Липиды для композиций для доставки терапевтических агентов - RU2658007C2
Код документа: RU2658007C2
Чертежи




Описание
РОДСТВЕННЫЕ ЗАЯВКИ НА ПОЛУЧЕНИЕ ПАТЕНТА
Настоящая заявка испрашивает приоритет согласно предварительной заявке на патент США №61/657,480, поданной 8 июня 2011 г., которая включена в настоящую заявку в полном объеме посредством ссылки.
Область изобретения
Настоящее изобретение относится к ионизируемым липидам для улучшения доставки терапевтических агентов.
УРОВЕНЬ ТЕХНИКИ
Ряд методик доступен для доставки терапевтического агента в клетку, например, siPHK, нуклеиновые кислоты и т.д. Эти системы включают вирусные и невирусные трансфекционные системы. Невирусные трансфекционные системы могут включать, например, полимеры, липиды, липосомы, мицеллы, дендримеры и наноматериалы. Примеры полимеров, которые ранее были изучены для клеточной трансфекции включают катионные полимеры, такие как поли(L-лизин), полиэтиленимин ("PEI"), хитозан и поли(2-диметиламино)этилметакрилат ("pDMAEMA").
Однако вирусная и невирусная трансфекционная методика имеют недостатки. Например, вирусные системы могут давать высокую эффективность трансфекции, но могут не быть полностью безопасными. Кроме того, вирусные системы могут быть сложными и/или дорогими для получения.
Невирусные трансфекционные системы, например, применяющие катионные полимеры, были предложены для трансфекции плазмидной ДНК в клетки. Однако катионные полимеры могут быть нестабильными и могут быть токсичными для клеток.
Существует потребность в новых соединениях, композициях и способах для применения катионной композиции для улучшения доставки терапевтических агентов в клетки, ткани и организмы.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к ионизируемым липидным соединениям Формулы I

где n и m независимо равны 1, 2, 3 или 4; R1 и R2 независимо представляют собой C10-18 алкил или C12-18 алкенил; X представляет собой -CH2-, S, O, N или отсутствует; L представляет собой C1-4 алкилен; -S-C1-4 алкилен; -O-C1-4 алкилен; -O-C(O)-C1-4 алкилен; -S(O)2-C1-4 алкилен;
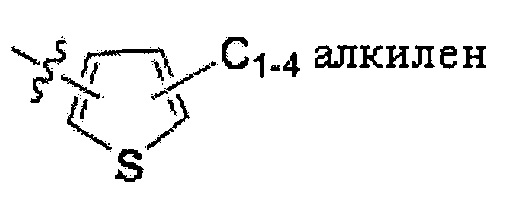


КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На Фиг. 1 показана in vitro синергетическая эффективность вариантов выполнения настоящего изобретения ионизируемый липид: ионизируемый липид.
На Фиг. 2 показана in vitro синергетическая эффективность вариантов выполнения настоящего изобретения ионизируемый липид: ионизируемый липид.
На Фиг. 3 показана in vitro синергетическая эффективность вариантов выполнения настоящего изобретения ионизируемый липид: катионный липид.
На Фиг. 4 показана in vitro синергетическая эффективность вариантов выполнения настоящего изобретения ионизируемый липид: катионный липид.
ПОДРОБНОЕ ОПИСАНИЕ ИЛЛЮСТРАТИВНЫХ ВАРИАНТОВ ВЫПОЛНЕНИЯ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к ионизируемым липидным соединениям, а также их применениям для доставки терапевтических агентов в клетки, ткани и организмы.
В объем настоящего изобретения входят ионизируемые липидные соединения формулы I:

где
n и m независимо равны 1, 2, 3 или 4;
R1 и R2 независимо представляют собой С10-18 алкил или С12-18 алкенил;
X представляет собой -СН2-, S, О, N или отсутствует;
L представляет собой С1-4 алкилен; -S-C1-4 алкилен; -О-С1-4 алкилен; -О-С(O)-С1-4 алкилен;
-S(O)2-C1-4 алкилен;



или их форме в виде фармацевтически приемлемой соли.
Согласно настоящему изобретению, n и m могут быть одинаковыми или различными. В предпочтительных вариантах выполнения настоящего изобретения, n и m являются одинаковыми. Особенно предпочтительными являются варианты выполнения настоящего изобретения, где n и m оба равны 1, или n и m оба равны 2.
В некоторых вариантах выполнения настоящего изобретения, X представляет собой связь. В других вариантах выполнения настоящего изобретения, X представляет собой -CH2-. В еще других вариантах выполнения настоящего изобретения, X представляет собой S. Также предпочтительными являются варианты выполнения настоящего изобретения, где X представляет собой О. Варианты выполнения настоящего изобретения, где X представляет собой N также входят в объем настоящего изобретения.
В некоторых вариантах выполнения настоящего изобретения, L представляет собой C1-4 алкилен. В других вариантах выполнения настоящего изобретения, L представляет собой -S-C1-4 алкилен. В еще других вариантах выполнения настоящего изобретения, L представляет собой -O-C1-4 алкилен. В еще других вариантах выполнения настоящего изобретения, L представляет собой -O-C(О)-C1-4 алкилен. Альтернативно, L представляет собой -S(O)2-C1-4 алкилен. Также в объем настоящего изобретения входят варианты выполнения настоящего изобретения, где L представляет собой

В тех вариантах выполнения настоящего изобретения, где X представляет собой связь, L предпочтительно представляет собой C1-4 алкилен. Такие примеры L включают -CH2-, -CH2-CH2-, -CH2CH2CH2-, и -CH2CH2CH2CH2-. В других вариантах выполнения настоящего изобретения, где X представляет собой связь,
L представляет собой

В тех вариантах выполнения настоящего изобретения, где X представляет собой -CH2-, L предпочтительно представляет собой -S-C1-4 алкилен. Такие примеры L включают -S-CH2-, -S-CH2-CH2-, -S-CH2CH2CH2-, и -S-CH2CH2CH2CH2-.
В других вариантах выполнения настоящего изобретения, где X представляет собой -CH2-, L предпочтительно представляет собой -S(O)2-C1-4 алкилен. Такие примеры L включают -S(O)2-CH2, -S(O)2-CH2-CH2, -S(O)2-CH2-CH2-CH2-, и S(O)2-CH2-CH2-CH2-CH2-.
В еще других вариантах выполнения настоящего изобретения, где X представляет собой -CH2-, L представляет собой -O-С1-4 алкилен. Такие примеры L включают -O-CH2-, -O-CH2-CH2-, -O-CH2CH2CH2-, и -O-CH2CH2CH2CH2-.
В тех вариантах выполнения настоящего изобретения, где X представляет собой S, L предпочтительно представляет собой C1-4 алкилен. Такие примеры L включают -CH2-, -CH2-CH2-, -CH2CH2CH2-, и -CH2CH2CH2CH2-.
В тех вариантах выполнения настоящего изобретения, где X представляет собой O, L предпочтительно представляет собой C1-4 алкилен. Такие примеры L включают -CH2-, -CH2-CH2-, -CH2CH2CH2-, и -CH2CH2CH2CH2-.
Согласно настоящему изобретению, R1 и R2 могут быть одинаковыми или различными. Предпочтительно R1 и R2 являются одинаковыми. Предпочтительно, R1 и R2 представляют собой C10-18 алкил. Также предпочтительными являются варианты выполнения настоящего изобретения, где С10-18 алкил представляет собой неразветвленный С10-18 алкил. Более предпочтительными являются варианты выполнения настоящего изобретения, где R1 и R2 представляют собой С12-18 алкил. Также предпочтительными являются варианты выполнения настоящего изобретения, где R1 и R2 представляют собой C13 алкил. В наиболее предпочтительных вариантах выполнения настоящего изобретения, R1 и R2 оба представляют собой C13 алкил.
В других вариантах выполнения настоящего изобретения, R1 и R2 представляют собой C12-18 алкенил. Предпочтительно, R1 и R2 представляют собой C13-17 алкенил. Более предпочтительно, R1 и R2 каждый представляет собой олеил:

В других вариантах выполнения настоящего изобретения, R1 и R2 представляют собой C12-18 алкенил. Предпочтительно, R1 и R2 представляют собой C13-17 алкенил. Более предпочтительно, R1 и R2 каждый представляет собой линолеоил.

Также в объем настоящего изобретения входят композиции, содержащие соединение формулы I в липосоме, где липосома содержит бислой липидных молекул. Тогда как соединение формулы I может содержать любой мольный процент липидных молекул в таких композициях, предпочтительно, когда соединение формулы I составляет от около 5 до около 50 мол. % от липидных молекул композиций согласно настоящему изобретению.
Композиции согласно настоящему изобретению, содержащие соединение формулы I в липосоме, могут содержать более одного соединения формулы I. В предпочтительных вариантах выполнения настоящего изобретения, такие композиции согласно настоящему изобретению включают два соединения формулы I. В этих вариантах выполнения настоящего изобретения, предпочтительно, когда мольное отношение двух соединений формулы I составляет от около 10:30 до около 30:10.
Композиции согласно настоящему изобретению, содержащие соединение формулы I в липосоме, могут, кроме того, сдержать катионный липид. В таких вариантах выполнения настоящего изобретения, катионный липид составляет от около 5 до около 40 мол. % от липидных молекул композиции. Также в этих вариантах выполнения настоящего изобретения, содержащих соединение формулы I в липосоме с катионным липидом, мольное отношение соединения формулы I к катионному липиду составляет от около 5:35 до около 35:5. Более предпочтительно, отношение составляет от около 10:30 до около 30:10.
Согласно настоящему изобретению, любые композиции согласно настоящему изобретению могут, кроме того, содержать жидкую среду. Предпочтительно, жидкая среда является подходящей для инъекции в живой организм. В некоторых вариантах выполнения настоящего изобретения, жидкая среда содержит органический растворитель. Альтернативно, жидкая среда, применяемая в определенных вариантах выполнения настоящего изобретения, содержит воду и органический растворитель. В других вариантах выполнения настоящего изобретения, жидкая среда может, кроме того, содержать неводную среду.
Также согласно настоящему изобретению, любые композиции согласно настоящему изобретению могут, кроме того, содержать по меньшей мере один фосфолипид.
В других вариантах выполнения настоящего изобретения, любые композиции согласно настоящему изобретению могут, кроме того, содержать по меньшей мере один ПЭГ-конъюгированный липид.
Также в объем настоящего изобретения входят специфичные к звездчатым клеткам лекарственные носители. Эти варианты выполнения настоящего изобретения включают любые вышеуказанные композиции, а также специфичное для звездчатых клеток количество нацеливающей молекулы, содержащей (ретиноид)n-линкер-(ретиноид)n, где n=0, 1, 2 или 3; и где линкер содержит полиэтиленгликоль (ПЭГ) или ПЭГ-подобную молекулу.
В предпочтительных вариантах выполнения настоящего изобретения лекарственные носители согласно настоящему изобретению дополнительно содержат молекулу siPHK.
Также в объем настоящего изобретения входят фармацевтические композиции. Фармацевтические композиции согласно настоящему изобретению включают любой вышеуказанный лекарственный носитель согласно настоящему изобретению и фармацевтически приемлемый носитель или разбавитель. Предпочтительно, когда в таких композициях siPHK инкапсулирована липосомой композиций согласно настоящему изобретению.
Также в объем настоящего изобретения входят способы доставки лекарственного средства пациенту, нуждающемуся в лечении. Эти способы содержат обеспечение фармацевтической композиции согласно настоящему изобретению и введение фармацевтической композиции пациенту.
Определения
Следующие термины применяются в описании настоящего изобретения.
Как применяется в описании настоящего изобретения, термин «катионный липид» относится к соединению, которое включает по меньшей мере одну липидную составляющую и положительно заряженный четвертичный азот, связанный с противоионом. «Липиды», как понимается в данной области техники, должны содержать гидрофобную алкильную или алкенильную составляющую и карбоновую кислоту или сложноэфирную составляющую. Предпочтительные катионные липиды для применения в настоящем изобретении включают:

и

Как применяется в описании настоящего изобретения, термин «ионизируемый липид» относится к соединению формулы I согласно настоящему изобретению. Эти соединения способны образовывать заряженные частицы при контакте с подходящими противоионами, как например, частицы, которые включают ионизируемый атом водорода.
Как применяется в описании настоящего изобретения, термин "алкил" относится к неразветвленной или разветвленной полностью насыщенной (нет двойных или тройных связей) углеводородной группе, например, группе, имеющей общую формулу -CnH2n+1. Алкильная группа может содержать от 1 до 50 атомов углерода (во всех случаях, когда она появляется в настоящем описании, числовой диапазон, такой как «1-50» относится к каждому целому числу в данном диапазоне; например, термин «1-50 атомов углерода» означает, что алкильная группа может состоять из 1 атома углерода, 2 атомов углерода, 3 атомов углерода и т.д., до 50 атомов углерода включительно, несмотря на то, что настоящее определение также предусматривает появление термина «алкил», где числовой диапазон не указан). Алкильная группа может также представлять собой алкил среднего размера, содержащий от 1 до 30 атомов углерода. Алкильная группа может также представлять собой низший алкил, содержащий от 1 до 5 атомов углерода. Алкильная группа соединений может быть обозначена «С1-С4 алкил» или схожими обозначениями. Только в качестве примера, термин «С1-С4 алкил» означает, что алкильная цепь содержит от одного до четырех атомов углерода, т.е. алкильная цепь выбрана из группы, включающей метил, этил, пропил, изопропил, н-бутил, изобутил, втор-бутил и трет-бутил. Типичные алкильные группы включают, но никоим образом не ограничиваются ими, метил, этил, пропил, изопропил, бутил, изобутил, третичный бутил, пентил, гексил и тому подобное.
Как применяется в описании настоящего изобретения, термин «алкилен» относится к алкандиильной функциональной группе, например, -CH2-, -CH2CH2-, -CH2CH2CH2-, -CH2CH2CH2CH2- и тому подобное.
Как применяется в описании настоящего изобретения, термин "алкенил" относится к алкильной группе, содержащей в линейной или разветвленной углеводородной цепи одну или более двойных связей. Алкенильная группа может не содержать заместители или содержать заместители. В случае, когда алкенильная группа содержит заместители, заместитель (заместители) может быть выбран из тех же групп, описанных выше относительно замещения алкильной группы, если не указано иное. Олеил является примером такой алкенильной группы.
Как применяется в описании настоящего изобретения, термин «фармацевтический носитель» относится химическому соединению, облегчающему введение соединения в клетки или ткани. Например, диметилсульфоксид (ДМСО) представляет собой широко используемый носитель, т.к. он облегчает поглощение многих органических соединений клетками или тканями организма.
Как применяется в описании настоящего изобретения, Как применяется в описании настоящего изобретения, термин «разбавитель» относится к химическим соединениям, разведенным в воде, которые будут растворять представляющую интерес композицию (например, композиция, которая может содержать соединение, ретиноид, второй липид, стабилизирующий агент и/или терапевтический агент), а также стабилизировать биологически активную форму соединения. Соли, растворенные в буферных растворах, используют в качестве разбавителей в данной области техники. Один из широко используемых буферных растворов представляет собой фосфатный буферный раствор, т.к. он имитирует солевые условия крови человека. Поскольку буферные соли могут контролировать pH раствора в низких концентрациях, буферный разбавитель редко модифицирует биологическую активность соединения. В настоящем описании термин «эксципиент» относится к инертному веществу, которое добавляют в композицию для придания, без ограничения, объема, консистенции, стабильности, связывающей способности, смазывания, способности к распаду и т.д. указанной композиции. «Разбавитель» представляет собой вид экциаиента.
«Органические растворители», применяемые согласно настоящему изобретению, по существу известны в данной области техники и включают, например, C1-4 алкиловые спирты, диметилсульфоксид ("ДМСО") и тому подобное.
Как применяется в описании настоящего изобретения, термин "терапевтический агент" относится к соединению, которое после введения млекопитающему в терапевтически эффективном количестве обеспечивает полезный терапевтический эффект для указанного млекопитающего. В настоящем описании терапевтический агент может называться лекарственным средством. Для специалиста в данной области техники очевидно, что термин «терапевтический агент» не ограничен лекарственными средствами, которые были одобрены регулирующим органом. «Терапевтический агент» может быть функционально связан с соединением согласно настоящему изобретению, ретиноидом и/или вторым липидом. Например, второй липид согласно настоящему изобретению может формировать липосому, и терапевтический агент может быть функционально связан с липосомой, например, как описано в настоящей заявке.
Как применяется в описании настоящего изобретения, термин "ретиноид" означает представителя класса соединений, состоящих из четырех изопреноидных звеньев, соединенных в конфигурации голова-к-хвосту, см. G.P. Moss, «Biochemical Nomenclature and Related Documents,» 2nd Ed. Portland Press, pp. 247-251 (1992). «Витамин A» представляет собой родовой дескриптор для ретиноидов, проявляющих в качественном отношении биологическую активность ретинола. Как применяется в описании настоящего изобретения, термин «ретиноид» относится к природным и синтетическим ретиноидам, включая ретиноиды первого поколения, второго поколения и третьего поколения.. Примерами встречающихся в природе ретиноидов являются, но без ограничения к этому, (1) 11-цис-ретиналь, (2) полностью транс-ретинол, (3) ретинилпальмитат, (4) полностью транс-ретиноевую кислоту и (5) 13-цис-ретиноевые кислоты. Кроме того, термин «ретиноид» включает ретинолы, ретинали, ретиноевые кислоты, рексиноиды и их производные.
Как применяется в описании настоящего изобретения термин "конъюгат ретиноида" относится к молекуле, которая включает по меньшей мере одну ретиноидную составляющую. В предпочтительных вариантах выполнения настоящего изобретения конъюгат ретиноида присутствует при концентрации от около 0.3 до около 30 мас. %, на основе общей массы композиции или состава, что эквивалентно от около 0.1 до около 10 моль. %, что эквивалентно мольному отношению от около 0.1 до около 10. Предпочтительно, конъюгат ретиноид представляет собой молекулу ретиноид-линкер-липид или молекулу ретиноид-линкер-ретиноид.
Пример конъюгата ретиноида включает соединения формулы II:

где q, r, и s каждый независимо равен 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10, и их энантиомеры и диастереомеры.
Предпочтительные соединения формулы II включают те, в которых q, r, и s каждый независимо равен 1, 2, 3, 4, 5, 6 или 7. Более предпочтительными являются соединения формулы II, в которых q, r, и s каждый независимо равен 3, 4 или 5. Наиболее предпочтительными являются соединения формулы II, в которых q равно 3, r равно 5, и s равно 3. Одним примером соединения формулы II является

DiVA-irar-DiVA включают стереоизомеры и все энантиомеры и диастереомеры, и должны рассматриваться в объеме настоящего изобретения.
Как применяется в описании настоящего изобретения, термин "молекула ретиноид-линкер-липид" относится к молекуле, которая включает по меньшей мере одну ретиноидную составляющую, прикрепленную к по меньшей мере одной липидной составляющей через по меньшей мере один линкер, такой как, например, ПЭГ составляющая.
Как применяется в описании настоящего изобретения, термин "молекула ретиноид-линкер-ретиноид" относится к молекуле, которая включает по меньшей мере одну ретиноидную составляющую, присоединенную к другой ретиноидной составляющей (которая может быть такой же или отличной) через по меньшей мере один линкер, такой как, например, ПЭГ составляющая.
Как применяется в описании настоящего изобретения, термины "липид" и "липофильный" применяются в описании настоящего изобретения согласно их общеизвестным значениям, как понимается специалистами в данной области техники. Неограничивающие примеры липидов и липофильных групп включают жирные кислоты, стиролы, С2-С50 алкил, С2-С50 гетероалкил, С2-С50 алкенил, С2-С50 етероалкенил, С5-С50 арил, С5-С50 гетероарил, С2-С50 алкинил, С2-С50 гетероалкинил, С2-С50 карбоксиалкенил и С2-С50 карбксигетероалкенил. Жирной кислотой является насыщенная или ненасыщенная длинноцепочечная монокарбоновая кислота, которая содержит, например, 12-24 атомов углерода Липид характеризуется как являющийся по существу не растворимым в воде, имеющим растворимость в воде менее около 0.01% (массовая основа). Согласно настоящему изобретению термины "липидная составляющая" и "липофильная составляющая" относятся к липиду или его части, которая присоединяется к другой группе. Например, липидная группа может присоединяться к другому соединению (например, мономеру) посредством химической реакции между функциональной группой (такой как группа карбоновой кислоты) липида и соответствующей функциональной группой мономера.
Как применяется в описании настоящего изобретения, термин «стволовая клетка» включает звездчатые клетки печени.
Как применяется в описании настоящего изобретения, термин "siPHК" относится к малой интерферирующей РНК, также известной в данной области техники как короткая интерферирующая РНК или сайлесинг РНК. siPHК является классом двухнитиевых молекул РНК, которые оказывают многообразие эффектов, известных в данной области техники, наиболее значимым является влияние на экспрессию специфических генов и экспрессию белка.
Термин "липосома" применяется в настоящей заявке в его общеизвестном значении, как понимается специалистами в данной области техники, и относится к структуре липидного бислоя, который содержит липиды, присоединенные к полярным, гидрофильным группам, которые образуют по существу закрытую структуру в водной среде. В некоторых вариантах выполнения настоящего изобретения, липосома может быть функционально связана с одним или более соединениями, такими как терапевтический агент и ретиноид или конъюгат ретиноида. Липосома может состоять из одного липидного бислоя (то есть униламеллярная), или она может состоять из двух или более сосредоточенных липидных бислоев (то есть мультиламеллярная). Тогда как внутренняя часть липомомы может состоять из множества соединений, внешняя часть липосомы доступна для водной композиции, содержащей липосому. Липосома может быть приблизительно сферической или эллипсоидной по форме.
В некоторых вариантах выполнения настоящего изобретения, siPHK инкапсулируется липосомой, так что siPHK недоступна для водной среды. При инкапсулировании siPHK, липосома будет иметь твердое ядро; такие липосомы, инкапсулирующие siPHK и имеющие твердое ядро, называются в настоящей заявке термином «липидные наночастицы». В других вариантах выполнения настоящего изобретения, siPHK неинкапсулирована липосомой. В таких вариантах выполнения настоящего изобретения, siPHK может быть объединена с внешней поверхностью липосомы путем смешивания предварительно образованных липосом с РНК в водном растворе. В этих вариантах выполнения настоящего изобретения, siPHK доступна для водной среды. Липосомы, имеющие siPHK, связанную только с их внешней поверхностью, называются в настоящей заявке термином «липоплексы».
Композиции согласно настоящему изобретению также могут включать ПЭГ-конъюгированные липиды. ПЭГ-конъюгированные липиды в объем настоящего изобретения включают, среди прочего, известные из уровня техники. Подходящие ПЭГ-липиды включают ПЭГ-фосфолипиды и ПЭГ-церамиды, такие как, например, ПЭГ2000-DSPE, ПЭГ2000-DPPE, ПЭГ2000-DMPE, ПЭГ2000-DOPE, ПЭГ1000-DSPE, ПЭГ1000-DPPE, ПЭГ1000-DMPE, ПЭГ1000-DOPE, ПЭГ550-DSPE, ПЭГ550-DPPE, ПЭГ-55DMPE, ПЭГ-1000DOPE, ПЭГ-ВМл, ПЭГ-холестерин. ПЭГ2000-Церамид, ПЭГ1000-Церамид, ПЭГ750-Церамид, ПЭГ550-Церамид.
Вышеизложенные композиции согласно настоящему изобретению могут включать один или более фосфолипидов, таких как, например, 1,2-дистеароил-sn-глицеро-3-фосфохолин ("DSPC"), дипальмитоилфосфатидилхолин ("DPPC"), 1,2-дипальмитоил-sn-глицеро-3-фосфоэтаноламин ("DPPE"), и 1,2-диолеоил-sn-глицеро-3-фосфоэтаноламин ("DOPE"). Предпочтительно, вспомогательным липидом является DOPE.
Также в объем настоящего изобретения входят фармацевтические композиции, которые включают любые вышеуказанные композиции, а также фармацевтически приемлемый носитель или разбавитель. Фармацевтические композиции согласно настоящему изобретению будут включать по меньшей мере один терапевтический агент. Предпочтительно, терапевтический агент представляет собой siPHK. Предполагается, что любая молекула siPHK может применяться согласно настоящему изобретению. Например, siPHK может включать:


и


В предпочтительных композициях согласно настоящему изобретению, включающих siPHK, siPHK инкапсулирована липосомой. В других вариантах выполнения настоящего изобретения, siPHK может находиться вне липосомы. В этих вариантах выполнения настоящего изобретения, siPHK может объединяться с внешней стороной липосомы.
Также в объем настоящего изобретения входят способы доставки терапевтического агента в организм пациента. Эти способы включают обеспечение фармацевтической композиции, включающей любую вышеуказанную композицию и фармацевтически приемлемый носитель или разбавитель; и введение фармацевтической композиции пациенту.
Другим объектом настоящего изобретения является фармацевтическая композиция, содержащая один или более физиологически приемлемых поверхностно-активных агентов, фармацевтических носителей, разбавителей, эксципиентов и суспендирующих агентов или их комбинацию; и к композиции (например композиция, которая может включать соединение, ретиноид, второй липид, стабилизирующий агент и/или терапевтический агент), как раскрывается в настоящей заявке. Приемлемые дополнительные фармацевтические носители или разбавители для терапевтического использования хорошо известны в фармации и описаны, например, в Remington's Pharmaceutical Sciences, 18th Ed., Mack Publishing Co., Easton, PA (1990), полностью включенном в настоящее описание посредством ссылки. Консерванты, стабилизаторы, красители и тому подобное, могут быть включены в фармацевтическую композицию. Например, бензоат натрия, аскорбиновая кислота и эфиры п-гидроксибензойной кислоты могут быть добавлены в качестве консервантов. Кроме того, могут быть использованы антиоксиданты и суспендирующие агенты. В различных вариантах выполнения настоящего изобретения спирты, эфиры, сульфатированные алифатические спирты и т.д. могут быть использованы в качестве поверхностно-активных агентов; сахароза, глюкоза, лактоза, крахмал, кристаллическая целлюлоза, маннит, легкий безводный силикат, алюминат магния, метасиликат-алюминат магния, синтетический силикат алюминия, карбонат кальция, кислый карбонат натрия, гидрофосфат кальция, карбоксиметилцеллюлоза кальция и т.д. могут быть использованы в качестве эксципиентов; кокосовое масло, оливковое масло, кунжутное масло, арахисовое масло, соевое масло могут быть использованы в качестве суспендирующих агентов или смазывающих веществ; ацетатфталат целлюлозы в качестве производного углевода, такого как целлюлоза или сахар, или сополимер метилацетат-метакрилат в качестве производного поливинила могут быть использованы в качестве суспендирующих агентов; и пластификаторы, такие как эфир-фталаты и т.д. могут быть использованы в качестве суспендирующих агентов.
Фармацевтические композиции согласно настоящему изобретению могут быть введены пациенту-человеку как таковые или в фармацевтических композициях, в которых их смешивают с другими активными компонентами, как в комбинированной терапии, или подходящими фармацевтическими носителями или эксципиентом (эксципиентами). Способы получения и введения соединений согласно настоящей заявке могут быть найдены в "Remington's Pharmaceutical Sciences," Mack Publishing Co., Easton, PA, 18th edition, 1990.
Подходящие пути введения могут включать, например, парентеральную доставку, включающую внутримышечные, подкожные, внутривенные, интрамедуллярные инъекции, а также интратекальные, прямые интравентрикулярные, интраперитонеальные, интраназальные или интраокулярные инъекции. Композиция (например, композиция, которая может включать соединение, ретиноид, второй липид, стабилизирующий агент и/или терапевтический агент) также может вводиться в лекарственных формах с замедленным или контролируемым высвобождением, включающих инъекции веществ замедленного всасывания, осмотические помпы и тому подобное для пролонгированного и/или рассчитанного по времени, импульсного введения с заданной скоростью. Кроме того, путь введения может быть местным или системным.
Фармацевтические композиции могут быть изготовлены известным способом, например, способами традиционного смешивания, растворения, гранулирования, получения драже, растирания в порошок, эмульгирования, инкапсулирования, «захвата» или таблетирования
Фармацевтические композиции могут быть изготовлены любым традиционным способом с использованием одного или более физиологически приемлемых фармацевтических носителей, включающих эксципиенты и вспомогательные вещества, облегчающие переработку активных соединений в препараты, которые могут быть фармацевтически использованы. Подходящий состав зависит от выбранного пути введения. Любые из хорошо известных способов, фармацевтических носителей и эксципиентов могут быть использованы в качестве подходящих и понятных в данной области техники; например, в Remington's Pharmaceutical Sciences, выше.
Лекарственные средства для инъекций могут быть получены в традиционных формах, либо в виде жидких растворов или суспензий, твердых форм, подходящих для растворения или суспендирования в жидкости перед инъекцией, либо в виде эмульсий. Подходящие эксципиенты представляют собой, например, воду, физиологический раствор, декстрозу, маннит, лактозу, лецитин, альбумин, глутамат натрия, гидрохлорид цистеина и т.д. Кроме того, при необходимости фармацевтические композиции для инъекций могут содержать небольшие количества нетоксичных вспомогательных веществ, таких как смачивающие агенты, pH буферные агенты и т.д. Физиологически совместимые буферы включают, но не ограничиваются следующими: раствор Хенкса, раствор Рингера или физиологический буферный раствор. При необходимости, могут быть использованы усиливающие абсорбцию препараты.
Фармацевтические составы для парентерального введения, например, путем болюсной инъекции или непрерывной инфузии, включают водные растворы активных композиций (например, композиция, которая может включать соединение, ретиноид, второй липид, стабилизирующий агент и/или терапевтический агент) в водорастворимой форме. Кроме того, суспензии активных соединений могут быть получены в виде подходящих масляных суспензий для инъекций. Водные суспензии для инъекций могут содержать вещества, повышающие вязкость суспензии, такие как натрий-карбоксиметилцеллюлоза, сорбит или декстран. Возможно, суспензия также может содержать подходящие стабилизаторы или агенты, повышающие растворимость соединений, что позволяет получать высококонцентрированные растворы. Составы для инъекции могут находиться в дозированной лекарственной форме, например в ампулах или в многодозовых контейнерах с добавленным консервантом. Композиции могут находиться в таких формах, как суспензии, растворы или эмульсии в масляных или водных растворителях, и могут содержать вспомогательные вещества, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. В качестве альтернативы активный компонент может находиться в форме порошка для разбавления подходящим растворителем, например, стерильной апирогенной водой перед применением.
В дополнение к составам, описанным ранее, композиции также могут быть изготовлены в виде депо-препарата. Указанные составы длительного действия могут быть введены путем имплантации (например, подкожно или внутримышечно) или путем внутримышечной инъекции. Таким образом, например, композиции (например, композиция, которая может включать соединение, ретиноид, второй липид, стабилизирующий агент и/или терапевтический агент) могут быть изготовлены совместно с подходящими полимерными или гидрофобными веществами (например, в виде эмульсии в подходящем масле), или ионообменными смолами, или в виде умеренно растворимых производных, например, в виде умеренно растворимой соли.
Композиции и составы согласно настоящему изобретению могут также быть приготовлены для местной доставки и могут наноситься на кожу субъекта с применением с применением любых подходящих способов нанесения средства для местной доставки. Например, композиция может наноситься вручную, с применением аппликатора или способом, который включает и то и другое. После нанесения композиция может вводиться в кожу субъекта, например, путем втирания. Нанесение может осуществляться множество раз каждый день или один раз каждый день. Например, композиция может наноситься на кожу субъекта один раз в день, дважды в день или множество раз в день или может наноситься один раз каждые два дня, один раз каждые три дня или около одного раза каждую неделю, один раз за каждые две недели или один раз за каждые несколько недель.
Некоторые варианты выполнения настоящего изобретения направлены на способ доставки терапевтического агента в клетку. Например, некоторые варианты выполнения настоящего изобретения направлены на способ доставки терапевтического агента, такого как siPHK, в клетку. Подходящие клетки для применения согласно способам, описанным в настоящей заявке, включают прокариоты, дрожи или высшие эукариотические клетки, включая растительные и животные клетки (например, клетки млекопитающих). В некоторых вариантах выполнения настоящего изобретения, клетками могут быть клетки фибросаркомы человека (например, НТ1080 клеточная линия). В других вариантах выполнения настоящего изобретения клетками могут быть раковые клетки. Клеточные линии, которые являются модельными системами для рака, могут применяться, включая, но без ограничения к этому, рак молочной железы (MCF-7, MDA-MB-438 клеточные линии), U87 клеточная линия глиобластомы, B16F0 клетки (меланома), HeLa клетки (рак шейки матки), А549 клетки (рак легких), и клеточная линия опухоли крысы GH3 и 9L. В этих вариантах выполнения настоящего изобретения композиции, описанные в настоящем изобретении, могут применяться для трансфекции клетки. Эти варианты выполнения настоящего изобретения могут включать контакт клетки с композицией, описанной в настоящем изобретении, которая включает терапевтического агента, для доставки терапевтического агента в клетку.
В настоящем описании указаны способы лечения состояния, характеризующегося аномальным фиброзом, которые могут включать введение терапевтически эффективного количества композиции согласно настоящему изобретению. Состояния, характеризующиеся аномальным фиброзом, могут включать раковое и/или фиброзное заболевание. Виды раковых заболеваний, которые можно лечить или облегчать композицией согласно настоящему изобретению включают, но без ограничения к этому, рак легкого, рак поджелудочной железы, рак молочной железы, рак печени, рак желудка и рак толстой кишки. В одном варианте выполнения настоящего изобретения раковое заболевание, которое можно лечить или облегчать, представляет собой рак поджелудочной железы. В другом варианте выполнения настоящего изобретения раковое заболевание, которое можно лечить или облегчать, представляет собой рак легкого. Виды фиброзного заболевания, которые можно лечить или облегчать терапевтической композицией, указанной в настоящем описании, включают, но не ограничиваются следующими: фиброз печени, цирроз печени, панкреатит, фиброз поджелудочной железы, кистозный фиброз, рубцевание голосовых связок, фиброз слизистой голосовых связок, фиброз гортани, фиброз легких, идиопатический фиброз легких, кистозный фиброз, миелофиброз, ретроперитонеальный фиброз и нефрогенный системный фиброз. В варианте выполнения настоящего изобретения состояние, которое можно лечить или облегчать, представляет собой фиброз печени.
Композиции или фармацевтические композиции согласно настоящему изобретению могут быть введены субъекту любыми подходящими способами. Неограничивающие примеры способов введения включают в том числе: (a) введение путем инъекции, подкожно, интраперитонеально, внутривенно, внутримышечно, внутрикожно, интраорбитально, интракапсулярно, интраспинально, интрастернально или тому подобное, включая доставку посредством инфузионной помпы; (b) локальное введение, например, путем инъекции напрямую в область почки или сердца, например, путем имплантации депо; а также местное введение; по усмотрению специалиста в данной области для приведения активного соединения в контакт с живой тканью.
Фармацевтические композиции, подходящие для введения, включают композиции (например композиция, которая может включать соединение, ретиноид, второй липид, стабилизирующий агент и/или терапевтический агент), в которых активные компоненты содержатся в количестве, эффективном для достижения намеченной цели. Терапевтически эффективное количество соединений, указанных в настоящем описании, необходимое в качестве дозы, будет зависеть от способа введения, вида животного, включая человека, которого лечат, и физических характеристик конкретного рассматриваемого животного. Доза может быть скорректирована для достижения необходимого эффекта, но будет зависеть от таких факторов, как масса, питание, сопутствующее лечение, и других факторов, очевидных для специалиста в области медицины. Более конкретно, «терапевтически эффективное количество» означает количество соединения, эффективное для предупреждения, уменьшения или облегчения симптомов заболевания или увеличения продолжительности жизни субъекта, которого лечат. Определение терапевтически эффективного количества находится в пределах возможностей специалиста в данной области техники, особенно в свете подробного описания, предложенного в настоящем документе.
Для специалиста в данной области техники очевидно, что подходящая дозировка in vivo для введения и конкретный способ введения будут варьировать в зависимости от возраста, массы и вида млекопитающего, которого лечат, конкретных используемых соединений и конкретного назначения, для которого используют данные соединения. Определение эффективных уровней дозировки, т.е. уровней дозировки, необходимых для достижения необходимого результата, может быть выполнено специалистом в данной области техники с использованием стандартных способов фармакологии. Как правило, клиническое применение продуктов у человека начинают на более низких уровнях дозировки и уровень дозировки увеличивают до достижения необходимого эффекта. В качестве альтернативы, подходящие исследования in vitro могут быть использованы для определения подходящих дозировок и способов введения композиций, идентифицированных настоящими способами с использованием известных способов фармакологии.
В исследованиях с участием животных, не относящихся к человеку, применение потенциальных продуктов начинают на более высоких уровнях дозировки и дозировку уменьшают до тех пор пока становится уже невозможным достичь необходимого эффекта или пока не исчезают неблагоприятные побочные эффекты. Дозировка может широко варьировать в зависимости от необходимых эффектов и терапевтического показания. Как правило, дозы могут составлять от примерно 10 микрограмм/кг до примерно 100 мг/кг массы тела, предпочтительно от примерно 100 мк/кг до примерно 10 мг/кг массы тела. В качестве альтернативы дозы могут быть основаны и рассчитаны исходя из площади поверхности тела пациента, как известно специалисту в данной области техники.
Точный состав, способ введения и дозировка фармацевтических композиций могут быть выбраны лечащим врачом с учетом состояния пациента, (смотрите, например, Fingl et al. 1975, in "The Pharmacological Basis of Therapeutics", полностью включенный в настоящее описание посредством ссылки, в частности гл. 1, стр. 1). Как правило, диапазон доз композиции, вводимой пациенту, может составлять от примерно 0.5 до примерно 1000 мг/кг массы тела пациента. Доза может представлять собой однократную дозу или серию двух или более доз, вводимых в течение одного или более дней, в зависимости от того, что необходимо пациенту. В тех случаях, когда человеческие дозы соединений были определены по меньшей мере для какого-либо состояния, дозы будут приблизительно такими же или составлять от примерно 0.1% до примерно 500%, более предпочтительно от примерно 25% до примерно 250% от определенной человеческой дозы. В случае, когда человеческая доза не определена, как будет в случае вновь открываемых фармацевтических композиций, подходящая человеческая доза может быть получена из значений ED50 или ID50 или других подходящих значений, полученных в результате исследований in vitro или in vivo, определенных исследованиями токсичности и исследованиями эффективности у животных.
Следует отметить, что лечащему врачу известно, как и когда прекращать, прерывать или корректировать введение из-за токсичности или дисфункций органов. Напротив, лечащему врачу также известно, как перевести лечение на более высокие уровни, если не был получен достаточный клинический ответ (исключая токсичность). Величина вводимой дозы в лечении представляющего интерес расстройства будет варьировать в зависимости от тяжести состояния, которое лечат, и способа введения. Тяжесть состояния может, например, быть оценена отчасти стандартными прогностическими способами оценки. Кроме того, доза и, возможно, частота введения дозы, также будут варьировать в зависимости от возраста, массы тела и реакции конкретного пациента. Программа, сопоставимая с описанной выше, может быть использована в ветеринарии.
Несмотря на то, что точная дозировка будет определена в зависимости от конкретного лекарственного средства, в большинстве случаев можно сделать некоторые обобщения относительно дозировки. Ежедневный режим приема для взрослого пациента-человека может представлять собой, например, пероральную дозу, равную от примерно 0.1 мг до примерно 2000 мг каждого активного компонента, предпочтительно от примерно 1 мг до примерно 500 мг, например, от 5 до 200 мг. В других вариантах реализации используют внутривенную, подкожную или внутримышечную дозу каждого активного компонента, равную от примерно 0.01 мг до примерно 100 мг, предпочтительно от примерно 0.1 мг до примерно 60 мг, например, от примерно 1 до примерно 40 мг. В случаях введения фармацевтически приемлемой соли, дозы могут быть рассчитаны в виде свободного основания. В некоторых вариантах реализации композицию вводят от 1 до 4 раз в сутки. В качестве альтернативы, композиции могут быть введены путем непрерывной внутривенной инфузии, предпочтительно в дозировке каждого активного компонента до примерно 1000 мг в сутки. Для специалиста в данной области техники очевидно, что в некоторых ситуациях может быть необходимым введение соединений, указанных в настоящем описании, в количествах, превышающих или даже значительно превышающих вышеприведенный предпочтительный диапазон доз для эффективного и «агрессивного» лечения наиболее агрессивных заболеваний или инфекций. В некоторых вариантах реализации соединения будут вводить на протяжении непрерывной терапии, например, в течение недели или больше, или в течение нескольких месяцев или лет.
Величина дозы и интервал могут быть скорректированы индивидуально для обеспечения уровней активного вещества в плазме, достаточных для поддержания модулирующих эффектов или минимальной эффективной концентрации (МЭК). МЭК будет варьировать для каждого соединения, но может быть определена исходя из данных in vitro. Дозы, необходимые для достижения МЭК, будет зависеть от индивидуальных особенностей и способа введения. Однако анализы ВЭЖХ или биоанализы могут быть использованы для определения концентраций в плазме.
Интервалы между введением лекарственного средства также могут быть определены с использованием значения МЭК. Композиции должны быть введены с использованием режима, при котором поддерживаются уровни в плазме выше МЭК в течение 10-90% времени, предпочтительно 30-90% и наиболее предпочтительно 50-90%.
В случаях местного введения или селективного поглощения, эффективная местная концентрация лекарственного средства может быть не связана с концентрацией в плазме.
Количество вводимой композиции может зависеть от подлежащего лечению субъекта, массы субъекта, тяжести заболевания, способа введения и решения лечащего врача.
Композиции согласно настоящему изобретению (например композиция, которая может включать соединение, ретиноид, второй липид, стабилизирующий агент и/или терапевтический агент) могут быть оценены на предмет эффективности и токсичности с использованием известных способов. Например, токсикология конкретного соединения или подмножества соединений, обладающих некоторыми общими химическими группами, может быть установлена путем определения токсичности in vitro в отношении линии клеток, такой как линия клеток млекопитающего и предпочтительно человека. Результаты указанных исследований часто прогнозируют токсичность у животных, таких как млекопитающие или, в частности, люди. В качестве альтернативы токсичность конкретных соединений в модели у животных, таких как мыши, крысы, кролики или обезьяны, может быть определена с использованием известных способов. Эффективность конкретного соединения может быть определена с использованием некоторых известных способов, таких как способы in vitro, моделей у животных или клинических испытаний с участием человека. Существуют известные модели in vitro почти для каждой группы состояния, включая, но не ограничиваясь следующими: раковое заболевание, сердечнососудистое заболевание и различные виды иммунной дисфункции. Подобным образом подходящие модели у животных могут быть использованы для определения эффективности химических соединений для лечения указанных состояний. При выборе модели для определения эффективности специалист может руководствоваться существующим уровнем техники для выбора подходящей модели, дозы и способа введения, и режима. Конечно, клинические испытания с участием человека также могут быть использованы для определения эффективности соединения у людей.
При необходимости, композиции могут быть предложены в упаковке или устройстве для распыления, которое может содержать одну или более дозированных лекарственных форм, содержащих активный компонент. Указанная упаковка может, например, включать металлическую фольгу или пластиковую пленку, как, например, блистерная упаковка. К упаковке или устройству для распыления может прилагаться инструкция по применению. К упаковке или устройству для распыления также может прилагаться памятка, содержащаяся в контейнере, в форме, предусмотренной государственным органом по контролю производства, применения или продажи фармацевтической продукции, отражающая разрешение государственным органом указанной формы лекарственного средства для введения человеку или использования в ветеринарии. Указанная памятка, например, может представлять собой маркировку, разрешенную Управлением по контролю за качеством пищевых продуктов и лекарственных средств США для лекарственных средств, отпускаемых по рецепту, или вкладыш разрешенного продукта. Композиции, содержащие соединение, полученное в совместимом фармацевтическом носителе, также могут быть получены, помещены в подходящий контейнер и помечены как предназначенные для лечения указанного состояния.
Понятно, что в любом соединении, описанном в настоящем изобретении, имеющем один или более стереоцентров, если абсолютная стереохимия точно не указана, тогда каждый центр может независимо иметь R-конфигурацию или S-конфигурацию или их смесь. Таким образом, соединения согласно настоящему изобретению могут быть энантиомерно чистыми или могут представлять собой стереоизомерные смеси. Кроме того, необходимо понимать, что в любом соединении, имеющем одну или более двойных связей, создающих геометрические изомеры, которые могут быть определены как E или Z, каждая двойная связь может независимо быть E или Z или их смесью. Подобным образом, все таутомерные формы также рассматриваются как включенные.
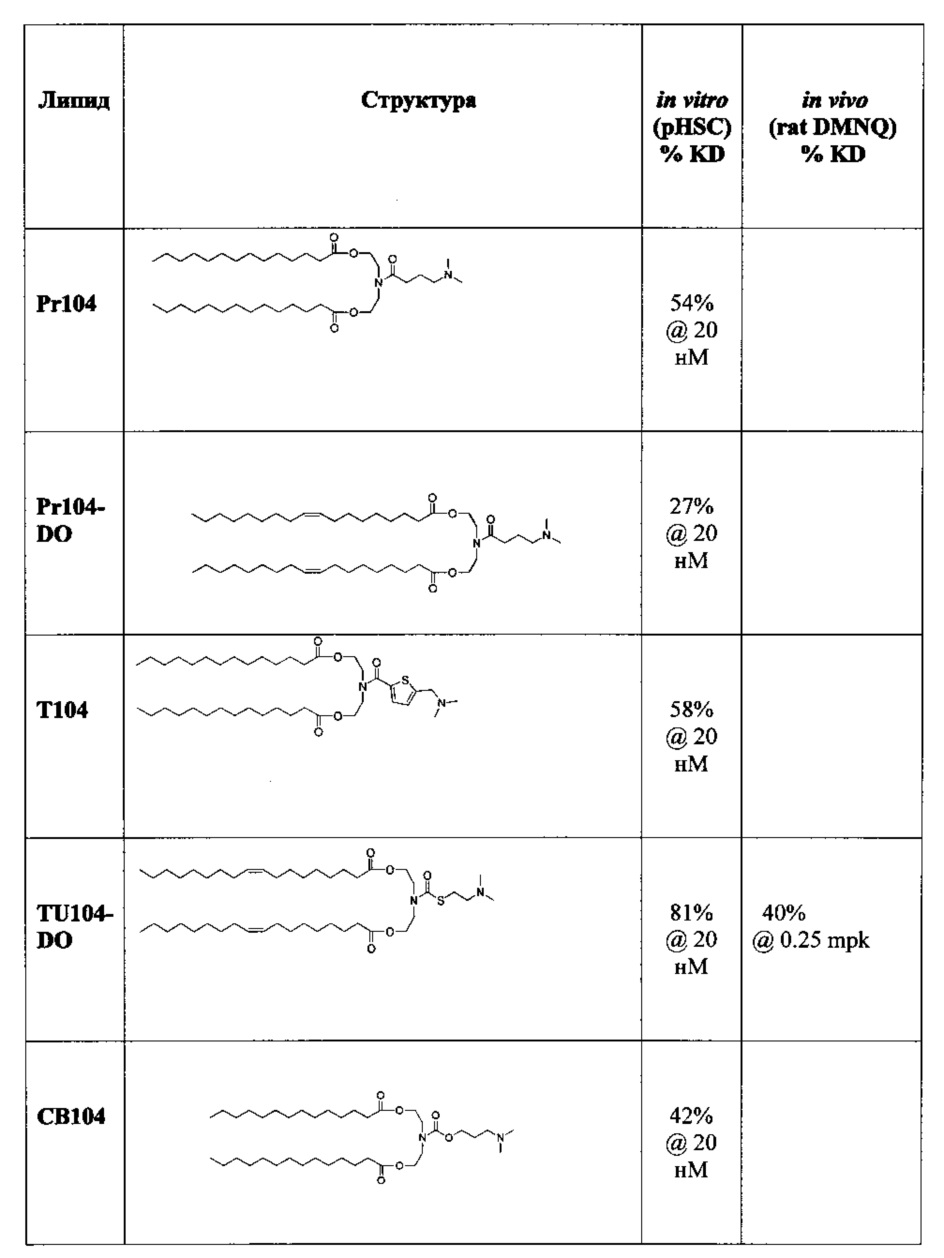
Предпочтительные соединения формулы I в объем настоящего изобретения приведены в Таблице 1. In vitro и in vivo данные (смотрите ниже) также приведены в Таблице 1.





Настоящее изобретение может быть далее проиллюстрировано посредством следующих примеров. Эти примеры являются только иллюстративными и предназначены для ограничения настоящего изобретения.
Примеры
Получение ((2-(диметиламино)ацетил)азанедиил)бис(пропан-3,1-диил)дитетрадеканоата (i-Pr-DC)

Стадия 1: Получение промежуточного соединения 1: 3,3'-азанедиилбис(пропан-1-ол)

Смесь 3-амино-1-пропанола (14.5 мл, 19.0 ммоль), 1-хлор-3-гидрокси пропана (8.00 мл, 95.6 ммоль) и H2O (~50 мл) нагревали до кипения с обратным холодильником в течение 24 часов. Гидроксид калия (5.40 г) затем добавили. После растворения, всю воду выпарили с получением вязкого масла и больших количеств хлорида калия. Отфильтровали и промыли сухим ацетоном и дихлорметаном. Органическую фазу высушили над Na2SO4, отфильтровали и выпарили с получением масла. Очищение посредством хроматографии на силикагеле при элюировании с помощью градиента DCM/MeOH дало 3,3'-азанедиилбис(пропан-1-ол) (12.5 г).
Стадия 2: Получение промежуточного соединения 2: трет-бутил бис(3-гидроксипропил)карбамат

3,3'-азанедиилбис(пропан-1-ол) (12.5 г, 95.4 ммоль) разбавили DCM (25 мл). Раствор ди-трет-бутил дикарбоната (26.0 г, 119 ммоль) в DCM (25 мл) медленно добавили при перемешивании под слоем газа аргона. Реакционную смесь перемешивали всю ночь. Реакционную смесь концентрировали. Очищение посредством хроматографии на силикагеле при элюировании с помощью градиента DCM/MeOH дало трет-бутил бис(3-гидроксипропил)карбамат.
Стадия 3: Получение промежуточного соединения 3: ((трет-6утоксикарбонил)азанедиил)бис(пропан-3,1-диил)дитетрадеканоат

Трет-бутил бис(3-гидроксипропил)карбамат (4.00 г, 17.3 ммоль), Et3N (4.8 мл, 34.6 ммоль) и DMAP (529 мг, 4.33 ммоль) растворили в хлороформе (50 мл). При перемешивании на ледяной бане, раствор миристоилхлорида добавили в течение 15 минут. Добавление осуществляли таким образом, что температура реакции не превышала 30°C. Реакционную смесь перемешивали при комнатной температуре всю ночь. На следующий день МеОН (50 мл) и 0.9% соляной раствор (50 мл) добавили, чтобы погасить реакцию. Органический слой отделили и промыли с помощью 1М NaHCO3. Растворитель высушили с помощью Na2SO4, отфильтровали и концентрировали в вакууме с получением ((трет-бутоксикарбонил)азанедиил)бис(пропан-3,1-диил)дитетрадеканоата в виде масла, которое применяли далее без дальнейшей очистки.
Стадия 4: Получение промежуточного соединения 4: азанедиилбис(пропан-3,1-диил)дитетрадеканоат TFA соли

((Трет-бутоксикарбонил)азанедиил)бис(пропан-3,1-диил)дитетрадеканоат (11.3 г, 17.3 ммоль) растворили в TFA/CHCl3 (1:1, 20 мл), и смесь перемешивали при комнатной температуре в течение 15 минут. Вещество затем концентрировали в вакууме. Это повторили еще раз. Вещество затем растворили в DCM и промыли с помощью H2O, высушили с помощью Na2SO4, концентрировали в вакууме и высушивали полностью всю ночь. Реакционную смесь концентрировали. Очищение посредством хроматографии на силикагеле при элюировании с помощью градиента DCM/MeOH дало азанедиилбис(пропан-3,1-диил)дитетрадеканоат TFA соль (7.5 г).
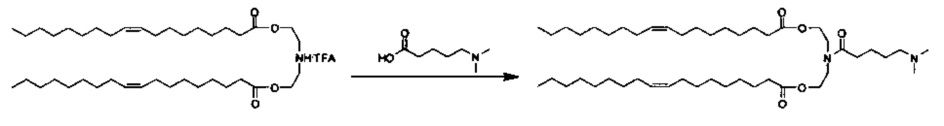
Стадия 5: Получение i-Pr-DC: ((2-(диметиламино)ацетил)азанедиил)бис(пропан-3,1-диил)дитетрадеканоат

Азанедиилбис(пропан-3,1-диил)дитетрадеканоат TFA соль (750 мг, 1.35 ммоль) разбавили DCM (5 мл) и добавили к предварительно активизированной смеси N,N-диметилглицина (154 мг, 1.49 ммоль), HATU (616 мг, 1.62 ммоль) и DIEA (495 мкл, 2.84 ммоль) в DCM (5 мл). Колбу продули аргоном и перемешивали при комнатной температуре всю ночь. Реакционную смесь концентрировали. Очищение посредством хроматографии на силикагеле при элюировании с помощью градиента DCM/MeOH дало ((2-(диметиламино)ацетил)азанедиил)бис(пропан-3,1-диил)дитетрадеканоат (465 мг). QTOF MS ESI+: m/z 639.6 (М+Н).
Получение (Z)-((2-(диметиламино)ацетил)азанедиил)бис(пропан-3,1-диил)диолеата (i-Pr-DODC)

Стадия 1: Получение промежуточного соединения 1: 3,3'-азанедиилбис(пропан-1-ол

Смесь 3-амино-1-пропанола (14.5 мл, 19.0 ммоль), 1-хлор-3-гидрокси пропана (8.00 мл, 95.6 ммоль) и воды (50 мл) нагревали до кипения с обратным холодильником в течение 24 часов. Гидроксид калия (5.40 г) затем добавили. После растворения, всю воду выпарили с получением вязкого масла и больших количеств хлорида калия. Отфильтровали и промыли сухим ацетоном и дихлорметаном. Органическую фазу высушили над Na2SO4, отфильтровали и выпарили с получением масла. Очищение посредством хроматографии на силикагеле при элюировании с помощью градиента DCM/MeOH дало 3,3'-азанедиилбис(пропан-1-ол) (12.5 г).
Стадия 2: Получение промежуточного соединения 2: трет-бутил бис(3-гидроксипропил)карбамат

3,3'-азанедиилбис(пропан-1-ол) (12.5 г, 95.4 ммоль) разбавили DCM (25 мл). Раствор ди-трет-бутил дикарбоната (26.0 г, 119 ммоль) в DCM (25 мл) медленно добавили при перемешивании под слоем газа аргона. Реакционную смесь перемешивали всю ночь. Реакционную смесь концентрировали. Очищение посредством хроматографии на силикагеле при элюировании с помощью градиента DCM/МеОН дало трет-бутил бис(3-гидроксипропил)карбамат.
Стадия 3: Получение промежуточного соединения 3: (Z)-((трет-бутоксикарбонил)азанедиил)бис(пропан-3,1-диил)диолеат

Трет-бутил бис(3-гидроксипропил)карбамат, триэтиламин и DMAP растворили в хлороформе. При перемешивании на ледяной бане, раствор олеилхлорид добавили в течение 15 минут. Добавление осуществляли таким образом, что температура реакции не превышала 30°C. Реакционную смесь перемешивали при комнатной температуре всю ночь. На следующий день МеОН (50 мл) и 0.9% соляной раствор (50 мл) добавили, чтобы погасить реакцию. Органический слой отделили и промыли с помощью 1М NaHCO3. Растворитель высушили с помощью Na2SO4, отфильтровали и концентрировали в вакууме с получением (Z)-((трет-бутоксикарбонил)азанедиил)бис(пропан-3,1-диил)диолеат в виде масла, которое применяли далее без дальнейшей очистки.
Стадия 4: Получение промежуточного соединения 4: (Z)-азанедиилбис(пропан-3,1-диил)диолеат TFA соль

(Z)-((трет-бутоксикарбонил)азанедиил)бис(пропан-3,1-диил)диолеат (13.2 г, 17.3 ммоль) растворили в TFA/CHCl3 (1:1, 20 мл), и смесь перемешивали при комнатной температуре в течение 15 минут. Вещество затем концентрировали в вакууме. Это повторили еще раз. Вещество затем растворили в DCM и промыли с помощью H2O, высушили с помощью Na2SO4 и концентрировали в вакууме. Очищение посредством хроматографии на силикагеле при элюировании с помощью градиента DCM/MeOH дало (Z)-азанедиилбис(пропан-3,1-диил)диолеат TFA соль.
Стадия 5: Получение i-Pr-DODC: (Z)-((2-(диметиламино)ацетил)азанедиил)бис(пропан-3,1-диил)диолеат

(Z)-азанедиилбис(пропан-3,1-диил)диолеат TFA соль (750 мг, 1.13 ммоль) разбавили DCM (5 мл) и добавили к предварительно активизированной смеси N,N-диметилглицина (128 мг, 1.24 ммоль), HATU (517 мг, 1.36 ммоль) и DIEA (413 мкл, 2.37 ммоль) в DCM (5 мл). Колбу продули аргоном и перемешивали при комнатной температуре всю ночь. Реакционную смесь концентрировали. Очищение посредством хроматографии на силикагеле при элюировании с помощью градиента DCM/MeOH дало (Z)-((2-(диметиламино)ацетил)азанедиил)бис(пропан-3,1-диил)диолеат (450 мг). QTOF MS ESI+: m/z 747.7 (М+Н).
Получение ((2-(диметиламино)ацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата, (i-DC)

Стадия 1: Получение промежуточного соединения 1: ((трет-бутоксикарбонил)азанедиил)бис(этан-2,1-диил)дитетрадеканоат

N-Boc диэтаноламин (MW 205.25; 8.4 г, 0.041 моль), триэтиламин (MW 101.19; 11.5 мл, 0.083 моль) и 4-(диметиламино)пиридин (MW 122.17; 1.3 г, 0.011 моль) растворили в хлороформе (170 мл). При перемешивании на ледяной/водной бане, раствор миристоилхлорида (MW 246.82; 22 мл, 80.9 ммоль) в 100 мл хлороформа добавили по каплям. Реакционную смесь затем перенесли на ледяную баню, и перемешивание продолжали при комнатной температуре в течение двух часов. Смесь 200 мл метанола и 200 мл 0.9% соляного раствора добавили, чтобы погасить реакцию. Перемешивание остановили, и органический слой отделили. Растворитель удалили путем роторного испарения с получением ((трет-бутоксикарбонил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата в виде бесцветного масла (25.7 г), которое применяли далее без дальнейшей очистки.
Стадия 2: Получение промежуточного соединения 2: азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соль

К раствору ((трет-бутоксикарбонил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата (33.0 г, 0.053 моль) в 100 мл хлороформа добавили трифторуксусную кислоту (150 мл, 2.02 моль). Реакционную смесь перемешивали при комнатной температуре всю ночь. После того как растворитель удалили путем роторного испарения, полученное полутвердое вещество перекристаллизовали из 80 мл метанола с получением азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соли (16.6 г) в виде твердого вещества белого цвета.
Стадия 3: Получение i-DC: ((2-(диметиламино)ацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоат

Азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соль (10 г, 16 ммоль) разбавили диметилглицином (42.5 g, 25 ммоль), DCC (4.7 г, 23 ммоль) и DIEA (6.33 мл, 40 ммоль) в пиридине (20 мл). Круглодонную колбу продули газом аргоном, и реакционную смесь нагревали до 55°C всю ночь. На следующий день реакционную смесь концентрировали. После очищения посредством хроматографии на силикагеле при элюировании с помощью градиента DCM/MeOH, объединенные фракции концентрировали с получением ((2-(диметиламино)ацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата.
Получение ((3-(диметиламино)пропаноил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата (i-Et-DC, также упоминается в настоящей заявке как Et104)

Получение i-Et-DC: ((3-(диметиламино)пропаноил)азанедиил)бис(этан-2,1-диил)дитетрадеканоат

Синтез азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соли описан ранее. Азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соль (1.50 г, 2.85 ммоль) разбавили DCM (10 мл) и добавили к предварительно активизированной смеси 3-(диметиламино)пропионовой кислоты HCl соли (482 мг, 3.14 ммоль), HATU (1.30 г, 3.42 ммоль) и DIEA (1.04 мл, 5.98 ммоль) в DCM (10 мл). Кругло донную колбу продули аргоном газом, и реакционную смесь перемешивали при комнатной температуре всю ночь. Реакционную смесь концентрировали. После очищения посредством хроматографии на силикагеле при элюировании с помощью градиента DCM/MeOH, объединенные фракции концентрировали и перемешивали в DCM (20 мл) и 10% K2CO3 (20 мл) при 0-5°C в течение 30 минут. Органический слой отделили, и водный слой далее экстрагировали с помощью DCM (2×10 мл). Объединенные органические фазы перемешивали с MgSO4 в течение 30 минут при 0-5°C, отфильтровали, промыли с помощью DCM, и концентрировали с получением ((3-(диметиламино)пропаноил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата (1.01 г). QTOF MS ESI+: m/z 625.6 (М+Н).
Получение (Z)-((3-(диметиламино)пропаноил)азанедиил)бис(этан-2,1-диил)диолеата i-Et-DODC

Стадия 1: Получение промежуточного соединения 1: (Z)-((трет-бутоксикарбонил)азанедиил)бис(этан-2,1-диил)диолеат
N-Boc диэтаноламин (17.8 г, 0.087 моль), триэтиламин (24.4 мл, 0.176 моль) и 4-(диметиламино)пиридин (2.76 г, 0.023 моль) растворили в 350 мл хлороформа. При перемешивании раствор олеилхлорида (61.6 г, 0.174 моль) в 100 мл хлороформа добавили в течение 10 минут (Альтернативно, раствор хлороформа N-Boc диэтаноламина погружали в баню лед/вода при добавлении олеилхлорида). Добавление осуществляли таким образом, что температура реакционной смеси не превышала 50°C. Реакционную смесь перемешивали при комнатной температуре в течение двух часов. Смесь 200 мл метанола и 200 мл 0.9% соляного раствора добавили, чтобы погасить реакцию. Органический слой отделили и промыли с помощью 2×100 мл разбавленного водного бикарбоната натрия. Растворитель удалили путем роторного испарения с получением (Z)-((трет-бутоксикарбонил)азанедиил)бис(этан-2,1-диил)диолеата в виде масла бледно-желтого цвета (59.5 г). Это вещество применяли на следующей стадии без дальнейшей очистки. 1H ЯМР (400 МГц, CDCl3) 0.87 (т, 6Н, CH3), 1.20-1.40 (м, 40Н, CH2), 1.45 (с, 9Н, tBu CH3), 1.59 (м, 4Н, CH2CH2C(=O)), 2.00 (м, 8Н, CH2CH=СН), 2.33 (т, 4Н, CH2C(=O)), 3.48 (м, 4Н, NCH2CH2O), 4.18 (м, 4H, NCH2CH2O), 5.33 (м, 4Н, СН=СН).
Стадия 2: Получение промежуточного соединения 2: (Z)-азанедиилбис(этан-2,1-диил)диолеата TFA соль

(Z)-((трет-бутоксикарбонил)азанедиил)бис(этан-2,1-диил)диолеат (59.5 г, 0.081 моль) дважды обработали с помощью 100 мл трифторуксусной кислоты (100 мл, 1.35 моль) и 100 мл хлороформа. Каждый раз перемешивали при комнатной температуре в течение 10 минут, и растворитель удалили путем роторного испарения в конце каждой обработки. Остаток растворили в 200 мл метиленхлорида, и смесь промыли с помощью 100 мл воды дважды. Остаток очистили с помощью хроматографии на силикагеле с помощью смеси метанола и метиленхлорида в качестве элюента с получением (Z)-азанедиилбис(этан-2,1-диил)диолеат TFA соли (44.0 г). 1H ЯМР (400 МГц, CDCl3) 0.87 (т, 6Н, СН3), 1.20-1.40 (м, 40Н, CH2), 1.59 (м, 4Н, CH2CH2C(=О)), 2.00 (м, 8Н, CH2CH=СН), 2.33 (т, 4Н, CH2C(=O)), 3.31 (м, 4Н, NCH2CH2O), 4.38 (м, 4Н, NCH2CH2O), 5.33 (м, 4Н, СН=СН).
Стадия 3: Получение i-Et-DODC: (Z)-((3-(диметиламино)пропаноил)азанедиил)бис(этан-2,1-диил)диолеат

(Z)-азанедиилбис(этан-2,1-диил)диолеат TFA соль (1.50 г, 2.37 ммоль) разбавили DCM (10 мл) и добавили к предварительно активизированной смеси 3-(диметиламино)пропионовой кислоты HCl соли (383 мг, 2.49 ммоль), HATU (1.03 г, 2.72 ммоль) и DIEA (831 мкл, 4.77 ммоль) в DCM (10 мл). Круглодонную колбу продули аргоном газом, и реакционную смесь перемешивали при комнатной температуре всю ночь. Реакционную смесь концентрировали. После очищения посредством хроматографии на силикагеле при элюировании с помощью градиента DCM/MeOH, объединенные фракции концентрировали и перемешивали в DCM (20 мл) и 10% К2СО3 (20 мл) при 0-5°C в течение 30 минут. Органический слой отделили, и водный слой далее экстрагировали с помощью DCM (2×10 мл). Объединенные органические фазы перемешивали с MgSO4 в течение 30 минут при 0-5°C, отфильтровали, промыли с помощью DCM и концентрировали с получением (Z)-((3-(диметиламино)пропаноил)азанедиил)бис(этан-2,1-диил)диолеата. QTOF MS ESI+: m/z 733.6 (М+Н).
Получение ((4-(диметиламино)бутаноил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата [i-Prop-DC (также упоминается в настоящей заявке как Pr104)]


Синтез азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соли описан ранее. Азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соль (1.00 г, 1.90 ммоль) разбавили DCM (5 мл) и добавили к предварительно активизированной смеси 4-(Диметиламино) масляной кислоты HCl соли (382 мг, 2.28 ммоль), HATU (867 мг, 2.28 ммоль) и DIEA (728 мкл, 4.18 ммоль) в DCM (5 мл). Кругло донную колбу продули аргоном газом, и реакционную смесь перемешивали при комнатной температуре всю ночь. Реакционную смесь концентрировали. После очищения посредством хроматографии на силикагеле при элюировании с помощью градиента DCM/MeOH, объединенные фракции концентрировали и перемешивали в DCM (20 мл) и 10% K2CO3 (20 мл) при 0-5°C в течение 30 минут. Органический слой отделили и водный слой далее экстрагировали с помощью DCM (2×10 мл). Объединенные органические фазы перемешивали с MgSO4 в течение 30 минут при 0-5°C, отфильтровали, промыли с помощью DCM, и концентрировали с получением ((4-(диметиламино)бутаноил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата. LCMS ESI+: m/z 639.6 (М+Н).
Получение (Z)-((4-(диметиламино)бутаноил)азанедиил)бис(этан-2,1-диил)диолеата [i-Prop-DODC (также упоминается в настоящей заявке как Pr104-DO)]


Синтез (Z)-азанедиилбис(этан-2,1-диил)диолеат TFA соли описан ранее. (Z)-азанедиилбис(этан-2,1-диил)диолеат TFA соль (1.00 г, 1.58 ммоль) разбавили DCM (5 мл) и добавили к предварительно активизированной смеси 4-(Диметиламино) масляной кислоты HCl соли (317 мг, 1.89 ммоль), HATU (719 мг, 1.89 ммоль) и DIEA (606 мкл, 3.48 ммоль) в DCM (5 мл). Круглодонную колбу продули аргоном газом, и реакционную смесь перемешивали при комнатной температуре всю ночь. Реакционную смесь концентрировали. После очищения посредством хроматографии на силикагеле при элюировании с помощью градиента DCM/MeOH, объединенные фракции концентрировали и перемешивали в DCM (20 мл) и 10% K2CO3 (20 мл) при 0-5°C в течение 30 минут. Органический слой отделили, и водный слой далее экстрагировали с помощью DCM (2×10 мл). Объединенные органические фазы перемешивали с MgSO4 в течение 30 минут при 0-5°C, отфильтровали, промыли с помощью DCM, и концентрировали с получением (Z)-((4-(диметиламино)бутаноил)азанедиил)бис(этан-2,1-диил)диолеата. LCMS ESI+: m/z 747.7 (М+Н).
Получение (Z)((2-((2-(диметиламино)этил)тио)ацетил)азанедиил)бис(этан-2,1-диил)диолеата (S104-DO)


Синтез (Z)-азанедиилбис(этан-2,1-диил)диолеат TFA соли описан ранее. (Z)-азанедиилбис(этан-2,1-диил)диолеат TFA соль (4.06 г, 6.41 ммоль) перемешивали в DCM (60 мл) с 10% K2CO3 (30 мл) при 0-5°C. Через 30 минут, органическую фазу отделили, и водную фазу далее экстрагировали с помощью DCM (30 мл). Объединенные органические фазы перемешивали с MgSO4 в течение 30 минут при 0-5°C, отфильтровали и промыли с помощью DCM (~30 мл). К объединенным фильтратам добавили 2-((2-(диметиламино)этил)тио)уксусную кислоту (1.26 г, 7.70 ммоль), EDC HCl соль (1.84 г, 9.62 ммоль), DMAP (78.3 мг, 0.64 ммоль), и тонкую суспензию перемешивали всю ночь при комнатной температуре. На следующий день H2O (60 мл) и МеОН (30 мл) добавили, и после перемешивания в течение 10 минут, отделили прозрачную органическую фазу. Мутную водную фазу экстрагировали с помощью DCM. Объединенные органические экстракты концентрировали. Неочищенное вещество отфильтровали через слой силикагеля и растворили в DCM (40 мл) и добавили PBS (pH=11, 50 мл). Смесь перемешивали при комнатной температуре в течение ~10 минут. Затем органическую фазу отделили, и водную фазу снова экстрагировали с помощью DCM (15 мл). Объединенные органические фазы высушили (MgSO4) в течение 30 минут, отфильтровали, промыли с помощью DCM, и концентрировали с получением (Z)-((2-((2-(диметиламино)этил)тио)ацетил)азанедиил)бис(этан-2,1-диил)диолеата (3.44 г). LCMS ESI+: m/z 780.2 (М+Н).
Получение ((5-(диметиламино)пентаноил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата (C104)


Синтез азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соли описан ранее. Азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соль (730 мг, 1.14 ммоль) перемешивали в DCM (20 мл) с 10% K2CO3 (10 мл) при 0-5°C. Через 30 минут, органическую фазу отделили, и водную фазу далее экстрагировали с помощью DCM (10 мл). Объединенные органические фазы перемешивали с MgSO4 в течение 30 минут при 0-5°C, отфильтровали и промыли с помощью DCM (10 мл). К объединенным фильтратам добавили, 5-(диметиламино)пентановую кислоту (248 мг, 1.37 ммоль), EDC HCl соль (328 мг, 1.71 ммоль), DMAP (14 мг, 0.114 ммоль), и тонкую суспензию перемешивали всю ночь при комнатной температуре, после чего раствор стал прозрачным. На следующий день H2O (20 мл) и МеОН (10 мл) добавили, и после перемешивания в течение 10 минут, отделили прозрачную органическую фазу. Мутную водную фазу экстрагировали с помощью DCM. Объединенные органические экстракты концентрировали. После очищения посредством хроматографии на силикагеле посредством элюирования с помощью 100% этилацетата, а затем 10% MeOH/DCM, очищенный остаток растворили в DCM (25 мл) и PBS (pH=11, 25 мл). Смесь перемешивали при комнатной температуре в течение 15 минут. Затем органическую фазу отделили, и водную фазу снова экстрагировали с помощью DCM (15 мл). Объединенные органические фазы высушили (MgSO4) в течение 30 минут, отфильтровали, промыли с помощью DCM, и концентрировали с получением ((5-(диметиламино)пентаноил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата (405 мг). LCMS ESI+: m/z 654.1 (М+Н).
Получение ((2-((2-(диметиламино)этил)сульфонил)ацетил)азанедиил)бис(этан-2,1-диил)дитетра-деканоата (SO2-S104)


Синтез ((2-((2-(диметиламино)этил)тио)ацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата, aka S104, описан. К ((2-((2-(диметиламино)этил)тио)ацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоату в круглодонной колбе, продутой аргоном, добавили DCM (10 мл). Раствор охладили с помощью ледяной бани. К нему добавили mCPBA (раствор в DCM) медленно в течение 5 минут. Ледяную баню удалили после добавления, и реакционную смесь перемешивали всю ночь при комнатной температуре. Через 3.5 часа, 2М DMA/THF (4.55 мл) медленно добавили, и реакционную смесь перемешивали всю ночь. Реакционную смесь затем разбавили DCM до 75 мл. Промыли с помощью H2O (2×50 мл) и 10% K2CO3 (50 мл). Все водные отмывки подвергли обратной экстракции с применением DCM (40 мл). Объединенные органические фазы высушили (MgSO4), отфильтровали, и концентрировали с получением бесцветного масла. Реакционную смесь концентрировали. Очищение посредством хроматографии на силикагеле при элюировании с помощью градиентаэтил ацетата/МеОН дало ((2-((2-(диметиламино)этил)сульфонил)ацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоат (540 мг). LCMS ESI+: m/z 704.0 (М+Н).
Получение ((((2-(диметиламино)этил)тио)карбонил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата (TU104)

Стадия 1: Получение промежуточного соединения 1: азанедиилбис(этан-2,1-диил)дитетрадеканоат

Синтез азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соли описан ранее. Азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соль растворили в DCM (50 мл), и PBS (рН=11, 50 мл) добавили. Смесь перемешивали при комнатной температуре в течение 15 минут. Затем органическую фазу отделили, и водную фазу снова экстрагировали с помощью DCM (25 мл). Объединенные органические фазы высушили (MgSO4) в течение 30 минут, отфильтровали, промыли с помощью DCM, и концентрировали с получением азанедиилбис(этан-2,1-диил)дитетрадеканоата в виде свободного основания.
Стадия 2: Получение TU104: ((((2-(диметиламино)этил)тио)карбонил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата

Трихлорметил хлорформиат (aka дифосген) (257 мкл, 2.13 ммоль) добавили к раствор 2-(диметиламино)этантиол HCl соли (302 мг, 2.13 ммоль) в сухом DCM (20 мл) и перемешивали под слоем аргона при комнатной температуре в течение 4 часов. Затем DCM и избыток дифосген удалили в вакууме. Азанедиилбис(этан-2,1-диил)дитетрадеканоат свободное основание (1068 мг, 2.03 ммоль), DCM (20 мл) и триэтиламин (580 мкл, 4.16 ммоль) затем добавили. Через 16 часов при комнатной температуре реакционную смесь разбавили DCM и промыли с помощью 1М HCl (75 мл), H2O (75 мл) и PBS (рН=11, 75 мл), высушили (MgSO4), отфильтровали и концентрировали. Очищение посредством хроматографии на силикагеле при элюировании этилацетатом, а затем DCM/MeOH дало ((((2-(диметиламино)этил)тио)карбонил)азанедиил)бис(этан-2,1-диил)дитетрадеканоат (120 мг). LCMS ESI+: m/z 657.5 (М+Н).
Получение ((2-(2-(диметиламино)этокси)ацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата (O104)

Стадия 1: Получение промежуточного соединения 1: ((2-бромацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоат

Синтез азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соли описан ранее. Азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соль (1500 мг, 2.34 ммоль) растворили в DCM (20 мл) и поместили на ледяную баню. Добавили бромацетил бромид (214 мкл, 2.46 ммоль), а затем триэтиламин (685 мкл, 4.91 ммоль). Ледяную баню удалили, и реакционную смесь перемешивали всю ночь при комнатной температуре под слоем инертного газа. На следующий день разбавили DCM до 100 мл. Промыли с помощью 1М HCl (75 мл), H2O (75 мл), насыщенного раствора NaHCO3 (75 мл) и насыщенного соляного раствора (75 мл). Все водные отмывки подвергли обратной экстракции с DCM (25 мл). Высушили органические фазы с помощью MgSO4, отфильтровали и концентрировали в вакууме. Очистили посредством хроматографии на силикагеле и элюирования с применением 100% этилацетата. Объедини и концентрировали фракции с получением ((2-бромацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата (1220 мг).
Стадия 2: Получение О104: ((2-(2-(диметиламино)этокси)ацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата

В круглодонную колбу, оборудованную мешалкой добавили ((2-бромацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоат (1.22 г, 1.87 ммоль), N,N-диметилэтаноламинуте (197 мкл, 1.96 ммоль), иодид калия (6.2 мг, 0.0374 ммоль) и сухой THF (25 мл). Полученный раствор охладили до -40°C. DBU (588 мкл, 3.93 ммоль) добавили по каплям в течение 5 минут, и реакционную смесь нагревали до 0°C в течение двух часов. Реакционную смесь концентрировали. Остаток разбавили DCM, 1М HCl (12 мл) добавили, и двухфазную смесь перемешивали в течение 15 минут. Затем превратили в основание с применением PBS (pH=11). Органический слой отделили, высушили (MgSO4), отфильтровали и концентрировали. Очищение посредством хроматографии на силикагеле при элюировании с применением 100% этилацетата, а затем DCM/MeOH дало ((2-(2-(диметиламино)этокси)ацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоат (53 мг). LCMS ESI+: m/z 655.6 (М+Н).
Получение ((2-((4-(диметиламино)бутаноил)окси)ацетил)азанедиил)бис(этан-2,1-диил)дитетра-деканоата (HEDC-M1)
Стадия 1: Получение промежуточного соединения 1: ((2-(бензилокси)ацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата

Синтез азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соли описан ранее. Азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соль перемешивали в DCM (25 мл) с 10% K2CO3 (12.5 мл) при 0-5°C. Через 30 минут, органический слой отделили, и водный слой далее экстрагировали с помощью DCM (12 мл). Объединенные органические фазы перемешивали с MgSO4 в течение 30 минут при 0-5°C, отфильтровали, промыли с помощью DCM (12 мл). К объединенным фильтратам добавили, бензилоксиуксусную кислоту (402 мкл, 2.81 ммоль), EDC HCl соль (673 мг, 3.51 ммоль), и DMAP (29 мг, 0.234 ммоль). Суспензию перемешивали при комнатной температуре всю ночь. На следующий день H2O (25 мл) и МеОН (12 мл) добавили, и после перемешивания в течение 10 минут отделили прозрачную органическую фазу. Мутную водную фазу экстрагировали с помощью DCM (25 мл). Объединенные органические экстракты высушили с помощью MgSO4, отфильтровали и концентрировали. Очищение посредством хроматографии на силикагеле при элюировании с помощью градиента гексаны/этилацетат дало ((2-(бензилокси)ацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоат (1.28 г).
Стадия 2: Получение промежуточного соединения 2: ((2-гидроксиацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата

((2-(бензилокси)ацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоат (1.28 г, 1.80 ммоль) растворили в круглодонной колбе с МеОН (20 мл). Колбу накрыли и продули аргоном,. 10% Pd/C (135 мг) добавили, и колбу еще раз продули аргоном. Весь воздух удалили посредством вакуумного насоса, и затем добавили 8" баллонов H2 газа. Реакционную смесь интенсивно перемешивали при комнатной температуре. Через 30 минут реакционную смесь отфильтровали (целит), промыли с помощью метанола, концентрировали до получения остатка, растворили в DCM (25 мл) и 10% K2CO3 (25 мл). Смесь перемешивали в течение 15 минут, и затем органический слой отделили. Водную отмывку подвергли обратной экстракции с применением DCM (15 мл). Объединенные органические слои высушили с помощью MgSO4, отфильтровали и концентрировали с получением ((2-гидроксиацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата (990 мг).
Стадия 3: Получение HEDC-M1: ((2-((4-(диметиламино)бутаноил)окси)ацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата

((2-гидроксиацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоат (990 мг, 1.70 ммоль) перемешивали в DCM (20 мл) и 4-диметиламино-масляной кислоте (268 мг), EDC HCl соль (487 мг) и DMAP (21 мг) добавили. Суспензию перемешивали при комнатной температуре всю ночь. На следующий день H2O (20 мл) и МеОН (10 мл) добавили, и после перемешивания в течение 10 минут отделили прозрачную органическую фазу. Мутную водную фазу экстрагировали с помощью DCM (20 мл). Объединенные органические экстракты высушили с помощью MgSO4, отфильтровали и концентрировали. Неочищенное вещество очистили посредством хроматографии на силикагеле при элюировании с помощью градиента DCM/MeOH. Объединенные и концентрированные фракции концентрировали и разбавили DCM (25 мл) и PBS (pH=11, 25 мл). Смесь перемешивали при комнатной температуре в течение 15 минут. Затем органическую фазу отделили, и водную фазу снова экстрагировали с помощью DCM (25 мл). Объединенные органические фазы высушили (MgSO4), отфильтровали, и концентрировали с получением ((2-((4-(диметиламино)бутаноил)окси)ацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата (672 мг). LCMS ESI+: m/z 697.6 (М+Н).
Получение (Z)-((5-(диметиламино)пентаноил)азанедиил)бис(этан-2,1-диил)диолеата (C104-DO)

Синтез (Z)-азанедиилбис(этан-2,1-диил)диолеат TFA соли описан ранее. (Z)-азанедиилбис(этан-2,1-Диил)диолеат TFA соль (1.50 г, 2.37 ммоль) перемешивали в DCM (20 мл) с 10% K2CO3 (10 мл) при 0-5°C. Через 30 минут органическую фазу отделили, и водную фазу далее экстрагировали с помощью DCM (10 мл). Объединенные органические фазы перемешивали с MgSO4 в течение 30 минут при 0-5°C, отфильтровали и промыли с помощью DCM (15 мл). К объединенным фильтратам добавили 5-(диметиламино)пентановую кислоту (516 мг, 2.84 ммоль), EDC HCl соль (681 мг, 3.55 ммоль), DMAP (29 мг, 0.237 ммоль), и суспензию перемешивали всю ночь при комнатной температуре, после чего образовался прозрачный раствор. На следующий день H2O (20 мл) и МеОН (10 мл) добавили, и после перемешивания в течение 10 минут, отделили прозрачную органическую фазу. Мутную водную фазу экстрагировали с помощью DCM. Объединенные органические фазы высушили (MgSO4), отфильтровали и концентрировали. После очищения посредством хроматографии на силикагеле при элюировании с помощью градиента DCM/MeOH, объединенные и концентрированные фракции разбавили DCM (25 мл) и PBS (pH=11, 25 мл). Смесь перемешивали при комнатной температуре в течение ~10 минут. Затем отделили органическую фазу, и водную фазу снова экстрагировали с помощью DCM (15 мл). Объединенные органические фазы высушили (MgSO4), отфильтровали, и концентрировали с получением (Z)-((5-(диметиламино)пентаноил)азанедиил)бис(этан-2,1-диил)диолеата (1.10 г). LCMS ESI+: m/z 761.7 (М+Н).
Получение ((5-((диметиламино)метил)тиофен-2-карбонил)азанедиил)бис(этан-2,1-диил)ди-тетрадеканоата (T104)


Синтез азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соли описан ранее. Азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соль (1004 мг, 1.57 ммоль) перемешивали в DCM (20 мл) с 10% K2CO3 (20 мл) при 0-5°C. Через 30 минут органическую фазу отделили, и водную фазу далее экстрагировали с помощью DCM (10 мл). Объединенные органические фазы высушили с помощью MgSO4 в течение 30 минут при 0-5°C, отфильтровали и промыли с помощью DCM (10 мл). К объединенным фильтратам добавили ((диметиламино)метил)тиофен-2-карбоновую кислоту (350 мг, 1.89 ммоль), EDC HCl соль (452 мг, 2.36 ммоль) и DMAP (19.2 мг, 0.157 ммоль). Суспензию перемешивали при комнатной температуре всю ночь. На следующий день Н2О (20 мл) и МеОН (10 мл) добавили, и после перемешивания в течение 10 минут, отделили прозрачную органическую фазу. Мутную водную фазу экстрагировали с помощью DCM (25 мл). Объединенные органические фазы высушили (MgSO4), отфильтровали, и концентрировали. После очищения посредством хроматографии на силикагеле при элюировании с помощью градиента гексаны/этилацетат, объединенные и концентрированные фракции разбавили DCM (20 мл) и PBS (pH=11, 20 мл). Смесь перемешивали при комнатной температуре в течение ~10 минут. Затем органическую фазу отделили, и водную фазу снова экстрагировали с помощью DCM (15 мл). Объединенные органические фазы высушили (MgSO4) в течение 30 минут, отфильтровали, промыли с помощью DCM, и концентрировали с получением ((5-((диметиламино)метил)тиорпепе-2-карбонил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата (482 мг). LCMS ESI+: m/z 693.6 (М+Н).
Получение (Z)-((((2-(диметиламино)этил)тио)карбонил)азанедиил)бис(этан-2,1-диил)диолеата (TU104-DO)


Синтез (Z)-((трет-бутоксикарбонил)азанедиил)бис(этан-2,1-диил)диолеата описан ранее. (Z)-((трет-бутоксикарбонил)азанедиил)бис(этан-2,1-диил)диолеат (4.20 г, 5.72 ммоль) растворили в DCM (20 мл) и охладили до 0°C на ледяной бане. TFA (20 мл) добавили, и смесь перемешивали под слоем инертного газа в течение 20 минут. Затем реакционную смесь концентрировали в вакууме. Остаток распределили между 10% K2CO3 (20 мл) и DCM (20 мл), и перемешивали на ледяной бане в течение 20 минут. Органический слой отделили, высушили (MgSO4) и отфильтровали. Дифосген (1.38 мл, 11.4 ммоль) добавили к (Z)-азанедиилбис(этан-2,1-диил)диолеату в DCM и перемешивали под слоем инертного газа при комнатной температуре. На следующий день DCM и избыток дифосгена удалили в вакууме. 2-(диметиламино)этантиол HCl соль (4.05 г, 28.6 ммоль) растворили в DCM (50 мл) и триэтиламине (5.2 мл, 37.2 ммоль) и добавили к (Z)-((хлоркарбонил)азанедиил)бис(этан-2,1-диил)диолеатному остатку. Вещество перемешивали всю ночь при комнатной температуре. На следующий день разбавили DCM и промыли с помощью 0.3М HCl (75 мл), H2O (75 мл) и 10% K2CO3 (75 мл). Водные отмывки подвергли обратной экстракции с применением DCM (25 мл). Объединенные органические фазы высушили над MgSO4, отфильтровали и концентрировали. Очищение посредством хроматографии на силикагеле при элюировании с применением DCM/MeOH дало (Z)-((((2-(диметиламино)этил)тио)карбонил)азанедиил)бис(этан-2,1-диил)диолеат (1.90 г). LCMS ESI+: m/z 765.7 (М+Н).
Получение (((3-(диметиламино)пропокси)карбонил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата (CB104)

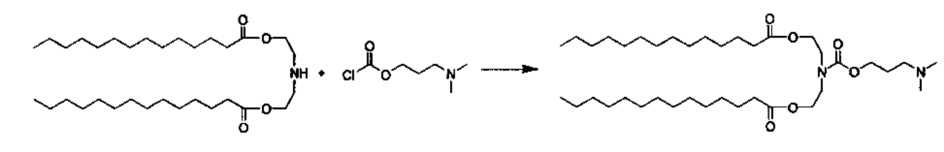
Синтез азанедиилбис(этан-2,1-диил)дитетрадеканоата описан ранее. Дифосген (266 мкл, 2.2 ммоль) добавили к диметиламинопропанолу (413 мг, 4.00 ммоль) в DCM (10 мл) и перемешивали под слоем инертного газа при комнатной температуре в течение 4 часов. DCM и избыток дифосгена удалили в вакууме и азанедиилбис(этан-2,1-диил)дитетрадеканоат добавили. Круглодонную колбу продули аргоном и DCM (10 мл) и триэтиламин (859 мкл, 6.16 ммоль) добавили. Вещество перемешивали всю ночь при комнатной температуре. Реакционную смесь затем разбавили DCM и промыли с помощью 0.3М HCl (75 мл), H2O (75 мл) и 10% K2CO3 (75 мл). Все водные отмывки подвергли обратной экстракции с применением DCM (25 мл). Объединенные органические фазы высушили над MgSO4, отфильтровали и концентрировали. Очищение посредством хроматографии на силикагеле при элюировании с применением DCM/MeOH дало (((3-(диметиламино)пропокси)карбонил)азанедиил)бис(этан-2,1-диил)дитетрадеканоат (87 мг).). LCMS ESI+: m/z 655.59 (М+Н).
Получение (((2-(диметиламино)этокси)карбонил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата (СА104)

Стадия 1: Получение промежуточного соединения 1: (((4-нитрофенокси)карбонил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата

Синтез азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соли описан ранее. Азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соль растворили в сухом DCM (10 мл) и триэтиламин (654 мкл, 4.69 ммоль) добавили. Реакционный сосуд продули инертным газом, и 4-нитрофенилхлорформиат добавили. Вещество перемешивали при комнатной температуре всю ночь. Реакционную смесь погасили водой (50 мл) и DCM (50 мл). Органический слой отделили, и водный слой далее экстрагировали с применением DCM (2×50 мл). Объединенные органические фазы высушили над MgSO4, отфильтровали и концентрировали. Очищение посредством хроматографии на силикагеле при элюировании с применением гексанов/этил ацетата дало (((4-нитрофенокси)карбонил)азанедиил)бис(этан-2,1-диил)дитетрадеканоат.
Стадия 2: Получение CA104: (((2-(диметиламино)этокси)карбонил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата

К (((4-нитрофенокси)карбонил)азанедиил)бис(этан-2,1-диил)дитетрадеканоату добавили 2-диметиламиноэтанол (2 мл) и нагревали до 140°C с конденсационной колонной в течение 20 минут. Затем неочищенное вещество очистили посредством хроматографии на силикагеле при элюировании с помощью градиента DCM/MeOH с получением (((2-(диметиламино)этокси)карбонил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата (38 мг). LCMS ESI+: m/z 641.7 (М+Н).
Получение ((2-(диметиламино)ацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата (INT-4)


Синтез (Z)-((2-(диметиламино)ацетил)азанедиил)бис(этан-2,1-диил)диолеата был проведен подобно i-Prop-DODC с замещением диметилглицина на 3-(диметиламино)пропионовую кислоту. QTOF MS ESI+: m/z 720.1 (М+Н).
Получение ((2-((2-(диметиламино)этил)тио)ацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата (S104)
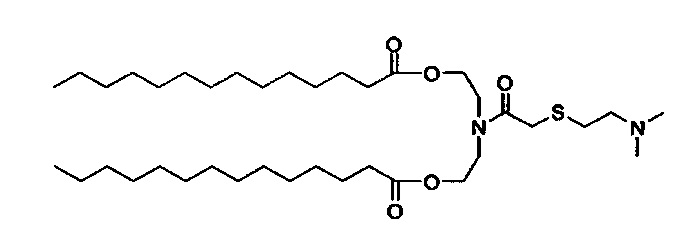
Стадия 1: Получение промежуточного соединения 1: 2-((2-(диметиламино)этил)тио)уксусной кислоты гидрохлорида:

Этанол (500 мл) трижды дегазировали вакуумированием до 60 мБар в течение 1-2 минут и повторно подкачали азотом. Хлоруксусную кислоту (36.9 г, 0.391 моль) добавили, и после перемешивания в течение 5 минут при 17-20°C образовался прозрачный раствор. 2-(Диметиламино)-этантиол гидрохлорид (52.7 г, 0.372 моль) добавили, и после перемешивания в течение 20 минут при 25°C образовался прозрачный раствор. Твердый гидроксид натрия (47.7 г, 1.19 моль) добавили по частям в течение 20 минут при охлаждении для сохранения температуры ниже 35°C - наблюдался непродолжительный период при 44°C. Наблюдалось практически немедленное осаждение. Реакционную смесь со временем нагрели и перемешивали при 40°C в течение двух часов, после чего ТСХ показала завершение реакции. Целит 545 (74 г) добавили, и смесь отфильтровали через G3 стеклянную-спеченную пластину в течение 6 минут, промыли этанолом (2×105 мл). Объединенные мутные фильтраты выпарили на 50°C бане с получением 110 г твердого вещества белого цвета. Твердое вещество растворили в воде (250 мл), затем pH установили от 13.1 (температура 30°C) до 10.5 (температура 31°C), с применением концентрированной HCl (5.5 мл) с получением очень бледного желтоватого раствора. Водную фазу промыли с помощью DCM (3×100 мл) для удаления дисульфидной примеси (потребовалось 3 промывки). Концентрированную HCl добавляли к водной фазе (pH 10.7, температура 22°C) пока значение pH не достигло 1.4 (57.5 мл добавили, температура 35°C). Водную фазу промыли с помощью DCM (100 мл) и затем концентрировали до сухости (температура бани 55°C). Толуол (250 мл) добавили, смесь концентрировали до сухости (температура бани 55°C), и это повторили еще раз с получением влажного твердого вещества белого цвета (98 г). Ацетонитрил (750 мл) добавили к твердому веществу, смесь перемешивали при 55°C в течение 45 минут, и затем отфильтровали через G3 стеклянную-спеченную пластину. Ацетонитрил (250 мл) добавили к остатку на фильтре, смесь перемешивали при 55°C в течение 25 минут и затем отфильтровали через G3 стеклянную-спеченную пластину, промыли ацетонитрилом (50 мл). Объединенные фильтраты концентрировали до 300 мл с получением большого осадка белого цвета. Смесь охладили в атмосфере азота и перемешивали при 0°C в течение 30 минут. Осадок отделили посредством фильтрации через G3 стеклянную-спеченную пластину, и остаток на фильтре промыли с помощью холодного ацетонитрила (100 мл). Высушивание при пониженном давлении в течение 3 дней дало 47.0 г (63%) 2-((2-(диметиламино)этил)тио)уксусной кислоты гидрохлорида.
Стадия 2: Получение S104: ((2-((2-(диметиламино)этил)тио)ацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата

Синтез азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соли описан ранее. Азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соль (152 г, 238 ммоль) перемешивали с DCM (2.3 L) и 10% бикарбонатом калия (1.15 L) при 0-5°C. Органическую фазу отделили, и водную фазу далее экстрагировали с помощью DCM (1.15 L). Объединенные органические фазы перемешивали с гидратом сульфата магния (236 г) в течение 30 минут при 0-5°C, отфильтровали и промыли с помощью DCM (1.15 L). К объединенным фильтратам добавили 2-((2-(диметиламино)этил)тио)уксусной кислоты гидрохлорид (57.0 г, 285 ммоль), EDC гидрохлорид (68.4 г, 357 ммоль) и DMAP (2.91 г, 23.8 ммоль), и тонкую суспензию перемешивали всю ночь при температуре окружающей среды, после чего образовался прозрачный раствор. Воду (2.3 L) и метанол (460 мл) добавили, и после перемешивания в течение 10 минут отделили прозрачную органическую фазу. Мутную водную фазу (pH 3.0) экстрагировали с помощью DCM (575 мл). Объединенные органические экстракты концентрировали с получением 143 г неочищенного вещества в виде гидрохоридной соль. Неочищенное вещество (142.6 г) перенесли в лабораторную перегонную колбу с DCM (500 мл), и этилацетат (1 L) добавили. Раствор нагревали до дистилляции при атмосферном давлении, и дистилляцию проводили в течение 70 минут, чтобы получить температуру остатка 76°C. Общий объем 1.4 л получили путем добавления этилацетата (800 мл), и этанол (70 мл) добавили. Прозрачный раствор при 50°C охладили до 37°C, и добавили затравочные кристаллы. Через 10 минут при 37-35°C наблюдали начало существенной кристаллизации, суспензию охладили и перемешивали при 0°C всю ночь, и остаток отделили путем фильтрации, и промыли с помощью холодного этилацетата (210 мл). Сушка при постоянной массе до температуры окружающей среды в масляном насосе при вакууме в течение 4.5 часов дала 134 г перекристаллизованного вещества в виде гидрохлоридной соли, кристаллическое твердое вещество белого цвета.
Трикалия фосфат (85 г, 0.40 моль) и дикалия гидрофосфат (226 г, 1.30 моль) добавили к очищенной воде (1.7 L), и образованный раствор с pH 10.9 охладили до 18-20°C. DCM (1.3 L) и перекристаллизованный S104 гидрохлорид (133.0 г, 0.188 моль) добавили, и смесь перемешивали в течение 10 минут. Прозрачную органическую фазу отделили при умеренной скорости (за 35 минут), и мутную водную фазу далее экстрагировали с помощью DCM (650 мл). Объединенные органические фазы перемешивали с гидратом сульфата магния (65 г) в течение 40 минут, и смесь отфильтровали, промыли с применением DCM (200 мл). Объединенные фильтраты выпарили на 50°C водяной бане при пониженном давлении (понижено до 20 мБар, выпаривание при этом давлении наблюдалось в течение одного часа). Дополнительное выпаривание на 15-20°C водяной бане с масляным насосом при вакууме дало 126 г частично отвержденного масла. Охлаждение на -20°C охлаждающей бане дало полное охлаждение, и после сушки при -20°C под вакуумом в течение двух дней получили 126 г ((2-((2-(диметиламино)этил)тио)ацетил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата, также известного как S104. ВЫЖХ показала чистоту 98.1%. QTOF MS ESI+: m/z 671.6 (М+Н).
Получение (9Z,9'Z)-((2-((2-(диметиламино)этил)тио)ацетил)азанедиил)бис(этан-2,1-диил)бис(тетрадец-9-еноата) (S104-DMO)

Стадия 1: Получение промежуточного соединения 1: (9Z,9'Z)-((трет-бутоксикарбонил)азанедиил)бис(этан-2,1-диил)бис(тетрадец-9-еноата)

N-Boc-диэтаноламин (454 мг, 2.21 ммоль), миристоленовую кислоту (1000 мг, 4.42 ммоль) и DMAP (54 мг, 0.442 ммоль) растворили в DCM (25 мл) и поместили на водяную баню при температуре окружающей среды в круглодонной колбе, и продули инертным газом. EDC HCl соль (932 мг, 4.86 ммоль) добавили в виде 3 частей за 5 минут. Реакционную смесь перемешивали всю ночь при комнатной температуре под слоем инертного газа. На следующий день добавили H2O (25 мл) и перемешивали в течение 10 минут. Органический слой отделили, и водный слой далее экстрагировали с помощью DCM (50 мл). Объединенные органические фазы высушили (MgSO4) в течение 10 минут, отфильтровали и концентрировали. Очищение посредством хроматографии на силикагеле при элюировании с помощью градиента гексан/этилацетат дало (9Z,9'Z)-((трет-бутоксикарбонил)азанедиил)бис(этан-2,1-диил)бис(тетрадец-9-еноат) (1.10 г).
Стадия 2: Получение промежуточного соединения 2: (9Z,9'Z)-азанедиилбис(этан-2,1-диил)бис(тетрадец-9-еноат)

К (9Z,9'Z)-((трет-бутоксикарбонил)азанедиил)бис(этан-2,1-диил)бис(тетрадец-9-еноату) (1100 мг, 1.77 ммоль) в круглодонной колбе добавили DCM (10 мл) и поместили на ледяную баню. TFA (10 мл) добавили и смесь перемешивали в течение 20 минут. Реакционную смесь затем концентрировали. Толуол добавили к остатку, чтобы обеспечить азеотропную перегонку избытка TFA. Остаток пометили назад на ледяную баню, и PBS (pH=11, 25 мл) и DCM (25 мл) добавили. Смесь перемешивали в течение 15 минут, и органический слой затем отделили. Мутный водный слой экстрагировали с помощью DCM (10 мл). Объединенные органические фазы высушили (MgSO4) при 0°C в течение 15 минут, отфильтровали и концентрировали с получением (9Z,9'Z)-азанедиилбис(этан-2,1-диил)бис(тетрадец-9-еноата) (923 мг).
Стадия 3: Получение S104-DMO: (9Z,9'Z)-((2-((2-(диметиламино)этил)тио)ацетил)азанедиил)бис(этан-2,1-диил)бис(тетрадец-9-еноат)

Смесь (9Z,9'Z)-азанедиилбис(этан-2,1-диил)бис(тетрадец-9-еноата) (923 мг, 1.77 ммоль), 2-((2-(диметиламино)этил)тио)уксусной кислоты (346 мг, 2.12 ммоль) и EDC HCl соли (509 мг, 2.66 ммоль) суспендировали в DCM (10 мл). DMAP (21.6 мг, 0.177 ммоль) добавили, и смесь перемешивали при комнатной температуре всю ночь. На следующий день H2O (10 мл) и МеОН (10 мл) добавили, и после перемешивания в течение 10 минут отделили прозрачную органическую фазу. Мутную водную фазу экстрагировали с помощью DCM (2×20 мл). Объединенные органические экстракты концентрировали и высушили с помощью MgSO4, отфильтровали и концентрировали. После очищения посредством хроматографии на силикагеле при элюировании с помощью градиента DCM/MeOH, объединенные и концентрированные фракции растворили в DCM (25 мл) и PBS (pH=11, 25 мл). Смесь перемешивали при комнатной температуре в течение ~10 минут. Затем органическую фазу отделили, и водную фазу снова экстрагировали с помощью DCM (2×15 мл). Объединенные органические фазы высушили (MgSO4), отфильтровали, и концентрировали с получением (9Z,9'Z)-((2-((2-(диметиламино)этил)тио)ацетил)азанедиил)бис(этан-2,1-диил)бис(тетрадец-9-еноата) (589 мг). LCMS ESI+: m/z 667.6 (М+Н).
Получение (R)-((1-метилпирролидин-2-карбонил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата (Pro-DC)

Стадия 1: Получение Pro-DC: (R)-((1-метилпирролидин-2-карбонил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата

Синтез азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соли описан ранее. Азанедиилбис(этан-2,1-диил)дитетрадеканоат TFA соль (1000 мг, 1.56 ммоль) перемешивали с DCM (10 мл) и N-Метил-L-пролином (228 мг, 1.77 ммоль), HOBtH2O (239 мг, 1.77 ммоль) добавили. NMM (365 мкл, 3.32 ммоль) добавили, и раствор стал в основном прозрачным. Суспензию EDC гидрохлорида (518 мг, 2.70 ммоль), NMM (257 мкл, 2.34 ммоль) и DMAP (19 мг, 0.156 ммоль) в DCM (10 мл) добавили, и смесь перемешивали в течение около 12 часов при температуре окружающей среды, после чего образовался прозрачный раствор. Затем смесь разбавили DCM (50 мл) и промыли с помощью 10% водного раствора K2CO3 (60 мл). Органические фазы высушили с помощью MgSO4, отфильтровали и концентрировали. Полученное соединение очистили посредством хроматографии на силикагеле, элюируя градиентом (0-10)% метанола в DCM с получением (R)-((1-метилпирролидин-2-карбонил)азанедиил)бис(этан-2,1-диил)дитетрадеканоата. LCMS ESI+: m/z 637.6 (М+Н).
Получение (9Z,9'Z,12Z,12'Z)-((2-((2-(диметиламино)этил)тио)ацетил)азанедиил)бис(этан-2,1-диил)бис(октадека-9,12-диеноата) (S104-DLin)

Стадия 1: Получение промежуточного соединения 1: (9Z,9'Z,12Z,12'Z)-((трет-бутоксикарбонил)азанедиил)бис(этан-2,1-диил)бис(октадека-9,12-диеноата)

N-Boc-диэтаноламин (5 г, 24.4 ммоль), линолевую кислоту (14.4 г, 51.2 ммоль) растворили в DCM (100 мл). Добавили EDC HCl соль (10.3 г, 53.7 ммоль), а затем DMAP (596 мг, 4.88 ммоль). Реакционную смесь перемешивали в течение около 12 часов при комнатной температуре под слоем инертного газа. Затем 50 мл воды и 50 мл метанола добавили, и смесь перемешивали в течение 10 минут. Органический слой отделили, и водный слой далее экстрагировали с помощью DCM (150 мл). Объединенные органические фазы высушили (MgSO4) в течение 10 минут, отфильтровали и концентрировали. Очищение посредством хроматографии на силикагеле при элюировании с помощью градиента гексан/эти л ацетат дало (9Z,9'Z,12Z,12'Z)-((трет-бутоксикарбонил)азанедиил)бис(этан-2,1-диил)бис(октадека-9,12-диеноат) (15.9 г).
Стадия 2: Получение промежуточного соединения 2: (9Z,9'Z,12Z,12'Z)-азанедиилбис(этан-2,1-диил)бис(октадека-9,12-диеноат)

К (9Z,9'Z,12Z,12'Z)-((трет-бутоксикарбонил)азанедиил)бис(этан-2,1-диил)бис(октадека-9,12-диеноату) (5.33 г, 7.30 ммоль) в круглодонной колбе добавили DCM (50 мл) и поместили на ледяную баню. TFA (50 мл) добавили, и смесь перемешивали в течение 30 минут. Реакционную смесь затем концентрировали. Толуол добавили к остатку для обеспечения азеотропной перегонки избытка TFA. Остаток поместили обратно на ледяную баню, и 10% K2CO3 (50 мл) и DCM (50 мл) добавили. Смесь перемешивали в течение 15 минут, и органический слой отделили. Мутный водный слой экстрагировали с помощью DCM (20 мл). Объединенные органические фазы высушили (MgSO4), отфильтровали и концентрировали с получением (9Z,9'Z,12Z,12'Z)-азанедиилбис(этан-2,1-диил)бис(октадека-9,12-диеноата) (количественный выход).
Стадия 3: Получение S104-DLin: (9Z,9'Z,12Z,12'Z)-((2-((2-(диметиламино)этил)тио)ацетил)азанедиил)бис(этан-2,1-диил)бис(октадека-9,12-диеноата)

Смесь (9Z,9'Z,12Z,12'Z)-азанедиилбис(этан-2,1-диил)бис(октадека-9,12-диеноата) (4.68 г, 7.30 ммоль), 2-((2-(диметиламино)этил)тио)уксусной кислоты (1.43 г, 8.76 ммоль) и EDC HCl соли (2.10 г, 10.95 ммоль) суспендировали в DCM (100 мл). DMAP (89 мг, 0.73 ммоль) добавили, перемешивали при комнатной температуре в течение 12 часов. Добавили по пятьдесят миллилитров воды и метанола, и, после перемешивания в течение 10 минут, отделили прозрачную органическую фазу. Мутную водную фазу экстрагировали с помощью DCM (2×20 мл), и промыли объединенные органические экстракты с помощью PBS (рН=11, 100 мл). Продукт высушили с помощью MgSO4, отфильтровали и концентрировали. Очистили с помощью хроматографии на силикагеле при элюировании с помощью градиента МеОН в DCM. Объединенные и концентрированные фракции дали (9Z,9'Z,12Z,12'Z)-((2-((2-(диметиламино)этил)тио)ацетил)азанедиил)бис(этан-2,1-диил) бис(октадека-9,12-диеноат) (4.3 г). LCMS ESI+: m/z 775.9 (М+Н).
Получение ((((2-(диметиламино)этил)тио)карбонил)азанедиил)бис(этан-2,1-диил) (9Z,9'Z,12Z,12'Z)-бис(октадека-9,12-диеноат) (TU104-DLin)

Стадия 1: Получение TU104-Dlin: ((((2-(диметиламино)этил)тио)карбонил)азанедиил)бис(этан-2,1-диил) (9Z,9'Z,12Z,12'Z)-бис(октадека-9,12-диеноата)

Синтез (9Z,9'Z,12Z,12'Z)-азанедиилбис(этан-2,1-диил) бис(октадека-9,12-диеноата) описан ранее. Трихлорметил хлорформиат (aka дифосген) (740 мкл,) добавили к раствору (9Z,9'Z,12Z,12'Z)-азанедиилбис(этан-2,1-диил) бис(октадека-9,12-диеноата) (2.6 г) в сухом DCM (40 мл) и перемешивали под слоем аргона при комнатной температуре в течение 12 часов. DCM и избыток дифосгена удалили в вакууме. 2-(диметиламино)этан тиол HCl соль (2.9 г), DCM (40 мл) и триэтиламин (3.7 мл) затем добавили. Через 16 часов при комнатной температуре реакционную смесь разбавили DCM и промыли с помощью 10% K2CO3 (75 мл), высушили (MgSO4), отфильтровали, и концентрировали. Очищение осуществили с помощью хроматографии на силикагеле, элюируя с помощью этилацетата, а затем градиента DCM/MeOH с получением ((((2-(диметиламино)этил)тио)карбонил)азанедиил)бис(этан-2,1-диил) (9Z,9'Z,12Z,12'Z)-бис(октадека-9,12-диеноата) (850 мг). LCMS ESI+: m/z 761.9 (М+Н).
Образование липосом, содержащих siPHК без-diVA. Ионизируемые липид, DOPE, холестерин и ПЭГ-конъюгированный липид солюбилизировали в абсолютном EtOH (200 градусов) при конечной массовой концентрации ~4.4 мг/мл. siPHК солюбилизировали в цитратном буфере при концентрации ~0.26 мг/мл, и установили температуру 35-40°С. Смесь этанол/липид затем добавили к siPHК-содержащему буферу при перемешивании до самопроизвольного образования липасом, нагруженных siPHК. Липиды объединили с siPHК до достижения конечного общего отношения липида и siPHК, равного от 7:1 до 14:1 (мас. : мас.). Липосомы, нагруженные siPHК, 1 подвергли диафильтрации с 10х объемами PBS для удаления этанола и замены буфера. Конечный продукт отфильтровали через 0.22 мкм, стерильной марки, фильтр для предварительной фильтрации. Этот процесс позволил получить липосомы со средним диаметром частиц 40-100 нм, PDI <0.2, и >85% эффективностью инкапсулирования РНК (захват).
Образование липосом, содержащих siPHК, сосолюбилизированных с diVA. siPHК-diVA-липосомальные композиции были получены с применением вышеописанного способа. DiVA-ПЭГ-diVA были сосолюбилизированы в абсолютном этаноле с другими липидами (ионизируемый липид, DOPE, холестерин, и ПЭГ-конъюгированные липиды) до добавления к siPHК содержащему буферу. Молярное содержание diVA-ПЭГ-diVA составляло от 0.1 до 5 моль %. Этот процесс позволил получить липосомы со средним диаметром частиц 40-100 нм, PDI <0.2, и >85% эффективность захвата.
Образование siPHК содержащих липосом с ионизируемыми и катионными липидами. siPHК-diVA-липосомальные композиции и siPHК-липосомальные композиции получили с применением вышеописанного способа. Катионный липид сосолюбилизировали в абсолютном этаноле с другими липидами (ионизируемый липид, DOPE, холестерин, ПЭГ-конъюгированные липиды и diVA-ПЭГ-diVA) до добавления к siPHK содержащему буферу. Молярное содержание катионного липида составляло от 5 до 40 моль %. Этот процесс позволил получить липосомы со средним диаметром частиц 40-100 нм, PDI <0.2, и >85% эффективность захвата.
In vitro (pHSC, gp46 KD @ 20 нМ) эффективность
PHSCs в 96-луночном планшете инкубировали с композицией, которая состояла либо из композиции ионизируемого липида С104, либо композиции ионизируемого липид Tu104, либо комбинации композиций с различным отношением ионизируемых липидов (C104:Tu104). Через 30 минут среду заменили на свежую среду для роста. Через сорок восемь часов клетки лизировали, и уровни gp46 и GAPDH mPHK измерили посредством анализа количественной ПЦР с обратной транскрипцией (TaqMan®), и уровни gp46 были нормализованы до GAPDH уровней. Нормализованные уровни gp46 выражались как процент от контрольных клеток mock. Усы показывают стандартные отклонения (n=3). Результаты показаны на Фиг. 1. Как показывают результаты, комбинация двух ионизируемых липидов согласно настоящему изобретению привела к наблюдаемому синергетическому эффекту уменьшения генной экспрессии.
Подобные эксперименты осуществили с другими комбинациями ионизируемый липид : ионизируемый липид и ионизируемый липид : атионный липид. Результаты для комбинаций S104-DO:Tu104-DO показаны на Фиг. 2. Результаты для комбинаций HEDODC:Tu104 показаны на Фиг. 3. Комбинации как ионизируемых липидов, так и ионизируемый липид : катионный липид снова привели к синергетическому эффекту уменьшения генной экспрессии.
In vivo (DMNQ) эффективность:
In vivo активность целевой композиции была оценена на модели непродолжительного повреждения печени (упоминается как Quick Model, DMNQ). В этой модели непродолжительное повреждение печени индуцируется обработкой гепатотоксическим агентом, таким как диметилнитрозамин (DMN) и сопровождается оценкой gp46 мРНК уровней. Чтобы индуцировать эти изменения самцов крыс Sprague-Dawley интраперитонеально инъецировали DMN в течение 6 последовательных дней. В конце периода обработки DMN животных случайным образом распредели по группам на основе индивидуальной массы тела животного. Композиции вводили в виде единичной внутривенной дозы и давали через час после последней инъекции DMN. Через двадцать два часа доли печени разрезали, и уровни мРНК gp46 и MRPL19 определили посредством анализа количественной ПЦР с обратной транскрипцией (TaqMan®). уровни мРНК для gp46 были нормализованы к уровням MRPL19. Результаты показана на Фиг. 4. Некоторые комбинации ионизируемый: катионный липиды достигли 50% уменьшения генной экспрессии за одну, 0.5 мг на кг массы тела животного, дозы инкапсулированной siPHK.
In vitro токсикологические данные, in vitro цитотоксичность (HepG2 @ 200 нМ)
Добавление 20 мол. % S104 в композиции согласно настоящему изобретению улучшило жизнеспособность клеток в HepG2 анализе цитотоксичности, улучшение от 27% до 52%.

Описание анализа HepG2 цитотоксичности:
HepG2 клетки, линии адгезивных клеток, полученную из гепатоцеллюлярной карциномы человека, культивировали в MEM/EBSS (Hyclone, Logan, Utah, Cat # SH30024.01) с добавкой 10% FBS (Hyclone, Logan, Utah Cat # SH30910). HepG2 клетки высеивали в 96-луночных черных планшетах Optilux (BD Falcon, Cat # BD353220) при плотности 5000 клеток/лунка всю ночь. Композиции добавили в каждую луну до конечной указанной концентрации siPHK (n=3). Через 48 часов после добавления композиции, жизнеспособность клеток определили с применением CellTiter-Glo Luminescent Cell Viability Assay Kit (Promega, Cat # G7572) согласно инструкциям производителя. Хемилюминисцентный сигнал измерили на Clarity Luminescence Micro plate Reader (502-Biotek, Winooski, Vermont). Жизнеспособность вычислили на основе % хемилюминесцентного сигнала в лунке, обработанной композицией, нормализованного к лункам с имитацией обработки.
In vivo токсикологические данные
HEDC:S104 (20:20) согласно настоящему изобретению особенно хорошо переносились, как показано при исследовании токсичности. Никакой токсичности не наблюдалось, когда композиция вводилась внутривенно крысам и обезьянам при дозах до 25 мг/кг и 12 мг/кг, соответственно, что специалистами в данной области техники считается превосходным.
Трансфекции композициями согласно настоящему изобретению:
Способ трансфекции является одинаковым для LX-2 и pHSC клеток. Композиции липосом или композиции липоплекса согласно настоящему изобретению смешали со средой для роста при желательных концентрациях. 100 мкл смеси добавили в клетки в 96-луночном планшете, и клетки инкубировали в течение 30 минут при 37°C в инкубаторе с 5% CO2. Через 30 минут среду заменили на свежую среду для роста. Через 48 часов трансфекции клетки обработали с применением Cell-to-Ct® лизисных реагентов (Applied Biosystems) согласно инструкциям производителей.
Количественная (q) ПЦР с обратной транскрипцией для измерения экспрессии HSP47 мРНК.
HSP47 и GAPDH TaqMan® анализы и одностадийную ПЦР с обратной транскрипцией master mix получили у Applied Biosystems. Каждая ПЦР реакция включала следующую композицию: одностадийная ПЦР с обратной транскрипцией mix 5 мкл, TaqMan® RT фермент mix 0.25 мкл, зонд анализа экспрессии гена TaqMan® (HSP47) 0.25 мкл, TaqMan® зонд анализа экспрессии гена (GAPDH) 0.5 мкл, свободная от рибонуклеазы вода 3.25 мкл, клеточный лизат 0.75 мкл, общий объем 10 мкл. GAPDH применяли в качестве эндогенного контроля для относительной количественной оценки уровней HSP47 мРНК. Количественную ПЦР с обратной транскрипцией осуществляли в системе ПЦР в реальном времени ViiA 7 (Applied Biosciences) с применением внутреннего способа относительной количественной оценки. Все значения были нормализованы до средней экспрессии HSP47 трансфектированных клеток mock и выражали как процент HSP47 экспрессии по сравнению с mock.
Реферат
Изобретение относится к ионизируемым липидным соединениям формулы I или их формам в виде фармацевтически приемлемых солей, которые могут быть полезны для улучшения доставки терапевтических агентов пациентам. В формуле I n и m независимо равны 1, 2, 3 или 4; Rи Rнезависимо представляют собой Салкил или Салкенил; когда X представляет собой S, L представляет собой Салкилен, -S-Cалкилен, -О-Салкилен, -O-С(O)-Салкилен, -S(O)-Салкилен илиили когда X представляет собой -СН-, О или отсутствует, L представляет собой -S-Салкилен, -O-С(O)-Салкилен, -S(O)-Салкилен или. Изобретение относится также к фармацевтической композиции для улучшения доставки терапевтических агентов, содержащей липидные соединения формулы I, лекарственному носителю, содержащему указанную фармацевтическую композицию, фармацевтической композиции на основе данного лекарственного носителя и способу доставки лекарственного средства. 5 н. и 40 з.п. ф-лы, 4 ил., 1 табл., 24 пр.
Формула









Комментарии