3:: URA3, разорванный геном, обрабатывали смесью рестриктаз для образования линеаризованной разорванной кассеты his3::URA3
и использовали для трансформации штамма 12930-190-S1-l ацетатно-литиевым методом (Методы энзимологии, 194:290, 1991). Ura + трансформанты отбирали на синтетической агаровой среде без урацила, повторно
штриховали клоновые изоляты на той же среде и затем
получали реплику на среде без урацила или без гистадина для скринирования изолятов Ura+ и His+. Отобрали один из них (штамм 12930-230-1) для
последующего разрыва геном РRВ1. Вектор (pUC18-prb1:: HS13, штамм # l245), разорванный геном РRВ1, обрабатывали смесью рестриктаз SacI и XbaI для получения линеаризованной разорванной кассеты рrb1::
HIS3 и использовали для трансформации штамма 12930-230-1 ацетатно-литиевым методом. His+ трансформанты отбирали на агаровой среде без гистидина и штриховали на той же среде для клонирования изолятов.
Из ряда полученных His+ изолятов выделяли геномную ДНК, расщепляли ее рестриктазой ЕсоRI и подвергали электрофорезу в 0,8% агарозном геле. Затем проводили Саузерн-блот анализ, используя меченый зонд
размером 617 п. н. для гена PRB1, который был приготовлен с помощью ПЦР с применением нижеследующих праймеров:
5' ТGG ТСА ТСС САА АТС ТТG ААА 3' (SEQ ID NO:3)
5' САС СGТ АGТ GTT ТGG
ААG CGA 3' (SEQ ID NО:4)
Получили девять изолятов, которые показали ожидаемую гибридизацию зонда с prbl: : HIS3 - фрагментом ДНК размером 2,44 т.п.н. Это контрастировало с гибридизацией зонда
с фрагментом PBBl-гена дикого типа размером 1,59 т.п.н. Один из этих изолятов, содержащий желаемый разрыв prb1::HIS3, отобрали для дальнейшего использования и обозначили как штамм #1558.
Пример 7
Конструирование вектора для разрыва гена РЕР4 дрожжей
Ген РЕР4 S. cerevisiae, клонировали из геномной библиотеки дрожжей следующим образом. Клетки Е. соli,
содержащие геномную библиотеку дрожжей pLS101 (Schultz and Friesen, 1983, J. Bacteriol., 155:8-14) размножали в 5 мл LB-среды, содержащей 100 мкг/мл ампициллина. Эту культуру с разведением 10-4 и 10-5 помещали на чашках с LB-средой и ампициллином. Колонии извлекали из чашек, используя нитроцеллюлозные фильтры. Зонд размером 600 п.н. для дрожжевого гена РЕР4 готовили с
помощью ПЦР, используя Таq-ДНК-полимеразу, тотальную плазмидную ДНК из библиотеки pLS 101 дрожжей и нижеприведенные олигодезоксинуклеотидные праймеры, обозначенные на основании опубликованной
последовательности ДНК гена РЕР4 (С.A. Woolford et al., Mol. Cell Biol., 6: 2500 (1986)).
Смысловой праймер:
5'-GAG GCT ACC АGC GAG CCG GGC-3' (SEQ ID NO:5)
Антисмысловой праймер:
5'- GGC CAC TCG GCC AAC AGG TTC-3' (SEQ ID NO:6)
Проводили ПЦР с 25 циклами амплификации (94oС 1 мин, 37oС 2 мин, 72oС 3 мин).
ПЦР-пробу извлекали из геля, метили и гибридизовали с вышеуказанными колониями на фильтрах. Несколько колоний были позитивными по гибридизации с пробой РЕР4 и их повторно штриховали на чашках,
содержащих LB-среду с ампициллином, для получения одиночных колоний. Плазмидную ДНК выделяли путем лизиса щелочным SDS (Sambrook et al., как указано выше) из нескольких изолятов и расщепляли
рестриктазой ВаmHI. Наблюдали ожидаемую полосу вектора размером 14 т. п. н. и инсерционную полосу РЕР4 размером 6,9 т.п.н. При двойном расщеплении смесью рестриктаз EcoRl и ХhoI получали ожидаемую
полосу размером 1,5 т. п.н. для гена РЕР4. Один из изолятов, показавший ожидаемый результат, отобрали для дальнейшего использования. Плазмидную ДНК из штамма 860 расщепляли рестриктазой ВаmHI и
ВаmHI-фрагмент ДНК размером 6,9 т.п.н., несущий хромосомный ген РЕР4, субклонировали в ВаmHI-сайте pUC13 для создания плазмиды р890. Плазмиду р890 затем расщепляли рестриктазой NсоI (которая разрезала
последовательность, кодирующую ген РЕР4), извлекали из геля, тупили концы в результате обработки Т4 ДНК-полимеразой и лигировали с затупленными концами фрагмента размером 1,1 т.п.н., несущим
функциональный ген URA3 (приготовленный, как в примере 2). Полученную плазмиду, содержащую ген РЕР4, разорванную геном URA3, обозначили pUC13-pep4:: URA3 (штамм #906).
Пример 8
Конструирование дрожжевого штамма #1569, который является производным штамма U9, содержащего мутации prb1 и рер4.
С целью разрыва геном HIS3 в штамме U9, разорванный вектор pUCl8-his3:: URA3 расщепляли смесью рестриктаз EcoRI и XbаI и затем использовали для трансформации штамма U9 с помощью ацетатно-литиевого метода. Ura+ трансформанты отбирали на агаровой среде без урацила и клонировали на той же среде повторным штрихованием. Ряд полученных Ura+ изолятов реплицировали затем на агаровую среду, не содержащую урацила или гистидина, и скринировали на наличие мутантов Ura+ и His-. Один из мутантов (штамм #1524) отбирали для последующего разрывания геном PRB1. Вектор pUC18-prb1:: HIS3, разорванный геном PRBl, расщепляли смесью рестриктаз SacI и XbaI и затем использовали для трансформации штамма # 1524 с помощью ацетатно-литиевого метода. His+ трансформанты отбирали на агаровой среде, не содержащей гистидина, и клонировали изоляты на той же среде повторным штрихованием. Геномную ДНК выделяли из ряда His+ изолятов и оценивали с помощью гибридизации Саузерн-блот с меченым зондом РRB1. Один из изолятов, обнаруживающий желаемый отрыв PRpl-гена HIS3-геном (т. е. prbl: : НIS3) был отобран для последующего разрыва геном РЕР4. Штамм #1537 пассировали на FOA-чашки с целью получения urа3-изолятов и один из них (штамм #1541) отобрали для дальнейшего использования.
Для получения разрыва геном РЕР4 в штамме #1541, вектор рuc13-рер4:: URA3, разорванный геном РЕР4, обрабатывали рестриктазой XhoI, для получения линеаризованной разорванной кассеты рер4:: URA3, и использовали для трансформации штамма # 1541 с помощью ацетатно-литиевого метода. Ura+ трансформанты отбирали на агаровой среде, не содержащей урацила, и клоновые изоляты пересевали на ту же среду повторным штрихованием. Геномную ДНК выделяли из нескольких Ura+ трансформантов и оценивали Саузерн-блотом, используя меченый зонд для гена РЕР4. Один из изолятов (штамм #1569), обнаруживающий желаемый отрыв гена РЕР4 геном URA3 отобрали для последующего использования.
Пример 9
Конструирование экспрессионного вектора
CRPV L1
Ген L1 вируса CRPV амплифицировали с помощью ПЦР из плазмиды pLA 11, содержащей полный вирусный геном CRPV (Dr. Peter Howley, NCI), используя Vent - полимеразу (Ntw Еngland Biolabs,
Inc.), 35 циклов амплификации (94oС 1 мин, 50oС 1 мин, 72oС 2 мин) и нижеследующие олигонуклеотидные праймеры, которые содержали фланкирующие Bgl II - сайты
(подчеркнуты):

Смысловой праймер интродуцирует дрожжевую нетранслируемую лидерную последовательность выше инициирующего метионин кодона гена L1 вируса CRPV (высвечивал на темном фоне). Продукт ПЦР гена L1 обрабатывали рестриктазой Вg1 II, извлекали из геля и субклонировали в Bg1 II-сайта вектора pSP 72 (Promega) для создания плазмиды pSP72-CRPV-L1 (pl2930-314-4-l). Один из субклонов полностью секвенировали и обнаружили разницу с опубликованной последовательностью CRPV L1 по 4 нуклеотидам. Данные замены не приводили к аминокислотным заменам. Ген L1 вырезали из плазмиды pSP 72-CRPV-L1 в виде Bg1 II-фрагмента и субклонировали в уникальном BamHi-сайте, расположенном между GAL10-промотором дрожжей и АDH1 - терминаторе транскрипции в дрожжевом экспрессионном векторе pC1/1-GAL10p-ADH1t, содержащем GAL10-промотор из YЕр52 (Broach еt al., Exp. Manipulation of Gene Expression, 1983, 83:81-116) и АDН1-терминатор транскрипции в остове вектора pCl/1). Созданную плазмиду обозначили р12930-323-6-1.
Пример 10
Конструирование экспрессионного
вектора CRPV L2
Ген L2 вируса CRPV амплифицировали ПЦР, используя Vent-полимеразу (New England Biolabs, Inc. ) из плазмиды pLAII после расщепления плазмиды рестриктазой Sa1I, гель для
выделения фрагмента размером 7,9 т.п.н. и лигировали его с самим собой. Проводили амплификацию из тридцати пяти циклов (90oС 1 мин, 50oС 1 мин, 72oС 2 мин) и
использовали олигонуклеотидные праймеры, содержащие фланкирующие ЕсоRI-сайты (подчеркнуты):

Смысловой праймер интродуцирует дрожжевую нетранслируемую лидерную последовательность выше инициирующего метионин кодона гена L1 вируса CRPV (высвечивает на темном фоне). Продукт ПЦР размером 1,5 т.п.н. извлекали из геля и субклонировали в EcoRI-сайте двунаправленного промоторного вектора pUC18-GAL10p, содержащего дивергентный дрожжевой GAL1/GAL10-промотор. Этот вектор содержит уникальный BamHI-сайт между GAL1-промотором и первой копией ADН1-терминатора транскрипции, уникальные ЕсоRI- и SmаI-сайты, расположенные между GAL10-промотором и второй копией АDН1-транскрипционного терминатора. Созданная экспрессионная кассета несет SphI - фрагмент размером 1,4 т.п.н. Один из клонов (р12930-295-2-2), содержащий желаемую вставку L2, примыкающую к GAL10-промотору, полностью секвенировали и обнаружили, что он содержит 6 нуклеотидных замен, отличающихся от опубликованной последовательности, 4 из которых приводили к аминокислотным заменам. Анализ последовательности исходной ДНК-матрицы подтверждает, что замены найдены также в pLAII, а не интродуцируются с помощью ПЦР. Вектор pUC18-GAL1p-GAL10p, содержащий ген L2, был вырезан рестриктазой SphI и фрагмент размером 2,9 т.п.н., содержащий экспрессионную кассету АDH1t-GAL1р-GAL10p-L2-ADH1t, был лигирован с Sph1-фрагментом дрожжевого челночного вектора рС1/1. Созданную плазмиду обозначили р12930-323-2-3 (рС1/1-GAL1p-GAL10p-CRPV-L2).
Пример 11
Экспрессия CRPV L1 и CRPV L2-капсидных белков в дрожжах
Плазмиды р12930-323-6-1 и р12930-323-2-3 (рС1/1-GАL10р-CRPV/L1 и
pCl/1-GAL10p-CRPVIL2) использовали для трансформации штаммов # 15696 5462 S. cerevisiae [E.W. Jones, Методы в энзимологии, 194 (1991), 428-453, и BJ 1995 (Jones, там же)]. Клоновые изоляты выращивали
при 30oС в YEHD-среде, содержащей 2% галактозы, в течение 48-72 часов. Клетки собирали, осадок клеток разрушали стеклянными шариками, добавляли тритон Х-100 до конечной концентрации 0,5% и
полученный клеточный лизат оценивали для экспрессии генов L1 и L2 вируса CRPV с помощью иммуноблотного анализа. Образцы, содержащие 40 мкг суммарного клеточного белка подвергали электрофорезу в 12%
трис-глициновом геле (Novex) в восстанавливающих и денатурирующих условиях и электроблоттированию в PVDЕ-мембранах (Novex). Детектировали L1- и L2-белки CRPV, используя поликлональную кроличью анти-L1
или анти-L2 антисыворотку (дар от Dr. John Кrеder, Медицинский Центр Hershey) в качестве первичных антител, а протеин А, присоединенный к пероксидазе хрена (Amersham, Inc.), в качестве вторичного
антитела. Мембраны обрабатывали, применяя хемилюминесцентный набор для детектирования EGL ТМ (Amersham, Inc.). Полоса белка L1, соответствующая молекулярной массе 55-61 кД, была обнаружена во всех
образцах, содержащих плазмиду, экспрессирующую белок L1, а полоса белка L2, соответствующая молекулярной массе около 90 кД, была обнаружена во всех образцах из клонов дрожжей, содержащих
L2-экспрессионную плазмиду.
В образцах из клонов дрожжей, содержащих экспрессионную плазмиду с анти- L2 антисывороткой сигнал не обнаруживали или vice versa.
Пример
12
А. Выделение рекомбинантного капсидного белка L1 CRPV.
Все операции, за исключением специфичности, выполняли при 4oС. Клетки, хранившиеся при -70oC, оттаивали и суспендировали в равном объеме "L1-буфера" (20 мМ натрийфосфат, рН 7,2, 100 мМ NaC1, 1,7 мМ ЭДТА). Ингибиторы протеаз PMSF и пепстатин А добавляли к полужидкой массе до конечных концентраций соответственно 2 мМ и 1,7 мкМ. Клетки лизировали в результате 10-кратного пропускания через микрофлуидизатор. Лизат осветляли центрифугированием при 5000 g в течение 10 минут. Супернатант наслаивали на вершину 5 см подушки из 45% сахарозы (в/о) в L1-буфере и белок L1 осаждали центрифугированием при 100000 g в течение 4 часов. Осадок ресуспендировали в 1/10 объема L1-буфера и осветляли центрифугированием при 5000 g в течение 10 минут. К супернатанту добавляли твин-80 до конечной концентрации 0,01% (в/о) и супернатант фракционировали при комнатной температуре с помощью хроматографии по размеру молекул в 1700 мл колонке (внутренний диаметр 5 см) со смолой сефакрилом S-1000 (Pharmacia). Для этой колонки использовали 10 мМ натрийфосфатный элюирующий буфер, рН 7,2, 150 мМ NаСl, 0,01 (о/о) твин-80. Фракции, содержащие иммунореактивный материал, определяемый с помощью иммунодот блота, объединяли и концентрировали до 1/6 объема ультрафильтрацией, используя ячейку с перемешиванием фирмы Amicon, снабженную 76 мм пластинчатой многослойной мембраной YМ-100 (100000 MWCO). Продукт стерилизовали фильтрованием через мембрану Millex-GV с размером пор 0,22 мкм (Мillipore). Очищенный CRPV L1-капсидный белок адсорбировали гидроокисью алюминия до концентрации 100 мкг/мл.
В. Характеристика рекомбинантного капсидного белка L1 CRPV.
Подлинность конечного продукта подтверждали Вестерн-блоттингом и с помощью N-концевого секвенирующего анализа. Степень чистоты оценивали SDS/ПАГ-электрофорезом с последующим окрашиванием Кумасси и серебром и с помощью просеивающего капиллярного электрофореза в растворе (SSCE) с оптической детекцией при 215 нм. Согласно SSCE препарат белка L1 достигал 75% чистоты.
Пример 13
Выделение
рекомбинантного капсидного белка L2 CRPV
Все операции, за исключением специфичности, выполняли при 4oС. Клетки, хранившиеся при -70oС, оттаивали и суспендировали в
равном объеме "Ll-буфера" (20 мМ натрийфосфат, рН 7,2, 100 мМ NaCl, 1,7 мМ ЭДТА). Ингибиторы протеаз PMSF и пепстатин А добавляли к полужидкой массе до конечных концентраций соответственно 2 мМ и 1,7
мкМ. Клетки лизировали путем 10-кратного пропускания через микрофлуидизатор. Лизат осветляли центрифугированием при 5000 g, в течение 10 минут. Супернатант наслаивали на вершину 5 см подушки из 45%
сахарозы (в/о) в L1-буфере и белок L2 осаждали центрифугированием при 100000 g в течение 4 часов. Осадок ресуспендировали в 1/10 объема L1-буфера. Ресуспендированный осадок осветляли
центрифугированием при 5000 g в течение 10 минут. К супернатанту добавляли твин-80 до конечной концентрации 0,01% (о/о) и супернатант фракционировали при комнатной температуре с помощью хроматографии
по размеру молекул в 1700 мл колонке (внутренний диаметр 5 см) со смолой Сефакрил S-1000 (Pharmacia). Проточный буфер для этой колонки был 10 мМ натрийфосфат, рН 7,2, 150 мМ NaCI, 0,01% (v/v) твин-80.
Фракции, содержащие иммунореактивный материал, определяли иммунно-дот блотом и объединяли. Объединенные фракции анализировали SDS/ПАГ-электрофорезом (окрашивание серебром) и с помощью
Вестерн-блоттинга.
Пример 14
А. Выделение рекомбинантного капсидного белка L1 CRPV (схема 1).
Клетки, хранившиеся при -70oC, оттаивали и суспендировали в равном объеме лизирующего буфера (20 мМ натрийфосфат, рН 7,2, 100 мМ NaC1, 1,7 мМ ЭДТА). Ингибиторы протеаз PMS F и пепстатин А добавляли к полужидкой массе до конечных концентраций соответственно 2 мМ и 1,7 мкМ. Клетки лизировали путем 10-кратного пропускания через микрофлуидизатор. Лизат осветляли центрифугированием при 5000 g в течение 10 минут. Супернатант наслаивали на вершину 5 см подушки из 45% сахарозы (в/о) в L1-буфере и белок L1 осаждали центрифугированием при 100000 g в течение 10 минут. Осадок ресуспендировали в 1/10 объема L1-буфера. Ресуспендированный осадок осветляли центрифугированием при 5000 g в течение 10 минут. К супернатанту добавляли твин-80 до конечной концентрации 0,01% и супернатант фракционировали при комнатной температуре с помощью хроматографии по размеру молекул в 1700 мл колонке (внутренний диаметр 5 см) со смолой Сефакрил S-1000 (Pharmacia). Проточный буфер для этой колонки был 10 мМ натрийфосфат, рН 7,2, 150 мМ NаСl, 0,01% твин-80. Фракции, содержащие иммунореактивный материал и определяемые иммунодот-блотом, объединяли и концентрировали до 1/6 объема с помощью ультрафильтрации, используя ячейку с перемешиванием фирмы Amicon, снабженную 76 мм пластинчатой многослойной мембраной YМ-100 (100000 MWCO). Продукт стерилизовали фильтрацией через мембрану Millex-GV с размером пор 0,22 мкм (Мillipore).
В. Характеристика рекомбинантного капсидного белка L1 CRPV.
Подлинность конечного продукта подтверждали Вестерн-блоттингом и N-концевым секвенирующим анализом. Степень чистоты оценивали SDS/ПАГ-электрофорезом с последующим окрашиванием Кумасси и серебром и просеивающим капиллярным электрофорезом в растворе (SSCE) с оптической детекцией при 215 нм. Электронная микроскопия показала, что VLP представлены размерным рядом 50-55 нм в диаметре.
С. Аналитическая HPLС по размеру молекул.
Для анализа VLP образцы фракционировали по размеру хроматографией по размеру молекул. Хроматографию осуществляли с Perkin-Elmer бионасосом серии 410 для HPLC, снабженного автоинжектором с 200 мкл петлей впрыскивания. Использовали колонку для TSK-геля размером 7, 5•600 мм (TOSOHAAS, Montgomeryville, PA).
Контроль за элюированием белка из колонки осуществляли с помощью оптической детекции при 280 нм, которая достигалась благодаря использованию детекторной матрицы на диоде Perkin-Elmer LС-235. Подвижная фаза содержала 0,5 М NaCl в 10 мМ натрийфосфатном буфере, рН 7,2. Скорость потока равнялась 0,5 мл/мин и собирали фракции объемом один миллилитр. Колонку калибровали белковыми стандартами фирмы Sigma и рекомбинантным поверхностным антигеном гепатита В (Rесоmbivax, Merck & Co., West Point, PA). Детекцию антигена во фракциях, собираемых во время элюирования осуществляли с помощью иммунодот-блот-анализа.
D. Иммунодот-блот-анализ.
10 мкл каждого образца наносили на полоску РVDF-мембраны (Immobilon-P, Millipore Corp. , Bedford, МА), которую предварительно смачивали и помещали на увлажненную бумагу для блоттинга. Образец впитывался в мембрану, мембрану помещали в останавливающий раствор (5% (в/о) обезжиренного сухого молока, растворенного в 0,15 М NaCl, 0,02% (в/о) азид натрия и 0,01 М натрийфосфат, рН 7,2) и инкубировали при комнатной температуре при легком встряхивании в течение, как минимум, трех часов. Останавливающий раствор декантировали и замещали на первичный раствор антител.
Первичные антитела менялись в зависимости от
обнаруживаемого антигена:
CRPV L1 испытывали с кроличьей анти-CRPV сывороткой "позднего ответа" (Bodesign International, Kennebunk, ME). CRPV L2 испытывали с кроличьей анти-CRPV
L2-сывороткой. HPV6a L1 испытывали с моноклональными антителами МАВ 837 (Chemicon International, Inc, Temecula, СА). HPV6a L2 испытывали с мышиной антисывороткой слитого белка L2-trpE HPV типа 6а.
Визуализацию иммунных комплексов осуществляли стандартными способами, используя конъюгацию вторичных антител со щелочной фосфатазой и хромогенным субстратом NBT/BCIP.
Е. Просеивающий капиллярный электрофорез в растворе (SSCE).
Образцы CRPV VLP (около 0,2 миллиграм/миллилитр) нагревали при 100oС в течение 15 минут в 1% 2-меркаптоэтаноле и 1% (в/о) SDS. Образец наносили электрокинетически вместе с маркером-свидетелем, меллитовой кислотой, в капилляр из плавленного кварца размером 42 см (22 см для детектора) х 0,05 мм внутреннего диаметра, предварительно уравновешенным с 10 облученными объемами 0,1 N NаОН, водой и просеивающим реагентом ProSort (Аррliеd Biosystems, Foster Citu, CA). Подавали градиент напряжения 300 В•сек/см от источника питания Applied Biosystems Model 270A-HT СЕ. Элюируемый образец отслеживали абсорбцией при 215 нм и данные обрабатывали, используя программное обеспечение Nelson Turbochrom 3.
F. Приготовление вакцины из рекомбинантного капсидного белка L1 CRPV.
Очищенный капсидный белок L1 CRPV адсорбировали Аl(ОН)3 при концентрации 100 мкг/мл.
Пример 15
Защита от появления папиллом, вызванного CRPV, вакцинацией VLPs L1 CRPV, полученных в дрожжах
5 новозеландских белых кроликов иммунизировали внутримышечно либо 135 мг VLPs
(75% чистоты, см. пример 17), адсорбированных квасцами, либо 100 мг рекомбинантного поверхностного антигена гепатита В (99% чистоты), адсорбированного гидроокисью алюминия, в качестве негативного
контроля. Животные получили более двух стимуляций с интервалом в 8 недель перед введением им CRPV в течение 10 дней после последней стимуляции. Сыворотку брали перед иммунизацией, при каждой
стимуляции, и перед контрольным заражением ее анализировали L1-VLP специфичной ELISA. Непрямое испытание ELlSA применяли для определения реакции антител сыворотки на VLPs L1 CRPV, экспрессированных
дрожжами. Антигенами, писпользованными в ELISA, являлись VLPs L1 CRPV, экспрессированные в клетках насекомых с рекомбинантными бакуловирусами, фактически, как описано Christensen et al. (J. Virol. ,
64:3151-3156, 1990, J. Gen. Virol., 75: 2271-2276, 1994). В качестве вторичных антител использовали конъюгат антикроличий IgG-щелочная фосфатаза козла и п-нитрофенилфосфат как субстрат (Kirkegaard and
Rerry Labs. , Inc.). Абсорбцию измеряли при 405 нм. Титр определяли разведением (3-кратное разведение сыворотки, начиная от 1:100) и считали положительным, если L-специфическая абсорбция превышала
показания средней абсорбции плюс 2 стандартных отклонения кроличьей сыворотки, полученной до иммунизации. Точные титры вычисляли по этим данным, используя SOFTmax-программу, версию 2.3 (Molecular
Devices Inc., Menlo Park. CA).
Титры анти-L1 VLP-антител были 4-недельными после первой иммунизации и они увеличивались с каждой дополнительной стимуляцией. Титры контрольных животных были отрицательными. 6 недель спустя после введения CRPV вакцинированные животные не имели бородавок в 15 из 15 участков (вирусный штамм имел разведение 1: 2 или 1:12), тогда как у контрольных животных образование бородавок наблюдали в 12 из 15 участков (разведение вируса 1:2) или в 9 из 15 участков (разведение вируса 1:12). Спустя 15 недель вакцинированные животные все еще не имели бородавок.
Опыты по нейтрализации вируса были выполнены (фактически в соответствии с методом Christensen et al., 1991, Virology, 181:572-579) путем смешивания кроличьей сыворотки с CRPV и последующим введении кроликам, зараженных CRPV. Стандартом для определения нейтрализации антителами была полная нейтрализация вируса (3/3 участков, не образующие бородавок). Анализировали образцы сыворотки от 5 вакцинированных животных и 5 контрольных животных. Сыворотка, собранная после введения 1 дозы VLPs L1 CRPV, содержала антитела, которые полностью нейтрализовали неразведенный препарат вируса у 80% кроликов (4/5). После введения 2 или 3 доз VLPs L1 СRРV 100% кроликов имели антитела, нейтрализующие вирус. Контрольная сыворотка не обнаруживала какой-либо активности, нейтрализующей вирус. В зависимости от конечного титра сыворотка отобранных вакцинированных животных могла бы быть разведена в 10-1000 раз и все еще обладала бы 100% нейтрализующей способностью.
Для выяснения, действительно ли нейтрализующая способность антител является специфичной для нативных VLРs L1 CRPV, отбирали образец иммунной сыворотки и инкубировали с квадратными кусками нитроцеллюлозы, насыщенной нативными или денатурированными VLPs L1 CRPV, полученными в дрожжах. Квадраты нитроцеллюлозы (1 см2) насыщали нативными или денатурированными (редуцировали и алкилировали иодуксусной кислотой в 8 М мочевине) VLPs L1 CRPV, экспрессированными в дрожжах. Нативные или денатурированные дрожжевые экстракты использовали в качестве контроля. Кроличью иммунную сыворотку инкубировали серийно 4 раза с нитроцеллюлозными квадратами в течение 8-14 часов при 4oС, до их испытания на способность нейтрализовать вирус, как это описано выше. Обнаружили, что только нативные VLPs L1 CRPV абсорбировали антитела, ответственные за нейтрализацию CRPV, тогда как денатурированные или контрольные дрожжевые белки не абсорбировали этиантитела. Кроме того, нативные VLPs L1 CRPV удаляли реактивность ELISА к VLPs L1 CRPV, экспрессированные в клетках насекомых (данные не представлены).
Пример 16
Конструирование вектора для коэкспрессии
L1/L2 СRPV из одиночной плазмиды
Плазмиду pSP72-CRPV-L1 (pl2930-314-4-l) обрабатывали рестриктазой Вg1 II и Bg1 II-фрагмент размером 1,5 т.п.н., несущий ОRF L1 CRPV с дрожжевым
5'-нетранслируемым лидером, извлекали из геля. Плазмиду р12930-323-2-3 (pCl/1-GAL1p-GАL1-p-CRPV-L2) обрабатывали рестриктазой ВаmHI, которая режет между GAL1-промотором и ADH1-транскрипционным
терминатором. Фрагмент линеаризованного вектора извлекали из геля и лигировали с вышеупомянутым Bg1 II-фрагментом CRPV-L1 для создания плазмиды pl2930-366-l-2 (pCl/l-GAL1/10р-CRPV/L1+L2). Эта
результирующая плазмида содержала ОRF CRPV-L1 под контролем GAL1-промотора и ORF CRPV-L2 под контролем GAL10-промотора.
Пример 17
Конструирование вектора для коэкспрессии
L1/L2 CRPV из двух плазмид
Вектор pUCl8-GAL1p-GAL10p, содержащий ген L2 разрезали рестриктазой SphI и фрагмент размером 2,9 т. п. н., несущий экспрессионную кассету
ADH1t-GAL1p-GAL10p-L2-ADH1t, извлекали из геля и концы тупили обработкой Т4 ДНК-полимеразой. Дрожжевой челночный вектор YEp24 (Botstein et al., Gene, 8:17, 1979) обрабатывали рестриктазой ВаmHI, концы
тупили обработкой Т4 ДНК-полимеразой, дефосфорилировали щелочной фосфатазой из кишечника теленка и затем лигировали с вышеуказанной экспрессионной кассетой L2 с затупленными концами для создания
плазмиды р1594.
Пример 18
Коэкспрессия L1 и L2 CRPV в дрожжах
Плазмиду р12930-366-1-2(рС1/1-GAL1/10р-CRPV/L1+L2 использовали для трансформации штаммов # 1569 и BJ
5462 S. сеrevisiae (одна плазмидная система) и полученные трансформанты отбирали на безлейциновой синтетической агаровой среде. В параллельном эксперименте штамм 1569 котрансформировали экспрессионным
вектором pCl/1-GAL10р-CRPV-L1 плазмиды р12930-323-6-1 и экспрессионным вектором YЕр24-GAL10p-L2 плазмиды р1594 (две плазмидные системы) и полученные трансформанты, содержащие оба вектора, отбирали на
синтетической агаровой среде, не содержащей лейцина и урацила. Клоновые изоляты с одной плазмидной системой и с двумя плазмидными системами выращивали при 30oС в течение 48-72 часов на
комплексной среде УЕНD), содержащей 2% галактозы. После сбора клеток клетки разрушали с помощью стеклянных шариков. Добавляли тритон Х-100 до конечной концентрации 0,5% и клеточные лизаты
анализировали на экспрессию L1 и L2 СRPV с помощью иммуноблот-анализа. Образцы, содержащие 50 мкг суммарного клеточного белка, подвергали электрофорезу в 8-16% трис-глициновом градиентном геле (Novex)
в восстанавливающих и денатурирующих условиях и электроблоттировали в РVDF-мембраны (Novex). Белки L1 и L2 CRPV детектировали, используя поликлональные кроличьи анти-L1 или анти-L2 антисыворотки (дар
от и Dr. John Kreider, Медицинский Центр Hershey) в качестве первичных антител, а протеин А, присоединенный к пероксидазе хрена, как вторичные антитела. Мембраны обрабатывали, используя
хемилюминисцентный набор для детекции ECL ТМ (Amersham, Inc.). Полосу белка L1, соответствующую молекулярной массе 55-61 кД, и полосу белка L2, соответствующую молекулярной массе около 90 кД,
обнаруживали во всех образцах, полученных из дрожжевых клонов, содержащих L1+L2 -экспрессионную плазмиду. В образцах, полученных из дрожжевых клонов, содержащих экспрессионную плазмиду L1, сигналы для
L2 не обнаруживались. Сигналы для L1 не обнаруживались в образцах из клеток, содержащих только L2-экспрессионный вектор.
Пример 19
Выделение VLPs L1 CRPV и L1+L2 CRPV для
электронно-микроскопических исследований
Дрожжи, экспрессирующие белки L1 CRPV и L1 и L2 CRPV, частично очищали и концентрировали для электронно-микроскопических (ЕМ) исследований. От одного
до 1,5 литров YEHD-среды, содержащей 2% галактозы, использовали для инокуляции штаммов # 1569 или BJ 5462 S. cеrevisiae, трансформированных коэкспрессионным вектором для L1+L2 - р12930-366-1-2
(pCl/1-GAL1/10р-СRPV/L1+L2), и выращивали при 30oС в течение 48-72 часов. В параллельном эксперименте клетки штамма # 1569, котрансформированные экспрессионным вектором GAL10p-CRPV-L1
плазмиды pl2930-323-6-1 и экспрессионным вектором YЕр24-GAL10р-L2 плазмиды р1594, выращивали подобным образом. Клетки собирали и осадок клеток замораживали при -70oС. Все последующие
операции выполняли при 4oС. Осадок клеток оттаивали и суспендировали в равном объеме "L-буфера" (20 мМ натрийфосфата, рН 7,2, 100 мМ NaCl, 1,7 ЭДТА). Ингибиторы протеаз PMSF и пепстатин А
добавляли к полужидкой массе до конечной концентрации соответственно 2 мМ и 1,7 мкМ. Клетки лизировали 3-5-кратным пропусканием через микрофлуидизатор. Лизаты осветляли и наслаивали на вершину 5 см
подушки 45% (о/о) сахарозы в L1-буфере, а белки Ll, L2 или L1 и L2 осаждали центрифугированием при 100000 g в течение 4 часов. Осадок ресуспендировали в 1/10 объема L1-буфера. Ресуспендированный
осадок осветляли центрифугированием при 5000 g в течение 10 минут.
Для ЕМ-анализа (Structure Probe, West Chester, PA) аликвот каждого образца помещали на покрытую медью угольную сетку, 200 меш. Каплю 2% фосфорновольфрамовой кислоты (РТА), рН 7,0, помещали на сетку на 20 секунд. Сетку высушивали на воздухе перед ТЕМ-анализом. Микроскопирование осуществляли, используя трансмиссионный электронный микроскоп JEOL 100CX (JEOL USA, Inc.) при быстром повышении напряжения 100 кВ. Все микрофотографии получали при увеличении 100000х.
Все дрожжевые образцы, содержащие экспрессионную плазмиду Ll CRPV или плазмиды для коэкспрессии VLP L1 и L2 CRPV, наблюдали в размерном ряду диаметром 50-55 нм. В контрольных образцах дрожжей, содержащих одну лишь экспрессионную плазмиду L2, VLР не наблюдали (фигуры 7, 8, 9).
Пример 20
Выделение рекомбинантного капсидного белка L1 CRPV (схема 2).
Все операции, за исключением специфичности, выполняли при 4oС. Клетки, хранившиеся при -70oС, оттаивали и суспендировали в равном объеме лизирующего буфера (20 мМ натрийфосфата, рН 7,2, 100 мМ NaCl, 1,7 мМ ЭДТА). Ингибиторы протеаз PMSF и пепстатин А добавляли к полужидкой массе до конечной концентрации соответственно 2 мМ и 1,7 мкМ. Клетки лизировали, используя систему разрушения клеток SioNeb (Glas-Col Apparatus Co., Terra Haute, IN). Лизат осветляли центрифугированием при 5000 g в течение 10 минут.
Супернатант наслаивали на вершину 5 см подушки 45% сахарозы (в/о) в L1-буфере и белок L2 осаждали центрифугированием при 100000 g в течение 4 часов. Осадок ресуспендировали в 1/10 объема L1-буфера. Ресуспендированный осадок осветляли центрифугированием при 5000 g в течение 10 минут.
Супернатант разбавляли 1:5 PBS, повторно осветляли центрифугированием в роторе Sorvall SA-600 при 6500 об/мин в течение 10 минут при 4oС. Супернатант фильтровали через шприцевый фильтр с размером пор 0,22 мкм и фракционировали с помощью анионообменной хроматографии.
Анионообменную хроматографию осуществляли с помощью HPLC-бионасоса серии 410 Perkin-Elmer, снабженного 5- миллилитровой инъекционной петлей. Хроматографической средой была смола Fractogel EMF TMAE-650 (S) с размером пор 25-40 мкм (ЕМ Separations, Gibbstown, NJ), загруженная в стеклянную колонку с размерами 150 мм x 10 мм внутреннего диаметра. Для слежения за элюированием белка из колонки осуществляли оптическую детекцию при 280 нм, которая достигалась с помощью детекторной матрицы на диоде Perkin-Elmer LС-235. Колонку предварительно уравновешивали 0,15 М NаС1 в 0,01 М натрийфосфатном буфере, рН 7,2 (буфер А). Колонку разгоняли при скорости потока 0, 75 мл/мин. Образец нагружали на колонку и колонку промывали буфером А для удаления несвязавшегося материала. Связавшийся материал элюировали линейным концентрационным градиентом хлорида натрия, 0,15 М - 0,65 М в течение 5 минут, с последующим 0,65 М - 1,15 М линейным градиентом в течение 30 минут. Детекцию антигена в собираемых фракциях во время элюирования осуществляли с помощью иммунодот-блот-анализа. Фракции, содержащие иммунореактивный материал (т.е. фракции, элюированные между 0,81 М и 1,05 М NaCl), объединяли.
Объединенные фракции концентрировали на центробежной концентрационной установке Macrosep (Filtron, Technology Corp., Northborough, MA) до 1/5 объема.
Концентрат фракционировали с помощью хроматографии по размеру молекул, используя смолу Сефакрил S-10000 SF (Рhаrmаcia, Piscataway, NJ) в колонке с размерами 87 см x 27 мм внутреннего диаметра. Колонку разгоняли при скорости потока 2,5 мл/мин. Элюируемый белок отслеживали абсорбцией при 280 нм. Антиген детектировали иммуноблоттом.
Фракции, содержащие иммунореактивный материал, объединяли и концентрировали ультрафильтрацией под азотом при давлении 10 фунтов на квадратный дюйм, используя ячейку с перемешиванием (Amicon, Inc., Beverly, MA), с 43 мм пластинчатой многослойной мембраной УМ-100 диа (Amicon, Inc. Beverly, MA).
Конечный продукт характеризовали SDS/ПАГ-электрофорезом с окрашиванием Кумасси и с помощью просеивающего капиллярного электрофореза в растворе (SSCE). Согласно SSE конечный продукт из cхемы 2 имел 88% чистоты.
Пример 21
А. Выделение рекомбинантного капсидного белка L1 CRPV (cхема 3).
Все операции, за исключением специфичности, выполняли при 4oС.
Клетки, хранившиеся при -70oС, оттаивали и суспендировали в равном объеме лизирующего буфера (20 мМ натрийфосфата, рН 7,2, 100 мМ NaCl, 1,7 мМ ЭДТА). Ингибиторы протеаз PMSF и пепстатин А добавляли к полужидкой массе до конечной концентрации соответственно 2 мМ и 1,7 мкМ. Клетки лизировали, используя систему разрушения клеток BioNeb (Glas-Col Apparatus Co., Terra Haute, IN). Лизат осветляли центрифугированием при 5000 g в течение 10 минут.
Супернатант наслаивали на вершину 5 см подушки 45% сахарозы (в/о) в L1-буфере и белок L1 осаждали центрифугированием при 100000 g в течение 4 часов. Осадок ресуспендировали в 1/10 объема L1-буфера. Ресуспендированный осадок осветляли центрифугированием при 5000 g в течение 10 минут.
Супернатант экстрагировали 1/2 объема хлороформа. Водный слой удаляли и осветляли центрифугированием при 12000 об/мин в микроцентрифуге Beckman в течение 5 минут при комнатной температуре.
Супернатант разбавляли PBS, повторно осветляли центрифугированием в роторе Sorvall SA-600 при 6500 об/мин в течение 10 минут при 4oС. Супернатант фильтровали через шприцевый фильтр с размером пор 0,22 микрон и фракционировали с помощью анионообменной хроматографии.
Анионообменную хроматографию осуществляли с помощью HPLC-бионасоса серии 410 Perkin-Elmer, снабженного 5- миллилитровой инъекционной петлей. Хроматографической средой была смола Fractogel EMF ТМАЕ-650 с размером пор 25-40 микрон (ЕМ Separations, Gibbstown, NJ), загруженная в стеклянную колонку с размерами 150 мм х 10 мм внутреннего диаметра. Для слежения за элюированием белка из колонки осуществляли оптическую детекцию при 280 нм, которая достигалась с помощью детекторной матрицы на диоде Perkin-Еlmer LC-235. Колонку предварительно уравновешивали 0,15 М NаС1 в 0,01 М натрийфосфатном буфере, рН 7,2 (буфер А). Колонку разгоняли при скорости потока 0,75 мл/мин. Образец нагружали на колонку и колонку промывали буфером А для удаления несвязавшегося материала. Связавшийся материал элюировали линейным концентрационным градиентом хлорида натрия 0,15 М - 0, 65 М в течение 5 минут с последующим линейным градиентом 0,65 М - 1,15 М в течение 30 минут. Детекцию антигена во фракциях, собираемых во время элюирования, осуществляли с помощью иммунодот-блот-анализа. Фракции, содержащие иммунореактивный материал (т.е. фракции, элюированные между 0,81 М и 1,05 М NаСl), объединяли.
Объединенные фракции концентрировали на центробежной концентрационной установке Macrosep (Filtron Technology Corp., Northborouogh, MA) до 1/5 объема.
Концентрат фракционировали с помощью хроматографии по размеру молекул, используя смолу Сефакрил S-1000 SF (Pharmacia, Piscataway, NJ) в колонке с размерами 87 см х 27 мм внутреннего диаметра. Колонку разгоняли при скорости потока 2,5 мл/мин. Элюируемый белок отслеживали при А280 нм. Антиген детектировали с помощью иммуноблота.
Фракции, содержащие иммунореактивный материал, объединяли и концентрировали ультрафильтрацией под азотом при давлении 10 фунтов на квадратный дюйм, используя ячейку с перемешиванием (Amicon, Inc. Beverly, MA) с 43 мм пластинчатой многослойной мембраной YМ-100 (Amicon, Inc. Beverly, MA).
Конечный продукт характеризовали SDS/ПАГ-электрофорезом и с помощью просеивающего капиллярного электрофореза в растворе (SSCE). Согласно SSCE конечный продукт из cхемы 3 имел 95% чистоты.
Пример 22
Экспрессирование L1 CRPV и L1 HPV типа 6а (штамм 1644) в богатой комплексной и химически точной среде
Инокулят из этих штаммов выращивали на безлейциновой синтетической среде, описанной
выше, для культивирования во встряхиваемых колбах. Встряхиваемые колбы имели большую аэрируемую емкость (70 мл жидкости на 300 мл объема колбы, Tunair Labware) и в среду добавляли около 0,5 мл/л
пеногасителя (UCON LB-625, Union Carbide). Богатая комплексная среда содержала (на 1 л): 40 г дрожжевого экстракта Difco, 20 г соевого пептона Sheffield, 30 г глюкозы, 50 г галактозы; перед
стерилизацией рН среды доводили до 5,3. Использовали химически точную среду, подобную той, которую описал Oura (Biotechnol. Bioengineer. 16:1197-1212 (1974)), но с добавлением (на 1 л): 0,1 г хлорида
холина, 0,4 г аденина, 30 г глутамата (мононатриевая соль) в качестве источника азота; 0,2 г урацила, 20 г глюкозы, 40 г галактозы. В каждую колбу вносили по 3 мл инокулята и инкубировали в течение 66
ч при 28oС при 250 оборотах на качалке с круговым вращением. Из колб с интервалами отбирали образцы для обнаружения экспрессии с помощью иммуноблота, используя антисыворотки анти-CRPV L1 и
анти-HPV 6а L1.
Пример 23
Клонирование генома HPV-6а
Ткань от пациента с диагнозом множественная кондилома остроконечная (переданная Dr. Darron Brown,
Университетская школа по медицине, штат Индиана) типировали ферментами рестрикции и полимеразной цепной реакцией (PCR) и показали, что она содержит HPV типа 6а. Из тканевого образца экстрагировали ДНК
и обрабатывали рестриктазой Hind III. Затем фракционировали по размеру через 0,8% препаративный гель легкоплавкой агарозы, вырезали из геля агарозную область, содержащую фрагмент размером 8 т.п.н., и
агарозный гель обрабатывали ферментом Gelase ТМ (Epicentre Technologies, Inc.). Образец лигировали с плазмидой pUC18, которую обрабатывали рестриктазой Hind III и дефосфорилировали (Pharmacia, Inс.).
После трансформации компетентных клеток Е. соli DH5 (BRL), скринировали плазмидную библиотеку на наличие положительных клонов HPV-6а, используя 32Р-меченый олигодезоксинуклеотид, комплементарный гену
L1 HPV-6а. Плазмиду pUC18, содержащую участок генома HPV-6а размером 8 т.п.н., выделяли и характеризовали с помощью ферментов рестрикции и Саузерн-блот анализом. Эту плазмиду обозначили рUC18-HPV-6а.
Фрагмент генома HPV-6а размером 8 т.п.н. полностью секвенировали, используя автоматический секвенатор ABI (#373А) и руководство для пользователей.
У 25-летней женщины после родов взяли участок с наружных половых органов, пораженный кондиломой остроконечной. Фрагмент этого участка заморозили в жидком азоте, а затем обработали Брауновским микродисмембратором (В. Braun Instruments, Melsungen, Germany). Результирующий материал растворяли с 0,6% (в/о) натрийдодецилсульфатом (SDS), обрабатывали протеиназой К (50 мкг/мл) и экстрагировали фенол/хлороформ/изоамиловым спиртом. ДНК осаждали этанолом и количественно оценивали на УФ-спектрофотометре. Наличие высокомолекулярной ДНК определяли электрофорезом в агарозном геле и последующим окрашиванием этидийбромидом.
Тип ДНК HPV определили, используя гибридную ловушку, помеченную как Vira Tyре Plus (Digene Diagnostics, Beltsville, MD). Используемые пробы HPV делили на две группы, чей состав обосновывали ассоциацией каждого типа со злокачественной трансформацией половых путей. Проба группы А содержала типы HPV "пониженного риска" - типы 6, 11, 42, 43 и 44, тогда как выборка В содержала типы "повышенного риска" - типы 16, 18, 31, 33, 35, 45, 51, 52 и 56. Тотальную ДНК обрабатывали рестриктазами PstI, BamHI и Hind III, а для определения субтипов HPV Саузерн-блоты осуществляли в очень строгих условиях (Тm -15oС).
Для определения полной последовательности HPV-6а секвенирующие праймеры синтезировали на основании опубликованной последовательности НРV-6b. Обе нити всего геномного участка НРV-6a размером 8,1 т.п.н. секвенировали методом дидезокситерминирования, используя набор PRISM TM и автоматический секвенатор [(# 373A) Applied Biosystems (АВI)] , согласно инструкциям для пользователей (АВI, Inс. , Foster Сity, СА). В случаях когда смысловая и антисмысловая последовательности не сочетались, синтезировали дополнительные специфические HPV6а-праймеры для ресеквенирования в обоих направлениях через сомнительную область для достижения консенсуса.
Последовательности ДНК HPV6a и HPV6b проявляли более 97% сходства по суммарному количеству замен 229 п.н., идентифицированных среди 8010 п.н. Наиболее значительные различия при сравнении с HPV6b-последовательностью обнаружены в протяженной регулируемой области (LCR; nt 7205 - nt 106). Кроме нескольких одиночных нуклеотидных (nt) замен в LCR НРV6a, обнаружены 94 п.н. вставка по 7350 нуклеотиду, а другая 19 п.н. вставка - по 7804 нуклеотиду. По 7615 нуклеотиду генома HPV6a делегировано шесть пар оснований.
Пример 24
Конструирование дрожжевого экспрессионного вектора L1 HPV6а
ДНК HPV типа 6а использовали в качестве матрицы для
ПЦР. Ген L1 HPV6a амплифицировали ПЦР, используя Vent-полимеразу (New England Biolabs, Inc.), 35 циклов амплификации (94oС 1 мин, 48oС 1 мин, 72oС 1 мин 45 сек) и
нижеследующие олигодезоксинуклеотидные праймеры, которые содержали фланкирующие Bg1 II-сайты (подчеркнуто):

Смысловой праймер вводит дрожжевую нетранслируемую лидерную последовательность непосредственно выше инициирующего метионин кодона L1 HPV6a (высвечивает на темном фоне). ПЦР-продукт L1 размером 1,5 т.п.н. обрабатывали рестриктазой Bg1 III и извлекали из геля. Сконструировали экспрессионный вектор pCl/1-GАL в результате выделения фрагмента SphI размером 1,4 т. п. н. из двунаправленного промоторного вектора pUC18-GAL1p - GAL10p, который содержит дивергентный промотор GAL1/GAL10 из плазмиды рВМ272 (предоставил Dr. Mark Jonston, Washington Universiti, St. Louis), фланкированый с каждой стороны копией дрожжевого терминатора транскрипции ADH1 (Bennetzen J.L. and Hall B.D. (1982), J. Biol. Chem., 257:3018-3025). В результирующей экспрессионной кассете Bam HI-сайт расположен между GAL1-промотором и первой копией ADH-терминатора транскрипции, a SmaI-клонируемый сайт расположен между дивергентным GAL10 -промотором и второй копией ADН1-терминатора транскрипции. Дрожжевой челночный вектор рС1/1 (Rоsеnbеrg et al., Nature, 312 (1984), 77-80) обрабатывали рестриктазой SphI и лигировали с SphI-фрагментом GAL-ромотора. Результирующий вектор pCl/1-GAL линеаризировали рестриктазой BamHI, которая резала между GAL1-промотором и ADH1-терминатором транскрипции. Вектор pCl/1-GAL, обработанный рестриктазой BamHI, и ПЦР-фрагмент Ll HPV6a, обработанный рестриктазой Вg1 III, лигировали и использовали для трансформации клеток DН5 Е. coli (BRL). Выделяли плазмиду pCl/1-GAL, которая содержала ген Ll НРV-6а, и обозначили ее р13173-357-6. Ген L1 в плазмиде р13173-357-6 секвенировали (ABI Seguencer # 373A) и установили его идентичность гену L1 в клоне рUC18-HPV 6а.
Пример 25
Конструирование дрожжевого экспрессионного
вектора L1 и L2 HPV 6а
Плазмиду р13173-357-6 (pCl/1-GAL+HPV6a L1) обрабатывали рестриктазой SmaI, которая резала между GAL10-промотором и АDН1-терминатором транскрипции. Ген L2 HPV6a размером
1,4 т.п.н. амплифицировали ПЦР, используя ДНК клона pUC18-НРV6a в качестве матрицы. Vent-полимеразу (New England Biolabs, Inc. ), 10 циклов амплификации ПЦР (94oС 1 мин, 48oС 1
мин, 72oС 1 мин 45 сек) и нижеследующие олигодезоксинуклеотидные праймеры, которые содержали фланкирующие SmaI-сайты (подчеркнуто):

Смысловой праймер вводит дрожжевую нетранслируемую лидерную последовательность непосредственно выше инициирующего метионин кодона L2 HPV 6а (высвечивает на темном фоне). ПЦР-фрагмент обрабатывали рестриктазой SmaI, извлекали из геля и лигировали с плазмидой р131713-357-6, обработанной рестриктазой SmaI. Выделили плазмиду рС1/1-GАL, содержащую гены L1 и L2, и обозначили ее р14049-7-2. Ген L2 секвенировали и обнаружили его идентичность гену L2 клона pU C18-HPV 6а.
Пример 26
Экспрессия L1 НРV6a и
коэкспрессия L1 и L2 HPV6a в дрожжах
Плазмиды р13173-357-6 (pCl/1-GAL+HPV6a L1) и р14049-7-2 (pCl/1-GAL+НPV6a L1 и L2) использовали для трансформации штаммов # 1569 (МАТа, lеu2-04, рrb 1, ade
1, рер 4, cir) и #1558 (МАТа, leu-04, prb1, mnn, ade 1, сir) S. cerevisiae. Результирующими рекомбинантными штаммами, которые получили, используя штамм-хозяин #1558, были штаммы #1644 (НРV6а L1) и
#1670 (HPV6a L1+L2), как показано в таблице. Клоновые изоляты росли при 30oC в YЕНD - среде, содержащей 2% галактозы, в течение 68-78 часов.
После сбора клеток осадок клеток разрушали со стеклянными шариками и клеточные лизаты анализировали на экспрессию белков L1 HPV 6а или L2 HPV6a с помощью иммуноблотного анализа. Образцы, содержащие 40 мкг суммарного клеточного белка, подвергали электрофорезу в 10% трис-глициновом геле в восстанавливающих и денатурирующих условиях и электроблоттировали на нитроцеллюлозные фильтры. Белок L1 иммунодетектировали, используя кроличью антисыворотку против слитого белка trpE-HPV 11 (Brown D.R. et al., Virology, 201:46-54) и ослиные антикроличьи IgG связанные с пероксидазой хрена (НRР) целые антитела (Amersham, Inc.) в качестве вторичных антител. Полоса белка, соответствующая молекулярной массе 50-55 кД, обнаруживалась во всех образцах за исключением отрицательного контроля (рС1/1 без гена L1 или гена L2).
Белок L2 обнаруживали Вестерн-анализом как белковую полосу, соответствующую молекулярному весу 70 кД, используя сыворотку мышей, разведенную 1: 250, которой иммунизировали 3 раза со слитым белком L2 trpE-HPV6а, приготовленным фактически по методу Сarter et al. в качестве первичных антител и НRР-связанные бараньи антимышиные IgG (Amersham, Inc.) в качестве вторичных антител (разведение 1:1000).
Для ЕМ-анализа (Structure Probe, West Chester, PA) аликвот каждого образца помещали на покрытую медью угольную сетку 200 меш. Каплю 2% фосфорновольфрамовой кислоты (РТА), рН 7,0, помещали на сетку на 20 секунд. Сетку высушивали на воздухе перед ТЕМ-анализом. Микроскопирование осуществляли, используя трансмиссионный электронный микроскоп JEOL 100CX (JEOL USA, Inc.) при быстром повышении напряжения до 100 кВ. Микрофотографии получали при увеличении 100000х.
VLP наблюдали в размерном ряду с диаметром 50-55 нм во всех дрожжевых образцах, содержащих L1 HPV6a или L1 и L2 HPV6a коэкспрессионные плазмиды. В контрольных образцах дрожжей VLP не наблюдали.
Пример 27
Ферментация L1+L2 НРV6а (штамм # 1670)
Поверхностно
растущую чашечную культуру штамма 1670 асептически переносили в безлейциновую среду, содержащую (на 1 л): 8,5 г основного дрожжевого азота Difco без аминокислот и сульфата аммония, 0,2 г аденина, 0,2
г урацила, 10 г янтарной кислоты, 5 г сульфата аммония и 0,25 г L-тирозина, перед стерилизацией эту среду доводили до рН 5,0-5,3 с помощью NаОН. По окончании выращивания в течение 25 часов при 28oС, 250 об/мин на качалке с круговым вращением культуру замораживали в ампулах с добавлением стерильного глицерина до конечной концентрации 17% (в/о) перед помещением на хранение при -70oС (1 мл на замораживаемую ампулу). Инокулюм для ферментации штамма 1670 помещали в такую же среду (500 мл на 2 л колбу) путем переноса оттаянного содержимого двух культу-ральных ампул в 2 л
колбы и начинали инкубацию при 28oС, 250 об/мин на качалке с круговым вращением в течение 25 часов. Ферментацию штамма 1670 вели в ферментере New Brunswick SF-116 с рабочим объемом 10 л
после инокуляции. Используемая питательная среда содержала (на 1 л): 20 г дрожжевого экстракта Difco, 10 г соевого пептона Sheffield, 20 г глюкозы, 20 г галактозы; перед стерилизацией рН среды
доводили до 5,3. 500 мл содержимого 2 л колбы переносили в качестве инокулюма в ферментер и инкубировали при 28oС, подаче воздуха 5 л/мин, 400 об/мин и давлении 3,5 фунтов на квадратный
дюйм. При необходимости увеличивали интенсивность перемешивания для поддержания уровня растворенного кислорода более 40% насыщения. Течение ферментации отслеживали автономными измерениями глюкозы
(Beckman Glucose 2 Analyzer) и оперативной масс-спектрометрией (Perkin-Elmer 1200). После 69 ч инкубирования плотность клеток составляла 9,9 г/л в расчете на вес сухих клеток. Культуру концентрировали
фильтрованием через полые волокна (патрон Amicon H5MPО1-43 в фильтрационной системе Avicon DС-10) объемом 2 л, диализовали фильтрацией 2 л забуференного фосфатом физиологического раствора и продолжали
концентрирование (до объема 1 л) для последующего перенесения в 500 мл центрифужные стаканы. Клетки осаждали центрифугированием при 8000 об/мин (ротор Sorval GS3) в течение 20 мин при 4oС.
После декантирования супернатанта осадок (225 г влажных клеток) хранили при -70oС перед использованием.
Пример 28
Выделение рекомбинантных капсидных белков L1+L2
HPV6a
Все операции выполняли при 4oС, кроме оговоренных особо.
Клетки штамма #1670 хранили замороженными при -70oС. Замороженные клетки (влажный вес 38, 0 г) оттаивали при 20-23oС и ресуспендировали в 50 мл "L1-буфера" (20 мМ натрийфосфата, рН 7,2, 100 мМ NaCl, 1,7 мМ ЭДТА). Ингибиторы протеаз PMSF и пепстатин А добавляли до конечной концентрации соответственно 2 мМ и 1,7 мкМ. Клеточную кашицу разрушали под давлением приблизительно 8000 фунтов на квадратный дюйм 3-кратным пропусканием через микрофлуидизатор M110 (Microffuidics Corp., Newton, MA). Разрушенную клеточную кашицу центрифугировали при 5000 g в течение 10 мин для удаления клеточных оболочек. Получали надосадочную жидкость, содержащую антиген L1+L2.
Надосадочную жидкость разбавляли 1:5 добавлением буфера А (20 мМ MOPS, рН 7,0) и наносили на анионообменную колонку (5,0 см внутреннего диаметра х 4 см) со смолой Fractogel EMD TMAE-650 (S) (ЕМ Separations, Gibbstown, NJ) уравновешенную в буфере А. После промывания буфером А антиген элюировали градиентом 0-1,0 М NаС1 в буфере А. Проводили иммунно-дот-блоттинг для обнаружения элюированных фракций, содержащих белок L1.
Во фракциях, содержащих белок L1, обнаруженный иммунн-дот-блотом, определяли суммарный белок по методу Bradford с последующим SDS/ПАГ-электрофорезом и применением серебряного окрашивания и Вестерн-блоттинга.
Фракции ТМАЕ, обнаруживающие сопоставимую степень чистоты и обогащенность белком L1, объединяли. Антиген концентрировали постепенным насыщением сульфатом аммония. Раствор доводили до 63% насыщения сульфатом аммония, добавляя сухой реагент при осторожном перемешивании каждые 30 мин. Образец помещали на лед и преципитировали в течение 30 мин. Образец центрифугировали при 12000 g. Осадок ресуспендировали в 20 мл PBS (6,25 мМ Na-фосфата, рН 7,2, 150 мМ NaCl).
Ресуспендированный осадок хроматографировали на эксклюзионной колонке (2,6 внутреннего диаметра х 89 см) со смолой Сефакрил 500 НR (Pharmacia, Piscataway, NS). Проточный буфер был PBS. Хроматографию осуществляли при 20-23oС. Фракции анализировали SDS/ПАГ-электрофорезом, используя окрашивание серебром и вестерн-блот-детектирование. Очищенные фракции объединяли и концентрировали.
Итоговый продукт анализировали SDS/ПАГ-электрофорезом, используя окрашивание коллоидальным Кумасси. Белки L1 и L2 имели 85: гомогенности. Подлинность белков L1 и L2 подтверждали вестерн-блоттингом, используя соответствующую антисыворотку. Конечный продукт фасовали и хранили при -70oС. При такой обработке получали 3,0 мг белка.
Электронно-микроскопический анализ осуществляли с помощью Structure Рrоbe (West Chester, PA). Аликвот образца помещали на покрытую медью угольную сетку с 200 меш. Каплю 20% фосфорновольфрамовой кислоты, рН 7,0 помещали на сетку на 20 секунд. Сетку высушивали на воздухе перед проведением ТЕМ-анализа. Микроскопирование осуществляли, используя трансмиссионный электронный микроскоп JEOL 100 СХ (JEOL USA, Inc.) при быстром повышении напряжения до 100 кВ. Полученные микрофотографии имели увеличение 100000х. Подтверждено наличие вирусоподобных частиц в размерном ряду 50-55 нм.
Пример 29
Выделение очисткой VLPs L1 НРV-6a
и L1/L2 для ЕМ исследований
Экспрессированные дрожжами белки L1 HPV6a и L1+L2 HPV-6a подвергали частичной очистке и концентрированию для электронно-микроскопических (ЕМ) исследований. От 1 до
1,5 л YЕНD-среды, содержащей 2% галактозы и 0,1 М сорбитола, использовали для инокуляции и выращивания при 30oС в течение 68-78 часов клеток штамма #1558 S. сerevisiae, содержащих плазмиду
р13173-357 (Ll-вектор экспрессии) или плазмиду р14049-7-2 (L1- и L2-вектор коэкспрессии). Клетки собирали центрифугированием при 4000 g в течение 10 мин и осадок клеток замораживали при -70o
С. Все последующие операции выполняли при 4oС. Осадок клеток оттаивали и суспендировали в равном объеме "L-буфера" (20 мМ натрийфосфат, рН 7,2, 100 мМ NaCl, 1,7 ЭДТА). Ингибиторы протеаз
РМSF и пепстатин А добавляли к полужидкой массе до конечной концентрации соответственно 2 мМ и 1,7 мкМ. Клетки лизировали 3-5-кратным пропусканием через микрофлуидизатор. Лизат осветляли
центрифугированием при 5000 g в течение 10 минут. Супернатант наслаивали на вершину 5 см подушки 45% сахарозы (в/о) в L-буфере и белки L1 или L1+L2 осаждали центрифугированием при 100000 g в течение 4
часов. Осадок ресуспендировали в 1/10 объема L-буфера и осветляли центрифугированием при 5000 g в течение 10 минут. Образцы изучали электронно-микроскопически и обнаружили наличие вирусоподобных
частиц.
Пример 30
Клонирование генов L1 HPV16 и L2 HPV 16
Тотальную геномную ДНК экстрагировали из Caski - клеток (АТСС # CRL 1550) по стандартной методике (Sambrook
et al., как указано выше). ДНК обрабатывали рестрицирующими эндонуклеазами Bst1 1071 и SphI и электрофорезировали в 0,8% препаративном геле из легкоплавкой агарозы. Область, соответствующую ДНК с
примерным размером 3,5 т.п.н., вырезали из геля и агарозу обрабатывали ферментом ТМ (Epicentre Techologies, Inс). У извлеченной из геля ДНК затупляли концы с помощью Т4 ДНК-полимеразы и лигировали с
тупыми концами фосфорилированных олигодезоксинуклеотидных линкеров, которые содержали вставленный Hind III-сайт. Лигированную смесь окончательно расщепляли рестриктазой Hind III и фрагменты ДНК
размером около 3,5 т.п.н. фракционировали по размеру в агарозном геле, как описано выше. Извлеченную из геля ДНК лигировали с ДНК-плазмидой pUC18, которую обрабатывали рестриктазой Нind III и
дефосфорилировали. После трансформации компетентных клеток DH5 Е. coli скринировали плазмидную библиотеку на наличие НРV16-положительных клонов путем гибридизации колоний, используя антисмысловой
олигодезоксинуклеотид, меченный 32 Р, комплементарный 3'-концу гена L1 HPV 16
(5' - GAG AGA ТСТ TАС AGC TTА CGT ТТТ TTG CGT ТТА GC-3').
Выделяли плазмиду pUC18, содержащую геномный фрагмент HPV 16 размером 3,3 т. п. н. , и характеризовали с помощью ферментов рестрикции и Саузерн-блот-анализом. Эта плазмида, обозначенная pUC18-HPV16 L1/L2, содержала все последовательности ДНК, кодирующие L1 b L2. Плазмидную ДНК выделяли, используя Qiagen ТМ Plasmid Maxi kit (Qiagen, Inс.).
Пример 31
Конструирование дрожжевого экспрессионного
вектора L1 HPV16
Клон pUC18-HPV 16 L1/L2 использовали в качестве матрицы для ПЦР. Ген L1 HPV 16 амплифицировали ПЦР, используя Vent - полимеразу (New England Biolabs, Inc. ) 10 циклов
амплификации (94oС 1 мин, 48oС 1 мин, 72oC 1 мин 45 сек) и нижеследующие олигодезоксинуклеотидные праймеры, которые содержали фланкирующие Bg 1 II-сайты
(подчеркнуто):

Смысловой праймер вводит дрожжевую нетранслируемую лидерную последовательность выше инициирующего метионин кодона L1 HPV 16 (высвечивает на темном фоне). ПЦР-продукт L1 размером 1,5 т.п.н. обрабатывали рестриктазой Bg1 II, извлекали из геля и лигировали с вектором pCl/l-GAL, обработанным рестриктазой BamHI. Выделенную плазмиду pCl/1-GAL, содержащую ген L1 HPV 16, обозначили р14049-37-1. Ген L1 в плазмиде 14049-37-1 секвенировали, используя набор PRIS M TM (ABI, Inc.) и секвенатор ABI модели #373A, в соответствии с указаниями для пользователя. Показано, что данный изолированный ген Ll содержит 3 нуклеотидные замены, в отличие от опубликованной последовательности прототипа (Kirnbauer R. еt al., (1993), J. Virol., 67:6929-6936), приводящие к двум аминокислотным заменам: His - 202 на Аsp, Thr - 266 на Ala. Анализ последовательности исходной ДНК-матрицы подтверждает, что эти замены также присутствовали в геноме клона рUС18 - HPV 16 L1/ L2, а не интродуцированы ПЦР.
Пример 32
Конструирование дрожжевого экспрессионного вектора
L1 и L2 HPV16
Плазмиду р14049-37-1 (pCl/1-GAL+HPV16 L1) обрабатывали рестриктазой SmaI, которая резала между GAL10-промотором и АDH1-терминатором транскрипции. Ген L2 HPV16 амплифицировали
ПЦР, используя ДНК pUC18-HPV 16 L1/L2 в качестве матрицы, Vent -полимеразу (New England Biolabs, Inc.), 10 циклов ПЦР-амплификации (94oС 1 мин, 48oС 1 мин, 72oС 1 мин
45 сек) и нижеследующие праймеры, которые содержали фланкирующие SmaI - сайты (подчеркнуто):

Смысловой праймер вводит дрожжевую нетранслируемую лидерную последовательность выше инициирующего метионин кодона L2 HPV 16 (высвечивает на темном фоне). ПЦР-продукт L2 размером 1,4 т.п.н. обрабатывали рестриктазой SmaI, извлекали из геля и лигировали с вектором р14049-37-1, обработанным рестриктазой SmaI. Выделенную плазмиду рС1/1-GAL, содержащую гены L1 и L2 HPV 16, обозначили р14049-42-2. Ген L2 в плазмиде р14049-42-2 секвенировали, используя набор PRIS M (ABI, Inc.) и секвенатор ABI модели # 373, в соответствии с указаниями для пользователей. Показано, что данный изолированный ген содержал 5 нуклеотидных замен, в отличие от опубликованной
последовательности прототипа (Kirnbauer R. et al. (1993), как указано выше), приводящих к одной аминокислотной замене: Ser-269 на Pro. Анализ последовательности генома клона pUC18-HPV16 L1/L2 подтвердил, что эта замена присутствует также в исходной ДНК-матрице, а не интродуцирована ПЦР.
Пример
33
А. Экспрессия L1 HPV16 и коэкспрессия L1 и L2 HPV16 в дрожжах.
Плазмиды р14049-37-1 (pCl/1-GAL+НPV16 L1) и р14049-42-2 (pCl/1- GAL+HPV16 L1 и L2) использовали для трансформации штамма # 1558 S. cerevisiae. Получили рекомбинантные штаммы # 1678 (HPV16 - L1) и # 1679 (HPV16 L1+L2), как показано в таблице. Клоновые изоляты выращивали при 30oС в YEHD - среде, содержащей 2% галактозы, в течение 68-78 часов. После сбора клеток осадки клеток разрушали со стеклянными шариками и клеточные лизаты анализировали на экспрессию белков L1 HPV16 или L2 HPV16 иммуноблотным анализом. Образцы, содержащие 40 мкг суммарного клеточного белка подвергали электрофорезу в 10% трис-глициновом геле в восстанавливающих и денатутрирующих условиях и электроблоттировали в нитроцеллюлозные фильтры. Белок L1 HPV16 иммунодетектировали, используя кроличью поликлональную антисыворотку, полученную против слитого белка L1 trpE-HPV11 (D. Brown et al., Virology, 201:46-54), как первичные антитела и HRP-связанные антикроличьи антитела IqG осла (Amersham, Inc.) как вторичные антитела. Фильтры обрабатывали, используя хемилюминисцентный набор для детекции ЕСL (Amersham, Inc.). Белковая полоса L1, соответствующая молекулярной массе 50-55 кД, была обнаружена во всех образцах, за исключением отрицательного контроля (рС1/1 без генов L1 или L2). Белок L2 обнаружили с помощью иммуноблота как белковую полосу, соответствующую молекулярной массе 70 кД иммуноблота, используя мышиную анти-HPV16 L2 сыворотку, полученную против слитого белка trpE-L2, экспрессированного в Е. coli как первичные антитела. HRP-связанные антимышиные IqG козла (Amersham, Inc. ) использовали, как вторичные антитела, а фильтры обрабатывали, как описано выше.
В. Выделение очисткой рекомбинантных капсидных белков L1+L2 НРV типа 16.
Все операции выполняли при 4oС, за исключением оговоренных особо. Клетки хранили замороженными при -70oС. Замороженные клетки (влажный вес 27,6 г) оттаивали при 20-23oС и ресуспендировали в 40 мл "L-буфера" (20 мМ натрийфосфата, рН 7,2, 100 мМ NaCl, 1,7 мМ ЭДТА). Ингибиторы протеаз PMSF и пепстатин А добавляли до конечной концентрации соответственно 2 мМ и 1,7 мМ. Кашицу из клеток разрушали под давлением приблизительно 8000 фунтов на квадратный дюйм 3-кратным пропусканием через микрофлуидизатор (Microfluidics Corp., Newton, MA). Разрушенную клеточную кашицу центрифугировали при 5000 g в течение 10 минут для удаления клеточных оболочек. Получали надосадочную жидкость, содержащую антиген L1+L2.
Надосадочную жидкость разбавляли 1:5 добавлением буфера А (20 мМ MOPS, рН 7,0) и нагружали на анионообменную колонку (5,0 см внутреннего диаметра х 4,8 см) со смолой Fractogel EMD ТМАЕ-650 (S) (ЕМ Separations, Gibbstown, NJ), уравновешенную в буфере А. После промывки буфером А антиген элюировали 0-1,0 М NаС1 градиентом в буфере А. Иммунно-дот-блоттинг проводили для выявления фракций, элюируемых с колонки, содержащих белок L1.
Во фракциях, содержащих белок L1, определенный с помощью иммуноблотинга, методом Bradford определяли общий белок с последующим SDS/ПАГ-электрофорезом, используя окрашивание серебром и Вестерн-блоттинг.
Фракции ТМАЕ, обнаруживающие обогащенность белком L1 сопоставимой чистоты, объединяли. Антиген концентрировали сульфатом аммония по фракциям. Образцы доводили до 48% насыщения сульфатом аммония, добавляя сухой реагент при осторожном перемешивании каждые 30 минут. Образцы помещали на лед и оставляли на ночь для осаждения. Образцы центрифугировали при 12000 g. Осадок ресуспендировали в 20,0 мл PBS (6,25 мМ Na-фосфат, рН 7,2, 150 мМ NaCl).
Ресуспендированные осадки хроматографировали раздельно на эксклюзионной колонке (2,6 см внутреннего диаметра х 89 см) со смолой Сефакрил 500 НR (Pharmacia, Piscataway, NJ) при 20-23oС. Проточный буфер был PBS. Фракции анализировали с помощью SDS/ПАГ-электрофореза, используя окрашивание серебром и Вестерн-блот-детекцию. Самые очищенные фракции белка объединяли и концентрировали в 50 мл ячейке с перемешиванием, при давлении N2 4-6 фунтов на квадратный дюйм, используя 43 мм пластинчатые многослойные мембраны YM (Amicon, Beverly, МA).
Итоговый продукт анализировали SDS/ПАГ-электрофорезом, применяя окрашивание коллоидальным Кумасси. Белки L1 и L2 имели 70% гомогенности. Идентичность белков L1 и L2 подтвердили Вестерн-блоттингом, используя соответствующую антисыворотку. Конечный продукт фасовали и хранили при -70oС. В результате такой обработки получали 0,53 мг белка.
Электронно-микроскопический анализ выполняли с помощью Structure Probe (West Chester, PA). Аликвот образца помещали на покрытую медью угольную сетку, 200 меш. Каплю 2% фосфорновольфрамовой кислоты, рН 7,0, помещали на сетку на 20 секунд. Перед ТЕМ-анализом сетку высушивали на воздухе. Микроскопирование осуществляли, используя трансмиссионный электронный микроскоп JEOL 100 СХ (JEOL USA, Inc.) при быстром увеличении напряжения до 100 кВ. Микрофотографии получали при увеличении 100000х. Было подтверждено наличие вирусоподобных частиц в 50-55 нм размерном ряду.
С. Выделение очисткой рекомбинантных капсидных белков L1+L2 НРV типа 16.
Все операции выполняли при 4oС, за исключением оговоренных особо.
Клетки хранили замороженными при -70oС. Замороженные клетки (влажный вес 92,8 г) оттаивали при 20-23oС и ресуспендировали в 105 мл "L-буфера" (20 мМ натрийфосфат, рН 7,2, 100 мМ NаСl, 1,7 мМ ЭДТА). Ингибиторы протеаз PMSF и пепстатин А добавляли до конечной концентрации, соответственно, 2 мМ и 1,7 мкМ. Клеточную кашицу разрушали при давлении приблизительно 16000 фунтов на квадратный дюйм 3-кратным пропусканием через микрофлуидизатор М110-Y (Microfluidics Corp., Newton, MA). Полужидкую массу разрушенных клеток центрифугировали при 6100 g в течение 15 минут для удаления клеточных оболочек. Получали надосадочную жидкость, содержащую антиген L1+L2.
Надосадочную жидкость разбавляли 1:5 добавлением буфера А (20 мМ MOPS, pH 7,0) и нагружали на анионообменную колонку (5,0 см внутреннего диаметра x 4,2 см) со смолой Frасtоgеl ЕМD ТМАЕ-650 (S) (ЕМ Separations, Gibbstown, NJ), уравновешенную в буфере А. После промывки буфером А антиген элюировали 0-1,0 М градиентом NаС1 в буфере А. Проводили иммунно-дот-блоттинг для определения фракций, содержащих белок L1.
Во фракциях, содержащих белок L1, идентифицированный с помощью иммунно-дот блота, определяли общий белок по методy Bradford с последующим SDS/ПАГ-электрофорезом, применяя окрашивание серебром и Вестерн-блоттинг.
Фракции ТМАЕ, обнаруживающие обогащенность белком L1 сопоставимой чистоты, объединяли. Антиген концентрировали сульфатом аммония по фракциям. Образцы доводили до 35% насыщения сульфатом аммония добавлением сухого реагента при осторожном перемешивании каждые 10 минут. Образцы помещали на лед и оставляли для преципитации на 4 часа. Образцы центрифугировали при 12000 g. Осадок ресуспендировали в 20,0 мл PBS (6,25 мМ Na-фосфата, pH 7,2, 150 мМ NaCl), который содержал 1 мМ ЭДТА.
Ресуспендированный осадок хроматографировали раздельно на эксклюзионной колонке (2,6 см внутреннего диаметра х 89 см) со смолой Сефакрил 500 НR (Pharmacia, Piscataway, NJ). Проточный буфер был PBS+1 мМ ЭДТА. Фракции анализировали SDS/ПАГ-электрофорезом, применяя окрашивание серебром и вестерн-блот-детекцию. Самые очищенные фракции белка объединяли и концентрировали в 50 мл ячейке с перемешиванием при давлении N2 4-6 фунтов на квадратный дюйм, используя 43 мм пластинчатые многослойные мембраны YМ-100 (Amicon, Beverly, МА).
Конечный продукт анализировали с помощью SDS/ПАГ-электрофореза, применяя окрашивание коллоидальным Кумасси. Белки L1 и L2 имели почти 70% гомогенности. Подлинность белков L1 и L2 подтверждали Вестерн-блоттингом, используя соответствующую антисыворотку. Конечный продукт фасовали и хранили при -70oС. В результате такой обработки получали 3,8 мг суммарного белка.
Пример 34
А. Ферментация штамма 1679
(L1+L2 HPV типа 16).
Операции по приготовлению замороженных маточных культур, созданию инокулюма, оживлению клеток штамма 1679 были фактически такими же, как описано выше. После 67 ч инкубации плотность клеток в расчете на сухой вес/л достигала 4,2 г и составляла 93 г влажного осадка оживленных клеток.
В. Ферментация штамма 1678 (L1 HPV типа 16).
Операции по приготовлению замороженных маточных культур, созданию инокулюма, оживлению клеток штамма 1678 были фактически такими же, как описано выше. После 70,5 ч инкубации содержимое двух 10 л ферментаций объединили (плотность клеток составила 8,7 г в расчете на сухой вес/л) и получили в целом 258 г влажного осадка оживленных клеток.
С. Выделение очисткой рекомбинантного капсидного белка L1 HPV типа 16.
Все операции выполняли при 4oС, за исключением оговоренных особо.
Клетки штамма #1678 хранили замороженными при -70oС. Замороженные клетки (влажный вес 128 г) оттаивали при 20-23oС и ресуспендировали в 140 мл "модифицированного L1-буфера" (20 мМ натрийфосфат, рН 7,2, 100 мМ NaCl). Ингибиторы протеаз PMSF и пепстатин А добавляли до конечной концентрации соответственно 2 мМ и 1,7 мкМ. Клетки в виде полужидкой массы разрушали при давлении приблизительно 16000 фунтов на квадратный дюйм 3-кратным пропусканием через микрофлуидизатор М110-Y (Microfluidics Corp., MA). Кашицу из разрушенных клеток центрифугировали при 11000 g в течение 40 минут для удаления клеточных оболочек. Получали надосадочную жидкость, содержащую антиген L1.
Надосадочную жидкость разбавляли 1:5 добавлением буфера А (20 мМ MOPS, рН 7,0) и нагружали на анионообменную колонку (5,0 см внутреннего диаметра x 6,4 см) со смолой Fractogel ЕМD ТМАЕ-650 (S) (ЕМ Separations, Gibbstown, NJ), уравновешенную в буфере А. После промывки буфером А антиген элюировали 0-1,0 М градиентом NaCl в буфере А. Проводили иммуно-дот-блоттинг для определения фракций, элюированных с колонки, которые содержали белок Ll.
Во фракциях, содержащих белок L1, идентифицированный иммуно-дот-блотом, определяли общий белок по методу Bradford с последующим SDS/ПАГ-электрофорезом, применяя окрашивание серебром и Вестерн-блоттинг.
Фракции ТМАЕ, обнаруживающие обогащенность белком сопоставимой чистоты, объединяли. Антиген концентрировали сульфатом аммония по фракциям. Образцы доводили до 35% насыщения сульфатом аммония, добавляя сухой реагент при осторожном перемешивании каждые 10 мин. Образцы помещали на лед и оставляли для преципитации на 5 часов. Образцы центрифугировали при 12000 g. Осадок ресуспендировали в 20,0 мл PBS (6,25 мМ Na-фосфата, рН 7,2, 150 мМ NaCl).
Ресуспендированный осадок хроматографировали раздельно на эксклюзионной колонке (2,6 см внутреннего диаметра x 89 см) со смолой Сефакрил 500 HR (Pharmaсia, Piscataway, NJ). Проточный буфер был PBS. Фракции анализировали SDS/ПАГ-электрофорезом, применяя окрашивание серебром и Вестерн-блот-детекцию. Самые чистые фракции белка объединяли и концентрировали в 50 мл ячейке с перемешиванием при давлении N2 4-6 фунтов на квадратный дюйм, используя 43 мм пластинчатые многослойные мембраны YM-100 (Amicon, Beverly, МA).
Конечный продукт анализировали SDS/ПАГ-электрофорезом, применяя окрашивание коллоидальным Кумасси. Белок L1 имел почти 70% гомогенности. Подлинность белка L1 подтверждали Вестерн-блоттингом с использованием соответствующей антисыворотки. Конечный продукт фасовали и хранили при -70oС. При такой обработке получали 7,4 мг суммарного белка.
D. Аналитические методы.
Метод
получения-дот-блота
Образцы разбавляли (при необходимости) 1:10 в деионизованной воде Milli-Q и 10 мкл образца капали на PVDF-мембраны PolyScreen (NEN Research Products, Boston, MA). После
высушивания пятен мембраны отмывали водой и высушивали. Готовили раствор первичных антител, разбавляя соответствующую антисыворотку в блоттинговом буфере (5% обезжиренного молока в 6,25 мМ
Na-фосфатном буфере, рН 7,2, 150 мМ NаСl, 0,02 NaN3). Инкубацию осуществляли не менее 1 часа при 20-23oС. Блот отмывали по 1 мин в каждой из трех смен РBS (6,25 мМ Na-фосфатном
буфере, рН 7,2, 150 мМ NaCl). Раствор вторичных антител готовили разбавлением соответствующего конъюгата щелочная фосфатаза-антисыворотка в буфере для переноса. Инкубацию осуществляли при тех же
условиях не менее 1 часа. Блоты отмывали, как указано выше, и детектировали, используя 1 порцию NBT/BCIP субстрата (Pierce, Rockford, IL).
Для детекции использовали следующие антитела.
Белок L1 HPV типа 6а детектировали с помощью МАВ 837 (Chemicon International, Inс., Temecula, CA). Белок L2 HPV типа 6а детектировали объединенной из #641 и 647 антисывороткой слитого белка L2-trpE HPV типа 6а. Белок L1 НРV типа 16 детектировали с помощью МАВ 885 (Chemicon International, Inc., Temecula, CA). Белок L2 HPV типа 16 детектировали мышиной объединенной из #611 антисывороткой слитого белка L2-trрЕ HPV типа 16.
Определение общего белка по Bradford
Общий белок определяли, используя коммерческий набор Кумасси плюс
(Pierce, Rockford, IL). Образцы разбавляли деионизованной водой Milli-Q до требуемого разведения. Использовали 0,1 мл и 1,0 мл объемы соответственно для стандартного и микроопределения. В обоих
случаях использовали BSA (Рiеrсe, Rockford, IL) для получения стандартной кривой. Анализ осуществляли в соответствии с рекомендациями для пользователя. Стандартную кривую строили, используя
программное обеспечение CriketGraph на компьютере Macintosh IIci.
Пример 35
Выделение очисткой рекомбинантного капсидного белка L1 HРV типа 11
Все операции выполняли
при 4oС, за исключением оговоренных особо.
Клетки хранили замороженными при -70oС. Замороженные клетки (влажный вес 180 г) оттаивали при 20-23oС и ресуспендировали в 900 мл "лизирующего буфера" (50 мМ MOPS, рН 7,2, 500 мл NaCl, 1 мМ CaCl2). Ингибиторы протеаз AEBSF и пепстатин А добавляли до конечной концентрации соответственно 1 мМ и 1,7 мкМ. Кашицу из клеток разрушали при давлении приблизительно 16000 фунтов на квадратный дюйм 4-кратным пропусканием через микрофлуидизатор М110-У (Microfludics Corp., Newton, MА). Добавляли необходимый объем 10% детергента тритон Х100 (Pierce, Rockford, IL) для разрушения полужидкой массы клеток, доводя концентрацию ТХ100 до 0,5%. Кашицу перемешивали в течение 20 часов. Обработанный тритоном Х100-лизат центрифугировали при 12000 g в течение 40 мин для удаления клеточных оболочек. Получали надосадочную жидкость, содержащую белок L1.
Надосадочную жидкость диафильтровали против пяти объемов 20 мМ натрийфосфата, рН 7,2, 0,5 М NaCl, используя тангенциальную проточную мембранную кассету 300К (Filtron, Northborough, MА). Материал задержанный мембраной проявлял радиоиммуночувствительность, а Вестерн-блоттинг показывал наличие белка L1.
Удержанный мембраной материал нагружали на высокоразрешающую афинную колонку (11 см внутреннего диаметра x 5,3 см) со смолой Spherodex (M) (1BF, Villeneuve-la-Garenne, France), уравновешенную в 20 мМ натрийфосфате, рН 7,2, 0,5 М NaCl. После промывания уравновешивающим буфером и однократной промывкой 20 мМ натрийфосфатом, рН 7,2, 1,0 М NaCl, белок L1 элюировали пропусканием одной порции 20 мМ натрийфосфата, рН 7,2, 2,5 М NаС1. Фракции собирали во время промывки и элюирования. Колоночные фракции анализировали на общий белок по методу Вradford. Затем фракции анализировали Вестерн-блоттингом и SDS/ПАГ-электрофорезом с детекцией коллоидальным Кумасси. Фракции подвергали также радиоиммунному анализу.
SР Spherоdex-фракции, обнаруживающие сопоставимую чистоту и обогащенность белком L1, объединяли.
Конечный продукт анализировали Вестерн-блоттингом и SDS/ПАГ-электрофорезом с детекцией коллоидальным Кумасси. Денситометрия гелей, окрашенных Кумасси, показала почти 90% гомогенность белка L1. Подлинность белка L1 подтвердили Вестерн-блоттингом. Конечный продукт стерилизовали фильтрованием через мембрану с размером пор 0,22 мкм и хранили при 4oС. При такой обработке получали 202 мг суммарного белка.
Электронно-микроскопический анализ осуществляли с помощью Structure Probe (West Chrster, РA).
Аликвот образца помещали на покрытую медью угольную сетку, 200 меш. Каплю 2% фосфорновольфрамовой кислоты, рН 7,0, помещали на сетку на 20 секунд. Перед проведением ТЕМ-анализа сетку высушивали на воздухе. Микроскопию осуществляли, используя трансмиссионный электронный микроскоп Jeol 100 СХ (Jeol USA, Inc.) при быстром увеличении напряжения до 100 кВ. Полученные микрофотографии имели увеличение 100000х.
SDS/ПАГ-электрофорез и Вестерн-блот-анализ
Все гели, буферы и электрофоретические аппараты получены от NOVEX (San Diеgо, Сa), а разгонки осуществляли в соответствии с рекомендациями для пользователя. Вкратце, образцы разбавляли до
одинаковой концентрации белка деионизованной водой Milli-Q и смешивали 1:1 с инкубационным буфером, содержащем 200 мМ DТТ. Образцы инкубировали 15 мин при 100oС и загружали в лунки 12%
трис-глицинового геля. Образцы подвергали электрофорезу при 125 В в течение 1 ч 45 мин. Гели окрашивали коллоидальным Кумасси, используя коммерческий набор (Integrated Separation Systems, Natick,
MA).
Для получения Вестерн-блотов белки переносили на РVDF-мембраны при 25 В в течение 40 мин. Мембраны ополаскивали деионизованной водой Milli-Q и высушивали на воздухе. Первичные антитела были поликлональной кроличьей антисывороткой, полученной против слитого белка L1-trpE НPV типа 11 (дар Dr. D. Brown). Раствор антител готовили разбавлением антисыворотки буфером для переноса (5% обезжиренное молоко в 6,25 мМ Na-фосфате, рН 7,2, 150 мМ NаС1, 0,02% NaN3). Инкубацию осуществляли не менее 1 часа при 20-23oС. Блот отмывали по 1 мин, трижды меняя PBS (6,25 мМ Na-фосфат, рН 7,2, 150 мМ NаСl). Раствор вторичных антител готовили в буфере для переноса разбавлением антисыворотки козла, конъюгированной со связанными с щелочной фосфатазой антикроличьими IgG, в буфере для переноса. Инкубацию осуществляли в тех же условиях не менее 1 часа. Блоты отмывали, как указано выше, и детектировали, используя 1 порцию NBT/BCIP-субстрата (Pierce, Rockford, IL).
Пример 36
Экспрессия L1 HPV типа 18 и L2 HPV типа 18 в дрожжах
Плазмиды р191-6 (pGAL1-10+HPV 18 L1) и р195-11 (pGAL1-10+HPV18 L1+L2) использовали для трансформации штамма
#1558 (МАТа, leu 2-04, prb::HIS3, mnn: : URA3, adel) S. cerevisiae. Клоновые изоляты выращивали при 30oС в YЕND-среде, содержащей 2% галактозы в течение 88 часов. После сбора клеток осадки
клеток разрушали со стеклянными шариками и все клеточные лизаты анализировали на экспрессию L1 HPV типа 18 и/или L2 НРV типа 18 с помощью иммунноблот-анализа. Образцы, содержащие 25 мкг суммарного
клеточного белка подвергали электрофорезу в 10% трис-глициновом геле (Novex, Inc.) в денатурирующих условиях и электроблоттировали в нитроцеллюлозные фильтры. Белок L1 иммунодетектировали, используя
кроличью антисыворотку, полученную против слитого белка L1 trpE-НРV типа 11, в качестве первичных антител (Brown et al., Virology 201:46-54) и связанные с пероксидазой хрена (НRР) антикроличьи IgG
осла (Аmеrsham, Inc.) в качестве вторичных антител. Фильтры обрабатывали, используя хемилюминесцентный набор для детекции ECL ТМ (Amersham, Inc. ). Белковую полосу L1, соответствующую молекулярной
массе 50-55 кД, обнаруживали в дрожжевых клонах коэкспрессирующих L1 и L1+L2 (соответственно штаммы 1725 и 1727) и не обнаруживали в отрицательном контроле (pGAL1-10 без генов L1 или L2).
Белок L2 HРV типа 18 детектировали Вестен-анализом, используя поликлональную антисыворотку козла, полученную против слитого белка L2-trpE HPV типа 18 в качестве первичных антител, а затем НРP-конъюгированные кроличьи антикозлиные IgG (Kirkegaard and Perry Laboratories, Gaithersburg, MD). Фильтры обрабатывали, как описано выше. Белок L2 обнаружили в виде белковой полосы, соответствующей молекулярной массе 75 кД в дрожжевом клоне коэкспрессирующего L1+L2 (штамм 1727) и не обнаружили ни в отрицательном контроле, ни в клоне экспрессирующем L1.
Пример 37
Ферментация HРV 18 L1 (штамм 1725) и 18 L1+L2 (штамм 1727)
Поверхностно растущую чашечную культуру штаммов 1725 и 1727 асептически переносили в безлейциновую жидкую среду, содержащую (на 1
л): 8,5 г дрожжевого азотного основания без аминокислот и сульфата аммония, 0,2 г аденина, 0,2 г урацила, 10 г янтарной кислоты, 5 г сульфата аммония, 40 г глюкозы, 0,25 г L-тирозина, 0,1 г L
-аргинина, 0,3 г L-изолейцина, 0,05 г L-метионина, 0,2 г L-триптофана, 0,05 г L-гистидина, 0,2 г лизина, 0,3 г L-фенилаланина, перед стерилизацией рН этой среды доводили NаОН до 5,0-5,3. После
выращивания при 28oC, 250 об/мин на качалке с круговым вращением культуру замораживали в ампулах, предварительно добавляя глицерин до конечной концентрации 17% (в/о) (1 мл на замораживаемую
ампулу) и замораживали при -70oС. Инокулирование осуществляли в ту же среду (500 мл на 2 л колбу), перенося оттаянное содержимое замороженной ампулы и инкубируя при 28oС, 250
об/мин на качалке с круговым вращением в течение 29 ч. Ферментацию каждого штама осуществляли на ферментере Brunswick SF-116 с рабочим объемом 10 л, включая инокулят. Питательная среда содержала (на 1
л): 20 г дрожжевого экстракта Dofco, 10 г соевого пептона Sheffield, 20 г глюкозы, 20 г галактозы, 0,3 мл пеногасителя UCON LB-625 фирмы UNION CARBID, перед стерилизацией рН среды доводили до 5,3.
Содержимое (500 мл) 2 л инокулированной колбы полностью переносили в ферментер и инкубировали при 28oС, 5 л воздуха в мин, 400 об/мин, давлении 3,5 фунтов на квадратный дюйм. Скорость
перемешивания увеличивали по необходимости для поддержания уровня растворенного кислорода, более 40% насыщения. Течение ферментации отслеживали с помощью автономного измерения глюкозы (анализатор
глюкозы 2, Beckman) и оперативной масс-спектрометрии (Perkin-Elmer 1200). После инкубации в течение 66 ч плотность клеток достигала значения 9,5-9,7 г веса сухих клеток на 1 л среды. Культуры
концентрировали фильтрацией через полые волокна (патрон Amicon H5MP01-43 в фильтрационной системе Amicon DC-10) до объема 2 л, диафильтровали с 2 л физиологического раствора, забуференного фосфатом, и
продолжали концентрировать (до объема 1 л) перед разливкой в 500 мл центрифужные склянки. Осадки клеток собирали центрифугированием при 8000 об/мин (ротор GS-3 Sorval в течение 20 мин при 4o
С. После декантирования супернатанта осадок (итого 191-208 г влажных клеток) хранили при -70oС до использования.
Пример 38
Выделение очисткой рекомбинантного
капсидного белка L1 HPV типа 18
За исключением особо оговоренных, все операции выполняли при 4oС.
Клетки хранили замороженными при -70oС. Замороженные клетки (влажный вес 126 г) оттаивали при 20-23oС и ресуспендировали в 70 мл "лизирующего буфера" (20 мМ натрийфосфата, рН 7,2, 100 мМ NaCl). Ингибиторы протеаз PMSF и пепстатин А добавляли до конечной концентрации соответственно 2 мМ и 1,7 мкМ. Клетки в виде кашицы разрушали под давлением приблизительно 16000 фунтов на квадратный дюйм 4-кратным пропусканием через микрофлуидизатор М110-У (Microfluidics Corp., Newton, МА). Кашицу разрушенных клеток центрифугировали при 12000 g в течение 40 мин для удаления клеточных оболочек. Получали надосадочнутю жидкость, содержащую антиген L1.
Надосадочную жидкость разбавляли 1:5 с помощью буфера А (20 мМ МОРS, рН 7,0) и нагружали на анионообменную колонку (9 см внутреннего диаметра х 3,9 см) со смолой Fractogel ЕМD ТМАЕ-650 (М) (ЕМ Separations, Gibbstown, NJ) уравновешенную в буфере А. После промывки буфером А антиген элюировали 0-1,0 М градиентом NаСl в буфере А. Колоночные фракции анализировали на общий белок методом Badford. Уравненные по общему белку фракции анализировали Вестерн-блоттингом и SDS/ПАГ-электрофорезом с детекцией окрашиванием серебром.
ТМАЕ-фракции, обнаруживающие сопоставимую чистоту и обогащенность белком L1, объединяли. Антиген концентрировали сульфатом аммония по фракциям. Раствор доводили до 35% насыщения сульфатом аммония, добавляя сухой реагент при осторожном перемешивании каждые 10 мин. Образцы помещали на лед и оставляли для преципитации в течение 4 часов. Образцы центрифугировали при 16000 g в течение 45 мин. Осадок ресуспендировали в 20 мл PBS (6,25 мМ Nа-фосфат, рН 7,2, 150 мМ NaCl).
Ресуспендированный осадок хроматографировали в эксклюзионной колонке (2,6 см внутреннего диаметра x 89 см) со смолой Сефакрил 500 НR (Pharmacia, Piscataway, NJ). Проточный буфер был PBS. Фракции анализировали Вестерн-блоттингом и SDS/ПАГ-электрофорезом с детекцией окрашиванием серебром. Наиболее чистые фракции объединяли и концентрировали под давлением N2 4-6 фунтов на квадратный дюйм в 50 мл ячейке с перемешиванием, используя 43 мм пластинчатую многослойную мембрану УМ-100 (Amicon., Beverly, MA).
Конечный продукт анализировали Вестерн-блоттингом и SDS/ПАГ-электрофорезом с детекцией коллоидальным Кумасси. Белок L1 имел почти 50-60% гомогенности. Подлинность белка L1 подтвердили Вестерн-блоттингом. Конечный продукт фасовали и хранили при -70oС. При такой обработке получали 12,5 мг суммарного белка.
Пример 39
Создание иммуногенных веществ
Очищенные
VLP получали согласно известным способам, таким как смешивание фармацевтически приемлемых носителей, стабилизаторов или адъюванта для вакцины. Иммуногенные VLP настоящего изобретения могут быть
изготовлены для создания вакцины путем комбинирования физиологически переносимых веществ таких, как, например, PBS, физиологический раствор или дистиллированная вода. Иммуногенные VLP назначали к
применению при дозировке от 0,1 до 100 мкг, предпочтительно от 1 до 20 мкг, с целью получения желаемого иммуногенного эффекта. Количество VLP по технологии приготовления лекарственного средства можно
варьировать в зависимости от разных факторов, включающих в себя, но не ограничивающих, состояние индивидуума, вес, возраст и пол. Введение технологического препарата VLP можно осуществлять разными
способами, включающими в себя, но не ограничиваемые ими, через рот, подкожно, местно, через слизистую и внутримышечно. Такие технологические VLP могут включать единственный тип VLP (т.е. VLP из HPV
типа 6a, HPV типа 11, HPV типа 16 и HPV типа 18).
Противомикробный консервант, например тимеросал, может присутствовать факультативно. Иммуногенные антигены настоящегоизобретения можно использовать по желанию в комбинации со стабилизаторами вакцины и адъювантами вакцины. Типичными стабилизаторами являются специфические вещества, например полианионы, такие как гепарин, гексасульфат инозита, сульфатированный бета-циклодекстрин, менее специфичные наполнители, например аминокислоты, сорбит, маннит, ксилит, глицерин, сахароза, декстроза, трегалоза, и изменения характеристик раствора, например нейтральный рН, высокая ионная сила (0,5-2,0 М соли), двухвалентные катионы (Са2+, Mg2+). Примерами адъювантов являются Аl(ОН)3 и Al(РO4). Вакцину настоящего изобретения можно хранить в замороженной или лиофилизированной форме.
Пример 40
Получение антител к VLP
Выделенные очисткой
VLP использовали для производства антител. Термин "антитело", используемый здесь, подразумевает поликлональные и моноклональные антитела, также как и их фрагменты такие, как Fv-, Fab- и
G(ab)2-фрагменты, которые обладают способностью связывать антиген или гаптен. Антитела использовали для разных целей, включающие, но не ограниченные ими, для выделения рекомбинантных VLP, выделения
нативных белков L1 и L2 и создания тары. Тара должна быть представлена изолированным несущим элементом, пригодным для удержания в ограниченном пространстве, как минимум, одного контейнера. Далее,
несущий элемент должен содержать реагенты, такие как антитела анти-VLP или соответствующие VLР для обнаружения НРV или фрагментов HPV или антитела к HPV. Несущий элемент должен также содержать
средства для обнаружения таких веществ, как меченые антигены или ферментные субстраты и т. п. Антитела или VLP или наборы полезны для разнообразных целей, включающие, но не ограниченные ими, для
судебного анализа или эпидемиологических исследований.
Реферат
Изобретение относится к биотехнологии, а именно к способам получения вирусоподобных частиц (VLP) папилломавируса человека, используемых для создания вакцин. Способ включает трансформацию дрожжевой клетки молекулой ДНК, кодирующей L1 HPV или L1+L2 HPV, культивирование трансформированной клетки, сбор VLP из трансформированной клетки и очистку VLP с помощью стадии хроматографии. 2 з.п.ф-лы, 14 ил.

Комментарии