Фармацевтическая композиция, представляющая микроэмульсию с биологически активным макромолекулярным материалом и способ ее получения - RU2122403C1
Код документа: RU2122403C1
Чертежи







Описание
Изобретение относится к технологии приготовления лекарственных средств. Более конкретно настоящее изобретение относится к перорально или ректально вводимым композициям биологически активных веществ, в частности белковых веществ.
Медицинская практика на протяжении многих лет предписывает или советует вводить многие биологически активные вещества для лечения или профилактики широкого круга заболеваний или состояний. Одним из наиболее хорошо известных, однако ни в коем случае не единственным прописываемым биологически активным белковым веществом является инсулин, который используется для лечения диабета.
Возможно, более простым способом приема любого лекарственного средства является пероральный прием. Такой путь введения, осуществляемый при помощи сиропа, таблеток, капсул, эликсира, гранул, порошков или любой другой приемлемой композиции, как правило, является легким и удобным и часто с точки зрения пациента является наименее неудобным и неприятным путем приема лекарственного средства. Однако с точки зрения медицинского лечения и профилактики предпочтительный путь введения белковых лекарственных средств и других биологически активных веществ заключает в себе прохождение вещества через желудок, который является враждебной средой для многих веществ, включая белки. Поскольку кислотная, гидролитическая и протеолитическая окружающая среда желудка эффективно эволюционирует с перевариванием белковых веществ в аминокислоты и олигопептиды для последующего анаболизма (ассимиляции), нет ничего удивительного в том, что очень незначительная часть из числа широкого круга биологически активных веществ при их пероральном приеме сохранится при прохождении через желудок для того, чтобы поглотиться в тонкой кишке.
Результатом сего (и многие больные диабетом могут подтвердить) является то, что многие белковые лекарственные средства необходимо принимать парентерально, часто путем подкожной, внутримышечной или внутривенной инъекции, со всеми вытекающими отсюда неудобствами, дискомфортом и трудностями соблюдения больным режима и схемы лечения.
Данная проблема не является изолированной, поскольку заболевания, нуждающиеся в извлечении путем введения белкового вещества, могут распространяться очень широко. Сахарный диабет, например, поражает большое число людей во многих странах мира. Сахарный диабет представляет собой хроническое расстройство, воздействующее на углеводный, жировой и белковый обмен. Данное заболевание отличается гипергликемией и гликозурией, происходящим в результате неправильной или недостаточной инсулинсекреторной реакции. Существуют два основных варианта данного заболевания.
Один вариант, который наблюдается приблизительно у 10% всех идиопатических больных диабетом, известен как юношеский (ювенильный) диабет или инсулин-зависимый сахарный диабет ("ID D M"). Этот вариант часто проявляется потерей инсулин-секреторной функции бета-клетками поджелудочной железы, а следовательно, прогрессирующей зависимостью от экзогенного инсулина для поддержания углеводородного обмена. Эту характеристику разделяют те неидиопатические или "вторичные" больные диабетом, чьи расстройства находят свое происхождение в панкреатической болезни. Второй вариант идиопатического сахарного диабета известен как поздний диабет или неинсулин-зависимый сахарный диабет ("NID DM").
Отчасти вследствие большого числа пациентов, страдающих диабетом одной или другой формы, существует необходимость разработки композиций инсулина для перорального введения, которые в известной степени защищены от воздействия враждебной окружающей среды желудка. Хотя предпринимаются многочисленные попытки разработать такие композиции, заявитель не знает ни одной, которая на сегодняшний день поставлялась бы в значительной мере на рынок. Заявитель осведомлен лишь о следующих предложениях.
Заявка на патент PCT N 8701035 относится к парентерально вводимым композициям жирорастворимых лекарственных средств и витаминов, причем композиции содержат "псевдомицеллы".
Заявка на патент PCT N 8705505 раскрывает перорально принимаемые композиции инсулина, нанесенного на твердые частицы из водного препарата, причем покрытые инсулином частицы затем покрывают липидной оболочкой.
Заявка на патент США N 4849405, опубликованная 18 июля 1989 года, раскрывает перорально принимаемые композиции инсулина. Описывается, что композиции представляют собой двухфазовые препараты и, оказывается, что обе фазы являются водными, причем фазы эффективно поддерживают раздельными системой коацервата.
Заявка на Европатент N 0140085 раскрывает получение липидных везикул, содержащих лекарственное средство.
Шичири и др. (Acta diabet lat., 15, 175-183 (1978)) раскрывает инсулиновые мицелы типа вода - в масле - в воде.
Заявка на патент США N 4784845, опубликованная 15 ноября 1988 года, и заявка на патент N 4816247 США, опубликованная 28 марта 1989 года, раскрывает эмульсионные композиции для парентерального введения гидрофобных лекарственных препаратов.
Заявка на патент Японии N 55017328 описывает содержащие инсулины эмульсии типа вод - в масле - в воде для перорального приема.
Изобретение предлагает усовершенствованные фармацевтические составы, которые могут вводиться перорально или ректально. Более конкретно, обнаружено, что даже белковые активные вещества, вводимые только парентерально, могут подаваться посредством более предпочтительного перорального или ректального способа применения в качестве компонента в двухфазную систему, которая включает гидрофобную фазу, содержащую хиломикроны или материал, из которого хиломикроны образуются in vivo в слизистой выстилке. Оказывается, не только проявляется биодоступность активного вещества, но также и эффективность доставки активного материала может быть повышена по крайней мере в некоторых случаях. Хотя основные принципы этих эффектов остаются неясными, полагают, что биологически активное вещество, водимое в сочетании с хиломикронами или составляющими хиломикронов, нацелено в ворсинки кишечной стенки (или микроворсинки), откуда оно секретирует в млечные сосуды и кишечную лимфу, а затем дренирует в грудной (лимфатический) проток и, наконец, в циркулирующий кровоток.
В соответствии с первым вариантом настоящего изобретения предлагается фармацевтическая композиция, содержащая микроэмульсию, имеющую гидрофильную фазу и гидрофобную фазу, причем (A) гидрофильную фазу диспергируют в гидрофобной фазе, (B) гидрофильная фаза содержит биологически активное вещество и (C) гидрофобная фаза содержит хиломикроны или материал, из которого хиломикроны образуются in vivo. Гидрофильная фаза может содержать физиологически совместимый растворитель для биологически активного вещества, такой как воды.
Следовательно, настоящее изобретение предлагает перорально или ректально вводимый состав биологически активного вещества, причем данный состав содержит водомасляную микроэмульсию, в которой водная или гидрофильная фаза биологически активного вещества, причем данный состав содержит биологически активное вещество и масляная или гидрофобная фаза содержит хиломикроны или материал, способный образовывать хиломикроны в кишечной слизистой оболочке после введения.
Биологически активное вещество в составах настоящего изобретения абсорбируется. Перорально вводимые составы являются предпочтительными, однако в некоторых случаях подходят ректально вводимые составы.
Составы в соответствии с настоящим изобретением фундаментально отличаются от описанных выше известных технических решений. Составы "псевдомицелл" в соответствии с заявкой на патент PCT N 8701035, хотя и описаны как аналогичные натуральным хиломикронам, на самом деле не приводят к получению перорально принимаемой композиции. Активное вещество, как оказывается, не будет биологически приемлемым, если композицию придется принимать через рот, из-за отсутствия поверхностно-активного вещества с низким гидрофильно-липофильным балансом (ГЛБ). Твердые композиции в оболочке в соотношении с заявкой на патент PCT N 8705505 не образуют абсорбируемые хиломикроны, поскольку опять они не содержат поверхностно-активное вещество с низким ГЛБ; причем в данном случае активный компонент, как полагают, абсорбируется пиноцитозом. Композиции в соответствии с заявкой на патент США N 4849405 являются водными, а не двухфазовыми системами (например, масляная и водная фазы), так что они совершенно отличаются по характеру. Ни одно из этих или оставшихся от числа вышеприведенных известных технических решений не раскрывает композиции, способные образовывать хиломикроны.
Термин "биологически активное вещество" включает, в частности, фармацевтически активные белковые вещества. Белковый материал может быть чистым белком или он может содержать белок таким образом, что гликопротеид содержит и белковые и сахарные остатки. Биологически активное вещество может быть полезно в медицине человека или ветеринарии как при лечении, так и при профилактике болезней и их симптомов, или может быть полезно косметически или диагностически. Примеры белкового биологического вещества, которые могут быть использованы в качестве перорально или ректально вводимых композиций в соответствии с настоящим изобретением, включают белковые гормоны, такие как инсулин, кальцитонин и гормон роста, имеющие как человеческое, так и животное происхождение, или полученные полу- или полностью синтетическим путем, эритропоэтин, активаторы плазминогена и их предшественника, такие как АПТ (тканевый активатор плазминогена), урокиназа, проурокиназа и стрептокиназа, интерфероны, включающие человеческий интерферон альфа, интерлейкины, включая ИЛ-1, ИЛ-2, ИЛ-3, ИЛ-4 и ИЛ-5, а также антигены (факторы) крови, включая фактор VIII.
Хотя и не полагают, что не существует какой-либо определенный молекулярный размер относительно биологически активных веществ, которые могут быть составлены в соответствии с настоящим изобретением, из примерных, однако неограничивающих вариантов биологически активных веществ, приведенных выше, станет понятно, что настоящее изобретение особенно пригодно для микромолекул. Молекулярная масса таких макромолекул может составлять около 1 кД или свыше 5 кД, около 10 кД свыше, или даже около 15 кД или свыше. И вновь, хотя и не полагают, что решающее значение имеет гидрофильность или гидрофобность (липофильность) биологически активного вещества, изобретение позволит получать гидрофильные молекулы, такие как инсулин, кальцитонин (особенно кальцитонин лосося) и гормоны роста или самототропин (особенно свиной самототропин), каждая из которых (в частности, кальцитонин лосося) представляет такое гидрофильное вещество, что оно может быть гигроскопичным.
Количество биологически активного вещества, присутствующего в композиции изобретения, будет зависеть от природы вещества и будет таким, чтобы приготовить лекарственное средство на основе традиционно вводимых количеств при практическом использовании. Учитывая все эти моменты, следует отметить, что композиции в соответствии с настоящим изобретением могут содержать от 1 мкг, 10 мкг, 0,1 мг или 1 мг на литр до 1, 10 г или 100 г на литр.
Микроэмульсии как таковые известны, в частности, для составления таких простых органических молекул, как гербициды. Как и макроэмульсии, микроэмульсии имеют две фазы: гидрофильную фазу и гидрофобную или липофильную фазу. Следует понять, что в данном описании изобретения термин "гидрофобная фаза" не должна рассматриваться так, будто бы вода присутствует, исключая все другие ингредиенты в данной фазе; напротив, фаза просто является гидрофильной. Аналогично, термин "гидрофобная фаза" или "неводная фаза" не должен рассматриваться так, будто бы присутствует только масло или будто бы данная фаза должна содержать любое углеводородное вещество, обычно известное под термином "масло"; напротив, данная фаза, как правило, является гидрофильной фазой. Обе фазы, однако, являются, как правило, в основном жидкими.
Микроэмульсии характеризуются размером капель и повышенной стабильностью. Микроэмульсии имеют размеры капель, средний диаметр которых обычно составляет менее чем 10 мкм и часто менее чем 1 или 2 мкм. Фактически, некоторые микроэмульсии могут иметь средние размеры капель в диапазоне 200 нм или менее. Термин "микроэмульсия", как он используется в данном описании, означает любую достаточно стабильную двухфазовую систему, в которой одну фазу диспергируют в другой. Некоторые двухфазовые системы иногда классифицируют как "эмульсия" или "макроэмульсия", и поэтому они могут подпадать в объем термина "микроэмульсии", как он используется в данном описании. Размер капелек в основном дисперсной фазы может составлять менее чем 2 мкм. Размер капелек может быть измерен сканирующей электронной микроскопией, темно-фазово-контрастной микроскопией, кондуктометрией, светорассеянием (например, лазерного света) или любым другим традиционным методом. "Капелька" означает структурные единицы, которые составляют дисперсную фазу.
Стабильность микроэмульсий, как этот термин используется в данном описании, продемонстрирован тем фактом, что микроэмульсии не имеют склонность к разделению при отстаивании; стабильность является "достаточной", если она позволяет осуществлять дальнейшую обработку, если желательно или необходимо, и/или обеспечивают достаточный срок годности. Кроме того, определенные микроэмульсии могут быть полупрозрачными или прозрачными, часто имеющими легкую окраску.
Отношение объемов гидрофильной фазы к гидрофобной фазе, как правило, составляет интервал от 0,1:1 до 10:1, например от 0,2:1 до 5:1, типично от 0,5:1 до 2:1.
Гидрофильная фаза может содержать смешивающийся с водой растворитель, например, с тем, чтобы содействовать при составлении рецептуры. Может присутствовать этанол или другой пригодный органический растворитель. Природа используемого растворителя будет зависеть от активного вещества. Гидрофильная фаза может быть в форме смеси вода:растворитель, например, при объемных соотношениях от 0,5:1 до 2:1.
Выше упомянуто, что гидрофобная фаза содержит либо хиломикроны, либо вещество, способное образовывать хиломикроны в кишечнике слизистой оболочки. Хиломикроны встречаются в природе в качестве мелких частиц, с преобладающим содержанием жира, обычно присутствующего в плазме крови, особенно после переваривания жирной пищи. Каждый хиломикрон можно рассматривать как белково-липидный комплекс, основной липидный компонент которого содержит триглицериды, которые имеют плотность около 0,95-1,006 и скорость флотации после ультрацентрифугирования свыше 400. Комплекс обычно содержит около 80 - 90%, более предпочтительно 85 - 88% моно-, ди- и триглицеридов; 5 - 19%, более предпочтительно 6 - 9% фосфолипидов; 1 - 3%, более предпочтительно 2% сложных эфиров холестерина; 0,1 - 2%, более предпочтительно 1%, свободных жирных кислот; 1 - 3%, более предпочтительно менее 2%, белка. Белковыми компонентами являются апопротеины, в частности апопротеины A, B, C и E. Следует отметить, что для образования хиломикронов в кишке нет необходимости поставлять экзогенно все компоненты, поскольку некоторые из них могут поставляться самим телом.
Хиломикроны образуются в слизистой оболочке кишечной стенки во время поглощения триглицеридов животными. После проглатывания и гидролиза жирных кислот в моноглицериды и их взаимодействия с желчью с образованием смешанных мицелл жирные кислоты и моноглицериды диффундируют в слизистую оболочку, причем жирные кислоты и моноглицериды, происходящие из длинноцепочечных триглицеридов, повторно этерифицируют в триглицеридов, повторно этерифицируют в триглицериды, которые взаимодействуют с холестерином и фосфолипидами (причем оба этих вида могут абсорбироваться или вновь синтезироваться). Полученный шарик покрывается белковой оболочкой (преобладающие, апопротеином А), что приводит к образованию хиломикронов. Сложный эфир холестерина, или холестерин, как полагают, действует в качестве матрикса или основания для других небелковых компонентов хиломикронов. Хиломикрон обходит печень и секретирует через лимфатические сосуды в грудной проток для системы кровообращения.
Хиломикроны могут преципитировать из человеческой, свиной или бычьей сыворотки с виниловыми полимерами, например поливинилпирролидоном (ПВП), экстрагируемыми из жидкости лимфы в грудном потоке или получаемыми синтетическим путем. Например, при получении из свежей человеческой, свиной или бычьей сыворотки к каждым 10 мл свежей сыворотки прибавляют по крайней мере 1,25 г NaCl и 2,5 мл ПВП, после чего смесь центрифугируют при 2500 об/мин в течение 30 мин. Полученный супернатант содержит ПВП - хиломикронный комплекс.
Альтернативно, хиломикроны могут быть получены из анестезированных, голодных свиней путем помещения трахеальных и желудочных трубок под общую анестезию и канюлирования грудного протока полиэтиленовым катетером. Около 250 г разрушенной эмульсии нагнетают в желудок животного через желудочную трубку каждые три часа и лимфатическую жидкость собирают в лабораторный стакан при температуре 4oC при одновременном вливании физиологического раствора через канюлированную вену. После сбора лимфатической жидкости в течение 12 - 18 ч из канюлированного грудного протока собранную лимфатическую жидкость разбавляют двумя объемами 0,9%-ного раствора NaCl и центрифугируют при 25000 g в течение 3 ч при температуре 4oC. К супернатантному раствору хиломикронов прибавляют половину первоначально разбавленного объема 0,9%-ного раствора NaCl и сохраняют в холоде (4oC) вплоть до использования (модификация работы Сагами и др. , Белок, Нуклеиновые Кислоты, Ферменты (Япония), 10, с. 443, 1965).
В качестве альтернативы хиломикронам гидрофобная фаза может содержать вещество, которое образует хиломикроны в кишечной слизистой оболочке. Такое вещество содержит холестерин или иной материал, который образуют матрикс хиломикронов, лецитин или любой другой пригодный фосфолипид, а также липофильное поверхностно-активное вещество, такое как длинноцепочечная жирная кислота (например, C16-C24-насыщенная или ненасыщенная), по выбору этерифицированная как сложный эфир глицерина, который может быть моно-, ди- или триглицеридом.
В качестве необязательного дополнительного компонента может присутствовать пригодный сложный эфир холестерина (например, образованный из длинноцепочечной жирной кислоты). В качестве альтернативы лецитину (который представляет собой тривиальное наименование для фосфатидилхолина) могут быть использованы другие фосфатидиламинокислоты, такие как фосфатидилэтаноламин (цефалин), фосфатидилсерин или фосфатидил-инозитол. Другими пригодными вариантами могут быть производные фосфатидилглицеринов, такие как сам фосфатидилглицерин, 3'-O-лизилфосфатидилглицерин и дифосфатидилглицерин (кардиолипин). И, конечно, могут быть использованы смеси фосфолипидов. В качестве липофильного поверхностно-активного вещества (сурфактанта) предпочитают использовать жирную кислоту или кислоты, необязательно этерифицированные с образованием глицеридов, причем они будут предпочтительно C18-C24 -насыщенными или ненасыщенными кислотами, такими как олеиновая кислота, линолевая кислота, линоленовая кислота или какая-либо другая пригодная кислота. Хотя апопротеины можно добавлять в хиломикронообразующее вещество, их присутствие не является обязательным. Хиломикроны могут быть образованы in vivo, даже если апопротеины не добавляют в хиломикронообразующее вещество; хотя заявитель не желает быть связанным этой теорией, все же весьма вероятно, что апопротеины либо уже имеются в наличии, либо синтезированы de novo для использования в том случае, если присутствуют хиломикронобразующие вещества.
Можно без труда определить экспериментированием, имеет ли композиция в соответствии с настоящим изобретением для своей гидрофобной фазы вещество, способное образовывать хиломикроны в кишечной слизистой оболочке после введения. Определение можно осуществить путем вливания испытуемой композиции в двенадцатиперстную кишку свиньи и регистрации уровней инсулина (или другого биологически активного вещества) в лимфатической жидкости, печеночной воротной крови и периферической венозной крови. Значительное повышение уровня содержания инсулина в лимфатической жидкости, а не в печеночной воротной крови, подтверждает то, что активное вещество абсорбирует через лимфатическую систему, а не через воротную вену. Уровень содержания инсулина в жидкости лимфы может быть в два раза, в пять раз, в десять раз, в пятьдесят раз или даже в сто раз выше, чем в печеночной воротной крови. Подробный протокол такого определения представлен в примерах, и ему можно следовать в точности или с подходящими модификациями.
Гидрофобная фаза, которая способна
образовывать хиломикроны в кишечной слизистой оболочке, как обсуждено выше,
содержит в качестве своих минимально-существенных ингредиентов:
холестерин или любое другое вещество, которое
образует матрикс хиломикронов;
лецитин или любой другой пригодный
фосфолипид;
липофильное поверхностно-активное вещество.
В принципе существуют три пути, по которым вещества могут абсорбировать через кишечную мембрану. Малые гидрофильные, водорастворимые химические вещества, такие как сахар, как известно, абсорбируют через "систему пор" кишечной мембраны, проникают в капиллярное кровообращение и затем в печеночную воротную вену человека. Липиды и липофильные вещества, с другой стороны, как известно, абсорбируют посредством двух совершенно различных механизмов. Те жирные кислоты, которые имеют относительно короткие углеродные цепи (например, C2-C6 или C8-кислоты, такие как капроновая и каприловая кислоты), абсорбируют через кишечную мембрану с ферментативной и физиохимической "помощью" от солей желчных кислот и панкреатической липазы. В конечном счете такие абсорбированные короткоцепочечные жирные кислоты дренируют в капиллярную кровь и проходят в печеночную воротную вену. Липиды и жирные кислоты, имеющие относительно длинные цепи, например олеиновая кислота и ди-олеат- и три-олеат-глицериды, а также холестерин и фосфолипиды, среди других соединений, образующих хиломикроны в пределах мембраны, абсорбируют через кишечную мембрану при помощи механизмов, которые еще не поняты до конца. В кишечной мембране они участвуют в образовании хиломикронов и затем "всасываются" в ворсинки кишечной системы, дренируют в жидкость лимфы, собираются в грудном протоке и в конце концов высвобождаются в большой круг кровообращения.
Широкие и предпочтительные процентные составы (которые обычно упоминаются как соотношения масс, однако могут быть соотношениями массы/объема или даже соотношениями объемов) хиломикронообразующего вещества для общих целей приведены в табл. 1, при условии всегда, что общее количество не должно превышать 100%.
Определено, что в пределах этих широких и предпочтительных соотношений гидрофобная фаза может иметь некоторые предпочтительные композиционные характеристики для определенных биологически активных веществ. Например, для инсулина (и также для интерферонов, таких как человеческие интерфероны бета и гамма) предпочтительными являются приведенные в табл. 2 более узкие пропорции (на той же основе и при тех же условиях).
Для лососевого кальцитонина (а также для этитропоэтина) предпочтительными являются приведенные в табл. 3 пропорции (на той же основе и при таких же условиях).
Для свиного соматотропина (а также для тканевого активатора плазминогена и Фактора VIII) предпочтительными являются приведенные в табл. 4 пропорции.
Может присутствовать некоторое количество органического растворителя, смешивающегося с гидрофобной фазой, и вновь для того, чтобы содействовать при составлении рецептуры. Природа растворителя зависит от присутствия другого вещества. Часто пригоден в качестве растворителя этанол. Количество растворителя может составлять, например, от 5 до 50 об.% на основе объема масляной фазы.
Для образования микроэмульсий иногда необходимо использовать два различных поверхностно-активных вещества, одно из которых является гидрофильным и имеет высокий гидрофильно-липофильный баланс (ГЛБ) и другое является более липофильным (как описано выше) и имеет низкий ГЛБ. Величина ГЛБ представляет собой относительное содержание гидрофильной группы поверхностно-активного вещества, выраженная как ее массовый процент молекулы поверхностно-активного вещества, деленный на пять. Полностью гидрофильная молекула, такая как полиэтиленгликоль, имеет поэтому теоретическое максимальное значение ГЛБ, равное 20.
Гидрофильные поверхностно-активные вещества, пригодные в практике настоящего изобретения, когда они присутствуют в композиции, имеют очень высокое значение ГЛБ, по крайней мере 17 и, возможно, 20. Липофильные поверхностно-активные вещества, используемые в настоящем изобретении, имеют низкий ГЛБ, например менее чем 10. Предпочтительно липофильное поверхностно-активное вещество имеет значение ГЛБ менее чем 7 или даже менее чем 4.
Предпочтительно, если каждый из сурфактантов, используемых при получении композиций настоящего изобретения, выбирают из тех поверхностно-активных веществ, которые классифицируются как анионогенные или неионогенные. Эти поверхностно-активные вещества особенно пригодны в фармацевтических системах по причине их совместимости, стабильности и нетоксичности. Поверхностно-активные вещества, как правило, пригодные для различных целей в соответствии с настоящим изобретением, включают длинноцепочечные (C16-C24) жирные кислоты, такие как, например, пальмитиновая кислота, стеариновая кислота и олеиновая кислота, сложные эфиры длинноцепочечных (C16-C24) жирных кислот, например пальмитат натрия, стеарат натрия и олеат натрия, натрийлаурилсульфат, полиэтиленликоль, полиэтиленгликолевые алкиловые эфиры, сложные жирнокислотные эфиры полиэтиленгликоля, например полиэтиленгликовый моно- или дистеарат, пропиленгликоль, сложные жирнокислотные эфиры пропиленгликоля, например моностеарат пропиленгликоля, глицерин, моно- или полиглицериды жирных кислот, такие как глицерилмоностеарат, сложные жирнокислотные эфиры полиоксиэтилена, простые эфиры данного соединения, а также его амины, например полиоксиэтилен-моно- и дистеарат, а также лауриловый эфир полиоксиэтилена, сложные полиоксиэтиленсорбитановые эфиры, например полиоксиэтиленсорбитанмонолаурат, монопальмитат, моностеарат или моноолеат, полиоксиэтиленовые алкилфенолы и алкил-фениловые эфиры, полиоксиэтиленовое касторовое масло, сложные сорбитановые жирнокислотные эфиры, полисорбаты, стеариламин, триэтаноламинолеат, растительные масла, например кунжутное масло или маисовое масло, холестерин, а также трагакант.
Поверхностно-активные вещества, подлежащие отбору, конечно, те, которые в настоящее время коммерчески доступны для фармацевтического применения и имеют приблизительно низкие значения ЛД50. Ниже приведен перечень определенных примерных поверхностно-активных веществ вместе с их значениями ГЛБ и там, где известно, ЛД50-значениями.
Примеры пригодных поверхностно-активных веществ с высоким ГЛБ приведены в табл. 5.
Примеры пригодных поверхностно-активных веществ с низким ГЛБ приведены в табл. 6.
Следует отметить, что в соответствии с настоящим изобретением вместо одиночных поверхностно-активных веществ часто используют смеси поверхностно-активных веществ. Например, вместо одиночного гидрофильного поверхностно-активного вещества может быть использована смесь двух или более относительно гидрофильных поверхностно-активных веществ, причем эффективный ГЛБ смеси должен, однако, быть выше, чем 17. Под термином "эффективный ГЛБ" подразумевают то, что гидрофильно-липофильный баланс смеси поверхностно-активных веществ должен быть эквивалентным балансу одиночного поверхностно-активного вещества, имеющего ГЛБ выше, чем 17. Аналогично, смеси липофильных поверхностно-активных веществ могут быть использованы вместо одиночного липофильного поверхностно-активного вещества. И вновь эффективный ГЛБ смеси липофильных поверхностно-активных веществ должен быть меньше 10.
Выбор количества поверхностно-активного вещества для использования в фармацевтических композициях настоящего изобретения является прерогативой специалиста в данной области техники. Точные количества, которые будут оптимальными в каждом случае, зависят от точной природы используемых поверхностно-активных веществ и других компонентов фармацевтических композиций настоящего изобретения. Тем не менее, в качестве основного руководства следует отметить, что количество гидрофильного поверхностно-активного вещества, как правило, составляет диапазон (на основе общего объема композиции) от 0,1 до 50 г на литр, предпочтительно от 0,5 до 25 г на литр и оптимально от 1 г на 10 г на литр. Липофильные поверхностно-активные вещества обсуждены выше касательно масляной фазы микроэмульсии. Они присутствуют в композиции в количестве от 0,1 г до 100 г на литр, предпочтительно от 0,5 г до 50 г на литр и оптимально от 2 г до 25 г на литр, при этом указанные значения приведены на основе общего объема фармацевтической композиции.
Несмотря на то, что присутствие любых других ингредиентов в композиции настоящего изобретения не является существенным, на практике обычно весьма удобно добавлять другие ингредиенты. Одним таким компонентом, который часто является очень желательным, может быть ингибитор протеазы, который может присутствовать в форме одного или более отдельных ингибиторов протеазы. Ингибиторы протеазы, пригодные в соответствии с настоящим изобретением, в широком смысле могут быть разделены на две категории. Первая категория представляет собой ингибиторы протеазы, которые пригодны для ограничения или предотвращения деградации биологически активного вещества, если оно является белковым. Такие ингибиторы протеазы должны иметь эффект ингибирования протеолитических ферментов, обнаруживаемых в желудочно-кишечном тракте, таких как трипсин, химотрипсин и карбоксипептидаза. В случае инсулина ингибиторы протеазы, как правило, являются факторами торможения такого класса ферментов, известного как инсулиназа, которая включает фермент транс-сульфатаза. Пригодные источники ингибиторов трипсина могут быть экстрагированы из сои культурной или яичного белка (ovomucoid). Во-вторых, если в фармацевтических композициях в соответствии с настоящим изобретением присутствует апопротеин, желательно прибавлять ингибиторы протеазы с тем, чтобы снизить величину деградации апопротеина перед тем, как он достигнет кишечную слизистую оболочку. Вообще говоря, аналогичные ингибиторы протеазы могут быть использованы для защиты белковых биологически активных веществ, и поэтому единственный ингибитор протеазы может выполнять обе функции. Выбор количества ингибитора протеазы зависит от квалификации специалиста в данной области техники, однако обычно данное количество составляет вплоть до 0,1% в отношении массы к объему или даже 0,5% в отношении массы к объему.
Другим необязательным ингредиентом является стабилизатор для биологически активного вещества. Точная природа стабилизатора, если такой присутствует, конечно, будет зависеть от природы самого биологически активного вещества. Например, есть целый ряд хорошо определенных стабилизаторов для инсулина, которые вводятся в инсулинсодержащие фармацевтические составы в соответствии с настоящим изобретением. Примеры включают гидроксипропилцеллюлозу (ГПЦ), кальциевые соли и цитратные соли. Кальций известен не только как стабилизатор инсулина, но также как вещество, имеющее дополнительное благоприятное воздействие по повышению пористости клеточных мембран, что способствует вхождению активного вещества в клетки кишечных стенок. Количество присутствующего стабилизатора вновь будет зависеть от его природы, а также от природы биологически активного вещества; выбор количества находится в пределах компетентности специалиста в данной области техники, однако часто составляет значение около 1 или 2% в отношении массы к объему.
Хотя композиции в соответствии с настоящим изобретением представляют собой микроэмульсии, как определено в описании данного изобретения, в некоторых случаях желательно вводить эмульгирующие добавки, которые могут быть традиционными эмульгирующими добавками, используемыми при получении макроэмульсий. Некоторые эмульгирующие добавки представляют собой поверхностно-активные вещества, и поверхностно-активные вещества, пригодные для этой цели, не ограничиваются никакими определенными значениями ГЛБ. Пригодные эмульгирующие добавки включают холестерин, стеариновую кислоту, стеарат натрия, пальмитиновую кислоту, пальмитат натрия, олеиновую кислоту, олеат натрия, глицерилмоностеарат, полиоксиэтилен 50 стеарат, полиоксиэтилен 40 стеарат, полисорбат 20, полисорбат 40, полисорбат 60, полисорбат 80, пропиленгликоль диацетат и пропиленгликоль моностеарат.
Количество эмульгирующей добавки, если таковая присутствует, составляет значение, достаточное для того, чтобы содействовать при получении устойчивой микроэмульсии. Точное количество может быть определено специалистом в данной области; как правило, эмульгирующие добавки используют в количестве от 0 до 10% в отношении массы к объему общей композиции.
По желанию в композицию в соответствии с настоящим изобретением можно добавлять один или более стабилизаторов и/или пластификаторов с целью придания большей устойчивости при хранении. Как упомянуто выше, микроэмульсии не имеет склонность к разделению при хранении в нормальных условиях, однако более высокая степень устойчивости может быть полезна при определенных обстоятельствах. Вещества, пригодные в качестве стабилизаторов и/или пластификаторов, включают декстрин, акацию, карбоксиполиметилен и коллоидную гидроокись алюминия. При добавлении стабилизаторов/пластификаторов их количества составляют вплоть до 10% в отношении массы к объему, предпочтительно около 0,5 - 6,5% в отношении массы к объему всего препарата.
Фармацевтические композиции в соответствии с настоящим изобретением могут содержать различные консерванты. Антиоксиданты (антиокислители) и противомикробные средства представляют собой две особенно пригодные категории консервантов. Антиоксиданты чрезвычайно полезны, поскольку хиломикроны и вещество, способное образовывать хиломикроны (включая апопротеины), подвергаются разрушению при самоокислении. Хотя данную проблему можно обойти путем получения композиций в соответствии с настоящим изобретением в инертной атмосфере, такой как азот, это в некоторой степени неудобно и дорого, и поэтому часто предпочитают добавлять антиоксиданты. Пригодные фармацевтически приемлемые антиоксиданты включают пропилгаллат, бутилированный гидроксианизол, бутилированный гидрокситолуол, аскорбиновую кислоту или натрий-аскорбат, DL- или D-альфа-токоферол и DL- или D-альфа-токоферилацетат. Антиоксидант можно прибавлять в композиции в соответствии с настоящим изобретением в количестве, например, от 0,1% в отношении массы к объему, предпочтительно от 0,0001 до 0, 3%.
Кунжутное масло, предпочтительно в виде рафинированного химического масла, может быть прибавлено в композиции настоящего изобретения, поскольку оно имеет активность антиоксиданта. Кунжутное масло имеет дополнительное преимущество, заключающееся в том, что оно придает приятный вкус фармацевтическим композициям (особенно для пациентов из восточных стран), тем самым улучшая соблюдение больными режима и схемы лечения. Кунжутное масло может присутствовать в количестве от 0,1 до 3% в отношении массы к объему, предпочтительно от 5 до 20% в отношении массы к объему конечной жидкой композиции. Могут присутствовать и другие вещества, повышающие вкусовые качества композиций.
Фармацевтические композиции в соответствии с настоящим изобретением могут быть получены и сохранены в стерильных условиях, которые препятствуют микробному заражению. Однако эта методика является расточительной для перорально принимаемого препарата, и было бы более приемлемо включать в композиции противомикробный консервант. Противомикробные средства, используемые, как правило, в количествах от 3% в отношении массы к объему, предпочтительно от 0,5 до 2,5%, приблизительно включают метилпарабен, этилпарабен, пропилпарабен, бутилпарабен, фенол, дегидроуксусную кислоту, фенилэтиловый спирт, бензоат натрия, сорбиновую кислоту, тимол, тимерозал, дегидроацетат натрия, бензиловый спирт, крезол, n-хлоро-m-крезол, хлорбутанол, фенилртутьацетат, фенилртутьборат, фенилртутьнитрат и бензилалконийхлорид.
Вследствие имманентной термодинамической устойчивости микроэмульсий жидкие фармацевтические композиции в соответствии с настоящим изобретением могут быть получены путем простого смешивания водной и масляной фаз, которые, в свою очередь, могут быть получены смешиванием их соответствующих ингредиентов.
В соответствии с вторым вариантом настоящего изобретения предлагается способ получения перорально принимаемой фармацевтической композиции в соответствии с первым вариантом настоящего изобретения, при этом данный способ предусматривает смешивание ингредиентов композиции.
Кинетические соображения предполагают, однако, что для обеспечения быстрого и эффективного образования микроэмульсионных композиций в соответствии с настоящим изобретением следует предпринять определенные операции. В частности, во время или после добавления гидрофильной и гидрофобной фаз микроэмульсию можно быстро образовать с использованием гомогенизатора, такого как АВТОГОМОМИКСЕР (слово АТВОГОМОМИКСЕР является товарным знаком фирмы Токушу Кика, Токио). Может быть полезным дополнительное или альтернативное использование микрофлюидизатора.
Вообще, предпочтительно добавлять по крайней мере некоторые (или по крайней мере один) компоненты гидрофильной фазы по крайней мере к некоторым (или по крайней мере одному, однако предпочтительно всем) компонентам гидрофобной фазы при осуществлении быстрого смешивания; и соответствующим образом могут добавляться остальные компоненты.
Один предпочтительный способ получения фармацевтических
композиций настоящего изобретения, содержащих и гидрофильное (высокий ГЛБ), липофильное (низкий ГЛБ) поверхностно-активные вещества, заключается в том, что
(а) осуществляют быстрое
смешивание
биологически активного вещества в пригодном водном растворителе с гидрофобной фазой, которая содержит поверхностно-активное вещество с низким ГЛБ;
(б) прибавляют
поверхностно-активное
вещество с высоким ГЛБ при дальнейшем быстром перемешивании;
(в) необязательно покрывают твердый носитель полученной таким образом композицией.
Ингибитор протеазы может быть прибавлен к биологически активному веществу перед смешиванием с гидрофобной фазой. Антиоксидант может быть прибавлен перед осуществлением быстрого смешивания на стадии (а). Стабилизатор для биологически активного вещества может быть прибавлен в одно время с поверхностно-активным веществом с высоким ГЛБ, так же, как и дополнительный или альтернативный ферментный ингибитор (ингибиторы).
Следует понять, что поскольку композиции в соответствии с настоящим изобретением представляют собой микроэмульсии, они, вероятно, являются жидкими. Однако жидкие композиции в некоторых случаях могут быть менее пригодными, нежели твердые композиции, и поэтому существует целый ряд способов, в соответствии с которыми композиции предлагаемого изобретения можно изготовить или превратить в твердые составы. Одним способом получения твердого состава является просто выбор подходящих ингредиентов так, чтобы при температурах хранения составы в соответствии с настоящим изобретением были твердыми. Такие препараты, как правило, реверсируют в их жидкое состояние при физиологических температурах и, следовательно, ведут себя как жидкости вскоре после их перорального введения. Однако данный подход может и не быть удобным или вполне пригодным во многих случаях. Поэтому, если необходим твердый состав, обычно предпочтительно наносить жидкий состав в соответствии с настоящим изобретением на твердый носитель, который может быть в форме гранул или частиц (корпускул). Следует отметить, что корпускулы можно компаундировать в гранулы после покрытия оболочкой. Жидкая композиция может адсорбировать или абсорбировать в носитель. Сам носитель для определенных (особенно человеческих) применений предпочтительно будет физиологически неабсорбируемым и поэтому будет экскрецировать как фекальное вещество после прохождения через желудочно-кишечный тракт. В качестве носителя особенно пригодно использовать вещество, которое набухает в желудочно-кишечном тракте (в частности, в тонкой кишке), например, до размера от 10 до 200 своих объемов. Особенно предпочтительным является быстро расширяющееся вещество, и такие вещества включают кальций карбоксиметилцеллюлозу или гидроксипропилцеллюлозу, альгинат натрия, желатину, поливинилпирролидон с поперечными связями, "эруптивный" рис и полистирол.
Особенно пригодный твердый носитель, включающий быстро расширяющийся материал, содержит кальцийкарбоксиметилцеллюлозу (например, от 20 до 60 мас.%, предпочтительно от 35 до 45 мас.%), альгиновую кислоту или альгинат натрия (например, от 5 до 25 мас.%, предпочтительно от 10 до 20 мас.%), желатину (например, от 2 до 20 мас.%, предпочтительно от 5 до 15 мас.%), гидроксипропилцеллюлозу (например, от 20 до 60 мас.%, предпочтительно от 30 до 40 мас. %) и лаурилсульфат натрия или иной подходящий сурфактант (например, от 0,1 до 20 мас. %, предпочтительно от 1 до 10 мас.%). Когда данные ингредиенты являются единственными, что предпочтительно, процентные содержания составляют 100%.
При использовании для ветеринарных целей носитель может приниматься внутрь и может содержать полезный диетический компонент (например, белок, углевод, жир или минерал) для животного, подлежащего лечению. В данном случае предпочтительными являются белковые носители, причем особенно пригоден порошок сои культурной, поскольку композиция может быть добавлена в пищу для животных (например, свиней).
Жидкую композицию можно наносить на носитель множеством различных способов, и многие из них хорошо известны в данной области знаний. Особенно пригодным способом является, например, нанесение покрытия распылением в псевдоожиженном слое. Носитель предпочтительно покрывают в размере от 50 до 500% его массы жидким составом.
Следует соблюдать осторожность при нанесении покрытия на носитель распылением жидкого состава микроэмульсии в соответствии с настоящим изобретением, как описано выше. По причине природы общих компонентов в гидрофобной фазе (холестерин или иной матрикс, лецитин или иной фосфолипид и липофильное поверхностно-активное вещество) температура жидкой композиции или частиц в слое флюидизатора не должна подниматься очень высоко, иначе масляная фаза может стать слишком легкосыпучей. Наоборот, если температура падает слишком низко, композиция становится очень вязкой для того, чтобы ее можно было распылять в псевдоожиженный слой. Кроме того, следует соблюдать осторожность с тем, чтобы покрытые частицы носителя не смогли спекаться чрезмерно в флюидизаторе.
Оптимальные результаты могут быть достигнуты при покрытии частиц носителя с использованием следующих методов, которые сами по себе составляют часть настоящего изобретения. В соответствии с одним вариантом настоящего изобретения предлагается способ нанесения покрытия на частицы носителя жидкостью, содержащей гидрофобное вещество, в котором частицы носителя псевдоожижают в слое флюидизатора, распыляют жидкость на псевдоожиженные частицы, нагревают псевдоожижающий газ (который обычно представляют собой воздух), когда температура в псевдоожиженном слое является очень низкой, и охлаждают псевдоожижающий газ, когда температура в псевдоожиженном слое является очень высокой.
В соответствии с другим вариантом настоящего изобретения температуру в псевдоожиженном слое можно поддерживать в приемлемых интервалах. Какие это интервалы и каковы границы этих интервалов, по-видимому, определит природа масла в распыляемой жидкости, а также природа частиц носителя и любых других компонентов жидкости и, кроме того, другие параметры. При распылении композиций в соответствии с первым вариантом температуру следует поддерживать на уровне 9± 5oC, предпочтительно ± 2oC, с тем, чтобы получить наилучшие результаты. Настоящее изобретение также относится к устройству для осуществления данного аспекта изобретения.
В соответствии с другим вариантом настоящего изобретения предлагается способ покрытия частиц носителя жидкостью, содержащей масло, в котором псевдоожижают частицы носителя в флюидизаторе и распыляют жидкость в псевдоожиженные частицы, причем распыление осуществляют периодически.
Временные интервалы между распылением могут быть более длительными, нежели продолжительность периодов самого распыления. Периоды распыления могут составлять от 1 до 20 с, предпочтительно от 2 до 15 с и, как правило, от 5 до 10 с. Интервалы между периодами распыления могут составлять от 5 с до 40 с, предпочтительно от 10 с до 30 с и оптимально от 15 с до 20 с.
Особенно предпочтительно объединять данный признак периодического распыления с упомянутым выше признаком стабилизированной температуры. Другие предпочтительные признаки данного способа, отличающиеся от общепринятого процесса распылительной сушки, включают нерегулярную (например, каждый 1 - 10 с) пульсацию внутренней части камеры устройства с псевдоожиженным слоем псевдоожижающим газом для того, чтобы удалить частицы, которые могут сцепиться со стенками камеры и/или любыми фильтрами, которые могут быть использованы, осушение (обезвоживание) псевдоожижающего газа (например, воздуха), фильтрацию псевдоожижающего газа по крайней мере частично, для того, чтобы удалить масло, или микробы, или то и другое, и/или создание вращающихся средств дробления комков, которые вращаются вокруг оси в основном под прямым углом к направлению подачи псевдоожижающего газа, предпочтительно без вращающейся мешалки с механическим приводом, которая вращается параллельно направлению подачи псевдоожижающего газа. Настоящее изобретение также относится к устройству для осуществления данного способа.
В общем, следует отметить, что содержание воды в гидрофильной фазе может быть уменьшено или потеряно, когда твердые частицы носителя покрывают распылением. Это не уводит полученную композицию от объема настоящего изобретения. Композицию можно соответствующим образом повторно гидратировать при введении.
Твердые композиции в соответствии с настоящим изобретением могут содержать фармацевтически приемлемые наполнители и/или связующие в подходящих количествах. Пригодные наполнители включают лактозу, маннит, сульфат кальция, дикальцийфосфат, трикальцийфосфат и микрокристаллическую целлюлозу. Пригодными связующими являются акация, трагакант, желатина, альгинат натрия, аммоний-кальций-альгинат, метилцеллюлоза, натрий карбоксиметилцеллюлоза, гидроксипропилметилцеллюлоза, метилгидроксипропилцеллюлоза, желатина, сложные полиэтиленгликолевые эфиры жирных кислот, поливинилпирролидон, алюминийсиликат магния и полиакриламиды.
Значительные количества, если не полные, биологически активного вещества в твердых или жидких композициях настоящего изобретения остаются в сохранности при прохождении через гидролитическую и протеолитическую среду желудка. С целью дополнительной защиты можно составлять твердые или жидкие композиции в соответствии с настоящим изобретением в энтеросолюбильное покрытие таблетки или по-иному защищенную форму. В случае использования жидких композиций они могут быть смещены или просто совместно введены с защитным веществом, таким как жидкая смесь триглицеридов с цепями средней длины, или они могут быть вставлены в энтеросолюбильные капсулы (например, выполненные из мягкой или твердой желатины, которые сами по себе необязательно покрыты энтеросолюбильной оболочкой), тогда как твердые композиции можно обрабатывать более гибко: их можно либо покрывать энтеросолюбильным материалом с образованием таблеток, либо они могут быть введены в энтеросолюбильную капсулы. Толщина энтеросолюбильного покрытия на таблетках или капсулах может быть, например, от 5 до 4 мкм, хотя точную толщину покрытия сможет определить специалист в области приготовления лекарственных средств. Гранулы с энтеросолюбильной оболочкой (размер частиц которых может быть, например, от 5 до 2 мм) могут быть сами покрыты без того, чтобы быть компаундированными в таблетку для покрытия. Аналогично, микрокапсулы могут быть покрыты энтеросолюбильной оболочкой. Энтеросолюбильное покрытие может содержать любое из энтеросолюбильных веществ, традиционно используемых в технологии приготовления перорально вводимых фармацевтических препаратов. Пригодные материалы для изготовления энтеросолюбильных покрытий известны, например, из "Renungton's Pharmaceutical Sciences", 15-е издание, с. 1614 - 1615 (1975); 2-е издание, с. 116 - 117, 371 - 374 (1976); Hagers Horndbuch der Pharmazeutikhen Praxie 4-е издание, том 7а (Springer Verlag 1971), стр. 739 - 742 и 776 - 778.
Пример пригодных материалов для энтеросолюбильных покрытий включают ацетилфталат целлюлозы, гидроксипропилметилцеллюлозафталат (ГПМЦ-Ф), бензофенилсалицилат, ацетосукцинат целлюлозы, сополимеры стирола и малеиновой кислоты, желатину, каратин, стеариновую кислоту, миристинивую кислоту, полиэтиленгликоль, щеллак, клейковину, акрилатную и метакрилатную смолу и сополимеры производных малеиновой кислоты и фталевой кислоты. Материалы для энтеросолюбильных покрытий могут быть растворены в растворителях, таких как дихлорметан, этанол и вода, целлюлозафталат или поливинилацетатфталат. Предпочтительно использовать ГПМЦ-Ф, полиэтиленгликоль 6000 или щеллак в качестве энтеросолюбильного покрытия. Запатентованный препарат ГПМЦ-Ф, служащий при растворении или диссипации при pH 5,5, поставляется на рынок под товарным знаком НР5-5 и является особенно предпочтительным материалом.
Чрезвычайно удобным путем введения фармацевтических композиций в соответствии с настоящим изобретением является использование твердых желатиновых капсул с энтеросолюбильным покрытием. Хотя при покрытии твердых желатиновых капсул оболочкой из определенных энтеросолюбильных материалов нет значительных проблем, существует трудность в покрытии таких капсул предпочтительным материалом на основе ГПМЦ-Ф. Трудность заключается в том, что ГПМЦ-Ф обычно наносят в машине для нанесения покрытий со смазыванием форм из раствора хлористого метилена и данный раствор имеет тенденцию разрушать твердые желатиновые капсулы.
В соответствии с другим вариантом осуществления настоящего изобретения предлагается способ получения желатиновой капсулы с энтеросолюбильным покрытием, в котором капсулу покрывают материалом, способным защищать желатину капсулы от пагубных воздействий хлористого метилена, и затем покрывают защищенную таким образом капсулу гидроксипропилметилцеллюлозавталатом (ГПМЦ-Ф) при помощи раствора ГПМЦ-Ф в хлористом метилене.
Посредством защитного "подслоя" капсулу защищают таким образом от воздействия растворителя для оптимального покровного средства.
Пригодные защитные подслои включают ПВП-Ф, ГПМЦ (гидроксипропилметилцеллюлоза), AYICEL (кристаллическая целлюлоза) и ГПЦ (гидроксипропилцеллюлоза), причем ГПЦ не столь предпочтительна, как остальные вещества, и она не имеет такую хорошую пленкообразующую способность, как другие материалы покрытия. Могут быть также использованы любые другие защитные подслои, которые можно наносить таким методом, который не причиняет вреда желатиновой капсуле; пригодные методы нанесения покрытия включают осаждение из раствора (например, 5%-ного в отношении массы к объему) в растворителе (таком, как этанол), который не оказывает существенного пагубного воздействия на желатину в применяемых условиях. Можно увеличить диапазон пригодных растворителей путем снижения температуры операции нанесения покрытия (например, во вращающемся барабане для нанесения покрытий) от традиционной температуры 80oC и более низкого уровня, например 50oC или менее, 40oC или менее, предпочтительно около 35oC, относительно этанола.
Также могут быть использованы смеси материалов "подслоя". Особенно предпочтительной является смесь ПВП и ГПМЦ. Массовое отношение поливинилпирролидона (например, ПВП-Ф) к гидроксипропилметилцеллюлозе (ГПМЦ) может составлять от 0,1: 1 до 20:1, предпочтительно от 0,2:1 до 5:1 и оптимально около 0,5:1 на массовой основе. Нанесение покрытия можно осуществлять с использованием от 1 до 10% (в отношении массы от общей массы капсулы) ПВП-Ф и от 2 до 20% (на той же самой основе) ГПМЦ, причем предпочтительным являются количества 5 и 10% соответственно.
ГПМЦ-Ф затем можно наносить из раствора хлористого метилена (например, около 5% в отношении массы к объему), поскольку он является традиционно используемым. Данная операция, как и нанесение нижнего слоя, может происходить во вращающемся барабане для нанесения покрытий, предпочтительно при пониженной температуре. ГМПЦ-Ф предпочтительно представляет собой НР5-5 и может наноситься в количестве 5 - 40%, предпочтительно 15 - 25% и оптимально, около 20% в массовом отношении на основе массы капсул.
Фармацевтические композиции в соответствии с настоящим изобретением могут вводиться перорально, однако множеством различных путей. Преимуществом перорально вводимых композиций настоящего изобретения является то, что энтеросолюбильные покрытия обычно не являются необходимыми. Кроме того, высокие уровни сывороток указывают на то, что биологически активные вещества, вводимые в соответствии с настоящим изобретением, имеют высокую биологическую доступность. Помимо этого, физиологически существенные уровни сыворотки могут быть достигнуты очень быстро путем применения композиций в соответствии с настоящим изобретением.
Для ректального пути введения жидкие или твердые фармацевтические композиции могут быть введены как клизма или в форме суппозиториев. Основой суппозитория может быть какао-масло или другое пригодное вещество.
В соответствии с другим вариантом осуществления настоящего изобретения предлагается способ лечения человека или другого животного, в котором проводят пероральное или ректальное введение фармацевтической композиции в соответствии с первым вариантом настоящего изобретения. В частности, изобретение касается лечения диабета путем ректального или предпочтительно перорального введения фармацевтической композиции в соответствии с настоящим изобретением, причем биологически активным веществом данной композиции является инсулин.
Настоящее изобретение также касается использования ингредиентов композиций в соответствии с первым вариантом изобретения при получении для лечения или профилактики расстройств, излечимых или регулируемых биологически активным веществом.
В частности, инсулин может быть использован при получении фармацевтической композиции для лечения или регулирования диабета. Лососевый кальцитонин может быть использован при лечении высокого костного обмена (например, при болезни Педжета (деформирующий остеодистрофии кости), острой гиперкальциемии, ассоциируемой со злокачественностью и остеопорозом (разрежением кости). Свиной сомототропин можно вводить свиньям с тем, чтобы снизить период разведения свиней и, возможно, уменьшить толщину хребтового шпинга.
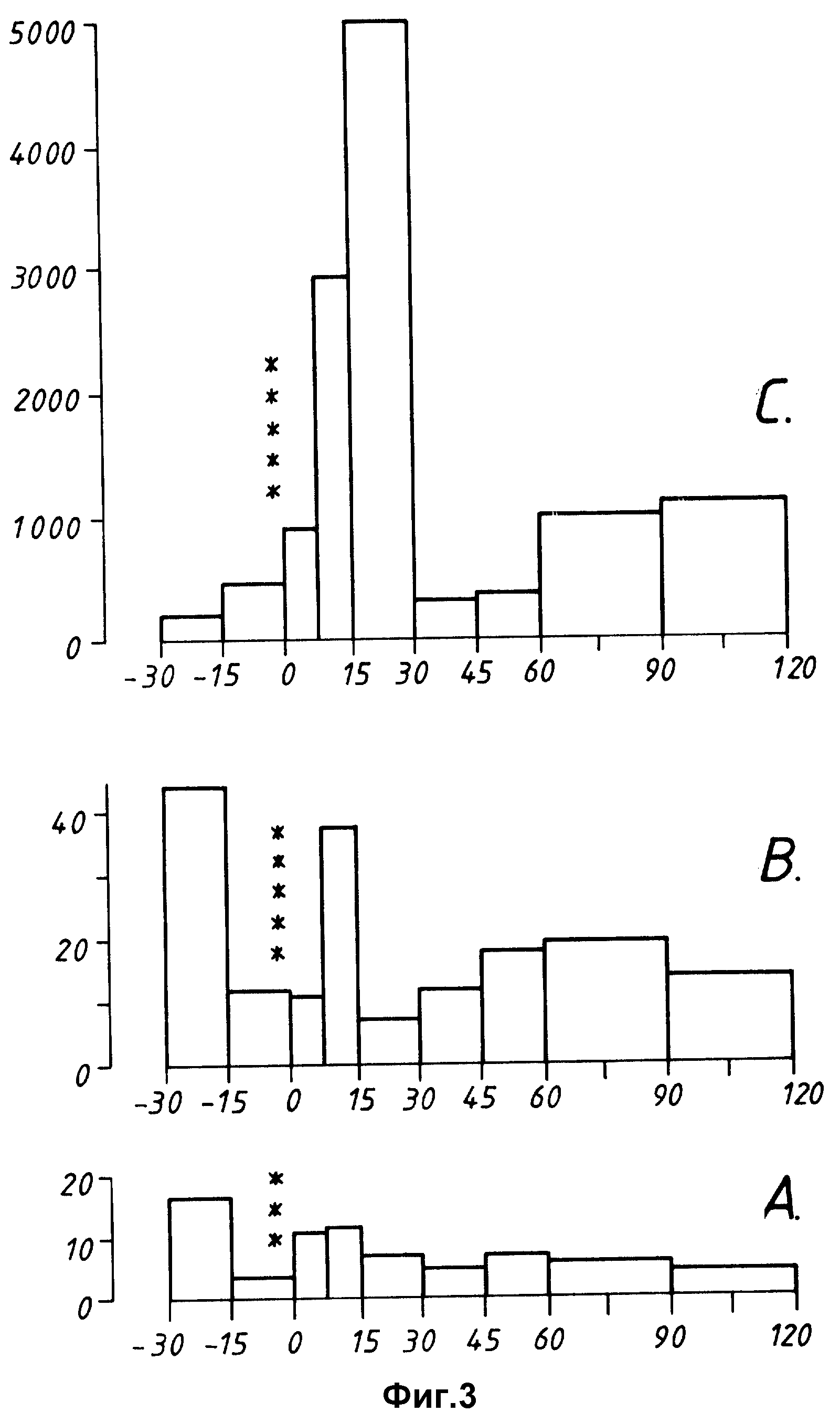
Теперь изобретение будет проиллюстрировано следующими неограничивающими примерами. Примеры относятся к предлагаемым чертежам, на которых фиг. 1 показывает частично вид в разрезе /частично схематический вид модифицированного устройства SPIP-A-FLOW, используемого в примере 8, фиг.2 представляет собой столбиковую диаграмму количества протекающей лимфы в зависимости от времени в биологическом примере 1, фиг.3A, 3B и 3C представляет собой столбиковые диаграммы уровней инсулина в периферической венозной крови, печеночной воротной крови и жидкости лимфы соответственно в зависимости от времени в биологическом примере 1.
Пример 1. Жидкую, перорально вводимую инсулинсодержащую композицию получают следующим образом.
Все химические вещества, используемые в данном и
других примерах, имеют
аналитическую и химическую чистоту. Вначале получают Под-Смесь A из следующих компонентов, г:
Липитин яичного желтка - 63,0
Глицерол моноолеат
(поверхностно-активное вещество с
низким ГЛБ) - 22,46
Холестерин - 30,00
Этанол (95%) - 100,00
путем нагревания этанола до температуры 75oC, прибавления
глицерола моноолеата, лецитина и
холестерина, перемешивания до тех пор, пока химические вещества растворятся, и охлаждения смеси до комнатной температуры (22oC).
Под-Смесь
антиоксиданта получают из
следующих компонентов:
Пропилгаллат - 37,5 г
Бутилированный гидроксианизол (БГА) - 25,0 г
Бутилированный гидрокситолуол (БГТ) - 37,5 г
Этанол (95%) - До 100,00
мл
путем растворения трех компонентов антиоксиданта в этаноле при комнатной температуре.
Под-Смесь B получают из следующих компонентов, г:
Олеиновая кислота
(эмульгатор) - 420
L-Альфа-токоферол (антиоксидант) - 30
Полисорбат 80 (эмульгатор) - 30
Под-смесь антиоксидант - 2,7
Аскорбиновая кислота
(антиоксидант) - 1,2
Пропилпарабен (противомикробное средство) - 1,2
Метилпарабен (противомикробное средство) - 6,8
Под-Смесь A - 300
Этанол (95%) - 750
путем смешивания данных
компонентов при комнатной температуре.
Под-Смесь C получают из следующих компонентов:
Инсулин (бычий, 24,6, международных единиц/мг, СР Фармасьютикалз,
Великобритания) - 2,5
г
Лимонная кислота (регулятор pH/ферментный ингибитор) - 2,6 г
Ингибитор протеиназы - 200000 международных единиц х 15
Этанол (95%) - До 300 мл
путем растворения
твердых компонентов в 100 мл этанола и прибавления остальной части этанола.
Под-Смесь D получают из следующих компонентов:
Полиоксиэтиловый (40)
стеарат (поверхностно-активное
вещество с высоким ГЛБ) - 6 г
Гидроксипропилцеллюлоза (стабилизатор) - 30 г
Бензоат натрия (противомикробное средство) - 6 г
Деионизированная
вода - До 400 мл
путем
растворения первых трех компонентов в воде при комнатной температуре.
При получении различных под-смесей инсулинсодержащие микроэмульсии типа вода в
масле получают из следующих
количеств под-смесей, мл:
Под-смесь B - 450
Под-смесь C - 150
Под-смесь D - 150
путем прибавления под-смеси C медленно к под-смеси D
при перемешивании с
использованием гомогенизатора АВТОГОМОМИКСЕР при 7500 об/мин при комнатной температуре 20oC. Полученную смесь медленно прибавляют к под-смеси B с использованием того же
миксера, при той же
самой температуре и скорости вращения. Полученную эмульсию пропускают последовательно пять раз через микрофлюидизатор (модель АРУ 15М8ВА) при следующих условиях:
Расход
воздуха: 2 дм3/мин
Давление воздуха: 5000 фунтов на кв.дюйм (35 Мн/м2)
Температура камеры охлаждения: 1,5oC.
Размер капелек полученной микроэмульсии составляет в среднем около 1 мкм.
Пример 2. Жидкую, перорально вводимую инсулинсодержащую фармацевтическую композицию получают следующим методом примера 1 со следующими изменениями.
1. Под-смесь A содержит 15 г холестерина вместо 30 г.
2. Под-смесь B содержит 200 г под-смеси A вместо 300 г и дополнительно содержит 150 г препарата ПВП - хиломикрон.
3. Под-смесь D содержит 6 г моностеарата полиэтиленгликоля в качестве поверхностно-активного вещества с высоким ГЛБ вместо полиоксиэтилен (40) стеарата.
Пример 3.
Твердую, перорально вводимую инсулинсодержащую фармацевтическую композицию получают следующим образом. Частицы носителя с твердым ядром получают смешиванием следующих
компонентов, г:
Кальцийкарбоксиметилцеллюлоза - 200
Альгиновая кислота - 75
Желатина - 50
Гидроксипропилцеллюлоза - 175
Лаурилсульфат натрия - 25
причем температура, при
которой осуществляют смешивание, составляет 22oC. Испытуемый образец показывает, что частицы набухают до 200 раз более своего первоначального объема при погружении
в воду при температуре
38oC.
Частицы ядра сушат в GL ATT (товарный знак), представляющем собой псевдоожиженный слой, при температуре 29oC в течение 45 мин. Затем 800 г частиц покрывают 1000 мл жидкого состава примера 1 в машине для нанесения покрытия в псевдоожиженном слое с одновременной сушкой типа SPHERONIZEP, модель 15 (слово PHEPOIII EP представляет собой товарный знак инофирмы Г.Б.Калева Лимитед, Эскот, Беркшир).
Пример 4. Корпускулярные твердые перорально вводимые инсулинсодержащие составы с энтеросолюбильным
покрытием получают из покрытых частиц,
полученных в примере 3, которые дополнительно покрывают следующим раствором:
Гидроксипропилметилцеллюлозафталат - 65 г
Этанол (95%) - 650
мл
Хлористый метилен - 650 мл
в центрифужном аппарате для нанесения покрытий распылением с поворотным столом.
Пример 5. Капсулы корпускулярного твердого, перорально вводимого инсулинсодержащего фармацевтического состава получают путем упаковки соответствующего количества корпускулярного твердого вещества, полученного в примере 3, в твердые желатиновые капсулы размерами 0-4.
Пример 6. Капсулы корпускулярного твердого, перорально вводимого инсулинсодержащего состава с энтеросолюбильным покрытием, получают путем упаковки соответствующего количества корпускулярного твердого вещества с энтеросолюбильным покрытием в твердые желатиновые капсулы размерами 0-4.
Пример 7. Повторяют способ примера 1, за исключение того, что в под-смесь B прибавляют 16 г (20 мл) рафинированного (фармацевтическая чистота) кунжутного масла и количество олеиновой кислоты снижают на 16 - 404 г. Кунжутное масло обеспечивает создание повышенной антиокислительной активности и повышает вкусовые качества композиций (особенно для восточных людей), тем самым улучшая условия соблюдения больными режима и схемы лечения.
Пример 8. Твердую, перорально вводимую инсулинсодержащую композицию получают следующим образом. Твердые стержневые частицы носителя получают, как в примере 3. 800 г частиц покрывают 1000 мл жидкого состава примера 7 в модифицированном аппарате для нанесения покрытий в псевдоожиженном слое с одновременной сушкой SPIR-A-FLOW следующим образом: (PIP-A-FIO представляет собой товарный знак инофирмы Фреунд Интернэшнл Лимитед, Токио, Япония).
Аппарат для нанесения покрытий с одновременной сушкой в псевдоожиженном слое показан частично в разрезе и частично схематически на фиг. 1, где он представлен в общем позицией 1.
Аппарат для нанесения покрытий/сушки 1 содержит камеру 3, в которую подают флюидизированный воздух через входное отверстие 5 и щелевой воздух через входное отверстие 7. Флюидизированный воздух от входного отверстия 5 поступает во входную камеру с флюидизированным воздухом 9, из которой он поступает через кольцевидную металлическую сетку с пересекающимися нитями 11 в камеру 3. Кольцевидная металлическая сетка 11 установлена в роторе 13, который определяет в основном плоское днище камеры 3. Ротор 13 определяет кольцевую щель 15 с периферией нижней части камеры 3, и щелевой воздух от входного отверстия 7 подается в камеру 3 через щель 15. Хотя традиционные аппараты для нанесения покрытий с одновременной сушкой содержат мешалку, которая вращается коаксиально с ротором 13, такая мешалка не предусмотрена в аппарате для нанесения покрытий с одновременной сушкой 1. Вместо этого в основном коническое утолщение 17 расположено в том месте, где обычно устанавливают мешалки, оно служит для защиты подшипников ротора 13 от чрезмерной пенетрации под действием частиц из камеры.
В стенке камеры установлен радиально расположенный вращающийся измельчитель комков 19, в основном в виде множества вращающихся лезвий.
В верхней части камеры 3 предусмотрено сопло 21 для распыления вниз жидкого состава в камеру. Сопло 21 питается насосом 23 из резервуара 25 с жидким составом. Подача осуществляется при помощи питающей трубы 27 и обратной трубы 29 для удаления избыточной жидкости. Подача воздуха в направлении и от сопла обеспечивает необходимое распыление.
В самой верхней части камеры 3 расположена пара рукавных фильтров 31, через которые псевдоожижающий воздух фильтруют перед тем, как он покинет камеру 3. В каждом рукавном фильтре 31 установлена пульсационная форсунка 33 для подачи импульсов воздуха с тем, чтобы удалять частицы на каждом рукавном фильтре 31.
При применении устройства твердые частицы носителя вводят в камеру 3 при помощи окна (не показано). Затем окно закрывают и начинают подачу флюидизирующего воздуха в устройство для нанесения покрытий с одновременным высушиванием изделия. Подачу воздуха осуществляют под давлением 100 мм воды и осуществляют осушение и фильтрацию с тем, чтобы удалить микробы и любые частицы масла, которые могут переноситься из компрессора (не показан). Температура подаваемого воздуха составляет нормально 40oC. Флюидизирующий воздух входит через впускное отверстие 5 и вращающееся кольцевидное сито 11 со скоростью 4 л в минуту и под давлением 50 мм воды для того, чтобы флюидизировать частицы носителя в камере. Щелевой воздух входит через кольцевидную щель 15 со скоростью 4 л в минуту, однако при более низком давлении от 5 до 10 мм воды для того, чтобы удерживать частицы в отделении от камеры 3. Установленный измельчитель комков 19 вращается со скоростью 2500 об/мин, и ротор 13 вращается со скоростью 250 об/мин. Утолщение 17, которое занимает место вращающейся мешалки соосно с ротором 13, существенно не вращается, однако может слегка повернуть с тем, чтобы поддерживать подшипники в свободном состоянии.
Жидкий состав примера 7 помещают в резервуар 25 и нагнетают при помощи насоса 23 через питающий трубопровод 27 к соплу 21, где жидкий состав распыляют в псевдоожиженные частицы носителя. Жидкий состав нагнетают по питающему трубопроводу 27 со скоростью 12,2 мл в минуту и воздух подают к соплу по питающему и обратному трубопроводам (не показаны) для того, чтобы обеспечить соответствующее распыление. Струйный воздух подают со скоростью 2,3 л в минуту под давлением 1,2 кгс/см2.
Жидкий состав распыляют в течение 10 с и затем отключают на 15 с, однако флюидизирующий воздух и щелевой воздух подают непрерывно с тем, чтобы поддержать флюидизированными частицы носителя.
Если температура внутри камеры 3 начинает подниматься выше 30oC, температуру подаваемого воздуха снижают до 40oC. Для того, чтобы осуществить быстрое охлаждение, если это необходимо, установлены средства (не показаны) охлаждения подаваемого воздуха с тем, чтобы температура распыляемых частиц не превышала значительно уровня 30oC.
Способ продолжают до тех пор, пока 1000 мл жидкого состава примера 7 не нанесут на 800 г частиц носителя.
Пример 9. Капсулы корпускулярной твердой, перорально вводимой инсулинсодержащей композиции получают путем упаковки соответствующего количества корпускулярного твердого состава, полученного в примере 8, в твердые желатиновые капсулы, размеры 0-4.
Пример 10. Для получения одного литра
перорально
принимаемой композиции лососевого кальцитонина применяют следующую методику. Первый и следующие ингредиенты используют для получения гидрофильной фазы:
Полиоксиэтилен 40 стеарат
- 6,7 г
Бензоат натрия - 12,0 г
Гидроксипропилцеллюлоза - 6,0 г
Апротинин (TRA SYL OL SOL) - 200000 международных единиц х 15
Лимонная кислота - 4,3 г
Аскорбиновая
кислота - 3,2 г
Деионизированная вода - 166,7 мл
Используемая методика заключается в растворении гидроксипропилцеллюлозы в апротининовом растворе TRASYL OL (товарный
знак) и
смешивании данного раствора с полиоксиэтилен 40 стеаратом, бензоатом натрия, а также лимонной и аскорбиновой кислотами. Прибавляют воду и смесь перемешивают в AVTOHOMOMIXER для растворения
компонентов. pH смеси доводят до 3,0 - 3,25 путем медленного прибавления концентрированного раствора лимонной и аскорбиновой кислот.
К полученной гидрофильной водной фазе медленно прибавляют лососевый кальцитонин (поставляется Рорер; также может поставляться Сигма Кемикал Компани, Сент-Луис, Миссури, США) при постоянном перемешивании при комнатной температуре и относительной влажности менее чем 40%. Достаточное количество лососевого кальцитонина прибавляют для получения 600 - 1200 международных единиц на мл конечного состава; 1000 международных единиц составляет отобранное количество.
Отдельно получают гидрофобную фазу из следующих ингредиентов, г:
Лецитин яичного желтка - 32,8
Холестерин - 26,7
L-альфа-токоферол
- 1,3
Глицеролмоноолеат - 23,7
Олеиновая кислота - 212,0
Твин 80 - 157,0
Под-смесь антиоксиданта - 2,8
Пропилпарабен - 3,0
Метилпарабен - 20,0
Кунжутное масло (фармацевтическая чистота) - 6,7
Этанол (95%) - Сколько понадобится
Способ заключается в том, что смешивают холестерин, токоферол, глицерилмоноолеат и другие
ингредиенты в этаноле, объем которого выбирают так, чтобы объем гидрофобной фазы был равен объему гидрофильной фазы (раствор антиоксиданта получают, как в примере 1, однако по выбору опускают
бутилированный гидроксианизол и бутилированный гидрокситолуол). Полученный раствор смешивают с тщательностью. Может быть использован гомогенизатор АВТОГОМОМИКСЕР, работающий со скоростью 7500 об/мин
при температуре 20oC, однако достаточным может быть простое механическое или магнитное перемешивание. Затем гидрофильную фазу вливают в равный объем гидрофобной фазы при перемешивании. И
вновь достаточно использовать простое механическое перемешивание или гомогенизатор АВТОГОМОМИКСЕР, работающий при упомянутых выше условиях. Полученную эмульсию пропускают три последовательных раза
через микрофлюидизатор, используемый в примере 1, при аналогичных условиях эксплуатации.
Пример 11. Твердую перорально вводимую композицию, содержащую лососевый кальцитонин, получают в основном так, как описано в примере 8, за исключением того, что 500 мл жидкого состава примера 10 наносят на 400 г карбоксиметилцеллюлозы, кальциевой соли в модифицированном устройстве SPIR-A-FL OW.
Пример 12. Капсулы корпускулярной, перорально вводимой композиции лососевого кальцитонина получают путем упаковки соответствующего количества корпускулярного твердого вещества, полученного в примере 11, в твердые желатиновые капсулы, размеры 0-4.
Пример 13. Твердые желатиновые капсулы с энтеросолюбильным покрытием, содержащие перорально вводимую композицию лососевого кальцитонина, получают следующим образом. Капсулы примера 12 покрывают во вращающемся барабане для нанесения покрытий HI-COATER, используя 5% ПВП-Ф и 10% ГПМЦ в этаноле. Процентные содержания базируются на массе капсул (слово HI-COATER представляет собой товарный знак Фреунд Интернэшнл Лтд., Токио, Япония). Капсулы, покрытые таким образом подслоем, затем покрывают 20% (мас., на основе массы капсул) HP5-5 (что является композицией ГПМЦ-Ф при pH 5,5) в хлористом метилене, вновь во вращающемся барабане HI-COATER для нанесения покрытий. После этого капсулы готовы для перорального введения.
Пример 14. Перорально принимаемую композицию свиного соматотропина (ССТ) получают следующим образом.
Под-смесь A получают из следующих
компонентов:
Соевый лецитин - 150 г
Глицерилмоноолеат - 22,46 г
Холестерин - 30 г
Этанол - 50 мл
путем растворения первых трех компонентов в теплом
(75oC) этаноле и перемешивая до растворения других компонентов. Затем этанол выпаривают.
Под-смесь B получают из следующих компонентов, г:
Олеиновая кислота
- 420
D-альфа-токоферол - 30
Полисорбат 80 - 30
Под-смесь антиоксиданта (из примера 1) - 2,7
Пропилпарабен - 1,2
Метилпарабен - 6,8
Под-смесь A
- 300
Этанол (95%) - 750
путем смешивания их при комнатной температуре.
Под-смесь C получают из следующих компонентов:
Свиной соматотропин (из
Американского Цианамида;
также поставляется Сигма) - 50 мг
Апротинин - 200000 международных единиц х
Натрий карбонат Soln - 300 см3
путем смешивания
их при комнатной
температуре. pH смеси доводят до 5,0 фосфатным буферным раствором.
Под-смесь D получают, как в примере 1, за исключением того, что опускают полиоксиэтилен (40) стеарат.
После получения различных под-смесей микроэмульсию, содержащую свиной соматотропин, получают из следующих количеств под-смесей, мл:
Под-смесь B - 450
Под-смесь C - 160
Под-смесь - 150
путем прибавления под-смеси C к под-смеси D при перемешивании гомогенизатором АВТОГОМОМИКСЕР при 7500 об/мин и температуре 20oC.
Полученную смесь медленно
прибавляют в под-смесь B, используя тот же смеситель при такой же температуре и скорости вращения. Полученную эмульсию пропускают пять раз через микрофлюидизатор, как в
примере 1, при аналогичных
условиях работы.
Пример 15. Твердую, перорально вводимую ССТ-содержащую композицию получают следующим образом. Твердые частицы носителя получают путем
смешивания следующих компонентов,
г:
Порошок сои культурной - 300
Гидроксипропилцеллюлоза - 50
Альгиновая кислота - 50
при температуре 22oC. Частицы
сердцевины сушат в псевдоожиженном
слое G1 ATT (товарный знак) при температуре 29oC в течение 45 мин. Затем 500 мл жидкого состава, полученного в примере 14, распыляют на высушенные
частицы сердцевины в модифицированном
устройстве SPIR-A-FLOW, описанном в примере 8.
Пример 16. Частицы с нанесенным покрытием, полученные в результате примера 15, гранулируют в грануляторе CF (Фреунд Индастриз, Инк., Токио, Япония) до размера частиц 1,5-2 мм. В основном используют традиционные условия и/или те, которые рекомендуют изготовитель. Раствор (приблизительно 8% в отношении массы к объему) гидроксипропилцеллюлозы-1 (ГПЦ-Л) в этаноле используют для агглютинации частиц в гранулы. Затем гранулы покрывают энтеросолюбильной оболочкой, используя 8%-ную (по массе, на основе массы гранул) ГПМЦ-Ф, подаваемой из 5%-ного (в отношении массы к объему) раствора в хлористом метилене, во вращающемся барабане для нанесения покрытий. И, наконец, аппарат для нанесения покрытий используют для получения восковой оболочки на этеросолюбильных гранулах в количестве, достаточном для обеспечения флотации гранул в желудке свиней при проглатывании.
Пример 17. С использованием основной методики примера 7, но замещая бычий инсулин соответствующим количеством человеческого инсулина, получают перорально принимаемую композицию человеческого инсулина. Жидкий состав можно наносить на твердый носитель, как описано в примере 8.
Пример 18. С использованием основной методики примера 7, но замещая бычий инсулин соответствующим количеством человеческого интерферона-гамма, получают перорально принимаемую композицию человеческого интерферона-гамма. Жидкий состав можно наносить на твердый носитель, как описано в примере 8.
Пример 19. Используя основную методику примера 7, однако замещая бычий инсулин соответствующим количеством человеческого интерферона-гамма, получают перорально принимаемую композицию человеческого интерферона-бета. Жидкий состав можно наносить на твердый носитель, как описано в примере 8.
Пример 20. Используя основную методику примера 10, однако замещая бычий инсулин соответствующим количеством эритропоэтина, получают перорально вводимую композицию эритропоэтина. Жидкий состав можно наносить на твердый носитель, как описано в примере 8.
Пример 21. Используя основную методику примера 14, однако замещая бычий инсулин соответствующим количеством тканевого активатора плазминогена, получают перорально принимаемую композицию тканевого активатора плазминогена. Жидким составом можно покрывать твердый носитель, как описано в примере 8.
Пример 22. Используя основную методику примера 14, однако замещая бычий инсулин соответствующим количеством Фактора VIII, получают перорально принимаемую композицию Фактора VIII. Жидким составом можно покрывать твердый носитель, как описано в примере 8.
Биологический пример A. Клиническое испытание перорально вводимого препарата примера 5.
В течение ночи подвергают голодной выдержке общее число 17 больных диабетом (8 инсулин-зависимых и 9 не-инсулин-зависимых диабетиков), а также одного здорового добровольца мужского пола. Все пероральные гипогликемические средства и инъекции инсулина не принимаются этими пациентами по крайней мере за 12 ч перед осуществлением испытания. Каждый больной диабетом получает per os перорально вводимые композиции инсулина, полученные в примере 5 (твердые желатиновые капсулы, содержащие частицы носителя, покрытые методом распыления инсулинсвязанной микроэмульсии, но без энтеросолюбильной оболочки). Каждая капсула содержит приблизительно 10 единиц бычьего инсулина, и каждый пациент получает перорально дозу, эквивалентную приблизительно одной единице на кг веса тела, а также около 250 мл воды. Уровни содержания сахара в крови измеряют на пробах крови, полученных прокалыванием кончика пальца с использованием комплекта FAEMOGLSVKOTEST 20-800P и устройства REFI OLVX (Boehringer Mannheim GmbH, ФРГ. В нескольких случаях уровни сывороточного инсулина измеряют с использованием метода радиоиммуноанализа. Для анализа на сывороточный инсулин пробы сыворотки декантируют в TRASYL OL - содержание пробирки и сохраняют при температуре от -20 до -35oC до анализа (TRASYL OL представляет собой товарный знак Байера для ингибитора апротинин-протеиназы).
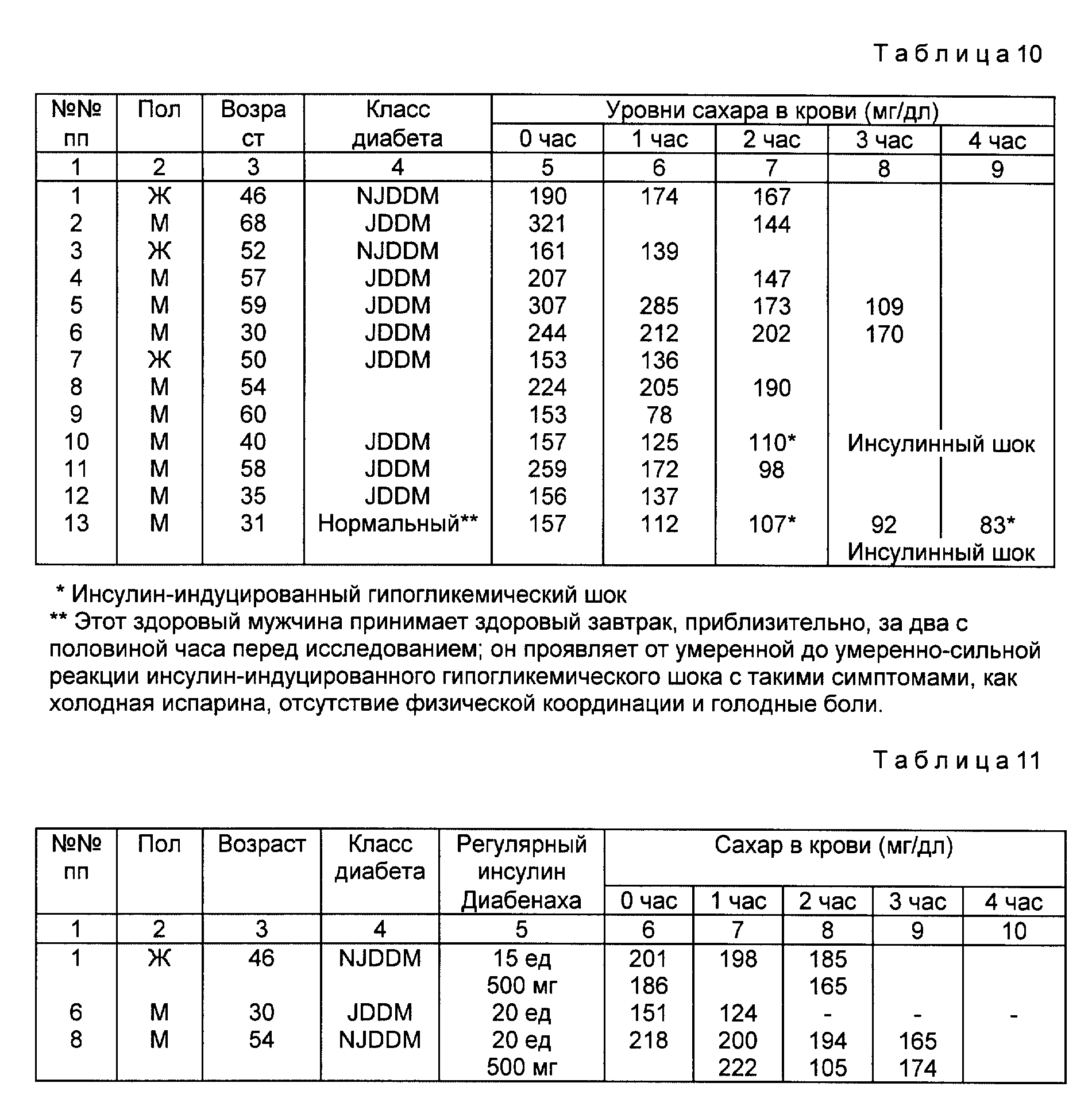
Уровни содержания сахара в крови приведены в табл. 7.
Некоторые пациенты плохо реагируют на подкожно вводимый инсулин (конкретно, NN 2, 5, 6, 10 и 14). Они показывают следующую реакцию, выражаемую уровнем содержания сахара в крови, на регулярно вводимый инсулин (см. табл. 8).
Перорально вводимый препарат примера 5 (без энтеросолюбильного покрытия частиц) является таким образом эффективным средством в снижении повышенных резистентных уровней сахара в крови до или по крайней мере в направлении нормальных уровней содержания сахара в крови у всех исследованных больных диабетом, за исключением одного случая (пациент N 11), в котором наблюдаемое понижение уровней содержания сахара в крови не рассматривается как клинически значительное. Здоровый доброволец не отвечает на перорально вводимую композицию примера 5. В двух случаях (пациенты N 11 и 18) развивается инсулининдуцированный гипогликемический шок на 75 и 120 мин соответственно после перорального введения композиции примера 5, который устраняется при приеме 100 г сахарной воды.
В нескольких случаях испытания ряд проб сыворотки собирают перед и после перорального приема композиции примера 5.
Результаты приведены в табл. 9.
Биологический пример В. Клиническое испытание перорально вводимого препарата примера 1.
Соответствующее количество микроэмульсии дают в жидкой форме рег os вместе с 10 мл МСТ (МСТ представляет собой товарный знак раствора триглицерида со средней цепью: изготовитель Мид-Джонсон энд Ко., Эвансвилл, Индиана, США). Препарат микроэмульсии МСТ действует, как если бы инсулинсодержащая микроэмульсия была покрыта энтеросолюбильной оболочкой. Каждый мл инсулинсодержащего состава содержит приблизительно 5 единиц бычьего инсулина.
В исследовании принимают участие двенадцать больных диабетом (9 IDDM и 3 NIDDM) и один здоровый доброволец мужского пола. Все пациенты голодают в течение ночи, а пероральные гипогликемические средства и инсулин не принимаются ими в течение 12 или более часов перед исследованием. Каждый субъект принимает одну единицу инсулина на кг веса тела в форме, описанной выше рег os. Результаты приведены в табл. 10.
Пациенты NN 1 и 8 являются плохими реагентами как на 500 мг таблетки, рег os диабеназы, так и на подкожно вводимые 20 единиц регулярного инсулина, как можно видеть из табл. 11.
Только один из двенадцати больных диабетом является плохим ответчиком на пероральное введение инсулинсодержащей микроэмульсии. Один здоровый доброволец отвечает хорошо и впадает в инсулин-индуцированный гипогликемический шок.
У нескольких обследуемых пациентов перед и после перорального приема композиции примера 5 собирают сывороточные пробы.
Результаты приведены в табл. 12.
Биологический пример C. Осуществляют введение той же смеси, которую перорально вводили в примере 8. Двум коротконогим гончим собакам, весящим 12 и 16 кг соответственно, вливают/инъецируют в течение 5 мин в двенадцатиперстную кишку 5 мл инсулинсодержащей микроэмульсии (каждый мл содержит 5 единиц бычьего инсулина). После введения измеряют уровни содержания сывороточной глюкозы и сывороточного инсулина (иммунореактивного инсулина, ИРИ).
Результаты приведены в табл. 13.
Интрадуоденальное введение инсулинсодержащей микроэмульсии индуцирует снижение уровней содержания сахара в крови и соответствующее повышение уровней содержания сывороточного инсулина у обеих исследуемых собак, что является показателем хорошей биологической доступности перорально-инградуоленально вводимого инсулина. Таким образом, инсулин является и биоактивным, и биодоступным.
Биологический пример 1. После ночной голодной выдержки в данном испытании принимают участие шесть добровольцев - мужчин в возрасте от 21 до 26 (среднее 23,1) лет, чей вес составляет от 58 до 78 (среднее 66) кг и рост от 171 до 187 (среднее 177,2) см. В 6 ч до полудня пять пациентов принимают перорально от 400 до 420 международных единиц лососевого кальцитонина в композиции примера 10, тогда как один оставшийся пациент получает подкожно 200 международных единиц лососевого кальцитонина (CALCYNAR зарегистрированный товарный знак) в условиях голодной выдержки. Системные выборки венозной крови собирают в течение 0 (перед введением лекарственного средства), а также 30, 60, 90, 120, 150, 180, 210, 240, 300, 360 и 480 мин после введения лекарственного средства. Сывороточный фосфорный уровень из собранных проб крови измеряют методом Фиско-Субарроу и уровень лососевого кальцитонина в ЭДТК-обработанной плазме определяют методом радиоиммуноанализа, используя125I и кроличьи иммутанные сыворотки лососевого кальцитонина. Все измерения осуществляют трижды.
Результаты приведены в табл. 14.
Можно заметить, что пероральное введение лососевого кальцитонина при концентрации от 400 до 420 международных единиц в виде композиции примера 7 вызывает в основном аналогичную степень снижения сывороточных фосфатных уровней и аналогичное повышение уровня лососевого кальцитонина в плазме, измеренного с использованием РИА, до такого уровня, который получают подкожным введением 200 международных единиц человеку.
Форма перорального введения лососевого кальцитонина, полученного в примере 10, вызывает образование резких максимумов содержания лососевого кальцитонина в человеческой плазме крови и снижение сывороточных фосфатных уровней у молодых мужчин-добровольцев. Перорально введенный лососевый кальцитонин при концентрации от 400 до 420 международных единиц индуцирует в основном аналогичную биологическую реакцию (измеренную как снижение в сывороточном фосфате) и биодоступность лососевого кальцитонина в ЭДТК-обработанной плазме (подвергнутой анализу РИА) у мужчин до 200 международных единиц лососевого кальцитонина, введенного подкожно. Лососевый кальцитонин, введенный в композицию примера 7, является биологически эффективным и биодоступным у мужчин после перорального приема.
Биологический пример E. Свинье (особь мужского пола) весом 75 кг через желудочную трубку вводят в желудок 500 мкг перорального свиного соматотропина (ССТ) (полученного в соответствии с примером 16) на кг веса тела, а другой свинье (особь мужского пола) весом 82 кг вводят в двенадцатиперстную кишку через энтеростомию 500 мкг перорального ССТ на кг веса тела.
Пробы крови отбирают посредством внутривенной канюли, помещенной в яремную вену, и сывороточный ССТ измеряют в каждой пробе радиоиммуноанализом.
У этих двух свиней, которые предварительно были обработаны ежедневными внутримышечными инъекциями 500 мг дексаметазона в течение трех последовательных дней (для подавления in vivo секреции СССТ), как внутрижелудочное, так и интрадуоденальное введение ССТ, 500 мкг на кг веса тела, показывают биодоступность ССТ, достигающего пиковых значений через 6 ч после интрадуоденального введения и через 10 ч после внутрижелудочного введения.
Результаты приведены в табл. 15.
ССТ, вводимый в желудок или двенадцатиперстную кишку, следовательно, является биодоступным.
Биологический пример F. Данный пример демонстрирует, что инсулин в композиции настоящего изобретения абсорбируется через лимфатический сосуд (сосуды), а не через "систему пор" мембраны (в данном случае не должен быть обнаружен в жидкости лимфы, и большая часть абсорбированного инсулина может быть обнаружена в воротной вене, дренирующей в печень).
Свинью (особь женского пола) весом 35 кг анестезируют и двенадцатиперстную кишку вскрывают. В двенадцатиперстную кишку вставляют канюлю; основной лимфатический сосуд, дренирующий из двенадцатиперстной кишки, канюлируют и жидкость лимфы собирают в цилиндр в течение каждого 15- минутного периода в продолжение полного периода испытания. Другую канюлю вставляют в воротную вену и ее открытый конец подвигают в печень; катетер помещают в правую яремную вену и канюлю помещают в левую вену предплечья и внутривенно вливают 10%-ную глюкозу в воде.
Жидкую инсулинсодержащую композицию (50 мл - каждый мл содержит 5 единиц бычьего инсулина), полученную в соответствии с примером 1, быстро вливают в двенадцатиперстную кишку через канюлю, помещенную в полость двенадцатиперстной кишки, во время - 0.
Инсулин сыворотки и жидкости лимфы анализируют радиоиммуноанализом. Жидкость лимфы разбавляют один к десяти из-за высоких уровней инсулина, обнаруженных в пробах и, как обнаружено, необходимо разбавлять еще до 1/50 в отношении пробы лимфы, отобранной в течение 15 - 30 мин исследования.
Основной сосуд лимфы, дренирующий из двенадцатиперстной кишки, после канюлирования и под анестезией проявляет незначительную тенденцию к снижению скорости тока жидкости. Исключение составляет временное повышение скорости тока жидкости, наблюдаемое перед интрадуоденальным введением ODDS-Инсулина, что может обусловливаться анестезирующим средством, применяемым в данном исследовании. Скорость протекания лимфы приведена на фиг. 2.
После интрадуоденального вливания инсулинсодержащей композиции временное повышение уровня сывороточного инсулина обнаруживают в печеночной воротной крови, пробы которой отбирают через 7,5 - 15 мин после введения лекарственного средства. В других случаях уровни сывороточного инсулина не изменяются в печеночных образцах крови воротной вены в продолжении исследования, как показано графически на фиг. 3B. Никаких изменений в уровнях сывороточного инсулина не обнаружено в системных пробах венозной крови, как показано графически на фиг. 3A.
Однако, как можно видеть на фиг. 3C, наблюдается заметное и продолжительное повышение уровня инсулина в жидкости лимфы, и уровень изменений составляет от 1000 до 5000 микроединиц на мл жидкости лимфы. Повышенные уровни содержания инсулина нельзя отнести за счет повышенной скорости тока лимфы, а нужно рассматривать с точки зрения повышенной концентрации.
Небольшие гидрофильные, водорастворимые химические соединения, такие как сахар, как известно, абсорбируют через "систему пор" кишечной мембраны, поставляются в капиллярное кровообращение и затем в печеночную воротную вену человека. Липиды и липофильные вещества, с другой стороны, как известно, абсорбируют посредством двух совершенно различных механизмов. Те жирные кислоты, которые имеют относительно короткие углеродные цепи (например, C2-C6 или C8-кислоты, такие как капроновая и каприловая кислота), абсорбируют через кишечную мембрану с ферментативной и физиохимической "помощью" от солей желчных кислот и панкреатической липазы. В конечном счете такие абсорбированные короткоцепочечные жирные кислоты дренируют в капиллярную кровь и проходят в печеночную воротную вену.
Липиды и жирные кислоты, имеющие относительно длинные цепи, например олеиновая кислота и ди-олеат и три-олеат-глицериды, а также холестерин и фосфолипиды, среди других соединений, образующих хиломикроны в пределах мембраны, абсорбируют через кишечную мембрану под действием механизмов, которые еще не поняты до конца. В кишечной мембране они участвуют в образовании хиломикронов и затем "всасываются" в ворсинки кишечной системы, дренируют в жидкость лимфы, собираются в грудном протоке и в конце концов высвобождаются в большой круг кровообращения.
Заметное и значительное повышение уровня содержания инсулина наблюдается главным образом в жидкости дуоденальной лимфы, и это подтверждает то, что интрадуоденально вводимая композиция инсулина (которая распространяется на хиломикроны или про-хиломикроны) абсорбируется через лимфатическую систему, а не через "систему воротной вены". Уровень инсулина, извлеченного из жидкости лимфы - до 5000 микроединиц на мл - является таким значительным, что подтверждает то, что инсулин абсорбируется посредством хиломикронов, а не системы воротной вены.
Реферат
Фармацевтическая композиция, представляющая микроэмульсию с биологически активным макромолекулярным материалом, состоит из гидрофильной фазы, содержащей биологически активный макромолекулярный материал, и гидрофобный фазы, содержащей хиломикроны или материал, из которого хиломикроны образуются in vivo, при этом гидрофильная фаза диспергирована в гидрофобной, состоящей из холестерина, лецитина и линофильного поверхностно-активного агента, взятых при определенных соотношениях ингредиентов в зависимости от используемого биологически активного макромолекулярного материала. Способ включает в себя перемешивание биологически активного макромолекулярного материала в подходящем гидрофильном растворителе с гидрофильной фазой, содержащей поверхностно-активный агент с высоким ГЛБ, и добавление полученной водной смеси к гидрофобной фазе, содержащей поверхностно-активный агент с низким ГЛБ, с дальнейшим перемешиванием. 2 с. и 48 з.п. ф-лы, 15 табл., 3 ил.

Формула
Холестерин - 0,5 - 5,0
Лецитин - 0,5 - 40,0
Липофильный поверхностно-активный агент - 0,5 - 95,0
2. Композиция по п.1, отличающаяся тем, что гидрофобная фаза содержит, мас.%:
Холестерин - 0,5 - 5,0
Лецитин - 0,5 - 10,0
Липофильный поверхностно-активный агент - 0,5 - 95,0
3. Композиция по п.1, отличающаяся тем, что гидрофобная фаза содержит, мас.%:
Холестерин - 0,5 - 5, 0
Лецитин - 4 - 10
Липофильный поверхностно-активный агент - 50 - 95
если макромолекула является инсулином.
Холестерин - 0,5 - 5,0
Лецитин - 0,5 - 7,0
Липофильный поверхностно-активный агент - 0,5 - 5,0
если макромолекула является кальцитонином.
Холестерин - 0,5 - 5,0
Лецитин - 5 - 40
Липофильный поверхностно-активный агент - 10 - 70
если макромолекула является свиным соматотропином.
Сложный эфир холестерина - Не более 5
Неэтерифицированная жирная кислота - Не более 50
Апопротеин - Не более 5.
Сложный эфир холестерина - Не более 5
Неэтерифицированная жирная кислота - Не более 2
Апопротеин - Не более 4
если макромолекула является инсулином.
Сложный эфир холестерина - Не более 5
Неэтерифицированная жирная кислота - Не более 4
Апопротеин - Не более 4
если макромолекула является кальцитонином.
Сложный эфир холестерина - Не более 5
Неэтерифицированная жирная кислота - Не более 5
Апопротеин - Не более 5
если макромолекула является свиным соматотропином.
Кальциевая соль карбоксиметилцеллюлозы - 20 - 60
Альгиновая кислота - 5 - 25
Гидроксипропилцеллюлоза - 20 - 60
Желатин - 2 - 20
Поверхностно-активное вещество - 0,1 - 20,0
19. Композиция по п.18, отличающаяся тем, что твердый носитель содержит компонент, имеющий пищевую ценность.
Холестерин - 0,5 - 5,0
Лецитин - 0,5 - 40,0
Липофильный поверхностно-активный агент - 0,5 - 95,0
24. Способ по п. 23, отличающийся тем, что гидрофобная фаза содержит, мас.%:
Холестерин - 0, 5 - 5,0
Лецитин - 0,5 - 10,0
Липофильный поверхностно-активный агент - 0,5 - 95,0
25. Способ по п. 23, отличающийся тем, что гидрофобная фаза содержит, мас.%:
Холестерин - 0,5 - 5,0
Лецитин - 4 - 10
Липофильный поверхностно-активный агент - 50 - 95
если макромолекула является инсулином.
Холестерин - 0,5 - 5,0
Лецитин - 0,5 - 7,0
Липофильный поверхностно-активный агент - 0,5 - 5,0
если макромолекула является кальцитонином.
Холестерин - 0,5 - 5,0
Лецитин - 5 - 40
Липофильный поверхностно-активный агент - 10 - 70
если макромолекула является свиным соматотропином.
Сложный эфир холестерина - Не более 5
Неэтерифицированная жирная кислота - Не более 50
Апопротеин - Не более 5.
Сложный эфир холестерина - Не более 5
Неэтерифицированная жирная кислота - Не более 2
Апопротеин - Не более 4
если макромолекула является инсулином.
Сложный эфир холестерина - Не более 5
Неэтерифицированная жирная кислота - Не более 4
Апопротеин - Не более 4
если макромолекула является кальцитонином.
Сложный эфир холестерина - Не более 5
Неэтерифицированная жирная кислота - Не более 5
Апопротеин - Не более 5
если макромолекула является свиным соматотропином.
Кальциевая соль карбоксиметилцеллюлозы - 20 - 60
Альгиновая кислота - 5 - 25
Гидроксипропилцеллюлоза - 20 - 60
Желатин - 2 - 20
Поверхностно-активный агент - 0,1 - 20,0
41. Способ по п.40, отличающийся тем, что твердый носитель содержит компонент, имеющий пищевую ценность.
Комментарии