Ферментативное получение 1-бутанола - RU2429295C2
Код документа: RU2429295C2
Чертежи






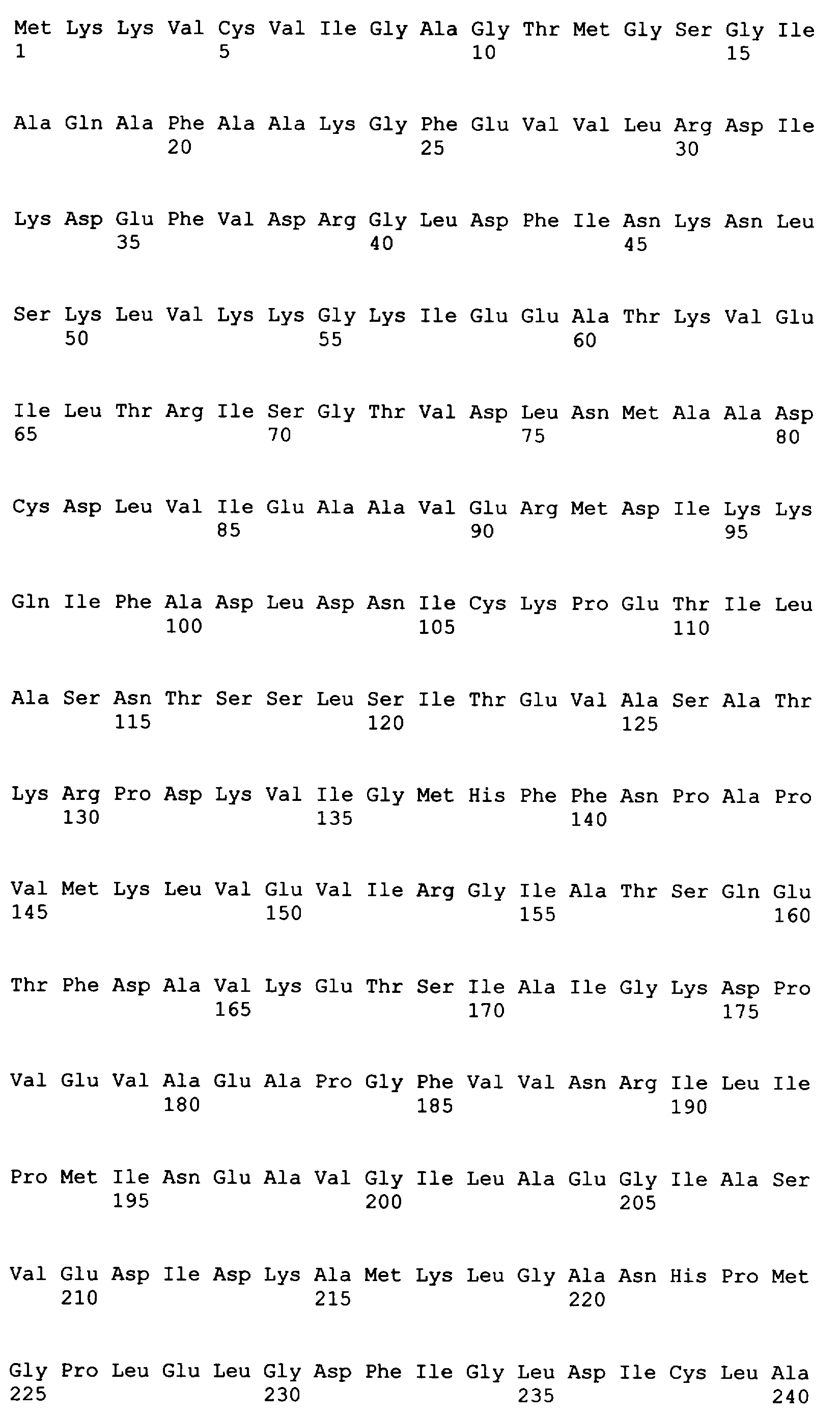










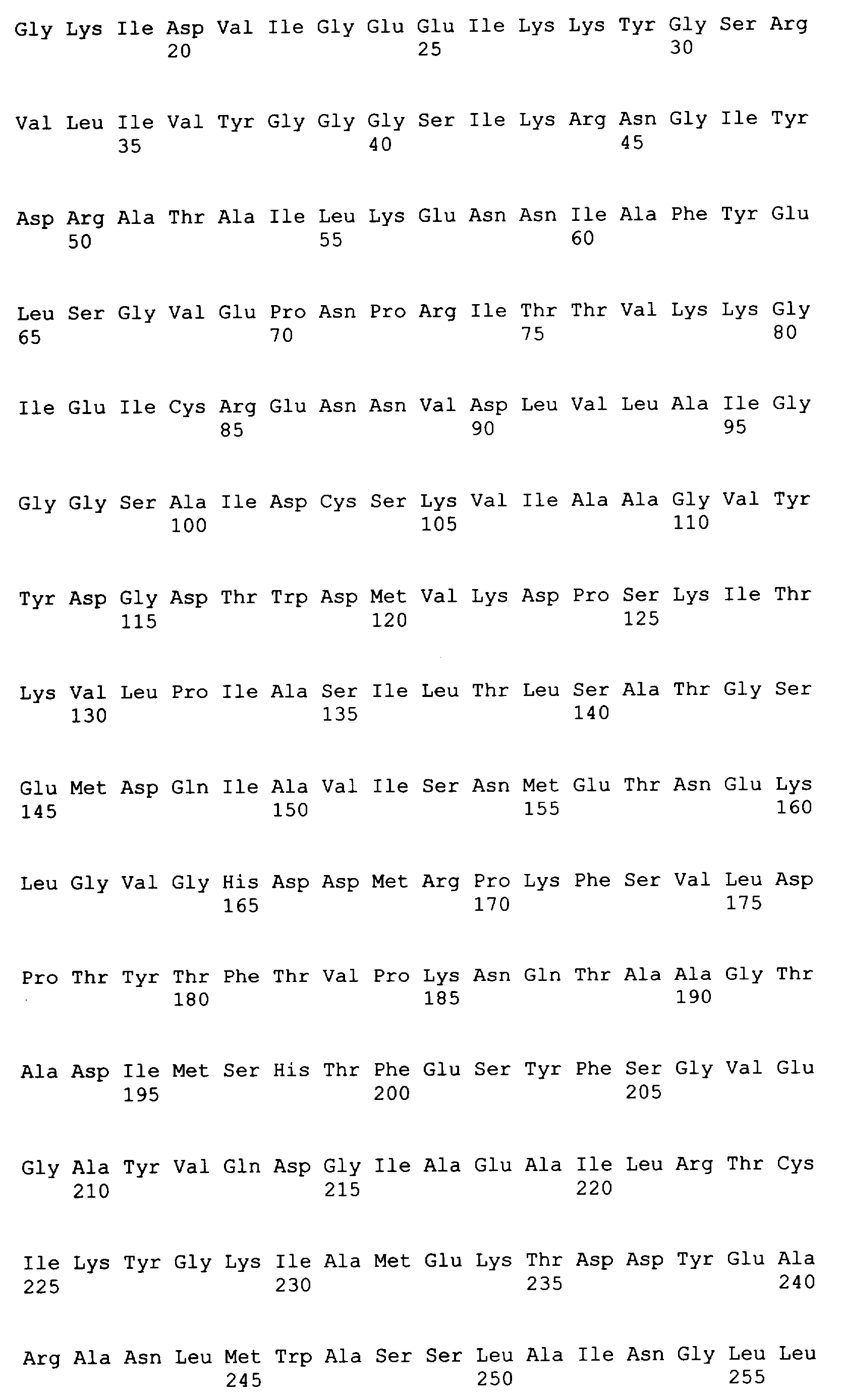













































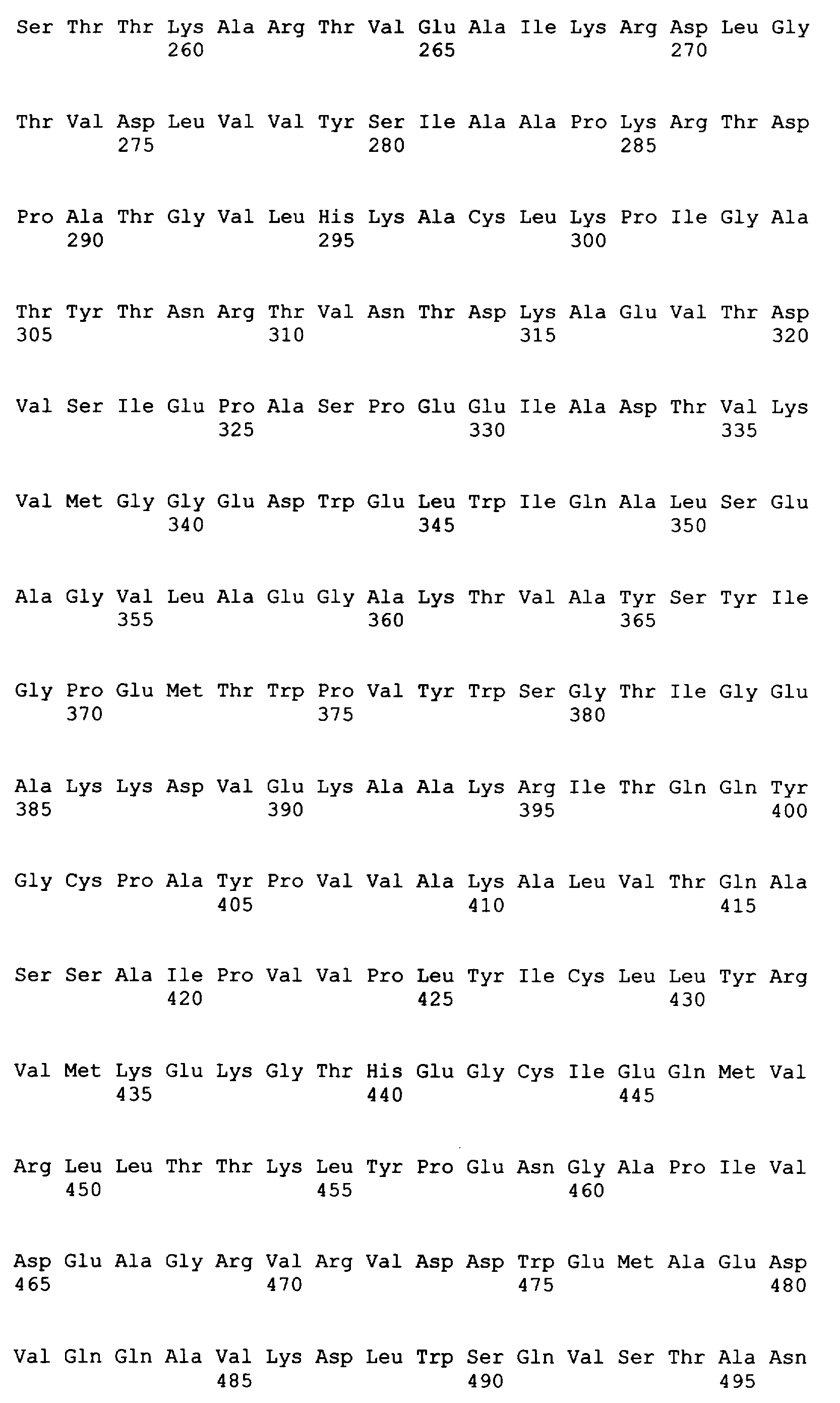

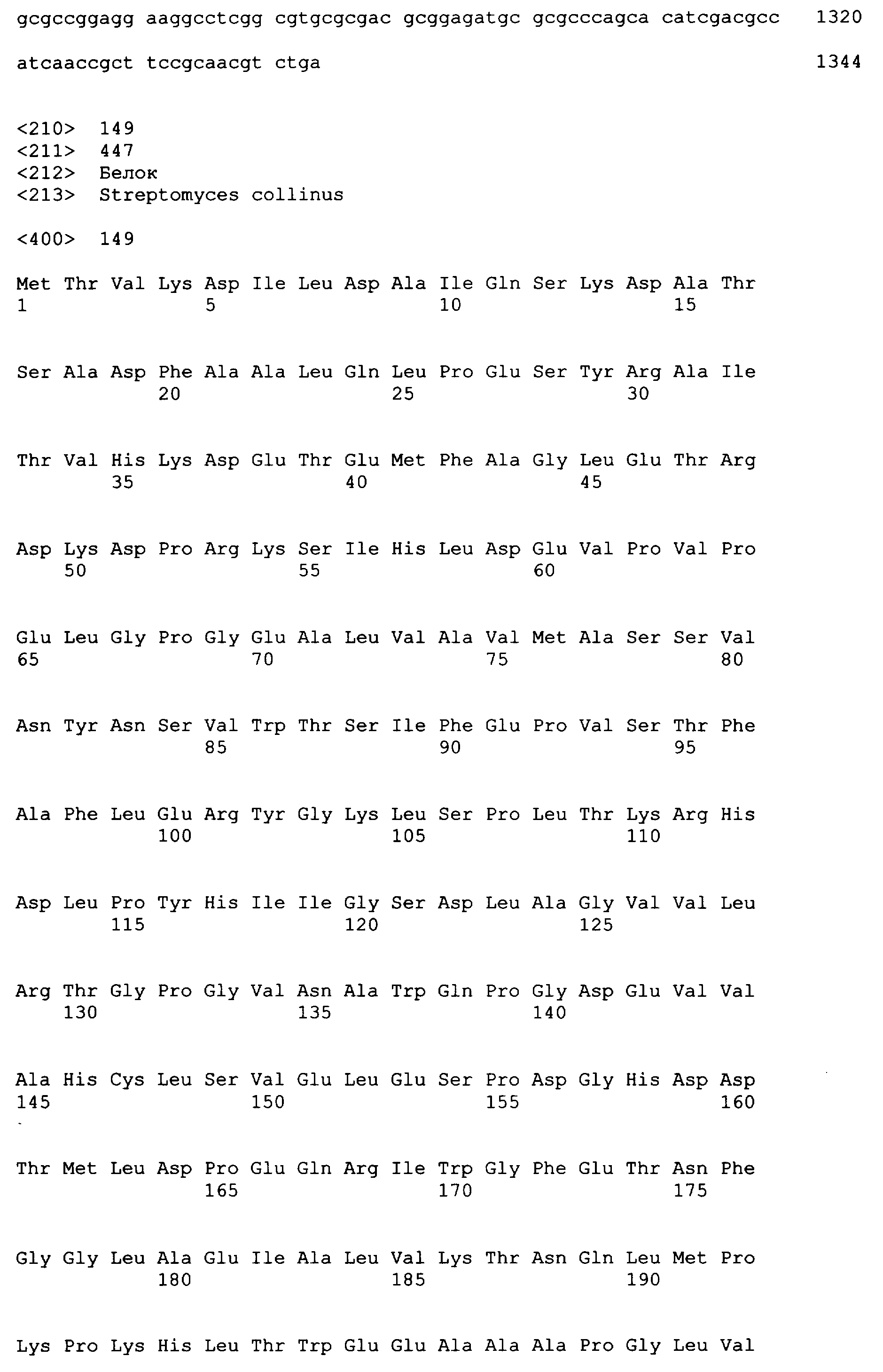


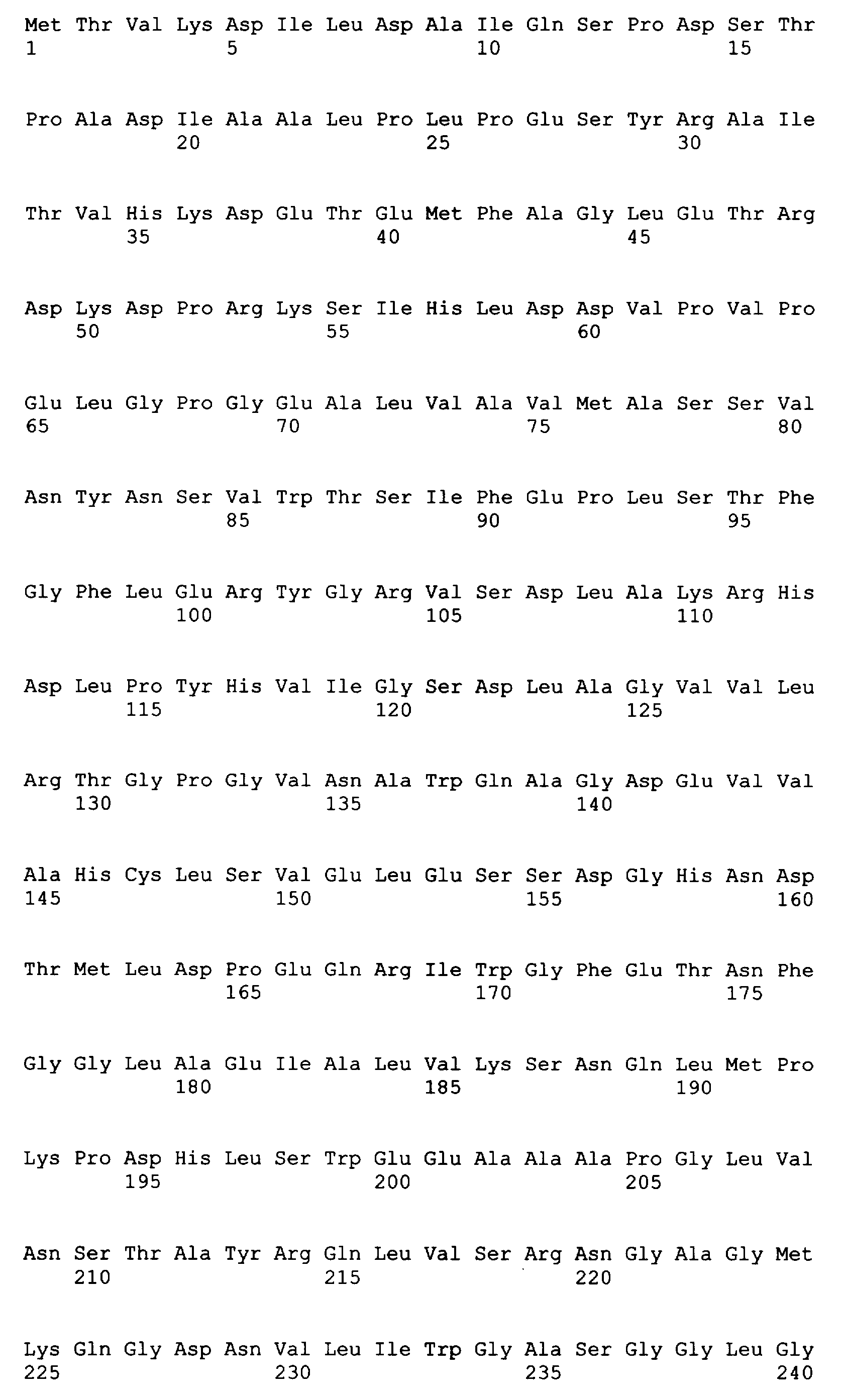











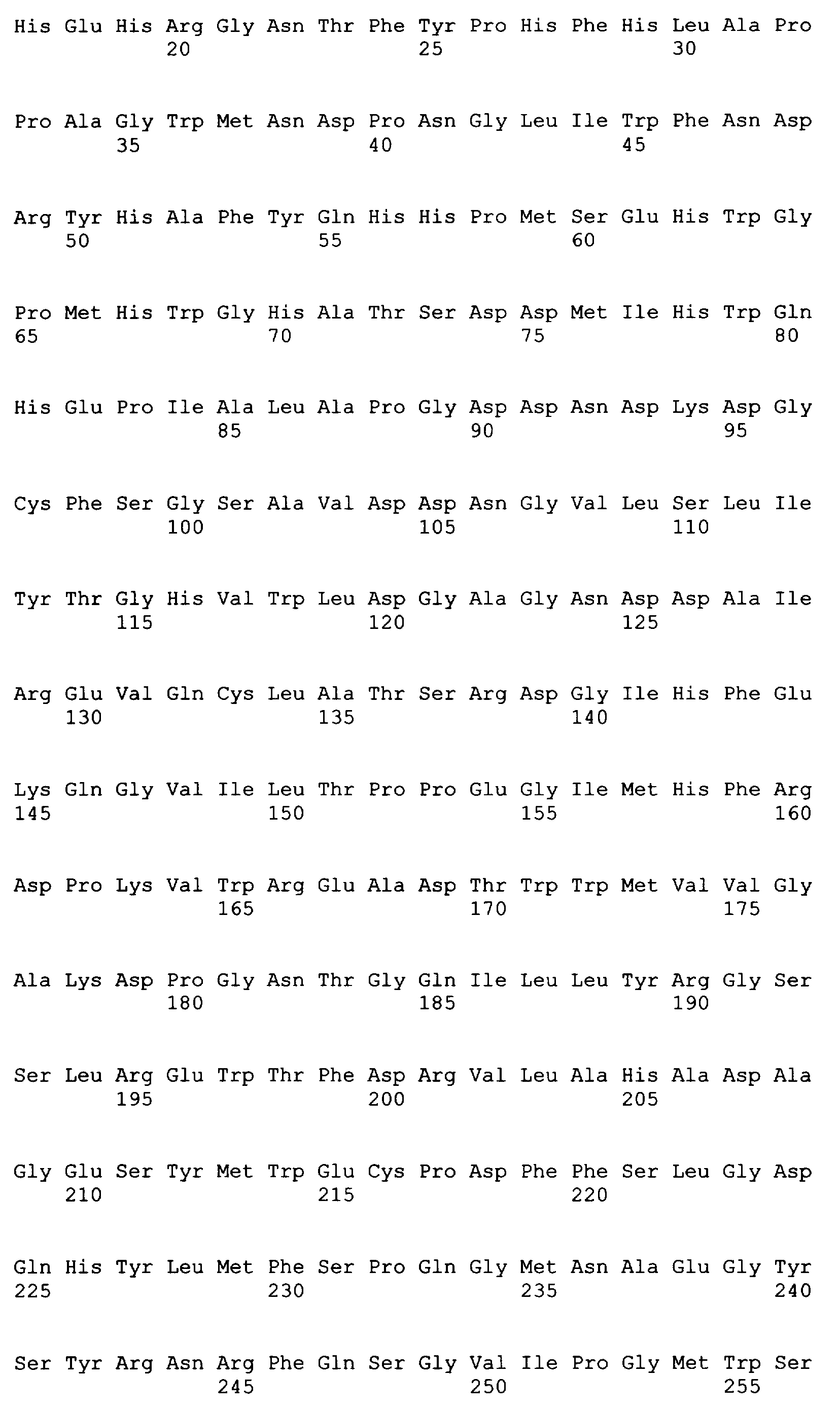









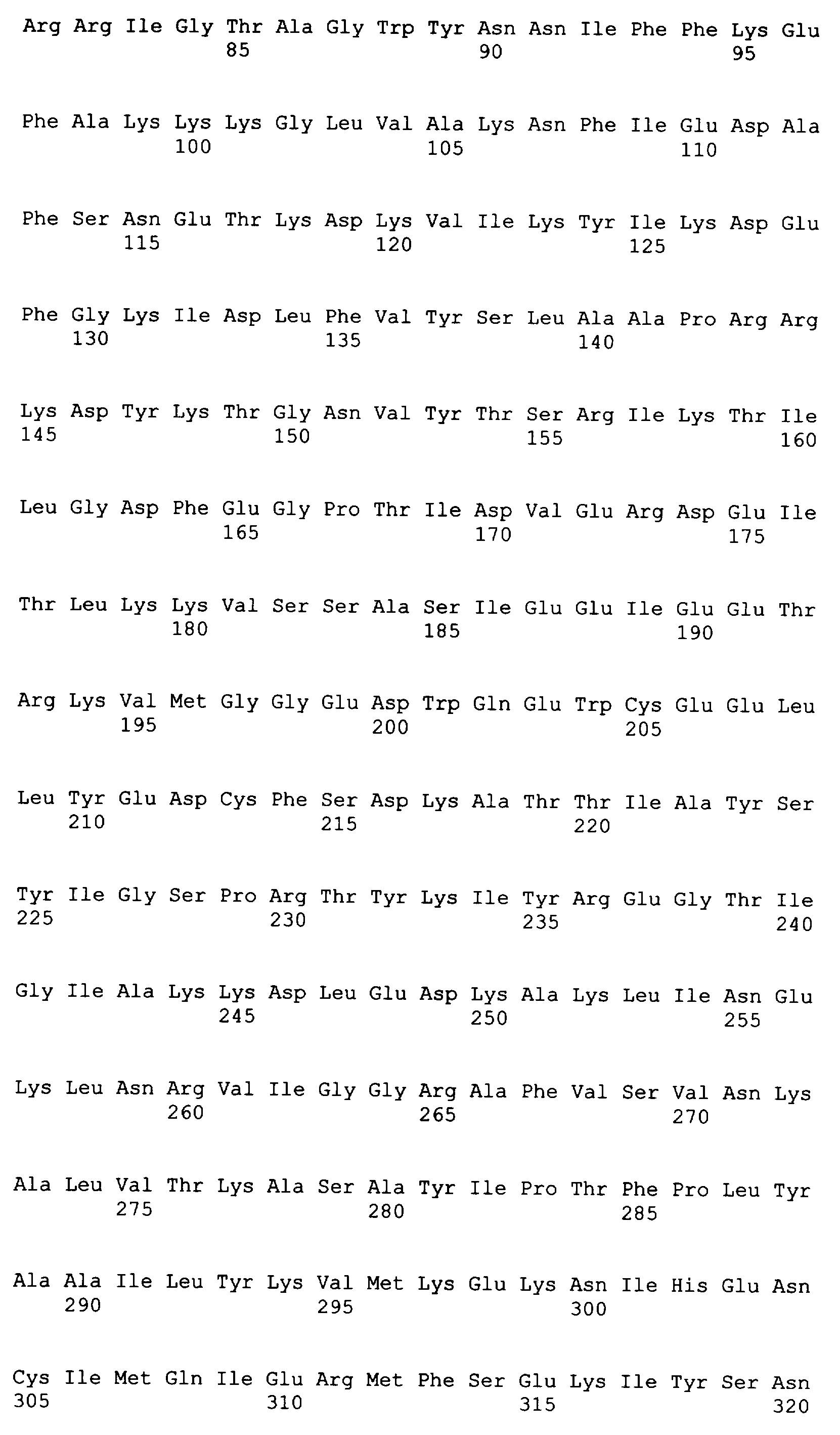








Описание
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННУЮ ЗАЯВКУ
В настоящей заявке испрашивается приоритет в соответствии с разделом 35 кодекса законов США §119 по предварительной заявке США № 60/721677, поданной 29 сентября 2005 года, и по предварительной заявке США № 60/814470, поданной 16 июня 2006 года.
ОБЛАСТЬ ПРИМЕНЕНИЯ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Изобретение относится к области промышленной микробиологии и получению спиртов. Конкретнее, 1-бутанол получают посредством промышленного брожения рекомбинантного микроорганизма.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Бутанол является важным промышленным химическим продуктом, который применим в качестве добавки к топливу, в качестве сырьевого химического вещества в производстве пластических масс и в качестве пищевых экстрагирующих агентов в пищевой промышленности и промышленности вкусовых и ароматических веществ. Каждый год от 10 до 12 миллиардов фунтов бутанола производится нефтехимическим способом, и потребность в этом химическом продукте, вероятно, будет увеличиваться.
Известны способы химического синтеза 1-бутанола, такие как оксопроцесс, процесс Реппе и гидрогенизация кротонового альдегида (Ullmann's Encyclopedia of Industrial Chemistry, 6th edition, 2003, Wiley-VCHVerlag GmbH and Co., Weinheim, Germany, Vol. 5, pp. 716-719). В этих процессах используют исходные вещества, которые получены из нефтехимических продуктов и в большинстве случаев являются дорогостоящими и вредными для окружающей среды. Получение 1-бутанола из сырьевых материалов, полученных из растений, могло бы свести к минимуму тепличные газовые выбросы и может представлять собой технический прогресс.
Также известны способы получения 1-бутанола посредством биотрансформации других органических химических веществ. Например, Muramoto et al. (JP 63017695) описывает способ получения спиртов, в том числе бутанола, из альдегидов, используя штаммы Pseudomonas. Кроме того, Kuehnle et al. (EP 1149918) описывает способ получения 1-бутанола и 2-бутанола в результате окисления углеводородов различными штаммами Rhodococcus ruber.
Также известны способы получения бутанола в результате брожения, где в самом популярном процессе получается смесь ацетона, 1-бутанола и этанола, который относится к АВЕ-процессам (Blaschek et al., патент США № 6358717). Ацетон-бутанол-этанольное брожение (АВЕ) посредством Clostridium acetobutylicum является одним из самых старых, известных среди промышленных брожений, и каскад реакций и гены, ответственные за продукцию этих растворителей, были описаны (Girbal et al., Trends in Biotechnology 16:11-16 (1998)). Однако фактическое брожение было достаточно сложным и трудно регулируемым. Использование АВЕ-брожения постоянно снижалось начиная с 1950-х годов, и в настоящее время почти весь бутанол получают посредством нефтехимических процессов, как описано выше. При обычном АВЕ-брожении сначала C.acetobutylicum продуцирует масляную, пропионовую, молочную и уксусную кислоты, уменьшается рН культуры и происходит метаболический сдвиг «бабочки», и затем образуются 1-бутанол, ацетон, изопропанол и этанол. В обычных процессах АВЕ-брожения уровень 1-бутанола, полученного из глюкозы, небольшой, обычно около 15 процентов и изредка превышает 25 процентов. Следовательно, концентрация 1-бутанола в обычных процессах АВЕ-брожения, как правило, ниже 1,3 процента.
Попытки максимизировать получение 1-бутанола в результате процесса АВЕ-брожения посредством удаления всех других побочных продуктов растворителей не были полностью успешными, и, таким образом, в этом процессе продуцируются значительные количества ацетона, который не пригоден в качестве добавки к бензину. Способ ферментативного получения бутанола, в котором 1-бутанол является единственным продуктом, представлял бы собой технический прогресс.
Таким образом, существует потребность в безопасном для окружающей среды, экономичном способе получения 1-бутанола в виде единственного продукта. Настоящее изобретение рассматривает указанную потребность посредством открытия рекомбинантного микробного продуцирующего организма-хозяина, экспрессирующего биосинтетический путь 1-бутанола.
КРАТКОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Настоящее изобретение представляет рекомбинантный микроорганизм, имеющий сконструированный биосинтетический путь 1-бутанола. Указанный сконструированный микроорганизм можно использовать для промышленного получения 1-бутанола. Соответственно настоящее изобретение представляет рекомбинантную микробную клетку-хозяина, содержащую по меньшей мере одну молекулу ДНК, кодирующую полипептид, который катализирует конверсию субстрата в продукт, выбранную из группы, состоящей из:
a) ацетил-CoA в ацетоацетил-CoA,
b) ацетоацетил-CoA в 3-гидроксибутирил-CoA,
c) 3-гидроксибутирил-CoA в кротонил-CoA,
d) кротонил-CoA в бутирил-CoA,
e) бутирил-CoA в бутиральдегид,
f) бутиральдегид в 1-бутанол;
где по меньшей мере одна молекула ДНК является гетерологичной указанной микробной клетке-хозяину и где указанная микробная клетка-хозяин продуцирует 1-бутанол.
В другом варианте осуществления настоящее изобретение относится к способу получения 1-бутанола, включающему:
i) получение рекомбинантной микробной клетки-хозяина, содержащей по меньшей мере одну молекулу ДНК, кодирующую полипептид, который катализирует конверсию субстрата в продукт, выбранную из группы, состоящей из:
a) ацетил-CoA в ацетоацетил-CoA,
b) ацетоацетил-CoA в 3-гидроксибутирил-CoA,
c) 3-гидроксибутирил-CoA в кротонил-CoA,
d) кротонил-CoA в бутирил-CoA,
e) бутирил-CoA в бутиральдегид,
f) бутиральдегид в 1-бутанол;
где по меньшей мере одна молекула ДНК является гетерологичной указанной микробной клетке-хозяину; и
ii) контактирование клетки-хозяина из (i) со способным к брожению углеродным субстратом в условиях, при которых продуцируется 1-бутанол.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖА И ОПИСАНИЯ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
Настоящее изобретение может быть более полно понято из следующих подробного описания, чертежа и сопровождающих описаний последовательностей, которые являются частью настоящей заявки.
На фиг.1 представлен биосинтетический путь 1-бутанола. Стадии, обозначенные «а», «b», «c», «d», «e» и «f», представляют собой конверсии субстрата в продукт, описанные далее.
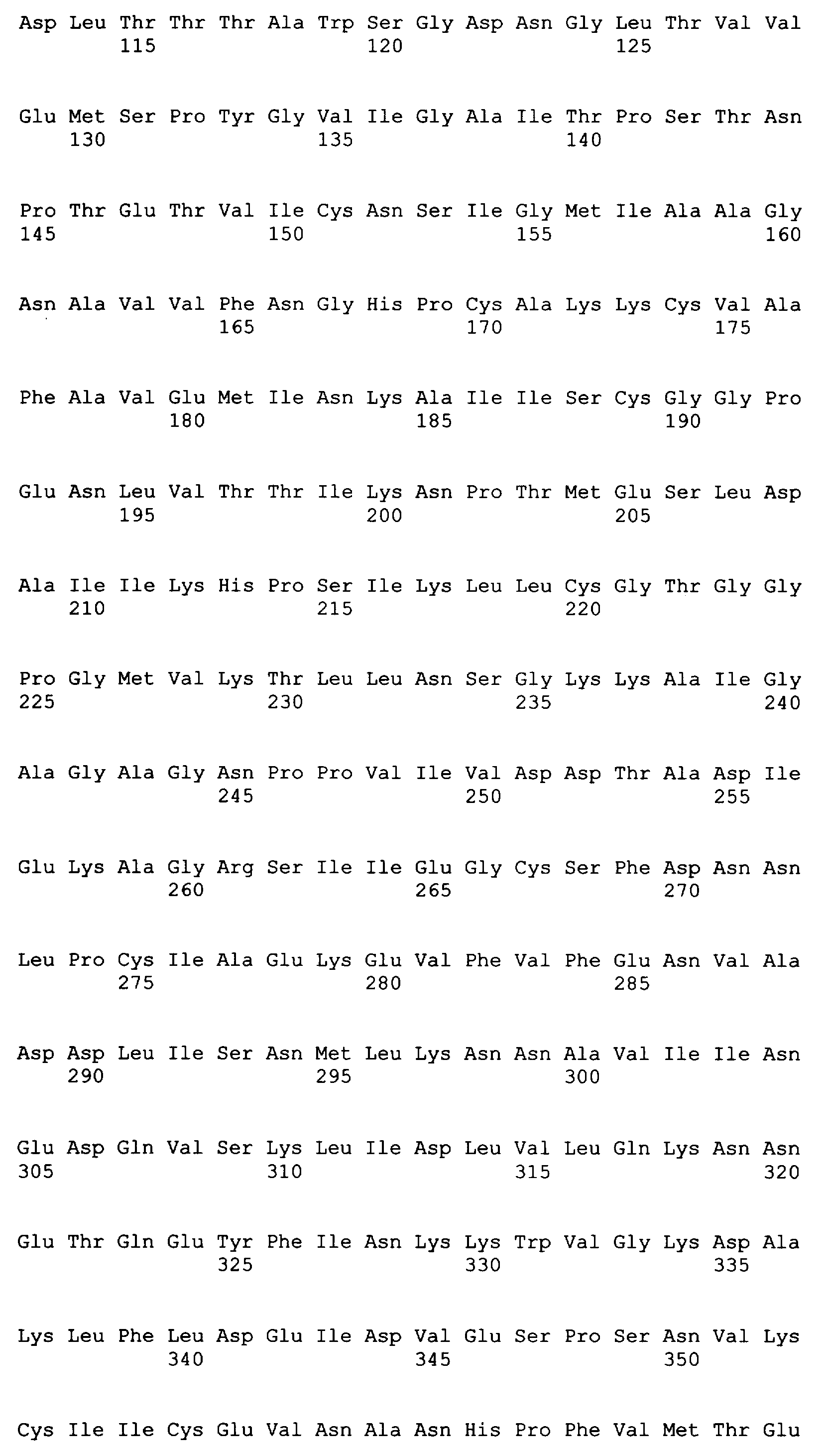
Следующие последовательности соответствуют 37 C.F.R. 1.821-1.825 («Требования, предъявляемые к патентным заявкам, содержащим описания нуклеотидных последовательностей и/или аминокислотных последовательностей - правила для последовательностей») и соответствуют стандарту ST.25 (1998) Всемирной организации интеллектуальной собственности (ВОИС) и условиям, которые необходимы для регистрации последовательностей EPO и PCT (правила 5.2 и 49.5(a-бис) и Раздел 208 и Приложение C Административной Инструкции). Символы и формат, использованные для характеристик нуклеотидной и аминокислотной последовательностей, подчиняются правилам, изложенным в 37 C.F.R. §1.822.
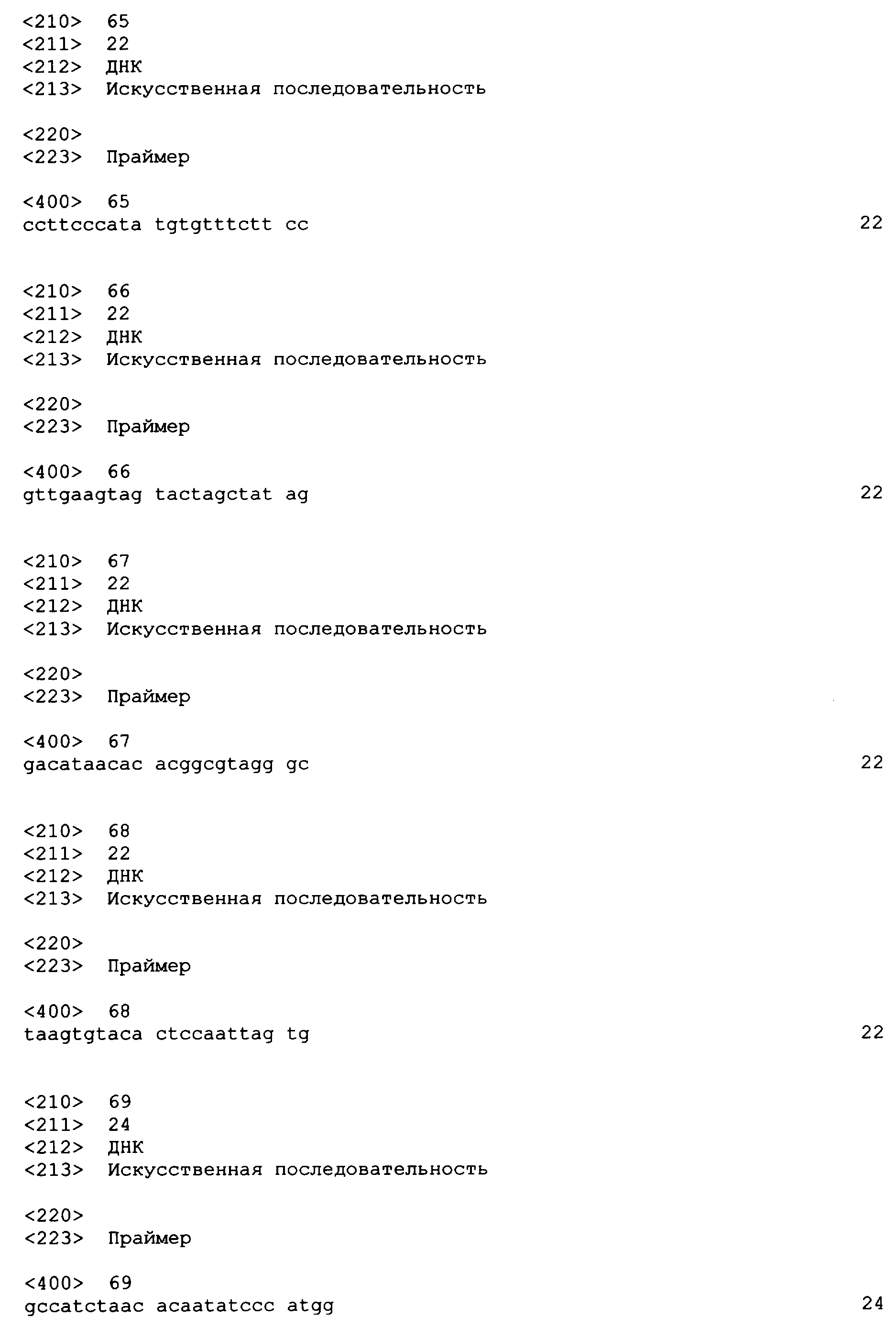
SEQ ID NO: 17-44 представляют собой нуклеотидные последовательности олигонуклеотидных праймеров, используемых для амплификации генов биосинтетического пути 1-бутанола.
SEQ ID NO: 45-72 представляют собой нуклеотидные последовательности олигонуклеотидных праймеров, используемых для секвенирования.
SEQ ID NO: 73-75 представляют собой нуклеотидные последовательности олигонуклеотидных праймеров, используемых для конструирования трансформационных векторов, описанных в примере 9.
SEQ ID NO: 76 представляет собой нуклеотидную последовательность кодон-оптимизированного гена САС0462, обозначенного в настоящем описании как CaTER.
SEQ ID NO: 77 представляет собой нуклеотидную последовательность кодон-оптимизированного гена EgTER, обозначенного в настоящем описании как EgTER(opt).
SEQ ID NO: 78 представляет собой нуклеотидную последовательность кодон-оптимизированного гена ald, обозначенного в настоящем описании как ald (opt).
SEQ ID NO: 79 представляет собой нуклеотидную последовательность плазмиды pFP988.
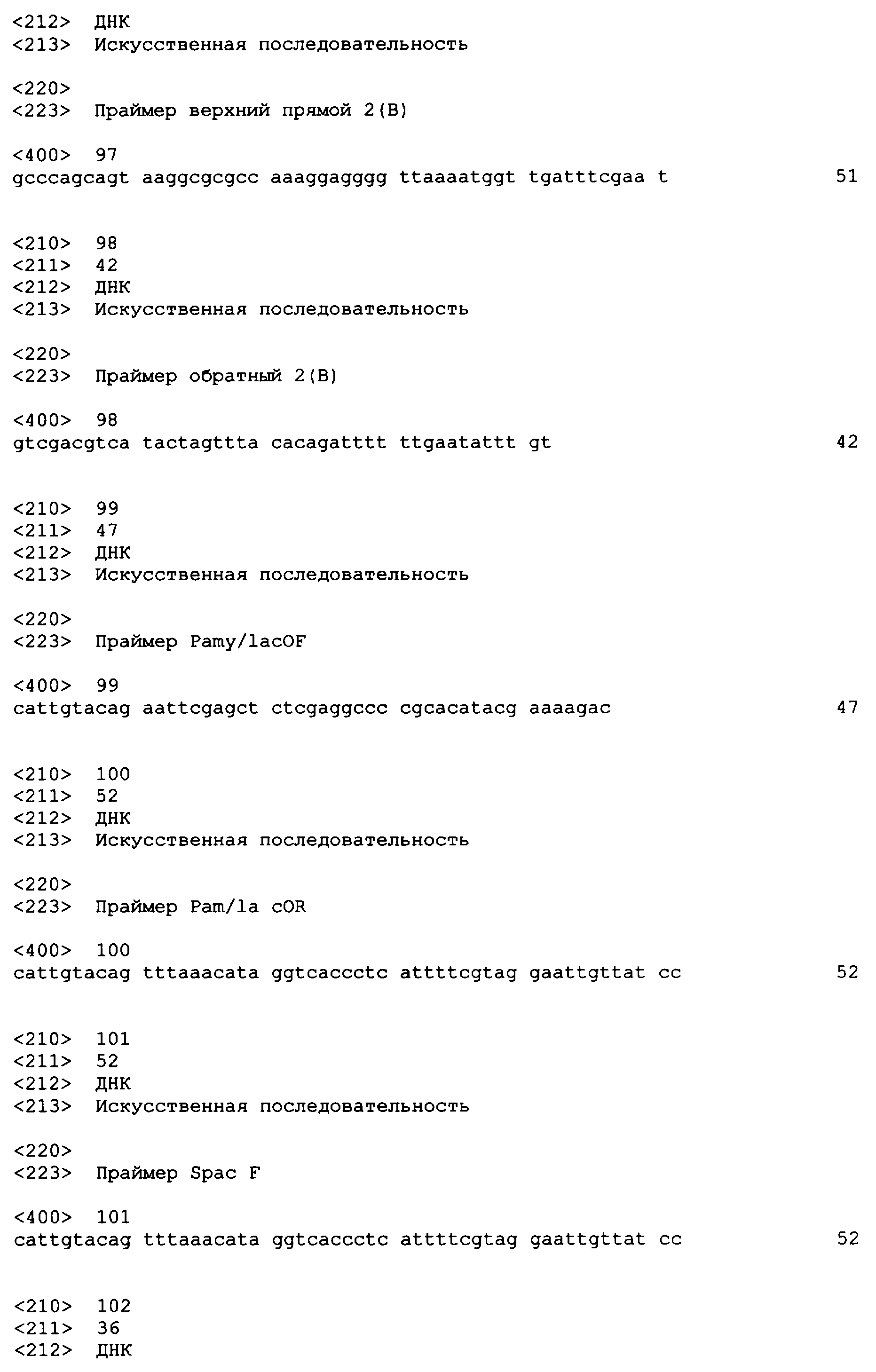
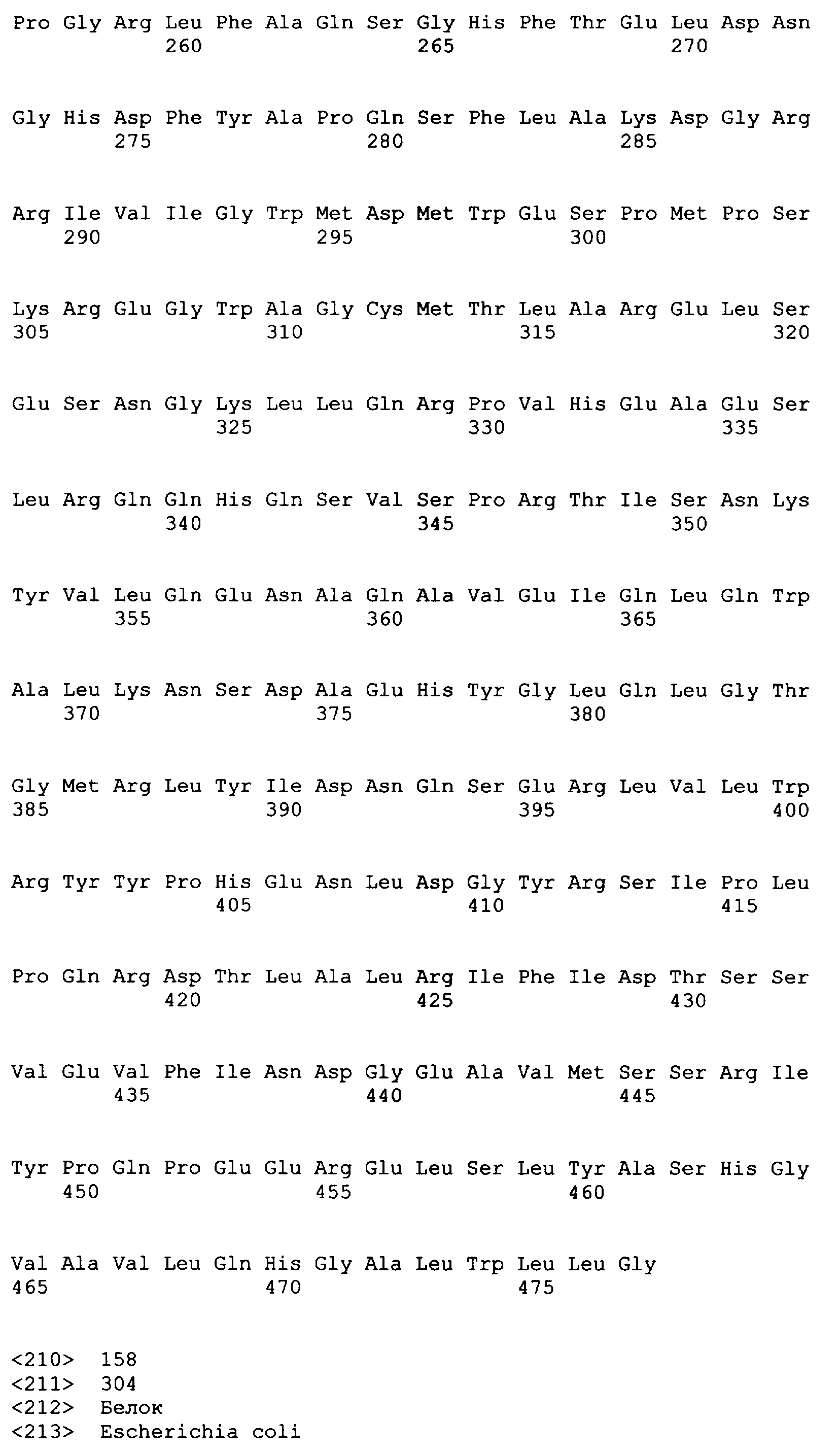
SEQ ID NO: 80-127, 160-185 и 190-207 представляют собой последовательности нуклеиновых кислот праймеров клонирования, секвенирования или скрининг-ПЦР, использованные для клонирования, секвенирования или скрининга генов биосинтетического пути 1-бутанола, описанного в настоящем описании, и они более подробно описаны в таблицах 4 и 5.
SEQ ID NO: 156 представляет собой нуклеотидную последовательность кластера гена cscBKA.
SEQ ID NO: 157 представляет собой аминокислотную последовательность гидролазы сахарозы (CscA).
SEQ ID NO: 158 представляет собой аминокислотную последовательность D-фруктокиназы (CscK).
SEQ ID NO: 159 представляет собой аминокислотную последовательность пермеазы сахарозы (CscB).
SEQ ID NO: 186 представляет собой нуклеотидную последовательность кодон-оптимизированного гена tery, описанного в примере 17.
SEQ ID NO: 187 представляет собой аминокислотную последовательность бутил-СоА-дегидрогеназы (ter), кодируемой кодон-оптимизированным геном tery (SEQ ID NO: 186).
SEQ ID NO: 188 представляет собой нуклеотидную последовательность кодон-оптимизированного гена aldy, описанного в примере 17.
SEQ ID NO: 189 представляет собой аминокислотную последовательность бутиральдегиддегидрогеназы (ald), кодируемой кодон-оптимизированным геном aldy (SEQ ID NO: 188).
SEQ ID NO: 208 представляет собой нуклеотидную последовательность матричной ДНК, используемой в примере 14.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам получения 1-бутанола с помощью рекомбинантных микроорганизмов. Настоящее изобретение отвечает целому ряду торгово-промышленных потребностей. Бутанол является важным промышленным химическим продуктом с целым рядом применений, среди которых его потенциал в качестве топлива или добавки к топливу является чрезвычайно значимым. Являясь только четырехуглеродным спиртом, бутанол имеет энергосодержание, аналогичное таковому бензина, и может быть смешан с любым природным топливом. Бутанол предпочтителен в качестве топлива или добавки к топливу, поскольку при сгорании в стандартном двигателе внутреннего сгорания он приводит к образованию только СО2 и небольшого количества или вовсе не приводит к образованию SOx или NOx. Кроме того, бутанол менее коррозийный, чем этанол, наиболее предпочтительная добавка к топливу на сегодняшний день.
В дополнение к его практичности в качестве биотоплива или добавки к топливу, бутанол имеет потенциал оказывать влияние на проблему распределения водорода в развивающейся индустрии тепловых элементов. В настоящее время проблемы безопасности, связанные с переносом водорода и распределением, ограничивают применение топливных элементов. Бутанол можно без труда преобразовать в его водородное состояние и можно распространять через существующие бензозаправочные станции в чистоте, требуемой либо для топливных элементов, либо для носителей.
Наконец, по настоящему изобретению бутанол получают из источников углерода растительного происхождения, что позволяет избежать отрицательного воздействия на окружающую среду, связанного со стандартными нефтехимическими способами получения бутанола.
Следующие определения и сокращения должны использоваться для интерпретации настоящей формулы изобретения и описания.
Используемый в настоящем описании термин «изобретение» или «настоящее изобретение» является неограничивающим термином и не предназначен для определения какого-либо единственного варианта осуществления данного конкретного изобретения, а охватывает все возможные варианты осуществления, как описано в настоящем описании и формуле изобретения.
«ABE» является сокращением для способа ацетон-бутанол-этанольного брожения.
Термин «биосинтетический путь 1-бутанола» означает каскад ферментативных реакций для получения 1-бутанола из ацетил-коэнзима А (ацетил-СоА).
Термин «ацетил-CoA-ацетилтрансфераза» означает фермент, который катализирует превращение двух молекул ацетил-СоА в ацетоацетил-СоА и коэнзим А (СоА). Предпочтительными ацетил-CoA-ацетилтрансферазами являются ацетил-CoA-ацетилтрансферазы с предпочтением субстрата (реакция в прямом направлении) для короткой цепи ацил-СоА и ацетил-СоА, и они классифицированы как Е.С. 2.3.1.9 [Номенклатура ферментов 1992, Academic Press, San Diego]; хотя ферменты с более широким диапазоном субстрата (E.С. 2.3.1.16) также будут эффективны. Ацетил-CoA-ацетилтрансферазы доступны из различных источников, например Escherichia coli (GenBank NO: NP_416728 (SEQ ID NO: 129), NC_000913 (SEQ ID NO: 128); аминокислотная последовательность NCBI (National Center for Biotechnology Information), нуклеотидная последовательность NCBI), Clostridium acetobutylicum (GenBank NO: NP_349476.1 (SEQ ID NO: 2), NC_003030 (SEQ ID NO: 1); NP_149242 (SEQ ID NO: 4), NC_001988 (SEQ ID NO: 3)), Bacillus subtilis (GenBank NO: NP_390297 (SEQ ID NO: 131), NC_000964 (SEQ ID NO: 130)) и Saccharomyces cerevisiae (GenBank NO: NP_015297 (SEQ ID NO: 133), NC_001148 (SEQ ID NO: 132)).
Термин «3-гидроксибутирил-СоА-дегидрогеназа» относится к ферменту, который катализирует превращение ацетоацетил-СоА в 3-гидроксибутирил-СоА. 3-гидроксибутирил-СоА-дегидрогеназы могут быть восстановленный никотинамидадениндинуклеотид (NADH)-зависимыми, с субстратным предпочтением для (S)-3-гидроксибутирил-СоА или (R)-3-гидроксибутирил-СоА, и классифицированы как Е.С. 1.1.1.35 и Е.С. 1.1.1.30 соответственно. Кроме того, 3-гидроксибутирил-CoA-дегидрогеназы могут быть восстановленный никотинамидадениндинуклеотидфосфат (NADHР)-зависимыми, с субстратным предпочтением для (S)-3-гидроксибутирил-СоА или (R)-3-гидроксибутирил-СоА, и классифицированы как Е.С. 1.1.1.157 и Е.С. 1.1.1.36 соответственно. 3-гидроксибутирил-CoA-дегидрогеназы доступны из различных источников, например C.acetobutylicum (GenBank NO: NP_349314 (SEQ ID NO: 6), NC_003030 (SEQ ID NO: 5)), В.subtilis (GenBank NO: AAB09614 (SEQ ID NO: 135), U29084 (SEQ ID NO: 134)), Ralstonia eutropha (GenBank NO: YP_294481 (SEQ ID NO: 137), NC_007347 (SEQ ID NO: 136)) и Alcaligenes eutrophus (GenBank NO: AAA21973 (SEQ ID NO: 139), J04987 (SEQ ID NO: 138)).
Термин «кротоназа» относится к ферменту, который катализирует превращение 3-гидроксибутирил-СоА в кротонил-СоА и Н2О. Кротоназы могут иметь субстратные предпочтения для (S)-3-гидроксибутирил-CoA или (R)-3-гидроксибутирил-CoA, и они классифицированы как E.C. 4.2.1.17 и E.C. 4.2.1.55 соответственно. Кротоназы доступны из различных источников, например E.coli (GenBank NO: NP_415911 (SEQ ID NO: 141), NC_000913 (SEQ ID NO: 140)), C.acetobutylicum (GenBank NO: NP_349318 (SEQ ID NO: 8), NC_003030 (SEQ ID NO: 6)), B.subtilis (GenBank NO: CAB13705 (SEQ ID NO: 143), Z99113 (SEQ ID NO: 142)) и Aeromonas caviae (GenBank NO: BAA21816 (SEQ ID NO: 145), D88825 (SEQ ID NO: 144)).
Термин «бутирил-CoA-дегидрогеназа» относится к ферменту, который катализирует превращение кротонил-СоА в бутирил-СоА. Бутирил-CoA-дегидрогеназы могут быть либо NADH-зависимыми, либо NADHP-зависимыми, и они классифицированы как E.С. 1.3.1.44 и E.C. 1.3.1.38 соответственно. Бутирил-CoA-дегидрогеназы доступны из различных источников, например C.acetobutylicum (GenBank NO: NP_347102 (SEQ ID NO: 10), NC_003030 (SEQ ID NO: 9)), Euglena gracilis (GenBank NO: Q5EU90 (SEQ ID NO: 147), AY741582 (SEQ ID NO: 146)), Streptomyces collinus (GenBank NO: AAA92890 (SEQ ID NO: 149), U37135 (SEQ ID NO: 148)) и Streptomyces coelicolor (GenBank NO: CAA22721 (SEQ ID NO: 151), AL939127 (SEQ ID NO: 150)).
Термин «бутиральдегиддегидрогеназа» относится к ферменту, который катализирует превращение бутирил-СоА в бутиральдегид с использованием NADH или NADPH в качестве кофактора. Бутиральдегиддегидрогеназы с предпочтением к NADH известны как E.C. 1.2.1.57 и доступны, например, из Clostridium beijerinckii (GenBank NO: AAD31841 (SEQ ID NO: 12), AF157306 (SEQ ID NO: 11)) и C.acetobutylicum (GenBank NO: NP_149325 (SEQ ID NO: 153), NC_001988 (SEQ ID NO: 152)).
Термин «бутанолдегидрогеназа» относится к ферменту, который катализирует превращение бутиральдегида в 1-бутанол с использованием либо NADH, либо NADPH в качестве кофактора. Бутанолдегидрогеназы доступны, например, из C.acetobutylicum (GenBank NO: NP_149325 (SEQ ID NO: 153), NC_001988 (SEQ ID NO: 152); примечание: этот фермент обладает действием и альдегид-, и алкогольдегидрогеназы; NP_349891 (SEQ ID NO: 14), NC_003030 (SEQ ID NO: 13) и NP_349892 (SEQ ID NO: 16), NC_003030 (SEQ ID NO: 15)) и E.coli (GenBank NO: NP_417484 (SEQ ID NO: 155), NC_000913 (SEQ ID NO: 154)).
Термин «факультативный анаэроб» относится к микроорганизму, который может расти и в аэробной, и в анаэробной среде.
Термин «углеродный субстрат» или «сбраживаемый углеродный субстрат» относится к источнику углерода, поддающемуся превращению в процессе обмена веществ организмами-хозяевами по настоящему изобретению, и определенным источникам углерода, выбранным из группы, состоящей из моносахаридов, олигосахаридов, полисахаридов и одноуглеродных субстратов или их смесей.
Термин «ген» относится к фрагменту нуклеиновой кислоты, который способен быть экспрессирован как специфический белок, при желании включающий регуляторные последовательности, предшествующие (5'-некодирующие последовательности) и последующие (3'-некодирующие последовательности) указанной кодирующей последовательности. «Нативный ген» означает ген, который встречается в природе, с его собственными регуляторными последовательностями. «Химерный ген» означает любой ген, который не является нативным геном, содержащий регуляторные и кодирующие последовательности, которые вместе не встречаются в природе. Соответственно химерный ген может содержать регуляторные последовательности и кодирующие последовательности, которые происходят из различных источников, или регуляторные последовательности и кодирующие последовательности, происходящие из одного и того же источника, но расположенные в порядке, отличном от того, который обнаружен в природе. «Эндогенный ген» относится к нативному гену при его природной локализации в указанном геноме организма. «Чужеродный» или «гетерологичный ген» означает ген, который обычно не обнаруживается в организме-хозяине, но который встроен в организм-хозяин посредством переноса гена. Чужеродные гены могут включать нативные гены, встроенные в ненативные организмы, или химерные гены. «Трансген» представляет собой ген, который был встроен в геном в результате процесса трансформации.
Используемый в настоящем описании термин «кодирующая последовательность» относится к последовательности ДНК, которая кодирует определенную аминокислотную последовательность. «Подходящие регуляторные последовательности» относятся к нуклеотидным последовательностям, располагающимся выше (5'-некодирующие последовательности), в пределах или ниже (3'-некодирующие последовательности) от кодирующей последовательности, которые влияют на транскрипцию, процессинг РНК, или стабильность, или трансляцию связанной кодирующей последовательности. Регуляторные последовательности могут включать промоторы, лидерные последовательности для трансляции, интроны, последовательности распознавания для полиаденилирования, участок процессирования РНК, участок связывания эффектора и структуру типа «стебель-петля».
Термин «промотор» относится к последовательности ДНК, способной регулировать экспрессию кодирующей последовательности или функциональной РНК. В большинстве случаев кодирующая последовательность расположена в 3'-положении от последовательности промотора. Промоторы могут происходить полностью из нативного гена, или могут состоять из различных элементов, полученных из различных промоторов, обнаруженных в природе, или даже включать сегменты синтетической ДНК. Специалисту в уровне техники понятно, что различные промоторы могут направлять экспрессию гена в различных тканях или клеточных типах, или на разных этапах развития, или в ответ на различные условия окружающей среды или физиологические состояния. Промоторы, которые обеспечивают экспрессию гена в большинстве клеточных типов в большинстве случаев, обычно называются «конститутивными промоторами». Дополнительно признано, что поскольку в большинстве случаев точные границы регуляторных последовательностей не были полностью определены, фрагменты ДНК различной длины могут иметь идентичные промоторные активности.
Термин «функционально связанный» относится к ассоциации последовательностей нуклеиновых кислот на отдельном фрагменте нуклеиновой кислоты таким образом, что на функцию одной влияет другая. Например, промотор функционально связан с кодирующей последовательностью, когда он способен влиять на экспрессию этой кодирующей последовательности (то есть указанная кодирующая последовательность находится под транскрипционным контролем указанного промотора). Кодирующие последовательности могут быть функционально связаны с регуляторными последовательностями в прямом или обратном направлении.
Используемый в настоящем описании термин «экспрессия» относится к транскрипции и стабильной аккумуляции смысловой (мРНК) или антисмысловой РНК, полученной из указанного фрагмента нуклеиновой кислоты по настоящему изобретению. Экспрессия также может относиться к трансляции мРНК в полипептид.
Используемый в настоящем описании термин «трансформация» относится к переносу фрагмента нуклеиновой кислоты внутрь организма-хозяина, что приводит к генетически стабильному наследованию. Организмы-хозяева, содержащие указанные трансформированные фрагменты нуклеиновых кислот, называются «трансгенными», или «рекомбинантными», или «трансформированными» организмами.
Термины «плазмида», «вектор» и «кассета» относятся к дополнительному хромосомному элементу, зачастую несущему гены, которые не являются частью центрального метаболизма указанной клетки и обычно находятся в виде кольцевых двуцепочечных фрагментов ДНК. Такими элементами могут быть автономно реплицирующиеся последовательности, геномные интегрирующие последовательности, фаговые или нуклеотидные последовательности линейной или кольцевой, одно- или двуцепочечной ДНК или РНК, полученные из любого источника, в которых ряд нуклеотидных последовательностей объединен или рекомбинирован в уникальную конструкцию, которая допускает встраивание промоторного фрагмента и последовательности ДНК для выбранного генного продукта наряду с соответствующей 3'-нетранслируемой последовательностью внутрь клетки. «Кассета для трансформации» относится к специфическому вектору, включающему чужеродный ген и имеющему элементы в дополнение к указанному чужеродному гену, что облегчает трансформацию конкретной клетки-хозяина. «Экспрессионная кассета» относится к определенному вектору, включающему чужеродный ген и имеющему элементы в дополнение к указанному чужеродному гену, которые делают возможной усиленную экспрессию указанного гена в чужеродном хозяине.
Используемый в настоящем описании термин «вырожденность кода» относится к природе генетического кода, допускающей изменение указанной нуклеотидной последовательности без влияния на аминокислотную последовательность кодируемого полипептида. Квалифицированный специалист хорошо осведомлен о «статистическом отклонении от равномерности в использовании кодонов», проявляемом определенной клеткой-хозяином при использовании нуклеотидных кодонов для определения указанной аминокислоты. Поэтому при синтезировании гена для улучшенной экспрессии в клетке-хозяине желательно сконструировать указанный ген таким образом, чтобы его частота использования кодона приближалась к частоте предпочтительного использования кодона указанной клетки-хозяина.
Термин «кодон-оптимизированный», когда он относится к генам или кодирующим областям молекул нуклеиновых кислот для трансформации различных организмов-хозяев, относится к изменению кодонов в указанном гене или кодирующих областях указанных молекул нуклеиновых кислот для отображения типичного использования кодонов указанного организма-хозяина без изменения указанного полипептида, кодируемого указанной ДНК.
Используемые в настоящем описании стандартные технологии рекомбинантных ДНК и молекулярного клонирования хорошо известны в уровне техники и описаны Sambrook, J., Fritsch, E.F. и Maniatis, T., Molecular Cloning: A Laboratory Manual, Second Edition, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY (1989) (в дальнейшем «Maniatis»); Silhavy, T.J., Bennan, M.L. и Enquist, L.W., Experiments with Gene Fusions, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY (1984); и Ausubel, F.M. et al., Current Protocols in Molecular Biology, published by Greene Publishing Assoc and Wiley-Interscience (1987).
Биосинтетический путь 1-бутанола
Микроорганизмы, утилизирующие углеводы, задействуют путь Эмбден-Мейергофа-Парнаса (EMP), путь Энтнера-Дудороффа и пентозофосфатный цикл в качестве центральных путей метаболизма для обеспечения энергии и клеточных предшественников для роста и поддержания. Указанные пути имеют главным образом промежуточное соединение глицеральдегид-3-фосфат, и в конечном счете пируват образуется непосредственно или в сочетании с путем ЕМР. Затем пируват трансформируется в ацетил-коэнзим А (ацетил-СоА) различными способами, в том числе в результате контактирования с пируватдегидрогеназным комплексом, пируват-формиатлиазой и пируват-ферредоксин оксидоредуктазой. Ацетил-СоА служит ключевым промежуточным звеном, например, при образовании жирных кислот, аминокислот и вторичных метаболитов. Указанные комбинированные реакции по превращению сахара в ацетил-СоА приводят к образованию энергии (например, аденозин-5'-трифосфат, АТФ) и восстановленных эквивалентов (например, восстановленный никотинамидадениндинуклеотид, NADH, и восстановленный никотинамидадениндинуклеотидфосфат, NADPH). NADH и NADPH должны быть возвращены в повторный цикл для получения их окисленных форм (NAD+ и NADP+ соответственно). В присутствии неорганических акцепторов электронов (например, O2, NO3- и SO42-), указанные восстановленные эквиваленты можно использовать для увеличения энергетического пула; альтернативно, может быть образован восстановленный углеродный побочный продукт. Продукция этанола и 1-бутанола в результате указанного углеводного брожения является примером последнего.
Настоящее изобретение делает возможным получение 1-бутанола из источников углевода с помощью рекомбинантных микроорганизмов, обеспечивая законченный биосинтетический путь 1-бутанола из ацетил-СоА до 1-бутанола, как показано на чертеже. Указанный биосинтетический путь, как правило, отсутствующий в указанном микробном сообществе по причине отсутствия генов или отсутствия соответствующей генной регуляции, включает следующие превращения (конверсии) субстрата в продукт:
a) ацетил-CoA в ацетоацетил-CoA, катализируется, например, ацетил-CoA-ацетилтрансферазой;
b) ацетоацетил-CoA в 3-гидроксибутирил-CoA, катализируется, например, 3-гидроксибутирил-CoA-дегидрогеназой;
c) 3-гидроксибутирил-CoA в кротонил-CoA, катализируется, например, кротоназой;
d) кротонил-CoA в бутирил-CoA, катализируется, например, бутирил-CoA-дегидрогеназой;
e) бутирил-CoA в бутиральдегид, катализируется, например, бутиральдегиддегидрогеназой;
f) бутиральдегид в 1-бутанол, катализируется, например, бутанолдегидрогеназой.
В указанном пути не требуется АТФ и продуцируется NAD+ и/или NADP+, таким образом, сохраняется равновесие с центральным путем метаболизма, в котором продуцировался ацетил-СоА. Способность природных организмов производить 1-бутанол посредством брожения существует редко и проявляется наиболее заметно у Clostridium beijerinckii и Clostridium acetobutylicum. Описана генная организация и генная регуляция для Clostridium acetobutylicum (L. Girbal и P. Soucaille, Trends in Biotechnology 216: 11-16 (1998)). Тем не менее, многие из указанных ферментативных действий связаны также с альтернативными путями, например использование углеводорода, окисление жирных кислот и метаболизм полигидроксиалканоата. Таким образом, в обеспеченном рекомбинантном пути из ацетил-СоА в 1-бутанол существует целый ряд возможностей для осуществления индивидуальных стадий реакций, и специалист в уровне техники будет способен использовать публично доступные последовательности для конструкции соответствующих путей. Список репрезентативного ряда генов, известных в уровне техники и пригодных для конструкции биосинтетического пути 1-бутанола, представлен ниже в таблице 2.
Микробные организмы-хозяева для получения 1-бутанола
Микробные организмы-хозяева для получения 1-бутанола могут быть выбраны из бактерий, цианобактерий, мицелиальных грибов и дрожжей. Указанный микробный организм-хозяин для получения 1-бутанола предпочтительно устойчив к 1-бутанолу, таким образом, выход продукта не ограничен токсичностью бутанола. Микробы, которые метаболически активны при высоких уровнях титра 1-бутанола, не известны в уровне техники. Несмотря на то что мутанты, устойчивые к бутанолу, были выделены из сольвентогенной Clostridia, в наличие имеется немного информации об указанной устойчивости к бутанолу других потенциально пригодных бактериальных штаммов. Большинство исследований по сравнению устойчивости к действию спирта у бактерий показывают, что бутанол более токсичен, чем этанол (de Cavalho et al., Microsc. Res. Tech. 64: 215-22 (2004), и Kabelitz et al., FEMS Microbiol. Lett. 220: 223-227 (2003)). Tomas et al. (J. Bacteriol. 186: 2006-2018 (2004)) сообщает, что выход бутанола в результате сбраживания в Clostridium acetobutylicum может быть ограничен токсичностью бутанола. Основным эффектом бутанола на Clostridium acetobutylicum является нарушение мембранных функций (Hermann et al., Appl. Environ. Microbiol. 50: 1238-1243 (1985)).
Микробные организмы-хозяева, выбранные для получения 1-бутанола, предпочтительно устойчивы к 1-бутанолу и способны превращать углеводы в 1-бутанол. Критерии отбора подходящих микробных организмов-хозяев включают следующие: природная устойчивость к 1-бутанолу, высокая скорость утилизации глюкозы, возможность использовать генетические механизмы для манипуляций с геном и способность к генерированию стабильных хромосомных изменений.
Подходящие штаммы-хозяева с устойчивостью к 1-бутанолу можно определить в результате скрининга исходя из природной устойчивости указанного штамма. Указанная природная устойчивость микробов к 1-бутанолу может быть измерена посредством определения концентрации 1-бутанола, которая приводит к 50% ингибированию скорости роста (IC50), при выращивании в минимальной среде. Значения IC50 можно определить с использованием методов, известных в уровне техники. Например, представляющие интерес микробы можно выращивать в присутствии различных количеств 1-бутанола и скорость роста контролировать посредством измерения оптической плотности при 600 нанометрах. Время удвоения можно вычислить по логарифмической составной части кривой роста и использовать в качестве меры скорости роста. Полученная концентрация 1-бутанола, которая приводит к 50% ингибированию роста, может быть определена по графику зависимости процента ингибирования роста от концентрации 1-бутанола. Предпочтительно, указанный штамм-хозяин должен иметь IC50 для 1-бутанола более приблизительно 0,5% вес./об.ъем.
Микробный хозяин для получения 1-бутанола также должен утилизировать глюкозу с высокой скоростью. Большинство микробов способно утилизировать углеводы. Тем не менее, некоторые экзогенные микроорганизмы не могут утилизировать углеводы с высокой эффективностью и вследствие этого не могут быть пригодными хозяевами.
Способность к генетическому изменению хозяина является необходимой для получения любого рекомбинантного микроорганизма. Принципом технологии передачи генов может быть электропорация, конъюгация, трансдукция или природная трансформация. В наличии имеется широкий спектр плазмид, сопрягающихся с организмом-хозяином, и маркеров, устойчивых к лекарственным средствам. Векторы для клонирования разработаны для организмов-хозяев исходя из природы маркеров, устойчивых к антибиотикам, которые могут функционировать в указанном организме-хозяине.
Микробный хозяин также должен быть обработан для того, чтобы инактивировать конкурирующие пути для расхода углерода посредством удаления различных генов. Для этого требуется наличие либо транспозонов для непосредственной инактивации, либо векторов для интегрирования в хромосомы. Кроме того, указанныйанный продуцирующий хозяин должен поддаваться химическому мутагенезу, для того чтобы получить мутации для повышения природной устойчивости к 1-бутанолу.
Основываясь на вышеописанных критериях, подходящие микробные хозяева для производства 1-бутанола включают, но не ограничиваются, членов родов Clostridium, Zymomonas, Escherichia, Salmonella, Rhodococcus, Pseudomonas, Bacillus, Lactobacillus, Enterococcus, Alcaligenes, Klebsiella, Paenibacillus, Arthrobacter, Corynebacterium, Brevibacterium, Pichia, Candida, Hansenula и Saccharomyces. Предпочтительные организмы-хозяева включают: Escherichia coli, Alcaligenes eutrophus, Bacillus licheniformis, Paenibacillus macerans, Rhodococcus erythropolis, Pseudomonas putida, Lactobacillus plantarum, Enterococcus faecium, Enterococcus gallinarium, Enterococcus faecalis, Bacillus subtilis и Saccharomyces cerevisiae.
Конструирование продуцирующего организма-хозяина
Рекомбинантные организмы, содержащие необходимые гены, которые будут кодировать ферментативный каскад реакций для превращения поддающегося ферментации углеродного субстрата в 1-бутанол, можно сконструировать с использованием технологий, хорошо известных в уровне техники. В настоящем изобретении гены, кодирующие ферменты биосинтетического пути 1-бутанола, а именно ацетил-CoA-ацетилтрансферазу, 3-гидроксибутирил-CoA-дегидрогеназу, кротоназу, бутирил-CoA-дегидрогеназу, бутиральдегиддегидрогеназу и бутанолдегидрогеназу, могут быть выделены из различных источников, как описано выше.
Способы получения требуемых генов из бактериального генома являются общепринятыми и хорошо известны в области молекулярной биологии. Например, если последовательность указанного гена известна, могут быть созданы соответствующие геномные библиотеки посредством расщепления рестрикционными эндонуклеазами и могут быть проверены с использованием зондов, комплементарных к требуемой последовательности гена. Как только указанная последовательность выделена, указанную ДНК можно амплифицировать с использованием стандартных методов амплификации с использованием праймеров, например полимеразной цепной реакции (Mullis, патент США № 4683202), для получения количества ДНК, подходящего для трансформации с использованием соответствующих векторов. Средства для оптимального выбора кодонов для экспрессии в гетерологичном организме-хозяине легкодоступны. Некоторые средства для оптимального выбора кодонов доступны исходя из содержания GC в указанном организме-хозяине. Содержание GC в некоторых типичных микробных хозяевах представлено в таблице 3.
Как только соответствующие гены каскада реакций идентифицированы и выделены, они могут быть трансформированы в соответствующие экспрессирующие организмы-хозяева способами, хорошо известными в уровне техники. Векторы или кассеты, используемые для трансформации различных клеток-хозяев, являются общепринятыми и коммерчески доступны в таких компаниях, как EPICENTRE® (Madison, WI), Invitrogen Corp. (Carlsbad, CA), Stratagene (La Jolla, CA) и New England Biolabs, Inc. (Beverly, MA). Обычно вектор или кассета содержит последовательности, направляющие транскрипцию и трансляцию соответствующего гена, селектируемый маркер и последовательности, обеспечивающие автономную репликацию или хромосомное интегрирование. Подходящие векторы включают 5'-область гена, которая содержит элементы управления инициацией транскрипции, и 3'-область фрагмента ДНК, которая контролирует терминацию транскрипции. Обе контролирующие области могут быть получены из генов, гомологичных трансформированной клетке-хозяину, хотя должно быть понятно, что такие контролирующие области также могут быть получены из генов, которые не являются нативными для определенных видов, выбранных в качестве продуцирующих хозяев.
Контролирующие области инициации или промоторы, которые используются для направления экспрессии областей, кодирующих соответствующий каскад реакций в требуемой клетке-хозяине, являются многочисленными и хорошо известны специалисту в уровне техники. В сущности, любой промотор, способный управлять указанными генетическими элементами, пригоден для настоящего изобретения, в том числе, но не ограничиваясь этим, CYC1, HIS3, GAL1, GAL10, ADH1, PGK, PHО5, GAPDH, ADC1, TRP1, URA3, LEU2, ENO, TPI, CUP1, FBA, GPD и GPM (эффективны для экспрессии в Saccharomyces); AОX1 (эффективен для экспрессии в Pichia) и lac, ara, tet, trp, IPL, IPR, Т7, tac и trc (эффективны для экспрессии в Escherichia coli, Alcaligenes и Pseudomonas); промоторы amy, apr, npr и различные фаговые промоторы, эффективные для экспрессии в Bacillus subtilis, Bacillus licheniformis и Paenibacillus macerans; nisA (эффективен для экспрессии в грамположительных бактериях, Eichenbaum et al. Appl. Environ. Microbiol. 64(8): 2763-2769 (1998)) и синтетический промотор P11 (эффективен для экспрессии в Lactobacillus plantarum, Rud et al., Microbiology 152: 1011-1019 (2006)).
Области, контролирующие терминацию, также могут быть получены из различных генов, нативных для предпочтительных хозяев. При желании, сайт терминации может быть излишним, тем не менее, наиболее предпочтительно, если он имеется в наличии.
Некоторые векторы допускают репликацию в широком диапазоне бактерий-хозяев и могут быть перенесены посредством конъюгации. Доступна полная и описанная последовательность pRK404 и трех родственных векторов pRK437, pRK442 и pRK442(H). Доказано, что указанные производные представляют собой чрезвычайно полезные средства для генетической манипуляции в грамотрицательных бактериях (Scott et al., Plasmid 50(1): 74-79 (2003)). Некоторое количество производных плазмиды широкого круга хозяев lnc P4 RSF1010 также доступно с промоторами, которые могут функционировать в ряде грамотрицательных бактерий. Плазмиды pAYC36 и pAYC37 имеют активный промотор наряду с множественными областями для клонирования, что делает возможной экспрессию гетерологичных генов в грамотрицательных бактериях.
Также широко доступны средства для хромосомной замены генов. Например, термочувствительный вариант репликона широкого круга хозяев pWV101 был модифицирован для конструирования плазмиды pVE6002, которую можно использовать для создания замены генов в ряде грамположительных бактерий (Maguin et al., J. Bacteriol. 174(17): 5633-5638 (1992)). Кроме того, транспозоны in vitro доступны для создания редких мутаций в различных геномах из различных коммерческих источников, например EPICENTRE®.
Экспрессия биосинтетического пути 1-бутанола в различных предпочтительных микробных хозяевах описана более подробно далее.
Экспрессия биосинтетического пути 1-бутанола в E.coli
Векторы или кассеты, пригодные для трансформации E.coli, являются общепринятыми и доступны для приобретения у компаний, перечисленных выше. Например, гены биосинтетического пути 1-бутанола могут быть выделены из различных штаммов Clostridium, клонированных в модифицированный вектор pUC19 и трансформированных в E.coli NM522, как описано в примере 11. Экспрессия биосинтетического пути 1-бутанола в некоторых других штаммах E.coli описана в примере 13.
Экспрессия биосинтетического пути 1-бутанола в Rhodococcus erythropolis
Ряд «челночных» векторов E.coli-Rhodococcus доступен для экспрессии в R.erythropolis, в том числе, но не ограничиваясь, pRhBR17 и pDA71 (Kostichka et al., Appl. Microbiol. Biotechnol 62: 61-68(2003)). Кроме того, ряд промоторов доступен для экспрессии гетерологичных генов в R.erythropolis (смотрите, например, Nakashima et al., Appl. Envir. Microbiol. 70: 5557-5568 (2004), и Tao et al., Appl. Microbiol. Biotechnol. 2005, DOI 10.1007/s00253-005-0064). Целенаправленное генное разрушение хромосомных генов в R.erythropolis может быть произведено посредством способа, описанного у Tao et al., выше, и Brans et al. (Appl. Envir. Microbiol. 66: 2029-2036 (2000)).
Гетерологичные гены, необходимые для продуцирования 1-бутанола, как описано выше, могут быть изначально клонированы в pDA71 или pRhBR71 и трансформированы в E.coli. Векторы затем могут быть трансформированы в R.erythropolis посредством электропорации, как описано Kostichka et al., выше. Рекомбинанты можно выращивать в синтетической среде, включающей глюкозу, и получение 1-бутанола может продолжаться с использованием способов, известных в уровне техники.
Экспрессия биосинтетического пути 1-бутанола в Bacillus Subtilis
Способы для экспрессии генов и создания мутаций в B.Subtilis также хорошо известны в уровне техники. Например, гены биосинтетического пути 1-бутанола могут быть выделены из различных штаммов Clostridium, клонированных в модифицированный вектор pUC19 и трансформированных в Bacillus subtilis BE1010, как описано в примере 12. Кроме того, шесть генов биосинтетического пути 1-бутанола могут быть разделены в два оперона для экспрессии, как описано в примере 14. Первые три гена пути биосинтеза (thl, hbd и crt) были интегрированы в хромосому Bacillus subtilis BE1010 (Payne и Jackson, J. Bacteriol. 173: 2278-2282 (1991)). Последние три гена (EgTER, ald и bdhB) были клонированы в экспрессирующие плазмиды и трансформированы в штамм Bacillus, несущий указанные встроенные гены 1-бутанола.
Экспрессия биосинтетического пути 1-бутанола в Bacillus licheniformis
Большинство плазмид и «челночных» векторов, которые реплицируют в B.subtilis, можно использовать для трансформации B.licheniformis либо посредством трансформации протопластов, либо посредством электропорации. Например, гены, необходимые для получения 1-бутанола, могут быть клонированы в производные плазмид pBE20 или pBE60 (Nagarajan et al., Gene 114: 121-126 (1992)). Способы трансформации B.licheniformis известны в уровне техники (например, смотрите Fleming et al. Appl. Environ. Microbiol., 61 (11): 3775-3780 (1995)). Плазмиды, сконструированные для экспрессии в B.subtilis, также могут быть трансформированы в B.licheniformis для получения рекомбинантного микробного хозяина, который продуцирует 1-бутанол.
Экспрессия биосинтетического пути 1-бутанола в Paenibacillus macerans
Плазмиды могут быть сконструированы, как описано выше для экспрессии в B.subtilis, и использованы для трансформации Paenibacillus macerans посредством трансформации протопластов для получения рекомбинантного микробного хозяина, который продуцирует 1-бутанол.
Экспрессия биосинтетического пути 1-бутанола в Alcaligenes (Ralstonia) eutrophus
Способы для экспрессии генов и создания мутаций в Ralstonia eutrophus хорошо известны в уровне техники (смотрите, например, Taghavi et al., Appl. Environ. Microbiol., 60(10): 3585-3591 (1994)). Гены биосинтетического пути 1-бутанола могут быть клонированы в любой вектор из широкого диапазона круга хозяев, описанных выше, и электропорированы для образования рекомбинантов, которые продуцируют 1-бутанол. Путь полигидроксибутирата в Ralstonia был описан подробно, и известны различные генетические технологии для изменения генома Ralstonia eutrophus, и эти средства можно использовать для конструирования биосинтетического пути 1-бутанола.
Экспрессия биосинтетического пути 1-бутанола в Pseudomonas putida
Способы экспрессии генов в Pseudomonas putida известны в уровне техники (смотрите, например, Ben-Bassat et al., патент США № 6586229, который включен в настоящее описание посредством ссылки). Например, гены пути биосинтеза бутанола могут быть встроены в pPCU18, и указанную лигированную ДНК можно электропорировать в электрокомпетентные клетки Pseudomonas putida DOT-T1 C5aAR1 для продуцирования рекомбинантов, которые производят 1-бутанол.
Экспрессия биосинтетического пути 1-бутанола в Saccharomyces cerevisiae
Способы экспрессии генов в Saccharomyces cerevisiae известны в уровне техники (смотрите, например, Methods in Enzymology, Volume 194, Guide to Yeast Genetics and Molecular and Cell Biology (Part A, 2004, Christine Guthrie and Gerald R. Fink (Eds.), Elsevier Academic Press, San Diego, CA)). Для экспрессии генов в дрожжах обычно требуется промотор, за которым следует представляющий интерес ген, и терминатор транскрипции. Целый ряд дрожжевых промоторов можно использовать при конструировании экспрессирующих кассет для генов, кодирующих биосинтетический путь 1-бутанола, в том числе, но не ограничиваясь, конститутивные промоторы FBA, GPD и GPM и индуцируемые промоторы GAL1, GAL10 и CUP1. Подходящие терминаторы транскрипции включают, но не ограничиваются, FBAt, GPDt, GPMt, ERG10t и GAL1t. Подходящие промоторы, терминаторы транскрипции и гены биосинтетического пути 1-бутанола могут быть клонированы в дрожжевые 2-микронные (2μ) плазмиды, как описано в примере 17.
Экспрессия биосинтетического пути 1-бутанола в Lactobacillus plantarum
Род Lactobacillus, принадлежащий к семейству Lactobacillales, и множество плазмид и векторов, применяемых для трансформации Bacillus subtilis и Streptococcus, можно использовать для lactobacillus. Неограничивающие примеры подходящих векторов включают pAMβ1 и его производные (Renault et al., Gene 183: 175-182 (1996); и O'Sullivan et al., Gene 137: 227-231 (1993)); pMBB1 и pHW800, производное pMBB1 (Wyckoff et al. Appl. Environ. Microbiol. 62: 1481-1486 (1996)); pMG1, конъюгативную плазмиду (Tanimoto et al., J. Bacteriol. 184: 5800-5804 (2002)); pNZ9520 (Kleerebezem et al., Appl. Environ. Microbiol. 63: 4581-4584 (1997)); pAM401 (Fujimoto et al., Appl. Environ. Microbiol. 67: 1262-1267 (2001)) и pAT392 (Arthur et al., Antimicrob. Agents Chemother. 38: 1899-1903 (1994)). Также сообщалось о нескольких плазмидах из Lactobacillus plantarum (например, van Kranenburg R, Golic N, Bongers R, Leer RJ, de Vos WM, Siezen RJ, Kleerebezem M. Appl. Environ. Microbiol. 2005 Mar; 71(3): 1223-1230). Например, экспрессия биосинтетического пути 1-бутанола в Lactobacillus plantarum описана в примере 18.
Экспрессия биосинтетического пути 1-бутанола в Enterococcus faecium, Enterococcus gallinarium и Enterococcus faecalis
Род Enterococcus, принадлежащий к семейству Lactobacillales, и множество плазмид и векторов, используемых для трансформации Lactobacillus, Bacillus subtilis и Streptococcus, можно использовать для Enterococcus. Неограничивающие примеры подходящих векторов включают pAMβ1 и его производные (Renault et al., Gene 183: 175-182 (1996); и O'Sullivan et al., Gene 137: 227-231 (1993)); pMBB1 и pHW800, производное pMBB1 (Wyckoff et al. Appl. Environ. Microbiol. 62: 1481-1486 (1996)); pMG1, конъюгативную плазмиду (Tanimoto et al., J. Bacteriol. 184: 5800-5804 (2002)); pNZ9520 (Kleerebezem et al., Appl. Environ. Microbiol. 63: 4581-4584 (1997)); pAM401 (Fujimoto et al., Appl. Environ. Microbiol. 67: 1262-1267 (2001)) и pAT392 (Arthur et al., Antimicrob. Agents Chemother. 38: 1899-1903 (1994)). Также можно использовать экспрессирующие векторы для E.faecalis, использующие ген nisA из Lactococcus (Eichenbaum et al., Appl. Environ. Microbiol. 64: 2763-2769 (1998)). Кроме того, можно использовать векторы для замены генов в хромосоме E.faecium (Nallaapareddy et al., Appl. Environ. Microbiol. 72: 334-345 (2006)). Например, экспрессия биосинтетического пути 1-бутанола в Enterococcus faecalis описана в примере 19.
Ферментационная среда
Ферментационная среда в настоящем изобретении должна включать подходящие углеродные субстраты. Подходящие углеродные субстраты могут включать, но не ограничиваются этим, моносахариды, например глюкозу и фруктозу, олигосахариды, например лактозу или сахарозу, полисахариды, например крахмал, или целлюлозу, или их смеси, и неочищенные смеси из возобновляемого сырья, например фильтрат сырной сыворотки, кукурузный экстракт, патоку сахарной свеклы и ячменный солод. Кроме того, указанным углеродным субстратом также могут быть одноуглеродные субстраты, такие как диоксид углерода или метанол, для которых была продемонстрирована биотрансформация в ключевые биохимические промежуточные продукты. В дополнение к одно- или двухуглеродным субстратам также известно, что метилотрофные организмы для метаболической активности используют некоторое количество других соединений, содержащих углерод, например метиламин, глюкозамин и различные аминокислоты. Например, известно, что метилотрофные дрожжи используют углерод из метиламина для образования трегалозы или глицерина (Bellion et al., Microb. Growth C1 Compd. [Int. Symp.], 7th (1993), 415-32. Editor(s): Murrell, J. Collin; Kelly, Don P. Publisher: Intercept, Andover, UK). Аналогично, различные виды Candida будут метаболизировать аланин или олеиновую кислоту (Sulter et al., Arch. Microbiol. 153: 485-489 (1990)). Таким образом, ожидается, что указанный источник углерода, используемый в настоящем изобретении, может охватывать большое разнообразие субстратов, содержащих углерод, и будет ограничен только выбором организма.
Хотя ожидается, что все вышеупомянутые углеродные субстраты и их смеси являются подходящими в настоящем изобретении, предпочтительными углеродными субстратами являются глюкоза, фруктоза и сахароза.
В дополнение к соответствующему источнику углерода, ферментационная среда должна содержать подходящие минералы, соли, кофакторы, буферы и другие компоненты, известные специалисту в уровне техники, подходящие для выращивания указанных культур и стимулирования указанного ферментативного пути, необходимого для получения 1-бутанола.
Условия культивирования
Обычно клетки выращивают при температуре в диапазоне приблизительно от 25 приблизительно до 40°С в соответствующей среде. Подходящими средами для выращивания по настоящему изобретению являются общепринятые коммерчески доступные среды для выращивания, такие как, например, питательная среда Лурия-Бертани (LB), декстрозная среда Сабуро (SD) или дрожжевая питательная среда (YM). Другие среды определенного состава или синтетические среды для выращивания также можно использовать, и соответствующая среда для выращивания конкретного микроорганизма известна специалисту в области микробиологии или культивирования. Также в указанной бродильной среде можно использовать агенты, про которые известно, что они модулируют катаболитическую репрессию напрямую или косвенно, например циклический аденозин 2':3'-монофосфат.
Подходящие значения рН для ферментации находятся в диапазоне между рН 5,0 и рН 9,0, где значения от рН 6,0 до рН 8,0 являются предпочтительными в качестве исходного состояния.
Ферментацию можно осуществлять в аэробных или анаэробных условиях с предпочтением анаэробных или микроаэробных условий.
Количество 1-бутанола, полученного в указанной бродильной среде, можно определить с помощью ряда методов, известных в уровне техники, например высокоэффективная жидкостная хроматография (ВЭЖХ) или газовая хроматография (ГХ).
Периодическая и непрерывная ферментация в промышленных условиях
В настоящем процессе используется периодический метод ферментации. Классическая периодическая ферментация представляет собой закрытую систему, в которой композиция указанной среды установлена в начале ферментации и не подвергается искусственным изменениям в процессе брожения. Таким образом, в начале ферментации, среду инокулируют требуемым организмом или организмами и делают возможным брожение без добавления чего-либо в систему. Однако типично «периодическая» ферментация является периодической относительно добавления источника углерода, и часто делают попытки контролировать такие факторы, как рН и концентрация кислорода. В периодических системах метаболит и композицию биомассы системы изменяют непрерывно вплоть до времени окончания ферментации. В течение периодического культивирования клетки постепенно переходят из статической лаг-фазы в фазу интенсивного логарифмического роста и в конце концов в фазу стационарной численности, в которой скорость роста замедляется или останавливается. Если не провести обработку, клетки в фазе стационарной численности с течением времени погибают. Клетки в фазе логарифмического роста, как правило, ответственны за основную часть продуцирования конечного продукта или промежуточного продукта.
Вариантом стандартной периодической системы является система с подпиткой. Процессы ферментации с подпиткой также подходят для настоящего изобретения и включают типичную периодическую систему за исключением того, что субстрат добавлен дополнительно для продолжения ферментации. Системы с подпиткой применяются, когда катаболитная регрессия имеет склонность к ингибированию метаболизма клеток и когда желательно иметь ограниченные количества субстрата в среде. Измерение достоверной концентрации субстрата в системах с подпиткой затруднено и вследствие этого производится исходя из изменений поддающихся измерению факторов, таких как рН, растворенный кислород и парционное давление газообразных продуктов, таких как CO2. Периодическая ферментация и ферментация с подпиткой являются общепринятыми и хорошо известны в уровне техники, и примеры можно найти у Thomas D. Brock в Biotechnology: A Textbook of Industrial Microbiology, Second Edition (1989) Sinauer Associates, Inc., Sunderland, MA., или Deshpande, Mukund V., Appl. Biochem. Biotechnol., 36: 227, (1992), которые включены в настоящее описание посредством ссылки.
Несмотря на то что настоящее изобретение осуществлено в периодическом режиме, предполагается, что указанный метод может быть адаптирован для способов непрерывной ферментации. Непрерывная ферментация представляет собой открытую систему, в которой бродильная среда определенного состава добавляется постоянно в биореактор и равное количество кондиционной среды удаляется одновременно для переработки. Непрерывная ферментация в основном поддерживает указанные культуры при постоянной высокой плотности, где клетки находятся главным образом в фазе логарифмического роста.
Постоянная ферментация учитывает модулирование одного фактора или любого количества факторов, которые влияют на клеточный рост или концентрацию конечного продукта. Например, в одном способе поддерживают лимитирующее питательное вещество, например источник углерода или уровень азота, при фиксированной скорости и позволяют уменьшаться всем остальным параметрам. В других системах множество факторов, влияющих на рост, можно постоянно изменять, в то время как концентрацию клеток, определенную по замутненности среды, сохраняют постоянной. В непрерывных системах стремятся поддерживать условия стационарного роста, и, таким образом, потеря клеток в результате извлечения среды должна быть уравновешена скоростью роста клеток при ферментации. Способы модулирования питательных веществ и факторов роста для процессов постоянной ферментации, так же как технологии для доведения до максимума скорости образования продукта, хорошо известны в области промышленной микробиологии, и целый ряд способов детально описан Brock, выше.
Предусмотрено, что настоящее изобретение на практике можно применять с использованием как периодических, так и периодических с подпиткой или постоянных процессов и что любой известный режим ферментации может быть пригодным. Кроме того, предусмотрено, что клетки могут быть иммобилизованы на субстрате в виде цельных клеточных катализаторов и подвергнуты ферментационным условиям для продуцирования 1-бутанола.
Способы выделения 1-бутанола из ферментационной среды
Полученный в результате биологического процесса 1-бутанол может быть выделен из ферментационной среды с использованием способов, известных в уровне техники. Например, твердые примеси могут быть удалены из ферментационной среды посредством центрифугирования, фильтрования, декантации или тому подобного. Затем 1-бутанол может быть выделен из ферментационной среды, которая была обработана для удаления твердых примесей, как описано выше, с использованием таких способов, как дистилляция, жидкость-жидкостная экстракция или мембранное разделение. Поскольку 1-бутанол образует азеотропную смесь с водой с низкой точкой кипения, можно использовать только дистилляцию для отделения данной смеси до ее азеотропной композиции. Дистилляцию можно использовать в сочетании с другим методом разделения для получения разделения вокруг данного азеотропа. Способы, которые можно использовать в сочетании с дистилляцией для выделения 1-бутанола, включают, но не ограничиваются, декантацию, жидкость-жидкостную экстракцию, адсорбцию и технологии, основанные на мембранах. Кроме того, 1-бутанол может быть выделен с использованием азеотропной дистилляции с помощью азеотропообразователя (смотрите, например, Doherty и Malone, Conceptual Design of Distillation Systems, McGraw Hill, New York, 2001).
Смесь 1-бутанол-вода образует гетерогенный азеотроп, так что дистилляцию можно использовать в сочетании с декантацией для выделения и очистки 1-бутанола. В указанном способе ферментационную среду, включающую 1-бутанол, дистиллируют до приближения к азеотропной композиции. Затем указанную азеотропную композицию конденсируют и 1-бутанол отделяют из ферментационной среды посредством декантации. Декантированную водную фазу можно вернуть в первую дистилляционную колонку в качестве орошения. Декантированную органическую фазу, богатую 1-бутанолом, можно дополнительно очистить посредством дистилляции во второй дистилляционной колонке.
1-бутанол также может быть выделен из ферментационной среды с использованием жидкость-жидкостной экстракции в сочетании с дистилляцией. В указанном способе 1-бутанол экстрагируют из ферментационной среды с использованием жидкость-жидкостной экстракции с помощью подходящего растворителя. Органическую фазу, содержащую 1-бутанол, затем дистиллируют для отделения 1-бутанола от растворителя.
Также для выделения 1-бутанола из ферментационной среды можно использовать дистилляцию в сочетании с адсорбцией. В указанном способе ферментационную среду, содержащую 1-бутанол, дистиллируют до приближения к азеотропной композиции и затем оставшуюся воду удаляют посредством адсорбента, например молекулярного сита (Aden et al. Lignocellulosic Biomass to Ethanol Process Design and Economics Utilizing Co-Current Dilute Acid Prehydrolysis and Enzymatic Hydrolysis for Corn Stover, Report NREL/TP-510-32438, National Renewable Energy Laboratory, июнь 2002 года).
Кроме того, для выделения и очистки 1-бутанола от ферментационной среды можно использовать дистилляцию в сочетании с диффузным испарением через мембрану. В указанном способе ферментационную среду, содержащую 1-бутанол, дистиллируют до приближения к азеотропной композиции и оставшуюся воду удаляют посредством диффузного испарения через гидрофильную мембрану (Guo et al., J. Membr. Sci. 245, 199-210 (2004)).
ПРИМЕРЫ
Настоящее изобретение дополнительно описано в следующих примерах. Следует понимать, что настоящие примеры, несмотря на то что в них указываются предпочтительные варианты осуществления настоящего изобретения, даны в настоящем описании только в качестве иллюстрации. Из вышеприведенного обсуждения и настоящих примеров специалист в уровне техники может установить необходимые характеристики настоящего изобретения и, не отклоняясь от его сущности и объема, может произвести различные изменения и модификации настоящего изобретения для адаптирования его к различным применениям и условиям.
ОБЩИЕ МЕТОДЫ
Стандартные технологии рекомбинантных ДНК и молекулярного клонирования, используемые в примерах, хорошо известны в уровне техники и описаны Sambrook, J., Fritsch, E. F. и Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, (1989) (Maniatis), T. J. Silhavy, M. L. Bennan и L. W. Enquist, Experiments with Gene Fusions, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. (1984), и Ausubel, F. M. et al., Current Protocols in Molecular Biology, опубликовано Greene Publishing Assoc. and Wiley-Interscience (1987).
Материалы и методы, пригодные для поддержания и выращивания бактериальных культур, хорошо известны в уровне техники. Технологии, пригодные для использования в следующих примерах, можно обнаружить в Manual of Methods for General Bacteriology (Phillipp Gerhardt, R. G. E. Murray, Ralph N. Costilow, Eugene W. Nester, Willis A. Wood, Noel R. Krieg и G. Briggs Phillips, eds), American Society for Microbiology, Washington, DC. (1994)) или у Thomas D. Brock в Biotechnology: A Textbook of Industrial Microbiology, Second Edition, Sinauer Associates, Inc., Sunderland, MA (1989). Все реагенты, рестрикционные ферменты и материалы, используемые для выращивания и поддержания бактериальных клеток, были приобретены у Aldrich Chemicals (Milwaukee, WI), BD Diagnostic Systems (Sparks, MD), Life Technologies (Rockville, MD) или Sigma Chemical Company (St. Louis, MO), если не указано особо.
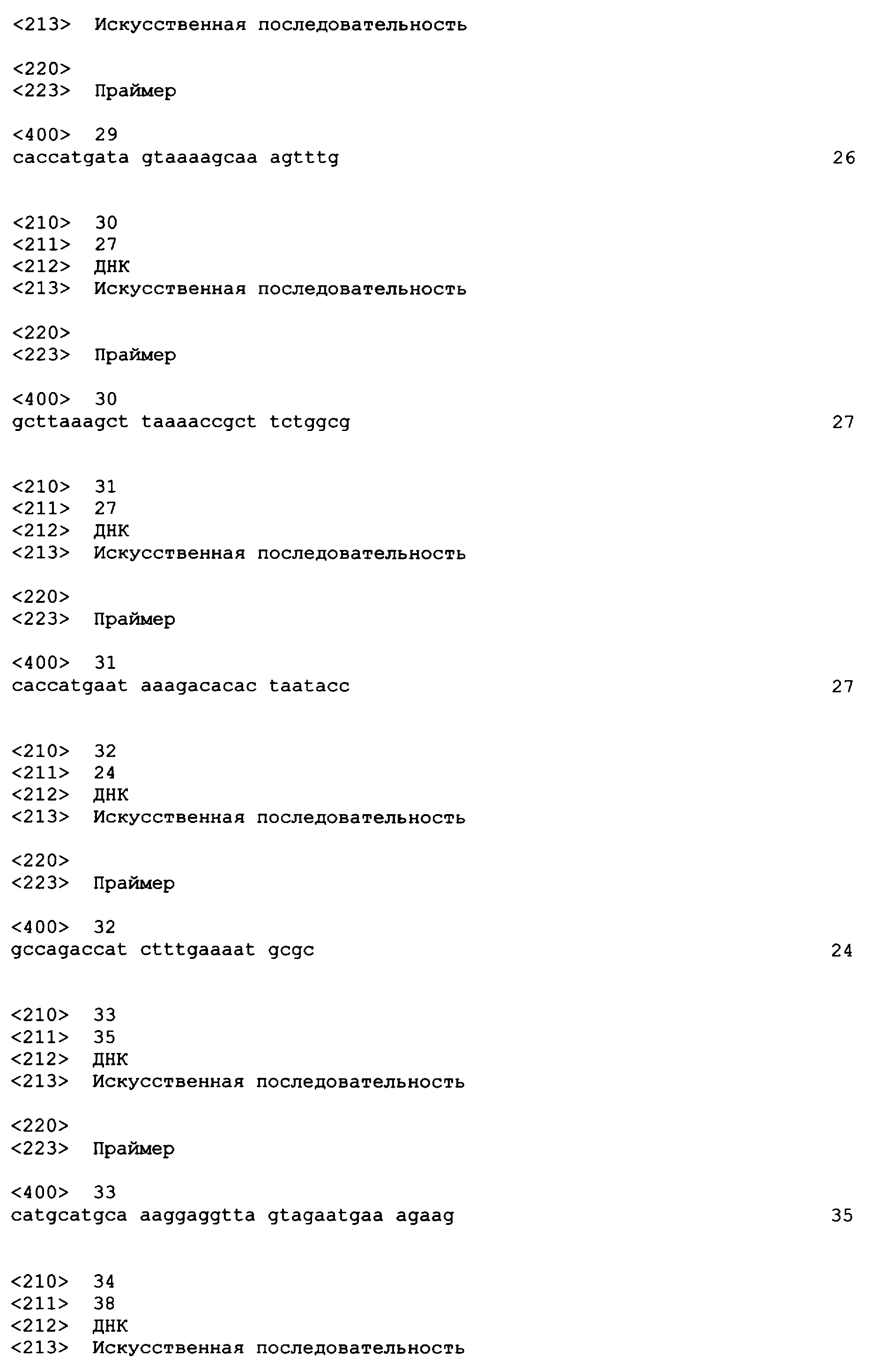
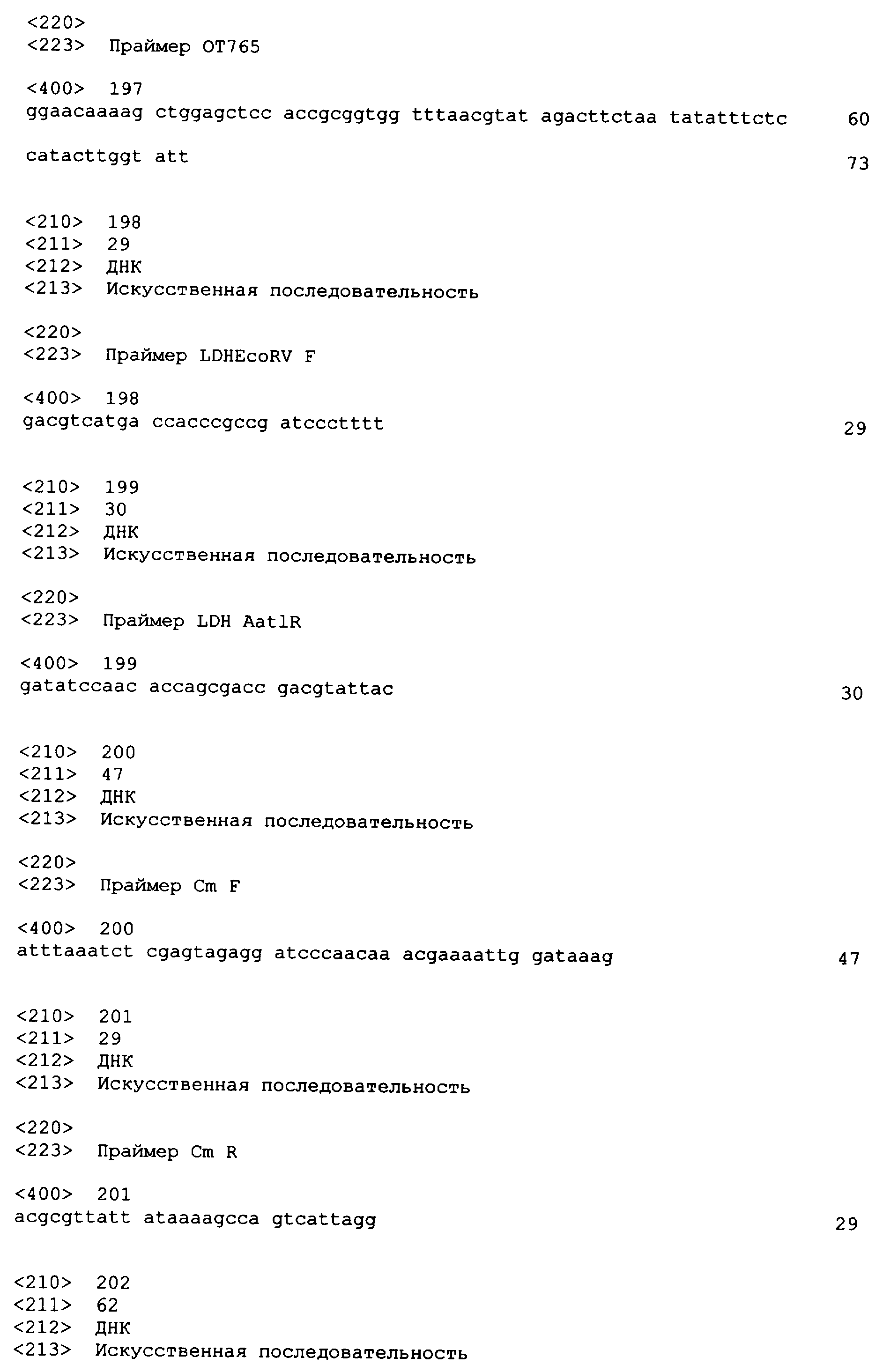
Олигонуклеотидные праймеры, используемые для клонирования в следующих примерах, приведены в таблице 4. Праймеры, используемые для секвенирования или скрининга клонированных генов, приведены в таблице 5. Все указанные олигонуклеотидные праймеры были синтезированы компанией Sigma-Genosys (Woodlands, TX).
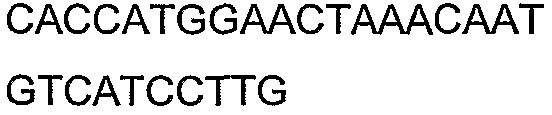

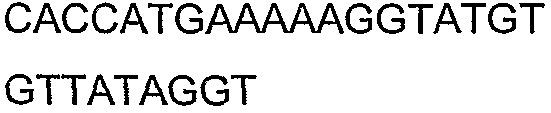



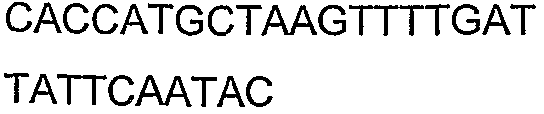
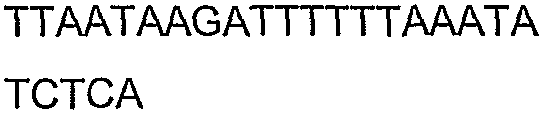

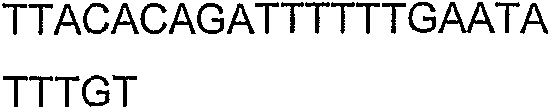
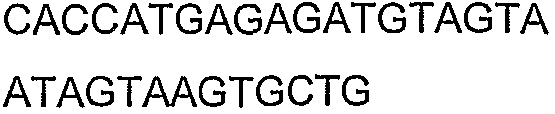

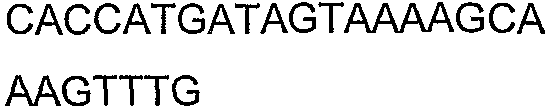



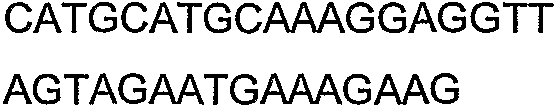
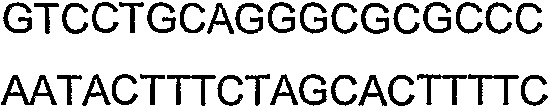
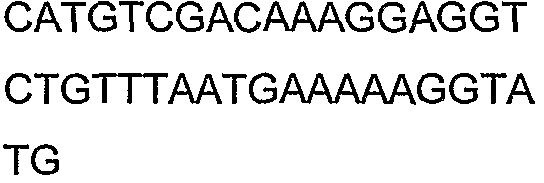
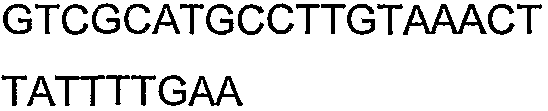


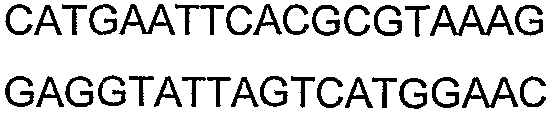
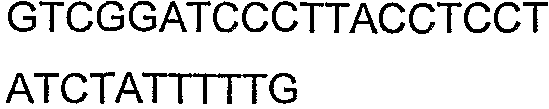
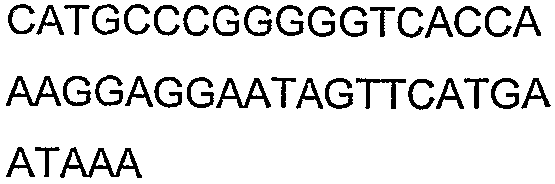
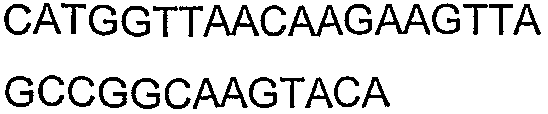

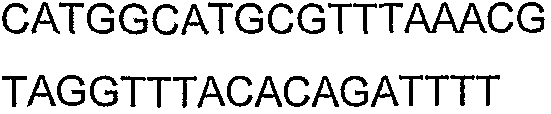
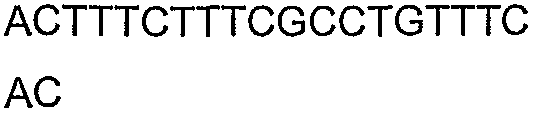
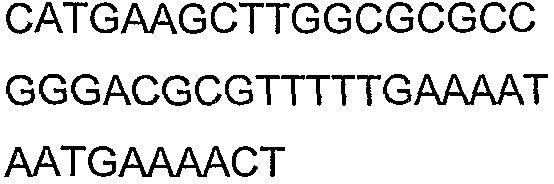

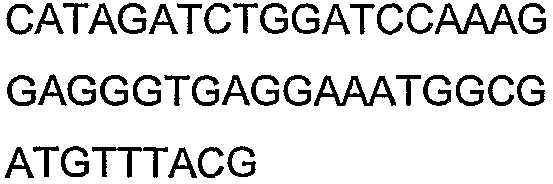



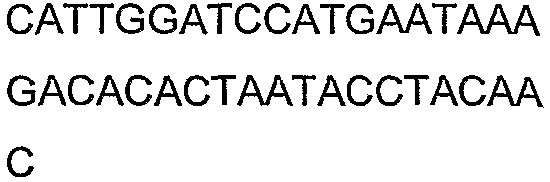
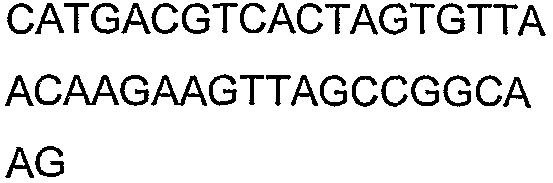

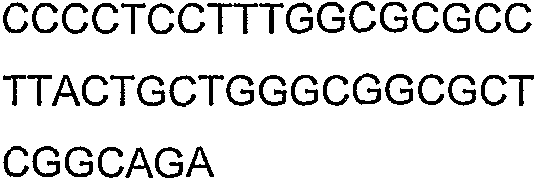
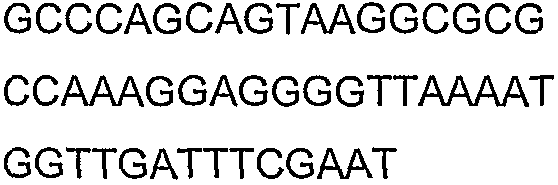


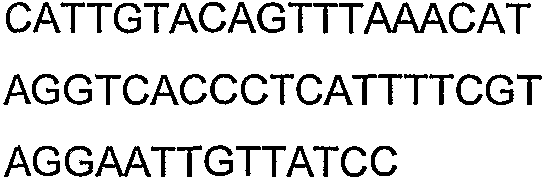
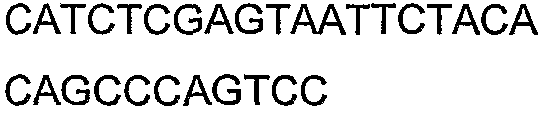
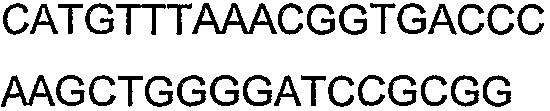

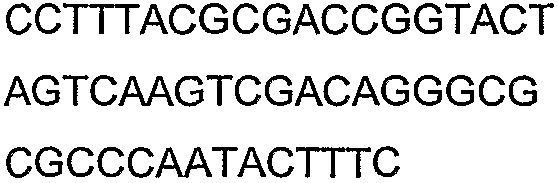

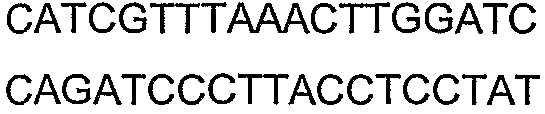
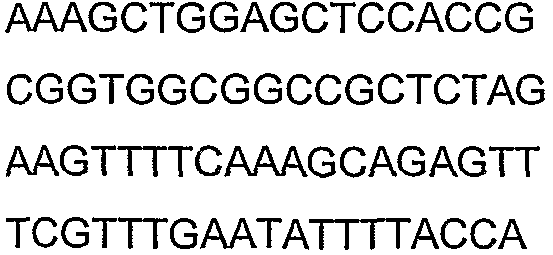
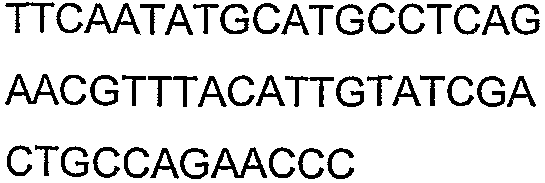
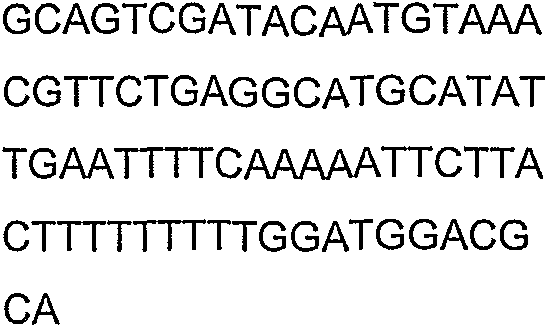
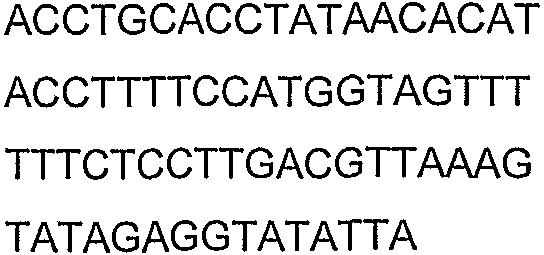
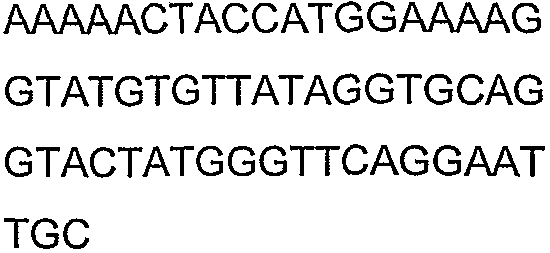
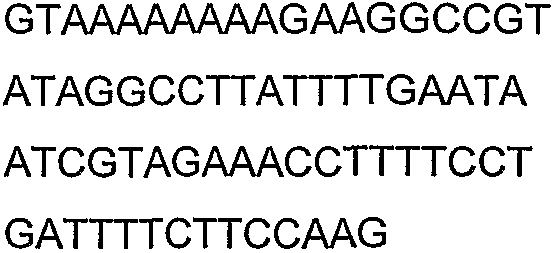
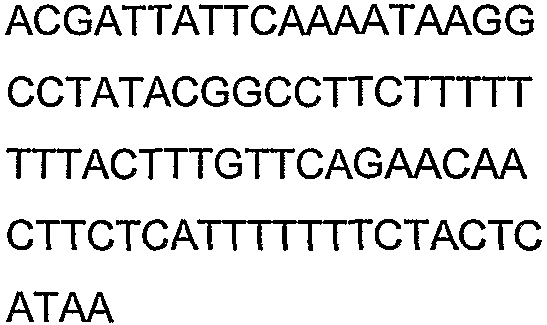
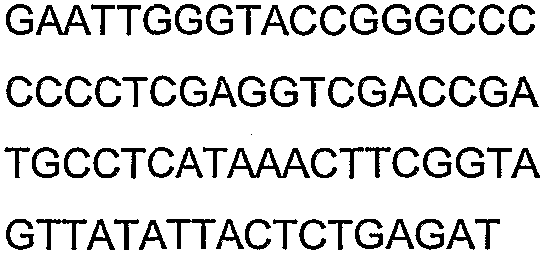
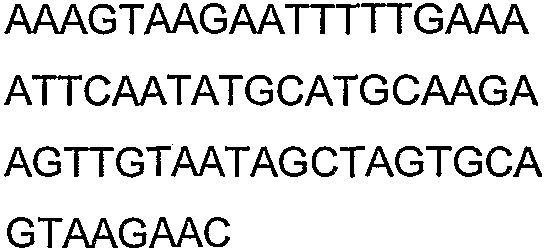

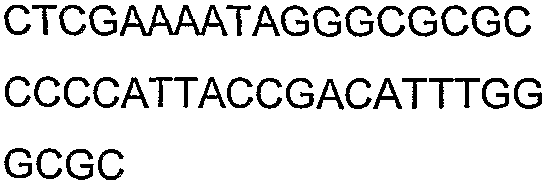
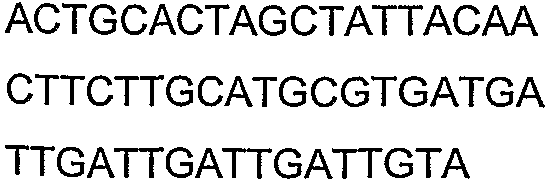
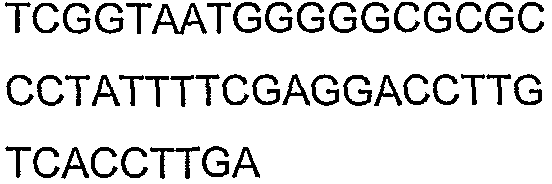


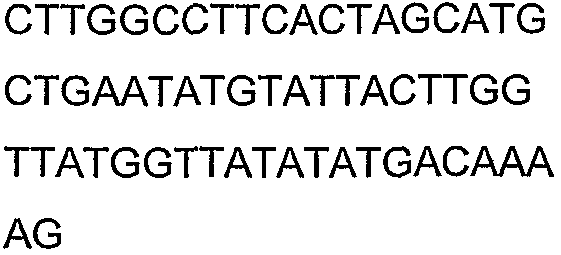






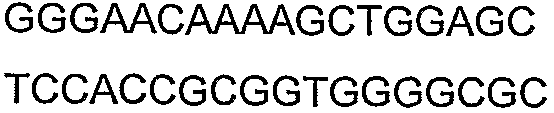




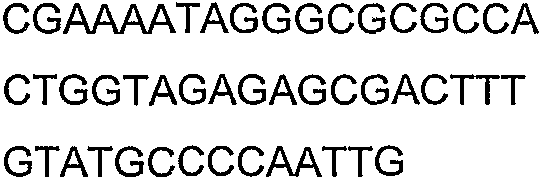


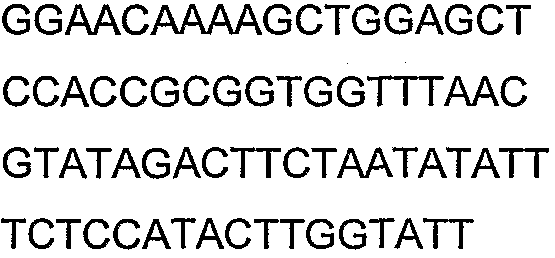

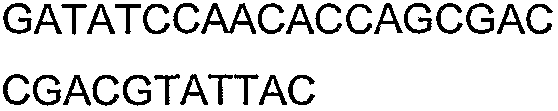

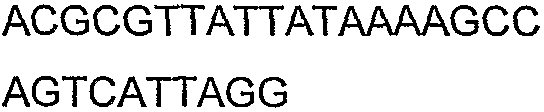



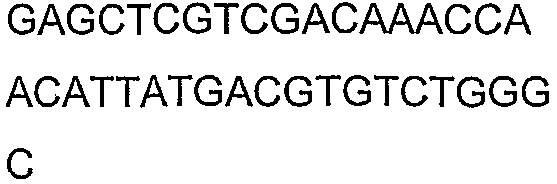
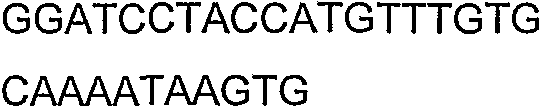
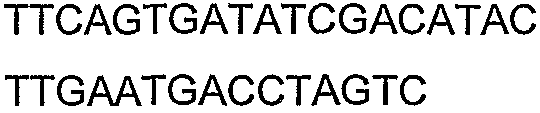
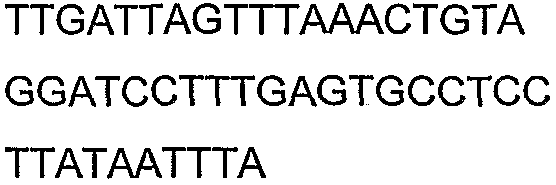


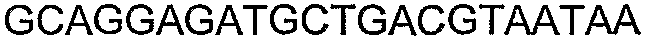
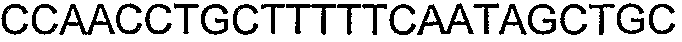


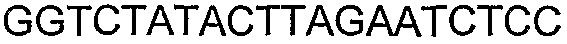


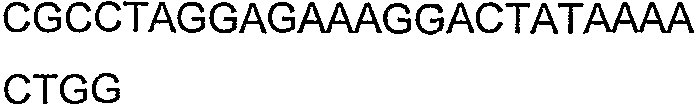


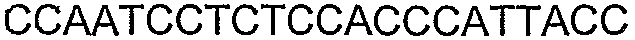
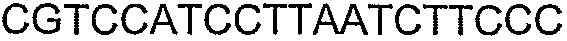

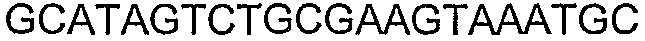
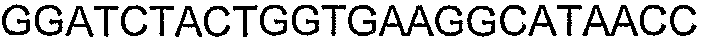
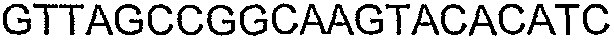
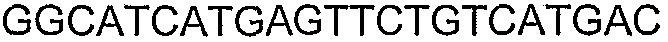

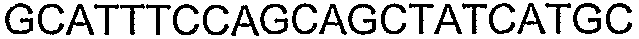


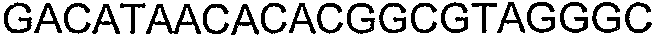

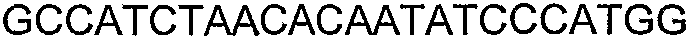
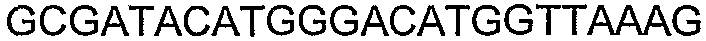


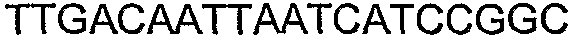

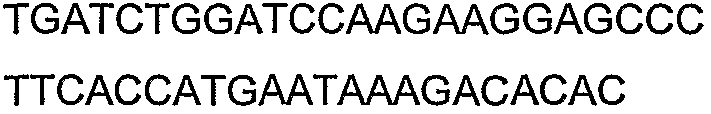

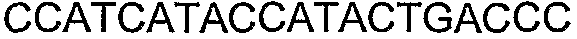
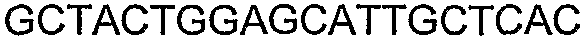

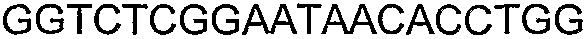




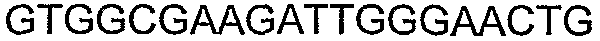
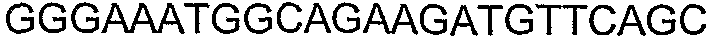

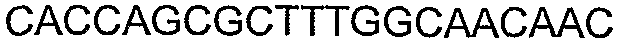


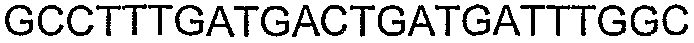
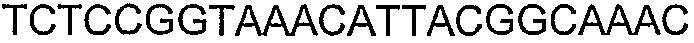






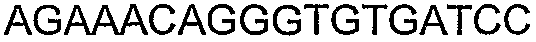


Способы определения концентрации 1-бутанола в культуральной среде
Концентрацию 1-бутанола в культуральной среде можно определить посредством ряда способов, известных в уровне техники. Например, в способе специфической высокоэффективной жидкостной хроматографии (ВЭЖХ) используется колонка Shodex SH-1011 с защитной колонкой Shodex SH-G, обе приобретены у Waters Corporation (Milford, MA), с детекцией коэффициента преломления (RI). Хроматографическое разделение достигается с использованием 0,01 М H2SO4в качестве мобильной фазы со скоростью потока 0,5 мл/мин и температурой колонки 50°С. При используемых условиях 1-бутанол имел время удержания 52,8 мин. Альтернативно доступны способы газовой хроматографии (ГХ). Например, в методе специфической ГХ используется колонка HP-INNOWax (30 м × внутренний диаметр 0,53 мм, толщина пленки 1 мкм, Agilent Technologies, Wilmington, DE) с плазменно-ионизационным детектором (FID). Газом-носителем был гелий при скорости потока 4,5 мл/мин, измеренной при 150°С с постоянным давлением нагнетания; деление потока составляло 1:25 при 200°С; температура в печи составляла 45°C в течение 1 мин, от 45 до 220°C при 10°C/мин и 220°C в течение 5 мин; и FID детекция использовалась при 240°С с нагнетаемым газом гелием при 26 мл/мин. Время удержания 1-бутанола составляло 5,4 мин. Также использовали схожий способ ГХ с использованием колонки Varian CP-WAX 58 (FFAP) CB (25 м × внутренний диаметр 0,25 мм, толщина пленки 0,2 мкм, Varian, Inc., Palo Alto, CA).
Смысл сокращений состоял в следующем: «с» означает секунда(ы), «мин» означает минута(ы), «ч» означает час(ы), «psi» означает фунты на квадратный дюйм, «нм» означает нанометры, «д» означает день (дни), «мкл» означает микролитр(ы), «мл» означает миллилитр(ы), «л» означает литр(ы), «мм» означает миллиметр(ы), «мМ» означает миллимолярный, «М» означает молярный, «ммоль» означает миллимоль(и), «мкмоль» означает микромоль(и), «г» означает грамм(ы), «мкг» означает микрограмм(ы) и «нг» означает нанограмм(ы), «ПЦР» означает полимеразную цепную реакцию, «OD» означает оптическую плотность, «OD600» означает оптическую плотность, измеренную при длине волны 600 нм, «OD550» означает оптическую плотность, измеренную при длине волны 550 нм, «кДа» означает килодальтоны, «g» означает гравитационную постоянную, «об/мин» означает число оборотов в минуту, «п.н.» означает пара (пары) нуклеотидов, «т.п.н.» означает тысяча(тысячи) пар нуклеотидов, «% вес./об.ъем» означает отношение веса к объему, «% об./об.» означает отношение объем к объему, «ВЭЖХ» означает высокоэффективную жидкостную хроматографию и «ГХ» означает газовую хроматографию.
ПРИМЕР 1
Клонирование и экспрессия ацетил-CoA-ацетилтрансферазы
Целью настоящего примера являлась экспрессия фермента ацетил-CoA-ацетилтрансфераза, также именуемого в настоящем описании как ацетоацетил-CoA-тиолаза, в E.сoli. Ген ацетоацетил-CoA-тиолазы thlA был клонирован из C.acetobutylicum (ATCC 824) и экспрессирован в E.coli. Ген thlA амплифицировали из геномной ДНК C.acetobutylicum (ATCC 824) с использованием ПЦР, и в результате получали продукт массой 1,2 т.п.н.
Геномная ДНК Clostridium acetobutylicum (ATCC 824) была либо приобретена в американской коллекции типовых культур (ATCC, Manassas, VA), либо была выделена из культур Clostridium acetobutylicum (ATCC 824), как описано ниже.
Геномную ДНК Clostridium acetobutylicum (ATCC 824) выделяли из культур, выращенных в анаэробных условиях. Штамм Clostridium выращивали в 10 мл среды для выращивания Clostridial (Lopez-Contreras et al., Appl. Env. Microbiol. 69(2), 869-877 (2003)) в закрытых притертой пробкой флаконах с сывороткой Bellco объемом 100 мл (Bellco Glass Inc., Vineland, NJ) в анаэробной камере при 30°С. Инокулюм представлял собой единичную колонию из чашки с 2 XYTG (Kishii, et al., Antimicrobial Agents & Chemotherapy, 47(1), 77-81 (2003)), выращенную в 2,5 л MGC AnaeroPak™ (Mitsubishi Gas Chemical America Inc, New York, NY) при 37°C.
Геномную ДНК выделяли с использованием набора Gentra Puregene® (Gentra Systems, Inc., Minneapolis, MN; каталожный № D-6000A) с модификациями рекомендаций фирмы-изготовителя (Wong et al., Current Microbiology, 32, 349-356 (1996)). Ген thlA амплифицировали из геномной ДНК Clostridium acetobutylicum (ATCC 824) посредством ПЦР с использованием праймеров N7 и N8 (смотрите таблицу 4), представленных как SEQ ID NO: 21 и 22 соответственно. Другие реагенты для ПЦР-амплификации входили в состав наборов фирмы-изготовителя, например Kod HiFi DNA Polymerase (Novagen Inc., Madison, WI; каталожный № 71805-3), и использовались в соответствии с протоколом фирмы-изготовителя. Амплификацию проводили в ДНК термоциклере GeneAmp 9700 (PE Applied Biosystems, Foster city, CA).
Для исследований экспрессии использовали технологию клонирования Gateway (Invitrogen Corp., Carlsbad, CA). Входной вектор pENTR/SD/D-TOPO делает возможным прямое клонирование и обеспечивает последовательность Шайн-Далгарно для гена, представляющего интерес. Целевой вектор pDEST14 использует промотор T7 для экспрессии указанного гена без тага. Прямой праймер содержал четыре основания (САСС), непосредственно прилегающие к инициирующему трансляцию кодону для обеспечения прямого клонирования в pENTR/SD/D-TOPO (Invitrogen) для образования плазмиды pENTRSDD-TOPOthlA. Указанная конструкция pENTR была трансформирована в клетках Top10 E.coli (Invitrogen), и ее высевали на планшеты согласно рекомендациям фирмы-изготовителя. Трансформанты выращивали в течение ночи и плазмидную ДНК выделяли с использованием набора QIAprep Spin Miniprep (Qiagen, Valencia, CA; каталожный № 27106) в соответствии с рекомендациями фирмы-изготовителя. Клоны были предоставлены для секвенирования с использованием прямого и обратного праймеров М13 (смотрите таблицу 5), представленных как SEQ ID NO: 45 и 46 соответственно, для подтверждения того, что указанные гены встроены в правильной ориентации, и подтверждения указанной последовательности. Дополнительные праймеры для секвенирования, N7SeqF1 и N7SeqR1 (смотрите таблицу 5), представленные как SEQ ID NO: 47 и 48 соответственно, были необходимы для полного секвенирования указанного ПЦР-продукта. Нуклеотидная последовательность открытой рамки считывания (ORF) для указанного гена и предсказанная аминокислотная последовательность указанного фермента представлены как SEQ ID NO: 1 и SEQ ID NO: 2 соответственно.
Для создания экспрессирующего клона ген thlA был перенесен в вектор pDEST 14 посредством рекомбинации для образования pDEST14thlA. Вектор pDEST14thlA трансформировали в клетках BL21-AI. Трансформанты инокулировали в среду LB, дополненную 50 мкг/мл ампициллина, и выращивали в течение ночи. Аликвоту культуры, выращиваемой в течение ночи, использовали для инокуляции 50 мл среды LB, дополненной 50 мкг/мл ампициллина. Культуру инкубировали при 37°С со встряхиванием до тех пор, пока OD600 не достигнет 0,6-0,8. Культуру разделяли в два 25-мл флакона и в один флакон добавляли арабинозу до конечной концентрации 0,2% по массе. Во флакон с отрицательным контролем не добавляли арабинозу. Флаконы инкубировали в течение 4 часов при 37°С со встряхиванием. Клетки собирали посредством центрифугирования и клеточные осадки ресуспендировали в 50 мМ буфере MOPS, рН 7,0. Клетки разрушали либо посредством обработки ультразвуком, либо пропуская через Френч-пресс. Цельноклеточный лизат центрифугировали, что приводило к образованию супернатанта, или бесклеточного экстракта, и осадка в пробирке, или нерастворимой фракции. Аликвоту каждой фракции (цельноклеточный лизат, бесклеточный экстракт и нерастворимая фракция) ресуспендировали в нагрузочном буфере SDS (MES) (Invitrogen), нагревали до 85°С в течение 10 минут и подвергали анализу в SDS-PAGE (NuPAGE 4-12% Bis-Tris Gel, каталожный № NP0322Box, Invitrogen). Белок ожидаемой молекулярной массы около 41 кДа, как было рассчитано по последовательности нуклеиновой кислоты, был обнаружен в индуцированной культуре, но не обнаружен в неиндуцированном контроле.
Активность ацетоацетил-CoA-тиолазы в бесклеточном экстракте была измерена как деградация комплекса Mg2+-ацетоацетил-СоА, контролируя уменьшения оптической плотности при 303 нм. Стандартные условия анализа представляли собой 100 мM Трис-HCl pH 8,0, 1 мM DTT (дитиотреитол) и 10 мM MgCl2. Указанный коктейль уравновешивали в течение 5 минут при 37°С, затем добавляли указанный бесклеточный экстракт. Реакцию инициировали добавлением 0,05 мМ ацетоацетил-СоА плюс 0,2 мМ СоА. Концентрацию белка определяли либо методом Бредфорда, либо с помощью набора с бицинхониновой кислотой (Sigma, каталожный № BCA-1). В качестве стандарта в обоих случаях использовали альбумин бычьей сыворотки (Bio-Rad, Hercules, CA). В одном стандартном исследовании специфическая активность белка ThlA в индуцированной культуре составляла 16,0 мкмоль·мг-1·мин-1 по сравнению с 0,27 мкмоль·мг-1·мин-1 в неиндуцированной культуре.
ПРИМЕР 2
Клонирование и экспрессия ацетил-CoA-ацетилтрансферазы
Целью настоящего примера являлась экспрессия фермента ацетил-CoA-ацетилтрансфераза, также именуемого в настоящем описании как ацетоацетил-CoA-тиолаза, в E.сoli. Ген ацетоацетил-CoA-тиолазы thlВ был клонирован из C.acetobutylicum (ATCC 824) и экспрессирован в E.coli. Ген thlВ амплифицировали из геномной ДНК C.acetobutylicum (ATCC 824) посредством ПЦР.
Ген thlB был клонирован и экспрессирован таким же способом, как ген thlA, описанный в примере 1. Геномная ДНК C.acetobutylicum (ATCC 824) была амплифицирована посредством ПЦР с использованием праймеров N15 и N16 (смотрите таблицу 4), представленных как SEQ ID NO: 27 и 28 соответственно, образуя продукт 1,2 т.п.н. Прямой праймер, содержащий четыре основания (ССАС), непосредственно прилегает к инициирующему трансляцию кодону для обеспечения прямого клонирования в pENTR/SD/D-TOPO (Invitrogen) для образования плазмиды pENTRSDD-TOPOthlВ. Клоны были предоставлены для секвенирования с использованием прямого и обратного праймеров М13, представленных как SEQ ID NO: 45 и 46 соответственно, для подтверждения того, что указанные гены встроены в правильной ориентации, и подтверждения указанной последовательности. Дополнительные праймеры для секвенирования, N15SeqF1 и N16SeqR1 (смотрите таблицу 5), представленные как SEQ ID NO: 49 и 50 соответственно, были необходимы для полного секвенирования указанного ПЦР-продукта. Нуклеотидная последовательность открытой рамки считывания (ORF) для указанного гена и предсказанная аминокислотная последовательность указанного фермента представлены как SEQ ID NO: 3 и SEQ ID NO: 4 соответственно.
Для создания экспрессирующего клона ген thlВ был перенесен в вектор pDEST 14 (Invitrogen) посредством рекомбинации для образования pDEST14thlВ. Вектор pDEST14thlB трансформировали в клетках BL21-AI и экспрессию с промотора T7 индуцировали добавлением арабинозы. Белок ожидаемой молекулярной массы около 42 кДа, как было рассчитано по последовательности нуклеиновой кислоты, был обнаружен в индуцированной культуре, но не обнаружен в неиндуцированном контроле. Ферментные анализы осуществляли, как описано в примере 1. В одном стандартном исследовании специфическая активность белка ThlВ в индуцированной культуре составляла 14,9 мкмоль·мг-1·мин-1 по сравнению с 0,28 мкмоль·мг-1·мин-1 в неиндуцированной культуре.
ПРИМЕР 3
Клонирование и экспрессия 3-гидроксибутирил-CoA-дегидрогеназы
Целью настоящего примера было клонировать ген hbd из C.acetobutylicum (ATCC 824) и экспрессировать его в E.coli. Ген hbd был амплифицирован из геномной ДНК C.acetobutylicum (ATCC 824) посредством ПЦР.
Ген hbd был клонирован и экспрессирован с использованием метода, описанного в примере 1. Ген hbd был амплифицирован из геномной ДНК C.acetobutylicum (ATCC 824) посредством ПЦР с использованием праймеров N5 и N6 (смотрите таблицу 4), представленных как SEQ ID NO: 19 и 20 соответственно, с образованием продукта 881 п.н. Прямой праймер, содержащий четыре основания (САСС), непосредственно прилегает к инициирующему трансляцию кодону для обеспечения прямого клонирования в pENTR/SD/D-TOPO (Invitrogen) для образования плазмиды pENTRSDD-TOPOhbd. Клоны были предоставлены для секвенирования с использованием прямого и обратного праймеров М13, представленных как SEQ ID NO: 45 и 46 соответственно, для подтверждения того, что указанные гены встроены в правильной ориентации, и подтверждения указанной последовательности. Дополнительные праймеры для секвенирования, N5SeqF2 и N6SeqR2 (смотрите таблицу 5), представленные как SEQ ID NO: 51 и 52 соответственно, были необходимы для полного секвенирования указанного ПЦР-продукта. Нуклеотидная последовательность открытой рамки считывания (ORF) для указанного гена и предсказанная аминокислотная последовательность указанного фермента представлены как SEQ ID NO: 5 и SEQ ID NO: 6 соответственно.
Для создания экспрессирующего клона ген hbd был перенесен в вектор pDEST 14 (Invitrogen) посредством рекомбинации для образования pDEST14hbd. Вектор pDEST14hbd трансформировали в клетках BL21-AI и экспрессию с промотора T7 индуцировали добавлением арабинозы, как описано в примере 1. Белок ожидаемой молекулярной массы около 31 кДа, как было рассчитано по последовательности нуклеиновой кислоты, был обнаружен в индуцированной культуре, но не обнаружен в неиндуцированном контроле.
Активность гидроксибутирил-CoA-дегидрогеназы определяли, измеряя степени окисления NADH, что было измерено по уменьшению оптической плотности при 340 нм. Стандартная аналитическая смесь включала 50 мM MOPS, pH 7,0, 1 мM DTT и 0,2 мM NADH. Указанный коктейль уравновешивали в течение 5 минут при 37°С и затем добавляли бесклеточный экстракт. Реакции инициировали добавлением субстрата, 0,1 мM ацетоацетил-CoA. В одном стандартном исследовании специфическая активность белка BHBD в индуцированной культуре составляла 57,4 мкмоль·мг-1·мин-1 по сравнению с 0,885 мкмоль·мг-1·мин-1 в неиндуцированной культуре.
ПРИМЕР 4
Клонирование и экспрессия кротоназы
Целью настоящего примера было клонирование гена crt из C.acetobutylicum (ATCC 824) и экспрессия его в E.coli. Ген crt был амплифицирован из геномной ДНК C.acetobutylicum (ATCC 824) посредством ПЦР.
Ген crt был клонирован и экспрессирован с использованием метода, описанного в примере 1. Ген crt был амплифицирован из геномной ДНК C.acetobutylicum (ATCC 824) посредством ПЦР с использованием праймеров N3 и N4 (смотрите таблицу 4), представленных как SEQ ID NO: 17 и 18 соответственно, с образованием продукта 794 п.н. Прямой праймер, содержащий четыре основания (САСС), непосредственно прилегает к инициирующему трансляцию кодону для обеспечения прямого клонирования в pENTR/SD/D-TOPO (Invitrogen) для образования плазмиды pENTRSDD-TOPOcrt. Клоны были предоставлены для секвенирования с использованием прямого и обратного праймеров М13, представленных как SEQ ID NO: 45 и 46 соответственно, для подтверждения того, что указанные гены встроены в правильной ориентации, и подтверждения указанной последовательности. Нуклеотидная последовательность открытой рамки считывания (ORF) для указанного гена и предсказанная аминокислотная последовательность указанного фермента представлены как SEQ ID NO: 7 и SEQ ID NO: 8 соответственно.
Для создания экспрессирующего клона ген crt был перенесен в вектор pDEST 14 (Invitrogen) посредством рекомбинации для образования pDEST14crt. Вектор pDEST14crt трансформировали в клетках BL21-AI и экспрессию с промотора T7 индуцировали добавлением арабинозы, как описано в примере 1. Белок ожидаемой молекулярной массы около 28 кДа, как было рассчитано по последовательности нуклеиновой кислоты, был обнаружен в индуцированной культуре, но не обнаружен в неиндуцированном контроле.
Активность кротоназы исследовали, как описано Stern (Methods Enzymol. 1, 559-566, (1954)). В одном стандартном исследовании специфическая активность белка кротоназы в индуцированной культуре составляла 444 мкмоль·мг-1·мин-1 по сравнению с 47 мкмоль·мг-1·мин-1 в неиндуцированной культуре.
ПРИМЕР 5
Клонирование и экспрессия бутирил-CoA-дегидрогеназы
Целью настоящего примера была экспрессия фермента бутирил-CoA-дегидрогеназа, также называемого в настоящем описании транс-2-еноил-CoA-редуктаза, в E.coli. Ген CAC0462, предполагаемый гомолог транс-2-еноил-CoA-редуктазы, был клонирован из C.acetobutylicum (ATCC 824) и экспрессирован в E.coli. Ген CAC0462 был амплифицирован из геномной ДНК C.acetobutylicum (ATCC 824) посредством ПЦР.
Ген CAC0462 был клонирован и экспрессирован с использованием метода, описанного в примере 1. Ген CAC0462 был амплифицирован из геномной ДНК C.acetobutylicum (ATCC 824) посредством ПЦР с использованием праймеров N17 и N21 (смотрите таблицу 4), представленных как SEQ ID NO: 29 и 30 соответственно, с образованием продукта 1,3 т.п.н. Прямой праймер, содержащий четыре основания (САСС), непосредственно прилегает к инициирующему трансляцию кодону для обеспечения прямого клонирования в pENTR/SD/D-TOPO (Invitrogen) для образования плазмиды pENTRSDD-TOPOСАС0462. Клоны были предоставлены для секвенирования с использованием прямого и обратного праймеров М13, представленных как SEQ ID NO: 45 и 46 соответственно, для подтверждения того, что указанные гены встроены в правильной ориентации, и подтверждения указанной последовательности. Дополнительные праймеры для секвенирования, N22SeqF1 (SEQ ID NO: 53), N22SeqF2 (SEQ ID NO: 54), N22SeqF3 (SEQ ID NO: 55), N23SeqR1 (SEQ ID NO: 56), N23SeqR2 (SEQ ID NO: 57) и N23SeqR3 (SEQ ID NO: 58) (смотрите таблицу 5), были необходимы для полного секвенирования указанного ПЦР-продукта. Нуклеотидная последовательность открытой рамки считывания (ORF) для указанного гена и предсказанная аминокислотная последовательность указанного фермента представлены как SEQ ID NO: 9 и SEQ ID NO: 10 соответственно.
Для создания экспрессирующего клона ген САС0462 был перенесен в вектор pDEST 14 (Invitrogen) посредством рекомбинации для образования pDEST14САС0462. Вектор pDEST14САС0462 трансформировали в клетках BL21-AI и экспрессию с промотора T7 индуцировали добавлением арабинозы, как описано в примере 1. Анализ в SDS-PAGE не выявил наличия сверхпродукции белка ожидаемой молекулярной массы в отрицательном контроле или в индуцированной культуре. Ген САС0462 C.acetobutylicum использовал много редких кодонов E.coli. Для того чтобы обойти проблемы с использованием кодонов, плазмида pRARE (Novagen) была трансформирована в клетках BL21-AI, несущих вектор pDEST14CAC0462. Исследования экспрессии с индукцией арабинозой повторяли с культурами, несущими вектор pRARE. Белок ожидаемой молекулярной массы около 46 кДа был обнаружен в индуцированной культуре, но не обнаружен в неиндуцированном контроле.
Активность транс-2-еноил-CoA-редуктазы исследовали, как описано Hoffmeister et al. (J. Biol. Chem. 280, 4329-4338 (2005)). В одном стандартном исследовании специфическая активность белка TER CAC0462 в индуцированной культуре составляла 0,694 мкмоль·мг-1·мин-1 по сравнению с 0,0128 мкмоль·мг-1·мин-1 в неиндуцированной культуре.
ПРИМЕР 6
Клонирование и экспрессия бутиральдегиддегидрогеназы (ацетилирование)
Целью настоящего примера было клонирование гена ald из C.beijerinckii (ATCC 35702) и экспрессия его в E.coli. Ген ald был амплифицирован из геномной ДНК C.beijerinckii (ATCC 35702) посредством ПЦР.
Ген ald был клонирован и экспрессирован с использованием метода, описанного в примере 1. Ген ald был амплифицирован из геномной ДНК C.beijerinckii (ATCC 35702) (выделенной из культур, выращенных в анаэробных условиях, как описано в примере 1) посредством ПЦР с использованием праймеров N27 F1 и N28 R1 (смотрите таблицу 4), представленных как SEQ ID NO: 31 и 32 соответственно, с образованием продукта 1,6 т.п.н. Прямой праймер содержит четыре основания (САСС), непосредственно прилегающие к инициирующему трансляцию кодону, для обеспечения прямого клонирования в pENTR/SD/D-TOPO (Invitrogen) для образования плазмиды pENTRSDD-TOPOald. Клоны были предоставлены для секвенирования с использованием прямого и обратного праймеров М13, представленных как SEQ ID NO: 45 и 46 соответственно, для подтверждения того, что указанные гены встроены в правильной ориентации, и подтверждения указанной последовательности. Дополнительные праймеры для секвенирования, N31SeqF2 (SEQ ID NO: 59), N31SeqF3 (SEQ ID NO: 60), N31SeqF4 (SEQ ID NO: 61), N32SeqR1 (SEQ ID NO: 72), N31SeqR2 (SEQ ID NO: 62), N31SeqR3 (SEQ ID NO: 63), N31SeqR4 (SEQ ID NO: 64) и N31SeqR5 (SEQ ID NO: 65) (смотрите таблицу 5), были необходимы для полного секвенирования указанного ПЦР-продукта. Нуклеотидная последовательность открытой рамки считывания (ORF) для указанного гена и предсказанная аминокислотная последовательность указанного фермента представлены как SEQ ID NO: 11 и SEQ ID NO: 12 соответственно.
Для создания экспрессирующего клона ген ald был перенесен в вектор pDEST 14 (Invitrogen) посредством рекомбинации для образования pDEST14ald. Вектор pDEST14ald трансформировали в клетках BL21-AI и экспрессию с промотора T7 индуцировали добавлением арабинозы, как описано в примере 1. Белок ожидаемой молекулярной массы около 51 кДа, как было рассчитано по последовательности нуклеиновой кислоты, был обнаружен в индуцированной культуре, но не обнаружен в неиндуцированном контроле.
Активность ацелирующей альдегиддегирогеназы была определена в результате контролирования образования NADH и измерена по повышению оптической плотности при 340 нм, как описано Husemann et al. (Appl. Microbiol. Biotechnol. 31: 435-444 (1989)). В одном стандартном исследовании специфическая активность белка Ald в индуцированной культуре составляла 0,106 мкмоль·мг-1·мин-1 по сравнению с 0,01 мкмоль·мг-1·мин-1 в неиндуцированной культуре.
ПРИМЕР 7
Клонирование и экспрессия бутанолдегидргеназы
Целью настоящего примера было клонирование гена bdhB из C.acetobutylicum (ATCC 824) и экспрессия его в E.coli. Ген bdhB был амплифицирован из геномной ДНК C.acetobutylicum (ATCC 824) посредством ПЦР.
Ген bdhB был клонирован и экспрессирован с использованием метода, описанного в примере 1. Ген bdhB был амплифицирован из геномной ДНК C.acetobutylicum (ATCC 824) посредством ПЦР с использованием праймеров N11 и N12 (смотрите таблицу 4), представленных как SEQ ID NO: 25 и 26 соответственно, с образованием продукта 1,2 т.п.н. Прямой праймер содержит четыре основания (САСС), непосредственно прилегающие к инициирующему трансляцию кодону, для обеспечения прямого клонирования в pENTR/SD/D-TOPO (Invitrogen) для образования плазмиды pENTRSDD-TOPObdhB. Инициирующий трансляцию кодон также был изменен с «GTG» на «ATG» посредством последовательности праймера. Клоны были предоставлены для секвенирования с использованием прямого и обратного праймеров М13, представленных как SEQ ID NO: 45 и 46 соответственно, для подтверждения того, что указанные гены встроены в правильной ориентации, и подтверждения указанной последовательности. Дополнительные праймеры для секвенирования, N11SeqF1 (SEQ ID NO: 66), N11SeqF2 (SEQ ID NO: 67), N12SeqR1 (SEQ ID NO: 68) и N12SeqR2 (SEQ ID NO: 69) (смотрите таблицу 5), были необходимы для полного секвенирования указанного ПЦР-продукта. Нуклеотидная последовательность открытой рамки считывания (ORF) для указанного гена и предсказанная аминокислотная последовательность указанного фермента представлены как SEQ ID NO: 13 и SEQ ID NO: 14 соответственно.
Для создания экспрессирующего клона ген bdhB был перенесен в вектор pDEST 14 (Invitrogen) посредством рекомбинации для образования pDEST14bdhB. Вектор pDEST14bdhB трансформировали в клетках BL21-AI и экспрессию с промотора T7 индуцировали добавлением арабинозы, как описано в примере 1. Белок ожидаемой молекулярной массы около 43 кДа, как было рассчитано по последовательности нуклеиновой кислоты, был обнаружен в индуцированной культуре, но не обнаружен в неиндуцированном контроле.
Активность бутанолдегидрогеназы была определена по степени окисления NADH, измеренной по уменьшению оптической плотности при 340 нм, как описано Husemann и Papoutsakis, выше. В одном стандартном исследовании специфическая активность белка BdhB в индуцированной культуре составляла 0,169 мкмоль·мг-1·мин-1 по сравнению с 0,022 мкмоль·мг-1·мин-1 в неиндуцированной культуре.
ПРИМЕР 8
Клонирование и экспрессия бутанолдегидрогеназы
Целью настоящего примера было клонирование гена bdhА из C.acetobutylicum 824 и экспрессия его в E.coli. Ген bdhА был амплифицирован из геномной ДНК C.acetobutylicum 824 посредством ПЦР.
Ген bdhА был клонирован и экспрессирован с использованием метода, описанного в примере 1. Ген bdhА был амплифицирован из геномной ДНК C.acetobutylicum 824 посредством ПЦР с использованием праймеров N9 и N10 (смотрите таблицу 4), представленных как SEQ ID NO: 23 и 24 соответственно, с образованием продукта 1,2 т.п.н. Прямой праймер содержит четыре основания (САСС), непосредственно прилегающие к инициирующему трансляцию кодону, для обеспечения прямого клонирования в pENTR/SD/D-TOPO (Invitrogen) для образования плазмиды pENTRSDD-TOPObdhА. Клоны были предоставлены для секвенирования с использованием прямого и обратного праймеров М13, представленных как SEQ ID NO: 45 и 46 соответственно, для подтверждения того, что указанные гены встроены в правильной ориентации, и подтверждения указанной последовательности. Дополнительные праймеры для секвенирования, N9SeqF1 (SEQ ID NO: 70) и N10SeqR1 (SEQ ID NO: 71) (смотрите таблицу 5), были необходимы для полного секвенирования указанного ПЦР-продукта. Нуклеотидная последовательность открытой рамки считывания (ORF) для указанного гена и предсказанная аминокислотная последовательность указанного фермента представлены как SEQ ID NO: 15 и SEQ ID NO: 16 соответственно.
Для создания экспрессирующего клона ген bdhА был перенесен в вектор pDEST 14 (Invitrogen) посредством рекомбинации для образования pDEST14bdhА. Вектор pDEST14bdhА трансформировали в клетках BL21-AI и экспрессию с промотора T7 индуцировали добавлением арабинозы, как описано в примере 1. Белок ожидаемой молекулярной массы около 43 кДа, как было рассчитано по последовательности нуклеиновой кислоты, был обнаружен в индуцированной культуре, но не обнаружен в неиндуцированном контроле.
Активность бутанолдегидрогеназы была определена по степени окисления NADH, измеренной по уменьшению оптической плотности при 340 нм, как описано Husemann и Papoutsakis, выше. В одном стандартном исследовании специфическая активность белка BdhА в индуцированной культуре составляла 0,102 мкмоль·мг-1·мин-1 по сравнению с 0,028 мкмоль·мг-1·мин-1 в неиндуцированной культуре.
ПРИМЕР 9
Конструкция трансформационного вектора для генов в биосинтетическом пути 1-бутанола - нижний путь биосинтеза
Для конструкции трансформационного вектора, включающего гены, кодирующие шесть этапов в биосинтетическом пути 1-бутанола, указанные 6 этапов пути были разделены на два оперона. Верхний путь биосинтеза включает первые четыре этапа, катализируемые ацетил-CoA-ацетилтрансферазой, 3-гидроксибутирил-CoA-дегидрогеназой, кротоназой и бутирил-CoA-дегидрогеназой. Нижний путь биосинтеза включает последние два этапа, катализируемые бутиральдегиддегидрогеназой и бутанолдегидрогеназой.
Целью настоящего примера было конструирование оперона нижнего пути биосинтеза. Конструкция оперона верхнего пути биосинтеза описана в примере 10.
Отдельные гены были амплифицированы посредством ПЦР с использованием праймеров, которые включают сайты рестрикции для последующего клонирования, и прямые праймеры содержали оптимизированный рибосомосвязывающий участок E.coli (AAAGGAGG). ПЦР-продукты были клонированы ТОРО в вектор pCR 4Blunt-TOPO и трансформированы в клетках Top10 E.coli (Invitrogen). Плазмидная ДНК была выделена из клонов ТОРО, и последовательность указанных генов была определена. Рестрикционные ферменты и Т4 ДНК-лигазу (New England Biolabs, Beverly, MA) использовали в соответствии с рекомендациями фирмы-изготовителя. Для экспериментов с клонированием фрагменты рестрикции очищали посредством электрофореза в геле с использованием набора QIAquick Gel Extraction (Qiagen).
После подтверждения указанной последовательности гены были субклонированы в модифицированный вектор pUC19 в качестве основы для клонирования. Вектор pUC19 был модифицирован посредством обработки с помощью HindIII/SapI с образованием pUC19dHS. Указанная обработка удалила промотор lac, расположенный рядом с MCS (множественные участки клонирования), предотвращая транскрипцию оперонов в указанном векторе.
Ген ald был амплифицирован из геномной ДНК C.beijerinckii ATCC 35702 посредством ПЦР с использованием праймеров N58 и N59 (смотрите таблицу 4), представленных как SEQ ID NO: 41 и 42 соответственно, с образованием продукта 1,5 т.п.н. Прямой праймер включал сайты рестрикции для AvaI и BstEII и RBS (рибосомосвязывающий участок). Обратный праймер включал сайт рестрикции для HpaI. ПЦР-продукт был клонирован в pCRBlunt II-TOPO с образованием pCRBluntII-ald. Плазмидная ДНК была выделена из клонов ТОРО, и последовательность указанных генов была определена с помощью праймеров M13 прямого (SEQ ID NO: 45), M13 обратного (SEQ ID NO: 46), N31SeqF2 (SEQ ID NO: 59), N31SeqF3 (SEQ ID NO: 60), N31SeqF4 (SEQ ID NO: 61), N32SeqR1 (SEQ ID NO: 72), N31SeqR2 (SEQ ID NO: 62), N31SeqR3 (SEQ ID NO: 63), N31SeqR4 (SEQ ID NO: 64) и N31SeqR5 (SEQ ID NO: 65) (смотрите таблицу 5).
Ген bdhB был амплифицирован из геномной ДНК C.acetobutylicum (ATCC 824) посредством ПЦР с использованием праймеров N64 и N65 (смотрите таблицу 4), представленных как SEQ ID NO: 43 и 44 соответственно, с образованием продукта 1,2 т.п.н. Прямой праймер включал сайты рестрикции для HpaI и RBS. Обратный праймер включал сайты рестрикции для PmeI и SphI. ПЦР-продукт был клонирован в pCRBlunt II-TOPO с образованием pCRBluntII-bdhB. Плазмидная ДНК была выделена из клонов ТОРО, и последовательность указанных генов была определена с помощью праймеров M13 прямого (SEQ ID NO: 45), M13 обратного (SEQ ID NO: 46), N11SeqF1 (SEQ ID NO: 66), N11SeqF2 (SEQ ID NO: 67), N12SeqR1 (SEQ ID NO: 68) и N12SeqR2 (SEQ ID NO: 69) (смотрите таблицу 5).
Для конструкции оперона нижнего пути биосинтеза фрагмент 1,2 т.п.н. SphI и HpaI из pCRBluntII-bdhB, фрагмент 1,4 т.п.н. HpaI и SphI из pCRBluntII-ald и большой фрагмент после расщепления pUC19dHS посредством AvaI и SphI были лигированы вместе. В результате указанного тройственного лигирования образуется pUC19dHS-ald-bdhB.
Вектор pUC19dHS-ald-bdhB был расщеплен с помощью BstEII и PmeI, в результате чего был высвобожден фрагмент 2,6 т.п.н, который был клонирован в pBenBP, «челночный» вектор E.coli-Bacillus subtilis. Плазмида pBenBP была образована посредством модификации вектора pBE93, который описан Nagarajan, WO 93/24631 (пример 4). Промотор нейтральной протеазы Bacillus amyloliquefaciens (NPR), сигнальная последовательность и ген phoA были удалены из pBE93 посредством обработки NcoI/HindIII. Промотор NPR был амплифицирован посредством ПЦР из pBE93 с использованием праймеров BenF и BenBPR, представленных как SEQ ID NO: 73 и 75 соответственно. Праймер BenBPR включает сайты для BstEII, PmeI и HindIII в 5'-3' направлении от указанного промотора. ПЦР-продукт был обработан с помощью NcoI и HindIII, и фрагмент был клонирован в соответствующих сайтах в векторе рВЕ93 для образования pBenBP. Указанный фрагмент нижнего оперона был субклонирован в участки BstEII и PmeI в pBenBP с образованием pBen-ald-bdhB.
Анализы активности бутиральдегиддегидрогеназы и бутанолдегидрогеназы проводили на первичных экстрактах с использованием методов, описанных выше. Для обеих ферментативных активностей были продемонстрированы уровни выше контрольного штамма, который содержал пустой вектор.
ПРИМЕР 10 (предсказательный)
Конструкция трансформирующего вектора для генов биосинтетического пути 1-бутанола - верхний путь биосинтеза
Целью настоящего предсказательного примера является описание того, как сконструировать оперон верхнего пути биосинтеза. Основной принцип является таким же, как описанный в примере 9.
Ген thlA был амплифицирован из геномной ДНК C.acetobutylicum (ATCC 824) посредством ПЦР с использованием пары праймеров N44 и N45 (смотрите таблицу 4), представленных как SEQ ID NO: 33 и 34 соответственно, с образованием продукта 1,2 т.п.н. Прямой праймер включает сайт рестрикции для SphI и рибосомосвязывающий сайт (RBS). Обратный праймер включает сайты рестрикции для AscI и PstI. ПЦР-продукт клонирован в pCR4 Blunt-TOPO с образованием pCR4 Blunt-TOPO-thlA. Плазмидная ДНК была выделена из клонов TOPO и последовательность указанных генов определена с помощью праймеров M13 прямого (SEQ ID NO: 45), M13 обратного (SEQ ID NO: 46), N7SeqF1 (SEQ ID NO: 47) и N7SeqR1 (SEQ ID NO: 48) (смотрите таблицу 5).
Ген hbd был амплифицирован из геномной ДНК C.acetobutylicum (ATCC 824) посредством ПЦР с использованием пары праймеров N42 и N43 (смотрите таблицу 4), представленных как SEQ ID NO: 35 и 36 соответственно, с образованием продукта 0,9 т.п.н. Прямой праймер включает сайт рестрикции для SalI и RBS. Обратный праймер включает сайт рестрикции для SphI. ПЦР-продукт клонирован в pCR4 Blunt-TOPO с образованием pCR4 Blunt-TOPO-hbd. Плазмидная ДНК была выделена из клонов TOPO, и последовательность указанных генов определена с помощью праймеров M13 прямого (SEQ ID NO: 45), M13 обратного (SEQ ID NO: 46), N5SeqF2 (SEQ ID NO: 51) и N6SeqR2 (SEQ ID NO: 52) (смотрите таблицу 5).
Ген CAC0462 является кодон-оптимизированным для экспрессии в E.coli в качестве первичного хозяина и B.subtilis в качестве промежуточного хозяина. Новый ген, называемый CaTER, данный как SEQ ID NO: 76, синтезирован в Genscript Corp (Piscataway, NJ). Ген CaTER клонирован в вектор pUC57 как фрагмент BamHI-SalI и включает RBS, образуя плазмиду pUC57-CaTER.
Ген crt был амплифицирован из геномной ДНК C.acetobutylicum (ATCC 824) посредством ПЦР с использованием пары праймеров N38 и N39 (смотрите таблицу 4), представленных как SEQ ID NO: 39 и 40 соответственно, с образованием продукта 834 п.н. Прямой праймер включает сайты рестрикции для EcoRI и MluI и RBS. Обратный праймер включает сайт рестрикции для BamHI. ПЦР-продукт клонирован в pCR4 Blunt-TOPO с образованием pCR4 Blunt-TOPO-crt. Плазмидная ДНК была выделена из клонов TOPO, и последовательность указанных генов определена с помощью праймеров M13 прямого (SEQ ID NO: 45) и M13 обратного (SEQ ID NO: 46) (смотрите таблицу 5).
После подтверждения указанной последовательности гены были субклонированы в модифицированный вектор pUC19 в качестве основы для клонирования. Вектор pUC19 был модифицирован посредством обработки с помощью SphI/SapI с образованием pUC19dSS. Обработка удалила промотор lac, расположенный рядом с MCS, предотвращая транскрипцию указанных оперонов в указанном векторе.
Для конструкции оперона верхнего пути биосинтеза pCR4 Blunt-TOPO-crt расщепляли с помощью EcoRI и BamHI, в результате чего высвобождался фрагмент crt размером 0,8 т.п.н. Вектор pUC19dSS также расщепляли с помощью EcoRI и BamHI, в результате чего высвобождался фрагмент вектора размером 2,0 т.п.н. Фрагмент crt и фрагмент вектора лигировали вместе с использованием T4 ДНК-лигазы (New England Biolabs) для образования pUC19dSS-crt. Ген CaTER был встроен в pCU19dSS-crt посредством обработки pUC57-CaTER с помощью BamHI и SalI, в результате чего высвобождался фрагмент CaTER размером 1,2 т.п.н. pUC19dSS-crt обрабатывали с помощью BamHI и SalI и большой фрагмент вектора лигировали с фрагментом CaTER, в результате чего образуется pUC19dSS-crt-CaTER. Для завершения оперона были лигированы фрагменты SalI и SphI из pCR4 Blunt-TOPO-hbd размером 884 п.н., фрагмент thlA SphI и PstI из pCR4 Blunt-TOPO-thlA размером 1,2 т.п.н. и большой фрагмент после обработки pUC19dSS-crt-CaTER с помощью SalI и PstI. Продукт тройственного лигирования представляет собой pUC19dSS-crt-CaTER-hbd-thlA.
Вектор pUC19dSS-crt-CaTER-hbd-thlA обрабатывали с помощью MluI и AscI, высвобождая фрагмент размером 4,1 т.п.н, который клонировали в производное вектора pBE93 (Caimi, WO2004/018645, pp. 39-40) «челночный» вектор E.coli-B.subtilis, называемый pBenMA. Плазмида pBenMA была создана в результате модификации данного вектора pBE93. Промотор нейтральной протеазы (NPR) Bacillus amyloliquefaciens, сигнальная последовательность и ген phoA удалены из pBE93 посредством обработки с помощью NcoI/HindIII. Промотор NPR был амплифицирован посредством ПЦР из pBE93 с использованием праймеров BenF и BenMAR, представленных как SEQ ID NO: 73 и 74 соответственно. Праймер BenMAR включает сайты для MluI, AscI и HindIII в 5'-3' направлении от указанного промотора. ПЦР-продукт был обработан с помощью NcoI и HindIII, и фрагмент был клонирован в соответствующих сайтах в векторе рВЕ93 для образования pBenMA. Фрагмент верхнего оперона был субклонирован в участки MluI и AscI в pBenMA с образованием pBen-crt-hbd-CaTER-thlA.
ПРИМЕР 11 (предсказательный)
Экспрессия биосинтетического пути 1-бутанола в E.coli
Целью данного предсказательного примера является описание того, как экспрессировать биосинтетический путь 1-бутанола в E.coli.
Плазмиды pBen-crt-hbd-CaTER-thlA и pBen-ald-bdhB, сконструированные, как описано в примерах 10 и 9 соответственно, трансформированы в E.coli NM522 (ATCC 47000), и экспрессию генов в каждом опероне контролировали посредством анализа в SDS-PAGE, ферментативного анализа и вестерн-анализа. Для вестерн-анализа выращивали антитела к синтетическим пептидам в Sigma-Genosys (The Woodlands, TX). После подтверждения экспрессии всех генов pBen-ald-bdhB обрабатывали с помощью EcoRI и PmeI для высвобождения фрагмента промотор NPR-ald-bdhB. Расщепление указанного фрагмента с помощью EcoRI является тупоконечным с использованием фрагмента Кленова ДНК полимеразы (New England Biolabs, каталожный № M0210S). Плазмиду pBen-crt-hbd-CaTER-thlA расщепляли с помощью PvuII для образования линеаризированного фрагмента вектора с тупыми концами. Вектор и фрагмент NPR-ald-bdhB лигировали с образованием p1B1 O.1 и p1B1 O.2, включающих полный биосинтетический путь 1-бутанола с фрагментом промотор NPR-ald-bdhB в противоположных ориентациях. Плазмиды p1B1 O.1 и p1B1 O.2 трансформированы в E.coli NM522, и экспрессия генов контролируется, как описано ранее.
Штамм E.coli NM522/p1B1 O.1 или NM522/p1B1 O.1 высевали в 250-мл встряхиваемую колбу, содержащую 50 мл среды, и встряхивали при 250 об/мин при 35°С. Указанная среда имела следующий состав: декстроза, 5 г/л; MOPS, 0,05 M; сульфат аммония, 0,01 M; фосфат калия, одноосновный, 0,005 M; смесь металлов S10, 1% (об./об.); дрожжевой экстракт, 0,1% (вес./об.); казаминовые кислоты, 0,1% (вес./об.); тиамин, 0,1 мг/л; пролин, 0,05 мг/л; и биотин, 0,002 мг/л, и была титруема до pH 7,0 с помощью KOH. Смесь металлов S10 содержит: MgCl2, 200 мM; CaCl2, 70 мM; MnCl2, 5 мM; FeCl3, 0,1 мM; ZnCl2, 0,1 мM; тиамин гидрохлорид, 0,2 мM; CuSO4, 172 мкM; CoCl2, 253 мкM; и Na2MoO4, 242 мкM. Через 18-24 часа определяли 1-бутанол посредством ВЭЖХ или ГХ анализа, как описано в разделе «Общие методы».
ПРИМЕР 12 (предсказательный)
Экспрессия биосинтетического пути 1-бутанола в Bacillus subtilis
Целью настоящего предсказательного примера является описание того, как экспрессировать биосинтетический путь 1-бутанола в Bacillus subtilis. Использовали тот же принцип, что и в примере 11.
Использовали верхний и нижний опероны, сконструированные, как описано в примерах 10 и 9 соответственно. Плазмиды p1B1 O.1 и p1B1 O.2 трансформировали в Bacillus subtilis BE1010 (J. Bacteriol. 173: 2278-2282 (1991)) и экспрессию генов в каждом опероне контролировали, как описано в примере 11.
Штамм B.subtilis BE1010/p1B1 O.1 или BE1010/p1B1 O.2 высевали в 250-мл встряхиваемую колбу, содержащую 50 мл среды, и встряхивали при 250 об/мин при 35°С в течение 18 часов. Указанная среда имела следующий состав: декстроза, 5 г/л; MOPS, 0,05 M; глутаминовая кислота, 0,02 М; сульфат аммония, 0,01 M; фосфат калия, одноосновный буфер, 0,005 M; смесь металлов S10 (как описано в примере 11), 1% (об./об.); дрожжевой экстракт, 0,1% (вес./об.); казаминовые кислоты, 0,1% (вес./об.); триптофан, 50 мг/л; метионин, 50 мг/л; и лизин 50 мг/л, и была титруема до pH 7,0 с помощью KOH. Через 18-24 часа определяли 1-бутанол посредством ВЭЖХ или ГХ анализа, как описано в разделе «Общие методы».
ПРИМЕР 13
Получение 1-бутанола из глюкозы с использованием рекомбинантной E.coli
Настоящий пример описывает продуцирование 1-бутанола в E.coli. Экспрессия генов, кодирующих 6 этапов биосинтетического пути 1-бутанола, была разделена на три оперона. Верхний путь биосинтеза содержал первые четыре этапа, кодируемые thlA, hbd, crt и EgTER, в одном опероне. Следующий этап, кодируемый ald, был обеспечен вторым опероном. Последний этап пути биосинтеза, кодируемый yqhD, был обеспечен в третьем опероне. Продуцирование 1-бутанола было продемонстрировано в штаммах E.coli, включающих указанные три оперона.
Если в тексте не указано иначе, праймеры для клонирования, описанные в настоящем примере, обозначены их последовательностями SEQ ID NO: в таблице 4, и праймеры для секвенирования обозначены их последовательностями SEQ ID NO: в таблице 5.
Ацетил-CoA-ацетилтрансфераза. Ген thlA был амплифицирован из геномной ДНК C.acetobutylicum (ATCC 824) посредством ПЦР с использованием пары праймеров N44 и N45 (смотрите таблицу 4), представленных как SEQ ID NO: 33 и 34 соответственно, с образованием продукта размером 1,2 т.п.н. Прямой праймер включает сайт рестрикции для SphI и рибосомосвязывающий сайт (RBS). Обратный праймер включает сайты рестрикции для AscI и PstI. ПЦР-продукт клонировали в pCR4Blunt-TOPO (Invitrogen Corp., Carlsbad, CA) с образованием pCR4Blunt-TOPO-thlA. Плазмидную ДНК выделяли из клонов ТОРО, и последовательность указанных генов определяли с использованием праймеров M13 прямого (SEQ ID NO: 45), M13 обратного (SEQ ID NO: 46), N7SeqF1 (SEQ ID NO: 47) и N7SeqR1 (SEQ ID NO: 48) (смотрите таблицу 5).
3-гидроксибутирил-CoA-дегидрогеназа. Ген hbd был амплифицирован из геномной ДНК C.acetobutylicum (ATCC 824) посредством ПЦР с использованием пары праймеров N42 и N43 (смотрите таблицу 4), предсталенных как SEQ ID NO: 35 и 36 соответственно, с образованием продукта 0,9 т.п.н. Прямой праймер включает сайт рестрикции для SalI и RBS. Обратный праймер включает сайт рестрикции для SphI. ПЦР-продукт клонировали в pCR4Blunt-TOPO с образованием pCR4Blunt-TOPO-hbd. Плазмидную ДНК выделяли из клонов ТОРО и последовательность указанных генов определяли с использованием праймеров M13 прямого (SEQ ID NO: 45), M13 обратного (SEQ ID NO: 46), N5SeqF2 (SEQ ID NO: 51) и N6SeqR2 (SEQ ID NO: 52) (смотрите таблицу 5).
Кротоназа. Ген crt был амплифицирован из геномной ДНК C.acetobutylicum (ATCC 824) посредством ПЦР с использованием пары праймеров N38 и N39 (смотрите таблицу 4), представленных как SEQ ID NO: 39 и 40 соответственно, с образованием продукта размером 834 п.н. Прямой праймер включает сайты рестрикции для EcoRI и MluI и RBS. Обратный праймер включает сайт рестрикции для BamHI. ПЦР-продукт клонировали в pCR4Blunt-TOPO с образованием pCR4Blunt-TOPO-crt. Плазмидную ДНК выделяли из клонов ТОРО и последовательность указанных генов определяли с использованием праймеров M13 прямого (SEQ ID NO: 45) и M13 обратного (SEQ ID NO: 46) (смотрите таблицу 5).
Бутирил-CoA-дегидрогеназа (транс-2-еноил-CoA-редуктаза). Ген CAC0462 был синтезирован для повышенной частоты встречаемости кодонов в E.coli в качестве первичного хозяина и в В.subtilis в качестве промежуточного хозяина. Новый ген (CaTER, SEQ ID NO: 76) был синтезирован и клонирован в компании Genscript Corporation (Piscataway, NJ) в вектор pUC57 в виде фрагмента BamHI-SalI и включает RBS.
Альтернативный ген бутирил-CoA-дегидрогеназы из Euglena gracilis (TER, GenBank NO: Q5EU90) был синтезирован для повышенной частоты встречаемости кодонов в E.coli и в Bacillus subtilis. Указанный ген был синтезирован и клонирован в компании GenScript Corporation в вектор pUC57 с образованием pUC57::EgTER. Праймеры N85 и N86 (SED ID NO: 80 и 81 соответственно) вместе с pUC57::EgTER в качестве матричной ДНК обеспечивают ПЦР-фрагмент, включающий 1224 п.н. из ДНК pUC57::EgTER. Последовательность размером 1224 п.н. представлена как SEQ ID NO: 77, где п.н. 1-1218 представляют собой кодирующую последовательность (cds) гена EgTER(opt). EgTER(opt) представляет собой кодон-оптимизированный ген TER, в котором отсутствует нормальная митохондриальная предпоследовательность, чтобы быть функциональным в E.coli (Hoffmeister et al., J. Biol. Chem. 280: 4329 (2005)).
EgTER(opt) был клонирован в pCR4Blunt-TOPO, и его последовательность была подтверждена с использованием праймеров M13 прямого (SEQ ID NO: 45) и M13 обратного (SEQ ID NO: 46). Дополнительные праймеры для секвенирования N62SeqF2 (SEQ ID NO: 114), N62SeqF3 (SEQ ID NO: 115), N62SeqF4 (SEQ ID NO: 116), N63SeqR1 (SEQ ID NO: 117), N63SeqR2 (SEQ ID NO: 118), N63SeqR3 (SEQ ID NO: 119) и N63SeqR4 (SEQ ID NO: 120) были необходимы для полного секвенирования ПЦР-продукта. Последовательность EgTER(opt) размером 1,2 т.п.н. затем была вырезана с помощью HincII и PmeI и была клонирована в pET23+ (Novagen), линеаризированный с помощью HincII. Расположение гена EgTER(opt) по отношению к промотору было подтверждено посредством ПЦР-скрининга колоний с использованием праймеров T7Primer и N63SeqR2 (SEQ ID NO: 82 и 118 соответственно). Полученная плазмида, pET23+::EgTER(opt), была трансформирована в BL21 (DE3) (Novagen) для исследований экспрессии.
Анализ активности транс-2-еноил-CoA-редуктазы проводили, как описано Hoffmeister et al., J. Biol. Chem. 280: 4329 (2005). В одном стандартном исследовании специфическая активность белка EgTER(opt) в индуцированной культуре BL21 (DE3)/Pet23+::EgTER(opt) составляла 1,9 мкмоль·мг-1·мин-1 по сравнению с 0,547 мкмоль·мг-1·мин-1 в неиндуцированной культуре.
Затем ген EgTER(opt) был клонирован в вектор pTrc99a под контролем промотора trc. Ген EgTER(opt) был выделен из pET23+::EgTER(opt) в виде фрагмента BamHI/SalI размером 1287 п.н. Вектор pTrc99a размером 4,2 т.п.н. был линеаризирован с помощью BamHI/SalI. Вектор и фрагмент были лигированы с побразованием pTrc99a-EgTER(opt) размером 5,4 т.п.н. Положительные клоны были подтверждены с помощью ПЦР колоний с использованием праймеров Trc99aF и N63SeqR3 (SEQ ID NO: 83 и 119 соответственно) с образованием продукта размером 0,5 т.п.н.
Конструкция плазмиды pTrc99a-E-C-H-T, включающей гены, кодирующие ацетил-CoA-ацетилтрансферазу (thlA), 3-гидроксибутирил-CoA-дегидрогеназу (hbd), кротоназу (crt) и бутирил-CoA-дегидрогеназу (транс-2-еноил-CoA-редуктаза, EgTER(opt)). Для начала конструирования оперона четырех генов, включающего верхний путь биосинтеза (EgTER(opt), crt, hbd и thlA), pCR4Blunt-TOPO-crt была подвергнута расщеплению с помощью EcoRI и BamHI, высвобождая фрагмент crt размером 0,8 т.п.н. Вектор pUC19dSS (описанный в примере 10) также был расщеплен с помощью EcoRI и BamHI, высвобождая фрагмент вектора размером 2,0 т.п.н. Фрагмент crt и фрагмент вектора лигировали вместе с использованием Т4 ДНК-лигазы (New England Biolabs) для образования pUC19dSS-crt. Ген CaTER был встроен в pUC19dSS-crt посредством расщепления pUC57-CaTER с помощью BamHI и SalI, в результате которого высвобождается фрагмент CaTER размером 1,2 т.п.н. pUC19dSS-crt расщепляли с помощью BamHI и SalI и большой фрагмент вектора лигировали с фрагментом CaTER, образуя pUC19dSS-crt-CaTER. Для завершения оперона были лигированы фрагмент SalI и SphI размером 884 п.н. из pCR4Blunt-TOPO-hbd, thlA фрагмент SphI и PstI размером 1,2 т.п.н. из pCR4Blunt-TOPO-thlA и большой фрагмент после расщепления pUC19dSS-crt-CaTER с помощью SalI и PstI. Продукт указанного трехстороннего лигирования называется pUC19dSS-crt-CaTER-hbd-thlA или pUC19dss::Operon1.
Более высокая активность бутирил-CoA-дегидрогеназы была получена от pTrc99a-EgTER(opt) по сравнению с конструкциями CaTER, следовательно, был сконструирован оперон, полученный из pTrc99a-EgTER(opt). Ген CaTER был удален из pUC19dss::Operon1 посредством обработки с помощью BamHI/SalI и очистки посредством электрофореза в геле фрагмента вектора размером 5327 п.н. Указанный вектор был обработан фрагментом Кленова и повторно лигирован с образованием pUC19dss::Operon 1 ΔCaTer. Фрагмент crt-hbd-thlA (C-H-T) размером 2934 п.н. затем был выделен в виде фрагмента EcoRI/PstI из pUC19dss:Operon 1 ΔCaTer. Фрагмент C-H-T обрабатывали фрагментом Кленова для затупления концов. Вектор pTrc99a-EgTER(opt) расщепляли с помощью SalI и концы обрабатывали фрагментом Кленова. Вектор с тупыми концами и фрагмент с тупыми концами С-Н-Т были лигированы для образования pTrc99a-E-C-H-T. ПЦР колоний проводили с праймерами N62SeqF4 и N5SeqF4 (SEQ ID NO: 116 и 84 соответственно) для подтверждения ориентации указанной вставки.
Конструкция плазмид pBHR T7-ald и pBHR-Ptrc-ald(opt), включающих гены, кодирующие бутиральдегиддегидрогеназу (ald и ald(opt)). Оперон PT7-ald был субклонирован из pDEST14-ald (пример 6) в плазмиду широкого круга хозяев pBHR1 (MoBitec, Goettingen, Germany) для образования pBHR1 PT7-ald. Плазмида pBHR1 сходна с плазмидами pUC19 или pBR322, следовательно, pBHR1 PT7-ald можно использовать в сочетании с производными pUC19 или pBR322, несущими оперон верхнего пути для получения 1-бутанола в E.coli. Плазмиду pDEST14-ald расщепляли с помощью BglII и обрабатывали фрагментом Кленова ДНК-полимеразы для образования тупых концов. Затем указанную плазмиду расщепляли с помощью EcoRI и фрагмент PT7-ald размером 2245 п.н. очищали посредством электрофореза в геле. Плазмиду pBHR1 расщепляли с помощью ScaI и EcoRI и фрагмент размером 4883 п.н. очищали посредством электрофореза в геле. Фрагмент PT7-ald лигировали с вектором pBHR1, образуя pBHR T7-ald. ПЦР-амплификция трансформантов с праймерами T-ald(BamHI) и B-ald(EgTER) (SEQ ID NO: 85 и 86 соответственно) подтвердила предполагаемый ПЦР-продукт размером 1,4 т.п.н. Рестрикционное картирование клонов pBHR T7-ald с использованием EcoRI и DrdI подтвердило предполагаемые фрагменты размерами 4757 и 2405 п.н.
Для проведения анализа активности бутиральдегиддегидрогеназы плазмида pBHR T7-ald была трансформирована в клетках BL21Star™(DE3) (Invitrogen), и экспрессию с промотора Т7 индуцировали посредством добавления L-арабинозы, как описано в примере 1. Активнность ацилирующей альдегиддегирогеназы определяли посредством контроля образования NADH, что измерено по повышению оптической плотности при 340 нм, как описано в примере 6.
Альтернативная последовательность ДНК для гена ald из Clostridium beijerinckii ATCC 35702 была синтезирована (оптимизация частоты встречаемости кодонов в E.coli и Bacillus subtilis) и клонирована в pUC57 в компании GenScript Corporation (Piscaway, NJ) с образованием плазмиды pUC57-ald(opt). Плазмиду pUC57-ald(opt) расщепляли с помощью SacI и SalI для высвобождения фрагмента размером 1498 п.н., уже включающего кодон-оптимизированный ген ald(opt) и RBS для E.coli. Последовательность фрагмента размером 1498 п.н. представлена как SEQ ID NO: 78.
pTrc99a расщепляли с помощью SacI и SalI, образуя фрагмент вектора размером 4153 п.н., который лигировали с фрагментом ald(opt) размером 1498 п.н., для получения pTrc-ald(opt). Экспрессия синтетического гена ald(opt) находится под контролем промотора Ptrc, индуцируемого IPTG.
Оперон Ptrc-ald(opt) был субклонирован в плазмиду широкого спектра хозяев pBHR1 (MoBitec) затем, чтобы быть сравнимым с плазмидой верхнего пути, описанной выше. Фрагмент Ptrc-ald(opt) был ПЦР-амплифицирован из pTrc99A::ald(opt) с T-Ptrc(BspEI) и B-aldopt(ScaI) (SEQ ID NO: 87 и 88 соответственно), включающие сайты рестрикции для BspEI и ScaI в пределах соответствующих праймеров. ПЦР-продукт расщепляли с помощью BspEI и ScaI. Плазмиду pBHR1 расщепляли с помощью SсaI и BspEI и фрагмент размером 4883 п.н. очищали с посредством электрофореза в геле. Фрагмент Ptrc-ald(opt) лигировали с вектором pBHR1 с образованием pBHR-PcatPtrc-ald(opt). Рестрикционное картирование клонов pBHR-PcatPtrc-ald(opt) с помощью ScaI и BspEI подтвердило предполагаемые фрагменты размерами 4883 и 1704 п.н. Для удаления данной области произведенного плазмидой промотора cat (Pсat) плазмиду pBHR-PcatPtrc-ald(opt) расщепляли с помощью BspEI и AatII, и фрагмент размером 6172 п.н. был очищен посредством электрофореза в геле. T-BspEIAatII и B-BspEIAatII (SEQ ID NO: 89 и 90 соответственно) смешивали в растворе, включающем 50 мM NaCl, 10 мM Трис-HCl и 10 мM MgCl2 (pH 7,9) до конечной концентрации 100 мкM, гибридизовали посредством инкубирования при 75°С в течение 5 минут и медленно охлаждали до комнатной температуры. Гибридизованные олигонуклеотиды лигировали с фрагментом размером 6172 п.н. с образованием pBHR-Ptrc-ald(opt).
Конструкция штаммов E.coli, экспрессирующих бутанолдегидрогеназу (vqhD). E.coli включает нативный ген (yqhD), который установлен как 1,3-пропандиолдегидрогеназа (патент США № 6514733). Ген yqhD имеет 40% идентичности с геном adhB Clostridium, предполагаемой бутанолдегилрогеназой, зависящей от NADH. Ген yqhD был помещен под контроль промотора конститутивной экспрессии варианта изомеразы глюкозы 1.6GI (SEQ ID NO:91) в штамме E.coli MG1655 1.6yqhD::Cm (WO 2004/033646) посредством технологии λ Red (Datsenko и Wanner, Proc. Natl. Acad. Sci. U.S.A. 97: 6640 (2000)). Схожим образом указанный нативный промотор был заменен промотором 1.5GI (WO 2003/089621) (SEQ ID NO: 92) с образованием штамма MG1655 1.5GI-yqhD::Cm, следовательно, замещая промотор 1.6GI из MG1655 1.6yqhD::Cm на промотор 1.5GI.
Лизат P1 был получен из MG1655 1.5GI yqhD::Cm, и данную кассету перемещали в экспрессирующие штаммы, MG1655 (DE3), полученные из штамма E.coli MG1655 с помощью набора для лизогенизации лямбда DE3 (Invitrogen), и BL21 (DE3) (Invitrogen) с образованием MG1655 (DE3) 1.5GI-yqhD::Cm и BL21 (DE3) 1.5GI-yqhD::Cm соответственно.
Демонстрация получения 1-бутанола из рекомбинантной E.coli. Штамм E.coli MG1655 (DE3) 1.5GI-yqhD::Cm трансформировали с использованием плазмид pTrc99a-E-C-H-T и pBHR T7-ald для получения штамма MG1655 (DE3) 1.5GI-yqhD::Cm/pTrc99a-E-C-H-T/pBHR T7-ald. Два независимых изолята изначально выращивали в среде LB, содержащей 50 мкг/мл канамицина и 100 мкг/мл карбенициллина. Указанные клетки использовали для высевания во встряхиваемые колбы (общий объем приблизительно 175 мл), содержащие 15, 50 и 150 мл среды ТМ3а/глюкоза (с соответствующими антибиотиками) для моделирования условий с высоким, средним и низким содержаниями кислорода соответственно. Среда TM3a/глюкоза содержала (на литр): 10 г глюкозы, 13,6 г KH2PO4, 2,0 г моногидрата лимонной кислоты, 3,0 г (NH4)2SO4, 2,0 г MgSO4·7H2O, 0,2 г CaCl2·2H2O, 0,33 г двойной соли лимоннокислого железа, 1,0 мг тиамин·HCl, 0,50 г дрожжевого экстракта и 10 мл раствора микроэлементов, значение pH доведено до 6,8 с помощью NH4OH. Указанный раствор микроэлементов содержал: лимонная кислота·H2O (4,0 г/л), MnSO4·H2O (3,0 г/л), NaCl (1,0 г/л), FeSO4·7H2O (0,10 г/л), CoCl2·6H2O (0,10 г/л), ZnSO4·7H2O (0,10 г/л), CuSO4·5H2O (0,010 г/л), H3BO3 (0,010 г/л) и Na2MoO4·2H2O (0,010 г/л). Флаконы инокулировали исходно при OD600≤0,01 единицы и инкубировали при 34°C со встряхиванием при 300 об/мин. Флаконы, содержащие 15 и 50 мл среды, были закрыты крышками с отверстиями; флаконы, содержащие 150 мл, были закрыты крышками без отверстий для сведения к минимуму воздухообмена. Добавляли IPTG до конечной концентрации 0,04 мМ; во время добавления во флаконах OD600было ≥0,4 единицы.
Приблизительно через 15 часов после индукции аликвоту указанной среды анализировали посредством ВЭЖХ (колонка Shodex Sugar SH1011) с определением коэффициента преломления (RI) и посредством ГХ (колонка Varian CP-WAX 58(FFAP)СВ, 25 м × внутренний диаметр 0,25 мм, толщина пленки 0,2 мкм) с определением плазменной ионизации (FID), для содержания 1-бутанола, как описано в разделе «Общие методы». Результаты по определению 1-бутанола представлены в таблице 6.
Два отдельных изолята MG1655 (DE3) 1.5GI-yqhD::Cm/pTrc99a-E-C-H-T/pBHR T7-ald были протестированы на продуцирование 1-бутанола в идентичной манере за исключением того, что в среде содержалось 5 г/л дрожжевого экстракта. Результаты представлены в таблице 7.
Штамм E.coli BL21 (DE3) 1.5GI-yqhD::Cm был трансформирован с помощью плазмид pTrc99a-E-C-H-T и pBHR T7-ald для получения штамма BL21 (DE3) 1.5GI-yqhD::Cm/pTrc99a-E-C-H-T/pBHR T7-ald. Два независимых изолята были протестированы на продуцирование 1-бутанола точно так же, как описано выше. Результаты представлены в таблицах 8 и 9.
Штамм E.coli MG1655 1.5GI-yqhD::Cm был трансформирован с помощью плазмид pTrc99a-E-C-H-T и pBHR-Ptrc-ald(opt) для получения штамма MG1655 1.5GI-yqhD::Cm/pTrc99a-E-C-H-T/pBHR-Ptrc-ald(opt). Два изолята изначально выращивали в среде LB, включающей 50 мкг/мл канамицина и 100 мкг/мл карбенициллина. Указанные клетки использовали для высевания во встряхиваемые колбы (общий объем приблизительно 175 мл), содержащие 50 и 150 мл среды ТМ3а/глюкоза (с соответствующими антибиотиками). Флаконы инокулировали исходно при OD550≤0,04 единицы и инкубировали, как описано выше, с и без индукции. Добавляли IPTG до конечной концентрации 0,04 мМ; во время добавления во флаконах OD550было от 0,6 до 1,2 единицы. В указанном случае индукция не была необходимой для экспрессии гена пути 1-бутанола из-за неплотности индуцибельных промоторов IPTG и конститутивного характера промотора 1.5GI; однако индукция обеспечивала более широкий диапазон экспрессии.
Приблизительно через 15 часов после индукции, аликвота среды была проанализирована на содержание 1-бутанола посредством ГХ с определением плазменной ионизации, как описано выше. Результаты представлены в таблице 10. Для рекомбинантных штаммов E.coli 1-бутанол продуцировался во всех случаях; в отдельных экспериментах показано, что штаммы E.coli дикого типа не продуцировали определеяемого количества 1-бутанола (данные не представлены).
ПРИМЕР 14
Получение 1-бутанола из глюкозы с помощью рекомбинантного B.subtilis
В настоящем примере описывается продуцирование 1-бутанола в Bacillus subtilis. Шесть генов биосинтетического пути 1-бутанола, кодирующие шесть ферментативных действий, были распределены в два оперона для экспрессии. Первые три гена указанного пути биосинтеза (thl, hbd и crt) были интегрированы в хромосому Bacillus subtilis BE1010 (Payne и Jackson, J. Bacteriol. 173: 2278-2282 (1991)). Последние три гена (EgTER, ald и bdhB) были клонированы в экспрессирующую плазимиду и трансформированы в штамме Bacillus, несущем интегрированные гены 1-бутанола.
Если в тексте не указано иначе, праймеры для клонирования, описанные в настоящем примере, представлены их SEQ ID NO: в таблице 4, а праймеры для секвенирования и ПЦР-скрининга представлены их SEQ ID NO: в таблице 5.
Интеграционная плазмида. Плазмида pFP988 представляет собой интеграционный вектор Bacillus, который содержит репликон E.coli из pBR322, маркер к антибиотику ампициллин для селекции в E.coli и два фрагмента гомологии к гену sacB в хромосоме Bacillus, что контролирует интегрирование указанного вектора и интрона посредством гомологичной рекомбинации. Между гомологичными областями sacB находится промотор Pamy и сигнальная последовательность, которые могут направлять синтез и секрецию клонированного гена, His-Tag и эритромицин в качестве селектируемого маркера для Bacillus. Промотор Pamy и сигнальная последовательность происходят из альфа-амилазы Bacillus amyloliquefaciens. Указанная промоторная область также включает последовательность lacO для регуляции экспрессии посредством репрессорного белка lacl. Последовательность pFP988 (6509 п.н.) представлена как SEQ ID NO: 79.
Поскольку указанные гены пути биосинтеза 1-бутанола подлежат экспрессии в цитоплазме, сигнальная последовательность амилазы была удалена. Плазмиду pFP988 амплифицировали с использованием праймеров Pamy/lacO F и Pamy/lacO R с образованием продукта размером 317 п.н. (0,3 т.п.н.), содержащего промотор Pamy/lacO. На 5'-конце праймер Pamy/lacO F содержит сайт рестрикции для BsrGI, за которым следует сайт для EcoRI. На 5'-конце праймер Pamy/lacO R содержит сайт рестрикции для BsrGI, за которым следует сайт для PmeI. ПЦР-продукт был клонирован ТОРО в pCR4Blunt-TOPO с образованием pCR4Blunt-TOPO-Pamy/lacO. Плазмидную ДНК выделяли из культур, выращиваемых в течение ночи, и проводили секвенирование с использованием праймеров M13 прямого и M13 обратного (SEQ ID NO: 45 и SEQ ID NO: 46 соответственно) для подтверждения того, что никакая мутация не была введена в промотор. Клон pCR4Blunt-TOPO-Pamy/lacO расщепляли с помощью BsrGI и фрагмент размером 0,3 т.п.н. очищали посредством электрофореза в геле. Вектор pFP988 расщепляли с помощью BsrGI, что приводит к делеции 11 п.н. из 5' гомологичной области sacB и удалению промотора Pamy/lacO и сигнальной последовательности и His-Tag. Вектор, расщепленный с помощью BsrGI, размером 6 т.п.н. был очищен посредством электрофореза в геле и лигирован со вставкой Pamy/lacO BsrGI. Проводили скрининг полученных плазмид с помощью праймеров Pamy SeqF2 и Pamy SeqR для определения ориентации указанного промотора. Надлежащий клон восстанавливал промотор Pamy/lacO в его изначальной ориентации и был назван pFP988Dss.
Кассета с генами thl-crt была сконструирована посредством SOE (сплайсинг посредством перекрывающихся вставок). Указанные гены были амплифицированы с использованием в качестве матрицы pUC19dss::Operon1. Праймерами к гену thl были Top TF и Bot TR, ампллифицировали продукт размером 0,9 т.п.н. Праймерами к гену crt были Top CF и Bot CR, амплифицировали продукт размером 1,3 т.п.н. Указанные два гена соединяли посредством SOE с помощью ПЦР-амплификации с использованием праймеров Top TF и Bot CR с образованием продукта размером 2,1 т.п.н., который был клонирован ТОРО в pCR4Blunt-TOPO, образуя pCR4Blunt-TOPO-T-C. Клоны подвергали секвенированию для подтверждения указанной последовательности. Плазмиду pCR4Blunt-TOPO-T-C расщепляли с помощью BstEII и PmeI с высвобождением фрагмента размером 2,1 т.п.н., который был очищен посредством электрофореза в геле. Указанную вставку обрабатывали фрагментом Кленова полимеразы для затупления сайта для BstEII. Вектор pFP988Dss расщепляли с помощью PmeI и обрабатывали щелочной фосфтазой кишечника теленка (New England BioLabs) для предотвращения самозамыкания. Фрагмент thl-crt размером 2,1 т.п.н. и расщепленный вектор pFP988Dss лигировали и трансформировали в клетках Top10 E.coli. Проводили скрининг трансформантов посредством ПЦР-амплификации с использованием Pamy SeqF2 и N7SeqR2 для продукта 0,7 т.п.н., надлежащий продукт назван pFP988Dss-T-C.
Конструкция кассеты thl-crt создала уникальные сайты для SalI и SpeI между указанными двумя генами. Для добавления гена hbd в указанную кассету ген hbd был субклонирован из pCR4Blunt-TOPO-hbd в виде фрагмента SalI/SpeI размером 0,9 т.п.н. Вектор pFP988Dss-T-C был расщеплен с помощью SalI и SpeI, и фрагмент вектора размером 8 т.п.н был очищен посредством электрофореза в геле. Указанный вектор и вставка hbd были лигированы и трансформированы в клетках Тор10 E.coli. Проводили скрининг трансформантов посредством ПЦР-амплификации с использованием праймеров Pamy SeqF и N3SeqF3 для фрагмента размером 0,3 т.п.н. Полученную плазмиду назвали pFP988Dss-T-H-C.
Затем промотор Pamy был заменен на промотор Pspac из плазмиды pMUTIN4 (Vagner et al., Microbiol. 144: 3097-3104 (1998)). Промотор Pspac был амплифицирован из pMUTIN4 с помощью праймеров Spac F и Spac R в виде продукта размером 0,4 т.п.н. и клонирован ТОРО в pCR4Blunt-TOPO. Проводили скрининг трансформантов посредством ПЦР-амплификации с использованием праймеров M13 прямого и M13 обратного на наличие вставки размером 0,5 т.п.н. Положительные клоны были подвергнуты секвенированию с теми же праймерами. Плазмиду pCR4Blunt-TOPO-Pspac расщепляли с помощью SmaI и XhoI, и фрагмент размером 0,3 т.п.н. был очищен посредством электрофореза в геле. Вектор pFP988Dss-T-H-C расщепляли с помощью SmaI и XhoI, и вектор размером 9 т.п.н. был выделен посредством электрофореза в геле. Указанный расщепленный вектор и вставка Pspac были лигированы и трансформированы в клетках Тор10 E.coli. Проводили скрининг трансформантов посредством ПЦР-амплификации с использованием праймеров SpacF Seq и N7SeqR2. Положительные клоны дают продукт размером 0,7 т.п.н. Плазмидную ДНК выделяли из положительных клонов и проводили дополнительный скрининг посредством ПЦР-амплификации с использованием праймеров SpacF Seq и N3SeqF2. Положительные клоны дают ПЦР-продукт размером 3 т.п.н. и названы pFP988DssPspac-T-H-C.
Интегрирование в B.subtilis BE1010 для образования B.subtilis ΔsacB::T-H-C::erm #28, включающей экзогенные гены thl, hbd и crt. Компетентные клетки B.subtilis BE1010 были получены, как описано Doyle et al., J. Bacteriol. 144: 957-966 (1980). Компетентные клетки были собраны посредством центрифугирования, и дебрисы были повторно суспендированы в малом объеме указанного клеточного супернатанта. К 1 объему компетентных клеток добавляли 2 объема среды SPII-EGTA (Methods for General and Molecular Bacteriology, P. Gerhardt et al., Eds, American Society for Microbiology, Washington, DC (1994)). Аликвоты клеток по 0,3 мл были помещены в пробирки, и в указанные пробирки добавляли плазмиду pFP988DssPspac-T-H-C. Клетки инкубировали в течение 30 минут при 37°С со встряхиванием, после чего в каждую пробирку добавляли 0,1 мл 10% дрожжевого экстракта и клетки далее инкубировали в течение 60 минут. Трансформанты высевали на планшеты для селекции на LB эритромицин с использованием метода двойного агарового покрытия (Methods for General and Molecular Bacteriology, выше). Изначально проводили скрининг трансформантов посредством ПЦР-амплификации с праймерами Pamy SeqF и N5SeqF3. Положительные клоны, которые давали ожидаемый ПЦР-продукт размером 2 т.п.н., были дополнительно скринированы посредством ПЦР-амплификации. Если вставка указанной кассеты внутрь хромосомы происходила в результате события двойного кроссинговера, тогда с парой праймеров sacB Up и N7SeqR2 и парой праймеров sacB Dn и N4SeqR3 будет амплифицироваться продукт размером 1,7 т.п.н. и 2,7 т.п.н. соответственно. Положительные клоны были идентифицированы и названы B.subtilis ΔsacB::T-H-C::erm #28.
Экспрессия генов EgTER, ald и bdhB плазмидой. Три оставшихся гена 1-бутанола были экспрессированы плазмидой pHT01 (MoBitec). Плазмида pHT01 представляет собой «челночный» вектор Bacillus-E.coli, который реплицируется посредством тета-механизма. Клонированные белки экспрессируются из промотора GroEL, слитого с последовательностью lacO. В направлении 5'-3' от lacO находится эффективный RBS из гена gsiB, за которым следует MCS. Ген ald был амплифицирован посредством ПЦР с помощью праймеров AF BamHI и AR Aat2, используя в качестве матрицы pUC19dHS-ald-bdhB (описана в примере 9), с образованием продукта размером 1,4 т.п.н. Продукт был клонирован ТОРО в pCR4-TOPO и трансформирован в клетках Top10 E.coli. Проводили скрининг трансформантов с использованием праймеров M13 прямого и M13 обратного. Положительные клоны давали продукт размером 1,6 т.п.н. Клоны подвергали секвенированию с использованием праймеров M13 прямого и M13 обратного, N31SeqF2, N31SeqF3, N32SeqR2, N32SeqR3 и N32SeqR4. Указанная плазмида названа pCR4TOPO-B/A-ald.
Вектор pHT01 и плазмиду pCR4TOPO-B/A-ald расщепляли с помощью BamHI и AatII. Фрагмент вектора размером 7,9 т.п.н. и фрагмент ald размером 1,4 т.п.н. лигировали вместе для создания pHT01-ald. Указанную сшивку трансформировали в клетках Тор10 E.coli и проводили скрининг трансформантов на продукт размером 1,3 т.п.н. посредством ПЦР-амплификации с помощью праймеров N31SeqF1 и HT R.
Для добавления двух последних этапов пути биосинтеза к вектору pHT01 были спланированы две системы клонирования. Для обеих систем гены EgTER и bdhB были амплифицированы вместе посредством SOE. Затем фрагмент EgTER-bdh был либо клонирован в pHT01-ald с образованием pHT01-ald-EB, либо клонирован в pCR4-TOPO-B/A-ald с образованием pCR4-TOPO-ald-EB. Фрагмент ald-EgTer-bdhB из вектора TOPO затем был клонирован в pHT01 с образованием pHT01-AEB.
Фрагмент EgTER-bdhB был амплифицирован посредством ПЦР с помощью праймеров прямого 1 (E) и обратного 2 (B) с использованием матричной ДНК, представленной как SEQ ID NO: 208. Полученный ПЦР-продукт размером 2,5 т.п.н. был клонирован ТОРО в pCR4Blunt-TOPO с образованием pCR4Blunt-TOPO-E-B. Реакция TOPO была трансформирована в клетках ТОР10 E.coli. Проводили скрининг колоний посредством ПЦР-амплификации с использованием праймеров M13 прямого и M13 обратного. Положительные клоны давали продукт размером 2,6 т.п.н. Клоны pCR4Blunt-TOPO-E-B подвергали секвенированию с использованием праймеров M13 прямого и обратного, N62SeqF2, N62SeqF3, N62SeqF4, N63SeqR1, N63SeqR2, N63SeqR3, N11SeqF1 и N11SeqF2, N12SeqR1 и N12SeqR2.
Плазмиду pCR4Blunt-TOPO-E-B расщепляли с помощью HpaI и AatII для высвобождения фрагмента размером 2,4 т.п.н. Фрагмент E-B обрабатывали фрагментом Кленова полимеразы для затупления концов и затем очищали посредством электрофореза в геле. Плазмиду pHT01-ald расщепляли с помощью AatII и обрабатывали фрагментом Кленова полимеразы для затупления концов. Затем вектор обрабатывали щелочной фосфатазой кишечника теленка и очищали посредством электрофореза в геле. Фрагмент E-B лигировали с указанным линиаризированным вектором pHT01-ald, трансформировали в клетках Тор10 E.coli и отбирали на планшетах с LB, содержащих 100 мкг/мл ампициллина. Проводили скрининг трансформантов посредством ПЦР-амплификации с использованием праймеров N3SeqF1 и N63SeqR1 для получения продукта размером 2,4 т.п.н. Полученная плазмида, pHT01-ald-EB, была трансформирована в клетках JM103 штамма recA+ E.coli. Плазмиды, полученные из штаммов recA+, формируют больше мультимеров, чем штаммы recA-. Bacillus subtilis трансформирует более эффективно с плазмидными мультимерами, чем с мономерами (Methods for General and Molecular Bacteriology, выше). Плазмидная ДНК была выделена из JM103 и трансформирована в компетентной В.subtilis ΔsacB::T-H-C::erm #28 с образованием штамма B.subtilis ΔsacB::T-H-C::erm #28/pHT01-ald-EB. Компетентные клетки были получены и трансформированы, как описано ранее. Трансформанты были отобраны на планшетах с LB, содержащих 5 мкг/мл хлорамфеникола, и проводили скрининг на продукт 1,3 т.п.н. посредством ПЦР колоний с использованием праймеров N31SeqF1 и N63SeqR4.
В альтернативной стратегии клонирования pCR4Blunt-TOPO-E-B расщепляли с помощью HpaI и AatII с высвобождением фрагмента размером 2,4 т.п.н., который был очищен посредством электрофореза в геле. Плазмиду pCR4-TOPO-B/A-ald расщепляли с помощью HpaI и AatII, и фрагмент вектора размером 5,4 т.п.н. был очищен посредством электрофореза в геле. Фрагмент вектора из pCR4-TOPO-B/A-ald был лигирован с данным фрагментом HpaI-AatII E-B с образованием pCR4-TOPO-ald-EB. Указанную сшивку трансформировали в клетках Тор10 E.coli и проводили скрининг указанных трансформантов на продукт размером 2,1 т.п.н. посредством ПЦР-амплификации с использованием праймеров N11SeqF2 и N63SeqR4. Плазмиду pCR4-TOPO-ald-EB расщепляли с помощью BamHI и AatII и SphI. Указанное расщепление с помощью BamHI/AatII высвобождает фрагмент ald-EB размером 3,9 т.п.н., который был очищен посредством электрофореза в геле. Целью указанного расщепления с помощью SphI было разрезать оставшийся вектор на более мелкие фрагменты так, чтобы они не могли в геле совместно мигрировать со вставкой ald-EB. Вектор pHT01 расщепляли с помощью BamHI и AatII, и фрагмент вектора размером 7,9 т.п.н. был очищен посредством электрофореза в геле. Указанный вектор и фрагменты вставки ald-EB были лигированы для образования плазмиды pHT01-AEB и трансформированы в клетках Тор10 E.coli. Проводили скрининг колоний на продукт размером 1,5 т.п.н. посредством ПЦР-амплификации с использованием праймеров N62SeqF4 и HT R. Плазмида была получена и трансформирована в JM103. Плазмидная ДНК была выделена из JM103 и трансформирована в компетентных В.subtilis ΔsacB::T-H-C::erm #28 с образованием штамма В.subtilis ΔsacB::T-H-C::erm #28/pHT01-AEB. Компетентные клетки BE1010 были получены и трансформированы, как описано ранее. Проводили скрининг трансформантов Bacillus на продукт размером 1,3 т.п.н. посредством ПЦР-амплификации с использованием праймеров N31SeqF1 и NT63SeqR4.
Демонстрация продуцирования 1-бутанола в рекомбинантном B.subtilis
Три независимых изолята каждого штамма В.subtilis ΔsacB::T-H-C::erm #28/pHT01-ald-EB и B.subtilis ΔsacB::T-H-C::erm #28/pHT01-AEB инокулировали во встряхиваемые колбы (общий объем приблизительно 175 мл), содержащие 15 мл среды. В качестве отрицательного контроля также инокулировали штамм B.subtilis BE1010, не содержащий экзогеннный 1-бутанол, шесть генов пути биосинтеза. Среда содержала (на литр): 10 мл 1 M (NН4)2SO4; 5 мл 1 M калийфосфатного буфера, pH 7,0; 100 мл 1 M буфера MOPS/KOH, pH 7,0; 20 мл 1 M L-глутаминовой кислоты, калиевая соль; 10 г глюкозы; 10 мл 5 г/л каждого из L-метионина, L-триптофана и L-лизина; 0,1 г каждого из дрожжевого экстракта и казаминовых кислот; 20 мл смеси металлов и соответствующие антибиотики (5 мг хлорамфеникола и эритромицина для указанных рекомбинантных штаммов). Смесь металлов содержала 200 мМ MgCl2, 70 мМ CaCl2, 5 мМ MnCl2, 0,1 мМ FeCl3, 0,1 мМ ZnCl2, 0,2 мМ тиамин гидрохлорид, 172 мкМ CuSO4, 253 мкМ CoCl2 и 242 мкМ Na2MоO4. Флаконы засевали при изначальной OD600≤0,1 единицы, плотно закрывали крышками без отверстий и инкубировали при 37°С со встряхиванием приблизительно при 200 об/мин.
Приблизительно через 24 часа после инокуляции аликвоту указанной среды анализировали посредством ВЭЖХ (колонка Shodex Sugar SH1011) с определением коэффициента преломления (RI) и посредством ГХ (колонка Varian CP-WAX 58(FFAP)СВ, 25 × внутренний диаметр 0,25 мм, толщина пленки 0,2 мкм) с определением плазменной ионизации (FID) для содержания 1-бутанола, как описано в разделе «Общие методы». Результаты по определению 1-бутанола представлены в таблице 11.
ПРИМЕР 15
Получение 1-бутанола из глюкозы или сахарозы рекомбинантной E.coli
Для наделения E.coli MG1655 способностью использовать сахарозу в качестве источника углерода и энергии для продуцирования 1-бутанола кластер генов утилизации сахарозы (cscBKA) из плазмиды pScrI (описано далее) был субклонирован в pBHR-Ptrc-ald(opt) (описано в примере 13) в организме. Гены утилизации сахарозы (cscA, cscK и cscB) кодируют гидролазу сахарозы (CscA), представленную как SEQ ID NO: 157, D-фруктокиназу (CscK), представленную как SEQ ID NO: 158, и пермеазу сахарозы (CscB), представленную как SEQ ID NO: 159. Для обеспечения возможности конститутивной экспрессии указанных трех генов с их природных промоторов сахарозо-специфический репрессорный ген, cscR, который регулирует кластер генов, не присутствует в конструкции.
Клонирование и экспрессия кластера генов утилизации сахарозы cscBKA в плазмиде pBHR-Ptrc-ald(opt). Кластер генов утилизации сахарозы cscBKA, представленный как SEQ ID NO: 156, был выделен из геномной ДНК штамма E.coli, утилизирующего сахарозу, происходящего от штамма E.coli ATCC 13281. Геномную ДНК до завершения расщепляли с помощью BamHI и EcoRI. Фрагменты, имеющие средний размер около 4 т.п.н., были изолированы из агарозного геля, лигированы в плазмиду pLitmus28 (New England Biolabs, Beverly, MA), которую затем расщепляли с помощью BamHI и EcoRI. Полученную ДНК трансформировали в ультракомпетентных клетках ТОР10F' E.coli (Invitrogen, Carlsbad, CA). Трансформанты высевали на планшеты с агаровой средой МакКонки, содержащей 1% сахарозу и 100 мкг/мл ампициллина, и проводили скрининг на колонии пурпурного цвета. Плазмидная ДНК была выделена из трансформантов пурпурного цвета, и проводили секвенирование с праймерами M13 прямым (SEQ ID NO: 45), M13 обратным (SEQ ID NO: 46), scr1 (SEQ ID NO: 160), scr2 (SEQ ID NO: 161), scr3 (SEQ ID NO: 162) и scr4 (SEQ ID NO: 163). Плазмида, включающая гены cscB, cscK и cscA (cscBKA), была обозначена как pScr1.
Плазмиду pScrI расщепляли с помощью XhoI и обрабатывали фрагментом Кленова ДНК-полимеразы для образования тупых концов. Затем плазмиду расщепляли с помощью AgeI, и фрагмент кластера генов cscBKA размером 4179 п.н. был очищен посредством электрофореза в геле. Плазмиду pBHR-Ptrc-ald(opt) получали, как описано в примере 13, и расщепляли с помощью AgeI и NaeI. Полученный фрагмент pBHR-Ptrc-ald(opt) размером 6003 п.н. был очищен посредством электрофореза в геле. Фрагмент cscBKA был лигирован с pBHR-Ptrc-ald(opt) с образованием pBHR-Ptrc-ald(opt)-cscAKB. Плазмида pBHR-Ptrc-ald(opt)-cscAKB была трансформирована в электрокомпетентных клетках E.coli NovaXG (Novagen, Madison, WI), и утилизация сахарозы была подтверждена посредством высевания указанных трансформантов в планшеты с агаровой средой МакКонки, содержащей 2% сахарозу и 25 мкг/мл канамицина. В конструкции pBHR-Ptrc-ald(opt)-cscAKB гены утилизации сахарозы были клонированы в 5'-3' направлении от Ptrc-ald(opt) в виде отдельного фрагмента в следующем порядке: cscA, cscK и cscB.
Альтернативно, указанные гены утилизации сахарозы были клонированы в противоположном направлении в pBHR-Ptrc-ald(opt). Плазмиду pBHR-Ptrc-ald(opt) расщепляли с помощью ScaI и AgeI, и фрагмент pBHR-Ptrc-ald(opt) размером 5971 п.н. был очищен посредством электрофореза в геле. Фрагмент cscBKA размером 4179 п.н., полученный, как описано выше, был лигирован с фрагментом pBHR-Ptrc-ald(opt), образуя pBHR-Ptrc-ald(opt)-cscBKA. Плазмида pBHR-Ptrc-ald(opt)-cscBKA была трансформирована в электрокомпетентных клетках E.coli NovaXG (Novagen, Madison, WI), и утилизация сахарозы была подтверждена посредством высевания указанных трансформантов в планшеты с агаровой средой МакКонки, содержащей 2% сахарозу и 25 мкг/мл канамицина. В конструкции pBHR-Ptrc-ald(opt)-cscВКА гены утилизации сахарозы были клонированы в 5'-3' направлении от Ptrc-ald(opt) в виде отдельного фрагмента в следующем порядке: cscВ, cscK и cscА.
Демонстрация получения 1-бутанола из глюкозы или сахарозы с использованием рекомбинантной E.coli. Штамм E.coli MG1655 1.5GI-yqhD::Cm (описанный в примере 13) был трансформирован с помощью плазмид pTrc99a-E-C-H-T (получена, как описано в примере 13) и pBHR-Ptrc-ald(opt)-cscAKB или pBHR-Ptrc-ald(opt)-cscBKA для получения двух штаммов, MG1655 1.5GI-yqhD::Cm/pTrc99a-E-C-H-T/pBHR-Ptrc-ald(opt)-cscAKB #9 и MG1655 1.5GI-yqhD::Cm/pTrc99a-E-C-H-T/pBHR-Ptrc-ald(opt)-cscBKA #1. Посевные культуры указанных двух штаммов были получены посредством выращивания клеток в среде LB, включающей 25 мкг/мл канамицина и 100 мкг/мл карбенициллина. Затем указанные клетки использовали для инокуляции встряхиваемых колб (общий объем приблизительно 175 мл), содержащих 50, 70 и 150 мл среды TM3a/глюкоза (с соответствующими антибиотиками) для моделирования условий с высоким, средним и низким содержаниями кислорода соответственно, как описано в примере 13. Третий штамм, E.coli MG1655/pScr1, выращенный в среде TM3a/глюкоза, содержащей 100 мкг/мл карбенициллина, использовали в качестве отрицательного контроля. Для каждого из указанных штаммов был приготовлен идентичный набор колб со средой TM3a/сахароза (с соответствующими антибиотиками). Среда TM3a/сахароза является идентичной среде TM3a/глюкоза за исключением того, что глюкоза заменена сахарозой (10 г/л). Флаконы инокулировали при начальной OD550≤0,03 единицы и инкубировали, как описано в примере 13. За исключением колб с отрицательным контролем, в указанные колбы добавляли IPTG (конечная концентрация 0,04 мМ), когда в указанных культурах OD550 достигало значения от 0,2 до 1,8 единицы. Клетки собирали, когда OD550 указанных культур увеличивалась по меньшей мере в три раза.
Приблизительно через 24 часа после инокуляции аликвоту указанной среды анализировали посредством ВЭЖХ (колонка Shodex Sugar SH1011) с определением коэффициента преломления (RI) и посредством ГХ (колонка HP-INNOWax, 30 м × внутренний диаметр 0,53 мм, толщина пленки 1 мкм) с определением плазменной ионизации (FID) содержания 1-бутанола, как описано в разделе «Общие методы».
Концентрации 1-бутанола в культурах после выращивания в средах, содержащих глюкозу и сахарозу, представлены в таблице 12 и таблице 13 соответственно. Оба рекомбинантных штамма E.coli, включающие биосинтетический путь 1-бутанола, продуцируют 1-бутанол из глюкозы и сахарозы при всех условиях содержания кислорода, в то время как штамм отрицательного контроля не продуцировал определяемых количеств 1-бутанола.
ПРИМЕР 16
Получение 1-бутанола из сахарозы посредством рекомбинантного В.subtilis
В настоящем примере описывается получение 1-бутанола из сахарозы с использованием рекомбинантного Bacillus subtilis. Два независимых изолята штамма B.subtilis ΔsacB::T-H-C::erm #28/pHT01-ald-EB (пример 14) были проверены на продуцирование 1-бутанола преимущественно, как описано в примере 14.
Указанные штаммы были инокулированы во встряхиваемые колбы (общий объем приблизительно 175 мл), содержащие либо 20 мл, либо 100 мл среды для моделирования условий высокого или низкого содержания кислорода соответственно. Среда А была точно такой же, как описано в примере 14, за исключением того, что глюкоза была заменена сахарозой 5 г/л. Среда В была идентичной среде TM3a/глюкоза, описанной в примере 13, за исключением того, что глюкоза была заменена сахарозой 10 г/л и указанная среда была дополнена (на литр) 10 мл 5 г/л раствора каждого из L-метионина, L-триптофана и L-лизина. Инокуляцию колб производили при начальной OD550≤0,1 единицы, колбы закрывали крышками с отверстиями и проводили инкубирование при 34°С со встряхиванием при 300 об/мин.
Приблизительно через 24 часа после инокуляции проводили анализ аликвоты указанной среды посредством ГХ (колонка HP-INNOWax, 30 м × внутренний диаметр 0,53 мм, толщина пленки 1 мкм) с определением FID содержания 1-бутанола, как описано в разделе «Общие методы». Результаты по определению 1-бутанола представлены в таблице 14. Рекомбинантный штамм Bacillus, включающий биосинтетический путь 1-бутанола, продуцировал определяемые уровни 1-бутанола при условиях высокого и низкого содержаний кислорода в обеих средах.
ПРИМЕР 17
Получение 1-бутанола из глюкозы и сахарозы с использованием рекомбинантного Saccharomyces cerevisiae
В настоящем примере описывается продуцирование 1-бутанола в дрожжевых Saccharomyces cerevisiae. Из шести генов, кодирующих ферменты, катализирующие стадии биосинтетического пути 1-бутанола, пять были клонированы в три схожие дрожжевые 2-микронные (2μ) плазмиды и совместно экспрессировались в Saccharomyces cerevisiae. «Верхний путь биосинтеза» определен как первые три ферментативных этапа, катализируемые ацетил-CoA-ацетилтрансферазой (thlA, тиолаза), 3-гидроксибутирил-CoA-дегидрогеназой (hbd) и кротоназой (crt). Нижний путь биосинтеза определен как четвертый (бутил-CoA-дегидрогеназа, ter) и пятый (бутилальдегиддегидрогеназа, ald) ферментативные этапы указанного пути биосинтеза. Последний ферментативный этап пути биосинтеза 1-бутанола катализируется алкогольдегидрогеназой, которая может кодироваться эндогенными генами дрожжей, например adhI и adhII.
Для экспрессии генов в дрожжах обычно требуется промотор, за которым следует вставка гена, и терминатор транскрипции. Целый ряд конститутивных дрожжевых промоторов использовали при конструировании экспрессионных кассет для генов, кодирующих биосинтетический путь 1-бутанола, в том числе промоторы FBA, GPD и GPM. Некоторые индуцибельные промоторы, например GAL1, GAL10, CUP1, также использовали при конструкции промежуточной плазмиды, но не для окончательной демонстрации штамма. Использовали несколько терминаторов транскрипции, в том числе FBAt, GPDt, GPMt, ERG10t и GAL1t. Гены, кодирующие биосинтетический путь 1-бутанола, сначала были субклонированы в дрожжевую плазмиду, фланкируемую промотором и терминатором, образуя экспрессирующие кассеты для каждого гена. Экспрессирующие кассеты были при желании скомбинированы в единый вектор клонированием посредством восстановления разрывов, как описано далее. Например, указанные три кассеты генов, кодирующих верхний путь биосинтеза, были субклонированы в дрожжевую плазмиду 2μ. Гены ter и ald, каждый по отдельности, были экспрессированы в указанных плазмидах 2μ.
Совместная трансформация всех трех плазмид в единственный дрожжевой штамм приводит к функциональному биосинтетическому пути 1-бутанола. Альтернативно, несколько фрагментов ДНК, кодирующих промоторы, гены и терминаторы, были напрямую скомбинированы в единственный вектор клонированием посредством восстановления разрывов.
Способы конструирования плазмид и штаммов в дрожжевых Saccharomyces cerevisiae. Фундаментальные протоколы молекулярной биологии дрожжей, в том числе трансформация, рост клеток, экспрессия генов, рекомбинация посредством восстановления разрывов и тому подобное, описаны в Methods in Enzymology, Volume 194, Guide to Yeast Genetics and Molecular and Cell Biology (Part A, 2004, Christine Guthrie and Gerald R. Fink (Eds.), Elsevier Academic Press, San Diego, CA).
Плазмидами, используемыми в настоящем примере, были «челночные» векторы E.coli-S.cerevisiae, pRS423, pRS424, pRS425 и pRS426 (американская коллекция типовых культур, Rockville, MD), которые содержат начало репликации E.coli (например, pMB1), начало репликации дрожжей 2μ и маркер для питательной селекции. Маркерами селекции для указанных четырех векторов были His3 (вектор pRS423), Trp1 (вектор pRS424), Leu2 (вектор pRS425) и Ura3 (вектор pRS426). Указанные векторы делают возможным воспроизведение и штаммов E.coli и штаммов дрожжей. Гаплоидный дрожжевой штамм BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) (Research Genetics, Huntsville, AL, также доступен из ATCC 201388) и диплоидный штамм BY4743 (MATa/alpha his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 lys2Δ0/LYS2 MET15/met15Δ0 ura3Δ0/ura3Δ0) (Research Genetics, Huntsville, AL, также доступен из ATCC 201390) использовали в качестве хозяев для клонирования и экспрессии генов. Конструкцию экспрессирующих векторов для генов, кодирующих ферменты биосинтетического пути 1-бутанола, осуществляли либо посредством стандартных технологий молекулярного клонирования в E.coli, либо используя способ рекомбинации посредством восстановления разрывов в дрожжах.
В указанном методе клонирования посредством восстановления разрывов используется высокоэффективная гомологичная рекомбинация в дрожжах. Обычно ДНК дрожжевого вектора расщепляют (например, в ее множественных сайтах клонирования) для образования «двухцепочечных разрывов» в его последовательности. Создан целый ряд представляющих интерес вставок ДНК, которые содержат последовательность ≥21 п.н. с обоих концов 5' и 3', которые последовательно перекрываются друг с другом, и с 5' и 3' концами указанной векторной ДНК. Например, для конструкции дрожжевого экспрессирующего вектора для «Гена Х» дрожжевой промотор и дрожжевой терминатор отобраны для экспрессирующей кассеты. Промотор и терминатор амплифицированы из геномной ДНК дрожжей, и Ген Х либо ПЦР-амплифицирован из его исходного микроорганизма, либо получен из клонирующего вектора, включающего последовательность Гена Х. Существует по меньшей мере 21 п.н. перекрывающаяся последовательность между 5'-концом линеаризированного вектора и промоторной последовательностью, между указанным промотором и Геном Х, между Геном Х и последовательностью терминатора и между терминатором и 3'-концом линеаризованного вектора. «Рваный» вектор и указанные вставки ДНК затем совместно траснформировали в дрожжевом штамме и высевали на планшеты с минимальной селективной средой SD и колонии отбирали для выращивания культур и для минипрепов для плазмидных ДНК. Наличие правильных комбинаций вставок может быть подтверждено посредством картирования с использованием ПЦР. Плазмидная ДНК, выделенная из дрожжей (обычно низкая концентрация), затем может быть трансформирована в штамме E.coli, например ТОР10, с последующим образованием минипрепов и рестрикционным картированием для дополнительного подтверждения конструкции плазмиды. В конце концов, указанная конструкция может быть проверена посредством секвенирования. Дрожжевые трансформанты положительных плазмид выращивали в среде SD для осуществления ферментативного анализа, чтобы охарактеризовать активности ферментов, экспрессированных представляющими интерес генами.
Дрожжевые культуры выращивали в комплексной среде YPD или синтетической минимальной селективной среде, включающей глюкозу (среда SD) и соответствующие смеси соединений, которые делают возможной комплементацию питательных маркеров селекции в указанных плазмидах (Methods in Enzymology, Volume 194, Guide to Yeast Genetics and Molecular and Cell Biology, 2004, Part A, pp.13-15). Сахарным компонентом в указанной селективной среде была 2% глюкоза. Для продуцирования 1-бутанола дрожжевые культуры также выращивали в синтетической минимальной селективной среде с 2% сахарозой (среда SS).
Для анализа ферментативной активности одиночную колонию каждого штамма высевали штрихами на свежие планшеты, содержащие минимальную селективную среду SD, и инкубировали при 30°С в течение 2 дней. Клетки с указанного планшета использовали для инокуляции 20 мл селективной среды SD во встряхиваемые колбы объемом 125 мл и выращивали в течение ночи при 30°С со встряхиванием при 250 об/мин. Измеряли оптическую плотность (OD600) культуры, выращиваемой в течение ночи, указанную культуру разбавляли до OD600=0,1 в 250 мл этой же среды во встряхиваемой колбе объемом 1,0 л и выращивали при 30°С со встряхиванием при 250 об/мин до OD600 от 0,8 до 1,0. Клетки затем были собраны посредством центрифугирования при 2000×g в течение 10 минут и ресуспендированы в 20 мл 50 мМ буфера Трис-HCl, рН 8,5. Анализ ферментативных активностей осуществляли, как описано выше.
Конструкция плазмиды pNY102 для совместной экспрессии генов thlA и hbd. Для совместной экспрессии генов thlA и hbd был сконструирован целый ряд дуальных экспрессирующих векторов. Ген ERG10 Saccharomyces cerevisiae представляет собой функциональный ортолог гена thlA. Изначально дуальный вектор генов ERG10 и hbd был сконструирован с использованием дрожжевого дивергентного дуального промотора GAL1-GAL10, терминатора GAL1 (GAL1t) и терминатора ERG10 (ERG10t). Фрагмент ДНК ген ERG10-ERG10t был ПЦР-амплифицирован из геномной ДНК штамма Saccharomyces cerevisiae BY4743 с использованием праймеров OT731 (SEQ ID NO: 164) и OT732 (SEQ ID NO: 165). Дрожжевой дивергентный промотор GAL1-GAL10 также был амплифицирован посредством ПЦР из геномной ДНК штамма BY4743 с использованием праймеров OT733 (SEQ ID NO: 166) и OT734 (SEQ ID NO: 167). Ген hbd был амплифицирован из плазмиды E.coli pTrc99a-E-C-H-T (описана в примере 13) с использованием праймеров для ПЦР OT735 (SEQ ID NO: 168) и OT736 (SEQ ID NO: 169). GAL1t был амплифицирован из геномной ДНК штамма BY4743 с использованием праймеров OT737 (SEQ ID NO: 170) и OT738 (SEQ ID NO: 171). Четыре ПЦР-фрагмента, erg10-ERG10t, промоторы GAL1-GAL10, hbd и GAL1t, были, таким образом, получены приблизительно с 25 п.н. перекрывающимися последовательностями между каждым смежным ПЦР-фрагментом. Фрагменты GAL1t и ERG10-ERG10t каждый содержал приблизительно 25 п.н. перекрывающиеся последовательности с данным дрожжевым вектором pRS425. Для объединения указанных последовательностей с помощью рекомбинации посредством восстановления разрывов указанные фрагменты ДНК были совместно трансформированы в дрожжевом штамме BY4741 вместе с вектором pRS425, который был расщеплен с помощью ферментов BamHI и HindIII. Колонии были отобраны с планшетов с минимальной средой SD-Leu, и клоны со вставками были определены посредством ПЦР-амплификации. Новая плазмида названа pNY6 (pRS425.ERG10t-erg10-GAL10-GAL1-hbd-GAL1t). Дальнейшее подтверждение было осуществлено посредством рестрикционного картирования.
Дрожжевой штамм BY4741 (pNY6), полученный трансформацией плазмиды pNY6 в штамме BY4741 S.cerevisiae, показал хорошую активность Hbd, но не проявлял тиолазной активности. Вследствие отсутствия тиолазной активности ген ERG10 был заменен геном thlA с помощью рекомбинации посредством восстановления разрывов. Ген thlA был амплифицирован из вектора E.coli pTrc99a-E-C-H-T посредством ПЦР с использованием праймеров OT797 (SEQ ID NO: 172), который добавляет сайт рестрикции для SphI, и OT798 (SEQ ID NO: 173), который добавляет сайт рестрикции для AscI. Плазмиду pNY6 расщепляли с помощью рестрикционных ферментов SphI и PstI, очищали посредством электрофореза в геле и совместно с ПЦР-продуктом thlA трансформировали в дрожжевом штамме BY4741. Из-за 30 п.н. перекрывающихся последовательностей между ПЦР-продуктом thlA и расщепленной pNY6 ген thlA был рекомбинирован в pNY6 между промотором GAL10 и терминатором ERG10t. Это приводит к образованию плазмиды pNY7 (pRS425.ERG10t-thlA-GAL10-GAL1-hbd-GAL1t), которая была проверена посредством ПЦР и рестрикционного картирования.
В последующей стадии клонирования, основанного на рекомбинации посредством восстановления разрывов, промотор GAL10 в pNY7 был заменен промотором CUP1, и промотор GAL1 был заменен активным промотором GPD. Указанная плазмида, pNY10 (pRS425.ERG10t-thlA-CUP1-GPD-hbd-GAL1t), делает возможной экспрессию гена thlA под контролем CUP1, медьсодержаащего индуцибельного промотра, и экспрессию гена hbd под контролем промотора GPD. Последовательность промотора CUP1 была ПЦР-амплифицирована из геномной ДНК дрожжевого штамма BY4743 с использованием штаммов OT806 (SEQ ID NO: 174) и OT807 (SEQ ID NO: 175). Промотор GPD был амплифицирован из геномной ДНК штамма BY4743 с использованием праймеров OT808 (SEQ ID NO: 176) и OT809 (SEQ ID NO: 177). ПЦР-продукты промоторов CUP1 и GPD были объединены с плазмидой pNY7, расщепленной с помощью рестрикционных ферментов NcoI и SphI. На этапе клонирования посредством восстановления разрывов была сконструирована плазмида pNY10, которая была проверена посредством ПЦР и рестрикционного картирования. Дрожжевой штамм BY4741, включающий pNY10, имел активность Hbd, но не имел активность ThlA. Активность Hbd под контролем промотора GPD была значительно улучшенной по сравнению с активностью Hbd, контролируемой промотором GAL1 (1,8 Ед/мг против 0,40 Ед/мг). Анализ посредством секвенирования показал, что ген thlA в pNY10 имел делецию одного основания рядом с 3'-концом, что привело к процессированию белка. Это объясняет отсутствие тиолазной активности в указанном штамме.
Плазмида pNY12 была сконструирована с правильной последовательностью гена thlA. Ген thlA был вырезан из вектора pTrc99a-E-C-H-T посредством расщепления с помощью SphI и AscI. Промотор FBA1 был ПЦР-амплифицирован из геномной ДНК штамма BY4743 с использованием праймеров OT799 (SEQ ID NO: 178) и OT761 (SEQ ID NO: 179) и расщеплен с помощью рестрикционных ферментов SalI и SphI. Фрагмент гена thlA и фрагмент промотора FBA1 были лигированы в плазмиду pNY10 в сайтах для AscI и SalI с образованием плазмиды pNY12 (pRS425.ERG10t-thlA-FBA1), которая была подтверждена посредством рестрикционного картирования. pNY12 была трансформирована в дрожжевом штамме BY4741, и полученный трансформант проявлял активность ThlA 1,66 Ед/мг.
Фрагмент промотор FBA1-ген thlA был повторно субклонирован в pNY10. Вектор pNY10 был разрезан с помощью рестрикционного фермента AscI и лигирован с расщепленным с помощью AscI фрагментом промотор FBA1-ген thlА, выделенным из плазмиды pNY12. Это привело к образованию новой плазмиды с двумя возможными ориентациями вставки. Были отобраны клоны с промоторами FBA1 и GPD, расположенными смежно друг с другом в противоположном направлении, и плазмида, названная pNY102. pNY102 (pRS425.ERG10t-thlA-FBA1-GPD-hbd-GAL1t) была проверена посредством рестрикционного картирования. Штамм DPD5206 был получен посредством трансформирования pNY102 в дрожжевом штамме BY4741. Активность ThlA штамма DPD5206 составляла 1,24 Ед/мг, и активность Hbd составляла 0,76 Ед/мг.
Конструкция плазмиды pNY11 для экспрессии crt. Экспрессирующая кассета для гена crt была сконструирована посредством объединения промотора GPM1, гена crt и терминатора GPM1t в вектор pRS426 с использованием рекомбинации посредством восстановления разрывов в дрожжах. Промотор GPM1 был ПЦР-амплифицирован из геномной ДНК дрожжевого штамма BY4743 с использованием праймеров OT803 (SEQ ID NO: 180) и OT804 (SEQ ID NO: 181). Ген crt был амплифицирован с использованием праймеров для ПЦР OT785 (SEQ ID NO: 182) и OT786 (SEQ ID NO: 183) из плазмиды E.coli pTrc99a-E-C-H-T. Терминатор GPM1t был ПЦР-амплифицирован из геномной ДНК дрожжевого штамма BY4743 с использованием OT787 (SEQ ID NO: 184) и OT805 (SEQ ID NO: 185). Дрожжевой вектор pRS426 был расщеплен с помощью BamHI и HindIII и был очищен посредством электрофореза в геле. Указанная ДНК была совместно трансформирована с ПЦР-продуктами промотра GPM1, гена crt и терминатора GPM1 в компетентных клетках дрожжевого штамма BY4741. Клоны с надлежащими вставками были проверены посредством ПЦР и рестрикционного картирования, и полученный дрожжевой штамм BY4741 (pNY11:pRS426-GPM1-crt-GPM1t) имел активность Crt 85 Ед/мг.
Конструкция плазмиды pNY103 для совместной экспрессии генов thlA, hbd и crt. Для совместной экспрессии ферментов верхнего пути 1-бутанола кассета гена crt из pNY11 была субклонирована в плазмиду pNY102 для создания экспрессирующего вектора hbd, thlA и crt. Фрагмент ДНК размером 2347 п.н., включающий промотор GPM1, ген crt и терминатор GPM1, был вырезан из плазмиды pNY11 с помощью рестрикционных ферментов SacI и NotI и клонирован в вектор pNY102, который был расщеплен с помощью NotI и частично расщеплен с помощью SacI, образуя экспрессирующий вектор pNY103 (pRS425.ERG10t-thlA-FBA1-GPD-hbd-GAL1t-GPM1t-crt-GPM1). После подтверждения расщеплением с помощью HindIII наличия всех трех кассет в pNY103 указанная плазмида была трансформирована в дрожжевых клетках BY4743, и трансформированный дрожжевой штамм назван DPD5200. При выращивании при стандартных условиях DPD5200 проявлял ферментативные активности ThlA, Hbd и Crt, равные 0,49 Ед/мг, 0,21 Ед/мг и 23,0 Ед/мг соответственно.
Конструкция плазмиды pNY8 для экспрессии ald. Кодон-оптимизированный ген, названный tery (SEQ ID NO: 186), кодирующий белок Ter (SEQ ID NO: 187), и кодон-оптимизированный ген, названный aldy (SEQ ID NO: 188), кодирующий белок Ald (SEQ ID NO: 189), были синтезированы с использованием предпочтительных кодонов Saccharomyces cerevisiae. Плазмида pTERy, включающая кодон-оптимизированный ген ter, и pALDy, включающая кодон-оптимизированный ген ald, были созданы с помощью DNA2.0 (Palo Alto, CA).
Для сборки pNY8 (pRS426.GPD-ald-GPDt) три фрагмента вставок, в том числе ПЦР-продукт промотора GPD (синтезированный из геномной ДНК штамма BY4743 с праймерами OT800 (SEQ ID NO: 190) и OT758 (SEQ ID NO: 191)), фрагмент гена aldy, вырезанный из pALDy посредством расщепления с помощью NcoI и SfiI (SEQ ID NO: 188), и ПЦР-продукт терминатора GPD (синтезированный из геномной ДНК штамма BY4743 с праймерами OT754 (SEQ ID NO: 192) и OT755 (SEQ ID NO: 193)), были рекомбинированы с помощью вектора pRS426, расщепленного с помощью BamHI и HindIII, клонированием посредством восстановления разрывов. Трансформационные клоны дрожжей BY4741 были анализированы посредством ПЦР-картирования. Таким образом, сконструированная новая плазмида, pNY8, была дополнительно подтверждена рестрикционнным картированием. Дрожжевые трансформанты BY4741, включающие pNY8, были проанализированы на активность Ald, и специфическая активность для бутирил-СоА была приблизительно 0,07 Ед/мг.
Контрукция плазмид pNY9 и pNY13 для экспрессии ter. Кодон-оптимизированный ген tery был клонирован в вектор pRS426 под контролем промотора FBA1 посредством восстановления разрывов. Промотор FBA1 был ПЦР-амплифицирован из геномной ДНК дрожжевого штамма BY4743 с использованием праймеров OT760 (SEQ ID NO: 194) и OT792 (SEQ ID NO: 195). Ген tery был получен в результате расщепления плазмиды pTERy с помощью рестрикционных ферментов SphI и NotI, что в результате давало фрагмент, обозначенный как SEQ ID NO: 186. ПЦР-фрагмент терминатора FBA1 был образован посредством ПЦР из геномной ДНК дрожжевого штамма BY4743 с использованием праймеров OT791 (SEQ ID NO: 196) и OT765 (SEQ ID NO: 197). Три фрагмента ДНК, промотор FBA1, ген ter и терминатор FBA1, были объединены с вектором pRS426, расщепленным с помощью BamHI и HindIII, и трансформированы в дрожжевом штамме BY4741 с использованием рекомбинации посредством восстановления разрывов. Полученная плазмида, pNY9 (pRS426-FBA1-tery-FBA1t), была подтверждена посредством ПЦР-картирования, а также рестрикционным расщеплением. Дрожжевой трансформант BY4741 плазмиды pNY9 продуцирует активность Ter, равную 0,26 Ед/мг.
Для создания финального штамма биосинтетического пути 1-бутанола было необходимо сконструировать дрожжевой экспрессирующий штамм, который включает несколько плазмид, каждая с уникальным питательным маркером селекции. Поскольку исходный вектор pRS426 содержал маркер селекции Ura, экспрессирующая кассета ter была субклонирована в вектор pRS423, который содержит маркер His3. Фрагмент размером 3,2 т.п.н., включающий кассету FBA1-tery-FBA1t, был выделен из плазмиды pNY9 посредством расщепления с помощью рестрикционных ферментов SacI и XhoI и лигирован в вектор pRS423, который был разрезан с помощью этих же двух ферментов. Новая плазмида, pNY13 (pRS423-FBA1-tery-FBA1t), была картирована посредством рестрикционного расщепления. pNY13 была трансформрована в штамме BY4741, и трансформант был культивирован в среде SD-His, производя штамм с активностью Ter, равной 0,19 Ед/мг.
Конструкция дрожжевого штамма, содержащего гены биосинтетического пути 1-бутанола, для демонстрации получения 1-бутанола. Как описано выше, дрожжевой штамм DPD5200 был сконструирован посредством трансформации плазмиды pNY103 в штамме S.cerevisiae BY4743, которая обеспечивает совместную экспрессию генов thlA, hbd и crt. Компетентные клетки дрожжевого штамма DPD5200 были получены, как описано выше, и плазмиды pNY8 и pNY13 были совместно трансформированы в DPD5200 с образованием штамма DPD5213. DPD5213 делает возможной одновременную конститутивную экспрессию пяти генов биосинтетического пути 1-бутанола, thlA, hbd, crt, ter и ald. В качестве отрицательного контроля использовали штамм DPD5212 (штамм S.cerevisiae BY4743, трансформированный с пустыми плазмидами, pRS425 и pRS426). Четыре независимых изолята штамма DPD5213 выращивали в минимальной селективной среде SD-Ura,-Leu,-His в присутствии либо 2% глюкозы, либо 2% сахарозы для обеспечения комплементации роста всех трех плазмид. Единственный изолят штамма DPD5212 аналогично выращивали в соответствующей среде.
Для демонстрации продуцирования 1-бутанола аэробными культурами одиночную колонию каждого штамма высевали штрихами на планшет со свежим агаром, содержащим минимальную селективную среду для выращивания SD (содержащую 2% глюкозу) или минимальную селективную среду для выращивания SS (содержащую 2% сахарозу), и инкубировали при 30°С в течение 2 дней. Клетки с указанных планшетов использовали для инокулирования 20 мл минимальной селективной среды (либо SD, либо SS) в пластиковых встряхиваемых колбах объемом 125 мл и выращивали в течение ночи при 30°С со встряхиванием при 250 об/мин. Измеряли оптическую плотность (OD600) указанной культуры, выращиваемой в течение ночи, культуру разбавляли до OD600, равной 0,1, в 25 мл той же самой среды во встряхиваемой колбе объемом 125 мл и выращивали при 30°С со встряхиванием при 250 об/мин.
Аликвоты указанной культуры были отобраны через 24 часа и 48 часов для анализа продуцирования 1-бутанола посредством ГХ (колонка HP-INNOWax, 30 м × внутренний диаметр 0,53 мм, толщина пленки 1 мкм) с определением FID, как описано в разделе «Общие методы». Результаты анализа посредством ГХ представлены в таблице 15.
ПРИМЕР 18 (предсказательный)
Экспрессия биосинтетического пути 1-бутанола в Lactobacillus plantarum
Целью настоящего предсказательного примера является описание того, как экспрессировать биосинтетический путь 1-бутанола в Lactobacillus plantarum. Шесть генов пути биосинтеза 1-бутанола, кодирующие активности шести ферментов, разделены на два оперона для экспрессии. Первые три гена указанного пути биосинтеза (thl, hbd и crt, кодирующие ферменты ацетил-CoA-ацетилтрансфераза, 3-гидроксибутирил-CoA-дегидрогеназа и кротоназа соответственно) интегрированы в хромосому Lactobacillus plantarum посредством гомологичной рекомбинации с использованием способа, описанного Hols et al. (Appl. Environ. Microbiol. 60: 1401-1413 (1994)). Последние три гена (EgTER, ald и bdhB, кодирующие ферменты бутирил-CoA-дегидрогеназа, бутиральдегиддегидрогеназа и бутанолдегидрогеназа соответственно) клонированы в экспрессирующую плазмиду и трансформированы в штамме Lactobacillus, несущем интегрированные гены верхнего пути 1-бутанола. Lactobacillus выращивали в среде MRS (Difco Laboratories, Detroit, MI) при 37°С. Хромосомная ДНК выделена из Lactobacillus plantarum, как описано Moreira et al. (BMC Microbiol. 5: 15 (2005)).
Интегрирование. Кассета thl-hbd-crt под контролем синтетического промотора P11 (Rud et al., Microbiology 152:1011-1019 (2006)) интегрирована в хромосому Lactobacillus plantarum ATCC BAA-793 (NCIMB 8826) в локус ldhL1 посредством гомологичной рекомбинации. Для конструирования интегрирующего целевого вектора ldhL фрагмент ДНК из Lactobacillus plantarum (Genbank NC_004567) с гомологией к ldhL ПЦР-амплифицирован с использованием праймеров LDH EcoRV F (SEQ ID NO: 198) и LDH AatIIR (SEQ ID NO: 199). ПЦР-фрагмент размером 1986 п.н. клонировали в pCR4Blunt-TOPO и секвенировали. Клон pCR4Blunt-TOPO-ldhL1 расщепляли с помощью EcoRV и AatII с высвобождением фрагмента ldhL1 размером 1982 п.н., который был очищен посредством электрофореза в геле. Интегрирующий вектор pFP988, описанный в примере 14, расщепляли с помощью HindIII и обрабатывали фрагментом Кленова ДНК-полимеразы для затупления концов. Линеаризированную плазмиду затем расщепляли с помощью AatII и фрагмент вектора размером 2931 п.н. очищали посредством электрофореза в геле. Фрагмент EcoRV/AatII ldhL1 лигировали с фрагментом вектора pFP988 и трансформировали в клетках Тор10 E.coli. Трансформанты отбирали на планшетах с агаровой средой LB, содержащей ампициллин (100 мкг/мл), и проводили скрининг посредством ПЦР колоний для подтверждения конструкции pFP988-ldhL.
Для добавления селектируемого маркера к интегрируемой ДНК ген Cm с его промотором ПЦР-амплифицировали из pC194 (Genbank NC_002013) с использованием праймеров Cm F (SEQ ID NO: 200) и Cm R (SEQ ID NO: 201) с образованием ПЦР-продукта размером 836 п.н. Ампликон был клонирован в pCR4Blunt- TOPO и трансформирован в клетках Тор10 E.coli, образуя pCR4Blunt-TOPO-Cm. После секвенирования для подтверждения того, что никаких ошибок не внесено посредством ПЦР, кассету Cm вырезали из pCR4Blunt-TOPO-Cm в виде фрагмента MluI/SwaI размером 828 п.н. и очищали посредством электрофореза в геле. Указанный интегрирующий вектор pFP988-ldhL, включающий гомологию ldhL, расщепляли с помощью MluI и SwaI, и фрагмент вектора размером 4740 п.н. был очищен посредством электрофореза в геле. Фрагмент кассеты Cm лигировали с вектором pFP988-ldhL, образуя pFP988-DldhL::Cm.
В заключение кассета thl-hbd-crt из pFP988Dss-T-H-C, описанная в примере 14, была модифицирована для замены амилазного промотора на синтетический промотор Р11. Затем целый оперон переносили в pFP988-DldhL::Cm. Промотор Р11 был сконструирован посредством отжига олигонуклеотидных праймеров P11 F (SEQ ID NO: 202) и P11 R (SEQ ID NO: 203). Отожженный олигонуклеотид был очищен посредством электрофореза в 6% геле Ultra PAGE (Embi Tec, San Diego, CA). Плазмиду pFP988Dss-T-H-C расщепляли с помощью XhoI и SmaI, и фрагмент вектора размером 9 т.п.н. был очищен посредством электрофореза в геле. Изолированный фрагмент Р11 был лигирован с расщепленной плазмидой pFP988Dss-T-H-C для образования рFP988-P11-T-H-C. Плазмиду pFP988-P11-T-H-C расщепляли с помощью XhoI и BamHI, и фрагмент P11-T-H-C размером 3034 п.н. был очищен посредством электрофореза в геле. pFP988-DldhL::Cm расщепляли с помощью XhoI и BamHI и выделяли фрагмент вектора размером 5558 п.н. Оперон верхнего пути лигировали с интегрирующим вектором для создания pFP988-DldhL-P11-THC::Cm.
Интегрирование pFP988-DldhL-P11-THC::Cm в L.plantarum BAA-793 для образования L.plantarum ΔldhL.1::T-H-C::Cm, включающего экзогенные гены thl, hbd и crt. Электрокомпетентные клетки L.plantarum получали, как описано Aukrust, T.W., et al. (Electroporation Protocols for Microorganisms; Nickoloff, J. A., Ed.; Methods in Molecular Biology, Vol. 47; Humana Press, Inc., Totowa, NJ, 1995, pp.201-208). После электропорации клетки избыточно выращивали в среде MRSSM (среда MRS, дополненная 0,5 M сахарозой и 0,1 M MgCl2), как описано Aukrust et al., выше, в течение 2 часов при 37°С без встряхивания. Электропорированные клетки высевали на планшеты со средой MRS, содержащей хлорамфеникол (10 мкг/мл), и инкубировали при 37°С. Изначально проводили скрининг трансформантов посредством ПЦР-амплификации колоний для подтверждения интегрирования и затем проводили более точный скрининг предварительно положительных клонов посредством ПЦР-амплификации с группой праймеров.
Плазмидная экспрессия генов EgTER, ald и bdhB. Три оставшихся гена 1-бутанола экспрессировали из плазмиды pTRKH3 (O'Sullivan DJ and Klaenhammer TR, Gene 137: 227-231 (1993)) под контролем промотора ldhL L.plantarum (Ferain et al., J. Bacteriol. 176: 596-601 (1994)). Промотор ldhL ПЦР-ампдифицировали из генома L.plantarum ATCC BAA-793 с использованием праймеров PldhL F (SEQ ID NO: 204) и PldhL R (SEQ ID NO: 205). ПЦР-продукт размером 369 п.н. клонировали в pCR4Blunt-TOPO и секвенировали. Полученную плазмиду, pCR4Blunt-TOPO-PldnhL, расщепляли с помощью SacI и BamHI, высвобождая фрагмент PldhL размером 359 п.н.
pHT01-ald-EB, описанный в примере 14, расщепляли с помощью SacI и BamHI и фрагмент вектора размером 10503 п.н. извлекали посредством очистки электрофорезом в геле. Фрагмент PldhL и вектор лигировали, образуя pHT01-Pldhl-ald-EB.
Для субклонирования кассеты промотор ldhL-ald-EgTER-bdh, pHT01-Pldhl-ald-EB расщепляли с помощью MluI и концы обрабатывали фрагментом Кленова ДНК-полимеразы. Линеаризированный вектор расщепляли с помощью SalI и фрагмент размером 4270 п.н., включающий фрагмент PldhL-AEB, очищали посредством электрофореза в геле. Плазмиду pTRKH3 расщепляли с помощью SalI и EcoRV и фрагмент вектора, очищенный в геле, лигировали с фрагментом PldhL-AEB. Указанную смесь для лигирования трансформировали в клетках Тор10 E.coli и трансформанты высевали на планшеты со средой Brain Heart Infusion (BHI, Difco Laboratories, Detroit, MI), включающей эритромицин (150 мг/л). Проводили скрининг трансформантов посредством ПЦР для подтверждения конструкции pTRKH3-ald-E-B. Данную экспрессирующую плазмиду, pTRKH3-ald-E-B, трансформировали в L.plantarum ΔldhL1::T-H-C::Cm посредством электропорации, как описано выше.
L.plantarum ΔldhL1::T-H-C::Cm, включающую pTRKH3-ald-E-B, инокулировали во встряхиваемые колбы объемом 250 мл, содержащие 50 мл среды MRS с эритромицином (10 мкг/мл) и выращивали при 37°С в течение 18-24 часов без встряхивания. Через 18-24 часа проводили определение 1-бутанола посредством ВЭЖХ или ГХ анализа, как описано в разделе «Общие методы».
ПРИМЕР 19 (предсказательный)
Экспрессия биосинтетического пути 1-бутанола в Enterococcus faecalis
Целью настоящего предсказательного примера является описание того, как экспрессировать биосинтетический путь 1-бутанола в Enterococcus faecalis. Последовательность полного генома штамма Enterococcus faecalis V583, который использовали в настоящем примере в качестве штамма-хозяина для экспрессии биосинтетического пути 1-бутанола, опубликована (Paulsen et al., Science 299: 2071-2074 (2003)). Плазмиду pTRKH3 (O'Sullivan DJ и Klaenhammer TR, Gene 137:227-231 (1993)), E.coli/грамположительный «челночный» вектор, использовали для экспрессии шести генов (thlA, hbd, crt, EgTER, ald, bdhB) пути биосинтеза 1-бутанола в одном опероне. pTRKH3 содержит начало репликации E.coli плазмиды p15A и репликон pAMβ1 и два устойчивых к антибиотикам селектируемых маркера, устойчивый к тетрациклину и устойчивый к эритромицину. Устойчивость к тетрациклину экспрессируется только в E.coli, и устойчивость к эритромицину экспрессируется и в E.coli, и в грамположительных бактериях. Производные плазмиды pAMβ1 могут реплицироваться в E.faecalis (Poyart et al., FEMS Microbiol. Lett. 156: 193-198 (1997)). Индуцибельный промотор nisA (PnisA), который использовали для эффективного контроля экспрессии генов посредством низина в различных грамположительных бактериях, в том числе Enterococcus faecalis (Eichenbaum et al., Appl. Environ. Microbiol. 64: 2763-2769 (1998)), применяется для контроля экспрессии шести требующихся генов, кодирующих ферменты биосинтетического пути 1-бутанола.
Линейный фрагмент ДНК (215 п.н.), включающий промотор nisA (Chandrapati et al., Mol. Microbiol. 46(2):467-477 (2002)), ПЦР-амплифицировали из геномной ДНК Lactococcus lactis с использованием праймеров F-PnisA(EcoRV) (SEQ ID NO: 206) и R-PnisA(PmeI BamHI) (SEQ ID NO: 207). ПЦР-фрагмент размером 215 п.н. расщепляли с помощью EcoRV и BamHI, и полученный фрагмент PnisA был очищен посредством электрофореза в геле. Плазмиду pTRKH3 расщепляли с помощью EcoRV и BamHI, и фрагмент вектора был очищен посредством электрофореза в геле. Линеаризированную pTRKH3 лигировали с фрагментом PnisA. Указанную смесь для лигирования трансформировали в клетках Тор10 E.coli посредством электропорации и трансформанты отбирали после выращивания в течение ночи при 37°С на планшетах с агаровой средой LB, содержащей эритромицин (25 мкг/мл). Затем проводили скрининг трансформантов посредством ПЦР колоний с использованием праймеров F-PnisA(EcoRV) и R-PnisA(BamHI) для подтверждения соответствующего клона pTRKH3-PnisA.
Плазмиду pTRKH3-PnisA расщепляли с помощью PmeI и BamHI, и указанный вектор был очищен посредством электрофореза в геле. Плазмиду pHTO1-ald-EgTER-bdhB конструировали, как описано в примере 14, и расщепляли с помощью SmaI и BamHI, и фрагмент ald-EgTER-bdhB размером 2973 п.н. был очищен посредством электрофореза в геле. Фрагмент ald-EgTER-bdhB размером 2973 п.н. лигировали в вектор pTRKH3-PnisA в сайтах PmeI и BamHI. Указанную смесь для лигирования трансформировали в клетках Тор10 E.coli посредством электропорации и трансформанты отбирали после выращивания в течение ночи при 37°С на планшетах с агаровой средой LB, содержащей эритромицин (25 мкг/мл). Затем проводили скрининг трансформантов посредством ПЦР колоний с использованием ald прямого праймера N27F1 (SEQ ID NO: 31) и bdhB обратного праймера N65 (SEQ ID NO: 44). Полученная плазмида названа pTRKH3-PnisA-ald-EgTER-bdhB (=pTRKH3-A-E-B).
Плазмиду pTRKH3-A-E-B очищали от трансформанта и использовали для дальнейшего клонирования оставшихся генов (thlA, hbd, crt) в сайте BamHI, расположенном в 5'-3' направлении от гена bdhB. Плазмиду pTRKH3-A-E-B расщепляли с помощью BamHI и обрабатывали фрагментом Кленова ДНК-полимеразы для образования тупых концов. Плазмиду pFP988Dss-thlA-hbd-crt (=pFP988Dss-T-H-C) конструировали, как описанов в примере 14, и расщепляли с помощью SmaI и BamHI. Полученный фрагмент thlA-hbd-crt размером 2973 п.н. обрабатывали фрагментом Кленова ДНК-полимеразы для образования тупых концов и очищали посредством электрофореза в геле. Фрагмент thlA-hbd-crt размером 2973 п.н. лигировали с линеаризированной pTRKH3-A-E-B. Указанную смесь для лигирования трансформировали в клетках Тор10 E.coli посредством электропорации и трансформанты отбирали после выращивания в течение ночи при 37°С на планшетах с агаровой средой LB, содержащей эритромицин (25 мкг/мл). Затем проводили скрининг трансформантов посредством ПЦР колоний с использованием thlA прямого праймера N7 (SEQ ID NO: 21) и crt обратного праймера N4 (SEQ ID NO: 18). Полученная плазмида названа pTRKH3-PnisA-ald-EgTER-bdhB-thlA-hbd-crt (=pTRKH3-A-E-B-T-H-C). Плазмида pTRKH3-A-E-B-T-H-C получена из трансформантов E.coli и трансформирована в электрокомпетентных клетках E.faecalis V583 посредством электропорации с использованием способов, известных в уровне техники (Aukrust, T.W., et al., Electroporation Protocols for Microorganisms; Nickoloff, J.A., Ed.; Methods in Molecular Biology, Vol. 47; Humana Press, Inc., Totowa, NJ., 1995, pp.217-226), с образованием E.fаecalis V583/pTRKH3-A-E-B-T-H-C.
Вторую плазмиду, включающую регуляторные гены nisA, nisR и nisK, ген устойчивости к спектиномицину add9 и репликатор pSH71, трансформировали в E.faecalis V583/pTRKH3-A-E-B-T-H-C посредством электропорации. Плазмида, включающая репликатор pSH71, совместима с производными pAMβ1 в E.faecalis (Eichenbaum et al., выше). Трансформанты с двойной лекарственной устойчивостью отобраны на планшетах с агаровой средой LB, содержащей эритромицин (25 мкг/мл) и спектиномицин (100 мкг/мл).
Полученный штамм E.faecalis V583B, несущий две плазмиды, а именно экспрессирующую плазмиду (PTRKH3-A-E-B-T-H-C) и регуляторную плазмиду (pSH71-nisRK), инокулировали во встряхиваемые колбы объемом 250 мл, содержащие 50 мл бульона Тодда-Гевитта, дополненного дрожжевым экстрактом (0,2%) (Fischetti et al., J. Exp. Med. 161: 1384-1401 (1985)), низином (20 мкг/мл) (Eichenbaum et al., выше), эритромицином (25 мкг/мл) и спектиномицином (100 мкг/мл). Колбы инкубировали без встряхивания при 37°С в течение 18-24 часов, после чего продуцирование 1-бутанола определяли посредством ВЭЖХ или ГХ анализа, как описано в разделе «Общие методы».
Реферат
Изобретение относится к биотехнологии и генной инженерии и касается ферментативного способа получения 1-бутанола с использованием бактериальной клетки. Данная клетка содержит гетерологичные молекулы ДНК, кодирующие ферменты пути биосинтеза 1-бутанола: ацетил-СоА-ацетилтрансферазу, 3-гидроксибутирил-СоА-дегидрогеназу, кротоназу, бутирил-СоА-дегидрогеназу, бутиральдегиддегидрогеназу и, необязательно, бутанолдегидрогеназу. Бутанол получают посредством ферментативного выращивания рекомбинантной бактериальной клетки в среде с глюкозой или сахарозой. Изобретение позволяет получать 1-бутанол в виде единственного продукта безопасным для окружающей среды и экономичным способом. 2 н. и 16 з.п. ф-лы, 1 ил., 15 табл.
Формула
a) ацетил-СоА-ацетилтрансферазу, которая катализирует конверсию ацетил-СоА в ацетоацетил-СоА;
b) 3-гидроксибутирил-СоА-дегидрогеназу, которая катализирует конверсию ацетоацетил-СоА в 3-гидроксибутирил-СоА;
c) кротоназу, которая катализирует конверсию 3-гидроксибутирил-СоА в кротонил-СоА;
d) бутирил-СоА-дегидрогеназу, которая катализирует конверсию кротонил-СоА в бутирил-СоА;
e) бутиральдегиддегидрогеназу, которая катализирует конверсию бутирил-СоА в бутиральдегид; и
необязательно f) бутанолдегидрогеназу, которая катализирует конверсию бутиральдегида в 1-бутанол;
где указанная микробная клетка-хозяин продуцирует 1-бутанол.
i) получение рекомбинантной микробной клетки-хозяина по любому из п.1-10;
ii) контактирование клетки-хозяина из (i) с глюкозой или сахарозой в условиях, при которых продуцируется 1-бутанол.
Комментарии