Реагент (варианты), библиотека реагентов, способы проведения анализа - RU2158310C2
Код документа: RU2158310C2
Чертежи







Описание
В биологических и химических анализах часто используются анализируемые молекулы, меченные "репортерными" группами. Настоящее изобретение предлагает реагенты, имеющие по меньшей мере две группы аналитов, связанные с одной или более репортерными группами. Такие реагенты могут быть использованы способом, описанным ниже, что позволяет получить гораздо более полную информацию, чем с использованием простых меченных аналитов. Можно закодировать репортерные группы таким образом, чтобы реагенты, несущие множество групп аналитов и множество репортерных групп, можно было подвергать комбинаторному синтезу и использовать одновременно, а репортерные группы в процессе анализа отщепляются.
В патенте WO 93/06121 (Affymax) описана библиотека синтетических олигомеров, состоящая из множества различных членов, причем каждый член включает в себя олигомер, состоящий из последовательности мономеров, связанных с одной или более метками, идентифицирующими последовательности мономеров в олигомере. Связующее звено между олигомером и меткой-идентификатором, предпочтительно, включает в себя твердую частицу. Меткой-идентификатором, предпочтительно, является олигонуклеотид.
В работе Proc. Natl. Acad Sci., Vol. 89, N 12, 15 июня 1992 г., стр. 5381-5383 (S. Brenner и R.A.Lerner) описан химический способ комбинаторного кодирования для создания библиотеки реагентов, каждый из которых содержит генетическую олигонуклеотидную метку.
В работе "Rapid Communications in Mass Spectrometry", Vol. 6, стр. 369-372 (1992), авторов G.R.Parr и др. описана масс-спектрометрия на матрице с использованием лазерного десорбционного/ионизационного спектрометра, применяемая для анализа синтетических олигодезоксирибонуклеотидов.
В работе "Nucleic Acids Research", Vol. 21, N 15, от 25 июня 1993 г., стр. 3347-3357, авторы E.Nordhoff и др., описана ионная стабильность нуклеиновых кислот при проведении масс-спектрометрии на матрице с использованием лазерного десорбционного/ионизационного спектрометра ИК-диапазона.
В одном аспекте настоящее изобретение относится к реагенту, который включает в себя:
а) анализируемый компонент,
состоящий по меньшей мере из двух
анализируемых остатков, и связанный с
б) компонентом-меткой, который включает одну или более репортерных групп, которые можно обнаружить методом
масс-спектрометрии, за исключением
олигонуклеотидов, где репортерная группа обозначает анализируемый остаток, причем эта репортерная группа в каждом положении компонента-метки выбрана так, чтобы она
обозначала анализируемый остаток в
определенном положении анализируемого компонента.
Предпочтительно, анализируемый компонент связан с компонентом-меткой связью, которая может расщепляться, например, под действием света. При этом может присутствовать линкерная группа, к которой присоединены и анализируемый компонент, и компонент-метка. В предпочтительном варианте анализируемый компонент представляет собой цепь из n-го числа анализируемых остатков, а компонент-метка представляет собой цепь, содержащую до n-числа групп-репортеров, причем репортерная группа в каждом положении цепи метки выбрана так, чтобы эта группа могла обозначать анализируемый остаток в соответствующем положении цепи аналита. При этом "n" представляет собой целое число, равное, по крайней мере, 2, а предпочтительно от 3 до 20.
Настоящее изобретение может быть использовано для определения всех аналитов, представляющих интерес. Такими аналитами являются, но не ограничиваются ими, белковая пептидная цепь, где анализируемые остатки представляют собой остатки аминокислот; цепь нуклеиновых кислот/олигонуклеотидов, где анализируемые остатки представляют собой нуклеотидные остатки; углеводородные цепи, где анализируемые остатки представляют собой остатки сахаров. Помимо этого, аналит может представлять собой класс небольших молекул, обладающих биологической, фармакологической или терапевтической активностью. Например, это может быть центральная часть молекулы, в которой различные замещающие группы, например алкилы, сложные эфиры, амины, простые эфиры и т.п. могут быть замещены метками для проведения масс-спектрометрии, причем такое замещение производится на комбинаторной основе.
Компонент-метка и/или какая-либо одна или все репортерные группы в этой метке должны обладать такими свойствами, чтобы их можно было наблюдать/обнаружить/проанализировать с целью получить информацию о природе анализируемого компонента и/или анализируемых остатков в этом компоненте.
В одном из вариантов осуществления настоящего изобретения реагент имеет формулу A-L-R, где A представляет собой цепь, состоящую из n-го числа анализируемых остатков, образующих анализируемый компонент, L представляет собой линкер, R представляет собой цепь, содержащую до n-го числа репортерных групп, образующих компонент-метку, и n равно от 2 до 20, причем компонент-метка содержит информацию, указывающую на местонахождение анализируемых остатков в анализируемом компоненте.
Компонент-метка состоит из одной или более репортерных групп, различаемых по массе, что позволяет производить анализ этих групп с применением масс-спектрометрии. Репортерные группы могут иметь различную химическую природу, и поэтому их можно различать по молекулярной массе. Или же репортерные группы могут быть химически идентичными, но отличаться друг от друга тем, что содержат различные изотопы (например,12C/13C и1H/2H, о чем будет сказано ниже). Компонент-метка и/или репортерные группы пригодны для анализа методом масс-спектрометрии, например, после отщепления их от реагента фотохимическим или иным методом.
Преимущества масс-спектрометрии, как системы обнаружения, состоят в следующем: она обладает высокой чувствительностью, т.е. для получения хорошего сигнала достаточно несколько сотен молекул; широким динамическим диапазоном и высокой разрешающей способностью, т.е. при разрешении свыше 0,01 можно различать молекулы в диапазоне масс от 100 до 200000 Дальтон; разнообразием применения - можно легко проводить анализы химических структур самых разных молекул; возможностью исследовать аналиты путем сочетания масс-спектрометрии с другими методами, например с методом десорбции с лазерным сканированием; а также возможностью проводить не только качественный, но и количественный анализ.
Таким образом, метод использования масс-мечения сочетает в себе достоинства метода с использованием радиоактивных меток и метода флюоресценции, и обладает дополнительными свойствами, которые предполагают новые виды применения.
В другом аспекте настоящее изобретение включает в себя также библиотеку вышеуказанных реагентов, причем такая библиотека состоит из множества реагентов, каждый из которых включает в себя отличающийся от других анализируемый компонент из n-го числа анализируемых остатков.
Например, библиотека может состоять из 4n числа реагентов, каждый из которых включает в себя отличную от других олигонуклеотидную цепь из n-го числа нуклеотидных остатков. Реагенты данной библиотеки могут присутствовать в виде их смеси друг с другом в растворе.
В другом аспекте настоящее изобретение относится к методу проведения анализа, состоящего из следующих стадий: получение исследуемого вещества; инкубирование исследуемого вещества с указанной библиотекой реагентов в таких условиях, при которых, по меньшей мере, один реагент связывается с исследуемым веществом; удаление несвязанных реагентов; отщепление компонента-метки каждого связанного реагента; анализ отщепленных компонентов-меток, который позволяет получить информацию о природе анализируемых компонентов, связанных с исследуемым веществом.
Исследуемое вещество может быть иммобилизовано, что является удобным способом отделения связанных реагентов от несвязанных. В одном аспекте осуществления изобретения исследуемое вещество может представлять собой организм или ткань или группу клеток, а анализ может быть осуществлен с целью выявления семейства лекарственных препаратов-кандидатов. В другом аспекте осуществления изобретения исследуемое вещество может представлять собой нуклеиновую кислоту, и этот вариант будет подробно рассмотрен ниже.
Сущность изобретения поясняют иллюстрации.
Фиг. 1 демонстрирует общую схему синтеза реагентов настоящего изобретения.
Фиг. 2 иллюстрирует реагенты с тремя различными системами цепей-меток, содержащих репортерные группы.
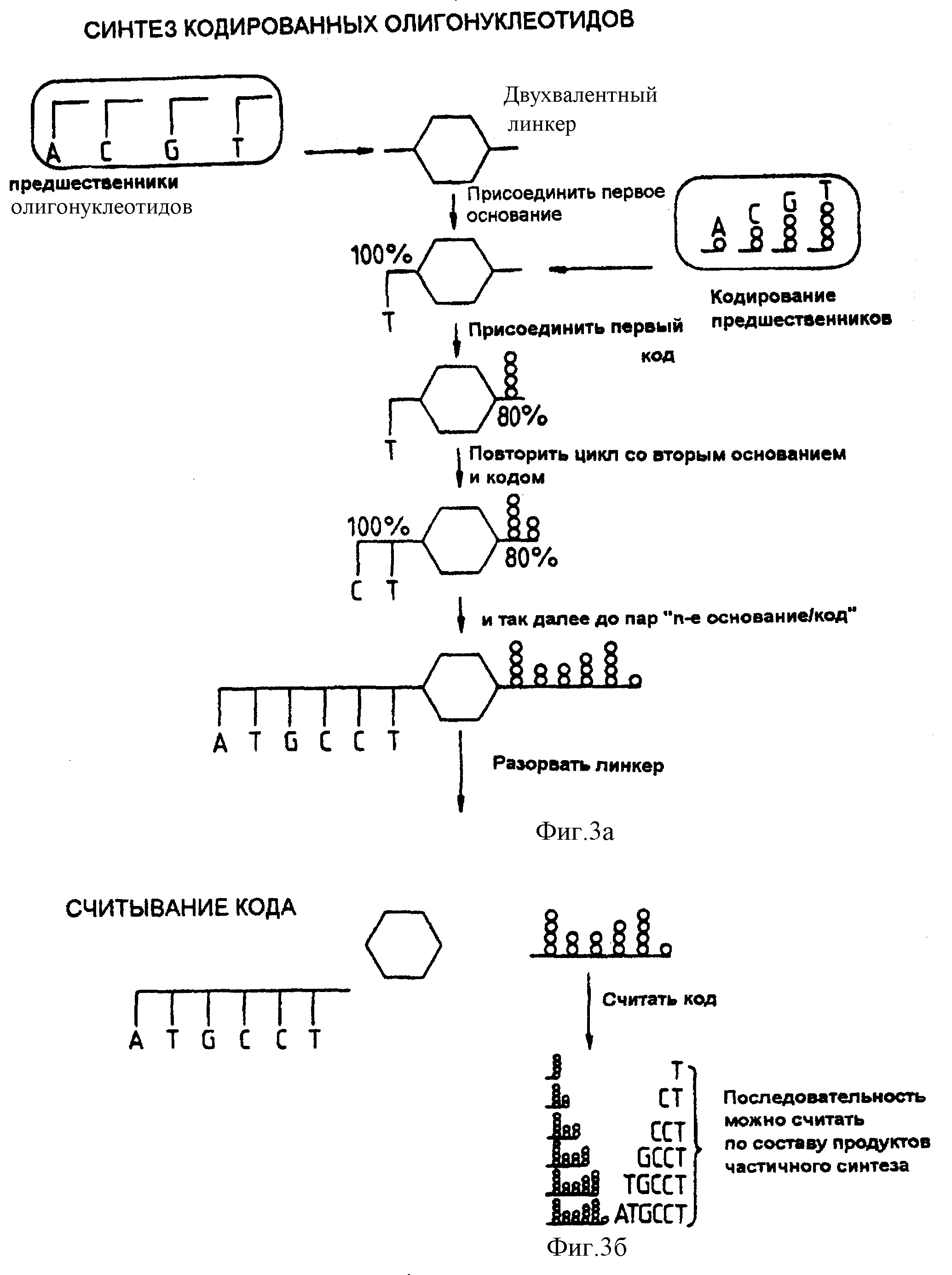
Фиг. 3a представляет собой схему синтеза кодированных олигонуклеотидов.
Фиг. 3b представляет собой схему считывания кода цепи-метки.
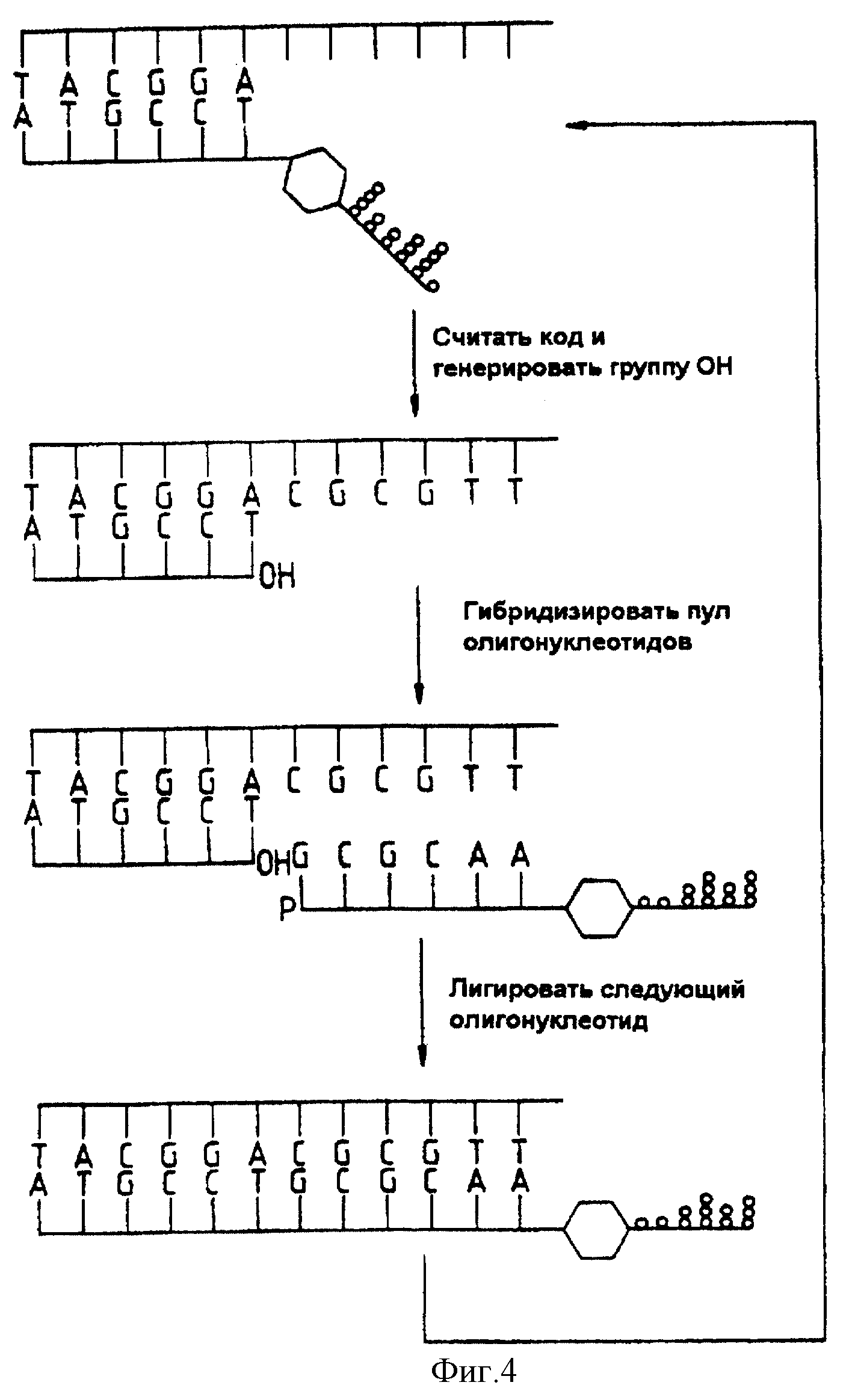
Фиг. 4 представляет собой диаграмму, иллюстрирующую анализ последовательности с применением постепенного лигирования.
Фиг. 5 представляет собой схему удлинения последовательности, считываемой с применением гибридизации с олигонуклеотидами.
Разъяснения к фиг. 1 и 2 даны в конце настоящего описания.
Ссылки относятся к приведенным ниже примерам осуществления изобретения, где описано, каким образом заявленный способ может быть применен для анализа последовательностей нуклеиновых кислот и для скрининга лекарственных препаратов-кандидатов.
Синтез кодированных меток
Принцип, на
котором основан способ одновременного мечения множества аналитов,
аналогичен принципу, предложенному в работе Brenner and Lerner (1992) для кодирования пептидов с присоединенными последовательностями
нуклеиновых кислот. Основная идея авторов указанной работы состоит
в том, чтобы добавить метку, которая может быть амплифицирована с помощью полимеразной цепной реакции и считана путем секвенирования
полученной молекулы ДНК.
Структура реагентов лучше всего может быть проиллюстрирована способом их получения. Синтез начинается с того, что двухвалентный или многовалентный линкер поэтапно удлиняют в одном направлении, чтобы присоединить к аналиту остаток, и в другом направлении, чтобы присоединить специфичные к остатку репортерные группы (фиг. 1). Предположим, мы хотим получить смесь органических соединений, вводя различные остатки на каждом этапе синтеза. Например, смесь может включать в себя набор пептидов с различными последовательностями аминокислот или олигонуклеотиды с различными последовательностями оснований, или набор вариантов, обладающих различной фармакологической активностью и различными группами, присоединенными к структуре кора; в каждом случае мы хотим пометить каждый структурный вариант уникальной меткой. Это достигается за счет разделения синтеза на каждом этапе, когда различные остатки добавляют к интересующему соединению, и за счет добавления соответствующих остатков к метке.
Для примера предположим, что мы хотим получить смесь 4096 гексануклеотидов, каждый из которых будет иметь уникальную метку. Четыре образца двухвалентных линкеров должны быть соединены с каждым основанием и с уникальной для каждого основания репортерной группой (фиг. 3a). После этого четыре образца перемешивают, разделяют на четыре части и процесс повторяют. В результате получают набор динуклеотидов, каждый из которых имеет уникальную метку. Процесс повторяют до тех пор, пока не будут завершены шесть этапов присоединения.
Линкерные и репортерные группы
Линкер должен иметь
одну группу, которая совместима с синтезом аналита, а именно гидроксильную группу, аминогруппу или
сульфидрильную группу, пригодные для инициации синтеза олигонуклеотида; аналогичные группы можно
использовать для инициации другого вида синтеза - например, синтеза полипептидов. Для некоторых
классов соединений желательно начать с "центрального" соединения, которое образует часть аналита. Выбор
группы (групп) для инициации присоединения репортерных групп зависит от природы этих групп и от
типа химических реакций, используемых для их присоединения. Тип химических реакций должен быть совместим
с типом реакций, используемых для синтеза аналита. Например, для синтеза олигонуклеотидов
можно использовать ряд альтернативных вариантов. При обычном способе синтеза используются бензоильная и
изопропильная группы для защиты оснований, кислотолабильные тритильные группы для временной
защиты 5'-OH-групп в процессе присоединения, и бета-цианоэтильные группы для защиты фосфатов. Способ,
используемый для присоединения репортерных групп, не должен оказывать неблагоприятное воздействие
на эти защитные группы или на другие связи в олигонуклеотиде, а на синтез меток не должны оказывать
влияние реакции присоединения, окисления и отщепления защитных групп, используемые при удлинении
олигонуклеотида.
Присоединение мономеров-репортеров или блокирование цепи может оставаться незавершенным на каждом этапе (фиг. 2, B и C), что позволяет присоединить аналит к гнездовому множеству структур-репортеров. Это позволяет легче вычислить структуру аналита, исходя из композиции метки (фиг. 1; фиг. 3). Для облегчения синтеза желательно, чтобы линкер был присоединен к твердому носителю посредством связи, которая может разрушаться, не приводя к разрушению аналита или репортерных групп. В альтернативном варианте линкер может нести группу, такую как заряженная группа или липофильная группа, которая позволяет отделять промежуточные соединения и конечный продукт от реагентов.
Репортерные группы могут быть самыми различными, главное, чтобы можно было считывать композицию или последовательность метки методом масс-спектрометрии. Можно использовать группы с различной атомной или формульной массой, такие как алифатические цепи различной длины или с различным изотопным составом. Используя метиленовые группы, меченные изотопами, можно присвоить группу с уникальной формульной массой каждому из четырех различных репортеров (таблица 1).
Если взять в качестве примера олигонуклеотиды, то эти метки образуют набор, который позволяет считывать основание в каждом положении олигонуклеотида по возрастанию массы продуктов, являющихся частью серии (таблица 2). Все последовательности олигонуклеотидов будут давать уникальные серии масс фрагментов-меток, если при добавке репортерной группы самое малое увеличение массы превышает разницу между самой малой и самой крупной группой-репортером.
Для масс-спектрометрии желательно иметь простой способ отщепления цепи метки от аналита. Для этого существует несколько путей. Для олигонуклеотидных и пептидных аналитов применяют расщепление фотолабильных связей под действием света; ферментативное расщепление, например, эфирной связи; расщепление с применением свободных радикалов.
Еще одно условие заключается в том, что метки должны быть совместимы с химическими и биохимическими процессами, применяемыми в процессе анализа: в примере с олигонуклеотидами, используемыми для молекулярной гибридизации, или в примере с предлагаемыми методами секвенирования, метки должны быть растворимыми и не должны ингибировать определенные ферментные реакции, которые применяются в ходе анализа. Как показывает практика, олигоэтиленгликольные связи, схожие с метиленовыми аналогами, показанными в таблице 1, совместимы с молекулярной реассоциацией олигонуклеотидов. Более того, такие связи совместимы с, по меньшей мере, некоторыми ферментными реакциями, поскольку, как мы показали, олигонуклеотиды, связанные на стекле гексаэтиленгликольным линкером, можно преобразовать в 5'-фосфомономерный эфир, обработав их полинуклеотид-киназой и аденозин-5'-трифосфатом.
Желательные свойства линкера
Для
целей настоящего изобретения желательно, чтобы молекула линкера обладала следующими свойствами.
Должна существовать возможность связать эту молекулу с твердым носителем так, чтобы в результате циклов синтеза получать аналит и соответствующие метки, не прибегая при этом к сложным процедурам очистки промежуточных соединений. После завершения циклов синтеза линкер должен удаляться из твердого носителя так, чтобы аналит и метки оставались в целости. В качестве функциональной группы для синтеза метки следует использовать такую группу, которая позволит осуществить прямой синтез меток, различаемых с помощью масс-спектрометрии.
Линкер должен иметь защищенные функциональные группы, что позволит удлинять аналит и метки по-отдельности в условиях, при которых химические реакции, используемые для удлинения одного из этих компонентов, не оказывают действия на другой компонент.
Желательно, чтобы линкер имел заряженную группу, благодаря чему масс-спектрометрию можно будет проводить при отсутствии матрицы. Для этой цели желательно, чтобы метки образовывали достаточно летучие соединения, которые можно будет выпаривать в масс-спектрометре без применения сложных технологий, таких как электрораспыление. Метки должны давать либо стабильные ионы, либо такие ионы, которые образуют характерные структуры, так чтобы их можно было использовать для идентификации соответствующего аналита.
Связь между меткой и аналитом должна быть расщепляемой под действием света, чтобы метки можно было непосредственно отщеплять в масс-спектрометре с помощью облучения лазером, а последующее отщепление до полного удаления меток (что необходимо для биохимических процессов, таких как лигирование) можно было бы произвести просто с помощью облучения лампой.
Предпочтительно, чтобы связанные продукты реакций растворялись в водных растворителях, что позволит использовать их в биохимических реакциях.
Примеры, описанные в настоящей заявке, иллюстрируют линкеры, обладающие вышеуказанными свойствами.
Фотоотщепляемая группа
Фотоотщепляемая группа основана на известной
фотолабильной орто-нитробензильной группе. Эта группа используется в качестве защитной для фосфатной группы и
2'-гидроксильной группы в синтезе олигонуклеотидов /см. работу Pillai "Синтез 1" (1980)/.
Сама по себе орто-нитробензильная группа не обладает достаточной функциональностью для последующего соединения
с линкером между метками и аналитом. Подходящими источниками является выпускаемый
промышленностью 5-гидрокси-2-нитробензиловый спирт. Известно, что группы OMe можно ввести в 5,4-положение без
существенного снижения фотолабильных свойств (см. работу Pillai). Поэтому
5-гидрокси-2-нитробензиловый спирт был использован в качестве исходного вещества для удлинения ДНК путем синтеза из
бензилового спирта и цепи линкера до присоединения меток из эфира путем
присоединения к 5-гидроксильной группе.
Требование, предъявляемое к функциональной группе, состоит в том, что эта группа должна быть подходящей для комбинаторного синтеза аналитов и меток. Таким образом, для синтеза метки требуется, чтобы линкер имел ответвление, идущее от фотоотщепляемой группы до нужной функциональной группы. Желательно также проводить комбинаторный синтез на твердом носителе. Таким образом, ответвление линкера должно быть двухвалентным в функциональных группах и иметь ортогональные защитные группы, чтобы можно было в процессе синтеза осуществлять селективные преобразования. Предпочтительные реагенты-метки имеют гликольные/эфирные связи. С целью проведения синтеза олигонуклеотиды обычно присоединяют к носителю из стекла с контролируемой пористостью, дериватизированного длинноцепочечными аминогруппами посредством 3'-гидрокси-связи и сложного эфира янтарной кислоты. Таким образом, нужные функциональные группы представляют собой спирты.
Было синтезировано нижеследующее промежуточное соединение:
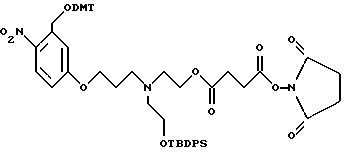
Это соединение включает ароматический линкер, несущий:
- метокситритильную группу (-CH2ODMT) для синтеза аналита;
- орто-нитрогруппу для фотоотщепления;
- O-трет-бутилдифенилсилильную группу (OTBDPS) для синтеза меток;
- группу третичного амина для преобразования в положительно заряженную группу для анализа методом масс-спектрометрии;
- N-гидроксисукцинимидильную группу для присоединения к носителю.
В тех случаях, когда аналитом является пептид, то вышеуказанные условия претерпевают лишь незначительные изменения. 2-нитробензильная группа является стабильной в условиях почти всех реакций, применяемых для синтеза этой группы и пептидов, поэтому для синтеза пептидов в качестве фотолабильных групп были использованы аналогичные группы (см. работу Pillai и приведенные в ней ссылки). Имеется уже несколько смол, пригодных для синтеза пептидов с различными типами расщепления. Защитные группы с прямоугольной цепью для синтеза аналита и метки должны быть основаны на трет-бутоксикарбонильной и 2-метоксиэтоксиметильной группах. Трет-бутоксикарбонильная группа может быть использована для защиты аминогруппы в аминокислотах, причем расщепление можно производить путем обработки трифторуксусной кислотой.
2-метоксиэтоксиметил можно использовать для защиты групп-меток и меток, основанных на 1,n-алкилдиольных производных с различными массами, как описано выше. Как было показано, отщепление трет-бутоксикарбонильных групп совместимо с защитными 2-метоксиэтоксиметильными группами. 2-метоксиэтоксиметильные группы можно селективно отщеплять с помощью бромида цинка в дихлорметане. Хотя приведенное выше описание иллюстрирует способ, однако специалистам должно быть ясно, что вышеуказанный список ортогональных защитных групп не является исчерпывающим и приведен здесь в качестве примера.
Обнаружение и анализ
групп-репортеров
Фотоотщепление
является предпочтительным способом отсоединения меток от аналитов: оно занимает мало времени и может быть проведено на веществах в сухом состоянии, а кроме
того, в этом случае может быть использован
сканирующий лазер для получения изображений очень малого масштаба, достаточного для определения внутриклеточных характеристик (см. de Vries et al., 1992),
поэтому предлагаемый способ можно
использовать для обнаружения положений конкретных аналитов, использованных для "мечения" поверхности или внутренних частей клеток, или же различных клеток в срезе
ткани, например, если требуется
получить изображение взаимодействий между лигандами, напр., лекарств-кандидатов и их рецепторов.
Фоточувствительные защитные группы можно получить для
самых различных остатков
химических соединений (см. Pillai, 1980). Фотолабильная орто-нитробензильная группа, которую можно использовать в качестве защитной группы для широкого круга соединений,
является идеальной исходной
точкой для создания линкера для многих аналитов, в том числе пептидов и олигонуклеотидов. В примере с олигонуклеотидами эта группа обеспечивает фоточувствительную связь,
которая может быть разрушена
с использованием определенного количества вещества, с образованием гидроксильной группы. Это позволит олигонуклеотидам, лишенным защитных групп, принять участие в
удлинении посредством лигирования,
как указано ниже в описании способа секвенирования. Более того, известно, что такие группы стабильны в процессе синтеза олигонуклеотидов. Необходимо модифицировать
бензильное кольцо, чтобы получить
группу, которую можно использовать для инициации синтеза меток; при этом репортерные группы, такие как олигоэтиленгликольные группы, описанные выше, не оказывают
влияния на реакцию фотохимического
отщепления орто-нитробензоильной группы (Pillai, цит. работа). В ароматическое кольцо можно ввести другие группы, которые повысят способность к отщеплению; такие
группы можно использовать и для того,
чтобы присоединить заряженную группу (группы), что позволит упростить анализ в масс-спектрометре. Современные масс-спектрометры способны проводить измерения
нескольких сотен молекул с разрешением
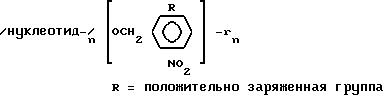
более одного Дальтона в сотне, с максимальной суммарной массой в 200 кДа. Предпочитаемый фотолабильный линкер, в котором положительно заряженная группа R может
быть непосредственно соединена с
ароматическим кольцом, или может присутствовать в одном из ответвлений линкера, может иметь следующий вид:
Методы измерения
Анализ предлагаемых молекулярных меток может быть проведен одним из нескольких методов масс-спектроскопии. Несмотря на то, что во многих случаях желательно отщеплять метки от аналита, не обязательно делить метки на фрагменты; а в ряде случаев это даже нежелательно, т.к. может вызвать путаницу. Последние достижения в области масс-спектрометрии позволяют измерять очень крупные молекулы без фрагментации; а поскольку можно создать такой линкер, который легко расщепляется, причем в таких условиях, когда остальная часть метки остается стабильной, то следует избегать фрагментации метки в процессе измерения. Группа аналита в большинстве случаев будет обладать меньшей летучестью, чем метка, а часто эта группа будет связана с твердым носителем, благодаря чему она не будет вступать в реакции в процессе масс-спектрометрии.
Описанный выше линкер очень лабилен к облучению фотонами в условиях, которые не вызывают расщепления большинства ковалентных химических связей. Подходящий измерительный инструмент описан в литературе /de Vries и др., 1992/. Этот инструмент использует лазер, который может быть сфокусирован вниз на точку менее 1 мкм. Изображения до 250 мм сканируются путем перемещения платформы, которую можно выставить на 0,1 мкм. Этот инструмент позволяет также производить ионизацию образцов, подлежащих измерению, путем освещения ионизирующим лазером поперек поверхности платформы так, чтобы лазер взаимодействовал с образцами, инициированными десорбционным лазером. Эта способность инструмента могла бы оказаться полезной для осуществления способа настоящего изобретения, если было бы невозможно включить заряженный остаток в метки, или если для считывания меток требовалась бы фрагментация.
В другом варианте осуществления изобретения предлагается способ секвенирования исследуемой нуклеиновой кислоты, который включает в себя следующие
стадии:
a) получение
олигонуклеотида, иммобилизованного на носителе,
b) гибридизация исследуемой нуклеиновой кислоты с иммобилизованным олигонуклеотидом,
c) инкубирование
гибрида (b) с библиотекой, в
которой реагенты смешаны вместе в растворе так, чтобы олигонуклеотидная цепь первого реагента библиотеки образовала гибрид с исследуемой нуклеиновой кислотой,
соседствующей с иммобилизованным
олигонуклеотидом,
d) лигирование соседних олигонуклеотидов до образования лигированного первого реагента,
e) удаление прочих нелигированных реагентов,
f) отщепление и
анализ фрагмента-метки лигированного первого реагента в качестве индикатора последовательности первой части исследуемой нуклеиновой кислоты.
Примеры
применения
Чтобы
проиллюстрировать возможные применения изобретения, мы хотим показать, каким образом кодированные олигонуклеотиды можно использовать для анализа нуклеиновых кислот.
1. Определение последовательности нуклеиновой кислоты путем поэтапного лигирования (фиг. 4).
Последовательность, которую необходимо определить, сначала гибридизировали на этапе (b) с получением олигонуклеотида, присоединенного к твердому носителю. Если ДНК, подлежащая секвенированию, была клонирована в однонитевой вектор, такой как бактериофаг М 13, то "затравочный" олигонуклеотид на твердом носителе может стать частью последовательности этого вектора. На этапе (c) твердый носитель, несущий гибриды из этапа (b), инкубировали с раствором кодированных олигонуклеотидных реагентов, например с вышеуказанной библиотекой, включающей все последовательности данной длины, скажем, 4096-гексануклеотиды (в общем, 4n n-меры). На этапе (d) лигаза вводится таким образом, чтобы гексануклеотид, комплементарный к первым шести основаниям в исследуемой ДНК, соединялся с иммобилизованным "затравочным" олигонуклеотидом. На этом этапе первый кодированный олигонуклеотидный реагент из библиотеки присоединяется путем лигирования своей олигонуклеотидной цепи к иммобилизованному "затравочному" олигонуклеотиду; этот реагент далее будет называться "лигированным первым реагентом".
На этапе (e) нелигированные реагенты удаляют, например, промывкой. На этапе (f) линкер лигированного первого реагента разрушают, чтобы отщепить цепь-метку, которую отделяют и анализируют в качестве индикатора последовательности первой части исследуемой ДНК.
Предпочтительно, чтобы после удаления линкера на конце первой олигонуклеотидной цепи оказалась гидроксильная или фосфатная группа, которую можно лигировать с олигонуклеотидной цепью второго реагента. Можно воспользоваться несколькими способами разрушения линкера, включая фотохимический и ферментный способы, а также химический гидролиз; в результате такого разрушения получают 3'-гидроксильную или 5'-фосфатную группу, которая необходима для последующего лигирования. После этого повторяют этапы (c), (d), (e), (f). На этих этапах производится гибридизация второго реагента из библиотеки, лигирование, отделение и анализ цепи-метки лигированного второго реагента, в результате чего получают следующую 3'-гидроксильную или 5'-фосфатную группу, необходимую для последующего лигирования. Этот процесс можно повторять до тех пор, пока вся последовательность ДНК не будет считана, или пока выход реакции не станет слишком малым для последующих операций.
Четыре этапа этой последовательности схематично показаны на фиг. 4. Первая схема соответствует ситуации в конце этапа (e) первого цикла. Вторая схема соответствует ситуации в конце этапа (f). Третья схема соответствует положению в конце этапа (c) второго цикла. Четвертая схема соответствует ситуации в конце этапа (d) второго цикла. Показано, что метод носит циклический характер.
2. Секвенирование нуклеиновых кислот множества кодирующих нитей ДНК путем
секвенциального лигирования
При удлинении,
описанном в первом примере, предполагается, что анализы множества последовательностей будут производиться одновременно. Например, индивидуальные
клоны ДНК, подлежащие секвенированию, могут быть
иммобилизованы следующим образом:
a) можно использовать подставку с иглами, на конце каждой из которых иммобилизован один и тот же
олигонуклеотидный вектор. Индивидуальный клон исследуемой
ДНК гибридизируют с олигонуклеотидом, иммобилизованным на конце каждой отдельной иглы. Затем подставку с иглами, на которых находятся
указанные гибриды, ингибируют с библиотекой кодированных
олигонуклеотидных реагентов в растворе, который содержит также ингредиенты для лигирования. В результате этого этапа каждая игла несет
различный лигированный реагент. В заключение цепь-метку каждого
лигированного реагента отделяют и анализируют вышеуказанным способом. Если иглы на подставке расположены подходящим образом, то их можно
погрузить в лунки планшетов для микротитрования; причем первый
планшет содержит матрицы, подлежащие секвенированию, второй планшет содержит библиотеку реагентов и раствор для лигирования, а третий
- реагент для отщепления цепей-меток от игл.
b) Альтернативно поверхность можно покрыть "затравочным" олигонуклеотидом, предпочтительно ковалентно присоединенным своим 5'-концом или в какой-либо другой точке. Индивидуальные клоны ДНК, подлежащей секвенированию, наносят на расположенные на расстоянии друг от друга участки носителя, покрытого олигонуклеотидом, так, чтобы каждый индивидуальный клон исследуемой ДНК гибридизировался с олигонуклеотидом, иммобилизованным в одном из местоположений носителя. Затем носитель инкубируют с раствором, содержащим библиотеку реагентов и ингредиентов для лигирования. Нелигированные реагенты удаляют. После этого линкер каждого лигированного реагента на каждом положении носителя отщепляют, метки удаляют и анализируют.
Отщепление желательно осуществлять таким образом, как лазерная десорбция, которая может быть направлена на малые участки поверхности. Преимущество этого подхода состоит в том, что он позволяет одновременно проводить анализ большого числа последовательностей ДНК.
3. Расширение способов определения последовательностей за счет гибридизации олигонуклеотидов
a) Схема
1.
Способы нанесения ДНК с высокой плотностью на мембраны хорошо известны /Hoheisel и др. , 1992; Ross и др., 1992/. Для того, чтобы получить фингерпринт молекул и определить последовательность, олигонуклеотиды следует наносить либо поодиночке, либо небольшими группами, чтобы не затруднять интерпретацию характеристик гибридизации; следовательно, только небольшая часть кодирующих нитей дает сигналы в каждом цикле анализов. Если сигнал от каждой гибридизации, содержащий кодированную информацию, позволяет обнаружить ее последовательность, то можно применить более сложные измерения и получить больше информации по каждому циклу гибридизации. Состав смеси будет зависеть от длины кодирующих нитей ДНК и от того, насколько точно метод анализа позволяет определять последовательности в смешанных нуклеотидах.
Нуклеиновокислотные зонды, кодированные указанными метками для масс-спектрометрии или группами-репортерами, будут очень эффективны в тех случаях, когда желательно использовать множество зондов, например, при определении "отпечатков пальцев" ДНК или при анализе мутации. Метки для проведения масс-спектрометрии позволяют проводить анализ сложных структур.
Несколько различных зондов, каждый из которых имеет свою уникальную метку, пригодную для проведения масс-спектрометрии, можно использовать вместе в типичных анализах методом гибридизации нуклеиновых кислот. Последовательность каждого отдельного зонда, подвергнутого гибридизации, можно определить в присутствии других зондов, благодаря разделению и разрешению меток в масс-спектре.
В данном
аспекте настоящее изобретение предлагает способ секвенирования исследуемой нуклеиновой кислоты, включающий в себя следующие этапы:
i) получение
исследуемой нуклеиновой кислоты,
иммобилизованной на носителе. Предпочтительно, индивидуальные клоны исследуемой нуклеиновой кислоты иммобилизуют на носителе на расстоянии друг от друга;
ii)
инкубирование иммобилизованной
исследуемой нуклеиновой кислоты из этапа i) со множеством вышеописанных кодированных олигонуклеотидных реагентов так, чтобы олигонуклеотидные цепи разных реагентов
гибридизировались с исследуемой
нуклеиновой кислотой на носителе;
iii) удаление негибридизированных реагентов;
iv) отделение и анализ фрагмента-метки каждого реагента, которые
являются индикаторами
последовательности части исследуемой нуклеиновой кислоты.
После этого предпочтительно используют библиотеку реагентов, причем гибридизацию, лигирование, отщепление и анализ повторяют несколько раз для получения дополнительной информации о последовательностях исследуемой нуклеиновой кислоты.
b) Схема 2.
Можно установить последовательности нуклеиновой кислоты по набору дуплексов, образующихся при их гибридизации с массивом олигонуклеотидов. Длина последовательности, которую можно определить, приблизительно равна корню квадратному из размера массива: если используется массив из всех 65536 октануклеотидов, то последовательность, подлежащая определению, должна составлять около 200 п. о. /Southern и др., 1992/. Такое ограничение размера обусловлено тем, что в последовательности, подлежащей определению, ни один набор из восьми оснований не должен встречаться более одного раза. Массив и его применение для определения последовательности описаны в Заявке на международный патент N WO 89/10977; а способ получения массива олигонуклеотидов, иммобилизованных, например, своими 5'- или 3'-концами на поверхности, описан в Заявке на международный патент N WO 90/03382.
Способ настоящего изобретения позволяет определить последовательности гораздо большей длины. В этом аспекте осуществления
изобретения
способ включает в себя следующие этапы:
a) получение массива олигонуклеотидов, иммобилизованных на носителе на определенном расстоянии друг от друга, причем олигонуклеотид на
одном участке
должен отличаться от олигонуклеотидов на других участках. Желательно, чтобы была известна последовательность олигонуклеотида, иммобилизованного посредством ковалентной связи на каждом
разнесенном
участке носителя,
b) инкубирование исследуемой нуклеиновой кислоты с массивом иммобилизованных олигонуклеотидов так, чтобы на одном или более из разнесенных участков носителя
образовались
гибриды,
c) инкубирование гибридов из этапа b) с библиотекой кодированных олигонуклеотидных реагентов так, чтобы олигонуклеотидная цепь реагента из библиотеки гибридизировалась с
исследуемой
нуклеиновой кислотой, соседней с каждым иммобилизованным олигонуклеотидом,
d) лигирование соседних олигонуклеотидов с образованием лигированных реагентов на одном или нескольких
разнесенных
участках носителя,
e) удаление нелигированных реагентов,
f) выделение и анализ фрагмента-метки каждого лигированного реагента, как индикаторов последовательности,
являющейся частью
исследуемой нуклеиновой кислоты.
Желательно, чтобы отщепление цепи-метки в каждом положении проводилось фотохимическим способом с помощью лазера. Желательно производить анализ цепи-метки с помощью масс-спектрометрии. Желательно повторить несколько раз этапы гибридизации, лигирования, отщепления и анализа, как описано выше, чтобы получить дополнительную информацию о последовательности в исследуемой нуклеиновой кислоте.
Предпочтительный порядок действий показан на четырех схемах, изображенных на фиг. 5. На первой схеме показано начало этапа b). На второй схеме показано положение в конце этапа b) - часть исследуемой нуклеиновой кислоты гибридизировалась со связанным олигонуклеотидом, образующим часть массива. На третьей схеме показано положение в конце этапа c), а на четвертой схеме показано положение в конце этапа d); реагент из библиотеки гибридизирован с исследуемой нуклеиновой кислотой и лигирован с иммобилизованным олигонуклеотидом.
Такой усовершенствованный способ дает прекрасные результаты. При условии, что способ, используемый для считывания меток, позволяет производить анализ смеси, одно только увеличение длины на длину, равную длине олигонуклеотидов в массиве, позволяет увеличить считываемую длину до величины, равной квадрату этого значения. В этом случае длина, которую можно считать из массива октануклеотидов, удлиненных на восемь оснований, составляет около 60000 оснований.
Сравнение анализа по методу гибридизации с меченными олигонуклеотидами с другими
методами
a) Методы, основанные на применении геля
Наиболее совершенный прибор для автоматического анализа последовательности позволяет считывать около 40000 оснований в день. Сюда не
входит время,
затрачиваемое на биологические и биохимические процессы, необходимые для проведения реакций соединений, загружаемых на гель. Если предположить, что матрицы можно наносить на поверхность
с плотностью
одна на квадратный миллиметр /Hoheisel и др., 1992; Ross и др., 1992/, то 10000 матриц можно нанести на площадь в 100 х 100 мм. После гибридизации в каждой ячейке будет несколько фмоль
меченных
олигонуклеотидов, так что один импульс лазера в 2 нс может высвободить достаточно меток для считывания, но даже если мы предположим, что нам потребуется 100 импульсов, то суммарное время для
считывания ячейки составит несколько мсекунд, так что все 10000 ячеек можно будет считать за несколько минут. Если олигонуклеотиды представляют собой гексамеры, то можно будет получить необработанные
данные по 60000 оснований. Для определения последовательности эти данные будут не столь информативными, как эквивалентные необработанные данные, полученные способом, основанным на применении геля,
потому что последний способ позволяет считывать непрерывные последовательности гораздо большей длины. Но данное преимущество способов, основанных на использовании гелей, исчезнет, если мы сможем
считывать из массива последовательности, длину которых можно увеличить с помощью нескольких циклов анализа. Но основное преимущество способа, основанного на использовании массивов, состоит в том, что
он позволяет параллельно считывать тысячи матриц одновременно; число матриц, которое можно проанализировать способом, основанным на использовании гелей, ограничено шириной геля и составляет менее
пятидесяти.
b) Применяемые в настоящее время способы, основанные на использовании массивов.
Основные недостатки применяемых в настоящее время способов, основанных на использовании массивов, состоят в следующем.
a) Последовательность, которую можно считать из массива размером N, равна только около

b) Длина последовательности, которую считывают в результате каждого взаимодействия с олигонуклеотидами при гибридизации, обязательно ограничена длиной олигонуклеотида. Это вызывает проблемы при считывании повторяющихся последовательностей, таких как серии из одного основания. Если мы расширим рамки считывания, используя лигирование, то это позволит производить считывание до последующего лигирования.
c) Из существующих в настоящее время способов определения последовательностей способ, основанный на использовании радиоактивности, обладает высокой чувствительностью и высоким разрешением; способ, основанный на флуоресценции, обладает высокой чувствительностью и высоким разрешением; но оба эти способа не позволяют производить анализы с достаточной скоростью. Предлагаемое использование масс-спектрометрии может повысить разрешающую способность, скорость и чувствительность анализа, а также дополнительно предоставляет возможность считывать последовательности меток.
Аналиты, обладающие потенциальной фармакологической активностью
Многие лекарственные препараты специфичны к определенным тканям. Их
действие часто зависит от взаимодействия с рецептором на поверхности клетки. Есть семейства лекарственных препаратов, основанные на минимальных структурах; например, некоторые структуры, состоящие из
коротких пептидов. Желательно получить возможность выявлять лекарства-кандидаты для того, чтобы пронаблюдать, на какие клетки или ткани они могут влиять. Желательно иметь возможность выявлять
множество лекарств-кандидатов одновременно. Используя библиотеки аналитов, меченных метками с кодированной массой, можно выявлять взаимодействия путем изучения клеток или тканей в масс-спектрометре.
Если метки присоединены посредством фотолабильных защитных групп, то можно получать изображения целого животного или участка ткани, используя отщепление методом лазерного сканирования в сочетании с
масс-спектрометрией.
Приведенные ниже примеры иллюстрируют настоящее изобретение.
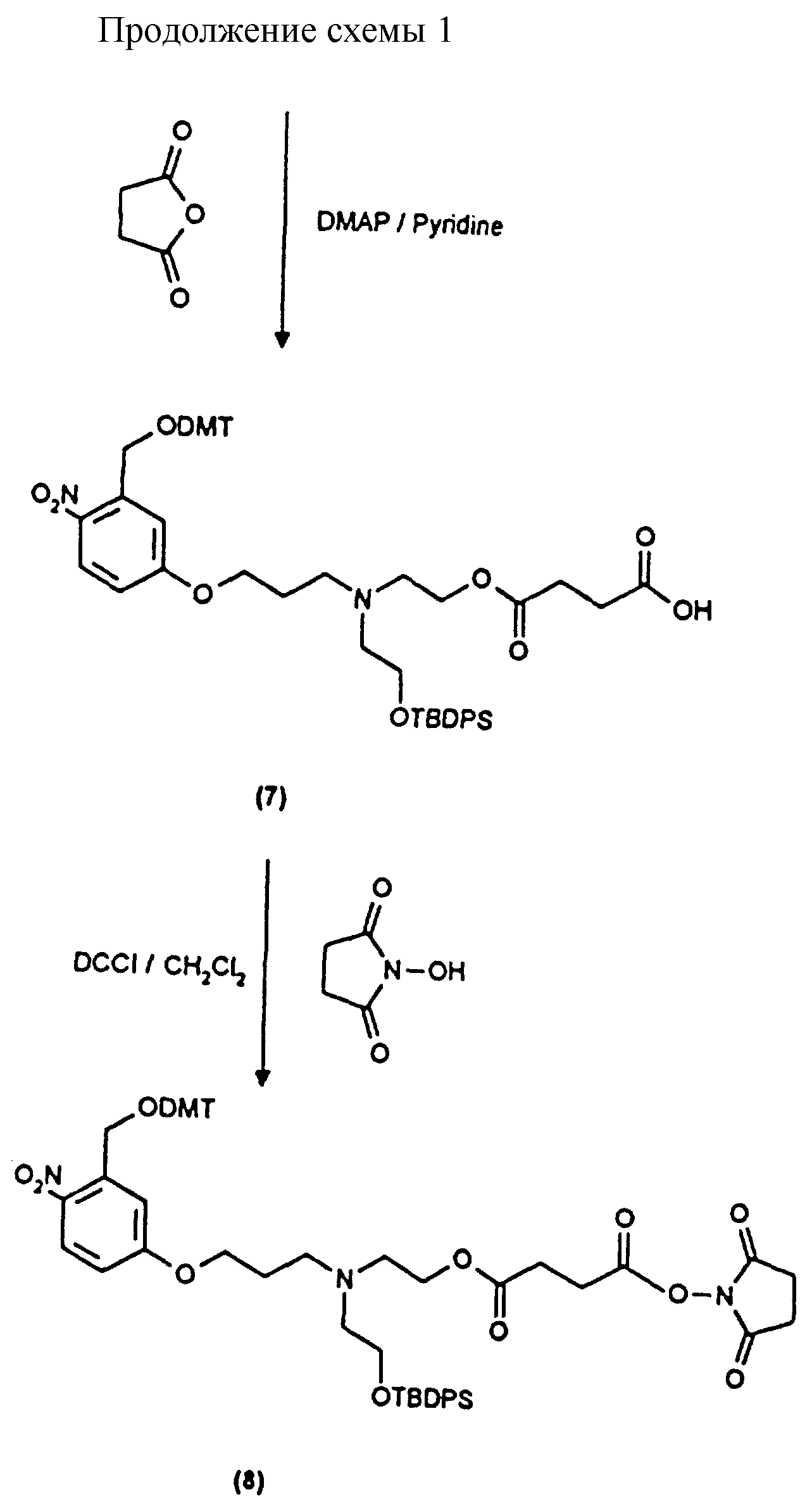
Примеры 1-6 показывают этапы в соответствии с приведенной ниже Схемой реакций 1, синтеза соединения (8), включающего в себя ароматический линкер, несущий: метилокситритильную группу (-CH2ODMT) для синтеза аналита; орто-нитрогруппу для фотоотщепления; O-трет-бутил-дифенил-силильную группу (OTBDPS) для синтеза метки; группу третичного амина для преобразования в положительно заряженную группу для анализа методом масс-спектрометрии; а также N-гидроксисукцинимидильную группу для присоединения к носителю.
Примеры 7 и 8 показывают последующие этапы в соответствии с приведенной ниже Схемой реакций 2.
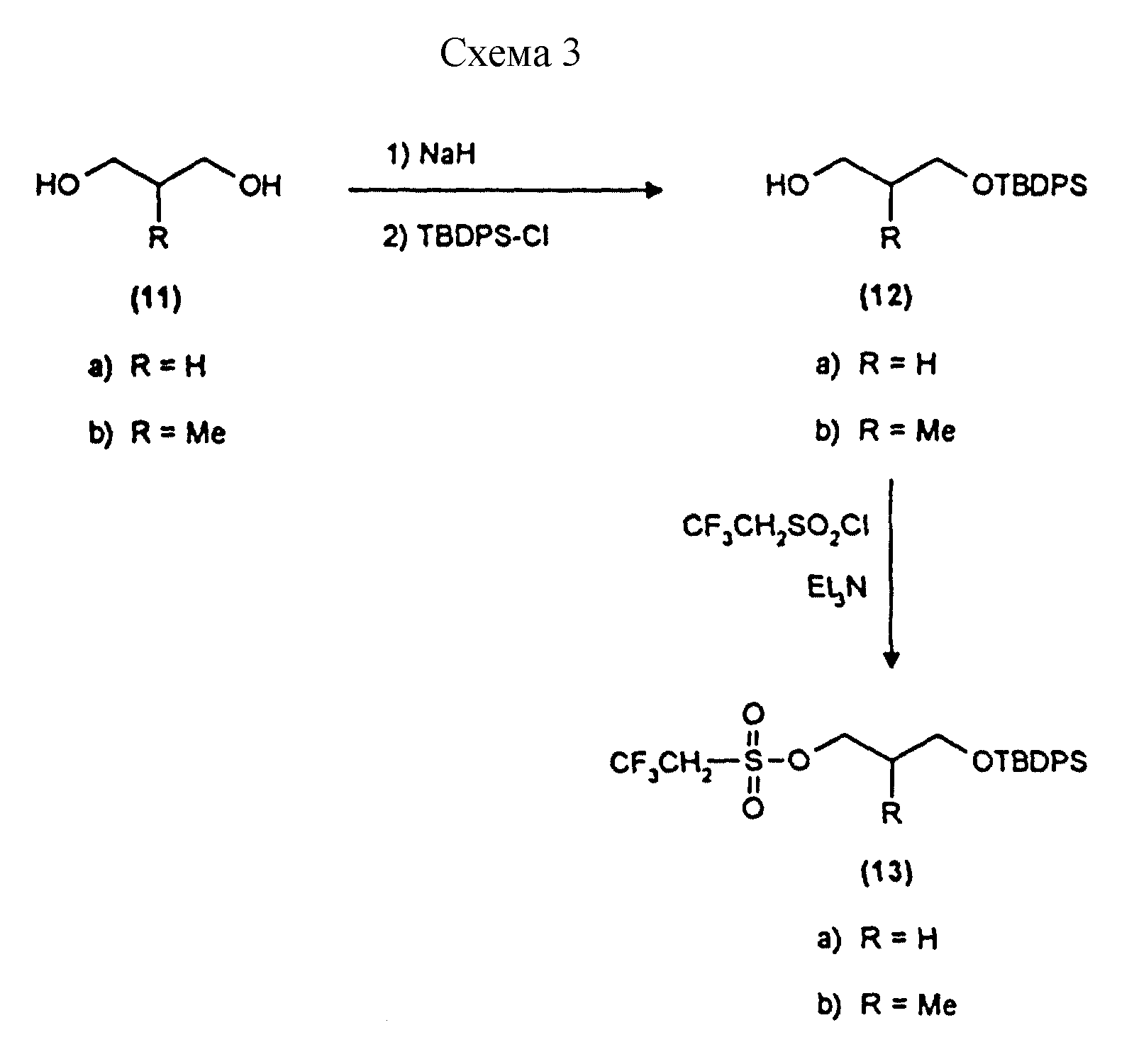
Примеры 9 и 10 показывают этапы в соответствии с приведенной ниже Схемой реакций 3 получения репортерных групп (13), основанных на пропан-1,3-диоле.
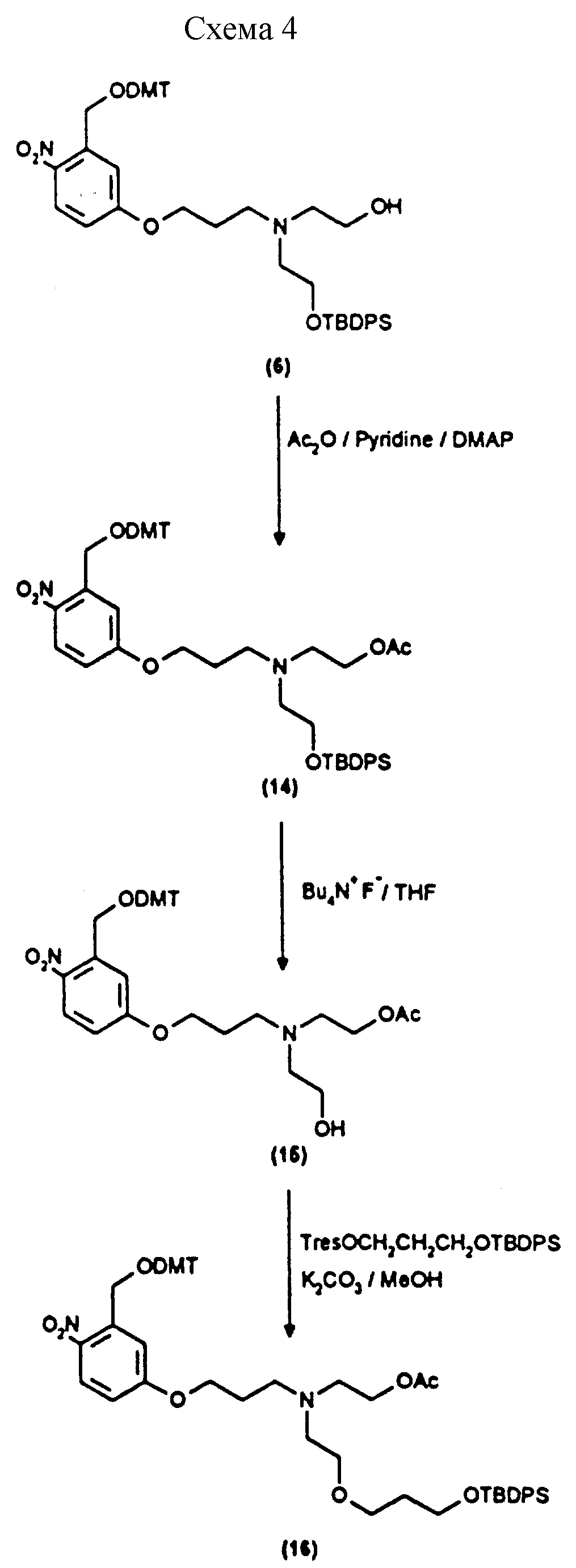
Примеры 11-13 показывают этапы в соответствии с приведенной ниже Схемой реакций 4, предусматривающие присоединение к соединению (6) защищенного пропан-1,3-диольного остатка в качестве репортерной группы.
Примеры 14-19 описывают получение, характеризацию и использование различных реагентов настоящего изобретения.
Общие положения
5-гидрокси-2-нитробензиловый спирт был приобретен у
фирмы Aldrich, стекло
с контролируемой пористостью, дериватизированное длинноцепочечными алкиламинами, было закуплено у фирмы Sigma. Безводные растворители относятся к материалам класса "Aldrich Sure
Seal" и упакованы в
атмосфере азота. Триэтиламин перед использованием был подвергнут предварительной перегонке из гидрида кальция и хранился в атмосфере азота. Другие растворители и реагенты могут
быть закуплены у
различных производителей.
Спектры1H ЯМР были получены с помощью аппарата "Jeol" 270 МГц с использованием указанного в примерах растворителя, и тетраметилсилана в качестве стандарта.
Инфракрасные спектры были получены на приборе "Nicolet SDXC F.T. IR" при использовании либо таблетки из бромида калия, либо раствора хлороформа, как указано.
Температуру плавления определяли с помощью аппарата "Gallenkamp", причем полученные данные были некорректированными.
Тонкослойную хроматографию проводили на пластинках "Kieselgel 60F254" с алюминиевой подложкой, используя указанную систему растворителей. Пластины визуализировали ультрафиолетовым облучением и/или погружением в 3% раствор молибдофосфорной кислоты в этаноле с последующим нагревом струей горячего воздуха. Образцы, содержащие тритил, выглядели ярко-оранжевыми пятнами, спирты - голубыми пятнами.
Хроматографию на силикагеле производили методом флэш-хроматографии, размеры частиц 40 --> 63 мкм.
Аббревиатуры, используемые в схемах реакций и в тексте:
DMT ДМТ 4,
41-диметокситритил
THF ТГФ тетрагидрофуран
TBDPS ТБДФС трет-бутилдифенилсилан
DMAD ДМАД 4-диметиламинопиридин
DCCI ДЦКИ дициклогексилдикарбодиимид
CH2Cl2
дихлорметан
CPG стекло с контролируемым размером пор
MeI иодометан
Tresyl трезил 2,2,2-трифторэтилсульфонил
ПРИМЕР 1
Синтез 5-гидрокси-O-(4,41
-диметокситритил)-2-нитробензилового спирта (соединение 2, схема 1).
В 5-гидрокси-2-нитробензиловый спирт (5,11 г, 30,2 ммоля), растворенный в безводном пиридине (40 ммоль), добавили 4, 41-диметокситритилхлорид (10,25 г, 30,2 ммоль) и закрыли колбу пробкой. Затем реакционную смесь оставляли, перемешивая, на 72 часа при комнатной температуре. Тонкослойная хроматография (эфир/петролейный эфир 40-60 градусов Цельсия, 65%/35%) показала присутствие нового материала, содержащего положительно заряженные тритильные группы с RF в 0,27, и исчезновение исходного спирта. Затем пиридин удаляли выпариванием в роторном испарителе, причем последние следы удаляли совместным выпариванием с толуолом (х2). Полученную смолу растворяли в этилацетате и раствор промывали водой (х1) и соляным раствором (х1). Затем раствор этилацетата высушивали безводным сульфатом магния и выпаривали до получения красновато-коричневой смолы. Смолу растворяли в CH2Cl2 (20 мл) и подвергали хроматографии на колонке с силикагелем (14 см х 6,5 см), которую элюировали смесью эфир/петролейный эфир при 40-60 градусах Цельсия, 65%/35%. Полученные фракции объединяли, а растворитель удаляли выпариванием в роторном испарителе с получением белого твердого вещества (13,49 мг, 95%, температура плавления 80-82 градуса Цельсия с разложением). Аналитический образец был получен перекристаллизацией из смеси хлороформ/петролейный эфир при 40-60 градусах Цельсия, температура плавления 134-7 градусов Цельсия с разложением.
1H ЯМР (270 МГц, CDCl3, δ): 3,79 (s, 6H, DMT-OCH3), 4,63 (s, 2H, CH2-ODMT), 6,77-6,85 (m, 5H, арил), 7,22-7,49 (m, 9H, арил), 7,63 (s, 1H, арил), 8,06 (d, 1H, J = 9,06 Гц, арил).
IR (диск KBr) 1610, 1509, 1447, 1334, 1248, 1090, 1060, 1033. 828 см-1.
ПРИМЕР 2
Синтез
O-(4,41
-диметокситритил)-5-/1-(3-бром-1-оксипропил)/-2- нитробензилового спирта (соединение 3, схема 1).
В соединение 2 (10,18 г, 21, 6 ммоль), растворенное в ацетоне (150 мл), добавили 1, 3-дибромпропан (11 мл, 108 ммоль) и карбонат калия (4,47 г, 32,3 ммоль). Затем реакционную смесь нагревали до 80 градусов Цельсия в течение трех часов, после чего перемешивали при комнатной температуре еще в течение 16 часов. Тонкослойная хроматография (эфир/петролейный эфир, 40-60 градусов Цельсия, 60%/40%) показала полное исчезновение исходного материала и образование двух новых соединений, содержащих тритил; основной продукт RF 0,48, минорный продукт RF 0,23. Затем ацетон удаляли высушиванием в роторном испарителе, а полученный остаток распределяли между водой и дихлорметаном. Раствор дихлорметана отделяли и промывали соляным раствором. Затем раствор дихлорметана высушивали безводным сульфатом магния и выпаривали до получения смолы. Смолу растворяли в дихлорметане (20 мл) и наносили на колонку с силикагелем (6,5 см х 14 см), которую элюировали смесью эфира/петролейного эфира, 40-60 градусов Цельсия, 60%/40%. Полученные очищенные фракции объединяли, а растворитель удаляли высушиванием в роторном испарителе до получения соединения 3 в виде белого твердого вещества (8,18 г, 64%, температура плавления 132-4 градуса Цельсия, RF 0,48 эфир/петролейный эфир 40-60 градусов Цельсия, 60%/40%). Небольшой образец перекристаллизовывали из смеси этилацетат/петролейный эфир для проведения анализа, температура плавления 132-4 градуса Цельсия.
1H ЯМР (270 МГц, CDCl3, δ): 2,40 (m, 2H,

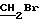
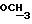
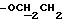

IR (диск KBr) 1608, 1577, 1511, 1289, 1253, 1230, 1174, 1065, 1030 см-1.
ПРИМЕР 3
Синтез
N-/O-(трет-бутилдифенилсилил)-2-оксиэтил)/-N-(2- гидроксиэтил)амина (соединение 5, схема 1)
В гидрид натрия (0,76 г 60% дисперсии в масле, 19 ммоль) в атмосфере N2 добавили
безводный ТГФ (15 мл), а затем суспензию диэтаноламина (2 г, 19 ммоль) в ТГФ (30 мл); добавление производили с такой скоростью, с какой позволяло выделение водорода. Затем реакционную смесь
перемешивали при комнатной температуре в течение 30 мин в атмосфере N2 и в течение этого времени образовывался серый осадок. Реакцию образования алкоксида останавливали путем добавления
трет-бутилхлордифенилсилана (4,95 мл, 19 ммоль), после чего реакционную смесь перемешивали при комнатной температуре в течение двух часов в атмосфере N2. Тонкослойная хроматография
(этилацетат) показала образование двух новых УФ-позитивных пятен, относящихся к исходному материалу, основной продукт с RF 0,05, минорный продукт с RF 0,60. ТГФ удаляли
высушиванием в роторном испарителе, остаток растворяли в 0,1 М растворе бикарбоната натрия. Полученный продукт затем экстрагировали этилацетатом (х2). Экстракты этилацетата объединяли и промывали
соляным раствором (х1). После этого раствор этилацетата высушивали безводным сульфатом магния и выпаривали с получением масла. Полученное масло наносили на колонку с силикагелем, которую элюировали
раствором хлороформа/метанола, 90%/10%. Фракции с RF 0,33 объединяли и высушивали в роторном испарителе с получением соединения 5 в виде белого кристаллического твердого вещества (3,93 г,
60%, температура плавления 73->75 градусов Цельсия). Небольшое количество вещества перекристаллизовывали из раствора этилацетата/петролейного эфира, 40-60 градусов Цельсия, с целью проведения
анализа; температура плавления 76->77 градусов Цельсия.
1H ЯМР (270 МГц, CDCl3, δ): 1,06 (s, 9H,tBu), 2,13 (brs, 1H,
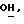



IR (диск, KBr) 3100, 1430, 1114, 1080, 969, 749, 738, 707 см-1.
ПРИМЕР 4
Синтез N-/O-(трет-бутилдифенилсилил)-2-оксиэтил/-N-/O-(3-(O-(4,41-диметокситритил)-1-оксиметил)-4-нитрофенил)-3-оксипропил/- N-(2-гидроксиэтил)амина
(соединение 6, схема 1).
В соединение 3 (7,46 г, 12,6 ммоль), растворенное в 1-метил-2-пирролидиноне (65 мл), добавили соединение 5 (8,63 г, 25,2 ммоль). Реакционную смесь нагревали до 80 градусов Цельсия в течение 5 часов, а затем оставили охлаждаться с перемешиванием при комнатной температуре еще в течение 16 часов. Тонкослойная хроматография (этилацетат) показала образование новых продуктов, содержащих тритил, с RF 0,51, и остаточные количества двух исходных материалов. Реакционную смесь вылили в смесь воды (600 мл) и соляного раствора (100 мл), а затем продукт экстрагировали этилацетатом (3х200 мл). Экстракты этилацетата объединяли и высушивали безводным сульфатом магния. Затем этилацетат удаляли высушиванием в роторном испарителе с получением коричневой смолы, из которой медленно образовывался кристаллический продукт. Добавили минимальное количество этилацетата, чтобы растворить оставшуюся смолу так, чтобы кристаллический продукт можно было отфильтровать; получили бромистоводородную соль соединения 5. Затем раствор этилацетата поместили на колонку с силикагелем (13 см х 6,5 см), которую элюировали этилацетатом. В результате недостаточного разделения остатка соединения 3 и нужного продукта на этой колонке фракции, содержащие продукт, объединили и выпаривали с получением смолы. Смолу растворяли в минимальном количестве этилацетата и наносили на другую колонку с силикагелем (14 см х 6,5 см), используя градиентное элюирование, сначала смесью этилацетата/петролейного эфира, 40-60 градусов Цельсия, 50%/50%, затем - этилацетатом. Полученные фракции объединяли, а растворитель удаляли путем осушки в роторном испарителе с получением соединения 6 в виде смолы. Следовые количества растворителя удалили, вместив смолу на 1 час в высокий вакуум. Выход продукта составил 7,64 г, 71%.
1H ЯМР (270 МГц, CDCl3, δ): 1,04 (s, 9H,tBu), 1,79 (m, 2H,
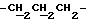

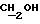
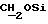
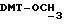
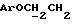

IR (диск KBr), 1608, 1579, 1509, 1287, 1251, 1232, 1112, 1092, 1064, 1035, 826, 754, 703, 613 см-1.
ПРИМЕР 5
Синтез
N-/O-(трет-бутилдифенилсилил)-2-оксиэтил/-N-/O-(3-(O-(4,41-диметокситритил)-1-оксиметил)-4-нитрофенил)-3-оксипропил/- N-/O-(3-карбоксилатропионил))-2-оксиэтил/амина (соединение 7, схема
1).
В соединение 6 (5,64 г, 6,59 ммоль), растворенное в безводном дихлорметане (40 мл) и безводном пиридине (50 мл), добавили ангидрид винной кислоты (2,06 г, 20,6 ммоль) и диметиламинопиридин (210 мг, 1,72 ммоль), после чего колбу закрыли пробкой. Реакционную смесь перемешивали при комнатной температуре в течение 72 часов. Тонкослойная хроматография (метанол/этилацетат, 10%/90%) показала образование новых продуктов, содержащих тритил, RF 0,45, и исчезновение исходного материала. Растворитель удаляли высушиванием в роторном испарителе, а последние следы пиридина удаляли совместным выпариванием с толуолом (х2). Полученную смолу разделили между хлороформом и водой. Органическую фазу отделили, а водную фазу экстрагировали хлороформом (х1). Затем органические фазы объединили и промыли соляным раствором (х1). После этого раствор хлороформа высушили безводным сульфатом магния и выпаривали до получения смолы. Последние следы растворителя удаляли, поместив смолу под высокий вакуум на 1 час, в результате чего получали соединение 7, 6,75 г. Продукт использовали в последующей стадии без дальнейшей очистки.
1H ЯМР (270 МГц, CDCl3, δ): 1,0 (s, 9H,tBu), 1,9 (m, 2H,
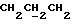



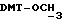
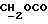
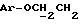
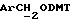
IR (раствор CHCl3), 1736, 1608, 1579, 1509, 1288, 1251, 1232, 1175, 1158, 1112, 1093, 1065, 1035, 755, 703 см-1.
ПРИМЕР 6
Синтез
N-/O-(трет-бутилдифенилсилил)-2-оксиэтил/-N-/O-(3-(O-(4,4-диметокситритил)-1-оксиметил)-4-нитрофенил)-3-оксипропил/-N- /O-(сукцинил-(3-карбоксилатпропионил)))-2-оксиэтил/амина (соединение 8, схема
1).
В соединение 7 (2,99 г, 3,13 ммоль), растворенное в безводном дихлорметане (30 мл), добавили дициклогексилкарбодиимид (0,710 г, 3,45 ммоль) и N-гидроксисукцинимид (0,396 г, 3,44 ммоль) и закрыли колбу пробкой. После этого реакционную смесь перемешивали при комнатной температуре в течение 18 часов, в течение которых образовывался белый осадок. Белый осадок отфильтровывали, а раствор дихлорметана промывали водой (х1) и соляным раствором (х1). Затем раствор дихлорметана высушивали безводным сульфатом магния и удаляли растворитель путем выпаривания в роторном испарителе до получения пены, 3,26 г (99%). Тонкослойная хроматография (этилацетат) обнаруживала присутствие только лишь продуктов, содержащих тритил, RF 0,74, и отсутствие каких-либо существенных загрязнений. Попытки получить аналитический образец путем обработки небольшого количества материала на колонке с силикагелем привели к разложению активного эфира до кислоты (соединение 7). Поэтому материал использовали на всех последующих стадиях без дальнейшей очистки.
1H ЯМР: (270 МГц, CDCl3, δ): 1,04 (s, 9H,tBu), 1,97 (m, 2H,
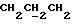
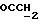
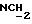


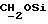
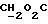
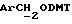
IR (диск KBr), 1742, 1713, 1509, 1288, 1251, 1211, 1175, 1090, 1067 см-1.
ПРИМЕР 7
Получение стекла с контролируемым размером пор,
дериватизированного длинноцепочечными алкиламино-группами (соединение 9, схема 2).
Стекло с контролируемым размером пор, дериватизированное длинноцепочечными алкиламино-группами (фирмы "Sigma Chemical Co, 3,5 г), предварительно обрабатывали трихлоруксусной кислотой (1,5 г), растворенной в дихлорметане (50 мл) в течение 2,5 часов, промывали аликвотами дихлорметана (100 мл всего) и безводного эфира (100 мл всего) и высушивали под вакуумом. К носителю из пористого стекла с контролируемым размером пор добавили безводный пиридин (35 мл), диметиламинопиридин (42 мг, 344 мкмоль), триэтиламин (280 мкл, 201 ммоль) и соединение 8 (см. схему 1) (736 мг, 700 мкмоль). Затем смесь осторожно взбалтывали в течение 18 часов, после чего шарики многократно промыли пиридином (7 х 10 мл), метанолом (5 х 15 мл) и хлороформом (5 х 15 мл), а затем высушили под вакуумом.
ПРИМЕР 8
Метилирование третичных аминогрупп, присоединенных к носителю из стекла с
контролируемым размером пор (соединение 10, схема 2).
К стеклу с контролируемым размером пор, дериватизированному длинноцепочечными алкиламино-группами (соединение 9, схема 2) (1,01 г) добавили безводный ТГФ (10 мл) и иодометан (0,5 мл, 8 ммоль). Затем смесь осторожно взбалтывали в течение 18 часов, после чего шарики многократно промыли безводным ТГФ (5 х 10 мл), а затем высушили под вакуумом.
ПРИМЕР 9
Синтез монозащищенных производных 1,3-пропандиола (соединения 12a и 12b, схема 3) - общая процедура
В гидрид натрия (1,05 г 60%
дисперсии в
масле, 26,3 ммоль) в атмосфере N2 добавили безводный ТГФ (10 мл), после чего по капле добавили производное 1,3-пропандиола (26,3 ммоль), растворенного
в безводном ТГФ (20 мл).
После
перемешивания еще в течение 30 минут в атмосфере N2 образовался алкоксид, о чем свидетельствовало появление серого осадка. Реакцию прекратили, по капле добавив
трет-бутилхлордифенилсилан (7,
24 г, 26,3 ммоль), растворенный в безводном ТГФ (20 мл), после чего реакционную смесь перемешивали в атмосфере N2 еще в течение 40 минут. После этого ТГФ
удаляли высушиванием в роторном
испарителе, а остаток распределяли между дихлорметаном и 0,1 М раствором бикарбоната натрия. Раствор дихлорметана отделили и промыли соляным раствором (х1). Затем
раствор дихлорметана высушили
сульфатом магния и выпарили до получения масла. Это масло нанесли на колонку с силикагелем (16 см х 5 см), которую элюировали смесью эфира/петролейного эфира, 40-60
градусов Цельсия, 30%/70%.
Полученные фракции объединили и выпарили в роторном испарителе с получением целевого производного 1,3-пропандиола.
Более подробно соединения описаны ниже.
12a 1-O-трет-бутилдифенилсилил-1,3-пропандиол, белое кристаллическое вещество, RF 0,21 эфир/петролейный эфир 40->60 градусов Цельсия, 30%/70%, 7,61 г, 92%, температура плавления 40->42 градуса Цельсия.
IR (диск KBr) 3400, 1448, 1112, 822, 734, 702, 689, 506, 488 см-1.
1H ЯМР (270 МГц, CDCl3,



12b 2-метил-1-O-трет-бутилдифенилсилил-1,3-пропандиол. Бесцветное масло, RF 0,21 эфир/петролейный эфир 40->60 градусов Цельсия, 30%/70%, 6,60 г, 77%.
IR (тонкая пленка) 3400, 1472, 1428, 1087, 1040, 823, 740, 702 см-1.
1H ЯМР (270 МГц, CDCl3, δ): 0,82 (d, 3H, J = 6,87 Гц,
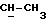
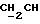

См. работу P.G.McDougal et al., JOC, 51, 3388 (1986), где описаны общие процедуры моносилилирования симметричных 1,n-диолов.
ПРИМЕР 10
Синтез производных треслата (соединения 13a и 13b, схема 3) - общие процедуры.
В спиртовое производное (4,94 ммоль), растворенное в безводном дихлорметане (10 мл) и сухом триэтиламине (0,84 мл, 6,03 ммоль), в атмосфере N2 и охлажденное до температуры с -15 градусов Цельсия до -30 градусов Цельсия, добавили по капле в течение 20-40 минут 2,2,2-трифторэтилсульфонилхлорид (1 г, 5,48 ммоль) в безводном дихлорметане (5 мл). Реакция завершилась после дополнительного перемешивания в течение 30 минут в атмосфере N2 при температуре от -15 до -30 градусов Цельсия. Затем реакционную смесь переносили в делительную воронку и промыли охлажденными льдом 1,0 М соляной кислотой (х1), водой (х1) и соляным раствором (х1). После этого раствор дихлорметана осушали сульфатом магния, а растворитель выпарили в роторном испарителе с получением треслата. Треслаты хранили при -20 градусах Цельсия в атмосфере N2.
Подробное описание соединений приведено ниже.
13a 1-O-трет-бутилдифенилсилил-3-O-трезил-1,3-пропандиол. Белое кристаллическое твердое вещество, 1,74 г, 77%, температура плавления 34-35 градусов Цельсия. 3 мл этой реакционной смеси удалили до проведения дополнительных реакций.
1H ЯМР (270 МГц, CDCl3, δ): 1,06 (s, 9H,tBu), 1,97 (m, 2H,
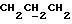
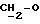
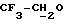
IR (диск KBr) 1386, 1329, 1274, 1258, 1185, 1164, 1137, 1094, 941, 763, 506 см-1.
13b 2-метил-1-O-трет-бутилдифенилсилил-3-O-трезил-1,3-пропандиол. Бесцветное масло, 2,57 г, 99%
1H ЯМР (270 МГц, CDCl3, δ): 0,97 (d, 3H,
J = 6,87 Гц, CH3), 1,06 (s, 9H,tBu), 2,10 (m, 1H,
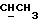
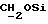
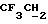
Более подробное описание общего процесса получения треслатов см. в работе R.K.Crossland et al., JACS, 93, 4217 (1971).
ПРИМЕР 11
Синтез N-/ацетокси-2-оксиэтил/-N-/O-(3-(O-(4,41
- диметокситритил)-1-оксиметил)-4-нитрофенил)-3-оксипропил/-N-/2- гидроксиэтил/амина (соединение
15, схема 4).
В соединение 11 (1,72 г, 1,92 ммоль), растворенное в безводном ТГФ (20 мл), добавили фторид тетрабутиламмония (0,55 мл раствора 1М в ТГФ, 1,92 ммоль). Затем реакционную смесь перемешивали в течение двух часов при комнатной температуре. После этого реакционную смесь разбавляли водой (50 мл), а ТГФ удаляли высушиванием в роторном испарителе. Затем водный раствор экстрагировали хлороформом (х1). Органический раствор высушивали безводным сульфатом натрия и выпаривали до получения смолы. Полученный продукт очищали хроматографией на силикагеле, элюируя колонку этилацетатом. Фракции полученного продукта объединяли и выпаривали в роторном испарителе, в результате чего получали соединение 12 в виде бесцветной смолы, которая медленно кристаллизовывалась: 0,73 г, 58%, температура плавления 95->97 градусов Цельсия, RF 0,26 этилацетат.
1H ЯМР (270 МГц, CDCl3, δ): 1,75 (brs, 1Н,

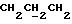

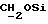


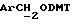
IR (диск KBr) 3459, 1738, 1608, 1577, 1506, 1444, 1313, 1288, 1250, 1230, 1175, 1154, 1070, 1035, 984 см-1.
ПРИМЕР 12
Синтез
N-/O-(трет-бутилдифенилсилил)-2-оксиэтил/-N-/O-(3-(O- (4,41-диметокситритил)-1-оксиметил)-4-нитрофенил)-3-оксипропил/- N-/ацетокси-2-оксиэтил/амина
(соединение 14, схема 4).
В соединение 6 (1,73 г, 2,02 ммоль), растворенное в безводном пиридине (10 мл), добавили уксусный ангидрид (0,5 мл, 4,54 ммоль) и 4-диметиламинопиридин (55 мг, 0,45 ммоль), после чего колбу закрыли пробкой. Затем реакционную смесь перемешивали при комнатной температуре в течение 16 часов, после чего провели анализ с применением тонкослойной хроматографии (метанол/этилацетат 5%/95%), который показал полное исчезновение исходного материала и образование нового тритил-содержащего пятна, RF 0,80. Пиридин удаляли высушиванием в роторном испарителе, а оставшиеся следы удаляли совместным выпариванием с толуолом (х2). Полученную смолу разделяли между хлороформом и водой. Раствор хлороформа отделяли и промывали соляным раствором (х1). Затем раствор хлороформа высушивали безводным сульфатом магния и растворитель выпаривали в роторном испарителе до получения бесцветной смолы, 1,94 г. Полученный материал был достаточно чистым для того, чтобы использовать его в следующих реакциях без дополнительной очистки.
1H ЯМР (270 МГц, CDCl3, δ): 1,04 (s, 9H,tBu), 1,9 (m, 2H,

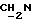



ПРИМЕР 13
Синтез N-/ацетокси-2-оксиэтил/-N-/O-(3-(O-(4,41
- диметокситритил)-1-оксиметил)-4-нитрофенил)-3-оксипропил/- N-/O-(трет-бутилдифенилсилил)-3-оксо-6-оксигексил/амина (соединение 16, схема 4).
В соединение 12 (66 мг, 0,10 ммоль), растворенное в безводном ацетонитриле (5 мл), добавили карбонат калия (55 мг, 0,4 ммоль) и соединение 13a (1 мл реакционной смеси, приблизительно 0,30 ммоль); после чего колбу закрыли трубкой, через которую производили осушение хлоридом кальция. Затем реакционную смесь перемешивали в течение 22 часов, после чего карбонат калия отфильтровывали, а растворитель удаляли высушиванием в роторном испарителе. Полученное масло помещали на колонку с силикагелем (14 х 1 см), продукт элюировали смесью эфира/петролейного эфира 40->60 градусов Цельсия, 75%/25%. Очищенные фракции продукта соединяли и выпаривали до получения прозрачной смолы, 6 мг, 6%, RF 0,47 в смеси эфира/петролейного эфира 40->60 градусов Цельсия, 80%/20%.
1H ЯМР (270 МГц, CDCl3, δ): 1,05 (s, 9H,tBu), 1,8 (m, 2H,
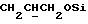
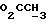


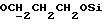
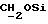
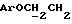
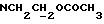

Указанное ниже соединение было синтезировано аналогичным способом с использованием треслата 13b.
N-/ацетокси-2-оксиэтил/-N-/O-(3-(O-(4, 4'-диметокситритил)-1- оксиметил)-4-нитрофенил)-3-оксипропил/-N-/O-(трет-бутилдифенилсилил)-5- метил-3-оксо-6-оксигексил/амин.
Соединение представляло собой прозрачную смолу, RF 0,53 в смеси эфира/петролейного эфира, 40->60 градусов Цельсия, 80%/20%.
1H ЯМР (270 МГц, CDCl3, δ): 0,88 (d, 3H,

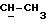


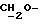
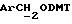
ПРИМЕР 14
Синтез олигонуклеотидов на твердом носителе
Стекло с контролируемой пористостью, несущее линкеры 9 и 10 (соединения 9 и 10
по
схеме 2) загружали в колонки, используемые в автоматическом синтезе олигонуклеотидов (AB1 381A); использованные количества вещества обеспечивали синтез в масштабе 0,2 или 1 мкмоль. Колонки
вставляли в
автоматический синтезатор, который программировали на соответствующие циклы. Использовали два различных типа нуклеотидных предшественников: нормальные фосфорамидиты с диметокситритильными
защитными
группами на 5'-гидроксилах; "переполяризованные синтоны"*) с 5'-фосфорамидитами и диметокситритильными защитными группами на 3'-гидроксилах. Список олигонуклеотидов,
синтезированных на этих
носителях, показан в таблице 4, где R9 и R10 получают из соединений 9 и 10 соответственно. Выходы контролировали по количеству диметокситритильных групп, высвобождаемых при
каждом взаимодействии. Эти
выходы соответствовали выходам, полученным на носителях из стекла с контролируемым размером пор, использованных для обычного синтеза олигонуклеотидов.
ПРИМЕР 15
Синтез меток
в условиях, при которых аналит остается интактным.
После синтеза 5'R9T5 на носителе 9 твердый носитель разделяли, часть обрабатывали раствором 5 мМ фторида тетрабутиламмония в ТГФ в течение 10 минут при комнатной температуре для удаления трет-бутилдифенилсилильной защитной группы. Оба образца обрабатывали 29% аммиаком при комнатной температуре в течение ночи до удаления продуктов реакции из твердого носителя. Аммиак удаляли под вакуумом, а твердый остаток растворяли в воде. ВЭЖХ указывала на успешное удаление силильной защитной группы с удерживанием 4,41-диметокситритильной группы. Этот пример показывает, что две защитные группы можно удалить в условиях, когда другая защитная группа остается; более того, что при удалении защитных групп не повреждается цепь олигонуклеотида.
ПРИМЕР 16
Биохимические реакции с меченными аналитами.
16a. Ферментное фосфорилирование меченных олигонуклеотидов.
Во многих случаях желательно иметь олигонуклеотиды, имеющие фосфатную группу на 5'-конце. Такая группа необходима в том случае, если олигонуклеотид предполагается использовать в качестве донора в реакции лигирования; она может быть так же использована для введения радиоактивной группы с целью определить биохимические свойства вещества. Олигонуклеотиды A5, A10 и T5 получили с метками R9 и R10 на 3'-концах, причем в одних соединениях защитные силильные группы были удалены, а в других - нет (это достигалось благодаря тому, что олигонуклеотиды, еще находящиеся на твердом носителе, обрабатывали раствором 5 мМ фторида тетрабутиламмония в ацетоне; при комнатной температуре, в течение 15 минут). Эти олигонуклеотиды фосфорилировали с использованием полинуклеотидкиназы T4 и гамма-33P-аденозин-5'-трифосфата с применением стандартных протоколов, рекомендованных поставщиком. Тонкослойная хроматография продуктов реакции на целлюлозе, пропитанной полиэтиленимином (PEl), и проявленной в 0,5 М растворе бикарбоната аммония, показала в каждом случае, что меченный фосфор почти полностью был перенесен на олигонуклеотид.
16b. Ферментное лигирование меченных олигонуклеотидов.
В некоторых видах применения меченных олигонуклеотидов они могут
быть успешно лигированы к рецепторам. В приведенных ниже тестах
мы показали, что меченные олигонуклеотиды могут принимать участие в ферментном лигировании:
(1) Олигонуклеотиды, имеющие метку
на 5'-конце.
В этом тесте матрица представляла собой 5'-ATCAAGTCAGAAAAATATATA (последовательность N 1). Эту матрицу гибридизировали с донором - 3'-TAGTTCAGTC (последовательность N 2), который фосфорилировали на конце 5', используя радиоактивный фосфор. Провели четыре реакции лигирования, каждую с модификацией последовательности T5, которую можно лигировать с фосфорилированным 5' концом донора, после гибридизации с серией 5 A's в матрице. Использованные в реакции четыре олигоT'a отличалась по природе своих концов 5'. Один имел диметокситритильную группу, присоединенную через гидроксил. Второй и третий имели метки R9 и R10, присоединенные к концам 5' посредством фосфодиэфирной связи. Четвертый представлял собой позитивный контроль, с нормальной 5'OH. Негативный контроль не имел олигоT'a. Реакции лигирования осуществляли с помощью T4-лигазы в соответствии с инструкцией поставщика. Реакции контролировали методами тонкослойной хроматографии на полиэтиленимин-целлюлозе, проявленной в растворе 0,75 М бикарбоната аммония. Все четыре реакции дали дополнительное пятно на хроматограмме, которое обладало меньшей мобильностью по сравнению с донором; при этом, как и ожидалось, негативный контроль не дал дополнительного пятна. Это показывает, каким образом олигонуклеотиды с различными метками могут участвовать в реакциях лигирования, специфичных к последовательностям.
В работе Cozzarelli и др. (1967) показано, что полинуклеотиды, присоединенные к твердым носителям, можно лигировать к акцептору в присутствии комплементарной матрицы.
ПРИМЕР 17
Гибридизация меченных
олигонуклеотидов с олигонуклеотидами, связанными с твердым носителем.
Пример 16b показывает, что меченные олигонуклеотиды могут участвовать в реакциях лигирования, в результате чего можно сделать вывод, что они могут участвовать также в формировании дуплексов в растворе, поскольку от этого процесса зависит лигирование. Описанный ниже эксперимент показывает, что они могут образовывать дуплексы также с олигонуклеотидами, связанными с твердым носителем. T10 был синтезирован на поверхности листа аминированного полипропилена в соответствии с инструкцией фирмы-производителя. Известно, что этот процесс дает около 10 пмоль олигонуклеотида на кв. мм. На поверхность дериватизированного полипропилена нанесли раствор A10 (65 ммоль на микролитр), меченного на 5'-конце33P, а на 3'-конце - меткой R10, в 3,5 М хлориде тетраметиламмония, и оставили на ночь при температуре 4 градуса Цельсия. После промывки в гибридизирующем растворителе было обнаружено, что около третьей части зонда было гибридизировано со связанным олиго-dT. Это приближается к теоретическому пределу гибридизации и свидетельствует о том, что олигонуклеотиды, содержащие метки, могут с высокой степенью эффективности принимать участие в реакциях гибридизации.
ПРИМЕР 18
Фотолиз меток.
Возможность удалять метки фотолизом намного повышает эффективность метода: это позволяет проводить непосредственный анализ путем лазерной десорбции в масс-спектрометре; это позволяет простым способом удалять метки для проведения других биохимических или химических реакций.
18a. Объемный фотолиз.
Известно, что нитробензильная группа лабильна к облучению при 305 нм. Растворы R10A10 и R10T5 в воде облучали на расстоянии 2 см от трансиллюминатора в течение 20 минут в условиях, при которых, как известно, не происходит разрушения нуклеиновых кислот. ВЭЖХ анализ показал наличие предполагаемых продуктов фоторасщепления и отсутствие обнаружимых остатков исходного соединения.
18b. Фотолиз, вызванный облучением лазером в масс-спектрометре.
Образцы R10T5 и T5R10 осаждали на металлической мишени масс-спектрометра при масс-спектроскопии по времени пролета ("Finnigan Lasermat") без добавления матрицы. Спектр показал наличие одного насыщенного пика вблизи массы 243 в режиме положительно заряженных ионов, который отсутствовал в других образцах.
ПРИМЕР 19
Идентификация различных меток, присоединенных к разным аналитам, с использованием методов
масс-спектрометрии.
Последовательность из пяти тимидиновых остатков, содержащих диметокситритильную группу, присоединенную в качестве метки к 3'-концу, синтезировали обычным способом на твердой фазе, но с применением "переполяризованных синтонов". В масс-спектрометре это соединение дало большой и яркий пик у массы 304 в режиме положительно заряженных ионов. Напротив, последовательность из десяти аденазиновых остатков, несущих метку, обозначенную выше как R10, дала большой и очевидный пик на массе 243 в режиме положительно заряженных ионов. В обоих случаях лазерную десорбцию проводили в отсутствии матрицы. В обоих случаях пики отсутствовали при анализе олигонуклеотидов, не имеющих меток. Эти примеры показывают, что очень просто идентифицировать последовательность аналита по наличию пика в спектрограмме, который дает метка, введенная в процессе синтеза аналита, а также что характерные метки легко идентифицировать по различиям в их массе.
РАЗЪЯСНЕНИЯ К ФИГУРАМ
Фиг. 1. Общая схема синтеза молекул с специфичными
метками.
Синтез начинается с линкера (L), который имеет по меньшей мере один сайт для присоединения групп для синтеза аналита, и один сайт для синтеза метки. (Линкер может быть также обратимо присоединен к твердому носителю в процессе синтеза, и этот линкер может иметь сайты для генерации групп, таких как заряженные группы, которые также могут использоваться при анализе). Pa и Pr представляют собой временные защитные группы, используемые, соответственно, для защиты предшественника аналита и репортерных групп. Эти защитные группы можно удалить различными способами обработки. Например, Pa может представлять собой кислотную или основную группу, такую как тритил, F-MOC или t-BOC, а Pr представляет собой группу, которую можно удалить обработкой фторидом. Например, такую группу, как силильный остаток. Группы U-Z также могут иметь защитные группы, которые должны быть стабильными к реагентам, используемым для удаления групп Pa и Pr. Для присоединения различных типов аналитов применяются разные способы: для синтеза олигонуклеотидов и пептидов можно использовать стандартные способы.
На фиг. 2 описаны три различных типа меток. На первой схеме каждое удлинение метки осуществляется с применением репортерной группы, которая специфична для позиции и для типа остатка, добавленного к аналиту. Для этой схемы блокировка не обязательна.
На второй и третьей схемах положение определяется суммарной массой репортера, достигаемой на стадии синтеза в тот момент, когда к аналиту добавляется остаток. В этом случае важно частично прекратить удлинение метки путем блокировки части молекулы. Вторая и третья схемы отличаются друг от друга способом добавления репортерных групп. На второй схеме эти группы в агентах удлинения; на третьей - в "кэпах".
Фиг. 2. Три типа молекуло-специфичных меток.
A. Иллюстрируются метки из репортерных групп (E), которые определяют как положение (подстрочные знаки), так и идентичность (надстрочные знаки) групп в аналите (U-Z). Такой набор может образовывать серию алифатических цепей с возрастающей формульной массой, которая определяет положение: например, метилен для положения 1, этилен для положения 2, пропилен для положения 3 и т. д. Их можно разделять на группо-специфичные типы по различным составам изотопов углерода и водорода: например, есть шесть различных изотопных составов CH2, как показано в таблице 1. Четыре из них отличаются на одну массовую единицу и их легко различить методом масс-спектрометрии. Можно предложить и другие способы дифференцированного мечения, например, либо положение, либо группу можно метить репортерными группами с разными зарядами. Такие группы можно отделять и распознавать разными способами, включая масс-спектрометрию.
B. Показанные метки, полученные частичным синтезом, при котором любая структура аналита присоединяется к серии меток; первый член серии имеет репортерную группу, специфичную к первой группе аналита; второй член имеет первую репортерную группу плюс вторую репортерную группу, специфичную ко второй группе аналита, и т.д. Такие серии очень просто получить, используя два вида предшественников для удлинения метки: один предшественник защищен обратимо блокирующей группой, а второй предшественник предотвращает дальнейшее удлинение. Например, смесь RX и P-(CH2)nX, где R представляет собой нереакционноспособную алифатическую группу, такую как метил или этил, P представляет собой обратимо защитную группу, а X является активированным остатком, который может взаимодействовать с группой, защищенной P. Те молекулы, которые были "блокированы" нереакционноспособными алифатическими группами, не будут принимать участия в следующем цикле деблокирования и удлинения.
На схеме B группо-специфичная информация заключена в остатках, использованных для удлинения. Как и в A, информацию можно получить с помощью масс-изотопов. Например, каждое добавление остатка CH2, меченного изотопами C и H, к p-(CH2)nX, дает дополнительные сайты, которые могут давать различие в массе репортеров. Массы групп (CH2)n могут варьироваться в пределах от 14 н до 17 н, и в этом диапазоне имеется 4 + 3(n-1) значений массы. Для этиленовой группы имеется семь различных значений массы в пределах от 28 до 34, а для пропиленовой группы таких значений десять - от 42 до 51.
C. Показан другой способ введения группо-специфичной информации; в данном случае эта информация содержится в терминаторе цепи - "кэпе", как указано в B. Здесь снова различия в массе можно добиться путем мечения алифатического остатка. Информацию о положениях можно получить, определив, на какой длине был добавлен терминатор. Предположим, что E представляет собой (CH2)2-O, а терминаторами являются меченные изотопами метильные группы с формульными массами от 15 до 19. Каждое удлинение будет добавлять 44 массовых единицы к репортеру. Диапазон масс для самого короткого репортера будет от 44 + 15 = 59 до 44 + 19 = 63. Диапазон масс для второго положения будет от 88 + 15 = 103 до 88 + 19 = 107, и так далее до шестой позиции, где диапазон масс будет составлять от 284 + 15 = 299 до 284 + 19 = 303. В этом диапазоне значения не перекрывают друг друга, и видно, что количество репортеров и диапазон можно расширить, используя терминаторы и удлинение с большим числом атомов.
Список
литературы
1.
Brenner, S. and Lerner, R.A. (1992). Encoded combinatorial chemistry. Proc. Natl. Acad. Sci. USA 89: 5381-5383.
2. Drmanac, R., Labat, I., Brukner, I., and Crkvenjakov, R. (1989). Sequencing of megabase plus DNA by hybridization: Theory of the method. Genomics 4: 114-128.
3. Pillai, V. N. R. (1980). Photoremovable protecting groups in organic chemistry. Synthesis 39: 1-26.
4. Hoheisel, J.D., Maier, E., Mott, R., McCarthy, L., Grigoriev, A.V., Schalkwyk, L.C., Nitzetic, D., Francis, F. and Lehrach, H. (1993) High resolution cosmid and P1 maps spanning the 14 Mbp genome of the fission yeast Schizosaccharomvces pombe. Cell 73: 109-120.
5. Khrapko, K. R. , Lysov, Yu. P., Khorlyn, A. A., Shick, V. V., Florentiev, V. L. , and Mirzabekov. (1989). An oligonucleotide hybridization approach to DNA sequencing. FEBS Lett. 256: 118-122.
6. Patchornik, A., Amit, B. and Woodward, R. B. (1970). Photosensitive protecting groups. J. AMER. Chem. Soc. 92:21: 6333-6335.
7. Ross, M. T., Hoheisel, J.D., Monaco, A.P., Larin, Z., Zehetner, G., and Lehrach, H. (1992) High density gridded Yac filters; their potential as genome mapping tools. In "Techniques for the analysis of complex genomes". Anand, R. ed. (Academic Press) 137-154.
8. Southern, E. M. (1988). Analyzing Polynucleotide Sequences. International Patent Application PCT GB 89/00460.
9. Southern, E.M., Maskos, U. and Elder, J.K. (1992). Analysis of Nucleic Acid Sequences by Hybridization to Arrays of Oligonucleotides: Evaluation using Experimental Models. Genomics 12: 1008-1017.
10. de Vries, M. S. , Elloway, D.J., Wendl, R.H., and Hunziker, H.E. (1992). Photoionisation mass spectrometer with a microscope laser desorption source, Rev. Sci. Instrum. 63(6): 3321-3325.
11. Zubkov, A. M., and Mikhailov, V. G. (1979). Repetitions of s-tuples in a sequence of independent trials. Theory Prob. Appl. 24, 269-282.
12. Cozzarelli, N. R. , Melechen, N.E., Jovin, T.M. and Kornberg, A. (1967). BBRC, 28, 578-586.
Реферат
Изобретение относится к биотехнологии и может быть использовано для определения аналитов. Реагент включает в себя а) анализируемый компонент, состоящий, по меньшей мере, из двух анализируемых остатков, и связанный с b) компонентом-меткой, включающим в себя, по меньшей мере, одну или более репортерных групп, подходящих для проведения масс-спектрометрии, где репортерная группа обозначает анализируемый остаток, причем репортерная группа в каждом положении компонента-метки выбирается так, чтобы она обозначала анализируемый остаток в определенном положении анализируемого компонента. Множество таких реагентов, каждый из которых включает в себя отличный от других анализируемый компонент, образуют библиотеку реагентов, которую можно использовать для анализов исследуемого вещества. Анализ компонентов-меток позволяет установить природу анализируемых компонентов, связанных с исследуемым веществом. Секвенирование нуклеиновых кислот с применением библиотеки реагентов позволяет установить последовательность исследуемой нуклеиновой кислоты. 7 с. и 15 з.п. ф-лы, 5 ил. 4 табл.
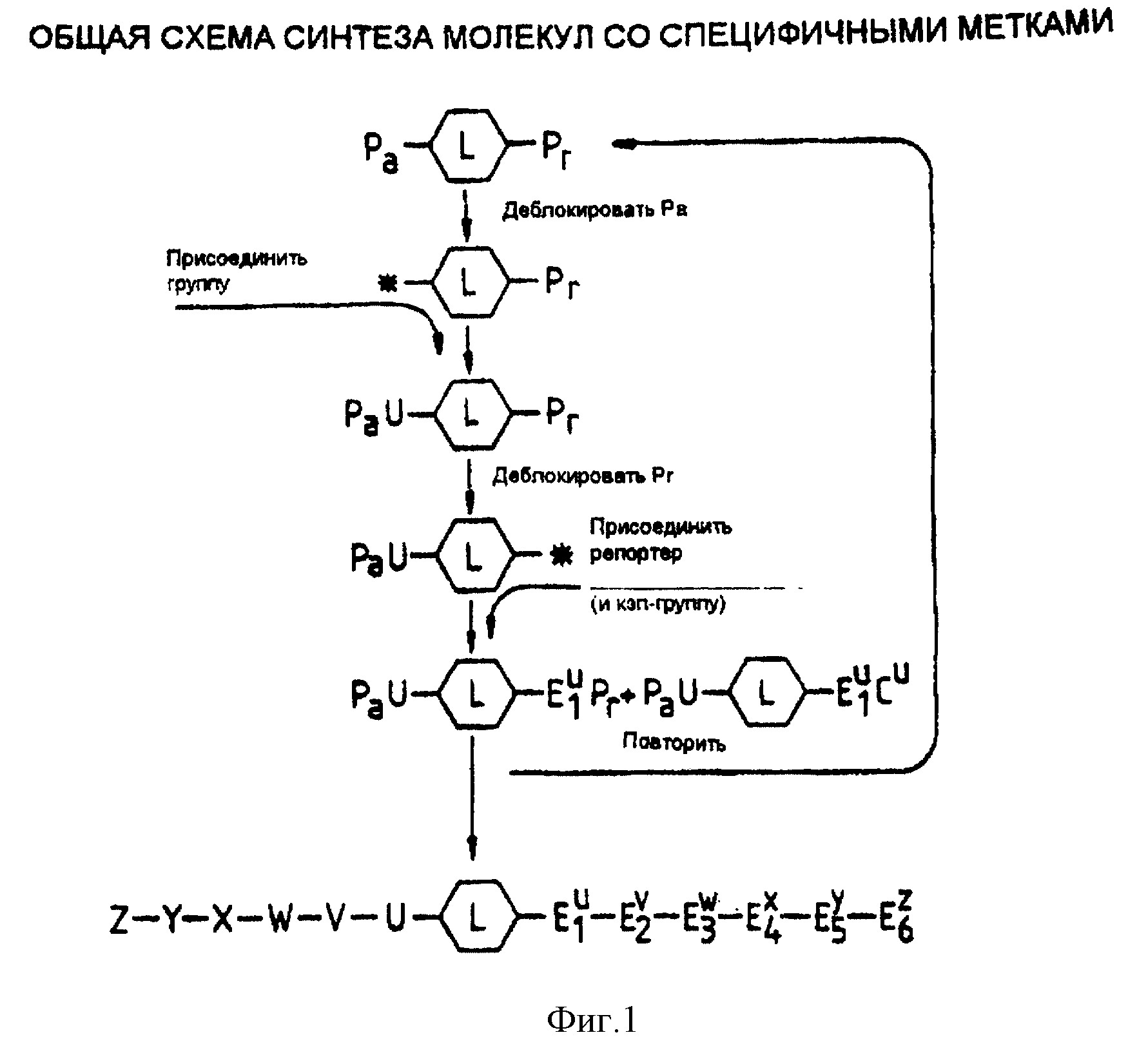
Формула
A - L - R,
где A - олигонуклеотид, состоящий, по меньшей мере, из 2 анализируемых нуклеотидных остатков;
R - компонент-метка, содержащий одну или более репортерных групп;
L - линкер, содержащий ароматическую группу, несущую гидроксильную группу, аминогруппу или сульфогруппу, предназначенную для синтеза анализируемого олигонуклеотида, реакционноспособную группу для синтеза компонента-метки и ортонитрогруппу, предназначенную для фоторасщепления.
Комментарии