Фармацевтические композиции - RU2159107C2
Код документа: RU2159107C2
Чертежи

Описание
Настоящее изобретение относится к пероральным композициям в виде твердой дисперсии, содержащим макролиды, например рапамицин или аскомицин.
Рапамицин является иммунодепрессивным лактамным макролидом, продуцируемым, например, Streptomyces hygroscopicus. Структура рапамицина известна (Kesseler Н. , et аl., 1993; Helv. Chim. Acta; 76, 117). Рапамицин является чрезвычайно сильнодействующим иммунодепрессантом, а также обладает противоопухолевой и противогрибковой активностью. Его применимость в качестве фармацевтического препарата, однако, ограничена из-за его очень низкой и неустойчивой биологической доступности. Более того, рапамицин почти нерастворим в водной среде, например воде, что затрудняет образование стабильных галеновых композиций. Известны многочисленные производные рапамицина. Конкретные 16-О-замещенные рапамицины раскрыты в WO 94/02136, содержание которой включено в данные материалы в качестве ссылки.
40-O-замещенные рапамицины описаны, например, в US 5258389 и WO 94/09010 (О-арил и О-алкил рапамицины); WO 92/05179 (сложные эфиры карбоновой кислоты); US 5118677 (сложные эфиры амида); US 5118678 (карбаматы); US 5100883 (фторированные сложные эфиры); US 5151413 (ацетали); US 5120842 (силиловые сложные эфиры); WO 93/11130 (метиленрапамицин и производные); WO 94/02136 (метокси производные); WO 94/02385 и WO 95/14023 (алкенил производные), каждый из которых включен в данные материалы в качестве ссылки, 32-O-дигидро или замещенный рапамицин описаны, например, в US 5256790, введенном в данные материалы в качестве ссылки.
Далее, производные рапамицина описаны в заявке PCT N ЕР 96/02441, например, 32-дезоксирапамицин, как описано в примере 1, и 16-пент- 2-инолокси-32(S)-дигидрорапамицин, как описано в примерах 2 и 3. Содержание заявки PCT N ЕР 96/02441 включено в данные материалы в качестве ссылки.
Рапамицин и его структурные аналоги и производные все наименованы здесь как "рапамицины".
ЕР 240773 раскрывает твердую дисперсную композицию, содержащую вещество FR- 900506 и водорастворимый полимер.
При пероральном введении людям твердые рапамицины, например рапамицин, могут не абсорбироваться до какой-либо значительной степени в кровоток. Известны простые смеси для рапамицинов, например рапамицин с традиционными фармацевтическими эксципиентами; однако недостатки, связанные с этими композициями, включают в себя непредсказуемые скорости растворения, нестабильные показатели биологической доступности и нестабильность. До настоящего времени не существует подходящих для перорального введения твердых препаратов, приемлемых для рапамицина и его производных.
Задача настоящего изобретения заключается в устранении вышеуказанных недостатков.
Поставленная задача достигается разработкой фармацевтической композиции для орального введения в форме твердой дисперсии, содержащей рапамицин и среду-носитель.
Композиции по изобретению обеспечивают высокую биологическую доступность лекарственного препарата, удобны для введения и стабильны.
Рапамицин, используемый в композициях по изобретению, может быть любым рапамицином или его производным, например, как описано выше или в вышеупомянутых патентных заявках.
Таким образом, рапамицин, используемый в твердых дисперсных композициях по изобретению, может быть рапамицином или О-замещенным производным, в котором гидроксильная группа в циклогексильном кольце рапамицина замещена на -OR1, где R1 является гидроксиалкилом, гидроксиалкоксиалкилом, ациламиноалкилом и аминоалкилом; например, как описано в WO 94/09010, например, 40-O-(2-гидрокси)этилрапамицин, 40-O-(3- гидрокси)пропилрапамицин, 40-O-[2-(2-гидрокси)этокси] этилрапамицин и 40-O-(2-ацетаминоэтил)рапамицин. Производное рапамицина может быть 26- или 28-замещенным производным.
Предпочтительные рапамицины для использования в твердых дисперсных композициях по изобретению включают в себя рапамицин, 40-O-(2-гидрокси)этилрапамицин, 32-дезоксирапамицин и 16-пент-2-инилокси-32(S)-дигидрорапамицин. Более предпочтительным рапамицином является 40-О-(2-гидрокси)этилрапамицин (далее называемый соединение X).
Многочисленные производные рапамицина, которые используются в композициях по настоящему изобретению, относятся к структуре, описанной как Формула А на странице 4 опубликованной заявки WO 96/13273, содержание которое включено в данные материалы в качестве ссылки.
Термин твердая дисперсия, который используется здесь, следует понимать в значении со-осадок рапамицина, например 40-O-(2-гидрокси)этилрапамицина или рапамицина со средой-носителем. В твердой дисперсии рапамицин находится в аморфной или практически аморфной форме и физически связан со средой-носителем.
Композиции по изобретению могут вводиться в любой удобной форме, например в таблетках, капсулах, в форме гранул или порошка, например, в пакетиках.
Рапамицин может присутствовать в композиции в количестве от примерно 0,01 до примерно 30 мас.% в расчете на массу композиции ( мас.% /мас.), и предпочтительно в количестве от 1 до 20 мас.%/мас. в расчете на общую массу композиции.
Среда-носитель присутствует в количестве вплоть до 99,99 мас.%, например от 10 до 95 мас.% в расчете на общую массу композиции.
Среда-носитель может содержать водорастворимый полимер или циклодекстрин.
В одном из вариантов согласно настоящему изобретению среда-носитель может содержать водорастворимый полимер, предпочтительно производное целлюлозы, такое как гидроксипропилметилцеллюлозу (HPMC), фталат гидроксипропилметилцеллюлозы или поливинилпирролидон (PVP). Хорошие результаты могут быть получены с использованием HPCM с низкой кажущейся динамической вязкостью, например ниже 100 сП при 20oC для 2 мас.% водного раствора, например ниже 50 сП, предпочтительно ниже 20 сП, например HPCM 3 сП. HPCM хорошо известен и описан, например, в Handbook of Pharmaceutical Excipients, Second Edition, pub. Pharmaceutical Society of Great Britain and American Pharmaceutical Association, 1994, pp.229-232, содержание которой включено здесь в качестве ссылки. HPМС, включая HPМС 3 сП, коммерчески доступен под товарным наименованием Pharmacoat 603 от компании Shinetsu.
Предпочтительно водорастворимый полимер представляет собой гидроксипропилметилцеллюлозу в количестве до примерно 95 мас.%.
Предпочтительно также соотношение рапамицина и полимера составляет менее чем 1:4.
PVP доступен, например, под наименованием Povidone (Handbook of Pharmaceutical Excipients), и предпочтителен PVP, имеющий среднюю молекулярную массу между примерно 8000 и примерно 50000 Да.
В другом варианте среда-носитель содержит:
- гидроксипропилцеллюлозу (HPC) или ее производное. Примеры производных HPC включают в себя те из них, которые имеют низкую
динамическую вязкость в водной среде, например в воде, например, ниже примерно 400 сП, например ниже 150 сП при измерении в 2%-ном водном растворе при 25oC. Предпочтительные производные HPC
имеют низкую степень замещения и среднюю молекулярную массу ниже примерно 200000 Да, например между 50000 и 150000 Да. Примеры коммерчески доступных HPC включают в себя Klucel LF, Klucel EF и Klucel
JF от компании Aqualon; и Nisso HPC-L, поставляемый Nippon Soda Ltd.;
- полиэтиленгликоль (PEG). Примеры включают в себя PEG, имеющие среднюю молекулярную массу между 1000 и 9000 Да, например
между примерно 1800 и 7000, например PEG 2000, PEG 4000 или PEG 6000 (Handbook of Pharmaceutical Excipients);
- насыщенный полигликолизированный глицерид, доступный, например, под торговой
маркой Gelucir, например Gelucir 44/14, 53/10, 50/13, 42/12 или 35/10 от компании Gattefosse; или/,
- циклодекстрин, например β -циклодекстрин или α-циклодескстрин.
Примеры подходящих β -циклодекстринов включают в себя метил- β -циклодекстрин; диметил- β -циклодекстрин; гидроксипропил- β -циклодекстрин; гликозил- β -циклодекстрин; мальтозил- β -циклодекстрин; сульфо- β -циклодекстрин, сульфоалкиловые сложные эфиры β -циклодекстрина, например, сложные сульфо-C1-4-алкиловые эфиры. Примеры α -циклодекстринов включают в себя гликозил- α -циклодекстрин и мальтозил- α -циклодекстрин.
Среда-носитель может дополнительно содержать водорастворимую или водонерастворимую сахарозу или другой подходящий носитель или наполнитель, такой как лактозы или микрокристаллическая целлюлоза. Наполнитель, если он есть, обычно присутствует в количестве до примерно 30 мас.%, например от 0,5 до 20 мас. %, предпочтительно от примерно 5 до примерно 15% от массы композиции. Микрокристаллическая целлюлоза коммерчески доступна под торговым наименованием Avicel, например от FMC Corporation.
Среда-носитель может дополнительно содержать одно или более, чем одно, поверхностно-активное вещество, например неионное, ионное, анионное или
амфотерное поверхностно-активное вещество. Примеры подходящих поверхностно-активных веществ включают в себя:
- полиоксиэтилен-полиоксипропиленовые сополимеры и блок- сополимеры, известные,
например, под торговым наименованием Pluronic или Poloxamer, например, как описано в Fiedler Н.Р. "Lexikon der Hilfsstoffe fur Pharmazie, Kosmetik und angrenzende Gebiete", Editio Cantor, D-7960
Aulendorf, 3rd revised and expanded edition (1989), содержание которого включено в данные материалы в качестве ссылки. Предпочтительным полиоксиэтилен- полиоксипропилен блок-полимером является
Poloxamer 188, поставляемый компанией BASF;
- этоксилированные холестерины, известные, например, под торговым наименованием Solulan, например, Solulan C24, коммерчески доступный от компании
Amerchol;
- производные витаминов, например производные витамина E, такие как сукцинат полиэтиленгликольтокоферола (TPGS), поставляемый компанией Eastman;
- додецилсульфат натрия или
лаурилсульфат натрия;
- желчная кислота или ее соль, например холевая кислота, гликолевая кислота или соль, например холат натрия; или
- лецитин.
Если в композициях по изобретению присутствует поверхностно-активное вещество (вещества), то оно обычно присутствует в количестве до примерно 20%, например от 1 до 15 мас.%.
В композицию по изобретению может быть включен один или более, чем один, разрыхлитель. Примеры разрыхлителей включают в себя Polyplasdone (Handbook of Pharmaceutical Excipients), коммерчески доступный от компании ISP; гликолят крахмала натрия, коммерчески доступный от компании Generichem; и кросскармелозу натрия под торговой маркой Ac-di-sol от FMC Corporation. В композицию данного изобретения могут быть дополнительно включены одно или более, чем одно, смазывающее вещество, например стеарат магния или коллоидный диоксид кремния в количестве до примерно 5 мас.%, например, от 0,5 до 2 мас. %, в расчете на массу композиции.
Может быть полезным включать в композицию данного изобретения одно или более, чем одно, ароматизирующее вещество.
Согласно настоящему изобретению получены хорошие результаты с использованием композиций рапамицина, не содержащих поверхностно-активные вещества. Поэтому в данном изобретении описывается не содержащая поверхностно-активные вещества твердая дисперсная композиция, содержащая рапамицин, как это представлено в данном описании.
В композицию по изобретению могут быть включены антиоксиданты и/или стабилизаторы в количестве до примерно 1 мас.%, например между 0,05 и 0,5 мас. %. Примеры антиоксидантов включают в себя: бутилированный гидрокситолуол, DL- α -токоферол, пропилгаллат, аскобилпальмитат и фумаровую кислоту. Малоновая кислота является подходящим стабилизатором.
В одном воплощении данного изобретения композиция содержит до 30 мас.%, например от 1 до 20 мас.% 40-O-(2-гидрокси)этилрапамицина и до 95%, например от 30 до 90 мас.% HPCM.
Массовое соотношение рапамицина и среды-носителя в композициях по изобретению составляет обычно не более чем 1:3, предпочтительно менее чем 1: 4.
Описывается также способ получения композиции в форме твердой дисперсии.
Композиции по изобретению могут быть получены растворением или суспендированием рапамицина и среды-носителя в растворителе или смеси растворителей. Растворителем может быть один растворитель или смесь растворителей, и порядок растворения и суспендирования рапамицина и среды-носителя в растворителе может варьироваться. Подходящими растворителями для использования в приготовлении твердых дисперсных композиций по изобретению могут быть органические растворители, такие как спирты, например метанол, этанол или изопропанол; сложный эфир, например, этилацетат; простой эфир, например диэтиловый эфир; кетон, например ацетон; или галогенированные углеводороды, например дихлорэтан. Подходящей смесью растворителей является смесь этанол/ацетон, имеющая массовое соотношение этанола к ацетону между от примерно 1:10 до примерно 10:1, например от 1:5 до 5:1.
Обычно рапамицин и среда-носитель присутствуют в массовом соотношении к растворителю от 1:0,1 до 1:20. Растворитель может быть выпарен, а рапамицин соосажден со средой-носителем. Полученный в результате остаток может быть высушен, например, при пониженном давлении, просеян и измельчен. Измельченная дисперсия может быть объединена с другими эксципиентами и, например, спрессована в таблетку или помещена в пакетики или желатиновые капсулы.
Твердые дисперсные композиции могут быть получены плавлением среды-носителя с образованием расплава и объединением расплава с рапамицином, например, путем перемешивания, возможно в присутствии растворителя или смеси растворителей.
Альтернативно твердые дисперсии по изобретению могут быть получены путем сухого распыления, как описано, например, в Theory and Practice of Industrial Pharmacy, Lachmann et al., 1986. Суспензию, полученную, как описано выше, диспергируют через насадку в камеру при температуре, поддерживаемой, например, от 20 до 80oC. Растворитель испаряется при пропускании через насадку, а тонкодисперсные частицы собирают.
Композиции по изобретению после измельчения обычно имеют средний размер частиц менее чем примерно 0,5 мм, например менее чем примерно 350 мкм, например от примерно 100 до примерно 300 мкм.
Пероральные композиции по
изобретению пригодны для известных показаний рапамицина, например, для следующих состояний:
а) лечение и профилактика отторжения алло- или ксено-трансплантата органов и тканей, например, для
лечения реципиентов, например, сердца, легкого, совместно сердца и легкого, печени, почек, панкреатических, кожных или роговичных трансплантатов. Они также показаны для профилактики заболевания
"трансплантат против хозяина", такого как последствие трансплантации костного мозга;
б) лечение и профилактика аутоиммунных заболеваний и воспалительных состояний, в частности воспалительных
состояний с этиологией, включая аутоиммунный компонент, таких как артриты (например, ревматоидный артрит, хронический прогрессирующий артрит и деформирующий артрит) и ревматические заболевания.
Специфические аутоиммунные заболевания, для которых могут применяться соединения по изобретению, включают в себя аутоиммунные гематологические нарушения (включая, например, гемолитическую анемию,
гипопластическую анемию, эритроцитную анемию и идиопатическую тромбоцитопению), системную узловую эритему, полихондроз, склеродермию, гранулематоз Вегенера, дерматомиозит, хронический активный гепатит,
миастению, псориаз, синдром Стивена-Джонсона, идиопатическое спру, аутоиммунный неспецифический колит (включая, например, язвенный колит и болезнь Крона), эндокринную офтальмопатию, болезнь Грейвза,
саркоидоз, рассеянный склероз, первичный биллиарный цирроз, юношеский диабет (диабет меллитус тип I), увеит (передний и задний), сухой кератоконъюктивит и истинный кератоконъюктивит, внутритканевый
легочный фиброз, псориатический артрит, гломерулонефрит (с нефротическим синдромом и без него, например, включая идиопатический нефротический синдром или минимальный уровень нефропатии) и юношеский
дерматомиозит;
в) лечение и профилактика астмы;
г) лечение резистентности ко многим лекарствам (MDR). MDR, в частности, является проблемой у раковых пациентов и пациентов со СПИДом,
на которых традиционная химиотерапия не сможет повлиять, поскольку лекарства выталкиваются из клеток посредством Pgp. Поэтому композиции полезны для увеличения эффективности других
химиотерапевтических агентов при лечении и контроле состояний резистентности ко многим лекарствам, таких как резистентный ко многим лекарствам рак и резистентный ко многим лекарствам СПИД;
д)
лечение пролиферативных нарушений, например, опухолей, гиперпролиферативных кожных заболеваний и т.п.;
е) лечение грибковых инфекций;
ж) лечение и профилактика воспалений, особенно
потенциирующих активность стероидов;
з) лечение и профилактика инфекций, особенно инфекций патогенами, имеющими Mip или Mip-подобные факторы;
и) лечение передозировок FK-506 и других
макрофилин связывающих иммунодепрессантов.
Когда фармацевтическая композиция по изобретению находится в форме стандартной дозы, например, в виде таблетки, капсулы, гранулы или порошка, каждая стандартная доза будет соответственно содержать между 1 мг и 100 мг лекарственного вещества, более предпочтительно между 10 и 50 мг, например, 15, 20, 25 или 50 мг. Такая форма стандартной дозы пригодна для введения от 1 до 5 раз в сутки в зависимости от конкретного назначения терапии, стадии терапии и т.п.
Точное количество композиции, которое должно быть введено, зависит от нескольких факторов, например от желаемой продолжительности лечения и скорости высвобождения рапамицина.
Применимость фармацевтических композиций можно наблюдать по стандартным клиническим тестам, например по известным показателям доз активного агента, дающих эквивалентные уровни активного агента в крови; например, с использованием доз в интервале от 1 мг до 1000 мг, например, от 5 мг до 100 мг активного агента в день на 75 кг веса взрослого человека или на стандартных моделях животных. Возросшую биодоступность лекарственного вещества, обеспечиваемую с помощью композиций, можно наблюдать в стандартных опытах с животными и в клинических испытаниях.
Используемая дозированная форма, например таблетка, может быть покрыта оболочкой, например энтеросолюбильной оболочкой. Подходящие оболочки могут содержать фталат ацетата целлюлозы, фталат гидроксипропилметилцеллюлозы, сополимер полиметилакриловой кислоты, например, Eudragit L, S; или сукцинат гидроксипропилметилцеллюлозы.
Рапамицин, используемый в композициях по изобретению, например, 40-O-(2-гидрокси)этилрапамицин или рапамицин, может находиться в кристаллической или аморфной форме до образования твердой дисперсии. Поэтому преимуществом данного изобретения является то, что нет необходимости, чтобы рапамицин был кристаллическим. Таким образом, рапамицин может быть использован непосредственно в комбинации, например, с растворителем и не требует предварительного выделения. Другим преимуществом данного изобретения является то, что скорость растворения твердой дисперсии выше, чем скорость растворения, обнаруженная для кристаллического рапамицина или аморфного рапамицина в простой смеси.
Другим объектом изобретения является фармацевтическая композиция в форме твердой дисперсии, содержащая аскомицин и среду-носитель.
До настоящего времени не существовало подходящих для перорального введения твердых препаратов для 33-эпихлор-33- дезоксиаскомицина. Для устранения указанного недостатка в данном изобретении предлагается фармацевтическая композиция в виде твердой дисперсиии, содержащая 33-эпихлор-33-дезоксиаскомицин и среду-носитель, причем эта среда-носитель включает водорастворимый полимер или циклодекстрин.
Соединение 33-эпихлор-33-дезоксиаскомицин описано в опубликованной Европейской заявке ЕР 427680 в примере 66a.
33-Эпихлор-33-дезоксиаскомицин здесь будет именоваться как Соединение Y.
Содержащие соединения Y композиции по изобретению обусловливают высокую биодоступность лекарственного вещества, удобны для введения и стабильны.
Соединение Y может присутствовать в композиции в количестве от примерно 0, 01 до примерно 30 мас.% /масс, и предпочтительно в количестве от 1 до 20 мас.%/масс.
Среда-носитель может содержать любой из вышеупомянутых компонентов в вышеуказанных количествах, мас. %. Подходящие водорастворимые полимеры, циклодекстрины и другие эксципиенты, например, поверхностно-активные вещества, для использования в содержащих 33-эпихлор-33-дезоксиаскомицин композициях по изобретению аналогичны вышеописанным.
В предпочтительном аспекте данного изобретения композиция в форме твердой дисперсии, содержащая соединение Y, содержит дополнительно поверхностно-активное вещество.
Массовое соотношение соединения Y к среде-носителю составляет обычно не более чем 1:3, предпочтительно менее чем 1:4.
Аскомициновые, содержащие соединения Y, твердые дисперсные композиции могут быть приготовлены способами, аналогичными вышеописанным.
Описанные пероральные композиции, содержащие соединения Y, полезны,
например, при лечении воспалительных и гиперпролиферативных кожных заболеваний и кожных проявлений иммунологических заболеваний. Более конкретно композиции по изобретению полезны как
противовоспалительные, иммунодепрессантные и антипролиферативные агенты для применения при профилактике и лечении воспалительных состояний и состояний, требующих иммуносупрессии, таких как:
а) профилактика и лечение
- отторжения трансплантата органа или ткани, например сердца, почек, печени, костного мозга и кожи,
- заболевания "трансплантат против хозяина", такого как
последствие трансплантации костного мозга,
- аутоиммунных заболеваний, таких как ревматоидный артрит, системная узловая эритема, тиреоидит Хашимото, рассеянный склероз, миастения, диабет тип
I и увеит,
- кожных проявлений иммунологических заболеваний;
б) лечение воспалительных и гиперпролиферативных кожных заболеваний, таких как псориаз, диффузный нейродермит, контактный
дерматит и другие экзематозные дерматиты, себорейная экзема, Lichen planus, Pemphigus, bullous Pemphigoid, врожденный буллезный эпидермолиз, крапивница, ангионевротический отек, васкулит, эритема,
кожная эозинофилия, Lupus erythematosus и акне; и
в) гнездная алопеция
Когда фармацевтическая композиция по изобретению находится в форме единичной дозы, например в форме таблетки,
капсулы, гранулы или порошка, каждая единичная доза будет соответственно содержать между 1 мг и 100 мг лекарственного вещества, более предпочтительно между 10 и 50 мг, например 15, 20, 25 или 50 мг.
Такая форма единичной дозы пригодна для введения от 1 до 5 раз в сутки в зависимости от конкретного назначения терапии, стадии терапии и т. д.
В одном воплощении данного изобретения композиция содержит 30 мас.% соединения Y и 70 мас.% HPMC в дозе, например, от 10 до 50 мг в день для применения при, например, псориазе, диффузном нейродермите или контактном дерматите.
Точное количество композиций, которое должно быть введено, зависит от нескольких факторов, например, от желаемой продолжительности лечения и скорости высвобождения соединения Y.
Применимость фармацевтических композиций, содержащих соединение Y, можно наблюдать по стандартным клиническим тестам, например по известным показателям доз активного агента, дающим эквивалентный уровень активного агента в крови; например, используя дозы в интервале от 1 до 1000 мг, например, от 5 до 100 мг, активного агента в день на 75 кг веса взрослого человека и на стандартных моделях животных. Возросшую биодоступность лекарственного вещества с помощью композиций можно наблюдать в стандартных опытах с животными и в клинических испытаниях.
Ниже представлены примеры твердых дисперсных композиций по изобретению.
Пример 1
Твердую дисперсную композицию получают с содержанием следующих ингредиентов, мас.ч.:
Соединение X - 9,1
HPMC 3 сП - 81,8
Лактоза 200 меш - 9,1
Композицию (форма А) получают растворением рапамицина и среды-носителя в смеси этанол/ацетон. Абсолютный этанол
используется в массовом соотношении к ацетону 1:1. Затем растворители выпаривают и полученный сухой остаток измельчают в тонкий порошок со средним размером частиц < 0,5 мм.
Пример 2
Твердую дисперсную композицию получают с содержанием следующих ингредиентов, мас.ч.:
Соединение X - 16,7
HPMC 3 сП - 66,7
Poloxamer 188 (от BASF) - 16,7
Композицию (форма В) получают как в примере 1.
Пример 3
Твердую дисперсную композицию получают с содержанием следующих ингредиентов, мас.ч.:
Соединение X - 16,
7
HPMC 3 сП - 66,7
TGPS* - 16,7
Композицию (форма С) получаю/как в примере 1.
* сукцинат полиэтиленгликольтокоферола
Пример
4
Твердую дисперсную композицию получают с содержанием следующих ингредиентов, мас.ч.:
Соединение X - 10
HPMC 3 сП - 80
Solulan C24 (от Amerchol) - 10
Композицию (форма D) получают как в примере 1.
Вышеуказанные композиции, формы от А до D, могут быть сформованы в таблетки, помещены в капсулы или измельчены в порошок и упакованы в пакетики.
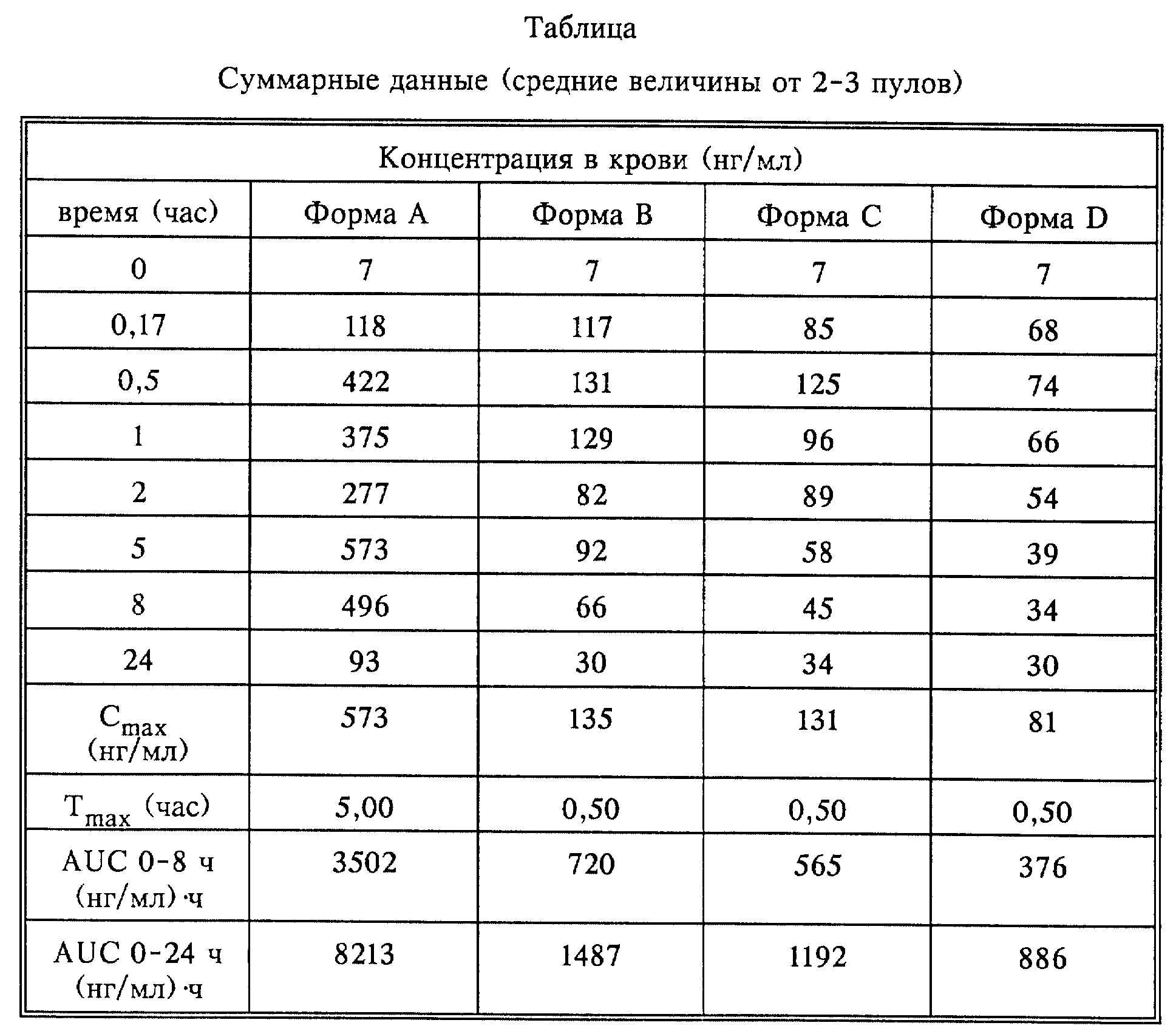
Фармакокинетика после введения 40-O-(2-гидрокси)этилрапамицина крысам
а) Введение лекарства
0,5 мл водной дисперсии композиции Соединения X (что
соответствует 4,0 мг активного ингредиента на крысу) вводили через зонд в желудок во время непродолжительного ингаляционного наркоза с помощью шприца на 1 мл, присоединенного к полиэтиленовой трубке.
Для каждой композиции формы А, В, С и D было использовано по 6 животных.
б) Взятие проб крови
За один день до эксперимента животным вставляли постоянную канюлю в яремную вену.
0,5 мл венозной крови (яремная вена) собирали от каждой крысы и хранили в 2,5 мл EDTA пробирках. Образцы крови 2 животных (1 и 2, 3 и 4, 5 и 6) пулировали в общий фонд и хранили при -80oC
до проведения лекарственного анализа. Образцы отбирали перед введением и через 10 мин, 30 мин, 60 мин, 120 мин, 300 мин, 480 мин и 1440 мин после введения лекарства.
в) Биоаналитика
Образцы крови анализировали с использованием HPLC (жидкостная хроматография высокого разрешения) с обращенной фазой.
Таблица показывает фармакокинетические данные, полученные после введения соединения X крысам.
Форма А дает результат по уровню в крови выше, чем при введении композиций, содержащих поверхностно-активные вещества.
Исследования
на собаках
Исследование биологической доступности было проведено на гончих собаках с применением дозы 1 мг/кг массы тела. Твердые желатиновые капсулы с содержанием в каждой 10 мг соединения X
вводили 8 собакам по схеме 4-числового латинского квадрата; собак кормили через 6 ч после введения капсулы, а уровень соединения X в крови определялся через 48 ч. Аналогичные показатели концентрации
соединения X в крови наблюдались у всех собак с окончательным полупериодом жизни соединения X в крови между 10 и 40 ч. Наблюдался срединный пик уровня 140 нг/мл и срединный AUC уровень 0-48 ч
приблизительно 1600 нг•ч/мл.
Пример 5
Твердую дисперсную композицию получают с содержанием следующих ингредиентов, мас.ч.:
Соединение Y - 20
HPMC 3 сП
- 80
Композицию (форма Е) получают растворением соединения Y и среды-носителя в смеси этанол/ацетон. Затем растворители выпаривают, а полученный сухой остаток измельчают.
Пример 6
Твердую дисперсную композицию получают с содержанием следующих ингредиентов, мас.ч:
Соединение Y - 20
HPMC 3 сП - 70
Poloxamer 188 - 10
Композицию
(форма F) получают как в примере 5.
Пример 7
Твердую дисперсную композицию получают с содержанием следующих ингредиентов, мас.ч.:
Соединение Y - 20
HPMC 3 сП
- 75
Лаурилсульфат натрия - 5
Композицию (форма G) получают как в примере 5.
Вышеуказанные композиции, формы от E до G могут быть сформованы в таблетки, помещены в капсулы или измельчены в порошок и упакованы в пакетики.
Фармакокинетика после введения 33-эпихлор-33-дезоксиаскомицина крысам
а) Введение лекарства
0,5 мл водной
дисперсии лекарственной композиции (что соответствует 4,0 мг активного ингредиента на крысу) вводили через зонд в желудок во время непродолжительного ингаляционного наркоза с помощью шприца на 1 мл,
присоединенного к полиэтиленовой трубке. Для каждой композиции формы E, F и G использовали по 6 животных.
б) Взятие проб крови
За один день до эксперимента животным вставляли
постоянную канюлю в яремную вену. 0,5 мл венозной крови (яремная вена) отбирали от каждой крысы и хранили в 2,5 мл EDTA пробирках. Образцы крови 2 животных (1 и 2, 3 и 4, 5 и 6) пулировали в общий
фонд и хранили при -80oC до проведения лекарственного анализа. Образцы отбирали перед введением и через 10 мин, 30 мин, 60 мин, 120 мин, 300 мин, 480 мин и 1440 мин после введения.
в) Биоаналитика
Образцы крови анализировали с использованием HPLC с обращенной фазой.
Результаты представлены на фиг. 1 и 2, на которых по вертикальной оси указаны данные по уровню в крови соединения Y в нг/мл, по горизонтальной оси указаны показатели времени в часах. Фиг. 1 показывает, что форма F дает результат по уровню в крови значительно выше, чем уровень крови, наблюдаемый после введения формы E или формы G. Фиг.2 показывает, что форма F дает высокий результат уровня в крови при введении с пищей.
Соединение Y находится в аморфной форме в композициях E, F и G сразу после образования и после 6 месяцев хранения, как определено дифракцией рентгеновских лучей.
Формы E, F и G проверяли на соответствующие скорости растворения. При перемешивании в растворе 0,2 мас.% додецилсульфата натрия в воде при температуре 37oC было обнаружено, что свыше 80% доступного соединения Y высвобождается и растворяется из каждой измельченной композиции, содержащей 10 мг соединения Y через 30 мин 92% доступного соединения Y высвобождается из формы Е. Это сравнимо с примерно 5% высвобождением через 30 мин из эквивалентного количества кристаллического соединения Y.
Реферат
Изобретение относится к химико-фармацевтической промышленности и касается фармацевтической композиции для перорального введения в форме твердой дисперсии. Изобретение заключается в том, что композиция содержит рапамицин или 33-эпихлор-33-дезоксиаскомицин и среду-носитель. Изобретение обеспечивает пероральное введение твердых препаратов, приемлемых для рапамицина и его производных, и стабильность композиции. 2 с. и 13 з.п. ф-лы, 2 ил., 1 табл.

Комментарии