Липосомальная композиция - RU2476216C1
Код документа: RU2476216C1
Чертежи


Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к новой липосомальной композиции, содержащей эрибулин или его фармакологически приемлемую соль. Настоящее изобретение также относится к способу производства липосомальной композиции.
Уровень техники изобретения
Липосомы представляют собой микроскопические замкнутые везикулы, имеющие внутреннюю фазу, окруженную одним или несколькими липидными бислоями, и способные удерживать водорастворимый материал во внутренней фазе, а липофильный материал - в липидном бислое. При заключении активного соединения в липосому и доставке его в ткани-мишени важными задачами являются захват активного соединения в липосому с высокой эффективностью и обеспечение устойчивого удержания активного соединения липосомой.
При заключении липофильных соединений в липосому высокий коэффициент захвата может быть достигнут относительно легко, за исключением случаев с соединениями, имеющими очень высокое сродство к мембранам, такими как амфотерицин B (главное действующее вещество в липосомальном лекарственном средстве AmBisome), стабильность удержания в сыворотке крови обычно низкая, и трудно добиться достаточного улучшения фармакокинетики. Что касается методов заключения водорастворимых соединений в липосомы, существуют различные методы, такие как метод липидной пленки (метод вихрей), метод обращенно-фазового выпаривания, метод удаления поверхностно-активных веществ, метод замораживания-оттаивания и методы дистанционной загрузки (метод градиента рН, метод ионного градиента). Однако только дистанционные методы загрузки обеспечивают близкий к 100% коэффициент захвата; коэффициент захвата в случае других методов составляет лишь порядка 5-30%.
В качестве методов дистанционной загрузки известны методы с использованием градиента pH и градиента ионов сульфата аммония. Метод градиента pH, который представляет собой метод дистанционной загрузки с использованием градиента pH, является методом заключения соединений в липосомы при помощи изменения равновесия молекулярной/ионной диссоциации из-за рН целевого соединения.
В качестве одного из примеров соединения, заключенного в липосоме методом градиента рН, можно привести, например, доксорубицин (DOX, pKa: 8,2). После получения липосомального раствора с буферным раствором, имеющим pH 4, внешнюю фазу липосом заменяют на буферный раствор с pH 7. В случае когда DOX добавляют к этому раствору липосом, то поскольку молекулярный DOX в растворе с pH 7 является липофильным, он мигрирует в мембраны липосом, а не в водную фазу. В случае когда DOX, который мигрировал в мембраны липосом, далее контактирует с внутренней фазой липосом с pH 4, он становится ионным и включается во внутреннюю фазу липосом. Таким образом, DOX переносится из внешней фазы во внутреннюю фазу липосом за счет изменения равновесия диссоциации (см. непатентный литературный источник 1, непатентный литературный источник 2 и патентный литературный источник 1).
Сообщалось о множестве способов усовершенствования этого типа методов удаленной загрузки. В непатентном литературном источнике 3 описан способ улучшения коэффициента захвата активных соединений путем добавления этанола вместе с активным соединением во внешнюю фазу липосом, когда метод градиента рН осуществляют в липосомах специального состава, называемых бесхолестериновыми липосомами.
В патентном литературном источнике 2, в дополнение к градиенту pH описан способ улучшения коэффициента захвата активных соединений за счет наличия во внутренней фазе липосом ионов меди.
Вместо градиента pH в методе с градиентом pH метод с сульфатом аммония, представляющий собой метод удаленной загрузки с использованием градиента ионов сульфата аммония, является методом заключения активных соединений во внутренней фазе липосом с помощью градиента ионов, таких как двухвалентный сульфат аммония (см. непатентный литературный источник 1 и патентный литературный источник 3).
В дополнение к методу градиента ионов, основанному на сульфате аммония, в патентном литературном источнике 4 описан метод заключения активных соединений в липосомы путем добавления бороновой кислоты вместе с активным соединением во внешнюю фазу липосом.
Вместо ионного градиента на основе сульфата аммония в патентном литературном источнике 5 описан метод, в котором по сравнению с ситуацией когда используют сульфат аммония скорость высвобождения активного соединения улучшается благодаря заключению активного соединения в липосомы с помощью ионного градиента анионов глюкуроновой кислоты.
Таким образом, с точки зрения коэффициента захвата методы удаленной загрузки являются прекрасными методами захвата. Однако в случае когда используют методы удаленной загрузки, за исключением особых случаев, например с Доксилем (липосомальный препарат DOX), в котором активное соединение, заключенное во внутренней фазе липосом, кристаллизованно, существует проблема, состоящая в том, что активное соединение имеет тенденцию к утечке из липосом в сыворотку крови и стабильность удержания активного соединения низка.
Как описано выше, при использовании обычных технических методов ситуация в настоящее время такова, что трудно достичь одновременно высокого коэффициента захвата активного соединения в липосомы и стабильности удержания активного соединения в липосомах.
Литературные источники предшествующего уровня техники
Патентные литературные источники
Патентный литературный источник 1: Патент Соединенных Штатов Америки №5192549, спецификация.
Патентный литературный источник 2: PCT Международная публикация WO 2006/037230, брошюра.
Патентный литературный источник 3: Патент Соединенных Штатов Америки №5316771, спецификация.
Патентный литературный источник 4: Патент Соединенных Штатов Америки №6051251, спецификация.
Патентный литературный источник 5: PCT Международная публикация WO 2005/046643, брошюра.
Непатентные литературные источники
Непатентный литературный источник 1: Yasuyuki Sazuka, «Liposome Preparation Method», «New Developments in Liposome Application: Toward the Development of Artificial Cells» (Kazunari Akiyoshi, Shigeru Tsujii, редакционная статья)» NTS, (2005), pp.33-37.
Непатентный литературный источник 2: Mayer LD et al., Biochimica et Biophysica Acta, (1986), 857: pp.123-126.
Непатентный литературный источник 3: N. Dos Santos et al., Biochimica et Biophysica Acta, (2004), 1661(1): pp.47-60.
Раскрытие изобретения
Проблема, решаемая с помощью изобретения
Задачей настоящего изобретения является создание липосомальной композиции с высоким коэффициентом захвата и стабильностью удержания активного соединения.
Способы решения проблемы
В результате всестороннего исследования, направленного на решение вышеуказанных проблем, авторы настоящего изобретения установили в отношении липосомальной композиции, активным соединением которой является эрибулин или его фармакологически приемлемая соль, что коэффициент захвата и стабильность удержания активного соединения в липосомальной композиции чрезвычайно высоки, что привело к завершению настоящего изобретения.
В частности, настоящее изобретение представляет собой следующее.
(1)
Липосомальная композиция, содержащая липосомы и содержащая активное соединение во внутренней фазе липосом, где активное соединение представляет собой эрибулин или его фармакологически приемлемую соль.
(2)
Липосомальная композиция по 1, находящаяся в твердой или жидкой форме.
(3)
Липосомальная композиция по 1 или 2, в которой внутренняя фаза липосом дополнительно содержит соль аммония.
(4)
Липосомальная композиция по 3, в которой концентрация вышеуказанной соли аммония составляет 10 мМ или выше.
(5)
Липосомальная композиция по любому из 1-4, в которой внутренняя фаза липосом дополнительно содержит соль, кислоту, основание и/или аминокислоту.
(6)
Липосомальная композиция по 5, в которой концентрация вышеуказанной соли составляет 1-300 мМ.
(7)
Липосомальная композиция по 5 или 6, в которой концентрация вышеуказанной кислоты составляет 1-300 мМ.
(8)
Липосомальная композиция по любому из 5-7, в которой концентрация вышеуказанной аминокислоты составляет 1-300 мМ.
(9)
Липосомальная композиция по любому из 5-8, в которой концентрация вышеуказанного основания составляет 1-300 мМ.
(10)
Липосомальная композиция по любому из 1-9, в которой концентрация вышеуказанного активного соединения составляет 0,01-300 мг/мл.
(11)
Липосомальная композиция по любому из 1-10, в которой вышеуказанное активное соединение представляет собой эрибулин мезилат.
(12)
Липосомальная композиция по любому из 1-11, в которой внутренняя фаза липосом дополнительно содержит сульфат аммония, лимонную кислоту и активное соединение.
(13)
Липосомальная композиция по любому из 1-12, в которой внешняя фаза липосом содержит сахар, электролит и/или аминокислоту.
(14)
Липосомальная композиция по любому из 1-13, в которой внешняя фаза липосом содержит сахар или электролит и аминокислоту.
(15)
Липосомальная композиция по 13 или 14, в которой концентрация вышеуказанного сахара составляет 2-20%.
(16)
Липосомальная композиция по любому из 13-15, в которой концентрация вышеуказанной аминокислоты составляет 1-300 мМ.
(17)
Липосомальная композиция по любому из 1-16, в которой внешняя фаза липосом содержит сахарозу или хлорид натрия и гистидин.
(18)
Липосомальная композиция по любому из 1-17, в которой вышеуказанная внутренняя фаза липосом по существу не содержит циклодекстрин.
(19)
Липосомальная композиция по любому из 1-18, в которой липосомы содержат гидрогенизированный фосфатидилхолин.
(20)
Липосомальная композиция по любому из 1-19, в которой липосомы содержат холестерин.
(21)
Липосомальная композиция по любому из 1-20, в которой липосомы содержат конденсат метоксиполиэтиленгликоля.
(22)
Липосомальная композиция по 21, в которой вышеуказанный конденсат метоксиполиэтиленгликоля представляет собой конденсат дистеароилфосфатидилэтаноламин-полиэтиленгликоля.
(23)
Липосомальная композиция по любому из 1-22, в которой липосомы содержат гидрогенизированный фосфатидилхолин, холестерин и конденсат дистеароилфосфатидилэтаноламин-полиэтиленгликоля.
(24)
Липосомальная композиция по 23, которая содержит от 10 до 80% вышеуказанного гидрогенизированного фосфатидилхолина, от 1 до 60% вышеуказанного холестерина и от 0 до 50% вышеуказанного конденсата дистеароилфосфатидилэтаноламин-полиэтиленгликоля.
(25)
Липосомальная композиция по любому из 1-24, в которой липосомы содержат гидрогенизированный соевый фосфатидилхолин, холестерин и полиэтиленгликоль 2000-фосфатидилэтаноламин.
(26)
Способ производства липосомальной композиции по любому из 1-25, включающий:
этап, на котором получают липосомальную дисперсионную жидкость, содержащую липосомы;
этап, на котором вышеуказанную липосомальную дисперсионную жидкость смешивают с вышеуказанным активным соединением; и
этап, на котором вышеуказанное активное соединение вводится во внутреннюю фазу липосом вышеуказанной липосомальной дисперсионной жидкости.
(27)
Способ по 26, в котором вышеуказанная липосомальная дисперсионная жидкость по существу не содержит соль аммония во внешней фазе липосом.
(28)
Способ по 26 или 27, в котором pH внешней фазы липосом вышеуказанной липосомальной дисперсионной жидкости равен 3-10.
(29)
Способ по любому из 26-28, в котором pH внешней фазы липосом вышеуказанной липосомальной дисперсионной жидкости равен 7-10.
(30)
Способ по 28 или 29, в котором вышеуказанный pH представляет собой pH внешней фазы липосом вышеуказанной липосомальной дисперсионной жидкости на этапе, на котором вышеуказанную липосомальную дисперсионную жидкость и вышеуказанное активное соединение смешивают.
(31)
Способ по любому из 26-30, в котором этап, на котором получают вышеуказанную липосомальную дисперсионную жидкость, включает этап, на котором получают предварительный липосомальный раствор, содержащий липосомы и содержащий соль аммония во внутренней фазе липосом и внешней фазе липосом; и этап, на котором внешнюю фазу липосом вышеуказанного предварительного липосомального раствора заменяют или разбавляют.
(32)
Способ по 31, в котором этап, на котором вышеуказанную внешнюю фазу липосом заменяют или разбавляют, является этапом, на котором pH внешней фазы липосом делают выше, чем рН внутренней фазы липосом.
(33)
Способ по 31 или 32, в котором этап, на котором вышеуказанную внешнюю фазу липосом заменяют или разбавляют, является этапом, на котором разница между pH внутренней фазы липосом и pH внешней фазы липосом составляет 1-5.
(34)
Способ по любому из 26-33, в котором pH вышеуказанной внутренней фазы липосом равен 3-9.
(35)
Способ по любому из 26-34, в котором pH вышеуказанной внутренней фазы липосом равен 4-9.
(36)
Способ по любому из 26-35, в котором pH вышеуказанной внутренней фазы липосом равен 5-8.
(37)
Способ по любому из 26-36, в котором внешняя фаза липосом представляет собой раствор, содержащий электролит на этапе, в котором вводят вышеуказанное активное соединение.
(38)
Способ по любому из 26-37, в котором вышеуказанная липосомальная дисперсионная жидкость по существу не содержит циклодекстрин во внутренней фазе липосом.
(39)
Способ по любому из 26-38, дополнительно включающий этап, на котором pH внешней фазы липосом делают нейтральным.
Эффект изобретения
По настоящему изобретению предложена новая липосомальная композиция. Липосомальная композиция по настоящему изобретению захватывает активное соединение во внутреннюю фазу липосом с высокой степенью эффективности и имеет высокую стабильность удержания активного соединения.
Краткое описание чертежей
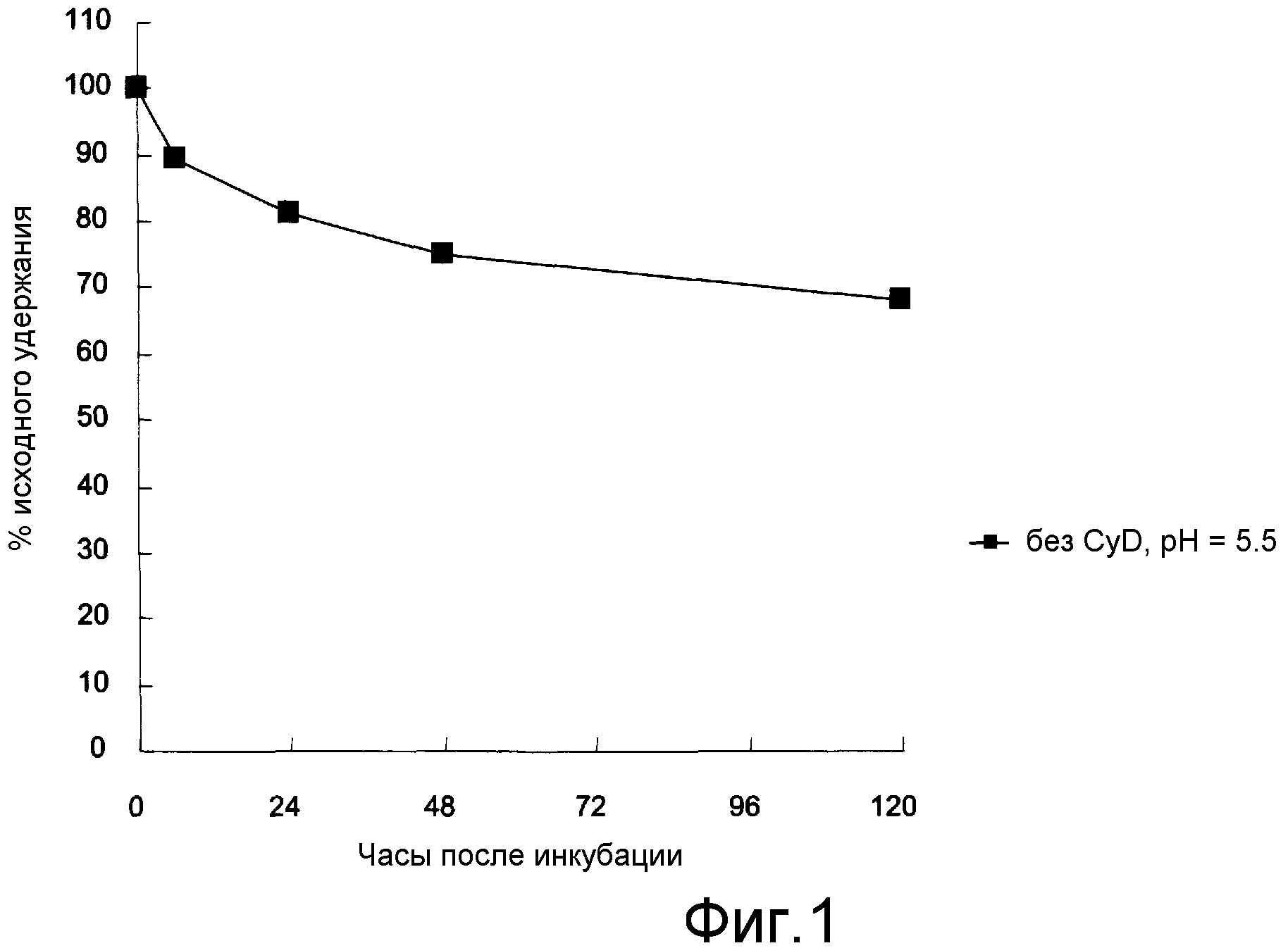
На фигуре 1 представлены изменения концентрации эрибулин мезилата в липосомальной композиции в сыворотке крови крыс (37°C) in vitro.
На фигуре 2 представлена противоопухолевая активность in vivo эрибулин мезилата из липосом у «голых» мышей с раковой опухолью FaDu.
На фигуре 3 представлена противоопухолевая активность in vivo эрибулин мезилата из липосом у «голых» мышей с раковой опухолью ACHN.
Лучший способ осуществления изобретения
Настоящее изобретение конкретно описано с помощью вариантов осуществления изобретения, однако настоящее изобретение не ограничивается следующими вариантами осуществления изобретения и может осуществляться с различными модификациями.
Содержание литературных источников, приведенных в виде ссылок в настоящем изобретении, включено в настоящее изобретение посредством ссылок.
Определения
«Липосомы» означают микроскопические замкнутые везикулы, имеющие внутреннюю фазу, окруженную липидным бислоем. В настоящем изобретении липосомы включают небольшие одномембранные липосомы (SUV: небольшие однослойные везикулы), большие одномембранные липосомы (LUV: большие однослойные везикулы), одномембранные липосомы еще большего размера (GUV: гигантские однослойные везикулы), многослойные липосомы, имеющие несколько концентрических мембран (MLV: многослойные везикулы), липосомы, имеющие несколько мембран, не концентрических, а имеющих неправильную форму (MVV: мультивезикулярные везикулы), и так далее.
«Внутренняя фаза липосом» означает водный слой, заключенный в липидный бислой липосом, и используется в том же значении, что и «внутренняя водная фаза» и «внутренняя водная фаза липосом». «Внешняя фаза липосом» означает пространство, не заключенное в липидный бислой липосом (то есть пространство за пределами внутренней фазы и липидного бислоя), в случае когда липосомы диспергированы в жидкости.
«Липосомальная композиция» означает композицию, содержащую липосомы и дополнительно содержащую эрибулин мезилат во внутренней фазе липосом. В настоящем изобретении липосомальная композиция включает как твердые, так и жидкие формы.
«Липосомальная дисперсионная жидкость» означает композицию, содержащую липосомы, и представляет собой композицию до введения активного соединения во внутреннюю фазу липосом.
«Предварительный липосомальный раствор» означает композицию, содержащую липосомы, и представляет собой композицию до корректировки внешней фазы липосом с целью захвата эрибулин мезилата во внутреннюю фазу липосом.
«Липосомальный реагент» означает липосомальную дисперсионную жидкость в случае когда он находится в жидком виде. В случае когда он находится в твердом виде, это означает реагент, из которого липосомальную дисперсионную жидкость можно получать путем растворения или суспендирования в установленном растворителе. Растворитель описан ниже. Как описано ниже, твердый липосомальный реагент можно получать, например, путем высушивания липосомальной дисперсионной жидкости.
В настоящем описании «смешивание твердого и жидкого» включает растворение и суспендирование твердого вещества в жидкости, и смешивание, растворение и суспендирование используют взаимозаменяемым образом. Аналогично, растворитель и дисперсионную среду также используют взаимозаменяемым образом.
Кроме того, липосомальная композиция, липосомальная дисперсионная жидкость, предварительный липосомальный раствор и липосомальный реагент по настоящему изобретению по существу не содержат циклодекстрин. «По существу не содержать циклодекстрин» означает, что циклодекстрин не добавляют. Достаточно, если циклодекстрин не содержится в количестве, при котором улучшение растворимости (номинальной растворимости) активного соединения из-за циклодекстрина наблюдается в значительной степени, и даже в случае когда его добавляют в количестве, при котором улучшение растворимости активного соединения не наблюдается в значительной степени, это не должно быть исключением из вариантов осуществления настоящего изобретения.
Кроме того, в качестве предпочтительного способа осуществления настоящего изобретения «липосомальная дисперсионная жидкость, по существу не содержащая соль аммония во внешней фазе липосом», означает, что соль аммония не добавляют во внешнюю фазу липосомальной дисперсионной жидкости. Добавление соли аммония в количестве, находящемся в пределах диапазона, который позволяет достичь цели настоящего изобретения, не должно быть исключением из вариантов осуществления настоящего изобретения. В случае когда соль аммония содержится во внешней фазе липосом предварительного липосомального раствора, возможно получать липосомальную дисперсионную жидкость, которая по существу не содержит соль аммония, путем замены или разбавления внешней фазы липосом предварительного липосомального раствора, используя раствор, который по существу не содержит соль аммония.
Активное соединение
Активным соединением по настоящему изобретению является эрибулин или его фармакологически приемлемая соль (далее в данном документе иногда называемый «эрибулин и так далее»). Не существует конкретных ограничений в отношении фармакологически приемлемой соли при условии, что образуются эрибулин и соль, будь то соль неорганической кислоты или соль органической кислоты. Например, можно упомянуть соль соляной кислоты, соль серной кислоты, цитрат, соль бромистоводородной кислоты, соль йодистоводородной кислоты, соль азотной кислоты, бисульфат, соль фосфорной кислоты, соль суперфосфорной кислоты, соль изоникотиновой кислоты, соль уксусной кислоты, соль молочной кислоты, соль салициловой кислоты, соль винной кислоты, соль пантотеновой кислоты, соль аскорбиновой кислоты, соль янтарной кислоты, соль малеиновой кислоты, соль фумаровой кислоты, соль глюконовой кислоты, соль сахариновой кислоты, соль муравьиной кислоты, соль бензойной кислоты, соль глютаминовой кислоты, соль метансульфоновой кислоты, соль этансульфоновой кислоты, соль бензолсульфоновой кислоты, соль п-толуолсульфоновой кислоты, соль памовой кислоты (памоат) и так далее. Предпочтительными среди них являются соль соляной кислоты, соль серной кислоты, соль уксусной кислоты, соль фосфорной кислоты, цитрат и соль мезиловой кислоты, и наиболее предпочтительной из всех является соль мезиловой кислоты. То есть предпочтительным активным соединением по настоящему изобретению является эрибулин мезилат.Более того, в качестве фармакологически приемлемой соли эрибулина допустимо использовать эрибулин и соли алюминия, кальция, лития, магния, кальция [sic], натрия, цинка и диэтаноламина. Эрибулин или его фармакологически приемлемая соль представляет собой соединение или его соль, указанные в брошюре РСТ международной публикации WO 99/65894 или патенте Соединенных Штатов Америки 6214865 (содержание этих патентов включено в данный документ посредством ссылок) и обладающие фармакологическим действием, включая противоопухолевое действие и антимитотическое действие. Эрибулин или его фармакологически приемлемая соль оказывает противоопухолевое действие в отношении меланомы, фибросаркомы, моноцитарного лейкоза, рака толстой кишки, рака яичников, рака молочной железы, рака костей, рака простаты, рака легких и ras-трансформированных фибробластов.
Однако в качестве активных соединений, которые можно комбинировать с эрибулином, и так далее можно выбирать среди соединений, используемых в области медицины (в том числе диагностических препаратов), косметической продукции, пищевых продуктов и так далее. Что касается активных соединений, допустимо объединять одно или несколько соединений, отличных от эрибулина, и так далее.
В качестве активных соединений можно упомянуть низкомолекулярные соединения и так далее. Среди них подходящими являются соединения, используемые в качестве противоопухолевых средств, антибактериальных средств, противовоспалительных средств, средств против инфаркта миокарда и контрастных веществ.
Что касается молекулярного веса активного соединения, более предпочтительным является диапазон от 100 до 2000, диапазон от 200 до 1500, и еще более предпочтительным является диапазон от 300 до 1000. В пределах этих диапазонов проницаемость липосомальной мембраны для активного соединения в целом удовлетворительная, и настоящее изобретение может быть соответствующим образом применено.
Активные соединения включают водорастворимые соединения и липофильные соединения, и настоящее изобретение может быть применено при условии, что они более или менее растворимы в воде или водных растворителях.
В настоящем изобретении не существует конкретных ограничений в отношении противоопухолевых средств и можно упомянуть, например, производные камптотецина, такие как иринотекан гидрохлорид, ногитекан гидрохлорид, экзатекан, RFS-2000, луртотекан, BNP-1350, Bay-383441, PNU-166148, IDEC-132, BN-80915, DB-38, DB-81, DB-90, DB-91, CKD-620, T-0128, ST-1480, ST-1481, DRF-1042, DE-310; производные таксана, такие как доцетаксел гидрид, доцетаксел, паклитаксел, IND-5109, BMS-184476, BMS-188797, T-3782, TAX-1011, SB-RA-31012, SBT-1514 и DJ-927; ифосфамид, нимустин гидрохлорид, карвокон, циклофосфамид, дакарбазин, тиотепа, бусульфан, мелфалан, ранимустин, эстрамустин натрия фосфат, 6-меркаптопурин-рибозид, эноцитабин, гемцитабин гидрохлорид, кармфур, цитарабин, цитарабин окфосфат, тегафур, доксифлуридин, гидроксикарбамид, фторурацил, метотрексат, меркаптопурин, флударабин фосфат, актиномицин D, акларубицин гидрохлорид, идарубицин гидрохлорид, пирарубицин гидрохлорид, эпирубицин гидрохлорид, даунорубицин гидрохлорид, доксорубицин гидрохлорид, эпирубицин, пирарубицин, даунорубицин, доксорубицин, пирарубицин гидрохлорид, блеомицин гидрохлорид, зиностатин стималамер, неокарциностатин, митомицин С, блеомицин сульфат, пепломицин сульфат, этопозид, винорельбин тартрат, винкристин сульфат, виндезин сульфат, винбластин сульфат, амрубицин гидрохлорид, гефитиниб, экземестан, капецитабин, TNP-470, TAK-165, KW-2401, KW-2170, KW-2871, KT-5555, KT-8391, TZT-1027, S-3304, CS-682, YM-511, YM-598, TAT-59, TAS-101, TAS-102, TA-106, FK-228, FK-317, E7070, (8E, 12E, 14E)-7-[(4-циклогептипиперазин-1-ил)карбонил]окси-3,6,16,21-тетрагидрокси-6,10,12,16,20-пентаметил-18,19-эпокситрикоза-8,12,14-триен-11-олид (E7107), KRN-700, KRN-5500, J-107088, HMN-214, SM-11355, ZD-0473 и так далее. Что касается соединений, записанных в виде солей, среди вышеупомянутых противоопухолевых средств любая соль является приемлемой и свободные основные вещества также являются приемлемыми. Что касается соединений, записанных в виде свободных основных веществ, любая их соль является приемлемой.
Не существует конкретных ограничений в отношении антибактериальных средств и можно упомянуть, например, амфотерицин B, цефотиам гексил, цефалоспорин, хлорамфеникол, диклофенак и так далее. Что касается соединений вышеупомянутых антибактериальных средств, любая их соль является приемлемой.
Не существует конкретных ограничений в отношении противовоспалительных средств и можно упомянуть, например, простагландины (PGE1, PGE2), дексаметазон, гидрокортизон, пироксикам, индометацин, преднизолон и так далее. Что касается соединений вышеупомянутых противовоспалительных средств, любая их соль является приемлемой.
Не существует конкретных ограничений в отношении средств против инфаркта миокарда и можно упомянуть, например, аденозин, атенолол, пилсикаинид и так далее, а в качестве контрастных веществ можно упомянуть, например, йопамидол, йоксагловую кислоту, йогексол, йомепрол и так далее. Что касается соединений вышеупомянутых средств против инфаркта миокарда и контрастных веществ, любая их соль является приемлемой.
Липиды
Предпочтительно, чтобы мембранные составляющие липосом по настоящему изобретению включали фосфолипиды и/или производные фосфолипидов. В качестве фосфолипидов и производных фосфолипидов можно упомянуть, например, фосфатидилэтаноламин, фосфатидилхолин, фосфатидилсерин, фосфатидилинозитол, фосфатидилглицерин, кардиолипин, сфингомиелин, церамид фосфорилэтаноламин, церамид фосфорилглицерин, церамид фосфорилглицеринфосфат, 1,2-димиристоил-1,2-дезоксифосфатидилхолин, плазмалоген, фосфатидную кислоту и так далее. Можно комбинировать один или несколько этих фосфолипидов и производных фосфолипидов.
Не существует конкретных ограничений в отношении остатков жирных кислот в фосфолипидах и производных фосфолипидов и можно упомянуть, например, остатки насыщенных или ненасыщенных жирных кислот с числом атомов углерода от 12 до 20. Конкретно можно упомянуть ацильные группы из жирных кислот, таких как лауриновая кислота, миристиновая кислота, пальмитиновая кислота, стеариновая кислота, олеиновая кислота и линолевая кислота. Можно также использовать фосфолипиды, полученные из природных веществ, такие как лецитин яичного желтка и соевый лецитин, частично гидрогенизированный лецитин яичного желтка, в котором остаток ненасыщенной жирной кислоты частично или полностью гидрогенизирован, (полностью) гидрогенизированный лецитин яичного желтка, частично гидрогенизированный соевый лецитин, (полностью) гидрогенизированный соевый лецитин и так далее.
Не существует конкретных ограничений в отношении смешиваемого количества (мольной доли) фосфолипидов и/или производных фосфолипидов, которые используют при получении липосом, однако предпочтительно использовать от 10 до 80% по отношению ко всему составу мембран липосом, и более предпочтительно - от 30 до 60%.
Что касается компонентов мембран, помимо фосфолипидов и/или производных фосфолипидов липосомы по настоящему изобретению могут также включать стерины, такие как холестерин и холестенол, в качестве стабилизаторов мембран - жирные кислоты, содержащие насыщенные или ненасыщенные ацильные группы с числом атомов углерода от 8 до 22, и антиоксиданты, такие как α-токоферол.
Не существует конкретных ограничений в отношении смешиваемого количества (мольной доли) этих стеринов, используемых при получении липосом, однако предпочтительно использовать от 1 до 60% по отношению ко всему составу мембран липосом, более предпочтительно - от 10 до 50% и еще более предпочтительно - от 30 до 50%.
Кроме того, не существует конкретных ограничений в отношении смешиваемого количества (мольной доли) жирных кислот, однако предпочтительно использовать от 0 до 30% по отношению ко всему составу мембран липосом, более предпочтительно - от 0 до 20% и еще более предпочтительно - от 0 до 10%. Что касается смешиваемого количества (мольной доли) антиоксидантов, достаточно если добавляют количество, способное обеспечить антиоксидантный эффект, однако предпочтительно использовать от 0 до 15% по отношению ко всему составу мембран липосом, более предпочтительно - от 0 до 10% и еще более предпочтительно - от 0 до 5%.
Липосомы по настоящему изобретению могут также содержать функциональные липиды и модифицированные липиды в качестве мембранных компонентов.
В качестве функциональных липидов можно упомянуть производные липидов, сохраняющиеся в крови, термочувствительные производные липидов, рН-чувствительные производные липидов и так далее. В качестве модифицированных липидов можно упомянуть ПЭГ-липиды, сахарные липиды, антитело-модифицированные липиды, пептид-модифицированные липиды и так далее.
В качестве производных липидов, сохраняющихся в крови, можно упомянуть, например, гликофорин, ганглиозид GM1, ганглиозид GM3, производные глюкуроновой кислоты, производные глютаминовой кислоты, производные полиглицеринфосфолипидов, производные полиэтиленгликоля (конденсаты метоксиполиэтиленгликоль и так далее), такие как N-[карбонил-метоксиполиэтиленгликоль-2000]-1,2-дипальмитоил-sn-глицеро-3-фосфоэтаноламин, N-[карбонил-метоксиполиэтиленгликоль-5000]-1,2-дипальмитоил-sn-глицеро-3-фосфоэтаноламин, N-[карбонил-метоксиполиэтиленгликоль-750]-1,2-дистеароил-sn-глицеро-3-фосфоэтаноламин, N-[карбонил-метоксиполиэтиленгликоль-2000]-1,2-дистеароил-sn-глицеро-3-фосфоэтаноламин, (MPEG 2000-дистеароилфосфатидилэтаноламин) и N-[карбонил-метоксиполиэтиленгликоль-5000]-1,2-дистеароил-sn-глицеро-3-фосфоэтаноламин, которые представляют собой конденсаты фосфоэтаноламина и метоксиполиэтиленгликоля. При наличии липосом, содержащих производные липидов со способностью сохраняться в крови, становится возможным улучшать способность сохраняться в крови липосом, поскольку захват липосом как посторонних примесей, в печени и так далее становится затруднительным.
В качестве термочувствительных производных липидов можно упомянуть, например, дипальмитоилфосфатидилхолин и так далее. При наличии липосом, содержащих термочувствительные производные липидов, становится возможным вызывать разрушение липосом при определенных температурах и вызывать изменения в поверхностных свойствах липосом. Более того, при сочетании этого с повышением температуры в целевой зоне опухоли и так далее становится возможным разрушать липосомы в целевой зоне и высвобождать активное соединение в целевой зоне.
В качестве pH-чувствительных производных липидов можно упомянуть, например, диолеоилфосфатидилэтаноламин и так далее. При наличии липосом, содержащих pH-чувствительные производные липидов, становится возможным стимулировать мембранное слияние липосом и эндосом, когда липосомы попадают в клетку за счет эндоцитоза, и улучшать перенос активного соединения в клеточную ткань.
В качестве сахарных липидов антитело-модифицированных липидов и пептид-модифицированных липидов можно упомянуть липиды, связанные с сахарами, антителами или пептидами, которые совместимы с клетками-мишенями или тканями-мишенями. Благодаря использованию модифицированных липидов липосомы могут активно переноситься к клеткам-мишеням или тканям-мишеням.
Не существует конкретных ограничений в отношении смешиваемого количества (мольной доли) используемых производных липидов со способностью сохраняться в крови при получении липосом, однако предпочтительно использовать от 0 до 50% относительно всего количества липидов, составляющих мембраны липосом, более предпочтительно - от 0 до 30% и еще более предпочтительно - от 0 до 20%.
Липосомы
Как упоминалось выше, липосомы представляют собой микроскопические замкнутые везикулы, имеющие внутреннюю фазу, окруженную липидным бислоем.
В идеале, что касается липосом, предпочтительно, чтобы для эрибулина и так далее существовала барьерная функция, препятствующая его утечке во внешнюю фазу липосом после того как эрибулин и так далее был заключен во внутреннюю фазу липосом. В случае когда их используют в качестве лекарственного средства, желательно, чтобы липосомы обладали стабильностью in vivo и чтобы для эрибулина и так далее существовала барьерная функция, препятствующая его утечке во внешнюю фазу липосом в крови, когда липосомы вводят in vivo.
Состав мембранных компонентов для липосом, обладающих такой мембранной проницаемостью на уровне, допускающем практическое применение, могут должным образом выбирать специалисты в данной области в зависимости от активного соединения, ткани-мишени и тому подобного, сверяясь при необходимости с вариантами осуществления, описанными ниже (Hiroshi Kikuchi, et. al., «Liposome I-Preparation Method and Assay Method-», Cell Technology (1983), 2(9): pp.1136-1149, и с литературными источниками, приведенными в указанной публикации).
При использовании в качестве лекарственного средства желательно, чтобы эрибулин и так далее высвобождался из липосом после того как липосомы достигнут ткани-мишени, клеток или внутриклеточных органелл. Что касается липосом, сами мембранные компоненты, как правило, являются биодеградируемыми и в конечном счете разлагаются в ткани-мишени и тому подобном. Считается, что захваченный эрибулин и так далее высвобождается таким образом. Более того, также приемлемо, если липосомы сами включаются в клетки.
Липосомальная композиция не только может быть нацелена на ткань-мишень, такую как солидный рак, но ее также можно использовать для переноса активных соединений для гематологического рака и так далее. Ее также можно использовать в крови в качестве препарата с замедленным высвобождением, препарата с контролируемым высвобождением и так далее.
Размер частиц липосом можно устанавливать в зависимости от задач. Например, если необходимо доставить липосомы к раковой ткани или воспаленной ткани при помощи эффекта EPR (повышенная проницаемость и удержание) в виде инъекционного продукта или тому подобного, предпочтительно, чтобы размер частиц липосом составлял от 30 до 400 нм, и более предпочтительно, чтобы размер частиц составлял от 50 до 200 нм. В случае когда нужно доставить липосомы к макрофагам, предпочтительно, чтобы размер частиц липосом составлял от 30 до 1000 нм, и более предпочтительно, чтобы размер частиц составлял от 100 до 400 нм. В случае когда липосомальную композицию предстоит использовать в качестве перорального препарата или трансдермального препарата, размер частиц липосом можно устанавливать в несколько микрон. Следует отметить, что (1) в нормальной ткани стенки сосудов служат барьером (потому что стенки сосудов состоят из плотно прилегающих сосудистых эндотелиальных клеток) и микрочастицы, такие как супермолекулы и липосомы заданного размера, не могут распространяться в ткани. Однако в пораженной ткани стенки сосудов рыхлые (потому что между клетками эндотелия сосудов существуют промежутки), что увеличивает проницаемость сосудов, и супермолекулы и микрочастицы могут распространяться в ткани за пределами сосудов (повышенная проницаемость). Кроме того, (2) в нормальной ткани лимфатическая система хорошо развита, однако известно, что в пораженной ткани лимфатическая система не развита и что супермолекулы или микрочастицы после их включения не перерабатываются в рамках общей системы, а сохраняются в пораженной ткани (повышенное удержание) - это называется эффект EPR (Matsumura, Maeda, Cancer Research, (1986), 46: pp.6387-6392). Следовательно, можно контролировать фармакокинетику путем регулирования размеров частиц липосом.
В настоящем изобретении размер частиц липосом означает средневзвешенный размер частиц, определенный методом динамического рассеяния света (метод квазиупругого рассеяния света). В данном документе приведен размер частиц, измеренный на приборах для динамического рассеяния света (например, модель Zetasizer Nano ZS производства Malvern Instruments Ltd. и ELS-8000 производства Otsuka Electronics Co., Ltd.). Приборы измеряют броуновское движение частиц, и размер частиц определяют на основе устоявшейся методологической теории динамического рассеяния света.
Не существует конкретных ограничений в отношении растворителя во внутренней фазе липосом и можно упомянуть, например, буферные растворы, такие как фосфатный буферный раствор, цитратный буферный раствор и фосфатно-солевой буферный физиологический раствор, физиологический солевой раствор, культуральную среду для культивирования клеток и так далее. Что касается растворителя в случае когда используют буферный раствор, предпочтительно, чтобы концентрация буферного средства составляла 5-300 мМ, и более предпочтительной является концентрация 10-100 мМ. Не существует конкретных ограничений в отношении pH внутренней фазы липосом, однако предпочтительными являются значения от 3 до 11, и более предпочтительными - от 4 до 9.
Липосомальная композиция
По настоящему изобретению предложена липосомальная композиция. Липосомальная композиция содержит липосомы, а также содержит эрибулин и так далее во внутренней фазе липосом. Как упоминалось выше, липосомальная композиция включает как твердую форму, так и жидкую форму. В случае когда липосомы находятся в твердой форме, их можно переводить в жидкую форму путем растворения или суспендирования их в установленном растворителе, как описано ниже. В случае когда липосомальная композиция находится в замороженной твердой форме, ее можно переводить в жидкую форму, оставляя ее для оттаивания при комнатной температуре.
Концентрацию липосом и концентрацию активного соединения в липосомальной композиции можно соответствующим образом регулировать в соответствии с назначением липосомальной композиции, препаратом и так далее. В случае когда липосомальная композиция представляет собой жидкий препарат, концентрацию липосом как концентрацию всех липидов, составляющих липосомы, можно устанавливать в пределах от 0,2 до 100 мМ, и предпочтительно - от 1 до 30 мМ. Концентрация (доза) активного соединения в случае когда липосомальную композицию используют в качестве лекарственного средства, описана ниже. Что касается количества циклодекстрина в липосомальной композиции, предпочтительно, чтобы оно было меньше, чем 0,1 моль-эквивалент относительно эрибулина и так далее, и наиболее предпочтительно, чтобы оно было меньше, чем предел обнаружения.
В липосомальной композиции по настоящему изобретению эрибулин и так далее может быть распределен в липидный бислой.
Не существует конкретных ограничений в отношении растворителя (дисперсионной среды) в липосомальной композиции в случае когда липосомальная композиция представляет собой жидкий препарат, и можно упомянуть, например, буферные растворы, такие как фосфатный буферный раствор, цитратный буферный раствор и фосфатно-солевой буферный физиологический раствор, физиологический солевой раствор и культуральную среду для культивирования клеток. Не существует конкретных ограничений в отношении pH внешней фазы липосом, однако предпочтительными являются значения от 3 до 11, и более предпочтительными - от 4 до 9.
В липосомальную композицию можно также добавлять следующее: моносахариды, такие как глюкоза, галактоза, манноза, фруктоза, инозит, рибоза и ксилоза; дисахариды, такие как лактоза, сахароза, целлобиоза, трегалоза и мальтоза; трисахариды, такие как раффиноза и мелицитоза; полисахариды, такие как циклодекстрин; и сахарные спирты, такие как эритрит, ксилит, сорбит, маннит и мальтит; поливалентные спирты, такие как глицерин, диглицерин, полиглицерин, пропиленгликоль, полипропиленгликоль, этиленгликоль, диэтиленгликоль, триэтиленгликоль, полиэтиленгликоль, моноалкиловый эфир этиленгликоля, моноалкиловый эфир диэтиленгликоля, 1,3-бутиленгликоль. Можно также использовать сочетания сахара и спирта.
В целях стабильного долгосрочного хранения липосом, диспергированных в растворителе (дисперсионной среде), с точки зрения физической стабильности, включая коагуляцию и так далее, желательно, насколько возможно, удалять электролит из растворителя (дисперсионной среды). Более того, с точки зрения химической стабильности липидов желательно устанавливать значения рН растворителя (дисперсионной среды) от кислых до близких к нейтральным (pH 3,0-8,0) и удалять растворенный кислород барботированием азотом.
Не существует конкретных ограничений в отношении концентрации сахара или поливалентного спирта, содержащегося в липосомальной композиции, однако в ситуации, когда, например, липосомы диспергированы в растворителе, предпочтительно, чтобы концентрация сахара составляла от 2 до 20% (вес/объем), и более предпочтительно - от 5 до 10% (вес/объем). Что касается концентрации поливалентного спирта, предпочтительными значениями являются от 1 до 5% (вес/объем), и более предпочтительными - от 2 до 2,5% (вес/объем). Эти растворители можно также использовать в качестве внешней фазы липосом в липосомальной дисперсионной жидкости и, заменяя или разбавляя внешнюю фазу липосом предварительного липосомального раствора этими растворителями, можно заменять растворы внешней фазы липосом на эти растворы.
Предпочтительно, чтобы твердые препараты липосомальной композиции включали, например, моносахариды, такие как глюкоза, галактоза, манноза, фруктоза, инозит, рибоза и ксилоза; дисахариды, такие как лактоза, сахароза, целлобиоза, трегалоза и мальтоза; трисахариды, такие как раффиноза и мелицитоза; полисахариды, такие как циклодекстрин, и сахарные спирты, такие как эритрит, ксилит, сорбит, маннит и мальтит.Более предпочтительными являются смеси глюкозы, лактозы, сахарозы, трегалозы и сорбита. Еще более предпочтительными являются смеси лактозы, сахарозы и трегалозы. В этом случае твердые препараты могут стабильно храниться в течение длительного периода времени. При замораживании предпочтительно, чтобы твердые препараты содержали поливалентные спирты (водные растворы), такие как глицерин, диглицерин, полиглицерин, пропиленгликоль, полипропиленгликоль, этиленгликоль, диэтиленгликоль, триэтиленгликоль, полиэтиленгликоль, моноалкиловый эфир этиленгликоля, моноалкиловый эфир диэтиленгликоля и 1,3-бутиленгликоль. Что касается поливалентных спиртов (водных растворов), предпочтительными являются глицерин, пропиленгликоль и полиэтиленгликоль, и более предпочтительными - глицерин и пропиленгликоль. В этом случае возможно стабильно хранить твердые препараты в течение длительных периодов времени. Сахара и поливалентные спирты можно использовать в сочетании.
Способ производства липосомальной композиции
По настоящему изобретению предложен способ производства липосомальной композиции, содержащей эрибулин или его фармакологически приемлемую соль. Способ производства липосомальной композиции включает этап, в котором получают липосомальную дисперсионную жидкость, содержащую липосомы; этап, в котором вышеуказанную липосомальную дисперсионную жидкость смешивают с вышеуказанным активным соединением (эрибулин или его фармакологически приемлемая соль); и этап, в котором вышеуказанное активное соединение вводится во внутреннюю фазу липосом вышеуказанной липосомальной дисперсионной жидкости.
Предпочтительно, если этап, в котором получают липосомальную дисперсионную жидкость, содержащую липосомы, включает этап, в котором получают предварительный липосомальный раствор, и этап, в котором внешнюю фазу липосом вышеуказанного предварительного липосомального раствора заменяют или разбавляют.
Предварительный липосомальный раствор можно получать, например, создавая липосомы в растворе, содержащем соль аммония. Получая предварительный липосомальный раствор в растворе, содержащем соль аммония, можно создавать липосомальную дисперсионную жидкость, которая также содержит соль аммония во внутренней фазе липосом.
Не существует конкретных ограничений в отношении раствора, содержащего соль аммония, используемого при приготовлении предварительного липосомального раствора, и можно использовать любой раствор, содержащий соль аммония.
В качестве соли аммония можно упомянуть, например, хлорид аммония, борат аммония, сульфат аммония, формиат аммония, ацетат аммония, цитрат аммония, тартрат аммония, сукцинат аммония и фосфат аммония. Среди них предпочтительными являются сульфат аммония, ацетат аммония, цитрат аммония, тартрат аммония и фосфат аммония; более предпочтительными являются сульфат аммония, цитрат аммония и тартрат аммония; и сульфат аммония является наиболее предпочтительным.
Можно использовать эти соли аммония в комбинации из двух или более.
Концентрацию соли аммония в растворе, содержащем соль аммония, можно соответствующим образом устанавливать в зависимости от количества эрибулина и так далее, подлежащего захвату, и чем выше, тем лучше; предпочтительно 10 мм или более; более предпочтительно 20 мм или более; и еще более предпочтительно 50 мм или более. Что касается рН раствора, содержащего соль аммония, предпочтительно значение от 3 до 9, значение от 4 до 9 более предпочтительно с точки зрения баланса коэффициента захвата и стабильности, и еще более предпочтительно значение от 5 до 8.
Для регулирования рН раствора, содержащего соль аммония, можно использовать регулятор рН. Не существует конкретных ограничений в отношении концентрации отдельных регуляторов рН в растворе, содержащем соль аммония, однако концентрация от 1 до 300 мМ является предпочтительной, и более предпочтительной - концентрация от 5 до 100 мМ.
В качестве регулятора pH можно упомянуть, например, аминокислоты, такие как аргинин, гистидин и глицин; кислоты, такие как аскорбиновая кислота, бензойная кислота, лимонная кислота, глютаминовая кислота, фосфорная кислота, уксусная кислота, пропионовая кислота, винная кислота, углекислота, молочная кислота, борная кислота, малеиновая кислота, фумаровая кислота, яблочная кислота, адипиновая кислота, соляная кислота и серная кислота; соли вышеуказанных кислот, такие как соль натрия, соль калия и соль аммония; а также щелочные соединения (основания), такие как трис-гидроксиметиламинометан, аммиачная вода (аммиак), гидрид натрия и гидрид калия. В качестве регуляторов pH предпочтительными являются гидрид натрия, соляная кислота, аммиачная вода, уксусная кислота, молочная кислота, винная кислота, янтарная кислота, лимонная кислота и фосфорная кислота; более предпочтительными являются гидрид натрия, аммиачная вода, соляная кислота, уксусная кислота, лимонная кислота и фосфорная кислота; и еще более предпочтительными являются гидрид натрия, аммиачная вода, соляная кислота, лимонная кислота и фосфорная кислота. В качестве регуляторов pH можно использовать две или более из солей аммония в сочетании. Кроме того, в качестве регуляторов рН можно также использовать буферные растворы, фосфатный буферный раствор, цитратный буферный раствор и фосфатно-солевой буферный физиологический раствор.
В качестве предварительного липосомального раствора лучше всего использовать раствор, который получен путем изготовления липосом без существующего включения циклодекстрина. В предварительном липосомальном растворе внутренняя фаза липосом также может содержать соль, кислоту, основание и/или аминокислоту. В этом случае предпочтительно, чтобы внутренняя фаза липосом содержала активное соединение, соль аммония и кислоту. В качестве соли аммония предпочтительным примером может являться сульфат аммония; в качестве кислоты предпочтительным примером может являться лимонная кислота.
Что касается получения липосом, можно упомянуть метод липидной пленки (метод вихрей), метод обращенно-фазового выпаривания, ультразвуковой метод, предвезикулярный метод, метод инъекции этанола, метод френч-пресса, метод удаления желчной кислоты, метод обработки в объеме с Тритоном X-100, метод слияния с Ca2+, метод инъекции эфира, метод отжига, метод замораживания-оттаивания и так далее.
Различные условия (количество мембранных компонентов, температуру и так далее) при получении липосом можно соответствующим образом выбирать в зависимости от метода получения липосом, состава целевых липосом, размера частиц и так далее (см. Op.cit, Kikuchi (1983), и так далее).
Размер липосомальных частиц можно дополнительно корректировать по мере необходимости. Размер частиц можно корректировать, например, путем проведения экструзии (экструзионной фильтрации) под высоким давлением с помощью мембранного фильтра с обычным диаметром пор. Корректировку размеров частиц можно проводить в любое время в процессе производства липосомальной композиции по настоящему изобретению. Например, ее можно проводить до корректировки внешней фазы липосом в предварительном липосомальном растворе, после корректировки внешней фазы липосом в предварительном липосомальном растворе или после введения активного соединения во внутреннюю фазу липосом. Предпочтительно проводить корректировку размера частиц до введения активного соединения во внутреннюю фазу липосом и более предпочтительно проводить ее до корректировки внешней фазы липосом в предварительном липосомальном растворе.
Липосомальную дисперсную жидкость можно получать путем замены или разбавления внешней фазы полученного предварительного липосомального раствора. Замену или разбавление внешней фазы липосом можно проводить один раз, либо различные виды методов замены или разбавления в сочетании можно применять несколько раз.
В качестве метода замены внешней фазы липосом предварительного липосомального раствора можно упомянуть диализ, центрифугирование и гель-фильтрацию. Путем замены внешней фазы липосом настоящее изобретение можно реализовать таким образом, чтобы внешняя фаза липосом по существу не содержала циклодекстрин или соль аммония. Более того, путем замены или разбавления внешней фазы липосом можно добиться эффективного захвата эрибулина или его фармакологически приемлемой соли во внутреннюю фазу липосом.
Диализ можно проводить, например, с помощью диализной мембраны. В качестве диализной мембраны можно упомянуть мембрану с номинально отсекаемой молекулярной массой, такую как целлюлозная трубка или Spectra/Por.
Что касается разделения центрифугированием, центробежное ускорение можно доводить предпочтительно до 100000 g или выше, и более предпочтительно до 300000 g или выше. Заменяя внешнюю фазу липосом при помощи центрифугирования, можно также проводить концентрирование липосом одновременно с заменой внешней фазы липосом.
Гель-фильтрацию можно осуществлять, например, путем проведения фракционирования на основе молекулярной массы с использованием, например, колонки с сефадексом или сефарозой.
В качестве растворителя (дисперсионной среды), используемого при замене и/или разбавлении внешней фазы липосом, можно упомянуть, например, раствор сахарозы, физиологический раствор и культуральную среду для культивирования клеток. С помощью этих растворителей можно получать стабильную липосомальную композицию.
Не существует конкретных ограничений в отношении pH указанного растворителя, однако можно устанавливать диапазон значений от 2 до 11; предпочтительно - от 3 до 10, более предпочтительно - от 6 до 10, и еще более предпочтительно - от 7 до 10. Как описано ниже, градиент рН можно использовать для введения эрибулина и так далее во внутреннюю фазу липосом. В этом случае рН растворителя можно устанавливать таким образом, чтобы во внешней фазе липосом было достигнуто целевое значение pH.
Для регулирования pH указанного растворителя можно использовать регулятор pH. Не существует конкретных ограничений в отношении используемой концентрации, однако предпочтительной является концентрация от 1 до 300 мМ, и более предпочтительной - концентрация от 5 до 100 мМ.
В качестве регулятора pH можно упомянуть, например, аминокислоты, такие как аргинин, гистидин, глицин; кислоты, такие как аскорбиновая кислота, бензойная кислота, лимонная кислота, глютаминовая кислота, фосфорная кислота, уксусная кислота, пропионовая кислота, винная кислота, углекислота, молочная кислота, борная кислота, малеиновая кислота, фумаровая кислота, яблочная кислота, адипиновая кислота, соляная кислота и серная кислота; соли вышеуказанных кислот, такие как соль натрия, соль калия и соль аммония; а также щелочные соединения, такие как трис-гидроксиметиламинометан, аммиачная вода, гидрид натрия и гидрид калия. Предпочтительными являются гидрид натрия, соляная кислота, гистидин, винная кислота, янтарная кислота, лимонная кислота и фосфорная кислота; более предпочтительными являются гидрид натрия, соляная кислота, гистидин, винная кислота, лимонная кислота и фосфорная кислота; и еще более предпочтительными являются гидрид натрия, соляная кислота, гистидин и фосфорная кислота.
В целях совершенствования коэффициента захвата эрибулина или его фармакологически приемлемой соли в липосомах коэффициент захвата можно увеличивать путем добавления раствора (солевого раствора), содержащего электролит, во внешнюю фазу липосом для увеличения интенсивности ионов. Не существует конкретных ограничений в отношении электролита (соли), содержащегося во внешней фазе липосом, однако предпочтительными являются хлорид натрия и хлорид калия, и более предпочтительным является хлорид натрия. Можно также использовать физиологический солевой раствор. Более того, в качестве внешней фазы липосом липосомальной дисперсионной жидкости или тому подобного можно включать сахар, электролит и/или аминокислоту, и также можно включать сахар или электролит и аминокислоту. В качестве предпочтительного примера сахара можно упомянуть сахарозу; в качестве предпочтительного примера электролита можно упомянуть физиологический солевой раствор и хлорид натрия; и в качестве предпочтительного примера аминокислоты можно упомянуть гистидин.
Предпочтительно, чтобы полученная липосомальная дисперсионная жидкость по существу не содержала циклодекстрин или соль аммония во внешней фазе липосом и внутренней фазе липосом, однако в настоящем изобретении эрибулин или его фармакологически приемлемую соль можно вводить во внутреннюю фазу липосом даже в случае, если по каким-то причинам циклодекстрин или соль аммония были добавлены во внешнюю фазу липосомальной дисперсионной жидкости, и даже если внешняя фаза липосом липосомальной дисперсионной жидкости содержит циклодекстрин или соль аммония.
Что касается липидной концентрации липосом в липосомальной дисперсионной жидкости, предпочтительной концентрацией является 1-100 мМ, и более предпочтительной - 1-50 мМ. В таких диапазонах можно соответствующим образом сформировать большее число липосомальных частиц без ухудшения физических свойств липосомальной дисперсионной жидкости.
Липосомальную композицию можно получать путем смешивания полученной липосомальной дисперсионной жидкости и активного соединения эрибулина и так далее и путем введения активного соединения во внутреннюю фазу липосом липосомальной дисперсионной жидкости. Предпочтительно, чтобы этап введения включал этап, в котором проницаемость мембран липосом увеличена в смешанном растворе липосомальной дисперсионной жидкости и активного соединения. Благодаря этому захват эрибулина и так далее в липосомы может происходить за более короткий период времени. Однако даже если никаких конкретных действий не предпринято для усиления проницаемости мембран липосом после смешивания липосомальной дисперсионной жидкости и эрибулина и так далее возможно осуществлять захват эрибулина и так далее в липосомы, если затратить необходимое время.
На этапе, в котором примешивают эрибулин или его фармакологически приемлемую соль, можно использовать вещество, растворенное в растворителе, либо твердое вещество, такое как эрибулин и так далее. Не существует конкретных ограничений в отношении растворителя и можно использовать, например, вещество, идентичное внешней фазе липосом липосомальной дисперсионной жидкости.
При необходимости можно использовать градиент рН для введения эрибулина и так далее во внутреннюю фазу липосом. В этом случае что касается рН внутренней фазы липосом липосомальной дисперсионной жидкости, предпочтительными значениями являются от 3 до 9, более предпочтительными - от 4 до 9, и еще более предпочтительными - от 5 до 8.
Более того, можно сделать pH внешней фазы липосом более высоким, чем pH внутренней фазы липосом, для создания градиента pH. Предпочтителен градиент pH от 1 до 5, и более предпочтителен - от 2 до 3.
Кроме того, можно увеличивать коэффициент захвата в липосомах путем доведения pH внешней фазы липосом до значений, более близких к области pKa эрибулина и так далее, предпочтительны значения от 7,5 до 12,5, более предпочтительны - от 8,5 до 11,5, и еще более предпочтительны - от 9 до 10,5 (pKa эрибулин мезилата составляет 9,6).
В качестве предварительного липосомального раствора оптимально использовать раствор, полученный путем создания липосом без существующего включения циклодекстрина.
В качестве метода повышения проницаемости мембран липосом в полученном смешанном растворе можно упомянуть метод нагревания смешанного раствора, метод добавления флюидизатора к смешанному раствору и так далее.
В случае когда смешанный раствор нагревают, активное соединение, как правило, может быть более эффективно введено во внутреннюю фазу липосом путем нагревания до более высоких температур. В частности, предпочтительно устанавливать температуру нагрева с учетом термической стабильности активного соединения и используемых компонентов мембран липосом. Особенно предпочтительно, чтобы температура нагрева была установлена на температуру фазового перехода мембран липидного бислоя или выше.
«Температура фазового перехода» мембран липидного бислоя липосом означает температуру, при которой начинается поглощение тепла (температуру, при которой начинается эндотермическая реакция) в дифференциально-термическом анализе условий повышенных температур. Дифференциально-термический анализ представляет собой метод, позволяющий анализировать тепловые свойства образцов путем измерения разности температур образца или препарата сравнения в зависимости от времени или температуры при изменении температуры образца или препарата сравнения. В случае когда дифференциально-термический анализ проводится по отношению к компонентам мембран липосом, компоненты мембран липосом разжижаются при повышении температуры, и наблюдается эндотермическая реакция. Как широко известно в данной области техники, диапазон температур, в котором наблюдается эндотермическая реакция, значительно варьируется в зависимости от компонентов мембран липосом. Например, в случае, когда компоненты мембран липосом состоят из чистого липида, диапазон температур, в котором наблюдается эндотермическая реакция, чрезвычайно узок, и эндотермическая реакция часто наблюдается в диапазоне ±1°C относительно температуры эндотермического пика. С другой стороны, в случае когда компоненты мембран липосом состоят из нескольких липидов, и, в частности, в случае когда компоненты мембран липосом состоят из липидов, полученных из природных материалов, диапазон температур, в котором наблюдается эндотермическая реакция, имеет тенденцию к расширению, и эндотермическая реакция наблюдается, например, в диапазоне ±5°C относительно температуры эндотермического пика (то есть наблюдается широкий пик и так далее). В соответствии с настоящим изобретением считается, что флюидизация мембран липосом увеличивается и проницаемость мембран для активного соединения увеличивается за счет повышения температуры выше температуры фазового перехода мембран липидного бислоя липосом.
Например, хотя и с учетом зависимости от термической стабильности и так далее активного соединения и используемых компонентов липосомальных мембран, предпочтительно иметь температуру в диапазоне от температуры фазового перехода мембран липидного бислоя липосом до +20°C от температуры фазового перехода; более предпочтительно - температуру в диапазоне от температуры фазового перехода до +10°C от температуры фазового перехода; и еще более предпочтительно - температуру в диапазоне от +5°C от температуры фазового перехода до +10°C от температуры фазового перехода.
Температура нагрева, как правило, составляет от 20 до 100°C; предпочтительно - от 40 до 80°C; и более предпочтительно - от 45 до 65°C.
В частности, в случае липосомальных мембран, основными ингредиентами которых являются дипальмитоилфосфатидилхолин (температура фазового перехода как простого вещества 41°C) и холестерин, хотя также будучи зависимой от их состава, температура нагрева от 40 до 60°C обычно является предпочтительной, и более предпочтительна температура от 45 до 50°C. Кроме того, в случае липосомальных мембран, основными ингредиентами которых являются гидрогенизированный соевый фосфатидилхолин (HSPC; температура фазового перехода как простого вещества 50-60°C) и холестерин, хотя также будучи зависимой от их состава, температура нагрева от 50 до 70°C обычно является предпочтительной, и более предпочтительна температура от 55 до 65°C. Однако эти температуры нагрева никоим образом не ограничивают настоящее изобретение.
На этапе нагревания не существует конкретных ограничений в отношении времени, в течение которого температура поддерживается на уровне или выше температуры фазового перехода, и оно может быть надлежащим образом установлено в диапазоне, например, от нескольких секунд до 30 минут. С учетом термической стабильности активного соединения и липидов, а также эффективного массового производства желательно проводить обработку в течение короткого промежутка времени. То есть предпочтительно, чтобы период поддержания повышенной температуры составлял от 1 до 30 минут, и более предпочтительно - от 2 минут до 5 минут. Однако эти времена поддержания температуры никоим образом не ограничивают настоящее изобретение.
Более того, как было указано выше, также возможно повышать проницаемость мембран липосом путем добавления флюидизатора мембран к полученному смешанному раствору (то есть добавляя его к стороне внешней фазы липосом). В качестве флюидизатора мембран можно упомянуть органические растворители, сурфактанты, ферменты и так далее, которые растворяются в водных растворителях. Более конкретно в качестве органических растворителей можно упомянуть, например, моновалентные спирты, такие как этиловый спирт и бензиловый спирт; поливалентные спирты, такие как глицерин и пропиленгликоль; апротонные полярные растворители, такие как диметилсульфоксид (ДМСО). В качестве сурфактантов можно упомянуть, например, анионные сурфактанты, такие как жирные кислоты натрия, моноалкилсульфат и моноалкилфосфат; катионные сурфактанты, такие как соль алкилтриметиламмония; амфолитические сурфактанты, такие как алкилдиметиламиноксид; и неионные сурфактанты, такие как алкилэфир полиоксиэтилена, алкилмоноглицерил эфир, сложный эфир жирной кислоты и сорбитана. В качестве ферментов можно упомянуть, например, холинэстеразу и холестериноксидазу. Специалисты в данной области могут установить количество флюидизатора мембран в зависимости от состава компонентов мембран липосом, флюидизатора мембран и так далее, а также принимая во внимание степень эффективности захвата активного соединения благодаря добавлению флюидизатора мембран, стабильность липосом и так далее.
Способ производства липосомальной композиции по настоящему изобретению может включать этап регулирования pH внешней фазы липосом полученной липосомальной композиции после вышеуказанного этапа введения вещества.
Не существует конкретных ограничений в отношении pH внешней фазы, но предпочтительными могут быть значения от 4 до 10, более предпочтительными - от 5 до 9, и еще более предпочтительными - нейтральные от 6 до 8, с точки зрения химической стабильности фосфолипидов, составляющих липосомы.
Кроме того, можно дополнительно включать этап высушивания полученной липосомальной композиции. То есть при использовании липосомальной композиции в виде жидкого препарата липосомальную композицию в жидком виде, полученную на вышеуказанном этапе введения вещества, можно использовать без изменений как конечную липосомальную композицию, либо внешнюю фазу липосом в жидкой липосомальной композиции, полученной на вышеуказанном этапе введения вещества, можно корректировать (заменять и так далее) для получения конечной липосомальной композиции. При осуществлении этого корректировку внешней фазы липосом можно проводить аналогично корректировке внешней фазы липосом в предварительной липосомальной жидкости. В случае когда липосомальная композиция представляет собой жидкий препарат, ее можно использовать без дальнейших модификаций.
Кроме того, в случае когда липосомальную композицию предстоит преобразовать в твердый препарат, жидкую липосомальную композицию, полученную на вышеуказанном этапе введения вещества, можно высушивать для получения конечной твердой липосомальной композиции. Сублимационную сушку и распылительную сушку можно привести в качестве примеров методов высушивания липосомальной композиции. В случаях когда липосомальная композиция представляет собой твердый препарат, ее можно растворять или суспендировать в подходящем растворителе и использовать как жидкий препарат. Растворитель для использования можно соответствующим образом выбирать в зависимости от цели использования и так далее липосомальной композиции, и в случае использования липосомальной композиции в качестве, например, инъекционного продукта растворитель предпочтительно представляет собой стерильную дистиллированную воду. В случае использования липосомальной композиции в качестве лекарственного средства врач или пациент может вводить растворитель во флакон, в котором находится твердый препарат, например, чтобы получать препарат непосредственно в момент использования. В случае когда жидкая липосомальная композиция представляет собой замороженный твердый препарат, ее можно использовать в качестве жидкого препарата, храня ее в замороженном виде и вновь переводя в жидкое состояние, оставляя для оттаивания при комнатной температуре или нагревая ее для быстрого оттаивания в момент использования.
Фармацевтические композиции и так далее.
Липосомальную композицию по настоящему изобретению можно использовать в качестве лекарственного препарата в медицинской сфере. В частности, липосомальную композицию по настоящему изобретению можно использовать в качестве противоопухолевой фармацевтической композиции.
В случае когда липосомальную композицию по настоящему изобретению используют в качестве фармацевтической композиции, липосомальную композицию можно вводить с помощью инъекции (внутривенной, внутриартериальной или местной инъекции), перорально, через нос, подкожно, в легкие или с помощью глазных капель и, в частности, местная инъекция в целевую группу клеток или орган или другая подобная инъекция является предпочтительной в дополнение к внутривенной инъекции, подкожной инъекции, внутрикожной инъекции и внутриартериальной инъекции. Таблетки, порошок, гранулы, сироп, капсулы, жидкость и тому подобное можно привести в качестве примеров препарата липосомальной композиции в случае перорального введения. Инъекционный продукт, капельную инъекцию, глазные капли, мази, суппозиторий, суспензию, горячий компресс, лосьон, аэрозоль, пластырь и тому подобное можно привести в качестве примеров препаратов липосомальной композиции в случае неперорального введения, и особенно предпочтительными являются инъекционный продукт и средство для капельного введения.
Дозировка фармацевтической композиции по существу различается в зависимости от типа целевой болезни, типа активного соединения, а также возраста, пола и веса пациента, тяжести симптомов, наряду с другими факторами, но, как правило, суточная доза эрибулина или его фармакологически приемлемой соли для взрослых не имеет особых ограничений, хотя для эрибулин мезилата, который является подходящей солью, она составляет, как правило, от 0,1 до 10 мг.Кроме того, введение можно разделить на более чем одну дозу в сутки. Липосомальную композицию, содержащую, например, 0,01-300 мг/мл эрибулина или его фармакологически приемлемой соли во внутренней фазе липосом, можно вводить как липосомальную композицию по настоящему изобретению.
Набор
По настоящему изобретению предложен набор для получения липосомальной композиции. Набор можно использовать для получения липосомальной композиции в качестве лекарственного средства, которое может быть использовано врачом в клинических условиях или пациентом.
Набор включает липосомальный реагент. Липосомальный реагент может быть как в твердой, так и в жидкой форме. Если липосомальный реагент находится в жидкой форме, то в качестве липосомального реагента можно использовать вышеуказанную липосомальную дисперсионную жидкость. Кроме того, если липосомальный реагент находится в твердой форме, липосомальный реагент можно растворять или суспендировать в соответствующем растворителе для получения липосомальной дисперсионной жидкости, и вышеуказанную липосомальную дисперсионную жидкость можно высушивать для получения липосомального реагента. Высушивание можно проводить аналогично вышеуказанному высушиванию липосомальной композиции. При использовании набора, если липосомальный реагент находится в твердой форме, липосомальный реагент можно растворять или суспендировать в соответствующем растворителе для получения липосомальной дисперсионной жидкости. При этом растворитель аналогичен внешней фазе липосом в вышеуказанной липосомальной дисперсионной жидкости.
Набор по настоящему изобретению дополнительно содержит эрибулин или его фармакологически приемлемую соль (эрибулин мезилат является подходящей солью). Эрибулин или его фармакологически приемлемая соль может находиться либо в твердой либо в жидкой форме (в растворенном или суспендированном в растворителе состоянии). При использовании набора, если эрибулин, и так далее, находится в твердой форме, предпочтительно его растворять или суспендировать в соответствующем растворителе для получения жидкой формы. Растворитель можно соответствующим образом выбирать в зависимости от физических свойств, и тому подобного, эрибулина, и тому подобного, и делать его аналогичным, например, внешней фазе липосом в вышеуказанной дисперсионной жидкости. Набор по настоящему изобретению может включать активное соединение, отличное от эрибулина или его фармакологически приемлемой соли.
В наборе липосомальный реагент и активное соединение могут быть упакованы раздельно либо они могут находиться в твердой форме и быть смешаны.
В случае когда липосомальный реагент находится в твердой форме, за исключением случаев растворения или суспендирования для получения вышеуказанной липосомальной дисперсионной жидкости, набор можно использовать, выполняя этап, аналогичный этапу смешивания липосомальной дисперсионной жидкости и активного соединения, а также введения активного соединения во внутреннюю фазу липосом липосомальной дисперсионной жидкости в способе производства вышеуказанной липосомальной композиции. Таким образом можно производить липосомальную композицию, в которой активное соединение вводят во внутреннюю фазу липосомального реагента.
В случае когда липосомальный реагент и активное соединение оба находятся в твердой форме и упакованы вместе, смесь липосомального реагента и активного соединения соответствующим образом растворяют или суспендируют в растворителе. При этом растворитель аналогичен внешней фазе липосом в вышеуказанной липосомальной дисперсионной жидкости. Тем самым можно создать состояние, в котором липосомальная дисперсионная жидкость и активное соединение смешиваются, после чего использование становится возможным в результате осуществления других этапов при введении активного соединения во внутреннюю фазу липосом липосомальной дисперсионной жидкости в способе производства вышеуказанной липосомальной композиции.
Варианты осуществления
Настоящее изобретение конкретно описано с помощью вариантов осуществления и сравнительных примеров, но не ограничено приведенными ниже вариантами осуществления.
Вариант осуществления 1
Получение водного раствора для внутренней фазы липосом
396,4 мг сульфата аммония и 189,1 мг лимонной кислоты моногидрата растворяли в чистой воде, а затем разбавляли до 15 мл для получения водного раствора 200 мМ сульфата аммония/60 мМ лимонной кислоты. После доведения 2,5 мл водного раствора 200 мМ сульфата аммония/60 мМ лимонной кислоты водным раствором аммиака до рН 5,5 водный раствор для внутренней фазы липосом разбавляли до 5 мл чистой водой.
Получение предварительной липосомальной жидкости
После растворения 317,9 мг гидрогенизированного соевого фосфатидилхолина (производства Lipoid), 116,0 мг холестерина (производства Sigma) и 130,4 мг полиэтиленгликоль 2000-фосфатидилэтаноламина (производства Genzyme, MPEG 2000-дистеароилфосфатидилэтаноламин) в 10 мл хлороформа раствор точно распределяли в три флакона, после чего хлороформ из одного флакона удаляли при пониженном давлении в ротационном испарителе для создания липидной пленки. 5 мл водного раствора для внутренней фазы липосом нагревали до примерно 60°C, добавляли к полученной липидной пленке и все перемешивали для получения предварительной липосомальной жидкости. После обработки предварительной липосомальной жидкости ультразвуковыми волнами в течение 20 минут она было гранулирована экструдером (производства Lipex Biomembranes), нагретым до примерно 65°C, для получения предварительной липосомальной жидкости. Размер частиц липосом в полученной предварительной липосомальной жидкости измеряли с помощью метода динамического рассеяния света и у всех он составлял 90-100 нм.
Получение липосомальной дисперсионной жидкости
При помощи колонок с сефадексом G-50 полученную предварительную липосомальную жидкость элюировали водным раствором 0,9% хлорида натрия/10 мМ гистидина (pH 7,6), заменяя внешнюю фазу липосом водным раствором 0,9% хлорида натрия/10 мМ гистидина. После замены внешней фазы липосом проводили центрифугирование в течение 30 минут при 400000 × g. После центрифугирования осадок повторно диспергировали и водный раствор 0,9% хлорида натрия/10 мМ гистидина использовали для получения 5 мл липосомальной дисперсионной жидкости.
Получение раствора активного соединения
Эрибулин мезилат растворяли в водном растворе 0,9% хлорида натрия/10 мМ гистидина, получая 1 мг/мл эрибулин мезилат.
Получение липосомальной композиции
0,5 мл липосомальной дисперсионной жидкости и 0,5 мл раствора эрибулин мезилата смешивали в 10 мл стеклянном флаконе и инкубировали в течение 3 минут в воде с температурой 55°C для получения липосомальной композиции с введенным в липосомы эрибулин мезилатом.
Измерение коэффициента захвата
Коэффициент захвата определяли, как описано ниже.
Липосомальную композицию с захваченным активным соединением подвергали ультрацентрифугированию в течение 30 минут при 400000 × g. Концентрацию активного вещества в фильтрате измеряли методом ВЭЖХ, определяя количество активного соединения, не заключенного в липосомы. Коэффициент захвата рассчитывали при помощи приведенной ниже формулы.
Формула 1

Коэффициент захвата эрибулин мезилата составлял 90,9%.
Вариант осуществления 2
Получение водного раствора для внутренней фазы липосом
Аналогично варианту осуществления 1, 264,3 мг сульфата аммония и 126,1 мг лимонной кислоты моногидрата растворяли в чистой воде и, используя градуированную колбу, разбавляли этот раствор до 10 мл для получения водного раствора 200 мМ сульфата аммония/60 мМ лимонной кислоты. Из него отбирали 1 мл и доводили до рН 5,5 аммиачной водой, после чего разбавляли чистой водой до 2 мл, получая водный раствор для внутренней фазы липосом.
Получение предварительной липосомальной жидкости
80 мг каждого компонента липидной смеси (гидрогенизированный соевый фосфатидилхолин: холестерин: полиэтиленгликоль 2000-фосфатидилэтаноламин=58,6:19,2:22,2 (по весу)) взвешивали, 2 мл водного раствора для внутренней фазы липосом нагревали примерно до 80°C и добавляли к ним, и все это перемешивали для получения предварительной липосомальной жидкости. Эту предварительную липосомальную жидкость гранулировали экструдером (производства Lipex Biomembranes), нагретым до примерно 80°C, для получения предварительной липосомальной жидкости.
Получение липосомальной дисперсионной жидкости
Полученную предварительную липосомальную жидкость разбавляли до 10 мл водным раствором 0,9% хлорида натрия/10 мМ гистидина (pH 7,6) и центрифугировали в течение 30 минут при 400000 × g. После центрифугирования весь фильтрат удаляли. Осадок повторно диспергировали в водном растворе 0,9% хлорида натрия/10 мМ гистидина и, используя градуированную колбу, получали 1 мл липосомальной дисперсионной жидкости.
Получение раствора лекарственного средства
Эрибулин мезилат (эрибулин мезилат) растворяли в водном растворе 0,9% хлорида натрия/10 мМ гистидина и получали 5 мг/мл раствор эрибулин мезилата.
Получение липосомальной композиции
0,96 мл липосомальной дисперсионной жидкости и 0,24 мл раствора эрибулин мезилата смешивали в 10 мл стеклянном флаконе, и смесь инкубировали в течение 3 минут в воде с температурой 60°C для получения липосомальной композиции с введенным в липосомы эрибулин мезилатом.
Стабильность в плазме крови крыс
0,2 мл полученных липосом с захваченным эрибулин мезилатом и 1,8 мл плазмы крови крыс смешивали и встряхивали при 37°C, используя жидкофазный инкубатор. Сразу же после подготовки отбор проб проводили через 6 часов, 12 часов, 24 часов, 48 часов и 72 часов после начала встряхивания, и остаточное количество эрибулин мезилата в липосомах измеряли методом ВЭЖХ.
Результаты измерений представлены на фигуре 1. Как видно на фигуре 1, эрибулин мезилат стабильно сохранялся в плазме крови даже в течение длительного промежутка времени в течение 120 часов, и было возможным постепенное высвобождение.
Вариант осуществления 3
Получение водного раствора для внутренней фазы липосом
264,3 мг сульфата аммония и 126,1 мг лимонной кислоты моногидрата растворяли в чистой воде, чтобы получить примерно 15 мл. После доведения рН до 7,0 с помощью водного раствора гидроксида натрия раствор разбавляли чистой водой до 20 мл для получения водного раствора для внутренней фазы липосом (100 мМ сульфат аммония/30 мМ лимонная кислота).
Получение предварительной липосомальной жидкости
378 мг липидной смеси (гидрогенизированный соевый фосфатидилхолин: холестерин: полиэтиленгликоль 2000-фосфатидилэтаноламин=58,6:19,2:22,2 (по весу)) взвешивали, 10 мл вышеуказанного водного раствора для внутренней фазы липосом нагревали примерно до 80°C и добавляли к ним, и все это перемешивали для получения предварительной липосомальной жидкости. Эту предварительную липосомальную жидкость гранулировали экструдером (производства Lipex Biomembranes), снабженным 50 нм поликарбонатным мембранным фильтром, и нагревали примерно до 80°C для получения предварительной липосомальной жидкости с размером частиц примерно 80 нм.
Получение липосомальной дисперсионной жидкости
При помощи колонок с сефадексом G-50 полученную предварительную липосомальную жидкость элюировали водным раствором 0,9% хлорида натрия/10 мМ гистидина (pH 7,6), заменяя внешнюю фазу липосом водным раствором 0,9% хлорида натрия/10 мМ гистидина. После замены внешней фазы липосом проводили центрифугирование в течение 30 минут при 400000 × g. После центрифугирования осадок повторно диспергировали в водном растворе 96 мг/мл сахарозы/10 мМ гистидина (pH 7,6) и 10 мл раствора разбавляли до 10 мл, получая липосомальную дисперсионную жидкость.
Получение раствора лекарственного средства
Эрибулин мезилат растворяли в водном растворе 96 мг/мл сахарозы/10 мМ гистидина (pH 7,6) и получали 5 мг/мл раствор эрибулин мезилата.
Получение липосомальной композиции
9,6 мл липосомальной дисперсионной жидкости и 1,2 мл раствора эрибулин мезилата смешивали в 10 мл стеклянном флаконе и использовали гидроксид натрия для доведения рН до 9,5. Раствор инкубировали в течение 3 минут в воде с температурой 60°C для получения липосомальной композиции с введенным в липосомы эрибулин мезилатом. После охлаждения использовали хлорид для доведения рН до 7,5. Как и в варианте осуществления 1, измеряли коэффициент захвата и установили, что он соответствовал 99%.
Вариант осуществления 4
Получение водного раствора для внутренней фазы липосом
Как и в варианте осуществления 1, получали водный раствор 100 мМ сульфата аммония/30 мМ лимонной кислоты (рН 5,5).
Получение предварительной липосомальной жидкости
Гидрогенизированный соевый фосфатидилхолин, холестерин и полиэтиленгликоль 2000-фосфатидилэтаноламин взвешивали в количествах, указанных в таблице 1. После растворения каждого в 3 мл хлороформа хлороформ удаляли при пониженном давлении в ротационном испарителе для создания липидной пленки. 10 мл полученного водного раствора для внутренней фазы липосом нагревали до примерно 80°C, добавляли к полученной липидной пленке и перемешивали для получения предварительной липосомальной жидкости. Ее гранулировали с помощью экструдера (производства Lipex Biomembranes), нагретого примерно до 80°C, для получения гранулированной предварительной липосомальной жидкости. Размер частиц липосом в полученной предварительной липосомальной жидкости измеряли с помощью метода динамического рассеяния света и Rp.1 составлял 77 нм, Rp.2 - 95 нм, Rp.3 - 79 нм и Rp.4 - 128 нм.
Получение липосомальной композиции
Как и в варианте осуществления 1, получали липосомальную дисперсионную жидкость. Также эрибулин мезилат растворяли в водном растворе 0,9% хлорида натрия/10 мМ гистидина и получали 5 мг/мл эрибулин мезилата.
4,8 мл каждой из липосомальных дисперсионных жидкостей и 0,6 мл раствора эрибулин мезилата смешивали в 10 мл стеклянных флаконах, которые инкубировали в течение 3 минут в воде с температурой 60°C для получения липосомальной композиции с введенным в липосомы эрибулин мезилатом. 24,6 мл водного раствора 0,9% хлорида натрия/10 мМ гистидина добавляли в каждую из липосомальных композиций и использовали 0,22 мкм фильтр из поливинилиденфторида (PVDF) для фильтрования и стерилизации, получая образец для введения (концентрация эрибулин мезилата 0,1 мг/мл).
Как и в варианте осуществления 1, измеряли коэффициент захвата и подтверждали, что он составляет как минимум 90% в каждой из прописей.
Самкам «голых» мышей (NU/NU, Charles River Laboratories Japan, Inc.) подкожно инокулировали клетки LOX меланомы человека и через 11 или 12 дней образцы вводили в хвостовые вены из расчета 10 мл/кг массы тела (1,0 мг/кг массы тела для эрибулин мезилата). Забор образцов крови и извлечение опухолевой ткани проводили путем пункции сердца в установленные сроки после введения (15 минут, 30 минут, 1, 2, 4, 8, 12, 24, 36 и 48 часов) (n=3). Кровь собирали в пробирки с гепарином и в течение 30 минут после отбора проб кровь разделяли центрифугированием при 1500 × g в течение 10 минут при 4°C для получения плазмы крови. Все опухолевые ткани извлекали, промывали PBS и вытирали промокательной бумагой, а затем немедленно определяли и записывали вес ткани. Ткань помещали в пробирки и охлаждали в ледяной воде, а затем хранили при −80°C до проведения анализов.
Эрибулин мезилат в плазме крови и в опухолевой ткани измеряли с помощью ЖХ/МС/МС.
Параметры PK рассчитывали, используя программное обеспечение для анализа не-камерной модели (WinNonlin версия 5.0.1). Результаты определения параметров PK в плазме крови и параметров PK в опухолевой ткани для эрибулин мезилата представлены, соответственно, в таблице 2 и таблице 3.
Из таблицы 2 и таблицы 3 можно увидеть, что AUC в плазме крови и в ткани опухоли увеличена по сравнению со свободным эрибулин мезилатом во всех четырех липосомальных композициях Rp.1-4, и вследствие этого количество мигрировавшего к опухоли и сохранение эрибулин мезилата улучшены.
Вариант осуществления 5
Получение водного раствора для внутренней фазы липосом
Как и в варианте осуществления 1, получали водный раствор 100 мМ сульфата аммония/30 мМ лимонной кислоты (рН 5,5).
Получение предварительной липосомальной жидкости
Взвешивали 221,8 мг гидрогенизированного соевого фосфатидилхолина, 72,5 мг холестерина и 86,9 мг полиэтиленгликоль 2000-фосфатидилэтаноламина. После растворения их в 3 мл хлороформа хлороформ удаляли при пониженном давлении в ротационном испарителе и получали липидную пленку. 10 мл полученного водного раствора для внутренней фазы липосом нагревали до примерно 80°C, добавляли к полученной липидной пленке и перемешивали для получения предварительной липосомальной жидкости. Ее гранулировали с помощью экструдера (производства Lipex Biomembranes), нагретого примерно до 80°C, и получали гранулированную предварительную липосомальную жидкость. Когда размеры частиц липосом в полученной предварительной липосомальной жидкости измеряли с помощью метода динамического рассеяния света, они составили примерно 90 нм.
Получение липосомальной дисперсионной жидкости
При помощи колонок с сефадексом G-50 полученную предварительную липосомальную жидкость элюировали водным раствором 0,9% хлорида натрия/10 мМ гистидина (pH 7,6), заменяя внешнюю фазу липосом водным раствором 0,9% хлорида натрия/10 мМ гистидина. После замены внешней фазы липосом проводили центрифугирование в течение 30 минут при 400000 × g. После центрифугирования осадок повторно диспергировали и водный раствор 0,9% хлорида натрия/10 мМ гистидина использовали для получения 10 мл липосомальной дисперсионной жидкости.
Получение раствора лекарственного средства
Эрибулин мезилат растворяли в водном растворе 0,9% хлорида натрия/10 мМ гистидина и получали 1 мг/мл раствор эрибулин мезилата. Кроме того, в качестве образцов свободного основного вещества для введения раствор эрибулин мезилата разбавляли водным раствором 0,9% хлорида натрия/10 мМ гистидина и использовали 0,22 мкм PVDF фильтр для фильтрования и стерилизации, чтобы получить образцы для введения (концентрации эрибулин мезилата 0,3 мг/мл и 0,4 мг/мл).
Получение липосомальной композиции
1,8 мл липосомальной дисперсионной жидкости и 1,2 мл раствора эрибулин мезилата смешивали в 10 мл стеклянном флаконе, который инкубировали в течение 3 минут в воде с температурой 60°С для получения липосомальной композиции с введенным в липосомы эрибулин мезилатом. Полученную липосомальную композицию разбавляли водным раствором 0,9% хлорида натрия/10 мМ гистидина и использовали 0,22-мкм PVDF фильтр для фильтрования и стерилизации, чтобы получить образец для введения (концентрация эрибулин мезилата 0,2 мг/мл). Как и в варианте осуществления 1, измеряли коэффициент захвата и подтверждали, что он составляет как минимум 90%.
Клетки FaDu (полученные из Американской коллекции типовых культур), которые представляют собой линию фарингеальной плоскоклеточной карциномы человека, культивировали и выращивали в культуральной среде MEM, содержащей 10% бычьей фетальной сыворотки. Клетки отделяли от матраса с помощью 0,05% раствора трипсина-ЭДТА и собирали. После промывания PBS клетки суспендировали в PBS до плотности 5×107 клеток/мл и хранили на льду. 0,1 мл жидкой клеточной суспензии подкожно вводили в правую часть брюшной области 6-недельных «голых» мышей (Charles River Laboratories Japan, Inc.). Каждую мышь ежедневно осматривали и делали соответствующие пометки в тех случаях, когда обнаруживали аномальное состояние. Для измерения размеров опухоли с течением времени использовали штангенциркули и размер опухоли рассчитывали на основе формулы расчета: большая ось × (малая ось в квадрате) ÷2. В момент когда размер опухоли составлял от 100 до 200 мм3, мышей разделяли на группы так, чтобы средние значения размеров опухоли и вес тела мышей были одинаковыми среди экспериментальных групп (пять мышей в экспериментальной группе), и лекарственное средство вводили в хвостовую вену (0,2 мл/20 г, 3 раза через 7-дневные интервалы).
Полученные изменения в среднем объеме опухоли после введения образца представлены на фигуре 2.
Как показано на фигуре 2, эффект уменьшения опухолей не был получен даже при дозе 4 мг/кг массы тела, которая является максимально переносимой дозой для свободного основного вещества, поскольку FaDu является линией клеток с низкой чувствительностью к эрибулин мезилату. Между тем, в случае липосомального композита четкий эффект уменьшения опухолей наблюдали даже при введении дозы 2 мг/кг массы тела, что ниже максимально переносимой дозы, это указывало на возможность достижения чрезвычайно высокого фармакологического эффекта даже для тех видов рака, против которых эрибулин мезилата не эффективен.
Вариант осуществления 6
Получение водного раствора для внутренней фазы липосом
Как и в варианте осуществления 1, получали водный раствор 100 мМ сульфата аммония/30 мМ лимонной кислоты (рН 5,5).
Получение раствора лекарственного средства
Как и в варианте осуществления 5, получали образцы для введения (концентрации эрибулин мезилата: 0,2 мг/мл, 0,3 мг/мл и 0,4 мг/мл) свободных основных веществ.
Получение липосомальной композиции
За исключением использования водного раствора внутренней фазы липосом [sic: водного раствора для внутренней фазы липосом], полученного, как описано выше, липосомальную композицию (концентрация эрибулин мезилата 0,3 мг/мл) получали как в варианте осуществления 5. Как и в варианте осуществления 1, измеряли коэффициент захвата и подтверждали, что он составляет как минимум 90%.
Клетки ACHN (полученные из Американской коллекции типовых культур), которые представляют собой линию клеток человеческого рака почки, культивировали и выращивали в культуральной среде MEM, содержащей 10% бычьей фетальной сыворотки. Клетки отделяли от матраса с помощью 0,05% раствора трипсина-ЭДТА и собирали. После промывания PBS клетки суспендировали в PBS до плотности 5×107 клеток/мл и хранили на льду. 0,1 мл жидкой клеточной суспензии подкожно вводили в правую часть брюшной области 6-недельных «голых» мышей (Charles River Laboratories Japan, Inc.). Каждую мышь ежедневно осматривали и делали соответствующие пометки в тех случаях, когда обнаруживали аномальное состояние. Для измерения размеров опухоли с течением времени использовали штангенциркули и размер опухоли рассчитывали на основе формулы расчета: большая ось × (малая ось в квадрате) ÷2. В момент когда размер опухоли составлял от 150 до 200 мм3, мышей разделяли на группы так, чтобы средние значения размеров опухоли и вес тела мышей были одинаковыми среди экспериментальных групп (пять мышей в экспериментальной группе), и лекарственное средство вводили в хвостовую вену (0,2 мл/20 г, 3 раза через 7-дневные интервалы).
Полученные изменения в среднем объеме опухоли после введения образца представлены на фигуре 3.
Как показано на фигуре 3, поскольку ACHN является клеточной линией, устойчивой к эрибулин мезилату, не было обнаружено значительной разницы между любой из групп с введением 2 мг/кг, введением 3 мг/кг и введением 4 мг/кг массы тела (максимально переносимая доза) свободного основного вещества и группой, не получавшей лечение, через 45 дней после начала введения образца. Между тем, в группе с введением липосомальной композиции в дозе 3 мг/кг массы тела наблюдали эффект подавления роста опухоли, и значительно меньшее значение объема опухоли имело место по сравнению с группой, не получавшей лечение, и группами с введением свободного основного вещества через 45 дней после начала введения образца. Как следует из полученных данных, возможно задерживать рост опухоли, создавая липосомальный препарат для опухоли, для которой никогда прежде не был получен терапевтический эффект при применении эрибулин мезилата.
Вариант осуществления 7
Получение водного раствора для внутренней фазы липосом
Готовили 12 типов водных растворов для внутренней фазы липосом, приведенных ниже в таблице 4.
Получение предварительной липосомальной жидкости
120 мг липидной смеси (гидрогенизированный соевый фосфатидилхолин: холестерин: полиэтиленгликоль 2000-фосфатидилэтаноламин=58,6:19,2:22,2 (по весу)) взвешивали в пробирки, и 3 мл каждого образца водного раствора для внутренней фазы липосом нагревали до 80°C.
Эту предварительную липосомальную жидкость гранулировали с помощью экструдера, нагретого примерно до 80°C, и получали гранулированную предварительную липосомальную жидкость.
Получение липосомального дисперсионного раствора
При помощи колонок с сефадексом G-50 полученную предварительную липосомальную жидкость элюировали водным раствором 0,9% хлорида натрия/10 мМ гистидина, заменяя внешнюю фазу липосом водным раствором 0,9% хлорида натрия/10 мМ гистидина.
После замены внешней фазы липосом проводили центрифугирование в течение 1 часа при 400000 × g и фильтрат полностью удаляли. Осадок повторно суспендировали в водном растворе 96 мг/мл сахарозы/10 мМ гистидина (pH 7,5) так, чтобы получить примерно 2 мл.
Размер частиц в полученной липосомальной дисперсионной жидкости измеряли с помощью метода динамического рассеяния света и у всех он составлял примерно 80 нм.
Получение раствора лекарственного средства
Эрибулин мезилат растворяли в водном растворе 96 мг/мл сахарозы/10 мМ гистидина и получали 5 мг/мл раствор эрибулин мезилата.
Получение липосомальной композиции
Липосомальную дисперсионную жидкость и раствор эрибулин мезилата смешивали в 10 мл стеклянном флаконе так, чтобы концентрация эрибулин мезилата составляла 0,2 мг/мл и общая концентрация липидов составляла 16 мкмоль/мл. Смесь нагревали в течение 5 минут при 60°С для получения липосомальной композиции с введенным в липосомы эрибулин мезилатом.
Измерение коэффициента захвата
Коэффициент захвата определяли как в варианте осуществления 1 и результаты приведены в таблице 4. Как видно из таблицы 4, независимо от того какую соль аммония использовали во внутренней фазе, коэффициент захвата эрибулин мезилата явно улучшился. В частности, улучшение коэффициента захвата отмечали при использовании сульфата аммония, цитрата аммония, фосфата аммония и тартрата аммония.
Вариант осуществления 8
Получение водного раствора для внутренней фазы липосом
Как и в варианте осуществления 7, готовили водный раствор для внутренней фазы липосом из раствора 100 мМ сульфата аммония/30 мМ водной лимонной кислоты (pH 7,5).
Получение предварительной липосомальной жидкости
Как и в варианте осуществления 7, вышеуказанный водный раствор для внутренней фазы липосом использовали для получения предварительной липосомальной жидкости.
Получение липосомальной дисперсионной жидкости
При помощи колонок с сефадексом G-50 полученную предварительную липосомальную жидкость элюировали водным раствором 0,9% хлорида натрия/10 мМ гистидина, заменяя внешнюю фазу липосом водным раствором 0,9% хлорида натрия/10 мМ гистидина.
После замены внешней фазы липосом проводили центрифугирование в течение 1 часа при 400000 × g, полностью удаляя фильтрат. Осадок повторно суспендировали в водном растворе 96 мг/мл сахарозы/10 мМ гистидина (pH 7,5), внешнюю фазу липосом заменяли водным раствором 96 мг/мл сахарозы/10 мМ гистидина (pH 7,5) и получали липосомальную дисперсионную жидкость. Размер частиц в полученной липосомальной дисперсионной жидкости измеряли с помощью метода динамического рассеяния света и он составлял примерно 80 нм.
Липосомальную дисперсионную жидкость разливали в семь флаконов и известное количество сульфата аммония (доведенного до рН 7,5 при помощи водного раствора гидроксида натрия) добавляли к внешней фазе липосом так, чтобы во флаконах были концентрации, приведенные в таблице 5, и получали липосомальную дисперсионную жидкость, в которой сульфат аммония во внешней фазе липосом был известной концентрации.
Получение раствора лекарственного средства
Эрибулин мезилат растворяли в водном растворе 96 мг/мл сахарозы/10 мМ гистидина и получали 5 мг/мл раствор эрибулин мезилата.
Получение липосомальной композиции
Липосомальную дисперсионную жидкость и раствор эрибулин мезилата смешивали в 10 мл стеклянном флаконе так, чтобы концентрация эрибулин мезилата составляла 0,2 мг/мл и общая концентрация липидов составляла 16 мМ. Смесь нагревали в течение 5 минут при 60°С для получения липосомальной композиции с введенным в липосомы эрибулин мезилатом.
Измерение коэффициента захвата
Коэффициент захвата определяли как в варианте осуществления 1 и результаты приведены в таблице 5. Они свидетельствуют, что если даже 0,4 мМ сульфат аммония присутствует во внешней фазе липосом, коэффициент захвата заметно падает, и в случае присутствия 10 мМ сульфата аммония захвата почти не происходит.
Вариант осуществления 9
Получение водного раствора для внутренней фазы липосом
Как и в варианте осуществления 7, готовили водный раствор для внутренней фазы липосом из раствора 100 мМ сульфата аммония/30 мМ водной лимонной кислоты (pH 7,5).
Получение предварительной липосомальной жидкости
Как и в варианте осуществления 7, вышеуказанный водный раствор для внутренней фазы липосом использовали для получения предварительной липосомальной жидкости.
Получение липосомальной дисперсионной жидкости
При помощи колонок с сефадексом G-50 полученную предварительную липосомальную жидкость элюировали водным раствором 0,9% хлорида натрия/10 мМ гистидина, заменяя внешнюю фазу липосом водным раствором 0,9% хлорида натрия/10 мМ гистидина.
После замены внешней фазы липосом проводили центрифугирование в течение 1 часа при 400000 × g и фильтрат полностью удаляли. Осадок повторно суспендировали в водном растворе 96 мг/мл сахарозы/10 мМ гистидина (pH 7,5), внешнюю фазу липосом заменяли водным раствором 96 мг/мл сахарозы/10 мМ гистидина (pH 7,5) и получали липосомальную дисперсионную жидкость. Размер частиц в полученной липосомальной дисперсионной жидкости измеряли с помощью метода динамического рассеяния света и он составлял примерно 80 нм.
Получение раствора лекарственного средства
Эрибулин мезилат растворяли в водном растворе 96 мг/мл сахарозы/10 мМ гистидина и получали 5 мг/мл раствор эрибулин мезилата.
Получение липосомальной композиции
Липосомальную дисперсионную жидкость и раствор эрибулин мезилата смешивали в 10 мл стеклянном флаконе так, чтобы концентрация эрибулин мезилата составляла 0,2 мг/мл и общая концентрация липидов составляла 16 мМ. Как показано в таблице 6, в каждом случае pH внешней фазы липосом доводили при помощи 1M водного раствора гидроксида натрия. Раствор нагревали в течение 5 минут при 60°C для получения липосомальной композиции с введенным в липосомы эрибулин мезилатом. Затем использовали соляную кислоту для доведения pH внешней фазы до 7,5.
Измерение коэффициента захвата
Коэффициент захвата определяли как в варианте осуществления 1 и результаты приведены в таблице 6. Вместе с ростом рН внешней фазы липосом коэффициент захвата эрибулина по существу увеличивался, достигая значения около 100%.
Вариант осуществления 10
Получение водного раствора для внутренней фазы липосом
Как и в варианте осуществления 7, готовили водный раствор для внутренней фазы липосом из раствора 100 мМ сульфата аммония/30 мМ водной лимонной кислоты (pH 7,5).
Получение предварительной липосомальной жидкости
Как и в варианте осуществления 7, вышеуказанный водный раствор для внутренней фазы липосом использовали для получения предварительной липосомальной жидкости.
Получение липосомальной дисперсионной жидкости
При помощи колонок с сефадексом G-50 полученную предварительную липосомальную жидкость элюировали водным раствором 0,9% хлорида натрия/10 мМ гистидина, заменяя внешнюю фазу липосом водным раствором 0,9% хлорида натрия/10 мМ гистидина.
Липосомальную дисперсионную жидкость разливали в четыре флакона, которые центрифугировали в течение 1 часа при 400000 × g, и фильтрат полностью удаляли. Осадок из двух флаконов повторно суспендировали в водном растворе 96 мг/мл сахарозы/10 мМ гистидина (pH 7,5) и внешнюю фазу липосом заменяли водным раствором 96 мг/мл сахарозы/10 мМ гистидина (pH 7,5). Осадок в оставшихся двух флаконах повторно суспендировали в водном растворе 0,9% натрия хлорида/10 мМ гистидина (pH 7,5) и внешнюю фазу липосом заменяли водным раствором 0,9% натрия хлорида/10 мМ гистидина (pH 7,5). Размер частиц в полученной липосомальной дисперсионной жидкости измеряли с помощью метода динамического рассеяния света и у всех он составлял примерно 80 нм.
Получение раствора лекарственного средства
Эрибулин мезилат растворяли в водном растворе 96 мг/мл сахарозы/10 мМ гистидина и получали 5 мг/мл раствор эрибулин мезилата. Аналогичным образом, эрибулин мезилат растворяли в водном растворе 0,9% натрия хлорида/10 мМ гистидина и получали 5 мг/мл раствор эрибулин мезилата.
Получение липосомальной композиции
Липосомальную дисперсионную жидкость и раствор эрибулин мезилата смешивали в 10 мл стеклянном флаконе так, чтобы концентрация эрибулин мезилата составляла 0,2 мг/мл и общая концентрация липидов составляла 16 мМ. Значение pH внешней фазы липосом в одном из двух флаконов с водным раствором 96 мг/мл сахарозы/10 мМ гистидина (pH 7,5) доводили до 9,5, добавляя гидроксид натрия. Аналогично, значение pH внешней фазы липосом в одном из двух флаконов с водным раствором 0,9% натрия хлорида/10 мМ гистидина (pH 7,5) доводили до 9,5, добавляя гидроксид натрия. Растворы нагревали в течение 5 минут при 60°C для получения липосомальной композиции с введенным в липосомы эрибулин мезилатом.
Измерение коэффициента захвата
Коэффициент захвата определяли как в варианте осуществления 1, и результаты приведены в таблице 7. По сравнению со случаем, когда внешняя фаза липосом представляет собой сахарозу, которая не является электролитом, в случае хлорида натрия, который является электролитом, четко достигается чрезвычайно высокий коэффициент захвата. В дополнение к эффекту электролита применение градиента pH для защелачивания внешней фазы липосом приводит к 100% коэффициенту захвата.
Настоящая заявка основана на японской патентной заявке (Патентная заявка Японии 2009-082521), поданной 30 марта 2009 г., и предварительной патентной заявке Соединенных Штатов Америки (61/164653), и их содержание включено в данный документ посредством ссылки.
Промышленная применимость
Настоящее изобретение способно предложить способ производства липосом с высокой стабильностью удержания активного соединения в сочетании с высоким коэффициентом захвата.
Липосомальную композицию по настоящему изобретению полезно использовать в терапевтических целях за счет фармакологического эффекта эрибулина или его фармакологически приемлемой соли.
Реферат
Настоящее изобретение относится к области фармацевтики и касается липосомальной композиции, содержащей эрибулин или его фармакологически приемлемую соль, а также способа ее получения. Изобретение обеспечивает высокую стабильность липосомальной композиции. 2 н. и 37 з.п. ф-лы, 3 ил., 7 табл., 10 пр.
Формула
этап, на котором получают липосомальную дисперсионную жидкость, содержащую липосомы;
этап, на котором вышеуказанную липосомальную дисперсионную жидкость смешивают с вышеуказанным активным соединением; и этап, на котором вышеуказанное активное соединение вводится во внутреннюю фазу липосом вышеуказанной липосомальной дисперсионной жидкости.
Комментарии