Биоразлагаемые полукристаллические термопластичные мультиблочные сополимеры с разделенными фазами для контролируемого высвобождения биологически активных соединений - RU2662818C2
Код документа: RU2662818C2
Чертежи
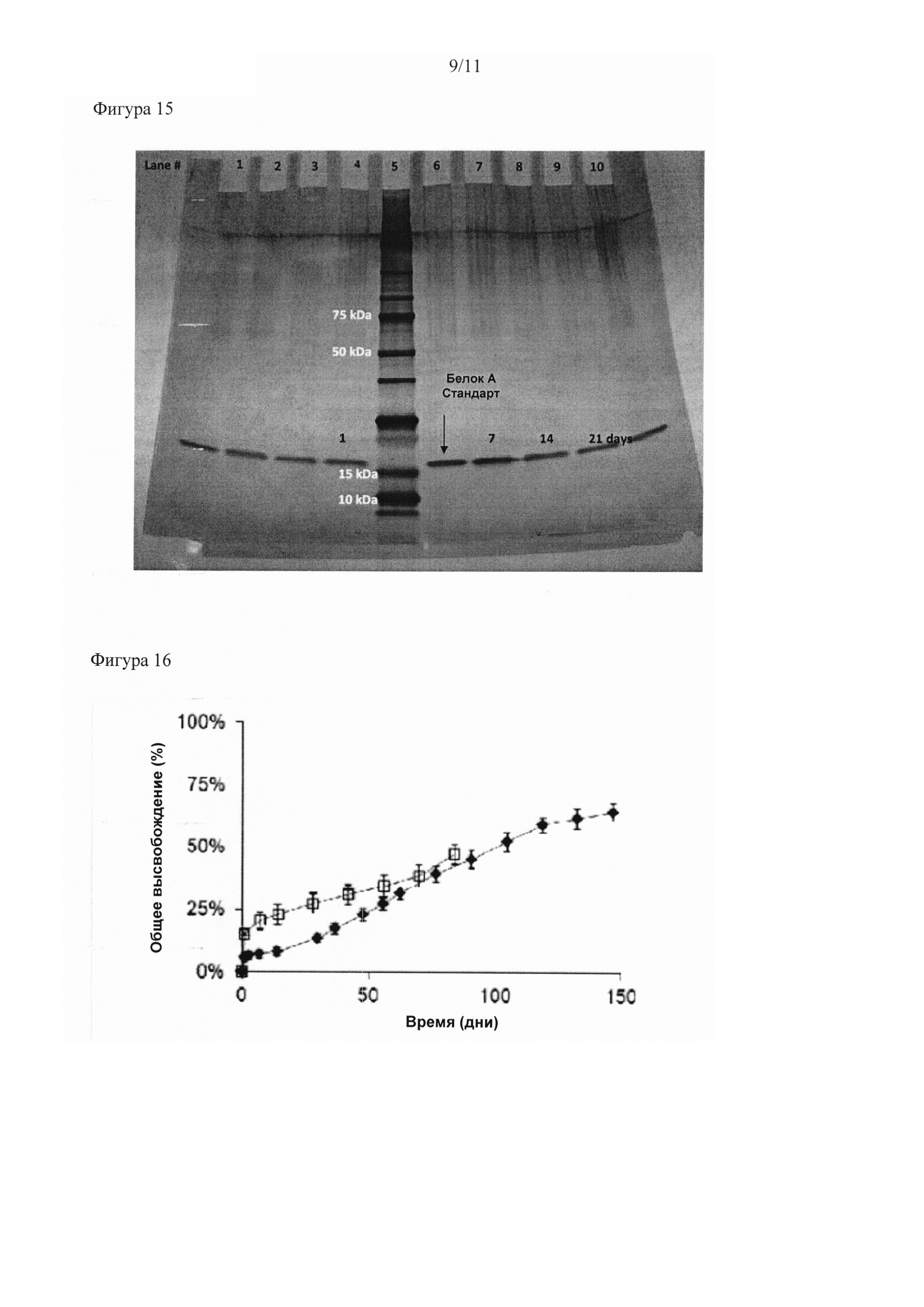
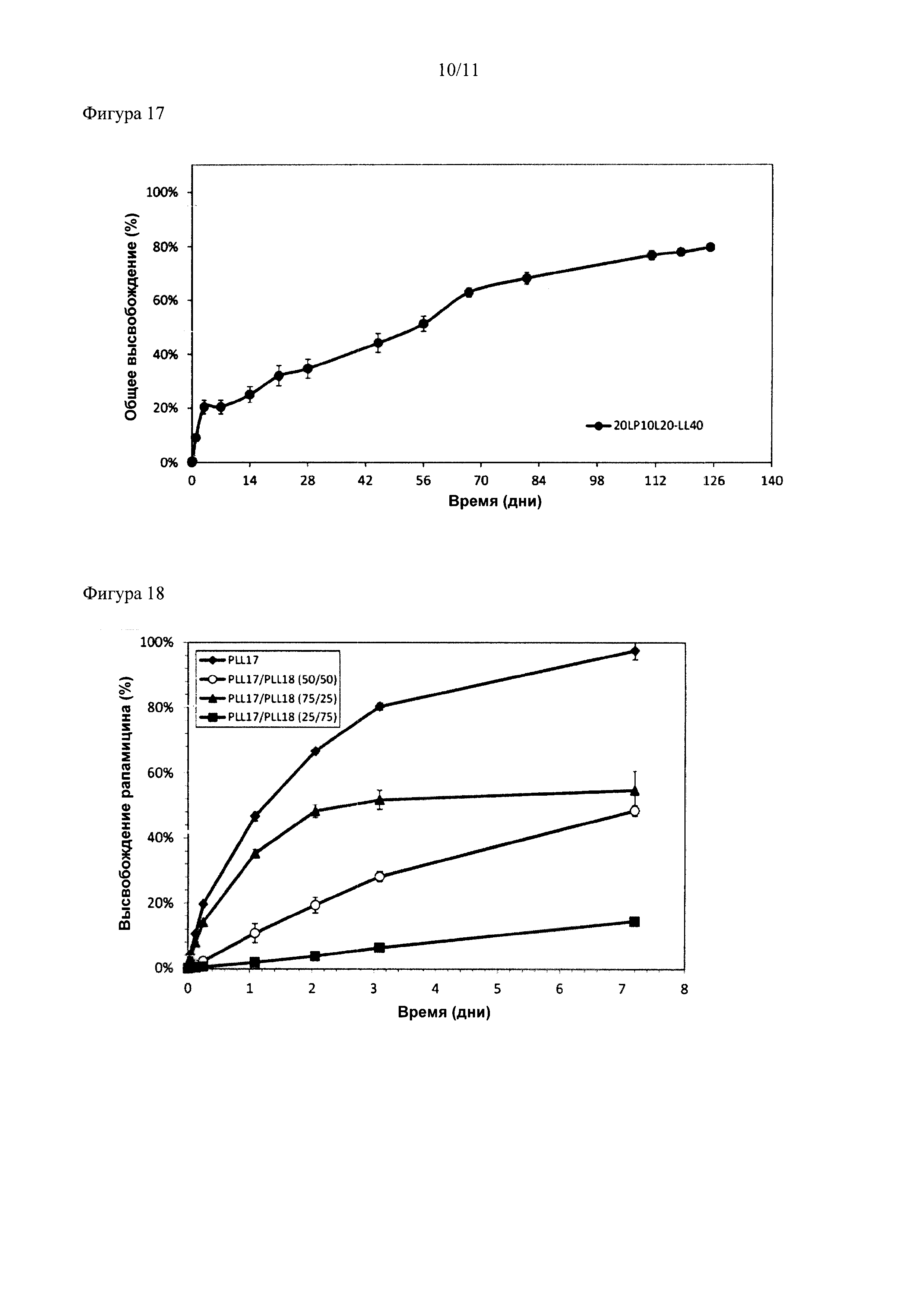
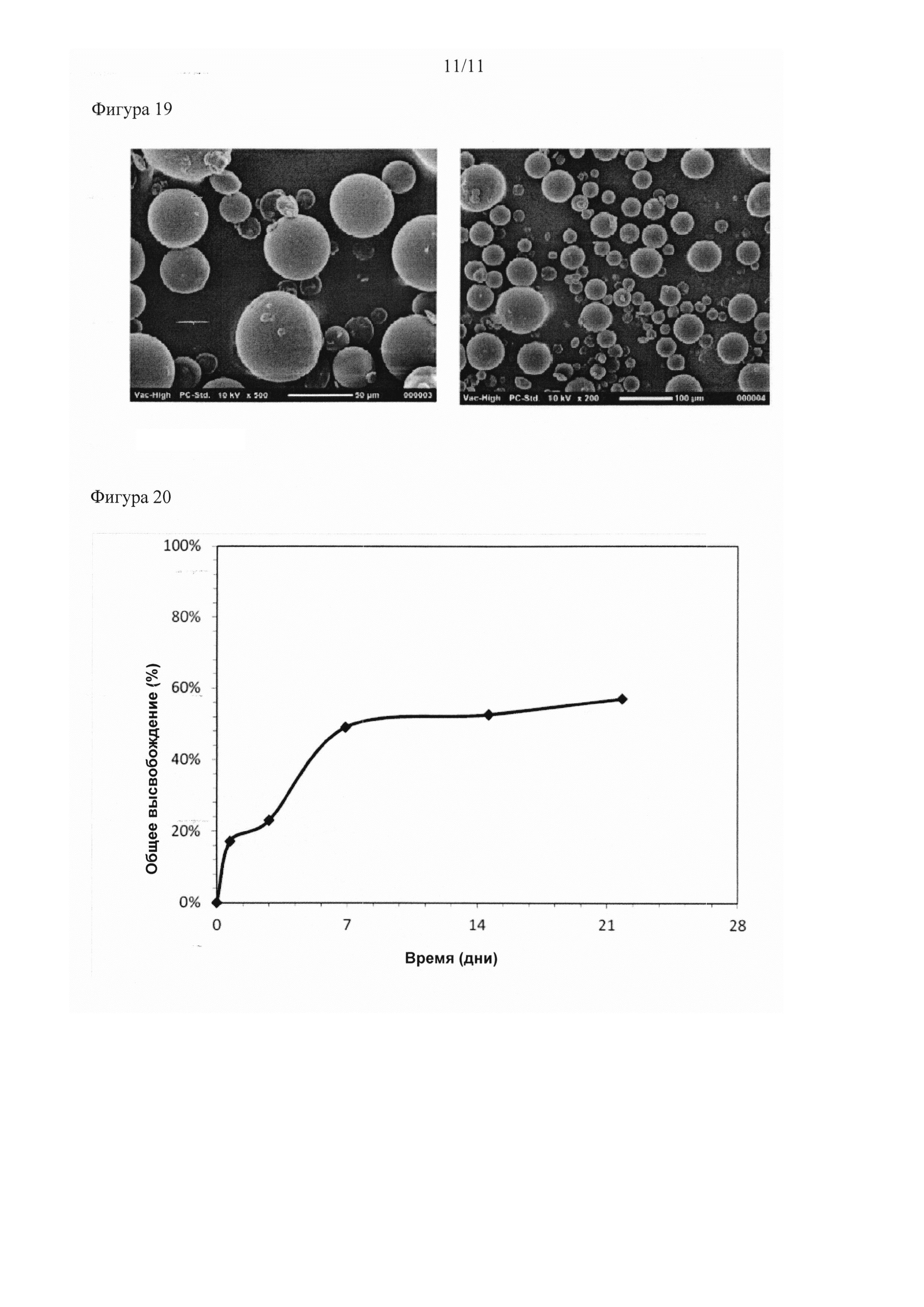
Описание
Изобретение относится к биоразлагаемым полукристаллическим термопластичным мультиблочным сополимерам с разделенными фазами, к способу получения указанных мультиблочных сополимеров, к композиции для доставки по меньшей мере одного биологически активного соединения и к способам доставки биологически активного соединения субъекту, нуждающемуся в этом.
Пептиды и белки, вместе называемые полипептидами, играют жизненно важную роль во всех биологических процессах и в последние годы привлекают к себе все больше внимания в качестве потенциальных лекарственных средств. Быстрое развитие фармакологии пептидов и белков наряду с крупномасштабным производством этих соединений при помощи технологии рекомбинантных ДНК, помимо прочих технологий, вызвало огромный интерес к указанным соединениям. К сожалению, разработка пептидов и белков намного опережает возможности системной или местной доставки указанных соединений с использованием удобных и эффективных систем доставки.
В последнее десятилетие повышенное внимание уделяется биоразлагаемым полимерам для применения в парентеральных системах длительного действия с контролируемым высвобождением для системной или точечной доставки лекарственных средств. Биоразлагаемые составы с контролируемым высвобождением могут значительно улучшать фармакокинетику лекарственных соединений. Это особенно важно при лечении хронических заболеваний, а также для соединений с узким «терапевтическим окном», поскольку в результате снижения системной концентрации в плазме снижаются и нежелательные побочные эффекты. Кроме того, множество новых биологически активных соединений имеют короткий период полувыведения, что делает необходимым частое введение инъекций для достижения терапевтически эффективного уровня соединения в плазме. Требования к соблюдению пациентами схемы лечения, а также большие затраты, связанные с необходимостью частого дозирования вводимых парентерально биологически активных соединений, повысили интерес к биоразлагаемым парентеральным лекарственным формам с контролируемым высвобождением.
Поли(D,L-молочная кислота) (PDLLA) и сополимеры молочной кислоты и гликолевой кислоты, также известные как сополимеры PLGA, являются наиболее часто используемыми биоразлагаемыми полимерами для применения в парентеральных составах в виде депо с контролируемым высвобождением. Сополимеры PLGA успешно применяют для разработки составов в виде депо с замедленным высвобождением для доставки небольших молекул, таких как рисперидон, и терапевтических пептидов, таких как лейпролид, госерелин или октреотид.
Полимеры PLGA, тем не менее, имеют несколько недостатков, которые ограничивают их применение и делают менее подходящими для доставки полипептидов. Во-первых, сополимеры PLGA представляют собой относительно гидрофобные полимеры и не обеспечивают оптимальные условия для инкапсулированных белков. Белки могут адсорбироваться на полимере, что приводит к замедлению и неполноте высвобождения, развертыванию и/или агрегации белков. Во-вторых, возможность воздействовать на высвобождение более крупных биологически активных соединений, таких как инкапсулированный полипептид, ограничена, так как диффузия указанных соединений в относительно жесткие и не поддающиеся разбуханию матрицы PLGA является незначительной. Высвобождение белков из сополимеров PLGA, таким образом, зависит от диффузии через поры, присутствующие в матрице, а также от времени разложения или растворения матрицы. Как правило, инкапсулированный белок остается внутри полимерной матрицы до того момента, когда матрица разлагается до такой степени, что теряет целостность или растворяется, это приводит к двухфазным или трехфазным профилям высвобождения, зависящим от разложения, как правило, получаемым для составов в виде депо на основе PLGA. Наконец, при разложении сополимеров PLGA образуются кислотные фрагменты, которые накапливаются в жесткой и не поддающейся набуханию матрице PLGA, что приводит к получению кислотной микросреды в полимерной матрице, где pH in situ может составлять не более 1-2. В указанных кислотных условиях инкапсулированные белки могут образовывать агрегаты, что приводит к неполному высвобождению белка. Кроме того, низкий pH может оказывать отрицательное действие на структурную целостность и биологическую активность инкапсулированного пептида или белка и может приводить к снижению терапевтической эффективности и повышению иммуногенности. Сообщалось о химической модификации белков и пептидов, такой как ацилирование и образование аддуктов.
Таким образом, существует необходимость в получении биоразлагаемых полимеров, которые лучше подходят для доставки белков. Тем не менее, одним из преимуществ PLGA и родственных полимеров является их успешное клиническое применение, а также то, что их считают биосовместимыми, и вследствие или по причине снижения риска они часто используются фармацевтическими компаниями для разработки составов в виде депо для доставки активных соединений. Таким образом, желательна разработка новых биоразлагаемых полимерных систем доставки белков из полимеров, состоящих из хорошо известных биологически безопасных и клинически приемлемых мономеров.
Для обеспечения гидрофильной матрицы с улучшенной совместимостью с белковыми лекарственными средствами, обеспечивающей их контролируемое высвобождение, Киссел с соавторами (Kissel et al, J. Contr. Rel. 1996, 39(2), 315-326) синтезировали трехблочные ABA сополимеры, содержащие гидрофильные В блоки поли(этиленоксида) и гидрофобные биоразлагаемые A блоки, состоящие из поли(L-молочной-гликолевой кислоты). Киссел с соавторами сообщали о замедленном высвобождении различных белковых соединений из микросфер, состоящих из сополимеров поли(L-молочной-гликолевой кислоты)-поли(этиленгликоля)-поли(L-молочной-гликолевой кислоты) и поли(L-молочной кислоты)-поли(этиленгликоля)-поли(L-молочной кислоты), т.е. полимеров типа ABA, где A представляет собой гидрофобный блок, а B представляет собой полиэтиленгликоль. Указанные сополимеры, тем не менее, имеют ограниченное соотношение A/B, т.е. содержание поли(этйленгликоля) (ПЭГ). Для предотвращения проблемы почечного клиренса, связанной с применением высокомолекулярного ПЭГ молекулярная масса ПЭГ-фрагмента, применяемого в указанных трехблочных ABA сополимерах, предпочтительно не должна превышать 5000 г/моль. Таким образом, для достижения высокого содержания ПЭГ в трехблочных полимерах, описанных Кисселом с соавторами, при сохранении низкой молекулярной массы ПЭГ гидрофобные блоки также должны быть короткими. Это приводит к получению полимеров со свойствами, нежелательными для биоматериалов, поскольку при низкой длине блоков температура стеклования (Тст) ниже комнатной температуры (что определили авторы изобретения), а кристалличность (в случае поли(L-молочной кислоты) (PLLA)) является очень низкой или отсутствует (De Jong, Macromolecules, 1998, 57(19), 6397-6402), что, таким образом, приводит к получению вязкого материала и (слишком) быстрому и плохо контролируемому высвобождению включенного активного вещества.
Примеры сегментированных/блок-сополимеров с разделенными фазами приведены, например, в патентах США №5554170 A, №5066772 A, №5236444 A, №5133739 A и №4429080 A. Эти известные материалы представляют собой биоресорбируемые сополимеры сложных эфиров, где жесткие блоки, главным образом, состоят из кристаллического полигликолида и/или полилактида. Указанные полимеры являются жесткими и не поддаются набуханию и, таким образом, страдают от тех же недостатков и ограничений, что и PLGA и PDLA, это делает их не подходящими для замедленного высвобождения белков.
Проводились исследования содержания лекарственных средств и способности высвобождения биоразлагаемых мультиблочных сополимеров, содержащих один гидролизуемый сегмент сложного полиэфира и один гидрофильный гидролитически стабильный сегмент (например, мультиблочные сополимеры на основе сегментов ε-капролактона и сегментов поли(этиленгликоля) описаны Ли с соавторами (Lee et al, J. Control. Rel, 2001, 75(2), 315-327)). Указанные полимеры содержат только один разлагаемый сегмент, что, таким образом, ограничивает возможность управления свойствами их разложения и высвобождения.
Известные мультиблочные сополимеры, состоящие из двух типов биоразлагаемых преполимеров (сегментов), с другой стороны, можно получать исключительно с чередующейся последовательностью преполимеров, что приводит к ограничению диапазона возможных переменных (Penco et al, J. Appl. Polym. Set 2000, 75(10), 1721-1728).
Примеры биоразлагаемых мультиблочных сополимеров, содержащих гидролизуемые сегменты сложного полиэфира с различным составом, описаны в международной публикации WO-A-2004/007588. Эти мультиблочные сополимеры содержат биоразлагаемые сополимеры с разделенными фазами, содержащие сегменты аморфного «мягкого» биоразлагаемого преполимера (А), характеризующегося Тст (температурой стеклования) ниже 37°C, и сегменты полукристаллического «жесткого» биоразлагаемого преполимера (B), характеризующегося температурой фазового перехода 40-100°C, в которых сегменты связаны посредством полифункционального удлинителя цепи. Для получения мультиблочных сополимеров с Тпл 40-100°C, описанных в публикации WO-A-2004/007588, выбор преполимеров, которые можно применять в качестве сегментов В, ограничен преполимерами, состоящими из поли(ε-капролактона) (ПКЛ) (WO-A-2004/007588), поли(валеролактона) (ПВЛ) и/или полидиоксанона (ПДС). В случае применения ПДС в качестве сегмента B получают мультиблочные сополимеры с Тпл 80-90°C (патент США №5711958 А). В случае использования ПКЛ в качестве сегмента B получают мультиблочные сополимеры с Тпл 40-60°C (WO-A-2004/007588). Гомополимеры ПВЛ имеют Тпл, схожую с гомополимерами ПКЛ (т.е. ~60°C). Таким образом, в случае использования ПВЛ в качестве сегмента B получают мультиблочные сополимеры с Тпл 40-60°C.ПДС, ПКЛ и ПВЛ имеют относительно низкие значения Тст, которые составляют -10, -60 и -60°C, соответственно. Низкие сегментов ПДС, ПКЛ и ПВЛ ограничивают диапазон Тст получаемого мультиблочного сополимера (где Тст определяется перемешиванием фаз аморфного сегмента A и аморфной части полукристаллического сегмента B), что, таким образом, ограничивает возможность управления свойствами высвобождения и разложения.
В международной публикации WO-A-99/02168 описаны биоразлагаемые мультиблочные сополимеры для биомедицинских применений, где преполимеры типа ABA или АВ получены с использованием удлинителей цепи. Удлинение цепей преполимеров типа ABA или АВ может приводить исключительно к получению чередующихся мультиблочных сополимеров. Чередующийся блоксополимер может быть представлен формулой ABABABABAB в случае удлинения цепи AB-преполимеров или ABAABAABAABA в случае удлинения цепи ABA-преполимеров.
Биоразлагаемые мультиблочные сополимеры с разделенными фазами, содержащие жесткий и мягкий сегмент, описаны в патенте США №6160084 А. В этом документе описано применение мультиблочных сополимеров ПКЛ-PLLA, состоящих из преполимеров, связанных посредством триметилгексан-1,6-диизоцианата (THDI). Указано, что эти материалы подходят для применения в системах доставки лекарственных средств, в которых требуется память формы. В заявке на патент США №2006/0140999 описано применение схожих полимеров с памятью формы для применения в системах доставки лекарственных средств, где материал с памятью формы содержит звенья, полученные из мономеров, выбранных из группы, состоящей из капролактона, лактида, гликолида и диоксанона. Примеры включают мультиблочные сополимеры ПДС-ПКЛ и ПДС-PLGA. Эти материалы не могут набухать в значительной степени в (имитируемых) физиологических условиях, поскольку набухание приведет к потере механических свойств, а, таким образом, к потере «запомненной» формы.
Другие сегментированные мультиблочные сополимеры с разделенными фазами включают сополимеры простых и сложных полиэфиров, такие как описаны в патенте США №5980948 A. Указанные сополимеры состоят их кристаллических ароматических сегментов и мягких сегментов, содержащих ПЭГ, связанных гидролизуемыми сложноэфирными связями. Сополимеры имеют характерный недостаток, заключающийся в том, что композиции с низкой набухаемостью, т.е. композиции с высоким содержанием гидрофобных ароматических сегментов плохо разлагаются вследствие высокой кристалличности и гидрофобности ароматических сегментов. Композиции с высокой набухаемостью, т.е. композиции с высоким содержанием ПЭГ, также плохо разлагаются, но уже из-за низкой концентрации сложноэфирных связей. В противоположность этому, мультиблочные сополимеры согласно настоящему изобретению разлагаются при любых соотношениях сегмент A/сегмент B вследствие присутствия сложноэфирных связей как в сегменте A, так и в сегменте B. Кроме того, в отличие от мультиблочных сополимеров согласно настоящему изобретению сополимеров простых и сложных полиэфиров невозможно регулировать, и она всегда является низкой и примерно равна Тст ПЭГ, т.е. -30°C.
Задачей настоящего изобретения является устранение одного или более недостатков, присущих уровню техники.
Согласно первому аспекту настоящее изобретение относится к биоразлагаемому полукристаллическому термопластичному мультиблочному сополимеру с разделенными фазами, причем сополимер характеризуется тем, что:
a) содержит по меньшей мере один сегмент гидролизуемого преполимера (А) и по меньшей мере один сегмент гидролизуемого преполимера (В);
b) указанный мультиблочный сополимер в физиологических условиях имеет Тст 37°C или менее и Тпл 110-250°C;
c) сегменты связаны посредством полифункционального удлинителя цепи;
d) сегменты случайным образом распределены по полимерной цепи; и
e) по меньшей мере часть преполимера (А) получена из водорастворимого полимера.
Мультиблочный сополимер согласно настоящему изобретению может состоять по меньшей мере из двух различных сегментов, каждый из которых имеет различные физические характеристики, включая характеристики разложения и набухания. Неожиданно оказалось, что вследствие своего уникального состава и полукристаллической структуры с разделенными фазами материалы согласно настоящему изобретению имеют множество применений и особенно подходят для создания матриц для доставки лекарственных средств и покрытий, через которые выделяются лекарственные средства, которые можно применять для инкапсулирования определенных терапевтических агентов и для замедленного высвобождения инкапсулированного терапевтического агента местно или в системный кровоток. Согласно приведенному ниже описанию композиция согласно настоящему изобретению является важной с учетом контролируемого высвобождения биологически активного соединения, такого как биологически активный полипептид, в организм хозяина.
Термин «разделенные фазы», используемый в настоящем описании, относится к системе, в частности к сополимеру, образованной двумя или более различными преполимерами, по меньшей мере два из которых (частично) несовместимы друг с другом при температуре тела или ниже (в физиологических условиях, таких как организм человека). Таким образом, преполимеры не образуют гомогенную смесь при объединении ни в виде физической смеси преполимеров, ни при объединении преполимеров в одном химическом соединении в виде «химической смеси», т.е. в сополимере.
Термин «преполимер», используемый в настоящем описании, относится к полимерным сегментам, которые случайным образом связаны посредством полифункционального удлинителя цепи и совместно образуют мультиблочный сополимер согласно настоящему изобретению. Каждый преполимер можно получать путем полимеризации подходящих мономеров, и указанные мономеры, таким образом, являются химическими звеньями каждого преполимера. Желаемые свойства преполимеров, а следовательно и мультиблочного сополимера согласно настоящему изобретению можно контролировать путем выбора преполимера с подходящим составом и молекулярной массой (в частности Mn), в результате чего достигаются требуемые значения Тпл или Тст.
Термин «мультиблочный», используемый в настоящем описании, относится к наличию по меньшей мере двух сегментов преполимеров в полимерной цепи.
Термин «термопластичный», используемый в настоящем описании, относится мультиблочному сополимеру без перекрестной сшивки. При нагревании термопластичный полимер становится текучим, после чего отверждается при (повторном) охлаждении. Термопластичные полимеры растворимы в подходящих растворителях.
Термин «гидролизуемый», используемый в настоящем описании, относится к способности взаимодействовать с водой, в результате чего молекула расщепляется. Гидролизуемые группы включают сложноэфирную, карбонатную, фосфазеновую, амидную и уретановую группы. В физиологических условиях только сложноэфирные, карбонатные и фосфазеновые группы взаимодействуют с водой в течение приемлемого времени.
Термин «полифункциональный удлинитель цепи», используемый в настоящем описании, относится к присутствию в удлинителе цепи по меньшей мере двух реакционноспособных групп, которые обеспечивают химическое связывание реакционноспособных преполимеров и образование, тем самым, мультиблочного сополимера.
Термин «случайный мультиблочный сополимер», используемый в настоящем описании, относится к мультиблочному сополимеру, в котором различные сегменты случайным образом распределены по полимерной цепи.
Термин «водорастворимый полимер», используемый в настоящем описании, относится к полимеру, который имеет хорошую растворимость в водной среде, предпочтительно в воде, в физиологических условиях. Такой полимер при сополимеризации с более гидрофобными фрагментами позволяет получаемому сополимеру набухать в воде. Водорастворимый полимер может быть получен из диола, диамина или двухосновной кислоты. Для инициирования полимеризации циклических мономеров с раскрытием цикла подходят диол или двухосновная кислота.
Термин «набухаемый», используемый в настоящем описании, относится к захвату воды полимером. Степень набухания можно вычислять по соотношению массы набухшего в воде сополимера и массы сухого сополимера.
Термин «полукристаллический», используемый в настоящем описании, относится к структуре мультиблочного сополимера, содержащей две различные фазы, аморфную фазу и кристаллическую фазу. Предпочтительно мультиблочный сополимер состоит из аморфной фазы и кристаллической фазы.
Термин «биологически активное соединение», используемый в настоящем описании, имеет широкое определение и относится к любым агентам, обеспечивающим терапевтическое или профилактическое действие. Указанные агенты включают, но не ограничиваются ими, противомикробные агенты (включая антибактериальные и противогрибковые агенты), противовирусные агенты, противоопухолевые агенты, гормоны и иммуногенные агенты.
Термин «биологически активный полипептид», используемый в настоящем описании, относится к пептидам и белкам, которые обладают биологической активностью в организме млекопитающего, более конкретно в организме человека.
Применение полукристаллических мультиблочных сополимеров с разделенными фазами согласно настоящему изобретению позволяет преодолеть один или более указанных выше недостатков или ограничений. В результате наличия сегментов, полученных из водорастворимого полимера (такие как гидрофильные сегменты ПЭГ), мультиблочный сополимер с разделенными фазами набухает в водной среде с образованием набухшего гидрогеля, обеспечивая естественную среду для биологически активных соединений, таких как белки. При применении мультиблочных сополимеров согласно настоящему изобретению в качестве полимерных матриц в составе с контролируемым высвобождением для доставки биологически активного соединения способность мультиблочных сополимеров к набуханию может предотвращать накопление продуктов кислотного разложения, образующихся в результате гидролиза полимерных цепей, в полимерной матрице. Вместо этого указанные продукты разложения высвобождаются из матрицы, что, таким образом, предотвращает образование кислотной микросреды в полимерной матрице, которая негативно воздействует на инкапсулированное биологически активное соединение. Кроме того, в результате способности мультиблочных сополимеров с разделенными фазами согласно настоящему изобретению к набуханию любые инкапсулированные соединения могут высвобождаться постепенно за счет диффузии, что, таким образом, предотвращает появление двухфазных или трехфазных профилей высвобождения, как правило, наблюдаемых для ненабухающих биоразлагаемых сложных полиэфиров, таких как поли(D,L-лактид) или поли(молочная-гликолевая кислота).
В физиологических условиях Тпл мультиблочных сополимеров согласно настоящему изобретению составляет 110-250°C. Это происходит вследствие наличия сегмента B преполимера. Сегмент B состоит из кристаллизуемых полимеров, таких как PLLA, поли(D-молочная кислота) (PDLA), полигликолевая кислота (PGA) или полигидроксибутират (ПГБ), или комбинации кристаллизуемых полимеров. Наиболее предпочтительно сегмент B состоит из преполимера, состоящего из PLLA. Аморфная фаза мультиблочных сополимеров с разделенными фазами согласно настоящему изобретению, главным образом, состоит из мягких сегментов А. Неожиданно, авторы настоящего изобретения обнаружили, что аморфная часть жестких сегментов В также дополняет аморфную фазу мультиблочных сополимеров согласно настоящему изобретению.
Выбор преполимеров, применяемых в качестве сегментов В в мультиблочных сополимерах, описанных в публикации WO-A-2004/007588, ограничен преполимерами, состоящими из поли(ε-капролактона) (ПКЛ), поли(валеролактона) (ПВЛ) и поли(диоксанона) (ПДС), так как Тпл преполимера (В) находится в диапазоне 40-100°C (если рассматривать традиционные полимеры, используемые для биомедицинских применений). Согласно настоящему изобретению Тпл преполимера (В) предпочтительно находится в диапазоне 110-250°C. В результате преполимер (В) может быть выбран из списка преполимеров, имеющих различные химические свойства, которые ранее даже не рассматривали. Авторы настоящего изобретения обнаружили, что другие химические свойства преполимера (В) обеспечивают получение мультиблочных сополимеров с более эффективными свойствами, которые невозможно достичь в случае сополимеров, описанных в WO-A-2004/007588.
При использовании ПДС в качестве сегмента B получают мультиблочные сополимеры с Тпл 80-90°C (патент США №5711958 A). При использовании ПКЛ в качестве сегмента B получают мультиблочные сополимеры с Тпл 40-60°C (патент США №5711958 А). Гомополимер ПВЛ имеет Тпл 60°C, схожую с Тпл гомополимера ПКЛ. При использовании сегментов ПВЛ в качестве сегмента B получают мультиблочные сополимеры с Тпл примерно 40-60°C.ПДС, ПКЛ и ПВЛ являются полукристаллическими и, таким образом, помимо Тпл характеризуются также Тст. Тст аморфных фаз ПДС, ПКЛ и ПВЛ являются низкими и составляют -10°C, -60°C и -70°C, соответственно. Повышение диапазона температур блока B до 110-250°C открывает возможность применения PLLA, PDLA, PGA и ПГБ. Эти полимеры характеризуются более высокими Тст, которые составляют примерно 50°C, 35°C и 0°C, соответственно. Вне зависимости от полимера, применяемого в качестве жестких сегментов B, указанные жесткие сегменты B всегда сами по себе являются полукристаллическими, т.е. отчасти аморфными. Неожиданно было обнаружено, что аморфная часть жестких сегментов B (отчасти) смешивается с аморфной фазой мягких сегментов A, и, таким образом, оба сегмента влияют на итоговую Тст мультиблочного сополимера. Таким образом, TCT аморфной фазы определяется Тст сегмента A и Тст сегмента B, а также молярным соотношением сегментов A/B. Тст может изменяться от Тст, близкой к значению преполимера (А) (если используют соотношение преполимеров A/B, близкое к 1), до Тст, близкой к значению преполимера (В) (если используют соотношение преполимеров A/B, близкое к нулю). Важно отметить, что высвобождение активных веществ, инкапсулированных в полимерной матрице, зависит, главным образом, от Тст аморфной фазы, поскольку диффузия активных веществ происходит через аморфную фазу, а не через плотную кристаллическую фазу. Скорость разложения полимера также зависит, главным образом, от Тст аморфной фазы, поскольку она влияет на скорость захвата воды и, таким образом, на скорость гидролиза. Применение преполимера (В) с Тпл 110-250°C и относительно высокой Тст позволяет покрывать значительно более широкий диапазон Тст по сравнению с диапазоном для преполимера (В), имеющего Тпл 40-100°C и относительно низкую Тст. Вследствие этого, применение указанных преполимеров (В) для получения мультиблочных сополимеров с Тпл в диапазоне 110-250°C позволяет получать полимеры со значительно более широким диапазоном свойств высвобождения и разложения, и, таким образом, более эффективно контролировать высвобождение различных биологически активных соединений.
Кроме того, более высокая Тпл мультиблочных сополимеров согласно настоящему изобретению позволяет получать невязкие микросферы при помощи способа двойной эмульсии в условиях окружающей среды, даже с использованием коротких сегментов B. Ограничение длины кристаллизуемого сегмента B является важным для того, чтобы мультиблочные сополимеры хорошо разлагались в физиологических условиях в отличие от кристаллических полимеров PLLA с высокой молекулярной массой. В противоположность этому, невозможно получать микросферы с использованием мультиблочных сополимеров, в которых сегмент B состоит из коротких цепей ПКЛ, так как короткие блоки ПКЛ не образуют кристаллические домены при получении микросфер. Как следствие полимер остается аморфным. Вследствие низкой TCT аморфного полимера он остается вязким, вследствие чего микросферы образуют агломераты и слипаются друг с другом во время стадии экстракции/выпаривания. Так как ПВЛ имеет Тпл, схожую с ПКЛ, можно ожидать, что с использованием микроблочных сополимеров, в которых сегмент B состоит из коротких цепей преполимера ПВЛ, микросферы также получать нельзя. В литературе отсутствуют упоминания о микросферах, состоящих из ПДС или сополимеров ПДС. Из литературы известно, что кристаллизация ПДС при высоких скоростях охлаждения и/или при низкой молекулярной массе ПДС является медленной и неполной. Эти результаты позволяют предположить, что получение микросфер при помощи способа двойной эмульсии с использованием мультиблочных сополимеров, в которых сегмент B состоит из короткого блока ПДС, является нецелесообразным.
В теории, стабильность микросфер, полученных из преполимера (В), имеющего Тпл 110-250°C, при хранении в условиях окружающей среды является более высокой по сравнению с преполимером (В), имеющим Тпл 40-100°C.Повышение Тпл приводит к повышению Ткр и, таким образом, повышает степень кристалличности микросфер. Увеличение кристалличности снижает подвижность молекул инкапсулированного биологически активного соединения в полимерной матрице и улучшает стабильность продукта при хранении. Из литературы известно, что увеличенная кристалличность увеличивает стабильность частиц при хранении. Также, преполимеры B, имеющие Тпл 110-250°C, имеют более высокую Тст по сравнению с преполимерами (В), имеющими Тпл 40-100°C. Из литературы известно, что для полукристаллических, а также аморфных частиц, повышение Тст приводит к увеличению стабильности при хранении.
Кроме того, мультиблочные сополимеры согласно настоящему изобретению имеют улучшенную скорость разложения по сравнению с мультиблочными сополимерами, в которых кристаллизуемый сегмент состоит из ПКЛ, так как сегменты B в мультиблочных сополимерах согласно настоящему изобретению являются менее гидрофобными по сравнению с ПКЛ.
Синтез мультиблочных сополимеров, в которых кристаллизуемый сегмент состоит из ПДС, затруднен вследствие ограниченной полимеризации мономера ПДС, п-диоксанона, а также ограниченной растворимости ПДС в традиционных растворителях. Хорошо известно, что п-диоксанон имеет относительно низкую температуру разложения, в результате чего максимальная степень конверсии составляет примерно 80%. В противоположность этому, мономеры, применяемые для получения мультиблочных сополимеров согласно настоящему изобретению, такие как лактид и гликолид, легко можно полимеризовать со степенями конверсии более 95%. Ограниченная растворимость полимеров, содержащих ПДС, также ограничивает их применимость для получения составов с контролируемым высвобождением.
Мультиблочные сополимеры согласно настоящему изобретению, состоящие из сегмента B на основе PLLA, имеют дополнительное преимущество, заключающееся в том, что в качестве дополнительного сегмента B может быть добавлен PDLA, что приводит к получению мультиблочных сополимеров с повышенной кристалличностью и сниженной скоростью разложения за счет образования кристаллов стереокомплекса PLLA/PDLA с Тпл, составляющей до 220°C, что примерно на 50°C выше Тпл кристаллических сегментов PLLA, которые состоят исключительно из одного энантиомера L-лактида.
В мультиблочных сополимерах согласно настоящему изобретению содержание сегментов, полученных из водорастворимого полимера, можно изменять независимо от длины блока гидрофобного (кристаллического) сегмента. Таким образом, можно добиваться высокого содержания сегментов, полученных из водорастворимого полимера, сохраняя необходимую степень кристалличности. Кроме того, в отличие от трехблочных ABA сополимеров, описанных Кисселом с соавторами, характеристическую вязкость (IV) мультиблочных сополимеров согласно настоящему изобретению можно изменять независимо от состава. Широкие возможности изменения мультиблочных сополимеров согласно настоящему изобретению позволяют легко регулировать длину, соотношение и состав сегментов для получения желаемых характеристик разложения и кинетики высвобождения лекарственного средства.
Мультиблочные сополимеры согласно настоящему изобретению обладают дополнительными преимуществами по сравнению с блоксополимерами со структурой ABA, указанными в примерах, приведенных во введении. Несмотря на то, что за счет применения блоксополимеров с блоками из различных сополимеров вместо гомополимеров или случайных сополимеров можно значительно улучшать свойства полимера, указанные ABA сополимеры сохраняют определенные недостатки.
Для получения ABA сополимера с минимальной молекулярной массой последовательности A и B должны иметь определенную длину. Блоки, входящие в состав одного полимера, могут иметь не зависящие друг от друга характеристики гомополимеров. Свойства сополимеров типа ABA можно регулировать исключительно путем изменения состава блоков A и B. Другим недостатком является то, что блоксополимеры необходимо получать при относительно высоких температурах (>100°C) в инертных условиях для достижения полной конверсии всех мономеров и достаточной молекулярной массы. Первый недостаток можно устранить за счет применения мультиблочных сополимеров со значительно более короткими блоками или сегментами, связанными друг с другом в результате химической реакции, проводимой при температурах ниже 100°C. Свойства, такие как характеристики разложения, можно регулировать значительно лучше за счет выбора подходящей комбинации длин, соотношения и состава сегментов.
Кроме того, вследствие относительно высоких температур, применяемых в способе получения ABA блоксополимеров (и их производных) всегда существует вероятность переэтерификации, что приводит к смешению фаз в той или иной степени. Мультиблочные сополимеры согласно настоящему изобретению не страдают от такого недостатка, поскольку их можно получать путем связывания преполимеров, имеющий предварительно определенный состав мономеров, при относительно низких температурах (<100°C), что, таким образом, позволяет предотвращать переэтерификацию и другие побочные реакции, которые могут инициировать нежелательное разложение и получение других побочных продуктов. Это означает, что длина последовательности мономеров в сополимере определяется выбором составных компонентов и в меньшей степени продолжительностью и температурой проведения реакции, что обычно свойственно синтезу случайных сополимеров. Другим преимуществом мультиблочных сополимеров согласно настоящему изобретению, полученных путем связывания преполимеров с использованием полифункционального удлинителя цепи, является то, что сегменты преполимера случайным образом распределены в сополимере, и это, таким образом, предоставляет существенно больше возможностей для регулирования свойств. Случайным мультиблочным сополимером является, например, АВВВВАВАААВВААААА и т.д. Случайные мультиблочные сополимеры согласно настоящему изобретению обеспечивают ряд преимуществ, которых невозможно добиться, применяя чередующиеся мультиблочные сополимеры.
Во-первых, случайные мультиблочные сополимеры, полученные путем удлинения цепи блоков A и B, имеют неограниченный диапазон соотношений А к В. A:B может, например, составлять 10:90, но также может составлять и 90:10. В противоположность этому, соотношение блоков в чередующемся мультиблочном сополимере ограничено соотношением, используемым в полимере, подвергающемся удлинению цепи. Например, в случае удлинения АВ полимера соотношение A:B в мультиблочном сополимере составляет 50:50. Случайная природа мультиблочных сополимеров согласно настоящему изобретению значительно повышает число различных составов материала и, таким образом, улучшает контроль его физических и химических свойств. Эти улучшения включают улучшенный контроль способности набухания в воде, структуры (разделения фаз, аморфности/кристалличности) и разложения полимера.
Во-вторых, способ синтеза случайных мультиблочных сополимеров согласно настоящему изобретению является значительно менее трудоемким по сравнению с синтезом чередующихся мультиблочных сополимеров. В чередующихся мультиблочных сополимерах перед удлинением цепи необходимо связывать сегменты A и B в случае двухблочных АВ сополимеров или сегменты A и C в случае трехблочных АСА сополимеров (или необходимо синтезировать макромолекулярный удлинитель цепи). В случайных мультиблочных сополимерах проводят удлинение цепи отдельно блоков A и B с использованием, например, коммерчески доступного удлинителя цепи.
Другим преимуществом мультиблочных сополимеров согласно настоящему изобретению является то, что они содержат полифункциональный (предпочтительно алифатический) удлинитель цепи. Выбор типа и количества удлинителя цепи может влиять на свойства полимера (например, удлинитель цепи может выступать в качестве пластификатора или же он может влиять на степень разделения фаз). Общее число степеней свободы при получении полимеров с желаемыми свойствами, таким образом, является более высоким по сравнению с полимерами, известными в уровне техники.
Согласно настоящему изобретению предложены мультиблочные сополимеры с разделенными фазами, которые обладают достаточной набухаемостью в водной среде и в физиологических условиях при введении и, таким образом, обеспечивают водную микросреду для инкапсулированного пептида или белка, а также контролируемое диффузией высвобождение пептидов и белков. Материалы, таким образом, имеют значительно сниженную механическую прочность. Несмотря на то, что указанные материалы можно использовать в качестве материалов с памятью формы в сухих условиях в отсутствие значительного снижения механической прочности перед переходом в запомненную форму, например, под воздействием внешних источников тепла или света, в гидратированном состоянии указанные материалы претерпевают значительные изменения размеров, кроме того происходит существенное снижение их механической прочности, очевидно, вследствие того, что указанные материалы адсорбируют значительное количество воды из-за своей гидрофильной природы, что приводит к существенному набуханию и пластификации материала. В результате этого, в гидратированных условиях, таких как физиологические условия организма человека или животного, размер полимерных конструкций, получаемых из указанных материалов, значительно изменяется, а механические свойства указанных материалов изменяются на порядок. В противоположность мультиблочным сополимерам согласно настоящему изобретению материалы с памятью формы, описанные в патенте США №5711958 A, незначительно набухают в гидратированных условиях, таких как физиологические условия организма человека или животного.
Сложные полиэфиры или полиэфиркарбонаты с разделенными фазами согласно настоящему изобретению входят в группу перспективных биоматериалов, их можно применять для доставки различных лекарственных средств, так как они обеспечивают превосходный контроль высвобождения лекарственного средства и высвобождение биологически активных соединений, таких как полипептиды.
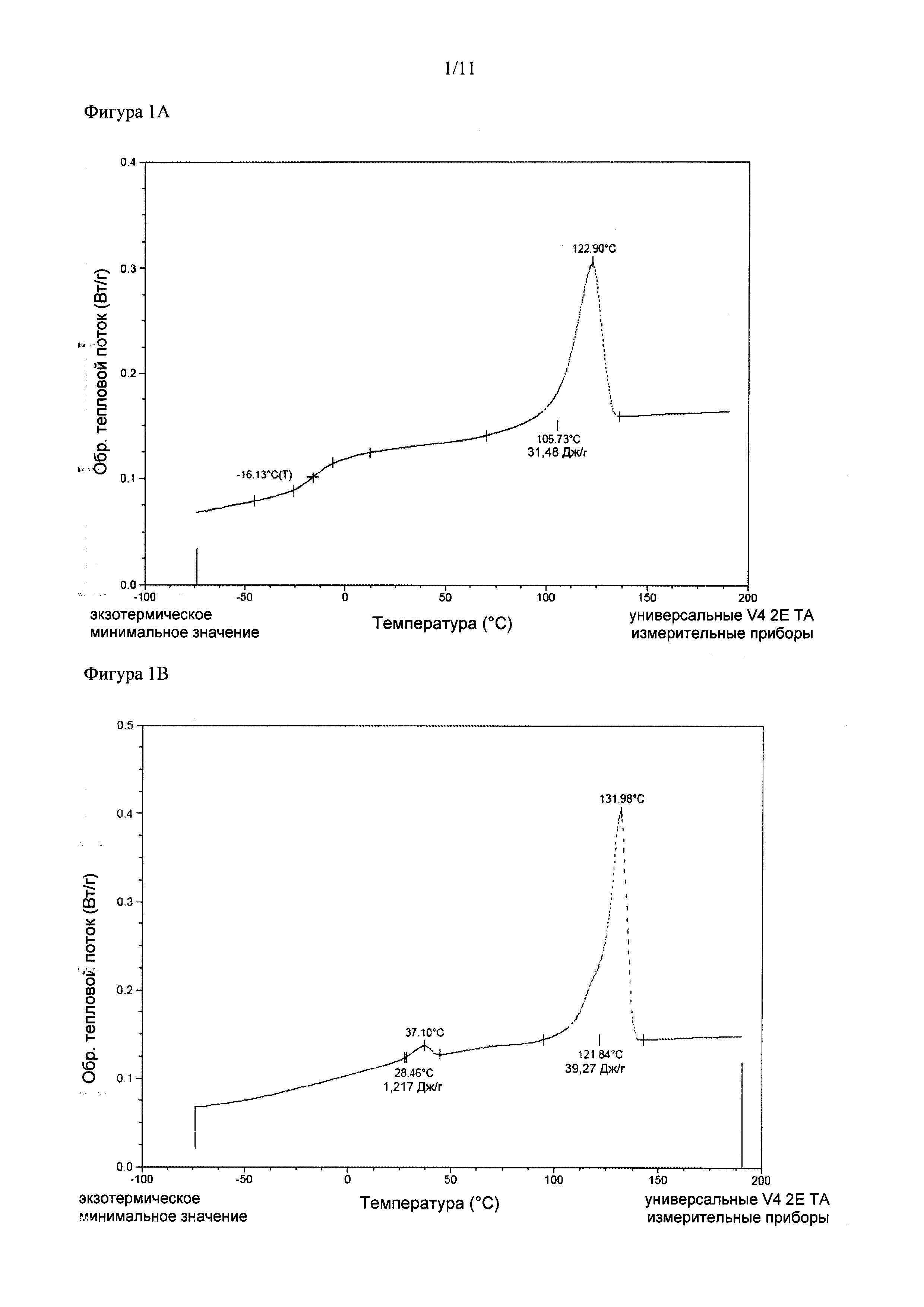
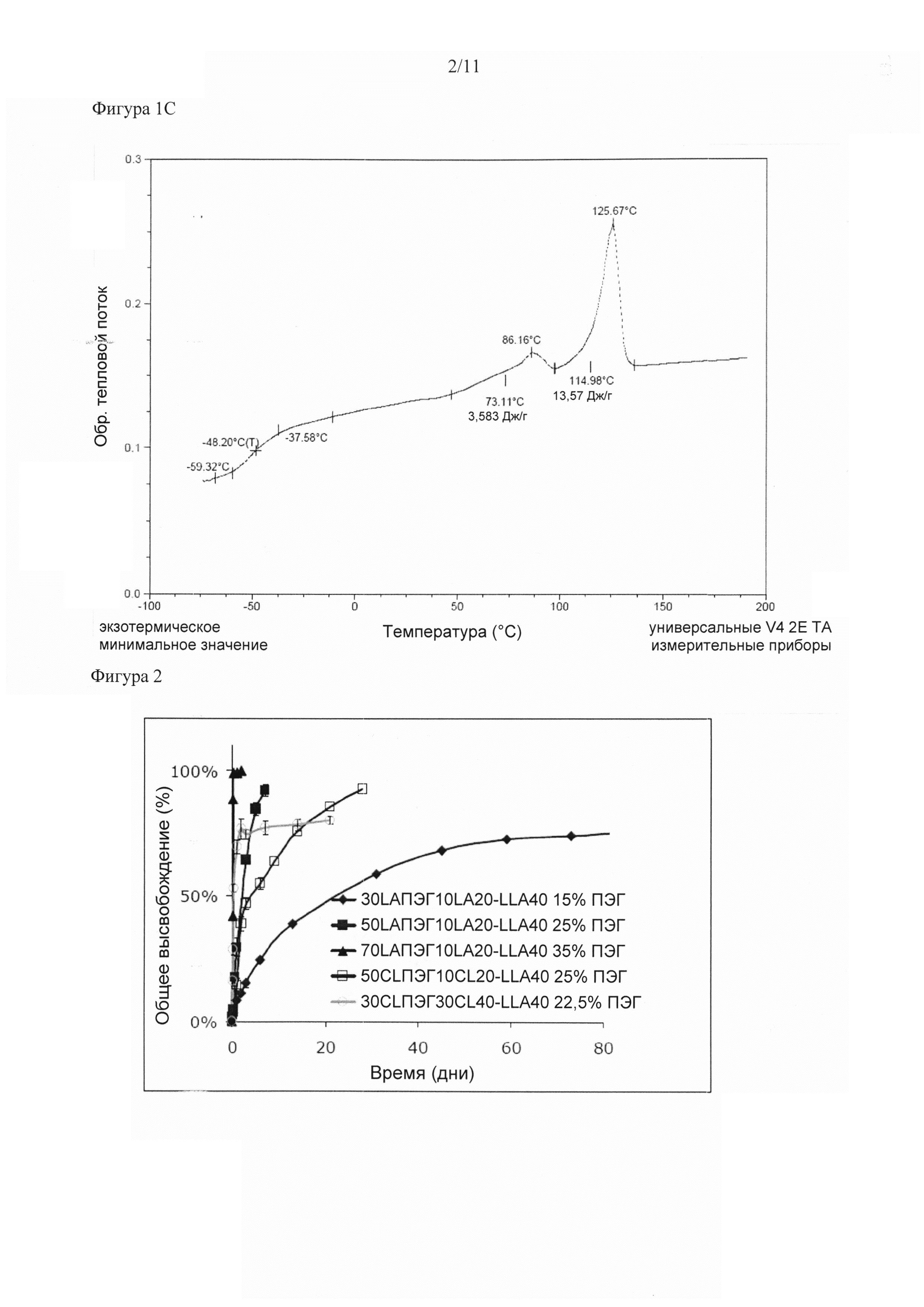
Структура мультиблочного сополимера (или конструкта, полученного из него) зависит от условий среды: измерения ДСК (дифференциальной сканирующей калориметрии) можно проводить в инертных (сухих) условиях, и полученные результаты можно использовать для определения термодинамических свойств сухих материалов. Тем не менее, структура и свойства в физиологических условиях (т.е. в организме) могут отличаться от структуры и свойств в условиях окружающей среды (сухая среда, комнатная температура). Также следует понимать, что температуры фазовых переходов, Тст и Тпл, используемые в настоящем описании, относятся к соответствующим значениям для материала, находящегося in vivo, т.е. в равновесной атмосфере, насыщенной парами воды при температуре тела. Ее можно имитировать in vitro посредством проведения измерений ДСК после установления равновесия материала и атмосферы, насыщенной парами воды. В сухом состоянии материалы, применяемые согласно настоящему изобретению, могут иметь значения Тст, которые несколько превышают значения в условиях организма млекопитающего, т.е. при исследовании сухих материалов путем ДСК первая точка перегиба может появляться при более высоких температурах, например, при 42°C или 50°C или более. В результате применения in vivo, тем не менее, Тст и/или Тпл сухого материала снижаются за счет поглощения воды, которая придает пластичность полимеру, и итоговая Тст согласно настоящему изобретению должна быть примерно равна температуре тела или ниже. Итоговая Тпл в физиологических условиях должна составлять от 110°C до 250°C.
Например, полимер, содержащий ПЭГ в мягком сегменте, может быть кристаллическим в сухих условиях при температуре окружающей среды, но аморфным во влажных условиях, что дает непостоянные значения Тст или два различных значения Тст мягкого сегмента, полученного из аморфного пластифицированного ПЭГ и полиэфиркарбоната. Присутствие разделенных фаз в сополимерах согласно настоящему изобретению отражается на профиле Тст или Тпл. Сополимеры с разделенными фазами характеризуются по меньшей мере двумя фазовыми переходами, каждый из которых происходит при температурах, схожих (но, в целом, не равных) с соответствующими значениями Тст или Тпл преполимеров, из которых состоит сополимер. Т^ определяется как среднее значение скачка теплоемкости, которое можно измерить, например, путем ДСК. Тпл является максимумом пика плавления, что схематически изображено на Фигуре 1, на которой показана эндотерма теплового потока для сополимера, характеризующегося Тст и Тпл. По определению значения Тст и Тпл для определенного преполимера отражают значения, как если бы их измеряли в сополимере. В случае полного разделения фаз преполимеров Тст сополимера определяется исключительно Тст аморфного «мягкого» преполимера. Фактически, тем не менее, составы кристаллической и аморфной фаз мультиблочного сополимера не полностью соответствует составам мягких сегментов A и полукристаллических сегментов В. Аморфная часть преполимера, образующего исходный жесткий сегмент, смешивается с преполимером (А), образующим мягкий сегмент, и, таким образом, становится составляющей частью аморфной фазы. Итоговое значение Тст аморфной фазы отличается от значения применяемого преполимера. Степень разделения фаз (а таким образом и степень отклонения значений Тст и/или Тпл от значений соответствующих преполимеров) зависит от состава и соотношения преполимеров и длины сегментов в сополимере Тст сегментов сополимера, в целом, находится между значением Тст сополимера со смешанными фазами и значения Тст отдельных преполимеров.
Физико-химические свойства (такие как свойства разложения, набухания и термодинамические свойства) мультиблочных сополимеров можно легко регулировать путем изменения типа мономеров, применяемых для получения преполимеров, образующих мягкие и жесткие сегменты, а также длины цепи и соотношения длин цепей, кроме того за счет выбора типа и количества удлинителя цепи. Кроме того, температуры фазовых переходов являются достаточно низкими, что позволяет проводить обработку полимера в расплаве. Соотношение и распределение мономеров в сополимере можно легко контролировать путем изменения условий полимеризации.
Обычно для получения невязких материалов желательно использовать кристаллический сегмент В. Кроме того, структура с разделенными фазами, содержащая аморфные и кристаллические домены, должна сохраняться в физиологических условиях (т.е. при воздействии водной среды при температуре тела) для обеспечения контролируемого набухания полимерной матрицы. Контроль степени набухания является существенно важным для контроля высвобождения инкапсулированных соединений. Кристаллические сегменты B выступают в качестве физических сшивающих групп, которые контролируют набухание более гидрофильных мягких сегментов. Помимо уровня жесткого сегмента B степень набухания полимеров зависит от содержания и молекулярной массы/длины водорастворимого полимера, входящего в состав мягкого сегмента A.
Как отмечалось ранее, необходимым требованием к сегментированному сополимеру Сложного полиэфира с разделенными фазами является то, что Тпл полиэфирного сегмента B должна находиться в диапазоне 110-250°C, а Тст сегмента A должна быть ниже 37°C в физиологических условиях. Тпл сегмента B в мультиблочном сополимере, в целом, ниже температуры непрореагировавшего преполимера (B) вследствие снижения гибкости цепи после встраивания преполимера в мультиблочный сополимер, а также из-за возможного смешения фаз других компонентов мультиблочного сополимера с кристаллической фазой. Важным классом сегментированных сополимеров сложных полиэфиров с хорошим разделением фаз являются сополимеры на основе жестких сегментов B, состоящих из кристаллической PLLA. Авторы настоящего изобретения показали, что мультиблочные сополимеры, содержащие сегменты B на основе PLLA, в физиологических условиях характеризуются Тпл, составляющей по меньшей мере 110°C. Указанные мультиблочные сополимеры обеспечивают несколько преимуществ. Можно добиваться широкого диапазона скоростей разложения. Преполимер (В), образующий жесткий сегмент B, состоит из кристаллической PLLA, а, как известно, такие полимеры разлагаются очень медленно. В противоположность этому, преполимер (A) представляет собой полимер на основе водорастворимого полимера и аморфного сложного полиэфира. Известно, что такие полимеры разлагаются относительно быстро. Итоговая скорость разложения определяется соотношением сегмент A/сегмент B, таким образом, ее можно легко регулировать. Поскольку высвобождение помимо прочих факторов зависит от скорости разложения мультиблочного сополимера, его можно регулировать также путем изменения соотношения сегмент A/сегмент В. Также, степень кристалличности можно легко повышать путем смешения PLLA и PDLA с образованием стереокомплекса. Образование стереокомплекса приводит к повышению степени кристалличности по сравнению с отдельным энантиомером, а также к повышению Тпл (на ~50°C выше по сравнению с отдельным энантиомером). Кроме того, Тст мультиблочных сополимеров, содержащих сегменты B на основе PLLA, можно изменять в широком диапазоне от примерно -40 до 40°C (при измерении в сухих условиях). Так как скорость разложения и высвобождения помимо прочих факторов зависят от Тст матрицы, то обеспечение широкого диапазона Тст позволяет более широко регулировать свойства высвобождения и разложения.
В целом, структуру с желаемым разделением фаз (имеющую одну температуру плавления и по меньшей мере одно значение Тст) можно получать путем изменения состава, например, за счет выбора среднечисловой молекулярной массы, Mn, преполимеров A и B. На структуру с разделенными фазами также можно влиять путем изменения соотношения сегмент A/сегмент В.
Сегментированные мультиблочные сополимеры согласно настоящему изобретению содержат мягкий сегмент A, полученный из преполимера (А), который поддается гидролизу и, как правило, является полностью аморфным в физиологических условиях (в организме). Кроме того, преполимер (А) предпочтительно имеет по меньшей мере одну температуру фазового перехода, где Тст, измеренная в физиологических условиях (в условиях организма), составляет 37°C или менее, предпочтительно 25°C или менее. Этот сегмент является частью аморфной фазы мультиблочного сополимера, при этом аморфную фазу в настоящем описании называют фазой (А). Сополимеры согласно настоящему изобретению также содержат жесткий сегмент B, полученный из преполимера (В), содержащего полукристаллический гидролизуемый полимер, как правило, имеющий Тпл, измеренную в физиологических условиях (в условиях организма), составляющую 110-250°C. Сегмент B, главным образом, составляет фазу (В). Преполимеры A и B, которые образуют «мягкие» и «жесткие» сегменты, соответственно, связаны посредством полифункционального удлинителя цепи. Как правило, кристаллическая(ие) фаза(ы) содержит(ат) жесткие сегменты В, а аморфная(ые) фаза(ы) содержит(ат) мягкие сегменты А и аморфную часть сегментов В. Кристаллическая и аморфная фаза(ы) являются несовместимыми или только частично совместимыми в условиях организма, т.е. они имеют разделенные фазы. Полифункциональный удлинитель цепи предпочтительно представляет собой алифатическую молекулу.
Получаемые мультиблочные сополимеры согласно настоящему изобретению предпочтительно имеют структуру формулы (I):
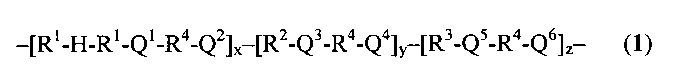
где R1 представляет собой часть сегмента A, который в свою очередь является частью фазы (А), и может представлять собой аморфный сложный полиэфир, аморфный полиэфир-эфирный сополимер или аморфный поликарбонат; или аморфный преполимер, который получают из объединяемых сложноэфирных, простых эфирных и/или карбонатных групп. H представляет собой средний блок сегмента A и получен из водорастворимого полимера. При комнатной температуре блок, полученный из водорастворимого полимера, может быть аморфным или полукристаллическим. Тем не менее, блок H, введенный в сегмент A, становится аморфным в физиологических условиях. Этот водорастворимый полимер выбран из группы, состоящей из простых полиэфиров, таких как полиэтиленгликоль (ПЭГ), политетраметиленоксид (ПТМО) и полипропиленгликоль (ППГ); поливинилового спирта (ПВС), поливинилпирролидона (ПВП), поливинилкапролактама, поли(гидроксиэтилметакрилата) (поли-(ГЭМА)), полифосфазенов, сложных полиортоэфиров, полиортоэфирамидов или сополимеров приведенных выше полимеров. Предпочтительно, Н представляет собой ПЭГ, который является инициатором полимеризации с раскрытием цикла циклического мономера, который образует R1.
R2 представляет собой сегмент B и по существу или полностью составляет фазу (В). R2 может представлять собой кристаллический или полукристаллический сложный полиэфир, полиэфир-эфирный сополимер, поликарбонат или полиангидрид; или преполимеры из объединяемых сложноэфирных, простых эфирных, ангидридных и/или карбонатных групп. Часть фазы R может быть аморфной, в этом случае эта часть R2 входит в состав фазы (A). R1 и R2 предпочтительно не являются одинаковыми. Переменная z равна нулю или положительному целому числу. Переменные x и y обе представляют собой положительные целые числа.
Возможно присутствие сегмента R3. Этот сегмент получен из водорастворимого полимера, выбранного из группы полимеров, указанных в определении H. В физиологических условиях R3 является частью аморфной фазы (А). В случае присутствия R3 мультиблочный сополимер согласно настоящему изобретению содержит водорастворимый полимер в качестве дополнительного преполимера. Предпочтительно этот водорастворимый полимер выбран из группы, состоящей из простых полиэфиров, таких как полиэтиленгликоль (ПЭГ), политетраметиленоксид (ПТМО) и полипропиленгликоль (ППГ); поливинилового спирта (ПВС), поливинилпирролидона (ПВП), поливинилкапролактама, поли(гидроксиметилметакрилата) (поли-(ГЭМА)), полифосфазенов, сложных полиортоэфиров, полиортоэфирамидов или сополимеров указанных выше полимеров. Например, указанный сегмент водорастворимого полимера получен из ПЭГ, имеющего Mn 150-5000 г/моль.
R4 получен из удлинителя цепи и состоит из алифатической C2-C8 алкиленовой группы, возможно замещенной C1-С10алкиленом, где алифатическая группа является линейной или циклической. R4 представляет собой бутиленовую, -(CH2)4-группу. C1-C10 алкиленовая боковая группа может содержать защищенные S, N, P или O-фрагменты. Удлинители цепи, содержащие ароматические группы, в целом, не подходят, так как удлинители цепи, содержащие ароматические группы, могут приводить к получению нежелательных продуктов разложения. Таким образом, предпочтительными являются алифатические удлинители цепи.
Q1-Q6 представляют собой линкерные звенья, получаемые в результате взаимодействия преполимеров с полифункциональным удлинителем цепи. Каждый из Q1-Q6 может быть независимо выбран из амина, уретана, амида, карбоната, сложного эфира и ангидрида. Все линкерные группы Q редко являются одинаковыми, и, как правило, это не является предпочтительным.
Как правило, можно применять один тип удлинителя цепи совместно с тремя преполимерами с одинаковыми концевыми группами, что приводит к получению сополимера формулы (1) с шестью схожими линкерными группами.
В случае если преполимеры R1 и R2 имеют различные концевые группы, в сополимере будут присутствовать два типа групп Q: например, между двумя связываемыми сегментами R1 Q1 и Q2 будут одинаковыми, но при связывании R1 и R2 Q1 и Q2 будут различными. В примере формулы (1) показан продукт взаимодействия бифункционального удлинителя цепи и бифункциональных преполимеров.
Что касается формулы (1), сложные полиэфиры согласно настоящему изобретению также могут быть представлены в виде мультиблочных или сегментированных сополимеров со случайным распределением сегментов (AB)r, где «A» соответствует сегменту A, а «B» соответствует сегменту B (если z=0). В (AB)r соотношение A/B (соответствующее x/y в формуле (1)) может быть равно единице или отличаться от единицы. Преполимеры можно смешивать в любых желаемых количествах, связывание можно проводить с использованием полифункционального удлинителя цепи, т.е. соединения, имеющего по меньшей мере две функциональные группы, за счет чего их можно применять для химического связывания преполимеров. Предпочтительно полифункциональный удлинитель цепи представляет собой бифункциональный удлинитель цепи. В случае если z≠0, случайное распределение всех сегментов может быть отражено формулой (ABC)r, где три различных преполимера (один из которых составляет сегмент, полученный из водорастворимого полимера, такого как ПЭГ) случайным образом распределены во всех возможных соотношениях.
Преполимеры, из которых получают сегменты a и b (и возможно c) в (AB)r и (ABC)r, связаны посредством полифункционального удлинителя цепи. Этот удлинитель цепи предпочтительно представляет собой диизоцианатный удлинитель цепи, но также может представлять собой двухосновную кислоту или диольное соединение. Если все преполимеры содержат гидроксильные концевые группы, а применяют диизоцианатный удлинитель цепи, то линкерные звенья представляют собой уретановые группы. Если преполимеры (или один из них) имеют(ет) карбоксильные концевые группы, то линкерные звенья представляют собой амидные группы. Мультиблочные сополимеры со структурой (AB)r и (ABC)r также можно получать путем взаимодействия преполимеров с концевыми группами двухосновных карбоновых кислот и диольного удлинителя цепи, или наоборот (преполимера с диольными концевым группами и удлинителя цепи, представляющего собой двухосновную кислоту) с применением агента сочетания, такого как DCC (дициклогексилкарбодиимид), с образованием сложноэфирных линкерных групп.
Как отмечалось выше случайные сегментированные сополимеры относятся к сополимерам со случайным распределением (т.е. не чередующимся) сегментов A и B. В случае сегментов A и B это можно выразить формулой (AB)r, а в случае сегментов A, B и C - формулой (ABC)r.
Гидролизуемый сегмент R1-H-R1 в формуле (1) получают путем взаимодействия с применением преполимера (A).
Преполимер (А), например, можно получать путем полимеризации с раскрытием цикла. Таким образом, преполимер (А) может представлять собой гидролизуемый сополимер, получаемый путем полимеризации с раскрытием цикла, инициируемой с использованием диольного соединения или двухосновной кислоты, предпочтительно сополимер со случайным распределением мономеров. Диольное соединение предпочтительно представляет собой алифатический диол или низкомолекулярный простой полиэфир, такой как ПЭГ. Простой полиэфир является частью преполимера (А) за счет того, что его применяют в качестве инициатора, его можно дополнительно смешивать с преполимером (А), что, таким образом, приводит к получению дополнительного гидрофильного сегмента R3 в формуле (1). Преполимер (А) может представлять собой гидролизуемый сложный полиэфир, полиэфир-эфирный сополимер, поликарбонат, полиэфиркарбонат, полиангидрид или их сополимеры. Например, преполимер (А) содержит продукты взаимодействия мономеров, образующих сложные эфиры, выбранных из диолов, двухосновных карбоновых кислот и гидроксикарбоновых кислот. Преполимер (А) может содержать продукты взаимодействия циклических мономеров и/или нециклических мономеров. Типовые циклические мономеры включают гликолид, лактид, ε-капролактон, δ-валеролактон, триметиленкарбонат, тетраметиленкарбонат, 1,5-диоксепан-2-он, 1,4-диоксан-2-он (пара-диоксанон) и/или циклические ангидриды, такие как оксепан-2,7-дион. В одном из вариантов реализации применяют L-лактид, D-лактид и/или D,L-лактид.
По уровню соответствия требуемой Тст, которая составляет менее 37°C, некоторые из указанных выше мономеров или комбинаций мономеров являются более предпочтительными по сравнению с остальными. Например, преполимер (А), содержащий в качестве мономеров лактид и/или гликолид, предпочтительно объединяют с любыми другими указанными циклическими сомономерами (с ε-капролактоном, δ-валеролактоном, триметиленкарбонатом, 1,4-диоксан-2-оном и из комбинациями). В результате может снижаться Тст. В качестве альтернативы инициатором получения преполимера является ПЭГ с достаточной молекулярной массой, что снижает Тст мультиблочного сополимера.
Если преполимер (А) состоит из поли(D,L-лактида), то соотношение L/D лактидов может отличаться от единицы (отличаться от 50/50). Например, использование соотношения L/D от 85/15 до 15/85 приводит к получению полностью аморфного гомополимера. Кроме того, известно, что избыток одного изомера (L или D) по отношению к другому повышает Тст поли(D,L-лактида). Незначительные количества каких-либо других из указанных выше мономеров, которые составляют аморфную фазу, также могут присутствовать в преполимере или блоке, образующем кристаллическую фазу.
Кроме того, преполимер (А) может быть получен из (смесей) мономеров конденсационного типа (нециклических), таких как гидроксикислоты (например, молочная кислота, гликолевая кислота, гидроксимасляная кислота), двухосновные кислоты (например, глутаровая, адипиновая или янтарная кислота, себациновая кислота) и диолы, такие как этиленгликоль, диэтиленгликоль, 1,4-бутандиол или 1,6-гександиол, с образованием сложноэфирных и/или ангидридных гидролизуемых фрагментов.
Сегмент R2 в формуле (1) можно получать путем взаимодействия преполимеров (В), полученных из применяемых в качестве мономеров L-лактида, D-лактида, гидроксибутирата, гликолида или комбинации указанных мономеров, что приводит к образованию стереокомплекса, имеющего температуру фазового перехода от 110°C до 250°C в физиологических условиях. Предпочтительно сегмент B получают в результате взаимодействия L-лактидных мономеров.
Как правило, преполимер (В) имеет Mn 1000 г/моль или более, предпочтительно 2000 г/моль или более, более предпочтительно 3000 г/моль или более. В целом, Mn преполимера (В) составляет 10000 г/моль или менее. Содержание преполимера (В) в сополимере предпочтительно составляет 10-90 масс. % от общей массы мультиблочного сополимера, более предпочтительно 25-70 масс. %, наиболее предпочтительно 30-50 масс. %.
Преполимеры предпочтительно представляют собой линейные и случайные сложные полиэфиры (или их сополимеры), полиэфиркарбонаты, полиэфир-эфирные сополимеры или полиангидриды с реакционноспособными концевыми группами. Указанные концевые группы могут представлять собой гидроксил или карбоксил. Предпочтительно использовать сополимер с двумя гидроксильными концевыми группами, но также можно применять полимеры с гидроксильной и карбоксильной или двумя карбоксильными концевыми группами. С учетом того, что полимер должен быть линейным, его можно получать с использованием бифункционального компонента (диола) в качестве исходного вещества, но в случае применения трехфункционального полиола или полиола с более высоким количеством функциональных групп можно получать звездообразные сложные полиэфиры. Диол, входящий в состав преполимера (А), может представлять собой алифатический диол или низкомолекулярный простой полиэфир.
Синтез преполимера путем полимеризации с раскрытием цикла предпочтительно проводят в присутствии катализатора. Подходящим катализатором является Sn(Oct)2, а M/I=5000-30000 (где M/I представляет собой соотношение мономера и инициатора). Синтез также можно проводить в отсутствие катализатора.
Условия получения сложных полиэфиров, поликарбонатов и полиангидридов хорошо известны в данной области техники.
Сополимеры согласно настоящему изобретению, в целом, являются линейными. Тем не менее, можно получать разветвленные сополимеры. Указанные нелинейные сополимеры согласно настоящему изобретению можно получать путем использования трехфункционального (или содержащего большее количество функциональных групп) удлинителя цепи, такого как триизоцианат. Разветвленные сополимеры могут иметь улучшенные характеристики ползучести.
В кристаллизуемом жестком сегменте длина (Mn) преполимера должна быть достаточно высокой, чтобы обеспечивать протекание кристаллизации сополимера. Например, преполимер PLLA, образующий жесткий сегмент, предпочтительно имеет Mn 700 г/моль или более, более предпочтительно 2000 г/моль или более, наиболее предпочтительно 3000 г/моль или более. Предполагается, что более высокая длина преполимера PLLA обеспечивает структуру с разделенными фазами при более низком содержании жесткого сегмента. Соотношение преполимеров, при котором происходит разделение фаз, таким образом, зависит от длин преполимеров. В целом, длины преполимеров, из которых получают мягкий и жесткий сегмент сополимера, должны быть такими, чтобы образовывалась структура с разделенными фазами, где степень разделения фаз (несовместимости) положительно влияет на желаемые свойства биомедицинского устройства.
Преполимер (А), образующий мягкий сегмент, может иметь Mn 500 г/моль или более, предпочтительно 1000 г/моль или более, более предпочтительно 2000 г/моль или более. Необходимо выбирать максимально возможную длину преполимеров, необходимую для получения структуры с хорошим разделением фаз, а также для обеспечения хороших механических и термических свойств получаемого сополимера. Длина преполимера должна быть достаточно низкой, чтобы обеспечивать возможность смешения с удлинителем цепи при температуре проведения полимеризации. Как правило, этого можно достигать при Mn, составляющей 10000 г/моль или менее.
В целом, содержание жесткого сегмента в диапазоне 10-90 масс. % относительно общей массы мультиблочного сополимера, предпочтительно 25-90 масс. %, обеспечивает получение гибких термопластичных материалов с хорошими свойствами разложения и набухания при температурах применения (т.е. примерно при 37°C в случае медицинских применений).
Согласно дополнительному аспекту изобретение относится к способу получения термопластичных мультиблочных сополимеров с разделенными фазами согласно настоящему изобретению, включающему реакцию удлинения цепи преполимера (А) и преполимера (В) в присутствии полифункционального удлинителя цепи с получением, тем самым, случайного сегментированного мультиблочного сополимера.
Сегментированные мультиблочные сополимеры со структурами (AB)r и (ABC)r можно получать путем удлинения цепи смеси преполимеров, содержащей желаемое соотношение мономеров, образующих жесткие и мягкие сегменты R1, H и R2 и возможно R3, с использованием эквивалентного количества полифункционального удлинителя цепи, предпочтительно алифатической молекулы, более предпочтительно диизоцианата, такого как 1,4-бутандиизоцианат (BDI). Сегментированные сополимеры со структурами (AB)r или (ABC)r предпочтительно получают в растворе. Соответственно, преполимер(ы) растворяют в инертном органическом растворителе, а удлинитель цепи добавляют в чистом виде или в виде раствора. Температура полимеризации может совпадать с максимальной температурой фазового перехода преполимеров или иметь меньшее значение. Реакции сочетания с дициклогексилкарбодиимидом (DCC) предпочтительно проводят в растворе. Два (или три) преполимера, все из которых содержат диольные концевые группы или концевые группы двухосновных кислот, можно смешивать в растворе с удлинителем цепи с концевыми группами двухосновных кислот или диольными концевыми группами, соответственно, после чего добавляют DCC.
Полимеризацию проводят в течение времени, достаточного для достижения характеристической вязкости сополимера, составляющей 0,1 дл/г или более (измеренной при 25°C в хлороформе). Низкая температура полимеризации и непродолжительное время полимеризации предотвращают переэтерификацию, таким образом, получают структуру с разделенными фазами, а распределение мономеров является таким же, как и в преполимерах, из которых состоит сополимер. В противоположность этому, высокомолекулярные случайные сополимеры необходимо получать при более высоких температурах (>100°C) и в течение значительно более продолжительного времени, чтобы обеспечить полное включение всех мономеров. За это время происходит переэтерификация, в результате чего происходит более случайное (т.е. менее блочное) распределение мономеров.
Материалы, синтезируемые путем удлинения цепи в массе, также можно получать in situ в экструдере.
Если удлинитель цепи является бифункциональным, а алифатическая молекула и преполимеры являются линейными, то образуется линейный сополимер; если один из реагентов (удлинитель цепи или по меньшей мере один из преполимеров) или оба имеют более чем две функциональные группы, можно получать разветвленные структуры с достаточно низкой степенью конверсии. Удлинитель цепи может представлять собой бифункциональный алифатический удлинитель цепи, предпочтительно диизоцианат, такой как 1,4-бутандиизоцианат.
Комбинацию преполимеров или мономеров, образующих кристаллическую или аморфную фазу, выбирают таким образом, чтобы получать сегментированные или блочные сополимеры полиэфиров или полиэфиркарбонатов с разделенными фазами с желаемыми свойствами разложения, набухания, физическими и термическими свойствами. Как правило, характеристическая вязкость (измеренная при 25°C в хлороформе) составляет более 0,1 дл/г и менее 10 дл/г, предпочтительно 0,1-2 дл/г, более предпочтительно 0,2-1 дл/г.
Мультиблочные сегментированные сополимеры можно получать в виде составов с различной формой и размерами при помощи любых известных способов, таких как, например, способы эмульсификации, экструзия, литье, формование путем заливки раствора, сушка распылением, распылительная сублимационная сушка, электроформование или лиофильная сушка. Последнюю технику применяют для получения пористых материалов. Пористость можно регулировать путем добавления сорастворителей, осадителей и/или выщелачивающих агентов. Сополимеры (твердые или пористые) можно обрабатывать с получением микросфер, микрочастиц, наносфер, стержней, пленок, покрытий, напылений, трубок, мембран, сетчатых имплантов, волокон, тампонов, оболочек и других изделий. Продукты могут быть твердыми, полыми или (микро)пористыми. Для применений, например, для ухода за ранами, восстановления кожи, восстановления нервов, в качестве сосудистых протезов, для доставки лекарственных средств, реконструкции мениска, тканевой инженерии, покрытия хирургических устройств, восстановления связок и сухожилий, протезирования зубов или ортопедического протезирования можно получать широкий диапазон биомедицинских имплантатов. Сополимеры можно применять по отдельности или можно смешивать и/или экструдировать совместно с другими абсорбируемыми или неабсорбируемыми полимерами.
Кроме того, их можно применять в фармацевтических целях, например, для доставки лекарственных средств, например, в виде микросфер, твердых имплантатов, гелей, оболочек, пленок, покрытий, напылений, трубок, мембран, сетчатых имплантов, волокон, тампонов и других форм.
Ниже в примерах будет показано, что материалы согласно настоящему изобретению имеют улучшенные свойства, включая термические, механические, технологические, по сравнению с сополимерами, описанными в уровне техники.
Согласно дополнительному аспекту изобретение относится к композиции для доставки по меньшей мере одного биологически активного соединения (например, биологически активной низкомолекулярной молекулы, белка или пептида) хозяину, содержащей по меньшей мере одно биологически активное соединение, инкапсулированное в матрице, причем указанная матрица содержит по меньшей мере один термопластичный мультиблочный сополимер с разделенными фазами, такой как определено в настоящем описании.
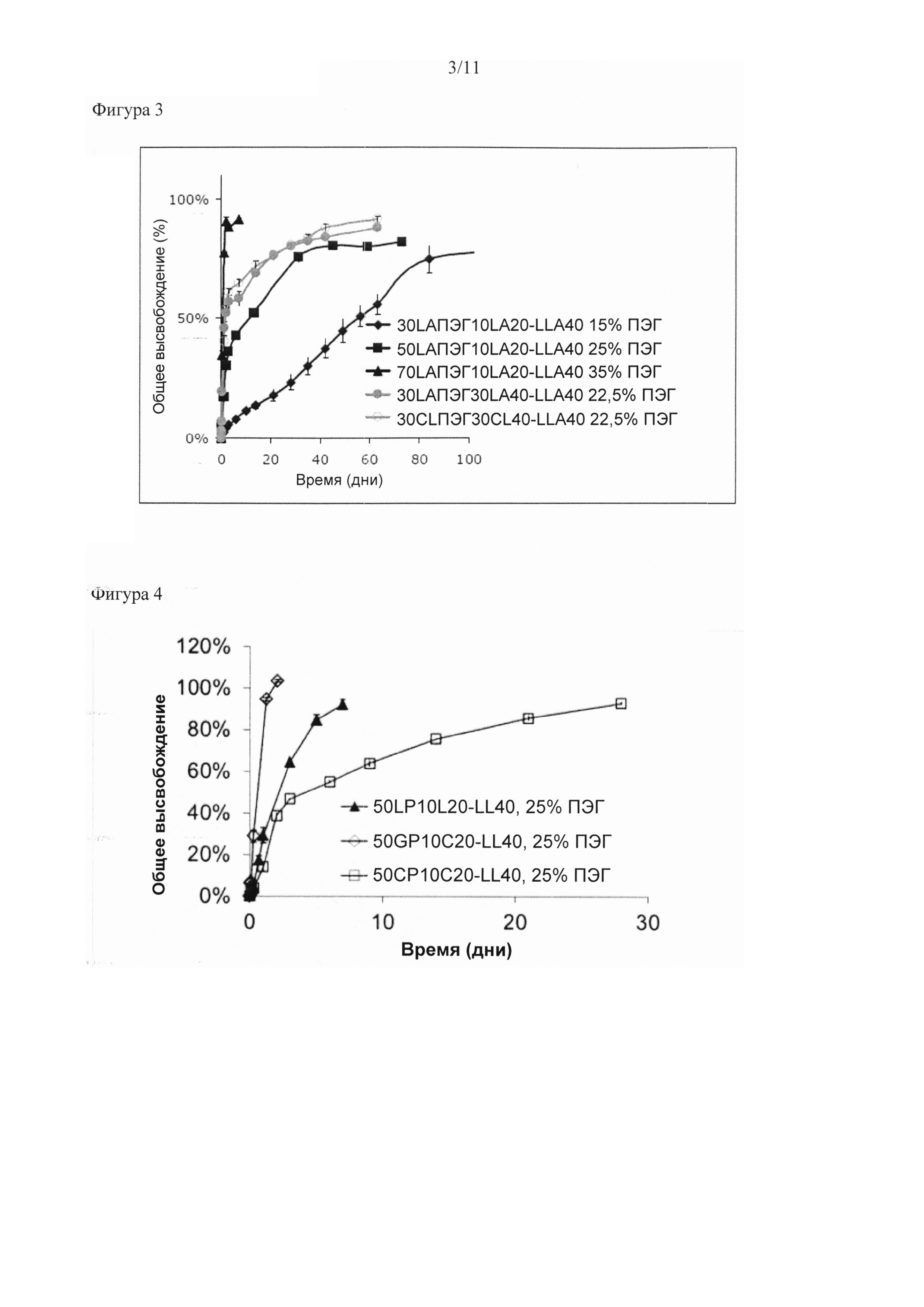
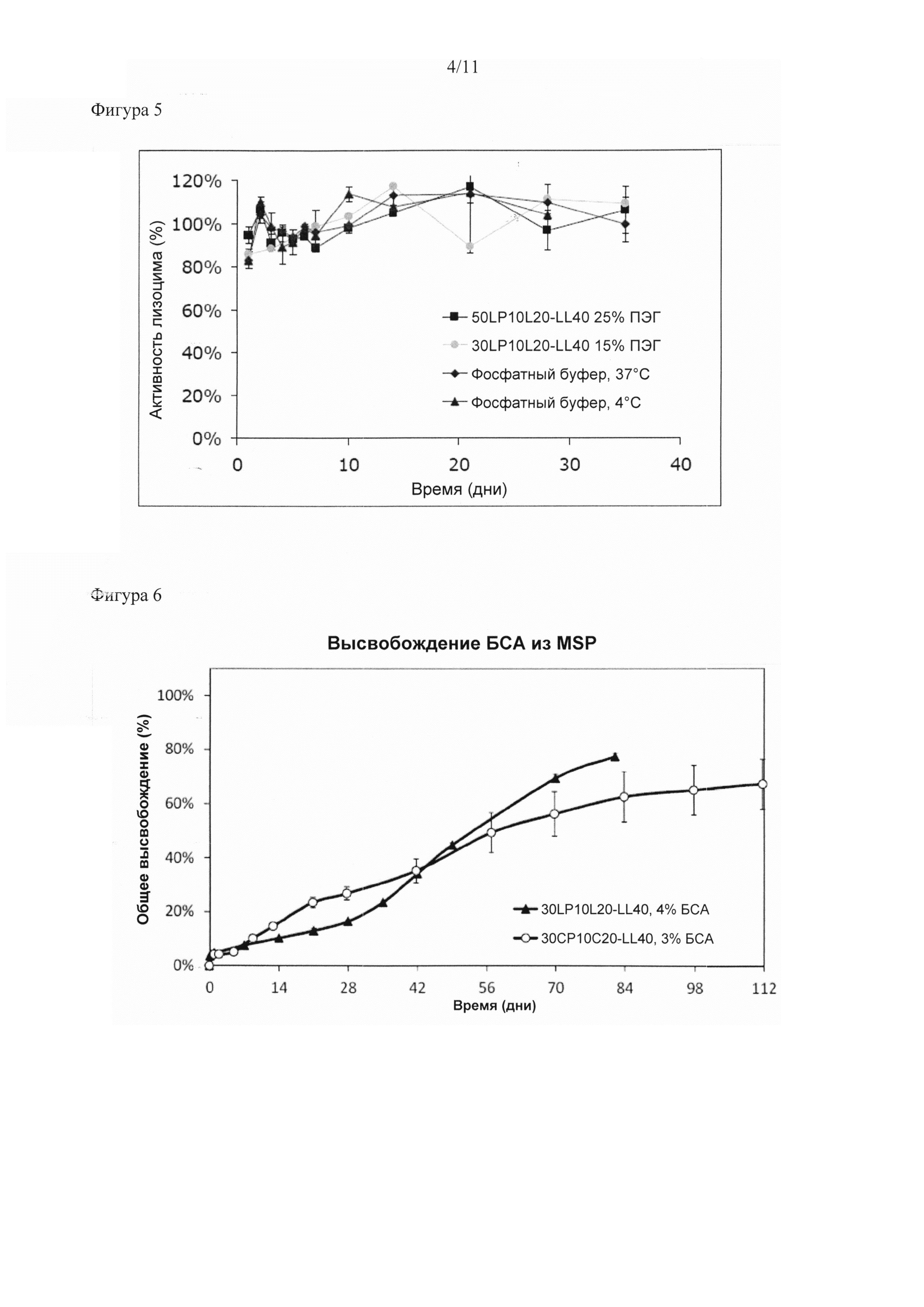
Было обнаружено, что биоразлагаемый мультиблочный сополимер согласно настоящему изобретению особенно подходит в качестве носителя для доставки полипептида, обеспечивающего контролируемое высвобождение полипептида из матрицы в окружающую среду, например, в организм субъекта.
Существует множество вариантов регулирования свойств высвобождения мультиблочных сополимеров согласно настоящему изобретению для доставки композиции для конкретного применения. Скорость высвобождения биологически активного соединения, например, можно повышать путем:
- увеличения молекулярной массы водорастворимого полимера в преполимере (А) при сохранении молекулярной массы преполимера (А);
- увеличения мольного отношения преполимера (А) к преполимеру (В);
- увеличения содержания мономера, обеспечивающего получение полимера в преполимере (А) с более высокой скоростью разложения, например, путем замены ε-капролактона на D,L -лактид или гликолид или путем замены D,L-лактида на гликолид;
- снижения молекулярной массы преполимера (В) при сохранении мольного отношения преполимера (А) к преполимеру (В) (это приводит к увеличению процентного массового содержания преполимера (А), а также снижает Тпл преполимера (В) и общее количество кристаллической фазы);
- снижения молекулярной массы преполимера (А) при сохранении молекулярной массы водорастворимого полимера и мольного отношения преполимера (А) к преполимеру (В); и/или
- применения дополнительного третьего сегмента, полученного из водорастворимого полимера, за счет чего повышается содержание водорастворимого полимера.
Скорость высвобождения можно снижать за счет изменений, противоположных указанным выше, а также путем
- увеличения Тпл сегмента B, например, за счет применения смеси PLLA и PDLA в качестве преполимера (В) (вместо одной PLLA) с таким соотношением, при котором происходит образование стереокомплекса PLLA и PDLA;
- применения дополнительного третьего сегмента, полученного из водорастворимого полимерного диола, где диизоцианат применяют в качестве удлинителя цепи, а содержание водорастворимого полимера сохраняется или снижается. Водорастворимый полимер в третьем сегменте состоит из мультиблочного сополимера с медленно разлагающейся уретановой связью в отличие от водорастворимого полимера с быстро разлагающейся сложноэфирной связью в преполимере (А).
Биологически активные соединения, которые могут содержаться в матрице мультиблочного сополимера, такой как матрица поли(D,L-молочная кислота)-ПЭГ-поли(D,L-молочная кислота)-блок-PLLA ((PDLLA-ПЭГ-PDLLA)-блок-PLLA) или матрица поли(ε-капролактон)-ПЭГ-поли(ε-капролактон)-блок-PLLA ((ПКЛ-ПЭГ-ПКЛ)-блок-PLLA), включают, но не ограничиваются ими, отличные от пептидов и белков Низкомолекулярные лекарственные средства с молекулярной массой, в целом, составляющей 1000 Да или менее, а также биологически активные пептиды.
Примеры отличных от пептидов и белков низкомолекулярных лекарственных средств, которые могут содержаться в сополимерной полиэфирэфирной полиуретановой матрице, такой как матрица (PDLLA-ПЭГ-PDLLA)-блок-PLLA или матрица ПКД-ПЭГ-ПКЛ-блок-PLLA, включают, но не ограничиваются ими, противоопухолевые агенты, противомикробные агенты, включая антибиотики, цефалоспорины, аминогликозиды; макролиды; тетрациклины, химиотерапевтические агенты, включая сульфонамиды; антисептики мочевыводящих путей; лекарственные средства против анаэробных инфекций; лекарственные средства от туберкулеза; лекарственные средства от проказы, противогрибковые агенты, противовирусные агенты, агенты от гельминтоза, противовоспалительные агенты, агенты от подагры, анальгетики центрального действия (опиоиды); местные анестетики, лекарственные средства от болезни Паркинсона, мышечные релаксанты центрального действия, гормоны и антагонисты гормонов, кортикостероиды, глюкокортикостероиды, андрогены, андрогенные стероиды, анаболические стероиды, антиандрогены, эстрогены, эстрогенные стероиды, антиэстрогены, прогестины; лекарственные средства для щитовидной железы и антитиреоидные средства.
При включении низкомолекулярного лекарственного средства, такого как описано выше, в. матрицу (PDLLA-ПЭГ-PDLLA)-блок-PLLA компонент ПЭГ сополимера предпочтительно имеет молекулярную массу от 200 до 1500 г/моль, более предпочтительно от 600 до 1000 г/моль, и его содержание в сополимере составляет от 5 масс. % до 20 масс. %) от массы сополимера, предпочтительно от 5 масс. %) до 10 масс. %о от массы сополимера. В целом, содержание PLLA в сополимере составляет от 20 масс. %) до 90 масс. % от массы сополимера, предпочтительно от 30 масс. % до 70 масс. % сополимера. Содержание по меньшей мере одной молекулы низкомолекулярного лекарственного средства в матрице может составлять от 0,1 масс. % до 80 масс. %, предпочтительно от 1,0 масс. %) до 40 масс. %, наиболее предпочтительно от 5 до 20 масс. %о. Если желательным является повышение гидрофильности мультиблочного сополимера и, таким образом, увеличение скорости разложения сополимера и скорости высвобождения включенного биологически активного соединения, сополимер можно модифицировать путем частичной или полной замены D,L-лактида в гидрофильном сегменте на гликолид и/или путем применения компонента ПЭГ с более высокой молекулярной массой или путем повышения массовой доли компонента ПЭГ в сегменте преполимера. Если желательным является снижение гидрофильности полимера и, таким образом снижение скорости разложения сополимера и скорости высвобождения включенного биологически активного соединения, сополимер можно модифицировать путем частичной или полной замены D,L-лактида в гидрофильном сегменте на ε-капролактон и/или путем использования компонента ПЭГ с более низкой молекулярной массой или путем снижения массовой доли компонента ПЭГ в сегменте преполимера.
Полипептид состоит из аминокислот, связанных пептидными связями. Короткие полипептиды также называют пептидами, тогда как более длинные полипептиды, как правило, называют белками. Одним из условий отнесения полипептидных цепей к пептидам, а не белкам, является возможность их синтеза из составляющих аминокислот. Тем не менее, в связи с появлением усовершенствованных способов синтеза можно получать полипептиды, содержащие сотни аминокислот, включая полные белки, такие как убиквитин. Другая условная граница разделения пептидов и белков составляет примерно 50 аминокислот в молекуле. Это определение до некоторой степени является субъективным. Длинные полипептиды, такие как амилоидный бета-пептид, связанный с болезнью Альцгеймера, можно рассматривать как белки; а низкомолекулярные белки, такие как инсулин, можно рассматривать как пептиды. В любом случае, для специалистов в данной области техники будет очевидно, что по существу любые типы полипептидов можно инкапсулировать, а затем высвобождать из матрицы сополимера.
В одном из вариантов реализации композиция согласно настоящему изобретению содержит биологически активный пептид или биологически активный белок. Инкапсулированные полипептиды предпочтительно содержат только природные аминокислоты, хотя в качестве альтернативы можно применять и синтетические аминокислоты (т.е. соединения, которые не содержатся в природе, но которые можно включать в полипептидную цепь) и/или аналоги аминокислот, известные в данной области техники. Кроме того, одну или более аминокислот в полипептиде можно модифицировать, например, путем добавления химического фрагмента, такого как углеводная группа, фосфатная группа, фарнезиловая группа, изофарнезиловая группа, группа жирной кислоты, линкерная группа для сопряжения, функционализации или иной модификации (например, альфа-амидирования) и т.д.
В предпочтительном варианте реализации модификации пептида приводят к получению более стабильного пептида (например, с более высоким периодом полувыведения in vivo). Указанные модификации могут включать циклизацию пептида, включение D-аминокислот и т.д. Модификации не должны по существу нарушать желаемую биологическую активность пептида. В конкретных вариантах реализации модификации пептида приводят к получению пептида с более высокой биологической активностью.
Биологически активный полипептид предпочтительно выбран из группы, состоящей из белковых/пептидных лекарственных средств, ферментов, лигандов рецепторов, нейротрансмиттеров, ингибиторных пептидов, регуляторных пептидов, активаторных пептидов, цитокинов, факторов роста, моноклональных антител, фрагментов моноклональных антител, противоопухолевых пептидов, антибиотиков, антигенов, вакцин и гормонов. Типовые полипептиды, которые можно инкапсулировать, указаны в патенте США №5980948 A и в Crommelin et al, Int. J. Pharm. 2003, 266(1-2), 3-16. Очевидно, предполагается, что можно инкапсулировать два или более различных (биологически активных) полипептидов.
Размер полипептида(ов) может быть различным. В одном из вариантов реализации полипептид имеет молекулярную массу 10000 Да или менее. Было обнаружено, что полипептиды указанного размера особенно подходят для инкапсулирования в матрицу сополимера, содержащего ПЭГ в качестве сегмента преполимера (А) и/или дополнительного преполимера, где указанный ПЭГ имеет среднечисловую молекулярную массу от 400 до 3000 г/моль, предпочтительно от 600 до 1500 г/моль. В качестве альтернативы или в дополнение содержание указанного ПЭГ составляет от 5 масс. % до 60 масс. %, от общей массы сополимера, предпочтительно от 5 масс. % до 40 масс. %).
В другом варианте реализации указанный полипептид представляет собой биологически активный белок с молекулярной массой 10000 Да или более. Указанные более крупные полипептиды предпочтительно инкапсулируют в матрицу из сополимера, содержащего ПЭГ в качестве сегмента преполимера (А) и/или дополнительного преполимера, где указанный ПЭГ имеет среднечисловую молекулярную массу от 600 до 5000 г/моль, предпочтительно от 1000 до 3000 г/моль, и/или где указанный ПЭГ содержится в количестве от 5 масс. %) до 70 масс. % от общей массы сополимера, более предпочтительно от 10 масс. % до 50 масс. %.
Композиция согласно настоящему изобретению может быть представлена в любом виде или форме. В одном из вариантов реализации мультиблочные сополимеры согласно настоящему изобретению перерабатывают в форму микросфер, микрочастиц, напылений, имплантата, оболочки, геля, пленки, фольги, покрытия, мембраны или стержня.
Один из конкретных аспектов относится к композиции в виде микросфер. В целом, микросферы представляют собой мелкодисперсные сферические частицы с диаметром менее 1000 мкм, содержащие биологически активное соединение. Микросфера может быть гомогенной или монолитной микросферой, где биологически активное соединение содержится в виде раствора или дисперсии в полимерной матрице. Также микросфера может представлять собой резервуар, где биологически активное соединение заключено внутри полимера в одноядерном или полиядерном состоянии. Если биологически активное соединение представляет собой низкомолекулярное водорастворимое лекарственное средство, то лекарственное средство сначала можно диспергировать в гидрофобном или липофильном наполнителе, после чего комбинацию диспергируют в виде частиц, капель или микросуспензий в полимерной матрице. Затем из эмульсии можно получать микросферы.
Микросферы можно получать при помощи способов, известных специалистам в данной области техники, включая, но не ограничиваясь ими, экстракцию/выпаривание растворителя, распылительную сушку или распылительную сублимационную сушку.
В одном из вариантов реализации микросферы получают при помощи способа экстракции/выпаривания растворителя, который включает растворение мультиблочного сополимера в органическом растворителе, таком как дихлорметан, и эмульсификацию раствора мультиблочного сополимера в водной фазе, содержащей эмульгатор, такой как поливиниловый спирт (способ описан помимо прочего в Okada, Adv. Drug Del. Rev. 1997, 28(1), 43-70).
Характеристики, такие как размер частиц, пористость и содержание лекарственного средства, в получаемых таким образом микросферах, зависят от параметров способа, таких как вязкость или концентрация водной фазы поливинилового спирта, концентрация раствора мультиблочного сополимера, соотношение раствора активного вещества в дихлорметане и водного раствора, соотношение первичной эмульсии и фазы поливинилового спирта и скорость перемешивания.
При получении микросфер при помощи способа распылительной сушки применяют мультиблочный сополимер с низкой концентрацией в органическом растворителе от 0,5 масс. % до 5 масс. %, предпочтительно примерно 2 масс. %. Распылительная сушка, в целом, приводит к получению пористых частиц неправильной формы.
После образования микросфер биологически активное соединение оказывается инкапсулированным в микросферах или микрочастицах. В целом, при использовании техники экстракции/выпаривания растворителя для инкапсулирования липофильных соединений соединение сначала растворяют в растворе мультиблочного сополимера в органическом растворителе, таком как дихлорметан или этилацетат. Органический раствор затем эмульгируют в водном растворе поливинилового спирта, что приводит к получению эмульсии типа масло-в-воде (М/В). Органический растворитель затем экстрагируют в водной фазе и выпаривают для отверждения микросфер.
В целом, при использовании техники выпаривания растворителя для инкапсулирования водорастворимого соединения водный раствор соединения сначала эмульгируют в растворе мультиблочного сополимера в органическом растворителе, таком как дихлорметан. Эту первичную эмульсию затем последовательно эмульгируют в водном растворе поливинилового спирта, что приводит к получению эмульсии типа вода-в-масле-в-воде (В/М/B). Органический растворитель, такой как дихлорметан или этилацетат, затем экстрагируют аналогично способу для эмульсии М/B для отверждения микросфер. В качестве альтернативы водорастворимые агенты можно диспергировать непосредственно в растворе мультиблочного сополимера в органическом растворителе. Получаемую дисперсию затем эмульгируют в водном растворе, содержащем поверхностно активное вещество, такое как поливиниловый спирт, что приводит к получению эмульсии типа твердое вещество-в-масле-в-воде (Т/М/В). Органический растворитель затем экстрагируют аналогично способу для эмульсии М/В для отверждения микросфер.
При использовании способов эмульсификации B/М/В и T/М/В для инкапсулирования водорастворимого соединения может возникать проблема получения микросфер с достаточной эффективностью инкапсулирования. Вследствие растворимости соединения в воде часть соединения может теряться и переходить в водную экстрагирующую среду, такую как водный раствор поливинилового спирта. Для снижения диффузии соединения из внутренней водной фазы во внешнюю водную фазу во внутренней водной фазе можно применять загуститель, такой как желатин. Кроме того, для снижения растворимости соединения во внешней водной фазе в нее можно вводить добавки. Для этого можно применять соли или изменять pH.
Способы эмульсификации по типу вода-в-масле-в-масле (В/М/М) или твердое вещество-в-масле-в-масле (Т/М/М) обеспечивают интересный альтернативный способ получения микросфер с достаточной эффективностью инкапсулирования. В способе B/М/М биологически активное соединение по аналогии со способом В/М/В растворяют в водном растворе и эмульгируют с использованием раствора полимера в органическом растворителе, как правило, таком как дихлорметан или этилацетат. Затем для получения зародышевых микрочастиц при перемешивании медленно добавляют полимерный осадитель, такой как силиконовое масло, затем зародышевые микрочастицы выливают в гептан или гексан для экстракции силиконового масла и органического растворителя и отверждения микросфер. Микрочастицы можно собирать путем вакуумного фильтрования, после чего их промывают дополнительным растворителем и сушат в вакууме. В способе эмульсификации T/М/М биологически активное соединение по аналогии со способом T/М/В в виде твердого порошка диспергируют в растворе полимера в органическом растворителе, таком как дихлорметан или этилацетат. Затем для получения зародышевых микрочастиц при перемешивании медленно добавляют полимерный осадитель, такой как силиконовое масло, затем зародышевые микрочастицы выливают в гептан или гексан для экстракции силиконового масла и дихлорметана и отверждения микросфер.
В водный раствор белка для предотвращения потери активности белка во время обработки с получением микросфер можно добавлять стабилизаторы. Примерами указанных стабилизаторов являются альбумин сыворотки человека, желатин и углеводы, такие как трегалоза, инулин и сахароза.
При использовании метода распылительной сушки водный раствор соединения эмульгируют в растворе сополимера в органическом растворителе, таком как метиленхлорид, как описано выше в настоящем описании. Эмульсию типа вода-в-масле затем сушат распылением с использованием распылительной сушилки.
В дополнительных вариантах реализации композиция согласно настоящему изобретению имеет форму оболочки, инъецируемого геля, имплантата (предпочтительно инъецируемого имплантата) или имплантата, покрытого оболочкой. Композицию в форме оболочки можно применять в качестве покрытий, через которые выделяются лекарственные средства, например, на медицинском имплантате, таком как сосудистый или мочевой стент, ортопедический протез или глазной имплантат.
Биологически активные соединения можно вводить в состав инъецируемых твердых имплантатов путем экструзии. Как правило, порошкообразные соединение и мультиблочный сополимер физически перемешивают, после чего получаемую смесь порошков вводят в экструдер, нагревают и обрабатывают с получением составов с желаемой формой и размерами, таких как цилиндрический стержень с низким диаметром. Вместо физического перемешивания порошкообразных соединения и мультиблочного сополимера соединение и полимер можно совместно растворять в подходящем растворителе или же можно получать дисперсию соединения в растворе полимера в подходящем растворителе, после чего проводить лиофильную сушку и экструзию лиофилизированного порошка. Последний способ, в целом, улучшает гомогенность смеси и однородность состава имплантата.
Согласно другому аспекту изобретение относится к способу доставки биологически активного соединения субъекту, нуждающемуся в этом, включающему введение эффективной дозы композиции, такой как определено в настоящем описании, указанному субъекту.
Субъект, как правило, представляет собой млекопитающее, предпочтительно человека. Тем не менее, также предполагается и ветеринарное применение. Способ можно использовать в терапевтических, профилактических и/или косметических целях. В зависимости от обстоятельств можно выбирать любой подходящий способ введения. Например, введение может включать парентеральное, пероральное, внутриартериальное, внутрисуставное, внутрипочечное, внутриглазное, эпидуральное, интратекальное, внутримышечное, интраперитонеальное, внутривенное, внутривагинальное, ректальное, местное или подкожное введение композиции. В одном из вариантов реализации в изобретении предложен способ доставки указанного биологически активного полипептида субъекту, нуждающемуся в этом, включающий введение эффективной дозы композиции согласно настоящему изобретению указанному субъекту, причем композиция имеет форму микросфер, инъецируемого имплантата или геля, образующегося in situ, при этом композицию вводят внутрь глаза, внутриартериально, внутримышечно или подкожно.
Для местного введения микросферы могут содержаться в геле, креме или мази, а также при желании могут быть покрыты защитным слоем. Таким образом, микросферы могут содержать одно или более биологически активных соединений, применяемых для лечения заболеваний кожи, таких как псориаз, экзема, себорея и дерматит.
В другом варианте реализации микросферы могут содержаться в геле, таком как гель гиалуроновой кислоты или гель макромолекулярного полисахарида. Указанные вариант реализации можно применять, в частности для парентерального введения, например, во время и после хирургии.
При введении путем инъекции микросферы могут содержаться в фармацевтическом носителе, таком как вода, солевой раствор (например, 0,9%) или раствор, содержащий поверхностно активное вещество в количестве от 0,1% (масс./об.) до 0,5% (масс./об.). Примеры поверхностно активных веществ, которые можно применять, включают, но не ограничиваются ими, поверхностно активное вещество Tween 80. Фармацевтический носитель может дополнительно содержать загуститель, такой как карбоксиметилцеллюлоза натрия.
При введении путем инъекции средний размер микросфер составляет от 1 мкм до 200 мкм, предпочтительно от 5 мкм до 100 мкм, наиболее предпочтительно от 10 мкм до 50 мкм. Указанные микросферы при введении в комбинации с приемлемым фармацевтическим наполнителем можно применять для лечения ряда заболеваний или нарушений в зависимости от инкапсулированного биологически активного соединения. Таким образом, инъецируемые составы, содержащие микросферы согласно настоящему изобретению, можно применять для лечения системных заболеваний, таких как ревматоидный артрит, гепатит, диабет или метаболические синдромы, и местно локализованных заболеваний, таких как остеоартрит, заболевания почек, воспаления, местные болезненные процессы, местные инфекции, местные заболевания кожи, опухолей (или их фрагментов, остающихся после хирургического удаления, в качестве послеоперационного лечения для уничтожения каких-либо возможно остающихся опухолевых клеток), рака простаты или молочной железы, агромегалии, заболеваний глаз, таких как возрастная макулярная дегенерация, местных заболеваний мозга (например, болезни Паркинсона) и сердечнососудистых заболеваний, таких как острый инфаркт миокарда, хроническая сердечная недостаточность или артросклероз. Указанные инъецируемые составы также можно применять для долгосрочного терапевтического лечения, такого как, например, лечение с использованием кортикостероидов, андрогенов, антиандрогенов, эстрогенов, антиэстрогенов, прогестагеновых агентов или гормонов щитовидной железы или лекарственных средств против туберкулеза, проказы или малярии.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1 A: Термограммы ДСК 50LP10L20-LL40.
Фиг.1 B: Термограммы ДСК 30LP30L40-LL40.
Фиг. 1C: Термограммы ДСК 50CP10C20-LL40.
Фиг. 2: Общее высвобождение лизоцима из пленок, состоящих из 30LP10L20-LL40, 50LP10L20-LL40, 70LP10L20-LL40, 50CP10C20-LL40 и 30CP30C40-LL40. Пленки содержали 10 масс. % лизоцима. Высвобождение измеряли при 37°C в фосфатном буфере с pH 7,4 (n=3).
Фиг. 3: Общее высвобождение бычьего сывороточного альбумина (БСА) из пленок, состоящих из 30LP10L20-LL40, 50LP10L20-LL40, 70LP10L20-LL40, 30LP30L40-LL40 и 30CP30C40-LL40. Пленки содержали 10 масс. % БСА. Высвобождение измеряли при 37°C в фосфатном буфере с pH 7,4 (n=3).
Фиг. 4: Влияние состава гидрофильного блока мультиблочных сополимеров на
общее высвобождение лизоцима из пленок. Пленки состояли из 50LP10L20-LL40, 50GP10C20-LL40 или 50CP10C20-LL40 (25 масс. % ПЭГ 1000) и содержали 10 масс. % лизоцима. Высвобождение измеряли при 37°C в фосфатном буфере с pH 7,4 (n=3).
Фиг. 5: Активность лизоцима, высвобождаемого из пленок, состоящих из 30LP10L20-LL40 или 50LP10L20-LL40, содержащих 10 масс. % лизоцима (37°C, фосфатный буфер с pH 7,4), и активность лизоцима в растворах лизоцима (0,01 масс. % в фосфатном буфере с pH 7,4) при хранении при 4 и 37°C в зависимости от времени (n=3).
Фиг. 6: Высвобождение БСА in vitro из микросфер, состоящих из 30LP10L20-LL40 и 50CP10C20-LL40, содержащих 3-4 масс. % БСА, при 37°C в фосфатном буфере с pH 7,4 (n=3).
Фиг. 7: Высвобождение ИФР-1 in vitro из пленок 50CP30C50-LL40 и 30CP30C40-LL40, содержащих ИФР-1, полученных путем формования B/M с использованием заливки раствора. Высвобождение измеряли при 37°C в фосфатном буфере с pH 7,2 (n=3). Твердая линия соответствует высвобождению ИФР-1, измеренному путем СВЭЖХ. Пунктирные линии соответствуют высвобождению ИФР-1, измеренному путем ELISA.
Фиг. 8: Фотография СЭМ микросфер 50CP10C20-LL40, полученных при помощи способа двойной эмульсии В/М/В, содержащих 0,2 масс. % ИФР-1.
Фиг. 9: Высвобождение ИФР-1 in vitro из микросфер, полученных из 50СР10С20-LL40 с различными значениями IV, содержащих 0,2% ИФР-1. Высвобождение измеряли при 37°C в фосфатном буфере с pH 7,2 (n=3).
Фиг. 10: Результаты ДСН-ПААГ для ИФР-1, высвобождаемого из микросфер 50CP10C20-LL40 с заданным 0,2 масс. % содержанием ИФР-1, полученных при использовании различных скоростей Ultra-Turrax, в течение 1 и 2 недель.
Фиг. 11: Высвобождение белка A (ММ 15000 Да) из пленок, состоящих из 20LP10L20-LL40, 30LP6L20-LL40 и 30CP10C20-LL40 (содержание белка A 5 масс. %; толщина пленки 80-120 мкм). Высвобождение измеряли при 37°C в фосфатном буфере с pH 7,4 (n=3).
Фиг. 12: Фотография СЭМ микросфер, состоящих из 30CP10C20-LL40 (IV 0,71 дл/г), содержащих 3-4 масс. % белка A, полученных с использованием различных количеств инулина во внутренней водной фазе A: 0% инулина, В: 2% инулина. 1: общий вид. 2: увеличение.
Фиг. 13: Высвобождение белка A in vitro из микросфер, состоящих из 30СР10С20-LL40 с заданным 3-4 масс. % содержанием белка A, инкапсулированного совместно с 2 или 5 масс. % инулина, при 37°C в фосфатном буфере с pH 7,4 (n=3).
Фиг. 14: Высвобождение белка A in vitro из микросфер, состоящих из 30СР10С20-LL40 с различными IV, с заданным 3-4 масс. % содержанием белка A, при 37°C в фосфатном буфере с pH 7,4 (n=3).
Фиг. 15: Результаты ДСН-ПААГ для белка A, высвобождаемого из микросфер 30CP10C20-LL40 с заданным содержанием белка A 4 масс. % и инулина 2 масс. %, через 1 (линия 4), 7 (линия 7), 14 (линия 8) и 21 (линия 9) день. Линия 5: маркеры молекулярных масс. Линия 6: стандарт белка A. Следует отметить, что темные пятна вызваны окрашиванием фосфатных буферных солей.
Фиг. 16: Высвобождение пептида A (ММ 2500) in vitro из пленок, состоящих из 20LP10L20-LL40 (содержание пептида 5 и 10 масс. %; толщина пленки 80-100 мкм). Высвобождение измеряли при 37°C в фосфатном буфере с pH 7,4 (n=3).
Фиг. 17: Высвобождение пептида А (ММ 2500) in vitro из микросфер, состоящих из 20LP10L20-LL40 (размер частиц 30 мкм; содержание пептида 10 масс. %). Высвобождение измеряли при 37°C в фосфатном буфере с pH 7,4 (n=3).
Фиг. 18: Высвобождение рапамицина in vitro из микросфер, состоящих из различных смесей 20LP1020-LL40 и 10LP10L20-LL40.
Фиг. 19: Фотографии СЭМ микросфер 20LP1020-LL40, содержащих госерелин, полученных при помощи способа B/М/М.
Фиг. 20: Высвобождение госерелина in vitro из микросфер 20LP1020-LL40, полученных при помощи способа B/М/М.
ПРИМЕРЫ
В следующих примерах синтезировали различные биоразлагаемые полукристаллические мультиблочные сополимеры с разделенными фазами и проводили оценку технологических характеристик и контролируемого высвобождения. Полимеры состояли из жесткого сегмента B на основе кристаллического L-лактида, имеющего температуру плавления (Тпл), и сегмента А на основе гидрофильного поли(этиленгликоля) (ПЭГ), имеющего температуру стеклования (Тст) ниже температуры тела в физиологических условиях. В следующих примерах ПЭГ имеет обозначение молекулярной массы (ММ). Например, ПЭГ1000 относится к ПЭГ с MM 1000 г/моль.
ПРИМЕР 1:
В данном примере предложены общие способы получения преполимера (А), поли(D,L-лактид-ПЭГ). Мономеры взвешивали в трехгорлой колбе в атмосфере азота и сушили при 50°C в случае гликолида и D,L-лактида в течение по меньшей мере 16 часов при пониженном давлении. ПЭГ сушили при 90°C при пониженном давлении в течение по меньшей мере 16 часов. ПЭГ добавляли в мономер(ы) в атмосфере азота. Затем добавляли октоат олова и смесь перемешивали с использованием магнитной мешалки, взаимодействие в смеси проводили при 140°C в течение нескольких дней. Анализ1H ЯМР проводили при 300 МГц на ЯМР спектрометре VXR Unity Plus. Время ожидания d1 устанавливали на 20 секунд, число сканов составляло 16. Спектры снимали в диапазоне от 0 до 14 ppm. Конверсию и Mn преполимера определяли по данным1H ЯМР. Образцы для1H ЯМР получали путем растворения 10 мг полимера в 1 мл дейтерированного хлороформа.
ПРИМЕР 2:
В данном примере описано получение поли(DL-лактид-ПЭГ1000) (pLP10L20) с Mn 2000 г/моль. Взвешивали 149,84 грамма (1,04 моль) D,L-лактида (Purac) и добавляли 149,21 г (0,149 моль) ПЭГ с ММ 1000 (Ineos, категория PU). Добавляли 71,6 мг октоата олова (Sigma Corp) (мольное соотношение мономер/катализатор = 5900) и смесь перемешивали с использованием магнитной мешалки, взаимодействие проводили при 140°C в течение 245 часов. Анализ1H ЯМР показывал 94,8% конверсию мономера. Расчетная молекулярная масса (Mn) составляла 2000 г/моль. Молекулярная масса, определенная путем1H ЯМР, составляла 1950 г/моль.
ПРИМЕР 3
В данном примере описано получение поли(D,L-лактид-ПЭГ3000) (pLP30L40) с Mn 4000 г/моль. Взвешивали 50,35 г (0,349 моль) D,L-лактида (Purac) и добавляли 151,08 г (50,4 ммоль) ПЭГ с ММ 3000 (Sigma Corp). Добавляли 37,5 мг октоата олова (Sigma Corp) (мольное соотношение мономер/катализатор = 4300) и смесь перемешивали с использованием магнитной мешалки, взаимодействие проводили при 140°C в течение 90 часов. Анализ1H ЯМР показывал 93,4% конверсию мономера. Расчетная молекулярная масса (Mn) составляла 4000 г/моль. Молекулярная масса, определенная путем 1И ЯМР, составляла 3940 г/моль.
ПРИМЕР 4
В данном примере описано получение преполимера, поли(ε-капролактон-ПЭГ1000) (рСР10С20) с Mn 2000 г/моль. 100,81 г (0,101 моль) ПЭГ с ММ 1000 (Ineos, категория PU) взвешивали в трехгорлой колбе в атмосфере азота и сушили при 90°C в течение по меньшей мере 16 часов при пониженном давлении. В ПЭГ в атмосфере азота добавляли 101,76 г (0,892 моль) ε-капролактона (Arcos, предварительно высушенного и перегнанного над CaH2 при пониженном давлении) и смесь нагревали до 135°C. Добавляли 57,9 мг октоата олова (Sigma Corp) (мольное соотношение мономер/катализатор=6200) и смесь перемешивали с использованием магнитной мешалки, взаимодействие проводили при 135°C в течение 76 часов. Анализ1H ЯМР показывал 100% конверсию мономера. Расчетная молекулярная масса (Mn) составляла 2010 г/моль. Молекулярная масса, определенная путем1H ЯМР, составляла 1950 г/моль.
ПРИМЕР 5
В данном примере описано получение преполимера, поли(ε-капролактон-ПЭГ3000) (рСР30С40) с Mn 4000 г/моль. 176,60 г (58,9 ммоль) ПЭГ с ММ 3000 (Ineos, категория PU) взвешивали в трехгорлой колбе в атмосфере азота и сушили при 90°C в течение по меньшей мере 16 часов при пониженном давлении. В ПЭГ при пониженном давлении добавляли 59,4 г (0,520 моль) s-капролактона (Acros, предварительно высушенного и перегнанного над CaH2 при пониженном давлении) и смесь нагревали до 135°C. Добавляли 69,6 мг октоата олова (Sigma Corp) (мольное соотношение мономер/катализатор=3000) и смесь перемешивали с использованием магнитной мешалки, взаимодействие проводили при 135°C в течение 243 часов. Анализ1H ЯМР показывал 100% конверсию мономера. Расчетная молекулярная масса (Mn) составляла 2010 г/моль. Молекулярная масса, определенная путем1H ЯМР, составляла 1950 г/моль.
ПРИМЕР 6
В данном примере описано получение преполимера, поли(L-молочной кислоты) (LL4000) с Mn=4000 г/моль, инициируемое 1,4-бутандиолом (BDO). 399,89 г (2,77 моль) L-лактида (Purac) взвешивали в трехгорлой колбе в атмосфере азота и сушили при 50°C в течение по меньшей мере 16 часов при пониженном давлении. В L-лактид в атмосфере азота добавляли 9,36 г (0,104 моль) BDO (Acros, предварительно перегнанного при пониженном давлении). Для растворения L-лактида и BDO добавляли 434 мл диоксана (Acros, предварительно высушенного и перегнанного над натриевой проволокой) и смесь нагревали до 80°C. Добавляли 87,8 мг октоата олова (Sigma Corp) (мольное соотношение мономер/катализатор=12800). Смесь перемешивали с использованием магнитной мешалки, взаимодействие проводили при 80°C в течение 50,6 часа. Полимер выделяли из диоксана путем лиофильной сушки в течение 72 часов до достижения конечной температуры 50°C. Если полимер растворяли в диоксане, то сначала при пониженном давлении при 50°C удаляли диоксан. Анализ1H ЯМР показывал 96,5% конверсию мономера. Расчетная молекулярная масса (Mn) составляла 3940 г/моль. Молекулярная масса, определенная путем1H ЯМР, составляла 3900 г/моль. После лиофильной сушки содержание диоксана определяли путем1H ЯМР (300 МГц, 50 мг полимера растворяли в 1 мл дейтерированного хлороформа, d1=30 сек, 32 скана). Для количественной оценки содержания диоксана в образце растворяли 5 мг дибромбензола (Acros). Обнаружили, что содержание диоксана составляло 1193 ppm.
ПРИМЕР 7
В данном примере описаны общие способы, применяемые для получения мультиблочных сополимеров. Преполимеры, ε-капролактон-ПЭГ-ε-капролактон (СРС) или D,L-лактид-ПЭГ-D,L-лактид (LPL) (Mn 2000 г/моль) нагревали до 50-80°C до тех пор, пока они не становились более текучими. Соответствующие количества преполимера LL4000 (Mn 4000 г/моль) и преполимера СРС или LPL взвешивали в стеклянной ампуле, оборудованной подводом азота, и сушили при 50°C в течение по меньшей мере 48 часов. Затем к стеклянной ампуле присоединяли механическую мешалку. 1,4-диоксан (Acros, перегнанный над натрием) добавляли до достижения 30 масс. % концентрации полимера, и для растворения преполимеров содержимое ампулы нагревали до 80°C. Добавляли 0,900-0,990 эквивалента (относительно количества гидроксильных групп в преполимере) 1,4-бутандиизоцианата (Bayer, перегнанного при пониженном давлении) и реакционную смесь перемешивали с использованием механической мешалки в течение 16-22 часов. Для гашения непрореагировавших изоцианатных групп неперегнанный диоксан добавляли до достижения 20 масс. % концентрации полимера. Реакционную смесь дополнительно разбавляли неперегнанным диоксаном до достижения 10 масс. % концентрации полимера. Ампулу охлаждали до комнатной температуры, реакционную смесь выливали в неглубокую емкость и замораживали при -18°C. Затем диоксан удаляли путем размещения замороженной реакционной смеси в вакууме при 30°C. Полимер хранили в герметичной упаковке при -18°C. Анализировали термические свойства (мДСК), содержание диоксана (газовая хроматография), характеристическую вязкость и состав полимера (1H ЯМР) небольшой части навески. Термический анализ проводили путем модулированной дифференциальной сканирующей калориметрии (мДСК). В кювете для ДСК взвешивали образцы по 5-10 мг. Измерение проводили на DSC Q1000 (TA Instruments) с использованием программы модуляции температуры. Амплитуду устанавливали на 1°C, период модуляции на 60 секунд, а скорость нагрева на 5°C/мин. Образцы нагревали от -80°C до 100-200°C (в зависимости от типа полимера). Характеристическую вязкость измеряли на вискозиметре Уббелоде (DIN) типа 0C, 0a или I, Schott Gerate, поставляемом с вискозиметром Schott AVS-450, оборудованным водяной баней. Измерения проводили в хлороформе при комнатной температуре. Концентрация полимера в хлороформе была такой, что относительная вязкость находилась в диапазоне от 1,2 до 2,0. Содержание диоксана определяли при помощи ГХ-ПИД анализа равновесной паровой фазы. Измерения проводили на ГХ-ПИД Combi Sampler, поставляемом с колонкой Agilent, DB-624/30 м/0,53 мм. Готовили образцы в ДМСО. Содержание диоксана определяли с использованием калибровочных стандартов диоксана.
ПРИМЕР 8
В данном примере описано получение 20(D,L-лактид-ПЭГ1000-D,L-лактид)2000-80(L-лактид)4000 (20LP10L20-LL40). Взвешивали 42,02 г преполимера LL40 (Mn 4040 г/моль, 10,40 ммоль) и 10,16 г преполимера D,L-лактид-ПЭГ1000-D,L-лактида (Mn 2000 г/моль, 5,08 ммоль) и растворяли в 100 мл 1,4-диоксана при 80°C. Добавляли 1,8466 г (13,2 ммоль) 1,4-бутандиизоцианата (0,851 эквивалента относительно количества гидроксильных групп в преполимере) и 20 мл 1,4-диоксана. Через 17 часов реакцию гасили 88 мл неперегнанного диоксана и смесь дополнительно разбавляли 255 мл неперегнанного диоксана. Диоксан удаляли путем введения замороженной реакционной смеси в вакуум при 30°C.
ПРИМЕР 9
В данном примере описано получение 30(D,L-лактид-ПЭГ1000-D,L-лактид)2000-7(L-лактид)4000 (30LP10L20-LL40). Взвешивали 34,44 г преполимера LL40 (Mn 4020 г/моль, 8,57 ммоль) и 14,95 г преполимера D,L-лактид-ПЭГ1000-D,L-лактида (Mn 2040 г/моль, 7,33 ммоль) и растворяли в 100 мл 1,4-диоксана при 80°C. Добавляли 2,7386 г (19,5 ммоль) 1,4-бутандиизоцианата (1,231 эквивалента относительно количества гидроксильных групп в преполимере) и 20 мл 1,4-диоксана. Через 20 часов реакцию гасили 85 мл неперегнанного диоксана и смесь дополнительно разбавляли 240 мл неперегнанного диоксана. Диоксан удаляли путем введения замороженной реакционной смеси в вакуум при 30°C.
ПРИМЕР 10
В данном примере описано получение 50(D,L-лактид-ПЭГ1000-D,L-лактид)2000-50(L-лактид)4000 (50LP10L20-LL40). Взвешивали 19,59 г преполимера LL40 (Mn 4060 г/моль, 4,83 ммоль) и 19,57 г преполимера D,L-лактид-ПЭГ1000-D,L-лактида (Mn 2040 г/моль, 9,59 ммоль) и растворяли в 78 мл 1,4-диоксана при 80°C. Добавляли 2,0018 г (14,3 ммоль) 1,4-бутандиизоцианата (0,991 эквивалента относительно количества гидроксильных групп в преполимере) в 20 мл 1,4-диоксана. Через 20 часов реакцию гасили 67 мл неперегнанного диоксана и смесь дополнительно разбавляли 189 мл неперегнанного диоксана. Диоксан удаляли путем введения замороженной реакционной смеси в вакуум при 30°C.
ПРИМЕР 11
В данном примере описано получение 70(D,L-лактид-ПЭГ1000-D,L-лактид)2000-30(L-лактид)4000 (70LP10L20-LL40). Взвешивали 8,59 г преполимера LL40 (Mn 4020 г/моль, 2,14 ммоль) и 19,96 г преполимера D,L-лактид-ПЭГ1000-D,L-лактида (Mn 2040 г/моль, 9,78 ммоль) и растворяли в 48 мл 1,4-диоксана при 80°C. Добавляли 1,648 г (11,8 ммоль) 1,4-бутандиизоцианата (0,986 эквивалента относительно количества гидроксильных групп в преполимере) и 20 мл 1,4-диоксана. Через 21 час реакцию гасили 49 мл неперегнанного диоксана и смесь дополнительно разбавляли 147 мл неперегнанного диоксана. Диоксан удаляли путем введения замороженной реакционной смеси в вакуум при 30°C.
В данном примере описано получение 30(D,L-лактид-ПЭГ3000-D,L-лактид)4000-70(L-лактид4000 (30LP30L40-LL40). Взвешивали 29,96 г преполимера LL40 (Mn 4030 г/моль, 7,43 ммоль) и 14,01 г преполимера D,L-лактид-ПЭГ1000-D,L-лактида (Mn 4000 г/моль, 3,50 ммоль) и растворяли в 83 мл 1,4-диоксана при 80°C. Добавляли 1,52 г (10,8 ммоль) 1,4-бутандиизоцианата (0,992 эквивалента относительно количества гидроксильных групп в преполимере) и 20 мл 1,4-диоксана. Через 21 час реакцию гасили 74 мл неперегнанного диоксана и смесь дополнительно разбавляли 222 мл неперегнанного диоксана. Диоксан удаляли путем введения замороженной реакционной смеси в вакуум при 30°C.
ПРИМЕР 13
В данном примере описано получение 50(ε-капролактон-ПЭГ1000-ε-капролактон)2000-50(L-лактид)4000 (50CP10C20-LL40). Взвешивали 24,34 г преполимера LL40 (Mn 4030 г/моль, 6,04 ммоль) и 23,87 г преполимера ε-капролактон-ПЭГ1000-ε-капролактона (Mn 2010 г/моль, 11,9 ммоль) и растворяли в 95 мл 1,4-диоксана при 80°C. Добавляли 2,4098 г (17,2 ммоль) 1,4-бутандиизоцианата (0,960 эквивалента относительно количества гидроксильных групп в преполимере) и 20 мл 1,4-диоксана. Через 18 часов реакцию гасили 82 мл неперегнанного диоксана и смесь дополнительно разбавляли 246 мл неперегнанного диоксана. Диоксан удаляли путем введения замороженной реакционной смеси в вакуум при 30°C.
ПРИМЕР 14
В данном примере описано получение 30(ε-капролактон-ПЭГ3000-ε-капролактон)4000-70(L-лактид)4000 (30CP30C40-LL40). Взвешивали 35,84 г преполимера LL40 (Mn 4030 г/моль, 8,89 ммоль) и 14,79 г преполимера ε-капролактон-ПЭГ3000-ε-капролактона (Mn 4010 г/моль, 3,69 ммоль) и растворяли в 100 мл 1,4-диоксана при 80°C. Добавляли 1,7428 г (12,4 ммоль) 1,4-бутандиизоцианата (0,988 эквивалента относительно количества гидроксильных групп в преполимере) и 20 мл 1,4-диоксана. Через 18 часов реакцию гасили 83 мл неперегнанного диоксана и смесь дополнительно разбавляли 240 мл неперегнанного диоксана. Диоксан удаляли путем введения замороженной реакционной смеси в вакуум при 30°C.
ПРИМЕР 15
Проводили анализ химического состава, молекулярной массы и остаточного содержания диоксана в синтезированных мультиблочных сополимерах. В Таблице 1 показаны результаты анализа 20LP10L20-LL40, 30LP10L20-LL40, 50LP10L20-LL40, 70LP10L20-LL40, 20LP30L40-LL40, 50CP10C20-LL40, 30CP30C40-LL40. Фактический состав сополимеров, определенный путем1H ЯМР по значениям мольных соотношений L/P и С/Р, хорошо соответствовал заявленным значениям. Все полимеры имели характеристическую вязкость от 0,7 до 1,1 дл/г. Содержание диоксана составляло менее 1000 ppm, что указывало на эффективное удаление диоксана путем вакуумной сушки.
Для подтверждения наличия структуры с разделенными фазами проводили анализ термических свойств мультиблочных сополимеров. Результаты показаны в Таблице 2. На Фигуре 1 показаны типовые термограммы ДСК мультиблочных сополимеров 50LP10L20-LL40 (Фигура 1А), 30LP30L40-LL40 (Фигура 1B) и 50CP10C20-LL40 (Фигура 1C). Все мультиблочные сополимеры имели температуру плавления (Тпл), обусловленную плавлением сегмента LL40, составляющую примерно 120-133°C. Как ожидалось, энтальпия плавления (ΔНпл) кристаллического сегмента LLA40 повышалась при увеличении количества сегмента. 70LP10L40-LL40, 50CP10C20-LL40 также имели Тпл примерно 85°C, что определяется плавлением менее идеальных кристаллов LL40. Сополимеры, содержащие ПЭГ3000, имели Тпл примерно 40°C, вызванную плавлением ПЭГ. Температура стеклования (Тст) мультиблочных сополимеров, в целом, находится между значениями Тст преполимера (А) и преполимера (В), что указывает на смешение фаз аморфного преполимера (А) и аморфной части преполимера (В). Тст мультиблочных сополимеров типа LP10L20-LL40 повышалась от -18 до 50°C при увеличении содержания сегмента LLA40 от 30 до 80%. Тст указанных мультиблочных сополимеров находится между значениями Тст преполимера (A) (pLP10L20, Тст -37°C) и преполимера (В) (LL40, Тст ~50°C), что связано, таким образом, со смешением аморфного полилактида полукристаллического блока LL40 и ПЭГ. 50CP10C20-LL40 имел Тст -48°C, что также связано со смешением аморфного ПЭГ, поликапролактона и полилактида. В Таблице 3 показаны значения степени набухания мультиблочных сополимеров. Для измерения характеристик набухания полимеров получали полимерные пленки путем заливки 13 масс. % раствора полимера в дихлорметане (примерно 300 мг полимера в 1,5 мл дихлорметана) на стеклянную пластинку и распределения раствора полимера с использованием формовочного ножа или путем литья в форму Teflon™. Дихлорметан оставляли для медленного испарения в течение ночи, остаточный дихлорметан удаляли путем вакуумной сушки при 20°C. Полученные пленки имели толщину 100-200 мкм. Для исследования набухания взвешивали 15-40 мг круглых пленок с диаметром примерно 25 мм и погружали их в колбу, содержащую 10 мл фосфатного буфера с pH 7,4 (ISO-15814). Образцы хранили в печи при 37°C. Для каждого момента времени отбора собирали образцы, с поверхности удаляли избыток буферного раствора, после чего образцы взвешивали на весах с точностью до 4 знака после запятой. Все испытания проводили в двух повторностях. Было показано, что степень набухания постепенно повышалась при увеличении содержания ПЭГ в сополимерах, а также при увеличении MM ПЭГ при практически неизменном содержании ПЭГ.
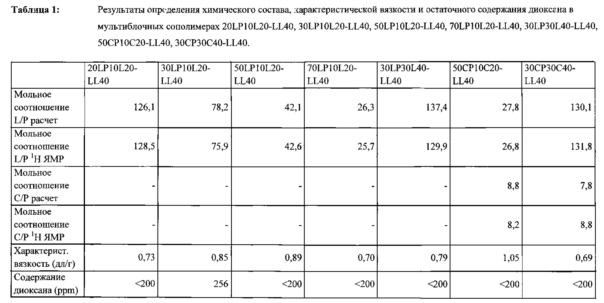
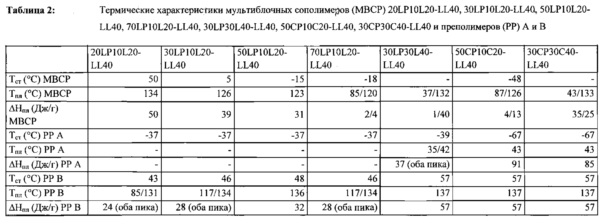
где МВСР представляет собой мультиблочный сополимер, РР А представляет собой преполимер А, РР В представляет собой преполимер В
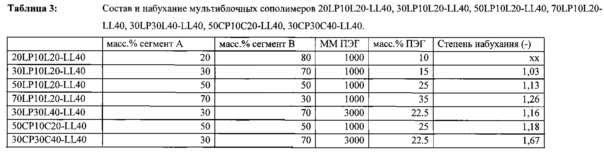
ПРИМЕР 16
В данном примере проводили оценку характеристик высвобождения белка из различных гидрофильных мультиблочных сополимеров с разделенными фазами с использованием бычьего сывороточного альбумина (БСА, 69 кДа) и лизоцима (14 кДа) в качестве модельных белков.
Пленки, содержащие 10 масс. % белка, получали путем смешения примерно 150 мкл 20 масс. % раствора белка с 1,5 мл дихлорметана, содержащего 300 мг полимера, в течение 30 секунд с использованием Ultra turrax при 18000 об./мин. Эмульсию распределяли по стеклянной пластине с использованием формовочного ножа или выливали в форму Teflon™. Дихлорметан оставляли для медленного испарения в течение ночи, остаточный дихлорметан удаляли путем вакуумной сушки при 20°C. Полученные пленки имели толщину 80-120 мкм.
Для исследования высвобождения взвешивали 20 мг пленки, содержащие белок, и погружали их в пробирки, содержащие 5 мл фосфатного буфера с pH 7,4, и хранили в печи при 37°C. В каждый момент времени отбора пробы собирали 1 мл среды высвобождения и помещали ее в 1 мл свежего буфера. Содержание белка в образцах среды высвобождения определяли путем исследования с использованием бицинхониновой кислоты (ВСА) (Pierce) на планшетном анализаторе Easys Expert 96.
Биологическую активность высвобождаемого лизоцима измеряли при помощи исследования лизиса бактерий. Пленки, содержащие лизоцим, получали согласно приведенному выше описанию. 0,01 масс. % растворы лизоцима, которые далее использовали в качестве контроля, получали путем взвешивания 2,1 мг лизоцима и добавления 20 мл фосфатного буфера. Пленки, содержащий лизоцим, взвешивали и погружали в пробирки, содержащие 5 мл фосфатного буфера pH 7,4. Пробирки, содержащие пленки с лизоцимом, а также свежеприготовленные растворы лизоцима, хранили в печи при 37°C. В каждый момент времени отбора пробы собирали 1 мл среды высвобождения и заменяли ее 1 мл свежего буфера. Содержание белка в образцах среды высвобождения определяли с использованием BCA согласно приведенному выше описанию. Активность (высвобождаемого) лизоцима определяли по изменению мутности дисперсии бактерий (Micrococcus lysodeikticus, Sigma, 0,21 мкг/мл) при 450 нм в течение 3 минут после добавления 10 мкл образца. Для этого использовали УФ-Вид. спектрометр (Varian). Образцы при необходимости разбавляли до достижения концентрации лизоцима 5-100 мкг/мл. Активность лизоцима в образцах рассчитывали путем сравнения коэффициента наклона получаемых кривых (коэффициент наклона определяет активность лизоцима) и коэффициента наклона кривой, полученной для свежего раствора лизоцима.
На Фигурах 2 и 3 показано высвобождение лизоцима и бычьего сывороточного альбумина из пленок, соответственно. Результаты показывают, что путем изменения содержания ПЭГ и MM ПЭГ можно изменять скорость и профиль высвобождения. Высвобождение лизоцима происходило в течение от нескольких дней до примерно 3 месяцев. Высвобождение БСА вследствие его более крупных размеров было более медленным и происходило в течение от нескольких дней до примерно 4 месяцев. Кроме того, высвобождение лизоцима можно регулировать путем введения различных (комбинаций) мономеров по соседству с ПЭГ-группой гидрофильного блока мультиблечных сополимеров. Получаемые мультиблочные сополимеры (50LP10L20-LL40, 50GP10C20-LL40 и 50CP10C20-LL40) содержали 25 масс. % ПЭГ 1000 и имели схожие значения степени набухания, но различную скорость разложения, что приводило к получению различных профилей высвобождения инкапсулированного лизоцима (Фигура 4).
На Фигуре 5 показана активность лизоцима, высвобождаемого из пленок 30LP10L20-LL40 или 50LP10L20-LL40, содержащих 10 масс. % лизоцима, (фосфатный буфер, pH 7,4, 37°C). В качестве контроля проводили измерения активности лизоцима в 0,01 масс. %) растворах лизоцима, которые хранили при 4 или 37°C (фосфатный буфер, pH 7,4). Результаты показывают, что высвобождение лизоцима из пленок при сохранении его биологической активности происходило в течение примерно одного месяца, это указывает на то, что структурная целостность и биологическая активность лизоцима сохранялись не только во время процесса инкапсулирования, но также во время продолжительного пребывания лизоцима в гидратированной и набухшей полимерной матрице при 37°C перед его высвобождением.
ПРИМЕР 17
В данном примере для введения БСА в состав микросфер применяли сополимеры 30LP10L20-LL40 (IV 0,85 дл/г) и 50CP10C20-LL40 (IV 1,06 дл/г) с разделенными фазами.
Микросферы, содержащие БСА, получали из гидрофильных мультиблочных сополимеров 50CP10C20-LL40 (IV 1,06 дл/г) и 30LP10L20-LL40 (IV 0,85 дл/г) с разделенными фазами при помощи способа выпаривания растворителя с использованием процедур, описанных в Kissel et al, J. Contr. Rel 1996, 39(2), 315-326 и Meinel et al, J. Contr. Rel 2001, 70(1-2), 193-202. БСА (25-50 мг) растворяли примерно в 150 мг ультрачистой воды и эмульгировали 2-3 мл раствора 50CP10C20-LL40 (15% (масс./об.)) или 30LP10L20-LL40 (23% (масс./об.)) в дихлорметане в течение 60 секунд на Ultra turrax IKA Т18 при 20000 об./мин с получением эмульсии типа вода-в-масле (В/М). Полученную таким образом первичную эмульсию затем эмульгировали примерно в 80-130 мл UP-воды, содержащей 4,0 масс. % ПВС в течение 30 секунд на Ultra turrax IKA Т18 при 14000 об./мин. с получением эмульсии типа вода-в-масле-в-воде (В/М/В). Полученную таким образом вторичную эмульсию осторожно перемешивали в течение 2 часов при 600 об./мин при комнатной температуре. В результате испарения дихлорметана полимер осаждался из раствора с образованием микросфер. Через 3 часа (время, необходимое для практически полного испарения дихлорметана) образовавшиеся микросферы собирали путем центрифугирования, промывали их трижды 100-200 мл 0,05 масс. % водного раствора Tween 20 в ультрачистой воде. На завершающей стадии микросферы лиофилизировал и.
Для исследования IVR (высвобождение in vitro) в 20 мг микросфер добавляли 2 мл 100 мМ фосфатного буфера (pH 7,4, 0,02 масс. % NaN3) в случае микросфер из 30LP10L20-LL40 и 25 мМ NaPi буфера (pH 7,2, 105 мМ NaCl, 0,01 масс. % Tween 80, 0,02 масс. % NaN3) в случае микросфер из 50CP10C20-LL40. Образец инкубировали при 37°C, во время каждого отбора пробы собирали 1,8 мл образца и заменяли буфером высвобождения. Содержание БСА измеряли путем белкового исследования ВСА в случае микросфер из 30LP10L20-LL40 и СВЭЖХ (элюент A: 1 масс. % ТФУ в UP-воде, элюент B: 0,085 масс. % ТФУ в ацетонитриле, градиент от 95/5 (об./об.) A/B до 5/95 A/B за 25 минут) в случае микросфер из 50CP10C20-LL40.
Распределение микросфер по размерам определяли с использованием анализатора Coulter. Примерно 1 мг микросфер диспергировали в 50-100 мл раствора Isotron II путем осторожного перемешивания, размер частиц измеряли на анализаторе Coulter, оборудованном 100 мкм измерительной ячейкой.
Содержание БСА в микросферах определяли путем растворения точно взвешенного количества 5-10 мг микросфер в 5,0 мл ацетонитрила. После центрифугирования удаляли 4 мл надосадочной жидкости и добавляли 5 мл PBS. Содержание БСА измеряли путем СВЭЖХ (элюент A: 0,1 масс. %) ТФУ в UP-воде, элюент В: 0,1% ТФУ в ацетонитриле, градиент от 90/10 (об./об.) A/B до 10/90 (об./об.) A/B за 4 минуты).
В Таблице 4 приведены размер частиц, эффективность инкапсулирования (EE) полученных микросфер, содержащих БСА. На Фигуре 6 показано высвобождение БСА in vitro из микросфер 30LP10L20-LL40 с заданным 5 масс. % содержанием БСА и микросфер 50CP10C20-LL40 с заданным 10 масс. %» содержанием БСА. Высвобождение БСА из микросфер 30LP10L20-LL40 происходило в течение примерно 3 месяцев линейным образом без значительных скачков. Высвобождение БСА из микросфер 50CP10C20-LL40 происходило в течение ~3 месяцев линейным образом без значительных скачков, после чего более медленное высвобождение продолжалось еще ~1,5 месяца.
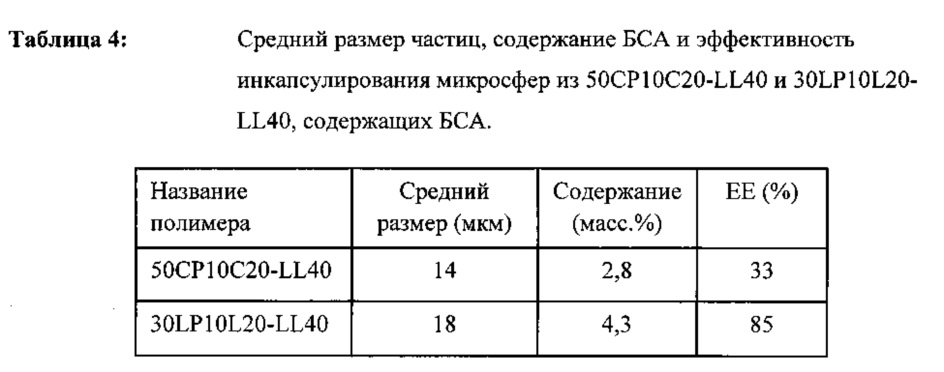
ПРИМЕР 18
В данном примере различные гидрофильные мультиблочные сополимеры с разделенными фазами, полученные согласно описанию приведенных выше примеров, применяли для получения составов в виде пленок и микросфер, содержащих инсулиноподобный фактор роста I (ИФР-1).
Пленки, содержащие ИФР-1, получали путем растворения 0,18 г полимера в 1,46 г дихлорметана и последующей эмульсификации путем обработки на Ultra turrax совместно с ИФР-1, растворенным в ультрачистой воде, при 18000 об./мин. в течение 30 секунд или с использованием 100 Вт ультразвука в течение 5 секунд. Эмульсию выливали в форму Teflon™. Дихлорметан оставляли для испарения в течение ночи, остаточный дихлорметан удаляли путем вакуумной сушки в течение ночи. Вырезали 20 мг пленки и оставляли для высвобождения при 37°C в 1 мл фосфатного буферного солевого раствора (PBS, 25М, pH 7,2, 105 мМ NaCl, 0,01 масс. % Tween 80 и 0,02 масс. % NaN3). В предварительно определенные моменты времени отбирали образцы, отобранный объем заменяли свежим буфером.
Микросферы, содержащие ИФР-1, получали при помощи способа эмульсификации B/M/B на основе экстракции/выпаривания растворителя. 2,78 мг ИФР-1 и 51,8 мг БСА растворяли в 143 мкл UP-воды в пробирке Eppendorf и эмульгировали в растворе 0,47 г 50CP10C20-LL40 (IV 1,05 дл/г) в 2,62 г дихлорметана на Ultra turrax (20000 об./мин., 60 сек.). Полученную таким образом первичную эмульсию затем эмульгировали в 81 мл UP-воды, содержащей 4,0 масс. % ПВС, на Ultra turrax (14000 об./мин. в течение 60 секунд) и перемешивали в течение 2 часов при 600 об./мин. при комнатной температуре. Полученные микросферы собирали на 5 мкм мембранном фильтре и промывали 1 л UP-воды, содержащей 0,05 масс. % Tween 80. На завершающей стадии микросферы лиофилизировали.
Примерно 1 мг микросфер диспергировали в 50-100 мл раствора Isotron II путем осторожного перемешивания, размер частиц измеряли на анализаторе Coulter, оборудованном 100 мкм измерительной ячейкой.
Содержание ИФР-1 и БСА определяли путем растворения точно взвешенного количества 5 мг микросфер в 0,3 мл ацетонитрила. Затем добавляли 1,2 мл PBS, осторожно встряхивали. После центрифугирования содержание ИФР-1 и БСА в надосадочной жидкости определяли путем СВЭЖХ. Процедуру проводили в трех повторностях.
Для подтверждения того, что микроинкапсулированный и высвобождаемый ИФР-1 сохранял способность связывания с антителами захвата и детекторными антителами после высвобождения, а следовательно того, что разложение белка на этом уровне не происходило, концентрацию инсулиноподобного фактора роста I (ИФР-1) человека в образце измеряли с использованием коммерческой вариации «сэндвич» ELISA (R&D Systems). Антитело захвата и детекторное антитело, входящие в состав набора, обладали специфичностью к природному и рекомбинантному ИФР-1, а также к стандарту рекомбинантного ИФР-1.
Для исследования структурной целостности высвобождаемого ИФР-1 100-300 нг ИФР-1, собранного из образцов среды высвобождения, денатурировали с использованием буфера Леммли/β-меркаптоэтанола и наносили в предварительно изготовленный «Any KD TGX» минигель и разделяли в условиях денатурации при 100-200 B с использованием 1х Tris/глицин/ДСН в качестве буфера разделения и окрашивали в течение ночи в коллоидном окрашивающем агенте СВВ. Для определения размера частиц выделенных белков использовали маркер Dual Xtra Protein (Bio-Rad).
На Фигуре 7 показано высвобождение ИФР-1 in vitro из полимерных пленок 50CP30C40-LL40 и 30CP30C40-LL40, содержащих 0,6 масс. % ИФР-1, определенное путем СВЭЖХ и ELISA. Высвобождение ИФР-1 из пленок 50CP30C40-LL40 проходило в течение 7 дней, тогда как высвобождение ИФР-1 из полимерных пленок 30CP30C40-LL40 проходило медленно, общее высвобождение через 28 дней составляло примерно 40%. Так как общее высвобождение ИФР-1, измеренное путем СВЭЖХ, практически совпадало с общим высвобождением ИФР-1, измеренным путем ELISA, был сделан вывод об отсутствии отрицательного воздействия на структуру и биологическую активность высвобождаемого ИФР-1.
Микросферы с заданным 0,5 масс. % содержанием ИФР-1 получали из 50СР10С20-LL40 с IV 1,05 и 0,68 дл/г при помощи способа двойной эмульсификации. Микросферы имели гладкую поверхность (Фигура 8), эффективность инкапсулирования находилась в диапазоне от 40 до 60%. Средний объемный диаметр частиц (d50), измеренный на анализаторе Coulter, оборудованном 100 мкм измерительной ячейкой, составлял 54,4 мкм, а CV (коэффициент вариаций) составлял 61%). На Фигуре 9 показано высвобождение ИФР-1 in vitro из указанных микросфер. Для микросфер, состоящих из 50CP10C20-LL40 с IV 0,68 дл/г, полное высвобождение ИФР-1 проходило за 2 дня. Высвобождение ИФР-1 из микросфер, состоящих из 50CP10C20-LL40 с IV 1,05 дл/г, было более медленным, полное высвобождение происходило за 6 дней. По результатам ДСН-ПААГ (Фигура 10), в которых отсутствовали какие-либо признаки разложения или агрегации белка, можно сделать вывод об отсутствии отрицательного воздействия на структуру высвобождаемого ИФР-1.
ПРИМЕР 19
В данном примере различные гидрофильные мультиблочные сополимеры с разделенными фазами (20LP10L20-LL40 (IV 0,58 дл/г), 30LP6L20-LL40 (IV 0,60 дл/г) и 30CP10C20-LL40 (IV 0,71 дл/г)), полученные согласно описанию приведенных выше примеров, использовали для получения составов в виде пленок, содержащих биологически активный полипептид с высокой растворимостью в воде, имеющий молекулярную массу 15 кДа (Белок A). Кроме того, мультиблочные сополимеры 30CP10C20-LL40 с различными IV (0,81, 0,71 и 0,65 дл/г) использовали для введения Белка A в состав микросфер.
Пленки, содержащие белок A, получали при помощи способа с использованием заливки раствора. 10 мг белка A растворяли в 123 мг UP-воды и эмульгировали в растворе 0,18 г полимера в 1,46 г дихлорметана с использованием Ultra turrax (18000 об./мин., 60 сек). Получаемую таким образом первичную эмульсию выливали в форму Teflon™ и оставляли для испарения дихлорметана в течение ночи. Остаточный дихлорметан удаляли путем вакуумной сушки.
Микросферы, содержащие белок A, получали при помощи способа эмульсификации В/М/В, основанного на экстракции/выпаривании растворителя. В пробирке Eppendorf 21 мг белка A (для заданного содержания 5 масс. %) растворяли в 156 мкл UP-воды, возможно содержащей инулин, и эмульгировали в растворе 0,4 г полимера в 2,1 г дихлорметана с использованием Ultra turrax (20000 об./мин., 60 сек.). Полученную таким образом первичную эмульсию затем эмульгировали в 70 мл UP-воды, содержащей 4,0 масс. % ПВС, с использованием Ultra turrax (14000 об./мин. в течение 60 секунд) и перемешивали в течение 2 часов при 600 об./мин. при комнатной температуре. Полученные микросферы собирали на 5 мкм мембранном фильтре и промывали трижды 100 мл UP-воды, содержащей 0,05 масс. % Tween 80. На завершающей стадии микросферы лиофилизировали.
Примерно 10 мг микросфер диспергировали в 50-100 мл раствора Isotron II путем осторожного перемешивания, размер частиц измеряли на анализаторе Coulter, оборудованном 100 мкм измерительной ячейкой.
Содержание белка A определяли путем растворения точно взвешенного количества 5 мг микросфер в 0,3 мл ацетонитрила. После центрифугирования удаляли надосадочную жидкость, а остаточный ACN выпаривали. Добавляли 1,95 мл PBS. Содержание белка A измеряли путем СВЭЖХ (элюент А: 0,1 масс. % ТФУ в UP-воде, элюент B: 0,1 масс. % ТФУ в ацетонитриле, градиент от 80/20 (об./об.) A/B до 10/90 A/B за 3 минуты).
Для визуализации на СЭМ небольшое количество микросфер приклеивали к угольной проводящей ленте и наносили покрытие из золота в течение 3 минут. Визуализацию образца проводили с использованием 10 кВ пучка электронов.
Кинетику высвобождения белка A in vitro из пленок и микросфер измеряли в 100 мМ фосфатном буфере с pH 7,4 (20 мг пленки в 2 мл). Образцы инкубировали при 37°C. Во время каждого отбора пробы собирали 1,8 мл образца и заменяли 1,8 мл фосфатного буфера. Содержание белка A измеряли путем СВЭЖХ (элюент А: 0,1 масс. % ТФУ в UP-воде, элюент B: 0,1 масс. %» ТФУ в ацетонитриле, градиент от 80/20 (об./об.) A/B до 10/90 A/B за 3 минуты).
ДСН-ПААГ проводили в восстанавливающих условиях с использованием 4-20%» гелей Tris-HCl. При исследовании образцов и стандартного образца белка A наносили 20 мкл раствора белка в щель. В случае маркера в щель наносили 2 мкл. Количество белка, добавляемое в щель, составляло 75 или 150 нг. Образцы получали путем разбавления 12 мМ PBS с pH 7,4 или UP-водой до достижения концентрации белка A 150 или 300 нг/20 мкл. Затем добавляли рабочий раствор Леммли (буфер Леммли, содержащий 1% меркаптоэтанола) в отношении 1:1 (об./об.). Образцы нагревали до ~90°C в течение 5 минут и наносили в гель. Гели фиксировали в электрофоретической ячейке и добавляли подвижный буфер (Tris/глицин/ДСН, pH 8,3). Образцы и стандарты наносили в гель, цикл проведения фореза геля при 100 кВ составлял 15 минут. Затем напряжение устанавливали на 200 кВ, и форез геля проводили до достижения хорошего разделения стандартов молекулярной массы. Гели промывали UP-водой и окрашивали серебряным реагентом.
На Фигуре 11 показано высвобождение белка A in vitro из 20LP10L20-LL40 (10 масс. % ПЭГ ММ 1000), 30LP6L20-LL40 (9 масс. % ПЭГ ММ 600) и 30CP10C20-LL40 (15 масс. % ПЭГ ММ 1000). Высвобождение белка A из пленок на основе 30CP10C20-LL40 проходило относительно быстро, общее высвобождение белка A достигало 100% через 3 месяца. Путем замены ПЭГ 1000 на ПЭГ600, что приводит к снижению степени набухания, можно замедлять высвобождение белка A, за счет этого добивались достижения кинетики высвобождения, контролируемой диффузией, практически первого порядка, что давало общее высвобождение ~75% через 4 месяца. Снижения скорости высвобождения белка A также можно добиваться за счет снижения массовой доли гидрофильного блока LP10L20 в полимере. За счет использования 20LP10L20-LL40 (10 масс. % ПЭГ1000) можно дополнительно замедлять высвобождение, и после начального небольшого мгновенного высвобождения, составляющего менее 15%, добивались хорошо контролируемой кинетики высвобождения белка A, общее высвобождение составляло ~65% за 6 месяцев. Данные явно указывают на то, что за счет выбора полимера можно контролировать кинетику высвобождения белка A.
Микросферы, содержащие белок A, получали из 30CP10C20-LL40, содержание белка A составляло 3-4 масс. %. Для увеличения скорости высвобождения белка A в капсулы необязательно добавляли 2 или 5 масс. % инулина. Влияние молекулярной массы полимера на кинетику высвобождения белка определяли путем исследования кинетики высвобождения белка A из микросфер, состоящих из полимеров 30CP10C20-LL40 с различной характеристической вязкостью. Все получаемые микросферы, содержащие белок A, имели сферическую форму. В микросферах, в которые дополнительно инкапсулировали инулин, пористость поверхности повышалась при увеличении содержания инулина, что показано на изображениях СЭМ на Фигуре 12. В Таблице 5 приведены размер частиц и эффективность инкапсулирования (ЕЕ) белка A в микросферах. На Фигуре 13 показано, что после небольшого начального мгновенного высвобождения высвобождение белка A происходило с постоянной скоростью. При снижении содержания инулина наблюдали уменьшение мгновенного высвобождения и повышение линейности высвобождения. В отсутствие инулина высвобождаемое за 3 месяца количество составляло ~70%, тогда как при инкапсулировании 2 или 5 масс. %) инулина высвобождение составляло 90-100%). Высвобождение белка A из пленок 30CP10C20-LL40, содержащих 2 или 5 масс. % инкапсулированного инулина, было схожим. Данные высвобождения показаны вплоть до ~4 месяцев. Предполагаемая продолжительность высвобождения белка A из микросфер 30CP10C20-LL40 составляет примерно 6 месяцев.
На Фигуре 14 показана кинетика высвобождения белка A из пленок 30СР10С20-LL40 с различной характеристической вязкостью (IV) полимера. Скорость высвобождения белка A повышалась при увеличении IV полимера. В случае полимеров 30CP10C20-LL40 с IV 0,71 или 0,81 дл/г наблюдали замедленное высвобождение белка A, общее высвобождение составляло 60-70% через 2 месяца. Кинетика высвобождения белка A из микросфер, состоящих 30CP10C20-LL40 с IV 0,58 дл/г, значительно отличалась. Начальная скорость высвобождения вплоть до одного месяца была ниже, но на интервале от 1 до 3 месяцев высвобождение белка A увеличивалось, после чего оно снова замедлялось, что давало общую продолжительность высвобождения примерно 5 месяцев. Данные явно указывают на то, что высвобождение белка A из микросфер может быть линейным на протяжении по меньшей мере 4 месяцев, а кинетику высвобождения можно контролировать путем инкапсулирования Сахаров, таких как инулин, а также изменяя характеристическую вязкость полимера.
Структурную целостность белка A, высвобождаемого из микросфер, исследовали путем ДСН-ПААГ. Анализ ДСН-ПААГ подтверждал, что белок A, высвобождаемый в течение по меньшей мере 21 дня, состоял, главным образом, из белка в исходном виде (Фигура 15). Эти результаты показывают, что микросферы 30CP10C20-LL40 обеспечивают подходящую матрицу для долгосрочного высвобождения белка A, не оказывающую отрицательное воздействие на его структуру.
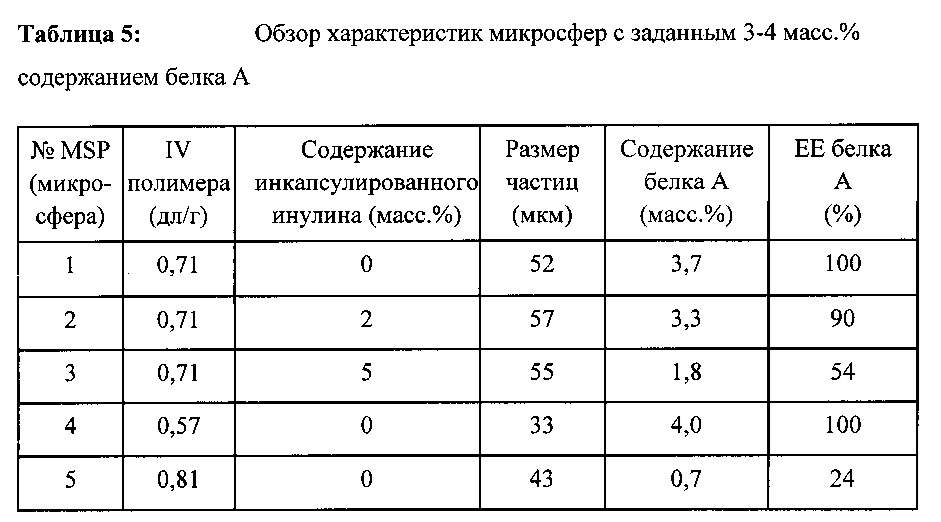
ПРИМЕР 20
В данном примере гидрофильные мультиблочные сополимеры 20LP10L20-LL40 (IV 0,73 дл/г) с разделенными фазами, полученные согласно описанию, приведенному выше в примерах, использовали для получения составов в виде пленок и микросфер, содержащих биологически активный полипептид с молекулярной массой 2,5 кДа (пептид А).
Пленки, содержащие пептид А, получали при помощи способа заливки раствора. 10 (в случае 5 масс. % содержания) или 20 мг (в случае 10 масс. % содержания) пептида А растворяли в 123 мг UP-воды и эмульгировали в растворе 0,18 г 20LP10L20-LL40 (IV 0,76 дл/г) в 1,46 г дихлорметана с использованием Ultra turrax (18000 об./мин., 30 сек.). Полученную таким образом первичную эмульсию выливали в форму Teflon и оставляли для испарения дихлорметана в течение ночи. Остаточный дихлорметан удаляли путем вакуумной сушки.
Микросферы, содержащие пептид А, получали при помощи способа двойной эмульсии, основанного на выпаривании растворителя. 50 мг пептида А растворяли в PBS и эмульгировали в растворе 0,5 г 20LP10L20-LL40 (IV 0,73 дл/г) в 2 г дихлорметана с использованием Ultra turrax (24000 об./мин., 60 сек.). Полученную таким образом первичную эмульсию затем эмульгировали в 200 мл UP-воды, содержащей 4,0 масс. % поливинилового спирта, с использованием Ultra turrax (14000 об./мин. в течение 30 секунд) и перемешивали в течение 3 часов при 600 об./мин. при комнатной температуре. Полученные микросферы центрифугировали, надосадочную жидкость удаляли и микросферы промывали трижды 200 мл UP-воды, содержащей 0,05 масс. % Tween 20. На завершающей стадии микросферы лиофилизировали. Распределение частиц по размерам измеряли на анализаторе Coulter. Примерно 10 мг микросфер диспергировали в 50-100 мл раствора Isotron II путем осторожного перемешивания, размер частиц измеряли в 100 мкм измерительной ячейке.
Содержание пептида A в микросферах определяли путем растворения точно взвешенного количества 5-10 мг микросфер в 5,0 мл ацетонитрила. После центрифугирования удаляли 4 мл надосадочной жидкости и добавляли 5 мл PBS. Содержание пептида A измеряли путем ВЭЖХ (элюент A: 1 масс. % ТФУ в UP-воде, элюент B: 0,085 масс. % ТФУ в ацетонитриле, градиент от 95/5 (об./об.) A/B до 5/95 A/B за 25 минут).
Кинетику высвобождения пептида A in vitro из пленок и микросфер измеряли в PBS при 37°C.Пленки или микросферы (5-20 мг), содержащие пептид A, взвешивали в пробирке и добавляли 2 мл PBS. Пробирки инкубировали при 37°C, образцы отбирали в предварительно определенные моменты времени. В каждый момент отбора пробы собирали 75-90% среды высвобождения и заменяли ее свежим PBS. Содержание пептида A в образцах среды высвобождения определяли путем ВЭЖХ (элюент A: 1 масс. % ТФУ в UP-воде, элюент В: 0,085 масс. % ТФУ в ацетонитриле, градиент от 95/5 (об./об.) A/B до 5/95 A/B за 25 минут).
На Фигуре 16 показано высвобождение пептида A in vitro из пленок 20LP10L20-LL40. Высвобождение пептида А из пленок 20LP10L20-LL40 с 5 масс. % содержанием пептида A происходило линейным образом без значительных скачков в течение по меньшей мере 5 месяцев. В случае пленок 20LP10L20-LL40 с более высоким содержанием пептида А (10 масс. %) мгновенное высвобождение повышалось до 15%. Через примерно 2 месяца высвобождение было аналогичным с пленками с 5 масс. % содержанием.
Микросферы 20LP10L20-LL40, содержащие пептид А, имели средний размер частиц 30 мкм и содержание пептида А 10,3 масс. %, а также эффективность инкапсулирования 100%. На фигуре 17 показано, что для MSP, содержащие пептид А, мгновенное высвобождение составляло примерно 10 масс. %, а затем кинетика высвобождения имела нулевой порядок в течение по меньшей мере 40 дней.
ПРИМЕР 21
В данном примере гидрофильные мультиблочные сополимеры 20LP10L20-LL40 (Пример 8) и 10LP10L20-LL40 с разделенными фазами использовали для получения микросфер, содержащих рапамицин (ММ 914 Да). Компонент полиэтиленгликоля в полимерах имел молекулярную массу 1000 г/моль.
Микросферы с заданным 20 масс. % содержанием рапамицина получали при помощи способа выпаривания растворителя с применением одной эмульсии типа масло-в-воде (М/В). Полимеры растворяли в дихлорметане в различных соотношениях до достижения концентрации примерно 20 масс. % и добавляли требуемое количество рапамицина. Раствор полимера/рапамицина затем эмульгировали в 200 мл UP-воды, содержащей 4,0 масс. % поливинилового спирта (ПВС), с использованием Ultra turrax (14000 об./мин. в течение 30 секунд), а затем перемешивали с использованием магнитной мешалки в течение 3 часов при 300 об./мин. при комнатной температуре. Дисперсию микросфер концентрировали путем центрифугирования и микросферы промывали трижды 50 мл водного 0,05 масс. % раствора Tween 20. На завершающей стадии микросферы лиофилизировали.
Распределение частиц по размерам определяли на анализаторе Coulter. Примерно 10 мг микросфер диспергировали в 50-100 мл раствора Isotron II путем осторожного перемешивания, размер частиц измеряли с использованием 100 мкм измерительной ячейки.
Содержание рапамицина в микросферах определяли путем растворения точно взвешенного количества 5-10 мг микросфер в 5,0 мл ацетонитрила. После центрифугирования удаляли 4 мл надосадочной жидкости и добавляли 5 мл PBS. Содержание рапамицина измеряли путем ВЭЖХ (элюенты: ацетонитрил/вода 70/30 (об./об.); 278 нм).
Кинетику высвобождения рапамицина in vitro из микросфер определяли при 37°C в 10 мМ PBS, pH 7,4, содержащем 0,5 масс. % ДСН. Микросферы (5-20 мг), содержащие рапамицин, взвешивали в пробирке и добавляли 2 мл среды высвобождения. Пробирки инкубировали при 37°C, отбор проб проводили в предварительно определенные моменты времени. В каждый момент отбора пробы собирали 75-90% среды высвобождения и заменяли ее свежим PBS. Содержание рапамицина в образцах среды высвобождения определяли путем ВЭЖХ.
Полученные таким образом микросферы с рапамицином имели средний размер 35 мкм, содержание рапамицина составляло от 17 до 20 масс. %, что соответствует эффективности инкапсулирования от 89%до 100%. На Фигуре 18 показано высвобождение рапамицина из микросфер, состоящих из различных смесей 20LP10L20-LL40 и 10LP10L20-LL40. Высвобождение рапамицина из микросфер на основе 20LP10L20-LL40 происходило относительно быстро, тогда как высвобождение рапамицина из микросфер на основе 10LP10L20-LL40 протекало очень медленно. Путем смешения двух полимеров получали микросферы с промежуточными профилями высвобождения.
ПРИМЕР 22
В данном примере микросферы, содержащие госерелина ацетат, получали из гидрофильного мультиблочного сополимера 20LP10L20-LL40 с разделенными фазами при помощи способа с использованием эмульсии типа вода-в-масле-в-масле. 62,6 мг госерелина ацетата растворяли в 150 мкл UP-воды (29,4 масс. %) и эмульгировали с использованием раствора 0,5 г полимера 20LP10-LLA40 в 7,4 г дихлорметана в сцинтилляционном флаконе (Ultra turrax, 20000 об./мин., 60 сек). Затем медленно добавляли 13,5 г полимерного осадителя (силиконовое масло, 350 сСт) (2-5 минут) при постоянном перемешивании (12000 об./мин.) с образованием зародышевых микрочастиц. Зародышевые микрочастицы затем выливали в 550 мл гептана при комнатной температуре (соотношение дихлорметана и гептана в растворителе составляло 13,5:1). Сосуд для экстракции закрывали для предотвращения избыточного испарения экстракционной среды. Примерно через 3 часа экстракции микрочастицы собирали путем вакуумного фильтрования, дополнительно промывали гептаном и сушили в вакууме. Микросферы имели средний размер 67 мкм, содержание госерелина составляло 8,3%), что соответствует эффективности инкапсулирования 88%).
Распределение частиц по размерам определяли на анализаторе Coulter. Примерно 10 мг микросфер диспергировали в 50-100 мл раствора Isotron II путем осторожного перемешивания, размер частиц измеряли с использованием 100 мкм измерительной ячейки.
Содержание госерелина в микросферах определяли путем растворения точно взвешенного количества 5-10 мг микросфер в 5,0 мл ацетонитрила. После центрифугирования удаляли 4 мл надосадочной жидкости и добавляли 5 мл PBS. Содержание госерелина измеряли путем ВЭЖХ (элюенты: вода/ацетонитрил/трифторуксусная кислота 72/28/0,1, 220 нм).
Кинетику высвобождения госерелина in vitro из микросфер определяли в PBS (192 мМ, pH 7,4, содержащем 0,01% Tween 80 и 0,02% азида натрия) при 37°C.Микросферы (5-20 мг), содержащие госерелин, взвешивали в пробирке и добавляли 2 мл среды высвобождения. Пробирки инкубировали при 37°C, отбор проб проводили в предварительно определенные моменты времени. В каждый момент отбора пробы собирали 75-90% среды высвобождения и заменяли ее свежим PBS. Содержание госерелина в образцах среды высвобождения определяли путем ВЭЖХ.
Полученные таким образом микросферы 20LP10-LLa40 с госерелином имели вид сферы с гладкой поверхностью (Фигура 19), средний размер 71 мкм (CV 47%), содержание госерелина составляло 8,3 масс. %, что соответствует эффективности инкапсулирования 88%. На Фигуре 18 показано высвобождение госерелина из микросфер.
ПРИМЕР 23
В данном примере микросферы, содержащие лизоцим, получали из гидрофильного мультиблочного сополимера 30CP10L20-LL40 с разделенными фазами при помощи способа с использованием эмульсии типа твердое вещество-в-масле-в-масле (Т/М/М). 0,43 г 30CP10L20-LL40 растворяли в 7,4 г дихлорметана в сцинтилляционном флаконе (5,4 масс. %), в раствор полимера добавляли 0,074 г высушенных распылением и стабилизированных инулином микрочастиц лизоцима (соотношение лизоцим/инулин: 1/2 (масс/масс.)) с размером 1-2 мкм, дисперсию гомогенизировали на Ultra turrax (20000 об./мин., 60 сек.). Затем для получения зародышевых микрочастиц при постоянном перемешивании (12000 об./мин.) медленно добавляли (2-5 минут) 11,46 г полимерного осадителя (силиконовое масло, 350 сСт). Зародышевые микрочастицы затем выливали в 550 мл гептана при комнатной температуре (соотношение дихлорметана и гептана в растворителе составляло 13,5:1). Сосуд для экстракции закрывали для предотвращения избыточного испарения экстракционной среды. Примерно через 3 часа экстракции микрочастицы собирали путем вакуумного фильтрования, дополнительно промывали гептаном и сушили путем вакуумного фильтрования. Микросферы имели средний размер 59 мкм, содержание госерелина составляло 4,1-5,6%, что соответствует эффективности инкапсулирования 80-100%.
Реферат
Настоящее изобретение относится к биоразлагаемому полукристаллическому термопластичному мультиблочному сополимеру с разделенными фазами. Описан биоразлагаемый полукристаллический термопластичный мультиблочный сополимер с разделенными фазами, характеризующийся тем, что: a) он содержит по меньшей мере один сегмент гидролизуемого преполимера (А), причем указанный сегмент преполимера (А) получен из лактида и/или ε-капролактона, и по меньшей мере один сегмент гидролизуемого преполимера (В), причем указанный сегмент преполимера (В) представляет собой кристаллический полимер, полученный из мономеров L-лактида, D-лактида, гликолида или комбинации указанных мономеров с Mn 1000 г/моль или более; причем указанный сегмент преполимера (А) образует аморфную фазу, а сегмент преполимера (В) образует кристаллическую фазу; b) указанный мультиблочный сополимер характеризуется Т37°С или менее и Т110-250°С в физиологических условиях; c) сегменты связаны посредством полифункционального удлинителя цепи, причем указанный полифункциональный удлинитель цепи представляет собой 1,4-бутандиизоцианат; d) сегменты случайным образом распределены по полимерной цепи; e) по меньшей мере часть сегмента преполимера (А) получена из водорастворимого полимера, причем указанный водорастворимый полимер представляет собой поли(этиленгликоль) (ПЭГ), имеющий Mn 150-5000 г/моль. Также описан способ получения указанного выше биоразлагаемого полукристаллического термопластичного мультиблочного сополимера с разделенными фазами, включающий: i) проведение реакции удлинения цепи преполимера (А) и преполимера (В) в присутствии полифункционального удлинителя цепи, причем указанный полифункциональный удлинитель цепи представляет собой 1,4-бутандиизоцианат, где преполимеры (А) и (В) оба имеют концевые группы диола или двухосновной кислоты. Описано применение указанного выше биоразлагаемого полукристаллического термопластичного мультиблочного сополимера для доставки лекарственных средств. Также описана композиция для доставки биологически активного соединения хозяину, содержащая биологически активное соединение, инкапсулированное в матрицу, причем указанная матрица содержит указанный выше биоразлагаемый полукристаллический термопластичный мультиблочный сополимер. Также описаны способы получения указанной выше композиции. 9 н. и 15 з.п. ф-лы, 22 ил., 5 табл., 23 пр.
Формула
Документы, цитированные в отчёте о поиске
Сложный полиэфир и конъюгат на его основе
Комментарии