Способ получения 2,4-дигидроксибутирата - RU2645260C2
Код документа: RU2645260C2
Чертежи

































































































Описание
Данное изобретение относится к новому способу получения 2,4-дигидроксибутирата (2,4-ДГБ) из гомосерина, включающему два этапа:
- первый этап замещения первичной аминогруппы гомосерина карбонильной группой для получения 2-оксо-4-гидроксибутирата, и
- второй этап восстановления полученного 2-оксо-4-гидроксибутирата (ОГБ) для получения 2,4-ДГБ.
Карбоновые кислоты, раскрытые в данной заявке, названы в соответствии с их солями (например, 2,4-дигидроксибутират) или кислотными формами (например, 2,4-дигидроксибутановая кислота).
2,4-дигидроксибутановая кислота (также 2,4-ДГБ или ДГБ) является соединением, представляющим значительный экономический интерес. ДГБ может быть легко превращена в α-гидрокси-γ-бутиролактон в водной среде путем регулировки соответствующего pH. α-гидрокси-γ-бутиролактон является известным предшественником для получения заместителя метионина 2-гидрокси-4-(метилтио)-бутирата (ГМТБ) (США 2009/318715), который имеет большой спрос в кормлении животных. В настоящее время α-гидрокси-γ-бутиролактон получают из γ-бутиролактона с помощью многоэтапного процесса, включающего галогенирование γ-бутиролактона в α-положении, и последующее замещение атома галогена гидроксильной группой в щелочной среде (США 2009/318715).
С ростом цен на нефть возникает необходимость в производстве ДГБ из возобновляемых ресурсов. Микроорганизмы способны к преобразованию полученного из биологической массы сырья, например, сахаров или органических кислот, в большое разнообразие различных химических соединений (Werpy & Petersen, 2004). С ростом биохимических и генетических данных становится возможным модифицировать микроорганизмы так, чтобы они производили в большом количестве естественно образующиеся промежуточные продукты метаболизма с высоким выходом и продуктивностью (Bailey, 1991). Оптимизация производственных микроорганизмов часто требует рационального проектирования метаболических сетей, которые гарантируют, среди прочего, повышенную экспрессию ферментов, необходимых для биосинтеза интересующего метаболита, и облегчение обратного ингибирования продукта. Другой возможностью является внедрение новейших ферментных систем, которые катализируют производство интересующего метаболита.
Подходы метаболической инженерии и ферментативных катализов требуют детального знания о биохимии и регулировании метаболических путей, приводящих к интересующему метаболиту. В случае производства ДГБ эта информация не доступна. Лишь несколько исследований показывают появление ДГБ у пациентов с дефицитом аспартат полуальдегид дегидрогеназы (Shinka и др., 2002), без выявления, однако, ферментативных реакций, вовлеченных в производство ДГБ. Зимотическое или ферментативное производство ДГБ, следовательно, требует (I) определение термодинамически возможного пути, для преобразования доступного предшественника в ДГБ, (II) идентификации или конструирования ферментов, которые способны катализировать индивидуальные стадии реакции в пути, и (III) функциональной экспрессии ферментов метаболического пути в соответствующем организме продуценте. Данное изобретение имеет в качестве цели, удовлетворение этих потребностей.
Соответственно, одним из объектов настоящего изобретения является способ получения 2,4-ДГБ из гомосерина, включающий два этапа (фиг. 1):
- первый этап замещения первичной аминогруппы гомосерина карбонильной группой для получения ОГБ, и
- второй этап восстановления полученного ОГБ до 2,4-ДГБ.
Первый и/или второй этап(ы) способа по изобретению можно катализировать ферментом, кодируемым эндогенно или гетерологичным геном.
В описании, ферментативные активности также определены путем ссылки на гены, кодирующие ферменты, имеющие такую же активность. Использование наименований генов не ограничивается конкретным организмом, а охватывает все соответствующие гены и белки в других организмах (например, микроорганизмы, функциональные аналоги, функциональные варианты и функциональные фрагменты, пока они сохраняют ферментативную активность).
В еще одном аспекте настоящего изобретения, ферментом, заменяющим первичную аминогруппу гомосерина карбонильной группой для получения ОГБ, может являться гомосерин трансаминаза, гомосерин дегидрогеназа или гомосерин оксидаза.
В еще одном аспекте настоящего изобретения, фермент, имеющий гомосерин трансаминазную активность, может быть обнаружен среди ферментов, обладающих аспартат трансаминазной (ЕС2.6.1.1) активностью, трансаминазной активностью (ЕС2.6.1.42) аминокислот с разветвленной цепью, или трансаминазной активностью (ЕС2.6.1.57) ароматических аминокислот.
В еще одном аспекте настоящего изобретения, гомосерин трансаминаза может являться трансаминазой аминокислот с разветвленной цепью из Escherichia coli, Ec-IIvE и Lactococcus lactis, LI-BcaT, трансаминазой ароматических аминокислот из E.coli, Ec-TyrB, L. lactis, LI-AraT и Saccharomyces cerevisiae, Sc-Aro8 или аспартаттрансаминазой из E.coli, Ec-AspC.
Второй этап способа по настоящему изобретению катализируют ферментом, обладающим ОГБ редуктазной активностью. В еще одном аспекте данного изобретения, фермент, обладающий ОГБ редуктазной активностью, может быть обнаружен среди ферментов, обладающих активностью дегидрогеназ 2-гидроксикислот, в частности, среди ферментов обладающих лактатдегидрогеназной (Ldh) (ЕС1.1.1.27, ЕС1.1.1.28), малатдегидрогеназной (Mdh) (ЕС1.1.1.37, ЕС 1.1.1.82, ЕС1.1.1.299) активностью, или активностью дегидрогеназы (D)-2-гидроксикислоты с разветвленной цепью (ЕС 1.1.1.272, ЕС1.1.1.345). Более конкретно, фермент, имеющий гомосерин трансаминазную активность, кодируется генами ilvE, tyrB, aspC, araT, bcaT или ARO8.
В еще более конкретном аспекте, фермент, обладающий гомосерин трансаминазной активностью, кодируется последовательностью SEQ ID No.59, SEQ ID No.61, SEQ ID No.63, SEQ ID No.65, SEQ ID No.67 или SEQ ID No.69 или любой последовательностью, гомологичной по меньшей мере на 50% указанным последовательностям, или соответствует последовательности SEQ ID No.60, SEQ ID No.62, SEQ ID No.64, SEQ ID No.66, SEQ ID No.68, SEQ ID No.70 или любой последовательности, гомологичной по меньшей мере на 50%, указанным последовательностям.
В еще одном аспекте данного изобретения ферментом редуктазой ОГБ может быть (L)-лактатдегидрогеназа Lactococcus lactis (LI-LdhA), Oryctalagus cuniculus (Oc-LldhA), Geobacillus stearothermophilus (Gs-LIdh), или Bacillus subtilis (Bs-Ldh), (D)-лактатдегидрогеназа Escherichia coli (Ec-LdhA), (L)-малатдегидрогеназа Escherichia coli (Ec-Ldh), дегидрогеназа (D)-2-гидроксикислот с разветвленной цепью Lactococcus lactis (Li-PanE).
В даже более конкретном аспекте изобретения, фермент редуктаза ОГБ представлен аминокислотными последовательностями SEQ ID No.2, SEQ ID No.4, SEQ ID No.6, SEQ ID No.8, SEQ ID No.10, SEQ ID No.12, SEQ ID No.14, SEQ ID No.288, SEQ ID No.30, SEQ ID No.32, SEQ ID No.102, SEQ ID No.104, SEQ ID No.106, SEQ ID No.108, SEQ ID No.110, SEQ ID No.112, SEQ ID No.114, SEQ ID No.116 или SEQ ID No.118 или любой последовательностью, гомологичной по меньшей мере на 50% указанным последовательностям, или кодируется последовательностями нуклеиновых кислот, представленными SEQ ID No.1, SEQ ID No.3, SEQ ID No.5, SEQ ID No.7, SEQ ID No.9, SEQ ID No.11, SEQ ID No.13, SEQ ID No.287, SEQ ID No.29, SEQ ID No.31, SEQ ID No.101, SEQ ID No.103, SEQ ID No.105, SEQ ID No.107, SEQ ID No.109, SEQ ID No.111, SEQ ID No.113, SEQ ID No.115 или SEQ ID No.117, или любой последовательностью, гомологичной по меньшей мере на 50% указанным последовательностям.
В дополнительном аспекте, изобретение также соотносится к применению фермента, восстанавливающего ОГБ до 2,4-ДГБ, как описано выше.
Белки, существенно гомологичные вышеуказанным ферментам, являются так же одним из аспектов данного изобретения, как и функциональные варианты или функциональные фрагменты.
Выражение "существенная гомология" охватывает гомологию по отношению к структуре, и/или аминокислотным компонентам и/или биологической активности.
В целом, в контексте настоящего изобретения, гомология между двумя последовательностями белков может быть определена способами, хорошо известными специалисту в данной области. Она, как правило, определяется как процент идентичности последовательностей между эталонной последовательностью и последовательностью интересующего белка.
Используемый здесь, "процент (%) идентичности последовательностей" по отношению к аминокислотным или нуклеотидным последовательностям идентифицированных здесь, определяют как процент аминокислотных остатков или нуклеотидов в последовательности кандидата, которые идентичны аминокислотным остаткам или нуклеотидам в последовательности фермента, после выравнивания последовательностей и введения пробелов, если это необходимо, для достижения максимального процента идентичности последовательностей, и без учета любых консервативных замен, как части идентичности последовательности. Способы осуществления выравнивания последовательностей и определения идентичности последовательностей известные специалистам в данной области, могут быть выполнены без излишнего экспериментирования, а расчеты значений идентичности могут быть получены с точностью. Смотри, например, Ausubel et al., eds. (1995) Current Protocols in Molecular Biology, Chapter 19 (Greene Publishing and Wiley-Interscience, New York); и программу ALIGN (Dayhoff (1978) в Atlas of Protein Sequence and Structure 5: Suppl.3 (National Biomedical Research Foundation, Washington, D.C.). Ряд алгоритмов доступны для выравнивания последовательностей и определения идентичности последовательностей и включают, например, алгоритм выравнивания гомологии Needleman et al. (1970), J. Mol Biol 48: 443; алгоритм локальной гомологии Smith et al. (1981) Adv. Appl Math. 2: 482; поиск сходства Pearson et al. (1988), Proc. Natl. Acad. Sci. 85:2444; алгоритм Smith-Waterman (Meth. Mol. Biol. 70:1 73-187 (1997); и BLASTP, BLASTN и BLASTX алгоритмы (см. Altschul et al. (1990), J. Mol Biol 215:403-410). Компьютерные программы, использующие эти алгоритмы, также доступны и включают, но не ограничиваются ими: ALIGN или обеспечение Megalign (DNASTAR), или WU-BLAST-2 (Altschul et al. Enzym., 266:460-480 (1996)); или GAP, BESTFIT, BLAST (Altschul et al.), supra, FASTA и TFASTA, доступны в пакете Genetics Computing Group (GCG), Version 8, Madison, Wis., США; и CLUSTAL в программе PC/Gene от Intelligenetics, Mountain View, Calif. Специалисты в данной области техники могут определить соответствующие параметры для измерения выравнивания, включая алгоритмы, необходимые для достижения максимального выравнивания по длине сравниваемых последовательностей. Предпочтительно, идентичность последовательностей может быть определена с использованием стандартных параметров заданных программой. В частности, идентичность последовательностей может быть определена с помощью алгоритма поиска гомологии Smith-Waterman (Meth. Mol. Biol. 70:173-187 (1997)), как реализовано в программе MSPRCH (Oxford Molecular) с использованием поиска аффинных пропусков со следующими параметрами поиска: штраф за открытый пропуск 12, и штраф за расширение пропуска 1. Предпочтительно, сравнение парных аминокислот может быть осуществлено с использованием GAP программы GCG программного обеспечения для анализа последовательности пакета Genetics Computer Group, Inc., Madison, Wis, с применением матрикса аминокислотных замещений blosum62, с весом пропуска 12 и весом длинны 2. В отношении оптимального выравнивания двух аминокислотных последовательностей, прилегающий участок варианта аминокислотной последовательности может иметь дополнительные аминокислотные остатки или удаленные аминокислотные остатки по отношению к эталонной аминокислотной последовательности. Смежный сегмент, используемый для сравнения с эталонной аминокислотной последовательностью будет включать по меньшей мере 20 смежных аминокислотных остатков, и возможно 30, 40, 50 или более аминокислотных остатков. Поправки на увеличенную идентичность последовательностей, связанную с включением пробелов в производную аминокислотную последовательность могут быть сделаны путем присвоения штрафов за пропуски.
Ферменты, в соответствии с настоящим изобретением, имеющие одинаковую активность (редуктаза ОГБ, или фермент заменяющий первичную аминогруппу гомосерина карбонильной группой для получения ОГБ), обладают по меньшей мере около 50%, 70% или 85% идентичности аминокислотной последовательности, предпочтительно по меньшей мере около 85% идентичности аминокислотной последовательности, более предпочтительно по меньшей мере около 90% идентичности аминокислотной последовательности, еще более предпочтительно по меньшей мере около 95% идентичности аминокислотной последовательности и наиболее предпочтительно 98% идентичности аминокислотной последовательности. Предпочтительно, любые аминокислотные замены являются "консервативными заменами аминокислот" с использованием L-аминокислот, в которых одна аминокислота заменена другой биологически аналогичной аминокислотой. Консервативными аминокислотными заменами являются те, которые сохраняют общий заряд, гидрофобность/гидрофильность и/или стерическую конформацию замещенной кислоты. Примерами консервативных замен являются замены, между следующими группами: Gly/Ala, Val/Lle/Leu, Lys/Arg, Asn/GIn, Glu/Asp, Ser/Cys/Thr и Phe/Trp/Tyr. Производное может, например, отличаться несколькими, от 1 до 10 аминокислотными остатками, такими как 6-10, лишь 5, всего лишь 4, 3, 2, или даже 1 аминокислотным остатком.
Термин «функциональный вариант» включает ферменты, которые могут представлять значительные модификации последовательности по сравнению с последовательностями, описанными, в частности, в настоящей заявке, но которые все еще сохраняют исходную ферментативную активность.
Это также означает, что последовательность фермента может содержать меньше аминокислот, чем оригинал, но указанный усеченный фермент по-прежнему сохраняет исходную ферментативную активность.
В соответствии с аспектом изобретения, активность фермента, катализирующего первый и/или второй этап способа по настоящему изобретению, усилена. Это усовершенствование может быть измерено путем ферментативного анализа, как описано в примерах 1 или 4.
Улучшение указанных ферментов может быть достигнуто, по меньшей мере, одной мутацией, указанная мутация(и) приводит к (i) повышению активности и/или сродства к субстрату мутированного фермента к гомосерину или ОГБ соответственно, и или (ii) уменьшению активности и/или сродства к субстрату мутированного фермента к их природному субстрату.
В настоящем изобретении выражение "улучшение активности и/или сродство к субстрату" означает, что фермент, до введения мутаций, и/или
- не в состоянии использовать субстрат, и/или
- синтезировал продукт реакции при максимальной специфической скорости по меньшей мере в три раза меньшей, и/или
- имел сродство к гомосерину или ОГБ, которое являлось, по меньшей мере в три раза меньше, и/или.
- имел максимальную специфическую активность на природном субстрате, которая была по меньшей мере в три раза выше, и/или
- имел сродство к природному субстрату, которое было, по меньшей мере в три раза выше.
В еще одном аспекте изобретение включает нуклеотидные последовательности, кодирующие ферменты, катализирующие первый и второй этап способа по настоящему изобретению.
В более конкретном аспекте изобретения, фермент редуктаза ОГБ кодируется последовательностями нуклеиновых кислот SEQ ID No.1, SEQ ID No. 3, SEQ ID No.5, SEQ ID No.7, SEQ ID No.9, SEQ ID No.11, SEQ ID No.13, SEQ ID No.287, SEQ ID No.29, SEQ ID No.31, SEQ ID No.101, SEQ ID No.103, SEQ ID No.105, SEQ ID No.107, SEQ ID No.109, SEQ ID No.111, SEQ ID No.113, SEQ ID No.115 или SEQ ID No.117 или любой последовательностью, гомологичной по меньшей мере на 50% указанным последовательностям.
Редуктаза ОГБ, в соответствии с изобретением соответствует в определенном аспекте (L)-лактатдегидрогеназе А, содержащей по меньшей мере одну мутацию, по сравнению с ферментом дикого типа, по меньшей мере в одной из позиций V17, Q85, Е89, I226 или А222. Эти позиции консервативны в семействе лактатдегидрогеназ, и они определяются в этом тексте путем ссылки на (L)-лактатдегидрогеназу A Lactococcus Lactis (SEQ ID NO: 6). Специалисту в данной области будет легко определить соответствующие аминокислотные остатки в других лактатдегидрогеназах выравниванием соответствующих аминокислотных последовательностей. Таким образом, изобретение также предусматривает изменения этих аминокислот в других лактатдегидрогеназных ферментах.
Редуктаза ОГБ, в соответствии с изобретением, соответствует в определенном аспекте (L)-малатдегидрогеназе, содержащей по меньшей мере одну мутацию по сравнению с ферментом дикого типа, по меньшей мере в одном из положений 112, R81, М85, D86, V93, G179, Т211 или М227. Эти позиции консервативны в семействе малатдегидрогеназ, и они определяются в данном тексте путем ссылки на последовательности (L)-малатдегидрогеназы E.coli (SEQ ID №. 2). Специалисту в данной области будет легко определить соответствующие аминокислотные остатки в других малатдегидрогеназах с помощью выравнивания соответствующих аминокислотных последовательностей. Таким образом, изобретение также предусматривает изменения этих аминокислот в других малатдегидрогеназных ферментах.
В соответствии с настоящим изобретением, "последовательностью нуклеиновой кислоты" называется ДНК или РНК молекула в одной или двуцепочечной форме, предпочтительно молекула ДНК. "Выделенной ДНК", как использовано здесь, является не природная ДНК или ДНК, которая более не находится в естественной среде, где она присутствовала первоначально, например, кодирующая последовательность ДНК, связанная с другими регулирующими элементами в химерном гене, ДНК перенесенная в другую клетку-хозяина, или искусственно, синтетически произведенная последовательность ДНК, имеющая отличающуюся последовательность нуклеотидов по сравнению с любой природной последовательностью ДНК.
Настоящее изобретение также относится к химерному гену, содержащему функционально связанные друг с другом по меньшей мере один промотор, который функционирует в организме-хозяине, полинуклеотид, кодирующий любой из ферментов, катализирующих первый и второй этап способа, как это определено в соответствии с изобретением, и терминаторный элемент, который является функциональным в том же организме-хозяине. Различными элементами, которые химерный ген может содержать, являются, во-первых, элементы, регулирующие транскрипцию, трансляцию и фолдинг белков, такие как промотор, последовательность, кодирующая сигнальный пептид или транзитный пептид, или терминаторный элемент, составляющий сигнал полиаденилирования и, во-вторых, полинуклеотид, кодирующий белок. Выражение "функционально связанные друг с другом" означает, что указанные элементы химерного гена связаны друг с другом таким образом, что функция одного из этих элементов, является зависимой от другого. В качестве примера, промотор функционально связан с кодирующей последовательностью, если он способен воздействовать на экспрессию указанной кодирующей последовательности. Конструкция химерного гена согласно настоящему изобретению и сборка различных его элементов может быть осуществлена с использованием техник, хорошо известных специалистам в данной области. Выбор регуляторных элементов, составляющих химерный ген, существенно зависит от организма-хозяина, в котором они должны функционировать, и специалисты в данной области способны выбрать регуляторные элементы, которые функциональны в данном организме хозяине. Термин "функциональный" означает способность функционировать в данном организме-хозяине.
Промоторы, которые химерный ген, согласно изобретению, может содержать, являются либо конститутивными либо индуцибельными. В качестве примера, промоторы, используемые для экспрессии в бактериях, могут быть выбраны из промоторов, указанных ниже. Для экспрессии в Escherichia coli можно упомянуть lac, trp, Ipp, phoA, recA, araBAD, prou, cst-I, tet-A, cadA, nar, tac, trc, Ipp-lac, Psyn, cspA, PL, PL-9G-50, PR-PL, 17, [лямбда] PL-PT7, T3-lac, T5-lac, T4 ген 32, nprM-lac, VHb и промотеры А-белка или еще промотер Ptrp (WO 99/64607). Для экспрессии в грам-положительных бактериях, таких как Corynebacteria или Streptomyces, можно упомянуть о промотерах PtipA или PS1 и PS2 (FR 91/09870) или промотерах, описанных в ЕР 0629699 А2. Для экспрессии в дрожжах и грибах могут быть упомянуты промоторы K. lactis PLAC4 или промоторы K. lactis Ppgk (заявка на патент FR 91/05294), промотеры tefl или cbhl Trichoderma reesei (WO 94/04673), промотеры his, csl или apf Penicillium funiculosum (WO 00/68401) и промотер gla Aspergillus niger.
В соответствии с изобретением, химерный ген может также содержать другие регуляторные последовательности, которые расположены между промотором и кодирующей последовательностью, такие как активаторы транскрипции (энхансеры).
Таким образом, химерный ген, согласно изобретению, содержит в данном воплощении, по меньшей мере для транскрипции, функционально связанную, промоторную регуляторную последовательность, которая является функциональной в организме-хозяине, последовательность нуклеиновой кислоты, кодирующей полинуклеотид, кодирующий любой из ферментов, катализирующих первый и второй этапы способа, как это определено в соответствии с изобретением, и терминаторную регуляторную последовательность, которая является функциональной в указанном организме-хозяине.
Настоящее изобретение также относится к клонированию и/или экспрессии вектора, содержащего химерный ген, согласно настоящему изобретению, или последовательности нуклеиновой кислоты, согласно изобретению. Вектор, согласно изобретению, используется для трансформации организма-хозяина и экспрессии в этом организме любого из ферментов, катализирующих первый и/или второй этап(ы) способа, в соответствии с настоящим изобретением. Этот вектор может являться плазмидой, космидой, бактериофагом или вирусом. Предпочтительно, вектор для трансформации, в соответствии с изобретением, представляет собой плазмиду. Как правило, основными свойствами этого вектора должна быть способность поддерживать себя и самореплицироваться в клетках организма-хозяина, в частности, в силу наличия ориджина репликации, а также экспрессировать любой из ферментов, катализирующих первый и/или второй этап(ы) способа по изобретению. С целью стабильной трансформации организма-хозяина, вектор может интегрироваться в геном. Выбор такого вектора, а также способы вставки химерного гена, согласно изобретению, в этот вектор являются частью общих знаний специалиста в данной области. Предпочтительно, чтобы вектор, используемый в настоящем изобретении, также содержал, в дополнение к химерному гену согласно данному изобретению, химерный ген, кодирующий селективный маркер. Это селективный маркер позволяет выбрать организмы-хозяева, которые эффективно трансформированы, т.е. те, которые содержат вектор. В соответствии с конкретным вариантом осуществления настоящего изобретения, организмом-хозяином для трансформации являются бактерии; дрожжи, грибы. Среди селективных маркеров, которые могут быть использованы, могут быть маркеры, содержащие гены устойчивости к антибиотикам, таким как, например, ген гигромицинфосфотрансфераза. Другими маркерами могут быть гены, дополняющие собой ауксотрофию, такие как pyrA, pyrB, pyrG, pyr4, arg4, argB и trpC гены, гены молибдоптерин-синтазы или ацетамидазы. Можно также может упомянуть гены, кодирующие легко идентифицируемые ферменты, такие как GUS ферменты, или гены, кодирующие пигменты или ферменты, регулирующие производство пигментов в трансформированных клетках. Такие селективные маркерные гены, в частности, описаны в патентных заявках WO 91/02071, WO 95/06128, WO 96/38567 и WO 97/04103.
Настоящее изобретение также относится к модифицированным микроорганизмам. Более конкретно, модифицированный микроорганизм, согласно изобретению, позволяет получать 2,4-ДГБ из гомосерина в два этапа, включающих:
- первый этап замещения первичной аминогруппы гомосерина карбонильной группой для получения 2-оксо-4-гидроксибутирата, и
- второй этап восстановления полученного 2-оксо-4-гидроксибутирата для получения 2,4-дигидроксибутирата.
Ферменты, участвующие в двух этапах, описаны выше.
Термин "микроорганизм" означает любой низший одноклеточный организм, в который химерный ген(ы), нуклеиновая кислота(ы) или вектор(ы), в соответствии с изобретением, могут быть введены для того, чтобы получить 2,4-ДГБ. Предпочтительно, организм-хозяин представляет собой микроорганизм, в частности грибы, например Penicillium, Aspergillus и, более конкретно, Aspergillusflavus, Chrysosporium или Trichoderma, дрожжи, в частности, Saccharomycetaceae, Pichiaceae или Schizosaccharomycetaceae, наиболее предпочтительно Saccharomyces cerevisiae, Schizosaccharomyces pombe, Kluyveromyces Lactis, Kluyveromyces marxianus или Pichia jadinii, Pichici stipitis или Pichia pastoris, бактерии, предпочтительно выбранные из Enterobacteriaceae, Clostridiaceae, Bacillaceae, Streptomycetaceae, Streptococcaceae, Methylobacteriacae и Corynebacteriaceae, наиболее предпочтительно Escherichia coli, Bacillus subtilis, Corynebacterium glutamicum, Clostridium acetobutylicum, Methylobacterium extorquens или Lactococcus lactis.
Настоящее изобретение также относится к модифицированным микроорганизмам, содержащим по меньшей мере один химерный ген согласно изобретению, либо интегрированный в их геном или внедренный на экстрахромосомном геномном элементе, например плазмиде. В более конкретном воплощении изобретения, трансформированный организм-хозяин содержит нуклеиновую кислоту, согласно изобретению, кодирующую полипептид, заменяющий первичную аминогруппу кислоты гомосерина карбонильной группой для получения ОГБ и/или нуклеиновую кислоту, кодирующую полипептид, преобразующий ОГБ в 2,4-ДГБ, или химерный ген, содержащий нуклеиновую кислоту, кодирующую полипептид, заменяющий первичную аминокислотную группу гомосерина карбонильной группой для получения ОГБ, и/или редуктазы ОГД, или вектор экспрессии, содержащий нуклеиновую кислоту, кодирующую полипептид, замещающий первичную аминокислотную группу гомосерина карбонильной группой для получения ОГБ, или полипептид, имеющий активность редуктазы ОГБ.
В еще одном аспекте настоящего изобретения, синтетический путь для превращения гомосерина в ДГБ осуществляется в микроорганизме с повышенным производством гомосерина. Усиление производства гомосерина в микроорганизмах может быть достигнуто путем (i) гиперэкспрессии ферментов аспартаткиназы, аспартат полуальдегид дегидрогеназы и гомосерин дегидрогеназы, (ii) путем перевода фермента аспартаткиназы в нечувствительное состояние к ингибированию продуктом, что может быть вызвано лизином, метионином или треонином, и (iii) путем устранения метаболических путей, которые отходят от пути биосинтеза гомосерина. Избыточная экспрессия аспартаткиназы, аспартат полуальдегид дегидрогеназы и гомосерин дегидрогеназы может быть достигнута путем экспрессии ферментов в многокопийной плазмиде под контролем соответствующего конститутивного или индуцируемого промотора. Кроме того, сверхэкспрессия указанных ферментов может быть достигнута путем устранения транскрипционных репрессоров, которые ограничивают транскрипцию генов, кодирующих аспартаткиназу, аспартат полуальдегид дегидрогеназу и гомосерин дегидрогеназу. Аспартаткиназа может быть нечувствительной к ингибированию аминокислотами производными аспартата путем введения соответствующих мутаций в их аминокислотные последовательности. Точки входа в метаболические пути, которые отходят от биосинтеза гомосерина, катализируются ферментами, имеющими О-сукцинил гомосерин или гомосерин О-ацетил синтазную активность (вступление в биосинтез метионина), гомосеринкиназную активность (вступление в биосинтез треонина), или диаминопимелат декарбоксилазную активность (вступление в биосинтез лизина). Устранение генов, кодирующих белки, имеющие указанные ферментативные активности позволяет избежать образования аминокислот, производных аспартата и, следовательно, помогает образованию гомосерина.
Соответственно, делетирование генов metA, thrB и lysA в E.coli ослабляет пути, которые отходят от пути биосинтеза гомосерина. Увеличение ферментативной активности пути гомосерина в E.coli может быть достигнуто, например, путем сверхэкспрессии бифункциональной аспартаткиназы мутантной гомосерин дегидрогеназы thrA S345F (нечувствительна к ингибированию треонином) и asd (оба гена E.coli); или сверхэкспрессией монофункциональных аспартаткиназ мутанта lysC E250K (нечувствителен к лизину), ask (оба гена из E.coli) и гена гомосерин дегидрогеназы НОМ6 S. cerevisiae.
Микроорганизм, по настоящему изобретению, может также иметь ослабленную способность к экспортированию гомосерина, что увеличивает внутриклеточную доступность этой аминокислоты. Для того чтобы достигнуть уменьшения экспорта гомосерина из клеток, могут быть удалены пермеазы, способные экспортировать гомосерин. Такие пермеазы могут быть идентифицированы путем сверхэкспрессии геномных библиотек в микроорганизме, и культивированием указанного микроорганизма на ингибирующих концентрациях гомосерина или структурно подобных аминокислот, таких как треонин, лейцин, или аспартат (Zakataeva др., 1999/FEBS Lett/452/228-232). Гены, сверхэкспрессия которых способствует росту при повышенных концентрациях указанных аминокислот, могут участвовать в экспорте гомосерина.
В дополнительном аспекте, микроорганизм Escherichia coli, согласно изобретению, имеет делеции в гене эффлюксного переносчика гомосерина rhtA, rhtB и/или rhtC.
Эффективное получение ДГБ может быть обеспечено за счет оптимизации перераспределения потока углерода в метаболической сети организма-хозяина по отношению к оптимизации поддержки кофактора для синтеза ДГБ и ослабления конкурирующих путей, которые вызывают образование продуктов метаболизма, отличных от ДГБ. Важный инструмент для улучшения штамма обеспечивает основанный на ограничении анализ баланса потоков. Этот способ позволяет вычислить теоретический выход данной метаболической сети в зависимости от условий культивирования, и облегчает идентификацию метаболических мишеней для экспрессии или устранения. Описаны экспериментальные способы, используемые для сверхэкспрессии и удаления целевой метаболической реакции (пример 8).
Соответственно, микроорганизм, по настоящему изобретению, может также обладать повышенной ферментативной активностью, выбранной из фосфоенолпируваткарбоксилазной, фосфоенолпируваткарбоксикиназной, изоцитратлиазной, пируваткарбоксилазной, и активности симпортера пермеазы гексозы, и/или по меньшей мере одной пониженной энзиматической активностью, выбранной из лактатдегидрогеназной, алкогольдегидрогеназной, ацетаткиназной, фосфат ацетилтрансферазной, пируват оксидазной, изоцитратлиазной, фумаразной, 2-оксоглутарат дегидрогеназной, пируваткиназной, активности малеинового фермента, фосфоглюкозо-изомеразной, фосфоенолпируваткарбоксилазной, фосфоенолпируваткарбоксикиназной, пируватформиат лиазной, аспартат полуальдегид дегидрогеназной, фосфотрансферазной транспорта сахаров, кетогидроксиглутарат альдолазной, гомосерин-О-сукцинил-трансферазной, гомосеринкиназной, активности эффлюксного переносчика гомосерина, диаминопимелатдекарбоксилазной и/или метилглиоксальсинтазной.
В дополнительном аспекте, микроорганизм, согласно изобретению, представляющий собой Escherichia coli сверхэкспрессирует по меньшей мере один ген, выбранный из ррс, pck, aceA, galP, asd, thrA, metL, lysC E.coli; pycA L lactis, и/или имеет по меньшей мере один делетированный ген, выбранный из IdhA, adhE, ackA, pta, рохВ, focA pfIB, sad, gabABC, sfcA, maeB, ррс, pykA, pykF, mgsA, sucAB, ptsI, ptsG, pgi, fumABC, aIdA, IIdD, icIR, metA, thrB, lysA, eda, rhtA, rhtB, rhtC.
Настоящее изобретение также относится к способу получения 2,4-ДГБ, включающему этапы:
- культивирование модифицированного микроорганизма по настоящему изобретению, в подходящей культуральной среде,
- выделение 2,4-ДГБ из культуральной среды.
Указанный 2,4-ДГБ может быть дополнительно очищен.
Разделение и очистка продукта являются очень важными факторами, чрезвычайно влияющими на общую эффективность способа и себестоимость продукции. Способы выделения продукта обычно включают этапы выделения клеток, а также очистку продукта, концентрирование и сушку, соответственно.
Отделение клеток
Ультрафильтрация и центрифугирование могут быть использованы для отделения клеток от ферментационной среды. Отделение клеток из ферментативной среды часто осложняется высокой вязкостью среды. Таким образом, можно внести добавки, такие как неорганические кислоты или соли щелочных металлов, или использовать нагревание бульона с культурой, чтобы оптимизировать отделение клеток.
Получение продукта
Разнообразие ионообменных хроматографических способов может быть применено для отделения ДГБ до или после удаления биомассы. Они включают в себя использование первичных катионообменных смол, которые облегчают разделение продуктов в соответствии с их изоэлектрической точкой. Как правило, раствор наносят на смолу, а сохраненный продукт элюируют отдельно с последующим увеличением pH (например, путем добавления гидроксида аммония) в элюенте. Другой возможностью является использование ионно-обменной хроматографии с использованием фиксированного или подвижного слоя смолы. Различные хроматографические этапы, могут быть объединены для достижения адекватной чистоты продукта. Эти способы очистки являются более экономичными по сравнению с дорогостоящими этапами кристаллизации, также обеспечивая дополнительные преимущества и гибкость в отношении формы конечного продукта.
Концентрирование продукта и сушка
Способ очистки может также включать стадию сушки, которая может включать любые подходящие сушильные средства, такие как распылительный гранулятор, распылительную сушилку, барабанную сушилку, вращающуюся сушилку и туннельную сушилку. Концентрированные растворы ДГБ могут быть получены путем нагревания ферментационного бульона, при пониженном давлении, паром при 130°С с использованием многофункционального концентратора или тонкопленочного испарителя.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
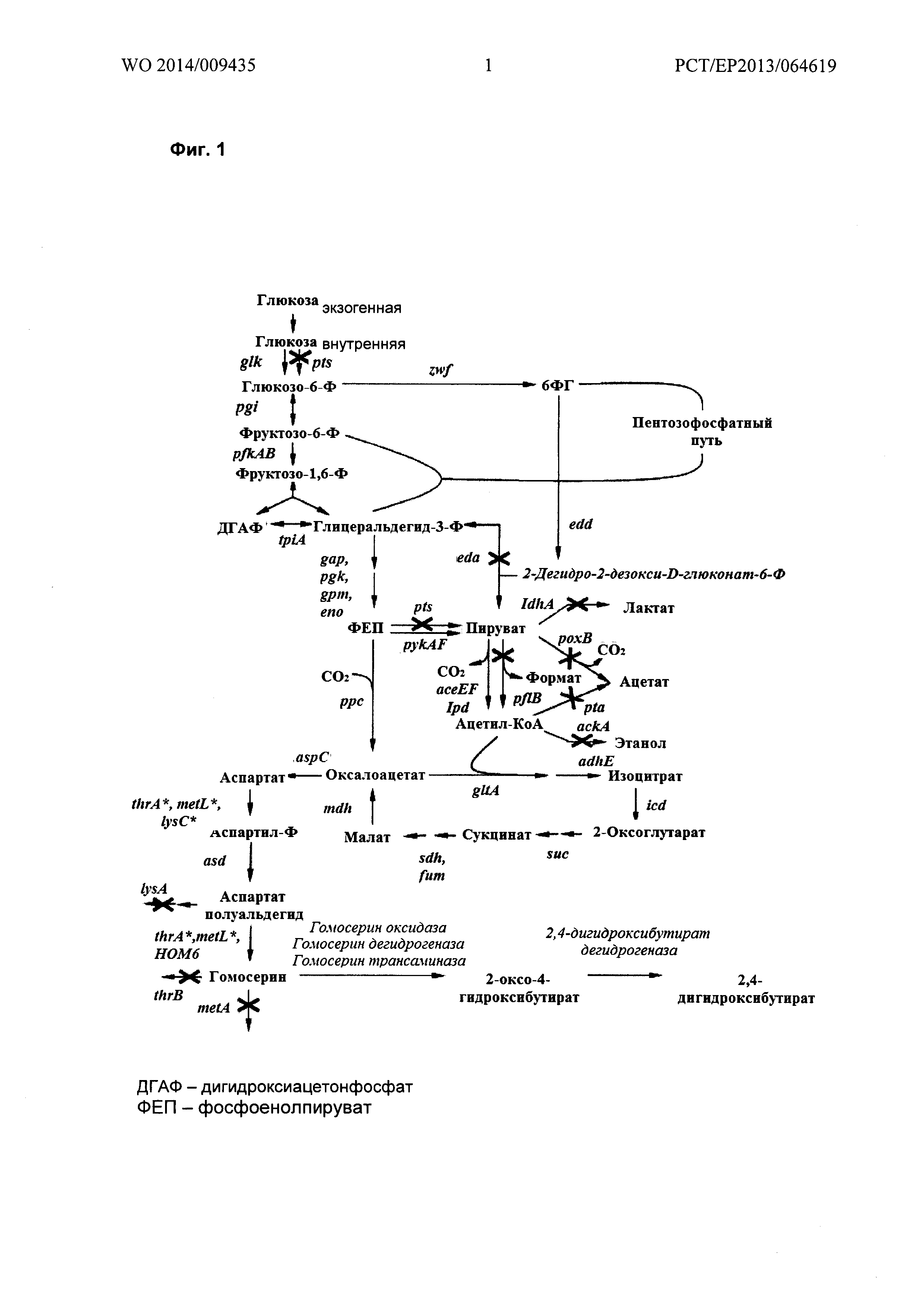
Фигура 1: способ получения 2,4-ДГБ из гомосерина, включающий два этапа, а именно, первый этап замещения первичной аминогруппы гомосерина карбонильной группой для получения ОГБ, и второй этап восстановления полученного ОГБ до 2,4-ДГБ.
Фигура 2: Специфическая активность очищенной лактатдегидрогеназы L Lactis, мутированной в положении Q85. (А) специфическая активность на ОГБ; (В) специфическая активность на пирувате; (С) специфичность субстрата выраженного, как отношение значений Vmax на ОГБ и на пирувате. Значения, выше чем 1, на графике С указывают предпочтение ОГБ (насыщение ферментативной активности не было получено для обоих субстратов для мутантных ферментов от 0 до 50 мМ ОГБ или пирувата). Активности были измерены при концентрации субстрата 20 мМ.
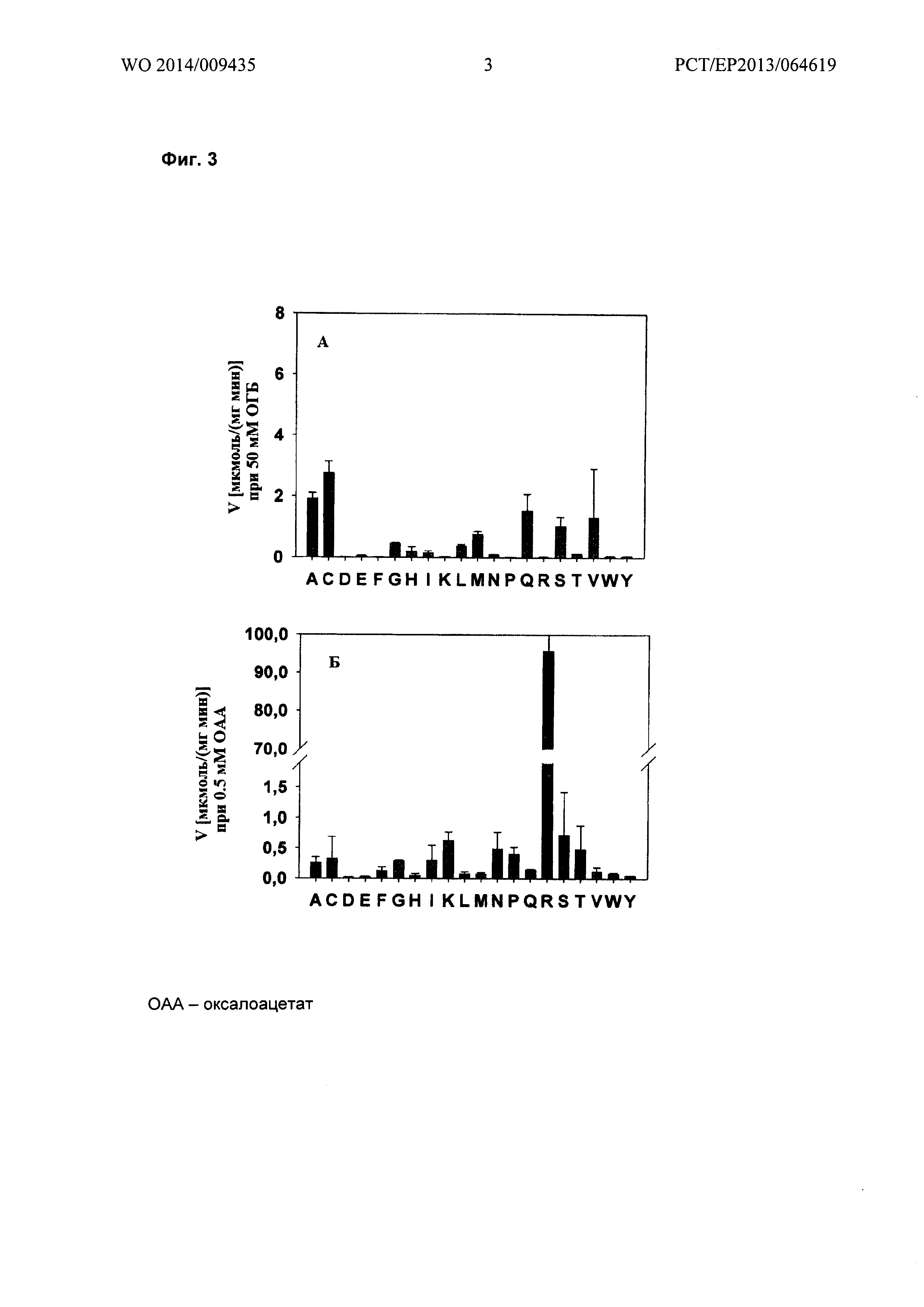
Фигура 3: Специфическая активность очищенной малатдегидрогеназы Е. Coli, мутированной в положении R81. (А) специфическая активность на ОГБ; (В) специфическая активность на оксалоацетате. Активности были измерены при концентрации субстрата 20 мМ ОГБ или 0,5 мМ оксалоацетата.
Следующие не ограничивающие примеры иллюстрируют изобретение.
ПРИМЕРЫ
Пример 1: Демонстрация активности редуктазы ОГБ
Конструирование плазмид, содержащих гены дикого типа, кодирующих лактатдегидрогеназу или малатдегидрогеназу:
Гены, кодирующие (L)-малатдегидрогеназу в Escherichia coli, Ec-MDH (SEQ ID No.1), (О)-лактатдегидрогеназу в E.coli, Ec-ldhA (SEQ ID No.3), (L)-лактатдегидрогеназу в Lactococcus lactis, LI-ldhA (SEQ ID No.5), (L)-лактатдегидрогеназу в Bacillus subtilis, Bs-Idh (SEQ ID No.7), (L)-лактатдегидрогеназу в Geobacillus stearothermophilus, Gs-ldh (SEQ ID No.9), две изоформы (L)-лактатдегидрогеназы Oryctalagus cuniculus, OC-ldhA (SEQ ID No.11 и SEQ ID No.13), амплифицировали методом ПЦР (полимеразная цепная реакция) с использованием высокоточной полимеразы Phusion™ (Fermentas) и праймеров, приведенных в Таблице 1. Геномные ДНК из штаммов E.coli MG1655, L. Lactis IL1403 и В.subtilis штамма 168 использовали в качестве матрицы. Гены Oc-IdhA и Gs-Idh оптимизировали в отношении кодонов для экспрессии в E.coli, и синтезировали с помощью MWG Eurofins. В праймеры были введены сайты рестрикции (Таблица 1) вверх от стартового кодона и вниз от стоп-кодона, соответственно, для облегчения лигирования расщепленных продуктов ПЦР по соответствующим сайтам в вектор экспрессии рЕТ28а+(Novagen) с помощью ДНК-лигазы Т4 (Fermentas), продукты лигирования трансформировали в клетки E.coli DH5a (NEB). Полученные pET28-Ec-mdh, pET28-Ec-IdhA, pET28-LI-IdhA, pET28-Bs-Idh, pET28-Gs-Idh, и pET28-Oc-IdhA плазмиды выделяли и, путем секвенирования ДНК, проверяли на наличие правильных полноразмерных последовательностей генов Е. coli mdh, Е. coli IdhA, L lactis IdhA, B. subtilis Idh, G. stearothermophilus Idh, и O.cuniculus IdhA, соответственно. Соответствующие белковые последовательности представлены в SEQ ID No.2, SEQ ID No.4, SEQ ID No.6, SEQ ID No.8, SEQ ID No.10, SEQ ID No.12 и SEQ ID No.14, соответственно.


Экспрессия ферментов: звездчатые клетки Е. coli BL21 (DE3) были трансформированы подходящими плазмидами с применением стандартных генетических протоколов (Sambrook, Fritsch, и Maniatis, 1989). Ферменты с N-концевой гекса-His меткой экспрессировали в 50 мл культур LB, которые были посеяны из ночной культуры до OD600 (оптическая плотность) 0,1 и выращивали до OD600 0,6 до этого экспрессию белка индуцировали путем добавления 1 мМ β-D-1-тиогалактопиранозид изопропила (IPTG) в культуральную среду. Через 15 ч экспрессии белка, клетки собирали центрифугированием при 4000 g при 4°С в течение 10 мин, а супернатант сливали. Клеточные осадки хранили при минус 20°С до дальнейшего анализа. Выращивание и экспрессию белка проводили при 25°С. Культуральные среды содержали 50 мкг/мл канамицина.
Очистка ферментов: замороженные клеточные осадки выделенных культур ресуспендировали в 0,5 мл лизисного буфера (50 мМ Hepes, 300 мМ NaCl, рН 7,5) и разрушались с помощью четырех последовательных этапов обработки ультразвуком (интервал обработки ультразвуком: 20 с, мощность 30%, соникатор: Bioblock Scientific, VibraCell™ 72437). Клеточные остатки удаляли центрифугированием неочищенных экстрактов в течение 15 мин при 4°С, при 4000 g, а прозрачный супернатант оставляли. РНК и ДНК удаляли из экстрактов путем добавления 15 мг/мл стрептомицина сульфата (Sigma), и центрифугировали образцы при 13000 g в течение 10 мин при 4°С, супернатант сохраняли. Чистый белковый экстракт инкубировали в течение 1 ч при 4°С с 0,75 мл (объем слоя) кобальта аффинной смолы Talon™ (Clontech). Суспензию центрифугировали при 700 g на настольной центрифуге, а супернатант удаляли. Смолу промывали 10 объемами слоя промывочного буфера (50 мМ Hepes, 300 мМ NaCl, 15 мМ имидазола, рН 7,5) перед тем как белки элюировали 0,5 мл буфером для элюции (50 мМ Hepes, 300 мМ NaCl, 250 мМ имидазола, рН 7,5). Чистота элюированных ферментов была проверена путем анализа SDS-PAGE. Концентрации белка оценивались с помощью метода Брэдфорда (Bradford (1976, Anal Biochem 72: 248-54). Для стабилизации фермента лактатдегидрогеназы, буфер для элюции систематически замещали 100 мМ фосфатного буфера до рН 7.0. Образец белка переносили на ультрацентрифужный фильтр Amicon™ (срез 10 кДа), и центрифугировали 8 мин при 4000 д, при 4°С для удаления буфера. Белок разбавляли в фосфатном буфере и процедуру повторяли 4 раза.
Ферментативный анализ: Реакционная смесь содержала 60 мМ Hepes (рН 7), 50 мМ хлорида калия, 5 мМ MgCl2, 0,25 мМ НАДН (никотинамидадениндинуклеотид Н), (необязательно 5 мМ фруктозо-1,6-бисфосфата) (все продукты от Sigma) и соответствующее количество очищенных малат- или лактатдегидрогеназ или клеточного экстракта. Реакции начинали добавлением соответствующих количеств 2-оксо-4-гидроксибутирата (ОГБ), пирувата, либо оксалоацетата (ОАА). Ферментативный анализ проводился при температуре 37°С в 96-луночных плоскодонных микротитрационных планшетах в конечном объеме 250 мкл. Реакции характеризовали характерным поглощением НАДН при 340 нм (εНАДН равно 6,22 мМ-1 см-1) в микропланшетном ридере (BioRad 680XR).
ОГБ синтезировали путем инкубации 125 мМ гомосерина с (L)-аминокислотной оксидазой змеиного яда (1,25 ед/мл, Sigma) и каталазой (4400 ед/мл, Sigma) в 100 мМ Трис-буфера при рН 7,8 в течение 90 мин при 37°С: Затем реакционную смесь очищали на ультрацентрифужном фильтре Amicon™ со срезом 10 кДа, для удаления ферментов (способ адаптирован Wellner и Lichtenberg, 1971).
ОГБ измерили количественно путем смешивания 100 мкл тестируемого раствора с 1 мл раствора, содержащего 1 М арсената натрия и 1 М борной кислоты при рН 6,5. Смесь инкубировали при комнатной температуре в течение 30 мин, а поглощение при 325 нм использовали для количественной оценки ОГБ. Соотношение между поглощением и концентрацией кетона было откалибровали с использованием растворов пирувата известных концентраций (способ адаптированный Wellner & Lichtenberg, 1971)). Типичный выход ОГБ данным способом составил 90%.
Результаты: Кинетические параметры приведены в Таблице 2 для тестированных ферментов на их природных субстратах и ОГБ. Существенная редуктазная активность ОГБ была обнаружена для всех лактатдегидрогеназ различного биологического происхождения. Малатдегидрогеназа Е. coli, Mdh, имела только очень незначительную активность на ОГБ. Дегидрогеназа 2-оксо-кислот с разветвленной цепью, РапЕ Llactis также имела значительную активность на ОГБ.
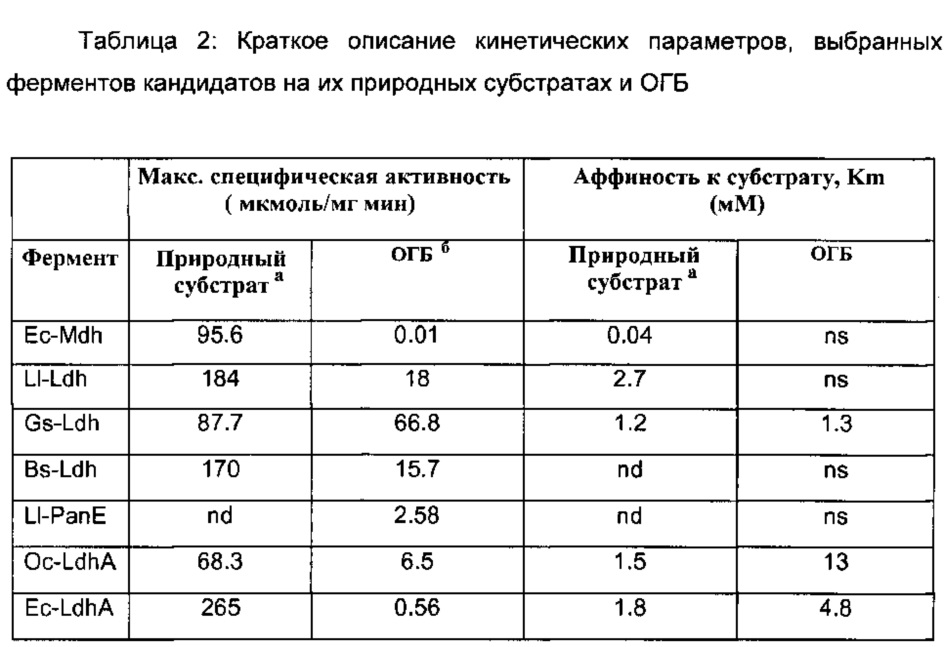

Пример 2: Конструирование фермента лактатдегидрогеназы с улучшенной активностью редуктазы ОГБ
Сайт-направленный мутагенез гена IdhA Llactis проводили с применением плазмиды pET28-LI-IdhA в качестве матрицы. Точечные мутации для изменения последовательности аминокислот были внесены с помощью ПЦР (Phusion 1 ед., HF буфер 20 об./об.%, dNTPs (дезоксинуклеозидтрифосфаты) 0,2 мМ, прямой и обратный праймеры 0,04 μМ каждый, матричная плазмида 30-50 нг, вода) с использованием пары олигонуклеотидов, перечисленых в Таблице 3. Гены, мутированные с помощью ПЦР, содержали новые сайты рестрикции, перечисленные в Таблице 3 (введенные с помощью молчащих мутации) в дополнение к функциональным мутациям, чтобы облегчить идентификацию мутантных клонов. ПЦР-продукт расщепляли DpnI при 37°С в течение 1 ч для удаления матричной ДНК, и трансформировали в компетентные клетки E.coli DH5α (NEB). Мутированные плазмиды идентифицировали с помощью анализа сайта рестрикции и проверяли на наличие требуемой мутации путем секвенирования ДНК.


Мутантные ферменты экспрессировали, очищали и тестировали на ОГБ и пируват редуктазную активность, как описано в Примере 1. Измерения активности для обоих субстратов приведены на Фигуре 2. Результаты показывают, что замена Gln85 предпочтительно аланином, цистеином, аспарагином, или метионином приводит к увеличению специфичности фермента к ОГБ, и/или увеличению максимальной специфической активности редуктазы ОГБ.
Мутация Q85N в LI-Ldh была объединена с мутацией 1226V. Было показано, что этот обмен имеет большое позитивное влияние на сродство к субстрату для ОГБ.

Пример 3: Конструирование фермента малатдегидрогеназы с улучшенной редуктазой ОГБ
Сайт-направленный мутагенез гена mdh E.coli проводился, как описано в примере 2, с использованием праймеров, приведенных в Таблице 5. Плазмида pET28-Ec-mdh была использована в качестве матрицы.
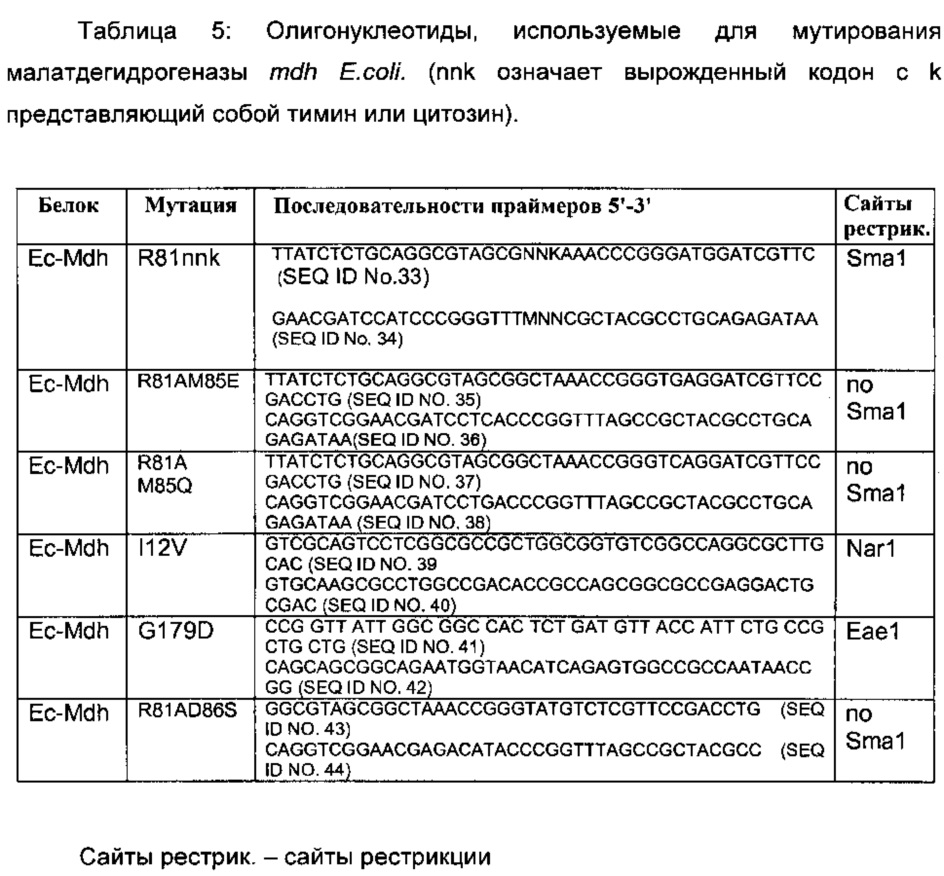
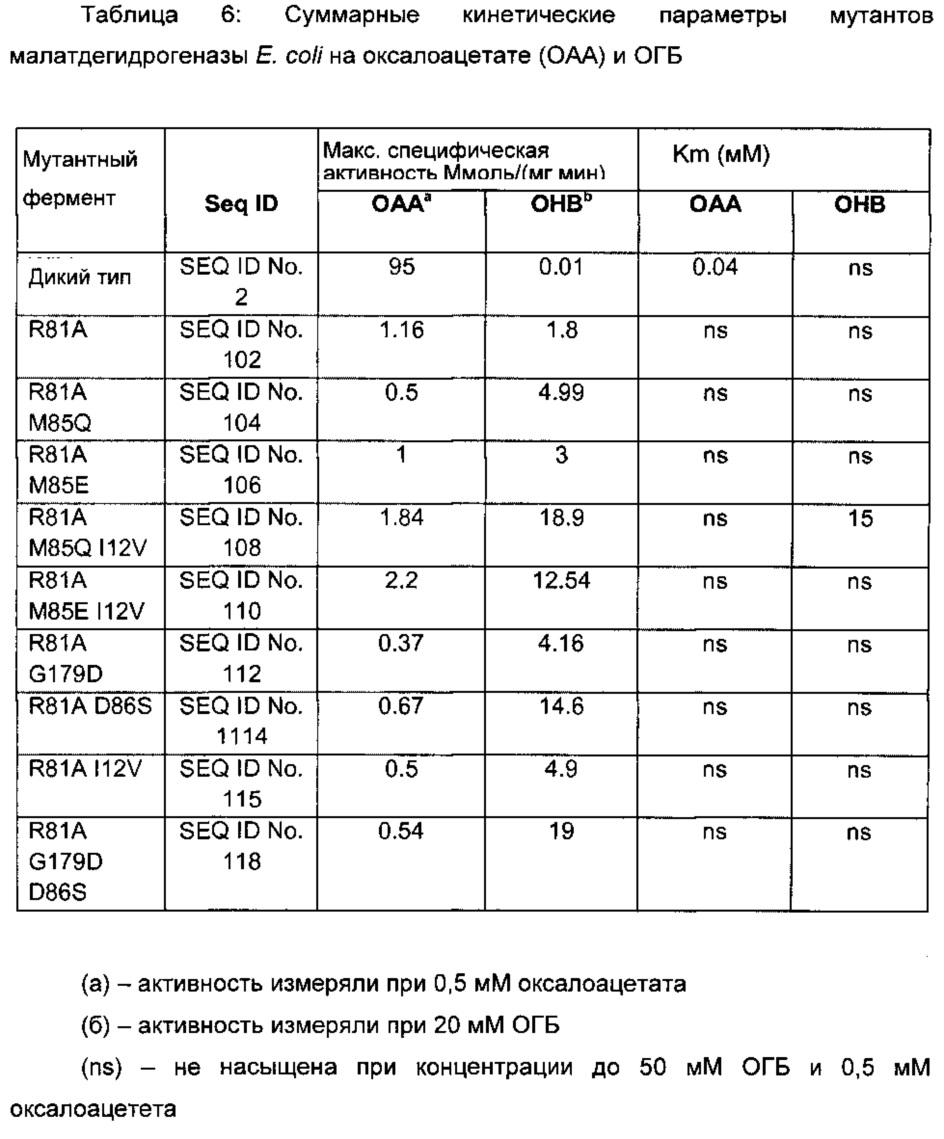
Мутантные ферменты выделяли, очищали и тестировали на активность ОГБ и оксалоацетат редуктаз, как описано в Примере 1. Измерения активности на ОГБ и оксалоацетате приведены на Фигуре 3. Результаты показывают, что замена Arg81 аланином, цистеином, глицином, гистидином, изолейцином, лейцином, метионином, аспарагином, глутамином, серином, треонином или валином, придает значительную активность редуктазе ОГБ и сопутствующее снижение активности оксалоацетат редуктазы.
Мутация R81A в Ec-Mdh была объединена с дополнительными изменениями в последовательности белка. Результаты приведены в Таблице 6. Было показано, что введение мутаций M85Q, М85Е, 112V, D86S или G179D приводят к увеличению активности на ОГБ.
Пример 4: Демонстрация трансаминазной активности гомосерина для выбранных трансаминаз.
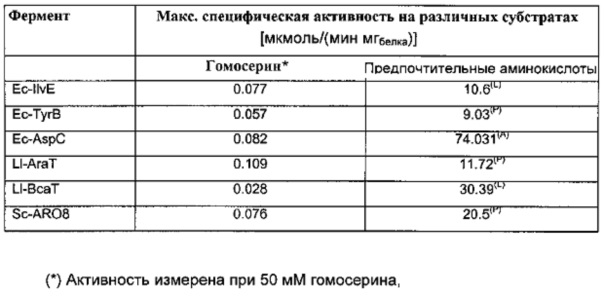
Гены, кодирующие различные трансаминазы в E.coli, S.cerevisiae, и L.lactis были амплифицированы с помощью ПЦР с использованием высоко точной полимеразы Phusion™ (Finnzymes) и праймеров, указанных в Таблице 7. Геномные ДНК E.coli MG1655, S. cerevisiae BY4741 и L. lactis IL1403 были использованы в качестве матриц. В праймеры были введены сайты рестрикции (Таблица 7) вверх от стартового кодона и вниз от стоп-кодона, соответственно, для облегчения лигирования расщепленных продуктов ПЦР по соответствующим сайтам вектора экспрессии рЕТ28а+(Novagen), с использованием ДНК-лигазы Т4 (Biolabs). Продукты лигирования трансформировались в клетки E.coli DH5a. Полученные плазмиды были выделены, путем секвенирования ДНК было показано наличие правильных последовательностей полной длины соответствующих генов. Ссылки на соответствующие белковые последовательностей приведены в Таблице 7.


Ферменты выделяли и очищали, как описано в Примере 1, и тестировали на гомосерин трансаминазную активность при условиях описанных ниже.
Ферментативный анализ: трансаминазная активность нескольких аминотрансфераз кандидатов была вычислена с 2-оксиглутаратом, применяемым в качестве акцептора аминогрупп. Трансаминазные реакции проводились с использованием гомосерина и предпочтительных аминокислот ферментов. За реакциями следовало аминокислотно-зависимое окисление НАДН в объединенных дегидрогеназных реакциях.

Дегидрогеназа: 2-оксо-кислота+НАДН →2-гидрокси-кислота+НАД+
Реакционная смесь содержала 60 мМ Hepes (рН 7), 50 мМ хлорида калия, 5 мМ MgCl2, 4 мМ 2-оксоглутарата, 0,1 мМ пиридоксаль-5'-фосфата (PLP), 0,25 мМ НАДН, (необязательно, 5 мМ фруктозо-1,6-бисфосфата) (все продукты от Sigma), 4 ед/мл вспомогательной дегидрогиназы 2-гидроксикислот, а также соответствующие количества очищенной аминотрансферазы или клеточного экстракта. Вспомогательный фермент дегидрогеназа очищали PanE L. Lactis в случае аминокислот фенилаланина и лейцина (Chambellon, Rijnen, Lorquet, Gitton, van HylckamaVlieg, Wouters, &Yvon, 2009), малатдегидрогеназы (Sigma) в случае аспартата, и мышечной (L)-лактатдегидрогеназы (Sigma) кролика, когда гомосерин применяли в качестве исходного субстрата. Реакции начинали добавлением 50 мМ аминокислоты.
Ферментативный анализ проводили при температуре 37°С в 96-луночных плоскодонных микротитрационных планшетах в конечном объеме 250 мкл. Реакция сопровождалась характерным поглощением НАД(Р)Н при 340 нм (εНАДРН=6,22 мМ-1 см-1) в микропланшетном ридере (BioRad 680XR)
Результаты: Кинетические параметры различных аминотрансфераз приведены в Таблице 8. Значительная активность гомосерин трансаминазы была найдена для перечисленных ферментов трансаминаз.

Пример 5: Конструирование плазмид для сверхэкспрессии ферментов метаболизма гомосерина
Конструирование плазмид pTAC-op-HMS1 и pACT3-op-HMS1
Плазмиду pET28-LYSCwt конструировали путем амплификации гена lysC методом ПЦР с использованием высокой точной полимеразы Phusion™ (Finnzymes) и прямого и обратного праймеров5'CACGAGGTACATATGTCTGAAATTGTTGTCTCC3' (SEQ ID No.71) и5'CTTCCAGGGGATCCAGTATTTACTCAAAC3' (SEQ ID No.72), с введением сайтов рестрикции NdeI и BamH1 выше стартового кодона и ниже стоп-кодона; соответственно. Геномная ДНК из штамма E.coli MG1655 использовалась в качестве матрицы. ПЦР продукт расщепляли NdeI и BamH1, лигировали в соответствующие сайты вектора экспрессии рЕТ28а (Novagen) с использованием ДНК-лигазы Т4 (Biolabs), и трансформировали в клетки E.coli DH5α. Полученную плазмиду pET28-LYSCwt выделяли и, путем секвенирования ДНК, проверяли наличие полноразмерного гена lysC, имеющего правильную последовательность (SEQ ID No.73).
Сайт-направленный мутагенез lysC, для облегчения ингибирования лизином, осуществляли с использованием плазмиды pET28-LYSCwt в качестве матрицы. Точечную мутацию для изменения аминокислотной последовательности в положении 250 глутамата в лизин (E250K, SEQ ID №36) вводили с помощью ПЦР (Phusion 1U, HF буфера 20 об./об.%, 0,2 мМ dNTPs, прямой и обратный праймер 0,04 мкМоль каждый, матричная плазмида 50 нг, вода) с использованием олигонуклеотидов5'GCGTTTGCCGAAGCGGCAAAGATGGCCACTTTTG3' (SEQ ID No.74) и5'CAAAAGTGGCCATCTTTGCCGCTTCGGCAAACGC3' (SEQ ID No.75). ПЦР продукт (SEQ ID No.35), расщепляли с помощью Dpnl при 37°C в течение 1 часа для удаления матричной ДНК, и трансформировали в компетентные клетки E.coli DH5a (NEB). Мутантную плазмиду pET28-LYSC* идентифицировали с помощью анализа сайта рестрикции и проверяли на наличие желаемой мутации путем секвенирования ДНК.
Плазмиду pET28-ASDwt конструировали путем амплификации гена asd E.Coli методом ПЦР с использованием высокой точной полимеразы Phusion™ (Finnzymes) и прямого и обратного праймеров5'TATAATGCTAGCATGAAAAATGTTGGTTTTATCGG3' (SEQ ID No. 76) и5'TATAATGGA-TCCTTACGCCAGTTGACGAAGC3' (SEQ ID No. 77), с введением сайтов рестрикции Nhel и BamHI вверх от стартового кодона и вниз от стоп-кодона, соответственно. Геномную ДНК E.coli DH5a использовали в качестве матрицы. ПЦР продукт расщепляли Ndel и BamHI, лигировали по соответствующим сайтам в вектор экспрессии рЕТ28а (Novagen), с использованием ДНК-лигазы Т4 (Biolabs), и трансформировали в клетки E.coli DH5a. Полученную плазмиду pET28-ASDwt выделяли и, путем секвенирования ДНК, проверяли наличие полноразмерного asd гена, имеющего правильную последовательность (SEQ ID No.98).
Плазмиду pET28-HOM6wt конструировали путем амплификации гена НОМ6 S.cerevisiae, методом ПЦР с использованием высокой точной полимеразы Phusion™ (Finnzymes) и прямого и обратного праймеров5'TATAATCATATGAGCACTAAAGTTGTTAATG3' (SEQ ID No.78) и5'TATAATGGATC-CCTAAAGTCTTTGAGCAATC3' (SEQ ID No.79), с введением сайтов рестрикции NdeI и BamHI вверх от стартового кодона и вниз от стоп-кодона, соответственно. Геномная ДНК S.cerevisiae BY4741 использовали в качестве матрицы. ПЦР продукт расщепляли NdeI и BamHI, лигировали по соответствующим сайтам в вектор экспрессии рЕТ28а (Novagen), с использованием лигазы Т4 (Biolabs), и трансформировали в клетки E.coli DH5α. Полученную плазмиду pET28-HOM6wt выделяли и, путем секвенирования ДНК, проверяли наличие полноразмерного гена НОМ6, имеющего правильную последовательность (SEQ ID No. 97).
Плазмида pET28-LYSC* была использована в качестве основы для конструирования плазиды pTAC-op-HMS, что сделало возможным экспрессию лизин-нечувствительной аспартаткиназы, аспартат полуальдегид дегидрогеназы и гомосерин дегидрогеназы под индуцибельным tac промотором.
Ген asd получали методом ПЦР из pET28-asdwt. Весь кодирующий регион и часть области выше, содержащую сайт связывания рЕТ28 с рибосомами (rbs) и внутрирамочную N-концевая His-Tag метку амплифицировали методом ПЦР с использованием полимеразы высокой точности Phusion™ (Finnzymes) и прямого и обратного праймеров5'TATAAGGATCCGTTTAACTTTAAGAAGGAGATATACCATGGG3' (SEQ ID No. 80) и5'TATAAGAATTCTTACGCCAGTTGACGAAG3' (SEQ ID No. 81), с введением сайтов рестрикции BamHI и EcoRI вверх по течению от rbs и вниз по течению от стоп-кодона, соответственно. ПЦР продукт расщепляли рестриктазами BamHI и EcoRI, лигировали по соответствующим сайтам pET28-LYSC*, с использованием ДНК-лигазы Т4 (Biolabs), и трансформировали в клетки E.coli DH5α. Полученную pET28-LYSC*-ASD плазмиду выделяли и, путем секвенирования ДНК, проверяли на наличие правильной последовательности.
Ген НОМ6 получали методом ПЦР из pET28-HOM6wt. Всю кодирующую область и часть области выше по течению, содержащую сайт связывания рЕТ28 с рибосомами и внутрирамочную N-концевую His-Tag метку, амплифицировали методом ПЦР с использованием высоко точной полимеразы Phusion™ (Finnzymes), прямого праймера5'TATAAGCGGCCGCGTTTAACTTTAAGAAGGAGATAT3' (SEQ ID No. 82), и обратного праймера5'TATAAACTCGAGCCTAAAGTCTTTGAGCAAT3' (SEQ ID No. 83), с введением сайтов для рестрикции NotI и PspXI вверх по течению от rbs и вниз по течению от стоп-кодона, соответственно. ПЦР продукт расщепляли NotI и PspXI, лигировали по соответствующим сайтам в pET28-LYSC*-ASD, с использованием ДНК-лигазы Т4 (Biolabs), и трансформировали в клетки E.coli DH5α. Полученную плазмиду pET28-op-HMS1 выделяли и, путем секвенирования ДНК, проверяли на наличие правильной последовательности.
5' регион вверх по течению промотора, одновременно регулирующий экспрессию трех генов (т.е. промотор Т7 в рЕТ28а+), может быть заменен на любой другой промотор, индуцируемый или конститутивный, расщеплением плазмиды SphI и XbaI и клонированием другой промоторной области с подходящими сайтами рестрикции.
В настоящем неисключительном примере, промотор Т7 основной рЕТ28а+был заменен искусственным IPTG-индуцируемым tac промотором (de Boer et al., 1983). Tac промотер был получен из плазмиды рЕХТ20 (Dykxhoorn et al., 1996) путем расщепления этой плазмиды Sphl и Xbal. Фрагмент ДНК, содержащий промотор, очищали и клонировали в pET28-op-HMS1, расщепленную Sphl и Xbal, с получением pTAC-op-HMS1. Полученную рТАС-op-HMS плазмиду выделяли и, путем секвенирования ДНК, проверяли на наличие правильной последовательности.
Оперон, содержащий кодирующие последовательности lysC*, asd, и НОМ6 амплифицировали методом ПЦР с плазмиды pTAC-op-HMS1 с использованием праймеров 5'-TATAAAGATCTTAGAAATAATTTTGTTTA-3' (SEQ ID No.84) и 5'-TATAATCTAGACTAAAGTCTTTGAGCAAT-3' (SEQ ID No.85), с введением сайтов рестрикции BgIII и XbaI на 5' и 3' концах ПЦР-фрагмента соответственно. Фрагмент очищали, расщепляли BgIII и XbaI и клонировали по соответствующим сайтам в рАСТ3 (Dykxhoorn et al., 1996), с получением вектора pACT3-op-HMS1. Полученную pACT3-op-HMS1 плазмиду выделяли и, путем ДНК секвенирования, проверяли на наличие правильной последовательности.
Конструирование плазмид pEXT20-op-HMS2 и pACT3-op-HMS2
Плазмиды pET28-thrAwt конструировали путем амплификации гена thrA E.coli кодирующего бифункциональный фермент аспартаткиназу/ гомосерин дегидрогеназу методом ПЦР с использованием высоко точной полимеразы Phusion™ (Finnzymes) и прямых и обратных праймеров 5'-TATAATCATATGCGAGTGTTGAAGTTCG-3' (SEQ ID No. 86) и 5'-TATAATGGATCCTCAGACTCCTAACTTCCA-3' (SEQ ID No. 87), с введением сайтов рестрикции NdeI и BamHI вверх по течению от стартового кодона и вниз по течению стоп-кодона, соответственно. Геномную ДНК из штамма E.coli MG1655 использовали в качестве матрицы. ПЦР продукт расщепляли NdeI и BamHI, лигировали по соответствующим сайтам в вектор экспрессии рЕТ28а+(Novagen) с помощью ДНК-лигазы Т4 (Biolabs), и трансформировали в NEB 5-альфа-компетентные клетки E.coli (NEB). Полученную плазмиду pET28-thrAwt выделяли и, путем секвенирования ДНК, проверяли на наличие полноразмерного гена thrA имеющего правильную последовательность (SEQ ID No.88). Соответствующий белок представлен SEQ ID No.89.
Аспартаткиназу/гомосерин дегидрогеназу с сильно пониженной чувствительностью к ингибированию треонином конструировали с помощью сайт-направленного мутагенеза, заменой серина в положении 345 на фенилаланин (S345F). Сайт-направленный мутагенез проводили с использованием прямого и обратного праймеров 5'-TGTCTCGAGCCCGTATTTTCGTGGTGCTG-3' (SEQ ID No.90) и 5'-CAGCACCACGAAAATACGGGCTCGAGACA-3' (SEQ ID No.91), и плазмиды pET28-thrAwt в качестве матрицы. Одиночная точечная мутация для изменения аминокислотной последовательности была введена с помощью метода ПЦР (Phusion 1 ед, HF буфер 20 об./об%, 0,2 мМ dNTPs, прямой и обратный праймеры 0,04 мкл каждый, матричная плазмида 30-50 нг, вода). Плазмиды, созданные с помощью ПЦР, содержали новый сайт рестрикции для XhoI (подчеркнуто), введенный молчащей мутацией в дополнение к функциональной мутации, для облегчения обнаружения мутантных клонов. Продукты ПЦР расщепляли DpnI при 37°С в течение 1 ч, для удаления матричной ДНК, и трансформировали в DH5a компетентные клетки E.coli (NEB). Мутантную плазмиду pET_Ec_thrA_S345F идентифицировали с помощью анализа сайта рестрикции и проверяли на наличие желаемой мутации путем секвенирования ДНК.
thrAS345F кодирующий регион бифункциональной аспартаткиназы/гомосерин дегидрогеназы E.coli был получен с использованием плазмиды pET_Ec_thrA_S345F в качестве матрицы (SEQ ID NO: 92) vtnjljv ПЦР. Всю кодирующую область амплифицировали методом ПЦР с использованием высоко точной полимеразы Phusion™ (Finnzymes) и прямого и обратного праймеров 5'-TATAATGAGCTCGTTTAACTTTAAGAAGGAGATATACCATGCGAGTGTTGAAGTTCGGCG-3' (SEQ ID No. 93) и 5'-TATAATCCCGGGTCAGACTCCTAACTTCCA-3' (SEQ ID No. 94), с введением сайтов для рестрикции SacI и XmaI (подчеркнуты) вверх по течению от стартового кодона и вниз от стоп-кодона, соответственно. Прямой праймер включает сайт связывания рибосом (жирным шрифтом) последовательности рЕТ28. ПЦР продукт расщепляли SacI и XmaI, лигировали по соответствующим сайтам в рЕХТ20 или рАСТ3 (Dykxhoorn, St Pierre, & Linn, 1996), с использованием ДНК-лигазы Т4 (Biolabs), и трансформировали в клетки DH5α E.coli. Полученные pEXT20-op-HMS2_этап1 и рАСТ3-ор-HMS2_3Tan1 плазмиды выделяли и путем секвенирования ДНК, проверяли на наличие правильной последовательности.
Аспартат полуальдегид дегидрогеназу Escherichia coli asd амплифицировали методом ПЦР с использованием высоко точной полимеразы Phusion™ (Finnzymes) и прямого и обратного праймеров 5'-TATAATCCCGGGGTTTAACTTTAAGAAGGAGATATACCATGAAAAATGTTGGTTTTATCGGC-3' (SEQ ID No. 95) и 5'-TATAATGGATCCTTACGCCAGTTGACGAAG-3'(SEQ ID No.96) с введением сайтов рестрикции XmaI и BamHI вверх от стартового кодона и вниз от стоп-кодона, соответственно (SEQ ID No. 98). Прямой праймер включает сайт связывания рибосом последовательности рЕТ28. Геномную ДНК Е. coli MG1655 использовали в качестве матрицы. ПЦР продукт расщепляли XmaI и BamHI, лигировали по соответствующим сайтам pEXT20-op-HMS2_этап1 и pACT3-op-HMS2_этап1, непосредственно вниз по течению гена E.coli thrA, с использованием ДНК-лигазы Т4 (Biolabs), и трансформировали в клетки DH5α Е. coli. Полученные pEXT20-op-HMS2 и pACT3-op-HMS2 плазмиды выделяли и, путем секвенирования ДНК, проверяли на наличие правильной последовательности.
Пример 6: Конструирование плазмиды для экспрессии фосфоенолпируват (ФЕП) карбоксикиназы, ФЕП-карбоксилазы, пируваткиназы, пируваткарбоксилазы, ферментов изоцитратлиазы и симпортера пермеазы галактозы:
Плазмиду рАСТ3-pck, несущая ген рек ФЕП карбоксикиназы E.coli, конструировали путем амплификации последовательности, кодирующей рек ген с использованием геномной ДНК E.coli MG1655 в качестве матрицы, и прямого и обратного праймеров, соответственно,5'TATAATCCCGGGATGCGCGTTAACAATGGTTTGACC3' (SEQ ID No. 119) и5'TATAATTCTAGATTACAGTTTCGGACCAGCCG3' (SEQ ID No. 120). Фрагмент ДНК расщепляли XmaI и XbaI, лигировали по соответствующим сайтам вектора экспрессии рАСТ3 (Dykxhoorn и et al., 1996) с использованием ДНК-лигазы Т4 (Biolabs), и трансформировали в клетки E.coli DH5α. Трансформанты отбирали на твердой LB среде (lysogeny broth - литическая среда), содержащей хлорамфеникол (25 мкг/мл). Полученную в результате плазмиду выделяли, и правильность вставки гена рек проверяли секвенированием. Плазмиды рАСТ3-асеА, рАСТ3-ррс, рАСТ3-galP, рАСТ3-pck и рАСТ3-русА несущие, соответственно, асеА, ррс, galP или рек (все Е. coli) или русА Lactococcus lactis конструировали аналогично, с использованием праймеров, перечисленных в Таблице 9.
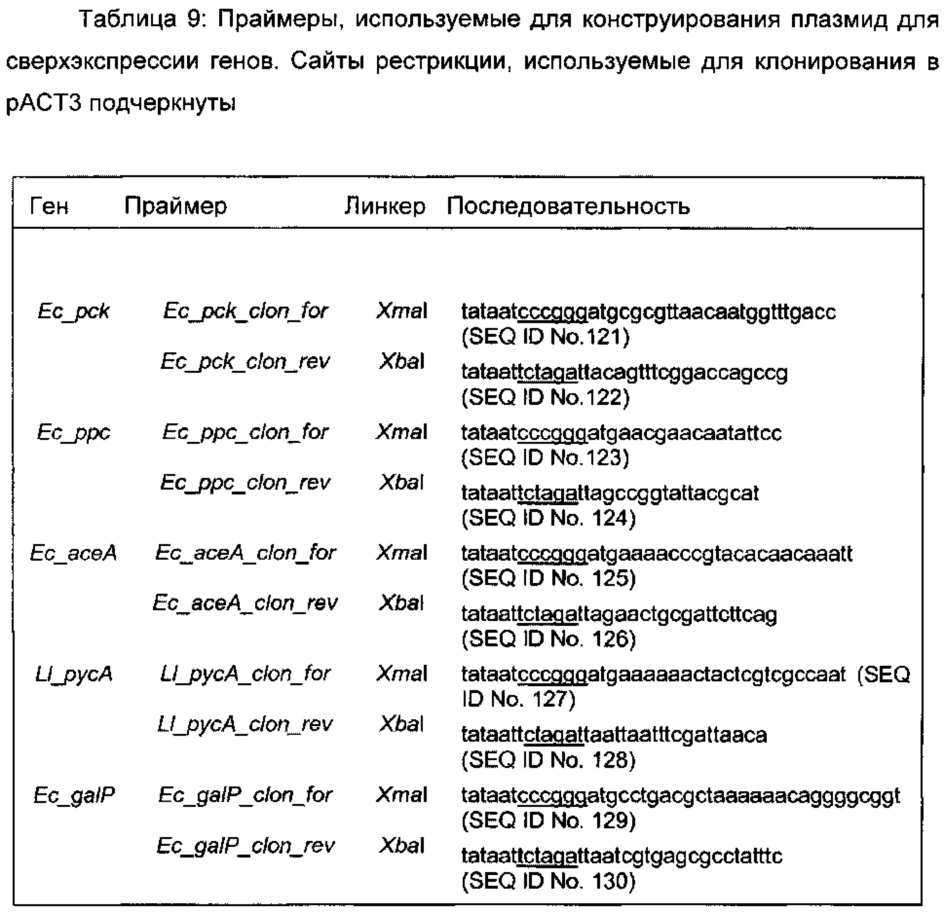
Пример 7: Конструирование плазмиды для сверхэкспрессии гомосерин трансаминазы и редуктазы ОГБ
Кодирующую последовательность аминотрансаминазы с разветвленной цепью NvE E.coli амплифицировали методом ПЦР с использованием прямого и обратного праймеров 5'-ACAATTTCACACAGGAAACAGAATTCGAGCTCGGTACCGTTTAACTTTAAGAAGGAGATATACCATGACCACGAAGAAAGCTGATTAC-3' (SEQ ID No. 131) и 5'-GGATAACTTTTTTACGTTGTTTATCAGCCATGGTATATCTCCTTCTTAAAGTTAAACGGATCCTTATTGATTAACTTG-3' (SEQ ID No. 132), соответственно, с плазмиды pET28-Ec-ilvE (Пример 4) в качестве матрицы. Кодирующую последовательность лактатдегидрогеназы, LdhA L. Lactis, амплифицировали методом ПЦР с использованием прямого и обратного праймеров 5'-TAATATGGATCCGTTTAACTTTAAGAAGGAGATATACCATGGCTGATAAACAACGTAAAAAAGTTATCC-3' (SEQ ID No. 133) и 5'-CAATGCGGAATATTGTTCGTTCATGGTATATCTCCTTCTTAAAGTTAAACTCTAGATTAGTTTTTAACTGCAGAAGCAAATTC-3' (SEQ ID No. 134), соответственно, с плазмиды pET28-LI-IdhA (Пример 1) в качестве матрицы. Амплифицированные ПЦР-фрагменты были объединены в ПЦР с перекрывающимися праймерами, путем добавления 150 нг каждого фрагмента в 50 мкл реакционной смеси и осуществлении ПЦР с использованием праймеров 5'-ACAATTTCACACAGGAAACAGAATTCGAGCTCGGTACCGTTTAACTTTAAGAAGGAGATATACCATGACCACGAAGAAAGCTGATTAC-3' (SEQ ID No. 135) и 5'-CAATGCGGAATATTGTTCGTTCATGGTATATCTCCTTCTTAAAGTTAAACTCTAGATTAGTTTTTAACTGCAGAAGCAAATTC-3'(SEQ ID No.136). Полученный ПЦР-фрагмент очищали, расщепляли с помощью KpnI и XbaI, и лигировали по соответствующим сайтам в рЕХТ20 (Dykxhoorn, St Pierre, & Linn, 1996) с использованием ДНК-лигазы ТА (Fermentas). Продукт лигирования трансформировали в E.coli DH5α. Полученную в результате плазмиду рЕХТ20-ДГБ выделяли, и, путем секвенирования ДНК, проверяли наличие правильной, кодирующей полноразмерной последовательности Ec-ilvE и LI-IdhA. Плазмиду затем трансформировали в полученный мутантный штамм E.coli MG1655 и тестировали в отношении получения ДГБ.
Пример 8: Конструирование оптимизированных штаммов для получения ДГБ
Несколько генов были нарушены в штамме E.coli MG1655 с целью оптимизации перераспределения потока углерода и добавления кофактора для получения ДГБ. Генные делеции были введены с использованием метода фаговой трансдукции, или метода рекомбиназы лямбда-Red в соответствии с Datsenko et al. (Datsenko & Wanner, 2000).
Протокол для внесения делеций генов с использованием метода фаговой трансдукции:
Штаммы, несущие желаемые одиночные делеции были получены из коллекции Keio (Baba et al., 2006). Фаговые лизаты отдельных мутантов с делецией получали путем посева на 10 мл среды LB, содержащей 50 мкг/мл канамицина, 2 г/л глюкозы и 5 мМ CaCl2, 100 мкл ночной прекультуры. После инкубации в течение 1 ч при 37°С, добавляли 200 мкл фагового лизата, полученного из дикого типа штамма MG1655, и культуры инкубировали в течение от 2 до 3 ч до тех пор, пока клеточный лизис не был завершен. После добавления 200 мкл хлороформа, клеточные препараты сначала энергично перемешивали, а затем центрифугировали в течение 10 мин при 4500×g. Прозрачный лизат собирали и хранили при 4°C.
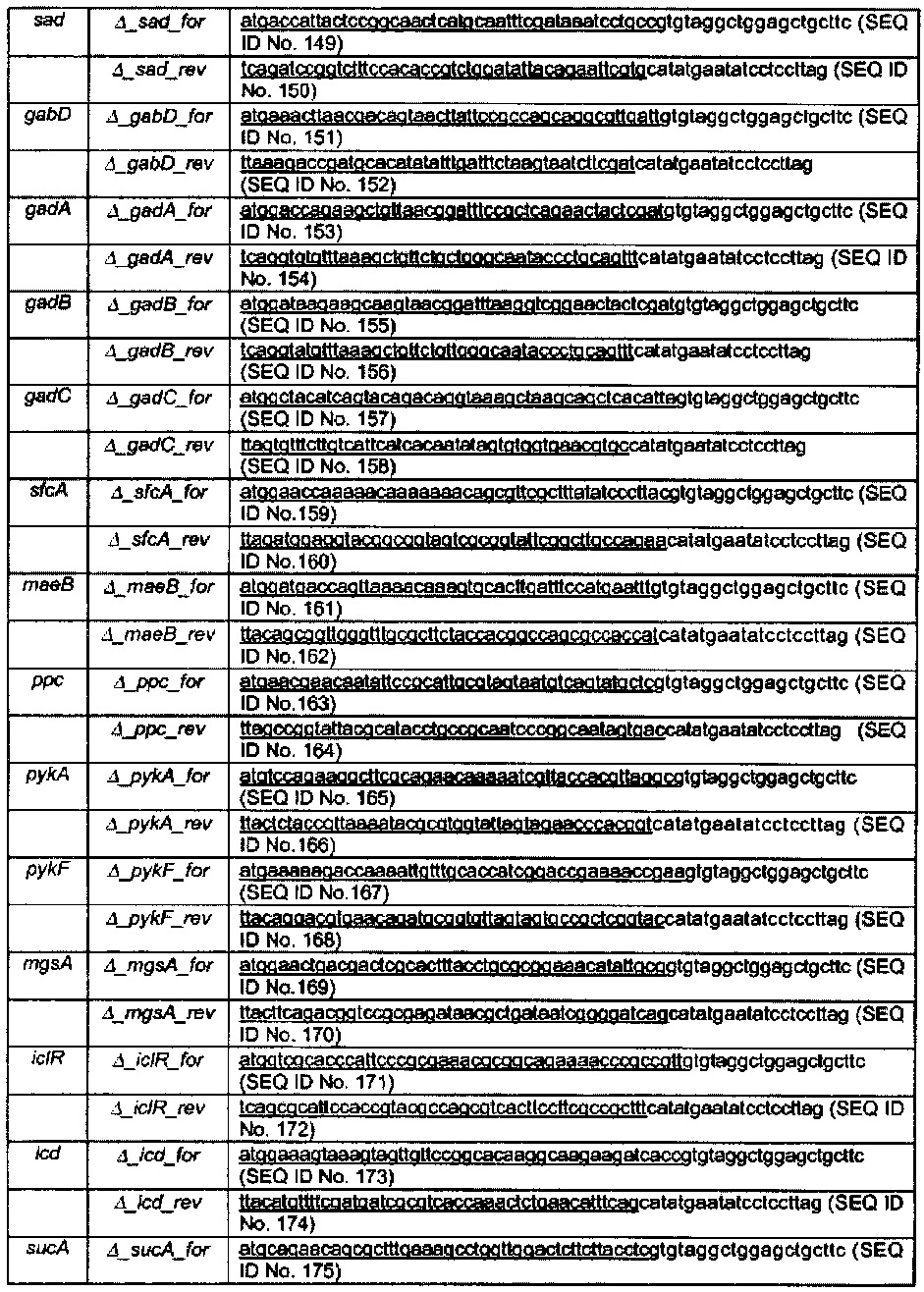
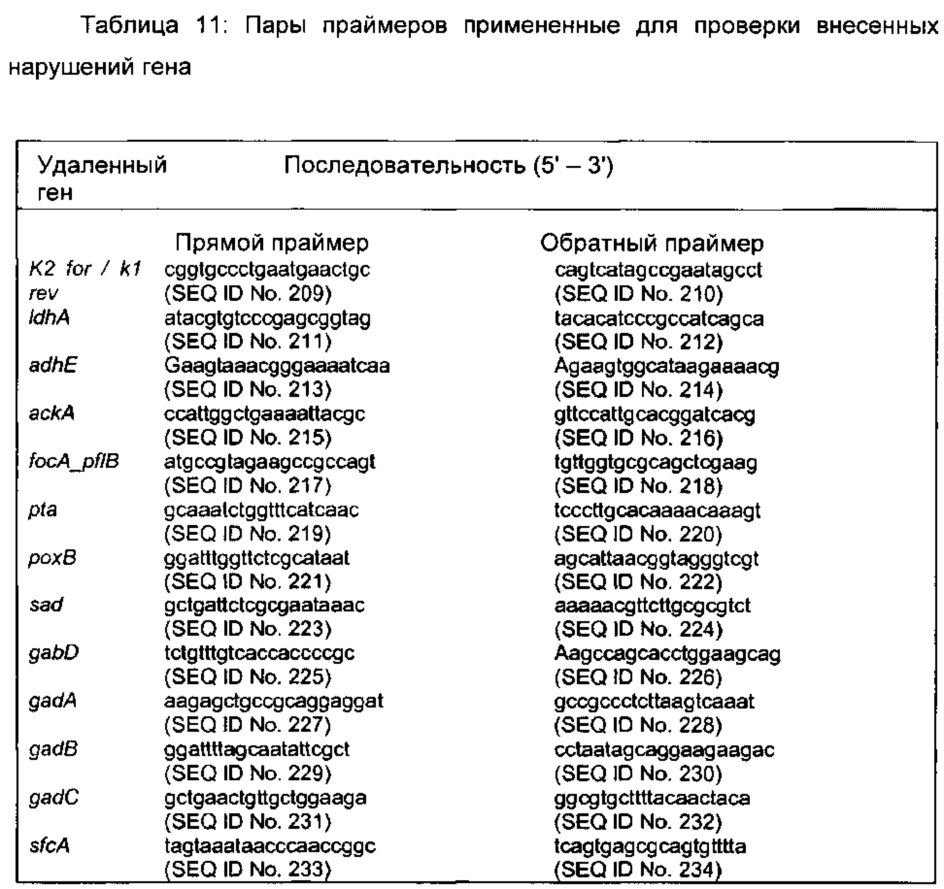
Рецепторный штамм был подготовлен для фаговой трансдукции путем культивирования в течение ночи при 37°С в LB среде. Объем 1,5 мл прекультуры центрифугировали при 1500×g в течение 10 мин. Супернатант отбрасывали, а осадок клеток ресуспендировали в 600 мкл раствора, содержащего 10 мМ MgSO4 и 5 мМ CaCl2. Трансдукцию проводили путем смешивания 100 мкл раствора, содержащего рецепторный штамм, с 100 мкл лизата, и инкубирования этой смеси при 30°С в течение 30 мин. После этого добавляли 100 мкл раствора 1М цитрата натрия с последующим энергичным встряхиванием. После добавления 1 мл среды LB, клеточную суспензию инкубировали при 37°С в течение 1 ч перед посевом клеток на чашки с LB агаром, содержащих 50 мкг/мл канамицина. Клоны, способные расти в присутствии антибиотика, проверяли методом молекулярных колоний ПЦР на содержание требуемой делеции с использованием праймеров, приведенных в Таблице 11. После введения каждой делеции гена, маркерный антибиотик удаляли, как описано выше, в соответствии со способом (Cherepanov & Wackernagel, 1995). Делеции ΔIdhA, ΔadhE, ΔmetA, ΔthrB, ΔrhtB и ΔIIdD последовательно вводили описанным способом.
Протокол о внесении делеций генов с использованием метода рекомбиназы лямбда-Red:
Делеционные кассеты были подготовлены методом ПЦР с использованием высокоточной полимеразы Phusion™ (Finzymes), и FRT-фланкированного гена резистентности к канамицину (кап) плазмиды pKD4 в качестве матрицы (Datsenko & Wanner, 2000). Смысловые праймеры содержали последовательности, соответствующие 5'-концу каждого гена-мишени (подчеркнуто), за которыми следовали 20 п.о. (пар оснований), соответствующих кассете FRT-kan-FRT pKD4. Анти-смысловые праймеры содержали последовательности, соответствующие 3'-концевым участкам каждого гена-мишени (подчеркнуты) с последующими 20 п.о., соответствующими кассете. Праймеры описаны в Таблице 10. ПЦР-продукт расщепляли DpnI и очищали до трансформации.
Штамм E.coli MG1655 был преобразован в электрокомпетентный путем выращивания клеток до CD600 0,6 в жидкой среде LB при 37°С, до концентрации клеток увеличенной в 100 раз, и промывания их дважды охлажденным на льду 10% глицеролом. Клетки трансформировали плазмидой pKD46 (Datsenko и Wanner, 2000) путем электропорации (2,5 кВ, 200 Ω, 25 μF, в кюветах с зазором в 2 мм). Трансформанты отбирали при 30°С на ампициллиновой (100 мкг/мл) твердой среде LB.
Кассеты с нарушениями были трансформированы в электрокомпетентные штаммы E.coli, несущие плазмиду экспрессирующую рекомбиназу лямбда-Red pKD46. Клетки выращивали при 30°С в жидкой SOB среде, содержащей ампициллин (100 мкг/мл). Система рекомбиназы лямбда-Red индуцировалась добавлением 10 мМ арабинозы до OD600 культур достигшей 0,1. Клетки выращивались до OD600 0,6, прежде чем их собирали центрифугированием, дважды промывали охлажденным 10% глицеролом, и трансформировали нарушенной кассетой путем электропорации. После ночной фенотипической экспрессии при 30°С в жидкой среде LB, клетки высеивались на твердую LB среду, содержащую 25 г/мл канамицина. Трансформантов отбирали после культивирования при 30°С.
Замена гена была подтверждена методом молекулярных колоний ПЦР с использованием Crimson Taq-полимеразы (NEB). Первую реакцию проводили с фланкирующими локус-специфическими праймерами (Таблица 11), чтобы проверить одновременную потерю родительского фрагмента и пропуск нового мутант-специфического фрагмента. Две дополнительных реакции были сделаны с помощью одного локус-специфического праймера вместе с одним из соответствующих k1 обратным, или k2 прямым праймерами (Таблица 11), что способствовало выравниванию кассеты FRT-канамициновой устойчивости (смысловой праймер локуса/ k1 обратный и k2 прямой/ обратный праймер локуса).
Ген устойчивости (FRT-kan-FRT) впоследствии вырезали из хромосомы с использованием FLP плазмиды несущей рекомбиназу рСР20 (Cherepanov & Wackernagel, 1995), оставляя регион, содержащий один FRT сайт. рСР20 представляет собой ампициллиновую и CmR плазмиду, которая проявляет, репликацию чувствительную к температуре и тепловой индукции синтеза FLP рекомбиназы. Устойчивые к канамицину мутанты трансформировали рСР20, и ампицилин устойчивые трансформанты были отобраны при 30°С. Трансформантов выращивали на твердой LB среде при 37°С и проверяли на потерю всех устойчивостей к антибиотикам. Вырезание FRT-канамициновой кассеты анализировали методом молекулярных колоний ПЦР с использованием Crimson Taq-полимеразы и фланкирующих локус-специфических праймеров (Таблица 11). Множественные делеции были получены путем повторения описанных выше шагов.





Плазмиды коэкспрессирующие аспартаткиназу, аспартат полуальдегид дегидрогеназу и гомосерин дегидрогеназу (pACT3-op-HMS1) были трансформированы вместе с плазмидой, экспрессирующей гомосериновую трансаминазу и редуктазу ОГБ (рЕХТ20-ДГБ), в оптимизированные штаммы-хозяева. Трансформанты были отобраны на твердой LB среде содержащей хлорамфеникол (25 мкг/мл) и ампицилин (100 г/мл). Неисключительные примеры сконструированных штаммов приведены в таблице 12.

Понятно, что удаление гена lacI из основы описанных выше плазмид вместе с геномной делецией lacI в штамме-хозяине может приводить к экспрессии белка из описанных выше плазмид конститутивно.
Пример 9: Демонстрация ферментативного получения ДГБ через путь гомосерин-ОГБ
Штаммы и условия культивирования: Эксперименты проводились со штаммами, перечисленными в Таблице 12. Культивирование проводили при 37°С на ротационном шейкере на скорости 170 об/мин. Ночные культуры (3 мл среды в пробирке) были посеяны из стоков глицерола и использовались для получения начальной OD600 0,05 в 100 мл растущей культуры в 500 мл качающихся флаконах. IPTG добавляли в концентрации 1 ммоль/л, когда OD600 в культурах роста достигала 0,8. Один литр культуральной среды содержит 20 г глюкозы, 18 г Na2HPO4*12 H2O, 3 г KH2PO4, 0,5 г NaCl, 2 г NH4Cl, 0,5 г MgSO4*7H2O, 0,015 CaCl2*2H2O, 1 мл 0,06 моль/л стокового раствора FeCl3 получаемого разведением в 100 раз концентрированной HCl, 2 мл 10 мМ стокового раствора тиамина HCl, 20 г MOPS (3-[N-Морфолино]пропансульфоновая кислота) и 1 мл раствора микроэлементов (содержание на литр: 0,04 г Na2ЭДTA*2H2O, 0,18 г CoCl2*6H2O, ZnSO4*7H2O, 0,04 г Na2MoO4*2H2O, 0,01 г H3BO3, 0,12 г MnSO4*H2O, 0,12 г CuCl2*H2O) (ЭДТА - этилендиаминтетрауксусная кислота). Средний рН доводили до 7, и среду стерилизовали фильтрацией. Добавляли антибиотики канамицин, сульфат, ампициллин, хлорамфеникол в концентрации 50 мг/л, 100 мг/л и 25 мг/л, соответственно, при необходимости.
Оценка концентрации ДГБ с помощью LC-MS анализа: жидкостная анионообменная хроматография проводилась на системе ICS-3000 Dionex (Sunnyvale, USA) оснащенной системой автоматического генератора элюента (KOH) (RFIC, Dionex), и автоматическим пробоотборником (AS50, Dionex) держащим пробы при 4°С. Аналиты разделяли на lonPac AS11 НС колонке (250×2 мм, Dionex) защищенной с помощью AG11 НС (50×2 мм, Dionex) предварительной колонкой. Температура колонки поддерживалась при 25°С, скорость потока была зафиксирована на уровне 0,25 мл/мин и аналиты элюировались с применением градиента KOH, описанным ранее (Groussac Е, Ortiz М & Francois J (2000): Improved protocols for quantitative determination of metabolites from biological samples using high performance ionic-exchange chromatography with conductimetric and pulsed amperometric detection. Enzyme. Microb. Technol. 26, 715-723). Введенный объем образца составил 15 мкл. Для уменьшения фона, на ASRS Ultra II (2 мм, внешний водного режима, 75 мА) был использован анионный супрессор. Аналиты были количественно посчитаны с помощью масс-чувствительного детектора (МСО Plus, Thermo), работающего в режиме ESI (раскол 1/3, давление азота 90 psi (фунтов на квадратный дюйм), напряжение в капилляре составило 3,5 кВ, датчик температуры 450°С).
Результаты:
После 24 ч культивирования, концентрацию ДГБ в супернатанте различных штаммов количественно посчитали с помощью LC-MS анализа.
Штаммы ЕСЕ73, ЕСЕ74, ЕСЕ75 и ЕСЕ76 продуцировали 0 мг/л, 3,7 мг/л, 0,67 мг/л и 11,9 мг/л ДГБ, соответственно.
Ссылки
Chambellon, Е., Rijnen, L., Lorquet, F., Gitton, С, van Hylckama Vlieg, J. E. Т., Wouters, J. A. & Yvon, M.(2009). The D-2-hydroxyacid dehydrogenase incorrectly annotated PanE is the sole reduction system for branched-chain 2-keto acids in Lactococcus lactis. J. Sacter/o/191, 873-881.
Cherepanov, P.P. & Wackernagel, W.(1995). Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene158, 9-14.
Datsenko, K.A. & Wanner, B.L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S. A97, 6640-6645.
Dykxhoorn, D.M., St Pierre, R. & Linn, T.(1996). A set of compatible tac promoter expression vectors. GeneMI, 133-136.
Hadicke, O. & Klamt, S. (2010). CASOP: a computational approach for strain optimization aiming at high productivity. J. Biotechnol'\47, 88-101.
Klamt, S., Saez-Rodriguez, J. & Gilles, E. D. (2007).Structural and functional analysis of cellular networks with CellNetAnalyzer. BMC Syst S/o/1, 2.
Rothman, S.C. & Kirsch, J.F. (2003). How does an enzyme evolved in vitro compare to naturally occurring homologs possessing the targeted function? Tyrosine aminotransferase from aspartate aminotransferase. J. Mol. S/o/327, 593-608.
Sambrook, J., Fritsch, E.F. & Maniatis, T. (1989). Mo/ecu/ar Cloning: A Laboratory Manual, 2 ed. Cold Spring Harbor: Cold Spring Harbor Laboratory Press.
Schuster, S., Dandekar, T. & Fell, D.A. (1999). Detection of elementary flux modes in biochemical networks: a promising tool for pathway analysis and metabolic engineering. Trends BiotechnolM, 53-60.
Wellner, D. & Lichtenberg, L.A. (1971). Assay of amino acid oxidase. Methods in EnzymologyW', Part B, 593-596.
Реферат
Группа изобретений относится к области биотехнологии. Предложен способ получения 2,4-дигидроксибутирата (2,4-ДГБ) из гомосерина, включающий два этапа: 1) замещение первичной аминогруппы гомосерина карбонильной группой для получения 2-оксо-4-гидроксибутирата (ОГБ), и 2) восстановление полученного ОГБ до 2,4-ДГБ. Предложен модифицированный микроорганизм для получения 2,4-ДГБ из гомосерина в два этапа, представляющий собой трансформированный организм-хозяин, включающий первый химерный ген, кодирующий полипептид с гомосеринтрансаминазной активностью для замещения первичной аминогруппы гомосерина карбонильной группой для получения ОГБ, и второй химерный ген, кодирующий полипептид с ОГБ-редуктазной активностью для восстановления ОГБ для получения 2,4-ДГБ. Предложен способ получения 2,4-ДГБ путем культивирования указанного микроорганизма. Группа изобретений позволяет осуществлять получение 2,4-ДГБ упрощенным ферментативным способом в две стадии из гомосерина. 3 н. и 16 з.п. ф-лы, 3 ил., 12 табл., 8 пр.
Комментарии