Высокопроизводительное секвенирование одиночной клетки со сниженной ошибкой амплификации - RU2744175C1
Код документа: RU2744175C1
Чертежи






















Описание
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке испрашивается приоритет предварительной патентной заявки США с серийным № 62/673023, поданной 17 мая 2018 года, и предварительной патентной заявки США с серийным № 62/821864, поданной 21 марта 2019 года, каждая из которых включена в настоящий документ в качестве ссылки в полном объеме.
БЮДЖЕТНОЕ ФИНАНСИРОВАНИЕ
Это изобретение было сделано при поддержке правительства путем гранта №. DP1 HG007811, присужденного Национальным Институтом здоровья. Правительство имеет определенные права на изобретение.
ОБЛАСТЬ ТЕХНИКИ
Варианты осуществления настоящего изобретения относятся к секвенированию нуклеиновых кислот. В частности, варианты осуществления способов и композиций, предлагаемых в настоящем документе, относятся к получению индексированных библиотек для секвенирования одиночных клеток и получению данных об их последовательностях для характеристики редких событий, в том числе кроссинговера и нарушенной сегрегации хромосом. В некоторых вариантах осуществления способы относятся к определению гетерогенности злокачественной опухоли на уровне одиночной клетки.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Современные технологии секвенирования генома одиночной клетки имеют два ключевых ограничения. Во-первых, большинство способов требуют компартментализации отдельных клеток, которая может ограничивать производительность. Во-вторых, большинство способов амплификации основаны на ПЦР и, таким образом, страдают от ошибок экспоненциальной амплификации. Чтобы решить первую проблему, мы с коллегами разработали комбинаторное индексирование одиночных клеток («sci-»), где вводят несколько раундов молекулярного штрихкодирования с разделением-объединением для уникального мечения содержимого нуклеиновых кислот в одиночных клетках, тем самым обеспечивая экспоненциальный прирост в производительности с каждым последующим раундом индексирования. Sci-способы были успешно разработаны для профилирования доступности хроматина (sci-ATAC-seq), транскриптомов (sci-RNA-seq), геномов (sci-DNA-seq), метилoмов (sci-MET), конформации хромосом (sci-Hi-C) на больших количествах одиночных клеток (Cao et al., 2017, Science 357:661-667; Cusanovich et al., 2015, Science, 348:910-914; Mulqueen et al., 2018, Nat. Biotechnol. 36:428-431; Ramani et al., 2017, Nat. Methods 14:263-266; Vitak et al., 2017, Nat. Methods 14:302-308). Для решения второй проблемы, линейная амплификация путем транскрипции на основе T7 обеспечивает потенциальное решение, которое ранее применяли для анализов одиночных клеток (Eberwine et al., 1992; Proceedings of the National Academy of Sciences 89:3010-3014; Hashimshony et al., 2012, Cell Rep. 2:666-673; Sos et al., 2016, Genom Biolol., 17:20). Например, недавно, Chen et al. разработали линейную амплификацию путем вставки транспозона («LIANTI»), которая использует транспозон Tn5 для фрагментации генома и одновременно вставляет РНК-промотор T7 для транскрипции in vitro (IVT). Копии РНК, полученные с ДНК-матрицы, не могут служить матрицей для дальнейшей амплификации; таким образом, все копии получены непосредственно с ДНК-матрицы. Избегая экспоненциальной амплификации, LIANTI поддерживает однородность и сводит к минимуму ошибки в последовательности. Однако способ имеет низкую производительность, потому что он требует получения серийных библиотек из каждой одиночной клетки (Chen et al., 2017, Science 356:189-194).
СУЩНОСТЬ ЗАЯВКИ
В настоящем документе предлагаются способы, которые интегрируют комбинаторное индексирование одиночных клеток и линейную амплификацию, чтобы свести к минимуму ошибки амплификации с одновременным экспоненциальным увеличением производительности. С помощью нескольких циклов молекулярного штрихкодирования способы улучшают производительность по меньшей мере, до тысяч и, возможно, миллионов клеток за эксперимент, сохраняя при этом преимущества линейной амплификации. Авторы изобретения демонстрируют генерализуемость способов с помощью демонстрации для подтверждения концепции полногеномного секвенирования одиночной клетки («sci-L3-WGS»), нацеленного секвенирования генома («sci-L3-target-seq»), и совместного анализа генома и транскриптома («sci-L3-RNA/DNA»). В качестве дополнительной демонстрации применяют полногеномное секвенирование одиночной клетки для картирования беспрецедентного количества мейотических кроссинговеров и редких случаев неправильной сегрегации хромосом у незрелых и зрелых мужских половых клеток от бесплодных, межвидовых (B6×Spretus) самцов мышей F1, а также фертильных внутривидовых (B6×Cast) самцов мышей F1.
Определения
Термины, используемые в данном документе, следует понимать как имеющие их обычное значение в соответствующей области техники, если не указано иное. Несколько терминов, используемых в настоящем документе, и их значения изложены в настоящем документе.
Как применяют в настоящем документе, термины «организм» и «индивидуум» используются взаимозаменяемо и относятся к микроорганизмам (например, прокариотическим или эукариотическим) животным и растениям. Пример животного представляет собой млекопитающего, такого как человек.
Как применяют в настоящем документе, термин «тип клеток» предназначен для идентификации клеток на основе морфологии, фенотипа, источника развития или других известных или распознаваемых отличительных клеточных характеристик. Разнообразные типы клеток могут быть получены из одного организма (или из одного и того же вида организма). Примеры типов клеток в качестве неограничивающих примеров включают гаметы (в том числе женские гаметы, например, яйцеклетки, и мужские гаметы, например, сперматозоид), эпителий яичника, фибробласт яичника, клетки яичка, клетки мочевого пузыря, иммунные клетки, B-клетки, T-клетки, клетки-естественные киллеры, дендритные клетки, злокачественные клетки, эукариотические клетки, стволовые клетки, клетки крови, мышечные клетки, жировые клетки, клетки кожи, нервные клетки, костные клетки, клетки поджелудочной железы, эндотелиальные клетки, эпителий поджелудочной железы, альфа-клетки поджелудочной железы, бета-клетки поджелудочной железы, эндотелий поджелудочной железы, лимфобласт костного мозга, B-лимфобласт костного мозга, макрофаг костного мозга, эритробласт костного мозга, дендритную клетку костного мозга, адипоцит костного мозга, остеоцит костного мозга, хондроцит костного мозга, промиелобласт, мегакариобласт костного мозга, клетку мочевого пузыря, B-лимфоцит головного мозга, глию головного мозга, нейрон, астроцит головного мозга, нейроэктодерму, макрофаг головного мозга, микроглию головного мозга, эпителий головного мозга, кортикальный нейрон, фибробласт головного мозга, эпителий молочной железы, эпителий толстого кишечника, лимфоцит толстого кишечника, миоэпителий молочной железы, фибробласт молочной железы, энтероцит толстого кишечника, эпителий шейки матки, эпителий протоков молочной железы, эпителий языка, дендритную клетку миндалины, B-лимфоцит миндалины, лимфобласт периферической крови, Т-лимфобласт периферической крови, T-лимфоцит кожи, клетку-естественного киллера периферической крови, В-лимфобласт периферической крови, моноцит периферической крови, миелобласт периферической крови, монобласт периферической крови, промиелобласт периферической крови, макрофаг периферической крови, базофил периферической крови, эндотелий печени, тучную клетку печени, эпителий печени, B-лимфоцит печени, эндотелий селезенки, эпителий селезенки, B-лимфоцит селезенки, гепатоцит печени, фибробласт печени, эпителий легкого, эпителий бронхов, фибробласт легкого, B-лимфоцит легкого, шванновскую клетку легкого, сквамозную клетку легкого, макрофаг легкого, остеобласт легкого, нейроэндокринную клетку, клетку альвеол легкого, эпителий желудка и фибробласт желудка.
Как применяют в настоящем документе, термин «ткань» предназначен для обозначения совокупности или скопления клеток, которые действуют вместе для выполнения одной или более специфических функций в организме. Клетки могут необязательно быть морфологически похожими. Примеры тканей в качестве неограничивающих примеров включают эпидидимий, глаз, мышцу, кожу, сухожилие, вену, артерию, кровь, сердце, селезенку, лимфоузел, кость, костный мозг, легкое, бронхи, трахею, кишечник, тонкий кишечник, толстую кишку, прямую кишку, слюнную железу, язык, желчный пузырь, аппендикс, печень, поджелудочную железу, головной мозг, желудок, кожу, почку, мочеточник, мочевой пузырь, уретру, гонаду, яичко, яичник, матку, фаллопиеву трубу, тимус, гипофиз, щитовидную железу, надпочечник, или паращитовидную железу. Ткань может быть получена из любого органа человека или другого организма. Ткань может быть здоровой тканью или нездоровой тканью. Примеры нездоровых тканей в качестве неограничивающих примеров включают злокачественные новообразования в репродуктивной ткани, легком, молочной железе, толстой кишке, предстательной железе, носоглотке, желудке, семенниках, коже, нервной системе, кости, яичнике, печени, кроветворных тканях, поджелудочной железе, матке, почке, лимфоидных тканях, и т.д. Злокачественные новообразования могут представлять собой ряд гистологических подтипов, например, карциному, аденокарциному, саркому, фиброаденокарциному, нейроэндокринный подтип или быть недифференнцированными.
Как применяют в настоящем документе, термин «нуклеосома» относится к основной повторяющейся единице хроматина. Геном человека состоит из нескольких метров ДНК, компактно упакованной в ядре клетки со средним диаметром 10 мкм. В ядре эукариот ДНК упакована в комплекс нуклеопротеинов, известный как хроматин. Нуклеосома (основная повторяющаяся единица хроматина), как правило, включает ~146 пар оснований ДНК, обернутых примерно 1,7 раза вокруг октамера из коровых гистонов. Гистоновый октамер состоит из двух копий каждого из гистонов H2A, H2B, H3 и H4. Нуклеосомы располагаются через равные промежутки вдоль ДНК в виде бус на нитке.
Как применяют в настоящем документе, термин «компартмент» предназначен для обозначения области или объема, который отделяет или изолирует что-то от других вещей. Примеры компартментов в качестве неограничивающих примеров включают в себя флаконы, трубки, лунки, капли, болюсы, гранулы, сосуды, элементы поверхности или области или объемы, разделенные физическими силами, такими как поток жидкости, магнетизм, электрический ток или т.п. В одном из вариантов осуществления компартмент представляет собой лунку из многолуночного планшета, такого как 96- или 384-луночный планшет. Как применяют в настоящем документе, капля может включать гранулу гидрогеля, которая представляет собой гранулу для инкапсулирования одного или более ядер или клетки, и включает гидрогелевую композицию или микрофлюидику на основе капли. В некоторых вариантах осуществления капля представляет собой гомогенную каплю гидрогельного материала или представляет собой полую каплю с оболочкой из полимера гидрогеля. Гомогенная или полая, капля способна инкапсулировать одно или более ядер или клеток.
Как применяют в настоящем документе, «транспозомный комплекс» относится к ферменту для интеграции и нуклеиновой кислоте, включая участок распознавания интеграции. «Транспозомный комплекс» представляет собой функциональный комплекс, образованный транспозазой и участком распознавания транспозазы, который способен катализировать реакцию транспозиции (см., Например, Gunderson et al., WO 2016/130704). Примеры ферментов для интеграции в качестве неограничивающих примеров включают интегразу или транспозазу. Примеры участков распознавания для интеграции в качестве неограничивающих примеров включают участок распознавания транспозазы.
Как применяют в настоящем документе, термин «нуклеиновая кислота» предназначен для использования в данной области и включает природные нуклеиновые кислоты или их функциональные аналоги. Особенно подходящие функциональные аналоги способны гибридизоваться с нуклеиновой кислотой в зависимости от последовательности или могут использоваться в качестве матрицы для репликации определенной нуклеотидной последовательности. Природные нуклеиновые кислоты, как правило, имеют остов, содержащий фосфодиэфирные связи. Аналогичная структура может иметь альтернативные связи в остове, включая любую из ряда известных в данной области. Природные нуклеиновые кислоты, в основном, содержат дезоксирибозный сахар (например, содержится в дезоксирибонуклеиновой кислоте (ДНК)) или рибозный сахар (например, содержится в рибонуклеиновой кислоте (РНК)). Нуклеиновая кислота может содержать любой из ряда аналогов этих сахарных групп, известный в данной области. Нуклеиновая кислота может включать природные или не-природные основания. Что касается этого, природная дезоксирибонуклеиновая кислота может иметь одно или более оснований, выбранных из группы, состоящей из аденина, тимина, цитозина или гуанина, и рибонуклеиновая кислота может иметь одно или более оснований, выбранных из группы, состоящей из аденина, урацила, цитозина или гуанина. Подходящие не-природные основания, которые могут быть включены в нуклеиновую кислоту, известны в данной области. Примеры не-природных оснований включают замкнутую нуклеиновую кислоту (ЗНК), мостиковую нуклеиновую кислоту (BNA) и псевдокомплементарные основания (Trilink Biotechnologies, San Diego, CA). Основания ЗНК и BNA могут быть включены в олигонуклеотид ДНК и повышают прочность и специфичность гибридизации олигонуклеотидов. Основания ЗНК и BNA и их использование известны специалистам в данной области и являются рутинными.
Как применяют в настоящем документе, термин «мишень» при использовании по отношению к нуклеиновой кислоте, предназначен в качестве семантического идентификатора для нуклеиновой кислоты в отношении способа или композиции, в настоящем документе, и не обязательно ограничивает структуру или функцию нуклеиновой кислоты за исключением того, если явно не указано иное. Нуклеиновой кислотой-мишенью может быть, по существу, любая нуклеиновая кислота с известной или неизвестной последовательностью. Это может быть, например, фрагмент геномной ДНК (например, хромосомной ДНК), внехромосомная ДНК, такая как плазмида, бесклеточная ДНК, РНК (например, РНК или некодирующая РНК), белки (например, клеточные белки или белки клеточной поверхности), или кДНК. Секвенирование может привести к определению последовательности целой молекулы-мишени или ее части. Мишени могут быть получены из первичного образца нуклеиновой кислоты, такого как ядро. В одном из вариантов осуществления мишени могут быть обработаны для получения матриц, подходящих для амплификации, путем размещения универсальных последовательностей на одном или обоих концах каждого фрагмента-мишени. Мишени также можно получить из первичного образца РНК путем обратной транскрипции в кДНК. В одном из вариантов осуществления «мишень» применяют по отношению к подгруппе ДНК, РНК или белков, присутствующих в клетке. Целевое секвенирование использует отбор и выделение генов или областей, или белков, представляющих интерес, как правило, с помощью или амплификации путем ПЦР (например, праймеры, специфичные к областям), или способа захвата на основе гибридизации (например, использование захватывающего зонда), или антител. Целевое обогащение может происходить на разных этапах способа. Например, целевое представление РНК можно получать с использованием целевых специфических праймеров в шаге обратной транскрипции или гибридизации на основе подмножества из более сложной библиотеки. Примером является секвенирование экзома или анализ L1000 (Subramanian et al., 2017, Клетка, 171; 1437-1452). Целевое секвенирование может включать в себя любой из процессов обогащения, известных специалисту в данной области.
Как применяют в настоящем документе, термин «универсальный», когда используется для описания нуклеотидной последовательности, относится к области последовательности, которая является общей для двух или более нуклеиновых кислот, где у молекулы также есть области последовательности, которые отличаются друг от друга. Универсальная последовательность, присутствующая у разных представителей совокупности молекул, может позволить захват нескольких различных нуклеиновых кислот с использованием популяции универсальных захватывающих нуклеиновых кислот, например, захватывающих олигонуклеотидов, которые комплементарны части универсальной последовательности, например универсальной последовательности для захвата. Неограничивающие примеры универсальной последовательности для захвата включают в себя последовательности, которые идентичны или комплементарны праймерам P5 и P7. Аналогично, универсальная последовательность, присутствующая у разных представителей совокупности молекул, может позволить репликацию (например, секвенирование) или амплификацию множества различных нуклеиновых кислот с использованием совокупности универсальных праймеров, которые комплементарны части универсальной последовательности, например, универсальной якорной последовательности. Неограничивающие примеры универсальной якорной последовательности включают в себя последовательности, которые идентичны или комплементарны спейсерным последовательностям, таким как sp1 и sp2. В одном из вариантов осуществления универсальные якорные последовательности используют в качестве участка, на котором отжигается универсальный праймер для секвенирования (например, сиквенсовый праймер для рида 1 или рида 2). Таким образом, захватывающий олигонуклеотид или универсальный праймер включает в себя последовательность, которая может специфически гибридизоваться с универсальной последовательностью.
Термины «P5» и «P7» можно использовать при ссылке на универсальную последовательность для захвата или захватывающий олигонуклеотид. Термины «P5’» (P5 штрих) и «P7’» (P7 штрих) относятся к последовательности, комплементарной P5 и P7, соответственно. Следует понимать, что любые подходящие универсальные последовательности для захвата или захватывающий олигонуклеотид можно использовать в способах, представленных в настоящем документе, и что использования P5 и P7 являются только иллюстративными вариантами. Использование олигонуклеотидов для захвата, таких как P5 и P7, или комплементарных им последовательностей на проточных ячейках известно в данной области, о чем свидетельствуют описания WO 2007/010251, WO 2006/064199, WO 2005/065814, WO 2015/106941, WO 1998/044151 и WO 2000/018957. Например, любой подходящий прямой праймер для амплификации, будь то иммобилизованный или растворенный, может быть полезен в способах, представленных в настоящем документе, для гибридизации с комплементарной последовательностью и амплификации последовательности. Аналогично, любой подходящий обратный праймер для амплификации, будь то иммобилизованный или растворенный, может быть полезен в способах, представленных в настоящем документе, для гибридизации с комплементарной последовательностью и амплификации последовательности. Специалист в данной области поймет, как разработать и использовать последовательности праймеров, подходящие для захвата и/или амплификации нуклеиновых кислот, как представлено в настоящем документе.
Как применяют в настоящем документе, термин «праймер» и его производные относятся в основном к любой нуклеиновой кислоте, которая может гибридизоваться с последовательностью-мишенью, представляющей интерес. Как правило, праймер функционирует как субстрат, на котором нуклеотиды могут быть полимеризованы с помощью полимеразы или с которым может быть лигирована нуклеотидная последовательность, такая как индекс; в некоторых вариантах осуществления, однако, праймер может стать частью синтезированной цепи нуклеиновой кислоты и обеспечить сайт, с которым другой праймер может гибридизоваться с первичным синтезом новой цепи, которая комплементарна синтезированной молекуле нуклеиновой кислоты. Праймер может включать любую комбинацию нуклеотидов или их аналогов. В некоторых вариантах осуществления праймером является одноцепочечный олигонуклеотид или полинуклеотид. Термины «полинуклеотид» и «олигонуклеотид» используются взаимозаменяемо в данном документе для обозначения полимерной формы нуклеотидов любой длины, и могут включать рибонуклеотиды, дезоксирибонуклеотиды, их аналоги или их смеси. Термины следует понимать как включающие в качестве эквивалентов аналоги любой из ДНК, РНК, кДНК или конъюгатов антитело-олигонуклеотид, полученных из нуклеотидных аналогов, и применимые к одноцепочечным (таким как смысловые или антисмысловые) и двухцепочечным полинуклеотидам. Термин, как применяют в настоящем документе, также охватывает кДНК, которая является комплементарной или копией ДНК, полученной из матрицы РНК, например, под действием обратной транскриптазы. Этот термин относится только к первичной структуре молекулы. Таким образом, термин включает в себя тройную, двойную и одноцепочечную дезоксирибонуклеиновую кислоту («ДНК»), а также тройную, двойную и одноцепочечную рибонуклеиновую кислоту («РНК»).
Как применяют в настоящем документе, термин «адаптер» и его производные, например, универсальный адаптер, относится, в основном, к любому линейному олигонуклеотиду, который можно лигировать с молекулами нуклеиновой кислоты по изобретению. В некоторых вариантах осуществления, адаптер, по существу, не является комплементарным к 3’-концу или 5’-концу любой последовательности-мишени, присутствующей в образце. В некоторых вариантах осуществления подходящие длины адаптера находятся в диапазоне приблизительно 10-100 нуклеотидов, приблизительно 12-60 нуклеотидов или приблизительно 15-50 нуклеотидов в длину. В основном, адаптер может включать любую комбинацию нуклеотидов и/или нуклеиновых кислот. В некоторых аспектах адаптер может включать одну или более расщепляемых групп в одном или более местах. В другом аспекте адаптер может включать в себя последовательность, которая по существу идентична или по существу комплементарна, по меньшей мере, части праймера, например универсального праймера. В некоторых вариантах осуществления адаптер может включать в себя штрихкод (также называемый в документе «метка или индекс») для помощи в исправлении последующих ошибок, идентификации или секвенирования. Термины «адаптор» и «адаптер» используют взаимозаменяемо.
Как применяют в настоящем документе, термин «каждый» при использовании по отношению к совокупности элементов предназначен для идентификации отдельного элемента в совокупности, но не обязательно относится к каждому элементу в совокупности, если из контекста явно не следует иное.
Как применяют в настоящем документе, термин «транспорт» относится к движению молекулы через жидкость. Термин может включать пассивный транспорт, такой как движение молекул вдоль градиента их концентрации (например, пассивная диффузия). Термин также может включать активный транспорт, посредством которого молекулы могут двигаться вдоль градиента концентрации или против градиента концентрации. Таким образом, транспорт может включать в себя применение энергии для перемещения одной или более молекул в желаемом направлении или в желаемое место, такое как сайт амплификации.
Как применяют в настоящем документе термины «амплифицировать», «амплифицированный» или «реакция амплификации» и их производные, в основном, относятся к любому действию или процессу, посредством которого, по меньшей мере, часть молекулы нуклеиновой кислоты реплицируется или копируется, по меньшей мере, в одну дополнительную молекулу нуклеиновой кислоты. Дополнительная молекула нуклеиновой кислоты необязательно включает последовательность, которая по существу идентична или по существу комплементарна, по меньшей мере, некоторой части матричной молекулы нуклеиновой кислоты. Матричная молекула нуклеиновой кислоты может быть одноцепочечной или двухцепочечной, а дополнительная молекула нуклеиновой кислоты может независимо быть одноцепочечной или двухцепочечной. Амплификация необязательно включает линейную или экспоненциальную репликацию молекулы нуклеиновой кислоты. В некоторых вариантах осуществления можно проводить такую амплификацию с использованием изотермических условий; в других вариантах осуществления такая амплификация может включать термоциклирование. В некоторых вариантах осуществления амплификация представляет собой мультиплексную амплификацию, которая включает одновременную амплификацию множества последовательностей-мишеней в одной реакции амплификации. В некоторых вариантах осуществления «амплификация» включает амплификацию, по меньшей мере, некоторой части нуклеиновых кислот на основе ДНК и РНК по отдельности или в комбинации. Реакция амплификации может включать любой из процессов амплификации, известных специалисту в данной области. В некоторых вариантах осуществления реакция амплификации включает в себя полимеразную цепную реакцию (ПЦР).
Как применяют в настоящем документе, термин «условия амплификации» и его производные, в основном относится к условиям, подходящим для амплификации одной или более последовательностей нуклеиновых кислот. Такая амплификация может быть линейной или экспоненциальной. В некоторых вариантах осуществления условия амплификации могут включать изотермические условия или альтернативно могут включать условия термоциклирования или комбинацию изотермических и термоциклирующих условий. В некоторых вариантах осуществления условия, подходящие для амплификации одной или более последовательностей нуклеиновой кислоты включают условия полимеразной цепной реакции (ПЦР). Как правило, условия амплификации относятся к реакционной смеси, которая достаточна для амплификации нуклеиновых кислот, например одной или более последовательностей-мишеней, фланкированных универсальной последовательностью, или для амплификации амплифицированной последовательности-мишени, лигированной с одним или более адаптерами. В основном, условия амплификации включают катализатор для амплификации или для синтеза нуклеиновой кислоты, например полимеразу; праймер, который обладает некоторой степенью комплементарности для нуклеиновой кислоты, которую необходимо амплифицировать; и нуклеотиды, такие как дезоксирибонуклеотид трифосфаты (dNTP), чтобы способствовать удлинению праймера после гибридизации с нуклеиновой кислотой. Условия амплификации могут потребовать гибридизации или отжига праймера с нуклеиновой кислотой, удлинения праймера и этапа денатурации, на котором удлиненный праймер отделяется от последовательности нуклеиновой кислоты, подвергающейся амплификации. Как правило, но не обязательно, условия амплификации могут включать термоциклирование; в некоторых вариантах осуществления условия амплификации включают множество циклов, в которых повторяются этапы отжига, удлинения и разделения. Как правило, условия амплификации включают катионы, такие как Mg2+ или Mn2+, и могут также включать различные модификаторы ионной силы.
Как применяют в настоящем документе, термин «повторная амплификация» и его производные относятся, в основном, к любому способу, посредством которого, по меньшей мере, часть амплифицированной молекулы нуклеиновой кислоты дополнительно амплифицируется с помощью любого подходящего способа амплификации (называемого в некоторых вариантах осуществления «вторичной» амплификацией), в результате чего образуется повторно амплифицированная молекула нуклеиновой кислоты. Вторичная амплификация не обязательно должна быть идентична первоначальному способу амплификации, в результате которого была получена амплифицированная молекула нуклеиновой кислоты; и при этом не требуется, чтобы повторно амплифицированная молекула нуклеиновой кислоты была полностью идентичной или полностью комплиментарной амплифицированной молекуле нуклеиновой кислоты; все, что требуется, - это, чтобы повторно амплифицированная молекула нуклеиновой кислоты включала, по меньшей мере, часть амплифицированной молекулы нуклеиновой кислоты или комплементарную ей последовательность. Например, повторная амплификация может включать использование разных условий амплификации и/или разных праймеров, в том числе мишень-специфичных праймеров, отличающихся от первичной амплификации.
Как применяют в настоящем документе, термин «полимеразная цепная реакция» («ПЦР») относится к способу, описанному у Мюллиса в патентах США №№ 4683195 и 4683202, который описывает способ увеличения концентрации сегмента полинуклеотида, представляющего интерес, в смеси геномной ДНК без клонирования или очистки. Этот способ амплификации полинуклеотида, представляющего интерес, состоит из введения большого избытка двух олигонуклеотидных праймеров в смесь ДНК, содержащую желаемый полинуклеотид, представляющий интерес, с последующей серией термоциклирования в присутствии ДНК-полимеразы. Два праймера комплементарны соответствующим цепям двухцепочечного полинуклеотида, представляющего интерес. Смесь сначала денатурируется при более высокой температуре, а затем праймеры отжигаются на комплементарных последовательностях в молекуле полинуклеотида, представляющего интерес. После отжига праймеры удлиняются с помощью полимеразы, образуя новую пару комплементарных цепей. Стадии денатурации, отжига праймера и удлинения при помощи полимеразы могут повторяться много раз (называются термоциклированием), чтобы получить высокую концентрацию амплифицированного сегмента желаемого полинуклеотида, представляющего интерес. Длина амплифицированного сегмента желаемого полинуклеотида, представляющего интерес, (ампликона) определяется относительными положениями праймеров относительно друг друга, и, таким образом, эта длина является контролируемым параметром. В силу повторения процесса способ обозначают ПЦР. Поскольку амплифицированные сегменты желаемого полинуклеотида, представляющие интерес, становятся преобладающими последовательностями нуклеиновой кислоты (в отношении концентрации) в смеси, их обозначают как «ПЦР-амплифицированные». В модификации вышеописанного способа молекулы-мишени нуклеиновых кислот могут быть амплифицированы с помощью ПЦР с использованием множества различных пар праймеров, в некоторых случаях одной или более пар праймеров на молекулу-мишень нуклеиновой кислоты, представляющую интерес, тем самым образуя мультиплексную реакцию ПЦР.
Как определено в настоящем документе, «мультиплексная амплификация» относится к селективной и неслучайной амплификации двух или более последовательностей-мишеней в образце с использованием, по меньшей мере, одного специфичного для мишени праймера. В некоторых вариантах осуществления мультиплексную амплификацию проводят таким образом, что некоторые или все последовательности-мишени амплифицируют в пределах одного реакционного сосуда. «Плексность» или «плекс» данной мультиплексной амплификации относится, в основном, к числу различных мишень-специфических последовательностей, которые амплифицируют во время этой одиночной мультиплексной амплификации. В некоторых вариантах осуществления пллексность может быть приблизительно 12-плексной, 24-плексной, 48-плексной, 96-плексной, 192-плексной, 384-плексной, 768-плексной, 1536-плексной, 3072-плексной, 6144-плексной или выше. Также возможно обнаружить амплифицированные последовательности-мишени, используя несколько различных способов (например, электрофорез в геле с последующей денситометрией, количественное определение с помощью биоанализатора или количественной ПЦР, гибридизацию с меченым зондом; включение биотинилированнных праймеров с последующим обнаружением конъюгата авидин-фермент; включение дезоксинуклеотидтрифосфатов, меченных32P, в амплифицированную последовательность-мишень).
Как применяют в настоящем документе, термин «амплифицированные последовательности-мишени» и его производные, в основном, относится к последовательности нуклеиновой кислоты, образующейся путем амплификации последовательностей-мишеней с использованием специфических для мишени праймеров и способов, представленных в настоящем документе. Амплифицированные последовательности-мишени могут быть смысловые (то есть быть положительной цепью) или антисмысловые (то есть быть отрицательной цепью) относительно последовательностей-мишеней.
Как применяют в настоящем документе, термины «лигированный», «лигирование» и их производные относятся, в основном, к процессу ковалентного связывания двух или более молекул друг с другом, например, ковалентного связывания двух или более молекул нуклеиновых кислот друг с другом. В некоторых вариантах осуществления лигирование включает в себя присоединение разрывов между соседними нуклеотидами нуклеиновых кислот. В некоторых вариантах осуществления лигирование включает формирование ковалентной связи между концом первой и концом второй молекулы нуклеиновой кислоты. В некоторых случаях лигирование может включать формирование ковалентной связи между 5'-фосфатной группой одной нуклеиновой кислоты и 3'-гидроксильной группой второй нуклеиновой кислоты, в результате чего образуется лигированная молекула нуклеиновой кислоты. В основном, для целей данного изобретения, амплифицированная последовательность-мишень может быть лигирована с адаптером, для получения лигированной с адаптером амплифицированной последовательности-мишени.
Как применяют в настоящем документе, термин «лигаза» и ее производные относится, в основном, к любому агенту, способному катализировать лигирование двух субстратных молекул. В некоторых вариантах осуществления лигаза включает фермент, способный катализировать соединение разрывов между соседними нуклеотидами нуклеиновой кислоты. В некоторых вариантах осуществления лигаза включает фермент, способный катализировать образование ковалентной связи между 5'-фосфатом одной молекулы нуклеиновой кислоты и 3'-гидроксилом другой молекулы нуклеиновой кислоты, тем самым образуя лигированную молекулу нуклеиновой кислоты. Подходящие лигазы могут в качестве неограничивающих примеров включать ДНК-лигазу Т4, РНК-лигазу Т4 и ДНК-лигазу Е. coli.
Как применяют в настоящем документе, термин «условия лигирования» и его производные, в основном, относится к условиям, подходящим для лигирования двух молекул друг с другом. В некоторых вариантах осуществления заявленные условия лигирования являются подходящими для лигирования разрывов или делеций между нуклеиновыми кислотами. Термин «разрыв» или «делеция» соответствует использованию термина в данной области. Как правило, разрыв или делеция могут быть лигированы в присутствии фермента, такого как лигаза, при соответствующей температуре и pH. В некоторых вариантах осуществления ДНК-лигаза Т4 может соединять разрыв между нуклеиновыми кислотами при температуре примерно 70-72°С.
Как применяют в настоящем документе, термин «проточная ячейка» относится к камере, содержащей твердую поверхность, через которую может протекать один или более жидких реагентов. Примеры проточных ячеек и связанных с ними флюидных систем и платформ для детекции, которые могут быть легко использованы в способах по настоящему изобретению, описаны, например, в Bentley et al., Nature 456: 53-59 (2008), WO 04/018497; US 7057026; WO 91/06678; WO 07/123744; US 7329492; US 7211414; US 7315019; US 7405281 и US 2008/0108082.
Как применяют в настоящем документе, термин «ампликон» при использовании по отношению к нуклеиновой кислоте означает продукт копирования нуклеиновой кислоты, где продукт имеет нуклеотидную последовательность, которая совпадает или комплементарна, по меньшей мере, части нуклеотидной последовательности нуклеиновой кислоты. Ампликон можно получать любым из ряда способов амплификации, использующих нуклеиновую кислоту или ее ампликон, в качестве матрицы, включая, например, удлинение с помощью полимеразы, полимеразную цепную реакцию (ПЦР), амплификацию по типу катящегося кольца (RCA), удлинение путем лигирования или лигазную цепную реакцию. Ампликоном может быть молекула нуклеиновой кислоты, имеющая одну копию конкретной нуклеотидной последовательности (например, продукт ПЦР) или более копий нуклеотидной последовательности (например, конкатамерный продукт RCA). Первый ампликон нуклеиновой кислоты-мишени является, как правило, комплементарной копией. Последующие ампликоны представляют собой копии, которые создаются после генерации первичного ампликона из нуклеиновой кислоты-мишени или первичного ампликона. Последующий ампликон может иметь последовательность, которая по существу комплементарна нуклеиновой кислоте-мишени или по существу идентична нуклеиновой кислоте-мишени.
Как применяют в настоящем документе, термин «сайт амплификации» относится к участку в чипе или на чипе, в котором можно получать один или более ампликонов. Сайт амплификации может быть дополнительно сконфигурирован для того чтобы содержать, сохранять или присоединять, по меньшей мере, один ампликон, который получают в этом сайте.
Как применяют в настоящем документе, термин «чип» относится к совокупности участков, которые можно различить друг от друга по относительному местоположению. Разные молекулы, которые находятся на разных участках чипа, могут различаться друг от друга в зависимости от местоположения участков на чипе. Отдельный участок чипа может включать одну или более молекул определенного типа. Например, участок может включать одну молекулу-мишень нуклеиновой кислоты, имеющую конкретную последовательность, или участок может включать несколько молекул нуклеиновой кислоты, имеющих одинаковую последовательность (и/или комплементарную ей последовательность). Участки чипа могут представлять собой разные элементы, расположенные на одном и том же субстрате. Типичные элементы в качестве неограничивающих примеров включают в себя лунки в субстрате, гранулы (или другие частицы) внутри или на субстрате, выступы из субстрата, борозды на субстрате или каналы в субстрате. Участки чипа могут быть отдельными субстратами, каждый из которых несет разную молекулу. Различные молекулы, прикрепленные к отдельным субстратам, могут быть идентифицированы в соответствии с местоположением субстратов на поверхности, с которой субстраты ассоциированы или согласно местоположениям субстратов в жидкости или геле. Примеры чипов, в которых отдельные субстраты расположены на поверхности, в качестве неограничивающих примеров включают в себя те, которые имеют гранулы в лунках.
Как применяют в настоящем документе, термин «емкость» при использовании в отношении к участку и материалу нуклеиновой кислоты означает максимальное количество материала нуклеиновой кислоты, которое может занимать участок. Например, термин может относиться к общему количеству молекул нуклеиновой кислоты, которые могут занимать участок в определенных условиях. Также можно использовать другие показатели, включая, например, общую массу материала нуклеиновой кислоты или общее количество копий конкретной нуклеотидной последовательности, которая может занимать участок в определенном состоянии. Как правило, емкость участка для нуклеиновой кислоты-мишени будет по существу эквивалентна емкости участка для ампликонов нуклеиновой кислоты-мишени.
Как применяют в настоящем документе, термин «захватывающий агент» относится к материалу, химическому веществу, молекулам или их группе, которые способны прикрепляться, удерживать или связываться с молекулой-мишенью (например, нуклеиновой кислотой-мишенью). Примеры захватывающих агентов в качестве неограничивающих примеров включают захватывающую нуклеиновую кислоту (также называемую в документе «захватывающим олигонуклеотидом»), которая комплементарна, по меньшей мере, части нуклеиновой кислоты-мишени, участника пары связывания рецептор-лиганд (например, авидин, стрептавидин, биотин, лектин, углевод, белок, связывающий нуклеиновую кислоту, эпитоп, антитело и т.д.), способного связываться с нуклеиновой кислотой-мишенью (или связывающая группа, присоединенная к нему), или химический реагент, способный формировать ковалентную связь с нуклеиновой кислотой-мишенью (или связывающая группа, присоединенная к нему).
Как применяют в настоящем документе, термин «репортерная группа» может относиться к любой идентифицируемой метке, индексам, штрихкодам или группе, которая позволяет определить состав, идентичность и/или источник исследуемого аналита. В некоторых вариантах осуществления репортерная группа может включать антитело, которое специфически связывается с белком. В некоторых вариантах осуществления антитело может включать детектируемую метку. В некоторых вариантах осуществления репортер может включать антитело или аффинный реагент, меченный меткой на основе нуклеиновой кислоты. Метка на основе нуклеиновой кислоты может быть обнаружена, например, с помощью способа близкого лигирования (PLA) или анализа расширения близости (PEA) или считывания на основе секвенирования (Shahi et al. Scientific Reports том 7, номер статьи: 44447, 2017) или CITE-seq (Stoeckius et al. Nature Methods 14: 865-868, 2017).
Как применяют в настоящем документе, термин «клональная популяция» относится к популяции нуклеиновых кислот, которая является гомогенной в отношении определенной нуклеотидной последовательности. Гомогенная последовательность имеет длину, как правило, по меньшей мере, 10 нуклеотидов, но может быть даже более длинной, включая, например, по меньшей мере, длину 50, 100, 250, 500 или 1000 нуклеотидов. Клональную популяцию можно получать из одной нуклеиновой кислоты-мишени или матричной нуклеиновой кислоты. Как правило, все нуклеиновые кислоты в клональной популяции будут иметь одинаковую нуклеотидную последовательность. Следует понимать, что небольшое количество мутаций (например, из-за артефактов амплификации) может происходить в клональной популяции без отхода от клональности.
Как применяют в настоящем документе, термин «уникальный молекулярный идентификатор» или «UMI» относится к молекулярной метке, случайной, неслучайной или полуслучайной, которая может быть присоединена к молекуле нуклеиновой кислоты. При включении в молекулу нуклеиновой кислоты UMI можно использовать для корректировки последующей ошибки амплификации путем прямого подсчета уникальных молекулярных идентификаторов (UMI), которые секвенировались после амплификации.
Как применяют в настоящем документе, «получение» в отношении композиции, изделия, нуклеиновой кислоты или ядра означает создание композиции, изделия, нуклеиновой кислоты или ядра, закупки композиции, изделия, нуклеиновой кислоты или ядра, или иным образом получения соединения, композиции, изделия или ядра.
Термин «и/или» означает один или все из перечисленных элементов или комбинацию любых двух или более из перечисленных элементов.
Слова «предпочтительный» и «предпочтительно» относятся к вариантам осуществления изобретения, которые могут давать определенные выгоды при определенных обстоятельствах. Однако другие варианты осуществления могут также быть предпочтительными при тех же или других обстоятельствах. Кроме того, изложение одного или более предпочтительных вариантов осуществления не подразумевает, что другие варианты бесполезны, и не предназначено для исключения других вариантов осуществления из объема изобретения.
Термин «содержит» и его вариации не имеют ограничивающего значения, когда эти термины появляются в описании и формуле изобретения.
Следует понимать, что где бы варианты не были описаны в документе с формулировкой «включают в себя», «включает в себя» или «включая» и т.д., также предлагаются иные аналогичные варианты, описанные в отношении «состоящий из» и/или «состоящий из по существу».
Если не указано иное, «a», «an», «the» и «по меньшей мере, один» используются взаимозаменяемо и означают один или более, чем один.
Также в настоящем документе, перечисление числовых диапазонов по конечным точкам включает все числа, отнесенные к этому диапазону (например, от 1 до 5 включает в себя 1, 1,5, 2, 2,75, 3, 3,80, 4, 5 и т.д.).
Для любого способа, описываемого в настоящем документе, который включает в себя отдельные этапы, этапы можно проводить в любом возможном порядке. И, при необходимости, любое сочетание двух или более этапов можно проводить одновременно.
Ссылка на всем протяжении этого описания на «один из вариантов осуществления», «вариант осуществления», «конкретные варианты осуществления» или «некоторые варианты осуществления» означает, что определенная особенность, конфигурация, композиция или характеристика, описанные в связи с вариантом осуществления, включены, по меньшей мере в один из вариантов осуществления. Таким образом, появление таких фраз в разных местах на всем протяжении настоящего описания не обязательно относится к одному и тому же варианту осуществления изобретения. Кроме того, конкретные особенности, конфигурации, композиции или характеристики можно комбинировать любым подходящим образом в одном или более вариантах осуществления.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Следующее подробное описание иллюстративных вариантов осуществления настоящего изобретения может быть лучше понято при чтении в сочетании со следующими чертежами.
На ФИГ. 1A-C показаны общие блок-схемы общих иллюстративных способов для комбинаторного индексирования одиночных клеток по настоящему изобретению.
На ФИГ. 2 показана общая блок-схема общего иллюстративного способа для комбинаторного индексирования одиночных клеток по настоящему изобретению.
На Фиг. 3А-F показано, что sci-L3-WGS обеспечивает высокопроизводительную линейную полногеномную амплификацию одиночной клетки. (A) Схема рабочего процесса sci-L3-WGS с тремя уровнями индексирования. (B) Вверху: структура штрихкода полученного амплифицированного ДНК-дуплекса, совместимая с различными способами получения библиотек. bc, штрихкод; sp, спейсер; гДНК, геномная ДНК. Посередине: пример структуры библиотеки для sci-L3-WGS. Адаптеры для секвенирования P5 и P7 добавляют посредством присоединения А-хвоста и лигирования. Следует отметить, что наличие P7 на конце UMI и P5 на конце гДНК одинаково возможно благодаря симметрии лигирования. Внизу: пример структуры библиотеки для sci-L3-target-seq. Адаптеры для секвенирования P5 и P7 добавляются путем праймирования из спейсера 2 (sp2) и локусов-мишеней в геноме, представляющих интерес, соответственно. Следует отметить, что новый третий раунд штрихкода bc3’также добавляют путем ПЦР, соответствующей каждому bc3 в библиотеке полногеномного секвенирования, а новый UMI’ добавляют вне bc3’. (C) Диаграмма рассеяния числа уникальных сайтов вставки Tn5 от клеток человека и мыши при низкой глубине секвенирования, 24 bc1×64 bc2×6 bc3 sci-L3-WGS, от 100 до 300 клеток, отсортированных на лунку. Синие, предполагаемые мышиные клетки (процент ридов для мыши >95%, медиана 98,7%, n=315); красные, предполагаемые клетки человека (процент ридов для человека >95%, медиана 99,8%, n=719); серые, предполагаемые столкновения (n=48,4%). (D) Коробчатые диаграммы, показывающие количество уникальных сайтов вставки Tn5 на клетку при среднем 2,4 миллиона необработанных ридов на клетку и глубине 1,78×. Глубина определяется как отношение количества уникальных транскриптов IVT к количеству уникальных сайтов вставки Tn5. Толстые горизонтальные линии, медианы; верхние и нижние края коробки, первый и третий квартили соответственно; усы, в 1,5 раза превышающие межквартильный размах; круги, выбросы). См. также Фиг. 5 и Пример 2, раздел «Способы и молекулярный дизайн sci-L3-WGS и sci-L3-target-seq», для характеристики библиотеки, созданной с улучшенными версиями протокола. (E) Пример графиков хромосомных CNV для отдельных клеток. Верхний, клетка HEK293T, 2,6 миллиона необработанных ридов, 2,4 миллиона уникальных молекул, 1,3 миллиона уникальных сайтов вставки Tn5 с MAPQ > 1. Нижний, клетка 3T3, 2,7 миллиона необработанных ридов, 2,4 миллиона уникальных молекул, 1,2 миллиона уникальных участков инсерции Tn5 с MAPQ > 1. (F) Коробчатые диаграммы для вариаций числа копий в 822 клетках 293T или 1453 клетках HAP1. Ось Y отображает доли ридов на хромосому, нормированные по длине хромосомы, так что ожидается, что эуплоидная хромосома без добавления или потери копии сегмента будет иметь значение 1.
На Фиг. 4A-F показаны молекулярные структуры для sci-LIANTI на каждом этапе. Пунктирная линия: РНК, сплошная линия: ДНК. (A) У адаптеров Tn5 оба 5’-конца фосфорилированы, один необходим для вставки, и один необходим для лигирования. «Липкий» конец отжигающегося транспозона содержит штрихкоды первого раунда («bc1») и спейсер («sp1») для лигирования. (B) Молекула для лигирования предварительно отжигается в виде шпилечной петли, которая снижает межмолекулярное лигирование от трех молекул до двух молекул; шпилечная структура также помогает улучшить эффективность ОТ в последующих этапах. Шпилька содержит 1) липкий конец, который отжигается с «sp1» для лигирования, 2) штрихкоды второго раунда («bc2») и спейсер («sp2»), который служит в качестве сайта прайминга на «стебле» для SSS в последующих этапах, и 3) промотор T7 в петле для IVT. (C) продление делеции превращает закольцованный промотор T7 в дуплекс. Следует отметить, что если лигирование успешно на обоих концах, промоторы T7 присутствуют на обеих сторонах; однако, если лигирование успешно на одном конце, часть, заключенная в рамку, будет отсутствовать. Однако, оба могут быть обратно транскрибированы на последующих этапах с различными праймерами для ОТ. (D) IVT генерирует одноцепочечные РНК-ампликоны ниже промотора Т7. (E) Если лигирование было успешным на обоих концах, ОТ предпочтительно запускается с самозакольцованными ОТ-праймерами, которые пришли от закольцованной молекулы для лигирования; если лигирование было успешным только на одном конце, RT запускается дополнительными РНК-праймерами для ОТ, добавленными в избытке. Избыток РНК-праймеров затем удаляют перед SSS, чтобы избежать вмешательства в последующую реакцию SSS. (F) Двухцепочечные молекулы ДНК производятся SSS, которая удаляет «sp2», чтобы одновременно добавить штрихкоды третьего раунда и UMI-метку каждого транскрипта. Более подробное объяснение приведено в разделе 2 «Способы и молекулярный дизайн sci-L3-WGS и sci-L3-target-seq».
На фиг.5А-G показаны количества ридов в разных экспериментах sci-L3-WGS и с различными концентрациями транспозомы Tn5. Коробчатые диаграммы показывают количество уникальных сайтов вставки Tn5 на клетку на указанных глубинах. Глубину определяют как отношение количества уникальных транскриптов IVT к количеству уникальных сайтов вставки Tn5. Толстые горизонтальные линии, медианы; верхние и нижние края ящика, первый и третий квартили соответственно; усы, в 1,5 раза превышающие межквартильный размах; круги, выбросы). Концентрированная транспозома Tn5: 0,2 мкМ, разбавленная транспозома Tn5: 0,1 мкМ. (A) yi128 (средняя глубина: 1,19×) уникальные риды человека по сравнению с мышью (медиана уникальных ридов человека: 215000, n=115 клеток; медиана уникальных ридов мыши: 169000, n=44) с концентрированным Tn5; уникальные риды человека с концентрированным Tn5 (медиана уникальных ридов: 215000) против разбавленного Tn5 (медиана уникальных ридов: 46000). (B) yi129 (средняя глубина: 1,78×) уникальные риды человека с концентрированным Tn5 (медиана уникальных ридов: 635000) против разбавленного Tn5 (медиана уникальных ридов: 183000). Уникальные риды мыши представлены на Фиг. 3D. (C) yi140 и yi141 (медиана глубины: 1,37×; медиана уникальных ридов человека: 660000) с концентрированным Tn5. См. также таблицу 2 и Пример 2. (D) yi144 и yi145 (медиана глубины: 1,05×; медиана уникальных ридов человека: 97300) с концентрированным Tn5. См. также таблицу 2. Следует заметить, что yi140, yi141, yi144 и yi145 представляют собой библиотеки с оптимизированным протоколом, обсуждаемые в Примере 2. (E) yi174 (медиана глубины: 1,06×) уникальные риды человек/мышь (медиана уникальных ридов человека: 100000, n=103; медиана уникальных ридов мыши: 23000, n=35) с концентрированным Tn5; уникальные риды человека с концентрированным Tn5 (медиана уникальных ридов: 100000) и разведенным Tn5 (медиана уникальных ридов: 54k) Tn5. (F) Библиотеки половых клеток мыши: yi186, yi187, yi188 получают с разведенным Tn5; yi190, yi192, yi193 получают с концентрированным Tn5. (G) Число уникальных сайтов вставки Tn5 как функция от глубины секвенирования. Синие и красные линии показывают sci-L3-WGS с РНК-праймерами для ОТ и без них, соответственно (Пример 2). yi129 (как на панели B, медиана глубины: 1,78×) уникальные вставки человека с концентрированным Tn5 (медиана уникальных вставок: 630000). При проектировании на глубину 5× и 10×, ожидаемое число уникальных вставок составило 1,9 миллиона и 2,6 миллиона, соответственно. Комбинация yi140 и yi141 имела медиану глубины 1,37×, медиану уникальных вставок 660000. При проектировании глубины до 1,78×, 5× и 10×, ожидаемое число уникальных вставок составило 1,5 миллиона, 4,2 миллиона и 6,0 миллиона, соответственно.
На фиг.6А-Е показан совместный анализ РНК/ДНК на основе Sci-L3, который позволяет совместно производить высокопроизводительную и линейную амплификацию генома и транскриптома из одной и той же одиночной клетки. (A) Схема рабочего процесса совместного анализа sci-L3-РНК/ДНК с тремя уровнями индексирования. Следует заметить, что как транспозон Tn5, так и праймер для синтеза кДНК содержат одну и ту же фосфорилированную посадочную площадку для лигирования (розовая) на 5'-липком конце вне штрихкодов первого раунда. (B) Структуры штрихкодов, полученных амплифицированных дуплексов, соответствующие геному и транскриптому (слева и справа соответственно), которые совместимы с различными способами получения библиотек. bc, штрихкод; sp, спейсер; гДНК, геномная ДНК. (C) Диаграмма рассеяния числа уникальных сайтов инсерции Tn5 из клеток человека и мыши с низкой и высокой глубиной секвенирования, нанесенных вместе, 24 bc1×64 bc2×6 bc3 совместный анализ sci-L3-РНК/ДНК, отсортировано от 100 до 300 клеток на лунку. Синие, предполагаемые мышиные клетки (процент чтения мыши> 95%, со средним значением 99,5%, n=2002); красные, предполагаемые клетки человека (процент чтения человека > 95%, со средним значением 99,8%, n=2419); серые, предполагаемые столкновения (n=149, 6,6% с малой и большой глубиной вместе; 5/270, 3,7% с большой глубиной). (D) То же, что в (С) для РНК. Синие, предполагаемые мышиные клетки (медиана чистоты ридов мыши составляет 95,1%); красные, предполагаемые клетки человека (медиана чистоты ридов человека составляет 91,5%); серые, предполагаемые столкновения (n=272, 12% с малой и большой глубиной вместе; 7/270, 5,2% с большой глубиной). (E) Seurat с сигналом РНК-seq показывает отличимые кластеры, соответствующие фибробласту кожи человека BJ-5ta (мужские) и клеткам HEK293T (женские). На основании наличия или отсутствия Y-хромосомы, 988/1024 клетки (96,5%) назначены правильно.
На фиг.7А-Е показана митотическая/эквационная и мейотическая/редукционная сегрегация хромосом с наличием и отсутствием кроссинговеров. Каждый вертикальный сегмент представляет одну хроматиду (цепи ДНК не показаны). Черный и синий представляют гомологи. Овалы представляют центромеры. Следует заметить, что мышиные хромосомы являются телецентрическими. Серые кресты изображают участки кроссинговера после репликации ДНК на стадии 4С. Красные прямоугольники указывают на дочерние клетки митоза, которые являются гетерозиготными, а черные и синие прямоугольники указывают на дочерние клетки мейоза I (MI), которые являются гомозиготными по соответствующей исходной цепи в центромерно-проксимальных областях. Области LOH в дочерних клетках отмечены фигурными скобками. (A) Митотическая/эквационная сегрегация без кроссинговера. Обе дочерние клетки сохраняют гетерозиготность. (Б) Митотическая/эквационная сегрегация с кроссинговером между гомологами. Рекомбинированные хроматиды сегрегируются раздельно, что приводит в области центромерно-дистальной LOH к кроссинговеру. (C) Митотическая/эквационная сегрегация с кроссинговером между гомологами. Рекомбинированные хроматиды сегрегируются вместе, так что обе дочерние клетки сохраняют гетерозиготность, но одна дочерняя клетка имеет переключатель сцепления. (D) Мейотическая/редукционная сегрегация с кроссинговером, приводящая в области центромерно-проксимальной LOH к кроссинговеру, в отличие от (B). (E) Мейотическая/редукционная сегрегация без кроссинговера, приводящая к реципрокной однородительской дисомии (UPD) в дочерних клетках. Обратите внимание, что MI с эквационной сегрегацией хромосом напоминает (B) и (C). В тексте, поскольку наше исследование в основном сфокусировано на MI, мы ссылаемся на ожидаемую мейотическую/редукционную сегрегацию во время MI, где сестринские хроматиды сегрегируют вместе, как на «редукционную сегрегацию», и на неожиданную митозоподобную/эквационную сегрегацию во время MI, когда сестринские хроматиды сегрегируют раздельно, как на «эквационную сегрегацию».
На Фиг. 8A-G показаны сперматозоиды и предшественники сперматозоидов и их плоидность по FACS. (A) Визуализация сперматозоидов B6. (B) Визуализация (B6×Spret) сперматозоидов F1. Мы наблюдаем небольшое количество округлых половых клеток неизвестной плоидности и крайне мало морфологически зрелых сперматозоидов (стрелки). (C) (B6×Spret). Сперматозоиды F1 и предшественники сперматозоидов, выделенные из придатка яичка, неожиданно включают большую долю клеток 2C. Напряжение DAPI составило 375. (D) Смесь HEK293/Patski, напряжение DAPI 350. Пик Patski (2C) слегка смещен влево относительно пика 2C в (C) из-за более низкого напряжения DAPI. (E) (B6×Cast) сперматозоиды F1, выделенные из придатка яичка, почти полностью состоят из клеток 1C. Напряжение DAPI 375. (F) (B6×Cast) Предшественники сперматозоидов F1, предварительная сортировка для клеток 2C из отделенных яичек; большое количество клеток 1С все еще присутствует. Напряжение DAPI 375. (G) (B6×Cast) Сперматозоиды F1 и предшественники сперматозоидов на этапе FACS во время sci-L3-WGS (после двух раундов штрихкодирования) все еще состоят в основном из клеток 1C. Основываясь на пропорциях загрязненных ядер 1C в предварительно отсортированных ядрах 2C из (F), мы оцениваем долю меченых ядер, которые являются 2C, равными 18%, 7,2-кратное обогащение по сравнению с 2,5% ядер 2C в гомогенизированных семенниках, мы отсортировали по популяции 2C (15,4% всех клеток, аналогично 18%, оцененным для этапа тагментации). Напряжение DAPI 375.
На фиг.9А-F показано sci-L3-WGS межвидовой гибридной мышиной самцовой зародышевой линии, которое выявляет многочисленные примеры независимой эквационной сегрегации при MI. В (A), (B) и (C) красной линией изображена подгонка перехода кроссинговера путем HMM. Центромера расположена слева для изображения каждой хромосомы. (A) Пример графика кроссинговеров для клеток 1С. Серая точка имеет значение 1 для аллеля Spret и 0 для аллеля B6. В (B) и (C) серая точка показывает частоту аллеля Spret, в среднем по 40 сайтам SNP. (B) Пример графика LOH для клетки M2 с редукционной сегрегацией (см. также фиг. 7D). LOH присутствует в центромерно-проксимальной области сайтов кроссинговера. (C) Пример графика LOH для клетки M2 с эквационной сегрегацией (см. также фиг. 7B). LOH присутствует в центромерно-дистальной области сайтов кроссинговера, в отличие от (B). (D-F) Количество редукционно (красный, розовый, черный) и эквационнно (синий, зеленый) сегрегированных хромосом для каждой клетки М2. Каждый столбец представляет одну клетку M2 (19 хромосом на клетку, распределенные по цветам). (D) Ожидаемое распределение редукционной и эквационной сегрегации на основе биномиального распределения и при условии, что вероятность редукционной сегрегации p равна 0,76, MLE из наблюдаемых данных. (E) Наблюдаемые данные в клетках M2. В редких случаях (27/5548 хромосомы) мы не смогли различить редукционную и эквационную сегрегацию из-за редкого покрытия SNP (пустое пространство в верхней части панели). Черный столбец изображает непересекающуюся область MI (NDJ, всего 40 хромосом), где мы наблюдали 0 или 4 копии хроматид. Обратите внимание, что NDJ рассматривается как редукционная сегрегация, потому что сестринские хроматиды сегрегируют вместе. (F) То же, что и (E), но далее с разбивкой по количеству хромосом с наличием или отсуствием кроссинговеров (сокращенно «CO»). Клетки сначала сортируют по количеству эквационно сегрегированных хромосом (светло-зеленый и синий, в порядке убывания), а затем по количеству наблюдаемых эквационно сегрегированных хромосом без кроссинговера (синий, в порядке убывания).
На Фиг. 10A-G показан мейотический кроссинговер и распределения однородительских хромосом в масштабе хромосомы. (A) После нормализации по размеру хромосомы число гаплоидных клеток, по меньшей мере, с одним кроссинговером на каждую хромосому отрицательно коррелирует с размером хромосомы (r=-0,87, p=2×10-6). Показано скрещивание (B6×Spret). См. Фиг. 14C для скрещивания (B6×Cast). (B) Аналогично (A) для клеток M2 (r=-0,91, p= 8×10-8). См. Фиг. 14D для скрещивания (B6×Cast). (C) Распределение количества кроссинговеров (CO) на хромосому на гаплоидную клетку (среднее=0,62 для (B6×Spret) и среднее=0,58 для (B6×Cast)). (D) Аналогично (C) для клеток M2 (среднее=0,92 для (B6×Spret) и среднее=1,03 для (B6×Cast)). (E) Для хромосом, по меньшей мере, с двумя кроссинговерами, расстояние кроссинговера для всех хромосом. Распределение ожидаемых чисел получают путем случайного размещения 2 кроссинговеров на хромосоме. Показано скрещивание (B6×Spret). См. Фиг. 14E для скрещивания (B6×Cast). (F) Количество (вверху) и распределение на хромосоме (внизу) событий UPD и LOH в клетках Patski. (G) Число митохондриальных копий (нормализованное) с разбивкой по клеткам М2, которые сегрегируют большинство хромосом редукционно, по сравнению с эквационным сегрегированием. Скрещивание (B6×Spret).
На Фиг. 11A-E показано sci-L3-WGS межвидовой гибридной мышиной самцовой зародышевой линии, которое также выявило примеры не-независимой эквационной сегрегации. (A-B) Количество редукционно (красный) и эквационно (голубой) сегрегированных хромосом для искусственных клеток «2C» из группы со штрихкодом 1, которые получены из дублетов двух случайных клеток 1C. Каждый столбец представляет одну клетку 2C (19 хромосом на клетку, распределенные по цветам). (A) ожидаемое распределение редукционной и эквационной сегрегации на основе биномиального распределения и предположения, что вероятность эквационной сегрегации p равна 0,5. (B) Наблюдаемые данные в клетках 2C, которые соответствуют ожидаемому распределению, показанному в (A). (C-E) Количество редукционно (красный, розовый, черный) и эквационно (синий, зеленый) сегрегировавших хромосом для не-1C-клеток из группы со штрихкодом 2, которые представляют собой смесь искусственных дублетов двух случайных ядер 1C и настоящих вторичных сперматоцитов 2C. Каждый столбец представляет одну не-1C клетку (19 хромосом на клетку, распределенные по цветам). (C) Все не-1C клетки из группы со штрихкодом 2. (D) Не-1C клетки только с нарушением сегрегации хромосом, т.е., по меньшей мере, с 15 хромосомами, сегрегировавшими либо эквационно, либо редукционно. Черный столбец изображает нерасхождение во время Мейоза I (NDJ, всего 2 из 2185 хромосом), где мы наблюдали 0 или 4 копии хроматид. (E) То же, что и (D), но далее с разбивкой по количеству хромосом с наличием или отсутствием кроссинговера (сокращенно «CO»). Клетки сортируют сначала по количеству эквационно сегрегированных хромосом (светло-зеленый и синий, в порядке убывания), а затем по количеству наблюдаемых эквационно сегрегированных хромосом без кроссинговера (синий, в порядке убывания).
На фигуре 12A-C показана подходящая модель конечной смеси с тремя биномиальными распределениями (вверху) по сравнению с данными наблюдений (внизу) из sci-L3-WGS мышиной самцовой зародышевой линии. См. Пример 2 для деталей моделирования смеси. (A) Моделирование смеси не-1C клеток из группы штрихкода 1 у гибрида (B6×Cast). (B) Моделирование смеси не-1C клеток из группы штрихкода 2 у гибрида (B6×Cast). (C) Моделирование смеси клеток 2C клеток из скрещивания (B6×Spret).
На Фиг. 13А-I показаны распределения мейотического кроссинговера и однородительских хромосом в масштабе хромосомы. (A) Количество кроссинговеров, нормализованных по размеру хромосомы (сМ/Мб), отрицательно коррелирует с размером хромосомы в гаплоидных клетках (r=-0,66, p=0,002). Показано скрещивание (B6×Spret). См. Фиг. 14A для скрещивания (B6×Cast). (B) То же, что (A) для клеток M2 (r=-0,83, p=1×10-5). Показано скрещивание (B6×Spret). См. Фиг. 14B для скрещивания (B6×Cast). (C) Распределение частоты кроссинговера (CO) на хромосому на гаплоидную клетку. См. Фиг. 10С для распределения подсчетов. (D) То же, что (C) для клеток M2. См. Фиг. S6D для распределения подсчетов. (E) Для хромосом, по меньшей мере, с двумя кроссинговерами расстояние (Мб) между кроссинговерами для хромосом 1, 2, 12 и 13. См. Фиг. S6E для всех хромосом. Показано скрещивание (B6×Spret). См. Фиг. 14E для скрещивания (B6×Cast). Распределение ожидаемого количества получают путем случайного размещения 2 кроссинговеров на хромосому. Коробчатая диаграмма показывает, что скрещивание (B6×Cast) имеет более сильное вмешательство кроссинговеров, чем скрещивание (B6×Spret) (p=5×10-91). (F) Гистограммы числа однородительских хромосом на гаплоид (медиана=8, среднее=8,1), клетка M2 (медиана=1, среднее=1,1) или другая клетка диплоид/4C (медиана=0, среднее=0, 4) клетка. Показано скрещивание (B6×Spret). См. Фиг. 14F для скрещивания (B6×Cast). (G) Распределение однородительских хромосом для гаплоида (r=-0,87, p=2×10-6), клетка M2 (r=-0,75, p=2×10-4) и других клеток диплоид/4C (r=-0,68, р=0,001). Показано скрещивание (B6×Spret). См. Фиг. 14G для скрещивания (B6×Cast). (H) Хромосомное распределение событий обратной сегрегации в скрещиваниях (B6×Spret) (слева) и (B6×Cast) (справа). (I) Количество митохондриальных ридов на клетку, нормализованное по глубине считывания, для гаплоидной, клетки М2 и других диплоидных/4C клеток. Скрещивание (B6×Spret).
На Фиг. 14A-G показано хромосомное распределение для мейотического кроссинговера и UPD, (B6×Cast). (A) Количество кроссинговеров, нормализованных по размеру хромосомы (сМ/Мб), отрицательно коррелирует с размером хромосомы в гаплоидных клетках (r=-0,65, p=0,003). Скрещивание (B6×Cast). (B) То же, что (A) в клетках M2 (r=-0,9, p=2×10-7). Скрещивание (B6×Cast). (C) После нормализации по размеру хромосомы число гаплоидных клеток, по меньшей мере, с одним кроссинговером на каждую хромосому отрицательно коррелирует с размером хромосомы (r=-0,85, p=5×10-6). Скрещивание (B6×Cast). (D) То же, что (C) для клеток M2 (r=-0,94, p=3×10-9). Скрещивание (B6×Cast). (E) Для хромосом, по меньшей мере, с двумя кроссинговерами, расстояние кроссинговера для всех хромосом. Распределение ожидаемых чисел получают путем случайного размещения 2 кроссинговеров на хромосоме. Скрещивание (B6×Cast). (F) Количество однородительских хромосом на гаплоидные клетки (медиана=8, среднее=8,9) и клетки M2 (медиана=0, среднее=0,54). Скрещивание (B6×Cast). (G) Распределение однородительских хромосом (корреляция с размером хромосомы, показанным в скобках), гаплоидная клетка (r=-0,8, p=4×10-5) и клетка M2 (r=-0,45, p=0,05). Скрещивание (B6×Cast).
На Фиг. 15А-С показан профиль наложения точек разрыва кроссинговера. (A) Сверху вниз: точка мейотического DSB по карте SSDS для B6, Cast и F1-гибрида (B6×Cast), карта кроссинговера у (B6 x Spret) и (B6×Cast), сгенерированная в этом исследовании). См. (B) и (C) для разрыва у гаплоидной клетки по сравнению с клеткой M2, а также Spol1-олиго-карта. (B) Сверху вниз: 1) карта горячих точек мейотического DSB по SSDS для гибрида F1 (B6×Cast), 2) карта кросинговеров для гаплоида (B6×Cast) и 3) карта кроссинговеров для клетки M2 у (B6×Cast). (C) Сверху вниз: 1) мейотическая горячая точка DSB по карте Spo11-oligo с «симметричными» точками, 2) мейотическая горячая точка DSB по карте Spo11-oligo со всеми горячими точками: мотивы PRDM9 не учитываются. 3) карта кросинговеров для гаплоида (B6×Spret), и 4 карта кроссинговеров для клетки M2 у (B6×Spret).
Фиг. 16A-F показывает горячие участки мейотического кроссинговера и объяснительные геномные признаки. (A) Маргинальная вероятность включения для признаков, связанных с кроссинговерной активностью по BMA. Ось X ранжирует модели по апостериорной вероятности, где серые прямоугольники показывают признаки, не включенные в каждую модель (вертикальная линия, показаны 20 верхних моделей), а оранжевая шкала отображает апостериорную вероятность моделей. Комбинированный набор данных из скрещиваний (B6×Spret) и (B6×Cast) показан здесь. См. Фиг. 15 для двух скрещиваний, проанализированных раздельно. (B) Распределение размеров для разрешения точки разрыва (логарифмически нормальное распределение). Слева: (B6×Spret), медиана 150 т.п.н. Справа: (B6×Cast), медиана 250 т.п.н. (C-D) Положение самого правого кроссинговера каждой хромосомы. Длина хромосомы указана самым правым SNP (черная полоса), а не протяженностью красной линии. (С) Клетка М2. Кроссинговеры для скрещивания (B6×Cast) (слева) предпочитают центромерно-дистальный конец хромосомы, в то время как кроссинговеры для скрещивания (B6×Spret) (справа) предпочитают среднюю область каждого плеча хромосомы. После учета изменчивости между хромосомами мы оцениваем, что кроссинговеры у скрещивания (B6×Spret) в среднем на 5,5 Мб более центромерно-проксимальные. См. Фиг. 20А, аналогично, но для клеток 1С. (D) Сравнение клеток 1C и M2, скрещивание(B6×Spret). После учета вариабельности между хромосомами, мы оцениваем, что кроссинговеры в клетках М2 (справа) в среднем на расположены 9,4 Мб более проксимально центромеры, чем в 1С (слева) в скрещивании (B6×Spret). Та же тенденция наблюдается в меньшей степени в скрещивании (B6×Cast) (см. Рис. 20B). (E) AUC 0,73 количественно определяет ожидаемую точность в прогнозировании, если область, взятая из мышиного генома, происходит из участка кроссинговера B6×Spret или равного числа случайно выбранных участков. Слева: все 76 признаков. Справа: подмножество 25 признаков из BMA с MIP> 0,5. (F) AUC 0,85 количественно определяет ожидаемую точность в прогнозировании, если область, взятая из мышиного генома, происходит из участка кроссинговера B6×Cast или равного числа случайно выбранных участков. Слева: все 69 признаков. Справа: подмножество 25 признаков из BMA с MIP> 0,5.
Фиг. 17A-B показывает маргинальную вероятность включения для характеристик, связанных с активностью кроссинговера по BMA. Ось X ранжирует модели по апостериорной вероятности. (A) Скрещивание (B6×Cast). (B) Скрещивание (B6×Spret).
На фиг. 18 представлена корреляционная матрица как для событий кроссинговера, так и для геномных особенностей для скрещивания (B6×Cast). Здесь мы показываем все возможные попарные корреляции между различными скоплениями участков кроссинговера и геномными характеристиками, рассчитанные на окна в 100 т.п.н. Участки кроссинговера представляют собой первые пять столбцов или строк (префикс «событие»; красные текстовые метки), а остальные - те же геномные элементы, которые использовались при моделировании (синие текстовые метки). Скопления участков кроссинговера с суффиксами «hp_m2», «hp», «m2», «mt» и «me» происходят из гаплоидов и клеток M2, гаплоидов, клеток M2, M2, которые имеют нарушенную эквационную сегрегацию, и клеток M2, которые имеют нарушенную редукционную сегрегацию, соответственно. Синие квадраты показывают положительную корреляцию, а красные квадраты - отрицательную корреляцию. Функции упорядочены иерархической кластеризацией. Открытые овалы выделяют функции «теломерный» и «квантиль_75_100», которые показывают разные тренды в двух скрещиваниях, как описано в тексте.
На фиг. 19 представлена корреляционная матрица как для событий кроссинговера, так и для геномных особенностей для скрещивания (B6×Spret). Тот же формат, как описано в легенде для Фиг. 18.
На Фиг. 20А-Е показаны положения самого правого кроссинговера на каждой хромосоме. (A) Гаплоидные клетки. В обоих скрещиваниях кроссинговеры предпочитают центромерно-дистальный конец хромосомы. (B) Сравнение гаплоидных клеток и клеток M2 (скрещивание B6×Cast). После учета вариабельности между хромосомами мы оцениваем, что кроссинговеры в клетках М2 в среднем расположены на 5,2 Мб более проксимально по центромере, чем в гаплоидах в скрещивании (B6×Cast). (С) Сравнение клеток М2 с нарушенной сегрегацией хромосом. После учета вариабельности между хромосомами мы оцениваем, что кроссинговеры в клетках M2 с нарушенной эквационной сегрегацией расположены в среднем на 13,7 Мб более центромерно-дистальнее, чем в клетках M2 с нарушенной редукционной сегрегацией в скрещивании (B6×Cast). (D) То же, что и в (C) для скрещивания (B6×Spret). Кроссинговеры в среднем на 8,7 Мб более центромерно-дистальные. (E) Модель влияния положений кроссинговера на правильную сегрегацию хромосом. Кроссинговер ближе к центромере (в середине двух квартилей, а не в последнем квартиле) может способствовать редукционной сегрегации благодаря более сильному сцеплению плеч; однако, кроссинговеры около конца хромосомного плеча могут облегчить сегрегацию MII, за счет более сильного сцепления CEN.
На фиг. 21 представлены основные компоненты анализа особенностей, выделяющих горячие точки кроссинговера в скрещивании B6×Spret. Обратите внимание, что «chr3_b.p. (точки разрыва)» и «chr1_upc (однородительские хромосомы)» представляют признаки, которые были включены для всех хромосом. Мы показываем 44 из 115 признаков. За исключением 36 опущенных других точек разрыва хромосом и признаков UPC, 35 других признаков не показаны из-за отсутствия очевидной тенденции.
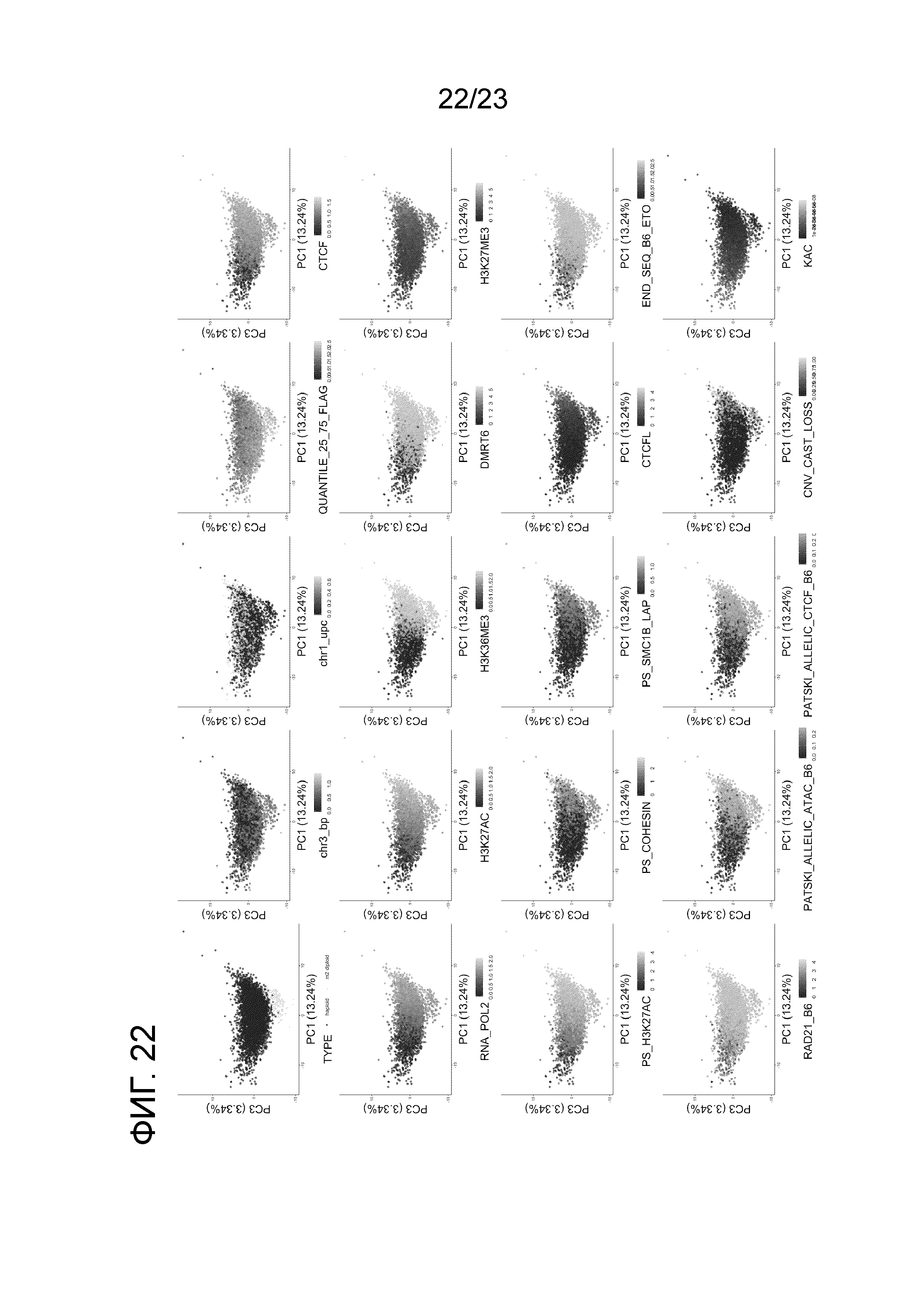
На фиг. 22 представлены основные компоненты анализа особенностей, выделяющих горячие точки кроссинговера в скрещивании B6×Cast. Обратите внимание, что «chr3_b.p. (точки разрыва)» и «chr1_upc (однородительские хромосомы)» представляют признаки, которые были включены для всех хромосом. Мы показываем 19 из 108 признаков. За исключением 36 опущенных других точек разрыва хромосом и признаков UPC, 53 других признака не показаны из-за отсутствия очевидной тенденции.
На фиг. 23 представлена модель для взаимосвязи мейотического кроссинговера и нарушенной сегрегации хромосом. «MI»: мейоз I, «CEN»: центромера (овальные или круглые кружки), «IH»: интер-гомолог. Следующее подробное описание иллюстративных вариантов осуществления настоящего изобретения будет понятно наилучшим образом при чтении в сочетании со следующими чертежами.
Схематические чертежи не обязательно выполнены в масштабе. Одинаковые числа, используемые на фигурах, относятся к одинаковым компонентам, этапам и т.п. Однако следует понимать, что использование номера для ссылки на компонент на данной фигуре не предназначено для ограничения компонента на другой фигуре, помеченного тем же номером. Кроме того, использование разных номеров для обозначения компонентов не предназначено для указания того, что пронумерованные по-разному компоненты не могут быть одинаковыми или похожими на другие пронумерованные компоненты.
ПОДРОБНОЕ ОПИСАНИЕ
Способ, предлагаемый в настоящем документе, можно использовать для получения библиотеки для комбинаторного индексированного секвенирования одиночных клеток (sci) из множества одиночных клеток или ядер, включая, например, секвенирование полных геномов (sci-WGS), транскриптомов (sci-RNA), совместный анализ генома и транскриптома (sci-DNA/RNA) и/или анализ метиломов (sci-MET). В одном из вариантов осуществления способ можно использовать для нацеленного секвенирования определенного области или областей, представляющих интерес. Например, для выборочного обогащения последовательности-мишени можно использовать праймер, который гибридизуется с конкретной областью (например, кодирующая область, некодирующая область и т.д.), направляющую РНК, или нуклеотидную последовательность, вставленную при помощи направляющей РНК. В одном из вариантов осуществления можно собирать и анализировать информацию для отдельных редактирований генов, ДНК, редактирования или маркера для редактирования, сигнатуры генов, отклонений и/или функциональных ридов (РНК, ДНК, белок или комбинация) из клеток или ядер (Perturb-seq). В других вариантах осуществления способ можно использовать для оценки доступности хроматина (sci-ATAC), конформации хроматина (Hi-C) и других способов комбинаторного индексирования одиночных клеток.
Способ включает в себя получение выделенных ядер или клеток, распределение субпопуляций ядер или клеток в компартменты, обработку ядер или клеток таким образом, чтобы они включали фрагменты нуклеиновой кислоты, добавление специфического индекса компартмента к фрагментам нуклеиновой кислоты и амплификацию фрагментов нуклеиновой кислоты путем линейной амплификации. Эти этапы могут происходить в разном порядке и их можно комбинировать по-разному. Три варианта предоставлены на ФИГ. 1А и 1B. В одном из вариантов осуществления способ включает получение распределенных субпопуляций выделенных ядер или клеток, которые содержат фрагменты нуклеиновой кислоты (фиг. 1A, блок 1, и ФИГ. 1B, блок 1). Как показано на ФИГ. 1AB, после амплификации фрагментов нуклеиновой кислоты с помощью линейной амплификации (фиг. 1A, блок 2) добавляют индекс к амплифицированным фрагментам нуклеиновой кислоты (фиг. 1A, блок 3). Как показано на ФИГ. 1B, фрагменты нуклеиновой кислоты в распределенных ядрах или клетках включают в себя индекс, и фрагменты нуклеиновой кислоты амплифицируют путем линейной амплификации (рис. 1B, блок 2). Этапы получения выделенных ядер или клеток, распределения субпопуляций выделенных ядер или клеток, обработки выделенных ядер или клеток для включения фрагментов нуклеиновой кислоты, добавления индекса специфического компартмента и амплификации фрагментов нуклеиновой кислоты с помощью линейной амплификации описаны в данном документе.
Получение выделенных ядер или клеток
В настоящем документе предлагается способ, который включает в себя получение клеток или выделенных ядер из множества клеток. Клетки и ядра могут быть из любого образца, например, из любого организма (организмов), и из любого типа клеток или любой ткани организма (организмов). В одном из вариантов осуществления клетки могут быть половыми клетками, например, сперматозоидами или яйцеклетками. В одном из вариантов осуществления ткань может быть репродуктивной тканью, например, придатком яичка. Ядро может быть из злокачественной опухоли или больной ткани. Способ может дополнительно включать диссоциацию клеток и/или выделение ядер. Способы выделения ядер из клеток известны специалистам в данной области и являются рутинными. Количество ядер или клеток может быть, по меньшей мере, равно двум. Верхний предел зависит от практических ограничений оборудования (например, многолуночных планшетов), используемых на других этапах способа, как описано в настоящем документе. Количество ядер или клеток, которые можно использовать, не ограничено и может исчисляться миллиардами. Например, число ядер или клеток может быть не более 100000000, не более 10000000, не более 1000000000, не более 100000000, не более 10000000, не более 1000000, не более 100000, не более 10000, не более 1000, не более 500 или не более 50. Можно получать один или более образцов. Например, образец может быть одним типом клеток или ткани из одного организма. Используя индексирование способами, описанными в данном документе, можно индексировать раздельно, а затем в сочетании первым индексом для идентификации образца несколько образцов, например, различные типы клеток из одного организма, один тип клеток или ткани из двух или более организмов, или различные типы клеток или ткани из двух или более организмов. Специалисту очевидно, что в некоторых вариантах осуществления молекулы нуклеиновой кислоты в каждом ядре представляют весь генетический комплекс организма (также обозначается как полный геном организма) и представляют собой молекулы геномной ДНК, которые включают как интронные, так и экзонные последовательности, а также некодирующие регуляторные последовательности установленным образом, такие как промоторные и энхансерные последовательности.
Выделение ядер можно осуществлять путем инкубации клеток в клеточном лизирующем буфере в течение от 1 до 20 минут, например, 5, 10 или 15 минут. Необязательно, клетки могут подвергаться воздействию внешней силы, способствующей лизису, такой как движение через пипетку. Пример клеточного лизисного буфера включает 10 мМ Трис-HCl, pH 7,4, 10 мМ NaCl, 3 мМ MgCl2, 0,1% IGEPAL CA-630 и 1% ингибитора РНКазы SUPERase In. Специалисту очевидно, что эти уровни компонентов могут быть несколько изменены без снижения полезности клеточного лизирующего буфера для выделения ядер. Специалисту очевидно, что ингибиторы РНКазы, БСА и/или поверхностно-активные вещества могут быть полезны в буферах, используемых для выделения ядер, и что другие добавки можно добавлять в буфер для других последующих применений комбинаторного индексирования одиночных клеток.
В одном из вариантов осуществления ядра выделяют из отдельных клеток, которые являются прикрепленными или находятся в суспензии. Способы выделения ядер из отдельных клеток известны специалисту в данной области. В одном из вариантов осуществления ядра отделяют от клеток, присутствующих в ткани. Способ получения выделенных ядер, как правило, включает подготовку ткани и выделение ядер из подготовленной ткани. В одном из вариантов осуществления все этапы выполняются на льду.
Получение ткани может включать мгновенное замораживание ткани в жидком азоте, а затем измельчение ткани или воздействие тупым предметом, чтобы уменьшить размер ткани до кусочков диаметром 1 мм или менее. Необязательно, можно использовать холодные протеазы и/или другие ферменты для разрушения межклеточных связей. Измельчать можно с помощью лезвия, чтобы разрезать ткань на мелкие кусочки. Применение тупой силы может быть достигнуто путем разбивания ткани молотком или подобным предметом, а полученная композиция из раздробленной ткани обозначается как порошок.
В общепринятых методах извлечения ядер из тканей обычно инкубируют ткани с тканеспецифическим ферментом (например, трипсином) при высокой температуре (например, 37°C) в течение от 30 минут до нескольких часов, а затем лизируют клетки с помощью буфера для лизиса клеток для экстракции ядер. Выделение ядер способом, описываемым в настоящем документе и во временной патентной заявке США 62/680259 имеет несколько преимуществ: (1) Не вводят никаких искусственных ферментов, и все шаги выполняют на льду. Это уменьшает потенциальное вмешательство в состояния клетки (например, состояние транскриптома, состояние хроматина или состояние метилирования). (2) Он был валидирован для большинства типов тканей, включая головной мозг, легкое, почку, селезенка, сердце, мозжечок и образцов при заболеваниях, таких как опухолевые ткани. По сравнению с общепринятыми методами извлечения ядер из тканей, в которых используются разные ферменты для разных типов тканей, новый способ может потенциально уменьшить ошибку при сравнении состояний клеток в разных тканях. (3) способ также снижает стоимость и повышает эффективность за счет удаления этапа ферментативного преобразования. (4) По сравнению с другими способами извлечения ядер (например, измельчитель ткани Даунса), этот способ является более надежным для разных типов тканей (например, способ Даунса требует оптимизации циклов Даунса для разных тканей) и позволяет обрабатывать большие куски образцов в высокопроизводительной манере (например, способ Даунса ограничен размером измельчителя).
Выделенные ядра или клетки могут включать нуклеосомы, могут быть свободны от нуклеосом или могут подвергаться воздействию условий, которые истощают ядра нуклеосом, генерируя обедненные нуклеосомами ядра. Нуклеосомно-обедненные ядра пригодны в способах определения последовательности ДНК всего генома клетки или его фракции.
В одном из вариантов осуществления, условия, используемые для истощения нуклеосом, поддерживают целостность выделенных ядер. Как правило, истощение нуклеосом применяют на осадке или суспензии отдельных клеток, таким образом, в тех вариантах, где в качестве источника клеток применяют прикрепленную клеточную культуру или ткань, источник обрабатывают для получения осадка или суспензии отдельных клеток.
Способы истощения нуклеосом известны и являются рутинными, и в качестве неограничивающих примеров, включают ферментативную обработку и химическую обработку. В одном из вариантов осуществления условия истощения нуклеосом включают химическую обработку хаотропным агентом, способным нарушать взаимодействия нуклеиновая кислота-белок. Пример подходящего хаотропного агента в качестве неограничивающих примеров включает 3,5-литий дииодосалициловую кислоту. Условия использования 3,5-литий дииодосалициловой кислоты включают добавление ее в осадок клеток и инкубацию на льду.
В предпочтительном варианте осуществления эти условия включают химическую обработку детергентом, способным нарушать взаимодействия нуклеиновая кислота-белок. Пример подходящего детергента в качестве неограничивающих примеров включает додецилсульфат натрия (SDS). Условия использования SDS включают добавление его в осадок клеток и инкубацию при повышенной температуре, примерно 42°C, а затем добавление неионного детергента, такого как Triton™ X-100, и инкубацию при повышенной температуре, такой как 42°C.
В некоторых вариантах осуществления, когда применяют детергент, такой как SDS, ядра подвергают воздействию сшивающего средства до истощения нуклеосом (WO 2018/018008). В одном из вариантов осуществления ядра подвергают воздействию сшивающего средства внутри клетки, и в другом варианте осуществления подвергают воздействию сшивающего средства выделенные ядра. Пример подходящего сшивающего средства в качестве неограничивающих примеров включает формальдегид (Hoffman et al., 2015, J. Biol. Chem., 290: 26404-26411). Обработка клеток формальдегидом может включать добавление формальдегида в суспензию клеток и инкубацию при комнатной температуре. В одном из вариантов осуществления после обработки формальдегидом ядра могут подвергаться воздействию глицина и неионогенного неденатурирующего детергента, такого как Igepal®.
Во время процесса истощения нуклеосом в выделенных ядрах сохраняется целостность выделенных ядер. Остаются ли ядра интактными после воздействия условий для истощения нуклеосом, можно определить, визуализируя состояние ядер обычными способами, такими как фазово-контрастная визуализация. В одном из вариантов осуществления число интактных ядер после истощения нуклеосом может составлять от 1 до 1000, от 1000 до 10000, от 10000 до 100000100000 до 1000000, от 1000000 до 10000000 или от 10000000 до 100000000.
Манипуляции с ядрами или клетками, включая этапы получения, объединения и распределения, описанные в настоящем документе, могут включать использование буфера для ядер. Пример буфера для ядер включает 10 мМ Трис-HCl, pH 7,4, 10 мМ NaCl, 3 мМ MgCl2, 1% ингибитора РНКазы SUPERase In (20 Ед/мкл, Ambion) и 1% BSA (20 мг/мл, NEB). Специалисту очевидно, что эти уровни компонентов могут быть несколько изменены без снижения полезности буфера для ядер, в котором суспендируют ядра. Специалисту также очевидно, что различные компоненты могут быть заменены без снижения полезности буфера для ядер, в котором суспендируют ядра.
В одном из вариантов осуществления клетки (включая клетки, из которых выделены ядра) подвергались воздействию различных предопределенных состояний. Например, субпопуляции клеток могут подвергаться различным предопределенным условиям. Различные условия могут включать, например, разные условия культивирования (например, разные среды, разные условия окружающей среды), разные дозы агента, разные агенты или комбинации агентов. Агенты описаны в настоящем документе. Ядра или клетки каждой субпопуляции клеток и/или образца или образцов индексируют с помощью одного или более индексов, объединяют и анализируют с помощью массового мультиплексного секвенирования одиночных ядер или одиночных клеток. По существу, можно использовать любой способ секвенирования одиночных ядер или одиночных клеток, включая в качестве неограничивающих примеров секвенирование транскриптома одиночных ядер (заявка на патент США № 62/680259 и Gunderson et al. (WO2016/130704)), полногеномное секвенирование одиночных ядер (публикация патентной заявки США 2018/0023119), или секвенирование транспозон-доступного хроматина одиночных ядер (патент США № 10059989), sci-HiC (Ramani et al., Nature Methods, 2017, 14: 263-266), DRUG-seq (Ye et al., Nature Commun., 9, статья № 4307), Perturb-seq (Dixit et al., Cell, 2016, 167 (7): 1853- 1866.e17) или любую комбинацию аналитов из ДНК, РНК и белков, например, sci-CAR (Cao et al., Science, 2018, 361 (6409): 1380-1385). Также можно применять анализ одиночных клеток на основе капель после первоначального разделения и смешанного индексирования (примеры включают систему 10× genomics Chromium™ или систему Biorad ddseq), включая использование индекса в качестве индекса образца. Хэширование ядер применяют для демультиплексирования и идентификации отдельных клеток или ядер из разных условий.
В одном из вариантов осуществления каждая субпопуляция клеток подвергается воздействию агента. Агент может быть по существу всем, что вызывает изменение в клетке. Например, агент может изменить транскриптом клетки, изменить структуру хроматина клетки, изменить активность белка в клетке, изменить ДНК клетки, изменить редактирование ДНК клетки или вызвать другие изменения. Примеры агентов в качестве неограничивающих примеров включают соединение, такое как белок (в том числе антитело), не рибосомальный белок, поликетид, органическую молекулу (включая органическую молекулу массой 900 дальтон или менее), неорганическую молекулу, молекулу РНК или РНКи, углевод, гликопротеин, нуклеиновую кислоту, или их сочетание. В одном из вариантов осуществления агент вызывает генетическое нарушение, например, ДНК-редактирующий белок и/или направляющая РНК, такая как CRISPR или Talen. В одном из вариантов осуществления агент является терапевтическим лекарственным средством. Клетки могут быть генетически модифицированы и включать генетическое нарушение, например, нокин генов или нокаут генов (Szlachta et al., Nat Commun., 2018). 9: 4275). Субпопуляции клеток могут подвергаться воздействию одного и того же агента, но различные переменные могут изменяться в компартментах многолуночного устройства, что позволяет тестировать несколько переменных в одном эксперименте. Например, разные дозы, разная продолжительность воздействия и разные типы клеток могут быть проверены на одном планшете. В одном из вариантов осуществления клетки могут экспрессировать белок, обладающий известной активностью, и влияние агента на активность оценивают при различных условиях. Использование индекса для маркировки фрагментов нуклеиновых кислот позволяет позднее идентифицировать нуклеиновые кислоты, происходящие из определенной субпопуляции ядер или клеток, например, из одной лунки многолуночного планшета.
Распределение субпопуляций
В настоящем документе предлагается способ, который включает в себя распределение субпопуляции ядер, например, обедненных нуклеосомами ядер или клеток, во множество компартментов. Способ может включать несколько этапов распределения, где популяция изолированных ядер или клеток (также называемая в документе «совокупность») делится на субпопуляции. Как правило, распределение субпопуляций выделенных ядер или клеток от совокупности к множеству компартментов происходит до добавления индекса к фрагментам нуклеиновой кислоты, присутствующим в субпопуляции выделенных ядер или клеток. Таким образом, способ включает в себя, по меньшей мере, один этап «разделения и смешения» из отбора объединенных выделенных ядер или клеток и их распределения, при этом число этапов «разделения и смешения» может зависеть от числа различных индексов, которые добавляют к фрагментам нуклеиновой кислоты. После индексирования субпопуляции могут быть объединены, разделены на субпопуляции, проиндексированы и снова объединены по мере необходимости, пока достаточное количество индексов не будет добавлено к фрагментам нуклеиновой кислоты.
Количество ядер или клеток, присутствующих в субпопуляции, и, таким образом, в каждом компартменте, может быть, по меньшей мере, равно 1. В одном из вариантов осуществления количество ядер или клеток, присутствующих в субпопуляции, составляет не более 100000000, не более 10000000, не более 1000000, не более 100000, не более 10000, не более 4000, не более 3000, не более 2000, или не более 1000, не более 500 или не более 50. В одном из вариантов осуществления число ядер или клеток, присутствующих в субпопуляции, может составлять от 1 до 1000, от 1000 до 10000, от 10000 до 100000100000 до 1000000, от 1000000 до 10000000 или от 10000000 до 100000000. В одном из вариантов осуществления количество ядер или клеток, присутствующих в каждой субпопуляции, примерно равно. Количество ядер, присутствующих в субпопуляции, и, следовательно, в каждом компартменте, частично основано на желании уменьшить столкновения индексов, которые представляют собой присутствие двух ядер с одинаковым индексом транспозазы, оказавшихся в одном и том же компартменте на этом этапе способа. Способы распределения ядер или клеток в субпопуляции известны специалистам в данной области и являются рутинными. Примеры в качестве неограничивающих примеров включают цитометрию с активируемой флуоресценцией сортировкой клеток (FACS) и простое разведение. Необязательно, ядра различной плоидности можно отбирать и обогащать путем окрашивания, например окрашивания DAPI (4’,6-диамидино-2-фенилиндол).
Количество компартментов на этапах распределения (и последующее добавление индекса) может зависеть от используемого формата. Например, количество компартментов может составлять от 2 до 96 компартментов (когда применяют 96-луночный планшет), от 2 до 384 компартментов (когда применяют 384-луночный планшет), или от 2 до 1536 компартментов (когда применяют 1536-луночный планшет). В одном из вариантов осуществления каждым компартментом может быть капля. Когда тип используемого компартмента представляет собой каплю, которая содержит два или более ядра или клетки, можно использовать любое количество капель, например, по меньшей мере, 10000, по меньшей мере, 100000, по меньшей мере, 1000000 или, по меньшей мере, 10000000 капель. В одном из вариантов осуществления количество компартментов составляет 24.
Обработка для получения фрагментов нуклеиновой кислоты
В одном из вариантов осуществления можно использовать обработку выделенных ядер или клеток для фрагментирования ДНК нуклеиновых кислот, например хромосом и/или плазмид, в выделенных ядрах или клетках в фрагменты нуклеиновой кислоты. Обработка, как правило, необходима, когда нуклеиновые кислоты-мишени, которые должны быть секвенированы, получены из ДНК, присутствующей в ядрах или клетках; однако, в некоторых вариантах осуществления обработка необязательна, когда нуклеиновые кислоты-мишени, которые должны быть секвенированы, получены из РНК (например, мРНК и/или некодирующей РНК), присутствующей в ядрах или клетках, поскольку молекулы РНК часто не нужно фрагментировать, Обработка нуклеиновых кислот в ядрах или клетках, как правило, добавляет нуклеотидную последовательность к одному или обоим концам фрагментов нуклеиновой кислоты, полученных в результате обработки, и нуклеотидная последовательность может включать, и, как правило, включает в себя одну или более универсальных последовательностей. Универсальная последовательность может использоваться в качестве, например, «посадочной площадки» на последующем этапе для отжига нуклеотидной последовательности, которую можно использовать в качестве праймера для добавления другой нуклеотидной последовательности, такой как индекс, к фрагменту нуклеиновой кислоты на последующем этапе лигирования, удлинения праймера или амплификации. Нуклеотидная последовательность такого праймера может необязательно включать индексную последовательность. Обработка нуклеиновых кислот в ядрах или клетках может добавлять один или более уникальных молекулярных идентификаторов к одному или обоим концам фрагментов нуклеиновых кислот, полученных в результате обработки.
Есть несколько точек в способе, в которые может происходить обработка нуклеиновых кислот в фрагменты нуклеиновых кислот. Например, в одном из вариантов осуществления отдельные ядра или клетки могут быть обработаны перед распределением субпопуляции выделенных ядер или клеток. В вариантах осуществления, обработка, как правило, включает добавление универсальной последовательности и/или универсального молекулярного идентификатора к фрагментам нуклеиновой кислоты, но не индекса, специфичного для компартмента, поскольку добавление индекса, специфичного для компартмента, когда объединяются все выделенные ядра или клетки, как правило, является бесцельным. В другом варианте осуществления, выделенные ядра или клетки могут быть обработаны после распределения субпопуляций в разные компартменты (например, ФИГ. 1А и ФИГ. 1Б). В одном из аспектов этого варианта осуществления обработка не добавляет индекс (фиг. 1A, блок 1), а в другом аспекте этого варианта обработка может включать добавление индекса, специфичного для компартмента (фиг. 1B, блок 1). Обработка в любой точке способа может включать добавление универсальной последовательности и/или универсального молекулярного идентификатора к одному или обоим концам фрагментов нуклеиновой кислоты.
Известны различные способы переработки нуклеиновых кислот в ядрах или клетках в фрагменты нуклеиновой кислоты. Примеры включают CRISPR и Talen-подобные ферменты, и ферменты, которые раскручивают ДНК (например, хеликазы), которые могут создавать одноцепочечные области, к которым могут гибридизоваться фрагменты ДНК и инициировать удлинение или амплификацию. Например, можно использовать амплификацию на основе хеликаз (Vincent et al., 2004, EMBO Rep., 5 (8): 795-800). В одном из вариантов осуществления удилинение или амплификация инициируется случайным праймером. В одном из вариантов осуществления используют транспозомный комплекс. Транспозомный комплекс представляет собой транспозазу, связанную с участком распознавания транспозазы, и может вставить участок распознавания транспозазы в нуклеиновую кислоту-мишень в ядре в процессе, иногда называемом «тагментацией». В некоторых таких событиях вставки одна цепь участка распознавания транспозазы может быть перенесена в нуклеиновую кислоту-мишень. Такая цепь обозначается как «перенесенная цепь». В одном из вариантов осуществления транспозомный комплекс включает димерную транспозазу, имеющую две субъединицы и две несмежных последовательности транспозона. В другом варианте осуществления транспозаза включает димерную транспозазу, имеющую две субъединицы и смежную последовательность транспозона. В одном из вариантов осуществления 5'-конец одной или обеих цепей участка распознавания транспозазы может быть фосфорилирован.
Некоторые варианты осуществления могут включать использование гиперактивной транспозазы Tn5 и участка распознавания для транспозазы типа Tn5 (Goryshin and Reznikoff, J. Biol. Chem., 273:7367 (1998)), или транспозазы MuA и участка распознавания для транспозазы Mu, включающего концевые последовательности R1 и R2 (Mizuuchi, K., Cell, 35: 785, 1983; Savilahti, H, et al.,EMBO J., 14: 4893, 1995). Также можно использовать мозаичные концевые (ME) последовательности Tn5, оптимизированные специалистами в данной области.
Дополнительные примеры систем транспозиции, которые можно использовать с определенными вариантами осуществления композиций и способов, предлагаемых в настоящем документе, включают Staphylococcus aureus Tn552 (Colegio et al.,J. Bacteriol., 183: 2384-8, 2001; Kirby C et al., Mol. Microbiol., 43: 173-86, 2002), Ty1 (Devine & Boeke, Nucleic Acids Res., 22: 3765-72, 1994 и международная публикация WO 95/23875), транспозон Tn7 (Craig, N L, Science. 271: 1512, 1996; Craig, N L, обзор в: Curr Top Microbiol Immunol., 204:27-48, 1996), Tn/O и IS10 (Kleckner N, et al.,Curr Top Microbiol Immunol., 204:49-82, 1996), транспозазу Mariner (Lampe D J, et al.,EMBO J., 15: 5470-9, 1996), Tc1 (Plasterk R H, Curr. Topics Microbiol. Immunol., 204: 125-43, 1996), P Element (Gloor, G B, MethodsMol. Biol., 260: 97-114, 2004), Tn3 (Ichikawa & Ohtsubo, J Biol. Chem. 265:18829-32, 1990), бактериальные инсерционные последовательности (Ohtsubo & Sekine, Curr. Top. Microbiol. Immunol. 204: 1-26, 1996), ретровирусы (Brown, et al.,Proc Natl Acad Sci USA, 86:2525-9, 1989), и ретротранспозон дрожжей (Boeke & Corces, Annu Rev Microbiol. 43:403-34, 1989). Дополнительные примеры включают IS5, Tn10, Tn903, IS911, и сконструированные версии ферментов семейства транспозаз (Zhang et al., (2009) PLoS Genet. 5:e1000689. Epub 2009 Oct 16; Wilson C. et al (2007) J. Microbiol. Methods 71:332-5).
Другие примеры интеграз, которые можно использовать со способами и составами, предлагаемыми в настоящем документе, включают ретровирусные интегразы и последовательности распознавания интегразы для таких ретровирусных интеграз, как, например, интегразы из ВИЧ-1, ВИЧ-2, SIV, PFV-1, RSV.
Последовательности транспозонов, подходящие для способов и композиций, описанных в настоящем документе, приведены в публикации патентной заявки США № 2012/0208705, публикации патентной заявки США № 2012/0208724 и публикации международной патентной заявки № WO 2012/061832. В некоторых вариантах осуществления последовательность транспозона включает первый участок распознавания транспозазы и второй участок распознавания транспозазы. В тех вариантах осуществления, где транспозомный комплекс используют для введения индексной последовательности, индексная последовательность может присутствовать между участками распознавания транспозазы или в транспозоне.
Некоторые транспозомные комплексы, используемые в настоящем документе, включают в себя транспозазу, имеющую два транспозона. В некоторых таких вариантах осуществления, две последовательности транспозонов не связаны друг с другом, другими словами, последовательности транспозонов не смежные друг с другом. Примеры таких транспозом известны в данной области (см., например, публикацию патентной заявки США № 2010/0120098).
В некоторых вариантах осуществления транспозомный комплекс включает последовательность нуклеиновой кислоты транспозона, которая связывает две субъединицы транспозазы с образованием «петлевого комплекса» или «петлевой транспозомы». В одном примере транспозома включает димерную транспозазу и последовательность транспозона. Петлевые комплексы могут гарантировать, что транспозоны будут вставлены в ДНК-мишень при сохранении информации о порядке исходной ДНК-мишени и без фрагментации ДНК-мишени. Понятно, что петлевые структуры могут вставлять нужные нуклеиновые кислоты, такие как индексы, в нуклеиновую кислоту-мишень, сохраняя при этом физическую связность нуклеиновой кислоты-мишени. В некоторых вариантах осуществления последовательность транспозонов петлевого транспозомного комплекса может включать сайт фрагментации, так что последовательность транспозонов может быть фрагментирована для создания транспозомного комплекса, включающего две последовательности транспозона. Такие транспозомные комплексы подходят для обеспечения того, чтобы соседние фрагменты ДНК-мишени, в которые вставляются транспозоны, получали кодовые комбинации, которые можно однозначно собрать на более поздней стадии анализа.
В одном из вариантов осуществления фрагментацию нуклеиновых кислот проводят с использованием сайта фрагментации, присутствующего в нуклеиновых кислотах. Как правило, сайты фрагментации вводят в нуклеиновые кислоты-мишени с помощью транспозомного комплекса. В одном из вариантов осуществления после фрагментации нуклеиновых кислот транспозаза остается прикрепленной к фрагментам нуклеиновой кислоты, так что фрагменты нуклеиновой кислоты, полученные из той же самой геномной молекула ДНК, остаются физически связанными (Adey et al., 2014, Genom Res., 24: 2041- 2049). Например, петлевой транспозомный комплекс может включать сайт фрагментации. Сайт фрагментации можно использовать для расщепления физических, но не информационных связей между индексами, которые были вставлены в нуклеиновую кислоту-мишень. Расщепление можно производить биохимическим, химическим или другим способом. В некоторых вариантах осуществления сайт фрагментации может включать нуклеотид или нуклеотидную последовательность, которая может быть фрагментирована различными способами. Примеры сайтов фрагментации в качестве неограничивающих примеров включают, в частности, рибонуклеотид, расщепляемый с помощью РНКазы, нуклеотидные аналоги, расщепляемые в присутствии определенного химического агента, диоловую связь, расщепляемую обработкой периодатом, дисульфидную группу, расщепляемую химическим восстановителем, расщепляемую группу, которая может подвергаться фотохимическому расщеплению, и пептид, расщепляемый пептидазным ферментомом или другими подходящими средствами (см., например, патентную заявку США, публикацию № 2012/0208705, патентную заявку США, публикацию № 2012/0208724 и WO 2012/061832).
Транспозомный комплекс необязательно может включать, по меньшей мере, одну индексную последовательность и может обозначаться как транспозазный индекс. Индексная последовательность присутствует в виде части последовательности транспозона. В одном из вариантов осуществления индексная последовательность может присутствовать на перемещаемой цепи, т.е. цепи с участком распознавания транспозазы, который переносят в нуклеиновую кислоту-мишень.
Транспозомный комплекс необязательно может включать в себя, по меньшей мере, одну нуклеотидную последовательность, которую можно использовать при помощи медиатора линейной амплификации. Примеры таких нуклеотидных последовательностей в качестве неограничивающих примеров включают РНК-полимеразу, когда фрагменты нуклеиновой кислоты включают промотор фага, такую как РНК-протеазу Т7 для использования с промотором Т7 и праймер для линейной амплификации. Примеры праймера для линейной амплификации включают в себя один праймер или медиатор для линейной амплификации для применения в амплификации типа ПЦР. Другие варианты осуществления нуклеотидной последовательности, которые можно использовать с помощью медиаторов для линейной амплификации, представляют собой последовательности, которые распознаются полимеразой, замещающей цепи. Медиатор может содержать сайт для одноцепочечного разрыва для инициации репликации. В некоторых случаях сайт для одноцепочечного разрыва восстанавливают для дополнительной амплификации.
Добавление компартмент-специфического индекса
Индексная последовательность, также обозначаемая как метка или штрихкод, пригодна в качестве маркера, характерного для компартмента, в котором присутствовала конкретная нуклеиновая кислота. Таким образом, индекс представляет собой метку для последовательности нуклеиновой кислоты, которая присоединена к каждой из нуклеиновых кислот-мишеней, присутствующих в конкретном компартменте, присутствие которой указывает на компартмент или ее наличие используют для идентификации компартмента, в котором присутствовала популяция выделенных ядер или клеток на определенном этапе способа. Добавление индекса к фрагментам нуклеиновой кислоты проводят с субпопуляциями выделенных ядер или клеток, распределенных по разным компартментам.
Индексная последовательность может иметь любое подходящее число нуклеотидов в длину, например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 или больше. Четыре нуклеотидных метки дают возможность мультиплексировать 256 образцов на одном чипе, а шесть меток позволяют обрабатывать 4096 образцов на одном чипе.
В одном из вариантов осуществления добавление индекса достигается при переработке нуклеиновых кислот в фрагменты нуклеиновой кислоты. Например, можно использовать транспозомный комплекс, включающий индекс. В других вариантах осуществления индекс добавляют после получения фрагментов нуклеиновой кислоты, содержащих нуклеотидную последовательность на одном или обоих концах. Способы добавления индекса в качестве неограничивающих примеров включают лигирование, удлинение (включая удлинение с использованием обратной транскриптазы), гибридизацию, адсорбцию, специфические или неспецифические взаимодействия праймера или амплификацию. Нуклеотидная последовательность, которая добавляется к одному или обоим концам фрагментов нуклеиновой кислоты, также может включать одну или более универсальных последовательностей и/или уникальных молекулярных идентификаторов. Универсальную последовательность можно использовать в качестве, например, «посадочной площадки» на последующем этапе отжига нуклеотидной последовательности к фрагменту нуклеиновой кислоты, которую можно использовать в качестве праймера для добавления другой нуклеотидной последовательности, такой как другой индекс и/или другая универсальная последовательность, к нуклеиновой последовательности, кислота фрагмент.
Например, в вариантах осуществления, которые включают использование фрагментов нуклеиновой кислоты, которые получены из мРНК, можно использовать различные способы для добавления индекса к мРНК в один или два этапа. Например, индекс можно добавлять с использованием типов способов, используемых для получения кДНК. Праймер с поли Т-последовательностью на 3'-конце можно отжигать на молекулах мРНК и удлинять с использованием обратной транскриптазы. Воздействие этими компонентами на выделенные ядра или клетки в условиях, подходящих для обратной транскрипции, приводит к добавлению индекса за один шаг, что приводит к образованию популяции индексированных ядер или клеток, где каждое ядро или клетка содержит индексированные фрагменты нуклеиновой кислоты. Альтернативно, праймер с поли Т-последовательностью включает в себя универсальную последовательность вместо индекса, а индекс добавляют путем последующего этапа лигирования, удлинения праймера, амплификации. Индексированные фрагменты нуклеиновой кислоты могут и, как правило, включаются в синтезированную цепь индексной последовательности, указывающую на конкретный компартмент.
В вариантах осуществления, которые включают использование фрагментов нуклеиновой кислоты, полученных из некодирующей РНК, можно использовать различные способы для добавления индекса к некодирующей РНК в один или два этапа. Например, индекс можно добавлять с использованием первого праймера, который включает в себя случайную последовательность и праймера с переключателем матрицы, где любой праймер может включать в себя индекс. Можно использовать обратную транскриптазу, обладающую терминальной трансферазной активностью, которая может привести к появлению не-матричных нуклеотидов на 3'-конце синтезированной цепи, а праймер с переключателем матрицы включает нуклеотиды, которые отжигаются с нематричными нуклеотидами, добавленными обратной транскриптазой. Примером подходящей обратной траскриптазы является обратная транскриптаза вируса лейкоза мышей Молони. В конкретном варианте осуществления реагент SMARTer™, доступный от Takara Bio USA, Inc. (каталожный номер 634926), который применяют для использования переключения матрицы для добавления индекса к некодирующим РНК, и мРНК при желании. Альтернативно, первый праймер и/или праймер с переключателем матрицы может включать в себя универсальную последовательность вместо индекса, а индекс добавляют путем последующего этапа лигирования, удлинения праймера, амплификации. Индексированные фрагменты нуклеиновой кислоты могут и, как правило, включаются в синтезированную цепь индексной последовательности, указывающую на конкретный компартмент. Последовательность, указывающая на конкретный отсек. Другие варианты включают 5'- или 3'-профилирование РНК или профилирование полноразмерной РНК.
Можно использовать другие способы для добавления индекса к фрагменту нуклеиновой кислоты, и способ добавления индекса не ограничен. Например, в одном из вариантов осуществления, вставка индексной последовательности включает лигирование праймера к одному или обоим концам фрагментов нуклеиновой кислоты. Лигированию лигирующего праймера может способствовать присутствие универсальной последовательности на концах фрагментов нуклеиновой кислоты. Неограничивающим примером праймера является шпилечный дуплекс для лигирования. Дуплекс для лигирования может быть лигирован к одному или, предпочтительно, к обоим концам фрагментов нуклеиновой кислоты. В одном из вариантов осуществления, праймер, такой как шпилечный дуплекс для лигирования может содержать нуклеотидную последовательность, которая распознается медиатором линенйной амплификации. Пример адаптера в виде шпильки, содержащего такие нуклеотиды, описан в примере 1, Фиг. 2. Желательна схема анализа, описанная в примере 1, в которой вводится медиатор амплификации, который требует успешного лигирования только на одном из двух концов молекул со штрихкодами, чтобы получить продукты амплификации этой молекулы, поскольку эта схема имеет преимущество в увеличении эффективности преобразования матрицы. Например, если отдельное событие лигирования имеет эффективность 50%, эта модификация дает 75% успеха на этапе лигирования амплифицированной молекулы вместо 25% (Пример 1, Фиг. 2).
В другом варианте осуществления, вставка индексной последовательности включает использование одноцепочечных фрагментов нуклеиновых кислот и синтез второй цепи ДНК. В одном из вариантов осуществления вторую цепь ДНК получают с использованием праймера, который включает в себя последовательности, комплементарные нуклеотидам, присутствующим на концах одноцепочечных фрагментов нуклеиновой кислоты.
В другом варианте осуществления включение индекса происходит в один, два, три или более раундов штрихкодирования разделения и смешения при штрихкодировании, что приводит к одиночному, двойному, тройному или множественному индексированию библиотек одиночных клеток.
В другом варианте осуществления включения индексов и нуклеотидных последовательностей можно использовать при помощи медиатора амплификации, разработаны как односторонне направленные, что позволяет подготовить библиотеку для секвенирования одиночной клетки-мишени (См. Пример 1, фигура 3b).
Линейная амплификация фрагментов нуклеиновых кислот
В настоящем документе предлагается способ, который включает линейную амплификацию фрагментов нуклеиновой кислоты. Большинство способов амплификации основаны на ПЦР и, таким образом, страдают от ошибок экспоненциальной амплификации. Линейная амплификация, как применяют в настоящем документе, может уменьшить или устранить ошибки экспоненциальной амплификации, что приведет к лучшей однородности и уменьшению ошибок последовательности. Во всех геномных способах для одиночной клетки, которые используют полногеномную амплификацию, продукты амплификации содержатся в компартменте (например, лунке или капле), и штриховой код прямо или косвенно прикрепляется к амплифицированным продуктам. Таким образом, только одна клетка присутствует в компартменте, что ограничивает производительность и увеличивает стоимость. Уникальным аспектом по настоящему изобретению является то, что библиотеки множества одиночных клеток можно амплифицировать в одном компартменте без ошибки экспоненциальной амплификации. Библиотеки из одиночных клеток можно определять на основе уникального штрихкода или штрихкодов для каждой уникальной отдельной клетки.
В одном из вариантов осуществления линейную амплификацию проводят путем добавления фагового промотора к одному или обоим концам фрагментов нуклеиновой кислоты. Когда промотор фага расположен выше фрагмента нуклеиновой кислоты, его можно использовать для управления транскрипцией с использованием соответствующей фаговой РНК-полимеразы путем транскрипции in vitro, продуцирующей одноцепочечную РНК. Копии РНК, сгенерированные из матрицы ДНК, не могут служить матрицей для дальнейшей амплификации; таким образом, все копии получены непосредственно из исходной матрицы ДНК и исключается экспоненциальная амплификация. В одном из вариантов осуществления последующие этапы могут включать обратную транскрипцию копий РНК для получения одноцепочечной ДНК, а затем синтез второй цепи для преобразования копий одноцепочечной ДНК в двухцепочечные молекулы. Синтез второй цепи, как правило, требует использования праймера, и этот праймер можно использовать для введения одного или более индексов, универсальной последовательности и/или универсального молекулярного идентификатора.
Можно использовать другие способы линейной амплификации. Например, ПЦР-амплификацию можно использовать с одним праймером, или двумя праймерами с избытком одного. В некоторых вариантах осуществления можно использовать линейную ПЦР для амплификации фланкирующих последовательностей, прилегающих к участкам вставки транспозона (Xianbo et al. AMB Express, 2017, 7: 195). Также можно использовать в некоторых вариантах осуществления связанную линейную амплификацию (Reyes et al., Clin. Chem., 2001, 47 (1): 31-40), линейное удлинение и линейное удлинение и лигирование, амплификацию со смещением цепи (SDA) (Walker et al., Nucl. Acids. Res., 1992, 20 (7): 1691-1696), и амплификацию по типу катящегося кольца (Ali et al., Chem. Soc. Rev., 2014, 43: 3324-3341. В некоторых вариантах осуществления индекс, универсальную последовательность и/или уникальный молекулярный идентификатор можно добавлять к фрагментам нуклеиновой кислоты во время линейной амплификации.
Как правило, линейная амплификация включает введение в выделенные ядра или клетки медиатора линейной амплификации. Примеры медиаторов линейной амплификации включают РНК-полимеразу, когда фрагменты нуклеиновой кислоты включают промотор фага, такую как РНК-полимеразу Т7 для использования с промотором Т7, и праймер для линейной амплификации. Примеры праймера для линейной амплификации включают в себя один праймер или медиатор для линейной амплификации для применения в амплификации типа ПЦР. Другие варианты осуществления медиаторов амплификации полимеразу, замещающую цепи, которая распознает нуклеотидную последовательность. Медиатор может содержать сайт для одноцепочечного разрыва для инициации репликации. В некоторых случаях сайт для одноцепочечного разрыва восстанавливают для дополнительной амплификации. Медиатор может содержать уникальный штрихкод или праймер, позволяющий копировать штрихкод во время амплификации или мечения продуктов амплификации.
Добавление универсальных последовательностей для иммобилизации
В одном из вариантов осуществления добавление нуклеотидов во время этапов обработки и/или индексирования добавляет универсальные последовательности, полезные для иммобилизации и секвенирования фрагментов. В другом варианте осуществления индексированные фрагменты нуклеиновой кислоты могут быть дополнительно обработаны для добавления универсальных последовательностей, полезных для иммобилизации и секвенирования фрагментов нуклеиновой кислоты. Специалисту будет очевидно, что в вариантах осуществления, где компартмент представляет собой каплю, последовательности для иммобилизации фрагментов нуклеиновой кислоты являются необязательными. В одном из вариантов осуществления включение универсальных последовательностей, полезных для иммобилизации и секвенирования фрагментов, включает лигирование идентичных универсальных адаптеров (также обозначаемых как «несовпадающие адаптеры», общие черты которых описаны в Gormley et al., US 7741463, и Bignell et al., US 8053192,) к 5'- и 3'-концам индексированных фрагментов нуклеиновой кислоты. В одном из вариантов осуществления универсальный адаптер включает в себя все последовательности, необходимые для секвенирования, в том числе для иммобилизации индексированных фрагментов нуклеиновых кислот на чипе.
В одном из вариантов осуществления можно использовать лигирование тупых концов. В другом варианте осуществления фрагменты нуклеиновой кислоты с единичными липкими нуклеотидами, например, при помощи активности определенных типов ДНК-полимеразы, например, Taq-полимеразы или полимеразы Кленова (экзо-минус), которая имеет не зависящую от матрицы терминальную трансферазную активность, которая добавляет один дезоксинуклеотид, например, дезоксиаденозин (A) к 3'-концам проиндексированных фрагментов нуклеиновой кислоты. В некоторых случаях липкий нуклеотид представляет собой более чем одно основание. Такие ферменты можно использовать для добавления одного нуклеотида «A» к тупому концу с 3‘-конца каждой цепи фрагментов нуклеиновой кислоты. Таким образом, «А» может быть добавлен к 3'-концу каждой цепи двухцепочечных фрагментов-мишеней путем реакции с Taq-полимеразой или полимеразой Кленова (экзо-минус), в то время как дополнительные последовательности, который будут добавлены к каждому концу нуклеиновой кислоты, могут включать совместимый липкий «T», присутствующий на 3'-конце каждой области двухцепочечной нуклеиновой кислоты, подлежащей добавлению. Эта конечная модификация также предотвращает самолигирование нуклеиновых кислот таким образом, что существует смещение в сторону образования индексированных фрагментов нуклеиновой кислоты, фланкированных последовательностями, которые добавляют в этом варианте осуществления.
В другом варианте осуществления, когда универсальный адаптер, лигированный с индексированными фрагментами нуклеиновой кислоты, не включает все последовательности, необходимые для секвенирования, тогда можно использовать этап амплификации, такой как ПЦР, для дальнейшей модификации универсальных адаптеров, присутствующих в каждом индексированном фрагменте нуклеиновой кислоты перед иммобилизацией и секвенированием. Например, начальную реакцию удлинения праймера можно проводить с использованием универсальной якорной последовательности, комплементарной универсальной последовательности, присутствующей в индексированном фрагменте нуклеиновой кислоты, в которой образуются продукты удлинения, комплементарные обеим цепям каждого индексированного фрагмента нуклеиновой кислоты. Как правило, ПЦР добавляет дополнительную универсальную последовательность, такую как универсальная захватывающая последовательность.
После добавления универсальных адаптеров либо путем одноэтапного способа лигирования универсального адаптера, включающего все необходимые для сиквенса последовательности, либо двухэтапным способом лигирования универсального адаптера, а затем амплификацией для дальнейшей модификации универсального адаптера, конечный индекс фрагментов будет включать в себя универсальную захватывающую последовательность и якорную последовательность. Результатом добавления универсальных адаптеров к каждому концу является множество индексированных фрагментов нуклеиновой кислоты или библиотека индексированных фрагментов нуклеиновой кислоты.
Полученные в результате индексированные фрагменты нуклеиновой кислоты вместе составляют библиотеку нуклеиновых кислот, которая может быть иммобилизована, а затем секвенирована. Термин «библиотека», также называемый в данном документе как «библиотека для секвенирования», относится к коллекции фрагментов нуклеиновой кислоты из единичных ядер или клеток, содержащих известные универсальные последовательности на 3'- и 5'-концах.
Индексированные фрагменты нуклеиновой кислоты могут подвергаться условиям для отбора по предопределенному диапазону размеров, например, от 150 до 400 нуклеотидов в длину, такие как от 150 до 300 нуклеотидов. Полученные в результате индексированные фрагменты нуклеиновой кислоты объединяют и необязательно их можно подвергать процессу очистки для повышения чистоты до молекул ДНК путем удаления, по меньшей мере, части невключенных универсальных адаптеров или праймеров. Можно использовать любой подходящий процесс очистки, такой как электрофорез, исключение размера хроматография с гель-фильтрацией, или т.п. В некоторых вариантах осуществления можно использовать парамагнитные гранулы для обратимой иммобилизации на твердой фазе для отделения желаемых молекул ДНК от неприкрепленных универсальных адаптеров или праймеров и для отбора нуклеиновых кислот в зависимости от размера. Парамагнитные гранулы для обратимой иммобилизации на твердой фазе являются коммерчески доступными у Beckman Coulter (Agencourt AMPure XP), Thermofisher (MagJet), Omega Biotek (Mag-Bind), Promega Beads (Promega) и Kapa Biosystems (Kapa Pure Beads).
Неограничивающий иллюстративный вариант осуществления настоящего изобретения показан на ФИГ. 2 и описан в примере 1. Этот способ предусматривает получение выделенных ядер из множества клеток (фиг. 2, блок 22). Выделенные ядра могут быть свободны от нуклеосом или могут быть подвергнуты условиям, которые истощают ядра по нуклеосомам, генерируя обедненные нуклеосомами ядра (рис. 2, блок 23).
В этом варианте осуществления способ включает распределение субпопуляции обедненных нуклеосомами ядер по первичному множеству компартментов (фиг. 2, блок 24). Количество компартментов на этапе первичного распределения (фиг. 2, блок 24) может зависеть от используемого формата. В одном из возможных вариантов количество компартментов составляет 24.
Каждый компартмент включает транспозомный комплекс. Транспозомный комплекс можно включать в каждый компартмент до, после или одновременно с субпопуляцией ядер, которые добавляют в компартмент. Транспозомный комплекс включает в себя, по меньшей мере, одну индексную последовательность и, по меньшей мере, одну универсальную последовательность. Универсальная последовательность, присутствующая в виде части транспозомного комплекса, может быть обозначена как спейсерная последовательность. Спейсерная последовательность присутствует в виде части последовательности транспозона. В одном из вариантов осуществления спейсерная последовательность может присутствовать на перенесенной цепи, т.е. цепи, где участок распознавания транспозазы переносится в нуклеиновую кислоту-мишень. Спейсерная последовательность пригодна в качестве сайта для отжига комплементарной последовательности. Например, спейсерная последовательность может быть универсальным праймером или последовательностью, комплементарной универсальному праймеру. Спейсерная последовательность транспозомного комплекса может быть одинаковой для каждого компартмента. В одном из вариантов осуществления индекс («bc1») и спейсер («sp1») присутствуют на липком конце, расположенном в ориентации, показанной на ФИГ. S2A из Примера 1.
Способ также включает получение индексированных ядер (фиг. 2, блок 25). В одном из вариантов осуществления получение индексированных ядер включает переработку нуклеиновых кислот, присутствующих в субпопуляции обедненных нуклеосомами ядер (например, нуклеиновых кислот, присутствующих в каждом компартменте), во множество фрагментов нуклеиновой кислоты. В одном из вариантов осуществления после фрагментации нуклеиновых кислот транспозаза остается прикрепленной к фрагментам нуклеиновой кислоты, так что фрагменты нуклеиновой кислоты, полученные из одной и той же геномной молекулы ДНК, остаются физически связанными (Adey et al., 2014, Genom Res., 24: 2041- 2049). Результатом фрагментации является популяция индексированных ядер, где каждое ядро содержит индексированные фрагменты нуклеиновой кислоты. Индексная последовательность транспозомного комплекса различна для каждого компартмента, таким образом, индексированные фрагменты нуклеиновой кислоты могут включать, и, как правило, включают, по меньшей мере, одну цепь индексной последовательности, указывающей на конкретный компартмент. Пример индексированного фрагмента нуклеиновой кислоты показан в рамке на ФИГ. S2A из Примера 1.
Индексированные ядра из множества компартментов можно комбинировать (фиг. 2, блок 26). Субпопуляции этих объединенных индексированных ядер затем распределяются во второе множество компартментов. Количество ядер, присутствующих в субпопуляции, и, следовательно, в каждом компартменте, частично основано на желании уменьшить столкновения индексов, которые представляют собой присутствие двух ядер с одинаковым транспозазным индексом в одном и том же компартменте на этом этапе способа. В одном из вариантов осуществления количество ядер, присутствующих в каждой субпопуляции, примерно одинаково.
Распределение ядер в субпопуляции сопровождается включением в индексированные фрагменты нуклеиновой кислоты в каждом отсеке второй индексной последовательности для создания фрагментов с двумя индексами. Это приводит к дальнейшему индексированию индексированных фрагментов нуклеиновой кислоты (фиг. 2, блок 27). В тех вариантах осуществления, где клетки перекрестно сшиты сшивающим средством, транспозазы, прикрепленные к индексированным фрагментам нуклеиновой кислоты, могут быть отделены от индексированных фрагментов нуклеиновой кислоты. Для диссоциации транспозаз можно использовать детергент, и в одном из вариантов осуществления детергент представляет собой додецилсульфат натрия (SDS).
В одном из вариантов осуществления включение второй индексной последовательности включает лигирование шпильки дуплекса для лигирования с индексированными фрагментами нуклеиновой кислоты в каждом компартменте. Дуплекс для лигирования может быть лигирован на одном конце или обоих концах двухиндексных фрагментов нуклеиновых кислот. В одном из вариантов осуществления дуплекс для лигирования включает в себя пять элементов: 1) последовательность, обратно комплементарную последовательности первого спейсера (например, «sp1» в ФИГ. S2B из примера 1), которая служит «посадочной площадкой» на этапе лигирования, описываемого в настоящем документе; 2) последовательность, обратно комплементарную штрихкоду для раунда 2; 3) последовательность, обратно комплементарную последовательности праймера для синтеза второй цепи (SSS); 4) промотор Т7, который предпочтительно является петлевой областью шпильки; 5) область праймера для синтеза второй цепи (SSS), начинающаяся с GGG, для усиления транскрипции T7 (вторая спейсерная последовательность, «sp2» на фиг. S2B в примере 1); и 6) вторую индексную последовательность штрихкода второго раунда, («bc2» на фиг. S2B из примера 1). Последовательности второго индекса уникальны для каждого компартмента, в который помещали распределенные индексированные ядра, (фиг. 2, блок 27) после добавления первого индекса путем тагментации.
Индексированные ядра из множества компартментов можно комбинировать (фиг. 2, блок 28). Субпопуляции этих комбинированных индексированных ядер затем распределяются в третье множество компартментов. Количество ядер, присутствующих в субпопуляции, и, следовательно, в каждом компартменте, частично основано на желании уменьшить столкновения индексов, которые представляют собой присутствие двух ядер с одинаковым транспозазным индексом в одном и том же компартменте на этом этапе способа. В одном из вариантов от 100 до 300 клеток распределяются по каждой лунке. В одном из вариантов осуществления в каждую лунку распределено до 300 клеток. В одном из вариантов осуществления количество ядер, присутствующих в каждой субпопуляции, примерно одинаково.
Распределение ядер с двумя индексами в субпопуляции сопровождается лизисом и дальнейшими манипуляциями (фиг. 2, блок 29). Способы лизиса ядер известны специалисту и являются рутинными. Дальнейшие манипуляции в качестве неограничивающих примеров включают в себя удлинение делеции, транскрипцию in vitro (IVT) и обратную транскрипцию.
Удлинение делеции превращает шпилечную структуру промотора T7 в дуплекс (рис. S2C из Примера 1). Как правило, для удлинения делеции используется полимераза, замещающая цепи. Доступны полимеразы, имеющие эту активность, например Bst-полимераза.
IVT создает линейные амплифицированные одноцепочечные молекулы РНК ниже промотора Т7 (рис. S2D из Примера 1). Способы для IVT известны и являются рутинными.
Обратная транскрипция может происходить по одному из двух маршрутов (фиг. S2E из Примера 1). Реакция лигирования, описываемая в настоящем документе приводит к двум типам фрагментов нуклеиновой кислоты: фрагментам нуклеиновой кислоты, имеющим лигирующий дуплекс на обоих концах, и фрагментам нуклеиновой кислоты, имеющим дуплекс для лигирования на одном конце. Если лигирование было успешным на обоих концах, обратная транскрипция может быть запущена самокольцующимися праймерами для обратной транскрипции, которые унаследовали свойства от петлевого дуплекса для лигирования; если лигирование было успешным только на одном конце, обратная транскрипция запускается дополнительными праймерами для обратной транскрипции РНК, добавленными в избытке.
Лизис ядер и обработка фрагментов нуклеиновой кислоты сопровождается включением в фрагменты нуклеиновой кислоты с двойным индексом в каждом компартменте третьей индексной последовательности для генерации фрагментов с тройным индексом, где третья индексная последовательность в каждом компартменте отличается от последовательностей первого и второго индекса в других компартментах, а третья индексная последовательность в каждом компартменте отличается от третьего индекса в других компартментах. Это приводит к дальнейшему индексированию индексированных фрагментов нуклеиновой кислоты (фиг. 2, блок 30; ФИГ. S2F из примера 1) до иммобилизации и секвенирования. Третий индекс может быть включен путем синтеза второй цепи ДНК. В одном из вариантов осуществления вторая цепь ДНК производится с использованием праймера, который включает в себя последовательности, комплементарные нуклеотидам, присутствующим на концах фрагментов нуклеиновой кислоты с двойным индексом. Например, праймер может включать в себя последовательность второго спейсера (sp2), которая будет отжигаться с последовательностью, обратно комплементарной последовательности второго спейсера (фиг. S2F из примера 1). Праймер дополнительно включает в себя третий индекс («bc3» на фиг. S2F из примера 1) и другие уникальные молекулярные идентификаторы (UMI). Полученную двухцепочечную ДНК можно очищать с помощью рутинных способов.
Для секвенирования можно получать множество трехиндексных фрагментов. После объединения трехиндексных фрагментов их обогащают перед секвенированием, как правило, путем иммобилизации и/или амплификации (фиг. 2, блок 31).
Получение иммобилизованных образцов для секвенирования
Для секвенирования можно получать множество индексированных фрагментов. Например, в тех вариантах, где создаются библиотеки фрагментов с тройным индексом, фрагменты с тройным индексом перед секвенированием, как правило, обогащают путем иммобилизации и/или амплификации (фиг. 2, блок 21). Способы прикрепления индексированных фрагментов из одного или более источников к субстрату известны в данной области. В одном из вариантов осуществления индексированные фрагменты обогащают с помощью множества захватывающих олигонуклеотидов, имеющих специфичность для индексированных фрагментов, и захватывающие олигонуклеотиды могут быть иммобилизованы на поверхности твердого субстрата. Например, захватывающие олигонуклеотиды могут включать первого участника универсальной пары связывания, где второй участник пары связывания иммобилизован на поверхности твердого субстрата. Аналогичным образом, способы для амплификации иммобилизованных фрагментов с двумя индексами в качестве неограничивающих примеров включают в себя мостиковую амплификацию и кинетическое исключение. Способы иммобилизации и амплификации перед секвенированием описаны, например, в Bignell et al. (US 8053192), Gunderson et al. (WO2016/130704), Shen et al. (US 8895249) и Pipenburg et al. (US 9309502).
Объединенный образец может быть иммобилизован при подготовке для секвенирования. Секвенирование можно проводить в виде чипа с одиночными молекулами или можно проводить амплификацию до секвенирования. Амплификацию можно проводить с использованием одного или более иммобилизованных праймеров. Иммобилизованный праймер/праймеры могут быть, например, в виде «газона» на плоской поверхности или на совокупности гранул. Совокупность гранул можно выделить в эмульсию с одной гранулой в каждом «компартменте» эмульсии. При концентрации только одной матрицы на «компартмент» только одну матрицу амплифицируют на каждой грануле.
Термин «твердофазная амплификация» относится к любой реакции амплификации нуклеиновой кислоты, проводимой на твердой подложке или в сочетании с твердой подложкой, так что все или часть амплифицированных продуктов иммобилизуются на твердой подложке по мере их образования. В частности, термин охватывает твердофазную полимеразную цепную реакцию (твердофазную ПЦР) и изотермическую твеердофазную амплификацию, которые являются реакциями, аналогичными стандартной амплификации в жидкой фазе, за исключением того, что один или оба из прямого и обратного праймеров для амплификации иммобилизованы на твердой подложке. Твердофазная ПЦР включает в себя системы, такие как эмульсии, где один праймер прикрепляется к грануле, а другой находится в свободном растворе, и колониеобразование в твердофазных гелевых матрицах, где один праймер прикреплен к поверхности, а один находится в свободном растворе.
В некоторых вариантах осуществления твердая подложка содержит поверхность с рисунком. «Поверхность с рисунком» относится к расположению различных областей в открытом слое или на открытом слое твердой подложки. Например, одна или более областей могут быть элементами, где присутствуют один или более праймеров для амплификации. Элементы могут быть разделены промежуточными областями, где праймеры для амплификации отсутствуют. В некоторых вариантах осуществления рисунок может представлять собой x-y формат элементов, которые находятся в строках и столбцах. В некоторых вариантах осуществления рисунок может представлять собой повторяющееся расположение элементов и/или промежуточных областей. В некоторых вариантах осуществления рисунок может быть случайным расположением элементов и/или промежуточных областей. Примеры поверхностей с рисунком, которые можно использовать в различных способах и композициях, изложенных в настоящем документе, описаны в патентах США №№ 8778848, 8778849 и 9079148 и публикации США № 2014/0243224.
В некоторых вариантах осуществления твердая подложка включает множество лунок или углублений на поверхности. Это может быть изготовлено, как это принято, в основном, в данной области, с использованием ряда способов, включая в качестве неограничивающих примеров, фотолитографию, техники штамповки, техники литья и техники микротравления. Как будет понятно специалистам в данной области, используемый способ будет зависеть от состава и формы субстрата чипа.
Элементами на поверхности с рисунком могут быть лунки на чипе с лунками (например, микроячейки или нанолунки) на стекле, кремнии, пластике или других подходящих твердых подложках с рисунком из ковалентно-связанного геля, такого как поли(N-(5-азидоацетамидилпентил)акриламид-со-акриламид) (PAZAM, см., например, публикацию США № 2013/184796, WO 2016/066586 и WO 2015/002813). Способ создает гелевые подушечки, используемые для секвенирования, которые могут быть стабильными в течение секвенирования с большим количеством циклов. Ковалентное связывание полимера с лунками полезно для сохранения геля в структурированных элементах на протяжении всего срока службы структурированного субстрата в течение ряда использований. Однако во многих вариантах осуществления гель не обязательно должен быть ковалентно связан с лунками. Например, в некоторых условиях акриламид без силана (SFA, см., например, патент США № 8563477), который ковалентно не присоединен к какой-либо части структурированного субстрата, можно использовать в качестве материала геля.
В определенных вариантах осуществления структурированный субстрат может быть изготовлен путем нанесения рисунка на материал твердой подложки с лунками (например, микролунками или нанолунками), покрытия подложки с рисунком материалом геля (например, PAZAM, SFA или его химически модифицированными вариантами, такими как азидолизированная версия SFA (азидо-SFA)) и полировка подложки с гелевым покрытием, например, посредством химической или механической полировки, таким образом, удерживая гель в лунках, но удаляя или инактивируя по существу весь гель из промежуточных областей на поверхности структурированного субстрата между лунками. Праймерные нуклеиновых кислот могут быть прикреплены к материалу геля. Затем раствор индексированных фрагментов может контактировать с полированным субстратом, так что отдельные индексированные фрагменты будут засеяны в отдельные лунки посредством взаимодействия с праймерами, прикрепленными к материалу геля; однако, нуклеиновые кислоты-мишени не будут занимать промежуточные области из-за отсутствия или неактивности материала геля. Амплификация индексированных фрагментов будет ограничена лунками, поскольку отсутствие или неактивность геля в промежуточных областях препятствует внешней миграции растущей колонии нуклеиновых кислот. Способ может быть удобно перенесен на производство, будучи масштабируемым и используя общепринятые микро- или нанотехнологические способы.
Хотя изобретение охватывает «твердофазные» способы амплификации, в которых иммобилизован только один праймер для амплификации (другой праймер, как правило, присутствует в свободном растворе), в одном из вариантов осуществления является предпочтительным, чтобы твердая подложка обеспечивала иммобилизацию обоих, прямого и обратного, праймеров. Практически будет «множество» идентичных прямых праймеров и/или «множество» идентичных обратных праймеров, иммобилизованных на твердой подложке, поскольку процесс амплификации требует избытка праймеров для поддержания амплификации. Ссылки в настоящем документе на прямые и обратные праймеры должны интерпретироваться таким образом, как охватывающие «множество» таких праймеров, если контекст не указывает на иное.
Как будет понятно квалифицированному читателю, любая данная реакция амплификации требует наличия, по меньшей мере, одного типа прямого праймера и, по меньшей мере, одного типа обратного праймера, специфичного для матрицы, подлежащей амплификации. Однако в определенных вариантах осуществления прямой и обратный праймеры могут включать специфичные для матрицы части идентичной последовательности и могут иметь полностью идентичную нуклеотидную последовательность и структуру (включая любые не нуклеотидные модификации). Другими словами, можно проводить твердофазную амплификацию с использованием только одного типа праймера, и такие однопраймерные способы включены в объем изобретения. В других вариантах осуществления можно использовать прямые и обратные праймеры, которые содержат идентичные, специфичные для матрицы последовательности, но отличаются некоторыми другими структурными особенностями. Например, один тип праймера может содержать ненуклеотидную модификацию, которой нет в другом.
Во всех вариантах осуществления изобретения праймеры для твердофазной амплификации предпочтительно иммобилизуются путем ковалентного прикрепления в одной точке к твердой подложке в 5'-конце или около 5'-конца праймера, оставляя специфическую для матрицы часть праймера свободной для отжига на его распознаваемой матрице и свободную 3'-гидроксильную группу для продления праймера. Любые подходящие способы ковалентного присоединения, известные в данной области, можно использовать для этой цели. Выбранная химия присоединения будет зависеть от природы твердой подложки и от любой применяемой к ней дериватизации или функционализации. Сам праймер может включать группу, которая может быть не нуклеотидной химической модификацией, для облегчения прикрепления. В конкретном варианте осуществления, праймер может включать серосодержащий нуклеофил, такой как фосфоротиоат или тиофосфат, на 5'-конце. В случае твердофазных полиакриламидных гидрогелей этот нуклеофил будет связываться с бромоацетамидной группой, присутствующей в гидрогеле. Более конкретным способом прикрепления праймеров и матриц к твердой подложке является присоединение 5'-тиофосфата к гидрогелю, состоящему из полимеризованного акриламида и N-(5-бромацетамидилпентил) акриламида (BRAPA), как описано в WO 05/065814.
В некоторых вариантах осуществления изобретения можно использовать твердые подложки, которые включают в себя инертный субстрат или матрицу (например, предметные стекла, полимерные шарики и т.д.), которые были «функционализированы», например, путем нанесения слоя или покрытия из промежуточного материала, включая реакционноспособные группы, которые позволяют ковалентно связываться с биомолекулами, такими как полинуклеотиды. Примеры таких подложек в качестве неограничивающих примеров включают полиакриламидные гидрогели, нанесенные на инертный субстрат, такой как стекло. В таких вариантах осуществления биомолекулы (например, полинуклеотиды) могут быть непосредственно ковалентно присоединены к промежуточному материалу (например, гидрогелю), но промежуточный материал сам может быть нековалентно присоединен к субстрату или матрице (например, стеклянному субстрату). Термин «ковалентное присоединение к твердой подложке» следует интерпретировать таким образом, как охватывающий этот тип конфигурации.
Объединенные образцы можно амплифицировать на гранулах, где каждая гранула содержит прямой и обратный праймер для амплификации. В конкретном варианте осуществления используют библиотеку индексированных фрагментов для подготовки кластерных массивов колоний нуклеиновой кислоты, аналогичных тем, которые описаны в публикации США № 2005/0100900, патенте США № 7115400, WO 00/18957 и WO 98/44151 путем твердофазной амплификации и более конкретно твердофазной изотермической амплификации. Термины «кластер» и «колония» используют взаимозаменяемо в данном документе для обозначения дискретного участка на твердой подложке, включающего множество идентичных иммобилизованных цепей нуклеиновой кислоты и множество идентичных иммобилизованных комплементарных цепей нуклеиновой кислоты. Термин «кластерный массив» относится к массиву, сформированному из таких кластеров или колоний. В этом контексте термин «массив» не следует понимать как требующий упорядоченного расположения кластеров.
Термин «твердая фаза» или «поверхность» применяется для обозначения либо плоского чипа, где праймеры прикреплены к плоской поверхности, например, стеклу, диоксиду азота или пластиковым микроскопным предметным стеклам или аналогичные устройствам с проточными ячейками; гранул, где один или два праймера прикреплены к гранулам, и гранулы амплифицируют; или чипу с гранулами на поверхности после того, как гранулы были амплифицированы.
Кластерные массивы можно получать, используя либо способ термоциклирования, как описано в WO 98/44151, либо способ, при котором поддерживается постоянная температура, а циклы удлинения и денатурирования проводят с использованием смены реагентов. Такие изотермические способы амплификации описаны в патентной заявке WO 02/46456 и публикации США № 2008/0009420. Из-за более низких температур, полезных в изотермическом процессе, это особенно предпочтительно в некоторых вариантах осуществления.
Следует понимать, что любой из способов амплификации можно использовать с универсальными или целевыми праймерами для амплификации иммобилизованных фрагментов ДНК. Подходящие способы амплификации в качестве неограничивающих примеров включают в себя полимеразную цепную реакцию (ПЦР), амплификацию с вытеснением цепи (SDA), амплификацию, опосредованную транскрипцией (TMA) и амплификацию на основе последовательности нуклеиновых кислот (NASBA), как указано в патенте США № 8003354. Вышеуказанные способы амплификации могут быть использованы для амплификации одной или более нуклеиновых кислот, представляющих интерес. Например, ПЦР, в том числе мультиплексную ПЦР, SDA, TMA, NASBA и т.п. можно использовать для амплификации иммобилизованных фрагментов ДНК. В некоторых вариантах осуществления в реакцию амплификации включены праймеры, направленные конкретно на полинуклеотид, представляющий интерес.
Другие подходящие способы амплификации полинуклеотидов могут включать удлинение и лигирование олигонуклеотидов, амплификацию по типу катящегося кольца (RCA) (Lizardi et al., Nat. Genet. 19: 225-232 (1998)) и анализ лигирования олигонуклеотидов (OLA) (См. в основном патенты США №№ 7582420, 5185243, 5679524 и 5573907; EP 0 320 308 B1; EP 0 336 731 B1; EP 0 439 182 B1; WO 90/01069; WO 89/12696; и WO 89/09835). Следует понимать, что эти способы амплификации могут быть разработаны для амплификации иммобилизованных фрагментов ДНК. Например, в некоторых вариантах осуществления способ амплификации может включать реакции амплификации лигированных зондов или анализа лигирования олигонуклеотида (OLA), которые содержат праймеры, направленные конкретно на интересующую нуклеиновую кислоту. В некоторых вариантах осуществления способ амплификации может включать реакцию удлинения-лигирования праймера, которая содержит праймеры, направленные конкретно на интересующую нуклеиновую кислоту. В качестве неограничивающего примера праймеров для реакции удлинения и лигирования праймеров, которые могут быть специально разработаны для амплификации интересующей нуклеиновой кислоты, амплификация может включать праймеры, используемые для анализа GoldenGate (Illumina, Inc., Сан-Диего, Калифорния), как показано в патентах США № 7582420 и 7611869.
Также можно использовать наносферы ДНК в комбинации со способами и композициями, описанными в настоящем документе. Способы создания и использования наносфер ДНК для геномного секвенирования можно найти, например, в патентах США и патентных публикациях США № 7,9103542009/0264299, 2009/0011943, 2009/0005252, 2009/0155781, 2009/0118488 и как описано, например, в Drmanac et al., 2010, Science 327 (5961): 78-81. В кратком изложении, после фрагментации библиотеки геномной ДНК адаптеры лигируют к фрагментам, фрагменты с лигированными адаптерами закольцовывают путем лигирования с кольцевой лигазой и проводят амплификацию по типу катящегося кольца (как описано в Lizardi et al., 1998. Nat. Genet. 19: 225-232 и US 2007/0099208 A1). Удлиненная конкатамерная структура ампликонов способствует скручиванию, создавая компактные наносферы ДНК. Наносферы ДНК могут быть захвачены на субстраты, чтобы создать упорядоченный чип или чип с рисунком таким образом, чтобы расстояние сохранялось между каждой наносферой, что позволяет секвенировать отдельные наносферы ДНК. В некоторых вариантах осуществления, таких как используемые в Complete Genomics (Маунтин-Вью, Калифорния), последовательные раунды лигирования адаптера, амплификации и расщепления выполняются перед циклизацией для получения конструкций голова к хвосту, имеющих несколько геномных фрагментов ДНК, разделенных адаптероными последовательностями.
Типичные изотермические способы амплификации, которые можно использовать в способе по настоящему изобретению в качестве неограничивающих примеров, включают в себя амплификацию с множественным вытеснением цепи (MDA), как показано, например, у Dean et al., Proc. Natl. Акад. Sci. США 99: 5261-66 (2002) или изотермическую амплификацию нуклеиновых кислот с вытеснением цепи, пример которой описан, например, в патенте США № 6214587. Другие, не основанные на ПЦР способы, которые можно использовать в настоящем изобретении, включают, например, амплификацию с вытеснением цепи (SDA), которая описана, например, у Walker et al., Molecular Methods for Virus Detection, Academic Press, Inc., 1995; патентах США №№ 5455166 и 5130238, и Walker et al., Nucl. Acids Res. 20: 1691-96 (1992) или амплификацию с вытеснением гиперразветвленной цепи, описанная, например, у Lage et al., Genom Res. 13: 294-307 (2003). Способы изотермической амплификации можно использовать, например, с вытесняющей цепи Phi 29-полимеразой или большим фрагментов Bst ДНК-полимеразы, с 5'-> 3' экзо- для амплификации геномной ДНК со случайным праймером. Использование этих полимераз имеет преимущество за счет их высокой процессивности и активности по вытеснению цепей. Высокая процессивность позволяет синтезировать фрагменты размером 10-20 т.п.н. в длину. Как указано выше, более мелкие фрагменты можно получать в изотермических условиях с использованием полимеразы, обладающей низкой процессивностью и активностью по вытеснению цепей, такой как полимераза Кленова. Дополнительное описание реакций амплификации, условий и компонентов подробно изложено в описании патента США № 7670810.
Другой способ амплификации полинуклеотидов, подходящий для применения в настоящем изобретении, представляет собой ПЦР с метками, которая использует популяцию праймеров из двух доменов, имеющих постоянную 5'-область, за которой следует случайная 3'-область, как описано, например, в Grothues et al. Nucleic Acids Res. 21 (5): 1321-2 (1993). Первые раунды амплификации проводят для обеспечения множества инициаций на ДНК, денатурированной высокой температурой, на основании индивидуальной гибридизации из случайно синтезированной 3'-области. Из-за природы 3'-области сайты инициации предполагаются случайными на всем протяжении генома. После этого несвязанные праймеры могут быть удалены, и дальнейшая репликация может осуществляться с использованием праймеров, комплементарных постоянной 5'-области.
Изотермическую амплификацию можно проводить с использованием амплификации с кинетическим исключением (KEA), также обозначаемой как исключающая амплификация (ExAmp). Библиотеку нуклеиновых кислот по настоящему изобретению можно получать при помощи способа, который включает в себя этап реакции реагента для амплификации для получения множества сайтов амплификации, каждый из которых включает по существу клональную популяцию ампликонов из отдельной нуклеиновой кислоты-мишени, которая содержит этот сайт. В некоторых вариантах осуществления реакция амплификации продолжается до тех пор, пока не будет получено достаточное количество ампликонов, чтобы заполнить емкость соответствующего сайта амплификации. Заполнение сайта до полной емкости, таким образом, препятствует посадке и амплификации нуклеиновых кислот-мишеней в этом сайте, создавая тем самым клональную популяцию ампликонов в этом сайте. В некоторых вариантах осуществления возможна очевидная клональность, даже если сайт амплификации не заполнен до полной емкости до того, как вторая нуклеиновая кислота-мишень прибудет на сайт. В некоторых вариантах осуществления амплификация первой нуклеиновой кислоты-мишени может продолжаться до такой степени, что получают достаточное количество копий, чтобы эффективно превзойти или преодолеть производство копий из второй нуклеиновой кислоты-мишени, которые транспортируются на сайт. Например, в варианте осуществления, который использует способ мостиковой амплификации на кольцевом элементе диаметром менее 500 нм, было установлено, что после 14 циклов экспоненциальной амплификации для первой нуклеиновой кислоты-мишени, загрязнение второй нуклеиновой кислотой-мишенью на этом же участке будет производить недостаточное количество загрязняющих ампликонов для неблагоприятного воздействия на анализ секвенирования путем синтеза на платформе для секвенирования Illumina.
В некоторых вариантах осуществления сайты на чипе могут быть, но не обязательно, полностью клональными. Скорее, для некоторых применений отдельный сайт амплификации может быть заполнен ампликонами из первичного индексированного фрагмента, а также может иметь низкий уровень загрязняющих ампликонов из второй нуклеиновой кислоты-мишени. Чип может иметь один или более сайтов амплификации, которые имеют низкий уровень загрязняющих ампликонов при условии, что уровень загрязнения не оказывает неприемлемого влияния на последующее использование чипа. Например, когда чип должен использоваться для детекции, приемлемым уровнем загрязнения будет уровень, который не влияет на сигнал/шум или разрешение способа детекции недопустимым образом. Таким образом, очевидная клональность будет, в основном, иметь отношение к конкретному использованию или применению чипа, созданного способами, изложенными в данном документе. Примерные уровни загрязнения, которые могут быть приемлемы на отдельном сайте амплификации для конкретных применений, включают в себя не более 0,1%, 0,5%, 1%, 5%, 10% или 25% загрязняющих ампликонов. Чип может включать один или более сайтов амплификации, имеющих эти типичные уровни загрязняющих ампликонов. Например, до 5%, 10%, 25%, 50%, 75% или даже до 100% сайтов амплификации на чипе могут содержать некоторые загрязняющие ампликоны. Следует понимать, что на чипе или другой совокупности сайтов, по меньшей мере, 50%, 75%, 80%, 85%, 90%, 95% или 99% или более сайтов могут быть клональными или, по-видимому, клональными.
В некоторых вариантах осуществления возможны кинетические исключения, когда процесс происходит с достаточно высокой скоростью, чтобы эффективно исключить другое событие или процесс. Возьмем, например, создание чипа с нуклеиновыми кислотами, где сайты на чипе случайным образом засеваются с помощью фрагментов с тройным индексом из раствора и копий фрагментов с тройным индексом в процессе амплификации, чтобы заполнить каждый из отобранных сайтов до полной емкости. В соответствии со способами кинетического исключения по настоящему изобретению процессы посева и амплификации могут протекать одновременно в условиях, когда скорость амплификации превышает норму высева. Таким образом, относительно высокая скорость, с которой копии делаются на сайте, который был посеян первичной нуклеиновой кислотой-мишенью, будет эффективно исключать вторую нуклеиновую кислоту из засева сайта для амплификации. Способы амплификации с кинетическим исключением можно проводить, как описано подробно в описании публикации заявки США № 2013/0338042.
Кинетическое исключение может использовать относительно медленную скорость для инициации амплификации (например, медленную скорость создания первой копии индексированного фрагмента) по сравнению с относительно быстрой скоростью для создания последующих копий фрагмента с тройным индексированием (или первой копии индексированного фрагмента). В примере из предыдущего абзаца кинетическое исключение происходит из-за относительно медленной скорости посева индексированного фрагмента (например, относительно медленной диффузии или транспорта) по сравнению с относительно быстрой скоростью, с которой происходит амплификация для заполнения сайта копиями посеянного индексированного фрагмента. В другом иллюстративном варианте осуществления кинетическое исключение может происходить из-за задержки в формировании первой копии индексированного фрагмента, который заполнил сайт (например, отложенная или медленная активация) по сравнению с относительно быстрой скоростью, с которой последующие копии создаются для заполнения сайта. В этом примере отдельный сайт мог быть заполнен несколькими различными проиндексированными фрагментами (например, несколько проиндексированных фрагментов могут присутствовать на каждом сайте до амплификации). Однако формирование первой копии для любого данного индексированного фрагмента может быть активировано случайным образом, так что средняя скорость формирования первой копии является относительно низкой по сравнению со скоростью, с которой получают последующие копии. В этом случае, хотя отдельный сайт мог быть засеян несколькими различными индексированными фрагментами, кинетическое исключение позволит амплифицировать только один из этих индексированных фрагментов. Более конкретно, как только первичный индексированный фрагмент был активирован для амплификации, сайт будет быстро заполнен до отказа своими копиями, тем самым предотвращая создание копий второго индексированного фрагмента на сайте.
В одном из вариантов осуществления способ проводят для одновременного (i) перемещения индексированных фрагментов в сайты амплификации со средней скоростью транспортировки, и (ii) амплификации индексированных фрагментов, которые находятся в сайтах амплификации, со средней скоростью амплификации, где средняя скорость амплификации превышает среднюю скорость транспортировки (патент США № 9169513). Таким образом, в таких вариантах осуществления кинетическое исключение может быть достигнуто путем использования относительно низкой скорости транспортировки. Например, можно выбрать достаточно низкую концентрацию индексированных фрагментов для достижения желаемой средней скорости транспортировки, более низкая концентрация приводит к более медленной средней скорости транспортировки. Альтернативно или дополнительно, раствор с высокой вязкостью и/или присутствие реагентов для молекулярного краудинга в растворе можно использовать для снижения скорости транспортировки. Примеры подходящих реагентов для молекулярного краудинга включают в себя полиэтиленгликоль (ПЭГ), фиколл, декстран или поливиниловый спирт. Типичные реагенты для молекулярного краудинга и составы приведены в патенте США № 7399590, включенном в настоящий документ в качестве ссылки. Другим фактором, который можно регулировать для достижения желаемой скорости транспортировки, является средний размер нуклеиновых кислот-мишеней.
Реагент для амплификации может включать дополнительные компоненты, которые облегчают образование ампликона, а, в некоторых случаях, увеличивают скорость образования ампликона. Примером является рекомбиназа. Рекомбиназа может облегчить формирование ампликона, обеспечивая повторную вставку/удлинение. Более конкретно, рекомбиназа может способствовать вставке индексированного фрагмента посредством полимеразы и удлинению праймера с помощью полимеразы, используя индексированный фрагмент в качестве матрицы для формирования ампликона. Этот процесс может быть повторен как цепная реакция, где ампликоны, произведенные от каждого раунда вставки/удлинения, служат матрицей в следующем раунде. Процесс может происходить быстрее, чем стандартная ПЦР, поскольку не требуется цикл денатурации (например, посредством нагревания или химической денатурации). Таким образом, амплификацию при содействии рекомбиназы можно проводить изотермически. Как правило, желательно включить АТФ или другие нуклеотиды (или в некоторых случаях их не гидролизуемые аналоги) в реагент для амплификации с содействием рекомбиназы для облегчения амплификации. Смесь рекомбиназы и одноцепочечного связывающего белка (SSB) особенно полезна, так как SSB может дополнительно облегчить амплификацию. Примеры составов для амплификации с содействием рекомбиназы включают те, которые продаются в виде наборов TwistAmp от TwistDx (Кембридж, Великобритания). Подходящие компоненты реагента для амплификации с содействием рекомбиназы описаны в US 5223414 и US 7399590.
Другим примером компонента, который может быть включен в реагент для амплификации для облегчения образования ампликона и, в некоторых случаях, для увеличения скорости образования ампликона, является хеликаза. Хеликаза может облегчить образование ампликонов, обеспечивая цепную реакцию образования ампликонов. Процесс может происходить быстрее, чем стандартная ПЦР, поскольку не требуется цикл денатурации (например, посредством нагревания или химической денатурации). Таким образом, амплификацию при содействии хеликазы можно проводить изотермически. Смесь хеликазы и одноцепочечного связывающего белка (SSB) особенно полезна, так как SSB может дополнительно облегчить амплификацию. Примеры составов для амплификации при содействии хеликазы включают те, которые продаются коммерчески в виде наборов IsoAmp от Biohelix (Beverly, MA). Кроме того, примеры подходящих составов, которые включают белок хеликазы, описаны в US 7399590 и US 7829284.
Еще одним примером компонента, который может быть включен в реагентдля амплификации для облегчения образования ампликона и, в некоторых случаях, увеличения скорости образования ампликона, является белок, связывающийся с точкой начала репликации.
Применение в секвенировании/способах секвенирования
После прикрепления индексированных фрагментов к поверхности, определяют последовательность иммобилизованных и амплифицированных индексированных фрагментов. Секвенирование можно проводить с использованием любой подходящей техники секвенирования, а способы определения последовательности иммобилизованных и амплифицированных индексированных фрагментов, включая повторный синтез цепей, известны в данной области и описаны, например, в Bignell et al. (US 8053192), Gunderson et al. (WO2016/130704), Shen et al. (US 8895249) и Pipenburg et al. (US 9309502).
Способы, описываемые в настоящем документе, можно использовать в сочетании с рядом способов секвенирования нуклеиновых кислот. Особенно применимыми способами являются те, где нуклеиновые кислоты присоединяются в фиксированных местах на чипе, так что их относительные положения не меняются, и где чип неоднократно визуализируют. Варианты осуществления, в которых изображения получают в разных цветовых каналах, например, совпадающих с разными метками, используемыми для различения одного типа нуклеотидного основания от другого, являются особенно применимыми. В некоторых вариантах осуществления способ определения нуклеотидной последовательности индексированного фрагмента может быть автоматизированным способом. Предпочтительные варианты включают способы секвенирования путем синтеза («SBS»).
Способы SBS в основном включают ферментативное удлинение зарождающейся цепи нуклеиновой кислоты путем итеративного добавления нуклеотидов к матричной цепи. В традиционных способах SBS один нуклеотидный мономер может быть предоставлен целевому нуклеотиду в присутствии полимеразы в каждой доставке. Однако в способах, описанных в настоящем документе, более чем один тип нуклеотидного мономера может быть предоставлен нуклеиновой кислоте-мишени в присутствии полимеразы при доставке.
В одном из вариантов осуществления нуклеотидный мономер включает в себя замкнутые нуклеиновые кислоты (LNA) или мостиковые нуклеиновые кислоты (BNA). Использование LNA или BNA в нуклеотидном мономере увеличивает силу гибридизации между нуклеотидным мономером и последовательностью праймера для секвенирования, присутствующей на иммобилизованном индексированном фрагменте.
SBS может использовать нуклеотидные мономеры, которые имеют терминирующую группу, или те, у которых нет терминирующих групп. Способы с использованием нуклеотидных мономеров, в которых отсутствуют терминаторы, включают, например, пиросеквенирование и секвенирование с использованием γ-фосфат-меченных нуклеотидов, как изложено более подробно в настоящем документе. В способах с использованием нуклеотидных мономеров, в которых отсутствуют терминаторы, количество нуклеотидов, добавляемых в каждом цикле, является, как правило, вариабельным и зависит от последовательности матрицы и способа доставки нуклеотидов. Для методов SBS, в которых используются нуклеотидные мономеры, имеющие терминирующую группу, терминатор может быть эффективно необратимым в используемых условиях секвенирования, как в случае традиционного секвенирования по Сэнгеру, в котором используются дидезоксинуклеотиды, или терминатор может быть обратимым, как в случае способов секвенирования, разработанных Solexa (сейчас Illumina, Inc.).
Способы SBS могут использовать нуклеотидные мономеры, которые имеют группу метки или те, которые не имеют группу метки. Таким образом, события включения можно детектировать на основе характеристики метки, такой как флуоресценция метки; характеристики нуклеотидного мономера, такой как молекулярная масса или заряд; побочного продукта включения нуклеотида, такого как высвобождение пирофосфата; или т.п. В вариантах осуществления, где два или более разных нуклеотида присутствуют в реагенте для секвенирования, разные нуклеотиды могут различаться друг от друга, или альтернативно две или более различных меток могут быть неразличимы при использовании методов детекции. Например, разные нуклеотиды, присутствующие в реагенте для секвенирования, могут иметь разные метки, и их можно различить с помощью соответствующей оптики, примером чего являются способы секвенирования, разработанные Solexa (в настоящее время Illumina, Inc.).
Предпочтительные варианты осуществления включают способы пиросеквенирования. Пиросеквенирование обнаруживает высвобождение неорганического пирофосфата (PPi), когда определенные нуклеотиды включаются в возникающую цепь (Ronaghi, M., Karamohamed, S., Pettersson, B., Uhlen, M. и Nyren, P. (1996) «Real-time DNA sequencing using detection of pyrophosphate release» Analytical Biochemistry 242 (1), 84-9; Ronaghi, M. (2001)«Pyrosequencing sheds light on DNA sequencing». Genome Res. 11 (1), 3-11; Ronaghi , M., Uhlen, M. и Nyren, P. (1998) «A sequencing method based on real-time pyrophosphate» Science 281 (5375), 363; патенты США №№ 6210891; 6258568 и 6274320). При пиросеквенировании высвобождаемый PPi может быть обнаружен путем немедленного превращения в аденозин трифосфат (АТФ) с помощью сульфуразы АТФ, а уровень генерируемого АТФ определяется с помощью фотонов, производимых люциферазой. Нуклеиновые кислоты, которые будут секвенироваться, могут быть присоединены кэлементам на чипе, а чип может быть визуализирован для захвата хемилюминесцентных сигналов, которые создаются в результате включения нуклеотидов в элементы чипа. Изображение можно получать после того, как чип обработали определенным типом нуклеотида (например, A, T, C или G). Изображения, полученные после добавления каждого типа нуклеотидов, будут отличаться в зависимости от того, какие элементы детектированы на чипе. Эти различия в изображении отражают различную последовательность содержимого элементов на чипе. Однако относительные местоположения каждого элемента на изображениях не изменятся. Изображения можно хранить, обрабатывать и анализировать с использованием способов, изложенных в настоящем документе. Например, изображения, полученные после обработки чипа каждым различным типом нуклеотида, можно обрабатывать так же, как и в качестве примера в настоящем документе для изображений, полученных из разных детектирующих каналов для способов секвенирования на основе обратимых терминаторов.
В другом иллюстративном типе SBS сиквенсовый цикл проводят путем поэтапного добавления нуклеотидов-обратимых терминаторов, содержащих, например, расщепляемую или фотообесцвеченную метку-краситель, как указано, например, в WO 04/018497 и патенте США № 7057026. Этот подход, который коммерциализируется Solexa (в настоящее время Illumina Inc.), также описан в WO 91/06678 и WO 07/123744. Наличие флуоресцентно-меченных терминаторов, в которых и терминация может быть обратима и флуоресцентная метка расщепляется, способствует эффективному секвенированию с циклической обратимой терминацией (CRT). Полимеразы также могут быть сконструированы таким образом, чтобы эффективно включать и удлинять эти модифицированные нуклеотиды.
В некоторых вариантах осуществления секвенирования на основе обратимых терминаторов метки по существу не ингибируют удлинение в условиях реакции SBS. Однако детектирующие метки могут быть удалены, например, путем расщепления или деградации. Изображения можно получать после включения меток в элементы нуклеиновых кислот на чипе. В конкретных вариантах осуществления каждый цикл включает в себя одновременную доставку четырех различных типов нуклеотидов на чип, и каждый тип нуклеотидов имеет спектрально отличную метку. Затем могут быть получены четыре изображения, каждое из которых использует детектирующий канал, который является селективным для одной из четырех различных меток. Альтернативно, различные типы нуклеотидов можно добавлять последовательно, а изображение чипа можно получать между каждым шагом добавления. В таких вариантах осуществления каждое изображение будет содержать элементы нуклеиновых кислот, в которые включены нуклеотиды определенного типа. Различные элементы будут присутствовать или отсутствовать на разных изображениях из-за различного содержания последовательности каждого элемента. Однако относительное положение элементов на изображениях останется неизменным. Изображения, полученные с помощью таких способов SBS с обратимыми терминаторами, можно хранить, обрабатывать и анализировать, как указано в настоящем документе. После этапа получения изображения метки можно удалить, а группы обратимых терминаторов можно удалить для последующих циклов добавления и детекции нуклеотидов. Удаление меток после того, как они были детектированы в определенном цикле и перед последующим циклом, может обеспечить преимущество уменьшения фонового сигнала и перекрестных помех между циклами. Примеры подходящих меток и способов удаления изложены в настоящем документе.
В конкретных вариантах осуществления некоторые или все нуклеотидные мономеры могут включать обратимые терминаторы. Обратимые терминаторы/расщепляемые флуорофоры могут включать флуорофоры, связанные с рибозной группой через 3' сложную эфирную связь (Metzker, Genom Res. 15: 1767-1776 (2005)). Другие подходы отделяют химию терминаторов от расщепления флуоресцентной метки (Ruparel et al., Proc Natl Acad Sci USA 102: 5932-7 (2005)). Ruparel et al. описали разработку обратимых терминаторов, которые использовали небольшую 3'-аллильную группу для блокировки удлинения, но могли легко быть разблокированы короткой обработкой палладиевым катализатором. Флуорофор был прикреплен к основанию с помощью фотоотверждаемого линкера, который можно легко отщепить 30-секундным воздействием длинноволнового ультрафиолетового излучения. Таким образом, либо дисульфидное восстановление, либо фоторасщепление можно использовать в качестве расщепляемого линкера. Другим подходом к обратимой терминации является использование естественной терминации, которая возникает после размещения объемного красителя на dNTP. Присутствие заряженного объемного красителя на dNTP может действовать как эффективный терминатор через стерические и/или электростатические помехи. Наличие одного события включения предотвращает дальнейшее включение, до тех пор пока краситель не удален. Расщепление красителя удаляет флуорофор и эффективно обращает терминацию. Примеры модифицированных нуклеотидов также описаны в патентах США №№ 7427673 и 7057026.
Дополнительные примеры систем SBS и способы, которые могут быть использованы со способами и системами, описываемыми в настоящем документе, описаны в публикациях США №№. 2007/0166705, 2006/0188901, 2006/0240439, 2006/0281109, 2012/0270305, и 2013/0260372, патенте США № 7057026, публикации PCT № WO 05/065814, публикации патентной заявки США №. 2005/0100900, и публикациях PCT №№. WO 06/064199 и WO 07/010251.
Некоторые варианты осуществления могут использовать детекцию четырех разных нуклеотидов с использованием менее четырех разных меток. Например, SBS можно проводить с использованием способов и систем, описанных во включенных материалах публикации США № 2013/0079232. Как и в первом примере, пару типов нуклеотидов можно деектировать на одной и той же длине волны, но различать на основе разницы в интенсивности для одного участника пары по сравнению с другим, или на основе изменения одного участника пары (например, путем химической модификации (фотохимической модификации или физической модификации), которая вызывает появление или исчезновение видимого сигнала по сравнению с сигналом, обнаруженным для другого участника пары. Как и во втором примере, три из четырех различных типов нуклеотидов могут быть обнаружены при определенных условиях, в то время как у четвертого типа нуклеотидов отсутствует метка, которая может быть обнаружена в этих условиях, или минимально обнаруживается при этих условиях (например, минимальное обнаружение из-за фоновой флуоресценции, и т.д.). Включение первых трех типов нуклеотидов в нуклеиновую кислоту можно определять на основе наличия их соответствующих сигналов, а включение четвертого типа нуклеотидов в нуклеиновую кислоту можно определять на основании отсутствия или минимального обнаружения какого-либо сигнала. В качестве третьего примера, один тип нуклеотидов может включать метки, которые обнаруживаются в двух разных каналах, тогда как другие типы нуклеотидов обнаруживаются не более чем в одном из каналов. Вышеуказанные три примерные конфигурации не считаются взаимоисключающими и могут использоваться в различных комбинациях. Иллюстративным вариантом, который объединяет все три примера, является метод SBS на основе флуоресценции, который использует тип первого нуклеотида, который обнаруживается в первом канале (например, dATP, имеющий метку, которая обнаруживается в первом канале при возбуждении первой длиной волны возбуждения), второй тип нуклеотида, который обнаруживается во втором канале (например, dCTP, имеющий метку, которая обнаруживается во втором канале при возбуждении второй длиной волны возбуждения), третий тип нуклеотида, который обнаруживается как в первом, так и во втором канале (например, dTTP, имеющий, по меньшей мере, одну метку, которая обнаруживается в обоих каналах при возбуждении первой и/или второй длиной волны возбуждения) и четвертый тип нуклеотида, в котором отсутствует метка, которая не детектируется или детектируется минимально в любом канале (например, dGTP без метки).
Далее, как описано во включенных материалах публикации США № 2013/0079232, данные последовательности можно получать с использованием одного канала. В таких так называемых подходах секвенирования с одним красителем тип первого нуклеотида метится, но метку удаляют после получения первого изображения, а тип второго нуклеотида метят только после получения первого изображения. Третий тип нуклеотидов сохраняет свою метку на и на первом, и на втором изображениях, а четвертый тип нуклеотидов остается без меток на обоих изображениях.
В некоторых вариантах осуществления можно использовать секвенирование с помощью способов лигирования. Такие способы используют ДНК-лигазу для включения олигонуклеотидов и определения включения таких олигонуклеотидов. Олигонуклеотиды, как правило имеют, разные метки, которые коррелируют с идентичностью конкретного нуклеотида в последовательности, с которой гибридизуются олигонуклеотиды. Как и в случае с другими способами SBS, изображения можно получать после обработки чипа с элементами нуклеиновоых кислот помеченными реагентами для секвенирования. Каждое изображение будет отображать элементы нуклеиновой кислоты, которые имеют метки определенного типа. Различные элементы будут присутствовать или отсутствовать на разных изображениях из-за различного содержания последовательности каждого элемента, но относительное положение элементов на изображениях останется неизменным. Изображения, полученные с помощью способов секвенирования на основе лигирования, можно хранить, обрабатывать и анализировать, как указано в настоящем документе. Примеры систем SBS и способов, которые могут быть использованы со способами и системами, описываемыми в настоящем документе, описаны в патентах США №№ 6969488, 6172218 и 6306597.
В некоторых вариантах осуществления можно использовать секвенирование с помощью нанопор (Deamer, DW & Akeson, M. «Nanopores and nucleic acids: prospects for ultrarapid sequencing». Trends Biotechnol. 18, 147-151 (2000); Deamer, D. и D. Branton, Characterization of nucleic acids by nanopore analysis», Acc. Chem. Res. 35: 817-825 (2002); Li, J., M. Gershow, D. Stein, E. Brandin, J.A. Golovchenko,«DNA molecules and configurations in a solid-state nanopore microscope» Nat. Mater. 2: 611-615 (2003). В таких вариантах осуществления индексированный фрагмент проходит через нанопоры. Нанопора может быть синтетической порой или белком биологической мембраны, таким как α-гемолизин. Когда индексированный фрагмент проходит через нанопоры, каждую пару оснований можно идентифицировать, измеряя флуктуации электропроводности поры (патент США № 7001792; Soni, GV & Meller, «A. Progress toward ultrafast DNA sequencing using solid-state nanopores». Clin. Chem. 53, 1996-2001 (2007); Healy, K. «Nanopore-based single-molecule DNA analysis» Nanomed. 2, 459-481 (2007); Cockroft, SL, Chu, J., Amorin, M. & Ghadiri, MR «A single-molecule nanopore device detects ДНК-polymerase activity with single-nucleotide resolution» J. Am. Chem. Soc. 130, 818-820 (2008)). Данные, полученные в результате секвенирования при помощи нанопор, можно хранить, обрабатывать и анализировать, как указано в настоящем документе. В частности, данные можно обрабатывать как изображения в соответствии с примерами обработки оптических изображений и других изображений, которые изложены в настоящем документе.
В некоторых вариантах осуществления можно использовать способы, включающие мониторинг активности ДНК-полимеразы в режиме реального времени. Включения нуклеотидов можно детектировать посредством резонансного переноса энергии флуоресценции (FRET) между флуорофорсодержащей полимеразой и меченными γ-фосфатом нуклеотидами, как описано, например, в патентах США №№ 7329492 и 7211414, или включения нуклеотидов можно детектировать при помощи волновода с нулевой модой, как описано, например, в патенте США № 7315019, и с использованием флуоресцентных нуклеотидных аналогов и сконструированных полимераз, как описано, например, в патенте США № 7405281 и публикации США № 2008/0108082. Освещение может быть ограничено объемом в цептолитровом масштабе вокруг поверхностно-связанной полимеразы, так что включение флуоресцентно меченных нуклеотидов можно наблюдать с низким фоном (Levene, M. J. et al. «Zero-mode waveguides for single-molecule analysis at high concentrations Science 299, 682-686 (2003); Lundquist, P. M. et al. «Parallel confocal detection of single molecules in real time.» Opt. Lett. 33, 1026-1028 (2008); Korlach, J. et al. «Selective aluminum passivation for targeted immobilization of single ДНК-polimerase molecules in zero-mode waveguide nano structures» Proc. Natl. Acad. Sci. 105 USA 1176-1181 (2008)). Изображения, полученные такими способами, можно хранить, обрабатывать и анализировать, как указано в настоящем документе.
Некоторые варианты осуществления SBS включают обнаружение протона, высвобождаемого при включении нуклеотида в продукт удлинения. Например, для секвенирования, основанного на обнаружении высвобожденных протонов, можно использовать электрический детектор и связанные с ним методы, которые являются коммерчески доступными от Ion Torrent (Guilford, CT, дочерняя компания Life Technologies), или методы секвенирования и системы, описанные в публикациях США. № 2009/0026082; 2009/0127589; 2010/0137143; и 2010/0282617. Способы, изложенные в настоящем документе для амплификации нуклеиновых кислот-мишеней с использованием кинетического исключения, могут быть легко применены к субстратам, используемым для обнаружения протонов. Более конкретно, способы, изложенные в настоящем документе, можно использовать для получения клональных популяций ампликонов, которые применяют для обнаружения протонов.
Вышеупомянутые способы SBS могут быть эффективно реализованы в мультиплексных форматах, таких, где множество различных индексированных фрагментов обрабатывают одновременно. В конкретных вариантах осуществления можно обрабатывать различные индексированные фрагменты в общем реакционном сосуде или на поверхности определенного субстрата. Это позволяет удобно доставлять реагенты для секвенирования, удалять непрореагировавшие реагенты и детектироватьь события включения в мультиплексной форме. В вариантах осуществления с использованием связанных с поверхностью нуклеиновых кислот-мишеней индексированные фрагменты могут быть в формате чипа. В формате чипа индексированные фрагменты могут быть, как правило, связаны с поверхностью пространственно различимым образом. Индексированные фрагменты могут быть связаны прямым ковалентным прикреплением, прикреплением к грануле или другой частице или связаны с полимеразой или другой молекулой, которая прикреплена к поверхности. Чип может включать в себя одну копию индексированного фрагмента на каждом участке (также обозначаемом как элемент) или более копий, имеющих одинаковую последовательность, могут присутствовать на каждом участке или элементе. Множество копий можно получить с помощью способов амплификации, такой мостиковая амплификация или эмульсионная ПЦР.
Способы, изложенные в настоящем документе, могут использовать чипы, имеющие элементы с любой из ряда плотностей, включая, например, по меньшей мере, приблизительно 10 элементов/см2, 100 элементов/см2, 500 элементов/см2, 1000 элементов/см2, 5000 элементов/см2, 10000 элементов/см2, 50000 элементов/см2, 100000 элементов/см2, 1000000 элементов/см2, 5000000 элементов/см2 или выше.
Преимущество способов, изложенных в данном документе, заключается в том, что они обеспечивают быстрое и эффективное обнаружение множества см2 параллельно. Таким образом, настоящее изобретение предлагает интегрированные системы, способные получать и обнаруживать нуклеиновые кислоты с применением известных в данной области способов, таких как способы, приведенные в качестве примеров в настоящем документе. Таким образом, интегрированная система по настоящему изобретению может включать в себя флюидные компоненты, способные доставлять реагенты для амплификации и/или реагенты для секвенирования к одному или более иммобилизованным индексированным фрагментам, при этом система включает в себя такие компоненты, как насосы, клапаны, резервуары, флюидные пути и т.п. Проточная ячейка может быть настроена и/или использована в интегрированной системе для обнаружения нуклеиновых кислот-мишеней. Примеры проточных ячеек описаны, например, в публикациях США № 2010/0111768 и США № 13/273666. Как показано в качестве примера для проточных ячеек, один или более флюидных компонентов интегрированной системы можно использовать для способа амплификации и для способа детекции. Принимая вариант осуществления секвенирования нуклеиновой кислоты в качестве примера, один или более флюидных компонентов интегрированной системы можно использовать для способа амплификации, изложенного в настоящем документе и для доставки реагентов секвенирования в способе секвенирования, таком как те, которые приведены в качестве примера выше. Альтернативно, интегрированная система может включать в себя отдельные флюидные системы для проведения способов амплификации и детекции. Примеры интегрированных систем секвенирования, которые способны создавать амплифицированные нуклеиновые кислоты, а также определять последовательность нуклеиновых кислот, в качестве неограничивающих примеров включают платформу MiSeqTM (Illumina, Inc., Сан-Диего, Калифорния) и устройства, описанные в US № 13/273666.
Также в настоящем документе предлагаются композиции. Во время практического осуществления способов, описываемых в настоящем документе, в результате могут быть получены различные композиции. Например, может быть получена композиция, включающая фрагменты нуклеиновой кислоты с тройным индексом. Также предлагается многолуночный планшет, где лунка многолуночного планшета включает фрагменты нуклеиновой кислоты с тремя индексами.
Также в настоящем документе предлагаются наборы. В одном из вариантов осуществления набор предназначен для подготовки библиотеки для секвенирования. Набор включает транспозому и/или медиатор линейной амплификации, описываемый в настоящем документе, в подходящем упаковочном материале в количестве, достаточном, по меньшей мере, для одного анализа или применения. Необязательно, могут быть включены другие компоненты, такие как одна или более нуклеиновых кислот, которые включают праймер, индекс, универсальную последовательность или их сочетание. Другие компоненты, которые могут быть включены, являются реагентами, такими как буферы и растворы. Инструкции по использованию упакованных компонентов также, как правило, включены. Как применяют в данном документе, фраза «упаковочный материал» относится к одному или более физическим структурам, используемым для размещения содержимого набора. Упаковочный материал конструируют обычными способами, в основном, чтобы обеспечить стерильную среду без загрязнителей. Упаковочный материал может иметь ярлык, который указывает, что компоненты можно использовать для создания библиотеки для секвенирования. Кроме того, упаковочный материал содержит инструкции, указывающие, как используются материалы в наборе. Как применяют в данном документе, термин «упаковка» относится к контейнеру, такому как стекло, пластик, бумага, фольга и т. п., способному удерживать в установленных пределах компоненты набора. «Инструкции по применению», как правило, включают в себя материальное выражение, описывающее концентрацию реагента или, по меньшей мере, один параметр способа анализа, такой как относительные количества реагента и образца, которые необходимо смешать, периоды времени обслуживания для смесей реагента/образца, температуру, буферные условия, и т.п.
ИЛЛЮСТРАТИВНЫЕ ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ
Вариант осуществления 1. Способ получения библиотеки для секвенирования, включающей нуклеиновые кислоты из множества одиночных ядер или клеток, включающий:
получение множества выделенных ядер или клеток в первом множестве компартментов, где каждый компартмент содержит субпопуляцию выделенных ядер или клеток, и где ядра или клетки содержат фрагменты нуклеиновой кислоты;
введение медиатора линейной амплификации в клетки или ядра;
амплификацию фрагментов нуклеиновой кислоты путем линейной амплификации;
обработку каждой субпопуляции ядер или клеток для получения индексированных ядер или клеток, где обработка включает добавление к фрагментам нуклеиновой кислоты, присутствующим в выделенных ядрах или клетках, индексной последовательности, специфической для первого компартмента, в результате чего индексированные нуклеиновые кислоты присутствуют в выделенных ядрах или клетках, где обработка включает в себя лигирование, удлинение праймера, гибридизацию, амплификацию или транспозицию; и
объединение индексированных ядер или клеток для создания объединенных индексированных ядер или клеток, тем самым создавая библиотеку для секвенирования из множества ядер или клеток.
Вариант осуществления 2. Способ по Варианту осуществления 1, где амплификация происходит перед обработкой.
Вариант осуществления 3. Способ по Варианту осуществления 1, где обработка происходит перед амплификацией.
Вариант осуществления 4. Способ получения библиотеки для секвенирования, включающей нуклеиновые кислоты из множества одиночных ядер или клеток, включающий:
получение множества выделенных ядер или клеток, где ядра или клетки содержат фрагменты нуклеиновой кислоты;
введение медиатора линейной амплификации в выделенные клетки или ядра;
распределение выделенных ядер или клеток в первое множество компартментов, где каждый компартмент содержит субпопуляцию выделенных ядер или клеток и;
амплификацию фрагментов нуклеиновой кислоты путем линейной амплификации;
обработку каждой субпопуляции ядер или клеток для получения индексированных ядер или клеток, где обработка включает добавление к фрагментам нуклеиновой кислоты, присутствующим в выделенных ядрах или клетках, индексной последовательности, специфической для первого компартмента, в результате чего индексированные нуклеиновые кислоты присутствуют в выделенных ядрах или клетках, где обработка включает в себя лигирование, удлинение праймера, гибридизацию, амплификацию или транспозицию;
объединение индексированных ядер или клеток для создания объединенных индексированных ядер или клеток, тем самым создавая библиотеку для секвенирования из множества ядер или клеток.
Вариант осуществления 5. Способ получения библиотеки для секвенирования, включающей нуклеиновые кислоты из множества одиночных ядер или клеток, включающий:
получение множества выделенных ядер или клеток в первом множестве компартментов, где каждый компартмент содержит субпопуляцию выделенных ядер или клеток, и где ядра или клетки содержат фрагменты нуклеиновой кислоты;
обработку каждой субпопуляции ядер или клеток для получения индексированных ядер или клеток, где обработка включает добавление к фрагментам нуклеиновой кислоты, присутствующим в выделенных ядрах или клетках (i) индексной последовательности, специфической для первого компартмента, в результате чего индексированные нуклеиновые кислоты присутствуют в выделенных ядрах или клетках и (ii) нуклеотидной последовательности, которую распознает медиатор линейной амплификации, где обработка включает в себя лигирование, удлинение праймера, гибридизацию, амплификацию или транспозицию;
введение медиатора линейной амплификации в клетки или ядра;
амплификацию фрагментов нуклеиновой кислоты путем линейной амплификации; и
объединение индексированных ядер или клеток для создания объединенных индексированных ядер или клеток, тем самым создавая библиотеку для секвенирования из множества ядер или клеток.
Вариант осуществления 6. Способ по одному из Вариантов осуществления 1-5, где медиатор линейной амплификации включает фаговую РНК-полимеразу или праймер линейной амплификации.
Вариант осуществления 7. Способ по одному из Вариантов осуществления 1-6, где фрагменты нуклеиновой кислоты включают промотор T7, и фаговая РНК-полимераза включает РНК-полимеразу T7.
Вариант осуществления 8. Способ по любому из Вариантов осуществления 1-7, где введение медиатора линейной амплификации включает добавление к фрагментам нуклеиновой кислоты, присутствующим в изолированных ядрах или клетках, медиатора линейной амплификации.
Вариант осуществления 9. Способ по любому из Вариантов осуществления 1-8, дополнительно включающий подвергание множества выделенных ядер или клеток каждого компартмента воздействию предопределенного условия.
Вариант осуществления 10. Способ по любому из Вариантов осуществления 1-9, дополнительно включающий выделение ядер из множества клеток после воздействия.
Вариант осуществления 11. Способ по любому из Вариантов осуществления 1-10, дополнительно включающий подвергание множества выделенных ядер или клеток воздействию предопределенного условия.
Вариант осуществления 12. Способ по любому из Вариантов осуществления 1-11, дополнительно включающий подвергание выделенных ядер воздействиям для получения обедненных нуклеосомами ядер при сохранении целостности выделенных ядер.
Вариант осуществления 13. Способ по любому из Вариантов осуществления 1-12, где обработка включает:
контакт каждой субпопуляции с транспозомным комплексом, где транспозомный комплекс в каждом компартменте содержит первую индексную последовательность, которая отличается от первого индекса в других компартментах; и
фрагментацию нуклеиновых кислот в субпопуляциях на множество нуклеиновых кислот и включение последовательностей первого индекса, по меньшей мере, в одну цепь нуклеиновых кислот для получения индексированных ядер или клеток, включающих индексированные нуклеиновые кислоты.
Вариант осуществления 14. Способ по любому из Вариантов осуществления 1-13, где обработка включает:
контакт каждой субпопуляции с обратной транскриптазой и праймером, который отжигается на молекулах РНК в выделенных ядрах, где праймер в каждом компартменте содержит первую индексную последовательность, которая отличается от первого индекса в других компартментах, для получения индексированных ядер или клеток, содержащих индексированные нуклеиновые кислоты.
Вариант осуществления 15. Способ по любому из Вариантов осуществления 1-14, где контакт дополнительно включает мишень-специфический праймер, который отжигается на конкретной нуклеотидной последовательности.
Вариант осуществления 16. Способ по любому из Вариантов осуществления 1-15, где обработка для добавления первой компартмент-специфической индексной последовательности включает двухэтапный процесс добавления нуклеотидной последовательности, включающей универсальную последовательность к фрагментам нуклеиновых кислот, с последующим добавлением первой компартмент-специфической индексной последовательности к фрагментам нуклеиновой кислоты.
Вариант осуществления 17. Способ по любому из Вариантов осуществления 1-16, где добавление включает в себя транспозомный комплекс, который содержит универсальную последовательность.
Вариант осуществления 18. Способ по любому из Вариантов осуществления 1-17, где обработка включает добавление первого индекса к ДНК-нуклеиновым кислотам, присутствующим в выделенных ядрах или клетках, первого индекса к РНК-нуклеиновым кислотам, присутствующим в выделенных ядрах или клетках, или их сочетание.
Вариант осуществления 19. Способ по любому из Вариантов осуществления 1-18, где добавление первой индексной последовательности к РНК-нуклеиновым кислотам включает:
контакт каждой субпопуляции с обратной транскриптазой и праймером, который отжигается на молекулы РНК в выделенных ядрах или клетках, где праймер в каждом компартменте содержит первую компартмент-специфическую индексную последовательность, для получения индексированных ядер или клеток, включающих индексированные нуклеиновые кислоты.
Вариант осуществления 20. Способ по любому из Вариантов осуществления 1-19, где добавление первой индексной последовательности к ДНК-нуклеиновым кислотам включает в себя:
связывание каждой субпопуляции с транспозомным комплексом, где транспозомный комплекс в каждом компартменте включает в себя первую компартмент-специфическую индексную последовательность; и
фрагментирование нуклеиновых кислот в субпопуляциях во множество нуклеиновых кислот и включение индексных последовательностей, специфических для первого компартмента, по меньшей мере, в одну из цепей нуклеиновых кислот для получения индексированных ядер или клеток, включающих индексированные нуклеиновые кислоты.
Вариант осуществления 21. Способ по любому из Вариантов осуществления 1-20, где первая индексная последовательность, добавленная к ДНК-нуклеиновым кислотам и первая индексная последовательность, добавленная к РНК-нуклеиновым кислотам в каждом компартменте, одинаковая.
Вариант осуществления 22. Способ по любому из Вариантов осуществления 1-21, где первая индексная последовательность, добавленная к ДНК-нуклеиновым кислотам и первая индексная последовательность, добавленная к РНК-нуклеиновым кислотам в каждом компартменте, не одинаковая.
Вариант осуществления 23. Способ по любому из Вариантов осуществления 1-22, дополнительно включающий экспоненциальную амплификацию фрагментов нуклеиновой кислоты, где экспоненциальная амплификация включает мишень-специфический праймер, который отжигается на конкретной нуклеотидной последовательности.
Вариант осуществления 24. Способ по любому из Вариантов осуществления 1-23, дополнительно включающий после объединения:
распределение субпопуляций объединенных индексированных ядер или клеток во второе множество компартментов; и
введение второй компартмент-специфической индексной последовательности для индексированных нуклеиновых кислот для получения ядер или клеток с двумя индексами, включающих в себя нуклеиновые кислоты с двумя индексами, где введение включает лигирование, удлинение праймера, амплификацию или транспозицию.
Вариант осуществления 25. Способ по любому из Вариантов осуществления 1-24, дополнительно включающий:
объединение ядер с двумя индексами для создания объединенных ядер или клеток с двумя индексами,
распределение субпопуляции объединенных ядер или клеток с двумя индексами в третье множество компартментов; и
введение третьей компартмент-специфической индексной последовательности для индексированных нуклеиновых кислот для получения трехиндексных ядер или клеток, включающих в себя трехиндексные нуклеиновые кислоты, где введение включает лигирование, удлинение праймера, амплификацию или транспозицию.
Вариант осуществления 26. Способ по любому из Вариантов осуществления 1-25, дополнительно включающий обработку индексированных ядер или клеток для анализа метилирования для получения фрагментов нуклеиновых кислот, подходящих для анализа метилирования.
Вариант осуществления 27. Способ по любому из Вариантов осуществления 1-26, дополнительно включающий лигирование индексированных ядер или клеток для получения фрагментов нуклеиновых кислот, подходящих для анализа конформации хроматина.
Вариант осуществления 28. Способ по любому из Вариантов осуществления 1-27, дополнительно включающий амплификацию фрагментов нуклеиновой кислоты из библиотеки для секвенирования для получения наношаров ДНК.
Вариант осуществления 29. Способ по любому из Вариантов осуществления 1-28, где компартмент содержит лунку или каплю.
Вариант осуществления 30. Способ по любому из Вариантов осуществления 1-29, где каждый компартмент из первого множества компартментов содержит от 50 до 100000000 ядер или клеток.
Вариант осуществления 31. Способ по любому из Вариантов осуществления 1-29, где каждый компартмент из второго множества компартментов содержит от 50 до 100000000 ядер или клеток.
Вариант осуществления 32. Способ по любому из Вариантов осуществления 1-31, дополнительно включающий:
получение поверхности, содержащей множество сайтов амплификации, где сайты амплификации включают по меньшей мере две популяции прикрепленных одноцепочечных захватывающих олигонуклеотидов, имеющих свободный 3'-конец, и
контакт поверхности, содержащей сайты амплификации, с индексированными фрагментами в условиях, подходящих для получения множества сайтов амплификации, каждый из которых содержит клональную популяцию ампликонов из отдельного фрагмента, содержащего множество индексов.
Вариант осуществления 33. Способ получения библиотеки для секвенирования, включающей нуклеиновые кислоты из множества отдельных клеток, включающий:
(а) получение выделенных ядер из множества клеток;
(b) химическую обработку выделенных ядер для получения обедненных нуклеосомами ядер с сохранением целостности выделенных ядер;
(c) распределение субпопуляции обедненных нуклеосомами ядер по первому множеству компартментов и контакт каждой субпопуляции с транспозомным комплексом, где транспозомный комплекс в каждом компартменте включает транспозазу и первую индексную последовательность, которая отличается от первого индекса в других компартментах ;
(d) фрагментацию нуклеиновых кислот в субпопуляции ядер, обедненных нуклеосомами, во множество фрагментов нуклеиновых кислот и включение первого индекса в одну из следующих последовательностей фрагментов нуклеиновых кислот для получения индексированных ядер, содержащих индексированные фрагменты нуклеиновых кислот, где индексированные фрагменты нуклеиновые кислоты остаются прикрепленными к транспозазам;
(d) объединение индексированных ядер для получения объединенных индексированных ядер;
(e) распределение субпопуляции объединенных индексируемых ядер во второе множество компартментов и контакт каждой субпопуляции со шпилечным дуплексом для лигирования в условиях, подходящих для лигирования шпилечного дуплекса для лигирования с одним или обоими концами индексируемых фрагментов нуклеиновой кислоты, для получения фрагментов нуклеиновой кислоты с двойным индексом, где шпилечный дуплекс для лигирования содержит вторую индексную последовательность, которая отличается от второй индексной последовательности в других компартментах;
(f) объединение ядер с двумя индексами для получения объединенных индексированных ядер;
(g) распределение субпопуляции объединенных двухиндексных ядер в третье множество компартментов;
(h) лизис двухиндексных ядер;
(i) обработку нуклеиновых фрагментов с двумя индексами для включения третьей индексной последовательности, которая отличается от третьих индексных последовательностей в других компартментах; и
(j) объединение фрагментов с тремя индексами, в результате чего получают библиотеку для секвенирования, включающую полногеномные нуклеиновые кислоты из множества одиночных клеток.
ПРИМЕРЫ
Настоящее изобретение иллюстрируется следующими примерами. Следует понимать, что конкретные примеры, материалы, количества и способы должны толковаться в широком смысле в соответствии с объемом и духом изобретения, как указано в настоящем документе.
Пример 1
Высокопроизводительное секвенирование одиночной клетки с линейной амплификацией
Общепринятые способы для секвенирования генома одиночной клетки ограничены в отношении единообразия и производительности. Здесь мы описываем «sci-L3», высокопроизводительный способ секвенирования одиночных клеток с высоким охватом, который сочетает в себе комбинаторное индексирование («sci») и линейную («L») амплификацию одиночных клеток. Способ sci-L3 использует однонаправленную трехуровневую («3») схему индексирования, которая сводит к минимуму ошибки амплификации и обеспечивает экспоненциальный прирост производительности. Мы демонстрируем генерализуемость каркаса sci-L3 через демонстрации экспериментальных доказательств концепции путем секвенирования полного генома одиночной клетки («sci-L3-WGS»), целевого секвенирования генома («sci-L3-target-seq»), и совместный анализ генома и транскриптома («sci-L3-RNA/DNA»). Мы применили sci-L3-WGS для профилирования геномов >10000 сперматозоидов и предшественников сперматозоидов от мышей-гибридов F1, картировав 86786 кроссинговеров и охарактеризовав редкие случаи нарушенной сегрегации хромосомы при мейозе у самцов, включая случаи полногеномной эквационной сегрегации хромосом. Мы ожидаем, что анализы sci-L3 можно будет применять для полной характеристики ландшафтов рекомбинации, для объединения отклонений CRISPR и параметров стабильности генома, а также для других целей, требующих высокопроизводительного секвенирования генома одиночной клетки.
Введение
Современные технологии секвенирования генома одиночной клетки имеют два основных ограничения. Во-первых, большинство способов требуют компартментализации отдельных клеток, что ограничивает производительность. Во-вторых, большинство способов амплификации основаны на ПЦР и, таким образом, страдают от ошибок экспоненциальной амплификации. Чтобы решить первую проблему, мы с коллегами разработали комбинаторное индексирование одиночной клетки («sci-»), где выполняется несколько циклов молекулярного штрихкодирования с разделением/объединением, чтобы ввести уникальную метку на нуклеиновые кислоты в отдельных клетках, тем самым обеспечивая экспоненциальный прирост производительности с каждым последующим раундом индексирования. Sci-способы успешно разработаны для профилирования доступности хроматина (sci-ATAC-seq), транскриптомов (sci-RNA-seq), генома (sci-DNA-seq), метилома (sci-MET), конформаций хромосом (sci-Hi-C) в большом количестве отдельных клеток (Cao et al., 2017; Cusanovich et al., 2015; Mulqueen et al., 2018; Ramani et al., 2017; Vitak et al., 2017). Для решения второй проблемы линейная амплификация путем транскрипции на основе Т7 предлагает потенциальное решение, которое применяли для анализов одиночных клеток (Eberwine et al., 1992; Hashimshony et al., 2012; Sos et al., 2016). Например, недавно Chen et al. разработали линейную амплификацию через транспозонную вставку («LIANTI»), которая использует транспозон Tn5 для фрагментирования генома и одновременно вставляет РНК-промотор T7 для транскрипции in vitro (IVT). Копии РНК, полученные из матрицы ДНК, не могут служить матрицей для дальнейшей амплификации; Таким образом, все копии получены непосредственно из исходной матрицы ДНК. Избегая экспоненциальной амплификации, LIANTI поддерживает единообразие и сводит к минимуму ошибки последовательности. Однако способ является низкопроизводительным, потому что он требует серийного получения библиотек из каждой отдельной клетки (Chen et al., 2017).
Чтобы минимизировать ошибки амплификации и в то же время обеспечить экспоненциальный прирост производительности, мы разработали sci-L3, который объединяет комбинаторное индексирование и линейную амплификацию одиночной клетки. Благодаря трем циклам молекулярного штрихкодирования sci-L3 улучшает производительность LIANTI до тысяч и потенциально до миллионов клеток за эксперимент, сохраняя при этом преимущества линейной амплификации. Мы демонстрируем генерализуемость каркаса sci-L3 через демонстрации экспериментальных доказательств концепции путем секвенирования полного генома одиночной клетки («sci-L3-WGS»), целевого секвенирования генома («sci-L3-target-seq»), и совместный анализ генома и транскриптома («sci-L3-RNA/DNA»). В качестве дополнительной демонстрации мы использовали sci-L3-WGS для картирования беспрецедентного числа мейотических кроссинговеров и редких случаев нарушенной сегрегации хромосомы у незрелых и зрелых мужских половых клеток от бесплодных, межвидовых (B6×Spretus) F1-самцов мышей, а также фертильных, внутривидовых (B6×Cast) F1-самцов мышей.
Дизайн
Потенциальный технический путь к минимизации ошибок амплификации при увеличении производительности заключается в простом сочетании способов «sci» и «LIANTI». Однако молекулярная структура LIANTI, где промотор T7 вводится с помощью транспозона Tn5, предоставляет возможности только для двух раундов клеточного штрихкодирования, что ограничило бы производительность тысячами отдельных клеток на эксперимент. Кроме того, способ ограничен профилированием геномной ДНК (Chen et al., 2017; Sos et al., 2016). При разработке sci-L3 мы интегрировали комбинаторное индексирование отдельных клеток, линейную амплификацию и три раунда клеточного штрихкода («трехуровневый»), вводя промотор T7 путем лигирования (рис. 3А). Подход sci-L3 имеет несколько основных преимуществ перед простым сочетанием «sci» и «LIANTI». Во-первых, потенциальная производительность экспоненциально увеличивается при трехуровневом индексировании до более одного миллиона клеток на эксперимент при значительно сниженной стоимости (Cao et al., 2019). Во-вторых, однонаправленный характер штрихкодирования одиночной клетки позволяет легко преобразовать sci-L3 в целевое секвенирование («target-seq») в дополнение к полногеномномугеномному секвенированию («WGS»), которое позволяет связывать отклонения CRISPR и полученную нестабильность генома, а также для других применения, где желательно секвенировать определенные геномные локусы в большом количестве одиночных клеток. В-третьих, в качестве обобщаемой схемы линейной амплификации и высокопроизводительного штрихкодирования клеток, sci-L3 обеспечивает гибкость для адаптации к другим анализам одиночных клеток и совместным анализам с небольшими модификациями протокола, что продемонстрировано нашим экспериментальным доказательством концепции sci-L3 на основе совместного анализа РНК/ДНК одиночной клетки.
Результаты
Экспериментальная проверка концепции sci-L3-WGS и sci-L3-target-seq
Схемы трехуровневого комбинаторного индексирования и амплификации для sci-L3-WGS и sci-L3-target-seq показаны на фиг. 3А: (i) Клетки фиксировали формальдегидом, а нуклеосомы истощали SDS (Vitak et al., 2017). Полученные ядра затем равномерно распределяли по 24 лункам. (ii) Штрихкоды первого раунда добавляют путем индексированной вставки Tn5 («тагментация») в каждую из 24 лунок. В отличие от LIANTI, где транспозон Tn5 содержит промотор T7 без штрихкода, в штрихкоды с 5'-стороны вставляли спейсерную последовательность, которая служила «посадочной площадкой» для последующего этапа лигирования (см. Фиг. 4 и Пример 2, «Способы и молекулярный дизайн sci-L3-WGS и sci-L3-target-seq», для подробностей о конструкции транспозона Tn5). (iii) все ядра объединяли и равномерно распределяли в 64 новых лунках; штрихкоды второго раунда добавляли путем лигирования, которое включало промоторную последовательность T7, расположенную снаружи обоих штрихкодов. (iv) Все ядра снова объединяли и сортировали с помощью цитометрии с активируемой флуоресценцией сортировкой клеток (FACS) и распределяли по последнему раунду лунок до 300 клеток на лунку. Следует отметить, что ядра различной плоидности могут быть отсортированы и обогащены путем окрашивания DAPI (4’,6-диамидино-2-фенилиндол). Кроме того, простое разведение является альтернативой FACS, которая может снизить уровень потерь. (v) отсортированные ядра лизируют и подвергают удлинению делеций in situ, чтобы сформировать дуплексный промотор T7. Затем следует IVT, обратная транскрипция (ОТ) и синтез второй нити (SSS) для линейной амплификации геномов. Штрихкоды третьего раунда добавляют на этапе SSS вместе с уникальными молекулярными идентификаторами (UMI) для метки отдельных транскриптов IVT. (vi) Дуплексные молекулы ДНК (фиг. 3B, вверху), каждая из которых содержит три штрихкода, определяющие их исходную клетку, совместимы с общепринятыми способами конструирования библиотек, если целью является WGS одиночной клетки (например, добавление последовательности адаптеров путем лигирования (фиг. 3B, посередине) или тегирование), или слегка модифицированными способами, если целью целевое ДНК-севенирование одиночной клетки (например, добавление шага ПЦР, где один из праймеров специфичен для мишени (фиг. 3B, внизу).
Для первоначального подтверждения концепции мы смешали клетки мыши и человека и выполнили sci-L3-WGS. Для более чем 95% полученных геномов одиночных клеток, подавляющее большинство ридов картировалось либо на геном мыши, либо на геном человека; случайные «столкновения» являются результатом случайного использования одной и той же комбинации штрихкодов двумя или более клетками (фиг. 3C). Производительность sci-L3-WGS сравнивается с LIANTI, а также нашим предыдущим способом sci-DNA-seq на основе ПЦР в таблице 1. Мы выделяем несколько преимуществ sci-L3-WGS: 1) Мы, в основном, восстановили 90% отсортированных клеток по сравнению с 60% восстановлением при помощи sci-DNA-seq на основе ПЦР (Vitak et al., 2017); 2) На 40% меньше необработанных считываний (329 миллионов по sci-L3-WGS против 549 миллионов по sci-DNA-seq), sci-L3-WGS произвел покрытие последовательности при ~97000 уникальных вставок Tn5 на клетку, по сравнению с ~30000 уникальных вставок с помощью sci-DNA-seq, улучшение в 3 раза. Секвенируя меньшее количество клеток на более высокую глубину, мы наблюдали ~660000 уникальных вставок Tn5 на клетку, сохраняя при этом более высокую сложность библиотеки, чем при sci-DNA-seq, что свидетельствует о дополнительном улучшении в 20 раз; 3) Уровень картируемых ридов улучшена с 61% для LIANTI до 86% для sci-L3-WGS. Это, вероятно, связано с тем, что LIANTI полностью находится в пробирке, и, таким образом, трудно удалить артефактные последовательности (например, вторичные по отношению к самопроизвольной вставке Tn5), тогда как с помощью sci-L3-WGS ядра осаждают несколько раз, чтобы удалить избыток свободной ДНК; 4) В отличие от амплификации на основе ПЦР, где повторные чтения не являются информативными для распознавания SNP, «дублированные» риды sci-L3-WGS почти всегда являются результатом независимых транскриптов IVT, полимеризованных из исходного матрицы, и, таким образом, полезны для обнаружения SNV de novo или для генотипирования известных SNP.
Таблица 1. Сравнение эффективности sci-ДНК-seq и sci-L3-WGS с LIANTI. Данные sci-DNA-seq из способа xSDS (Vitak et al., 2017). LIANTI из способа «в пробирке» (Chen et al., 2017). Для sci-L3-WGS мы показываем результаты для библиотек yi140 и yi141 (при высокой глубине секвенирования) и yi144 и yi145 (при низкой глубине секвенирования). В этих четырех библиотеках используется оптимизированный протокол, в котором мы использовали концентрированную транспозому Tn5 (0,2 мкМ) и улучшенную реакцию ОТ с дополнительными РНК-праймерами (см. фиг. 5 и пример 2 «Способы и молекулярный дизайн sci-L3-WGS и sci-L3-target-seq» для подробностей). Тот же цвет указывает на сравнения, представляющие интерес. Зеленый: процент восстановленных отдельных клеток из сортировки улучшен в 1,9 раза с sci-L3-WGS по сравнению с sci-DNA-seq; розовый: уровень картирования необработанных ридов улучшен в 1,4 раза с помощью sci-L3-WGS по сравнению с LIANTI; желтый: уникальные сайты вставки с различной глубиной секвенирования; строки 1 и 2 сравнивают при одинаковом количестве необработанных ридов с 3,3-кратным улучшением при 40% меньшем количестве необработанных ридов с sci-L3-WGS по сравнению с sci-DNA-seq, а строки 1 и 3 сравнивают при сходной сложности библиотеки с 22,4-кратным улучшением при 20% лучшей сложности введения Tn5 с sci-L3-WGS по сравнению с LIANTI; синий: средняя сложность библиотеки, показывающая, что способы, включая LIANTI и sci-L3-WGS, имеют минимальные повторы ПЦР; оранжевый: количество клеток с более чем 50 тысяч восстановленных уникальных ридов улучшено в 1,8 раза при использовании sci-L3-WGS по сравнению с LIANTI.

С использованием sci-L3-WGS вставка Tn5 происходит в среднем каждые 0,5-1,5 т.п.н. в геноме человека, а IVT дает около 1000 транскриптов. Это соответствует от 2 до 6 миллионов уникальных вставок Tn5, и, таким образом, от 2 до 6 миллиардов уникальных транскриптов IVT, полученных из генома, на одну клетку. Очевидно, что в настоящее время нецелесообразно секвенировать полученные библиотеки для насыщения в отношении количества уникальных транскриптов IVT. Здесь мы определяем «глубину секвенирования» для каждой библиотеки как отношение количества уникальных секвенированных транскриптов к количеству картированных уникальных сайтов вставок Tn5. В этом исследовании большинство библиотек секвенировали с глубиной от 1,1× до 2×, что приводит к покрытию генома каждой клетки от 0,5% до 5%. Распределение уникальных сайтов вставки Tn5 на клетку в эксперименте подтверждения концепции «человек/мышь» показано на фиг. 3D, а для других экспериментов на фиг. 5. Расчетные относительные числа хромосомных копий для репрезентативных одиночных клеток показаны на фиг. 3E, и их распределения по всем клеткам на фиг. 3F. Чтобы экстраполировать ожидаемое покрытие генома на одну клетку при более высокой глубине секвенирования, мы подбираем количество уникальных сайтов вставки как функцию глубины секвенирования (фиг. 5G). Мы ожидаем увидеть 4,2 миллиона и 6,0 миллионов уникальных вставок на клетку при глубине секвенирования 5× и 10× соответственно, что соответствует 16% и 22% покрытия генома отдельных клеток.
Как указано выше, двухцепочечные ампликоны, генерируемые sci-L3 (фиг. 3B, вверху), совместимы не только с WGS одиночной клетки (sci-L3-WGS; фиг. 3B, в середине), но также с целевым секвенированием ДНК одиночной клетки («sci-L3-target-seq»). В частности, для целевого секвенирования после синтеза второй цепи можно добавить адаптеры для секвенирования посредством ПЦР с одним праймером, несущим третий клеточный штрихкод, и с другим праймером, нацеленным на конкретную область генома (фиг. 3B, внизу). Чтобы количественно оценить эффективность восстановления с помощью sci-L3-target-seq, мы интегрировали лентивирусную библиотеку CRISPR с низким MOI (см. Пример 2, раздел «Способы и молекулярный дизайн sci-L3-WGS и sci-L3-target-seq» для деталей) и восстановленные последовательности ДНК, соответствующие спейсерам sgRNA по sci-L3-target-seq. Для 97 из 1003 отдельных клеток мы можем успешно восстановить одиночную интегрированную sgRNA. Эта 10% эффективность на гаплотип в целом согласуется с покрытием генома 22%, оцененным выше с помощью планирования глубины секвенирования (фиг. 5G).
Следует заметить, что на молекулярном уровне мы модифицировали способы «sci» и «LIANTI» несколькими способами. Подводя итог, мы: 1) изменили дизайн транспозона Tn5, чтобы он был совместим с лигированием, и, таким образом, обеспечили более двух раундов «sci», подхода, который потенциально приложим к другим анализам одиночных клеток, 2) добавили петлевую структуру промотора Т7 для облегчения внутримолекулярного лигирования, и 3) изменили схему ОТ так, что нам требуется успешное лигирование только на одном из двух концов молекул со штрихкодами первого раунда. Исходя из предположения, что одно событие лигирования имеет эффективность 50%, эта модификация дает 75% успеха на этапе лигирования вместо 25% (сравнение показано на фиг. 5). Мы показываем структуры молекул после каждого шага штрихкодирования на фиг. 4 и обсуждаем обоснования для этих конструкций в Примере 2 «Способы и молекулярный дизайн sci-L3-WGS и sci-L3-target-seq». Масштабируемость и стоимость также обсуждаются в Примере 2 и Таблице 2. Для библиотеки из 100, 1000, 10000 и 1 миллиона отдельных клеток мы оцениваем стоимость sci-L3-WGS в 14%, 1,5%, 0,26% и 0,014% от обработки эквивалентных количеств клеток LIANTI. Использование трех, а не двух уровней комбинаторного индексирования может быть использовано либо для увеличения производительности (например, стоимость создания библиотеки для 1 миллиона клеток при 5% вероятности столкновения с 3-уровневым sci-L3-WGS составляет ~8000 долларов США) или для уменьшения частоты столкновений (например, стоимость создания библиотеки для 10000 клеток при частоте столкновений 1% с 3-уровневым sci-L3-WGS составляет ~1500 долларов США).
Таблица 2. Расчет стоимости sci-L3-WGS. Текущий способ включает в себя три уровня индексирования, которые не только увеличивают производительность и уменьшают количество столкновений штрихкодов, но также значительно снижают затраты на клетку для получения библиотеки. Это связано с двумя причинами: 1) при двухуровневом индексировании нужно начинать с большего количества транспозомных комплексов Tn5, чтобы профилировать аналогичное количество клеток, что увеличивает затраты по существу; 2) при двухуровневом индексировании можно также ограничить сортировку гораздо меньшего числа ядер на лунку до IVT, ОТ и очисткой на колонках, что также существенно увеличивает затраты по существу. Для обработки ~10000 и ~1 миллиона клеток мы оцениваем, что sci-L3-WGS с 3 уровнями почти в 400 раз и в 7000 раз дешевле на клетку, чем LIANTI.



Использование sci-L3-WGS для совместного анализа РНК/ДНК одиночной клетки
Мы поняли, что схема sci-L3-WGS потенциально может быть адаптирована к другим аспектам молекулярной биологии с небольшими изменениями в протоколе. Чтобы продемонстрировать это, мы провели эксперимент для подтверждения концепции на совместимом анализе sci-L3-RNA/DNA. Вкратце, первый раунд штрихкодирования ДНК проводят путем вставки Tn5, как в sci-L3-WGS, но мы одновременно выполняем первый раунд штрихкодирования РНК, помечая мРНК через обратную транскрипцию штрихкодом и UMI-несущим праймером polyT (фиг. 6А). Как вставка Tn5, так и праймер для ОТ имеют липкие концы, которые могут опосредовать лигирование штрихкодов второго раунда, а также промотор T7, эффективно позволяющий индексирование на трех уровнях и последующую линейную амплификацию на основе IVT способом, в значительной степени идентичным sci-L3-WGS (фиг. 6A-6B, Пример 2, раздел «Способы и молекулярный дизайн совместного анализа sci-L3-RNA/DNA»). В качестве подтверждения концепции мы смешали клетки мыши с клетками из двух клеточных линий человека и провели совместный анализ sci-L3-RNA/DNA. Для подавляющего большинства клеток риды сопоставлялись либо с геномом мыши, либо с геномом человека, как для РНК (частота столкновений 5,2%), так и для ДНК (частота столкновений 6,6%) (фиг. 6C-6D). Кроме того, в соответствии с успешным совместным анализом, 100% клеток были отмечены одинаковыми видами по их профилям РНК и ДНК. В качестве дальнейшей проверки мы визуализировали клетки человека в оболочке t-SNE на основе их профилей РНК. Как и ожидалось, они разделились на два кластера. Маркировка отдельных клеток на основе наличия или отсутствия Y-хромосомы когерентно идентифицировала кластеры как соответствующие BJ-клеткам (мужчины) или HEK293T-клеткам (женщина) (фиг. 6E) с точностью 96,5%.
ДНК-профилирование одиночной клетки из половых клеток мыши путем sci-L3-WGS
В нормальных делениях митотических клеток диплоидные хромосомы подвергаются репликации с образованием четырех копий ДНК, а сестринские хроматиды сегрегируют в реципрокные дочерние клетки. Дочерние клетки получают по одной копии каждой наследуемой по материнской и отцовской линии последовательности ДНК и почти всегда поддерживают гетерозиготность в центромерно-проксимаьльных последовательностях (фиг. 7А). В редких случаях хромосомы подвергаются митотическому кроссинговеру между хромосомными гомологами, что иногда может приводить к диплоидным клеткам с потерей гетерозиготности (LOH) в последовательностях, расположенных центромерно-дистально по отношению к кроссинговеру, если две рекомбинировавших хроматиды сегрегируют в разные дочерние клетки (фиг. 7B-C).
При мейозе сестринские хроматиды сначала совместно сегрегируют в одну и ту же дочернюю клетку, а гомологи сегрегируют в реципрокные дочерние клетки на стадии мейоза I («MI»), также известной как «редукционная сегрегация», в результате чего образуются клетки 2C (содержание ДНК нереплицированной диплоидной клетки) с потерей гетерозиготности (LOH) в центромерно-проксимальных последовательностях (фиг. 7D-E). Для успешной редукционной сегрегации хромосом при MI (рис. 7D), кроссинговеры, инициированные Spo11-катализируемыми двухцепочечными разрывами (DSB) (Baudat et al., 2000; Keeney et al., 1997; Romanienko and Camerini-Otero, 2000), обеспечивают связь и необходимое напряжение (Hong et al., 2013) между хромосомными гомологами. В редких случаях хромосомы будут сегрегировать мейотическим образом без какого-либо межгомологичного кроссинговера, что приводит к однородительской дисомии (UPD). После MI эти 2C клетки затем подвергаются митозоподобной сегрегации хромосомы в мейозе II («MII»), также называемой «эквационной сегрегацией», так что сестринские хроматиды разделяются друг от друга с образованием гамет 1C (фиг. 7E). Далее, поскольку наше исследование в основном сфокусировано на MI, мы ссылаемся на мейотическую/редукционную сегрегацию во время MI, где сестринские хроматиды сегрегируют вместе, как «редукционную сегрегацию», и митозоподобную/эквационную сегрегацию во время MI, когда сестринские хроматиды разделяются друг от друга, как «эквационную сегрегацию».
На сегодняшний день большая часть работ по взаимосвязи между положением кроссинговера и сегрегацией хромосомы была выполнена с помощью визуализации (Wang et al., 2017a, 2017b), которая не в состоянии полностью охарактеризовать лежащие в основе геномные последовательности, которые имеют тенденцию к мейотическому кроссинговеру. Некоторые анализы позволяют детально картировать горячие точки мейотических DSB (Lange et al., 2016; Smagulova et al., 2011, 2016), но эти анализы не картируют напрямую мейотические кроссинговеры. Анализы, которые отделяют кроссинговер от некроссинговера в тонком разрешении, ограничены несколькими горячими точками (Cole et al., 2014). Таким образом, мы знаем гораздо меньше о взаимосвязи между кроссинговерами и характеристиками в хромосомном масштабе, таких как домены репликации, чем о мейотических горячих точках DSB (Baudat et al., 2013; Choi and Henderson, 2015; Yamada et al., 2017). Полногеномные карты мейотических кроссинговеров были получены путем картирования тетрад в дрожжах (Mancera et al., 2008; Zhang et al., 2017), одиночного сперматозоида человека и полных женских мейозов у человека (Hou et al., 2013; Lu et al., 2012; Ottolini et al., 2015; Wang et al., 2012). За исключением исследований мейоза у женсокй особи человека, которые в целом проанализировали 87 полных мейозов (Hou et al., 2013; Ottolini et al., 2015), большинство карт кроссинговера ограничены по меньшей мере в трех аспектах: 1) анализируют зрелые 1C гаметы, где клетки завершили оба раунда деления мейоза, что предотвращает непосредственное наблюдение за более информативным промежуточным соединением 2C клеток, чтобы оценить, насколько часто хромосомы подвергаются редукционной и эквационной сегрегации во время MI (фиг. 7); 2) для сравнения выбирают аномальные клетки из-за их неспособности перейти к зрелому гаметическому состоянию; 3) анализы с помощью секвенирования одного сперматозоида или яйцеклетки ограничены в производительности и составляют не более нескольких сотен клеток, и, как таковые, могут пропустить редкие события. Даже для фертильных скрещиваний количество потомков, которые могут быть разумно получены и генотипированы, весьма ограничено (Liu et al., 2014).
Чтобы устранить все эти ограничения сразу, мы применили sci-L3-WGS к бесплодному потомству межвидового скрещивания (самка Mus musculus domesticus C57BL/6 («B6») × самец Mus spretus SPRET/Ei (далее «Spret»)), а также к фертильному потомству внутривидового гибрида (самка B6 × самец Mus musculus castaneous CAST/Ei («Cast»)). Секвенируя сперму с помощью высоко масштабируемой технологии, мы можем картировать беспрецедентное количество событий кроссинговера для млекопитающей системы, как в бесплодных, так и в фертильных гибридах. Кроме того, используя производительность sci-L3-WGS для восстановления профилей из редких вторичных сперматоцитов 2C, мы также можем оценить кроссинговер и нарушенную сегрегацию хромосом одновременно из одних и тех же одиночных клеток.
В отличие от инбредных самцов, а также самцов F1 (B6×Cast), чьи придатки яичка хранят миллионы зрелых сперматозоидов, придатки яичка самцов F1 (B6×Spret) (Berletch et al., 2015) содержат крайне мало морфологически зрелых сперматозоидов и ограниченное количество круглых половых клеток неизвестной плоидности (фиг. 8А-Б). Интересно, что мы наблюдали гораздо более высокую долю клеток 2C во время FACS (фиг. 8C-D), чем можно было бы ожидать для «нормального» придатка яичка, в котором преобладает сперма 1C. Количество восстановленных клеток и их предполагаемая плоидность приведены в таблице 3. В отличие от этого, придатки яичка самцов F1 (B6×Cast) почти полностью содержали сперматозоиды 1C (фиг. 8E). Для этого скрещивания мы таким образом отсортировали клетки 1C и 2C из удаленных семенников (фиг. 8F).
Таблица 3. Количество восстановленных клеток и клеточная плоидность, придатки яичка (B6×Spret). Обратите внимание, что мы не создали библиотеку секвенирования для всех отсортированных клеток; например, библиотека 2C в Exp1 содержит только субпопуляцию клеток. Мы также расширенно отсортировали клетки 1C (до сигнала DAPI 58 для определенных лунок), и из-за обилия клеток 2C в этом скрещивании мы можем обогащать клетки 1C только примерно до 51-55%.
С клетками от самцов F1 из обоих скрещиваний (B6×Spret) и (B6×Cast) мы приступили к линейной амплификации, синтезу второй цепи для добавления штрихкода третьего раунда, получению библиотеки и секвенированию (подробности в Примере 2 «Схема эксперимента sci-L3-WGS для скрещиваний (B6×Spret) и (B6×Cast)». Важным моментом является то, что хотя клетки 1C и 2C можно различить информатически, их относительное содержание все еще влияет на наш анализ. В частности, в скрещивании (B6×Spret) клетки 1C встречаются редко, так что любые «дублеты» (например, две клетки 1C, которые слипаются или случайно получают одинаковые штрихкоды) по существу не вносят вклад в популяцию 2C. Напротив, в скрещивании (B6×Cast) большинство клеток имеют 1C (~85%, фиг. 8G), несмотря на обогащение, так что может быть много дублетов 1C, которые имитируют клетки 2C. Мы обсудим в последующих разделах, как информатически отличить дублеты 1C от аутентичных клеток 2C.
Клетки M2 демонстрируют кластерную редукционную или эквационную сегрегацию хромосом
Хромосомная сегрегация в клетках М2 из бесплодного скрещивания (B6×Spret)
Мы в первую очередь пытались проанализировать мейоз в клетках из придатков яичка у бесплодных (B6×Spretus) самцов F1, полученных, как описано выше. В двух экспериментах sci-L3-WGS мы профилировали геномы из 2689 (92% из 2919 отсортированных клеток с более чем 10000 необработанных ридов) и 4239 (94% из 4497 отсортированных клеток с более чем 30000 необработанных ридов) отдельных клеток. Количество уникально картироваенных ридов показано на фиг. 5F. При глубине секвенирования 1,6× и 1,4× для двух библиотек (подробности на фиг. 5) мы получили в среднем ~70000 и ~144000 уникальных сайтов Tn5 на клетку, что соответствует 0,7% и 1,4% среднего покрытия генома, соответственно.
Чтобы идентифицировать точки разрыва для кроссинговера, мы внедрили скрытую марковскую модель (HMM), которая опиралась на высококачественные риды, которые можно было четко отнести к B6 по сравнению со Spret (см. Пример 2, раздел «Способы биоинформатики и статистические анализы»). Мы охарактеризовали кроссинговеры в 1663 клетках 1С, типичный пример которых показан на фиг. 9А. Кроме того, мы обыскали ~5200 клеток 2C на события кроссинговера. Хотя большинство из этих 5200 могут быть просто соматическими клетками, к нашему удивлению, мы идентифицировали 292 клетки 2C со значительным количеством кроссинговеров, которые мы назвали «клетки M2» (фиг. 9B и 9C). Еще более удивительно, что значительная доля этих клеток демонстрирует скорее эквационную, чем редукционную сегрегацию.
После того, как кроссинговер происходит между двумя гомологами хромосомы, если хромосома сегрегирует редукционным образом, область между центромерой и положением кроссинговера станет гомозиготной, тогда как гетерозиготность будет сохраняться ниже от кроссинговера (фиг. 7D). Однако, если хромосома сегрегирует эквационно, дистальнее от центромеры по направлению к кроссинговеру наблюдают LOH, если рекомбинированные хроматиды сегрегируют раздельно (фиг. 7B). Мы показываем один пример клетки М2, чьи хромосомы подвергаются ожидаемой редукционной сегрегации на фиг. 9B (обратите внимание на постоянную гомозиготность между центромерой и точкой кроссинговера) и один пример клетки M2, чьи хромосомы неожиданно подвергаются эквационной сегрегации на фиг. 9C (обратите внимание на постоянную гетерозиготность между центромерой и точкой кроссинговера). В общей сложности на 292 клетках M2 мы наблюдали 4162 примера хромосом, подвергающихся редукционной сегрегации, среди которых 3740 содержали кроссинговеры (90%) и 1310 примеров хромосом, подвергающихся эквационной сегрегации, среди которых 636 содержали кроссинговеры (49%). Следует отметить, однако, что число событий кроссинговера в хромосомах, которые сегрегировали эквационно, может быть выше, так как мы не можем идентифицировать подмножество исходов кроссинговера (фиг. 7C); Между тем мы можем обнаружить все кроссинговеры для хромосом, сегрегировавших редукционно.
Хотя мы наблюдаем много примеров клеток, в которых некоторые хромосомы демонстрируют редукционную сегрегацию, а другие хромосомы демонстрируют эквационную сегрегацию, характер сегрегации отдельных хромосом в клетках M2, по-видимому, не является независимым. Если бы хромосомы в каждой клетке выбирали редукционную и эквационную сегрегацию независимо, мы ожидали бы биномиальное распределение редукционно и эквационно сегрегировавших хромосом, центрированное по оценке максимального правдоподобия (MLE) вероятности p, от редукционной сегрегации (p =0,76 по данным, 4162/5472), причем примерно три четверти хромосом сегрегируют редукционно, а одна четверть эквационно (фиг. 9D). Однако среди 292 клеток M2, которые мы профилировали, мы наблюдаем 202 клетки, которые имеют, по меньшей мере, 15 хромосом, которые сегрегировали редукционно, и 38 клеток, которые имеют, по меньшей мере, 15 хромосом, которые сегрегировали эквационно (фиг. 9E; это контрастирует с ожидаемыми 148 и 0 клетками, соответственно, при условии независимости; p=4×10-23, точный критерий Фишера). То, что отдельные клетки M2 смещены в сторону преимущественно редукционной или эквационной сегрегации, предполагает возможность клеточно-автономного глобального чувствительного механизма для принятия решения о том, продолжает ли клетка мейоз или возвращается к митозу.
Далее мы можем классифицировать клетки по наличию кроссинговера в хромосомах в клетках М2 (фиг. 9F). Редукционно сегрегировавшие хромосомы, по-видимому, имеют больше кроссинговеров (розовые на фиг. 9F), чем эквационные хромосомы (зеленые на фиг. 9F). Однако, в отличие от редукционно сегрегировавших хромосом, где мы можем обнаружить все кроссинговеры как центромерные LOH, эквационно сегрегировавшие хромосомы имеют LOH, только если две рекомбинированных хроматиды разделяются в реципрокные дочерние клетки (фиг. 7B). Если вместо этого рекомбинированные хроматиды совместно сегрегируют, гетерозиготность будет сохраняться на всем протяжении хромосомы, несмотря на неопределяемый переключатель сцепления (фиг. 7C). На Фиг. 9F, соотношение наличия (показано зеленым цветом) к отсутствию (показано синим цветом) наблюдаемого LOH в эквационно сегрегировавших хромосомах составляет примерно 1: 1. Это может означать либо то, что эквационно сегрегировавшие хромосомы имеют 50% шанс сегрегировать рекомбинированные хроматиды вместе, если эти полностью гетерозиготные хромосомы (показаны синим цветом) действительно имеют переключатель сцепления; или альтернативно, что эквационно сегрегировавшие хромосомы всегда сегрегируют рекомбинированные хроматиды отдельно, а частота кроссинговера уменьшается вдвое по сравнению с редукционно сегрегировавшими хромосомами.
Известно, что сегментарные или полнохромосомные LOH редки в млекопитающем митотических клетках млекопитающих. Однако, чтобы исключить митотическое происхождение таких событий, мы исследовали такие события в клеточной линии Patski, которая является спонтанно иммортализованной клеточной линией, происходящей от самки F1 мыши (B6×Spret). Мы анализировали 1107 одиночных клеток из линии Patski путем sci-L3-WGS, среди которых мы обнаружили в среднем 0,36 UPD хромосом и 0,098 сегментарных LOH-событий на клетку, что значительно ниже по сравнению с клетками M2. Также отметим, что эти события не обязательно независимы. Например, UPD, возникающая в начале пересева клеточной линии, может быть общей для большой части клеток-потомков, так что частота независимых событий LOH, вероятно, еще ниже. Распределение этих событий (относительно равномерное для хромосом, полученных из Spretus, и неоднородное для хромосом, полученных из B6), представлено на фиг. 10F.
В совокупности, контраст между низким уровнем митотической LOH (ожидаемым) и относительно высоким уровнем клеток 2C, проявляющих эквационную сегрегацию (неожиданным), измеренных по одной и той же технологии, подтверждает, что последние вряд ли соответствуют соматическим клеткам. В следующем разделе, анализируя фертильное скрещивание (B6×Cast), мы кроме того покажем: 1) что наблюдаемые здесь события эквационной сегрегации всего генома не являются артефактом дублетов из двух 1C клеток, и 2) что такие события сегрегации также встречаются у фертильного внутривидового гибрида, хотя и на сниженном уровне.
Хромосомная сегрегация в клетках М2 из фертильного скрещивания (B6×Cast)
Нам было интересно, происходит ли эквационная сегрегация также во время MI у фертильного потомства внутривидовых самцов F1 (B6×Cast). Как показано выше, придатки яичек от этого скрещивания почти полностью состоят из зрелых сперматозоидов 1С; мы, таким образом, провели обогащение для вторичных сперматоцитов 2С из целых яичек. Затем мы провели sci-L3-WGS на клетках как придатков яичек, так и яичек.
В первичном эксперименте sci-L3-WGS по этому скрещиванию, который в основном выполнялся для контроля качества для оценки восстановления и частоты столкновений штрихкодов, мы распределили круглые сперматиды 1С круглые равномерно и отсортировали только по клеткам 1С после двух раундов штрихкодирования. Дублеты, идентифицированные в силу того, что они не относятся к 1С, позволяют количественно определять частоту столкновений штрихкодов. Среди 2400 отсортированных клеток (200/лунку) мы восстановили 2127 (89%) с более чем 7000 ридов на клетку; 2008 из них представляют собой 1С с мейотическими кроссинговерами, что указывает на вероятность столкновения штрихкодов 5,5%. При глубине секвенирования 1,06× мы получили в среднем ~60000 уникальных вставок Tn5 на клетку, что соответствует ~ 0,6% среднего покрытия генома.
Во втором эксперименте sci-L3-WGS на этом скрещивании мы пометили круглые сперматиды 1С из яичек («группа штрихкодов 1»), клетки 2C из яичек («группа штрихкодов 2»; загрязненные большим количеством сперматид 1C, как показано на фиг. 8F), и 1C зрелые сперматозоиды из придатка яичка («группа штрихкодов 3», Пример 2, «Схема эксперимента sci-L3-WGS на скрещивании (B6×Spret) и скрещивании (B6×Cast)»), в отдельных лунках во время первого раунда штрихкодирования. В качестве дополнительного обогащения, на этапе FACS sci-L3-WGS для подмножества лунок мы специально провели сортировку по клеткам 2C (15,5% всех клеток, фиг. 8G). При глубине секвенирования 1,09× мы получили в среднем ~ 94000 уникальных вставок Tn5 на клетку, что соответствует ~ 0,9% среднего покрытия генома.
Всего из этого второго эксперимента sci-L3-WGS мы восстановили 3539 1C и 1477 не-1C клеток. Интересно, что> 97% клеток 1C происходят из групп штрихкода 1 (n=1853) и штрихкода 2 (n=1598), а не из группы 3 (n=88), что указывает на то, что зрелый сперматозоид из придатка яичка плохо восстанавливается с помощью L3-WGS. Это позволяет предположить, что клетки 1С, извлеченные из (B6×Spret) выше, вероятно, также не из зрелых сперматозоидов, а скорее из круглых сперматид, что согласуется с низким количеством сперматозоидов со зрелой морфологией на фиг. 8В. 1477 не-1C-клеток получены из обеих групп штрихкода 1 (n=1104; предположительно, дублетов круглых сперматид 1C) и группы штрихкода 2 (n=373; предположительно смесь аутентичных клеток M2 и дублетов 1C). Чтобы идентифицировать сигнатуру дублетов 1C, мы исследовали профили не-1C-клеток из группы штрихкодов 1 (которая была специально предварительно отсортирована по содержанию 1C, так что вряд ли она содержит аутентичные клетки M2). Центромерно-проксимальные SNP клеток 1C, которые завершили оба раунда мейотических делений, должны быть либо B6, либо получены из Cast. Для дублетов 1С эти области имеют одинаковую вероятность появления гетерозигот или гомозигот. Таким образом, в любом данном дублете 1C число хромосом, которые, по-видимому, сегрегировали по-разному, а также число, которое, по-видимому, сегрегировало редукционно, должно следовать биномиальному распределению с n=19 и p=0,5. Фактически, это то, что мы наблюдаем для дублетов 1C из группы штрихкодов 1 (p=0,53 для распределения пропорций эквационно сегрегировавших хромосом, отклоняющихся от биномиального (19, 0,5), критерий хи-квадрат, фиг. 11A-B). На самом деле, есть только 11 1C дублетных клеток с, по меньшей мере, 15 хромосомами, которые, по-видимому, сегрегируют непротиворечивым образом, будь то эквационно или редукционно.
Напротив, не-1C клетки из группы штрихкода 2 имеют очень различное распределение. Среди 373 таких клеток 258 похожи на дублеты 1С группы штрихкодов 1 в том, что у них одинаковое количество хромосом с эквационными или редукционными моделями сегрегации. Остальные 115 клеток смещены, причем, по меньшей мере, 15 хромосом сегрегируют постоянным образом, эквационно или редукционно (фиг. 11C-E; 115/373 для группы штрихкодов 2 против 11/1,104 для группы штрихкодов 1; p=3×10-70, критерий хи-квадрат), причем многие демонстрируют полностью эквационнные (n=6) или полностью редукционные (n=91) паттерны.
Модель конечной смеси для подгонки трех популяций не-1C клеток
Чтобы рассмотреть это более формально, мы подгоняем данные каждого эксперимента к байесовской конечной смеси трех биномиальных распределений. Подробности приведены в Примере 2, раздел «Модель конечных смесей для подгонки трех популяций не-1С клеток» и Фиг. 12, с основными выводами, кратко изложенными здесь. Во-первых, не-1C клетки из яичек внутривидовых самцов F1 (B6×Cast) (т.е. из группы штрихкодов 2), по оценкам, включают субпопуляцию клеток, сегрегирующих редукционно (28%), по сравнению с эквационной сегрегацией (2%), а также, вероятно, дублеты 1С (69%) (фиг. 12В). Пропорции для клеток M2 отличаются от межвидовых самцов F1 (B6×Spret), которые, по оценкам, включают субпопуляцию клеток, сегрегирующих редукционно (66%) по сравнению с эквационной сегрегацией (14%), а также, вероятно, дублеты 1С (20%) (фиг. 12C). Эти анализы подтверждают вывод о том, что в бесплодном скрещивании (B6×Spret) гораздо более высокая доля клеток, которые смещены в сторону эквационной, а не редукционной сегрегации.
Распределение мейотических кроссинговеров на хромосомном уровне
Затем мы попытались исследовать геномные корреляты событий кроссинговера. В общей сложности мы проанализировали 1663 клетки 1С, в которых есть 19601 кроссинговер, и 240 клеток М2 с 4184 точками разрыва для кроссинговера из скрещивания (B6×Spret), и 5547 клеток 1С, содержащих 60755 точек разрыва для кроссинговера и 115 клеток М2 с 2246 точками разрыва для кроссинговера из скрещивания (B6×Cast). Насколько нам известно, это беспрецедентный набор данных в отношении числа событий кроссинговера, выявленных в связи с мейозом млекопитающих.
Высокопроизводительный характер sci-L3-WGS позволил нам проанализировать большое количество незрелых половых клеток и выявить редкую популяцию клеток, у которой завершился MI, но не MII, и, таким образом, наблюдать события мейотического кроссинговера и нарушенной сегрегации хромосом в одной и той же клетке. При сравнении бесплодного межвидового гибрида (B6×Spret) с фертильным внутривидовым гибридом (B6×Cast) на хромосомном уровне мы наблюдаем следующие дефекты в MI: 1) доля клеток M2, которые имеют, по меньшей мере, один кроссинговер на всех 19 аутосомах уменьшена с ~2/3 у (B6×Cast) до ~1/2 у (B6×Spret); 2) среднее количество кроссинговеров на клетку М2 ниже у (B6×Spret), но среднее количество кроссинговеров на клетку 1С выше; 3) интерференция кроссинговера слабее у (B6×Spret), где среднее расстояние между соседними кроссинговерами уменьшено с 97 Мб до 82 Мб; 4) в клетках М2 (B6×Spret) кроссинговеры, как правило, встречаются в средней половине каждого плеча хромосомы, в отличие от 1C обоих скрещиваний, а также клетокМ2 (B6×Spret), где они отдают предпочтение наиболее центромерно-дистальному квартилю; 5) среди клеток M2 с нарушенной эквационной или редукционной сегрегацией хромосом, (B6×Spret) демонстрирует значительно более высокую долю от полногеномной эквационной сегрегации (38/240), чем (B6×Spret) (8/115); 6) среди клеток M2 сполногенмной редукционной сегрегацией при всего генома при MI среднее число спорадических эквационных сегрегаций (также называемых обратными сегрегациями (Ottolini et al., 2015)) увеличивается с 0,2 до 1,1. Эти результаты позволяют предположить механизмы, которые могут способствовать или отражать основные факторы, способствующие бесплодию самцов F1 (B6×Spret), включая дефекты в формировании и расположении кроссинговеров, дефектные механизмы для обеспечения, по меньшей мере, одного кроссинговера на хромосому, и увеличение как спорадической, так и полногеномной эквационной сегрегации. Детали эти анализов представлены на Фиг. 10, Фиг. 13, и Фиг. 14 и в Примере 2, раздел «Распределение мейотических кроссинговеров на хромосомном уровне».
Распределение событий мейотического кроссинговера в связи с ландшафтом генома
Геномные характеристики, регулирующие активность кроссинговера
Чтобы оценить распределение кроссинговеров в более мелком масштабе, мы свернули все события кроссинговера, чтобы сгенерировать «карты активности» вдоль каждой мышиной хромосомы. Мы сначала сравнили эти карты с картой секвенирования одноцепочечной ДНК (SSDS) (Brick et al., 2018; Smagulova et al., 2011, 2016) и картой олигонуклеотидного комплекса Spo11 (Lange et al., 2016), которые идентифицируют горячие точки мейотического DSB в самом высоком разрешении (фиг. 15А). Карты DSB в штамме B6 из этих двух способов картирования сильно коррелируют друг с другом вдоль окна в 100 т.п.н. (rho=0,87, p<2×10-308). Хотя наши скопления кроссинговеров в клетках 1C и M2 коррелируют друг с другом (rho=0,67 для скрещивания (B6×Spret) и rho=0,55 для (B6×Cast), p<2×10-308для обоих, фиг. 15B-C), оба отклоняются от карт DSB. Значимый ген PRDM9, важный игрок для описания горячих точек, был выделен для связывания различных мотивов между различными линиями мышей, даже между подвидами мышей (Davies et al., 2016; Gregorova et al., 2018). Мы обсуждаем его потенциальное влияние на различия между двумя скрещиваниями в Примере 2, раздел «Влияние PRDM9 на активность кроссинговера».
Только 10% специфических для мейоза DSB были восстановлены в виде кроссинговеров. Далее мы рассмотрели, какие факторы помимо разрывов Spo11 способствуют формированию кроссинговера, построив линейную модель с байесовским модельным усреднением (BMA) (Clyde et al., 2011). В данном случае BMA принимает средневзвешенное значение из более чем 15000 исследованных моделей выбора переменных и взвешивает их по апостериорной вероятности каждой модели, что объясняет неопределенность в выборе модели, в отличие от некоторых других методов выбора переменных, таких как регрессия Лассо. Мы количественно оценили предельную вероятность включения (MIP) для ~80 потенциально объясняющих переменных. Признаки, которые, как известно, имеют отношение к мейотическим кроссинговерам, такие как сайты разрыва Spo11, содержание GC и т.д. включены практически во все модели с высокими вероятностями (фиг. 16А, фиг. 17); Например, области с высоким содержанием GC являются более «горячими» для формирования кроссинговера. Мы также обнаружили еще несколько особенностей, которые ранее не были причастны к мейотическим кроссинговерам, таких как специфические семейства повторов и метки хроматина, и, особенно, домены ранней репликации. Матрицы корреляции между активностью кроссинговера и всеми признаками приведены на фиг. 18-19 для каждого скрещивания. Использованные признаки и обобщения простых линейных моделей и BMA также включены. Разрешение точек разрыва (медиана ~150 т.п.н. для (B6×Spret) и ~250 т.п.н. для (B6×Cast); Фиг. 16B) находится на одном уровне с предыдущими усилиями по картированию мейотических кроссинговеров с помощью секвенирования отдельных клеток (150-500 т.п.н.) (Lu et al., 2012; Ottolini et al., 2015; Wang et al., 2012); однако большая сложность библиотеки, предоставляемая sci-L3-WGS, позволила нам достичь этого с гораздо меньшей глубиной секвенирования.
Многие из признаков, которые коррелируют с образованием кроссинговера, согласуются между скрещиваниями (B6×Spret) и (B6×Cast), но некоторые - нет. Например, позиционные смещения формирования кроссинговера кажутся различными. В клетках 1С обоих скрещиваний, а также в клетках М2 в скрещивании (B6×Cast) кроссинговеры недостаточно представлены в пределах 10 Мб от центромеры и, скорее всего, встречаются вблизи теломер в крайнем правом позиционном «квартиле» (фиг. 18), Однако в клетках M2 в скрещивании (B6×Spret) кроссинговеры недостаточно представлены около центромеры, а также около теломер, и, скорее всего, встречаются в средних квартилях (фиг. 19). Эта тенденция сохраняется в линейных моделях, где мы учитываем вклад от всех других признаков.
Положение кроссинговера может сильно повлиять на величину напряжения, возникающего между гомологами хромосом, что, в свою очередь, способствует правильной сегрегации хромосома. Таким образом, мы исследовали это более подробно, взяв только самый правый кроссинговер для каждой хромосомы в каждой клетке и изучив его положение вдоль плеча хромосомы в каждом скрещивании (de Boer et al., 2015). Учитывая изменчивость между хромосомами с помощью линейной модели смешанного эффекта, мы оцениваем, что положения крайних правых кроссинговеров в скрещивании (B6×Spret) в среднем на 1,6 Мб больше центромерно-проксимальные, чем в скрещивании (B6×Cast) в клетках 1C (фиг. 20A, p=1×10-13, F-тест), но на 5,5 Мб более центромерно-проксимальные в клетках M2 (фиг. 16C, p=2,2×10-15). Обратите внимание, что самые правые кроссинговеры в клетках M2 имеют тенденцию быть более центромерно-проксимальными, чем в клетках 1C в обоих скрещиваниях, но в большей степени в скрещивании (B6×Spret) (фиг. 16D), чем в скрещивании (B6×Cast) (фиг. 20B). Эти различия позволяют предположить, что субпопуляция клеток M2 в скрещивании (B6×Spret), чьи кроссинговеры встречаются слишком близко к центромере, может не созреть в клетки 1C, возможно из-за дефектов сегрегации MII. Точно так же, хотя при ограниченном числе событий, мы также сравнили положения кроссинговеров в клетках M2, которые имеют нарушенную сегрегацию хромосом, и обнаружили, что в обоих скрещиваниях кроссинговеры в клетках с нарушенной эквационной сегрегацией являются более центромерно-дистальными, чем в клетках с нарушенной редукционной сегрегациейс разницей в 13,7 Мб в скрещивании (B6×Cast) (p=4×10-15) и 8,7 Мб в скрещивании (B6×Spret) (p=6×10-14) (фиг. 20C-D). Это говорит о возможных дефектах сегрегации MI в клетках, у которых кроссинговеры находятся слишком близко к теломере. Мы предлагаем предварительную модель для объяснения этого наблюдения на Фиг. 20E.
Гетерогенность клеток относительно точек разрыва для кроссинговера
Хотя клетки 1C и M2 выглядят в целом одинаково по скоплению кроссинговеров (фиг. 15), мы задавались вопросом, существует ли какая-либо структура в признаках, которые влияют на распределения кроссинговеров в субпопуляции отдельных клеток. Чтобы исследовать это, мы собрали в одно целое информацию, связанную с кроссинговером, для каждой отдельной клетки для каждого из 78 признаков (Пример 2, раздел «Способы биоинформатики и статистические анализы»). Затем мы использовали анализ основных компонентов (PCA) на матрице с каждой строкой как одной клеткой и каждым столбцом как одним суммарным значением признака. Для скрещивания (B6×Spret) первичные два основных компонента (PC) захватывают 26% дисперсии, а для пскрещивания (B6×Cast) PC1 и PC3 захватывают 17% дисперсии. В обоих скрещиваниях клетки 1C и M2 разделены на два кластера с помощью этих PC. На Фиг. 21 и Фиг. 22 мы нанесли на график каждый признак, спроецированный на эти PC. Хромосомное распределение кроссинговеров, однородительских хромосом и положения кроссинговеров в квартилях хромосомы являются признаками, которые, по-видимому, определяют разделение клеток 1С и М2.
Предсказание участков кроссинговера по геномным признакам
Наконец, мы стремились использовать большое количество событий, наблюдаемых здесь, чтобы построить предиктивную модель расположения кроссинговеров. В частности, мы построили линейную модель бинарного отклика, где 1 - это участки кроссинговера, а 0 - случайный участок, выбранный из генома из такого же распределения длины участка (подробности в примере 2, раздел «Способы биоинформатики и статистические анализы»). Используя те же 76 признаков, что и в анализах BMA, мы можем предсказать участки кроссинговера на удерживаемых данных со средним значением кривой ROC площади под кривой (AUC) 0,73 для скрещивания (B6×Spret). С подмножеством из 25 переменных высокой вероятности включения (MIP>0,5), определенных BMA, мы достигаем аналогичной средней AUC 0,72 (фиг. 16E). Аналогично, для скрещивания (B6×Cast) мы достигаем средней AUC 0,85, когда используем все признаки или подмножество из 25 признаков с MIP>0,5 (фиг. 16F).
Обсуждение
Здесь мы описываем sci-L3, концепция которого сочетает в себе 3-уровневое комбинаторное индексирование одиночных клеток и линейную амплификацию. Мы демонстрируем, что sci-L3 применим для секвенирования полного генома одиночной клетки (sci-L3-WGS), целевого секвенирования ДНК одиночной клетки (sci-L3-target-seq), и совместного анализа генома и транскриптома одиночной клетки (sci-L3-RNA/DNA). С sci-L3-WGS можно обработать за два дня эксперимента, по меньшей мере, десятки тысяч и, возможно, миллионы геномов одиночных клеток при стоимости создания библиотеки $ 0,14 за клетку для 10000 клеток и $ 0,008 за клетку для 1000000 клеток. Производительность sci-L3-WGS на несколько порядков выше, чем у альтернативных способов WGS одиночных клеток, основанных на линейной амплификации, таких как LIANTI (в пробирке) (Chen et al., 2017). Кроме того, улучшается число уникальных молекул, извлекаемых из каждой отдельной клетки, от нескольких тысяч (Pellegrino et al., 2018) или десятков тысяч (Vitak et al., 2017) до сотен тысяч.
Мы применили sci-L3-WGS для изучения мейоза самцов мышей и определили неожиданную популяцию клеток M2. Характер данных одиночных клеток также позволил нам одновременно характеризовать мейотический кроссинговер и нарушенную сегрегацию хромосом. События обратной сегрегации ранее наблюдались в полном анализе мейоза человека женского пола (Ottolini et al., 2015), и мы наблюдаем аналогичные события здесь в отношении мейоза самцов мыши (т.е. эквационную сегрегацию одной или более хромосом). Среди 292 клеток M2, которые мы проанализировали из скрещивания (B6×Spret), отдельные клетки были смещены в сторону эквационной или редукционной сегрегации хромосом, что предполагает глобальный механизм чувствительности для определения того, продолжает ли клетка мейоз или возвращается к митотической сегрегации ее хромосом. Кроме того, насколько нам известно, в первый раз для мейоза млекопитающих, мы наблюдали множественные случаи полной геномной эквационной сегрегации во время MI, что предполагает клеточно-автономный, а не хромосомо-автономный режим эквационной сегрегации. Мы идентифицировали такие события в обоих скрещиваниях, хотя реже в фертильном скрещивании (B6×Cast).
Высокая частота полногеномной обратной сегрегации по сравнению с тем, что можно ожидать для хромосомно-автономного механизма (частота 2-19), особенно в межвидовом скрещивании (B6×Spret), вызывает больше вопросов, чем дает ответов. Мы изобразили модель и выделили более нерешенных вопросов на фиг. 23. При нормальном MI сцепление центромер поддерживается при редукционной сегрегации, а сестринские хроматиды, расположенные центромерно- проксимальнее кроссинговера, не разделяются до MII (паттерн 1 на фиг. 23D). Эквационная сегрегация в MI указывает на преждевременное разделение центромерных когезинов (паттерн 2 и/или 3 на фиг. 23D). Предыдущая работа также показала, что спаривание гомологов может быть дефектным у F1 в этом скрещивании из-за эрозии участков связывания PRDM9 (Davies et al., 2016; Gregorova et al., 2018; Smagulova et al., 2016) и проблема спаривания, вероятно, становится более тяжелой при межвидовом скрещивании. В Примере 2, разделе «Размышления о причинах и следствиях обратной сегрегации», мы рассуждаем о том: 1) что может вызвать преждевременное разделение центромерных когезинов, 2) достаточно ли одного кроссинговера для правильной редукционной сегрегации, и 3) какие следствия может иметь эквационная сегрегация в MI.
Улучшенное покрытие генома позволило с высокой разрешающей способностью отображать точки разрыва кроссинговера по сравнению с другими способами секвенирования одиночных клеток, а производительность для картирования в общей сложности ~87000 кроссинговеров позволила нам лучше охарактеризовать геномные и эпигеномные особенности, связанные с активностью кроссинговера, с данными о накоплении. Мы обсуждаем, как континуум активности кроссинговера формируется многими факторами в Примере 2, разделе «Активность кроссинговера и связанные (эпи)геномные факторы».
Одно из ключевых отличий от простого сочетания схемы высокопроизводительного комбинаторного индексирования одиночных клеток («sci») с линейной амплификацией с помощью транспозонной вставки (LIANTI) в разработке sci-L3 заключается в том, что мы внедрили промотор T7 с помощью лигирования, который не только позволяет проводить более двух раундов штрихкодирования клеток и дополнительно увеличивает производительность при значительно сниженных затратах, но также обеспечивает гибкость, позволяющую генерализовать способ для других анализов одиночных клеток с небольшими изменениями протокола. В качестве первого примера мы демонстрируем, что sci-L3-WGS может быть легко адаптирован к sci-L3-target-seq. Хотя сообщалось о целевом секвенировании одиночной клетки на платформе 10X Genomics, насколько нам известно, это были РНК-транскрипты, а не локусы ДНК. Хотя текущая 10% «скорость восстановления» на гаплотип может не быть идеальной для целевого секвенирования, это смягчается большим количеством клеток, которые можно анализировать. В качестве второго примера мы демонстрируем, что sci-L3-WGS также может быть адаптирован к совместному анализу sci-L3-RNA/DNA. Мы ожидаем, что в дальнейшем возможно адаптировать sci-L3 к ATAC-seq, bisulphite-seq и Hi-C для профилирования отдельных клеток по доступности хроматина, метилому и конформации хроматина, соответственно, что может иметь преимущества перед опубликованными научными исследованиями (Cusanovich et al., 2015; Mulqueen et al., 2018; Ramani et al., 2017) для этих целей в отношении производительности и однородности амплификации.
Суммируя, sci-L3-WGS, sci-L3-target-seq и совместный анализ sci-L3-RNA/DNA расширяют набор инструментов для секвенирования одиночной клетки. В этом исследовании мы, кроме того, показываем, как sci-L3-WGS может обеспечить систематическое и количественное представление о мейотической рекомбинации и выявить редкое событие нарушенной полногеномной сегрегации хромосом с беспрецедентной комбинацией производительности. Мы ожидаем, что способы sci-L3 будут очень полезны в других контекстах, где секвенирование генома одиночной клетки оказывается трансформирующим, например, для изучения редких межгомологичных митотических кроссинговеров и для анализа генетической гетерогенности и эволюции злокачественных опухолей.
Ссылки
Baudat, F., Manova, K., Yuen, J.P., Jasin, M., and Keeney, S. (2000). Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol. Cell 6, 989-998.
Baudat, F., Imai, Y., and de Massy, B. (2013). Meiotic recombination in mammals: localization and regulation. Nat. Rev. Genet. 14, 794-806.
Berletch, J.B., Ma, W., Yang, F., Shendure, J., Noble, W.S., Disteche, C.M., and Deng, X. (2015). Escape from X inactivation varies in mouse tissues. PLoS Genet. 11, e1005079.
de Boer, E., Jasin, M., and Keeney, S. (2015). Local and sex-specific biases in crossover vs. noncrossover outcomes at meiotic recombination hot spots in mice. Genes Dev. 29, 1721-1733.
Brick, K., Pratto, F., Sun, C.-Y., Camerini-Otero, R.D., and Petukhova, G. (2018). Analysis of Meiotic Double-Strand Break Initiation in Mammals. Methods Enzymol. 601, 391-418.
Cao, J., Packer, J.S., Ramani, V., Cusanovich, D.A., Huynh, C., Daza, R., Qiu, X., Lee, C., Furlan, S.N., Steemers, F.J., et al. (2017). Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661-667.
Cao, J., Spielmann, M., Qiu, X., Huang, X., Ibrahim, D.M., Hill, A.J., Zhang, F., Mundlos, S., Christiansen, L., Steemers, F.J., et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature.
Chen, C., Xing, D., Tan, L., Li, H., Zhou, G., Huang, L., and Xie, X.S. (2017). Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI). Science 356, 189-194.
Choi, K., and Henderson, I.R. (2015). Meiotic recombination hotspots - a comparative view. Plant J. 83, 52-61.
Clyde, M.A., Ghosh, J., and Littman, M.L. (2011). Bayesian Adaptive Sampling for Variable Selection and Model Averaging. J. Comput. Graph. Stat. 20, 80-101.
Cole, F., Baudat, F., Grey, C., Keeney, S., de Massy, B., and Jasin, M. (2014). Mouse tetrad analysis provides insights into recombination mechanisms and hotspot evolutionary dynamics. Nat. Genet. 46, 1072-1080.
Cusanovich, D.A., Daza, R., Adey, A., Pliner, H.A., Christiansen, L., Gunderson, K.L., Steemers, F.J., Trapnell, C., and Shendure, J. (2015). Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910-914.
Davies, B., Hatton, E., Altemose, N., Hussin, J.G., Pratto, F., Zhang, G., Hinch, A.G., Moralli, D., Biggs, D., Diaz, R., et al. (2016). Re-engineering the zinc fingers of PRDM9 reverses hybrid sterility in mice. Nature 530, 171-176.
Eberwine, J., Yeh, H., Miyashiro, K., Cao, Y., Nair, S., Finnell, R., Zettel, M., and Coleman, P. (1992). Analysis of gene expression in single live neurons. Proceedings of the National Academy of Sciences 89, 3010-3014.
Gregorova, S., Gergelits, V., Chvatalova, I., Bhattacharyya, T., Valiskova, B., Fotopulosova, V., Jansa, P., Wiatrowska, D., and Forejt, J. (2018). Modulation of controlled meiotic chromosome asynapsis overrides hybrid sterility in mice. Elife 7.
Hashimshony, T., Wagner, F., Sher, N., and Yanai, I. (2012). CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2, 666-673.
Hong, S., Sung, Y., Yu, M., Lee, M., Kleckner, N., and Kim, K.P. (2013). The logic and mechanism of homologous recombination partner choice. Mol. Cell 51, 440-453.
Hou, Y., Fan, W., Yan, L., Li, R., Lian, Y., Huang, J., Li, J., Xu, L., Tang, F., Xie, X.S., et al. (2013). Genome analyses of single human oocytes. Cell 155, 1492-1506.
Keeney, S., Giroux, C.N., and Kleckner, N. (1997). Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88, 375-384.
Lange, J., Yamada, S., Tischfield, S.E., Pan, J., Kim, S., Zhu, X., Socci, N.D., Jasin, M., and Keeney, S. (2016). The Landscape of Mouse Meiotic Double-Strand Break Formation, Processing, and Repair. Cell 167, 695-708.e16.
Liu, E.Y., Morgan, A.P., Chesler, E.J., Wang, W., Churchill, G.A., and Pardo-Manuel de Villena, F. (2014). High-resolution sex-specific linkage maps of the mouse reveal polarized distribution of crossovers in male germline. Genetics 197, 91-106.
Lu, S., Zong, C., Fan, W., Yang, M., Li, J., Chapman, A.R., Zhu, P., Hu, X., Xu, L., Yan, L., et al. (2012). Probing meiotic recombination and aneuploidy of single sperm cells by whole-genome sequencing. Science 338, 1627-1630.
Mancera, E., Bourgon, R., Brozzi, A., Huber, W., and Steinmetz, L.M. (2008). High-resolution mapping of meiotic crossovers and non-crossovers in yeast. Nature 454, 479-485.
Mulqueen, R.M., Pokholok, D., Norberg, S.J., Torkenczy, K.A., Fields, A.J., Sun, D., Sinnamon, J.R., Shendure, J., Trapnell, C., O’Roak, B.J., et al. (2018). Highly scalable generation of DNA methylation profiles in single cells. Nat. Biotechnol. 36, 428-431.
Ottolini, C.S., Newnham, L., Capalbo, A., Natesan, S.A., Joshi, H.A., Cimadomo, D., Griffin, D.K., Sage, K., Summers, M.C., Thornhill, A.R., et al. (2015). Genome-wide maps of recombination and chromosome segregation in human oocytes and embryos show selection for maternal recombination rates. Nat. Genet. 47, 727-735.
Pellegrino, M., Sciambi, A., Treusch, S., Durruthy-Durruthy, R., Gokhale, K., Jacob, J., Chen, T.X., Geis, J.A., Oldham, W., Matthews, J., et al. (2018). High-throughput single-cell DNA sequencing of acute myeloid leukemia tumors with droplet microfluidics. Genome Res.
Ramani, V., Deng, X., Qiu, R., Gunderson, K.L., Steemers, F.J., Disteche, C.M., Noble, W.S., Duan, Z., and Shendure, J. (2017). Massively multiplex single-cell Hi-C. Nat. Methods 14, 263-266.
Romanienko, P.J., and Camerini-Otero, R.D. (2000). The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol. Cell 6, 975-987.
Smagulova, F., Gregoretti, I.V., Brick, K., Khil, P., Camerini-Otero, R.D., and Petukhova, G.V. (2011). Genome-wide analysis reveals novel molecular features of mouse recombination hotspots. Nature 472, 375-378.
Smagulova, F., Brick, K., Pu, Y., Camerini-Otero, R.D., and Petukhova, G.V. (2016). The evolutionary turnover of recombination hot spots contributes to speciation in mice. Genes Dev. 30, 266-280.
Sos, B.C., Fung, H.-L., Gao, D.R., Osothprarop, T.F., Kia, A., He, M.M., and Zhang, K. (2016). Characterization of chromatin accessibility with a transposome hypersensitive sites sequencing (THS-seq) assay. Genome Biol. 17, 20.
Vitak, S.A., Torkenczy, K.A., Rosenkrantz, J.L., Fields, A.J., Christiansen, L., Wong, M.H., Carbone, L., Steemers, F.J., and Adey, A. (2017). Sequencing thousands of single-cell genomes with combinatorial indexing. Nat. Methods 14, 302-308.
Wang, J., Fan, H.C., Behr, B., and Quake, S.R. (2012). Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell 150, 402-412.
Wang, S., Kleckner, N., and Zhang, L. (2017a). Crossover maturation inefficiency and aneuploidy in human female meiosis. Cell Cycle 16, 1017-1019.
Wang, S., Hassold, T., Hunt, P., White, M.A., Zickler, D., Kleckner, N., and Zhang, L. (2017b). Inefficient Crossover Maturation Underlies Elevated Aneuploidy in Human Female Meiosis. Cell 168, 977-989.e17.
Yamada, S., Kim, S., Tischfield, S.E., Jasin, M., Lange, J., and Keeney, S. (2017). Genomic and chromatin features shaping meiotic double-strand break formation and repair in mice. Cell Cycle 16, 1870-1884.
Zhang, K., Wu, X.-C., Zheng, D.-Q., and Petes, T.D. (2017). Effects of Temperature on the Meiotic Recombination Landscape of the Yeast. MBio 8.
Пример 2
Модель конечной смеси для подгонки трех популяций не-1C клеток
Клетки, не относящиеся к 1C, восстановленные из гибрида (B6×Cast) из группы штрихкодов 2, включают в себя дублеты 1C, клетки, которые, по-видимому, смещены в сторону эквационной сегрегации, и клетки, которые, по-видимому, смещены в сторону редукционной сегрегации. Чтобы количественно определить их относительные пропорции, мы подгоняем данные к смеси трех биномиальных распределений, с вероятностями того, что хромосомы сегрегируют эквационно 0,01, 0,48 и 0,95, и пропорциями смешивания 0,28, 0,69 и 0, 02 (фиг. 12А). Напротив, когда мы пытаемся подобным образом сопоставить не-1C клетки из группы штрихкода 1 со смесью из трех биномиальных распределений, мы получаем вероятности того, что хромосомы сегрегируют эквационно 0,46, 0,5 и 0,53 (все близко к 0,5) и пропорции смешивания 0,24, 0,44 и 0,31 (фиг. 12В).
Что касается вопроса о том, отличается ли доля клеток M2, которые смещены в сторону эквационной и редукционной сегрегации, между фертильным и бесплодным скрещиванием, мы можем аналогичным образом сопоставить хромосомные данные из скрещивания (B6×Spret) (фиг. 9E), которые дают вероятности хромосомы сегрегировать эквационно 0,05, 0,39 и 0,91, а пропорции смешивания 0,66, 0,2 и 0,14 (фиг. 12C). Эти пропорции позволяют предположить, что бесплодное скрещивание (B6×Spret) имеет более высокую долю клеток, которые смещены в сторону эквационной, а не редукционной сегрегации.
Распределение мейотических кроссинговеров на хромосомном уровне
Основываясь на 1663 клетках 1С, содержащих 19601 точек разрыва для кроссинговера, и 240 клеток клеток M2 с 4184 точками разрыва для кроссинговера (B6×Spret), и 5547 клетках 1C, содержащих 60755 точек разрыва для кроссинговера и 115 клетках M2 с 2246 точками разрыва для кроссинговера от (B6×Cast) мы сначала рассмотрели распределение мейотических кроссинговеров по хромосомам. Плотность кроссинговера определяется здесь как среднее количество кроссинговеров на клетку на деление на Мб, умноженное на 2 (в клетках 1C) или 1 (в клетках М2). В скрещивании (B6×Spret) мы наблюдали сильную отрицательную корреляцию между размером хромосомы и плотностью кроссинговера в клетках 1С (фиг. 13A, r=-0,66, p=0,002). В соответствии с предыдущими результатами (Lange et al., 2016), эта корреляция только частично объясняется плотностью олигонуклеотидного комплекса Spo11 (r=-0,46, p<0,05), что позволяет предположить, что более мелкие хромосомы содержат больше DSB, и эти DSB дают больше шансов породить кроссинговеры. Эта отрицательная корреляция еще сильнее проявляется в клетках M2 (фиг. 13B, r=-0,83, p=1×10-5). На Фиг. 10A-B, мы рассматриваем случаи множественных кроссинговеров на хромосому на клетку как одно событие, которое еще больше усиливает отрицательную корреляцию (r=-0,87, p= 2×10-6 для клеток 1С; r=-0,91, р=8×10-8 для клеток М2). Эти наблюдения позволяют предположить, что более мелкие хромосомы являются более горячими участками для кроссинговеров, и, в особенности, для того, чтобы иметь, по меньшей мере, один кроссинговер на клеточное деление. Такая же тенденция наблюдается в скрещивании (B6×Cast) (фиг. 14A-D). Клетки 1С клетки и в среднем 0,62 и 0,58 кроссинговеров на хромосому на клетку для меж- и внутривидовых скрещиваний соответственно, тогда как клетки М2 имели в среднем 0,92 и 1,03 на хромосому на клетку (фиг. 13C-D, 10C-D). Частота кроссинговера в межвидовых клетках М2 лишь на 9% ниже, чем количество кроссинговеров, измеренное по очагам Mlh1 в сперматоцитах 4С у инбредных мышей В6 (Froenicke et al., 2002), несмотря на расхождение в 2%. Уровень кроссинговера в клетках 1С на 45% ниже, чем при секвенировании сперматозоидов у одного человека (Lu et al., 2012; Wang et al., 2012). Последнее различие во многом может быть связано с телоцентрической природой мышиных хромосом. Хотя межвидовое скрещивание (B6×Spret) имеет более высокое среднее число кроссинговеров, обнаруженных в 1С, по сравнению со скрещиванием (B6×Cast) (p=7×10-26, тест Манна-Уитни), среднее число кроссинговеров в клетках M2 ниже (р= 2×10-10). Мы отмечаем, что доля клеток M2, которые сегрегировали все 19 аутосом редукционно, которые имеют кроссинговер на каждой хромосоме, выше для скрещивания (B6×Cast) (60/91 из 66%), чем для скрещивания (B6×Spret) (41/80 или 51%) (р=0,06, точный критерий Фишера), что может способствовать бесплодию последнего.
Чтобы исследовать интерференцию кроссинговеров, мы взяли хромосомы, по меньшей мере, с двумя кроссинговерами и нанесли на график расстояние между соседними кроссинговерами, и сравнили это распределение с ожидаемым на основе случайного моделирования (фиг. 13E, фиг. 10E, фиг. 14E). Среднее наблюдаемое расстояние между кроссинговерами составляло 82 Мб для (B6×Spret) и 97 Мб для (B6×Cast); и то, и другое намного превосходит ожидания 39 и 42 Мб (p=1×10-267 и p<2×10-308, соответственно, критерий Манна-Уитни). Это соответствует отталкиванию кроссинговеров в непосредственной близости. Обратите внимание, что интерференция кроссинговеров сильнее у (B6×Cast), чем у (B6×Spret) скрещивания, с более длинными расстояниями между соседними кроссинговерами (p=5×10-91).
Мы также проанализировали распределение одднородительских хромосом (т.е. без наблюдаемых кроссинговеров) в каждой отдельной клетке (фиг. 13F) и для каждой хромосомы (рис. 13G) в скрещивании (B6×Spret) (те же тенденции имеют место для скрещивания (B6×Cast), как показано на фиг. 14F-G). Хотя у более коротких хромосом наблюдается повышенная частота кроссинговера при нормализации по длине, частота однородительских хромосом (свернутых по всем классам клеток) все еще отрицательно коррелирует с размером хромосомы (фиг. 13G; r=-0,91, p=4,6×10-8).
В то время как мы показали, что клетки М2 сильно смещены в сторону эквационнойили редукционной сегрегации их хромосом, мы также наблюдали сотни случаев спорадической эквационной сегрегации среди клеток, которые имеют, по меньшей мере, 15 хромосом с редукционной сегрегацией. Это явление ранее наблюдали и назвали «обратной сегрегацией» (Ottolini et al., 2015). На фиг. 13H, мы показываем хромосомное распределение этих событий обратной сегрегации. Обратите внимание, что хотя коэффициент обратной сегрегации значительно выше при скрещивании (B6×Spret) (среднее=1,1), чем при скрещивании (B6×Cast) (среднее=0,2, p= 2×10-14, тест Манн-Уитни), хромосомы 7 и 11 имеют самые высокие показатели обратной сегрегации в обоих скрещиваниях.
Затем мы исследовали нормализованную пропорцию ридов на клетку, которые картируются на митохондриальный геном (фиг. 13I, фиг. 10G). Клетки 1С демонстрируют бимодальное распределение в отношении «числа копий» митохондриальной ДНК, наблюдение, для которого нам не хватает удовлетворительного объяснения. Мы наблюдали умеренную отрицательную корреляцию между соотношением митохондриальных ридов и числом кроссинговеров (rho=-0,11, p=3×10-6). Интересно, что хотя количество клеток М2, которые сегрегировали, по меньшей мере, 15 своих хромосом, либо эквационно, либо редукционно, у них были очень разные распределения пропорций митохондриальных ридов (фиг. 10G). В соответствии с этим пропорция митохондриальных ридов положительно коррелирует с числом редукционно сегрегировавших хромосом в клетках М2 (r=0,18, p=0,005). Обратите внимание, что мы не можем оценить это в скрещивании (B6×Cast), потому что более 90% секвенированных одиночных клеток не имеют каких-либо ридов, картирующихся на митохондриальный геном. Возможно, что различные способы, используемые для выделения ядер из яичек (B6×Cast) и придатка яичка (B6×Spret), в сочетании с предварительной сортировкой ядер из яичек фракционировали митохондрии вдали от основной массы ядер.
Воздействие PRDM9 на активность кроссинговера
На основании карты активности кроссинговера путем накапливания точек разрыва кроссинговера вдоль хромосом на всем протяжении генома (фиг. 15), мы обнаружили, что во внутривидовом скрещивании (B6×Cast) активность кроссинговера лучше коррелирует с горячими доменами DSB, картированными у самца Cast, чем у самца B6 (rho=0,28 и 0,12, p<2×10-308 и p=1×10-83, соответственно), возможно, в результате неполной доминантности аллеля PRDM9 Cast у гибрида F1. Корреляция сильнее с горячими доменами DSB, картированными у животных F1 (B6×Cast) (rho=0,3, p<2×10-308). Для скрещивания (B6×Spret) эрозия консенсусного участка связывания PRDM9 приводит к образованию четырех типов горячих точек DSB, определенных картой олигонуклеотидного комплекса Spo11: те, которые консервативны у B6 и Spret и называются «симметричными» горячими точками, те, которые присутствуют только у B6 или Spret и называются «асимметричными» горячими точками, и те, которые не содержат участок связывания PRDM9 ни у одного из видов. Все четыре типа горячих доменов DSB плохо коррелируют с кроссинговерами из скрещивания (B6×Spret) (rho=0,13, p=4×10-87 для использования всех горячих точек Spo11, картированных у B6; rho=0,11, p=3×10-63, если мы используем только «симметричные горячие точки»). Одна из возможностей состоит в том, что сайты DSB у скрещивания (B6×Spret) сильно доминируются аллелем PRDM9 Spret, таким образом, что горячие точки DSB, картированные в фоне штамма B6, не предсказывают сайты кроссинговеров.
Рассуждения о причинах и следствиях обратной сегрегации
Мы наблюдали высокую частоту обратной сегрегации, особенно в межвидовом скрещивании (B6×Spret). Ниже мы рассуждаем о том: 1) что может вызвать преждевременное разделение центромерных когезинов, 2) достаточно ли одного кроссинговера для правильной редукционной сегрегации, и 3) какие последствия у эквационной сегрегации при MI.
Во-первых, возможно, что из-за недостаточного спаривания гомологов между хромосомами B6 и Spret, DSB, которые должны были нормально репарироваться от гомолога во время мейоза, вместо этого часто репарируются с использованием сестринских хроматид в качестве матрицы. Это может вызвать разрушение когезинов (Storlazzi et al., 2008) и привести к преждевременному разделению центромерных когезинов.
Во-вторых, текущая модель позволяет предположить, что одного межгомологичного кроссинговера и правильной когезии сестринских хроматид достаточно для формирования хиазм (рис. 23), несмотря на первоначальное недостаточное спаривание гомологов в межвидовом скрещивании. Как только кроссинговер успешно сформирован, сегрегация хромосом не должна нарушаться. В нашем исследовании, на уровне отдельных хромосом, большое количество наблюдаемых эквационно сегрегировавших хромосом имеет нормальные кроссинговеры, о чем свидетельствует центромерно-дистальная LOH, что может указывать на то, что дефекты в первоначальном спаривании гомологов влияют на конечный результат. На уровне генома, однако, мы не можем с уверенностью оценить, имеют ли те клетки с с нарушенной эквационной сегрегацией такое же количество кроссинговеров, как и их аналоги с нарушенной редукционной сегрегацией, потому что мы можем обнаружить все кроссинговеры для хромосом, которые сегрегируют редукционно, но мы можем обнаружить только кроссинговеры в хромосомах, которые сегрегируют эквационно. когда две рекомбинированные хроматиды сегрегируют раздельно (фиг. 5B-C и фиг. 16D, схемы 2 и 3). Предполагая, что рекомбинированные хроматиды с одинаковой вероятностью будут сегрегировать вместе или раздельно, число кроссинговеров не меньше в тех случаях эквационной сегрегации на уровне генома, хотя мы не можем исключить возможность того, что сегрегация отклоняется от 50/50 из-за неразделенной рекомбинации промежуточных соединений (фиг. 23, паттерн 3).
В-третьих, каковы последствия этих эквационно сегрегировавших хромосом? Возвращаются ли они к митозу с обширной LOH, или они переходят к MII, и если да, способствуют ли образованию гамет 1C? У дрожжей было охарактеризовано явление, называемое «возвращение к росту», где клетки, которые инициируют программу мейоза, могут вернуться к нормальным митотическим делениям в присутствии правильных питательных веществ, что приводит к большому количеству событий LOH (Dayani et al., 2011). У человека при женском мейозе, хромосомы с обратной сегрегацией переходят в MII, приводя к одному эуплоидному ооциту и одному эуплоидному полярному телу 2, что соответствует нормальной сегрегации MII; авторы предполагают, что неразрешенная рекомбинация промежуточных соединений могла как вызвать обратную сегрегацию при MI, так и облегчить правильную сегрегацию MII путем связывания других неродственных гомологичных хроматид (фиг. 23, паттерн 3) (Ottolini et al., 2015). Mlh1 важен как для репарации ошибочно спаренных оснований (MMR), так и для разделения промежуточной структуры Холлидея при мейозе. Учитывая расхождение в 2% последовательности между B6 и Spret, возможно, что Mlh1 ограничен из-за интенсивной MMR, и Mlh1 может быть недостаточно для разделения рекомбинации промежуточных соединений. Однако мы подчеркиваем, что если рекомбинированные гомологичные хроматиды совместно сегрегируют, это не приведет к LOH (фиг. 5C). Таким образом, клетки M2 с LOH и эквационнная сегрегация не могут быть объяснены совместной сегрегацией нераспавшихся промежуточных соединений.
Наконец, на фиг. 23, мы также показываем возможный вклад в формирование гамет из хромосом без какого-либо межгомологичного кроссинговера, вероятно, из-за недостаточного спаривания гомологов, потому что один из паттернов (паттерн 4) не отличается от клеток, которые имеют кроссинговер, но совместно сегрегируют рекомбинированные хроматиды (паттерн 3). Однако, если эти клетки без кроссинговера вносят значительный вклад в клетки 1С, мы должны наблюдать большее количество хромосом без кроссинговера среди клеток 1С. Из клеток 1С, которые мы наблюдали в обоих скрещиваниях, количество хромосом с кроссинговерами и без кроссинговеров составляет примерно 50-50, что указывает на то, что они являются производными от некоторой комбинации паттернов 1-3 на фиг.23, и клетки 2C без внутригомологичных кроссинговеров (схемы 4 и 5) по существу не вносят вклад в клетки 1C, которые успешно завершают MII.
Активность кроссинговера и связанные (эпи)геномные факторы
Активность кроссинговера представляет собой пространственно-временной процесс и формируется многими факторами. Кроссинговеры в скрещивании (B6×Cast) сильнее коррелируют с с мейотическими горячими точками DSB, картированными у F1 скрещивания, чем на отдельных картах для двух родительских штаммов, что ожидалось бы на основании предыдущего открытия, что новые мейотические горячие точки могут образовываться в гибридах F1 (Smagulova et al. al., 2016). В скрещивании (B6×Spret) кроссинговеры слабо, но положительно коррелируют с разрывами Spo11. Обратите внимание, что карта Spo11 учитывает только сайты PRDM9, связывающие белок PRDM9 аллеля B6, и вполне вероятно, что Spret-копия PRDM9 связывается с разными сайтами и создает новые мейотические горячие точки DSB, которые не учитывались в наших анализах. Геномные признаки, которые, как мы наблюдали, положительно коррелируют с мейотическими кроссинговерами, включают в себя регионы, богатые GC (также в случае дрожжевого мейоза (Petes, 2001; Petes и Merker, 2002)), прибавление CNV между штаммами (Lilue et al., 2018), генные тела, псевдогенные транскрипты, участки связывания CTCF, домены репликации (Marchal et al., 2018), транспозоны ДНК, спутниковую ДНК и подмножество модификаций гистонов, включая H3K4me1, H3K27me3 и H3K36me3 (Mu et al., 2017). Интересно, что участки связывания Dmrt6, участвующие в регуляции перехода от митотического к мейотическому делению в мужских половых клетках (Zhang et al., 2014), тесно связаны с активностью мейотического кроссинговера. Геномные признаки, которые особенно отрицательно коррелируют с мейотическими кроссинговерами, включают 3'TR, LINE и области низкой сложности ДНК. В отличие от дрожжей, где рДНК чрезвычайно неактивна для мейотических кроссинговеров (Petes and Botstein, 1977), рДНК мыши, по-видимому, не подавляет кроссинговеры. С помощью этих геномных признаков мы можем отличить реальные места инициации мейотического кроссинговера от случайно выбранных путей в геноме мыши с точностью 0,73 и 0,85 по (B6×Spret) и (B6×Cast), соответственно, и точность прогноза 0, 85 в скрещивании (B6×Cast) поддерживается подмножеством из 25 геномных признаков. Мы подчеркиваем, что, хотя различные признаки ведут себя в значительной степени согласованно между подходами моделирования, мы не можем назначить причинно-следственную связь без дальнейших экспериментов.
Способы
Способы и молекулярный дизайн sci-L3-WGS и sci-L3-target-seq
Получение одиночных клеток и истощение нуклеосом
Клеточная суспензия получают путем трипсинизации из чашки Петри или гомогенизации из ткани. Самцов мышей F1 подвергали эвтаназии CO2 с последующим смещением шейных позвонков в соответствии с протоколами, утвержденными Вашингтонским университетом IACUC. Для выделения мужских половых клеток мы иссекали придаток яичка, разрезая канальцы внутри и инкубируя ткань в 1 мл 1×PBS с добавлением 10% FBS при комнатной температуре в течение 15 минут. После инкубации клеточную суспензию собирали с помощью пипетки. Клетки, выделенные из придатка яичка, применяли для экспериментов скрещивания (B6×Spret), а также в качестве источника зрелой спермы («группа штрихкода 3») в скрещивании (B6×Cast). Для выделения ядер из всего яичка в качестве способа обогащения для клеток 2C для скрещивания (B6×Cast) мы сначала перекрестно сшивали клетки яичка с 1% формальдегидом и извлекали ядра с использованием гипотонического буфера. Затем мы отсортировали путем FACS ядра 1C и 2C по содержанию ДНК, в основном, на основе сигнала DAPI. Культивированные клетки человека и мыши осаждают при 550g в течение 5 минут при 4°С, а мужские половые клетки осаждают при 2400 g в течение 10 минут при 4°С.
Истощение нуклеосом, в основном, следует за способами xSDS в sci-DNA-seq (Vitak et al., 2017), за исключением того, что буфер для лизиса модифицирован, чтобы быть совместимым с последующим протоколом LIANTI (Chen et al., 2017). Клетки перекрестно сшивают в 10 мл полной среды DMEM с 406 мкл 37% формальдегида (конечная концентрация 1,5%) при комнатной температуре в течение 10 минут (осторожно переворачивая пробирки). Затем добавляют 800 мкл 2,5 М глицина и инкубируют на льду в течение 5 минут. Клетки осаждают и промывают 1 мл лизирующего буфера (60 мМ Трис-Ас, рН 8,3, 2 мМ ЭДТА, рН 8,0, 15 мМ DTT). осадок ресуспендируют в 1 мл лизирующего буфера с 0,1% IGEPAL (I8896, SIGMA) и инкубируют на льду в течение 20 минут. Затем ядра осаждают, промывают 1×NEBuffer2.1 и ресуспендируют в 800 мкл 1×NEBuffer2.1 с 0,3% SDS для истощения нуклеосом при 42°C (энергичное встряхивание в течение 30 минут, 500 об./мин.). Затем мы добавляли 180 мкл 10% Triton-X и энергично встряхивали в течение 30 минут при 42°C (500 об./мин.). Пермеабилизированные ядра затем отмывают в 1 мл лизирующего буфера дважды и ресуспендируют в лизирующем буфере при 20000 ядер на мкл.
Дизайн и сборка транспозом
Транспозонную олигоДНК синтезируют с обеими цепями, фосфорилированными с 5' конца; одна из которых необходима для вставки Tn5 (5'/Phos/CTGTCTCTTATACACATCT, IDT, очистка PAGE (SEQ ID NO: 1)), аналогично LIANTI и Nextera, другая необходима для лигирования (5'/Phos/GTCTTG XXXXXXXX [штрихкод для раунда 1] AGATGTGTATAAGAGACAG, IDT, стандартное обессоливание (SEQ ID NO: 2)). После отжига 1:1 с постепенным охлаждением (95°C 5 мин, -0,1°C/цикл, 9 сек/цикл, 700 циклов до 25°C) в буфере для отжига (10 мМ Трис-HCl pH 8,0, 50 мМ NaCl, 1 мМ ЭДТА, рН 8,0), дуплекс Tn5 с 5’-липким концом разбавляют до 1,5 мкМ. Затем мы добавляли 7,2 мкл буфера для хранения (1×TE с 50% глицерина) к 12 мкл ~ 1 мкМ Tn5-транспозазы (Lucigen, TNP92110) и инкубируовали 0,79 мкл разбавленной транспозазы с 0,4 мкл 1,5 мкМ Tn5-дуплекса при комнатной температуре в течение 30 минут. Транспозома димеризуется до конечной концентрации 0,2 мкМ. Транспозомный комплекс может стабильно храниться при температуре -20°C до одного года. Мы настроили 24 реакции для штрихового кодирования 24 лунок в первом раунде, но в зависимости от применения может потребоваться больше лунок. Для каждого нового биологического применения мы дополнительно разбавляем транспозому до 0,1 мкМ для тестового эксперимента. Количество уникальных ридов и сложность библиотеки менее оптимальны (фиг. 5), но их можно использовать для картирования при низком разрешении.
На фиг. 7, мы показываем молекулярные структуры sci-L3-WGS на каждом этапе. В коммерческом получении библиотеки Nextera теряется, по меньшей мере, половина материала, пригодного для секвенирования ДНК, по причине того, что: 1) вставка Tn5 вводит симметричную последовательность транспозона на двух концах фрагментированной геномной ДНК, что может привести к образованию шпилечной петли при денатурировании и предотвратить амплификацию путем ПЦР; и 2) если два конца помечены как i5, так и i7 с вероятностью 50%, молекула не может быть секвенирована. Одно из ключевых преимуществ получения библиотеки LIANTI над Nextera заключается в том, что петлевой дизайн Tn5 нарушает симметрию, введенную транспозомным димером, и облегчает обратную транскрипцию (ОТ) с помощью внутримолекулярного праймера ОТ, также характерного для петлевого транспозона. Однако петлевой транспозон несовместим с более чем двумя раундами штрихкодирования, что ограничивает производительность и значительно увеличивает стоимость библиотеки (см. Таблицу 2 для сравнения). В изменениях, которые мы сделали для sci-L3-WGS, мы сохраняем преимущества, которые дает петлевой Tn5 на этапе лигирования.
Тагментация (первый раунд штрихкодирования) и лигирование (второй раунд штрихкодирования)
Затем мы распределяли 1,5 мкл ядер при концентрации 20000/мкл в каждую лунку в 96-луночном планшете с низким связыванием, добавляли 6,5 мкл H2O и 0,7 мкл 50 мМ MgCl2 (конечная концентрация 3,24 мМ с учетом ЭДТА в лизирующем буфере). Приготовленные выше 1,2 мкл транспозомы добавляют в каждую лунку, после чего планшет инкубируют при 55°С в течение 20 минут (термомиксер рекомендуется, но не является необходимым). Затем мы добавляли 5 мкл останавливающего раствора (40 мМ ЭДТА и 1 мМ спермидина) и совокупность ядер в лоток. Дополнительный 1 мл лизирующего буфера добавляют к суспензии ядер перед осаждением. После тщательного удаления супернатанта мы ресуспендировали ядра в 312 мкл буфера для ресуспендирования (24 мкл 10 мМ dNTP, 48 мкл 10× буфера для тагментации [50 мМ MgCl2, 100 мМ трис-HCl pH 8,0], 96 мкл H2O, 144 мкл лизирующего буфера), и распределяли 4,7 мкл смеси ядер в каждую лунку нового 96-луночного планшета с низким связыванием. Шпилечный дуплекс для лигирования (1. CAAGAC 2. Y'Y'Y'Y'Y'Y'Y'[последовательность, обратно комплементарная штрихкоду для раунда 2] 3. CAGGAGCGAGCTGCATCCC 4. AATTTAATACGACTCACTATA 5. GGGATGCAGCTCGCTCCTG 6. YYYYYYY [штрихкод для раунда 2] (SEQ ID NO: 3)) предварительно отжигали аналогично дуплексу транспозона Tn5 и разбавляли до 1,5 мкМ. Обратите внимание, что дуплекс для лигирования содержит пять элементов: 1) последовательность, обратно комплементарная лигирующему адаптеру на Tn5; 2) последовательность, обратно комплементарная штрихкоду для раунда 2; 3) последовательность, обратно комплементарная праймеру для синтеза второй цепи (SSS); 4) промотор Т7, обратите внимание, что это область петли шпильки; 5) область праймера синтеза второй цепи (SSS), начиная с GGG, для усиления транскрипции T7 («sp2» на фиг. 4B); 6) штрихкод для раунда 2 («bc2» на фиг. 4B). Мы добавляли 0,8 мкл этого дуплекса в каждую из 64 лунок с суспензией ядер и добавляли 1,18 мкл лигирующей смеси (0,6 мкл 10× буфера для лигазы Т4 NEB, 0,48 мкл PEG-4000, 0,1 мкл ДНК-лигазы T4 [Thermo EL0011]) в каждую лунку и инкубировали при 20°C в течение 30 минут. Обратите внимание, что после лигирования петлевая структура имитирует структуру LIANTI и повышает эффективность на этапе ОТ (обсуждается ниже), и что оба раунда штрихкодирования присутствуют на 3'-конце промотора T7 и, таким образом, будут включены в амплифицированную молекулу. Реакцию лигирования останавливали добавлением 4 мкл останавливающего раствора. Клетки затем объединяли в новый лоток (~ 630 мкл), окрашивали DAPI в конечной концентрации 5 мкг/мл и сортировали по 100-300 в каждую новую лунку с 3 мкл буфера для лизиса, добавленного до сортировки клеток. Обратите внимание, что каждое событие сортировки с FACS связано с ~3-5 нл буфера FACS в зависимости от размера носика, мы рекомендуем поддерживать общий объем жидкости, добавляемой в каждую лунку, <1 мкл, чтобы поддерживать низкую концентрацию соли.
Лизис клеток, удлинение делеций и линейная амплификация путем транскрипции in vitro
Затем мы продолжили с 3,5-4 мкл отсортированных ядер в каждой лунке для лизиса клеток путем инкубации при 75°С в течение 45 минут, охлаждения до 4°С и обработки свежеразведеннойпротеазой от Qiagen (конечная концентрация 2 мг/мл) при 55°С в течение 8 часов. Протеазу затем инактивировали нагреванием путем инкубации при 75°С в течение 30 минуь. Клеточный лизат может храниться при -80°С. Мы рекомендуем обрабатывать не более чем 32 лунки образцов (~9600 отдельных клеток) для каждого эксперимента, потому что последующий этап амплификации включает в себя РНК и чувствителен ко времени. Для удлинения делеций (фиг. 4C), используют полимеразу с активностью вытеснения цепей путем добавления смеси 2 мкл H2O, 0,7 мкл 10× буфера для тагментации, 0,35 мкл 10 мМ dNTP и 0,35 мкл Bst-полимеразы WarmStart 2.0 с вытеснением цепей, и инкубируют при 68°C в течение 5 минут. Обратите внимание, что если лигирование успешно на обоих концах, дуплекс симметричен с промотором T7 с обеих сторон, но если лигирование является успешным только на одном конце, область в пунктирной рамке отсутствует на одной стороне. Межмолекулярное лигирование, в основном, неэффективно. Хотя мы включили предварительно отожженную петлю шпильки, чтобы свести к минимуму необходимость межмолекулярного лигирования, двум молекулам (вместо трех без петли шпильки) все же нужно найти друг друга. Если эффективность лигирования составляет 50%, то лигирование на обоих концах имеет уровень 25%, но лигирование на обоих концах - 75%. Далее на этапе ОТ мы покажем, что успешное лигирование требуется только для одного конца. После удлинения делеций собирают транскрипционную систему T7 in vitro в объеме 20 мкл, добавляя 2 мкл H2O, 2 мкл смеси T7 Pol и 10 мкл смеси rNMP (NEB, набор для синтеза РНК HiScribe ™ T7 Quick High Yield). Смесь инкубируют при 37°С в течение 10-16 часов.
Очистка РНК, ОТ и SSS (или целевое секвенирование)
Транскрипцию прекращают добавлением 2,2 мкл 0,5М ЭДТА. Затем амплифицированную молекулу РНК очищают с помощью RCC-5 (Zymo Research, R1016) и элюируют 18 мкл 0,1× TE. Систему для ОТ объемом 30 мкл собирают путем первичного добавления 0,6 мкл РНК-праймера для ОТ (rArGrArUrGrUrGrUrArUrArArGrArGrArCrArG, IDT (SEQ ID NO: 4)), 2 мкл 10 мМ dNTP и 0,5 мкл ингибитора РНКазы SUPERase ⋅ In™ (20 Ед/мкл, Thermo Fisher AM2696). Затем инкубируют при температуре 70°С в течение 1 минуты и 90°С в течение 20 секунд для денатурирования и удаления вторичных структур и резко охлаждают на льду. Обратную транскриптазу SuperScript™ IV (SSIV, Thermo Fisher 18090050) используют для ОТ с буфером 6 мкл 5× RT, 1,5 мкл 0,1M DTT, 1 мкл SUPERase ⋅ In™ и 1 мкл SSIV. Реакцию ОТ инкубируют при 55°C в течение 15 минут, 60°C в течение 10 минут, 65°C в течение 12 минут, 70°C в течение 8 минут, 75°C в течение 5 минут и 80°C в течение 10 минут. Реакцию охлаждают до комнатной температуры перед добавлением 0,5 мкл РНКазы H (NEB) и 0,3 мкл РНКазы A (Life Technologies, AM2270) и инкубацией при 37°С в течение 30 минут. Обратите внимание, что Фиг. 4E изображает два сценария на этапе ОТ: 1) если оба конца имеют успешное лигирование, RT, вероятно, праймируется путем обратной петли, как в LIANTI; 2) если только один конец имеет успешное лигирование, RT, вероятно, праймируется РНК-праймером для ОТ, добавленным перед стадией денатурации. Избыточные праймеры РНК и транскрипты РНК разрушаются после синтеза кДНК. Наконец, синтезируют вторую цепь с помощью ДНК-полимеразы Q5, добавив 27 мкл H2O, 20 мкл 5× буфера Q5, 20 мкл энхансера Q5 GC, 1 мкл Q5 полимеразы и 1 мкл SSS-праймера (NNNN [UMI] ZZZZZZ [штрихкод для раунда 3] GGGATGCAGCTCGCTCCTG, IDT, стандартное обессоливание (SEQ ID NO: 5)). Полученную двухцепочечную ДНК можно очищать с помощью DCC-5 (Zymo Research, D4014) и приступать к работе набором для получения библиотек, таким как NEBNext Ultra II, с минимальными тремя циклами ПЦР для добавления адаптера секвенирования.
Стоит отметить, что шаг SSS может быть легко изменен для включения целевого секвенирования с помощью праймера для штрихкода одиночных клеток с концом P5 (AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGAC GCTCTTCCGATCT NNNNNNN ZZZZZZ [штрихкод для раунда 3] GGGATGCAGCTCGCTCCTG (SEQ ID NO: 6)) вместе с целевым праймером для одной области в геноме (рис. 3B). Например, в приложениях, где интегрируют библиотеку CRISPR на основе лентивируса (Shalem et al., 2014), направляющая последовательность РНК в каждой отдельной клетке может быть считана с использованием конца P7 с интегрированным в лентивирус праймером библиотеки CRISPR, CAAGCAGAAGACGGCATACGAGAT TCGCCTTG [индекс 1] GTGACTGGAGTTCAGACGTGTGCTCTT CCGATCTCCGACTCGGTGCCACTTTTTCAA (SEQ ID NO: 7)), таким образом, обходя необходимость в последовательности всего генома и обогащения для конкретной интересующей области. В этом случае, этап получения библиотеки и заменен очисткой на геле или гранулах для удаления димеров праймеров.
Способы и молекулярный дизайн совместного анализа sci-L3-RNA/DNA
Получение одиночных клеток и истощение нуклеосом
Клеточные суспензии получают по тому же протоколу, что и в sci-L3-WGS, кроме отличий, указанных ниже. Клетки HEK293T, BJ-5ta и 3T3 трипсинизировали из чашки Петри и фиксировали 2% PFA в 1× PBS при комнатной температуре в течение 10 минут при концентрации клеток 1 миллион/мл. Последующие этапы гашения (с помощью глицина), отмывки, выделения ядер (с 0,1% IGEPAL), истощения нуклеосом (способ xSDS) идентичны этапам sci-L3-WGS за исключением того, что мы добавляем 1% Superase-In ко всем лизирующим буферам и к 1× NEBuffer2.1. Ядра ресуспендируют в буфере для лизиса с 1% Superase-In при 20000 ядер на мкл.
Транспозома и конструирование праймеров для обратной транскрипции (ОТ)
Для компонента геномной амплификации одиночных клеток конструкция и сборка транспозомы идентичны sci-L3-WGS.
Для компонента профилирования транскриптома одиночных клеток праймеры для обратной транскрипции имеют сходную структуру с sci-РНК-seq у (Cao et al., 2017; Cusanovich et al., 2015; Mulqueen et al., 2018; Ramani et al., 2017; Vitak et al., 2017) для аспекта обратной транскрипции, т. е. праймирующую часть полиТ у олигонуклеотида, но с другой структурой штрихкода и посадочной площадкой для последующего этапа лигирования (/5Phos/GTCTTG [та же последовательность посадочной площадки, что и в sci-L3-WGS] NNNNNN [UMI1 для маркировки уникальных транскриптов] X'X'X'X'X'X'X'X'[штрихкоды для раунда 1 для транскриптома, которые отличаются от штрихкодов транспозона Tn5] TTTTTTTTTTTTTTTTTTTTTT TTTTTTTTVN , IDT, стандартное обессоливание (SEQ ID NO: 8)).
ОТ и тагментация (первый раунд штрихкодирования), лигирование (второйраунд штрихкодирования), FACS и лизис клеток
Затем мы распределяли 1,5 мкл ядер при концентрации 20000/мкл в каждую лунку в 96-луночном планшете с низким связыванием, добавляли 0,2 мкл H2O, 0,3 мкл 50 мМ MgCl2 (для нейтрализации ЭДТА в буфере для лизиса) 0,25 мкл 10 мМ dNTP и 1 мкл 25 мкМ ОТ-праймера, описанного выше, для подготовки к этапу ОТ. Затем смесь ядер инкубируют при 55°С в течение 5 минут для удаления вторичных структур и быстро охлаждают на льду. Затем добавляют1 мкл 5× буфера для ОТ, 0,03 мкл 100 мМ DTT (обратите внимание, что есть DTT из буфера для лизиса, конечная концентрация 5 мМ), 0,25 мкл SSIV, 0,25 мкл РНКазы OUT (Thermo Fisher, каталожный номер 10777019), инкубируют для реакции ОТ при 25°С 1 минуту, 37°С 1 минуту, 42°С 1 минуту, 50°С 1 минуту, 55°С 15 минут. Затем добавляют 0,4 мкл MgCl2 и 3,52 мкл H2O и 1,2 мкл транспозомы, приготовленной выше, в каждую лунку. Все последующие шаги до лизиса клеточной клетки идентичны sci-L3-WGS.
Удлинение делеций и линейная амплификация путем транскрипции in vitro
Для удлинения делеций мы используем случайный гептамер с частью праймера NEBNext Read 1 в качестве 5'-липкого конца (CACGACGCTCTTCCGATCT NNNNNNN (SEQ ID NO: 9)). Мы добавляем 1 мкл 20 мкМ олигонуклеотида, инкубируем при 95°С в течение 3 минуь, чтобы денатурировать ДНК, и постепенно охлаждаем до комнатной температуры (~ 5 минут) для отжига олигонуклеотида. Затем мы добавляем 2 мкл H2O, 0,8 мкл 10× NEBuffer2, 0,4 мкл 10 мМ dNTP, 0,4 мкл фрагмента Кленова (3'→ 5' экзо-, NEB M0212S) и инкубируем при 30°C в течение 8 минут и 75°С в течение 10 минут. После удлинения делеции транскрипционную систему T7 in vitro в объеме 20 мкл собирают по тому же протоколу sci-L3-WGS.
Очистка РНК, ОТ и SSS
Все этапы идентичны sci-L3-WGS, за исключением различных олигонуклеотидных последовательностей. На этапе ОТ после IVT вместо 0,6 мкл РНК ОТ-праймера мы используем 0,6 мкл праймера NEBNext Read 1 (AATGATACGGCGACCACCG AGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT, P5 конец секвенирования Illumina, IDT (SEQ ID NO: 10). Для праймера SSS мы используем AAGCAGAAGACGGCATACGAGAT [конец P7] NNNN [UMI2] Z’Z’Z’Z’Z’Z’ [штрихкод для раунда 3] CGTCTCTAC GGGATGCAGCTCGCTCCTG (SEQ ID NO: 11) для добавления адаптера для секвенирования. Обратите внимание, что полученная двухцепочечная ДНК теперь содержит оба конца P5 и P7 для секвенирования Illumina и может быть очищена с помощью 1,1× гранул AmpureXP и затем секвенирована. Этап получения библиотеки и минимальные 3 цикла ПЦР в sci-L3-WGS для добавления адаптера для секвенирования не подходят для совместного анализа.
Схема эксперимента sci-L3-WGS у скрещивания (B6×Spret) и скрещивания (B6×Cast)
Скрещивание(B6×Spret)
Мы объединили клетки, выделенные из 6 и 3 придатков яичек у самцов F1 (B6×Spret) в возрасте 70 суток и 88 суток, соответственно, в двух отдельных экспериментах и фиксировали их 1% формальдегидом. Для каждого эксперимента, после истощения нуклеосом, мы распределили 30000 клеток на лунку и выполнили инсерцию in situ индексированного Tn5 по 24 лункам, чтобы добавить штрихкоды первого раунда. Затем мы объединили все клетки и перераспределили их в 64 лунки, чтобы добавить штрихкоды второго раунда и промотор T7 путем лигирования. После повторного объединения всех клеток, мы разделили клеточную смесь 1:6, отсортировали путем FACS большинство клеток (6/7) и разбавили остальные (1/7). Полученные лунки содержали от 100 до 360 клеток на лунку с предполагаемой частотой столкновений 4-11%.
Скрещивание(B6×Cast)
Из 6 яичек было получено ~ 12 миллионов круглых сперматид 1С и ~ 0,5миллионов клеток 2С. Однако из-за более чем 20-кратного увеличения числа клеток 1C мы все же обнаружили много клеток 1C в популяции, отсортированной по клеткам 2C (фиг. 8F). В одном из экспериментов sci-L3-WGS, где мы пытались обогатить клетки 2C, мы оценили, что мы пометили ~ 160 тысяч сперматозоидов из придатка яичка, ~ 160 тысяч круглых сперматид и ~ 70 тысяч клеток 2C, и дополнительно обогатили клетки 2C во время этапа FACS sci-L3-WGS (фиг. 8G). Однако, несмотря на два раунда обогащения, клетки 1С все еще доминировали.
Таблица 4. Олигонуклеотиды для sci-L3.
Способы биоинформатики и статистические анализы
Обработка ридов, выравнивание и распознавание SNV
Распознавания оснований были преобразованы в файл fastq с помощью bcl2fastq с 1 несоответствием, допустимым для ошибок в индексе. Затем мы использовали настраиваемый сценарий оболочки «sci_lianti_v2.sh» для демультиплексирования (сценарии Python и файл R Markdown загружаются раздельно как «sci_lianti_inst.tar.gz»; пакет R, содержащий файлы данных промежуточного соединения для генерации всех основных и дополнительных фигур можно скачать и установить по следующей ссылке: https://drive.google.com/file/d/19NFubouHrahZ8WoblL-tcDrrTlIZEpJh/view?usp=sharing), которая вызывает сценарии python или инструменты NGS для следующих шагов: 1) упорядочить пары ридов таким образом, что все комбинаторные штрихкоды одиночных клеток находятся в риде 1 (R1); 2) демультиплексировать штрихкоды третьего раунда (SSS, 6nt, ошибка не допускается) и прикрепить штрихкоды и UMI для транскриптов к именам ридов, и разделить библиотеку по штрихкодам для раунда 3. Обратите внимание, что все последующие шаги выполняются параллельно для отдельных библиотек, разделенных штрихкодами для раунда 3, которые содержат 100-300 отдельных клеток; 3) использовать cutadapt для разделения штрихкодов 1-го (Tn5, 8nt, допускается 1 ошибка) и 2-го раундов (лигирование, 7nt, 1 ошибка допускается) штрихкодов в R1, ошибки рассчитываются по расстоянию Левенштейна, и прикрепить оба раунда штрихкодов к именам ридов. Этот шаг выполняется в режиме парных концов, то есть, если R1 не имеет правильного штрихкода и структуры спейсера, парный рид 2 (R2) отбрасывается; 4) использование cutadapt для очистки R2; 5) выравнивание в режиме парных концов для генома hg19 или mm10 с bwa mem (Li and Durbin, 2009). Для экспериментов, где мы оцениваем столкновение штрихкодов, мы используем соединенные референсные hg19 и mm10 и используем однозначно выровненные риды, чтобы определить относительный уровень картирования на геном человека или мыши; 6) разбить bam-файлы на bam-файлы отдельных клеток, используя штрихкоды 1-го и 2-го раундов, прикрепленных к имени рида; 7) преобразовать файл bam в файлы bed с помощью bedtools (Quinlan и Hall, 2010) и определить уникальные места вставки, если R1 или R2 имеют одинаковые конечные точки. Уникальный сайт вставки Tn5 определяется как фрагменты, где оба конца пары ридов должны быть разными; 8) использование функции «pileup» в пакете «lianti» (https://github.com/lh3/lianti/blob/master/pileup.c) (Chen et al., 2017) для распознавания вариантов в аллель-распознающем режиме. Обратите внимание, что мы включаем объединенный объемный файл bam (созданный слиянием samtools (Chen et al., 2017; Li and Durbin, 2009) из всех ~ 6900 отдельных клеток, более чем в 30×) с файлом bam каждой отдельной клетки на этом этапетаким образом, что порог глубины в каждом местоположении SNP необходимо превышать только в объединенном файле, чтобы распознавание SNP было включено в окончательный файл vcf, таким образом, необработанные подсчеты аллелей REF и ALT включаются в столбец отдельной клетки при условии, что вариант присутствует в виде гетерозиготного SNP в объединенном файле. Это обходит проблему высокого уровня ложных отрицательных результатов из-за низкой глубины секвенирования в отдельных клетках путем преобразования вопроса распознавания SNP de novo в вопрос о генотипировании; 9) аннотировать SNV, вызываемую в отношении качества SNP в каждой отдельной клетке, с помощью эталонного файла SNP vcf для Spret (SPRET_EiJ.mgp.v5.snps.dbSNP142.vcf.gz, загруженного из Mouse Genom Project). Аннотированный файл SNP затем используется в качестве входных данных для последующего анализа точек разрыва для кроссинговера.
HMM для распознавания точек разрыва
Генотип в сайте данного SNP определяют путем сравнения количества референсов, подтверждающих риды, и альтернативных аллелей. Для клеток 1С положение кроссинговера определяется путем подгонки скрытой марковской модели с тремя состояниями: референсное, альтернативное и гетерозиготное.
Матрица перехода указана в таблице 5.
Таблица 5. Матрица перехода.
Мы выбрали параметры вручную, основываясь на визуальной оценке того, насколько хорошо HMM отражает видимую структуру данных и что результаты не меняются заметно, когда мы меняем первичный параметр на два порядка. Для transprob требуется очень небольшое число [1×10-10/(общее количество SNP на данной хромосоме) в этом случае], чтобы отразить убеждение в том, что переход состояния в любом сайте отдельного SNP должен быть очень редким событием. Дальнейшая разбивка transprob на фракции 0,3 и 0,7 направлена на подавление быстрых последовательных переходов формы референсное-альтернативное-референсное или альтернативное-референсное-альтернативное.
Матрица эмиссии указана в таблице 6.
Таблица 6. Матрица эмиссии.
После того как скрытые состояния распознаются для каждого отдельного SNP, непрерывные блоки длинных состояний вызывают путем удаления блоков состояния короче, чем 50 КБ. Положение кроссинговера затем определяется тем, где длинный блок состояний переключается в другое состояние, где начальное положение участка точки разрыва является положением последнего SNP предыдущего блока состояния, а конечное положение участка является положением первого SNP следующего блока состояния.
Для клеток M2 среднюю частоту аллелей сначала получают путем усреднения по аллелям в пределах окна из 40 SNP. Затем сгруппированные частоты аллелей используют для вывода основных состояний хромосомы из скрытой марковской модели с единичными гауссовыми распределениями вероятностей.
Матрица перехода указана в таблице 7.
Таблица 7. Матрица перехода.
Матрица эмиссии указана в таблице 8.
Таблица 8. Матрица эмиссии.
Непрерывные блоки длинных состояний вызывают путем удаления блоков состояния короче, чем 50 КБ, а затем приблизительное положение точки разрыва определяется тем, где блоки длинных состояний переключаются в другое состояние. Затем приблизительное положение точки разрыва уточняют с помощью теста отношения правдоподобия, целью которого является нахождение вероятной точки разрыва в пределах 20 SNP выше и 20 SNP ниже около приблизительной точки разрыва. Для каждого SNP вероятность наблюдения наблюдаемого генотипа указана в таблице 9.
Таблица 9. Вероятность наблюдения наблюдаемого генотипа.
Error_prob указана как 1×10-3, котораяотражает вероятность того, что SNP определен неправильно. Для каждого SNP вокруг приблизительной точки разрыва вероятность того, что это фактическая точки разрыва, рассчитывают по приведенному выше распределению. Все SNP с вероятностью, превышающей 0,01×максимальную вероятность, считаются находящимися в диапазоне точки разрыва. Начало участка разрыва определяют как самый левый SNP в пределах этих SNP, а конец участка разрыва - самый правый SNP. Как и в случае 1C, все участки точек разрыва у клетки М2 дополнительно исследуют вручную для удаления артефактов, например, когда два непосредственно смежных переключателя присутствуют в пределах 50 КБ. Мы также выполнили такое же распознавание точек разрыва в митотически делящихся клетках Patski. Для клеток M2 и клеток Patski мы также вручную исследовали участки с точками разрыва, сравнивая размеры бинов 10 и 40 SNP для клеток с малым покрытием геномов.
Этот этап генерирует точки разрыва для кроссинговера. После обработки мы добавляем информацию о сегрегации хромосома на основе того, является ли центромерная область, т.е. начальная область каждой хромосомы, гетерозиготной («mt», митотическая сегрегация) или гомозиготной («me», мейотическая сегрегация).
Анализ однородительских хромосом
Этот этап берет файл rds из выходных данных HMM и генерирует рспознавание однородительских хромосом.
Анализы мейотического кроссинговера и сегрегации хромосом на хромосомном уровне
Этот этап генерирует характеристики хромосомного уровня мейотических кроссинговеров, показанных на рис. 10, 13 и 14.
Подгонка модели конечной смеси к клеткам 2C в группе штрихкодов 2 в скрещивании (B6×Cast)
Мы подгоняем данные к смеси трех биномиальных распределений, параметризованных p1, p2, p3, соответственно, обозначая вероятности хромосом сегрегировать эквационно. Относительный вклад этих трех биномиальных распределений обозначается длиной 3 вектора тета. Мы оцениваем p1, p2, p3 и также θ, вытягивая образцы из их апостериорных распределений, используя пакет R rstan (http://mc-stan.org/users/interfaces/rstan) с с однородным признаком Дирихле, предшествующим для θ:θ ~ Dir (K=3, α=1), и бета предшествует для p: p ~ Бета (a=5, b=5). Более подробно об описании модели, см. файл Stan mt_mi_model.stan.
Предварительная обработка наборов данных из других геномных исследований для построения линейных моделей активности кроссинговер жаркости и кластеризации клеток.
Мы обработали наборы данных из предыдущих геномных исследований и из загруженного файла аннотации для мыши в формате gff3 и RepeatMasker из UCSC Genom Browser (https: //геном.ucsc.edu/cgi-bin/hgTables) в отношении различных элементов генома. Наборы данных, основанные на mm9, сначала поднимали до mm10. Эти наборы данных условно делятся на две категории: подсчет данных в формате bed или сигнал о различных генетических или эпигенетических метках в формате bedGraph. Для кластеризации клеток и прогностического моделирования участки кроссинговера имеют разную длину. Мы нормализуем данные подсчета путем деления общего количества всех последовательностей, суммированных от всех кроссинговеров в каждой отдельной клетке, для кластерного анализа клеток, и мы нормализуем путем деления длины участка плюс 1 т.п.н. для каждого участка кроссинговера или случайно выбранных участков, так что чрезвычайно короткие участки не будут чрезмерно взвешенными. Обратите внимание, что средняя длина участка составляет 150 т.п.н., так что добавление 1 т.п.н. не включит много дополнительной последовательности. Для набора данных с непрерывным сигналом различных меток мы берем средний сигнал меток, которые пересекаются с кроссинговером или случайными участками. Набор данных кроссинговера мы не нормализовали для длины участка при использовании данных подсчета, так как мы использовали равноразмерные окна по100 т.п.н.
В дополнение к наборам данных, упомянутым в разделе «Обсуждение», где признаки имеют статистически значимую связь с появлением кроссинговера, мы также использовали следующие наборы данных: 1) расхождение последовательностей (Lilue et al., 2018); 2) ATAC-seq и H3K27ac, картированные из очищенных пахитенных сперматоцитов (Maezawa et al., 2018); 3) бисульфитное секвенирование из сперматогониев (Inoue et al., 2017); 4) позиционирование нуклеосом на основе MNase в сперматоцитах (Barral et al., 2017); 5) бутирилирование и ацетилирование H4K5 и H4K8 в сперматоцитах (Goudarzi et al., 2016); 6) убиквитинилирование H2A в сперматоцитах (Hasegawa et al., 2015); 7). участки связывания CTCFL, семенник-специфический паралог участков связывания CTCF (Sleutels et al., 2012); 8) карта 5-hmC в пахитенных сперматоцитах (Gan et al., 2013); 9) End-seq после лечения этопозидом и CTCF и RAD21 ChIP-seq в активированных B-клетках, TOP2A и TOP2B ChIP-seq в MEF (Canela et al., 2017); 10) аллельные данные ATAC-seq на клетках Patski (Bonora et al., 2018).
PCA для кластеризации клеток, BMA для линейных моделей активности кроссинговера и модель случайного леса для прогностических моделей участков кроссинговера и случайных участков
Метод главных компонент применяют для 2D-визуализации разделения клеток 1С и клеток на основе признаков их точек разрыва. Мы собрали информацию о кроссинговере для каждой отдельной клетки, в общей сложности 78 признаков, соответствующих трем типам. В качестве первого типа, мы просто рассчитали количество кроссинговеров или полнохромосомных событий LOH для каждой хромосомы в каждой клетке. Что касается второго типа, для функций, таких как содержание GC, расхождение последовательностей, интенсивность хроматиновых меток и т. д., мы рассчитали медианные значения для точек разрыва для кроссинговера в каждой клетке. В качестве третьего типа, мы вычислили нормализованные числа элементов генома, таких как генные тела, длинные концевые повторы (LTR), элементы LINE, которые перекрывались с точками разрыва для кроссинговера в каждой клетке.
Усреднение байесовской модели с использованием пакета «bas» (Clyde et al., 2011) применяют для построения линейных моделей, прогнозирующих активность кроссинговера (функция bas.lm, выборка 214 моделей с настройками по умолчанию), а переменные, важные для прогнозирования активности, определяются на основе их предельных вероятностей включения. Случайные леса обучаются отличать истинные участки кроссинговера от участков, случайно выбранных из генома, напоминающего «нулевое» распределение. Точность модели определяется полной вложенной 5-кратной перекрестной проверкой с 5 внешними сгибами и 5 сгибами в каждом обучающем наборе (см. Раздел «Модели» в sci-L3-WGS-figure.Rmd для кода R и аннотаций).
Чтобы оценить влияние линии (или типа клеток) на расположение самых правых кроссинговеров вдоль хромосомы, мы используем линейную модель смешанного эффекта с фиксированным эффектом для линии (или типа клеток) и случайным перехватом для хромосомы для учета изменчивости между хромосомами (см. раздел «Графики кариотипа» в sci-L3-WGS-figure.Rmd для R кода и аннотации).
Ссылки
Barral, S., Morozumi, Y., Tanaka, H., Montellier, E., Govin, J., de Dieuleveult, M., Charbonnier, G., Couté, Y., Puthier, D., Buchou, T., et al. (2017). Histone Variant H2A.L.2 Guides Transition Protein-Dependent Protamine Assembly in Male Germ Cells. Mol. Cell 66, 89-101.e8.
Bonora, G., Deng, X., Fang, H., Ramani, V., Qiu, R., Berletch, J.B., Filippova, G.N., Duan, Z., Shendure, J., Noble, W.S., et al. (2018). Orientation-dependent Dxz4 contacts shape the 3D structure of the inactive X chromosome. Nat. Commun. 9, 1445.
Canela, A., Maman, Y., Jung, S., Wong, N., Callen, E., Day, A., Kieffer-Kwon, K.-R., Pekowska, A., Zhang, H., Rao, S.S.P., et al. (2017). Genome Organization Drives Chromosome Fragility. Cell 170, 507-521.e18.
Cao, J., Packer, J.S., Ramani, V., Cusanovich, D.A., Huynh, C., Daza, R., Qiu, X., Lee, C., Furlan, S.N., Steemers, F.J., et al. (2017). Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661-667.
Chen, C., Xing, D., Tan, L., Li, H., Zhou, G., Huang, L., and Xie, X.S. (2017). Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI). Science 356, 189-194.
Clyde, M.A., Ghosh, J., and Littman, M.L. (2011). Bayesian Adaptive Sampling for Variable Selection and Model Averaging. J. Comput. Graph. Stat. 20, 80-101.
Cusanovich, D.A., Daza, R., Adey, A., Pliner, H.A., Christiansen, L., Gunderson, K.L., Steemers, F.J., Trapnell, C., and Shendure, J. (2015). Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910-914.
Dayani, Y., Simchen, G., and Lichten, M. (2011). Meiotic recombination intermediates are resolved with minimal crossover formation during return-to-growth, an analogue of the mitotic cell cycle. PLoS Genet. 7, e1002083.
Froenicke, L., Anderson, L.K., Wienberg, J., and Ashley, T. (2002). Male mouse recombination maps for each autosome identified by chromosome painting. Am. J. Hum. Genet. 71, 1353-1368.
Gan, H., Wen, L., Liao, S., Lin, X., Ma, T., Liu, J., Song, C.-X., Wang, M., He, C., Han, C., et al. (2013). Dynamics of 5-hydroxymethylcytosine during mouse spermatogenesis. Nat. Commun. 4, 1995.
Goudarzi, A., Zhang, D., Huang, H., Barral, S., Kwon, O.K., Qi, S., Tang, Z., Buchou, T., Vitte, A.-L., He, T., et al. (2016). Dynamic Competing Histone H4 K5K8 Acetylation and Butyrylation Are Hallmarks of Highly Active Gene Promoters. Mol. Cell 62, 169-180.
Hasegawa, K., Sin, H.-S., Maezawa, S., Broering, T.J., Kartashov, A.V., Alavattam, K.G., Ichijima, Y., Zhang, F., Bacon, W.C., Greis, K.D., et al. (2015). SCML2 establishes the male germline epigenome through regulation of histone H2A ubiquitination. Dev. Cell 32, 574-588.
Inoue, K., Ichiyanagi, K., Fukuda, K., Glinka, M., and Sasaki, H. (2017). Switching of dominant retrotransposon silencing strategies from posttranscriptional to transcriptional mechanisms during male germ-cell development in mice. PLoS Genet. 13, e1006926.
Lange, J., Yamada, S., Tischfield, S.E., Pan, J., Kim, S., Zhu, X., Socci, N.D., Jasin, M., and Keeney, S. (2016). The Landscape of Mouse Meiotic Double-Strand Break Formation, Processing, and Repair. Cell 167, 695-708.e16.
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754-1760.
Lilue, J., Doran, A.G., Fiddes, I.T., Abrudan, M., Armstrong, J., Bennett, R., Chow, W., Collins, J., Czechanski, A., Danecek, P., et al. (2018). Multiple laboratory mouse reference genomes define strain specific haplotypes and novel functional loci.
Lu, S., Zong, C., Fan, W., Yang, M., Li, J., Chapman, A.R., Zhu, P., Hu, X., Xu, L., Yan, L., et al. (2012). Probing meiotic recombination and aneuploidy of single sperm cells by whole-genome sequencing. Science 338, 1627-1630.
Maezawa, S., Yukawa, M., Alavattam, K.G., Barski, A., and Namekawa, S.H. (2018). Dynamic reorganization of open chromatin underlies diverse transcriptomes during spermatogenesis. Nucleic Acids Res. 46, 593-608.
Marchal, C., Sasaki, T., Vera, D., Wilson, K., Sima, J., Rivera-Mulia, J.C., Trevilla-García, C., Nogues, C., Nafie, E., and Gilbert, D.M. (2018). Genome-wide analysis of replication timing by next-generation sequencing with E/L Repli-seq. Nat. Protoc. 13, 819-839.
Mu, W., Starmer, J., Shibata, Y., Yee, D., and Magnuson, T. (2017). EZH1 in germ cells safeguards the function of PRC2 during spermatogenesis. Dev. Biol. 424, 198-207.
Mulqueen, R.M., Pokholok, D., Norberg, S.J., Torkenczy, K.A., Fields, A.J., Sun, D., Sinnamon, J.R., Shendure, J., Trapnell, C., O’Roak, B.J., et al. (2018). Highly scalable generation of DNA methylation profiles in single cells. Nat. Biotechnol. 36, 428-431.
Ottolini, C.S., Newnham, L., Capalbo, A., Natesan, S.A., Joshi, H.A., Cimadomo, D., Griffin, D.K., Sage, K., Summers, M.C., Thornhill, A.R., et al. (2015). Genome-wide maps of recombination and chromosome segregation in human oocytes and embryos show selection for maternal recombination rates. Nat. Genet. 47, 727-735.
Petes, T.D. (2001). Meiotic recombination hot spots and cold spots. Nat. Rev. Genet. 2, 360-369.
Petes, T.D., and Botstein, D. (1977). Simple Mendelian inheritance of the reiterated ribosomal DNA of yeast. Proc. Natl. Acad. Sci. U. S. A. 74, 5091-5095.
Petes, T.D., and Merker, J.D. (2002). Context dependence of meiotic recombination hotspots in yeast: the relationship between recombination activity of a reporter construct and base composition. Genetics 162, 2049-2052.
Quinlan, A.R., and Hall, I.M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841-842.
Ramani, V., Deng, X., Qiu, R., Gunderson, K.L., Steemers, F.J., Disteche, C.M., Noble, W.S., Duan, Z., and Shendure, J. (2017). Massively multiplex single-cell Hi-C. Nat. Methods 14, 263-266.
Shalem, O., Sanjana, N.E., Hartenian, E., Shi, X., Scott, D.A., Mikkelson, T., Heckl, D., Ebert, B.L., Root, D.E., Doench, J.G., et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84-87.
Sleutels, F., Soochit, W., Bartkuhn, M., Heath, H., Dienstbach, S., Bergmaier, P., Franke, V., Rosa-Garrido, M., van de Nobelen, S., Caesar, L., et al. (2012). The male germ cell gene regulator CTCFL is functionally different from CTCF and binds CTCF-like consensus sites in a nucleosome composition-dependent manner. Epigenetics Chromatin 5, 8.
Smagulova, F., Brick, K., Pu, Y., Camerini-Otero, R.D., and Petukhova, G.V. (2016). The evolutionary turnover of recombination hot spots contributes to speciation in mice. Genes Dev. 30, 266-280.
Storlazzi, A., Tesse, S., Ruprich-Robert, G., Gargano, S., Pöggeler, S., Kleckner, N., and Zickler, D. (2008). Coupling meiotic chromosome axis integrity to recombination. Genes Dev. 22, 796-809.
Vitak, S.A., Torkenczy, K.A., Rosenkrantz, J.L., Fields, A.J., Christiansen, L., Wong, M.H., Carbone, L., Steemers, F.J., and Adey, A. (2017). Sequencing thousands of single-cell genomes with combinatorial indexing. Nat. Methods 14, 302-308.
Wang, J., Fan, H.C., Behr, B., and Quake, S.R. (2012). Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell 150, 402-412.
Zhang, T., Murphy, M.W., Gearhart, M.D., Bardwell, V.J., and Zarkower, D. (2014). The mammalian Doublesex homolog DMRT6 coordinates the transition between mitotic and meiotic developmental programs during spermatogenesis. Development 141, 3662-3671.
Полное раскрытие всех патентов, патентных заявок, публикаций, и материалов, доступных в электронном виде (включая, например, подачу документов с нуклеотидной последовательностью, например, в GenBank и RefSeq, и подачу документов с аминокислотной последовательностью, например, в SwissProt, PIR, PRF, PDB, и трансляции из аннотированных кодирующих областей в GenBank и RefSeq), цитируемые в настоящем документе, в полном объеме включены в качестве ссылки. Дополнительные материалы, на которые есть ссылки в публикациях (такие как дополнительные таблицы, дополнительные фигуры, дополнительные материалы и способы, и/или дополнительные экспериментальные данные), также включены в качестве ссылки в полном объеме. В случае если существует какое-либо несоответствие между описанием настоящей заявки и описанием/описаниями любого документа, включенного в настоящий документ в качестве ссылоки, описание настоящей заявки имеет преимущественную силу. Вышеуказанное подробное описание и примеры даны только для ясности понимания. Из этого не следует ненужных ограничений. Изобретение не ограничено точными показанными и описанными деталями, поскольку очевидные для специалиста в данной области изменения будут включены в изобретение, определенное формулой изобретения.
Если не указано иное, все числа, выражающие количество компонентов, молекулярные массы и т.д., используемые в описании и формуле изобретения, следует понимать как модифицированные во всех случаях при помощи термина «приблизительно». Таким образом, если не указано иное, числовые параметры, изложенные в описании и формуле изобретения, являются приблизительными значениями, которые могут варьироваться в зависимости от желаемых свойств, которые должны быть получены путем настоящего изобретения. В крайнем случае, а не в виде попытки ограничить доктрину эквивалентов объемом формулы изобретения, каждый числовой параметр следует, по меньшей мере, интерпретировать в свете числа сообщаемых значащих цифр с применением обычных способов округления.
Несмотря на то, что числовые диапазоны и параметры, определяющие широкий объем изобретения, являются приблизительными, числовые значения, указанные в конкретных примерах, сообщаются с максимально возможной точностью. Все числовые значения, однако, по своей сути содержат диапазон, обязательно вытекающий из стандартного отклонения, найденного в их соответствующих тестовых измерениях.
Все заголовки предназначены для удобства читателя и не должны использоваться для ограничения значения текста, следующего за заголовком, если это не указано конкретно.
--->
СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> ILLUMINA, INC.
UNIVERSITY OF WASHINGTON
<120> ВЫСОКОПРОИЗВОДИТЕЛЬНОЕ СЕКВЕНИРОВАНИЕ ОДИНОЧНОЙ КЛЕТКИ СО СНИЖЕННОЙ ОШИБКОЙ АМПЛИФИКАЦИИ
<130> IP-1695-PCT
<140> PCT/US2019/032966
<141> 2019-05-17
<150> 62/821,864
<151> 2019-03-21
<150> 62/673,023
<151> 2018-05-17
<160> 176
<170> PatentIn version 3.5
<210> 1
<211> 19
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 1
ctgtctctta tacacatct 19
<210> 2
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(14)
<223> a, c, t, или g
<400> 2
gtcttgnnnn nnnnagatgt gtataagaga cag 33
<210> 3
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(13)
<223> a, c, t, или g
<220>
<221> модифицированное основание
<222> (73)..(79)
<223> a, c, t, или g
<400> 3
caagacnnnn nnncaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgnnnnnnn 79
<210> 4
<211> 19
<212> RNA
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 4
agauguguau aagagacag 19
<210> 5
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (1)..(4)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (5)..(10)
<223> a, c, t, или g
<400> 5
nnnnnnnnnn gggatgcagc tcgctcctg 29
<210> 6
<211> 90
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (59)..(65)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (66)..(71)
<223> a, c, t, или g
<400> 6
aatgatacgg cgaccaccga gatctacact ctttccctac acgacgctct tccgatctnn 60
nnnnnnnnnn ngggatgcag ctcgctcctg 90
<210> 7
<211> 89
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 7
caagcagaag acggcatacg agattcgcct tggtgactgg agttcagacg tgtgctcttc 60
cgatctccga ctcggtgcca ctttttcaa 89
<210> 8
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (13)..(20)
<223> a, c, t, или g
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 8
gtcttgnnnn nnnnnnnnnn tttttttttt tttttttttt tttttttttt vn 52
<210> 9
<211> 26
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (20)..(26)
<223> a, c, t, g, неизвестное или другое
<400> 9
cacgacgctc ttccgatctn nnnnnn 26
<210> 10
<211> 58
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 10
aatgatacgg cgaccaccga gatctacact ctttccctac acgacgctct tccgatct 58
<210> 11
<211> 61
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (24)..(27)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (28)..(33)
<223> a, c, t, или g
<400> 11
aagcagaaga cggcatacga gatnnnnnnn nnncgtctct acgggatgca gctcgctcct 60
g 61
<210> 12
<211> 19
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 12
ctgtctctta tacacatct 19
<210> 13
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 13
gtcttgtgat attgagatgt gtataagaga cag 33
<210> 14
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 14
gtcttggatc ccgtagatgt gtataagaga cag 33
<210> 15
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 15
gtcttgctcg attaagatgt gtataagaga cag 33
<210> 16
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 16
gtcttgcatc aaggagatgt gtataagaga cag 33
<210> 17
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 17
gtcttgtcct tgtgagatgt gtataagaga cag 33
<210> 18
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 18
gtcttgggtc atatagatgt gtataagaga cag 33
<210> 19
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 19
gtcttgatcg cgttagatgt gtataagaga cag 33
<210> 20
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 20
gtcttgcatg ccccagatgt gtataagaga cag 33
<210> 21
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 21
gtcttggtta cgcgagatgt gtataagaga cag 33
<210> 22
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 22
gtcttgccgc gcttagatgt gtataagaga cag 33
<210> 23
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 23
gtcttgtctt agtgagatgt gtataagaga cag 33
<210> 24
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 24
gtcttgtcgg cctaagatgt gtataagaga cag 33
<210> 25
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 25
gtcttgcttt ctctagatgt gtataagaga cag 33
<210> 26
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 26
gtcttgtcgc gtttagatgt gtataagaga cag 33
<210> 27
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 27
gtcttggtca gtagagatgt gtataagaga cag 33
<210> 28
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 28
gtcttgccat ggaaagatgt gtataagaga cag 33
<210> 29
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 29
gtcttgatgc tgcgagatgt gtataagaga cag 33
<210> 30
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 30
gtcttggagt ctttagatgt gtataagaga cag 33
<210> 31
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 31
gtcttgtacg atatagatgt gtataagaga cag 33
<210> 32
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 32
gtcttgacca tttaagatgt gtataagaga cag 33
<210> 33
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 33
gtcttgatcg ggacagatgt gtataagaga cag 33
<210> 34
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 34
gtcttggacg tcggagatgt gtataagaga cag 33
<210> 35
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 35
gtcttgcatt gtgtagatgt gtataagaga cag 33
<210> 36
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 36
gtcttgtttg actcagatgt gtataagaga cag 33
<210> 37
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 37
caagacaggt ggccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tggccacct 79
<210> 38
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 38
caagactaat agccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tggctatta 79
<210> 39
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 39
caagaccaac atacaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgtatgttg 79
<210> 40
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 40
caagaccggt taacaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgttaaccg 79
<210> 41
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 41
caagactgta ccccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tggggtaca 79
<210> 42
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 42
caagacaata gaacaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgttctatt 79
<210> 43
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 43
caagacatca agccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tggcttgat 79
<210> 44
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 44
caagacactt ggacaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgtccaagt 79
<210> 45
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 45
caagactagt tctcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgagaacta 79
<210> 46
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 46
caagacaaac cgacaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgtcggttt 79
<210> 47
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 47
caagacagtc tctcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgagagact 79
<210> 48
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 48
caagacttaa cagcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgctgttaa 79
<210> 49
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 49
caagacacta cctcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgaggtagt 79
<210> 50
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 50
caagacccaa gcccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgggcttgg 79
<210> 51
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 51
caagacaaca gtgcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgcactgtt 79
<210> 52
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 52
caagacacga cgtcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgacgtcgt 79
<210> 53
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 53
caagacttaa gcacaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgtgcttaa 79
<210> 54
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 54
caagacctat ggacaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgtccatag 79
<210> 55
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 55
caagacgcgg caccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tggtgccgc 79
<210> 56
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 56
caagacgacc tgccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tggcaggtc 79
<210> 57
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 57
caagaccggt gcacaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgtgcaccg 79
<210> 58
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 58
caagacagtc tctcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgagagact 79
<210> 59
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 59
caagaccttt tatcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgataaaag 79
<210> 60
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 60
caagactggg acccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgggtccca 79
<210> 61
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 61
caagacgtgc gaccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tggtcgcac 79
<210> 62
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 62
caagaccctt taccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tggtaaagg 79
<210> 63
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 63
caagaccaag tcgcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgcgacttg 79
<210> 64
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 64
caagactaag cggcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgccgctta 79
<210> 65
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 65
caagactgac catcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgatggtca 79
<210> 66
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 66
caagactgga tggcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgccatcca 79
<210> 67
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 67
caagacctcg ccccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tggggcgag 79
<210> 68
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 68
caagaccatg cagcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgctgcatg 79
<210> 69
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 69
caagacctgt aggcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgcctacag 79
<210> 70
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 70
caagacacct ctgcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgcagaggt 79
<210> 71
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 71
caagaccgtt ttgcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgcaaaacg 79
<210> 72
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 72
caagacgaag gtccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tggaccttc 79
<210> 73
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 73
caagacggct actcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgagtagcc 79
<210> 74
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 74
caagacccgg ctacaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgtagccgg 79
<210> 75
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 75
caagactaga ctacaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgtagtcta 79
<210> 76
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 76
caagacaaat taccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tggtaattt 79
<210> 77
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 77
caagactact cgacaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgtcgagta 79
<210> 78
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 78
caagactcct acccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgggtagga 79
<210> 79
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 79
caagaccccc gtccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tggacgggg 79
<210> 80
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 80
caagacgata cgacaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgtcgtatc 79
<210> 81
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 81
caagacgctg tgacaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgtcacagc 79
<210> 82
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 82
caagactata ggccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tggcctata 79
<210> 83
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 83
caagaccgac gcacaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgtgcgtcg 79
<210> 84
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 84
caagactcca tttcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgaaatgga 79
<210> 85
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 85
caagacaaga ccgcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgcggtctt 79
<210> 86
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 86
caagactaag taacaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgttactta 79
<210> 87
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 87
caagacctac tgccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tggcagtag 79
<210> 88
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 88
caagactctt atacaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgtataaga 79
<210> 89
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 89
caagacaacc caccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tggtgggtt 79
<210> 90
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 90
caagactacg gatcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgatccgta 79
<210> 91
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 91
caagacaatt ccacaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgtggaatt 79
<210> 92
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 92
caagacgtct ccgcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgcggagac 79
<210> 93
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 93
caagacatgc agtcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgactgcat 79
<210> 94
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 94
caagacgagc ttgcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgcaagctc 79
<210> 95
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 95
caagacgaga aaccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tggtttctc 79
<210> 96
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 96
caagactttg gcccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgggccaaa 79
<210> 97
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 97
caagactgcg agtcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgactcgca 79
<210> 98
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 98
caagactgca tcacaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgtgatgca 79
<210> 99
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 99
caagacggga tatcaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tgatatccc 79
<210> 100
<211> 79
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 100
caagactcgc ctccaggagc gagctgcatc ccaatttaat acgactcact atagggatgc 60
agctcgctcc tggaggcga 79
<210> 101
<211> 19
<212> РНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<400> 101
agauguguau aagagacag 19
<210> 102
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (1)..(4)
<223> a, c, t, g, неизвестное или другое
<400> 102
nnnnacgcga gggatgcagc tcgctcctg 29
<210> 103
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (1)..(4)
<223> a, c, t, g, неизвестное или другое
<400> 103
nnnncgcttg gggatgcagc tcgctcctg 29
<210> 104
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (1)..(4)
<223> a, c, t, g, неизвестное или другое
<400> 104
nnnngtccta gggatgcagc tcgctcctg 29
<210> 105
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (1)..(4)
<223> a, c, t, g, неизвестное или другое
<400> 105
nnnnaggatg gggatgcagc tcgctcctg 29
<210> 106
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (1)..(4)
<223> a, c, t, g, неизвестное или другое
<400> 106
nnnnttctcc gggatgcagc tcgctcctg 29
<210> 107
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (1)..(4)
<223> a, c, t, g, неизвестное или другое
<400> 107
nnnnaccact gggatgcagc tcgctcctg 29
<210> 108
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (1)..(4)
<223> a, c, t, g, неизвестное или другое
<400> 108
nnnntttcgc gggatgcagc tcgctcctg 29
<210> 109
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (1)..(4)
<223> a, c, t, g, неизвестное или другое
<400> 109
nnnncggtgg gggatgcagc tcgctcctg 29
<210> 110
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (1)..(4)
<223> a, c, t, g, неизвестное или другое
<400> 110
nnnntattct gggatgcagc tcgctcctg 29
<210> 111
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (1)..(4)
<223> a, c, t, g, неизвестное или другое
<400> 111
nnnnacttaa gggatgcagc tcgctcctg 29
<210> 112
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (1)..(4)
<223> a, c, t, g, неизвестное или другое
<400> 112
nnnntaaaga gggatgcagc tcgctcctg 29
<210> 113
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (1)..(4)
<223> a, c, t, g, неизвестное или другое
<400> 113
nnnngagttt gggatgcagc tcgctcctg 29
<210> 114
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (1)..(4)
<223> a, c, t, g, неизвестное или другое
<400> 114
nnnngggtgc gggatgcagc tcgctcctg 29
<210> 115
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (1)..(4)
<223> a, c, t, g, неизвестное или другое
<400> 115
nnnngggccg gggatgcagc tcgctcctg 29
<210> 116
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (1)..(4)
<223> a, c, t, g, неизвестное или другое
<400> 116
nnnnaattga gggatgcagc tcgctcctg 29
<210> 117
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (1)..(4)
<223> a, c, t, g, неизвестное или другое
<400> 117
nnnntaagcg gggatgcagc tcgctcctg 29
<210> 118
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (1)..(4)
<223> a, c, t, g, неизвестное или другое
<400> 118
nnnntaatgc gggatgcagc tcgctcctg 29
<210> 119
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (1)..(4)
<223> a, c, t, g, неизвестное или другое
<400> 119
nnnngtctat gggatgcagc tcgctcctg 29
<210> 120
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 120
gtcttgnnnn nnacccgaca tttttttttt tttttttttt tttttttttt vn 52
<210> 121
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 121
gtcttgnnnn nnaggctctc tttttttttt tttttttttt tttttttttt vn 52
<210> 122
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 122
gtcttgnnnn nntctaaact tttttttttt tttttttttt tttttttttt vn 52
<210> 123
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 123
gtcttgnnnn nntaccctcg tttttttttt tttttttttt tttttttttt vn 52
<210> 124
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 124
gtcttgnnnn nnctggtcat tttttttttt tttttttttt tttttttttt vn 52
<210> 125
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 125
gtcttgnnnn nnttataagc tttttttttt tttttttttt tttttttttt vn 52
<210> 126
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 126
gtcttgnnnn nnaatgtaga tttttttttt tttttttttt tttttttttt vn 52
<210> 127
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 127
gtcttgnnnn nncgcagacc tttttttttt tttttttttt tttttttttt vn 52
<210> 128
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 128
gtcttgnnnn nncgaatcaa tttttttttt tttttttttt tttttttttt vn 52
<210> 129
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 129
gtcttgnnnn nnccggaaag tttttttttt tttttttttt tttttttttt vn 52
<210> 130
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 130
gtcttgnnnn nngtttaaag tttttttttt tttttttttt tttttttttt vn 52
<210> 131
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 131
gtcttgnnnn nnaaagttga tttttttttt tttttttttt tttttttttt vn 52
<210> 132
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 132
gtcttgnnnn nncggaaact tttttttttt tttttttttt tttttttttt vn 52
<210> 133
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 133
gtcttgnnnn nntgagtacc tttttttttt tttttttttt tttttttttt vn 52
<210> 134
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 134
gtcttgnnnn nncgtagaat tttttttttt tttttttttt tttttttttt vn 52
<210> 135
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 135
gtcttgnnnn nncgacaccc tttttttttt tttttttttt tttttttttt vn 52
<210> 136
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 136
gtcttgnnnn nngtactgaa tttttttttt tttttttttt tttttttttt vn 52
<210> 137
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 137
gtcttgnnnn nncggaaaga tttttttttt tttttttttt tttttttttt vn 52
<210> 138
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 138
gtcttgnnnn nnatatcaat tttttttttt tttttttttt tttttttttt vn 52
<210> 139
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 139
gtcttgnnnn nntacccggc tttttttttt tttttttttt tttttttttt vn 52
<210> 140
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 140
gtcttgnnnn nngccatccc tttttttttt tttttttttt tttttttttt vn 52
<210> 141
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 141
gtcttgnnnn nnaccaacgc tttttttttt tttttttttt tttttttttt vn 52
<210> 142
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 142
gtcttgnnnn nntgcaagct tttttttttt tttttttttt tttttttttt vn 52
<210> 143
<211> 52
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (7)..(12)
<223> a, c, t, g, неизвестное или другое
<220>
<221> модифицированное основание
<222> (52)..(52)
<223> a, c, t, g, неизвестное или другое
<400> 143
gtcttgnnnn nngcaaccgg tttttttttt tttttttttt tttttttttt vn 52
<210> 144
<211> 26
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (20)..(26)
<223> a, c, t, g, неизвестное или другое
<400> 144
cacgacgctc ttccgatctn nnnnnn 26
<210> 145
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 145
caagcagaag acggcatacg agatnnnnga tccgcgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 146
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 146
caagcagaag acggcatacg agatnnnngg gtatcgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 147
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 147
caagcagaag acggcatacg agatnnnnca tggacgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 148
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 148
caagcagaag acggcatacg agatnnnntt gaagcgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 149
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 149
caagcagaag acggcatacg agatnnnnct gggtcgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 150
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 150
caagcagaag acggcatacg agatnnnnca ctaccgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 151
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 151
caagcagaag acggcatacg agatnnnnct tatacgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 152
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 152
caagcagaag acggcatacg agatnnnngt tggacgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 153
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 153
caagcagaag acggcatacg agatnnnnag cggtcgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 154
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 154
caagcagaag acggcatacg agatnnnncc gttccgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 155
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 155
caagcagaag acggcatacg agatnnnnac gttacgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 156
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 156
caagcagaag acggcatacg agatnnnnaa catacgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 157
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 157
caagcagaag acggcatacg agatnnnngc agaccgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 158
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 158
caagcagaag acggcatacg agatnnnnat tcgtcgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 159
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 159
caagcagaag acggcatacg agatnnnntg gggtcgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 160
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 160
caagcagaag acggcatacg agatnnnnct tccccgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 161
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 161
caagcagaag acggcatacg agatnnnntc cgtgcgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 162
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 162
caagcagaag acggcatacg agatnnnntt tgtacgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 163
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 163
caagcagaag acggcatacg agatnnnnga gatgcgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 164
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 164
caagcagaag acggcatacg agatnnnngg accacgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 165
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 165
caagcagaag acggcatacg agatnnnnta tgttcgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 166
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 166
caagcagaag acggcatacg agatnnnncg acgccgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 167
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 167
caagcagaag acggcatacg agatnnnngc tattcgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 168
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 168
caagcagaag acggcatacg agatnnnncg gctgcgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 169
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 169
caagcagaag acggcatacg agatnnnnca tctgcgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 170
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 170
caagcagaag acggcatacg agatnnnnaa gttccgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 171
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 171
caagcagaag acggcatacg agatnnnntt gttacgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 172
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 172
caagcagaag acggcatacg agatnnnnca ggcacgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 173
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 173
caagcagaag acggcatacg agatnnnngg tgagcgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 174
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 174
caagcagaag acggcatacg agatnnnnca aaagcgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 175
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 175
caagcagaag acggcatacg agatnnnnac tcctcgtctc tacgggatgc agctcgctcc 60
tg 62
<210> 176
<211> 62
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
олигонуклеотид
<220>
<221> модифицированное основание
<222> (25)..(28)
<223> a, c, t, g, неизвестное или другое
<400> 176
caagcagaag acggcatacg agatnnnntg cgggcgtctc tacgggatgc agctcgctcc 60
tg 62
<---
Реферат
Изобретение относится к биотехнологии. Описан способ получения библиотеки для секвенирования, содержащей нуклеиновые кислоты из множества отдельных ядер или клеток, включающий: получение множества выделенных ядер или клеток в первичных множествах компартментов, где каждый компартмент содержит субпопуляцию выделенных ядер или клеток, и где ядра или клетки содержат фрагменты нуклеиновой кислоты; введение медиатора линейной амплификации в клетки или ядра; амплификацию фрагментов нуклеиновой кислоты путем линейной амплификации; обработку каждой субпопуляции ядер или клеток для получения индексированных ядер или клеток, где обработка включает добавление к фрагментам нуклеиновых кислот, присутствующих в изолированных ядрах или клетках, первой компартмент-специфической индексной последовательности для получения индексированных нуклеиновых кислот, присутствующих в выделенных ядрах или клетках, где обработка включает лигирование, удлинение праймера, гибридизацию, амплификацию или транспозицию; объединение индексированных ядер или клеток для получения объединенных индексированных ядер или клеток, тем самым получая библиотеку для секвенирования из множества ядер или клеток. Изобретение расширяет арсенал методов секвенирования. 4 н. и 15 з.п. ф-лы, 9 табл., 2 пр., 23 ил.
Комментарии