Генетически модифицированный микроорганизм и способ получения макролидного соединения с гидроксильной группой в 16-положении с использованием таких микроорганизмов - RU2394906C2
Код документа: RU2394906C2
Чертежи



Описание
Описание
Область, к которой относится изобретение
Настоящее изобретение относится к рекомбинантным микроорганизмам, обладающим способностью продуцировать макролидные соединения с гидроксильной группой в 16-положении; и к способам получения макролидных соединений, имеющих гидроксильную группу в 16-положении, с использованием указанных микроорганизмов.
Описание прототипов
Среди различных метаболитов, продуцируемых актиномицетами, были выявлены важные биологически активные вещества. В частности, были обнаружены многие соединения, в каркасной структуре которых присутствует поликетид (далее называемые поликетидными соединениями). Известны соединения, имеющие различные биологические функции, включая, например, противомикробные средства, а именно эритромицин, жозамицин, тилозин, мидекамицин и мицинамицин; противогрибковые средства, а именно нистатин и амфотерицин; инсектициды, а именно милбемицин и авермектин; иммусупрессоры, а именно такролимус и рапамицин, и противоопухолевые средства, а именно дауномицин, адриамицин, аклациномицин и т.п.
Поликетидные соединения включают группу макролидных соединений, обладающих превосходной противоопухолевой активностью и называемых пладиенолидами. Общим термином “пладиенолиды” называют группу соединений, обнаруженных в культуре штамма Streptomyces sp. Mer-11107; причем известно более 50 родственных соединений, начиная с соединения 11107В (также называемого пладиенолидом В), представленного ниже формулой (А)(см. WO 2002/060890):

Из указанных пладиенолидов соединение, имеющее гидроксильную группу в 16-положении и представленное нижеследующей формулой (I), обладает превосходными свойствами, включая высокую противоопухолевую активность и т.п., однако, это соединение трудно поддается эффективному продуцированию, поскольку его продуцируемость уступает продуцируемости соединения 11107В.

(где R представляет собой атом водорода или гидроксильную группу).
Соединение, представленное формулой (I), может быть обозначено МЕ-282, если R представляет собой атом водорода, и оно может быть обозначено 11107D или названо пладиенолидом D, если R представляет собой гидроксильную группу.
Впоследствии группой исследователей, включая заявителей настоящего изобретения, были открыты актиномицеты, которые гидроксилируют 16-положение соединения 11107В, но не являются пладиенолид-продуцирующими микроорганизмами (см. WO 2003/099813 и WO 2004/050890), и из этих актиномицетов была получена и идентифицирована ДНК, участвующая в реакции гидроксилирования 16-положения соединения 11107В (ДНК, кодирующая фермент, гидроксилирующий 16-положение, и ферменты ферредоксины), и на основании полученных результатов авторами была представлена патентная заявка (см., WO-А 2005/052152, опубликованная 9 июня 2005.).
Авторами настоящего изобретения из пладиенолид-продуцирующего штамма Streptomyces sp. Mer-11107 были также получены и идентифицированы полипептиды, участвующие в биосинтезе 11107В, а также ДНК, кодирующая эти полипептиды, и на основании полученных результатов была представлена патентная заявка (см. заявку WO-А 2006/009276, опубликованную 26 января 2006 года).
В настоящее время появилась возможность более эффективного продуцирования соединений с гидроксильной группой в 16-положении, чем это было возможно ранее, и такое продуцирование предусматривает получение 11107В или т.п. с использованием штамма Streptomyces sp. Mer-11107 или его вариантов, а затем превращение полученного соединения 11107В в соединение, имеющее гидроксильную группу в 16-положении, с использованием других актиномицетов, способных осуществлять реакцию гидроксилирования в 16-положении. Однако поскольку этот способ требует проведения двухстадийной реакции, то трудности его осуществления возрастают вдвое, а поэтому желательно разработать более эффективный способ получения этих соединений.
Известны случаи, когда ген, кодирующий фермент, модифицирующий поликетидное соединение, был введен в штамм, продуцирующий поликетидное соединение, в целях его экспрессии в этом штамме, что приводило к прямому продуцированию поликетидного соединения, модифицированного введенным ферментом. Так, например, сообщалось, что прямое продуцирование 6-гидрокси-тетраценомицина С может быть достигнуто путем введения гена гидроксилирования, происходящего от Streptomyces fradiae Tu2717, в микроорганизм Streptomyces glaucescens GLA.0, который продуцирует тетраценомицин С (см. J. Bacteriol. 1995, Vol. 177, 6126-6136).
Известны также случаи, когда ген биосинтеза поликетидного соединения был введен для его экспрессии в штамме, имеющем фермент, модифицирующий поликетидное соединение, что позволяло осуществлять прямое продуцирование поликетидного соединения, модифицированного ферментом, изначально присутствующим в штамме-хозяине. Так, например, сообщалось, что прямое продуцирование 6-гидрокси-тетраценомицина С может быть достигнуто путем введения гена биосинтеза тетраценомицина С Streptomyces glaucescens GLA.0 в микроорганизм Streptomyces fradiae Tu2717, который содержит фермент, гидроксилирующий тетраценомицин С в 6-положении (см. J. Bacteriol. 1995, Vol. 177, 6126-6136).
В некоторых случаях продуцирование модифицированного поликетидного соединения может быть достигнуто с использованием штаммов различных видов путем введения в эти штаммы гена биосинтеза поликетидного соединения вместе с геном, кодирующим фермент, модифицирующий поликетидное соединение. Так, например, сообщалось, что прямое продуцирование 8,8а-дигидрокси-6-эритронолида В было достигнуто путем введения гена биосинтеза 6-эритронолида В Saccharopolyspora erythraeа и гена Р-450 Streptomyces antibioticus ATCC11891 в микроорганизм Streptomyces lividans K4-114, используемый в качестве хозяина (см. J. Antibiot. 2000, Vol. 53, 502-508).
Описание сущности изобретения
Целью настоящего изобретения является получение рекомбинантных микроорганизмов, обладающих способностью продуцировать макролидное соединение с гидроксильной группой в 16-положении, представленное вышеуказанной формулой (I). Другой целью настоящего изобретения является разработка способа продуцирования макролидного соединения с гидроксильной группой в 16-положении с использованием этих рекомбинантных микроорганизмов.
Для решения вышеуказанных проблем авторами настоящего изобретения были применены методы генной инженерии для получения (отбора) микроорганизмов, включающих ранее выделенную и идентифицированную ДНК, участвующую в биосинтезе 11107В, и ДНК, участвующую в реакции гидроксилирования 16-положения 11107В; при этом авторами было обнаружено, что макролидное соединение с гидроксильной группой в 16-положении аккумулируется в полученной культуральной среде в больших количествах. И на основании этого было осуществлено настоящее изобретение.
Таким образом, настоящее изобретение относится к нижеследующим аспектам (1)-(21):
(1) Рекомбинантный микроорганизм, который представляет собой микроорганизм, обладающий способностью продуцировать макролидные соединения с гидроксильной группой в 16-положении, представленные формулой (I):
(где R представляет собой атом водорода или гидроксильную группу), и который включает:
(а) ДНК, кодирующую полноразмерный или неполный полипептид, участвующий в биосинтезе макролидных соединений, представленных формулой (II):

(где R представляет собой атом водорода или гидроксильную группу), и
(b) ДНК, кодирующую полноразмерный или неполный полипептид, обладающий ферментативной активностью, направленной на гидроксилирование макролидных соединений в 16-положении, представленных вышеуказанной формулой (II).
(2) Рекомбинантный микроорганизм, описанный в п. (1), где ДНК, описанная выше в п. (а), представляет собой ДНК, кодирующую полноразмерный или неполный полипептид, обладающий поликетид-синтетазной активностью; полипептид, обладающий ацетилазной активностью в 7-положении; полипептид, обладающий эпоксидазной активностью в 18,19-положении; и полипептид, обладающий активностью фактора регуляции транскрипции.
(3) Рекомбинантный микроорганизм, описанный в п. (1) или (2), где ДНК, описанная выше в п.(а), представляет собой ДНК, включающую:
(а-1) непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 8340-27935 в последовательности NO:1;
(а-2) непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 28021-49098 в последовательности NO:1;
(а-3) непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 49134-60269 в последовательности NO:1;
(а-4) непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 60269-65692 в последовательности NO:1;
(а-5) непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 68160-66970 в последовательности NO:1;
(а-6) непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 69568-68270 в последовательности NO:1; и
(а-7) непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 72725-70020 в последовательности NO:1; или ее вариант.
(4) Рекомбинантный микроорганизм, описанный в п.(2) или (3), где ДНК, описанная выше в п.(а), также включает ДНК, кодирующую полноразмерный или неполный полипептид, обладающий ферментативной активностью, направленной на гидроксилирование 6-положения.
(5) Рекомбинантный микроорганизм, описанный в п.(4), где указанная ДНК, кодирующая полноразмерный или неполный полипептид, обладающий ферментативной активностью, направленной на гидроксилирование 6-положения, представляет собой ДНК, включающую непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 65707-66903 в последовательности NO:1, или ее вариант.
(6) Рекомбинантный микроорганизм, описанный в любом из п.п.(1)-(5), где ДНК, описанная выше в п.(b), представляет собой ДНК, выбранную из группы, состоящей из:
(b-1) непрерывной нуклеотидной последовательности, состоящей из нуклеотидов 1322-2548 в последовательности NO:2;
(b-2) непрерывной нуклеотидной последовательности, состоящей из нуклеотидов 420-1604 в последовательности NO:3; и
(b-3) непрерывной нуклеотидной последовательности, состоящей из нуклеотидов 172-1383 в последовательности NO:4; или ее вариант.
(7) Рекомбинантный микроорганизм, описанный в любом из п.п. (1)-(6), где ДНК, описанная выше в п. (b), представляет собой вариант ДНК, включающий непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 1322-2548 в последовательности NO:2, которая имеет 1 или 2 или более модифицированных сайтов, выбранных из группы, состоящей из нижеуказанных сайтов 1)-6):
1) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 1592-1594, заменена кодоном, кодирующим аминокислоту, не являющуюся аргинином;
2) модифицированного сайта, где последовательность аtg, состоящая из нуклеотидов 1655-1657, заменена кодоном, кодирующим аминокислоту, не являющуюся метионином;
3) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 1904-1906, заменена кодоном, кодирующим аминокислоту, не являющуюся аргинином;
4) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 2027-2029, заменена кодоном, кодирующим аминокислоту, не являющуюся аргинином;
5) модифицированного сайта, где последовательность atc, состоящая из нуклеотидов 2054-2056, заменена кодоном, кодирующим аминокислоту, не являющуюся изолейцином; и
6) модифицированного сайта, где последовательность сtg, состоящая из нуклеотидов 2528-2530, заменена кодоном, кодирующим аминокислоту, не являющуюся лейцином.
(8) Рекомбинантный микроорганизм, описанный в любом из п.п. (1)-(6), где ДНК, описанная выше в п. (b), представляет собой вариант ДНК, включающий непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 1322-2548 в последовательности NO:2, которая имеет 1 или 2 или более модифицированных сайтов, выбранных из группы, состоящей из нижеуказанных сайтов 1)-6):
1) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 1592-1594, заменена кодоном, кодирующим цистеин, метионин или пролин;
2) модифицированного сайта, где последовательность аtg, состоящая из нуклеотидов 1655-1657, заменена кодоном, кодирующим треонин или серин;
3) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 1904-1906, заменена кодоном, кодирующим лейцин, изолейцин, пролин, тирозин или фенилаланин;
4) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 2027-2029, заменена кодоном, кодирующим лейцин, изолейцин или пролин;
5) модифицированного сайта, где последовательность atc, состоящая из нуклеотидов 2054-2056, заменена кодоном, кодирующим фенилаланин или валин; и
6) модифицированного сайта, где последовательность сtg, состоящая из нуклеотидов 2528-2530, заменена кодоном, кодирующим метионин, цистеин или изолейцин.
(9) Рекомбинантный микроорганизм, описанный в любом из п.п. (1)-(6), где ДНК, описанная выше в п. (b), представляет собой вариант ДНК, включающий непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 1322-2548 в последовательности NO:2, которая имеет 1 или 2 или более модифицированных сайтов, выбранных из группы, состоящей из нижеуказанных сайтов 1)-6):
1) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 1592-1594, заменена кодоном, кодирующим цистеин;
2) модифицированного сайта, где последовательность аtg, состоящая из нуклеотидов 1655-1657, заменена кодоном, кодирующим треонин;
3) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 1904-1906, заменена кодоном, кодирующим лейцин или тирозин;
4) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 2027-2029, заменена кодоном, кодирующим лейцин;
5) модифицированного сайта, где последовательность atc, состоящая из нуклеотидов 2054-2056, заменена кодоном, кодирующим фенилаланин; и
6) модифицированного сайта, где последовательность сtg, состоящая из нуклеотидов 2528-2530, заменена кодоном, кодирующим метионин.
(10) Рекомбинантный микроорганизм, описанный в п. (1) и представляющий собой микроорганизм-хозяин, содержащий ДНК, описанную выше в п.(а), в которую была введена ДНК, описанная выше в п. (b) и происходящая от микроорганизма другого вида.
(11) Рекомбинантный микроорганизм, описанный в п. (1) и представляющий собой микроорганизм-хозяин, содержащий ДНК, описанную выше в п.(b), в которую была введена ДНК, описанная выше в п. (а) и происходящая от микроорганизма другого вида.
(12) Рекомбинантный микроорганизм, описанный в п. (1) и представляющий собой микроорганизм-хозяин, не содержащий ни одну из ДНК, описанных выше в п.п.(а) и (b), в которые была введена ДНК, описанная выше в п.п. (а) и (b) и происходящая от микроорганизма другого вида.
(13) Рекомбинантный микроорганизм, описанный в любом из п.п. (1)-(12), где указанным рекомбинантным микроорганизом, обладающим способностью продуцировать макролидное соединение с гидроксильной группой в 16-положении, представленное формулой (I), является штамм, принадлежащий к роду Streptomyces.
(14) Рекомбинантный микроорганизм, описанный в любом из п.п. (1)-(13), где 50 мг или более указанного макролидного соединения с гидроксильной группой в 16-положении, представленного формулой (I), на 1 л культуральной среды может быть продуцировано при посеве указанного рекомбинантного микроорганизма в 250 мл-колбу Эрленмейера, содержащую 30 мл описанной ниже среды, с последующим культивированием при 25°С в течение 4 дней при перемешивании на роторном шейкере (220 об/мин), экстрагированием путем добавления 270 мл ацетонитрила и анализом полученного экстракта с помощью ВЭЖХ в описанных ниже условиях анализа, проводимого для определения количества макролидного соединения с гидроксильной группой в 16-положении, представленного формулой (I).
Среда:
Растворимый 5% крахмал, 1% глюкоза, 3% Pharmamedia, 0,1% CaCО3, рН 7,5.
Условия измерения с помощью ВЭЖХ
Устройство: ВЭЖХ Shimadzu 10Avp
Колонка: Develosil ODS UG-3 (⌀ 4,6 мм × 50 мм × 3 мкм)
Подвижная фаза: 45%-55% метанол (0-5 мин), 55% метанол (5-13 мин), 55%-70% метанол (13-21 мин), 70% метанол (21-25 мин).
Скорость потока: 1,2 мл/мин
Детекция: УФ на 240 нм
Объем впрыска: 5 мкл
Температура колонки: 40°С.
(15) Способ получения макролидных соединений с гидроксильной группой в 16-положении, представленных формулой (I), или их фармакологически приемлемых солей или гидратов, где указанный рекомбинантный микроорганизм, описанный в любом из п.п. (1)-(14), культивируют в питательной среде, и из этой культуральной среды выделяют макролидные соединения с гидроксильной группой в 16-положении, представленные формулой (I).
(16) Способ, описанный в п.(15), где в указанной культуральной среде присутствует циклодекстрин.
(17) Способ, описанный в п.(16), где указанным циклодекстрином является циклодекстрин, выбранный из группы, состоящей из β-циклодекстрина, γ-циклодекстрина, частично метилированного β-циклодекстрина, диметил-β-циклодекстрина, триметил-β-циклодекстрина, гликозил-β-циклодекстрина и гидроксипропил-β-циклодекстрина.
(18) Вариант ДНК, включающий непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 1322-2548 в последовательности NO:2, которая имеет 1 или 2 или более модифицированных сайтов, выбранных из группы, состоящей из нижеуказанных сайтов 1)-6):
1) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 1592-1594, заменена кодоном, кодирующим аминокислоту, не являющуюся аргинином;
2) модифицированного сайта, где последовательность аtg, состоящая из нуклеотидов 1655-1657, заменена кодоном, кодирующим аминокислоту, не являющуюся метионином;
3) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 1904-1906, заменена кодоном, кодирующим аминокислоту, не являющуюся аргинином;
4) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 2027-2029, заменена кодоном, кодирующим аминокислоту, не являющуюся аргинином;
5) модифицированного сайта, где последовательность atc, состоящая из нуклеотидов 2054-2056, заменена кодоном, кодирующим аминокислоту, не являющуюся изолейцином; и
6) модифицированного сайта, где последовательность сtg, состоящая из нуклеотидов 2528-2530, заменена кодоном, кодирующим аминокислоту, не являющуюся лейцином.
(19) Вариант ДНК, включающий непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 1322-2548 в последовательности NO:2, которая имеет 1 или 2 или более модифицированных сайтов, выбранных из группы, состоящей из нижеуказанных сайтов 1)-6):
1) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 1592-1594, заменена кодоном, кодирующим цистеин, метионин или пролин;
2) модифицированного сайта, где последовательность аtg, состоящая из нуклеотидов 1655-1657, заменена кодоном, кодирующим треонин или серин;
3) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 1904-1906, заменена кодоном, кодирующим лейцин, изолейцин, пролин, тирозин или фенилаланин;
4) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 2027-2029, заменена кодоном, кодирующим лейцин, изолейцин или пролин;
5) модифицированного сайта, где последовательность atc, состоящая из нуклеотидов 2054-2056, заменена кодоном, кодирующим фенилаланин или валин; и
6) модифицированного сайта, где последовательность сtg, состоящая из нуклеотидов 2528-2530, заменена кодоном, кодирующим метионин, цистеин или изолейцин.
(20) Вариант ДНК, включающий непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 1322-2548 в последовательности NO:2, которая имеет 1 или 2 или более модифицированных сайтов, выбранных из группы, состоящей из нижеуказанных сайтов 1)-6):
1) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 1592-1594, заменена кодоном, кодирующим цистеин;
2) модифицированного сайта, где последовательность аtg, состоящая из нуклеотидов 1655-1657, заменена кодоном, кодирующим треонин;
3) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 1904-1906, заменена кодоном, кодирующим лейцин или тирозин;
4) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 2027-2029, заменена кодоном, кодирующим лейцин;
5) модифицированного сайта, где последовательность atc, состоящая из нуклеотидов 2054-2056, заменена кодоном, кодирующим фенилаланин; и
6) модифицированного сайта, где последовательность сtg, состоящая из нуклеотидов 2528-2530, заменена кодоном, кодирующим метионин.
(21) Полипептид, кодируемый вариантом ДНК, описанным в любом из п.п. (18)-(20).
Настоящее изобретение определено нижеследующими терминами.
Термин “рекомбинантный микроорганизм” означает микроорганизм (бактерии, актиномицеты, дрожжи, плесень или т.п.), содержащий ген, происходящий от другого организма и введенный в конкретный микроорганизм посредством генетической рекомбинации, однако, применяемые методы введения генов не ограничиваются генетической рекомбинацией с использованием плазмид и других векторов, и включают методы гомологичной рекомбинации и т.п.
“Вариант ДНК” означает:
(1) ДНК, которая гибридизуется с исходной ДНК в условиях высокой жесткости;
(2) ДНК, имеющую нуклеотидную последовательность, которая на 70% или более гомологична нуклеотидной последовательности исходной ДНК;
(3) ДНК, имеющую нуклеотидную последовательность, комплементарную нуклеотидной последовательности исходной ДНК; или
(4) ДНК, которая не гибридизуется в условиях высокой жесткости с исходной ДНК вследствие вырожденности генного кодона, но которая имеет нуклеотидную последовательность, кодирующую аминокислотную последовательность, аналогичную аминокислотной последовательности, кодируемой ДНК, определенной в любом из п.п. (1)-(3).
Термин “ДНК, которая гибридизуется в условиях высокой жесткости” означает, например, ДНК, полученную путем гибридизациии колоний, гибридизации методом бляшек, Саузерн-гибридизации или т.п., с использованием исходной ДНК в качестве зонда, а в частности, этот термин может означать ДНК, которая может быть идентифицирована с использованием фильтра, имеющего ДНК, выделенную из фиксированных на нем колоний или бляшек сначала путем гибридизации при 65°С в присутствии 0,7-1,0 М хлорида натрия, а затем промывки фильтра при 65°С с использованием 0,1 - 2х концентрации раствора SSC (раствора SSC, содержащего хлорид натрия в концентрации 1 × 150 мМ и цитрат натрия в концентрации 1 × 15 мМ).
Термин “родственное соединение” означает соединение, имеющее тот же самый остов, характеризующий исходную химическую структуру, но отличающееся тем, что оно имеет модификацию в боковой цепи или другую форму боковой цепи.
Термин “ферментативная активность, направленная на гидроксилирование 16-положения” означает активность, направленную на замену атома водорода в 16-положении макролидного соединения (II) в гидроксильную группу.
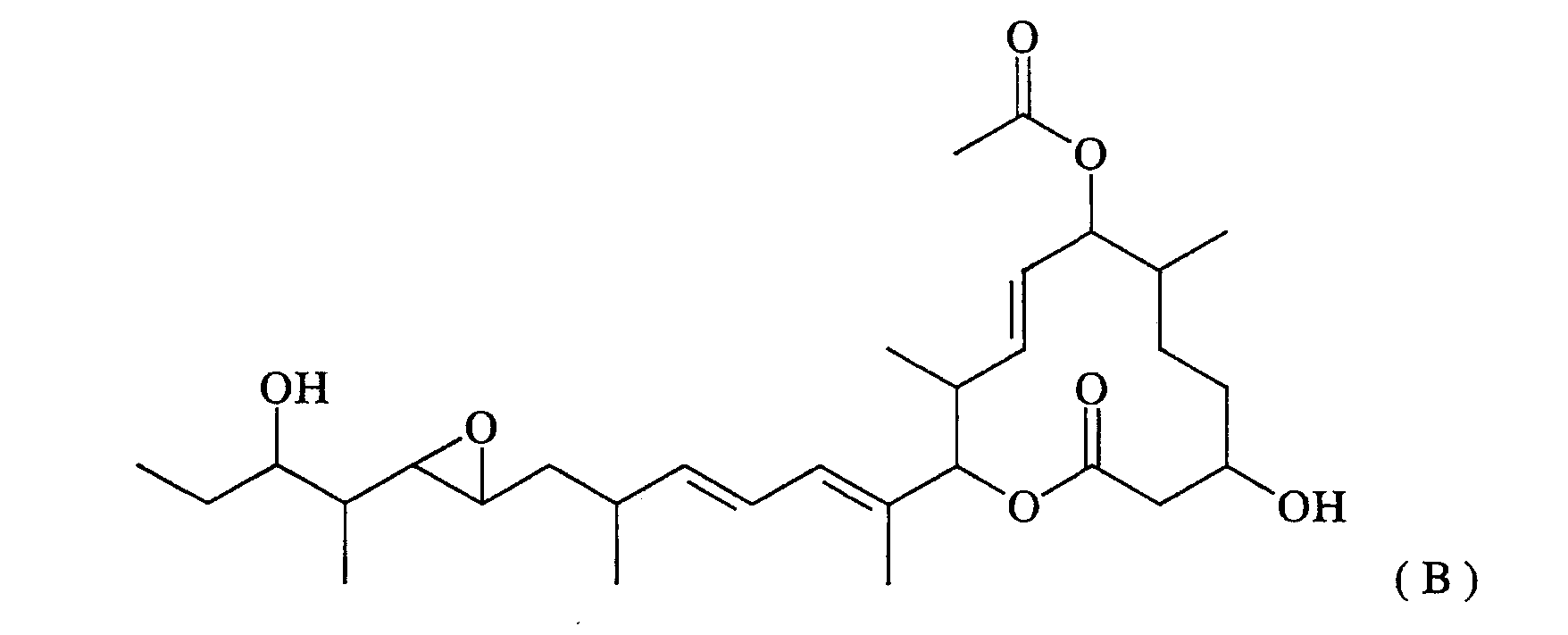
Термин “ферментативная активность, направленная на гидроксилирование 6-положения” означает активность, направленную на замену атома водорода в 6-положении макролидного соединения (иногда обозначаемого МЕ-265), представленного нижеследующей формулой (В), гидроксильной группой:
Термин “поликетид-синтетазная активность” означает активность, направленную на синтез макролидного соединения, представленного нижеследующей формулой (Е):

Термин “ацетилазная активность в 7-положении” означает активность, направленную на замену гидроксильной группы в 7-положении макролидного соединения, представленного нижеследующей формулой (С), ацетильной группой:

(где R представляет собой атом водорода или гидроксильную группу).
Термин “эпоксидазная активность в 18,19-положении” означает активность, направленную на замену двойной связи в 18,19-положении макролидного соединения, представленного нижеследующей формулой (D), эпоксигруппой:

(где R представляет собой атом водорода или гидроксильную группу).
Термин “активность регуляции транскрипции” означает активность, направленную на регуляцию транскрипции ДНК, кодирующей полипептид, участвующий в биосинтезе макролидного соединения (II).
В соответствии с настоящим изобретением может быть получен рекомбинантный микроорганизм, который имеет ДНК, кодирующую полипептид, участвующий в биосинтезе макролидного соединения (II), и ДНК, кодирующую полипептид, обладающий ферментативной активностью, направленной на гидроксилирование макролидного соединения (II) в 16-положении, и который способен к прямому ферментативному продуцированию макролидного соединения (I) с гидроксильной группой в 16-положении. Макролидное соединение (I) с гидроксильной группой в 16-положении может быть эффективно продуцировано путем культивирования указанного рекомбинантного микроорганизма и выделения этого соединения из культуральной среды.
В частности, макролидное соединение (I) с гидроксильной группой в 16-положении может быть продуцировано с большей эффективностью и селективностью с использованием ДНК, кодирующей полипептид, обладающий ферментативной активностью, направленной на гидроксилирование 16-положения, где аминокислотная последовательность данного полипептида была частично модифицирована вышеупомянутым методом.
Краткое описание графического материала
На фиг.1 представлен путь биосинтеза пладиенолидов в Mer-11107.
На фиг.2 представлено соответствие между космидами и каждой из ОРС ДНК, участвующей в биосинтезе пладиенолидов в Mer-11107.
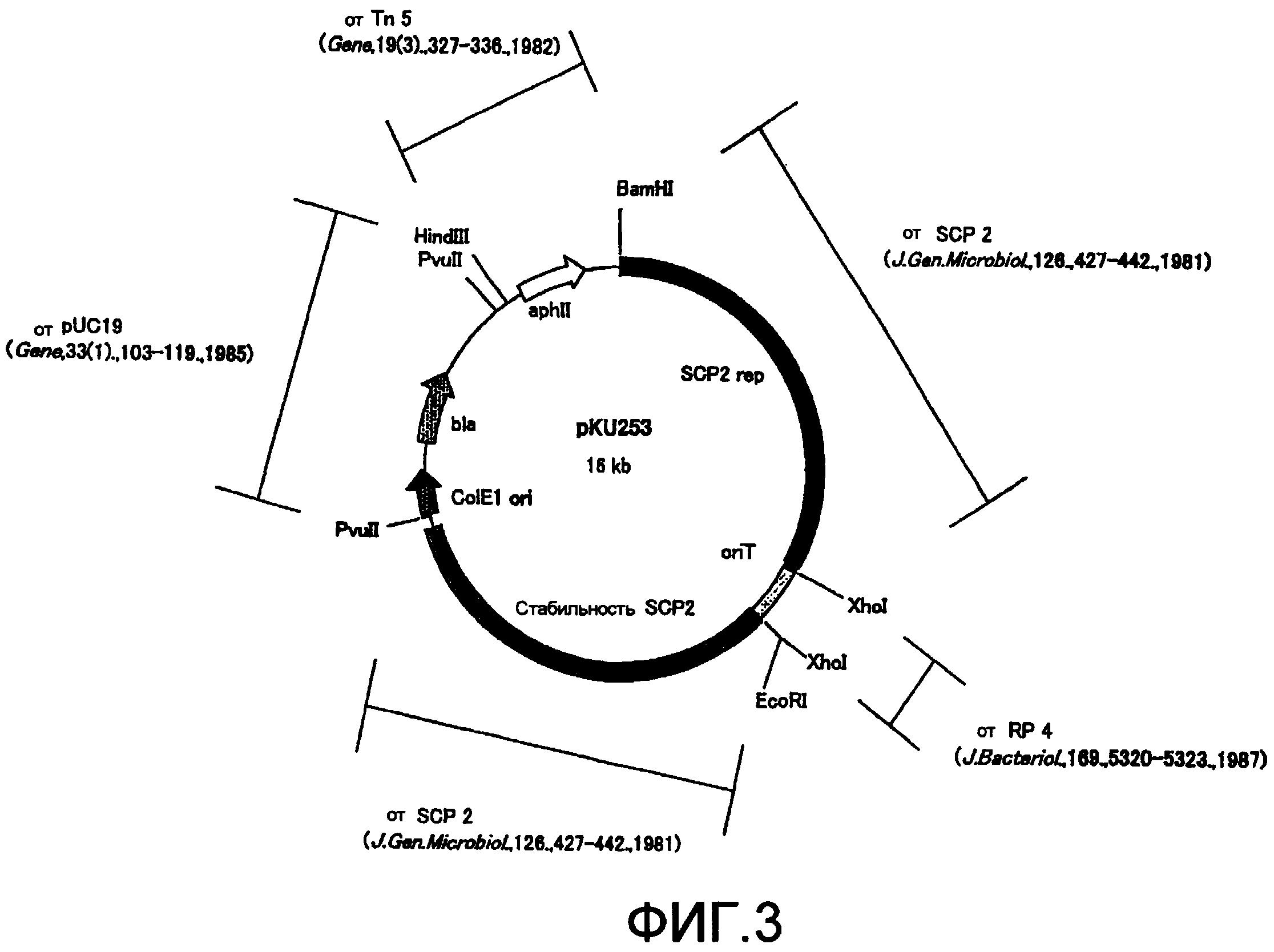
На фиг.3 представлена структура плазмиды pKU253.
На фиг.4 представлена структура плазмиды pUC19aph::oriT::intphiC31.
Подробное описание изобретения
Варианты настоящего изобретения более подробно описаны ниже.
ДНК, кодирующая полипептид, участвующий в биосинтезе макролидного соединения (II) согласно изобретению, может быть получена известным методом (таким, как метод гибридизации колоний, описанный в руководстве Molecular Cloning, 2-е издание) из культивированного мицелия микроорганизма, способного продуцировать указанное макролидное соединение. Любой из этих микроорганизмов может обладать способностью продуцировать макролидное соединение независимо от его вида или штамма, однако, предпочтительными являются штаммы, принадлежащие к актиномицетам рода Streptomyces. Одним из таких примеров является штамм Streptomyces sp. Mer-11107, который был выделен из почвы. Штамм Mer-11107 был депонирован 19 декабря 2000 под номером FERM P-18144 в депозитарии Национального института биологических наук и Центра промышленных исследования и технологий (National Institute of Bioscience and Human-Technology Agency of Industrial Science and Technology) (1-3, Higashi 1-chome Tsukuba-shi, Ibaraki-ken 305-8566 Japan), а затем 27 ноября 2001 года он был передан на международное депонирование в Международный депозитарий патентуемых микроорганизмов (IPOD) Национального института новых промышленных исследований и технологий под номером FERM BP-7812 (Tsukuba Central 6, 1-1, Higashi 1-Chome Tsukuba-shi, Ibaraki-ken 305-8566 Japan).
Процедуры получения нужной ДНК описаны ниже на примере штамма Mer-11107. Рекомбинантную ДНК, полученную путем присоединения геномной ДНК Mer-11107, которая была частично гидролизована подходящим рестриктирующим ферментом, к космидному вектору, способному реплицироваться в E.coli и гидролизованному подходящим рестриктирующим ферментом, вводят в E.coli с получением трансдуцированных штаммов. Затем множество трансдуцированных штаммов, полученных как описано выше, скринируют с использованием фрагмента известного гена гидроксилирующего фермента (фермента цитохорома Р450) в качестве зонда, а затем отбирают трансдуцированные штаммы, гибридизующиеся с этим зондом. После этого получают и секвенируют ДНК, которая гибридизуется с геном гидроксилирующего фермента, присуствующим в выбранных космидах. Эту ДНК вводят в E.coli, и после подтверждения присутствия в трансформированной E.coli ферментативной активности, направленной на гидроксилирование 6-положения, указанная ДНК, кодирующая фермент, гидроксилирующий 6-положение, может быть использована в качестве зонда в Саузерн-гибридизации для отбора космид, содержащих кластер генов биосинтеза макролидного соединения, смежный с ДНК, кодирующей фермент, гидроксилирующий 6-положение, из множества позитивных клонов (космид), полученных как описано выше, и нужную ДНК получают путем конструирования этих космид. Подробное описание всех указанных процедур приводится в сравнительном примере 1, а также в заявке WO-A 2006/009276, описание которой рассматривается заявителями в настоящей заявке и может быть использовано в настоящем описании посредством ссылки.
Полученная таким образом нуклеотидная последовательность (аминокислотная последовательность кодирующей последовательности), содержащая область, окружающую ДНК, кодирующую полипептид, участвующий в биосинтезе макролидного соединения (II), представлена последовательностью NO:1 в списке последовательностей.
ДНК, представленная последовательностью NO:1, включает 8 открытых рамок считывания (ОРС): pldA I (нуклеотиты 8340-27935), pldA II (нуклеотиды 28021-49098), pldA III (нуклеотиды 49134-60269), pldA IV (нуклеотиды 60269-65692), pldB (нуклеотиды 65707-66903), pldC (нуклеотиды 68160-66970), pldD (нуклеотиды 69568-68270) и pldR (нуклеотиды 72725-70020).
Из этих ДНК, pldA I, pldA II, pldA III и pldA IV имеют несколько транскрипционных рамок считывания, каждая из которых, так же как и другие известные гены биосинтеза поликетидов, включает одну или несколько повторяющихся единиц, называемых модулями. Как описано ниже, каждый модуль кодирует все или несколько доменов, выбранных из доменов ацил-переносящего белка (ACP), β-кетоацил-ACP-синтетазы (KS) и ацил-трансферазы (AT), которые участвуют в реакциях конденсации при синтезе поликетидов, а также доменов кетоацил-редуктазы (KR), дегидрогеназы (DH) и еноилредуктазы (ER), которые участвуют в реакциях модификации карбонильной группы β-положения, а конечный модуль включает домен тиоэстеразы (TE), который удаляет поликетидную цепь из поликетид-синтетазы.
На фиг.1 показан путь биосинтеза макролидного соединения в штамме Mer-11107. В отличие от других модулей в загрузочном модуле центральный цистеин заменен глутамином, что указывает на то, что pldA I участвует в исходной реакции. Модуль 10 включает домен тиоэстеразы (TE), что указывает на то, что pldA IV участвует в конечной реакции синтеза основного поликетидного остова. После образования основного остова поликетида таким путем его модифицируют в 18,19-положении эпоксидазой, кодируемой pldD, в 7-положении - ацетилазой, кодируемой pldC, и в 6-положении - гидроксилазой, кодируемой pldB, для биосинтеза макролидного соединения (II). Макролидное соединение (II), в котором R представляет собой атом водорода, может быть получено либо путем делеции pldB либо путем ее модификации так, чтобы функция, за которую ответственна эта рамка считывания, не экспрессировалась. PIdR, которая имеет высокую степень гомологии с геном aveR, кодирующим фактор регуляции транскрипции в биосинтезе авермектина, кодирует фактор регуляции транскрипции ДНК, участвующий в биосинтезе макролидных соединений.
В настоящем изобретении, ДНК, кодирующая полипептид, обладающий ферментативной активностью, направленной на гидроксилирование макролидного соединения (II) в 16-положении, может быть получена методом, хорошо известным специалисту в данной области (таким, как метод гибридизации колоний, описанный в руководстве Molecular Cloning, 2-е издание), из культивированного мицелия микроорганизма, обладающего способностью к гидроксилированию макролидного соединения в 16-положении. При этом может быть использован любой микроорганизм, обладающий способностью заменять атом водорода макролидного соединения в 16-положении гидроксильной группой, независимо от вида или штамма этого микроорганизма, однако, предпочтительными примерами являются штаммы, принадлежащие к актиномицетам. Примерами таких штаммов являются штамм Streptomyces sp. A-1544, штамм Streptomyces sp. Mer-11107 и штамм Streptomyces sp. A-1560. Штамм A-1544 был депонирован под рег. номером FERM P-18943 в Международном депозитарии патентуемых микроорганизмов Национального института новых промышленных исследований и технологий (Tsukuba Central 6, 1-1, Higashi 1-Chome, Tsukuba-shi, Ibaraki-ken 305-8566 Japan) 23 июля 2002 года, а затем он был передан на международное депонирование под номером FERM BP-8446 30 июля 2003 года, в Международный депозитарий патентуемых микроорганизмов (IPOD) Национального института новых промышленных исследований и технологий (Tsukuba Central 6, 1-1, Higashi 1-Chome, Tsukuba-shi, Ibaraki-ken 305-8566 Japan). Штамм A-1560 был депонирован под рег. номером FERM P-19585 в Международном депозитарии патентуемых микроорганизмов Национального института новых промышленных исследований и технологий (Tsukuba Central 6, 1-1, Higashi 1-Chome, Tsukuba-shi, Ibaraki-ken 305-8566 Japan) 13 ноября 2003 года, а затем 19 августа 2004 года он был передан на международное депонирование под в Международный депозитарий патентуемых микроорганизмов (IPOD) Национального института новых промышленных исследований и технологий (Tsukuba Central 6, 1-1, Higashi 1-Chome, Tsukuba-shi, Ibaraki-ken 305-8566 Japan) номером FERM BP-10102.
Процедуры получения нужной ДНК описаны ниже на примере штамма A-1544. Сначала получали геномную ДНК штамма A-1544 и подвергали ПЦР-реакции с использованием праймеров, созданных исходя из данных о последовательности известного гена гидроксилирующего фермента (цитохрома P450), в результате чего получали специфически амплифицированный ДНК-фрагмент, который затем секвенировали. Затем исходя из данных о полученной последовательности нуклеотидную последовательность смежной области, расположенной выше и ниже от фрагмента клонированной ДНК, амплифицировали с помощью обратной ПЦР (Cell Technology, Vol. 14, pp. 591-593, 1995) и клонировали, после чего эту последовательность анализировали. Детали всех этих процедур подробно описаны в сравнительном примере 2 (штамм A-1544), в сравнительном примере 3 (штамм Mer-11107) и в сравнительном примере 4 (штамм A-1560), а также в заявке WO-A 2005/052152, опубликованной 9 июня 2005 года, и могут быть указаны посредством ссылки.
Полученная таким образом ДНК (включая смежную область) из штамма A-1544, которая кодирует полипептид, обладающий ферментативной активностью, направленной на гидроксилирование макролидного соединения (II) в 16-положении, представлена последовательностью NO:2 в списке последовательностей. ДНК, представленная последовательностью NO:2, включает две открытые рамки считывания (ОРС), а именно psmA (нуклеотиды 1322-2548) и psmB (нуклеотиды 2564-2761), которые кодируют фермент, гидроксилирующий 16-положение, и ферредоксин соответственно.
Аналогичным образом ДНК (включая смежную область), кодирующая полипептид, обладающий ферментативной активностью, направленной на гидроксилирование макролидного соединения (II) в 16-положении, и выделенная из штамма Mer-11107, представлена последовательностью NO:3 в списке последовательностей. Эта ДНК, представленная последовательностью NO:3, включает две открытые рамки считывания (ОРС), а именно bpmA (нуклеотиды 420-1604) и bpmB (нуклеотиды 1643-1834), которые кодируют фермент, гидроксилирующий 16-положение, и ферредоксин соответственно.
ДНК (включая смежную область), кодирующая полипептид, обладающий ферментативной активностью, направленной на гидроксилирование макролидного соединения (II) в 16-положении, и выделенная из штамма А-1560, представлена последовательностью NO:4 в списке последовательностей. Эта ДНК, представленная последовательностью NO:4, включает две открытые рамки считывания (ОРС), а именно tpmA (нуклеотиды 172-1383) и tpmB (нуклеотиды 1399-1593), которые кодируют фермент, гидроксилирующий 16-положение, и ферредоксин соответственно.
Кроме того, варианты вышеупомянутых различных ДНК, кодирующих полипептид, обладающий ферментативной активностью, направленной на гидроксилирование соединения в 16-положении, могут быть получены в соответствии с нижеследующими процедурами. Сначала на основе данных об исходной последовательности ДНК проводят сайт-направленную модификацию, а затем ДНК-вариант, полученный посредством сайт-направленной модификации, вводят хозяину. Таким образом, нужные варианты могут быть отобраны и получены с использованием в качестве маркера, в высокой степени селективного и активного фермента, гидроксилирующего 16-положение и кодируемого ДНК-вариантом полученного рекомбинантного микроорганизма. Так, например, сайт-направленная модификация может быть проведена с использованием в качестве матрицы плазмиды, клонирующей исходную ДНК, и с использованием праймера, сконструированного исходя из данных об этой последовательности с помощью коммерчески доступного набора, такого как набор для сайт-направленного мутагенеза QuickChange™ (Stratagene Co.).
Из этих вариантов могут быть включены варианты, обладающие высокой ферментативной активностью, направленной на гидроксилирование соединения в 16-положении, а также высокой селективностью; так, например, в случае psmA, кодирующей фермент, гидроксилирующий 16-положение и выделенный из штамма A-1544, может быть включен вариант ДНК, имеющий непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 1322-2548 в последовательности NO:2, и 1 или 2 или более модифицированных сайтов, выбранных из группы, состоящей из сайтов 1) - 6), а именно:
1) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 1592-1594, заменена кодоном, кодирующим аминокислоту, не являющуюся аргинином, а предпочтительно цистеин;
2) модифицированного сайта, где последовательность аtg, состоящая из нуклеотидов 1655-1657, заменена кодоном, кодирующим аминокислоту, не являющуюся метионином, а предпочтительно треонин;
3) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 1904-1906, заменена кодоном, кодирующим аминокислоту, не являющуюся аргинином, а предпочтительно лейцин или тирозин;
4) модифицированного сайта, где последовательность сgc, состоящая из нуклеотидов 2027-2029, заменена кодоном, кодирующим аминокислоту, не являющуюся аргинином, а предпочтительно лейцин;
5) модифицированного сайта, где последовательность atc, состоящая из нуклеотидов 2054-2056, заменена кодоном, кодирующим аминокислоту, не являющуюся изолейцином, а предпочтительно фенилаланин; и
6) модифицированного сайта, где последовательность сtg, состоящая из нуклеотидов 2528-2530, заменена кодоном, кодирующим аминокислоту, не являющуюся лейцином, а предпочтительно метионин.
Рекомбинантом согласно изобретению является рекомбинантный микроорганизм, имеющий вышеупомянутую “ДНК, кодирующую полипептид, участвующий в биосинтезе макролидного соединения (II)”, и “ДНК, кодирующую полипептид, обладающий ферментативной активностью, направленной на гидроксилирование макролидного соединения (II) в 16-положении”, и этот микроорганизм может быть получен методами, описанными ниже в п.п. А)-С).
А) “ДНК, кодирующую полипептид, обладающий ферментативной активностью, направленной на гидроксилирование макролидного соединения (II) в 16-положении”, вводят хозяину, который по своей природе имеет “ДНК, кодирующую полипептид, участвующий в биосинтезе макролидного соединения (II)”;
В) “ДНК, кодирующую полипептид, участвующий в биосинтезе макролидного соединения (II),” вводят хозяину, который по своей природе имеет “ДНК, кодирующую полипептид, обладающий ферментативной активностью, направленной на гидроксилирование макролидного соединения (II) в 16-положении”;
С) “ДНК, кодирующую полипептид, участвующий в биосинтезе макролидного соединения (II)” и “ДНК, кодирующую полипептид, обладающий ферментативной активностью, направленной на гидроксилирование макролидного соединения (II) в 16-положении” вводят хозяину, который по своей природе не имеет ни одну из этих ДНК.
Хозяином для рекомбинанта согласно изобретению может быть любой микроорганизм, в который может быть введена нужная ДНК и который может продуцировать нужное макролидное соединение с гидроксильной группой в 16-положении, однако, предпочтительным примером такого микроорганизма является штамм, принадлежащий к актиномицетам рода Streptomyces sp. Так, например, Streptomyces sp. Mer-11107 или т.п. могут быть использованы в вышеописанном методе А), а штаммы Streptomyces sp. A-1544, Streptomyces sp. Mer-11107 или Streptomyces sp. A-1560 могут быть использованы в вышеописанном методе B). В методе C) может быть использован штамм Esсherichia coli или т.п.
Методы введения нужной ДНК и индуцирования ее экспрессии у хозяина не имеют конкретных ограничений и могут быть применены, например, методы, описанные в руководстве Molecular Cloning, 2nd Edition или Current Protocols in Molecular Biology. Выбор хозяина и системы плазмидных векторов не имеют конкретных ограничений, при условии, что нужная ДНК будет стабильно сохраняться и экспрессироваться в таком хозяине. Плазмида может включать, помимо нужной ДНК, самореплицирующиеся последовательности, промоторные последовательности, терминаторные последовательности и гены резистентности к лекарственным средствам и т.п., и такой плазмидой может быть либо самореплицирующаяся плазмида либо интегрированная плазмида, имеющая последовательность, гомологичную последовательности конкретной области генома выбранного хозяина. Нужная ДНК может быть введена в участок, находящийся либо в плазмиде либо в геноме микроорганизма-хозяина.
Если хозяином является актиномицет или родственный штамм, то может быть использован самореплицирующийся вектор pIJ6021 (Gene, 166, 133-137 (1995)) или интегрирующийся в геном вектор KC515 (The Bacteria Vol. 9, Antibiotic-producing Streptomyces (ed. Queener, S.E. and Day, L.E.), pp. 119-158, Academic Press, Orlando, FIa).
Примерами самореплицирующихся последовательностей являются rep pIJ101 (1988, J. Bacteriol., 170, 4634-4651), rep SCP2 (1981, J. Gen. Microbiol., 126, 427-442), pUC19 (Gene, 33 (1), 103-119 (1985)) и т.п.; примерами промоторных последовательностей являются ermEp (Mol. Microbiol., 1994, Nov; 14(3): 533-45), SnpAp (J. Bacteriol. 1992, May; 174(9): 2797-2808) и т.п.; примерами терминаторных последовательностей являются ter(mmr) (Gene, 1987; 58(2-3): 229-41), терминатор fd (Nucleic Acids Res., 5(12), 4495-4503 (1978) и т.п., а примерами генов резистентности к лекарственным средствам являются гены hyg (Nucleic Acids Res., 4(14), 1565-1581 (1986)), tsr (Mol. Gen. Genet., 199(1), 26-36 (1985)) и т.п. Микроорганизм согласно изобретению может быть легко получен специалистом в данной области исходя из описания настоящего изобретения.
Полученный таким образом рекомбинантный микроорганизм может быть культивирован и проанализирован на уровень продуцирования им макролидного соединения (I) с гидроксильной группой в 16-положении, после чего он может быть отобран для получения штамма с высоким уровнем продуцирования макролидного соединения. То есть может быть получен рекомбинантный микроорганизм, обладающий способностью продуцировать 50 мг или более макролидного соединения на 1 л культурального бульона с высокой продуктивностью, составляющей примерно 40%, и может быть получен рекомбинантный микроорганизм, обладающий способностью продуцировать 100 мг или более макролидного соединения на 1 л культурального бульона с высокой продуктивностью примерно 30%.
Способ оценки продуктивности
Рекомбинантный микроорганизм инокулируют в 250 мл-колбу Эрленмейера, содержащую 30 мл среды (растворимый 5% крахмал, 1% глюкоза, 3% Pharmamedia, 0,1% CaCО3, рН 7,5), а затем культивируют при 25°С в течение 4 дней при перемешивании на роторном шейкере (220 об/мин), экстрагируют путем добавления 270 мл ацетонитрила и полученный экстракт анализируют с помощью ВЭЖХ в нижеописанных условиях анализа, проводимого для определения концентрации аккумулированного макролидного соединения с гидроксильной группой в 16-положении, представленного формулой (I).
Условия измерения с помощью ВЭЖХ:
Устройство: ВЭЖХ Shimadzu 10Avp
Колонка: Develosil ODS UG-3 (φ 4,6 мм × 50 мм × 3 мкм)
Подвижная фаза: 45%-55% метанол (0-5 мин), 55% метанол (5-13 мин), 55%-70% метанол (13-21 мин), 70% метанол (21-25 мин)
Скорость потока: 1,2 мл/мин
Детекция: УФ на 240 нм
Объем впрыска: 5 мкл
Температура колонки: 40°С.
Рекомбинантный микроорганизм согласно изобретению может быть засеян в питательную среду и культивирован в аэробных условиях, и из полученного культурального бульона может быть выделено макролидное соединение (I) с гидроксильной группой в 16-положении или его фармакологически приемлемая соль или гидрат.
В принципе, рекомбинантный микроорганизм может быть культивирован стандартными методами культивирования микроорганизмов, но в случае жидкой культуры обычно предпочтительным является культивирование в аэробных условиях, такие как культивирование с аэрацией и с перемешиванием или встряхиванием культуры. В качестве среды, используемой для культивирования, может служить любая среда, при условии, что она содержит питательные вещества, которые могут утилизироваться этими микроорганизмами; при этом могут быть использованы все существующие различные синтетические среды, полусинтетические среды и органические среды. В своем составе такая среда может включать различные источники углерода, такие как глюкоза, сахароза, фруктоза, глицерин, декстрин, крахмал, меласса и соевое масло, используемые либо отдельно либо в комбинации. В качестве источника азота могут быть использованы органические источники азота, взятые либо отдельно либо в комбинации друг с другом, такие как реагент Pharmamedia, пептон, мясной экстракт, соевая мука, казеин, аминокислоты, дрожжевой экстракт или мочевина, и неорганические источники азота, такие как нитрат натрия или сульфат аммония. Кроме того, например, могут быть добавлены соли, такие как хлорид натрия, хлорид калия, карбонат кальция, сульфат магния, фосфат натрия, фосфат калия или хлорид кобальта; соли тяжелых металлов и витамины, такие как витамин В или биотин, если это необходимо. Кроме того, если во время культивирования образуется большое количество пены, то в такую среду могут быть добавлены соответствующие различные противовспенивающие средства. При желании могут быть также добавлены химические вещества, необходимые для сохранения генов резистентности к лекарственому средству. При добавлении противовспенивающего средства необходимо определить концентрацию этого средства, при которой оно не будет оказывать негативного влияния на продуцирование нужного соединения.
Концентрация аккумулированного макролидного соединения с гидроксильной группой в 16-положении может быть увеличена путем добавления циклодекстринов в культуральный бульон при продуцировании макролидного соединения (I) с гидроксильной группой в 16-положении путем культвирования рекомбинантного микроорганизма согласно изобретению. Примерами циклодекстринов, используемых в настоящем изобретении, являются β-циклодекстрин, γ-циклодекстрин, частично метилированный β-циклодекстрин, диметил-β-циклодекстрин, триметил-β-циклодекстрин, гликозил-β-циклодекстрин и гидроксипропил-β-циклодекстрин. Каждый из них может быть использован отдельно или в комбинации друг с другом.
Количество диклодекстрина, добавляемого в культуральный бульон, составляет предпочтительно 0,5%-5%. Добавление предпочтительно осуществляют в начале или во время культивирования.
Условия культивирования могут быть выбраны так, чтобы в этих условиях микроорганизмы хорошо росли и могли продуцировать макролидное соединение (I) с гидроксильной группой в 16-положении. Так, например, pH среды поддерживают при 5-9, то есть обычно в пределах значений рН, близких к нейтральному. Температура культивирования обычно составляет от 20°С до 40°C или предпочтительно от 23°С до 35°C. Культивирование проводят в течение 2-12 дней или обычно 3-10 дней. Само собой разумеется, что различные условия культивирования, описанные выше, могут быть изменены и могут быть использованы оптимальные условия, выбранные специально в зависимости от типа микроорганизма, от условий окружающей среды и т.п. Для выделения и очистки макролидного соединения (I) с гидроксильной группой в 16-положении, аккумулирующегося в культуральном бульоне, могут быть применены стандартные методы выделения и очистки микробных метаболитов из культурального бульона. Для этой цели могут быть использованы все известные методы, такие как экстракция органическим растворителем, таким как метанол, этанол, бутанол, этилацетат, хлороформ, ацетон, толуол или т.п., различные формы ионообменной хроматографии, гель-хроматография с использованием сефадекса LH-20 или т.п., адсорбционная хроматография с использованием активированного угля, хроматография на силикагеле, хроматография на смоле-адсорбенте (Diaion HP20) или т.п., адсорбция-десорбция, проводимая путем тонкослойной хроматографии, и высокоэффективная жидкостная хроматография, проводимая на колонке с обращенной фазой и т.п., и указанные методы не ограничиваются описанными здесь методами. Для выделения и очистки нужного макролидного соединения (I) с гидроксильной группой в 16-положении такие методы могут быть проведены отдельно или в виде комбинации двух или более методов, осуществляемых в произвольном порядке или повторно.
Примеры
Настоящее изобретение подробно описано на нижеследующих примерах, однако, настоящее изобретение не ограничивается этими примерами. В приведенном ниже описании концентрации компонентов среды выражены в процентах по массе, если это не оговорено особо.
Сравнительный пример 1: Получение ДНК, кодирующей полипептид, участвующий в биосинтезе макролидного соединения (II)
(1) Культивирование штамма Mer-11107 и выделение геномной ДНК
Гифы Streptomyces sp. Mer-11107 инокулировали в 25 мл триптиказо-соевого бульона и культивировали со встряхиванием при 28°C в течение 3 дней. Из полученного культурального бульона выделяли геномную ДНК в соответствии с методикой, описанной в работе D.A. Hopwood et al. "Isolation genomic DNA" (pp. 162-170) Practical Streptomyces Genetics (The John Innes Foundation, Norwich, England, 2000).
(2) Получение геномной библиотеки Mer-11107
160 мкл стерильной очищенной воды, 200 мкл раствора геномной ДНК Mer-11107 (1 мг/мл), 40 мкл 10x молярной концентрации буферного раствора (100 мМ трис-HCl (pH 7,5), 100 мМ MgCl2, 10 мМ дитиотреитола, 500 мМ NaCl) и 1 мкл рестриктирующего фермента Sau3AI (1 единица/мкл) смешивали и инкубировали при 37°C в течение 3 мин. Затем брали 50 мкл среды и экстрагировали 50 мкл смеси фенола-хлороформа (фенол:хлороформ:изоамиловый спирт = 25:24:1, отношение по объему), водный слой собирали и снова экстрагировали 50 мкл хлороформа, и водный слой снова собирали. К этой жидкости добавляли 5 мкл 3М ацетата натрия (pH 6,0) и 150 мкл этанола, после чего эту жидкость оставляли на 30 мин при -80°C и центрифугировали для сбора осажденной ДНК. После промывки 70% этанолом эту ДНК растворяли в 90 мкл стерильной очищенной воды и после добавления 10 мкл
10 х буферного раствора BAP (500 мМ трис-HCl (pH 9,0), 10 мМ MgCl2) и 5 мкл бактериальной щелочной фосфатазы (0,5 единиц/мкл, Takara Bio Inc.) инкубировали при 37°C в течение 3 ч. Реакционную жидкость экстрагировали 100 мкл смеси фенола-хлороформа (фенол:хлороформ:изоамиловый спирт = 25:24:1, отношение по объему), водный слой собирали, а затем снова экстрагировали 100 мкл хлороформа, и водный слой снова собирали. Затем к этой жидкости добавляли 10 мкл 3 M ацетата натрия (pH 6,0) и 300 мкл этанола, и полученную жидкость оставляли на 30 мин при -80°С и центрифугировали для сбора осажденной ДНК. После промывки 70% этанолом эту ДНК растворяли в 20 мкл буферного раствора ТЕ (10 мМ трис-HCl (pH 8,0), 1 мМ EDTA).
Кроме того, 10 мкг космидного вектора SuperCos (Stratagene Co.) гидролизовали рестриктирующим ферментом ХbaI в соответствии с руководством Stratagene, и концы ДНК дефосфорилировали кишечной щелочной фосфатазой теленка (Takara Bio Inc.), а затем после гидролиза рестриктирующим ферментом BamHI и очистки эту ДНК растворяли в 10 мкл буферного раствора ТЕ. 2,5 мкл раствора ДНК Mer-11107, частично гидролизованной ферментом Sau3A1, как описано выше, добавляли к 1 мкл полученного раствора космидной ДНК, и 1,5 мкл стерильно очищенной воды, 5 мкл раствора II, взятого из набора для лигирования ДНК (Takara Bio Inc.), и 10 мкл раствора I добавляли в указанном порядке и инкубировали при 23°C в течение 10 мин. 4 мкл реакционной жидкости упаковывали в фаг лямбда с использованием набора Gigapack III XL Kit (Stratagene Co.). Если полученную упакованную жидкость (всего 500 мкл) подвергали тесту на трансдукцию, то проводили анализ на способность к образованию колоний на уровне 380 к.о.е. (колониеобразующих единиц)/мкл.
(3) Получение различных зондов
(3-1) Получение зондов, содержащих области, кодирующие кетосинтетазу
Два нижеследующих праймера, KS-3F и KS-4R, синтезировали на основе последовательностей, которые обычно являются консервативными в доменах кетосинтеза поликетид-синтетаз (см. последовательности NO:5 и 6):
KS-3F: 5'-GACCGCGGCTGGGACGTGGAGGG-3'
KS-4R: 5'-GTGCCCGATGTTGGACTTCAACGA-3'
Эти праймеры использовали для проведения ПЦР в нижеследующих условиях:
Жидкая композиция для ПЦР-реакции:
Температуры реакции:
95°C, 3 мин
(98°C, 20 с; 63°C, 30 с, 68°C, 2 мин) - 30 циклов
72°C, 5 мин.
930 п.н.-ДНК-фрагменты, амплифицированые в результате такой реакции, подвергали электрофорезу на 0,8% агарозном геле, и выделенные 930 п.н.-ДНК-фрагменты вырезали и собирали, а затем очищали с использованием SUPREC-01 (Takara Bio Inc.). С использованием 10 нг полученных ДНК-фрагментов в качестве матрицы, 930 п.н.-ДНК-фрагменты, содержащие область, кодирующую кетосинтетазу, снова амплифицировали в тех же самых условиях ПЦР, описанных выше, за исключением того, что число циклов реакции составляло 20. Эти ДНК-фрагменты концентрировали и очищали с использованием фильтра SUPREC-02 (Takara Bio Inc.), и 50 мкл полученного раствора TE использовали в качестве раствора для зондирования.
(3-2) Получение зонда, содержащего область гена цитохрома Р450
Два известных гена цитохрома Р450 амплифицировали из актиномицетов в целях получения зонда для гена цитохрома Р450. То есть два праймера CB-1F и CB-2R, содержащие нижеследующие последовательности (см. последовательности 7 и 8), синтезировали в целях амплификации гена ORF-A, происходящего от Streptomyces thermotolerans ATCC11416 (Biosci. Biotechnol. Biochem. 59: 582-588, 1995):
CB-1F: 5'-ATGACAGCTTTGAATCTGATGGATCCC-3'
CB-2R: 5'-TCAGAGACGGACCGGCAGACTCTTCAGACG-3'
Кроме того, два праймера PKC-1F и PKC-2R, содержащие нижеследующие последовательности (см. последовательности NO:9 и 10), синтезировали в целях амплификации гена pik-C, происходящего от Streptomyces venezuelae ATCC15439 (Chem. Biol. 5: 661-667, 1998):
PKC-1F: 5'-GTGCGCCGTACCCAGCAGGGAACGACC-3',
PKC-2R: 5'-TCACGCGCTCTCCGCCCGCCCCCTGCC-3'.
Эти праймеры использовали для проведения ПЦР в нижеследующих условиях:
Жидкая композиция для ПЦР-реакции:
Температуры реакции:
95°C, 3 мин
(98°C, 20 с; 63°C, 30 с, 68°C 2 мин) - 30 циклов
72°C, 5 мин.
Два 1,2-т.п.н.-ДНК-фрагмента, амплифицированые в результате такой реакции, очищали с помощью набора для ПЦР-очистки QIAGEN (QIAGEN Co.), а затем получали смешанный раствор, содержащий 10 нг/мкл каждого ДНК-фрагмента, и использовали в качестве зонда.
(4) Скрининг с использованием зонда, содержащего область, кодирующую кетосинтетазу.
В хозяина E. coli XL-1Blue MR (Stratagene Co.) переносили раствор, содержащий библиотеку геномных ДНК Mer-11107 и полученный как описано выше в (2) в соответствии с руководством Stratagene. После трансдукции бактериальную суспензию разливали в десять чашек (внутренний диаметр 90 мм, высота 15 мм) со средой LB, содержащей 50 мкг/мл ампициллина-1,5% агара, и культивировали в течение 18 ч при 37°С. Колонии, выращенные на каждой чашке, переносили на фильтры HybondoN+ (Amersham Biosciences), обрабатывали щелочью и нейтральным веществом в условиях, описанных в руководстве по использованию фильтров HybondoN+, и сушили на фильтрах в течение 2 ч при 80°C для фиксации ДНК, выделенной из этих колоний.
Библиотеку геномных ДНК скринировали путем гибридизации колоний с помощью системы AlkPhos Direct (Amersham Biosciences) со 100 нг 930 п.н.-ДНК-фрагмента, содержащего область кетосинтетазы, полученную как описано выше в (3-1), в качестве зонда. Гибридизацию проводили в течение 2 ч при 65°C и при концентрации соли 0,5 M NaCl. Условия гибридизации и детекции описаны в указанном руководстве для системы AlkPhos Direct System. Из протестированных примерно 7600 колоний было отобрано 59 колоний, которые жестко гибризовались с зондом, меченным щелочной фосфатазой. Космиды экстрагировали и очищали от клонов E.coli, происходящих от этих колоний.
(5) Отбор и подтверждение космидных клонов, имеющих область гена биосинтеза пладиенолидов, с использованием зонда, содержащего область гена цитохрома Р450
Два микролитра каждого из растворов космидных ДНК, полученных как описано выше в (4), наносили пятнами на фильтр HybondoN+, обрабатывали щелочью и нейтральным веществом в условиях, описанных в прилагаемом руководстве, и сушили в течение 2 ч при 80°С для фиксации ДНК на фильтре. Затем осуществляли гибридизацию с этими фильтрами в условиях, описанных выше в (4), с использованием фрагмента гена цитохрома P450, описанного выше в (3), в качестве зонда. В результате была отобрана одна космида, которая жестко гибридизовалась с зондом, и эта космида была обозначена pKS58.
ДНК pKS58 частично гидролизовали рестриктирующим ферментом Sau3AI, лигировали с BamHI-CIAP-обработанным фаговым вектором Zap Express (Stratagene Co.) и упаковывали в фаг лямбда с использованием набора Gigapack III XL (Stratagene Co.). Штамм E. coli XL1-Blue MRF' инфицировали этим фаговым раствором с получением бляшек. Гибридизацию методом бляшек осуществляли с использованием зонда для гена цитохрома P450, полученного как описано выше в (3-2), в целях субклонирования фрагмента ДНК длиной приблизительно 2 т.п.н., содержащего ген цитохрома Р450:
Этот фрагмент ДНК гена цитохрома P450 секвенировали, и синтезировали два праймера, PDL58-1F и PDL58-2R (см. последовательности NO:11 и 12), имеющие у N- и С-концов нижеследующие последовательности, которые рассматриваются как области, кодирующие цитохром P450.
PDL58-1F: 5'-GCCCCGCATATGGATCTGGAAACCCAACTTCTC-3'
PDL58-2R: 5'-GCACTAGTCAGCCGCGCTCGACGAGGAGGTG-3'
Эти праймеры использовали для осуществления ПЦР в нижеследующих условиях:
Жидкая композиция для ПЦР-реакции:
Температуры реакции:
95°C, 3 мин
(98°C, 20 с; 63°C, 30 с; 68°C, 2 мин) - 20 циклов
72°C, 5 мин
1,2-т.п.н.-ДНК-фрагмент, амплифицированый в результате такой реакции, очищали с помощью набора для ПЦР-очистки QIAGEN (QIAGEN Co.), а затем гидролизовали рестриктирующими ферментами NdeI и SpeI. После проведения реакции ДНК подвергали электрофорезу на 0,8% агарозном геле, и выделенный 1,2-т.п.н.-ДНК-фрагмент вырезали, а затем ДНК собирали и очищали с использованием набора для гель-экстракции (QIAGEN Co.). Полученный ДНК-фрагмент встраивали в NdeI- и SpeI-сайты плазмиды pT7NS-camAB, экспрессирующей ген цитохрома P450 (см. сравнительный пример 5 и WO 03/087381), и получали конструкцию pPDL96.
BL21 E. coli (DE3) трансформировали с использованием этой плазмиды и культивировали в среде M9CG (1,28% Na2HPO4 · 7H2O, 0,3% KH2PO4, 0,05% NaCl, 0,1% NH4Cl, 1% казаминокислота, 0,4% глюкоза, 1 мМ MgCl2, 100 мкM CaCl2, 50 мкг/мл ампициллина) до достижения плотности 0,8 OD600 (оптическая плотность при 600 нм). Затем добавляли 5-аминолевулиновую кислоту до концентрации 80 мкг/мл и IPTG до концентрации 0,1 мМ, и культивирование продолжали при 22°C в течение 25 ч для индуцирования белка цитохрома P450. После индуцирования мицелий собирали и суспендировали в 5 мл буферного раствора CV (50 мМ NaPO4 (pH 7,3), 1 мМ EDTA, 10% глицерин, 1 мМ глюкоза). 1 мл этой суспензии вводили в тест-пробирку, а затем добавляли 5 мкл ДМСО-раствора (50 мг/мл) ME-265 (пладиенолида В, дезоксигенированного в 6-положении) и инкубировали при 28°C в течение 15 ч. Затем добавляли 1 мл ацетонитрила, и смесь перемешивали с этим реакционным раствором, после чего эту смесь центрифугировали и супернатант анализировали с помощью ВЭЖХ в описанных ниже условиях для подтверждения образования пладиенолида B. Эти результаты показали, что область гена, ответственная за биосинтез пладиенолидов, присутствует в pKS58.
Условия ВЭЖХ-анализа:
Устройство: ВЭЖХ Shimadzu 10Avp
Колонка: Develosil ODS UG-3 (φ 4,6 мм × 50 мм × 3 мкм)
Подвижная фаза: 45%-55% метанол (0-5 мин), 55% метанол (5-13 мин), 55%-70% метанол (13-21 мин), 45% метанол (21-25 мин).
Скорость потока: 1,2 мл/мин
Детекция: УФ на 240 нм
Объем впрыска: 5 мкл
Температура колонки: 40°С
Время анализа: 25 мин
Время удерживания: ME-265: 20 мин, пладиенолид B: 13 мин.
(6) Отбор космиды, содержащей кластер гена биосинтеза, смежного с геном цитохрома P450
Космиду, содержащую кластер гена биосинтеза, смежного с геном цитохрома P450, и полученную как описано выше в (5), отбирали из 59 образцов космидных ДНК, полученных как описано выше в (4).
59 образцов космидных ДНК гидролизовали рестриктирующими ферментами EcoRI и BamHI, и полученную в каждом случае ДНК подвергали электрофорезу на агарозном геле и Саузерн-гибридизации с использованием в качестве зондов ДНК домена KS (домена aveA2 KS6) и домена AT (домена aveA1 AT2) для гена биосинтеза агликон-содержащего авермектина (см. Proc. Natl. Acad. Sci. USA 96 (1999) 9509-9514; JP-A 2000-245457; или WO 00/50605) и для гена цитохрома P450, полученного как описано выше в (5).
Космиды, содержащие гибридизующиеся ДНК-фрагменты одной и той же длины, были разделены на группы исходя из электрофоретической картины ДНК, гидролизованной рестриктирующими ферментами EcoRI и BamHI, и картины гибридизации с использованием различных зондов. Из этих космид все без исключения космиды, обнаруживающие аналогичные картины, были удалены, а остальные космиды были систематизированы исходя из картин частичного соответствия полос. Прежде всего следует отметить, что космида pKS58 содержит ген цитохрома P450, полученный как описано в (5), космида pKS54 была выбрана как космида, смежная с областью, содержащей ген поликетид-синтетазы, расположенный со стороный гена цитохрома Р450, а космида pKS35 была выбрана как космида, смежная с pKS54. Космида pKS23 была также выбрана как космида, смежная с космидой pKS58, простирающейся от области гена цитохрома Р450 до области, не содержащей гена поликетид-синтетазы. В результате, как показано на фиг.2, pKS23, pKS58, pKS54 и pKS35 были выбраны как космидные клоны, содержащие кластер генов, ответственных за биосинтез пладиенолида.
(7) Определение нуклеотидной последовательности кластера генов, ответственных за биосинтез пладиенолида
Была определена нуклеотидная последовательность группы ДНК, содержащих ген биосинтеза пладиенолида. Каждую космиду, отобранную как описано выше в (6), сначала выделяли методом с использованием хлорида цезия, а затем разрезали на фрагменты длиной примерно 1 т.п.н. с использованием HydroShear (Genomic Solutions K.K.) и субклонировали с помощью набора BKL (Takara Bio Inc.).
Полученные субклоны подвергали реакции циклического секвенирования (Amersham Biosciences Co.) с использованием флуоресцентно меченых праймеров, и определяли нуклеотидные последовательности соответствующих фрагментов (MegaBACE 1000: Amersham Biosciences Co.). Так, например, была определена нуклеотидная последовательность длиной примерно 75 т.п.н., содержащая ДНК, ответственную за биосинтез пладиенолидов (см. последовательность NО: 1).
(8) Получение (pldB)-дефицитного штамма, не содержащего гена гидроксилазы, гидроксилирующей пладиенолид в 6-положении
Было показано, что пладиенолид биосинтезируется по пути биосинтеза, проиллюстрированного на фиг.1, из нуклеотидной последовательности длиной примерно 75 т.п.н., содержащей ДНК, которая участвует в биосинтезе пладиенолида и которая была секвенирована как описано выше в (7) (см. последовательность NО:1). Таким образом, получали pldB-дефицитный штамм описанными ниже методами, в основу которых была положена идея, что штамм, продуцирующий только ME-265 (пладиенолид В, дезоксигенированный в 6-положении), может быть получен путем деструкции только гена pldB, кодирующего цитохром P450.
Четыре праймера, pldB-L-Bgl2F, pldB-L-Hind3R, pldB-R-Hind3F и pldB-R-Bgl2R (см. последовательности NO:13, 14, 15 и 16), имеющие последовательности, представленные ниже, синтезировали исходя из нуклеотидной последовательности NO:1:
pldB-L-Bgl2F: 5'-GGGAGATCTAGAGGCCGGTTACCTCTACGAGTA-3'
pldB-L-Hind3R: 5'-GGGAAGCTTGCGATGAGCTGTGCCAGATAG-3'
pldB-R-Hind3F: 5'-GGGAAGCTTGAACTGGCGCGACAGTGTCTT-3'
pldB-R-Bgl2R: 5'-GGGAGATCTGCAGCGGATCGTCTTCGAGACCCTT-3'
Эти праймеры использовали для проведения ПЦР в нижеследующих условиях.
Жидкая композиция для ПЦР-реакции:
Температуры реакции:
95°C, 3 мин
(98°C, 20 с; 63°C, 30 с; 68°C, 2 мин) - 30 циклов
72°C, 5 мин.
В результате этого 1,57 п.н.-ДНК-фрагмент (ДНК-фрагмент L1), содержащий нуклеотиды 64756-66302 в последовательности NО: 1, амплифицировали с помощью реакции, проводимой с использованием pldB-L-Bgl2F и pldB-L-Hind3R, a 1,54-т.п.н.-ДНК-фрагмент (ДНК-фрагмент R1), содержащий нуклеотиды 66849-68368 в последовательности NO:1, амплифицировали с помощью реакции, проводимой с использованием pldB-R-Hind3F и pldB-R-Bgl2R. ДНК-фрагменты L1 и R1 очищали с помощью набора для ПЦР-очистки QIAGEN (QIAGEN Co.), и гидролизовали рестриктирующими ферментами BglII и HindIII.
ДНК-фрагменты, а именно L1 и R1, которые были гидролизованы рестриктирующими ферментами BglII и HindIII; 2,3 т.п.н.-ген резистентности к гигромицину B (происходящий от pHP45omegahyg: Gene 190, 315-317, 1997, иногда обозначаемый далее "hyg"), который был гидролизован рестриктирующим ферментом HindIII; и челночный вектор pKU253 (см. фиг. 3), который был гидролизован рестриктирующим ферментом BamHI, присоединяли друг к другу с помощью набора для лигирования ДНК Ver. 2.1 (Takara Bio Inc.). ДНК-фрагмент длиной примерно 5,4 т.п.н., имеющий ген резистентности к гигромицину B, встроенный между ДНК-фрагментами Ll и Rl, вводили в конструкцию pKU253 с получением плазмиды размером примерно 21,4 т.п.н., обозначенной pKU253-L1-hyg-R1.
Полученную плазмиду pKU253-L1-hyg-R1 вводили в E.coli S17-1 с помощью электропорации с получением S17-1/pKU253-L1-hyg-R1. Полученную плазмиду S17-1/pKU253-L1-hyg-R1 инокулировали на 10 мл LB-среды (1% бакто-триптона, 0,5% дрожжевого экстракта, 0,5% NaCl), содержащей 25 мкг/мл канамицина и 100 мкг/мл гигромицина B, а затем культивировали со встряхиванием при 30°С в течение 2 ч, после чего мицелий собирали, два раза промывали 10 мл LB-среды и суспендировали в 5 мл LB-среды. Таким образом была получена донорная суспензия.
Одновременно с получением донорной суспенизии Mer-11107 инокулировали на 10 мл среды TSB (триптиказо-соевый бульон: Nissui Seiyaku) и культивировали со встряхиванием при 30°С в течение 5 ч, затем мицелий собирали, два раза промывали 10 мл стерильной воды и суспендировали в 1 мл стерильной воды. Таким образом, была получена акцепторная суспензия. 500 мкл донорной суспензии S17-1/pKU253-L1-hyg-R1 смешивали с 10 мкл акцепторной суспензии Mer-11107 и наносили на агаровую среду Actino No. 4. После инкубирования при 30°C в течение 18 ч на эту среду наносили 2,5 мл среды SNA (0,8% питательная среда: Difco, 0,4% агар), содержащей 2 мг/мл рибостамицина, и инкубировали при 30°C в течение 7 дней с получением штамма-трансформанта pKU253-L1-hyg-R1, резистентного к рибостамицину.
Полученный штамм-трансформант pKU253-L1-hyg-R1 инокулировали на 10 мл среды TSB, не содержащей рибостамицина, и культивировали со встряхиванием при 30°С в течение 24 ч. Мицелий собирали из культурального бульона трансформанта pKU253-L1-hyg-R1, два раза промывали 10 мл стерильной воды и суспендировали в 10 мл стерилизованной воды. После соответствующего разведения полученную суспензию наносили на агаровую среду YMS (0,4% дрожжевой экстракт, 1% экстракт из семян пшеницы, 0,4% растворимый крахмал, 2% агар, 10 мМ хлорид кальция), содержащую 200 мкг/мл гигромицина B, и инкубировали при 30°C в течение 4 дней. Одиночные колонии, растущие на агаровой среде YMS, содержащей гигромицин B, переносили на агаровую среду YMS, содержащую 200 мкг/мл гигромицина В, и на агаровую среду YMS, содержащую 200 мкг/мл рибостамицина, а затем инкубировали при 30°C в течение 2 дней. После инкубирования отбирали штамм, резистентный к гигромицину В и чувствительный к рибостамицину. Полученный штамм, названный Mer-11107 pldB::hyg, представляет собой pldB-дефицитный штамм, в геноме которого отсутствуют 546 п.н. (нуклеотиды 66303-66848 в последовательности NO:1) гена pldB, а вместо этих нуклеотидов присутствует встроенный ген резистентности к гигромицину В.
(9) Тест на продуцирование пладиенолида (pldB)-дефицитным штаммом, не содержащим гена гидроксилазы, гидроксилирующей пладиенолид в 6-положении
200 мкл замороженного посевного материала штамма Mer-11107 pldB::hyg, полученного как описано выше в (8), инокулировали в 20 мл среды для посева (2% растворимый крахмал, 2% соевая мука (ESUSAN-MEAT, изготовленная Ajinomoto Co. Ltd.), 0,5% дрожжевой экстракт, 0,1% K2HPO4, 0,25% MgSO4 · 7H2O, 0,3% CaCO3, pH не корректировали) и инкубировали при 25°C в течение 2 дней. 300 мкл полученного культурального посевного бульона инокулировали в 30 мл посевной культуральной среды (5% растворимый крахмал, 1% глюкоза, 3% Pharmamedia, 2% β-циклодекстрин, 0,1% CaCO3, pH 7,5) и культивировали при 25°C в течение 4 и 5 дней. После завершения культивирования 20 мл полученной культуральной жидкости экстрагировали путем добавления равного количества ацетонитрила. Затем брали часть этого экстракта и 5 раз разводили ацетонитрилом, после чего уровни пладиенолида В и ME-265 измеряли с помощью ВЭЖХ в нижеследующих условиях. Результаты измерения приводятся в таблице 1.
Условия ВЭЖХ-анализа:
Устройство: ВЭЖХ Shimadzu 10Avp
Колонка: Develosil ODS UG-3 (φ 4,6 мм × 50 мм × 3 мкм)
Подвижная фаза: 45%-55% метанол (0-5 мин), 55% метанол (5-13 мин), 55%-70% метанол (13-17 мин), 70% метанол (17-35 мин), 45% метанол (35-40 мин)
Скорость потока: 1,2 мл/мин
Детекция: УФ на 240 нм
Объем впрыска: 10 мкл
Температура колонки: 40°С
Время анализа: 35 мин
Время удерживания: ME-265: 22 мин, пладиенолид B: 16 мин.
Сравнительный пример 2: ДНК-1, кодирующая полипептид, обладающий ферментативной активностью, направленной гидроксилированием макролидного соединения (II) в 16-положении
(1) Получение геномной ДНК штамма A-1544 Streptomyces sp.
Штамм A-1544 инокулировали в среду, содержащую 1% глюкозу, 0,4% солодовый экстракт и 1% дрожжевой экстракт, и инкубировали при 28°C в течение 3 дней. Полученный культуральный бульон центрифугировали при 3000 об/мин в течение 10 мин для сбора мицелия. Геномную ДНК получали из мицелия с использованием набора Blood & Cell Culture Kit (QIAGEN Co.).
(2) Клонирование неполной последовательности ДНК, кодирующей белок, обладающий активностью, направленной на гидроксилирование макролидного соединения 11107B в 16-положении
Были сконструированы и продуцированы нижеследующие смеси праймеров (5Dm-3F и 5Dm-3R), исходя из предполагаемой аминокислотной последовательности цитохрома P450 (CYP105D5) Streptomyces coelicolor A3 (2) (см. последовательности NO:17 и 18):
5Dm-3F: 5'-TTCGCSCTSCCSGTCCCSTCSATGGTSAT-3'
5Dm-3R: 5'-GTTGATSAYSGASGTSGAGAA-3'
Для стимуляции реактивности с учетом флуктуации кодона использовали смешанные основания S(=C+G) и Y(=C+T).
Затем праймеры этих двух типов (5Dm-3F и 5Dm-3R) и геномную ДНК штамма A-1544, взятую в качестве матрицы и полученную как описано выше в (1), использовали для проведения ПЦР. ПЦР проводили путем 35-кратного повторения трехстадийной реакции, включая денатурацию при 98°C в течение 20 с; отжиг при 50°C в течение 2 мин; и удлинение цепи при 68°C в течение 30 с с использованием полимеразы Taq Takara LA (Takara Bio Inc.) и ПЦР-амплификатора (T Gradient, Biometra Co.). В результате был амплифицирован ДНК-фрагмент (далее называемый ДНК-фрагментом-A1), имеющий размер примерно 500 п.н. По всей вероятности, этот ДНК-фрагмент-А1 является частью ДНК, кодирующей белок, обладающий гидроксилирующей активностью. ДНК-фрагмент-А1, амплифицированный посредством ПЦР, выделяли из реакционного раствора с помощью SUPREC PCR (Takara Bio Inc.).
Для получения ДНК-фрагмента А1 в количестве, достаточном для анализа нуклеотидной последовательности полученного ДНК-фрагмента А1, этот ДНК-фрагмент объединяли с плазмидым вектором pT7Blue T (Novagen Co.) с использованием набора для лигирования ДНК ver.2 (Takara Bio Inc.) в целях трансформации штамма JM109 E.coli. Затем трансформированную E.coli отбирали на LВ-среде с агаром (1,0% бакто-триптон, 0,5% дрожжевой экстракт, 0,5% NaCl, 1,5% агар), содержащей ампициллин (50 мкг/мл), X-gal (5-бром-4-хлор-3-индолил-β-D-галактозид, 40 мкг/мл) и IPTG (изопропил-β-D-тиогалактопиранозид; 100 мкМ). Выделенную таким образом колонию трансформированных E.coli культивировали в жидкой LВ-среде (1% бакто-триптон, 0,5% дрожжевой экстракт, 0,5% NaCl), содержащей ампициллин (50 мкг/мл). Плазмидную ДНК выделяли из мицелия размноженной трансформированной E.coli и очищали с помощью набора для очистки плазмид (QIA filter Plasmid Midi Kit, QIAGEN Co.), в результате чего получали достаточное количество ДНК-фрагмента A1.
(3) Анализ нуклеотидной последовательности клонированного ДНК-фрагмента A1
Нуклеотидную последовательность ДНК-фрагмента А1, полученного как описано выше в (2), анализировали с использованием анализатора нуклеотидной последовательности ДНК (PE Biosystems 377XL) методом проведения цикла реакций секвенирования с использованием красителя для терминации цепи. В результате проведения анализа нуклеотидной последовательности было выявлено, что ДНК-фрагмент А1, амплифицированный посредством ПЦР-реакции, имел точный размер 528 п.н., хотя в описанных выше измерениях, проводимых с помощью электрофореза, его размер составлял примерно 500 п.н. (см. нуклеотидную последовательность от нуклеотида 1775 до нуклеотида 2302 в последовательности NO:2). Поскольку было обнаружено, что последовательности ДНК, соответствующие двум типам праймеров, используемых в вышеописанной ПЦР, присутствуют на обоих концах описанной выше клонированной последовательности ДНК размером 528 п.н., то авторами настоящего изобретения был сделан вывод, что ДНК-фрагмент А1 был
1 х амплифицирован этими двумя типами праймеров (5Dm-3F и 5Dm-3R) в вышеуказанной ПЦР-реакции.
(4) Анализ смежной области ДНК-фрагмента А1
Как упоминалось выше, была определена неполная последовательность ДНК, кодирующая белок, обладающий гидроксилирующей активностью и происходящий от штамма A-1544. Поэтому амплификацию, клонирование и анализ последовательности нуклеотидов в смежной области, простирающейся с левой и с правой стороны от клонированного фрагмента, осуществляли методом обратной ПЦР (Cell Technology vol. 14, p.591-593, 1995). В частности, геномную ДНК штамма A-1544 (см. выше (1)) гидролизовали соответствующими рестриктирующими ферментами PstI и SalI в буферном растворе H (50 мМ трис-HCl, pH 7,5, 10 мМ MgCl2, 10 мМ дитиотреитол и 100 мМ NaCl). Каждый из полученных ДНК-фрагментов, гидролизованных рестриктирующими ферментами, подвергали аутоциркуляризации с использованием набора для лигирования ДНК ver.2 (Takara Bio Inc.).
Кроме того, нижеследующие праймеры (6PIN-2F и 6PIN-2R) конструировали и продуцировали на основе нуклеотидной последовательности ДНК-фрагмента А1 (см. последовательности NO:19 и 20):
6PIN-2F: 5'-GCTGCGCCTGGCCCTGGAGGACATCGAGAT-3'
6PIN-2R: 5'-CTGTTCCTCGAAGAACTCGTGGTCGGCGTA-3'
Затем праймеры этих двух типов (6PIN-2F и 6PIN-2R) и вышеуказанную аутоциркуляризованную геномную ДНК штамма A-1544, взятую в качестве матрицы, использовали для проведения ПЦР. В этой ПЦР-реакции цикл двухстадийной реакции, включающей денатурацию при 98°C в течение 20 с; и отжиг и удлинение цепи, при 68°C в течение 5 мин, повторяли 35 раз с использованием полимеразы Taq Takara LA (Takara Bio Inc.) и ПЦР-амплификатора (T Gradient, Biometra Co.).
В результате был амплифицирован ДНК-фрагмент (ДНК-фрагмент Вl), имеющий размер примерно 3,5 т.п.н., и ДНК-фрагмент (ДНК-фрагмент Сl), имеющий размер примерно 2,8 т.п.н. По всей вероятности, эти ДНК-фрагменты представляют собой ДНК, кодирующую белок, обладающий гидроксилирующей активностью, и ДНК, имеющую последовательность, включающую области, расположенные выше и ниже от первой ДНК.
ДНК-фрагмент В1 и ДНК-фрагмент С1 выделяли из ПЦР-амплифицированного реакционного раствора с помощью SUPREC PCR (Takara Bio Inc.). Затем для получения каждого из ДНК-фрагментов В1 и С1 в количестве, достаточном для проведения анализа нуклеотидной последовательности полученного ДНК-фрагмента, использовали плазмидый вектор pT7Blue T (Novagen Co.), набор для лигирования ДНК ver.2 (Takara Bio Inc.), штамм JM109 E.coli и набор для очистки плазмид (QIA filter Plasmid Midi Kit, QIAGEN Co.), как описано выше в (2), в результате чего получали достаточное количество каждого из указанных ДНК-фрагментов.
(5) Анализ нуклеотидной последовательности ДНК-фрагмента В1 (размером примерно 3,5 т.п.н.) и ДНК-фрагмента С1 (размером примерно 2,8 т.п.н.)
Нуклеотидные последовательности ДНК-фрагмента В1 и ДНК-фрагмента С1, полученные как описано выше в (4), анализировали с использованием анализатора нуклеотидной последовательности ДНК (PE Biosystems 377XL) методом проведения цикла реакций секвенирования с использованием красителя для терминации цепи. Эти нуклеотидные последовательности анализировали для получения данных о нуклеотидной последовательности размером 3793 п.н., представленной в последовательности NO:2 для каждой из последовательностей ДНК-фрагмента В1 и ДНК-фрагмента С1.
В этих 3793 п.н. была идентифицирована открытая рамка считывания (ОРС) для того, чтобы убедиться, что эти два фрагмента кодируют два белка. Каждую аминокислотную последовательность этих белков идентифицировали с помощью программы поиска BLAST и в результате было обнаружено, что ОРС (называемая далее PsmA), кодирующая белок, состоящий из 409 аминокислот, имеет высокую степень гомологии с цитохромом P450 и находится в положениях оснований 1322-2548 последовательности NO:2. psmA имеет самую высокую степень гомологии (гомология: 72,6%) с предполагаемой аминокислотной последовательностью цитохрома P450 (CYP105D5) штамма Streptomyces coelicolor A3 (2) и с предполагаемой аминокислотной последовательностью цитохрома P450 (CYP105D4) штамма Streptomyces lividans, а также было установлено, что эта рамка считывания имеет относительно высокую степень гомологии (гомология: 69,4%) с цитохромом P450 Soy (Soy C) Streptomyces griseus. Исходя из этого можно сделать вывод, что, по всей вероятности, psmA представляет собой ген, кодирующий гидроксилирующий фермент типа цитохрома P-450.
Кроме того, ОРС (далее называемая psmB), кодирующая белок, имеющий высокую степень гомологии с ферредоксином типа 2Fe-2S, находится непосредственно ниже (основания 2564-2761 последовательности NO:2) открытой рамки считывания psmA. Белок, кодируемый psmB, состоит из 66 аминокислот и имеет очень высокую степень гомологии (83,3%) с предполагаемой аминокислотной последовательностью ферредоксина, расположенный непосредственно ниже предполагаемой аминокислотной последовательности цитохрома P450 (CYP105D5) Streptomyces coelicolor A3(2) и относительно более высокую гомологию (гомология: 57,6%) с ферредоксином Soy (soyB) Streptomyces griseus. Поэтому можно сделать вывод, что psmB служит для переноса электронов и кодирует ферредоксин, участвующий в гидроксилировании вместе с psmA.
(6) Конструирование плазмиды pTC-DM
pT7NS-CamAB (см. сравнительный пример 5 и WO 03/087381) гидролизовали соответствующими рестриктирующими ферментами NdeI и SpeI в буферном растворе H (50 мМ трис-HCl, pH 7,5, 10 мМ MgCl2, 10 мМ дитиотреитол и 100 мМ NaCl) с получением гидролизованных плазмидных продуктов. Кроме того, исходя из нуклеотидной последовательности NO:2 конструировали и продуцировали праймер DM-NdeF (5'-GCCCCCATATGACGGAACTGACGGACATCA-3': см. последовательность NO:33), полученный путем добавления NdeI-сайта в 5'-конец, и праймер DM-SpeR (5'-GGGCCACTAGTCAGCCGGCCGGTTCGGTCA-3': см. последовательность NO:34), полученный путем добавления SpeI-сайта в 5'-конец. Затем праймеры этих двух типов (DM-NdeF и DM-SpeR) и геномную ДНК штамма A-1544, полученную как описано выше в (1) и взятую в качестве матрицы, использовали для проведения ПЦР. Эту ПЦР-реакцию осуществляли путем 30-кратного повторения циклов двухстадийной реакции, включающей денатурацию при 98°C в течение 20 с, отжиг и удлинение цепи при 68°C в течение 2 мин, с использованием полимеразы Taq Takara LA (Takara Bio HOLDINGS Inc.) и ПЦР-амплификатора (T Gradient, Biometra Co.). Из раствора реакции ПЦР-амплификации был выделен ДНК-фрагмент, имеющий размер примерно 1,5 т.п.н. и содержащий psmA и psmB, с помощью SUPREC PCR (TAKARA HOLDINGS INC.). Полученный ДНК-фрагмент гидролизовали рестриктирующими ферментами NdeI и SpeI, и этот гидролизат, а также гидролизат вышеупомянутой плазмиды pT7NS-camAB объединяли с использованием набора для лигирования ДНК Ver. 2 (Takara Bio Inc.). Таким образом была сконструирована плазмида размером примерно 9,5 т.п.н. (иногда называемая pTC-DM), включающая плазмиду pT7NS-camAB, присоединенную к ДНК-фрагменту, содержащему обе рамки считывания psmA и psmB.
Сравнительный пример 3: ДНК-2, кодирующая полипептид, обладающий ферментативной активностью, направленной на гидроксилирование макролидного соединения (II) в 16-положении
(1) Получение геномной ДНК штамма Mer-11107 Streptomyces sp.
Штамм Mer-11107 инокулировали в среду, содержащую 1% глюкозу, 0,4% солодовый экстракт и 1% дрожжевой экстракт, и инкубировали при 28°C в течение 3 дней. Полученный культуральный бульон центрифугировали при 3000 об/мин в течение 10 мин для сбора мицелия. Геномную ДНК получали из мицелия с использованием набора Blood & Cell Culture Kit (QIAGEN Co.).
(2) Клонирование неполной последовательности ДНК, кодирующей белок, обладающий активностью, направленной на гидроксилирование макролидного соединения 11107B в 16-положении
Были сконструированы и продуцированы нижеследующие смеси праймеров (5Dm-3F и 5D-1R), исходя из предполагаемой аминокислотной последовательности цитохрома P450 (CYP105D5) Streptomyces coelicolor A3 (2) (см. последовательности NO:17 и 21):
5Dm-3F: 5'-TTCGCSCTSCCSGTCCCSTCSATGGTSAT-3'
5D-1R: 5'-AGGTGCCCAGCGAGATCATGTT-3'
Для стимуляции реактивности с учетом флуктуации кодона использовали смешанные основания S(=C+G) и Y(=C+T).
Затем праймеры этих двух типов (5Dm-3F и 5D-1R) и геномную ДНК штамма Mer-11107, полученную как описано выше в (1) и взятую в качестве матрицы, использовали для проведения ПЦР. Реакцию ПЦР осуществляли путем 35-кратного повторения трехстадийной реакции, включая денатурацию при 98°C в течение 20 с; отжиг при 50°C в течение 2 мин; и удлинение цепи при 68°C в течение 30 с, с использованием полимеразы Taq Takara LA (Takara Bio Inc.) и ПЦР-амплификатора (T Gradient, Biometra Co.). В результате был амплифицирован ДНК-фрагмент (далее называемый ДНК-фрагментом A2), имеющий размер примерно 300 п.н. По всей вероятности, этот ДНК-фрагмент А2 является частью ДНК, кодирующей белок, обладающий гидроксилирующей активностью. ДНК-фрагмент А2, амплифицированный посредством ПЦР, выделяли из реакционного раствора с помощью SUPREC PCR (Takara Bio Inc.).
Для получения ДНК-фрагмента А2 в количестве, достаточном для анализа нуклеотидной последовательности полученного ДНК-фрагмента А2, этот ДНК-фрагмент А2 объединяли с плазмидым вектором pT7Blue T (Novagen Co.) с использованием набора для лигирования ДНК ver.2 (Takara Bio Inc.) в целях трансформации штамма JM109 E.coli. Затем трансформированную E.coli отбирали на LВ-среде с агаром (1,0% бакто-триптон, 0,5% дрожжевой экстракт, 0,5% NaCl, 1,5% агар), содержащей ампициллин (50 мкг/мл), X-gal (5-бром-4-хлор-3-индолил-β-D-галактозид; 40 мкг/мл) и IPTG (изопропил-β-D-тиогалактопиранозид; 100 мкМ). Выделенные таким образом колонии трансформированных E.coli культивировали в жидкой LВ-среде (1% бакто-триптон, 0,5% дрожжевой экстракт, 0,5% NaCl), содержащей ампициллин (50 мкг/мл). Плазмидную ДНК выделяли из мицелия размноженной трансформированной E.coli и очищали с помощью набора для очистки плазмид (QIA filter Plasmid Midi Kit, QIAGEN Co.), в результате чего получали достаточное количество ДНК-фрагмента A2.
(3) Анализ нуклеотидной последовательности клонированного ДНК-фрагмента A2
Нуклеотидную последовательность ДНК-фрагмента А2, полученного как описано выше в (2), анализировали с использованием анализатора нуклеотидной последовательности ДНК (PE Biosystems 377XL) методом проведения цикла реакций секвенирования с использованием красителя для терминации цепи. Хотя в анализе, проводимом с помощью электрофореза, было выявлено, что размер ДНК-фрагмента А2, амплифицированного посредством ПЦР-реакции, составлял примерно 300 п.н., однако, анализ нуклеотидной последовательности этого фрагмента показал, что его точная длина составляет 325 п.н. (см. нуклеотидную последовательность от нуклеотида 837 до нуклеотида 1161 в последовательности NO:3). Поскольку было обнаружено, что последовательности ДНК, соответствующие двум типам праймеров, используемых в вышеописанной ПЦР, присутствуют на обоих концах описанной выше клонированной последовательности ДНК, имеющей размер 325 п.н., то авторами настоящего изобретения был сделан вывод, что в вышеуказанной ПЦР-реакции, ДНК-фрагмент А2 был 1 х амплифицирован указанными двумя праймерами (5Dm-3F и 5D-1R).
(4) Анализ смежной области ДНК-фрагмента А2
После определения неполной последовательности ДНК, кодирующей белок, обладающий гидроксилирующей активностью и происходящий от штамма Mer-11107, как описано выше, нуклеотидную последовательность в смежной области, простирающейся с левой и с правой стороны от клонированного фрагмента, амплифицировали, клонировали и секвенировали методом обратной ПЦР (Cell Technology vol. 14, p.591-593, 1995). В частности, геномную ДНК штамма Mer-11107 (см. выше (1)) гидролизовали рестриктирующим ферментом BamHI в буферном растворе К (50 мМ трис-HCl, pH 8,5, 10 мМ MgCl2, 1 мМ дитиотреитол и 100 мМ КCl) и рестриктирующим ферментом SalI в буферном растворе Н (50 мМ трис-HCl, pH 7,5, 10 мМ MgCl2, 1 мМ дитиотреитол, 100 мМ NaCl). Каждый из полученных ДНК-фрагментов, гидролизованных рестриктирующими ферментами, подвергали аутоциркуляризации с использованием набора для лигирования ДНК Ver.2 (Takara Bio Inc.).
Кроме того, нижеследующие праймеры (7PIN-2F и 6PIN-2R) конструировали и продуцировали на основе нуклеотидной последовательности ДНК-фрагмента А2 (см. последовательности NO:22 и 20):
7PIN-2F: 5'-CCATGATCCTGCTGGTGGCCGGCCATGAGA-3'
6PIN-2R: 5'-CTGTTCCTCGAAGAACTCGTGGTCGGCGTA-3'
Затем праймеры этих двух типов (7PIN-2F и 6PIN-2R) и вышеуказанную аутоциркуляризованную геномную ДНК штамма Mer-11107, взятую в качестве матрицы, использовали для проведения ПЦР. В этой ПЦР-реакции цикл двухстадийной реакции, включающей денатурацию при 98°C в течение 20 с, отжиг и удлинение цепи при 68°C в течение 5 мин, повторяли 35 раз с использованием полимеразы Taq Takara LA (Takara Bio Inc.) и ПЦР-амплификатора (T Gradient, Biometra Co.).
В результате был амплифицирован ДНК-фрагмент (ДНК-фрагмент В2), имеющий размер примерно 1,3 т.п.н., и ДНК-фрагмент (ДНК-фрагмент С2), имеющий размер примерно 1,4 т.п.н. По всей вероятности, эти ДНК-фрагменты представляют собой соответственно ДНК, кодирующую белок, обладающий гидроксилирующей активностью, и ДНК, имеющую последовательность, включающую области, расположенные выше и ниже от первой ДНК.
ДНК-фрагмент В2 и ДНК-фрагмент С2 выделяли из ПЦР-амплифицированного реакционного раствора с помощью SUPREC PCR (Takara Bio Inc.). Затем для получения каждого из ДНК-фрагментов В2 и С2 в количестве, достаточном для проведения анализа нуклеотидной последовательности полученного ДНК-фрагмента, использовали плазмидый вектор pT7Blue T (Novagen Co.), набор для лигирования ДНК ver.2 (Takara HOLDING INC.), штамма JM109 E.coli и набор для очистки плазмид (QIA filter Plasmid Midi Kit, QIAGEN Co.), как описано выше в (2), в результате чего получали достаточное количество каждого из указанных ДНК-фрагментов.
(5) Анализ каждой нуклеотидной последовательности ДНК-фрагмента В2 (размером примерно 1,3 т.п.н.) и ДНК-фрагмента С2 (размером примерно 1,4 т.п.н.)
Нуклеотидную последовательность ДНК-фрагмента В2 и ДНК-фрагмента С2, полученную как описано выше в (4), анализировали с использованием анализатора нуклеотидной последовательности ДНК (PE Biosystems 377XL) методом проведения цикла реакций секвенирования с использованием красителя для терминации цепи. Эти нуклеотидние последовательности анализировали для получения данных о нуклеотидной последовательности размером 2329 п.н., представленной в последовательности NO:3 для каждой последовательности ДНК-фрагмента В2 и ДНК-фрагмента С2.
В этих 2329 п.н. была идентифицирована открытая рамка считывания (ОРС) для того, чтобы убедиться, что эти два фрагмента кодируют два белка. Каждую аминокислотную последовательность этих белков идентифицировали с помощью программы поиска BLAST, в результате чего было обнаружено, что ОРС (называемая далее bpmA), кодирующая белок, состоящий из 395 аминокислот, имеет высокую степень гомологии с цитохромом P450 и находится в положениях оснований 420-1604 последовательности NO:3. bpmA имеет самую высокую степень гомологии (гомология: 67,4%) с аминокислотной последовательностью psmA, выделенной из штамма А-1544, а также относительно высокую степень гомологии (гомология: 69,4%) с цитохромом P450 Soy (Soy C) Streptomyces griseus. Исходя из этого можно сделать вывод, что bpmA, по всей вероятности, представляет собой ген, кодирующий гидроксилирующий фермент типа цитохрома P-450.
Кроме того, ОРС (далее называемая bpmB), кодирующая белок, имеющий высокую степень гомологии с ферредоксином типа 2Fe-2S, находится непосредственно ниже (основания 1643-1834 последовательности NO:3) открытой рамки считывания bpmA. Белок, кодируемый bpmВ, состоит из 64 аминокислот и имеет очень высокую степень гомологии (81,0%) с аминокислотную последовательностью psmB, выделенной из штамма А-1544, и имеет относительно более высокую степень гомологии (гомология: 76,2%) с предполагаемой аминокислотной последовательностью ферредоксина, расположенной непосредственно ниже предполагаемой аминокислотной последовательности цитохрома P450 (CYP105D5) Streptomyces coelicolor A3(2). Поэтому можно сделать вывод, что bpmВ служит для переноса электронов и вместе с bpmA участвует в гидроксилировании.
Сравнительный пример 4: ДНК-3, кодирующая полипептид, обладающий ферментативной активностью, направленной на гидроксилирование макролидного соединения (II) в 16-положении
(1) Получение геномной ДНК штамма A-1560
Штамм A-1560 инокулировали в среду, содержащую 1% глюкозу, 0,4% экстракт из семян пшеницы и 1% дрожжевой экстракт, и инкубировали при 28°C в течение 3 дней. Полученный культуральный бульон центрифугировали при 3000 об/мин в течение 10 мин для сбора мицелия. Геномную ДНК штамма А-1560 получали из мицелия с использованием набора Blood & Cell Culture Kit (QIAGEN Co.).
(2) Клонирование неполной последовательности ДНК, кодирующей белок, обладающий активностью, направленной на гидроксилирование макролидного соединения 11107B в 16-положении
Исходя из предполагаемой аминокислотной последовательности цитохрома P450 (CYP105D5) Streptomyces coelicolor A3 (2) (см. последовательности NO:17 и 23) были сконструированы и продуцированы нижеследующие смеси праймеров (5Dm-3F и 5Dm-2R):
5Dm-3F: 5'-TTCGCSCTSCCSGTCCCSTCSATGGTSAT-3'
5Dm-2R: 5'-CTGGATSGTGTCSCCSGGYTT-3'
Для стимуляции реактивности с учетом флуктуации кодона использовали смешанные основания S(=C+G) и Y(=C+T).
Затем праймеры этих двух типов (5Dm-3F и 5Dm-2R) и геномную ДНК штамма A-1560, полученную как описано выше в (1) и взятую в качестве матрицы, использовали для проведения ПЦР. Реакцию ПЦР проводили путем 35-кратного повторения трехстадийной реакции, включая денатурацию при 98°C в течение 20 с; отжиг при 50°C в течение 2 мин; и удлинение цепи при 68°C в течение 30 с, с использованием полимеразы Taq Takara LA (Takara Bio Inc.) и ПЦР-амплификатора (T Gradient, Biometra Co.). В результате этого был амплифицирован ДНК-фрагмент (далее называемый ДНК-фрагментом A3), имеющий размер примерно 750 п.н. По всей вероятности, этот ДНК-фрагмент А3 является частью ДНК, кодирующей белок, обладающий гидроксилирующей активностью. ДНК-фрагмент А3, амплифицированный посредством ПЦР, выделяли из реакционного раствора с помощью SUPREC PCR (Takara Bio Inc.).
Для получения ДНК-фрагмента А3 в количестве, достаточном для анализа нуклеотидной последовательности полученного ДНК-фрагмента А3, этот ДНК-фрагмент А3 объединяли с плазмидным вектором pT7Blue T (Novagen Co.) с использованием набора для лигирования ДНК ver.2 (Takara Bio Inc.) в целях трансформации штамма JM109 colibacillus (Stratagene Со.). Затем трансформированную colibacillus отбирали на LВ-среде с агаром (1,0% бакто-триптон, 0,5% дрожжевой экстракт, 0,5% NaCl, 1,5% агар), содержащей ампициллин (50 мкг/мл), X-gal (5-бром-4-хлор-3-индолил-β-D-галактозид; 40 мкг/мл) и IPTG (изопропил-β-D-тиогалактопиранозид; 100 мкМ). Выделенную таким образом колонию трансформированных colibacillus культивировали в жидкой LВ-среде (1% бакто-триптон, 0,5% дрожжевой экстракт, 0,5% NaCl), содержащей ампициллин (50 мкг/мл). Плазмидную ДНК выделяли и очищали из мицелия размноженной трансформированной colibacillus с помощью набора для очистки плазмид (QIA filter Plasmid Midi Kit, QIAGEN Co.), в результате чего получали достаточное количество ДНК-фрагмента A3.
(3) Анализ нуклеотидной последовательности клонированного ДНК-фрагмента A3
Нуклеотидную последовательность ДНК-фрагмента А3, полученного как описано выше в (2), анализировали с использованием анализатора нуклеотидной последовательности ДНК (PE Biosystems 377XL) методом проведения цикла реакций секвенирования с использованием красителя для терминации цепи. Хотя в анализе, проводимом с помощью электрофореза, было выявлено, что размер ДНК-фрагмента А3, амплифицированного посредством ПЦР-реакции, составлял примерно 750 п.н., однако, анализ нуклеотидной последовательности этого фрагмента показал, что его точная длина составляет 741 п.н. (см. нуклеотидную последовательность от нуклеотида 616 до нуклеотида 1356 последовательности NO:4). Поскольку было обнаружено, что последовательности ДНК, соответствующие двум типам праймеров, используемых в вышеописанной ПЦР, присутствуют на обоих концах описанной выше клонированной последовательности ДНК размером 741 п.н., то авторами настоящего изобретения был сделан вывод, что ДНК-фрагмент А3 был специфически амплифицирован праймерами этих двух типов (5Dm-3F и 5Dm-2R) в вышеуказанной ПЦР-реакции.
(4) Анализ смежной области ДНК-фрагмента А3
После определения неполной последовательности ДНК, кодирующей белок, обладающий гидроксилирующей активностью и происходящий от штамма A-1560, как описано выше, нуклеотидную последовательность смежной области, простирающейся с левой и с правой стороны от клонированного фрагмента, амплифицировали, клонировали и секвенировали методом обратной ПЦР (Cell Technology vol. 14, p.591-593, 1995). В частности, геномную ДНК штамма A-1560 (см. выше (1)) гидролизовали рестриктирующим ферментом BamHI в буферном растворе К (50 мМ трис-HCl, pH 8,5, 10 мМ MgCl2, 1 мМ дитиотреитол и 100 мМ КCl), рестриктирующим ферментом KpnI в буферном растворе L (50 мМ трис-HCl, pH 7,5, 10 мМ MgCl2 и 1 мМ дитиотреитол) и рестриктирующим ферментом SalI в буферном растворе H (50 мМ трис-HCl, pH 7,5, 10 мМ MgCl2, 1 мМ дитиотреитол и 100 мМ NaCl). Каждый из полученных ДНК-фрагментов, гидролизованных рестриктирующими ферментами, подвергали аутоциркуляризации с использованием набора для лигирования ДНК ver.2 (Takara Bio Inc.).
Кроме того, нижеследующие праймеры (5PIN-2F и 6PIN-2R) конструировали и продуцировали на основе нуклеотидной последовательности ДНК-фрагмента А3 (см. последовательности NO:24 и 20):
5PIN-2F: 5'-CGGAATCCACCAGTGCCTCGGCCAGAACCT-3'
6PIN-2R: 5'-CTGTTCCTCGAAGAACTCGTGGTCGGCGTA-3'
Затем праймеры этих двух типов (5PIN-2F и 6PIN-2R) и вышеуказанную аутоциркуляризованную геномную ДНК штамма A-1560, взятую в качестве матрицы, использовали для проведения ПЦР. В этой ПЦР-реакции цикл двухстадийной реакции, включающей денатурацию при 98°C в течение 20 с; отжиг и удлинение цепи при 68°C в течение 5 мин, повторяли 35 раз с использованием полимеразы Taq Takara LA (Takara Bio Inc.) и ПЦР-амплификатора (T Gradient, Biometra Co.).
В результате был амплифицирован ДНК-фрагмент (ДНК-фрагмент В3), имеющий размер примерно 4,5 т.п.н., ДНК-фрагмент (ДНК-фрагмент С3), имеющий размер примерно 3,0 т.п.н., и ДНК-фрагмент (ДНК-фрагмент D3), имеющий размер примерно 1,7 т.п.н. По всей вероятности, эти ДНК-фрагменты представляют собой ДНК, кодирующую белок, обладающий гидроксилирующей активностью, и ДНК, имеющую последовательность, включающую области, расположенные выше и ниже от первой ДНК.
ДНК-фрагмент В3, ДНК-фрагмент С3 и ДНК-фрагмент D3 выделяли из ПЦР-амплифицированного реакционного раствора с помощью SUPREC PCR (Takara Bio Inc.). Затем в целях получения каждого из ДНК-фрагментов В3, С3 и D3 в количестве, достаточном для проведения анализа нуклеотидной последовательности полученного ДНК-фрагмента, использовали плазмидый вектор pT7Blue T (Novagen Co.), набор для лигирования ДНК ver.2 (Takara Bio Inc.), штамм JM109 E.coli и набор для очистки плазмид (QIA filter Plasmid Midi Kit, QIAGEN Co.), как описано выше в (2), в результате чего получали достаточное количество каждого из указанных ДНК-фрагментов.
(5) Анализ нуклеотидной последовательности ДНК-фрагмента В3 (размером примерно 4,5 т.п.н.), ДНК-фрагмента С3 (размером примерно 3,0 т.п.н.) и ДНК-фрагмента D3 (размером примерно 1,7 т.п.н.)
Каждую нуклеотидную последовательность ДНК-фрагмента В3, ДНК-фрагмента С3 и ДНК-фрагмента D3, полученную как описано выше в (4), анализировали с использованием анализатора нуклеотидной последовательности ДНК (PE Biosystems 377XL) методом проведения цикла реакций секвенирования с использованием красителя для терминации цепи. Эти нуклеотидние последовательности анализировали для получения данных о нуклеотидной последовательности размером 1860 п.н., представленной в последовательности NO:4 для каждой последовательности ДНК-фрагмента В3, ДНК-фрагмента С3 и ДНК-фрагмента D3.
В этих 1860 п.н. была идентифицирована открытая рамка считывания (ОРС) и было обнаружено, что она кодирует два белка. Каждую аминокислотную последовательность этих белков идентифицировали с помощью программы поиска BLAST, в результате чего было обнаружено, что ОРС (называемая далее tpmA), кодирующая белок, состоящий из 404 аминокислот, имеет высокую степень гомологии с цитохромом P450 и находится в положениях оснований 172-1383 последовательности NO:4. tpmA имеет самую высокую степень гомологии (гомология: 77,4%) с предполагаемой аминокислотной последовательностью цитохрома P450 (CYP105D5) штамма Streptomyces coelicolor A3 (2), а также высокую степень гомологии (гомология: 76,6%) с аминокислотной последовательностью psmA, выделенного из штамма А-1544. Исходя из этого можно сделать вывод, что tpmA, по всей вероятности, представляет собой ген, кодирующий гидроксилирующий фермент типа цитохрома P-450.
Кроме того, ОРС (далее называемая tpmB), кодирующая белок, имеющий высокую степень гомологии с ферредоксином типа 2Fe-2S, находится непосредственно ниже (основания 1399-1593 в последовательности NO:4) открытой рамки считывания tpmA. Белок, кодируемый tpmB, состоит из 65 аминокислот и имеет очень высокую степень гомологии (87,3%) с аминокислотной последовательностью psmB, выделенного из штамма А-1544, а также высокую степень гомологии (гомология: 82,5%) с аминокислотной последовательностью, которой предположительно, является последовательность ферредоксина, расположенная непосредственно ниже предполагаемой аминокислотной последовательности цитохрома P450 (CYP105D5) Streptomyces coelicolor A3(2). Поэтому можно сделать вывод, что tpmB вместе с tpmA служит для переноса электронов и кодирует ферредоксин, участвующий в гидроксилировании.
Сравнительный пример 5: Конструирование плазмиды pT7NS-camAB
Праймеры PRR-1F (см. последовательность NO:29) и PRR-2R (см. последовательность NO:30), имеющие последовательности, представленные ниже, использовали для проведения ПЦР в нижеследующих условиях с использованием геномной ДНК Pseudomonas putida ATCC 17453 в качестве матрицы:
PRR-1F: 5'-GCCCCCCATATGAACGCAAACGACAACGTGGTCATC-3'
PRR-2R: 5'-GCGGATCCTCAGGCACTACTCAGTTCAGCTTTGGC-5'
Жидкая композиция для реакции:
Температуры реакции:
95°C, 3 мин
(98°C, 20 с; 63°C, 30 с, 68°C, 2 мин) - 30 циклов
72°C, 5 мин
Полученный амплифицированный 1,5 т.п.н.-фрагмент (фрагмент camAB), содержащий ген путидаредоксин-редуктазы (camA) и расположенный непосредственно ниже ген путидаредоксина (camB), гидролизовали рестриктирующими ферментами NdeI и BamHI, а затем подвергали электрофорезу на 0,8% агарозном геле. После электрофореза фрагмент camAB собирали и выделяли с помощью SUPREC-01 (TAKARA HOLDINGS INC.) из гелевого среза, содержащего фрагмент, который был вырезан из этого геля. Этот фрагмент лигировали с помощью ДНК-лигазы Т4 с NdeI-сайтом и BamHI-сайтом плазмидного вектора E. coli pET11a (Stratagene Co.), а затем переносили в DH5α E.coli для конструирования плазмиды pT7-camAB. Затем молекулу линкера 1, полученную путем отжига двух синтетических олиго-ДНК SP-1 (см. последовательность NO:31) и SP-2 (см. последовательность NO:32) с последовательностями, представленными ниже, присоединяли к NdeI-сайту указанной плазмиды с помощью ДНК-лигазы T4 и переносили в DH5α E.coli для конструирования плазмиды pT7NS-camAB:
SP-1: 5'-TATGCGTCACTAGTCGGGAGTGCGTTA-3'
SP-2: 5'-TATAACGCACTCCCGACTAGTGACGCA-3'
Пример 1: Получение плазмиды pPapsmAB путем лигирования промотора pldA с psmAB
Промоторную область pldA гена биосинтеза пладиенолида В присоединяли к psmAB для осуществления экспрессии psmAB в комбинации с геном биосинтеза пладиенолида В. Два праймера, pdlApro-SpeNdeF (SpeI- и NdeI-сайты, присоединенные к 5'-концу: см. последовательность NO:25) и pldApro-NdeR (NdeI-сайт, присоединенный к 5'-концу: см. последовательность NO:26), содержащие последовательности, представленные ниже, синтезировали исходя из данных о нуклеотидной последовательности, представленной последовательностью NO:1, в целях амплификации промоторной области pldA:
pldApro-SpeNdeF: 5'-GGGCATATGACTAGTAGCCGTGTCCTGCCCGCC-3'
pldApro-NdeR: 5'-GGGCATATGTTCGGACGTGAATTCATTCG-3'
ПЦР проводили в нижеследующих условиях с использованием указанных праймеров.
Жидкая композиция для ПЦР-реакции: с использованием Toyo Boseki KOD plus-:
Условия реакции: с использованием Biometra T GRADIENT:
95°C, 5 мин
(98°C, 20 с; 63°C, 1 мин, 68°C, 1 мин) - 25 циклов
72°C, 5 мин
360 п.н.-ДНК-фрагмент, амплифицированный в результате этой реакции, собирали с использованием набора для ПЦР-очистки (QIAquick PCR Purification Kit (QIAGEN Co.)). Полученный 360 п.н.-ДНК-фрагмент гидролизовали рестриктирующим ферментом NdeI. Плазмиду pTC-DM (Сравнительный пример 2(6)) аналогичным образом гидролизовали рестриктирующим ферментом Ndel. Полученный гидролизат 360 п.н.-ДНК-фрагмента и гидролизат плазмиды присоединяли друг к другу с использованием набора для лигирования ДНК ver.2.1 (DNA Ligation Kit Ver. 2.1; Takara Bio Inc.). Аналогичным образом конструировали плазмиду pPapsmAB, имеющую размер примерно 9,9 т.п.н. и содержащую промотор pldA, присоединенный к psmAB.
Пример 2: Получение плазмиды для рекомбинации
Плазмиду pPapsmaAB, полученную как описано в примере 1, гидролизовали рестриктирующим ферментом SpeI и затупляли по концам с использованием набора BKL (Takara Bio Inc.). Полученную ДНК подвергали электрофорезу (Mupid-ex: Advance Co.) на 0,8% агарозе Molecular Biology Agarose™ (BIO-RAD), и выделенный 1,8- т.п.н.-ДНК-фрагмент, содержащий промотор pldA и psmAB (далее иногда называемый PapsmAB), вырезали, собирали и очищали с использованием набора для гель-экстракции QIAquick (QIAGEN Co.). Рекомбинационный вектор pUC19aph::oriT::intphiC31, представленный на фиг.4, также гидролизовали рестриктирующим ферментом BamHI и затупляли по концам с использованием набора BKL (Takara Bio Inc.). Полученный вектор и PapsmAB присоединяли друг к другу с использованием набора для лигирования ДНК Ver. 2.1 (Takara Bio Inc.). Таким образом была сконструирована рекомбинантная плазмида pAOC::PapsmAB, содержащая PapsmAB, встроенный в рекомбинантный вектор pUC19aph::oriT::intphiC31.
Пример 3: Введение рекомбинантной плазмиды pAOC::PapsmAB в Mer-11107
Полученную плазмиду pAOC::PapsmAB вводили в конъюгированный микроорганизм E. coli S17-1 (ATCC47055) посредством электропорации с получением S17-l/pA0C::PapsmAB. Полученный S17-1/pAOC::PapsmAB инокулировали в 10 мл LB-среды (1% бакто-триптон, 0,5% дрожжевой экстракт, 0,5% NaCl), содержащей 25 мкг/мл канамицина, и культивировали со встряхиванием при 30°С в течение 2 ч, после чего мицелий собирали, два раза промывали 10 мл LB-среды и суспендировали в 5 мл LB-среды. Таким образом, была получена донорная суспензия. Одновременно с получением донорной суспенизии Mer-11107 инокулировали на 10 мл среды TSB (триптиказо-соевый бульон: Nissui Seiyaku) и инкубировали со встряхиванием при 30°С в течение 5 ч, а затем мицелий собирали, два раза промывали 10 мл стерилизованной воды и суспендировали в 1 мл стерилизованной воды. Таким образом была получена акцепторная суспензия. 500 мкл донорной суспензии S17-1/pAOC::PapsmAB смешивали с 10 мкл полученной акцепторной суспензии Mer-11107 и наносили на агаровую среду Actino No. 4 (Nissui Pharmaceutical). После инкубирования при 30°C в течение 18 ч, на эту среду наносили 2,5 мл среды SNA (0,8% питательная среда: Difco, 0,4% агар), содержащей 2 мг/мл рибостамицина, и инкубировали при 30°C в течение 7 дней с получением резистентного к рибостамицину pAOC::PapsmAB-трансформированного штамма Mer-11107::pAOC::PapsmAB.
Пример 4: Тест на продуцирование пладиенолида штаммом Mer-11107::pAОC::PapsmAB
Mer-11107::pAОC::PapsmAB, полученный как описано в примере 3, и родительский штамм Mer-11107, используемый в качестве контроля, тестировали на продуцирование пладиенолида В и пладиенолида D с гидроксидом в 16-положении.
300 мкл замороженного посевного материала каждого из штаммов Mer-11107::pAOC::PapsmAB и Mer-11107 инокулировали в 60 мл среды для посева (2% растворимый крахмал, 2% соевая мука (ESUSAN-MEAT, изготовленная Ajinomoto Co. Ltd.), 0,3% дрожжевой экстракт, 0,1% K2HPO4, 0,25% MgSO4 · 7H2O, 0,3% CaCO3, pH не корректировали) и инкубировали при 25°C в течение 3 дней. 600 мкл полученного культурального посевного бульона инокулировали в 60 мл основной культуральной среды (5% растворимый крахмал, 1% глюкоза, 3% Pharmamedia, 0,05% адеканол LG-126, 0,1% CaCO3, pH 7,5), и культивировали при 25°C в течение 3, 4, 5 и 6 дней. После завершения культивирования полученный культуральный бульон экстрагировали путем 4-кратного добавления ацетонитрила. Количество пладиенолида В и пладиенолида D с гидроксидом в 16-положении, содержащееся в полученном экстракте, измеряли с помощью ВЭЖХ. Результаты измерения приводятся в таблице 2. Условия ВЭЖХ-измерения приводятся ниже:
Устройство: ВЭЖХ Shimadzu 10Avp
Колонка: Develosil ODS UG-3 (φ 4,6 мм × 50 мм × 3 мкм)
Подвижная фаза: 45%-55% метанол (0-5 мин), 55% метанол (5-13 мин), 55%-70% метанол (13-21 мин), 70% метанол (21-25 мин)
Скорость потока: 1,2 мл/мин
Детекция: УФ на 240 нм
Объем впрыска: 5 мкл
Температура колонки: 40°С
Время анализа: 25 мин
Время удерживания:
Пладиенолид В: 12,4 мин
Пладиенолид D: 4,3 мин.
Пладиенолид D почти не продуцировался родительским штаммом Mer-11107, но продуцировался штаммом Mer-11107::pAОC::PapsmAB, в который была введена pAOC::PapsmAB. Это указывало на то, что прямое продуцирование пладиенолида D может быть достигнуто путем введения гена psmAB, кодирующего гидроксилазу, гидроксилирующую пладиенолид В в 16-положении, в штамм Mer-11107, продуцирующий пладиенолид В.
Пример 5: Влияние введения β-CD на продуцирование пладиенолида D штаммом Mer-11107::pAOC::PapsmAB
Были проведены исследования на влияние введения β-циклодекстрина (β-CD) на продуцирование пладиенолида В и пладиенолида D с гидроксидом в 16-положении штаммом Mer-11107::pAOC::PapsmAB, полученным как описано в примере 3. 300 мкл замороженного посевного материала Mer-11107::pAOC::PapsmAB инокулировали в 30 мл среды для посева (2% растворимый крахмал, 2% соевая мука (ESUSAN-MEAT, изготовленная Ajinomoto Co. Ltd.), 0,3% дрожжевой экстракт; 0,1% K2HPO4, 0,25% MgSO4 · 7H2O, 0,3% CaCO3, pH не корректировали) и инкубировали при 25°C в течение 3 дней. 300 мкл полученного культурального посевного бульона инокулировали в 30 мл основной культуральной среды, в которую был добавлен 2% β-CD (5% растворимый крахмал, 1% глюкоза, 3% Pharmamedia, 0,05% адеканол LG-126, 2,0% β-CD, 0,1% CaCO3, pH 7,5), или 30 мл основной культуральной среды без β-CD (5% растворимый крахмал, 1% глюкоза, 3% Pharmamedia, 0,05% адеканол LG-126, 0,1% CaCO3, pH 7,5) и инкубировали при 25°C в течение 3, 4, 5 и 6 дней. После завершения культивирования полученный культуральный бульон экстрагировали путем 4-кратного добавления ацетонитрила. Количества пладиенолида В и пладиенолида D с гидроксидом в 16-положении, содержащиеся в полученном экстракте, измеряли с помощью ВЭЖХ. Результаты измерения приводятся в таблице 3.
Условия ВЭЖХ-измерения приводятся ниже:
Устройство: ВЭЖХ Shimadzu 10Avp
Колонка: Develosil ODS UG-3 (φ 4,6 мм × 50 мм × 3 мкм)
Подвижная фаза: 45%-55% метанол (0-5 мин), 55% метанол (5-13 мин), 55%-70% метанол (13-21 мин), 70% метанол (21-25 мин)
Скорость потока: 1,2 мл/мин
Детекция: УФ на 240 нм
Объем впрыска: 5 мкл
Температура колонки: 40°С
Время анализа: 25 мин
Время удерживания:
Пладиенолид В: 12,4 мин
Пладиенолид D: 4,3 мин.
В результате добавления 2% β-CD наблюдалось 1,4-кратное увеличение аккумуляции пладиенолида D, что свидетельствовало о том, что дабавление β-CD оказывает непосредственное воздействие на продуцирование пладиенолида D.
Пример 6: Продуцирование пладиенолида штаммом Mer-11107::pAOC::PapsmAB (2)
300 мкл замороженного посевного материала штамма Mer-11107::pAOC::PapsmAB, полученного как описано в примере 3, инокулировали в 30 мл культуральной среды для посева (2% растворимый крахмал, 2% соевая мука (ESUSAN-MEAT, изготовленная Ajinomoto Co. Ltd.), 0,3% дрожжевой экстракт, 0,1% K2HPO4, 0,25% MgSO4 · 7H2O, 0,3% CaCO3, pH не корректировали) и инкубировали при 25°C в течение 3 дней. 300 мкл полученного культурального посевного бульона инокулировали в 30 мл основной культуральной среды (5% растворимый крахмал, 1% глюкоза, 3% Pharmamedia, 1% соевая мука (J-Oil Mills: Honen SoyPro), 0,05% адеканол LG-126, 2,0% β-CD, 1,0% CaCO3, pH 7,5), и культивировали в течение 3, 4, 5, 6 и 7 дней при 25°C.
После завершения культивирования полученную культуральную жидкость экстрагировали путем 4-кратного добавления ацетонитрила. Количество пладиенолида В и пладиенолида D с гидроксидом в 16-положении, содержащееся в полученном экстракте, измеряли с помощью ВЭЖХ. Результаты измерения приводятся в таблице 4.
Условия ВЭЖХ-измерения приводятся ниже:
Устройство: ВЭЖХ Shimadzu 10Avp
Колонка: Develosil ODS UG-3 (φ 4,6 мм × 50 мм × 3 мкм)
Подвижная фаза: 45%-55% метанол (0-5 мин), 55% метанол (5-13 мин), 55%-70% метанол (13-21 мин), 70% метанол (21-25 мин)
Скорость потока: 1,2 мл/мин
Детекция: УФ на 240 нм
Объем впрыска: 5 мкл
Температура колонки: 40°С
Время анализа: 25 мин
Время удерживания:
Пладиенолид В: 12,4 мин
Пладиенолид D: 4,3 мин.
50 мл экстракта, полученного путем добавления в вышеуказанный культуральный бульон равного количества ацетонитрила, фильтровали, и мицелий промывали 30 мл воды. К фильтрату добавляли 70 мл воды, а затем экстрагировали один раз 100 мл и два раза 50 мл этилацетата. Этилацетатные слои объединяли, два раза промывали 50 мл солевого раствора и сушили над безводным сульфатом натрия. После удаления растворителя полученный остаток очищали тонкослойной хроматографией (силикагель MERCK 60 F254 0,5 мм; проявляющий раствор: толуол:ацетон=1:1) с получением 12,8 мг пладиенолида В и 23,1 мг пладиенолида D. Результаты1H-ЯМР-анализа для пладиенолида В и пладиенолида D приводятся ниже.
Пладиенолид B;
1H-ЯМР-спектр (CD3OD, 500 МГц): δ м.д. (сопряжение, мультиплетность, константа взаимодействия J (Гц) 0,93 (3H, д, J=7,0 Гц), 0,94 (3H, д, J=6,8 Гц), 0,98 (3H, т, J=8,0 Гц), 1,12 (3H, д, J=6,8 Гц), 1,23 (3H,с), 1,25 (1Н,м), 1,42 (2H,м), 1,53-1,70 (6H,м), 1,79 (3H, д, J=1,0 Гц), 2,10 (3H,с), 2,52 (1Н,м), 2,56 (2H,м), 2,60 (1H, м), 2,70 (1Н, дд, J=2,4, 8,3 Гц), 2,76 (1Н, дт, J=2,4, 5,7 Гц), 3,56 (1H, дт, J=8,3, 4,4 Гц), 3,82 (1Н, м), 5,08 (2H, д, J=9,8 Гц), 5,60 (1Н, дд, J=9,8, 15,2 Гц), 5,70 (1H, дд, J=8,3, 15,2 Гц), 5,74 (1H, дд, J=9,8, 15,2 Гц), 6,13 (1H, д, J=9,8 Гц), 6,36 (1Н, дд, J=9,8, 15,2 Гц).
Пладиенолид D;
1H-ЯМР-спектр (CD3OD, 500 MГц): δ м.д. (сопряжение, мультиплетность, константа взаимодействия J (Гц)):
0,93 (3H, д, J=7,0 Гц), 0,95 (3H, д, J=6,8 Гц), 0,98 (3H, т, J=8,0 Гц), 1,23 (3H,с), 1,30 (1Н,м), 1,36-1,66 (9H,м), 1,70 (1Н, дд, J=6,4, 14,2 Гц), 1,82 (3H, д, J=1,0 Гц), 1,90 (1Н, дд, J=6,4, 14,2 Гц), 2,10 (3H,с), 2,52 (2H,м), 2,62 (1Н,м), 2,72 (1Н, дд, J=2,4, 8,3 Гц), 2,94 (1Н, дт, J=2,4, 5,7 Гц), 3,55 (1Н, дт, J=8,3, 4,4 Гц), 3,82 (1Н, м), 5,10 (1Н, д, J=9,8 Гц), 5,11 (1H, д, J=I0,8 Гц), 5,60 (1Н, дд, J=9,8, 15,2 Гц), 5,74 (1Н, дд, J=8,3, 15,2 Гц), 5,92 (1Н, д, J=15,2 Гц), 6,18 (1Н, д, J=10,8 Гц), 6,57 (1Н, дд, J=10,8, 15,2 Гц)
Пример 7: Введение рекомбинантной плазмиды pAOC::PapsmAB
в штамм Mer-11107 pldB::hyg
Штамм S17-1/pAOC::PapsmAB, полученный как описано в примере 3, инокулировали в 10 мл LB-среды (1% бакто-триптон, 0,5% дрожжевой экстракт, 0,5% NaCl), содержащей 25 мкг/мл канамицина, и культивировали со встряхиванием в течение 2 ч при 30°С, а затем мицелий собирали, два раза промывали 10 мл LB-среды и суспендировали в 5 мл LB-среды. Таким образом, была получена донорная суспензия.
Одновременно с получением донорной суспенизии штамм Mer-11107 pldB::hyg, полученный как описано в сравнительном примере 1(8), инокулировали в 10 мл среды TSB (триптиказо-соевый бульон: Nissui Pharmaceutical) и инкубировали со встряхиванием при 30°С в течение 5 ч, а затем мицелий собирали, два раза промывали 10 мл стерилизованной воды и суспендировали в 1 мл стерилизованной воды. Таким образом была получена акцепторная суспензия. 500 мкл донорной суспензии S17-1/pAОC::PapsmAB смешивали с 10 мкл полученной акцепторной суспензии Mer-11107 pldB::hyg и наносили на агаровую среду Actino No. 4 (Nippon Seiyaku). После инкубирования при 30°C в течение 18 ч, на эту среду наносили 2,5 мл среды SNA (0,8% питательная среда: Difco, 0,4% агар), содержащей 2 мг/мл рибостамицина, и инкубировали при 30°C в течение 7 дней с получением резистентного к рибостамицину pAOC::PapsmAB-трансформированного штамма Mer-11107 pldB::hyg::pAOC::PapsmAB.
Пример 8: Тест на продуцирование пладиенолида штаммом Mer-11107 pldB::hyg::pAОC::PapsmAB
Штамм Mer-11107 pldB::hyg::pAOC::PapsmAB, полученный как описано в примере 7, и родительский штамм Mer-11107 pldB::hyg, используемый в качестве контроля, тестировали на продуцирование ME-265 и МЕ-282 с гидроксидом в 16-положении. 300 мкл замороженного посевного материала каждого из штаммов Mer-11107 pldB::hyg::pAOC::PapsmAB и Mer-11107 pldB::hyg инокулировали в 30 мл среды для посева (2% растворимый крахмал, 2% соевая мука (ESUSAN-MEAT, изготовленная Ajinomoto Co. Ltd.), 0,3% дрожжевой экстракт, 0,1% K2HPO4, 0,25% MgSO4 · 7H2O, 0,3% CaCO3, pH не корректировали) и инкубировали при 25°C в течение 3 дней. 300 мкл полученного культурального посевного бульона инокулировали в 30 мл основной культуральной среды, в которую добавляли 2% β-CD (5% растворимый крахмал, 1% глюкоза, 3% Pharmamedia, 0,05% адеканол LG-126, 2,0% β-CD, 0,1% CaCO3, pH 7,5), и инкубировали при 25°C в течение 3, 4, 5 и 6 дней. После завершения культивирования культуральный бульон экстрагировали путем 4-кратного добавления ацетонитрила. Количество МЕ-265 и МЕ-282 с гидроксидом в 16-положении, содержащееся в полученном экстракте, измеряли с помощью ВЭЖХ. Результаты измерения приводятся в таблице 5.
Условия ВЭЖХ-измерения приводятся ниже:
Устройство: ВЭЖХ Shimadzu 10Avp
Колонка: Develosil ODS UG-3 (φ 4,6 мм × 50 мм × 3 мкм)
Подвижная фаза: 45%-55% метанол (0-5 мин), 55% метанол (5-13 мин), 55%-70% метанол (13-17 мин), 70% метанол (1-35 мин)
Скорость потока: 1,2 мл/мин
Детекция: УФ на 240 нм
Объем впрыска: 5 мкл
Температура колонки: 40°С
Время анализа: 35 мин
Время удерживания:
МЕ-265: 21,0 мин
МЕ-282: 15,6 мин.
ME-282 почти не продуцировался родительским штаммом Mer-11107 pldB::hyg, но продуцировался штаммом Mer-11107 pldB::hyg::pAOC::PapsmAB, в который была введена pAOC::PapsmAB. Это указывало на то, что прямое продуцирование ME-282 может быть достигнуто путем введения гена psmAB, кодирующего фермент, гидроксилирующий МЕ-265 в 16-положении, в штамм Mer-11107 pldB::hyg, продуцирующий МЕ-265.
Примерно к 90 мл экстракта, полученного путем добавления равного количества ацетонитрила в вышеупомянутый культуральный бульон штамма Mer-11107 pldB::hyg::pAОC::PapsmAB, добавляли ускоритель фильтрации, а затем смесь перемешивали и фильтровали через воронку Кирияма φ 60 (фильтровальная бумага Кирияма No. 4) для выделения мицелия. Мицелий промывали 50 мл воды и получали фильтрат с выделенным мицелием. Этот фильтрат два раза экстрагировали 100 мл этилацетата. После сушки над безводным сульфатом натрия растворитель удаляли с получением остатка. Этот остаток очищали тонкослойной хроматографией (силикагель MERCK 60 F254 0,5 мм, проявляющий раствор: толуол:ацетон=1:1) с получением 0,5 мг МЕ-265 и 12,5 мг МЕ-282. Результаты1H-ЯМР-анализа для МЕ-265 и МЕ-282 приводятся ниже.
МЕ-265:
1H-ЯМР-спектр (CD3OD, 500 МГц): δ м.д. (сопряжение, мультиплетность, константа взаимодействия J (Гц)): 0,87 (3H, д, J=7,0 Гц), 0,90 (3H, д, J=7,0 Гц), 0,94 (3H, д, J=7,3 Гц), 0,97 (3H, д, J=7,0 Гц), 1,08 (3H, д, J=7,0 Гц), 1,17-1,21 (1Н, м), 1,24-1,36 (2H,м), 1,42-1,52 (3H,м), 1,61-1,66 (3H,м), 1,74 (3H, д, J=1,1 Гц), 1,89-1,96 (lH,м), 2,00 (3H,с), 2,41-2,47 (1H,м), 2,43 (1H, дд, J=5,5, 13,9 Гц), 2,51-2,58 (1Н,м), 2,56 (1H, дд, J=3,7, 13,9 Гц), 2,65 (1Н, дд, J=2,2, 8,1 Гц), 2,72 (1Н, дт, J=2,2, 5,9 Гц), 3,51 (1H, дт, J=4,4, 8,4 Гц), 3,75-3,80 (1Н,м), 4,91 (1H, дд, J=8,8, 10,6 Гц), 5,00 (1Н, д, J=10,6 Гц), 5,42 (1Н, дд, J=9,2, 15,0 Гц), 5,49 (1Н, дд, J=9,2, 15,0 Гц), 5,65 (lH, дд, J=8,4, 15,0 Гц), 6,08 (1Н, д, J=10,6 Гц), 6,32 (1Н, дд, J=10,6, 15,0 Гц).
ME-282:
1H-ЯМР-спектр (CD3OD, 500 МГц): δ м.д. (сопряжение, мультиплетность, константа взаимодействия J (Гц)): 0,87 (3H, д, J=7,0 Гц), 0,90 (3H, д, J=7,0 Гц), 0,94 (3H, т, J=7,3 Гц), 0,97 (3H, д, J=6,6 Гц), 1,21-1,26 (1H, м), 1,29-1,37 (3H, м), 1,34 (3H,с), 1,44-1,52 (2Н,м), 1,60-1,64 (1Н,м), 1,65 (1H, дд, J=6,2, 13,9 Гц), 1,77 (3H, д, J=1,1 Гц), 1,86 (1H, дд, J=5,4, 13,9 Гц), 1,89-1,94 (1Н,м), 2,00 (3H,с), 2,43 (1H, дд, J=5,5, 13,9 Гц), 2,50-2,60 (1Н,м), 2,56 (1Н, дд, J=3,3, 13,9 Гц), 2,66 (lH, дд, J=2,2, 7,7 Гц), 2,89 (1Н, дт, J=2,2, 6,2 Гц), 3,52 (1Н, дт, J=4,8, 8,4 Гц), 3,75-3,80 (1Н,м), 4,90 (1H, перекрывающийся с D2O), 5,01 (1Н, д, J=10,6 Гц), 5,42 (1H, дд, J=9,2, 15,0 Гц), 6,13 (1Н, д, J=10,6 Гц), 6,52 (1Н, дд, J=11,0, 15,0 Гц).
Пример 9: Получение плазмиды pPepsmAB, содержащей промотор ermE, присоединенный к psmAB
Промоторную область ermE лигировали с psmAB для индуцирования экспрессии psmAB с использованием промотора ermE. Два праймера, eDM-SphF (SphI-сайт, присоединенный к 5'-концу: см. последовательность NO:27) и eDM-SphR (SphI-сайт, присоединенный к 5'-концу: см. последовательность NO:28), содержащие последовательности, представленные ниже, синтезировали исходя из данных о нуклеотидной последовательности, представленной последовательностью NO:2, в целях амплификации psmAB:
eDM-SphF: 5'-TCCCCGCATGCCGGAACTGACGGACATCAC-3'
eDM-SphR: 5'-CCCGGGCATGCAGACGACGACGTAGAGGAA-3'
Эти праймеры использовали для осуществления ПЦР в нижеследующих условиях.
Жидкая композиция для ПЦР: с использованием полимеразы Takara LA (Takara Bio Inc.):
Условия реакции: с использованием T GRADIENT (Biometra):
95°C, 3 мин
(98°C, 20 сек; 68°C, 4 мин) - 30 циклов
68°C, 5 мин
1570 п.н.-ДНК-фрагмент, содержащий psmAB, амплифицированный в результате этой реакции, собирали с использованием набора для ПЦР-очистки (QIAquick PCR Purification Kit (QIAGEN Co.)). Полученный 1570 п.н.-ДНК-фрагмент гидролизовали рестриктирующим ферментом SphI. Плазмиду pSK117 (J. Bacteriol. Vol. 2002 184, 6417-6423) аналогичным образом гидролизовали рестриктирующим ферментом SphI. Полученный гидролизат ДНК-фрагмента, имеющего размер 1570 п.н. и содержащего psmAB, и гидролизат плазмиды присоединяли друг к другу с использованием набора для лигирования ДНК Ver. 2.1 (Takara Bio Inc.). Плазмиду pPepsmAB, содержащую промотор ermE, присоединенный к psmAB, конструировали аналогичным образом.
Пример 10: Получение рекомбинантной плазмиды, содержащей psmAB, присоединенный к промотору ermE
Плазмиду pPepsmAB, полученную как описано в примере 9, гидролизовали рестриктирующими ферментами KpnI и BglII, и затупляли по концам с использованием набора BKL (Takara Bio Inc.). Полученную ДНК подвергали электрофорезу (Mupid-ex: Advance) на 0,8% агарозе (Molecular Biology Agarose™) (BIO-RAD), и выделенный 2060 п.н.-ДНК-фрагмент (далее обозначаемый PepsmAB), содержащий промотор ermE и psmAB, вырезали, собирали и очищали с помощью набора для гель-экстракции QIAquick Gel Extraction Kit (QIAGEN Co.). Рекомбинантный вектор pUC19aph::oriT::intphiC31, представленный на фиг.4, также гидролизовали рестриктирующим ферментом BamHI и затупляли по концам с помощью набора BKL (Takara Bio Inc.). Полученный вектор лигировали с PepsmAB с использованием набора для лигирования ДНК Ver. 2.1 (Takara Bio Inc.). Таким образом была сконструирована рекомбинантная плазмида pAOC::PepsmAB, имеющая PepsmAB, встроенный в рекомбинантный вектор pUC19aph::oriT::intphaiC31.
Пример 11: Получение рекомбинантной плазмиды, содержащей природный psmAB
Праймер DM-Bg1F (5'-CGCATAGATCTTCACCCGAGCGGGTGATCA-3': см. последовательность NO:35), имеющий BglII-сайт, присоединенный к 5'-концу, и праймер DM-Bg1R (5'-TCCCGAGATCTTGAAGGTCCGCGTCACCGT-3': см. последовательность NO:36), имеющий BglII-сайт, присоединенный к 5'-концу, конструировали и продуцировали исходя из данных о нуклеотидной последовательности, представленной последовательностью NO:2. Затем эти два праймера (DM-Bg1F и DM-Bg1R) и геномную ДНК A-1544, полученную как описано в сравнительном примере 2(1) и взятую в качестве матрицы, использовали для проведения ПЦР. Реакцию ПЦР проводили путем 30-кратного повторения трехстадийной реакции, включающей денатурацию при 98°C в течение 20 с; отжиг при 63°C в течение 30 с; и удлинение цепи при 68°C в течение 4 мин, с использованием полимеразы Taq Takara LA (Takara Bio Inc.) и ПЦР-амплификатора (T Gradient, Biometra Co.). ДНК-фрагмент (далее называемый PopsmAB), имеющий размер примерно 3,5 т.п.н. и содержащий природные psmA и psmB, собирали из реакционной жидкости после ПЦР-амплификации с помощью электрофореза в агарозном геле и SUPREC 01 (Takara Bio Inc.). Полученный ДНК-фрагмент гидролизовали рестриктирующим ферментом BglII. Рекомбинантный вектор pUC19aph::oriT::intphiC31, представленный на фиг.4, также гидролизовали рестриктирующим ферментом BamHI. Полученный вектор и PopsmAB присоединяли друг к другу с использованием набора для лигирования ДНК Kit Ver. 2.1 (Takara Bio Inc.). Таким образом была сконструирована рекомбинантная плазмида pAOC::PopsmAB, содержащая PopsmAB, встроенный в рекомбинантный вектор pUC19aph::oriT::intphiC31.
Пример 12: Введение рекомбинантных плазмид pUC19aph::oriT::intphiC31, pAOC::PepsmAB и pAOC::PopsmAB в Mer-11107.
Каждый из рекомбинантных векторов pUC19aph::oriT::intphiC31, pAOC::PepsmAB и pAOC::PopsmAB, полученных как описано в примерах 10 и 11, переносили в конъюгированный штамм E.coli S17-1 (ATCC47055) путем электропорации с получением штаммов S17-1/pUC19aph::oriT::intphiC31, S17-1/pAOC::PepsmAB и S17- 1/pAOC::PoepsmAB. Полученные три штамма инокулировали в 10 мл LB-среды (1% бакто-триптон, 0,5% дрожжевой экстракт, 0,5% NaCl), соедржащей 25 мкг/мл канамицина, и культивировали со встряхиванием при 30°C в течение 2 ч, после чего мицелий собирали, два раза промывали 10 мл LB-среды и суспендировали в 5 мл LB-среды. Таким образом была получена донорная суспензия.
Одновременно с получением донорной суспенизии штамм Mer-11107 инокулировали в 10 мл среды TSB (триптиказо-соевый бульон: Nissui Seiyaku) и инкубировали со встряхиванием при 30°С в течение 5 ч, а затем мицелий собирали, два раза промывали 10 мл стерилизованной воды и суспендировали в 1 мл стерилизованной воды. Таким образом была получена акцепторная суспензия. 500 мкл каждой из донорных суспензий полученных трех штаммов, описанных выше, смешивали с 10 мкл полученной акцепторной суспензии Mer-11107 и наносили на агаровую среду Actino No. 4. После инкубирования при 30°C в течение 18 ч на эту среду наносили 2,5 мл среды SNA (0,8% питательная среда: Difco, 0,4% агар), содержащей 2 мг/мл рибостамицина, и инкубировали при 30°C в течение 7 дней с получением резистентного к рибостамицину pUC19aph::oriT::intphiC31-трансформированного штамма Mer-11107::pAOC, pAOC::PepsmAB-трансформированного штамма Mer-11107::pAОC::PepsmAB и pAOC::PopsmAB-трансформированного штамма Mer-11107::pAОC::PopsmAB.
Пример 13: Тест на продуцирование пладиенолида штаммом Mer-11107::pAOC, штаммом Mer-11107::pAOC::PepsmAB и штаммом Mer-11107::pAOC::PopsmAB
Штамм Mer-11107::pAOC, штамм Mer-11107::pAOC::PepsmAB и штамм Mer-11107::pAOC::PopsmAB, полученные как описано в примере 12, тестировали на продуцирование пладиенолида B и пладиенолида D с гидроксилатом в 16-положении. 300 мкл замороженного посевного материала каждого из штаммов Mer-11107::pAOC, Mer-11107::pAOC::PepsmAB и Mer-11107::pAОC::PopsmAB инокулировали в 20 мл среды для посева (2% растворимый крахмал, 2% соевая мука (ESUSAN-MEAT, изготовленная Ajinomoto Co. Ltd.), 0,3% дрожжевой экстракт, 0,1% K2HPO4, 0,25% MgSO4 · 7H2O, 0,3% CaCO3, pH не корректировали) и инкубировали при 25°C в течение 2 дней. 300 мкл полученного культурального посевного бульона инокулировали в 30 мл основной культуральной среды (5% растворимый крахмал, 1% глюкоза, 3% Pharmamedia, 0,1% CaCO3, pH 7,5), и инкубировали при 25°C в течение 4 дней. После завершения культивирования культуральный бульон экстрагировали путем 9-кратного добавления ацетонитрила. Количество пладиенолида В и пладиенолида D с гидроксидом в 16-положении, содержащееся в полученном экстракте, измеряли с помощью ВЭЖХ. Результаты измерения приводятся в таблице 6.
Условия ВЭЖХ-измерения приводятся ниже:
Устройство: ВЭЖХ Shimadzu 10Avp
Колонка: Develosil ODS UG-3 (φ 4,6 мм × 50 мм × 3 мкм)
Подвижная фаза: 45%-55% метанол (0-5 мин), 55% метанол (5-13 мин), 55%-70% метанол (13-21 мин), 70% метанол (21-25 мин)
Скорость потока: 1,2 мл/мин
Детекция: УФ на 240 нм
Объем впрыска: 5 мкл
Температура колонки: 40°С
Время анализа: 25 мин
Время удерживания:
Пладиенолид В: 12,4 мин
Пладиенолид D: 4,3 мин.
Пладиенолид D почти не продуцировался штаммом Mer-11107::pAOC, в который был введен только вектор, но продуцировался штаммами Mer-11107::pAOC::PepsmAB и Mer-11107::pAОC::PopsmAB, в которые были введены pAOC::PepsmAB и pAOC::PopsmAB. Это указывало на то, что прямое продуцирование пладиенолида D может быть достигнуто путем введения гена psmAB, кодирующего гидроксилазу, гидроксилирующую пладиенолид B в 16-положении, в штамм Mer-11107, продуцирующий пладиенолид B.
Пример 14: Получение плазмиды pPampsmAB, содержащей вариант psmA
Плазмиды pPampasmAB-CLM, pPampsmAB-CLLM, pPampsmAB-CLFM и pPampsmAB-TY, имеющие мутации, внесенные с использованием набора для сайт-направленного мутагенеза (QuickChange™ Site-Directed Mutagenesis Kit), включающего плазмиду, полученную как описано в примере 1 и используемую в качестве матрицы, были продуцированы в целях сайт-направленного введения мутаций в psmA.
Сначала были синтезированы праймеры R91C-F и R91C-R (см. последовательности NO:37 и 38), имеющие нижеследующие последовательности:
R91C-F: 5'-TTCGCGGCCGTCTGCGACCGGCGGGTG-3'
R9AC-R: 5'-CACCCGCCGGTCGCAGACGGCCGCGAA-3',
а затем была внесена мутациия с использованием набора для сайт-направленного мутагенеза (QuickChange™ Site-Directed Mutagenesis Kit), содержащего плазмиду pPapsmAB, используемую в качестве матрицы, где аргинин в положении #91 PsmA был заменен цистеином, в результате чего была получена плазмида pPampsmAB-C.
Затем были синтезированы праймеры R195L-F и R195L-R (см. последовательности NO:39 и 40), имеющие нижеследующие последовательности:
R195L-F: 5'-TCGCAAGGGGCGCTCGAGCGGCTCGAG-3'
R195L-R: 5'-CTCGAGCCGCTCGAGCGCCCCTTCCGA-3'
а затем была внесена мутация с использованием набора для сайт-направленного мутагенеза (QuickChange™ Site-Directed Mutagenesis Kit), содержащую плазмиду pPapsmAB-C, используемую в качестве матрицы, где аргинин в положении #195 PsmA был заменен лейцином, в результате чего была получена плазмида pPampsmAB-CL.
Затем были синтезированы праймеры L403M-F и L403M-R (см. последовательности NO:41 и 42), имеющие нижеследующие последовательности:
L403M-F: 5'-ACGATCCAGGGGATGATGGAACTCCCCGTGA-3'
L403M-R: 5'-TCACGGGGAGTTCCATCATCCCCTGGATCG-3'
а затем была внесена мутация с использованием набора для сайт-направленного мутагенеза (QuickChange™ Site-Directed Mutagenesis Kit), содержащего плазмиду pPapsmAB-CL, используемую в качестве матрицы, где лейцин в положении #403 PsmA был заменен метионином, в результате чего была получена плазмида pPampsmAB-CLM.
Затем были синтезированы праймеры R236L-F и R236L-R (см. последовательности NO:43 и 44), имеющие нижеследующие последовательности:
R236L-F: 5'-AGCTGGACCGACTCGACGTGGTGGCGCTGG-3'
R236L-R: 5'-AGCGCCACCACGTCGAGTCGGTCCAGCTC-3'
а затем была внесена мутация с использованием набора для сайт-направленного мутагенеза (QuickChange™ Site-Directed Mutagenesis Kit), содержащего плазмиду pPapsmAB-CLМ, используемую в качестве матрицы, где аргинин в положении #236 PsmA был заменен лейцином, в результате чего была получена плазмида pPampsmAB-CLLM.
Затем были синтезированы праймеры I244F-F и I244F-R (см. последовательности NO:45 и 46), имеющие нижеследующие последовательности:
I244F-F: 5'-TGGCGCTGGCCGTCTTCCTGCTCGTGG-3'
1244F-R: 5'-CCACGAGCAGGAAGACGGCCAGCGCCACCA-3'
а затем была внесена мутация с использованием набора для сайт-направленного мутагенеза (QuickChange™ Site-Directed Mutagenesis Kit), содержащего плазмиду pPapsmAB-CLМ, используемую в качестве матрицы, где изолейцин в положении #244 PsmA был заменен фенилаланином, в результате чего была получена плазмида pPampsmAB-CLFM.
Затем были синтезированы праймеры M112T-F и M112T-R (см. последовательности NO:47 и 48), имеющие нижеследующие последовательности:
M112T-F: 5'-CAGCGGCGGATGACGATCCCGTCGTTC-3'
M112T-R: 5'-GAACGACGGGATCGTCATCCGCCGCTG-3'
а затем была внесена мутация с использованием набора для сайт-направленного мутагенеза (QuickChange™ Site-Directed Mutagenesis Kit), содержащего плазмиду pPapsmAB, используемую в качестве матрицы, где метионин в положении #112 PsmA был заменен треонином, в результате чего была получена плазмида pPampsmAB-Т.
И наконец, были синтезированы праймеры R195Y-F и R195Y-R (см. последовательности NO:49 и 50), имеющие нижеследующие последовательности:
R195Y-F: 5'-AGGGGCGCGCGAGTACCTCGAGGAGTACCT-3'
R195Y-R: 5'-GGTACTCCTCGAGGTACTCGCGCGCCCCTT-3'
а затем была внесена мутация с использованием набора для сайт-направленного мутагенеза (QuickChange™ Site-Directed Mutagenesis Kit), содержащего плазмиду pPapsmAB-Т, используемую в качестве матрицы, где аргинин в положении #195 PsmA был заменен тирозином, в результате чего была получена плазмида pPampsmAB-TY.
Пример 15: Получение плазмиды для ее введения посредством двойной гомологичной рекомбинации
Для подтверждения того, что psmAB или его варианты могут быть введены посредством двойной гомологичной рекомбинации с использованием нуклеотидной последовательности, содержащей ДНК, участвующую в биосинтезе пладиенолида и описанную выше в сравнительном примере 1 (7), и для определения нуклеотидной последовательности кластера генов биосинтеза пладиенолида вместе со смежной с ним ДНК, была получена плазмида, предназначенная для введения и сконструированная методами, описанными ниже.
Четыре праймера, pldout-L-Bgl2F, pldout-L-Sph1R, pldout-R-Sph1F и pldout-R-Bgl2R (см. последовательности NO:51, 52, 53 и 54), имеющие последовательности, представленные ниже, синтезировали на основе нуклеотидной последовательности, расположенной ниже от ДНК, участвующей в биосинтезе пладиенолида, в целях проведения амплификации последовательности для осуществления двойной гомологичной рекомбинации:
pldout-L-Bgl2F: 5' -GGGAGATCTGCAGGTCATCGGGGGGAAGAACCACA-3'
pldout-L-Sph1R: 5'-TCTCCAGCCGCATGCGGTCCCGGGTCACCGT-3'
pldout-R-Sph1F: 5'-CCCGCATGCGGCTGGAGATCATCCAGAGCGCA-3'
pldout-R-Bgl2R: 5' -GGGAGATCTAGAACATGCCGGGCCAGAGGCTGAC-3'
Эти праймеры были использованы для проведения ПЦР в условиях, описанных ниже.
Жидкая композиция для ПЦР-реакции:
Температуры реакции:
95°C, 3 мин - 30 циклов
(98°C, 20 с; 65°C, 30 с, 68°C, 3 мин)
72°C, 5 мин
2,08-т.п.н.-ДНК-фрагмент (ДНК-фрагмент LZ) амплифицировали из реакционной смеси с использованием pldout-L-Bgl2F и pldout-L-Sph1R, а 2,73-т.п.н.-ДНК-фрагмент (ДНК-фрагмент RX) амплифицировали из реакционной смеси с использованием pldout-R-Sph1F и pldout-R-Bgl2R. ДНК-фрагменты LZ и RX очищали с использованием набора для ПЦР-очистки QIAGEN (QIAGEN Co.) и гидролизовали рестриктирующими ферментами BglII и SphI.
Затем два праймера, hyg-SpeSphF и hyg2-SphR (см. последовательности NO:55 и 56), имеющие последовательности, представленные ниже, синтезировали на основе нуклеотидных последовательностей в целях проведения амплификации гена резистентности к гигромицину:
hyg-SpeSphF: 5'-GGGGCATGCGGACTAGTACACCGTCGCCTCGGT-3'
hyg2-SphR: 5'-GCCGCATGCGTCAGGCGCCGGGGGCGGTGT-3'
Эти праймеры использовали для проведения ПЦР в нижеследующих условиях.
Жидкая композиция для ПЦР:
Температуры реакции
95°C, 6 мин
(98°C, 20 с; 63°C, 30 с, 68°C, 2 мин) - 30 циклов
72°C, 5 мин
1,21-т.п.н.-ДНК-фрагмент (ДНК-фрагмент hyg2), амплифицированный в результате этой реакции, очищали с помощью набора для ПЦР-очистки QIAGEN (QIAGEN Co.) и гидролизовали рестриктирующим ферментом SphI.
Все 4 последовательности, а именно ДНК-фрагменты LZ и RX, гидролизованные рестриктирующими ферментами BglII и SphI, ДНК-фрагмент hyg2, гидролизованный рестриктирующим ферментом SphI, и челночный вектор pKU253 (см. фиг.3), гидролизованный рестриктирующим ферментом BamHI, соединяли друг с другом с помощью набора для лигирования ДНК Ver. 2.1 (Takara Bio Inc.). Таким образом была сконструирована плазмида pKU253-LZ-hyg2-RX длиной примерно 22 т.п.н. для двойной гомологичной рекомбинации, где указанная плазмида включает вектор pKU253, имеющий встроенный в него ДНК-фрагмент длиной примерно 6.0 т.п.н., содержащий ДНК-фрагмент hyg2, встроенный между ДНК-фрагментами LZ и RX.
Пример 16: Включение psmAB или его вариантов в предназначенную для введения плазмиду pKU253-LZ-hyg2-RX посредством двойной гомологичной рекомбинации
Плазмиду pPapsmAB, сконструированную как описано в примере 1, и плазмиды pPampsmAB-CLM, pPampsmAB-CLLM, pPampsmAB-CLFM и pPampsmAB-TY, сконструированные как описано в примере 14, гидролизовали рестриктирующим ферментом SpeI. ДНК каждого вида, гидролизованные рестриктирующим ферментом SpeI, подвергали электрофорезу (Mupid-ex: Advance) на 0,8% агарозе (Certified™ Molecular Biology Agarose) (BIO-RAD), и выделенный 1,8 т.п.н.-ДНК-фрагмент, содержащий промотор pldA и psmAB или его варианты (далее иногда обозначаемые PapsmAB, PampsmAB-CLM, PampsmAB-CLLM, PapmsmAB-CLFM или PampsmAB-TY), вырезали и собирали, а затем очищали с помощью набора для гель-экстракции QIAquick (QIAGEN Co.). Полученные ДНК-фрагменты (PapsmAB, PampsmAB-CLM, PampsmAB-CLLM, PapmsmAB-CLFM и PampsmAB-TY) присоединяли с использованием набора для лигирования ДНК Ver. 2.1 (Takara Bio Inc.) к pKU253-LZ-hyg2-RX, полученному как описано в примере 15 и гидролизованному рестриктирующим ферментом SpeI. Таким образом, соответствующие ДНК-фрагменты (PapsmAB, PampsmAB-CLM, PampsmAB-CLLM, PapmsmAB-CLFM и PampsmAB-TY) были встроены выше от гена hyg2 вектора pKU253-LZ-hyg2-RX для конструирования плазмид pKU253-LZ-hyg2-PapsmAB-RX, pKU253-LZ-hyg2-PampsmAB-CLM-RX, pKU253-LZ-hyg2-PampsmAB-CLLM-RX, pKU253-LZ-hyg2-PampsmAB-CLFM-RX и pKU253-LZ-hyg2-PampsmAB-TY-RX длиной примерно 24 т.п.н. в целях введения psmAB или его вариантов посредством двойной гомологичной рекомбинации.
Пример 17: Введение psmAB или его вариантов в штамм Mer-11107 посредством двойной гомологичной рекомбинации
Каждый из полученных векторов pKU253-LZ-hyg2-PapsmAB-RX, pKU253-LZ-hyg2-PampsmAB-CLM-RX, pKU253-LZ-hyg2-PampsmAB-CLLM-RX, pKU253-LZ-hyg2-PampsmAB-CLFM-RX и pKU253-LZ-hyg2-PampsmAB-TY-RX были введены путем электропорации в конъюгированный штамм E.coli S17-1 с получением S17-1/pKU253-LZ-hyg2-PapsmAB-RX, S17-1/pKU253-LZ-hyg2-PampsmAB-CLM-RX, S17-1/pKU253-LZ-hyg2-PampsmAB-CLLM-RX, Sl7-1/pKU253-LZ-hyg2-PampsmAB-CLFM-RX и S17-1/pKU253-LZ-hyg2-PampsmAB-TY-RX. Полученные S17-1/pKU253-LZ-hyg2-PapsmAB-RX, S17-1/pKU253-LZ-hyg2-PampsmAB-CLM-RX, S17-1/pKU253-LZ-hyg2-PampsmAB-CLLM-RX, S17-1/pKU253-LZ-hyg2-PampsmAB-CLFM-RX и S17-1/pKU253-LZ-hyg2-PampsmAB-TY-RX инокулировали в LB-среду (1% бакто-триптон, 0,5% дрожжевой экстракт, 0,5% NaCl), содержащую 25 мкг/мл канамицина и 100 мкг/мл гигромицина В, и культивировали со встряхиванием в течение 2 ч при 30°С, а затем мицелий собирали, два раза промывали 10 мл LB-среды и суспендировали в 5 мл LB-среды. Таким образом были получены донорные суспензии.
Одновременно с получением донорных суспенизий штамм Mer-11107 инокулировали в 10 мл среды TSB (триптиказо-соевый бульон: Nissui Seiyaku) и инкубировали со встряхиванием при 30°С в течение 5 ч, а затем мицелий собирали, два раза промывали 10 мл стерилизованной воды и суспендировали в 1 мл стерилизованной воды. Таким образом была получена акцепторная суспензия. 500 мкл каждой из донорных суспензий S17-1/pKU253-LZ-hyg2-PapsmAB-RX, Sl7-1/pKU253-LZ-hyg2-PampsmAB-CLM-RX, S17-1/pKU253-LZ-hyg2-PampsmAB-CLLM-RX, S17-1/pKU253-LZ-hyg2-PampsmAB-CLFM-RX и S17-1/pKU253-LZ-hyg2-PampsmAB-TY-RX смешивали с 10 мкл полученной акцепторной суспензии Mer-11107 и наносили на агаровую среду Actino No. 4 (Nippon Seiyaku). После культивирования при 30°C в течение 18 ч на эту среду наносили 2,5 мл среды SNA (0,8% питательная среда: Difco, 0,4% агар), содержащей 2 мг/мл рибостамицина. Полученную культуру инкубировали при 30°C в течение 7 дней с получением резистентных к рибостамицину трансформантов pKU253-LZ-hyg2-PapsmAB-RX, pKU253-LZ-hyg2-PampsmAB-CLM-RX, pKU253-LZ-hyg2-PampsmAB-CLLM-RX, pKU253-LZ-hyg2-PampsmAB-CLFM-RX и pKU253-LZ-hyg2-PampsmAB-TY-RX.
Каждый из полученных трансформантов pKU253-LZ-hyg2-PapsmAB-RX, pKU253-LZ-hyg2-PampsmAB-CLM-RX, pKU253-LZ-hyg2-PampsmAB-CLLM-RX, pKU253-LZ-hyg2-PampsmAB-CLFM-RX и pKU253-LZ-hyg2-PampsmAB-TY-RX инокулировали в 10 мл среды TSB, не содержащей рибостамицина, и культивировали со встряхиванием при 30°С в течение 24 ч. Жидкую культуру каждого из трансформантов pKU253-LZ-hyg2-PapsmAB-RX, pKU253-LZ-hyg2-PampsmAB-CLM-RX, pKU253-LZ-hyg2-PampsmAB-CLLM-RX, pKU253-LZ-hyg2-PampsmAB-CLFM-RX и pKU253-LZ-hyg2-PampsmAB-TY-RX собирали, а затем два раза промывали 10 мл стерилизованной воды и суспендировали в 10 мл стерилизованной воды. Каждую соответствующим образом разведенную суспензию наносили на агаровую среду YMS (0,4% дрожжевой экстракт, 1% экстракт из семян пшеницы, 0,4% растворимый крахмал, 2% агар, 10 мМ хлорид кальция), содержащую 200 мкг/мл гигромицина B, и инкубировали при 30°C в течение 4 дней. Одиночные колонии, растущие на агаровой среде YMS, содержащей гигромицин B, переносили в агаровую среду YMS, содержащую 200 мкг/мл гигромицина B, и в агаровую среду YMS, содержащую 200 мкг/мл рибостамицина, а затем инкубировали при 30°C в течение 2 дней. После культивирования отбирали штаммы, резистентные к гигромицину В и чувствительные к рибостамицину. Полученные штаммы представляют собой штаммы, имеющие ген резистентности к гигромицину В hyg2 и ген psmAB или его варианты, встроенные ниже от геномной ДНК (между нуклеотидами 73362-73369 в последовательности NO:1), участвующей в биосинтезе пладиенолида, и эти штаммы были обозначены Mer-11107-ZX::hyg2::PapsmAB, Mer-11107-ZX::hyg2::PampsmAB-CLM, Mer-11107-ZX::hyg2::PampsmAB-CLLM, Mer-11107-ZX::hyg2::PampsmAB-CLFM и Mer-11107-ZX::hyg2::PampsmAB-TY.
Пример 18: Продуцирование пладиенолида штаммами Mer-11107-ZX::hyg2::PapsmAB, Mer-11107-ZX::hyg2::PampsmAB-CLM, Mer-11107-ZX::hyg2::PampsmAB-CLLM, Mer-11107-ZX::hyg2::PampsmAB-CLFM и Mer-11107-ZX::hyg2::PampsmAB-TY
500 мкл замороженного посевного материала каждого из штаммов Mer-11107-ZX::hyg2::PapsmAB, Mer-11107-ZX::hyg2::PampsmAB-CLM, Mer-11107-ZX::hyg2::PampsmAB-CLLM, Mer-11107-ZX::hyg2::PampsmAB-CLFM и Mer-11107-ZX::hyg2::PampsmAB-TY, полученных как описано в примере 17, инокулировали в 25 мл среды для посева (2% растворимый крахмал, 2% соевая мука (ESUSAN-MEAT, изготовленная Ajinomoto Co. Ltd.), 0,3% дрожжевой экстракт, 0,1% K2HPO4, 0,25% MgSO4 · 7H2O, 0,3% CaCO3, pH не корректировали) и инкубировали при 25°C в течение 3 дней. 300 мкл полученного культурального посевного бульона инокулировали в 30 мл основной культуральной среды (5% растворимый крахмал, 1% глюкоза, 3% Pharmamedia, 1% SoyPro, 0,05% адеканол LG-126, 2,0% β-CD, 0,1% CaCO3, pH 7,5), и инкубировали при 25°C в течение 6 дней.
После завершения культивирования полученный культуральный бульон экстрагировали путем 4-кратного добавления ацетонитрила. В полученном экстракте с помощью ВЭЖХ измеряли количество пладиенолида D, пладиенолида H13, соответствующего пладиенолиду D с гидроксильной группой в 20-положении; пладиенолида С2, соответствующего пладиенолиду D с карбонильной группой в 21-положении; и пладиенолида С3, соответствующего пладиенолиду D с гидроксильной группой в 20-положении и с карбонильной группой в 21-положении. Результаты измерения приводятся в таблице 7. Условия ВЭЖХ-анализа приводятся ниже.
Условия ВЭЖХ-анализа:
Устройство: ВЭЖХ Shimadzu 10Avp
Колонка: Develosil ODS UG-3 (φ 4,6 мм × 50 мм × 3 мкм)
Подвижная фаза: 20%-35% ацетонитрил (0-13 мин), 35%-100% ацетонитрил (13-17 мин), 20% ацетонитрил (17-22 мин)
Скорость потока: 1,0 мл/мин
Детекция: УФ на 240 нм
Объем впрыска: 5 мкл
Температура колонки: 40°С
Время анализа: 22 мин
Время удерживания:
Пладиенолид Н13: 5,5 мин
Пладиенолид С3: 8,3 мин
Пладиенолид D: 9,0 мин
Пладиенолид С2: 11,1 мин.
В результате штамм Mer-11107-Zx:hyg2::PapsmAB, в который был введен PapsmAB, продуцировал большое количество продуктов разложения пладиенолида Н13, пладиенолида С2 и пладиенолида С3 вместе с пладиенолидом D, а штаммы Mer-11107-ZX::hyg2::PampsmAB-CLM, Mer-11107-ZX::hyg2::PampsmAB-CLLM, Mer-11107-ZX::hyg2: :PampsmAB-CLFM и Mer-11107-ZX::hyg2::PampsmAB-TY, в которые были введены PampsmAB-CLM, PampsmAB-CLLM, PampsmAB-CLFM и PampsmAB-TY, также продуцировали пладиенолид D, однако, при этом уровень продуцирования продуктов разложения пладиенолида Н13, пладиенолида С2 и пладиенолида С3 снижался приблизительно на одну четверть. Полученные результаты дают основание предположить, что введение вариантов гена psmAB, кодирующего фермент, гидроксилирующий пладиенолид В в 16-положении, в штамм Mer-11107, продуцирующий пладиенолид В, может приводить к прямому продуцированию пладиенолида D при снижении уровня продуцирования продуктов разложения пладиенолида D.







































Реферат
Настоящее изобретение относится к области биотехнологии. Предлагается генетически модифицированный микроорганизм, способный продуцировать макролидное соединение, гидроксилированное в 16-положении. Описан способ получения макролидного соединения, гидроксилированного в 16-положении. В частности, предложены генетически модифицированные микроорганизмы, имеющие ДНК, кодирующую полипептид, участвующий в биосинтезе макролидного соединения пладиенолида, и ДНК, кодирующую полипептид, обладающий гидроксилазной активностью, направленной на гидроксилирование пладиенолидов в 16-положении. Изобретение позволяет эффективно продуцировать макролидное соединение, гидроксилированное в 16-положении, непосредственно в одном микроорганизме. Полученное соединение обладает высокой противоопухолевой активностью и может быть использовано для создания противоопухолевых препаратов. 2 н. и 15 з.п. ф-лы, 4 ил., 7 табл.
Формула

(где R представляет собой атом водорода или гидроксильную группу), и который включает:
(а) ДНК, кодирующую полноразмерный или неполный полипептид, участвующий в биосинтезе макролидных соединений, представленных формулой (II):

(где R представляет собой атом водорода или гидроксильную группу), и
(b) ДНК, кодирующую полноразмерный или неполный полипептид, обладающий ферментативной активностью, направленной на гидроксилирование макролидных соединений в 16-положении, представленных вышеуказанной формулой (II).
(а-1) непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 8340-27935 в последовательности NO:1;
(а-2) непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 28021-49098 в последовательности NO:1;
(а-3) непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 49134-60269 в последовательности NO:1;
(а-4) непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 60269-65692 в последовательности NO:1;
(а-5) непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 68160-66970 в последовательности NO:1;
(а-6) непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 69568-68270 в последовательности NO:1; и
(а-7) непрерывную нуклеотидную последовательность, состоящую из нуклеотидов 72725-70020 в последовательности NO:1;
или ее функциональный вариант.
(b-1) непрерывной нуклеотидной последовательности, состоящей из нуклеотидов 1322-2548 в последовательности NO:2;
(b-2) непрерывной нуклеотидной последовательности, состоящей из нуклеотидов 420-1604 в последовательности NO:3; и
(b-3) непрерывной нуклеотидной последовательности, состоящей из нуклеотидов 172-1383 в последовательности NO:4; или ее функциональный вариант.
1) модифицированного сайта, где последовательность cgc, состоящая из нуклеотидов 1592-1594, заменена кодоном, кодирующим аминокислоту, не являющуюся аргинином;
2) модифицированного сайта, где последовательность atg, состоящая из нуклеотидов 1655-1657, заменена кодоном, кодирующим аминокислоту, не являющуюся метионином;
3) модифицированного сайта, где последовательность cgc, состоящая из нуклеотидов 1904-1906, заменена кодоном, кодирующим аминокислоту, не являющуюся аргинином;
4) модифицированного сайта, где последовательность cgc, состоящая из нуклеотидов 2027-2029, заменена кодоном, кодирующим аминокислоту, не являющуюся аргинином;
5) модифицированного сайта, где последовательность atc, состоящая из нуклеотидов 2054-2056, заменена кодоном, кодирующим аминокислоту, не являющуюся изолейцином; и
6) модифицированного сайта, где последовательность ctg, состоящая из нуклеотидов 2528-2530, заменена кодоном, кодирующим аминокислоту, не являющуюся лейцином.
1) модифицированного сайта, где последовательность cgc, состоящая из нуклеотидов 1592-1594, заменена кодоном, кодирующим цистеин, метионин или пролин;
2) модифицированного сайта, где последовательность atg, состоящая из нуклеотидов 1655-1657, заменена кодоном, кодирующим треонин или серин;
3) модифицированного сайта, где последовательность cgc, состоящая из нуклеотидов 1904-1906, заменена кодоном, кодирующим лейцин, изолейцин, пролин, тирозин или фенилаланин;
4) модифицированного сайта, где последовательность cgc, состоящая из нуклеотидов 2027-2029, заменена кодоном, кодирующим лейцин, изолейцин или пролин;
5) модифицированного сайта, где последовательность atc, состоящая из нуклеотидов 2054-2056, заменена кодоном, кодирующим фенилаланин или валин; и
6) модифицированного сайта, где последовательность ctg, состоящая из нуклеотидов 2528-2530, заменена кодоном, кодирующим метионин, цистеин или изолейцин.
1) модифицированного сайта, где последовательность cgc, состоящая из нуклеотидов 1592-1594, заменена кодоном, кодирующим цистеин;
2) модифицированного сайта, где последовательность atg, состоящая из нуклеотидов 1655-1657, заменена кодоном, кодирующим треонин;
3) модифицированного сайта, где последовательность cgc, состоящая из нуклеотидов 1904-1906, заменена кодоном, кодирующим лейцин или тирозин;
4) модифицированного сайта, где последовательность cgc, состоящая из нуклеотидов 2027-2029, заменена кодоном, кодирующим лейцин;
5) модифицированного сайта, где последовательность atc, состоящая из нуклеотидов 2054-2056, заменена кодоном, кодирующим фенилаланин; и
6) модифицированного сайта, где последовательность ctg, состоящая из нуклеотидов 2528-2530, заменена кодоном, кодирующим метионин.
Устройство - ВЭЖХ Shimadzu 10 Avp;
Колонка - Develosil ODS UG-3 (φ4,6 мм×50 мм×3 мкм);
Подвижная фаза - 45-55% метанол (0-5 мин), 55% метанол (5-13 мин), 55-70% метанол (13-21 мин), 70% метанол (21-25 мин);
Скорость потока - 1,2 мл/мин;
Детекция - УФ на 240 нм;
Объем впрыска - 5 мкл;
Температура колонки - 40°С,
проводимого для определения количества макролидного соединения с гидроксильной группой в 16-положении, представленного формулой (I).
Комментарии