Обогащение активируемой флуоресценцией сортировки клеток (facs) для создания растений - RU2679510C2
Код документа: RU2679510C2
Чертежи















































































Описание
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННУЮ ЗАЯВКУ
Настоящая заявка притязает на приоритет на основании предварительной заявки на выдачу патента США с регистрационным номером 61/697890, поданной 7 сентября 2012.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Изобретение относится к области активируемой флуоресценцией сортировки клеток для создания растений. В предпочтительном варианте в описании раскрыто FACS-обогащение генетически отредактированных регенерируемых протопластов для создания фертильных подвергнутых редактированию растений.
УРОВЕНЬ ТЕХНИКИ
Активируемый флуоресценцией сортировщик клеток (FACS) был изобретен в конце 1960-х годов Bonner, Sweet, Hulett, Herzenberg и другими для осуществления проточной цитометрии и клеточной сортировки на живых клетках. Becton Dickinson Immunocytometry Systems начали производить коммерческие устройства в начале 1970-х годов. Активируемая флуоресценцией сортировка клеток (FACS) является специальным типом проточной цитометрии. Она обеспечивает осуществление способа сортировки гетерогенной смеси биологических клеток в две или большее количество емкостей, по одной клетке за один раз, на основе специфичного светорассеяния и характеристик флуоресценции каждой клетки. Это полезный научный инструмент, так как обеспечивает быструю, объективную и количественную регистрацию флуоресцентных сигналов от отдельных клеток, а также физическое разделение клеток, представляющий особый интерес.
Суспензия клеток захватывается в центр узкого, быстро протекающего потока жидкости. Поток распределяется таким образом, что имеется большое расстояние между клетками по сравнению с их диаметром. Механизм вибрации вызывает разбивание потока клеток на отдельные капельки. Систему регулируют таким образом, чтобы было маловероятным нахождение более одной клетки в капле. Непосредственно перед тем, как поток разбивается на капельки, поток проходит через устройство, измеряющее флуоресценцию, где происходит измерение представляющей интерес характеристики флуоресценции каждой клетки. Электрически заряжающее кольцо помещают непосредственно в точке, в которой поток разбивается на капельки. Заряд подают на кольцо на основе произведенного непосредственно перед этим измерения интенсивности флуоресценции, и противоположный заряд улавливается на капле, в то время как она отрывается от потока. Затем заряженные капельки проскакивают через систему, вызывающую электростатическое отклонение, которая отклоняет капельки по направлению к емкостям на основании их заряда. В некоторых системах заряд подают непосредственно на поток, и капля, отрывающаяся при разрыве, сохраняет заряд того же знака, что и поток. Затем поток возвращается в нейтральное состояние после того, как отрывается капля при разрыве.
В качестве меток в проточной цитометрии можно использовать широкий ряд флуорофоров. Флуорофоры или просто «флуоресцирующие средства» обычно связывают с антителом, которое распознает характерный элемент-мишень на клетке или в клетке; они также могут быть связаны с химической структурной единицей, обладающей аффинностью по отношению к клеточной мембране или другой клеточной структуре. Каждый флуорофор имеет характерную длину волны пика возбуждения и эмиссии, и спектры эмиссии часто перекрываются. Следовательно, сочетание меток, которое можно использовать, зависит от длины волны лампы (ламп) или лазера(-ов), используемых для возбуждения флуорохромов, и имеющихся детекторов.
Активируемая флуоресценцией сортировка клеток (FACS) обеспечивает быстрый способ выделения больших количеств флуоресцентно меченных клеток из гетерогенной смеси клеток. Коллекции трансгенных растений со специфичной для типа клеток экспрессией генов флуоресцирующих маркеров, таких как зеленый флуоресцирующий белок (GFP), идеально подходят для исследований отдельных типов клеток с помощью FACS.
Было показано, что проточный цитометрический анализ и флуоресцентно активируемая сортировка клеток (FACS) протопластов растений осуществимы на практике, кроме того, такая методика дала ценные результаты в нескольких разных областях исследований (Harkins и Galbraith, 1984; Galbraith et al., 1995; Sheen et al., 1995). Например, FACS протопластов из растений Arabidopsis, экспрессирующих тканеспецифичные флуоресцирующие белковые маркеры, применяли для исследования как базовых, так и стимулируемых факторами окружающей среды профилей транскрипции в конкретных типах клеток (Birnbaum et al., 2003; Brady et al., 2007; Gifford et al., 2008; Dinneny et al., 2008), и проточную цитометрию применяли для анализа продуцирования активных форм кислорода и запрограммированной клеточной гибели среди протопластов табака (Nicotiana tabacum; Lin et al., 2006). Широкий выбор флуоресцирующих средств доступен для исследования множества физиологических параметров в растениях, например, цис-регуляторных элементов, слитых с флуоресцирующими белками (Haseloff and Siemering, 2006), генетически кодируемые молекулярные сенсоры (Looger et al., 2005) или основанные на красителях сенсоры (Haugland, 2002) могут быть использованы в сочетании с цитометрией для измерения различных биологических процессов. Однако в таком способе имеются некоторые недостатки, связанные с чувствительностью анализов, и следовательно, есть место для усовершенствования.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Конкретный вариант изобретения относится к способу получения растения из популяции растительных клеток посредством выделения протопласта растения с использованием представляющего интерес полинуклеотида путем обеспечения популяции растительных протопластов, имеющей по меньшей мере один протопласт, содержащий представляющий интерес полинуклеотид и флуоресцирующий маркер, при этом популяция по существу не содержит растительных протопластов, содержащих флуоресцирующий маркер и не содержащих представляющий интерес полинуклеотид; при этом растительный протопласт инкапсулируют в альгинат натрия; отделения по меньшей мере одного протопласта, содержащего представляющий интерес полинуклеотид и флуоресцирующий маркер, от остальных растительных протопластов в популяции и, таким образом, выделения растительного протопласта, содержащего представляющий интерес полинуклеотид; регенерации растения из указанного выделенного растительного протопласта; и культивирования указанного растения.
Другим вариантом осуществления изобретения может быть растение, регенерированное посредством выделения растительного протопласта, содержащего представляющий интерес полинуклеотид, интегрированный в геном растительного протопласта, путем получения популяции растительных протопластов, имеющей по меньшей мере один протопласт, содержащий представляющий интерес полинуклеотид и флуоресцирующий маркер; при этом растительный протопласт инкапсулируют в альгинат натрия; извлечения микрокаллусов из популяции протопластов, содержащих представляющий интерес полинуклеотид и флуоресцирующий маркер, при этом по меньшей мере один протопласт, который содержит представляющий интерес полинуклеотид и флуоресцирующий маркер, был трансформирован представляющим интерес полинуклеотидом и полинуклеотидом, кодирующим флуоресцирующий маркер; регенерации растения из указанных микрокаллусов; и культивирования указанного растения.
Альтернативные варианты включают способы получения трансгенного растения, при этом способ может включат получение популяции растительных протопластов, имеющей по меньшей мере один протопласт, содержащий представляющий интерес полинуклеотид и флуоресцирующий маркер, при этом по меньшей мере один протопласт содержит сайт-специфичную нуклеазу, так что представляющий интерес полинуклеотид способен подвергаться интеграции в геном по меньшей мере одного растительного протопласта за счет гомологичной рекомбинации в сайте распознавания сайт-специфичной нуклеазой, и при этом растительный протопласт инкапсулируется в альгинат натрия; отделение по меньшей мере одного протопласта, содержащего представляющий интерес полинуклеотид и флуоресцирующий маркер, от остальных растительных протопластов в популяции; регенерацию трансгенного растения по меньшей мере из одного протопласта; и культивирование указанного трансгенного растения.
Вышеупомянутые и другие характерные признаки станут более понятными из следующего подробного описания нескольких вариантов, которое приведено со ссылками на прилагаемые фигуры.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фиг. 1A-1E: Показано выравнивание последовательностей гена FAD2, полученное с использованием ALIGNX®.
Фиг. 2: Показано филогенетическое древо последовательностей гена FAD2, созданное с использованием JALVIEW® V 2.3 на основе расстояний между ближайшими соседями.
Фиг. 3A-3M’: Показано выравнивание последовательностей гена FAD3, созданное с использованием ALIGNX®.
Фиг. 4: Показано филогенетическое древо последовательностей гена FAD3, созданное с использованием JALVIEW® V 2.3 на основе расстояний между ближайшими соседями. Отмеченные последовательности соответствуют следующему: FAD3A’/A” описана в настоящей заявке как FAD3A’; гаплотип 2 описан в настоящей заявке как FAD3C; гаплотип 1 описан в настоящей заявке как FAD3C”; и гаплотип 3 описан в настоящей заявке как FAD3A”.
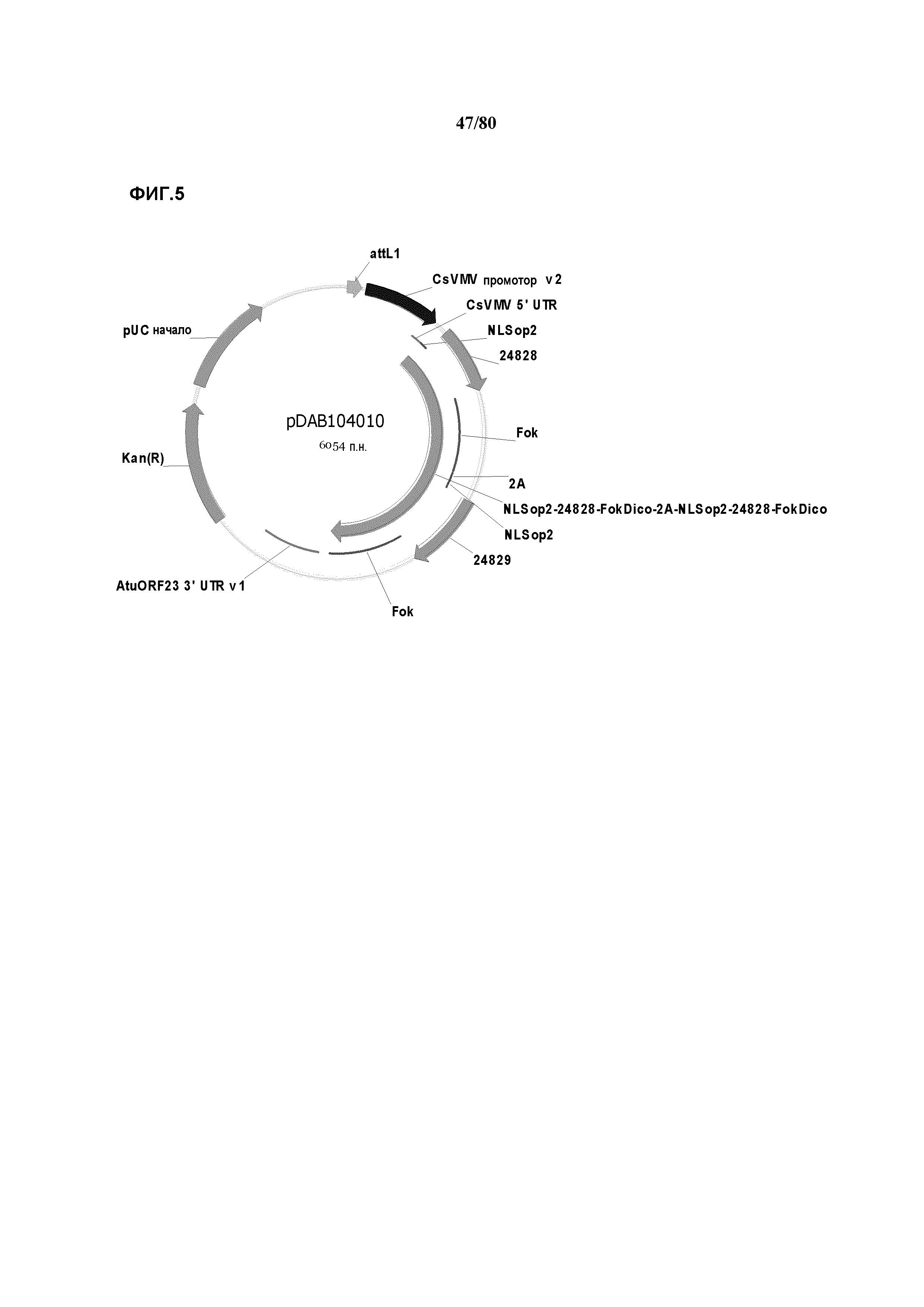
Фиг. 5: Показана плазмидная карта pDAB104010, которая является типичной кассетой экспрессии нуклеазы с цинковыми пальцами. Схема расположения в такой конструкции сходна со схемой других кассет экспрессии ZFN, где домены цинковых пальцев, 24828 и 24829, были заменены альтернативными доменами цинковых пальцев, которые описаны выше.
Фиг. 6: является иллюстративным многолинейным графиком, показывающим количество ридов последовательностей с делециями в сайте-мишени ZFN на 10000 ридов последовательностей. Ось X на графике означает количество делетированных оснований, ось Y означает количество ридов последовательностей, и ось Z означает кодируемое цветом наименование образцов, которые описаны справа от графика. В качестве конкретного примера показан локус 1 семейства генов FAD2, который содержит 3 сайта-мишени ZFN, A, B и C, с четырьмя представителями семейства генов и двумя контрольными трансфекциями, оцениваемыми в качестве контрольных образцов A и B.
Фиг. 7: (A) Подробное описание осей на фигуре такое же, как в случае фиг. 6. На фигуре представлены данные, полученные в результате ZFN-таргетинга локуса 4 семейства генов FAD2. Локус содержит два сайта ZFN и две необходимых контрольных трансфекции. Фиг. 7: (B) Специфичный контекст последовательностей (SEQ ID NO: 471-480), окружающих сайт-мишень ZFN, идентифицирующий FAD2A и C, содержащих трехнуклеотидные повторы C, T и G, приводящие к наблюдаемому увеличению количеств делеций одного основания при секвенировании локусов FAD2A и C.
Фиг. 8: Показана карта плазмиды pDAS000130.
Фиг. 9: Показана карта плазмиды pDAS000271.
Фиг. 10: Показана карта плазмиды pDAS000272.
Фиг. 11: Показана карта плазмиды pDAS000273.
Фиг. 12: Показана карта плазмиды pDAS000274.
Фиг. 13: Показана карта плазмиды pDAS000275.
Фиг. 14: Показана карта плазмиды pDAS000031.
Фиг. 15: Показана карта плазмиды pDAS000036.
Фиг. 16: Показана карта плазмиды pDAS000037.
Фиг. 17 иллюстрирует конфигурацию ETIP и нагружаемой нуклеиновой кислоты, а также продукт направленной в мишень нагрузки в сайте ETIP в геноме растительной клетки.
ФИГ.18 иллюстрирует трансформацию протопласта с последующей FACS-селекцией направленной в мишень ДНК-нагрузки в ETIP в линии хозяина с использованием реконструирования укороченных поддающихся подсчету и селектируемых маркеров на обоих 3’- и 5’-концах.
Фиг. 19A и 19B иллюстрируют зависимую от гомологии репарацию события ETIP канолы, которая возникает в результате двунитевого расщепления ДНК в геномном локусе нуклеазой с цинковыми пальцами (pDAS000074 или pDAS000075) и последующей интеграции донора (pDAS000068, pDAS000070 или pDAS000072) в локус ETIP в хромосоме канолы. Интеграция донора в геномный локус приводит к появлению полностью функционального высоко экспрессирующегося трансгена Ds-red.
Фиг. 20 демонстрирует FACS-сортировку протопластов канолы и рассчитанную эффективность трансфекции протопластов канолы, которые трансфицировали, используя pDAS000031 («pDAS31»). Кроме того, представлены результаты FACS-сортировки нетрансформированных протопластов канолы в качестве негативного контроля.
Фиг. 21 показывает FACS-сортировку протопластов канолы и рассчитанную эффективность трансфекции в случаях ETIP-протопластов канолы, которые трансфицировали, используя pDAS000064/pDAS000074 (верхний график) и pDAS000064/pDAS000075 (нижний график).
Фиг. 22 показывает FACS-сортировку протопластов канолы и рассчитанную эффективность трансфекции в случаях ETIP-протопластов канолы, которые трансформировали, используя pDAS000068/pDAS000074 (верхний график) и pDAS000068/pDAS000075 (нижний график).
Фиг. 23 показывает FACS-сортировку протопластов канолы и рассчитанную эффективность трансфекции в случаях ETIP-протопластов канолы, которые трансформировали, используя pDAS000070/pDAS000074 (верхний график) и pDAS000070/pDAS000075 (нижний график).
Фиг. 24 показывает FACS-сортировку протопластов канолы и рассчитанную эффективность трансфекции в случаях ETIP-протопластов канолы, которые трансформировали, используя pDAS000072/pDAS000074 (верхний график) и pDAS000072/pDAS000075 (нижний график).
Фиг. 25: Показана карта плазмиды pDAS000074.
Фиг. 26: Показана карта плазмиды pDAS000075.
Фиг. 27: Показана карта плазмиды pDAS000064.
Фиг. 28: Показана карта плазмиды pDAS000068.
Фиг. 29: Показана карта плазмиды pDAS000070.
Фиг. 30: Показана карта плазмиды pDAS000072.
Фиг. 31 представляет собой схему, показывающую сайты связывания праймеров трансгенной мишени и зонда для анализа с целью оценки количества копий трансгена.
Фиг. 32 демонстрирует файл SEQUENCHER®, показывающий домен распознавания ZFN ДНК FAD2A (bc12075_Fad2a-r272a2 и bc12075_Fad2a-278a2) и сайты связывания ZFN-специфичных праймеров (FAD2A.UnE.F1 и FAD2A.UnE.R1) и эндогенных праймеров (FAD2A/2C.RB.UnE.F1 и FAD2A/2C.RB.UnE.R1).
Фиг. 33 представляет собой схему, показывающую сайты связывания эндогенных праймеров и праймеров трансгенной мишени, используемых для выявления интеграции трансгена в FAD2A посредством точной HDR.
Фиг. 34 представляет собой схему, показывающую, где могут встречаться сайты рестрикции эндонуклеазой KpnI в точно отредактированном локусе FAD2A, и где связываются Саузерн-зонды FAD2a 5’, hph и FAD2A 3’.
Фиг. 35 показывает положение и размер KpnI-фрагментов, зондов FAD2A 5’, hph, FAD2A 3’ и ожидаемые результаты Саузерн-анализа в случае растений, в которых имеет место интеграция ETIP в локус FAD2A посредством HDR.
Фиг. 36 показывает типичные выходные данные, полученные в кПЦР при оценке количества копий. В левой колонке представлены данные, полученные для известного трансгенного растения T0 с одной случайной вставкой трансгена, используемого в качестве калибровочного образца, по отношению к которому «нормализуют» все другие образцы. Правая колонка относится к известному трансгенному растению T0 с 5 интеграциями трансгенов. Количество встроенных копий в случае обоих растений определяли, используя Саузерн-анализ. Остальные колонки представляют оценки количества копий в случае предполагаемых трансгенных растений. Обозначения под колонками соответствуют колонкам на графике и могут быть использованы для определения оцениваемого количества копий для каждого трансгенного растения. При использовании компьютерной программы для оценки количества копий, в случае растений дикого типа, нетрансформированных контрольных растений и только плазмидных контролей не получают количества копий, так как они не имеют Cq в случае мишени hph и HMG I/Y.
СПОСОБ(-Ы) ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Временная трансформация протопластов представляет собой широко используемый способ исследования растений, который является быстрым и несложным. Способ можно применять, например, для наблюдения за регуляцией промоторных элементов, чтобы анализировать экспрессию генов или ферментативную активность в ответ на различные стимулы, чтобы исследовать роль транскрипционных факторов или компонентов каскада сигнальной трансдукции или исследовать субклеточную локализацию белков (Sheen, 2001; Yoo et al., 2007). В противоположность стабильной трансформации растений (Arabidopsis thaliana является наиболее широко используемой платформой), на которую, как правило, затрачивают месяцы и которая требует использования трансфицирующего агента (обычно Agrobacterium tumefaciens), трансфекция протопластов может быть достигнута всего за один день и требует только ДНК в качестве сырья и использования любого химического или основанного на электропорации способа трансфекции. Кроме того, основанные на временной трансформации анализы могут преодолевать проблемы, с которыми сталкиваются в случае стабильной сверхэкспрессии, такие как плейотропное влияние на развитие или нежизнеспособность, когда целесообразным является основанный на клетках анализ. Однако вследствие того факта, что эффективность трансформации протопластов никогда не составляет 100%, результаты может быть трудно интерпретировать из-за нетрансформированных клеток.
Эффективности трансформации часто бывают низкими и вариабельными (например, Cummins et al., 2007; <10%) и зависят от применяемого способа, а также свойств используемых протопластов и ДНК. Настоящее изобретение относится к области биотехнологии растений, но может быть применено для всех биологических целей. В частности, варианты осуществления настоящего изобретения относятся к созданию нативных или трансгенных линий растительных клеток из гетерогенной популяции растительных клеток посредством проточно-цитометрической сортировки. Такая линия растительных клеток может быть либо из клеток однодольных, либо клеток двудольных. Как будет понятно специалисту, в изобретении также применяется линия растительных клеток для регенерации целых фертильных растений.
Один из вариантов осуществления настоящего изобретения относится к основанной на FACS чувствительной селекции, при этом происходит улучшенная селекция в отношении успешно трансформированных клеток. Ранее сообщалось, что трансформация популяции растительных клеток, таких как суспензионная культура растений, часто приводит к получению трансгенных культур, которые являются гетерогенными и имеют нестабильные уровни экспрессии. Настоящее изобретение, главным образом, относится к получению основанной на растениях системы. Возможность выделять и выращивать отдельные клетки имеет множество возможных применений. Например, способы, описанные в настоящей публикации, применимы для улучшения процессов, связанных с продуктивностью культур растительных клеток. Однако такое использование широко применимо для всех клеток.
Варианты осуществления настоящего изобретения включают применение проточно-цитометрической сортировки, такой как FACS-методика, для отделения или выделения одного, т.е. индивидуального протопласта, который получают из популяции растительных клеток, используя материалы и способы, известные в данной области. Такие протопласты могут быть трансформированы и способны 1) продуцировать флуоресцирующий маркерный белок или полипептид; 2) продуцировать требуемый продукт; и/или 3) выживать в присутствии агента для селекции. Критерии сортировки в случае FACS могут быть выбраны из группы, состоящей из генетического фона (например, плоидности, анеуплоидии), мутантов, трансгенных событий, продуктов обмена генов и флуоресценции (аутофлуоресценции (хлоропласты, метаболиты), флуоресценции, опосредованной флуоресцирующими белками или ферментами). Можно использовать любой флуоресцирующий белок. Агент для селекции может быть использован или не использован.
После отделения или выделения одиночных протопластов в результате проточно-цитометрической сортировки, каждый отдельный трансформированный протопласт подвергают регенерации вплоть до образования микроколонии (микрокаллуса) посредством совместного культивирования. Природа растительного источника не лимитирована, но ограничена такими линиями, сортами и видами, протопласты которых обладают способностью регенерировать до образования микроколонии или микрокаллуса. Следовательно, настоящее изобретение будет применимо по отношению ко всем сортам и видам растений, для которых был разработан или будет предоставлен протокол регенерации. Таким образом, настоящее изобретение может быть осуществлено с использованием всех сортов и видов растений, для которых разработан или будет представлен в будущем этап регенерации.
Микроколония, как таковая, может быть отделена или извлечена из материала питающих клеток и культивирована до образования линии растительных клеток.
Варианты осуществления настоящего изобретения также могут включать образование каллусной ткани посредством 1) переноса микроколонии или микрокаллуса на твердую среду для культивирования и 2) культивирования микроколонии или микрокаллуса в присутствии по меньшей мере одного агента для селекции вплоть до образования трансгенной каллусной ткани, из которой может быть получена линия трансгенных растительных клеток в результате переноса каллусной ткани в жидкую среду для культивирования. Микроколония также может быть извлечена или отделена от материала питающих клеток механическими способами, т.е. путем сбора клонов. В данном случае агент для селекции не требуется, и клетки, составляющие микроколонию, не должны проявлять резистентность к какому-либо агенту для селекции.
В некоторых вариантах клетки могут содержать гетерогенную популяцию растительных клеток, которые перед тем, как их подвергают проточно-цитометрической сортировке, являются нативными или нетрансформированными клетками и являются стабильно или временно трансформированными по меньшей мере одним экспрессирующим вектором, содержащим по меньшей мере одну гетерологичную последовательность нуклеиновой кислоты, которая может быть оперативно связана с функциональным промотором, при этом указанная по меньшей мере одна гетерологичная последовательность нуклеиновой кислоты кодирует требуемый продукт. В дополнительные вариантах по меньшей мере одна гетерологичная последовательность нуклеиновой кислоты оперативно связана с по меньшей мере одним функциональным промотором, при этом по меньшей мере одна гетерологичная последовательность нуклеиновой кислоты кодирует флуоресцирующий маркерный белок или полипептид, и по меньшей мере одна гетерологичная последовательность нуклеиновой кислоты кодирует резистентность к селектирующему агенту или требуемый продукт. Дополнительные варианты могут включать случаи, когда клетки могут дополнительно содержать гетерологичную последовательность нуклеиновой кислоты, которая кодирует требуемый продукт, который должен накапливаться в предлагаемой линии трансгенных растительных клеток.
В других вариантах геном клетки-хозяина может быть экспрессирован так, чтобы рекомбинантный белок или пептид могли быть модифицированы посредством рекомбинации, например, гомологичной рекомбинации или гетерологичной рекомбинации.
Любые полученные (трансгенные) моноклональные или биклональные линии растительных клеток могут быть обработаны и культивированы в присутствии предшественников, индукторов, гормонов, стабилизаторов, ингибиторов, молекул РНК-и/ми-РНК, соединений для передачи сигнала, ферментов и/или элиситоров дополнительно или вместо суспензии векторов для получения рекомбинантных белков или метаболитов.
Гетерологичные нуклеиновые кислоты могут кодировать гены бактериального, грибкового, растительного или нерастительного происхождения, такие как гены слитых белков и белков животного происхождения. Продуцируемые полипептиды могут быть использованы для получения полипептидов, которые могут быть очищены из них для применения в разных случаях. Белки, которые могут быть получены способом согласно изобретению, включают гетеродимеры, иммуноглобулины, слитые антитела и одноцепочечные антитела. Кроме того, указанные выше гены могут быть изменены для получения белков с измененными характеристиками.
Варианты осуществления настоящего изобретения включают возможность получать большое разнообразие белков и полипептидов. Такие варианты также могут включать способы получения по меньшей мере одного требуемого продукта, выбранного из группы, состоящей из гетерологичных белков или полипептидов, вторичных метаболитов и маркеров. Способ включает применение линии растительных клеток, которая создана согласно изобретению для продуцирования и накапливания по меньшей мере одного требуемого продукта, который затем получают или выделяют из продуцирующих клеток или из культуральной среды.
Дополнительные способы включают способы создания по меньшей мере одного внеклеточного гетерологичного белка, включающие стадии 1) стабильного введения в клетки-мишени, составляющие исходную популяцию растительных клеток, первой нуклеиновой кислоты, содержащей нуклеотидную последовательность, кодирующую гетерологичный белок или требуемый продукт; 2) получения протопластов из растительных клеток в суспензии, полученных из указанной суспензионной культуры растения, при этом протопласты дополнительно трансформированы и способны i) продуцировать флуоресцирующий маркерный белок или полипептид и ii) выживать в присутствии агента для селекции; 3) отделения отдельных трансформированных протопластов, для которого препарат протопластов подвергают FACS; 4) регенерации отдельного трансформированного протопласта вплоть до образования микроколонии или микрокаллуса в результате совместного культивирования в присутствии материала питающих клеток; 5) создания каллусной ткани посредством i) переноса микроколонии или микрокаллуса на твердую культуральную среду и ii) культивирования микроколонии или микрокаллуса в присутствии по меньшей мере одного агента для селекции вплоть до образования трансгенной каллусной ткани; 6) создания линии трансгенных растительных клеток путем переноса каллусной ткани в жидкую культуральную среду; 7) вызывания или обеспечения возможности экспрессии с нуклеиновой кислоты гетерологичного белка или требуемого продукта посредством обеспечения соответствующих условий культивирования; и 8) сбора накопленного гетерологичного белка или требуемого продукта из продуцирующих клеток. Такое выделение может быть осуществлено только обычными способами и может включать или не включать частичную или полную очистку.
В каждой конструкции можно использовать более одного гена. Множество векторов, каждый из которых содержит одну или несколько нуклеотидных последовательностей, кодирующих выбранный гетерологичный белок, могут быть введены в клетки-мишени, которые описаны в настоящей публикации или в других публикациях. Такой подход также может быть применимым для получения множественных субъединиц фермента.
Флуоресцирующим маркерным белком или полипептидом может быть белок, выявляемый по флуоресценции, такой как GUS, такие флуоресцирующие белки, как GFP или DsRed, люцифераза и т.д. Предпочтительно репортером является неинвазивный маркер, такой как DsRed или GFP.
Способы согласно настоящему изобретению можно применять для селекции в отношении определенных растений, которые необходимо выращивать. Выбор представляющего интерес гена можно проводить несколькими путями. Имеется большое количество способов встраивания ДНК в растительную клетку-хозяина. Такие способы включают трансформацию T-ДНК с использованием Agrobacterium tumefaciens или Agrobacterium rhizogenes в качестве трансформирующего агента, слияния, инъекции, биобаллистики (бомбардировки микрочастицами), нитевидных кристаллов карбида кремния, направленной подачи аэрозоля, ПЭГ или электропорации, а также других возможных способов. Если для трансформации используют Agrobacteria, встраиваемую ДНК необходимо клонировать в специальных плазмидах, а именно либо в промежуточном векторе, либо в бинарном векторе. Промежуточные векторы могут быть интегрированы в Ti- или Ri-плазмиду посредством гомологичной рекомбинации благодаря наличию последовательностей, которые гомологичны последовательностям в T-ДНК. Ti- или Ri-плазмида также содержит область vir, необходимую для переноса T-ДНК. Промежуточные векторы не могут реплицироваться сами в Agrobacteria. Промежуточный вектор может быть перенесен в Agrobacterium tumefaciens с помощью хелперной плазмиды (конъюгации). Бинарные векторы могут сами реплицироваться как в E. coli, так и в Agrobacteria. Они содержат селектируемый маркерный ген и линкер или полилинкер, которые обрамлены областями правой и левой границ T-ДНК. Ими можно непосредственно трансформировать Agrobacteria (Holsters, 1978). Agrobacterium, используемая в качестве клетки-хозяина, должна содержать плазмиду, несущую область vir. Область vir необходима для переноса T-ДНК в растительную клетку. Может содержаться дополнительная T-ДНК. Бактерию, трансформированную таким образом, используют для трансформации растительных клеток. Эксплантаты растений преимущественно можно культивировать с Agrobacterium tumefaciens или Agrobacterium rhizogenes для переноса ДНК в растительную клетку. Затем могут быть регенерированы целые растения из инфицированного растительного материала (например, кусочки листьев, участки стебля, корни, а также протопласты или культивируемые в суспензии клетки) в подходящей среде, которая может содержать антибиотики или биоциды для селекции. Затем полученные таким образом растения могут быть тестированы в отношении присутствия встроенной ДНК. Нет никаких специальных требований в отношении плазмид в случае инъекции и электропорации. Можно использовать обычные плазмиды, такие как, например, производные pUC.
Трансформированные клетки растут внутри растений обычным образом. Они могут образовывать зародышевые клетки и передавать трансформированный признак(-и) растениям-потомкам. Такие растения могут быть выращены обычным образом и скрещены с растениями, которые имеют такие же трансформированные наследственные факторы или другие наследственные факторы. Полученные гибридные особи обладают соответствующими фенотипическими свойствами.
В некоторых предпочтительных вариантах осуществления изобретения гены, кодирующие представляющие интерес белки, экспрессируются с транскрипционных единиц, встроенных в геном растения. Предпочтительно указанные транскрипционные единицы представляют собой рекомбинантные векторы, способные к стабильной интеграции в геном растения и позволяющие осуществлять селекцию линий трансформированных растений, экспрессирующих мРНК, кодирующую белки.
После интеграции встроенной ДНК в геном она оказывается относительно стабильной в геноме (и снова не выходит). В норме она содержит селектируемый маркер, который придает трансформированным растительным клеткам резистентность к биоциду или антибиотику, такому как наряду с прочими канамицин, G418, блеомицин, гигромицин или хлорамфеникол. Селектируемые маркеры растений также обычно могут обеспечивать резистентность к различным гербицидам, таким как глюфосинат (например, PAT/bar), глифосат (EPSPS), ALS-ингибиторам (например, имидазолинону, сульфонилмочевине, сульфонанилиду триазолопиримидина и другим), бромоксинилу, резистентность к ингибиторам HPPD, ингибиторам PPO, ингибиторам ACC-азы и многим другим. Отдельно используемый маркер должен соответствующим образом обеспечивать возможность отбора трансформированных клеток, но не клеток, которые не содержат встроенной ДНК. Представляющий интерес ген(-ы) предпочтительно экспрессируется либо конститутивными, либо индуцируемыми промоторами в растительной клетке. После экспрессии мРНК транслируется в белки, при этом включая представляющие интерес аминокислоты в белок. Гены, кодирующие белок, экспрессируемый в растительных клетках, могут быть под контролем конститутивного промотора, тканеспецифичного промотора или индуцируемого промотора.
Существует несколько способов введения чужеродных рекомбинантных векторов в растительные клетки и получения растений, которые стабильно содержат и экспрессируют введенный ген. Такие способы включают введение генетического материала с покрытием в виде микрокапсул непосредственно в клетки (патенты США № 4945050, Cornell, и 5141131, DowElanco, в настоящее время Dow AgroSciences, LLC). Кроме того, растения могут быть трансформированы с использованием основанной на Agrobacterium методики, см. патенты США № 5177010, Университет Толедо; 5104310 Texas A&M; заявку на выдачу Европейского патента 0131624B1; заявки на выдачу Европейского патента 120516, 159418B1 и 176112, Schilperoot; патенты США № 5149645, 5469976, 5464763 и 4940838 и 4693976, Schilperoot; заявки на выдачу Европейского патента 116718, 290799, 320500, все Max Planck; заявки на выдачу Европейского патента 604662 и 627752 и патент США № 5591616, Japan Tobacco; заявки на выдачу Европейского патента 0267159 и 0292435 и патент США № 5231019, все Ciba Geigy, в настоящее время Syngenta; патенты США № 5463174 и 4762785, оба Calgene; и патенты США № 5004863 и 5159135, оба Agracetus. Другая методика трансформации включает методику на основе нитчатых кристаллов. См. патенты США № 5302523 и 5464765, оба Zeneca, в настоящее время Syngenta. Другая методика трансформации посредством прямой доставки ДНК включает методику направленной подачи аэрозоля. См. патент США № 6809232. Методику электропорации также применяли для трансформации растений. См. WO 87/06614, Boyce Thompson Institute; патенты США № 5472869 и 5384253, оба Dekalb; и WO 92/09696 и WO 93/21335, обе Plant Genetic Systems. Кроме того, вирусные векторы также можно использовать для получения трансгенных растений, экспрессирующих представляющий интерес белок. Например, однодольные растения могут быть трансформированы вирусным вектором с использованием способов, описанных в патенте США № 5569597, Mycogen Plant Science и Ciba-Geigy (в настоящее время Syngenta), а также в патентах США № 5589367 и 5316931, оба Biosource, в настоящее время Large Scale Biology.
Как упоминалось ранее, способ, посредством которого вводят конструкцию ДНК в растение-хозяина, не является критическим для настоящего изобретения. Можно применять любой способ, который обеспечивает эффективную трансформацию. Например, различные способы трансформации растительных клеток описаны в настоящей публикации и включают использование Ti- или Ri-плазмид и тому подобных для осуществления опосредованной Agrobacterium трансформации. В многих случаях будет желательно иметь конструкцию, используемую для трансформации, ограниченную с одной или обеих сторон границами T-ДНК, более конкретно правой границей. Это особенно полезно в том случае, когда для конструкции используют Agrobacterium tumefaciens или Agrobacterium rhizogenes в качестве средства для трансформации, хотя границы T-ДНК могут быть применимы в случае других способов трансформации. В том случае, когда используют Agrobacterium для трансформации растительных клеток, можно применять вектор, который может быть введен в хозяина для гомологичной рекомбинации с T-ДНК или Ti-, или Ri-плазмидой, присутствующей в клетке хозяина. Введение вектора может быть осуществлено посредством электропорации, трехродительского скрещивания и других способов трансформации грамотрицательных бактерий, которые известны специалистам в данной области. Способ трансформации векторов в хозяина Agrobacterium не является критичным для настоящего изобретения. Ti- или Ri- плазмида, содержащая T-ДНК для рекомбинации, может быть способна или неспособна вызывать галлообразование и не является критичной для указанного изобретения, при условии, что гены vir присутствуют в указанном хозяине.
В некоторых случаях, когда Agrobacterium используют для трансформации, экспрессирующая конструкция, находящаяся в границах T-ДНК, будет встроена в вектор широкого спектра, такой как pRK2 или его производные, которые описаны в публикации Ditta et al. (1980) и EPO 0 120 515. В экспрессирующую конструкций и T-ДНК могут быть включены один или несколько маркеров, которые описаны в настоящей публикации, которые обеспечивают возможность селекции трансформированных Agrobacterium и трансформированных растительных клеток. Конкретный используемый маркер не важен для настоящего изобретения, при этом предпочтительный маркер зависит от используемого хозяина и конструкции.
Для трансформации растительных клеток с использованием Agrobacterium эксплантаты можно объединить и инкубировать с трансформированными Agrobacterium в течение периода времени, достаточного для того, чтобы обеспечить их трансформацию. После трансформации агробактерии убивают в ходе селекции с использованием подходящего антибиотика, и растительные клетки культивируют, используя подходящую селективную среду. После образования каллусов образование побегов может быть стимулировано благодаря использованию подходящих растительных гормонов согласно способам, хорошо известным в области культивирования тканей растений и регенерации растений. Однако промежуточная стадия каллусов не всегда является необходимой. После образования побегов указанные растительные клетки могут быть перенесены в среду, которая стимулирует образование корней, завершая таким образом регенерацию растений. Затем растения могут быть выращены до получения семян, и указанное семя можно использовать для получения будущих поколений. Независимо от способа трансформации ген, кодирующий бактериальный белок, предпочтительно включают в вектор для переноса генов, адаптированный для экспрессии указанного гена в растительной клетке за счет включения в вектор регуляторного промоторного элемента растений, а также 3’-нетранслируемых областей терминации транскрипции, таких как Nos и тому подобные.
Кроме многочисленных способов трансформации растений тип ткани, которую подвергают контакту с чужеродными генами, также может варьировать. Такая ткань может включать без ограничения эмбриогенную ткань, каллусную ткань типа I, II и III, гипокотиль, меристему, ткань корня, ткани для экспрессии в флоэме и тому подобные. Почти все растительные ткани могут быть трансформированы во время дифференцировки с использованием подходящих способов, описанных в настоящей публикации.
Как указано выше, при необходимости можно использовать множество селектируемых маркеров. Предпочтение конкретному маркеру отдается по решению специалиста в данной области, но любой из следующих селектируемых маркеров можно использовать вместе с любым другим геном, не указанным в настоящем описании, который может функционировать в качестве селектируемого маркера. Такие селектируемые маркеры включают без ограничения ген аминогликозидфосфотрансферазы транспозона Tn5 (Aph II), который кодирует резистентность к антибиотикам канамицину, неомицину и G41; резистентность к гигромицину; резистентность к метотрексату, а также такие гены, которые кодируют резистентность или толерантность к глифосату; фосфинотрицину (биалафосу или глюфосинату); ALS-ингибирующим гербицидам (имидазолинонам, сульфонилмочевинам и триазолопиримидиновым гербицидам), ингибиторам ACC-азы (например, арилоксипропионатам или циклогександионам) и другим, таким как бромоксинил, и ингибиторам HPPD (например, мезотриону) и тому подобным.
В дополнение к селектируемому маркеру может быть желательно использованием репортерного гена. В некоторых случаях репортерный ген может быть использован вместе или без селектируемого маркера. Репортерные гены представляют собой гены, которые обычно не присутствуют в организме или ткани реципиента и обычно кодируют белки, приводящие к определенному изменению фенотипа или ферментативному свойству. Примеры таких генов приведены в публикации Weising et al., 1988. Предпочтительные репортерные гены включают ген бета-глюкуронидазы (GUS) локуса uidA E. coli, ген хлорамфениколацетилтрансферазы из Tn9 E. coli, ген зеленого флуоресцирующего белка из биолюминесцирующей медузы Aequorea victoria и гены люциферазы светляка Photinus pyralis. Затем может быть осуществлен анализ для выявления экспрессии репортерного гена в подходящий момент времени после того, как указанный ген вводят в реципиентные клетки. Предпочтительный подобный анализ включает использование гена, кодирующего бета-глюкуронидазу (GUS) локуса uidA E. coli, как описано Jefferson et al. (1987), чтобы идентифицировать трансформированные клетки.
Кроме промоторных регуляторных элементов растений в растительных клетках эффективно можно использовать регуляторные промоторные элементы из множества источников, чтобы экспрессировать чужеродные гены. Например, можно использовать промоторные регуляторные элементы бактериального происхождения, такие как промотор октопинсинтазы, промотор нопалинсинтазы, промотор маннопинсинтазы; промоторы вирусного происхождения, такие как промотор вируса мозаики цветной капусты (35S и 19S), 35T (который представляет собой реконструированный промотор 35S, см. патент США № 6166302, в частности, пример 7E) и тому подобные. Промоторные регуляторные элементы растений включают без ограничения промотор малой субъединицы риболезо-1,6-дифосфат (RUBP)-карбоксилазы (ssu), промотор бета-конглицинина, промотор бета-фазеолина, промотор ADH, промоторы теплового шока и тканеспецифичные промоторы. Могут присутствовать другие элементы, такие как области присоединения к матриксу, области присоединения к каркасу, интроны, энхансеры, последовательности полиаденилирования и тому подобные, и при этом они могут повышать эффективность транскрипции или интеграцию ДНК. Такие элементы могут быть или не быть необходимыми для функционирования ДНК, хотя они могут обеспечивать улучшенную экспрессию или функционирование ДНК за счет влияния на транскрипцию, стабильность мРНК и тому подобное. Такие элементы при необходимости могут быть включены в ДНК, чтобы получить оптимальную эффективность трансформированной ДНК в растении. Типичные элементы включают без ограничения Adh-интрон 1, Adh-интрон 6, лидерную последовательность белка оболочки вируса мозаики люцерны, UTR-последовательности осмотина, лидерную последовательность белка оболочки вируса полосатости кукурузы, а также другие, известные специалисту в данной области. Также можно использовать конститутивные регуляторные промоторные элементы, управляя таким образом непрерывной экспрессией генов во всех типах клетках и во все периоды времени (например, актина, убиквитина, 35S CaMV и тому подобные). Тканеспецифичные регуляторные промоторные элементы ответственны за экспрессию генов в конкретных типах клеток или тканей, таких как листья или семена (например, зепина, олеозина, напина, ACP, глобулина и тому подобных), и их также можно использовать.
Промоторные регуляторные элементы также могут быть активными (или неактивными) во время определенной стадии развития растения, а также активными в тканях и органах растения. Примеры таких элементов включают без ограничения специфичные для пыльцы, специфичные для зародыша, специфичные для кукурузных рылец, специфичные для волокон хлопчатника, специфичные для корней, специфичные для эндосперма семян или специфичные для вегетативной стадии регуляторные промоторные элементы и тому подобные. В некоторых случаях может быть желательным использование индуцируемого регуляторного промоторного элемента, который ответственен за экспрессию генов в ответ на специфичные сигналы, такие как: физический стимул (гены теплового шока), свет (RUBP-карбоксилаза), гормон (Em), метаболиты, химические вещества (чувствительные к тетрациклину) и стресс. Можно использовать другие требуемые элементы для транскрипции и трансляции, которые функционируют в растениях. В данной области известны многочисленные векторы для переноса специфичных для растений генов.
Системы, основанные на РНК-вирусах растений, также могут быть использованы для экспрессии бактериального белка. При этом ген, кодирующий белок, может быть встроен в область промотора генов оболочки подходящего вируса растений, который будет инфицировать представляющее интерес растение-хозяина. Затем может быть экспрессирован белок, обеспечивающий при этом защиту растения от повреждения гербицидами. Системы, основанные на РНК-вирусах растений, описаны в патенте США № 5500360, Mycogen Plant Sciences, Inc., и в патентах США № 5316931 и 5589367, Biosource.
Средства дополнительно повышения уровней толерантности или резистентности. В данном описании показано, что растениям согласно настоящему изобретению могут быть приданы новые признаки резистентности к гербицидам без наблюдаемого неблагоприятного влияния на фенотип, включая урожайность. Такие растения входят в объем настоящего изобретения. Растения, приведенные в качестве примера и предлагаемые в настоящем изобретении, могут выдерживать 2-х, 3-х, 4-х и 5-кратные уровни внесения, например по меньшей мере одного испытываемого гербицида. Повышение уровней толерантности входит в объем настоящего изобретения. Например, в данной области известны разные способы, и они предположительно могут быть оптимизированы и дополнительно разработаны для повышения экспрессии данного гена.
Один из таких способов включает увеличение количества копий исследуемых генов (в кассетах экспрессии и тому подобном). События трансформации также могут быть подвергнуты селекции в отношении таких событии, когда имеется множество копий генов.
Могут быть использованы сильные промоторы и энхансеры для «нагнетания» экспрессии. Примеры таких промоторов включают предпочтительный промотор 35T, который использует энхансеры 35S. Промоторы 35S, убиквитина кукурузы, убиквитина Arabidopsis, актина A.t. и CSMV могут быть включены для таких применений. Также предпочтительны другие сильные вирусные промоторы. Энхансеры включают двойной энхансер 4 OCS и 35S. Также могут быть использованы области связывания с матриксом (MAR), например, для повышения эффективности трансформации и экспрессии трансгена.
Перетасовка (направленная эволюция) и факторы транскрипции также могут быть использованы в вариантах осуществления настоящего изобретения.
Также могут быть сконструированы варианты белков, которые отличаются на уровне последовательностей, но которые сохраняют такую же или сходную общую важную трехмерную структуру, распределение поверхностного заряда и тому подобное. См., например, патент США № 7058515; Larson et al., Protein Sci. 2002 11: 2804-2813, “Thoroughly sampling sequence space: Large-scale protein design of structural ensembles”; Crameri et al., Nature Biotechnology 15, 436-438 (1997), “Molecular evolution of an arsenate detoxification pathway by DNA shuffling”; Stemmer, W. P. C. 1994, DNA shuffling by random fragmentation and reassembly: in vitro recombination for molecular evolution, Proc. Natl. Acad. Sci. USA 91: 10747-10751; Stemmer, W. P. C. 1994, Rapid evolution of a protein in vitro by DNA shuffling, Nature 370: 389-391; Stemmer, W. P. C. 1995, Searching sequence space. Bio/Technology 13: 549-553; Crameri, A., Cwirla, S, and Stemmer, W. P. C. 1996, Construction and evolution of antibody-phage libraries by DNA shuffling, Nature Medicine 2: 100-103; and Crameri, A., Whitehorn, E. A., Tate, E. and Stemmer, W. P. C., 1996, Improved green fluorescent protein by molecular evolution using DNA shuffling, Nature Biotechnology 14: 315-319.
Активность рекомбинантных полинуклеотидов, встроенных в растительные клетки, может зависеть от влияния эндогенной растительной ДНК вблизи вставки. Таким образом, другой вариант заключается в том, чтобы воспользоваться преимуществом таких событий, которые, как известно, имеют прекрасные положения в геноме растения для инсерций. См., например, публикацию WO 2005/103266 A1, имеющую отношение к событиям cry1F и cry1Ac у хлопчатника; FAD2, FAD3, при этом такие гены, как AAD1 или AAD12 или другие могут быть использованы для замены в таких геномных локусах вместо указанных вставок. Следовательно, согласно настоящему изобретению можно применять целенаправленную гомологичную рекомбинацию. Указанный тип методики является предметом, например, заявки WO 03/080809 A2 и соответствующей опубликованной заявки на выдачу патента США (USPA 20030232410), которые относятся к использованию цинковых пальцев для целенаправленной рекомбинации. Использование рекомбиназ (например, cre-10xand flp-frt) также известно в данной области.
Компьютерное конструирование 5’- или 3’-UTR, наиболее подходящих для синтетических шпилек, также можно осуществлять в объеме настоящего изобретения. Компьютерное моделирование в общем, а также перетасовка генов и направленная эволюция обсуждается в настоящем описании в других разделах. Более конкретно, в отношении компьютерного моделирования и UTR, можно отметить, что способы компьютерного моделирования для применения с целью прогнозирования/оценки 5’- и 3’-UTR-производных согласно настоящему изобретению включают без ограничения: MFold, версию 3.1, доступную из Genetics Corporation Group, Madison, Wis. (см. Zucker et al., Algorithms and Thermodynamics for RNA Secondary Structure Prediction: A Practical Guide. In RNA Biochemistry and Biotechnology, 11-43, J. Barciszewski & B. F. C. Clark, eds., NATO ASI Series, Kluwer Academic Publishers, Dordrecht, NL, (1999); Zucker et al., Expanded Sequence Dependence of Thermodynamic Parameters Improves Prediction of RNA Secondary Structure. J. Mol. Biol. 288, 911-940 (1999); Zucker et al., RNA Secondary Structure Prediction. In Current Protocols in Nucleic Acid Chemistry, S. Beaucage, D. E. Bergstrom, G. D. Glick, and R. A. Jones eds., John Wiley & Sons, New York, 11.2.1-11.2.10, (2000)), COVE (анализ структуры РНК с использованием ковариационных моделей (способы стохастической бесконтекстной грамматики)) v. 2.4.2 (Eddy and Durbin, Nucl. Acids Res. 1994, 22: 2079-2088), который распространяется свободно в виде исходного текста и который можно загрузить, посетив веб-сайт genetics.wustl.edu/eddy/software/, и FOLDALIGN, также распространяемый свободно и доступный для загрузки с веб-сайта bioinf.au.dk. FOLDALIGN/(см. Finding the most significant common sequence and structure motifs in a set of RNA sequences. J. Gorodkin, L. J. Heyer and G. D. Stormo. Nucleic Acids Research, Vol. 25, no. 18 pp 3724-3732, 1997; Finding Common Sequence and Structure Motifs in a set of RNA Sequences. J. Gorodkin, L. J. Heyer, and G. D. Stormo. ISMB 5;120-123, 1997).
Варианты осуществления настоящего изобретения можно применять в сочетании с использованием естественно развивающихся или химически индуцированных мутантов (мутанты могут быть выбраны способами скрининга, затем трансформированы другими генами). В растения согласно настоящему изобретению можно вводить различные гены резистентности путем комбинации и/или вызывать развитие генов резистентности. Традиционные способы селекции также можно сочетать с настоящим изобретением, чтобы эффективно объединять, осуществлять интрогрессию и улучшать отбор признаков.
Все ссылки, включая публикации, патенты и заявки на выдачу патентов, цитированные и обсуждаемые в настоящем описании, приведены исключительно для их представления до даты подачи настоящей заявки. В настоящем описании ничего не следует истолковывать как признание того, что авторы изобретения не имеют права датировать задним числом такое раскрытие на основании предшествующего изобретения.
ПРИМЕРЫ
Следующие примеры приведены для иллюстрации некоторых конкретных признаков и/или аспектов. Такие примеры не следует считать ограничивающими раскрытие конкретных описанных признаков или аспектов.
ПРИМЕР 1: ИДЕНТИФИКАЦИЯ ПАРОЛОГИЧНЫХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ-МИШЕНЕЙ FAD2 и FAD3 ИХ БИБЛИОТЕКИ БАКТЕРИАЛЬНЫХ ИСКУССТВЕННЫХ ХРОМОСОМ
КОНСТРУИРОВАНИЕ BAC
Библиотеку бактериальных искусственных хромосом (BAC) получали от коммерческого поставщика (Amplicon Express, Pullman, WA). Библиотека BAC состояла из 110592 BAC-клонов, содержащих фрагменты геномной ДНК (гДНК) с высокой молекулярной массой, выделенные из Brassica napus L. сорта DH 10275. гДНК расщепляли ферментом рестрикции либо BamHI, либо HinDIII. Изолированные фрагменты гДНК длиной примерно 135 т.п.н. лигировали в вектор pCC1BAC (Epicentre, Madison, WI) и трансформировали Escherichia coli штамма DH10B (Invitrogen). Библиотеку BAC состояла из четного количества BAC-клонов, которые были сконструированы с использованием двух разных ферментов рестрикции. Фактически сконструированная с использованием Hind III BAC-библиотека состояла из 144 отдельных 384-луночных планшетов. Подобным образом, сконструированная с использованием BamHI BAC-библиотека состояла из 144 отдельных 384-луночных планшетов. Всего было выделено 110592 BAC-клонов, которые распределены в 288 отдельных 384-луночных планшетах. Каждый из 288 отдельных 384-луночных планшетов был представлен поставщиком для одной экстракции ДНК для быстрого основанного на ПЦР скрининга. Полученная в результате BAC-библиотека охватывает приблизительно 15 г. п. н. гДНК, что соответствует 12-кратному охвату генома Brassica napus L. сорта DH10275 (оценка генома Brassica napus L. составляет приблизительно 1,132 г. п. н., как описано Johnston et al. (2005), Annals of Botany 95: 229-235).
АНАЛИЗ КОДИРУЮЩИХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ FAD2, ВЫДЕЛЕННЫХ ИЗ BAC-БИБЛИОТЕКИ
Сконструированную BAC-библиотеку использовали для выделения кодирующих последовательностей гена FAD2. Осуществляли эксперименты по секвенированию, чтобы идентифицировать специфичные генные последовательности четырех паралогов гена FAD2 из Brassica napus L. сорта DH 10275.
Сначала идентифицировали последовательность гена FAD2 у модельного вида Arabidopsis thaliana. Последовательность гена указана в Genbank с маркировкой локуса: At3g12120. Ранее были описаны сравнительные геномные взаимосвязи между модельным видом растения Arabidopsis thaliana и диплоидным Brassica rapa, одним из предшественников тетраплоида Brassica napus (Schranz et al., (2006) Trends in Plant Science 11 (11): 535-542). Конкретно в отношении гена FAD2 сравнительный анализ предполагает, что 3-4 копии гена могут встречаться в диплоидном геноме Brassica. Дополнительные исследования по генетическому картированию были осуществлены Scheffler et al. ((1997) Theoretical and Applied Genetics 94: 583-591). Результаты таких исследований по генетическому картированию показали, что четыре копии гена FAD2 присутствовали в Brassica napus.
Основанный на секвенировании анализ BAC-библиотеки, которая была сконструирована из B. napus L. сорта DH 12075, привел к выделению четырех последовательностей BAC (SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3 и SEQ ID NO: 4), на основании которых определяли кодирующие последовательности генов FAD2A (SEQ ID NO: 5), FAD2-1 (SEQ ID NO: 6), FAD2-2 (SEQ ID NO: 7) и FAD2-3 (SEQ ID NO: 8). Идентифицировали и генетически картировали последовательности генов FAD2A, FAD2-1, FAD2-2 и FAD2-3. Анализ последовательностей четырех генов FAD2 проводили, применяя программу выравнивания последовательностей и построение филогенетического дерева способом ближайших соседей с использованием идентичности в процентах. Выравнивание последовательностей осуществляли с помощью программы ALIGNX® из компьютерной программы Vector NTI Advance 11.0 (Life Technologies, Carlsbad, CA), и такое выравнивание показано на фиг. 1. В программе ALIGNX® использован модифицированный алгоритм Clustal W для создания выравниваний множественных последовательностей либо белка, либо последовательностей нуклеиновых кислот для сравнения сходства и для пояснения. Филогенетическое древо, получаемое способом ближайших соседей, создавали с помощью компьютерной программы JALVIEW V2.3®, и древо показано на фиг. 2 (Waterhouse et al. (2009), Bioinformatics 25 (9): 1189-1191). Как показано на фиг. 2, анализ изолированных последовательностей показал, что последовательности FAD2A и FAD2-3 имеют высокие уровни сходства последовательностей, и что подобным образом FAD2-1 и FAD2-2 имеют высокие уровни сходства последовательностей. Четыре последовательности могут быть отнесены к двум кладам, при этом FAD2A и FAD2-3 составляют первую кладу, а FAD2-1 и FAD2-2 составляют вторую кладу.
Затем вновь выделенные последовательности FAD2 из Brassica napus использовали по отношению к геномным библиотекам BLAST, выделенным из геномной BAC-библиотеки Brassica rapa и ридов геномной последовательности Brassica oleracea, полученных методом «дробовика» (shotgun). Оба вида, Brassica rapa и Brassica oleracea, являются диплоидными предшественниками Brassica napus, который является амфиплоидным видом (AC-геном, n=19). Brassica napus возник в результате недавнего события гибридизации между Brassica rapa (субгеном A, n=10) и Brassica oleracea (субгеном C, n=9). Последовательности диплоидных предшественников сравнивали с четырьмя разными кодирующими последовательностями FAD2, выделенными из Brassica napus, используя анализ BLASTn. В таком анализе последовательностей идентифицировали специфичные аннотированные генные последовательности из Brassica rapa и Brassica oleracea, которые имеют наибольшее сходство последовательностей с вновь открытыми последовательностями FAD2 Brassica napus. В таблице 1 перечислены вновь идентифицированные кодирующие последовательности FAD2, и соответствующие номера для эталонных последовательностей предшественника, и организмы, из которых они происходят.
Гены FAD2 существуют в геноме Brassica napus в виде двух копий каждого гена на субгеном. Одна копия каждого гена локализована в субгеноме A, и подобным образом одна копия каждого гена локализована в субгеноме C. Описаны новые правила присвоения названия так, чтобы указать, в каком субгеноме локализован каждый ген. Высокие уровни сходства последовательностей между четырьмя разными кодирующими последовательностями FAD2, выделенными из BAC-библиотеки геномной ДНК Brassica napus, и данными о последовательностях предшественников свидетельствуют о том, что FAD2-3 является дубликатом последовательности FAD2 из субгенома C и может быть переименована как FAD2C; FAD2-1 является дубликатом последовательности FAD2 из субгенома A и поэтому может быть обозначена как FAD2A’; и наконец, FAD2-2 является второй копией, которая дуплицирована с последовательности FAD2 субгенома C и может быть обозначена как FAD2C’.
АНАЛИЗ ПОСЛЕДОВАТЕЛЬНОСТЕЙ КОДИРУЮЩИХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ FAD3, ВЫДЕЛЕННЫХ ИЗ BAC-БИБЛИОТЕКИ
Сконструированную BAC-библиотеку использовали для выделения кодирующих последовательностей гена FAD3. Проводили эксперименты по секвенированию, чтобы идентифицировать конкретные генные последовательности пяти паралогов гена FAD3 из Brassica napus L. сорта DH10275.
Последовательность гена FAD3 сначала идентифицировали в модельном виде Arabidopsis thaliana. Последовательность гена указана в Genbank с маркировкой локуса: At2g29980. Ранее были описаны сравнительные геномные взаимосвязи между модельным видом растения Arabidopsis thaliana и диплоидным Brassica rapa, одним из предшественников тетраплоида Brassica napus (Schranz et al. (2006), Trends in Plant Science 11 (11): 535-542). Конкретно в отношении гена FAD сравнительный анализ предполагает, что 3-4 копии гена могут встречаться в диплоидном геноме Brassica. Дополнительные исследования по генетическому картированию были осуществлены Scheffler et al. ((1997) Theoretical and Applied Genetics 94: 583-591). Результаты таких исследований по генетическому картированию показали, что шесть копий гена FAD3 присутствовали в Brassica napus.
Предыдущие попытки секвенирования, сфокусированные на генах FAD3 из Brassica napus, привели к идентификации и генетическому картированию специфичных копий обоих геномов A и C (Hu et al, (2006) Theoretical and Applied Genetics, 113(3): 497-507). Ранее была сконструирована и секвенирована коллекция EST-последовательностей из специфичных для семян библиотек кДНК для линии растений DH 12075 Andrew Sharpe, Agriculture and Agri-food Canada, 107 Science Place, Saskatoon, Saskatchewan. Что касается коллекции EST из растения канолы DH 12075 с двумя гаплоидными наборами, то не было полноразмерных последовательностей, кроме того также не было данных о качестве последовательностей и достоверности правильно названных нуклеотидов. Следовательно, вариации последовательностей между разными ридами последовательностей гена FAD нельзя было однозначно отнести к разными копиям генов разных паралогов семейства генов FAD3, не была доступна геномная последовательность. Однако когда осуществляли комбинированный анализ последовательностей с использованием EST, а также двух полноразмерных последовательностей генов FAD3A и FAD3C, описанных Hu et al. (2006), идентифицировали EST, которые соответствовали обоим генам, наряду с 3 дополнительными гаплотипами. В результате идентифицировали всего шесть уникальных гаплотипов FAD3. После сбора всех имеющихся данных о различных гаплотипах FAD3 идентифицировали высокие уровни дивергенции экзонных последовательностей в экзоне 1. Дивергенцию последовательности FAD3 в экзоне 1 расценили как возможность, которую можно использовать для конструирования специфичных для гена/аллеля ПЦР-праймеров. Кроме того, идентифицировали экзоны, которые либо имели минимальные различия в гаплотипах (например, экзоны 5, 6, 7 и 8 имели 1-3 п. н., которые варьировали в FAD3A и FAD3C), либо не имели изменений в последовательности (например, экзоны 2 и 3).
Анализ по определению последовательностей BAC-библиотеки, которая была сконструирована для B. napus L. сорта DH 12075, привел к выделению шести BAC-последовательностей (SEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 13 и SEQ ID NO: 14), на основании которых были определены кодирующие последовательности генов FAD3A (SEQ ID NO: 15), FAD3A’ (SEQ ID NO: 16), FAD3A” (SEQ ID NO: 17), FAD3C (SEQ ID NO: 18), FAD3C” (SEQ ID NO: 19) и FAD3C (SEQ ID NO: 20). Были идентифицированы и генетически картированы последовательности генов FAD3A, FAD3A’, FAD3A”, FAD3C, FAD3C” и FAD3C.
Анализ последовательностей шести генов FAD3 проводили, применяя программу выравнивания последовательностей и построение филогенетического дерева способом ближайших соседей с использованием идентичности в процентах. Выравнивание последовательностей осуществляли с помощью программы ALIGNX® из компьютерной программы Vector NTI Advance 11.0 (Life Technologies, Carlsbad, CA), и такое выравнивание показано на фиг. 3. В программе ALIGNX® использован модифицированный алгоритм Clustal W для создания выравниваний множественных последовательностей либо белка, либо последовательностей нуклеиновых кислот для сравнения сходства и для аннотации. Филогенетическое древо, получаемое способом ближайших соседей, создавали с помощью компьютерной программы JALVIEW V2.3®, и древо показано на фиг. 4 (Waterhouse et al. (2009), Bioinformatics 25 (9): 1189-1191). Идентифицированные контиги, которые содержали гены FAD3, использовали в качестве BLASTn-запросов для базы данных генов Arabidopsis thaliana. Область каждого из 6 контигов, содержащую ген FAD3, идентифицировали путем сравнения с геном FAD3 Arabidopsis thaliana (№ доступа в Genbank: At2g29980). Затем контиги FAD3 ориентировали таким образом, чтобы все гены FAD3 были в 5’-3’-ориентации. Контиги FAD3 урезали так, чтобы они содержали не более 2 вышерасположенных (5’) и 1 нижерасположенного (3’) гена Arabidopsis thaliana, где это было возможно. После ориентирования была извлечена полная кодирующая область генов FAD3 из каждого контига и использована для создания древа способом ближайших соседей, чтобы отобразить взаимосвязь между разными представителями семейства генов FAD3. 6 представителей семейства FAD3 были объединены в 3 пары генов FAD3 (фиг.4).
СКРИНИНГ НА ОСНОВЕ ПЦР
Конструировали группу ПЦР-праймеров для скрининга вышеуказанной BAC-библиотеки. Праймеры конструировали либо в виде универсальных праймеров, которые могут амплифицировать всех представителей семейства генов, либо в виде геноспецифичных праймеров для целенаправленной амплификации аллелей. ПЦР-праймеры конструировали так, чтобы они имели длину 20 п. н. (±1 п. н.) и имели содержание G/C 50% (±8%). В таблице 2 и таблице 3 перечислены праймеры, которые были сконструированы и синтезированы. Клоны BAC-библиотеки объединяли подвергали скринингу с использованием полимеразной цепной реакции (ПЦР).
Использовали два разных набора условий для полимеразных цепных реакций (ПЦР). Первую серию ПЦР-реакций проводили в следующих условиях: 1× буфер для ПЦР (содержащий dNTP); 1,5 мМ MgCl2; 200 мкМ 0,25 Ед ДНК-полимеразы IMMOLASE® (Bioline, London, UK); 250 нМ каждого праймера; и примерно 5-10 нг матричной ДНК. Вторая серия ПЦР-реакций была разработана для амплификации геномной ДНК, и реакционная смесь содержала: 5-10 нг геномной ДНК, 1× буфер для ПЦР, 2 мМ dNTP, 0,4 мкМ прямого и обратного праймеров, и 0,25 Ед ДНК-полимеразы IMMOLASE® (Bioline, London, UK). Смеси для амплификации объединяли в конечном объеме мкл и амплифицировали, используя термоциклер MJ PTC200® (BioRad, Hercules, CA) или систему ABI 9700 GENE AMP SYSTEM® (Life Technologies, Carlsbad, CA). Основанный на ПЦР скрининг конкретных планшетов проводили, используя способ четырехмерного скрининга на основе системы скрининга, описанной Bryan et al. (Scottish Crops Research Institute annual report: 2001-2002), и описанные выше условия для ПЦР. После основанного на ПЦР скрининга объединенных BAC-библиотек амплифицированный ПЦР-продукт секвенировали, используя способ прямого секвенирования по Сэнгеру. Амплифицированные продукты очищали, используя этанол, ацетат натрия и EDTA, следуя протоколу BIGDYE® V3.1 (Applied Biosystems), и осуществляли электрофорез на автоматизированной платформе для капиллярного электрофореза ABI3730x1®.
После основанного на ПЦР скрининга и конформационного секвенирования по Сэнгеру идентифицировали набор планшетов, которые содержали различные представители семейства генов FAD2 и FAD3. Всего идентифицировали четыре уникальных последовательности паралогичных генов FAD2 и FAD3 (таблица 4 и таблица 5). Было выбрано всего по два планшета для каждой последовательности паралогичных генов FAD2 и FAD3, чтобы подвергнуть скринингу планшетов с целью идентификации конкретной лунки и клона в планшете, который содержал ген FAD2 и FAD3 (таблица 4 и таблица 5). Конкретные лунки идентифицировали в обоих планшетах, и отбирали отдельный клон для каждого из представителей семейств генов FAD2 и FAD3.
Отдельный BAC-клон для каждого идентифицированного представителя семейства генов FAD дополнительно анализировали путем секвенирования. ДНК выделяли из BAC-клона и готовили для секвенирования с использованием набора LARGE CONSTRUCT KIT® (Qiagen, Valencia, CA), следуя инструкциям производителя. Экстрагированную BAC-ДНК подготавливали для секвенирования, используя методику GS-FLX® Titanium (Roche, Indianapolis, IN), следуя инструкциям производителя. Реакции секвенирования осуществляли, используя физически разделенный на секторы планшет для пико-титрования GS-FLX TI®, при этом BAC объединяли в пары для получения оптимальных данных. BAC объединяли в пары, при этом ген FAD2 объединяли в паре с геном FAD3. Все полученные данные о последовательностях собирали, используя NEWBLER V2.0.01.14® (454 Life Sciences, Branford, CT). Собранные контиги оценивали вручную в отношении присутствия соответствующего гена FAD, используя SEQUENCHER V3.7® (GeneCodes, Ann Arbor, MI).
После того, как была идентифицирована и полностью охарактеризована полная геномная последовательность всех четырех генов FAD2 и шести генов FAD3, конструировали нуклеазы с цинковыми пальцами для связывания с последовательностями каждого конкретного представителя семейства генов.
ПРИМЕР 2: КОНСТРУИРОВАНИЕ СВЯЗЫВАЮЩИХ ДОМЕНОВ ЦИНКОВЫХ ПАЛЬЦЕВ, СПЕЦИФИЧНЫХ ПО ОТНОШЕНИЮ К ГЕНАМ FAD2
Белки цинковых пальцев, направленные против последовательностей ДНК, кодирующих различные функциональные последовательности локуса гена FAD2, конструировали, как описано ранее. См., например, публикацию Urnov et al. (2005) (Nature 435: 646-651). Примеры последовательности-мишени и спиралей для распознавания показаны в таблице 6 и таблице 8 (конструкции областей спиралей распознавания) и таблице 7 и таблице 9 (сайты-мишени). В таблице 8 и таблице 9 нуклеотиды в сайте-мишени, с которыми контактируют спирали распознавания ZFP, указаны заглавными буквами; не подвергаемые контакту нуклеотиды указаны строчными буквами. Были сконструированы сайты-мишени нуклеаз с цинковыми пальцами (ZFN) так, чтобы связать пять сайтов-мишеней FAD2A и семь сайтов-мишеней FAD3. Конструкции цинковых пальцев FAD2 и FAD3 были включены в векторы экспрессии цинковых пальцев, кодирующие белок, имеющий по меньшей мере один палец со структурой CCHC. См. публикацию патента США № 2008/0182332. В частности, последний палец в каждом белке имел остов CCHC для спирали распознавания. Неканонические последовательности, кодирующие цинковые пальцы, сливали с нуклеазным доменом фермента рестрикции типа IIS FokI (аминокислоты 384-579 последовательности, описанной Wah et al. (1998), Proc. Natl. Acad. Sci. USA 95: 10564-10569) через состоящий из четырех аминокислот линкер ZC и сигналом ядерной локализации opaque-2, полученным из Zea mays, получая нуклеазы с цинковыми пальцами (ZFN) для FAD2A. Экспрессия слитых белков была под контролем относительно сильного конститутивного промотора, такого как промотор, полученный из промотора вируса мозаики прожилок маниока (CsVMV) и фланкированный 3’-нетранслируемой область ORF23 Agrobacterium tumefaciens (AtuORF23 3'UTR v1). Добавляли самогидролизующуюся кодирующую нуклеотидную последовательность 2A из вируса Thosea asigna (Szymczak et al., 2004) между двумя слитыми белками нуклеаз с цинковыми пальцами, которые клонировали в конструкции. Примеры векторов описаны ниже.
Оптимальные цинковые пальцы подтверждали в отношении активности расщепления, используя систему на основе почкующихся дрожжей, описанную ранее для идентификации активных нуклеаз. См., например, публикацию патента США № 20090111119; публикации Doyon et al. (2008) (Nat. Biotechnol. 26: 702-708), Geurts et al. (2009) (Science 325: 433). Цинковые пальцы для различных функциональных доменов отбирали для применения in-vivo. Из множества ZFN, которые были сконструированы, получены и тестированы в отношении связывания с предполагаемыми сайтами-мишенями геномного полинуклеотида FAD, были идентифицированы ZFN, обладающие активностью in vivo активность на высоких уровнях и отобраны для дальнейших экспериментов. Такие ZFN были охарактеризованы как способные эффективно связываться и расщеплять уникальные сайты-мишени геномных полинуклеотидов FAD2 in planta.



ПРИМЕР 3: ОЦЕНКА РАСЩЕПЛЕНИЯ НУКЛЕАЗОЙ С ЦИНКОВЫМИ ПАЛЬЦАМИ ГЕНОВ FAD2
СБОРКА КОНСТРУКЦИИ
Плазмидные векторы, содержащие экспрессирующие ZFN конструкции иллюстративных нуклеаз с цинковыми пальцами, которые были идентифицированы с использованием анализа на дрожжах, который описан в примере 2, конструировали и получали, используя приемы и методики, общеизвестные в данной области. Каждую последовательность, кодирующую цинковый палец, сливали с последовательностью, кодирующей сигнал ядерной локализации opaque-2 (Maddaloni et al. (1989) Nuc. Acids Res. 17(18): 7532), которую располагали выше нуклеазы с цинковыми пальцами.
Затем слитую последовательность сигнала ядерной локализации opaque-2::нуклеазы с цинковыми пальцами спаривали с комплементарной слитой последовательностью сигнала ядерной локализации opaque-2::нуклеазы с цинковыми пальцами. Как таковая, каждая конструкция состояла из одной открытой рамки считывания, состоящей из двух слитых последовательностей сигнала ядерной локализации opaque-2::нуклеазы с цинковыми пальцами, разделенных последовательностью 2A из вируса Thosea asigna (Mattion et al. (1996), J. Virol. 70: 8124-8127). Экспрессией слитых белков управлял относительно сильный конститутивный промотор, такой как промотор, полученный из промотора вируса мозаики прожилок маниока (CsVMV) и фланкированный 3’-нетраслируемой областью ORF23 Agrobacterium tumefaciens (AtuORF23 3’UTR).
Векторы собирали, используя методику IN-FUSION™ Advantage (Clontech, Mountain View, CA). Эндонуклеазы рестрикции получали из New England BioLabs (NEB; Ipswich, MA), и использовали ДНК-лигазу T4 (Invitrogen) для лигирования ДНК. Получение плазмид осуществляли, используя набор для плазмид NUCLEOSPIN® (Macherey-Nagel Inc., Bethlehem, PA) или набор Plasmid Midi Kit (Qiagen), следуя инструкциям поставщиков. Фрагменты ДНК выделяли, используя набор для экстракции из геля QIAquick (Qiagen) после электрофореза в агарозном геле в трис-ацетате. Колонии всех собранных плазмид сначала подвергали скринингу, используя рестрикционное расщепление минипрепаратов ДНК. Плазмидная ДНК выбранных клонов была секвенирована коммерческой фирмой по секвенированию (Eurofins MWG Operon, Huntsville, AL). Данные о последовательностях собирали и анализировали, используя компьютерную программу SEQUENCHER™ (Gene Codes Corp., Ann Arbor, MI). Перед доставкой протопластов B. napus готовили плазмидную ДНК из культур E. coli, используя систему Pure Yield Plasmid Maxiprep System® (Promega Corporation, Madison, WI) или набор Plasmid MAXI KIT® (Qiagen, Valencia, CA), следуя инструкциям поставщиков.
Полученные в результате одиннадцать плазмидных конструкций, pDAB 104008 (содержащая конструкцию ZFN24845 и ZFN24844), pDAB 104009 (содержащая конструкцию ZFN24820 и ZFN24821), pDAB104010 (содержащая конструкцию ZFN24828 и ZFN24829) (фиг.5), pDAB 104003 (содержащая конструкцию ZFN24810 и ZFN24811), pDAB104011 (содержащая конструкцию ZFN24832 и ZFN24833), pDAB104002 (содержащая конструкцию ZFN24794 и ZFN24795), pDAB 104006 (содержащая конструкцию ZFN24796 и ZFN24797), pDAB 104004 (содержащая конструкцию ZFN24814 и ZFN24815), pDAB 104001 (содержащая конструкцию ZFN24800 и ZFN24801), pDAB 104005 (содержащая конструкцию ZFN24818 и ZFN24819) и pDAB 104007 (содержащая конструкцию ZFN24836 и ZFN24837) проверяли посредством расщепления ферментами рестрикции и секвенированием ДНК. В таблице 10 перечислены различные конструкции и специфические FAD последовательности, где каждый ZFN конструировали для связывания и расщепления.
Полученные в результате плазмидные конструкции; pDAB107824 (ZFN 28025-2A-28026), pDAB107815 (ZFN 27961-2A-27962), pDAB107816 (ZFN 27969-2A-27970), pDAB107817 (ZFN 27973-2A-27974), pDAB107825 (ZFN 28035-2A-28036), pDAB107826 (ZFN 28039-2A-28040), pDAB107818 (ZFN 27987-2A-27988), pDAB107827 (ZFN 28051-2A-28052), pDAB107821 (ZFN 28004-2A-28005), pDAB107819 (ZFN 27989-2A-27990), pDAB107828 (ZFN 28053-2A-28054), pDAB107829 (ZFN 28055-2A-28056), pDAB107820 (ZFN 27991-2A-27992), pDAB107822 (ZFN 28021-2A-28022) и pDAB107823 (ZFN 28023-2A-28024 проверяли посредством расщепления ферментами рестрикции и секвенированием ДНК.
ПОЛУЧЕНИЕ ДНК ДЛЯ ТРАНСФЕКЦИИ
Плазмидную ДНК описанных выше векторов стерилизовали преципитацией и промывкой в 100% (об./об.) этаноле и сушили в ламинарном боксе. Осадок ДНК суспендировали в 30 мкл стерильной бидистиллированной воды в конечной концентрации 0,7 мкг/мкл для трансфекции в протопласты клеток, как описано ниже. Осуществляли получение плазмидной ДНК, чтобы получить суперспиральную плазмидную ДНК для временной трансфекции и линеаризованную плазмидную ДНК для стабильной трансфекции. Добавление ДНК-носителя (например, ДНК спермы рыб) к трансформирующей плазмиде не требовалось в случае временной трансфекция протопластов клеток. В случае временных исследований использовали примерно 30 мкг плазмидной ДНК на трансформацию 106 протопластов.
ТРАНСФЕКЦИЯ
Трансфекцию Brassica napus L. сорта DH 10275 осуществляли как описано Spangenberg et al. (1986) (Plant Physiology 66: 1-8), препараты сред описаны в публикации Spangenberg G. and Protrykus I. (1995) Polyethylene Glycol-Mediated Direct Gene Transfer in Tobacco Protoplasts. В: Gene Transfer to Plants. (Protrykus I. and Spangenberg G. Eds.) Springer-Verlag, Berlin. Семена Brassica napus поверхностно стерилизовали в 70% этаноле. Семена погружали в 12 мл 70% раствора этанола и перемешивали, осторожно покачивая смесь в течение 10 минут. 70% раствор этанола удаляли, сливая раствор и заменяя раствором для стерилизации семян, состоящим из 1% масс./об. гипохлорита кальция и 0,1% об./об. твина-20. Семена погружали в раствор для стерилизации семян и перемешивали, осторожно покачивая смесь в течение 25 минут. Раствор для стерилизации семян сливали, и стерильные семена промывали три раза в 50 мл стерильной воды. Наконец, семена переносили на стерильный диск из фильтровальной бумаги диаметром 80 мм WHATMAN® (Fisher-Scientific, St. Louis, MO), который был вложен в чашку Петри, и семена слегка смачивали стерильной водой. Чашку Петри герметично закрывали, используя парафильм (Fisher-Scientific, St. Louis, MO), и инкубировали при 25°C в полной темноте в течение одного-двух дней. После того как наблюдали признаки появления всходов из семян, проростки переносили в чашку Петри, содержащую отвержденную среду GEM, чтобы обеспечить дальнейшее прорастание семян. Проростки инкубировали на среде GEM при 25°C в течение четырех-пяти дней.
Объем жидкой среды PS (примерно 10 мл) сливали в стерильную чашку Петри. Используя стерильные пинцет и скальпель, наземную часть четырех-пятидневных проростков в стадии роста и развития, соответствующей появлению 4 листьев, извлекали и удаляли. Определяли участки гипокотиля длиной 20-40 мм для получения наибольшей популяции небольших богатых цитоплазмой протопластов. Асептически вырезали кусочки гипокотиля и переносили в жидкую среду PS. Вырезанные кусочки гипокотиля группировали вместе и разрезали поперечно на кусочки длиной 5-10 мм. Затем кусочки гипокотиля переносили в свежую среду PS и инкубировали при комнатной температуре в течение 1 часа. Плазмолизированные гипокотили переносили в чашку Петри, содержащую раствор фермента. Следили за тем, чтобы погрузить все кусочки гипокотилей в раствор. Чашки Петри герметично закрывали парафильмом и инкубировали в течение ночи от шестнадцати до восемнадцати часов при 20-22°C, осторожно покачивая.
Протопласты клеток высвобождали из кусочков гипокотилей. Подвергнутые расщеплению в течение ночи гипокотили осторожно встряхивали, чтобы высвободить протопласты в раствор фермента. Чашку Петри слегка наклоняли, чтобы легче перенести расщепляющую суспензию, которая состояла из раствора фермента и остатков растения. Используя пипетку объемом 10 мл, расщепляющую суспензию переносили в стерилизованный блок для фильтрования протопластов (фильтр с отверстиями 100 микрон), чтобы затем отделить протопласты от растительных остатков. Осторожно постукивали по фильтрационному блоку, чтобы высвободить избыток жидкости, который улавливался на сите. Суспензию протопластов, примерно от 8 до 9 мл, осторожно перемешивали и распределяли в стерильные пластиковые круглодонные центрифужные пробирки объемом 14 мл. На каждую суспензию наслаивали 1,5 мл раствора W5. Раствор W5 осторожно распределяли над суспензией протопластов под наклоном и распределяли по каплям с минимальным встряхиванием. Добавление раствора W5 к суспензии протопластов приводило к получению богатого протопластами промежуточного слоя. Такой промежуточный слой собирали, используя пипетку. Затем собранные протопласты переносили в новую центрифужную пробирку объемом 14 мл и осторожно перемешивали. Выход и получаемые протопласты определяли, используя гемоцитометр, чтобы определить количество протопластов в миллилитре. Способ повторяли, при этом расщепляли ткань листа, чтобы получить мезофильные протопласты.
Затем добавляли раствор W5 в объеме 10 мл, и протопласты осаждали при 70 g, затем раствор W5 удаляли. Остальную часть суспензии протопластов ресуспендировали, осторожно встряхивая. Каждую пробирку, содержащую суспензию протопластов, наполняли 5 мл раствора W5 и инкубировали при комнатной температуре от одного до четырех часов. Суспензии протопластов осаждали при 70 g, и весь раствор W5 удаляли. Затем добавляли по 300 мкл буфера для трансформации в каждую из суспензий осажденных протопластов, которые содержали изолированные протопласты. В каждую пробирку к суспензии протопластов добавляли 10 мкг плазмидной ДНК. Плазмидная ДНК состояла из конструкций нуклеаз с цинковыми пальцами, описанных выше (например, pDAB 104010). Затем к суспензии протопластов добавляли 300 мкг предварительно нагретого раствора ПЭГ 4000 и по пробиркам осторожно постукивали. Суспензии протопластов и смесь для трансформации инкубировали при комнатной температуре в течение пятнадцати минут без встряхивания. В каждую пробирку дополнительно добавляли 10 мл раствора W5, осуществляя добавления последовательно, используя аликвоты 1 мл, 1 мл, 1 мл, 2 мл, 2 мл и 3 мл, при этом осторожно переворачивая пробирки между каждыми добавлениями раствора W5. Протопласты осаждали окручивая на центрифуге при 70 g. Весь раствор W5 удаляли, оставляя чистую суспензию протопластов.
Затем к осажденным протопластам клеток добавляли 0,5 мл среды K3, и клетки ресуспендировали. Ресуспендированные протопласты клеток помещали в центр чашки Петри и добавляли 5 мл K3 и 0,6 мл агарозы Sea Plaque™ (Cambrex, East Rutherford, NJ) в концентрации 1:1. Чашки Петри встряхивали одним осторожным вихревым движением и оставляли для инкубации на 20-30 минут при комнатной температуре. Чашки Петри герметично закрывали парафильмом, и протопласты культивировали в течение двадцати четырех часов в полной темноте. После инкубации в темноте чашки Петри культивировали в течение шести дней при слабом освещении (5 мкмоль м-2сек-1, лампы белого свечения Lumilux Osram L36 W/21). После стадии культивирования использовали стерильный шпатель, чтобы разделить агарозу, содержащую протопласты, на квадранты. Разделенные квадранты помещали в пластиковый культуральный сосуд объемом 250 мл, содержащий 20 мл среды A, и инкубировали в роторном встряхивателе при 80 об/мин и амплитудой качания 1,25 см при 24°C и непрерывном тусклом освещении в течение 14 дней и затем анализировали, чтобы определить уровень активности каждой конструкции нуклеазы с цинковыми пальцами.
ВЫДЕЛЕНИЕ ГЕНОМНОЙ ДНК ИЗ ПРОТОПЛАСТОВ КАНОЛЫ
Трансфицированные протопласты получали в отдельных микроцентрифужных пробирках объемом 1,5 или 2,0 мл. Клетки были в осадке на дне пробирки в буферном растворе. Экстракцию ДНК осуществляли, мгновенно замораживая клетки в жидком азоте с последующим сушкой клеток вымораживанием в течение примерно 48 часов в устройстве LABCONCO FREEZONE 4.5® (Labconco, Kansas City, MO) при -40°C и давлении примерно 133×10-3 мбар. Лиофилизированные клетки подвергали экстракции ДНК, используя набор для растений DNEASY® (QIAGEN, Carlsbad, CA), следуя инструкция производителя, за исключением того, что разрушение ткани не требовалось, и протопласты клеток добавляли непосредственно в лизирующий буфер.
ТЕСТИРОВАНИЕ ZFN FAD2A и FAD3D В ОТНОШЕНИИ РАСЩЕПЛЕНИЯ ПОСЛЕДОВАТЕЛЬНОСТИ ГЕНОМНОЙ ДНК В ПРОТОПЛАСТАХ КАНОЛЫ
Дизайн сайтов-мишеней ZFN в случае локуса генов FAD2A и FAD3 был таким, чтобы сгруппировать сайты-мишени таким образом, чтобы несколько пар ZFN перекрывали сайты-мишени. Группирование сайтов-мишеней ZFN давало возможность сконструировать ПЦР-праймеры, которые могли бы амплифицировать окружающую геномную последовательность всех представителей семейства генов FAD2A и FAD3 в пределах окна в 100 п. н., чтобы инкапсулировать все перекрывающиеся сайты-мишени ZFN. Соответственно, можно использовать методику коротких последовательностей-ридов Illumina, чтобы оценить целостность сайта-мишени ZFN в трансфицированных протопластах. Кроме того, в сконструированные ПЦР-праймеры было необходимо включить специфичные нуклеотидные основания, которые позволяли бы отнести риды последовательностей к конкретному гену, представителю семейства FAD2A и FAD3. Таким образом, в случае всех ПЦР-праймеров требовалось связывание 5-10 нуклеотидов в сторону от любого сайта разрезания мишени ZFN, так как известно, что активность в негомологичном связывании концов (NHEJ) вызывает небольшие делеции, которые могут удалить сайт праймирования, ингибировать амплификацию и поэтому нарушить оценку активности NHEJ.
Конструировали праймеры так, чтобы они связывались со всеми локусами-мишенями ZFN для семейств генов FAD2A и FAD3 (таблица 11), и эмпирически тестировали в отношении амплификации всех представителей семейств генов, используя основанное на способе Сэнгера секвенирование продуктов ПЦР-амплификации. В нескольких случаях можно было не разрабатывать праймеры, которые могли бы различать всех представителей генного семейства (таблица 12 и таблица 13), однако нужно, чтобы во всех случаях можно было различить генные последовательности-мишени FAD2A и FAD3. После конструирования ПЦР-праймеров индивидуальные последовательности ДНК-штрих-кода включали в ПЦР-праймеры, которые использовали для того, чтобы отличить разные локусы-мишени ZFN и идентифицировать специфичные риды последовательностей для трансфекции и ZFN (таблицы 11, 12 и 13).


После экстракции ДНК протопластов канолы, трансфицированных ZFN, осуществляли ПЦР-амплификацию локусов-мишеней ZFN, чтобы создать необходимые специфичные для локусов молекулы ДНК в правильной форме для основанного на Illumina секвенирования с использованием методики синтеза. Каждый анализ оптимизировали так, чтобы он работал при использовании 25 нг исходной ДНК (примерно 12500 клеточных эквивалентов генома Brassica napus). Проводили несколько реакций на образец, чтобы обеспечить охват, необходимый для оценки эффективности и специфичности NHEJ на соответствующем уровне, примерно шестнадцать ПЦР-реакций, эквивалентных 200000 копиям генома Brassica napus из отдельных протопластов. Исходные смеси (мастер-микс) для ПЦР-амплификации готовили для всех образцов, которые тестировали в одном и том же анализе, и одну реакцию, осуществляемую в трех повторах, анализировали, используя способ количественной ПЦР, который применяли для определения оптимального количества циклов, осуществляемых на ткани-мишени, чтобы гарантировать, что ПЦР-амплификация не стала ограниченной по реагентам и все еще была в стадии экспоненциальной амплификации. Эксперименты с необходимыми негативными контрольными реакциями осуществляли в 96-луночном формате, используя MX3000P THE MOCYCLER® (Stratagene, LaJolla, CA). На основании данных, полученных на платформе количественной ПЦР, относительное увеличение флуоресценции наносили на график по циклам и определяли количество циклов на анализ, которое может обеспечивать достаточную амплификацию, при этом не позволяя реакции становиться ограниченной по реагентам и пытаясь снизить осуществление излишних циклов и амплификацию общих транскриптов или молекул. Неиспользованную исходную смесь оставляли на льду до тех пор, пока не был закончен анализ с использованием количественной ПЦР, и не было определено количество циклов, и затем делили на аликвоты в необходимое количество реакционных пробирок (примерно 16 на анализ ZFN) и осуществляли ПЦР-реакцию. После амплификации образцы для одного локуса ZFN объединяли вместе и 200 мкл объединенного продукта на одну ZFN очищали, используя набор для очистки ПЦР-продуктов MINELUTE® (Qiagen), следуя инструкциям производителя. Чтобы обеспечить секвенирование образца с использованием методики коротких ридов Illumina требовалось связать с создаваемыми фрагментами при амплификации дополнительные праймеры спаренных концов. Этого достигали путем ПЦР-амплификации с использованием праймеров, которые могут быть частично комплементарными последовательности, добавляемой в первом раунде амплификации, но также содержат необходимую парную концевую последовательность. Снова определяли оптимальное количество осуществляемых циклов ПЦР, которые могут добавлять парные концевые последовательности без сверхамплификации общих фрагментов, пропуская образец в анализе циклов количественной ПЦР, как описано ранее. После ПЦР-амплификации созданный продукт очищали, используя колонку MINELUTE® (Qiagen), следуя инструкциям производителя, и разделяли в 2,5% агарозном геле. Фрагменты ДНК, визуализированные с использованием SYBER SAFE® (Life Technologies, Carlsbad, CA) в виде полос правильного размера, экстрагировали из геля, чтобы удалить остаточные созданные в ПЦР димеры праймеров или другие ложные фрагменты, при этом ДНК экстрагировали из кусочка геля, используя набор для экстракции MINELUTE® (Qiagen), следуя инструкциям производителя. После завершения экстракции из геля проводили дополнительную очистку ДНК, используя магнитные шарики AMPURE® (Beckman-Coulter, Brea, CA) при соотношении ДНК к шарикам 1:1,7. Затем оценивали концентрацию ДНК, используя набор для количественного анализа библиотек на основе количественной ПЦР для секвенирования Illumina (KAPA) в разведении 1/40000 и 1/80000, при этом реакцию осуществляли в трех повторах. На основании результатов количественной ПЦР ДНК разбавляли до стандартной концентрации 2 нМ и объединяли со всеми библиотеками для секвенирования ДНК. Образцы готовили для секвенирования, используя набор cBOT CLUSTER® (Illumina, San Diego, CA), и секвенировали на ILLUMINA GA2X®, используя риды для секвенирования спаренных концов длиной 100 п. н., следуя инструкциям производителя.
СПОСОБ АНАЛИЗА ДАННЫХ ДЛЯ ВЫЯВЛЕНИЯ НЕГОМОЛОГИЧНОГО КОНЦЕВОГО СВЯЗЫВАНИЯ В САЙТАХ-МИШЕНЯХ ЦИНКОВЫХ ПАЛЬЦЕВ
После завершения реакции секвенирования и определения первичных данных, осуществляемого с использованием биоинформационного конвейера Illumina для распознавания оснований, осуществляли полный анализ, чтобы идентифицировать делетированные в сайте-мишени ZFN в каждом случае. Был создан собственный PERL-скрипт для того, чтобы с помощью вычислительных средств извлечь и отсортировать штрих-коды последовательностей ДНК, прослеживая список вводимых последовательностей. Штрих-код должен соответствовать эталонной последовательности с приемлемой оценкой Phred более 30, чтобы уменьшить их ошибочное приписывание ридам последовательностей. После того как риды последовательностей были отнесены в разные группы штрих-кодов, которые были использованы, все последовательности пропускали через фильтр качества. Фильтр качества был вторым специально разработанным PERL-скриптом. Риды последовательностей исключали, если было более трех оснований. обозначенных «N», или если медианная оценка Phred была меньше 20, или если было 3 следующих друг за другом основания с оценкой Phred менее 20, или если рид последовательности имел длину короче 40 п. н. Остальные последовательности объединяли, когда были доступны оба рида парных последовательностей, используя пакет NEXTGENE® (SoftGenetics, State College, PA). Оставшиеся риды объединенных последовательностей затем уменьшали до набора уникальных ридов последовательностей, используя третий специальный PERL-скрипт с использованием подсчета количества идентифицированных дублированных последовательностей, зарегистрированных на конце оставшегося идентификатора последовательностей. Уникальные риды последовательностей затем выравнивали с эталонной последовательностью FAD2 и FAD3, используя компьютерную программу NEXTGENE®, которая создавала файл выравнивания FASTA с пробелами.
Используя файл FASTA с пробелами, преобразование номера положения основания в пробеле в вводимый эталон осуществляли, используя четвертый специальный PERL-скрипт. Это позволяло идентифицировать в собранных данных такие основания, которые позволяют отличать разных представителей семейства генов (вариации либо гомеологичных, либо паралогичных последовательностей среди разных представителей семейств генов). После осуществления преобразования нумерации оснований можно было создавать отчеты о гаплотипах для ридов каждой уникальной последовательности и относить риды к конкретным представителям семейства генов. После группировки ридов в ген идентифицировали и оценивали окно длиной 10 п. н. в окружении сайта-мишени ZFN. Регистрировали количество последовательностей с делециями на ген, а также количество пропущенных оснований.
Затем данные представляли графически в виде многолинейного графика, показывающего количество последовательностей с делециями 1-10 оснований в сайте-мишени ZFN на 10000 ридов последовательностей (фиг.6). Такой анализ осуществляли в случае всех трансфекций ZFN и контрольных трансфекций. В нескольких случаях повторы в нативной последовательности ДНК приводили к увеличению ошибки секвенирования в сайте-мишени ZFN, такую ошибку обычно можно рассматривать как увеличение распространенности делеций отдельных оснований, о которых сообщалось в случае всех образцов, как трансфицированных ZFN, так и контрольных (фиг.7).
На основании полученных результатов наиболее высокий уровень активности ZFN в сайте-мишени FAD2, который определяли по наиболее высокой активности NHEJ, идентифицировали в локусе E. ZFN, которые были закодированы в плазмиде pDAB104010 (т.е., ZFN24828 и 24829) были выбраны для таргетинга in planta сконструированной платформы для интеграции трансгена (ETIP), учитывая их характеристики значимой активности в расщеплении геномной ДНК и минимальной активности, не связанной с мишенью.
ПРИМЕР 4: КОНСТРУКЦИИ ДНК ЛИНИЙ РАСТЕНИЙ КАНОЛЫ С СКОНСТРУИРОВАННОЙ ПЛАТФОРМОЙ ИНТЕГРАЦИИ ТРАНСГЕНОВ (ETIP)
Конструкции плазмидных векторов, описанные ниже, были построены с применением способов и методик, известных специалисту в данной области. Применение конкретных реагентов и методик, описанных в настоящем абзаце, хорошо известно специалистам в данной области, и они легко взаимозаменяемы другими реагентами и методиками для достижения требуемой цели создания конструкций плазмидных векторов. Эндонуклеазы рестрикции получали из New England BioLabs (NEB; Ipswich, MA). Лигирование осуществляли, используя ДНК-лигазу T4 (Invitrogen, Carlsbad, CA). Реакцию Gateway осуществляли, используя смесь ферментов GATEWAY® LR CLONASE® (Invitrogen) для встраивания путем сборки одного исходного вектора в один целевой вектор. Реакции INFUSION™ осуществляли, используя методику IN-FUSION™ Advantage Technology (Clontech, Mountain View, CA) для встраивания путем сборки одного исходного вектора в один целевой вектор. Получение плазмид осуществляли, используя набор для плазмид NUCLEOSPIN® (Macherey-Nagel Inc., Bethlehem, PA) или набор для плазмид Midi Kit® (Qiagen), следуя инструкциям поставщиков. Фрагменты ДНК выделяли, используя набор для экстракции из геля QIAquick (Qiagen) после гель-электрофореза в агарозе-трис-ацетате. Колонии всех собранных плазмид сначала подвергали скринингу, используя рестрикционное расщепление минипрепаратов ДНК. Плазмидная ДНК выбранных клонов была секвенирована коммерческой фирмой по секвенированию (Eurofins MWG Operon, Huntsville, AL). Данные о последовательностях собирали и анализировали, используя компьютерную программу SEQUENCHER™ (Gene Codes Corp., Ann Arbor, MI).
ВЕКТОРЫ, ОБЕСПЕЧИВАЮЩИЕ ПРЯМУЮ ДОСТАВКУ, ДЛЯ ТОЧНОЙ ИНТЕГРАЦИИ ETIP В ЛОКУС FAD2A КАНОЛЫ
Использовали стандартные способы клонирования для конструирования ETIP-содержащих векторов pDAS000130 (фиг.8, вставка T-нити в виде последовательности SEQ ID NO: 141) для специфичной интеграции в ген FAD2A B. napus. Такая конструкция была создана для доставки в протопласты канолы с конструкцией нуклеазы с цинковыми пальцами pDAB1004010. Конструкция нуклеазы с цинковыми пальцами будет расщеплять локус FAD2A, и затем конструкция pDAS000130 будет интегрирована в геном канолы посредством механизма зависимой от гомологии репарации. ETIP состоит из четырех кассет экспрессии (две неполных), разделенных дополнительными последовательностями распознавания ZFN, и сконструированной посадочной площадки (ELP), содержащей другие последовательности распознавания ZFN. Дополнительные последовательности распознавания ZFN являются уникальными и были сконструированы для целенаправленного введения полинуклеотидных последовательностей в трансгенные инсерции ETIP и ELP. Подобным образом последовательности распознавания ZFN могут быть использованы для вырезания полинуклеотидных последовательностей. Первая кассета экспрессии генов представляла собой неполную кассету экспрессии dsRED и содержала промотор, 5’-нетранслируемую область и интрон из гена полиубиквитина 10 Arabidopsis thaliana (промотор AtUbi) (Callis et al., (1990) J. Biol. Chem., 265: 12486-12493), за которыми следовали 210 п. н. гена dsRed из рифового коралла Discosoma sp. (Clontech, Mountain View, CA), оптимизированных по кодонам для экспрессии в двудольных растениях (экзон 1 dsRED (оптимизированный для двудольных)), с последующим интроном из гена подобного тиоредуктазе белка Arabidopsis thaliana (интрон 1 из тиоредуктазы At: номер доступа: NC_00374) и 3’-нетранслируемой областью, содержащей терминатор транскрипции и сайт полиаденилирования гена Viviparous-1 (Vp1) Zea mays (терминатор Zmlip: Paek et al., (1998) Molecules and Cells, 8(3): 336-342). Вторая кассета экспрессии содержала промотор 19S, включая 5’-UTR из вируса мозаики цветной капусты (CaMV 19S: Cook and Penon (1990) Plant Molecular Biology 14(3): 391-405), за которым следовал ген hph из E. coli, оптимизированный по кодонам для экспрессии в двудольных (hph(HygR): Kaster et al., (1983) Nucleic Acids Research 11 (19): 6895-6911), и 3’-UTR, содержащая терминатор транскрипции и сайт полиаденилирования открытой рамки считывания 1 A. tumefaciens pTi15955 (терминатор At-ORF1: Barker et al., (1983) Plant Molecular Biology 2(6): 335-50). Третья кассета экспрессии представляла собой неполную кассету экспрессии PAT и содержала первый интрон из гена 4-кумарил-CoA-синтазы Arabidopsis (интрон №2 4-кумарил-CoA-синтазы v: № доступа: At3g21320/NC003074), затем следуют последние 256 п. н. синтетического оптимизированного для растений варианта гена фосфинотрицинацетилтрансферазы, выделенного из Streptomyces viridochromogenes, который кодирует белок, который придает резистентность к ингибиторам глутаминсинтетазы, включая фосфинотрицин, глюфосинат и биалафос (3’-конец PAT(v6): Wohlleben et al., (1988) Gene 70(1): 25-37). Такая кассета заканчивалась областью 3’-UTR, содержащей терминатор транскрипции и сайты полиаденилирования открытой рамки считывания 23 A. tumefaciens pTi15955 (терминатор AtuORF23: Barker et al., (1983) Plant Molecular Biology 2(6): 335-50). Четвертая кассета экспрессии представляла собой кассету гена ipt и содержала укороченный вариант длиной 588 п. н. промотора и 5’-UTR из гена ДНК-связывающего белка MYB32 Arabidopsis (U26933) (промотор AtMYB32(T): Li et al., (1999) Plant Physiology 121: 313), за которым следовал ген изопентилтрансферазы (ipt) из A. tumefaciens и терминатор 35s, содержащий терминатор транскрипции и сайты полиаденилирования из вируса мозаики цветной капусты (терминатор CaMV 35S: Chenault et al., (1993) Plant Physiology 101 (4): 1395-1396). Для доставки к FAD2A каждый конец последовательности ETIP был фланкирован 1 т.п.н. геномной последовательности FAD2A с каждой стороны от положения двунитевого разрыва, индуцируемого доставкой ZFN, кодируемой в pDAB104010, к гену FAD2A B. napus.
Последовательность ETIP была синтезирована коммерческой фирмой, занимающейся синтезом генов (GeneArt, Life Technologies). Фрагменты длиной 1 т.п.н. геномной последовательности FAD2A амплифицировали с геномной ДНК, очищенной из ткани листа B. napus DH 12075 с использованием мининабора для растений Qiagen DNEASY® (Qiagen, Hilden), следуя инструкциям, предлагаемым производителем. Последовательности FAD2A длиной 1 т.п.н. лигировали в вектор ETIP, используя лигазу T4 (NEB, Ipswich, MA). Колонии всех собранных плазмид сначала подвергали скринингу, используя рестрикционное расщепление минипрепаратов ДНК. Эндонуклеазы рестрикции получали из New England BioLabs (NEB, Ipswich, MA) и Promega (Promega Corporation, WI). Приготовление плазмид осуществляли, используя набор QIAPREP SPIN MINIPREP® (Qiagen) или систему Pure Yield Plasmid MAXIPREP® (Promega Corporation, WI), следуя инструкциям поставщиков. Плазмидную ДНК выбранных клонов секвенировали, используя секвенирование по Сэнгеру ABI и протокол цикла секвенирования BIG DYE TERMINATOR® v3.1 (Applied Biosystems, Life Technologies). Данные о последовательностях собирали и анализировали, используя компьютерную программу SEQUENCHER™ (Gene Codes Corp., Ann Arbor, MI).
ВЕКТОРЫ, ОБЕСПЕЧИВАЮЩИЕ ПРЯМУЮ ДОСТАВКУ, ДЛЯ ТОЧНОЙ ИНТЕГРАЦИИ ETIP В ЛОКУС FAD3 КАНОЛЫ
Использовали стандартные способы клонирования для конструирования ETIP-содержащих векторов pDAS000271 (фиг.9, вставка T-нити в виде последовательности SEQ ID NO: 142), pDAS000272 (фиг.10, вставка T-нити в виде последовательности SEQ ID NO: 143), pDAS000273 (фиг.11, вставка T-нити в виде последовательности SEQ ID NO: 144), pDAS000274 (фиг.12, вставка T-нити в виде последовательности SEQ ID NO: 145) и pDAS000275 (фиг.13, вставка T-нити в виде последовательности SEQ ID NO: 146) для специфичной интеграции в ген FAD3A и FAD3C B. napus. Такая конструкция была создана для доставки в протопласты канолы с конструкцией нуклеазы с цинковыми пальцами pDAB107828 или pDAB107829. Специфичная конструкция нуклеазы с цинковыми пальцами будет расщеплять локус FAD3A, и затем конструкция pDAS000273 или pDAS275 будет интегрирована в геном канолы посредством механизма зависимой от гомологии репарации. Подобным образом, специфичная конструкция нуклеазы с цинковыми пальцами будет расщеплять локус FAD3C, и затем конструкция pDAS000271, pDAS000272 или pDAS000274 будет интегрирована в геном канолы посредством механизма зависимой от гомологии репарации. ETIP состоит из четырех кассет экспрессии (две неполных), разделенных дополнительными последовательностями распознавания ZFN, и сконструированной посадочной площадки (ELP), содержащей другие последовательности распознавания ZFN. Дополнительные последовательности распознавания ZFN являются уникальными и были сконструированы для целенаправленного введения полинуклеотидных последовательностей в трансгенные инсерции ETIP и ELP. Подобным образом последовательности распознавания ZFN могут быть использованы для вырезания полинуклеотидных последовательностей. Первая кассета экспрессии генов представляла собой неполную кассету экспрессии dsRED и содержала промотор, 5’-нетранслируемую область и интрон из гена полиубиквитина 10 Arabidopsis thaliana (промотор AtUbi) (Callis et al., (1990) J. Biol. Chem., 265: 12486-12493), за которыми следовали 210 п. н. гена dsRed из рифового коралла Discosoma sp. (Clontech, Mountain View, CA), оптимизированных по кодонам для экспрессии в двудольных растениях (экзон 1 dsRED (оптимизированный для двудольных)), с последующим интроном из гена подобного тиоредуктазе белка Arabidopsis thaliana (интрон 1 из тиоредуктазы At: номер доступа: NC_00374) и 3’-нетранслируемой областью, содержащей терминатор транскрипции и сайт полиаденилирования гена Viviparous-1 (Vp1) Zea mays (терминатор Zmlip: Paek et al., (1998) Molecules and Cells, 8(3): 336-342). Вторая кассета экспрессии содержала промотор 19S, включая 5’-UTR из вируса мозаики цветной капусты (CaMV 19S: Cook and Penon (1990) Plant Molecular Biology 14(3): 391-405), за которым следовал ген hph из E. coli, оптимизированный по кодонам для экспрессии в двудольных (hph(HygR): Kaster et al., (1983) Nucleic Acids Research 11 (19): 6895-6911), и 3’-UTR, содержащая терминатор транскрипции и сайт полиаденилирования открытой рамки считывания 1 A. tumefaciens pTi15955 (терминатор At-ORF1: Barker et al., (1983) Plant Molecular Biology 2(6): 335-50). Третья кассета экспрессии представляла собой неполную кассету экспрессии PAT и содержала первый интрон из гена 4-кумарил-CoA-синтазы Arabidopsis (интрон №2 4-кумарил-CoA-синтазы v: № доступа: At3g21320/NC003074), затем следовали последние 256 п. н. синтетического оптимизированного для растений варианта гена фосфинотрицинацетилтрансферазы, выделенного из Streptomyces viridochromogenes, который кодирует белок, который придает резистентность к ингибиторам глутаминсинтетазы, включая фосфинотрицин, глюфосинат и биалафос (3’-конец PAT(v6): Wohlleben et al., (1988) Gene 70(1): 25-37). Такая кассета заканчивалась областью 3’-UTR, содержащей терминатор транскрипции и сайты полиаденилирования открытой рамки считывания 23 A. tumefaciens pTi15955 (терминатор AtuORF23: Barker et al., (1983) Plant Molecular Biology 2(6): 335-50). Четвертая кассета экспрессии представляла собой кассету гена ipt и содержала укороченный вариант длиной 588 п. н. промотора и 5’-UTR из гена ДНК-связывающего белка MYB32 Arabidopsis (U26933) (промотор AtMYB32(T): Li et al., (1999) Plant Physiology 121: 313), за которым следовал ген изопентилтрансферазы (ipt) из A. tumefaciens и терминатор 35s, содержащий терминатор транскрипции и сайты полиаденилирования из вируса мозаики цветной капусты (терминатор CaMV 35S: Chenault et al., (1993) Plant Physiology 101 (4): 1395-1396). Для доставки к FAD3A и FAD3C каждый конец последовательности ETIP был фланкирован 1 т.п.н. геномной последовательности FAD3A или FAD3C с каждой стороны от положения двунитевого разрыва, индуцируемого доставкой кодируемой ZFN к гену FAD3A или FAD3C B. napus.
Последовательность ETIP была синтезирована коммерческой фирмой, занимающейся синтезом генов (GeneArt, Life Technologies). Фрагменты длиной 1 т.п.н. геномной последовательности FAD3A и FAD3C амплифицировали с геномной ДНК, очищенной из ткани листа B. napus DH 12075 с использованием мининабора для растений Qiagen DNEASY® (Qiagen, Hilden), следуя инструкциям, предлагаемым производителем. Последовательности FAD3A или FAD3C длиной 1 т.п.н. лигировали в вектор ETIP, используя лигазу T4 (NEB, Ipswich, MA). Колонии всех собранных плазмид сначала подвергали скринингу, используя рестрикционное расщепление минипрепаратов ДНК. Эндонуклеазы рестрикции получали из New England BioLabs (NEB, Ipswich, MA) и Promega (Promega Corporation, WI). Приготовление плазмид осуществляли, используя набор QIAPREP SPIN MINIPREP® (Qiagen) или систему Pure Yield Plasmid MAXIPREP® (Promega Corporation, WI), следуя инструкциям поставщиков. Плазмидную ДНК выбранных клонов секвенировали, используя секвенирование по Сэнгеру ABI и протокол цикла секвенирования BIG DYE TERMINATOR® v3.1 (Applied Biosystems, Life Technologies). Данные о последовательностях собирали и анализировали, используя компьютерную программу SEQUENCHER™ (Gene Codes Corp., Ann Arbor, MI).
КОНТРОЛЬНЫЕ ВЕКТОРЫ
Контрольный вектор использовали для разработки способа сортировки клеток на основе активируемой флуоресценцией сортировки клеток (FACS). Стандартные способы клонирования использовали для конструирования контрольного вектора, pDAS000031 (фиг.14: вставка T-нити в виде последовательности SEQ ID NO: 147), состоящего из двух кассет экспрессии генов. Первая кассета экспрессии генов содержала промотор 19s вируса мозаики цветной капусты (промотор 19S CaMV; Shillito et al., (1985) Bio/Technology 3; 1099-1103):: ген резистентности к гигромицину (hph(HygR); патент США № 4727028):: и 3’-нетранслируемую область открытой рамки считывания 1 Agrobacterium tumefaciens (терминатор AtORF1; Huang et al., (1990) J. Bacteriol. 1990 772: 1814-1822). Вторая кассета экспрессии генов содержала промотор убиквитина 10 Arabidopsis thaliana (промотор AtUbi10; Callis et al., (1990) J. Biol. Chem., 265: 12486-12493):: dsRED (dsRED(D); патент США № 6852849) и интрон из Arabidopsis (интрон № 1; GenBank: AB025639.1):: 3-нетранслируемую область открытой рамки считывания 23 Agrobacterium tumefaciens (терминатор AtORF23; патент США № 5428147) в виде слияния в рамке в транс-ориентации (например, в ориентации «голова к голове»). Плазмидный вектор собирали, используя методику IN-FUSION™ Advantage Technology (Clontech, Mountain View, CA).
КОНСТРУИРОВАНИЕ БИНАРНЫХ ВЕКТОРОВ ДЛЯ СЛУЧАЙНОЙ ИНТЕГРАЦИИ ETIP В РАСТЕНИЯХ КАНОЛЫ
Конструировали два бинарных вектора для случайной интеграции последовательности T-нити ETIP в геном Brassica napus. Использовали стандартные способы клонирования для конструирования ETIP-содержащих векторов pDAS000036 (фиг.15, вставка T-нити в виде последовательности SEQ ID NO: 148) и pDAS000037 (фиг.16, вставка T-нити в виде последовательности SEQ ID NO: 149). ETIP-векторы состоят из четырех кассет экспрессии (двух неполных кассет экспрессии), разделенных последовательностями распознавания ZFN, и сконструированной посадочной площадки (ELP), содержащей другие последовательности распознавания ZFN. Первая кассета экспрессии генов представляла собой неполную кассету экспрессии dsRED и содержала промотор, 5’-нетранслируемую область и интрон из гена полиубиквитина 10 Arabidopsis thaliana (промотор AtUbi) (Norris et al., (1993) Plant Molecular Biology, 21(5): 895-906), за которыми следовали 210 п. н. гена dsRed из рифового коралла Discosoma sp. (Clontech, Mountain View, CA), оптимизированных по кодонам для экспрессии в двудольных растениях, с последующим интроном из гена подобного тиоредуктазе белка Arabidopsis thaliana (№ доступа: NC_00374) и 3’-нетранслируемой областью, содержащей терминатор транскрипции и сайт полиаденилирования гена Viviparous-1 (Vp1) Zea mays (терминатор Zm Lip: Paek et al., (1998) Molecules and Cells, 8(3): 336-342). Вторая кассета экспрессии содержала промотор 19S, включая 5’-UTR из вируса мозаики цветной капусты (промотор 19 CsVMV 19S: Cook and Penon (1990) Plant Molecular Biology 14(3): 391-405), за которым следовал ген hph из E. coli, оптимизированный по кодонам для экспрессии в двудольных (hph(D): Kaster et al., (1983) Nucleic Acids Research 11 (19): 6895-6911), и 3’-UTR, содержащая терминатор транскрипции и сайт полиаденилирования открытой рамки считывания 1 (PRF1) A. tumefaciens pTi15955 (терминатор At-ORF1: Barker et al., (1983) Plant Molecular Biology 2(6): 335-50). Третья кассета экспрессии представляла собой неполную кассету экспрессии PAT и содержала первый интрон из гена 4-кумарил-CoA-синтазы Arabidopsis (№ доступа: At3g21320(NC003074)), затем следуют последние 256 п. н. синтетического оптимизированного для растений варианта гена фосфинотрицинацетилтрансферазы (PAT), выделенного из Streptomyces viridochromogenes, который кодирует белок, который придает резистентность к ингибиторам глутаминсинтетазы, включая фосфинотрицин, глюфосинат и биалафос (PATv6 (экзон2); Wohlleben et al., (1988) Gene 70(1): 25-37). Такая кассета заканчивалась областью 3’-UTR, содержащей терминатор транскрипции и сайты полиаденилирования открытой рамки считывания 23 (ORF23) A. tumefaciens pTi15955 (терминатор AtuORF23: Barker et al., (1983) Plant Molecular Biology 2(6): 335-50). Четвертая кассета экспрессии представляла собой кассету гена ipt и содержала укороченный вариант длиной 588 п. н. промотора и 5’-UTR из гена ДНК-связывающего белка MYB32 Arabidopsis (U26933) (промотор AtMYB32(T); Li et al., (1999) Plant Physiology 121: 313), за которым следовал ген изопентилтрансферазы (ipt) из A. tumefaciens и терминатор 35s, содержащий терминатор транскрипции и сайты полиаденилирования из вируса мозаики цветной капусты (терминатор CaMV 35S: Chenault et al., (1993) Plant Physiology 101 (4): 1395-1396).
Кассеты экспрессии и ELP были синтезированы с сайтами Multi-Gateway коммерческой фирмой, осуществляющей синтез генов (GeneArt, Life Technologies). Были сконструированы исходные клоны каждой кассеты экспрессии и ELP с использованием смеси ферментов BP Clonase II (Invitrogen, Life Technologies) и набора с вектором pDONR221 (Invitrogen, Life Technologies). Затем исходные клоны использовали в реакции Multi-Gateway со способным к Gateway бинарным вектором, используя смесь ферментов LR Clonase II Plus (Invitrogen, Life Technologies). Колонии всех собранных плазмид сначала подвергали скринингу, используя рестрикционное расщепление минипрепаратов ДНК. Эндонуклеазы рестрикции получали из New England BioLabs (NEB; Ipswich, MA) и Promega (Promega Corporation, WI). Получение плазмид осуществляли, используя набор QIAprep Spin Miniprep (Qiagen, Hilden) или систему Pure Yield Plasmid MAXIPREP® (Promega Corporation, WI), следуя инструкциям поставщиков. Плазмидную ДНК выбранных клонов секвенировали, используя протокол секвенирования по Сэнгеру ABI и протокол цикла секвенирования BIG DYE TERMINATOR® v3.1 (Applied Biosystems, Life Technologies). Данные о последовательностях собирали и анализировали, используя компьютерную программу SEQUENCHER™ (Gene Codes Corp., Ann Arbor, MI).
ПРИМЕР 5: СОЗДАНИЕ ETIP-ЛИНИЙ РАСТЕНИЙ КАНОЛЫ
ТРАНСФОРМАЦИЯ Brassica napus
Конструкции ETIP (pDAS000036, pDAS000037), контрольная конструкция DS-Red (pDAS000031) и специфичные для сайтов FAD2A, FAD3A и FAD3C конструкции (pDAS000130 и pDAS000271-pDAS000275) и сопутствующие нуклеазы с цинковыми пальцами (pDAB104010, pDAB10728 и pDAB10729) описаны в примере 4. Бинарными векторами трансформировали Agrobacterium tumefaciens штамма GV3101: PM90. Трансформацию протопластов клеток Brassica napus осуществляли, используя протокол трансфекции, описанный в примере 3, с некоторыми модификациями.
Модификации протокола включали использование альгината натрия вместо агарозы Sea PlaqueTM. Эксперименты по трансфекции, в которых конструкцию нуклеазы с цинковыми пальцами и конструкцию ETIP доставляли совместно в протопласты клеток Brassica napus, осуществляли при концентрациях ДНК, соответствующей плазмидной ДНК в молярном соотношении 5:1. Другими конструкциями ETIP и контрольных плазмид трансформировали в концентрациях 30 мкг плазмидной ДНК. Соответственно, pDAS000130 использовали в концентрации 27,8 мкг плазмидной ДНК, и pDAB104010 использовали в концентрации 2,2 мкг плазмидной ДНК. Другими конструкциями ETIP и контрольных плазмид трансформировали в концентрациях 30 мкг плазмидной ДНК.
Дополнительные модификации протокола включали выращивание целых растений из трансформированных протопластов клеток в среде, содержащей 1,5 мг/мл гигромицина. При выращивании целых растений требовалась замены среды A каждые две недели, и наблюдение за ростом полученных из протопластов колоний. После того, как полученные из протопластов колонии вырастали примерно до 2-3 мм в диаметре, колонии переносили в отдельные лунки 12-луночного планшета COSTAR® (Fisher Scientific, St. Louis, MO), содержащего отвержденную среду MS morpho. Планшеты инкубировали в течение одной-двух недель при 24°C при непрерывном тусклом освещении вплоть до того, как каллусы пролиферировали до размера 8-10 мм в диаметре. После того как протопласты клеток достигали 1-2 см в диаметре, протопласты клеток переносили в отдельные культуральные сосуды объемом 250 мл, содержащие среду MS morpho. Сосуды инкубировали при 24°C в условиях 16 часов освещения (20 мкмоль м-2сек-1, лампы белого свечения Lumilux Osram L36 W/21) и 8 часов темноты. В пределах периода времени одной-двух недель были видны множественные побеги. Побеги переносили в культуральные сосуды объемом 250 мл, содержащие среду MS, после того, как они достигали длины 3-4 см. Культуральные сосуды объемом 250 мл инкубировали при 24°C в условия 16 часов освещения (20 мкмоль м-2сек-1, лампы белого свечения Lumilux Osram L36 W/21) и 8 часов темноты. Побеги поддерживали в культуральных сосудах, пока они не развивались в растеньица, и в это время их переносили в теплицу для выращивания до достижения зрелости.
ПРИМЕР 6: МОЛЕКУЛЯРНОЕ ПОДТВЕРЖДЕНИЕ ИНТЕГРАЦИИ ETIP, СОДЕРЖАЩИХ T-ДНК, В КАНОЛЕ
Геномную ДНК экстрагировали из ткани листа всех предполагаемых трансгенных растений, используя набор для экстракции ДНК растений DNEASY® 96 или мини-набор для растений DNEASY® Plant Mini Kit™ (Qiagen). Геномную ДНК из каждого растения анализировали в ПЦР, используя праймеры, сконструированные для амплификации virC, прямой pTiC58 (SEQ ID NO: 150 CGAGAACTTGGCAATTCC) и обратный pTiC58 (SEQ ID NO:151 TGGCGATTCTGAGATTCC), чтобы тестировать сохранение A. tumefaciens, праймеры, сконструированные для амплификации актина из B. napus; прямой актина (SEQ ID NO: 152 GACTCATCGTACTCTCCCTTCG) и обратный актина (SEQ ID NO: 153 GACTCATCGTACTCTCCCTTCG), чтобы проверить качество геномной ДНК. Праймеры конструировали для амплификации гена hph; прямой HPH (SEQ ID NO: 154 TGTTGGTGGAAGAGGATACG) и обратный HPH (SEQ ID NO: 155 ATCAGCAGCAGCGATAGC), кодируемые ETIP. Растения, которые не давали продукта с праймеров virC, но в случае которых амплифицировали продукты правильного размера с использованием праймеров к актину и hph, классифицировали как трансгенные.
Осуществляли второй скрининг, при котором гДНК из каждого трансгенного растения анализировали в ПЦР, используя пять наборов праймеров, сконструированных для амплификации бинарного вектора вне области T-ДНК [(1F SEQ ID NO: 156 ATGTCCACTGGGTTCGTGCC; 1R SEQ ID NO: 157 GAAGGGAACTTATCCGGTCC) (2F SEQ ID NO: 158 TGCGCTGCCATTCTCCAAAT; 2R SE ID NO: 159 ACCGAGCTCGAATTCAATTC) (3F SEQ ID NO: 160 CCTGCATTCGGTTAAACACC; 3R SEQ ID NO: 161 CCATCTGGCTTCTGCCTTGC) (4F SEQ ID NO: 162 ATTCCGATCCCCAGGGCAGT; 4R SEQ ID NO: 163 GCCAACGTTGCAGCCTTGCT) (5F SEQ ID NO: 164 GCCCTGGGATGTTGTTAAGT; 5R SEQ ID NO: 165 GTAACTTAGGACTTGTGCGA)]. Считали, что растения, в случае которых амплифицировали ПЦР-продукты правильного и ожидаемого размера с использованием наборов праймеров 3 и 4, имеют интеграцию в остове.
ДНК из растений без интеграции в остове очищали из 20 г ткани листа, используя модифицированный способ CTAB (Maguire et al., (1994) Plant Molecular Biology Reporter, 12(2): 106-109). Изолированную гДНК расщепляли несколькими ферментами рестрикции, и 10 мкг гДНК разделяли электрофорезом в агарозном геле и переносили на мембрану, используя стандартный протокол Саузерн-блоттинга. Мембраны исследовали с помощью зондов, используя систему DIG Easy Hyb System™ (Roche, South San Francisco, CA), следуя инструкциям производителя. Зонды к каждой кассете экспрессии, к ELP и к эндогенному контрольному гену актина амплифицировали с конструкции ETIP, используя следующие праймеры: (IPT-F SEQ ID NO:166 TCTCTACCTTGATGATCGG; IPT-R SEQ ID NO:167 AACATCTGCTTAACTCTGGC; dsRED-F SEQ ID NO:168 ATGGCTTCATCTGAGAACG; dsRED-R SEQ ID NO:169 TTCCGTATTGGAATTGAGG; PAT-F SEQ ID NO:170 TTGCTTAAGTCTATGGAGGCG; PAT-R SEQ ID NO:171 TGGGTAACTGGCCTAACTGG; ELP-F SEQ ID NO:172 ATGATATGTAGACATAGTGGG; ELP-R SEQ ID NO:173 AGGGTGTAAGGTACTAGCC; Hph-F SEQ ID NO:174 TGTTGGTGGAAGAGGATACG; Hph-R SEQ ID NO:175 ATCAGCAGCAGCGATAGC; актин-F SEQ ID NO:176 GTGGAGAAGAACTACGAGCTACCC; актин-R SEQ ID NO:177 GACTCATCGTACTCTCCCTTCG).
Последовательность ETIP амплифицировали и секвенировали из всех растений, содержащих только одну копию ETIP. Последовательность каждой вставки T-ДНК анализировали прямым секвенированием продуктов ПЦР с использованием ABI3730xI™ (Applied Biosystems, Life Technologies). Вставку T-ДНК амплифицировали с геномной ДНК, используя полимеразу Phusion Hot Start II (Finnzymes, Thermo Fisher Scientific). Реакции амплификации T-ДНК осуществляли с использованием множественных пар праймеров, чтобы амплифицировать перекрывающиеся последовательности длиной приблизительно 2 т.п.н. Каждый продукт ПЦР секвенировали с использованием нескольких праймеров, чтобы обеспечить полное покрытие. Реакционные смеси для ПЦР обрабатывали щелочной фосфатазой креветки и экзонуклеазой I (Applied Biosystems, Life Technologies), чтобы инактивировать избыток праймера перед реакцией ПЦР для секвенирования. Последовательности, фланкирующие вставку T-ДНК каждой ETIP-линии с единичными копиями, идентифицировали, расщепляли очищенную геномную ДНК с использованием восьми эндонуклеаз рестрикции с последующим лигирование двунитевых адаптеров, специфичных для выступающих концов, создаваемых эндонуклеазами рестрикции. После такой стадии лигирования осуществляли ПЦР с использованием биотинилированного праймера либо к 3’-, либо к 5’-концу ETIP и праймера к каждому адаптеру. Продукты ПЦР улавливали и очищали на шариках для обратимой иммобилизации на твердой фазе (SPRI) Ampure (Agencourt Bioscience Coiporation, Beckman Coulter Company). Осуществляли гнездовую ПЦР, и все продукты секвенировали, используя секвенирование по Сэнгеру ABI и протокол цикла секвенирования BIG DYE TERMINATOR® v3.1 (Applied Biosystems, Life Technologies). Данные о последовательностях собирали и анализировали, используя компьютерную программу SEQUENCHER™ (Gene Codes Corp., Ann Arbor, MI).
САУЗЕРН-БЛОТ-АНАЛИЗ
Были выбраны специфичные ферменты рестрикции для расщепления образцов гДНК перед Саузерн-анализом. Предполагаемые трансгенные растения анализировали, расщепляя геномную ДНК EcoRI и SwaI. Затем образцы расщепленной гДНК и неразрезанной гДНК анализировали, используя в качестве зондов любой из полинуклеотидных фрагментов, содержащий элементы генов ΡΑΤv6, IPT или ELP, так как такие фрагменты полинуклеотидных зондов обеспечивают дифференциацию множественных вставок в продуктах расщепления EcoRI, а также продуктах расщепления SwaI. Идентифицированные линии растений с одиночными копиями трансгенов дополнительно анализировали с использованием всех шести зондов, чтобы идентифицировать присутствие всех важных элементов встроенного вектора.
Соответственно, было отобраны образцы для 67 независимых событий трансформации ETIP-pDAS000036 и тестированы в отношении присутствия трансгена (hph) и присутствия остова вектора. Из 67 тестированных растений в 47 обнаружено наличие трансгена, интегрированного в геном. Было обнаружено, что Из 47 трансгенных растений 17 растений содержали остов вектора (таблица 14). Из остальных 30 растений, которые не содержали значительной части остова вектора (отсутствие Ori или SpecR), отбирали образцы для Саузерн-анализа. Как правило, растения сначала подвергали скринингу с использованием зонда IPT, и линии растений, идентифицированные как предполагаемые линии с одной копией, дополнительно тестировали, используя зонды, содержащие элементы генов dsRED, PAT, ELP и hph, чтобы подтвердить присутствие полной кассеты.
Подобным образом, отбирали образцы для 52 независимых событий трансформации ETIP-pDAS000037 растений, выживших на почве, и тестировали в отношении присутствия трансгена (hph) и присутствия остова вектора. Было обнаружено, что из 52 тестированных растений 48 имели трансген, интегрированный в геном. Обнаружено, что из 48 трансгенных растений 23 растения также содержали остов вектора, и 3 растения не тестировали (таблица 14). Из остальных 22 растений, которые не содержали значительной части векторного остова (отсутствие Ori или SpecR), брали образцы для Саузерн-анализа. Такие трансгенные растения сначала подвергали скринингу с использованием зонда IPT, и линии растений, идентифицированные как предполагаемые линии с одной копией, дополнительно тестировали, используя зонды к dsRED, PAT, ELP, hph и актину, чтобы подтвердить результаты. После идентификации 5 независимых линий с одной копией осуществляли Саузерн-анализ на остальных растениях. Всего Саузерн-анализу подвергали 11 линий ETIP-pDAS000037.
РЕЗУЛЬТАТЫ, ПОЛУЧЕННЫЕ В СЛУЧАЕ ТРАНСГЕННОЙ ПО ETIP КАНОЛЫ, ТРАНСФОРМИРОВАННОЙ PDAS000036 и PDAS000037
Трансгенные события Brassica napus, которые получали в результате трансформации pDAS000036 и pDAS000037, приводили к получению однокопийных полноразмерных инсерций T-нити. Три-четыре события для каждого растения полностью характеризовали и предположительно картировали в конкретных хромосомах в геноме Brassica napus. Хотя происходило несколько перестроек отдельных пар оснований во время интеграции T-нити, в случае выбранных событий растения содержали полноразмерные кассеты экспрессии, которые способны давать устойчивую экспрессию трансгена. Отобранные растения T0 с такими событиями выращивали до стадии развития T1. T1 подвергали повторному скринингу, используя описанные выше ПЦР-анализы, чтобы определить зиготность интегрированной T-нити. Подвергнутые скринингу события классифицировали как гомозиготные, гемизиготные или нулевые.
Последовательность ETIP амплифицировали и секвенировали из всех растений с трансгенными событиями, содержащих только одну копию интегрированной последовательности ETIP. Последовательность каждой вставки T-ДНК анализировали прямым секвенированием продуктов ПЦР. Вставку T-ДНК амплифицировали с геномной ДНК, используя полимеразу Phusion Hot Start II (Finnzymes, Thermo Fisher Scientific). Затем T-ДНК амплифицировали с использованием множественных пар праймеров, чтобы амплифицировать перекрывающиеся последовательности длиной приблизительно 2 т.п.н. Каждый продукт ПЦР секвенировали с использованием нескольких праймеров, чтобы обеспечить полное покрытие. Реакционные смеси для ПЦР обрабатывали щелочной фосфатазой креветки и экзонуклеазой I (Applied Biosystems, Life Technologies), чтобы инактивировать избыток праймера перед реакцией ПЦР для секвенирования.
Последовательности, фланкирующие вставку T-ДНК каждой ETIP-линии с единичными копиями, идентифицировали, расщепляли очищенную геномную ДНК с использованием восьми эндонуклеаз рестрикции с последующим лигирование двунитевых адаптеров, специфичных для выступающих концов, создаваемых эндонуклеазами рестрикции. После такой стадии осуществляли ПЦР с использованием биотинилированного праймера либо к 3’-, либо к 5’-концу ETIP и праймера к каждому адаптеру. Продукты ПЦР улавливали и очищали на шариках для обратимой иммобилизации на твердой фазе (SPRI) Ampure (Agencourt Bioscience Coiporation, Beckman Coulter Company). Осуществляли гнездовую ПЦР, и все продукты секвенировали, используя секвенирование по Сэнгеру ABI и протокол цикла секвенирования BIG DYE TERMINATOR® v3.1 (Applied Biosystems, Life Technologies). Данные о последовательностях собирали и анализировали, используя компьютерную программу SEQUENCHER™ (Gene Codes Corp., Ann Arbor, MI). Восемь линий ETIP было идентифицировано и отобрано для анализа фланкирующих последовательностей (таблица 15). Левые и правые фланкирующие последовательности (также описанные как последовательности границ или соединительные последовательности) представлены в виде SEQ ID NO: 431 - SEQ ID NO: 446, подчеркнутые последовательности показывают плазмидный вектор, неподчеркнутые последовательности показывают геномную фланкирующую последовательность.
Подробное описание pDAS000036: левая фланкирующая граница em02-5788-1-1 (SEQ ID NO: 431)







КАРТИРОВАНИЕ ETIP
Для каждого трансгенного события, которое заключалось в инсерции одной копии ETIP, была взята фланкирующая последовательность после сборки вручную и использована в качестве запроса в локальном BLAST-анализе. Всего таким способом идентифицировали восемь растений, которые имели интеграции единичной копии (таблица 16 и таблица 17). Набор из 595478 последовательностей, полученных с использованием методики «дробовика» из генома Brassica oleracea, загружали из базы данных NCBI GSS и форматировали в виде базы данных нуклеотидных последовательностей BLAST. Затем фланкирующие ETIP последовательности сравнивали в BLASTn с базой данных, и все совпадения исследовали вручную. Затем выбирали наиболее значительное совпадение последовательности с фланкирующей последовательностью ETIP из базы данных для B. oleracea и выравнивали с доступной онлайн геномной последовательностью Brassica rapa (http://brassicadb.org/brad/blastPage.php), при этом также находили положение в геноме, которое имело наиболее значительное совпадение последовательности. В тех случаях, когда только 5’- или 3’-фланкирующие последовательности имели значительное совпадения с геномными последовательностями B. oleracea, полагали, что не поддающаяся выравниванию или несовпадающая последовательность либо связана с идентификацией отсутствия последовательности в базе данных, либо имели место значительные перестройки генома, возникающие во время интеграции ETIP. В случае образцов, которые дают значимые совпадения в BLASTn при анализе фланкирующей последовательности ETIP, наиболее значительно совпадающую последовательность B. oleracea GSS наряду с наиболее значимо совпадающей последовательностью из генома B. rapa затем выравнивали вручную в компьютерной программе SequencherTM (Gene Codes Corp., Ann Arbor, MI) для каждого из растений с единичной копией ETIP. Затем сравнивали три последовательности и наиболее сходную последовательность из любого из диплоидных видов Brassica, сравниваемую с фланкирующей ETIP, определяли как геном, в котором расположен ETIP. В случае большинства образцов существовала значимая вариабельность между двумя последовательностями диплоидного генома Brassica, и для полученной из B. napus фланкирующей последовательности ETIP наблюдали преобладающую ассоциацию с одной или другой из диплоидных последовательностей. Однако были случаи, когда была недостаточная вариабельность последовательностей между диплоидами, и оценка группы сцепления могла быть возможна, но оценка субгенома была невозможной. Затем предполагали специфичную локализацию в геноме на основании расположения геномной последовательности Brassica rapa. В случаях, когда ETIP идентифицировали как интегрированный в геном C B. oleracea, использовали сравнительную синтению между диплоидными геномами Brassica, описанную Parkin et al. (Genetics 2005, 171: 765-781), чтобы экстраполировать геномное положение в субгеноме C Brassica napus. Кроме того, идентифицированные последовательности сравнивали в BLASTn с кодирующими последовательностями геномов Arabidopsis thaliana (CDS TAIR 9, загруженными с сайта http://Arabidopsis.org/index.jsp) и идентифицировали любые нарушенные генные последовательности, а также подтверждали положения в геноме, следуя синтении Arabidopsis-Brassica, описанной Schranz et al. (Trend in Plant Science 2006, 11, 11: 535-542).
Гомозиготные события использовали для получения протопластов с применением описанного ранее способа. Затем протопласты котрансфицировали нуклеазой с цинковым пальцем, которая предназначена для целенаправленного воздействия на сайт связывания цинкового пальца, который включен в последовательность ETIP и донорную плазмиду, которая обладает гомологией с конкретными областями ETIP. Нуклеаза с цинковым пальцем расщепляет локус ETIP, и донорная плазмида интегрируется в геном клеток Brassica napus посредством зависимой от гомологии репарации. В результате интеграции донорной плазмиды неполный трансген DS-red репарируется до полноразмерного трансгена DS-red. После этого экспрессию полностью функционального трансгена DS-red использовали для сортировки протопластов клеток способом FACS. Предполагаемые трансгенные растения отсортировывали, используя способ FACS, описанный в примере 7, и изолированные протопласты регенерировали в зрелые растения. Интеграцию донорной плазмиды подтверждали в растениях с целенаправленно введенной ETIP, используя молекулярные способы подтверждения. Соответственно, локус ETIP служит в качестве сайт-специфичного локуса для целенаправленной интеграции в ген донорной полинуклеотидной последовательности.
Положения таргетинга в геном обеспечивают такие положения генома, которые не изменяют нормальный фенотип растения. Полученные в результате события, когда трансген целенаправленно вводят в ETIP, не приводят к агрономически значимым непредусмотренным различиям, которые могут быть выявлены при сравнении событий ETIP с контрольными растениями. Кроме того, уровни экспрессии белка с трансгенов, интегрированных в локус ETIP, устойчивы и сопоставимы и стабильны во множестве геномных положений. Раскрытые геномные последовательности SEQ ID NO: 431 - SEQ ID NO: 446 обеспечивают положения в геноме Brassica, которые могут быть мишенями для интеграции кассет экспрессии генов, содержащих трансген.
МОЛЕКУЛЯРНОЕ ПОДТВЕРЖДЕНИЕ ИНТЕГРАЦИИ ETIP В FAD2A КАНОЛЫ
Геномную ДНК экстрагировали из ткани листа всех предполагаемых трансгенных растений, используя набор DNeasy Plant Mini KitTM (Qiagen), следуя инструкциям производителя, за исключением того, что ткань элюировали в 80 мкл буфера AE. Тридцать миллиграмм ткани молодых листьев от регенерированных растений мгновенно замораживали в жидком азоте перед измельчением в порошок.
Молекулярную характеристику локуса FAD2A осуществляли, используя три независимых анализа. Анализы планировали и оптимизировали, используя следующие контроли: охарактеризованные трансгенные события с одним случайно интегрированным трансгеном, охарактеризованное трансгенное событие с пятью случайно интегрированными трансгенами, растения канолы дикого типа c.v. DH 12075 и нематричные контрольные реакции. Результаты трех следующих молекулярных анализов оценивали вместе, чтобы получить доказательство интеграции ETIP в FAD2A посредством HDR.
ИДЕНТИФИКАЦИЯ ИНТЕГРАЦИИ ТРАНСГЕНА С ИСПОЛЬЗОВАНИЕМ ПОЛИМЕРАЗНОЙ ЦЕПНОЙ РЕАКЦИИ В РЕАЛЬНОМ ВРЕМЕНИ
Четыре повтора каждого растения анализировали, используя праймеры, специфичные к гену-мишени hph (также описанный как hpt) (SEQ ID NO: 447, hpt F791 5’ CTTACATGCTTAGGATCGGACTTG 3’; SEQ ID NO: 448, hpt R909 5’ AGTTCCAGCACCAGATCTAACG 3’; SEQ ID NO: 449, hpt Taqman 872 5’ CCCTGAGCCCAAGCAGCATCATCG 3’ FAM) (фиг.31) и эталонному гену, кодирующему белок группы с высокой подвижностью I/Y (HMG I/Y) (SEQ ID NO: 450, F 5’ CGGAGAGGGCGTGGAAGG 3’; SEQ ID NO: 451, R 5’ TTCGATTTGCTACAGCGTCAAC 3’; SEQ ID NO: 452, зонд 5’ AGGCACCATCGCAGGCTTCGCT 3’ HEX). Реакции амплификации осуществляли, используя следующие условия: 95°C в течение 10 минут с последующими 40 циклами при 95°C в течение 30 секунд, при 60°C в течение 1 минуты, при этом данные об амплификация собирали в конце каждой стадии отжига. Количество копий вычисляли, используя способ ΔCq, где ΔCq = Cq(гена-мишени) - Cq(эталонного гена). Livak, K.J. and T.D. Schmittgen, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 2001. 25(4): p. 402-8. Растения с амплификацией hph и HMG I/Y и количеством копий 0,5 или больше считали трансгенными, тогда как растения с количеством копий ≥0,5 и ≤1,2 предположительно считал однокопийными. Амплификацию осуществляли, используя систему детекции ПЦР в реальном времени BioRad CFX96 Touch™ и набор FastStart Universal Probe Master (ROX) (Roche, Basel, Switzerland).
ВЫЯВЛЕНИЕ НАРУШЕННОГО САЙТА ZNF FAD2A
Каждое растение анализировали в отношении присутствия или отсутствия амплификации эндогенной мишени в тесте нарушенного локуса, который представляет собой анализ доминантов. Анализ представляет собой кПЦР-анализ с использованием SYBR® Green I и проводимый в синглплексном формате, но с проведением каждой реакцией одновременно в одном и том же планшете для ПЦР, и нацелен на эндогенный локус (FAD2A/2C.RB.UnE.F1, SEQ ID NO: 453, 5’ CTTCCACTCCTTCCTCCTCGT*C 3’ и FAD2A/2C.RB.UnE.R1, 5’ SEQ ID NO: 454, GCGTCCCAAAGGGTTGTTGA*G 3’) и локус ZFN (локус, в котором ZFN pDAB104010 связывается и разрезает геном) (FAD2A.UnE.F1, SEQ ID NO: 455, 5’ TCTCTACTGGGCCTGCCAGGG*C 3’ и FAD2A.UnE.R1, SEQ ID NO: 456, 5’ CCCCGAGACGTTGAAGGCTAAGTACAA*A 3’) (фиг.32). Обе пары праймеров амплифицировали, используя следующие условия: 98°C в течение 30 секунд с последующими 35 циклами (98°C в течение 10 секунд, 65°C в течение 20 секунд, 72°C в течение 90 секунд), затем 95°C в течение 10 секунд, затем анализ плавления от 50°C до 95°C с шагом 0,5°C в течение 0,05 секунд, и планшет считывали на каждом шаге. Условия реакции перечислены в таблице 18.
Растения, в которых имела место амплификация эндогенной мишени, не было амплификации мишени ZFN, оценивали как позитивные в тесте нарушенного локуса и считали имеющими нарушенный локус ZFN. Такой анализ оценивали как позитивный, когда сайт связывания ZFN в обоих аллелях в локусе FAD2A был нарушен.
ПЦР-ВЫЯВЛЕНИЕ ИНТЕГРАЦИИ ТРАНСГЕНА В FAD2A ПОСРЕДСТВОМ ЗАВИСИМОЙ ОТ ГОМОЛОГИИ РЕПАРАЦИИ
Каждый предполагаемый растительный трансформант анализировали, используя результат, полученный с использованием ПЦР-праймеров, сконструированных для амплификации трансгенной мишени hph (hph_ExoDigPC_F1, SEQ ID NO: 457, 5’TTGCGCTGACGGATTCTACAAGGA 3’ и hph_ExoDigPC_R1, SEQ ID NO: 458, 5’TCCATCAGTCCAAACAGCAGCAGA 3’), эндогенного локуса FAD2A (FAD2A.Out.F1, SEQ ID NO: 459, 5’ CATAGCAGTCTCACGTCCTGGT*C 3’ и FAD2A.Out.Rvs3, SEQ ID NO: 460, 5’ GGAAGCTAAGCCATTACACTGTTCA*G 3’), области, охватывающей 5’-конец любого трансгена, встроенного в локус FAD2A посредством HDR, выше трансгена в локусе FAD2 A (FAD2A.Out.F1, SEQ ID NO: 461, 5’ CATAGCAGTCTCACGTCCTGGT*C 3’ и QA520, SEQ ID NO: 462, 5’ CCTGATCCGTTGACCTGCAG 3’), и области, охватывающей 3’-конец любого трансгена, встроенного в локус FAD2A посредством HDR, ниже трансгена в локусе FAD2 A locus (QA558, SEQ ID NO: 463, 5’ GTGTGAGGTGGCTAGGCATC 3’ и FAD2A.Out.Rvs3, SEQ ID NO: 464, 5’ GGAAGCTAAGCCATTACACTGTTCA*G 3’) (фиг.33). Все пары праймеров амплифицировали, используя следующие условия: 98°C в течение 30 секунд с последующими 35 циклами (98°C в течение 10 секунд, 65°C в течение 20 секунд, 72°C в течение 90 секунд). Условия использования реагентов для реакции описаны в таблице 19.
Амплификация геномной 5’-фланкирующей трансген мишени и/или амплификация геномной 3’-фланкирующей трансген мишени показали предполагаемое событие инсерции. Необходимо отметить, что вследствие присутствия гомологичных плеч FAD2A длиной примерно 1000 п. н. в кассете pDAS000130 (содержащей полинуклеотидные последовательности со 100% идентичностью последовательности с областями FAD2A, расположенными непосредственно выше и ниже сайта разрезания ZFN), в реакционных смесях для ПЦР происходила ложноположительная амплификация продуктов ПЦР из-за химеризма ПЦР, возникающего в результате амплификации событий интеграции ETIP вне мишени. Амплификация мишени hph подтверждала тот факт, что происходила интеграция трансгена. Амплификация мишени FAD2A свидетельствует о том, что локус FAD2A является интактным или содержит только частичную инсерцию. Из-за размера ETIP (11462 п. н. для кассет ETIP или 13472 п. н., включая гомологичные плечи FAD2A и кассеты ETIP) ожидается, что праймеры FAD2A не могут амплифицировать продукт, когда интактная ETIP интегрирована в локус FAD2A.
ВЫЯВЛЕНИЕ РЕДАКТИРОВАНИЯ FAD2A В САУЗЕРН-АНАЛИЗЕ
Растения, которые либо имели амплификацию продукта геномной 5’-фланкирующей трансген мишени и/либо амплификацию 3’ геномной фланкирующей трансген мишени, либо не имели амплификации локуса-мишени ZFN, или и то и другое, подвергали Саузерн-анализу для выявления интеграции трансгена в локус FAD2A. Геномную ДНК очищали из 5 г ткани листа, используя модифицированный CTAB метод (Maguire, T.L., G.G. Collins, and M. Sedgley A modified CTAB DNA extraction procedure for plants belonging to the family proteaceae. Plant Molecular Biology Reporter, 1994. 12(2): p. 106-109). Затем 12 мкг геномной ДНК расщепляли, используя Kpn1-HF (New England BioLabs), и фрагменты расщепления разделяли электрофорезом в 0,8% агарозном геле перед переносом на мембрану с использованием стандартного протокола Саузерн-блоттинга. Праймеры к 5’-области мишени FAD2A (F, SEQ ID NO: 465, 5’ AGAGAGGAGACAGAGAGAGAGT 3’ и R, SEQ ID NO: 466, 5’ AGACAGCATCAAGATTTCACACA 3’), 3’-области мишени FAD2A (F, SEQ ID NO: 467, 5’ CAACGGCGAGCGTAATCTTAG 3’ и R, SEQ ID NO: 468, 5’ GTTCCCTGGAATTGCTGATAGG 3’) и hph (F, SEQ ID NO: 469, 5’ TGTTGGTGGAAGAGGATACG 3’ и R, SEQ ID NO: 470, 5’ ATCAGCAGCAGCGATAGC 3’) использовали для создания зондов, чтобы выявить присутствие ETIP в локусе FAD2A с использованием системы DIG EASY HYB SYSTEM® (Roche, South San Francisco, CA), следуя инструкциям производителя (фиг.34). Гибридизацию осуществляли при 42°C в случае 5’-области FAD2A, 45°C в случае 3’-области FAD2A и 42°C для выявления hph.
Связанную с мембраной геномную ДНК исследовали с использованием зондов в конкретном порядке: сначала исследовали 5’-последовательности FAD2A, затем исследовали 3’-последовательности FAD2A и, наконец, исследовали последовательности hph (фиг.35). Обоснование такого исследования заключалось в следующем. Первый зонд (5’-FAD2A) является диагностическим зондом, и если ETIP была интегрирована в FAD2A посредством точной HDR, то фрагмент длиной 5321 п. н. будет видно на мембране. Получаемый в результате размер полосы легко отличить во время электропорации, и он будет находиться вблизи фрагментов длиной 5148 п. н. DIG-меченных маркеров молекулярной массы ДНК Roche III (Roche, Indianapolis, IN). Второе исследование мембраны осуществляют с использованием зонда 3’-FAD2A, и подвергнутое генетическому редактированию растение будет иметь фрагмент длиной 22433 п. н., тогда как не подвергнутое редактированию растение будет иметь фрагмент 16468 п. н.. Такой же фрагмент длиной 22433 п. н., идентифицированный с использованием зонда 3’-FAD2A, также должен связываться и может быть идентифицирован с использованием зонда hph. Такие фрагменты трудно отличить в геле, так как они являются чрезвычайно крупными, и может быть трудно определить какое-либо различие между фрагментом, находящемся выше или ниже самого крупного фрагмент длиной 21226 п. н. среди DIG-меченных маркеров молекулярной массы ДНК Roche III®. Соответственно, такие зонды позволяют получать данные, которые могут подкрепить идентификацию интеграции ETIP в FAD2A посредством зависимой от гомологии репарации (HDR), за счет визуализации фрагмента длиной 5 т.п.н. с использованием зонда 5’ FAD2A. Фермент рестрикции, KpnI, был единственной подходящей эндонуклеазой рестрикции для применения в настоящем анализе, так как сайты KpnI находились в одном участке разрезания кассеты ETIP в одном локусе, и присутствовал в двух участках локуса ZNF FAD2A. Один участок располагался выше, а второй участок располагался ниже плеч гомологии FAD2A. Кроме того, KpnI не чувствительна к метилированию и доступна в виде рекомбинантного фермента повышенной точности (New England Biolabs).
РЕЗУЛЬТАТЫ МОЛЕКУЛЯРНОГО И САУЗЕРН-АНАЛИЗА
После трансфекции, культивирования и отбора трансгенные растения переносили в почву. В результате такого процесса выжили 139 растений, и из них брали образцы тканей для экстракции гДНК и анализа. Все 139 растений анализировали для оценки количества копий. Из указанных 139 растений 56 были позитивными в отношении ETIP, и 11 из 56 позитивных растений имели предполагаемую интеграцию одной копии (фиг.36) (таблица 20). Из 56 растений, которые были позитивными в отношении интеграции ETIP, амплификация геномной 5’фланкирующей трансген последовательности FAD2A происходила в 7 растениях. Амплификация геномной 3’-фланкирующей трансген последовательности FAD2A не происходила ни в одном из 56 растений, которые были позитивными в отношении интеграции ETIP. Кроме того, из 56 растений, которые были позитивными в отношении интеграции трансгена, 11 растений были позитивными в кПЦР-тесте нарушенного локуса. Четырнадцать растений, которые были позитивными в отношении амплификации геномной 5’-фланкирующей трансген последовательности FAD2A и/или позитивными в кПЦР-тесте нарушенного локуса, подвергали Саузерн-анализу с использованием 3 зондов, описанных выше. Из 14 растений, выбранных для Саузерн-анализа, во всех растениях наблюдали частичную интеграцию в локус FAD2A, но ни в одном из таких растений не наблюдали свидетельств полной интеграции полноразмерной ETIP в локус FAD2A посредством HDR при исследовании с использованием зонда 5’ FAD2A, зондов 3’ FAD2A и hph. Нет полос, которые выглядели i) крупнее WT и ii) идентичными полосам, наблюдаемым в случае таких образцов при исследовании с использованием зонда 3’ FAD2A (таблица 20).
Таблица 20
Краткое описание результатов анализа интеграции ETIP
РЕЗУЛЬТАТЫ ДЛЯ ТРАНСГЕННОЙ ПО ETIP КАНОЛЫ, ТРАНСФОРМИРОВАННОЙ PDAS000130 и PDAB104010
Трансгенные события Brassica napus, которые получали в результате трансформации pDAS000130 и pDAB104010, приводят к интеграции однокопийной полноразмерной вставки T-нити полинуклеотидной последовательности ETIP из pDAS000130 в локус FAD2A. Три-четыре события полностью характеризовали и подтвердили, что они содержат интегрированную ETIP. Подтверждение осуществляли, используя способ ПЦР-амплификации «внутри - за пределами» (in-out), и дополнительно подтверждали, используя Саузерн-блот. Отобранные растения T0, в которых произошли такие события, выращивали до стадии развития T1. Растения T1 подвергали повторному скринингу, чтобы определить зиготность интегрированной T-нити. Подвергнутые скринингу события классифицировали как гомозиготные, гемизиготные или нулевые.
Гомозиготные события использовали для получения протопластов с применением описанного ранее способа. Затем протопласты котрансфицировали нуклеазой с цинковым пальцем, которая предназначена для целенаправленного воздействия на сайт связывания цинкового пальца, который включен в последовательность ETIP и донорную плазмиду, которая обладает гомологией с конкретными областями ETIP, при этом донорная плазмида интегрируется в ETIP посредством механизма HDR. Затем подобным образом протопласты котрансформировали нуклеазой с цинковым пальцем, которая предназначена для целенаправленного воздействия на сайт связывания цинкового пальца, который включен в последовательность ETIP и донорную плазмиду, которая не обладает гомологией с конкретными областями ETIP, при этом донор интегрируется в ETIP посредством механизма связывания негомологичных концов. Нуклеаза с цинковым пальцем расщепляет локус ETIP, и донорная плазмида интегрируется в геном клеток Brassica napus посредством зависимой от гомологии репарации или связывания негомологичных концов. В результате интеграции донорной плазмиды неполный трансген DS-red репарируется до полноразмерного трансгена DS-red. После этого экспрессию полностью функционального трансгена DS-red используют для сортировки протопластов клеток способом FACS. Предполагаемые трансгенные растения отсортировывают, используя способ FACS, описанный в примере 7, и изолированные протопласты регенерируют в зрелые растения. Интеграцию донорной плазмиды подтверждают в растениях с целенаправленно введенной ETIP, используя молекулярные способы подтверждения. Соответственно, локус ETIP служит в качестве сайт-специфичного локуса для целенаправленной интеграции гена из донорной полинуклеотидной последовательности.
РЕЗУЛЬТАТЫ ДЛЯ ТРАНСГЕННОЙ ПО ETIP КАНОЛЫ, ТРАНСФОРМИРОВАННОЙ НУКЛЕАЗОЙ С ЦИНКОВЫМ ПАЛЬЦЕМ И ETIP-КОНСТРУКЦИЯМИ PDAS000271-PDAS000275
Трансгенные события Brassica napus, которые получали в результате трансформации конструкциями ETIP и нуклеазы с цинковым пальцем, приводят к интеграции однокопийной полноразмерной вставки T-нити полинуклеотидной последовательности ETIP из pDAS000273 или pDAS275 в локус FAD2A и из pDAS000271, pDAS000272 или pDAS000274 в локус FAD3C. Три-четыре события полностью характеризовали и подтвердили, что они содержат интегрированную ETIP. Подтверждение осуществляли, используя способ ПЦР-амплификации «in-out», и дополнительно подтверждали, используя Саузерн-блот. Отобранные растения T0, в которых произошли такие события, выращивали до стадии развития T1. Растения T1 подвергали повторному скринингу, чтобы определить зиготность интегрированной T-нити. Подвергнутые скринингу события классифицировали как гомозиготные, гемизиготные или нулевые.
Гомозиготные события использовали для получения протопластов с применением описанного ранее способа. Затем протопласты котрансфицировали нуклеазой с цинковым пальцем, которая предназначена для целенаправленного воздействия на сайт связывания цинкового пальца, который включен в последовательность ETIP и донорную плазмиду, которая обладает гомологией с конкретными областями ETIP. Нуклеаза с цинковым пальцем расщепляет локус ETIP, и донорная плазмида интегрируется в геном клеток Brassica napus посредством зависимой от гомологии репарации. В результате интеграции донорной плазмиды неполный трансген DS-red репарируется до полноразмерного трансгена DS-red. После этого экспрессию полностью функционального трансгена DS-red используют для сортировки протопластов клеток способом FACS. Предполагаемые трансгенные растения отсортировывают, используя способ FACS, описанный в примере 7, и изолированные протопласты регенерируют в зрелые растения. Интеграцию донорной плазмиды подтверждают в растениях с целенаправленно введенной ETIP, используя молекулярные способы подтверждения. Соответственно, локус ETIP служит в качестве сайт-специфичного локуса для целенаправленной интеграции гена из донорной полинуклеотидной последовательности.
ПРИМЕР 7: ОСНОВАННАЯ НА FACS СОРТИРОВКА КЛЕТОЧНЫХ ПРОТОПЛАСТОВ
Протопласты Brassica napus, которые были трансфицированы контрольной конструкцией DS-Red, pDAS000031, подвергали сортировке с применением опосредованной FACS сортировки клеток, используя сортировщик BD Biosciences Influx-Cell (San Jose, CA). Клеточные протопласты выделяли и трансфицировали, как описано в примере 3. После того, как клетки были трансфицированы pDAS000031, клетки отсортировывали, используя FACS-сортировщик, в условиях, описанных в таблице 21.
Протопласты, которые экспрессировали трансген DS-red, отсортировывали и выделяли. Выделенные в ходе FACS протопласты подсчитывали, используя сортировщик. Примерно от 1×105 до 1,8×105 клеток помещали в лунку 24-луночного планшета для микротитрования в первый день после FACS-выделения. Клетки переносили в культуру на шариках на 5-20 дней. Тестировали сходные условия, когда примерно 1×104 клеток помещали в лунку 2 или 4-луночного планшета для микротитрования на второй день после FACS-выделения. Различные условия, которые были тестированы, приводили к извлечению клеток с жизнеспособностью 95-98% от общего количества выделенных клеточных протопластов. Отсортированные в FACS клеточные протопласты переносили в культуру на шариках на 3-20 дней. Отсортированные с использованием FACS клеточные протопласты регенерировали в растения на среде, которая содержала 1,5 мг/мл гигромицина, используя описанный выше протокол. Подтверждали, что предполагаемые трансгенные растения содержат вставку интактной T-нити из pDAS000031, используя протоколы молекулярного подтверждения.
ТАРГЕТИНГ ETIP-ЛИНИЙ ПОСРЕДСТВОМ ОПОСРЕДОВАННОЙ ZFN ГОМОЛОГИЧНОЙ РЕКОМБИНАЦИИ DS-RED
Получали линию канолы, содержащую вставку T-нити из pDAS000036, и подтверждали, используя молекулярную характеристику, что она содержит полноразмерную однокопийную T-нить. Такое событие канолы обозначали pDAS000036-88 и использовали для получения протопластов ранее описанным способом. Выделяли протопласты и затем ~50000 протопластов клеток канолы трансформировали нуклеазой с цинковым пальцем, либо pDAS000074 (фиг.25), либо pDAS000075 (фиг.26), которая была предназначена для целенаправленного воздействия на сайты связывания цинкового пальца, включенные в последовательность ETIP и донорную плазмиду, pDAS000064, pDAS000068, pDAS000070 или pDAS000072 (фиг.27, фиг. 28, фиг. 29 и фиг. 30, соответственно), которая обладала гомологией с конкретными областями ETIP. На фиг. 19 и фиг. 20 представлены иллюстрации зависимой от гомологии репарации, которая приводит к сайт-специфичной интеграции трансгена Ds-red посредством опосредованной нуклеазой с цинковым пальцем гомологичной рекомбинации. Нуклеаза с цинковым пальцем была предназначена для расщепления локуса ETIP в определенной последовательности, связывающей цинковый палец и тем самым создания двунитевого разрыва в геноме. Затем донорная плазмида интегрировалась в геном клеточных протопластов Brassica napus посредством зависимой от гомологии репарации. Области интрона-1 и интрона-2 донорной плазмиды имеют гомологию с соответствующим областями интрона-1 и интрона-2 локуса ETIP. В результате интеграции донорной плазмиды неполный трансген DS-red репарируется до полноразмерного в высокой степени экспрессирующегося трансгена DS-red. Экспрессию полностью функционального трансгена DS-red использовали для сортировки клеточных протопластов описанным выше способом FACS. Соответственно, локус ETIP служит в качестве сайт-специфичного локуса для целенаправленной интеграции в ген донорной полинуклеотидной последовательности. Наконец, изолированные протопласты могут быть отсортированы и регенерированы в зрелые растения. Интеграция донорной плазмиды может быть подтверждена в растениях с целенаправленной введенной ETIP с использованием способов молекулярного подтверждения.
ДНК донорной плазмиды и ДНК ZFN-плазмиды смешивали в различных концентрациях и использовали для трансфекции клеточных протопластов канолы, содержащих событие pDAS000036 - 88, и трансгенные клеточные протопласты сортировали, используя FACS-сортировку, которая описана ранее. Таблица 22 описывает различные эксперименты по трансфекции и концентрации ДНК, которые были использованы для трансфекции протопластов канолы, содержащих событие pDAS000036-88. ДНК ZFN и донорной плазмиды выделяли и готовили для трансфекций ранее описанными способами.
После завершения экспериментов по трансфекции протопласты инкубировали при комнатной температуре в течение 48 часов и сортировали, используя описанный выше протокол FACS. Каждый эксперимент по сортировке проводили независимо, и опосредованную цинковыми пальцами интрогрессию трансгена подтверждали, идентифицируя отдельные события, при которых экспрессировался трансген DS-red. На фиг. 21-24 показаны результаты FACS-сортировки. Как показывают результаты, приведенные на графиках, были получены множественные события, которые содержали интактный полностью интегрированный трансген DS-red. Такие множественные события Ds-Red были результатом опосредованной нуклеазой с цинковым пальцем интеграции конструкции донорной ДНК в геномный локус ETIP. Такая сайт-специфичная интеграция приводила к появлению в высокой степени экспрессирующейся полной копии трансгена Ds-Red. Частота экспрессии трансгена Ds-Red была в диапазоне примерно 0,03-0,07% от общего количества протопластов клеток канолы (~50000). Однако частота эффективной трансфекции для опосредованной нуклеазой с цинковым пальцем интеграции конструкции донорной ДНК в геномный локус ETIP была намного больше, в диапазоне примерно 0,07-0,64%.
На фиг. 21 показаны результаты трансфекций, при которых осуществляли котрансформацию донорной плазмиды и ZFN-плазмиды. На верхнем графике показан случай, когда донор, pDAS000064, и нуклеазу с цинковым пальцем, pDAS000074, котрансфицировали в соотношении 26 мкг к 4 мкг плазмидной ДНК с получением опосредованной нуклеазой с цинковым пальцем интеграции конструкции донорной ДНК в геномный локус ETIP с частотой рекомбинации примерно 0,03% от количества протопластов клеток канолы, составляющего ~50000. В действительности, частота рекомбинации была намного выше. Из ~50000 протопластов клеток канолы, которые были использованы в эксперименте по трансфекции, только примерно 10-30% таких протопластов клеток канолы были действительно трансформированы. Соответственно, фактическая эффективность трансфекции в результате опосредованной нуклеазой с цинковым пальцем интеграции конструкции донорной ДНК в геномный локус ETIP находится в диапазоне примерно 0,22-0,07%. Подобным образом, на нижнем графике показан случай, когда донор, pDAS000064, и нуклеазу с цинковым пальцем, pDAS000075, котрансфицировали в соотношении 26 мкг к 4 мкг плазмидной ДНК с получением опосредованной нуклеазой с цинковым пальцем интеграции конструкции донорной ДНК в геномный локус ETIP с частотой рекомбинации примерно 0,03% от количества протопластов клеток канолы, составляющего ~50000. В действительности, частота рекомбинации была намного выше. Из ~50000 протопластов клеток канолы, которые были использованы в эксперименте по трансфекции, только примерно 10-30% таких клеток были действительно трансфицированы. Соответственно, фактическая эффективность трансфекции в результате опосредованной нуклеазой с цинковым пальцем интеграции конструкции донорной ДНК в геномный локус ETIP находится в диапазоне примерно 0,26-0,08%. Результаты опосредованной цинковым пальцем зависимой от гомологии репарации значимо превышают негативные контрольные эксперименты, когда идентифицировали только один протопласт из ~50000, который давал красную флуоресценцию, что в результате соответствует частоте рекомбинации 0,00%, как показано на фиг. 20.
На фиг. 22 показаны результаты трансфекций, при которых осуществляли котрансформацию донорной плазмиды и ZFN-плазмиды. На верхнем графике показан случай, когда донор, pDAS000068, и нуклеазу с цинковым пальцем, pDAS000074, котрансфицировали в соотношении 28 мкг к 2 мкг плазмидной ДНК с получением опосредованной нуклеазой с цинковым пальцем интеграции конструкции донорной ДНК в геномный локус ETIP с частотой рекомбинации примерно 0,03% от количества протопластов клеток канолы, составляющего ~50000. В действительности, частота рекомбинации была намного выше. Из ~50000 протопластов клеток канолы, которые были использованы в эксперименте по трансфекции, только примерно 10-30% таких протопластов клеток канолы были действительно трансформированы. Соответственно, фактическая эффективность трансфекции в результате опосредованной нуклеазой с цинковым пальцем интеграции конструкции донорной ДНК в геномный локус ETIP находится в диапазоне примерно 0,22-0,07%. Подобным образом, на нижнем графике показан случай, когда донор, pDAS000068, и нуклеазу с цинковым пальцем, pDAS000075, котрансфицировали в соотношении 28 мкг к 2 мкг плазмидной ДНК с получением опосредованной нуклеазой с цинковым пальцем интеграции конструкции донорной ДНК в геномный локус ETIP с частотой рекомбинации примерно 0,04% от количества протопластов клеток канолы, составляющего ~50000. В действительности, частота рекомбинации была намного выше. Из ~50000 протопластов клеток канолы, которые были использованы в эксперименте по трансфекции, только примерно 10-30% таких клеток были действительно трансфицированы. Соответственно, фактическая эффективность трансфекции в результате опосредованной нуклеазой с цинковым пальцем интеграции конструкции донорной ДНК в геномный локус ETIP находится в диапазоне примерно 0,38-0,12%. Результаты опосредованной цинковым пальцем зависимой от гомологии репарации значимо превышают негативные контрольные эксперименты, когда идентифицировали только один протопласт из ~50000, который давал красную флуоресценцию, что в результате соответствует частоте рекомбинации 0,00%, как показано на фиг. 20.
На фиг. 23 показаны результаты трансфекций, при которых осуществляли котрансформацию донорной плазмиды и ZFN-плазмиды. На верхнем графике показан случай, когда донор, pDAS000070, и нуклеазу с цинковым пальцем, pDAS000074, котрансфицировали в соотношении 28 мкг к 2 мкг плазмидной ДНК с получением опосредованной нуклеазой с цинковым пальцем интеграции конструкции донорной ДНК в геномный локус ETIP с частотой рекомбинации примерно 0,07% от количества протопластов клеток канолы, составляющего ~50000. В действительности, частота рекомбинации была намного выше. Из ~50000 протопластов клеток канолы, которые были использованы в эксперименте по трансфекции, только примерно 10-30% таких протопластов клеток канолы были действительно трансформированы. Соответственно, фактическая эффективность трансфекции в результате опосредованной нуклеазой с цинковым пальцем интеграции конструкции донорной ДНК в геномный локус ETIP находится в диапазоне примерно 0,64-0,21%. Подобным образом, на нижнем графике показан случай, когда донор, pDAS000070, и нуклеазу с цинковым пальцем, pDAS000075, котрансфицировали в соотношении 28 мкг к 2 мкг плазмидной ДНК с получением опосредованной нуклеазой с цинковым пальцем интеграции конструкции донорной ДНК в геномный локус ETIP с частотой рекомбинации примерно 0,04% от количества протопластов клеток канолы, составляющего ~50000. В действительности, частота рекомбинации была намного выше. Из ~50000 протопластов клеток канолы, которые были использованы в эксперименте по трансфекции, только примерно 10-30% таких клеток были действительно трансфицированы. Соответственно, фактическая эффективность трансфекции в результате опосредованной нуклеазой с цинковым пальцем интеграции конструкции донорной ДНК в геномный локус ETIP находится в диапазоне примерно 0,34-0,11%. Результаты опосредованной цинковым пальцем зависимой от гомологии репарации значимо превышают негативные контрольные эксперименты, когда идентифицировали только один протопласт из ~50000, который давал красную флуоресценцию, что в результате соответствует частоте рекомбинации 0,00%, как показано на фиг. 20.
На фиг. 24 показаны результаты трансфекций, при которых осуществляли котрансформацию донорной плазмиды и ZFN-плазмиды. На верхнем графике показан случай, когда донор, pDAS000072, и нуклеазу с цинковым пальцем, pDAS000074, котрансфицировали в соотношении 28 мкг к 2 мкг плазмидной ДНК с получением опосредованной нуклеазой с цинковым пальцем интеграции конструкции донорной ДНК в геномный локус ETIP с частотой рекомбинации примерно 0,07% от количества протопластов клеток канолы, составляющего ~50000. В действительности, частота рекомбинации была намного выше. Из ~50000 протопластов клеток канолы, которые были использованы в эксперименте по трансфекции, только примерно 10-30% таких протопластов клеток канолы были действительно трансформированы. Соответственно, фактическая эффективность трансфекции в результате опосредованной нуклеазой с цинковым пальцем интеграции конструкции донорной ДНК в геномный локус ETIP находится в диапазоне примерно 0,62-0,20%. Подобным образом, на нижнем графике показан случай, когда донор, pDAS000072, и нуклеазу с цинковым пальцем, pDAS000075, котрансфицировали в соотношении 28 мкг к 2 мкг плазмидной ДНК с получением опосредованной нуклеазой с цинковым пальцем интеграции конструкции донорной ДНК в геномный локус ETIP с частотой рекомбинации примерно 0,05% от количества протопластов клеток канолы, составляющего ~50000. В действительности, частота рекомбинации была намного выше. Из ~50000 протопластов клеток канолы, которые были использованы в эксперименте по трансфекции, только примерно 10-30% таких клеток были действительно трансфицированы. Соответственно, фактическая эффективность трансфекции в результате опосредованной нуклеазой с цинковым пальцем интеграции конструкции донорной ДНК в геномный локус ETIP находится в диапазоне примерно 0,44-0,14%. Результаты опосредованной цинковым пальцем зависимой от гомологии репарации значимо превышают негативные контрольные эксперименты, когда идентифицировали только один протопласт из ~50000, который давал красную флуоресценцию, что в результате соответствует частоте рекомбинации 0,00%, как показано на фиг. 20.
Отобранные эксплантаты переносили и культивировали в среде для регенерации, содержащей фосфинотрицин. После периода культивирования выжившие эксплантаты переносили на среду для роста в длину и среду для индукции корней с целью культивирования и развития растений. Целые растения, которые состояли из развитых структур корня и побега, переносили в почву и затем выращивали в теплице. В процессе культивирования тканей использовали среды и условия культивирования, которые описаны выше. Результаты получения растений способом культивирования тканей показаны в таблице 23 ниже.
Способ FACS-сортировки непосредственно применим для скрининга любой флуоресцирующей последовательности трансгена и используется для выделения части каких-либо протопластов, в данном случае протопластов клеток Brassica napus, в которые целенаправленно введен флуоресцирующий трансген посредством зависимой от гомологии репарации в конкретный сайт в области ETIP в геномном локусе.
Хотя в настоящем описании были раскрыты некоторые примеры вариантов осуществления изобретения, специалистам данной области будет понятно и учтено, что могут быть сделаны добавления, исключения и модификации приведенных в качестве примеров вариантов, не выходя за рамки объема следующей далее формулы изобретения. Кроме того, признаки одного варианта могут быть объединены с признаками другого варианта.
Реферат
Изобретение относится к области биотехнологии, в частности к способу получения растения из популяции клеток, включающему выделение при помощи активируемой флуоресценцией сортировки клеток (FACS) инкапсулированного в альгинат натрия протопласта растения, содержащего представляющий интерес полинуклеотид; регенерацию растения из выделенного растительного протопласта и культивирование данного растения. Изобретение позволяет эффективно отбирать протопласты с жизнеспособностью 95-98% от общего количества выделенных клеточных протопластов и регенерировать растения из вышеуказанных протопластов. 2 н. и 7 з.п. ф-лы, 23 табл., 80 ил., 7 пр.
Комментарии