Микроорганизм, продуцирующий о-фосфосерин, и способ получения l-цистеина или его производных из о-фосфосерина с его использованием - RU2536250C1
Код документа: RU2536250C1
Чертежи

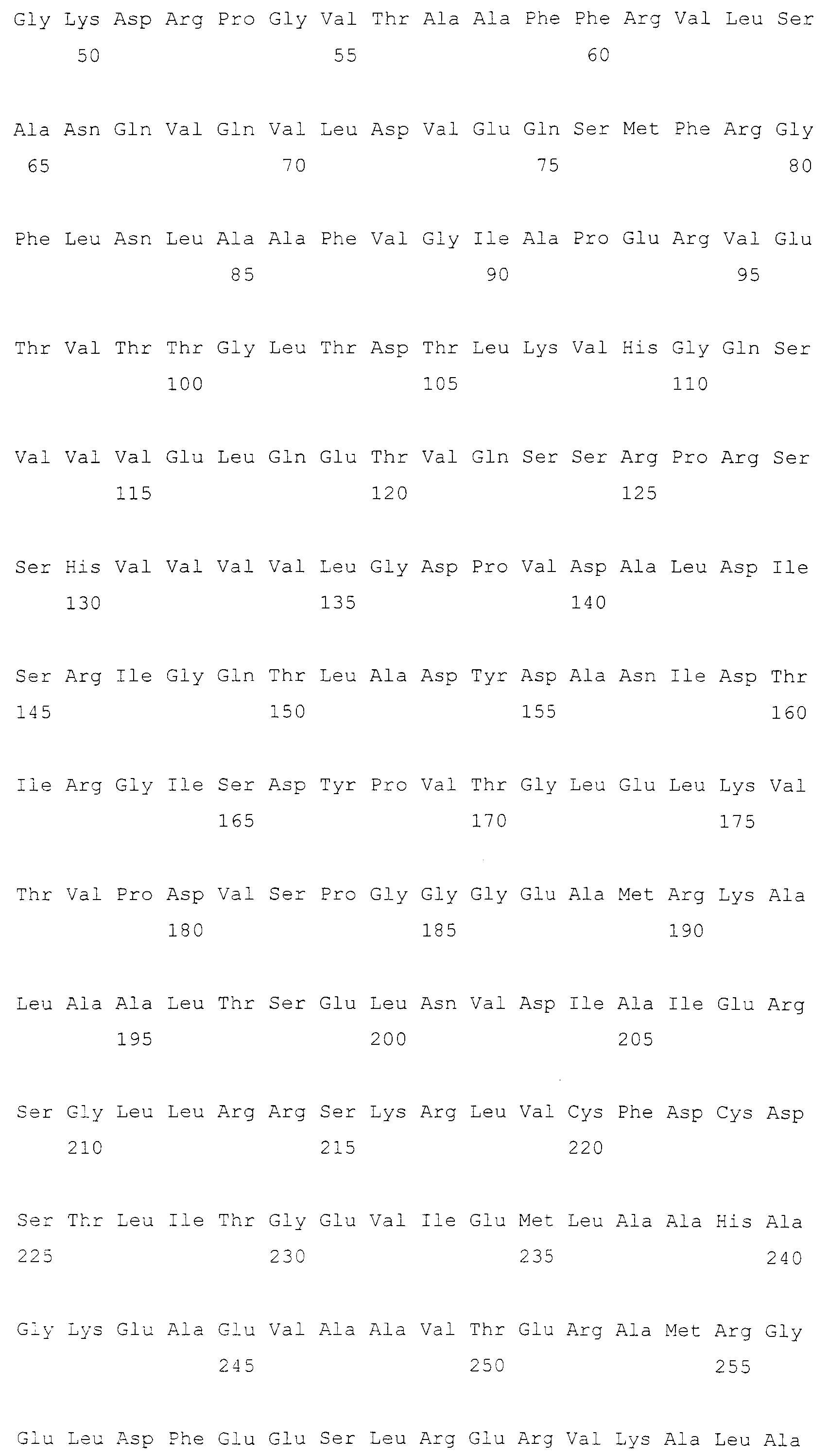
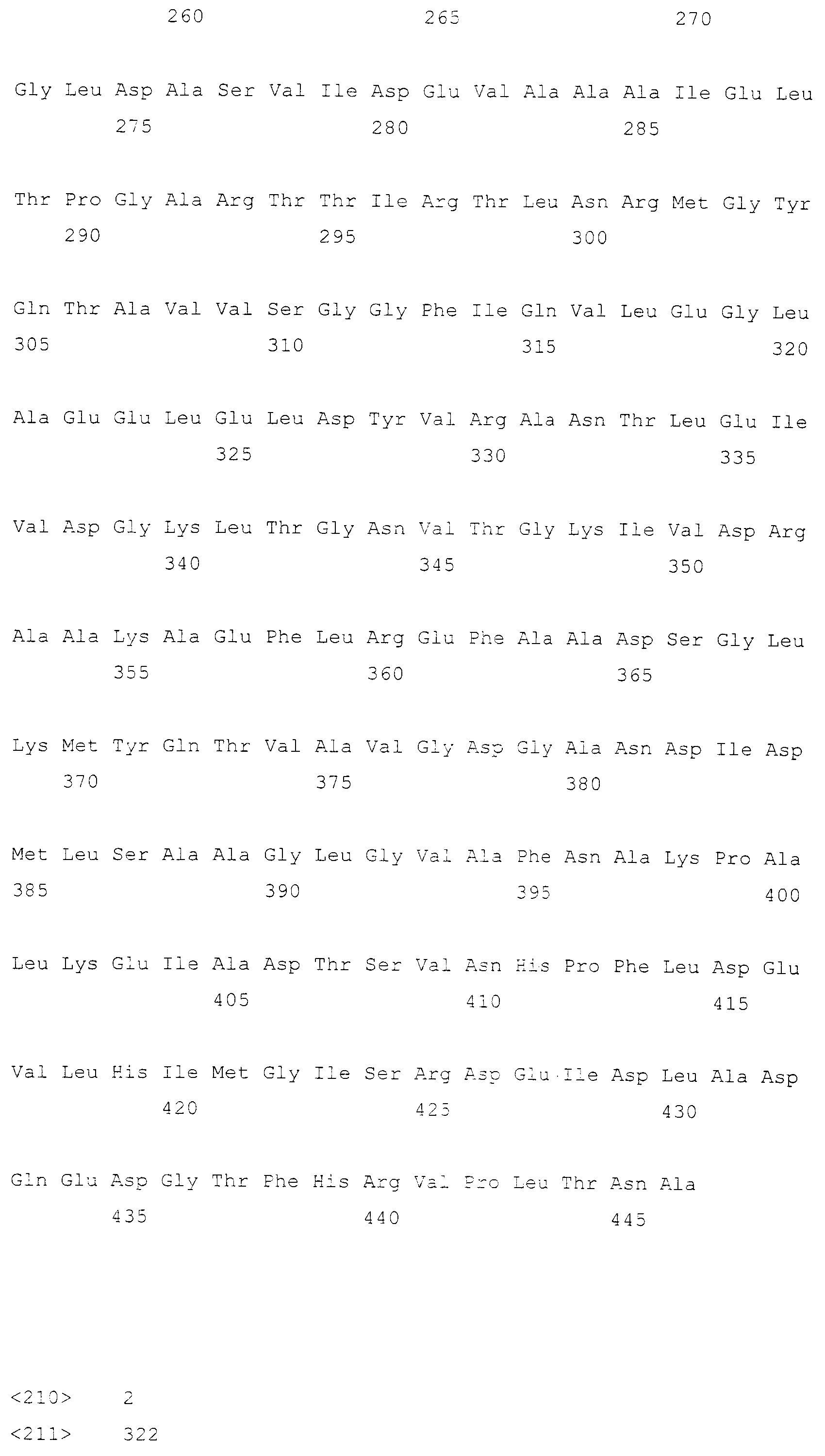

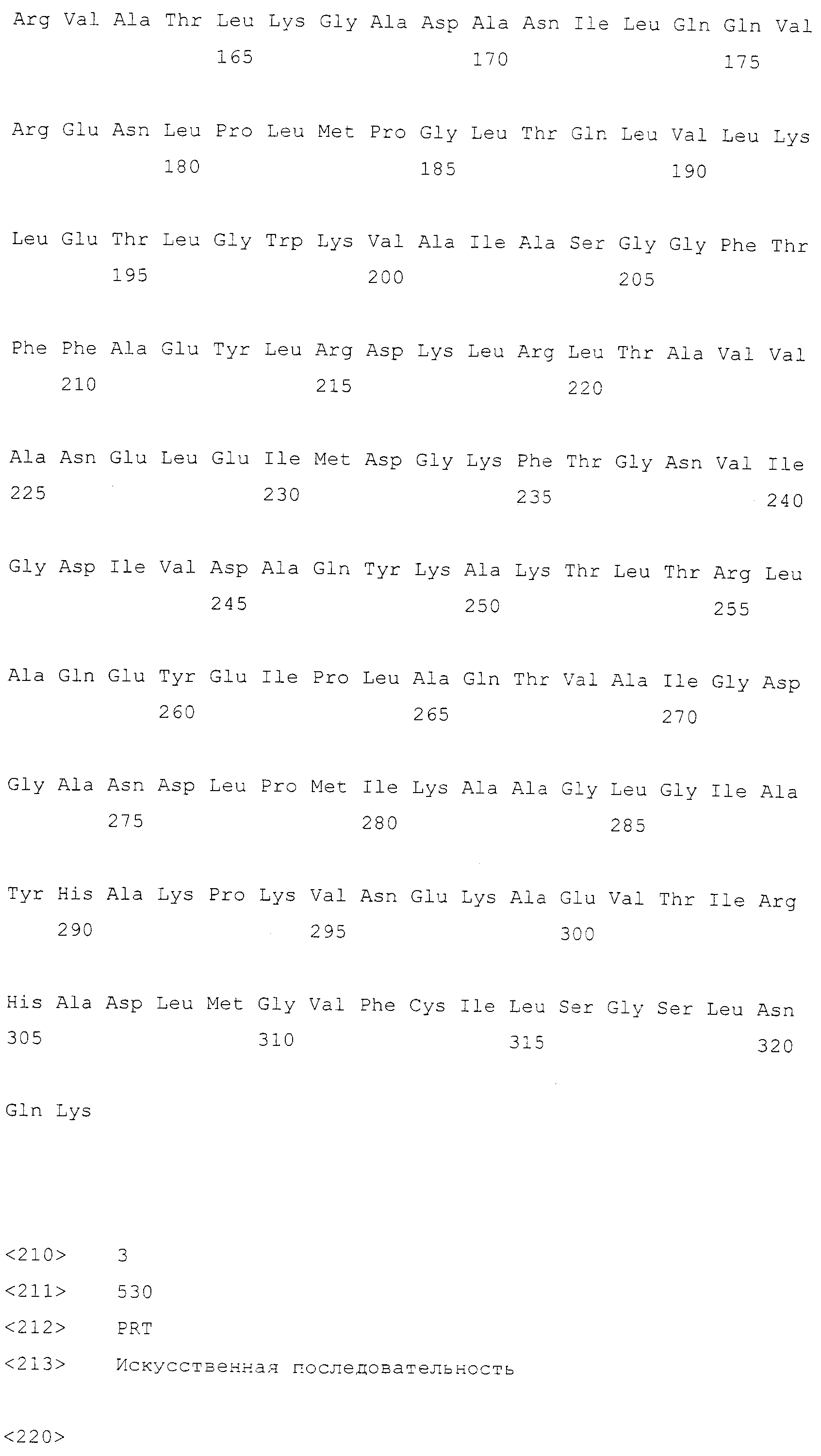

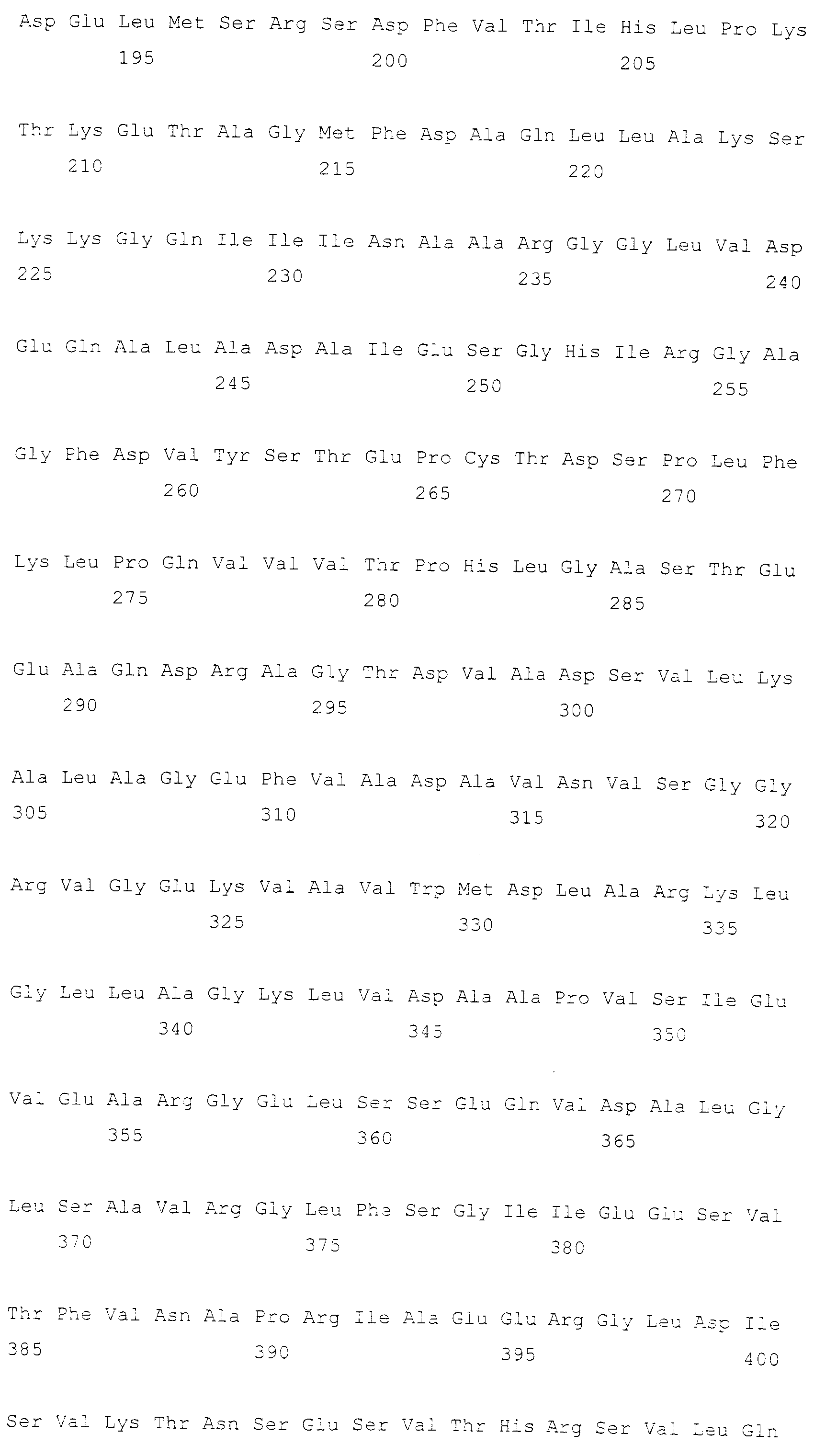

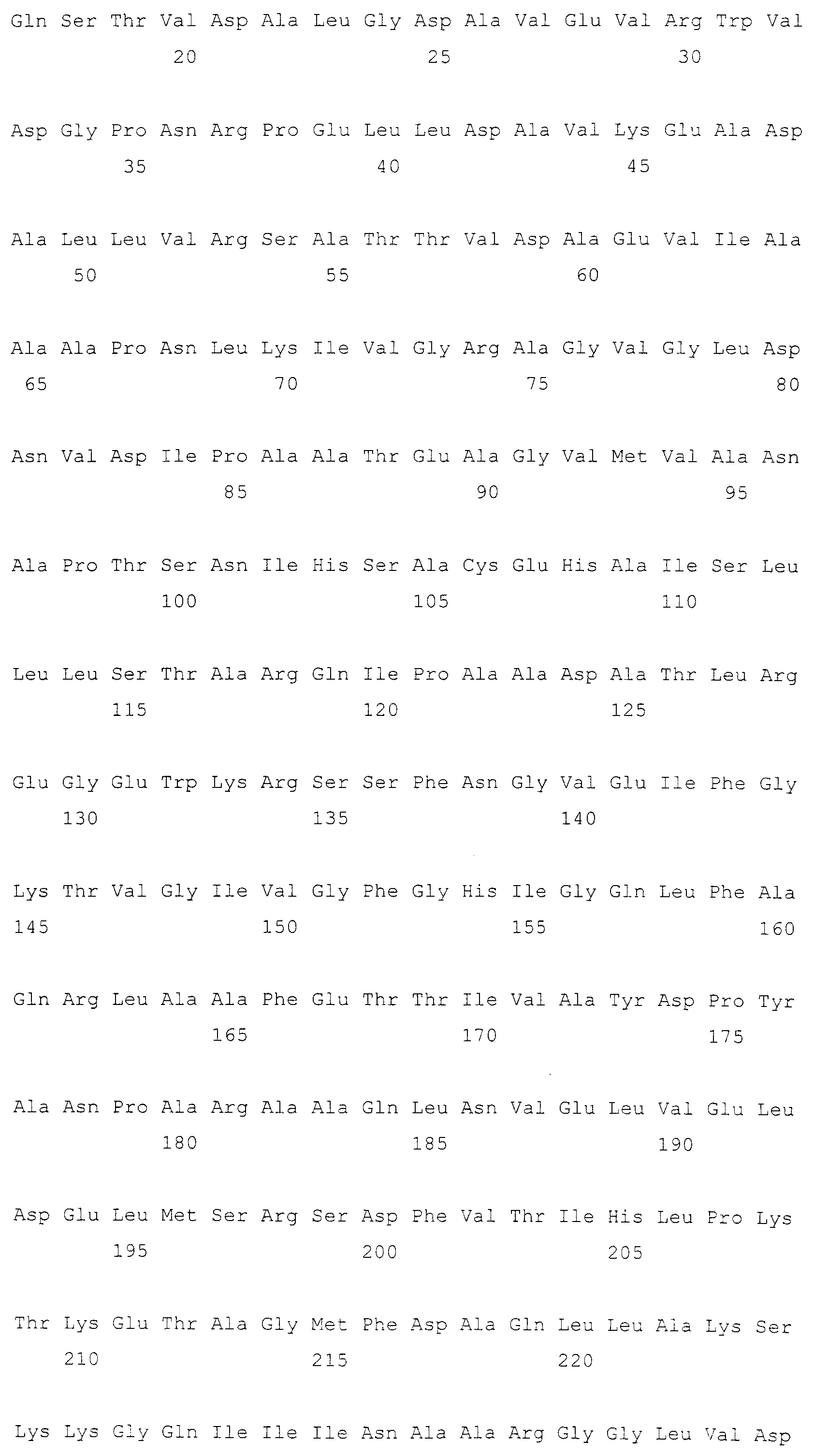
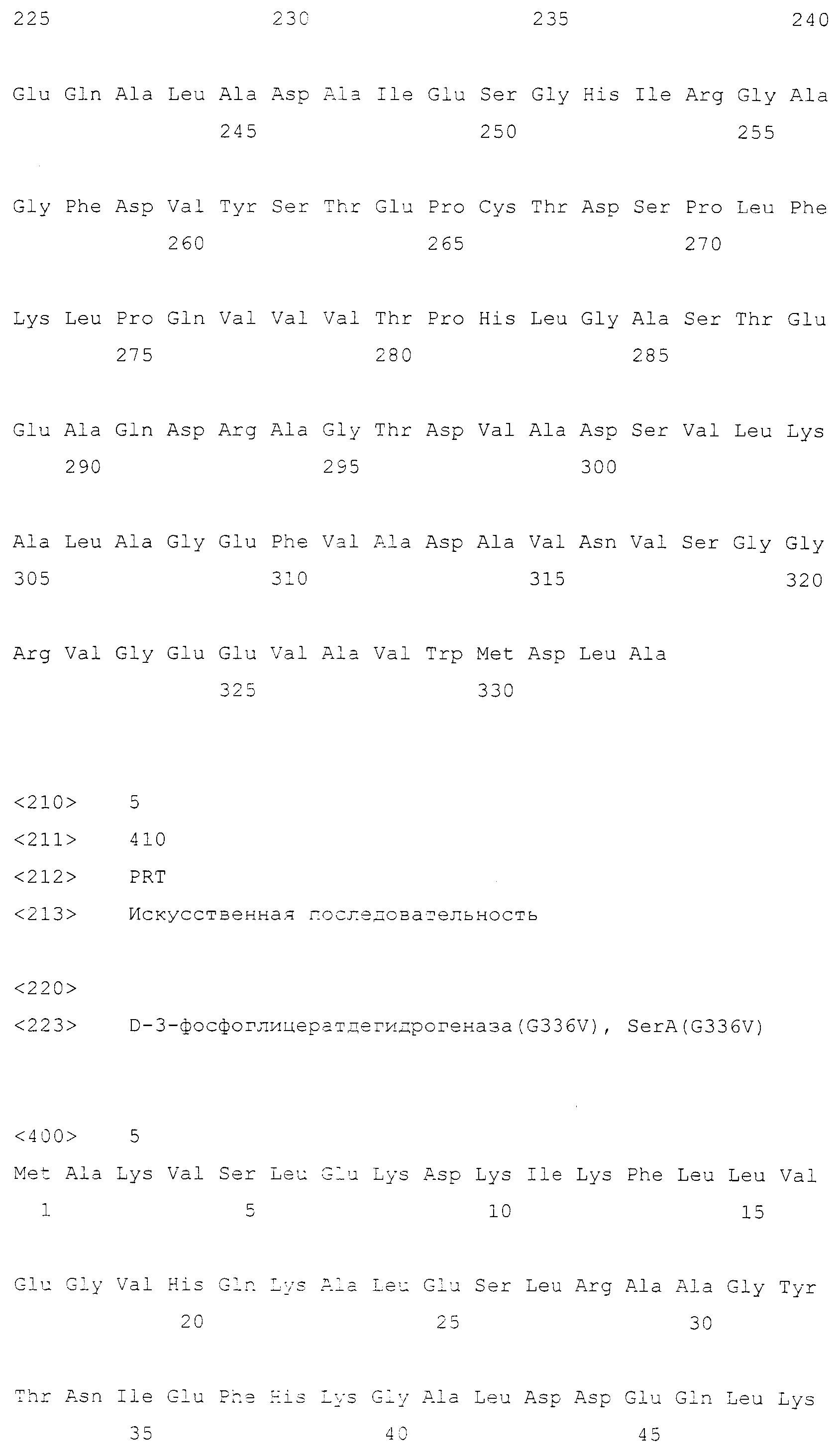
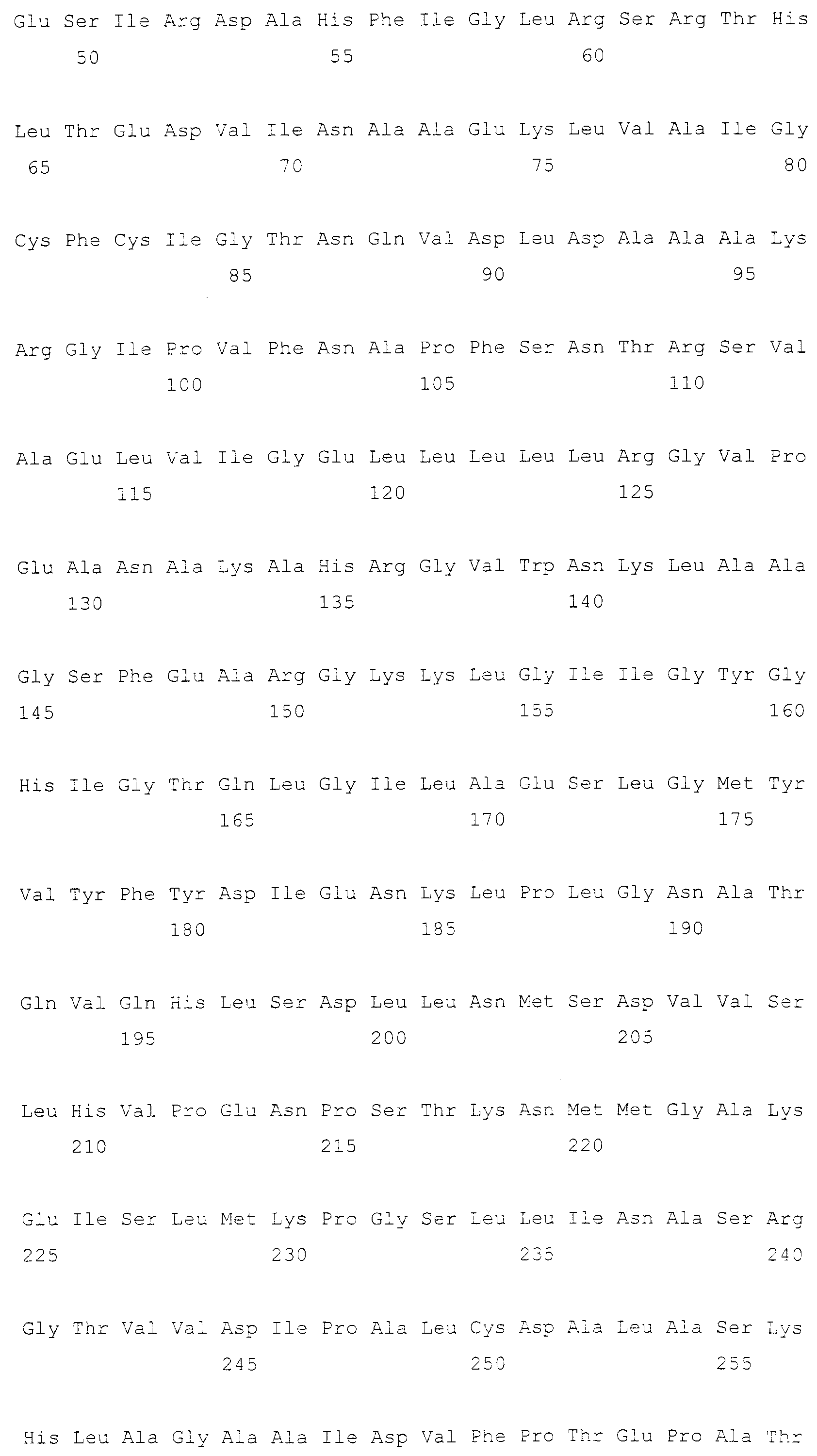
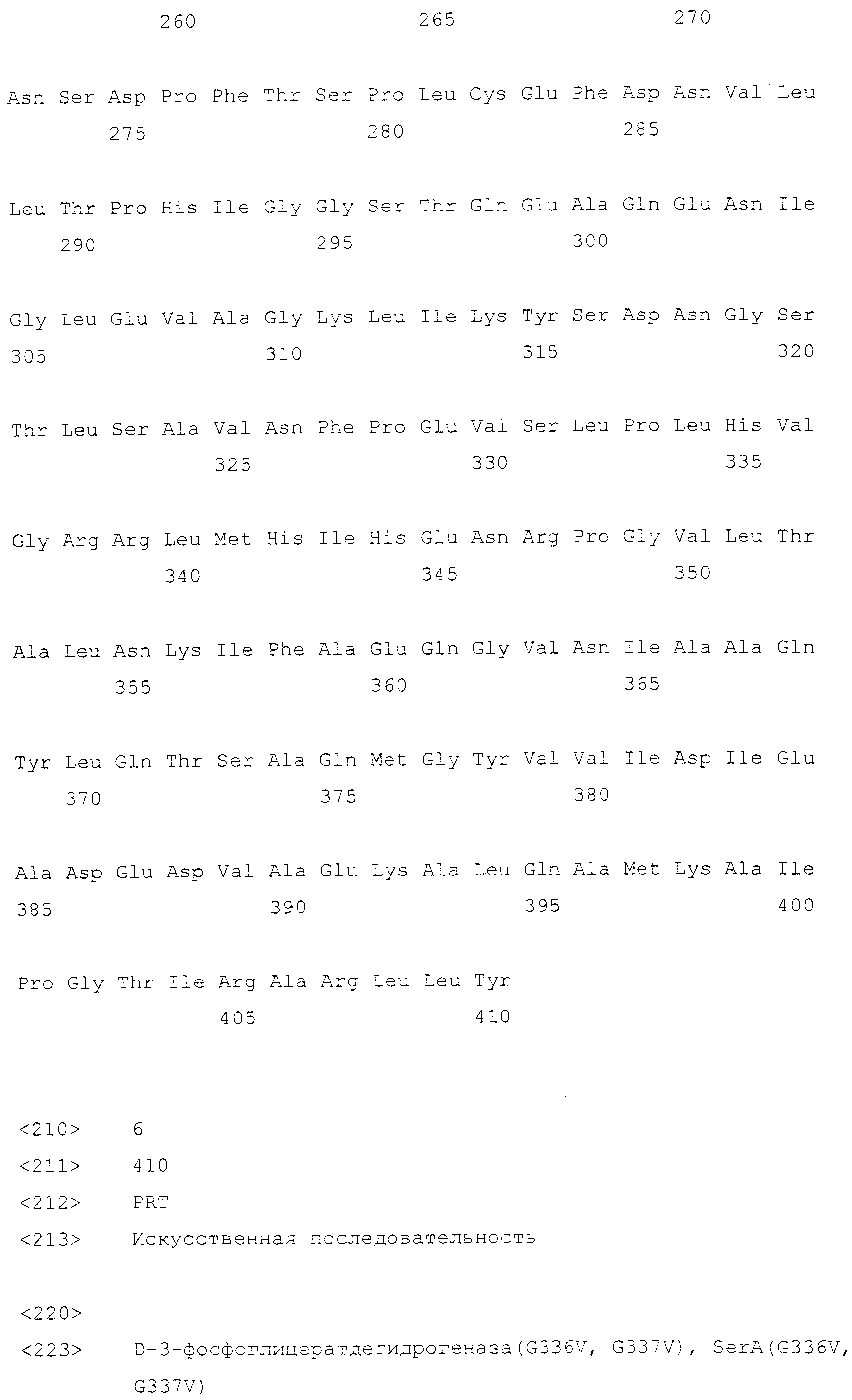
























































Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения цистеина или его производных с использованием O-фосфосерина в качестве промежуточного продукта и рекомбинантного микроорганизма, используемого для продукции O-фосфосерина.
Известный уровень техники
L-цистеин представляет собой аминокислоту, которая играет важную роль в метаболизме серы у всех живых организмов. Он используется в биосинтезе белков, таких как кератин волос, глутатиона, биотина, метионина, и других содержащих серу метаболитов, а также служит в качестве предшественника коэнзима A. Кроме того, как известно, биосинтез цистеина тесно связан с биосинтезом других аминокислот, включая L-серин, L-глицин и L-метионин. L-цистеин и его производные находят применение в различных отраслях промышленности, включая фармацевтическую промышленность (для лечения бронхиальных заболеваний), косметическую промышленность (в шампунях для волос, составах для перманентной завивки) и пищевую промышленность (для антиоксидантов, усилителей вкуса, в качестве вспомогательного агента при приготовлении теста и т.д.).
L-цистеин получали ранее промышленным способом с помощью кислотного гидролиза волос человека или перьев животных (Biotechnology of the Amino Acids Production под редакцией Ko Aida, p. 217-223, 1986). Однако получение цистеина из волос или перьев не только обеспечивало такой низкий выход, как 7-8%, но также из-за использования хлористоводородной кислоты или серной кислоты возникало большое количество отходов, что приводило к загрязнению окружающей среды. Кроме того, экстракция из волос или перьев может индуцировать у пользователя сильные неблагоприятные эффекты. Эти проблемы дали толчок к развитию способов получения L-цистеина, благоприятных для окружающей среды. Главный современный путь получения включает ферментацию с использованием микроорганизмов.
Среди вариантов микробной продукции L-цистеина примером является 1) биологическое превращение D, L-ATC с использованием микроорганизма (Ryu OH, Ju JY and Shin CS, Process Biochem., 32:201-209, 1997). Этот способ превращения является, однако, трудным для применения в промышленном масштабе из-за низкой растворимости предшественника D, L-ATC. 2) Другой способ получения L-цистеина представляет собой прямую ферментацию с использованием E. coli (патент № EP0885962B; Wada M and Takagi H, Appl. Microbiol. Biochem., 73:48-54, 2006). Избыточная аккумуляция L-цистеина в микроорганизмах вызывает внутриклеточную токсичность, создавая ограничение для получения L-цистеина в высокой концентрации. Для преодоления этого препятствия применяют белки, экспортирующие L-цистеин, но при этом не выявлено существенных улучшений продуктивности.
Относительно пути биосинтеза L-цистеина в микроорганизмах и растениях, O-ацетилсерин (OAS) выполняет функцию промежуточного продукта-предшественника, создавая углеродный каркас L-цистеина (Kredich NM and Tomkins GM, J. Biol. Chem., 241: 4955-4965, 1966). Фермент O-ацетилсеринсульфгидрилаза (OASS), используя сульфид водорода в качестве донора серы, катализирует превращение O-ацетилсерина в цистеин. Альтернативно, SO4 может быть восстановлен до тиосульфата для использования в качестве донора серы при получении цистеина (Nakamura T, Kon Y, Iwahashi H and Eguchi Y, J. Bacteriol., 156: 656-662, 1983). Следовательно, цистеин может быть получен с использованием микроорганизмов, аккумулирующих OAS и OASS, с использованием различных доноров серы (патент США 6579705). В пути биосинтеза цистеина через OAS используются два фермента серинацетилтрансфераза (CysE), которая катализирует превращение серина в OAS, и цистеинсинтаза (CysK), которая катализирует превращение OAS в цистеин. Среди них серинацетилтрансфераза (CysE) высокочувствительна к ингибированию конечным продуктом цистеином по механизму обратной связи (Wada M and Takagi H, Appl. Microbiol. Biochem., 73:48-54, 2006).
Раскрытие изобретения
Техническая задача
Авторы настоящего изобретения благодаря интенсивным исследованиям выявили в Aeropyrum pernix, Mycobacterium tuberculosis и Trichomonas vaginalis присутствие O-фосфосеринсульфгидрилазы (OPSS), которая использует O-фосфо-L-серин(OPS)-специфичный путь, а не OAS-специфичный путь для синтеза L-цистеина (Mino K and Ishikawa K, FEBS letters, 551: 133-138, 2003; Burns KE, Baumgart S, Dorrestein PC, Zhai H, McLafferty FW and Begley TP, J. Am. Chem. Soc., 127: 11602-11603, 2005; Westrop GD, Goodall G, Mottram JC and Coombs GH, J. Biol. Chem., 281: 25062-25075, 2006), и обнаружили что OPSS M. tuberculoses может использовать Na2S в качестве донора серы при превращении OPS в цистеин даже в отсутствие дополнительных ферментов, когда из нее удаляют пять C-концевых аминокислотных остатков (Argen D, Schnell R and Schneider G, FEBS letters, 583: 330-336, 2009), что привело к настоящему изобретению. В настоящем изобретении микроорганизм является мутантным для аккумуляции в нем OPS, затем происходит инкубация для превращения OPS в цистеин в присутствии фермента OPSS. Этот способ ранее нигде не был описан.
Техническое решение
Целью настоящего изобретения является разработка способа получения цистеина или его производного.
Другой целью настоящего изобретения является получение рекомбинантного микроорганизма для продукции O-фосфосерина.
Эффективные результаты изобретения
Способ по настоящему изобретению, в котором рекомбинантным микроорганизмом с высоким выходом продуцируется O-фосфосерин и используется для превращения в цистеин как таковой, является более благоприятным для окружающей среды и обеспечивает более высокую эффективность продукции цистеина по сравнению с методами химического синтеза. Цистеин и его производные, получаемые с помощью ферментации и биопревращения по настоящему изобретению, могут широко использоваться при изготовлении пищевых продуктов для человека и животных и пищевых добавок.
Краткое описание фигур
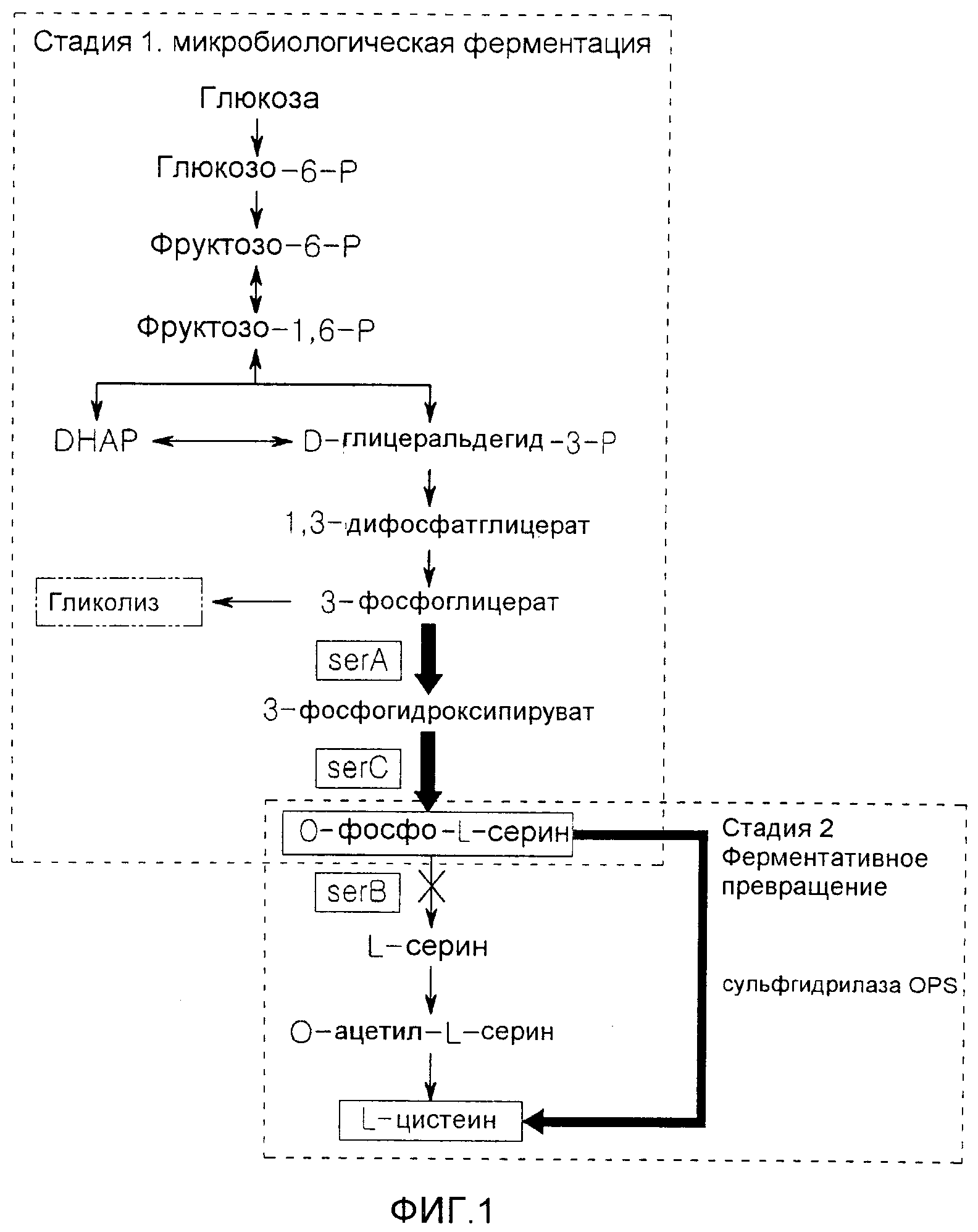
Фиг.1 представляет собой схематическую диаграмму, демонстрирующую аккумуляцию O-фосфосерина при микробной ферментации, и ферментативное превращение аккумулированного O-фосфосерина в L-цистеин.
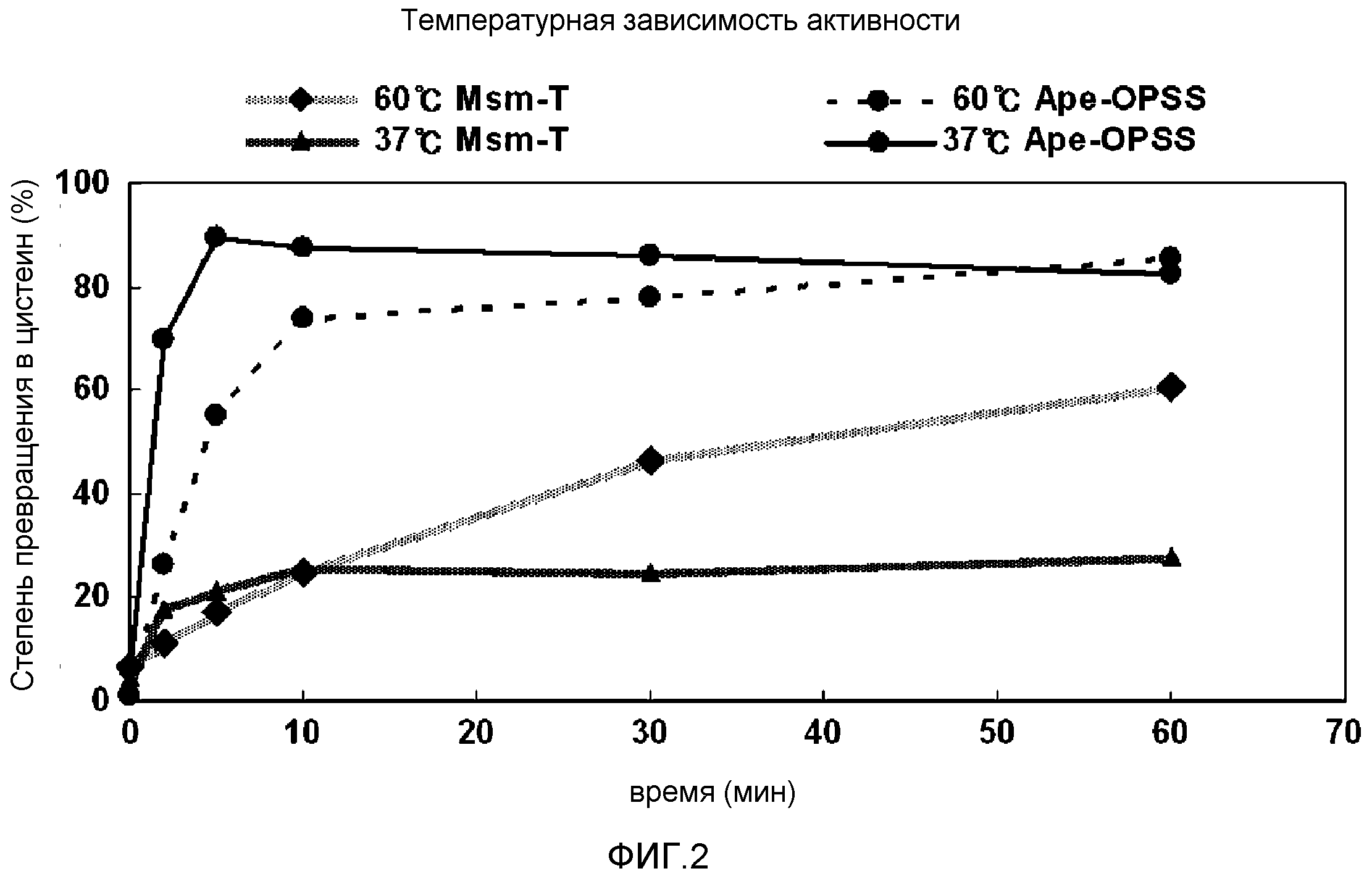
Фиг.2 представляет собой график, демонстрирующий активность сульфгидрилазы OPS при различной температуре.
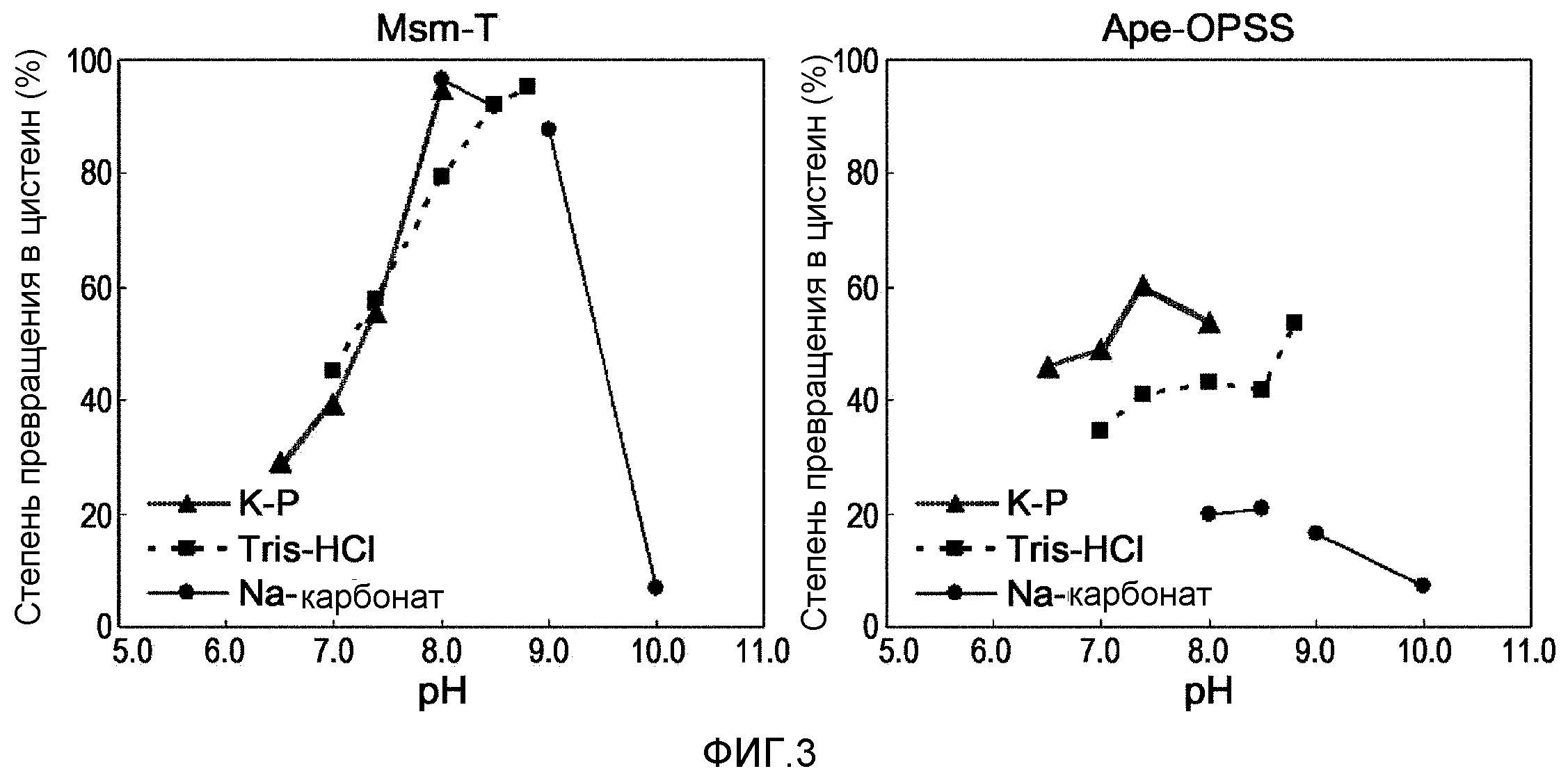
Фиг.3 представляет собой панель графиков, демонстрирующих чувствительность сульфгидрилазы OPS к рН.
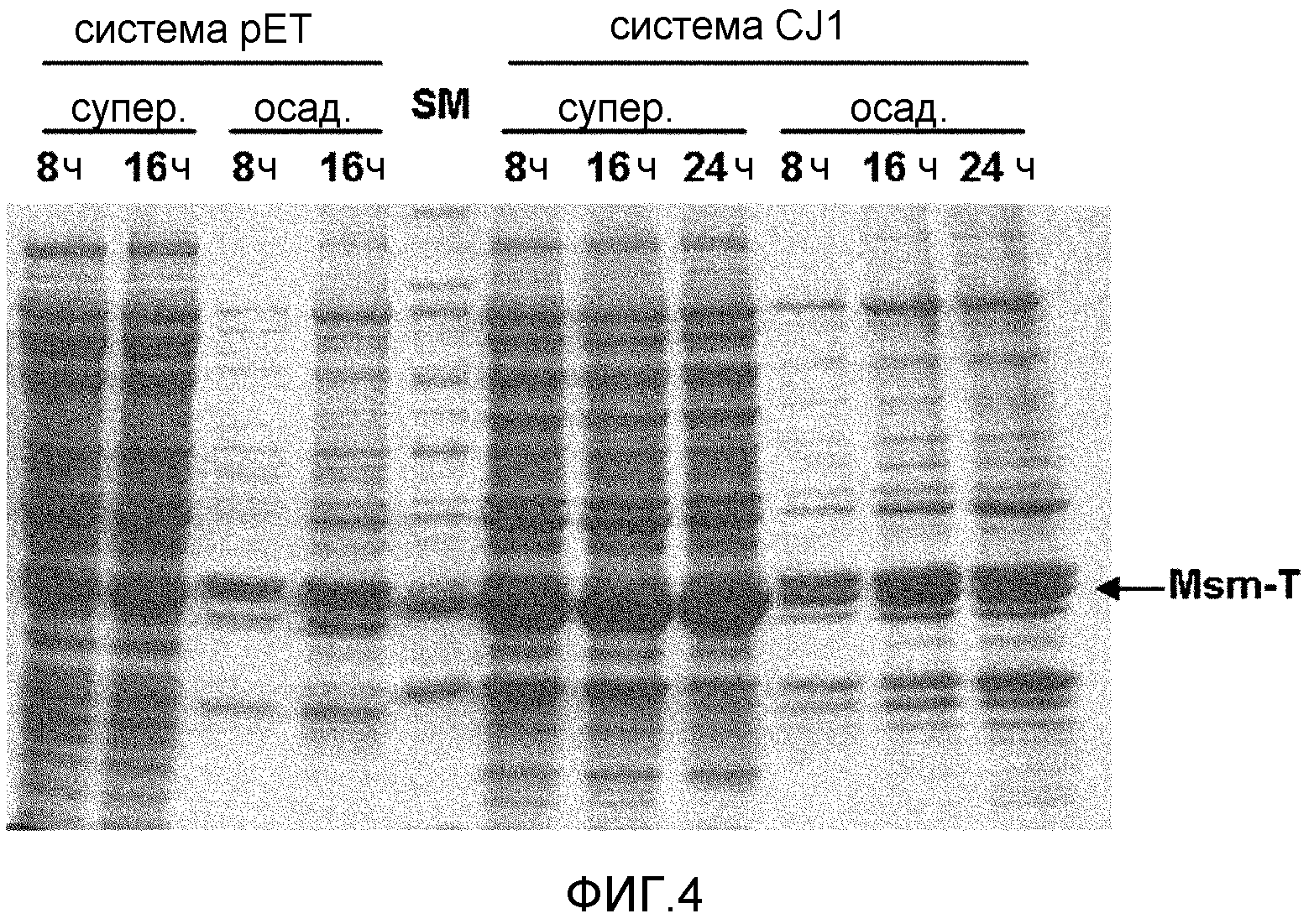
Фиг.4 представляет собой фотографию, демонстрирующую уровень экспрессии Msm-T в системе pET и системе pCL-Pcj1 при анализе с помощью SDS PAGE.
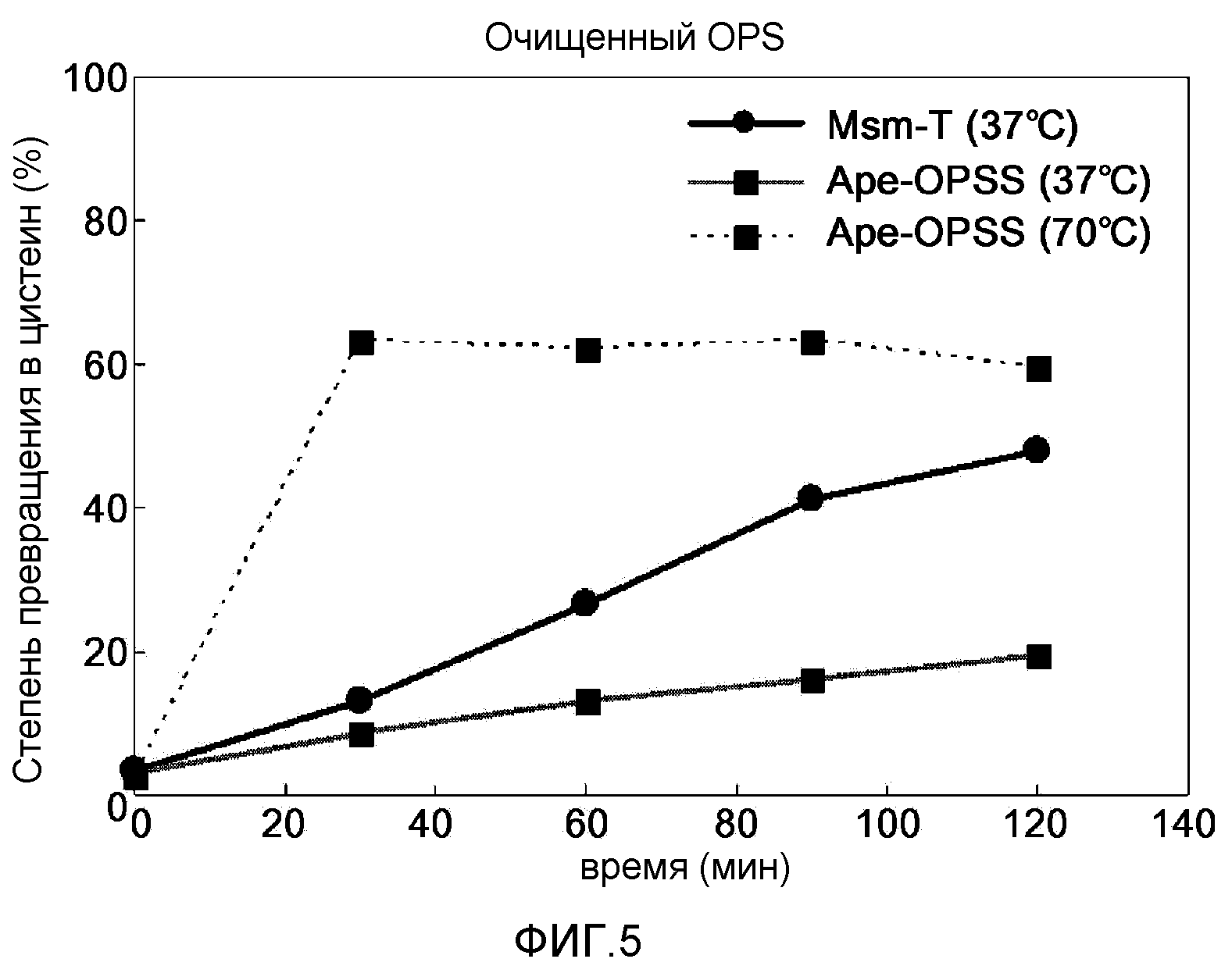
Фиг.5 представляет собой график, демонстрирующий ферментативную активность сульфгидрилазы OPS при превращении очищенного из ферментационного бульона OPS в цистеин.
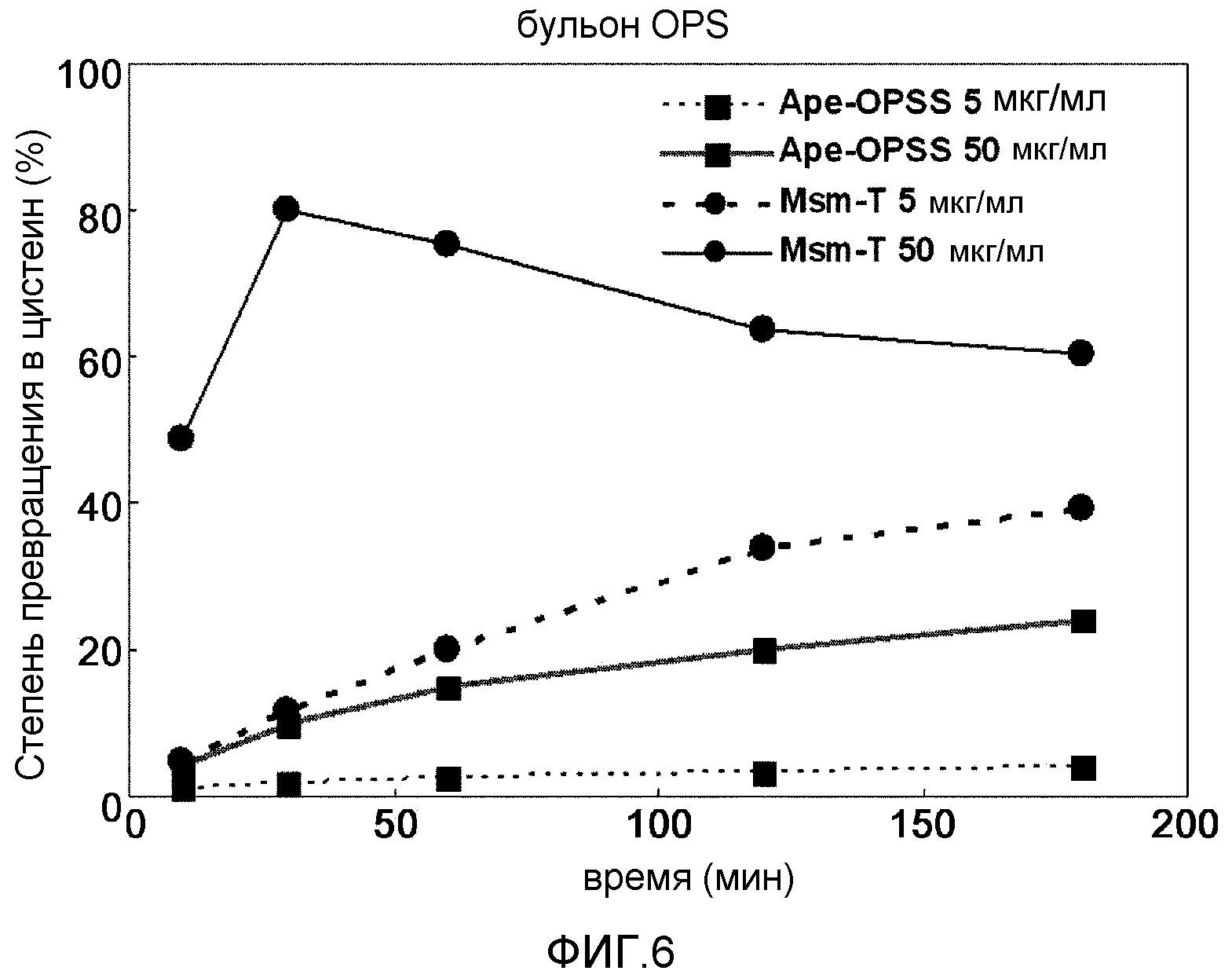
Фиг.6 представляет собой график, демонстрирующий ферментативную активность сульфгидрилазы OPS при превращении OPS ферментационного бульона в цистеин.
Лучший способ осуществления изобретения
В рамках изобретения термин «превращение в цистеин» обозначает каталитическую реакцию O-фосфосеринсульфгидрилазы (OPSS), которая приводит к превращению субстрата O-фосфосерина (OPS) в продукт цистеин, то есть он обозначает каталитическую реакцию превращения OPS в цистеин.
В рамках изобретения термин «степень превращения в цистеин» обозначает процентное отношение количества продукта цистеина к количеству исходного соединения OPS. При оптимальных условиях реакции 1 моль OPS превращается в 1 моль цистеина. Например, если 100 моль OPS превращается в 100 моль цистеина, степень превращения в цистеин составляет 100%.
В соответствии с одним из его аспектов настоящее изобретение относится к способу получения цистеина или его производного, включающему:
1) культивирование рекомбинантного микроорганизма, который модифицирован так, чтобы иметь пониженную активность эндогенной фосфосеринфосфатазы (SerB), для продукции O-фосфосерина (OPS); и 2) введение в реакцию OPS со стадии 1) с сульфидом в присутствии O-фосфосеринсульфгидрилазы (OPSS) или микроорганизма, экспрессирующего OPSS, для получения цистеина или его производного.
В способе стадия 1) относится к культивированию рекомбинантного микроорганизма, у которого активность эндогенной фосфосеринфосфатазы (SerB) понижена.
SerB представляет собой белок, который обладает гидролизирующей активностью, гидролизуя OPS до L-серина. Таким образом, микроорганизм, который имеет сниженную эндогенную активность SerB, характеризуется аккумуляцией в нем OPS. SerB без ограничения может включать любые аминокислотные последовательности, которые проявляют активность SerB, и предпочтительно может представлять собой аминокислотную последовательность SEQ ID NO: 1 или 2. Однако используется любая аминокислотная последовательность, до тех пор, пока она проявляет активность SerB, которая гомологична SEQ ID NO: 1 или 2 предпочтительно на 90% или более, более предпочтительно на 96% или более, еще более предпочтительно на 98% или более и наиболее предпочтительно на 99% или более. Сниженная активность SerB обозначает снижение активности SerB по сравнению с активностью ранее модифицированного штамма и охватывает разрушение SerB. Различные способы снижения активности SerB хорошо известны в данной области техники. Иллюстративные примеры включают делецию хромосомного serB, введение мутации в хромосомный serB для снижения эндогенной активности SerB, введение мутации в регуляторную область serB для снижения эндогенной активности SerB, замену хромосомного serB мутантным геном для снижения эндогенной активности SerB и введение антисмыслового олигонуклеотида, комплементарного транскрипту serB, для ингибирования трансляции мРНК, но методы снижения активности SerB не ограничиваются этими. Эти методы в настоящем изобретении могут быть применены для снижения активности других ферментов.
Разрушение эндогенной SerB ведет к введению сериновой ауксотрофии в рекомбинантном микроорганизме, так что среда должна быть дополнена глицином и серином. Глицин может быть предоставлен в форме очищенного глицина, содержащего глицин дрожжевого экстракта или триптона. Глицин содержится в концентрации от 0,1 до 10 г/л и предпочтительно в концентрации от 0,5 до 3 г/л. Что касается серина, он может быть предоставлен в форме очищенного серина, содержащего серин дрожжевого экстракта или триптона. Его концентрация в культуральной среде находится в диапазонах от 0,1 до 5 г/л и предпочтительно от 0,1 до 1 г/л.
В одном варианте осуществления настоящего изобретения при культивировании в среде, содержащей глицин или серин, мутантные Corynebacterium glutamicum или E. coli, в которых нарушена активность эндогенной SerB, как обнаружено, продуцируют более высокое количество OPS по сравнению с диким типом (смотри таблицы 2, 3, 6 и 7).
Кроме того, рекомбинантный микроорганизм по настоящему изобретению может иметь повышенную фосфоглицератдегидрогеназную (SerA) или фосфосеринаминотрансферазную (SerC) активность. SerA представляет собой белок, который активен в плане превращения 3-фосфоглицерата в 3-фосфогидроксипируват. SerA может иметь аминокислотную последовательность дикого типа или мутантную аминокислотную последовательность, которая проявляет устойчивость к ингибированию серином по механизму обратной связи, но не ограничивается этим. Предпочтительно SerA может иметь одну из последовательностей, выбранных из группы, состоящей из аминокислотных последовательностей SEQ ID NO: с 3 по 7. Любая аминокислотная последовательность может быть использована до тех пор, пока она проявляет активность SerA дикого типа или активность мутантной SerA, устойчивой к ингибированию серином по механизму обратной связи, хотя предпочтительно, чтобы она была гомологична одной из SEQ ID NO: с 3 по 7 на 90% или более, более предпочтительно на 96% или более, еще более предпочтительно на 98% или более и наиболее предпочтительно на 99% или более. «Мутантная SerA, устойчивая к ингибированию серином по механизму обратной связи», обозначает мутанта, проявляющего поддерживаемую или повышенную активность SerA безотносительно к ингибированию серином или глицином по механизму обратной связи. Мутанты, устойчивые к механизму обратной связи, хорошо известны в данной области техники (Grant GA et al., J. Biol. Chem., 39: 5357-5361, 1999; Grant GA et al., Biochem., 39: 7316-7319, 2000; Grant GA et al., J. Biol. Chem., 276: 17844-17850, 2001; Peters-Wendisch P et al., Appl. Microbiol. Biotechnol., 60: 437-441, 2002; патент EP0943687B). В одном варианте осуществления настоящего изобретения, когда serA, устойчивый к механизму обратной связи, был дополнительно введен в Corynebacterium glutamicum или E. coli, обладающих нарушенным serB, они, как обнаружено, продуцировали более высокое количество OPS по сравнению с диким типом (смотри таблицы 4 и 9).
SerC представляет собой белок, который активен в плане превращения 3-фосфогидроксипирувата в O-фосфосерин. SerC без ограничения может включать последовательности, которые проявляют активность SerC, и может предпочтительно иметь аминокислотную последовательность SEQ ID NO: 8. Однако может быть использована любая аминокислотная последовательность до тех пор, пока она проявляет активность SerC, но предпочтительно она должна быть гомологична SEQ ID NO: 8 на 90% или более, более предпочтительно на 96% или более, еще более предпочтительно на 98% или более и наиболее предпочтительно на 99% или более. Более того, в serC может быть введена мутация так, что ферментативная активность может быть повышена. В одном варианте осуществления настоящего изобретения, когда в них дополнительно был введен serC, Corynebacterium glutamicum или E. coli, обладающие нарушенным serB и устойчивым к механизму обратной связи serA, как обнаружено, продуцировали более высокое количество OPS по сравнению с диким типом (смотри таблицу 9).
Кроме того, может быть снижена способность рекомбинантного микроорганизма по настоящему изобретению осуществлять внутриклеточный захват O-фосфосерина или его разрушение. Конкретно, рекомбинантный микроорганизм может быть модифицирован для снижения активности ABC транспортера алкилфосфоната PhnC/PhnD/PhnE (оперона PhnCDE, который представляет собой АТФ-связывающий компонент транспорта фосфоната (PhnC; EG 10713)-компонента периплазматического связывающего белка транспортера Pn (PhnD; EG 10714)-интегрального мембранного компонента ABC транспортера алкилфосфоната (PhnE; EG 11283)), щелочной фосфатазы (PhoA) или кислой фосфатазы (AphA).
В одном варианте осуществления настоящего изобретения дополнительная делеция оперона phnCDE из рекомбинантного мутанта, как обнаружено, приводила к повышению продукции OPS (таблица 10). В рекомбинантном микроорганизме, в котором дополнительно нарушена активность PhoA или AphA, разрушение OPS начинает снижаться в то время, когда снижается концентрация фосфорной кислоты в культуральной среде (таблица 12). Более того, введение serA, устойчивого к механизму обратной связи, или serC повышало продукцию OPS (таблицы с 14 по 16).
Кроме того, рекомбинантный микроорганизм по настоящему изобретению может быть дополнительно охарактеризован по повышению активности пиримидиннуклеотидтрансгидрогеназы (PntAB; EC 1.6.1.1). Как описано ранее (Sauer U P et al., J Biol Chem. 20; 279(8):6613-9. Epub 2003), PntAB участвует в метаболизме НАДФН для регуляции внутриклеточного окислительно-восстановительного баланса. В одном варианте осуществления настоящего изобретения в рекомбинантном микроорганизме, в котором активность PntAB была дополнительно увеличена путем гиперэкспрессии pntAB, как обнаружено, увеличивалась продукция OPS (таблица 17).
Более того, рекомбинантный микроорганизм по настоящему изобретению может быть охарактеризован по повышению пермиазы оттока O-ацетилсерина/цистеина (YfiK), белка оттока гомосерина/гомосеринлактона (RhtB; EG 11469) или белка оттока треонина/гомосеринлактона (RhtC; EG11468). YfiK известен как экспортер для внеклеточного экспорта цистеина и OAS (Franke I et al., J. Bacteriology, 185: 1161-1166, 2003), а RhtB, как сообщалось, действует как экспортер из клетки гомосерина/гомосеринлактона, предшественника треонина. Кроме того, RhtC известен как экспортер треонина и гомосерина. Повышение активности YfiK, RhtC и RhtB выявило усиление роста и продукции OPS у штамма, аккумулирующего OPS (таблица 18).
Повышение ферментативной активности может быть достигнуто с использованием различных хорошо известных методов, включая, но не ограничиваясь этим, повышение количества копий гена, кодирующего фермент, представляющий интерес, введение мутации в регуляторную область гена для повышения ферментативной активности, замену хромосомного гена мутантным геном для повышения ферментативной активности и введение мутации в хромосомный ген для повышения ферментативной активности.
Кроме того, рекомбинантный микроорганизм по настоящему изобретению может быть дополнительно охарактеризован по сниженной активности фосфоглицератмутазы. Фосфоглицератмутаза существует в виде трех изозимов: GpmI, GpmA и GpmB и ответственна за превращение 3-фосфоглицерата в 2-фосфоглицерат в процессе гликолиза. При использовании 3-фосфоглицерата в качестве субстрата эти ферменты конкурируют с SerA, который катализирует синтез 3-фосфогидроксипирувата. Следовательно, пониженная активность каждого из ферментов, как обнаружено, является причиной избытка 3-фосфоглицерата, предшественника OPS, приводя к продукции повышенного уровня OPS (Таблица 19).
В рекомбинантном микроорганизме по настоящему изобретению может быть также снижена L-сериндегидратаза I (SdaA; EC 4.3.1.17) или 2-амино-3-кетобутират-коэнзим A-лигаза (Kbl). Таким образом, рекомбинантный микроорганизм характеризуется продукцией OPS, поддерживаемой или увеличенной, даже когда он культивируется в среде, содержащей низкую концентрацию глицина или серина (таблица 20).
Кроме того, рекомбинантный микроорганизм по настоящему изобретению может быть дополнительно охарактеризован по сниженной активности IclR. IclR представляет собой транскрипционный фактор, который функционирует как репрессор экспрессии aceBAK, оперона обхода глиоксилата (L Gui et al., J. Bacteriol., Vol. 178, №1, 321-324, 1996). Когда его удаляют делецией, продукция OPS, как показано, повышается (таблица 21).
Рекомбинантный микроорганизм по настоящему изобретению может быть также дополнительно охарактеризован по повышенной активности ферментов, выбранных из группы, состоящей из i) ацетил-CoA синтетазы (Acs), ii) киназы уксусной кислоты (AckA)-фосфотрансацетилазы (Pta), iii) малатсинтазы G (GlcB), iv) малатдегидрогеназы (MaeB), v) глутаматдегидрогеназы (GdhA), vi) глиоксалаткарболигазы (Glc), vii) тартронатсемиальдегидредуктазы 2 (GlxR), viii) глицераткиназы II (GlxK) и их сочетания.
В конкретном варианте осуществления настоящего изобретения, когда i) Acs (EC №6.2.1.1; J Bacteriol. 1995 May; 177(10):2878-86) или мономер пируватоксидазы (PoxB; EC 1.2.2.2) или ii) AckA и Pta (EC 2.3.1.8), целью которых у всех является эффективное повторное использование аккумулированного ацетата с сопутствующим потреблением продуцируемого НАДН, были дополнительно повышены, рекомбинантный микроорганизм по настоящему изобретению, как обнаружено, повышал продукцию OPS (таблица 22). Функционируя как катализаторы синтеза малата из глиоксилата и превращения малата в пируват iii) GlcB (EC No.2.3.3.9) и iv) MaeB (EC 1.1.137) могут ослаблять цикл TCA и таким образом использоваться для повышения потребления глюкозы и продукции O-фосфосерина (таблица 23). В соответствии с одним вариантом осуществления настоящего изобретения повышение v) GdhA; (EC 1.4.1.2), который катализирует синтез глутамата, субстрата SerC, из 2-оксоглутарата и НАДФН, придает намного более высокий потенциал для продукции OPS микроорганизмом (таблица 17). Все из vi) Glc (EC 4.1.1.47), vii) GlxR (EC 1.1.1.60) и viii) GlxK (EC 2.7.1.31), как известно, превращают гликоксилат в 3-фосфоглицерат, что означает повышение уровня субстрата фосфоглицератдегидрогеназы (Kim HJ et al., J. Bacteriol., 186(11), 3453-3460, 2004; Eva Cusa et al., J. Bacteriol., 181(24), 7479-7484, 1999; Chnag YY et al., J. Biol. Chem. 268(6): 3911-3919, 1993). Рекомбинантный микроорганизм по настоящему изобретению при дополнительном увеличении Glc, GlxR и GlxK был улучшен по потреблению сахара и росту (таблица 24).
Рекомбинантный микроорганизм по настоящему изобретению относится к любому микроорганизму, в котором существует снижение активности SerB таким образом, что продукция OPS находится на повышенном уровне. Если это условие удовлетворяется, любой микроорганизм, будет ли он прокариотным или эукариотным, попадает в объем настоящего изобретения. Иллюстративными примерами среди них являются энтеробактерии или дифтериеподобные бактерии. Примеры микроорганизмов, пригодных для настоящего изобретения, включают Escherichia sp., Erwinia sp., Serratia sp., Providencia sp., Corynebacterium sp. и Brevibacterium sp. Предпочтительными являются Escherichia sp. и Corynebacterium sp. при большей предпочтительности Escherichia sp. и наибольшей предпочтительности E. coli.
В одном варианте осуществления рекомбинантный штамм, способный продуцировать OPS, назван E. coli CA07-0012 и положен на хранение в Корейский центр культивируемых микроорганизмов, расположенный в 361-221, Hongje 1, Seodaemun, Seoul, Korea, 12 октября 2011 г. под номером поступления KCCM11212P.
Кроме того, в одном варианте осуществления рекомбинантный штамм, способный продуцировать OPS, назван E. coli CA07-0022/pCL-prmf-serA*(G336V)-serC и положен на хранение в Корейский центр культивируемых микроорганизмов, расположенный в 361-221, Hongje 1, Seodaemun, Seoul, Korea, 28 сентября 2010 г. под номером поступления KCCM11103P. В настоящем описании термин «CA07-0022/pCL-prmf-serA*(G336V)-serC» используется взаимозаменяемо с CA07-0022 serA*(G336V)/pCL-prmf-serA*(G336V)-serC.
После культивирования штамма в течение 80 часов в 1 л ферментере O-фосфосерин продуцировался в концентрации 19,5 г/л (пример 35).
В рамках изобретения термин «культивирование» обозначает растущие микроорганизмы в искусственно контролируемых условиях. Процедура культивирования может быть проведена с использованием подходящей среды и условий культивирования, хорошо известных в данной области техники. Специалисты в данной области техники могут легко контролировать процедуру культивирования для ее соответствия используемым штаммам. Например, без ограничения она может быть осуществлена в виде периодического культивирования, непрерывного культивирования или периодического подпитываемого культивирования.
Кроме того, культуральная среда содержит источник углерода. Примеры источника углерода включают сахариды и углеводы, такие как глюкоза, сахароза, лактоза, фруктоза, мальтоза, крахмал и целлюлоза, масла и жиры, такие как соевое масло, подсолнечное масло, касторовое масло и кокосовое масло, жирные кислоты, такие как пальмитиновая кислота, стеариновая кислота и линолевая кислота, спирты, такие как глицерин и этанол, и органические кислоты, такие как уксусная кислота. Эти источники углерода могут присутствовать в культуральной среде отдельно или в сочетании. В качестве источника азота в культуральной среде может содержаться органическое вещество, такое как пептон, дрожжевой экстракт, мясной бульон, солодовый экстракт, жидкий кукурузный экстракт, соя и белок злаков, или неорганическое азотистое вещество, такое как мочевина, сульфат аммония, хлорид аммония, фосфат аммония, карбонат аммония и нитрат аммония. Эти источники азота могут быть использованы отдельно или в сочетании. В качестве источника фосфора среда может содержать дигидрофосфат калия, фосфат калия или соответствующие соли натрия. Среда может содержать соли металлов, такие как сульфат магния или сульфат железа. Культуральная среда может также содержать аминокислоты, витамины и подходящие предшественники. К среде могут быть добавлены питательные вещества в виде периодического или непрерывного способа добавления.
Для доведения рН культуры к культуральной среде в течение культивирования может быть добавлено подходящим образом такое соединение, как гидроксид аммония, гидроксид калия, аммиак, фосфорная кислота и серная кислота. Кроме того, в течение культивирования используется противопенный агент, такой как полигликолевый эфир жирной кислоты, для подавления образования пены. Более того, для поддержания культуральной среды в аэробных условиях в культуральную среду можно вдувать кислород или содержащий кислород газ. Для анаэробных или микроаэробных условий азот, водород или углекислый газ подаются без аэрации. Культуральная среда может обычно поддерживаться при температуре от 27°С до 37°С и предпочтительно при температуре от 30°С до 35°С. Что касается периода культивирования, культивирование может поддерживаться до тех пор, пока представляющий интерес продукт будет получен в желаемом количестве, и предпочтительно этот период находится в диапазонах от 10 до 100 часов.
Для дальнейшего сбора и извлечения из культуральной среды OPS, продуцируемого в течение стадии культивирования, подходящий метод, известный в данной области техники, может быть выбран в зависимости от типа культуры и процесса культивирования, был ли он периодическим, непрерывным или периодическим подпитываемым.
В способе по настоящему изобретению стадия 2) направлена на взаимодействие OPS со стадии 1) с сульфидом в присутствии O-фосфосеринсульфгидрилазы (OPSS) или микроорганизма, экспрессирующего OPSS, для индукции превращения O-фосфосерина в цистеин или его производные.
Сульфид может быть использован в форме жидкости или газа, а также в твердой форме, обычно используемой в данной области техники, из-за различия в рН, давлении и/или растворимости. Любое соединение серы, такое как сульфид (S2-) или тиосульфат (S2O32-), может быть использовано в настоящем изобретении до тех пор, пока оно может превращаться в тиоловую группу (SH). Предпочтительно могут быть использованы Na2S, NaSH, H2S, (NH4)2S, NaSH и Na2S2O3, все они могут предоставлять тиоловую группу для OPS. В реакции одна тиоловая группа предоставляется одной молекуле OPS для получения одной молекулы цистеина или его производного. При этом ферментативном превращении сульфид может быть предпочтительно добавлен в молярной концентрации в от 0,1 до 3 раз и более предпочтительно от 1 до 2 раз более высокой по сравнению с используемой для OPS. В плане экономии предоставляющий тиоловую группу сульфид и OPS наиболее предпочтительно используются в молярном отношении 1:1. В одном варианте осуществления настоящего изобретения в качестве источника серы использовали Na2S. Na2S добавляли в молярной концентрации в от 1 до 3 раз более высокой, чем концентрация OPS, используемая в реакции превращения. Предпочтительно он подавался в молярной концентрации в два раза более высокой, чем концентрация OPS для эффективного превращения OPS в цистеин (таблица 34).
Используемый в настоящем описании термин «O-фосфосеринсульфгидрилаза (OPSS)» обозначает фермент, который катализирует перенос тиоловой группы (SH) на OPS (O-фосфосерин) с превращением OPS в цистеин. Фермент был впервые обнаружен в Aeropyrum pernix, Mycobacterium tuberculosis и Trichomonas vaginalis (Mino K and Ishikawa K, FEBS letters, 551: 133-138, 2003; Burns KE et al., J. Am. Chem. Soc., 127: 11602-11603, 2005).
Используемый в настоящем описании термин «мутантный» обозначает культуру или индивидуум, которые проявляют наследуемое или ненаследуемое изменение фенотипа. При использовании в отношении OPSS термин «мутантный» предназначен для обозначения фермента OPSS, который генетически изменен так, что его активность может быть эффективно увеличена по сравнению с ферментом дикого типа.
В настоящем изобретении мутантный OPSS может быть сконструирован путем делеции, замены или добавления части нуклеотидной последовательности, кодирующей OPSS. В соответствии с одним вариантом осуществления настоящего изобретения фермент OPSS с увеличенной активностью получали путем делеции пяти C-концевых аминокислотных остатков фермента OPSS Mycobacterium smegmatis.
Мутантный OPSS может быть получен в E. coli, широко используемой для экспрессии ферментов, с применением методов генного синтеза на основе оптимизации кодонов, с помощью которых представляющие интерес ферменты могут быть получены с высоким выходом. Альтернативно, для получения мутантного OPSS могут быть использованы методы скрининга пригодных источников фермента, основанные на анализе массивов генетической информации о микроорганизмах методами биоинформатики. В одном варианте осуществления настоящего изобретения ферменты OPSS, которые используют OPS в качестве субстрата для синтеза цистеина, были отобраны из различных микробов путем скрининга гомологии аминокислотных последовательностей. В этом случае осадки клеток, полученные с использованием среды и культуральных условий, которые подходят для данного уровня техники, лизировали с последующей очисткой супернатанта, содержащего фермент, с получением фермента OPSS (таблица 26).
Кроме того, для экономного получения фермента OPSS была разработана экспрессионная система, дающая высокий выход. Вектор pET с применением промотора T7 хорошо известен в данной области техники. Однако изобретатели настоящего изобретения разработали экспрессионную систему для фермента, названную системой CJ1 (корейский патент 10-0620092 B1), вместо применения обычной системы. В одном варианте осуществления настоящего изобретения сравнивали уровни экспрессии OPSS системой pET, включающей промотор T7, и системой CJ1, включающей промотор CJ1, при одних и тех же заданных условиях. В результате система CJ1 показала более высокий уровень экспрессии OPSS, чем система pET. Кроме того, для гиперэкспрессии OPSS в системе pET требовалась низкая температура (18°С) и длительный период времени, а в системе pCL-pCJ1 высокая температура (37°С) и короткий период времени. Для получения OPSS предпочтительно используется система pCL-pCJ1 (пример 46).
Увеличение ферментативной активности может быть достигнуто с использованием различных хорошо известных методов. Например, его можно добиться с помощью увеличения количества копий гена, кодирующего OPSS, использования сильного промотора или введения генетической мутации.
Оптимизация ферментативного превращения под действием OPSS может быть достигнута с использованием различных методов, известных в данной области техники. Например, оптимизация может быть основана на полном понимании характеристик OPSS, таких как оптимальная температура и рН, ингибирование субстратами, концентрация субстратов и т.д. Кроме того, оптимизация может быть определена по оптимальным условиям для ферментативного превращения, таким как оптимальная концентрация OPSS, оптимальные балансы используемых субстратов в плане концентраций, предпочтительные соединения серы, предоставляющие SH для ферментативного превращения, предпочтительность определенных буферов, влияние генерируемых ионов и кофакторы и их оптимальные концентрации.
В одном варианте осуществления настоящего изобретения был охарактеризован фермент OPSS, полученный с использованием указанного выше способа, и на основе определенных характеристик был разработан экономически выгодный способ ферментативного превращения, который характеризуется высокой степенью образования цистеина из OPS с гарантией стабильности фермента. В способе ферментативного превращения температура реакции может быть установлена от 37°С до 80°С. Подробнее, Ape-OPSS (SEQ ID NO: 12), принадлежащий Archea, проявляет повышенную ферментативную активность при 60°С по сравнению с 37°С и сам фермент высоко стабилен при нагревании с оптимальной реакционной способностью при 60°С. С другой стороны, Msm-T (SEQ ID NO: 10) проявляет оптимальную активность при 37°С и снижает активность при температурной обработке при 60°С. Фермент OPSS, как обнаружено, обладает ферментативной активностью на протяжении диапазона рН от 6,0 до 10,0. Ape-OPSS проявлял оптимальную активность при рН 7,4. При появлении оптимальной активности при рН от 8,0 до 9,0 Msm-T проявлял стабильность на протяжении более широкого диапазона рН по сравнению с Ape-OPSS (таблица 28 и 31 и фиг.2 и 3).
При ферментативном превращении в качестве кофактора может быть использован 0,001-2 мМ PLP (пиридоксаль-5'-фосфат) или 0,001-100 мМ DTT. В одном варианте осуществления настоящего изобретения степень превращения в цистеин была повышена в 2,3 раза в присутствии 25 мМ DTT или 0,2 мМ PLP. Как таковая обработка DTT или PLP вызывает улучшение степени превращения в цистеин на стадии 2). Добавление кофактора приводило к приемлемому уровню в плане рассмотрения повышенной стоимости продукции и увеличенной степени превращения (таблица 30).
Условия реакции для OPSS могут варьироваться в зависимости от типов и концентрации используемого OPS. В одном варианте настоящего изобретения чистый OPS (имеющийся в продаже), OPS, очищенный из культуры, полученной на стадии 1), и содержащую OPS культуру со стадии 1) использовали при различных условиях для обеспечения оптимальных степеней превращения. В результате степень превращения в цистеин варьировала в зависимости от типа и концентрации OPSS, температуры реакции и типа и концентрации OPS (фиг.5 и 6, таблица 35).
Способ по настоящему изобретению может дополнительно включать выделение и очистку цистеина, полученного на стадии 2). После ферментативного превращения цистеин может быть выделен и очищен из культуральной среды с использованием метода, хорошо известного в данной области техники.
Специалисты в данной области техники могут химически синтезировать из цистеина производные цистеина с использованием хорошо известного метода. Цистеин можно легко ввести в реакцию с ацетилирующим агентом с получением NAC (N-ацетилцистеина) и с галогенуксусной кислотой в основных условиях с получением SCMC (S-карбоксиметилцистеина). Эти производные цистеина используются в качестве агентов в медицине для лечения кашля, бронхита, бронхиальной астмы и больного горла.
В настоящем изобретении бульон OPS, полученный с помощью микробной ферментации, используется в качестве субстрата для синтеза цистеина. Бульон OPS, полученный с помощью микробной ферментации, имеет экономические преимущества над имеющимся в продаже чистым OPS, заключающиеся в том, что бульон OPS может быть использован без необходимости дополнительной очистки, и кофактор PLP, необходимый для превращения, может быть получен из ферментируемой культуры.
В одном варианте осуществления настоящего изобретения был разработан способ превращения, который обеспечивает степень превращения в цистеин настолько высокую, как 80%, при использовании 50 мкг/мл Msm-T при условиях реакции: 50 мМ бульона OPS или 60 мМ очищенного бульона OPS, 100 мМ Na2S или 120 мМ Na2S и 0,2 мМ PLP. Специалистам в данной области техники должно быть понятно, что ферментативное превращение с использованием высоко активных ферментов может быть легко оптимизировано и масштабировано.
В соответствии с другим его аспектом, настоящее изобретение относится к рекомбинантному микроорганизму, у которого снижена активность SerB, для продукции OPS. В одном варианте осуществления рекомбинантный микроорганизм демонстрирует повышение serA или serC, устойчивых к ингибированию серином по механизму обратной связи, или делецию, по меньшей мере, одного выбранного из PhnC/PhnD/PhnE ABC транспортера алкилфосфоната (оперона phnCDE), щелочной фосфатазы (phoA) и кислой фосфатазы (alpha). Предпочтительно рекомбинантный микроорганизм для получения OPS представляет собой микроорганизм, положенный на хранение под № поступления KCCM11103P или KCCM11212P. Более предпочтительно рекомбинантный микроорганизм для получения OPS представляет собой микроорганизм, положенный на хранение под № поступления KCCM11103P.
Способ осуществления изобретения
Лучшее понимание настоящего изобретения может быть достигнуто с помощью последующих примеров, которые предлагаются с целью иллюстрации, но не истолковываются как ограничивающие настоящее изобретение.
Получение O-фосфосерина, продуцируемого Corynebacterium, и продукция O-фосфосерина с ее использованием
Пример 1: Получение штамма Corynebacterium с недостаточностью фосфосеринфосфатазы (serB)
Corynebacterium glutamicum 13032 модифицировали путем делеции гена serB (SEQ ID NO: 13, EC 3.1.3.3), кодирующего фосфосеринфосфатазу, которая катализирует синтез L-серина из O-фосфосерина. С этой целью был сконструирован фрагмент для инактивации serB. Для этого были сконструированы праймеры для получения рекомбинантного штамма 13032-ДserB по настоящему изобретению. Прежде всего была получена последовательность serB Corynebacterium glutamicum 13032 со ссылкой на данные NIH GenBank, и на основе последовательности serB были синтезированы праймеры SEQ ID NO: с 22 по 27. Для сайт-специфического разрушения гена использовали вектор pDC, который не может реплицироваться в Corynebacterium glutamicum. Была сконструирована плазмида pDC-ДserB, в которой открытая рамка считывания serB была внутренне нарушена, и эта плазмида была адаптирована для получения сайт-специфической делеции гена serB в мутантном штамме Corynebacterium glutamicum. Внутреннее разрушение гена pDC-ДserB производилось с помощью кроссоверной ПЦР с использованием пар праймеров SEQ ID NO: 22 и 23 и SEQ ID NO: 24 и 25 с использованием геномной ДНК Corynebacterium glutamicum ATCC13032 в качестве матрицы и введением продукта ПЦР в вектор pDC. Полученной рекомбинантной плазмидой трансформировали Corynebacterium glutamicum дикого типа путем электропорации (van der Rest et al. 1999). Плазмиду вводили в хромосому путем первичной рекомбинации (кроссинговера) с последующей вторичной рекомбинацией (кроссинговером) для вырезания исходного serB из хромосомы.
По окончании вторичной рекомбинации трансформанты Corynebacterium glutamicum, содержащие делеционную мутацию serB, анализировали с помощью диагностической ПЦР с использованием специфичных для гена праймеров SEQ ID NO: 26 и 27. Рекомбинантный штамм был назван CB01-0047.
Пример 2: Анализ продукции O-фосфосерина в штамме Corynebacterium с недостаточностью фосфосеринфосфатазы
Мутантный штамм CB01-0047, полученный в результате делеции serB из Corynebacterium glutamicum 13032, который, как ожидалось, должен накапливать O-фосфосерин, наносили на чашки BHIS и инкубировали в течение ночи в инкубаторе при 30°С. Затем появившиеся на чашках BHIS колонии инокулировали с помощью платиновой петли в 25 мл среды для титрования, показанной в таблице 1, и инкубировали при 30°С в течение 48 часов со встряхиванием при 200 об/мин. Результаты суммированы в таблице 2 ниже.
Было обнаружено, что штамм CB01-0047 растет очень медленно в среде для титрования. Эта задержка роста не исправлялась даже после добавления L-глицина. Однако рост увеличивался в присутствии L-серина, но при этом наблюдалось слабое увеличение продукции O-фосфосерина по сравнению с диким типом. Результаты суммированы в таблице 3 ниже.
Пример 3: Конструирование мутантного гена фосфоглицератдегидрогеназы (SerA*) из Corynebacterium
Были сконструированы гены serA*(E235K) (SEQ ID NO: 14) и serA*(197Д) (SEQ ID NO: 15) из Corynebacterium glutamicum, которые кодируют соответствующие мутанты фосфоглицератдегидрогеназы, фермента, катализирующего синтез 3-фосфогидроксипирувата из 3-фосфоглицерата. Сообщалось, что эти мутанты резистентны к действию серина по механизму обратной связи (FBR) (Peters-Wendisch P et al., Appl. Microbiol. Biotechnol., 60: 437-441, 2002; EP0943687B). serA*(E235K) был получен с помощью сшивающей ПЦР на геномной ДНК из ATCC13032 с использованием праймеров SEQ ID NO: с 28 по 31, тогда как serA*(197Д) был сконструирован с помощью ПЦР с использованием пар праймеров SEQ ID NO: с 28 по 32. Полученные таким образом продукты ПЦР были вставлены в соответствующие T-векторы для конструирования рекомбинантных векторов, названных Tblunt-serA*(E235K) и Tblunt-serA*(197Д). Затем два вектора были обработаны ферментами рестрикции EcoRV и XbaI с получением двух фрагментов ДНК serA*(E235K) и serA*(197Д). Эти фрагменты были вставлены в соответствующие векторы pECCG117-Pcj7-GFP-terminator, которые гидролизовали теми же ферментами рестрикции. В результате было получено два рекомбинантных вектора pECCG117-Pcj7-serA*(E235K) и pECCG117-Pcj7-serA*(197Д).
Пример 4: Получение штамма Corynebacterium с гиперэкспрессией sera* и тестирование продукции O-фосфосерина
Две сконструированные в примере 3 FBR-serA* плазмиды, берущие начало от Corynebacterium, были введены в Corynebacterium glutamicum CB01-0047. Для оценки продукции O-фосфосерина трансформанты наносили на чашки BHIS и инкубировали в течение ночи при 30°С. Затем появившиеся на чашках BHIS колонии инокулировали с помощью платиновой петли в 25 мл среды для титрования, показанной в таблице 1 и дополнительно содержащей 2 г/л L-серина, и инкубировали при 30°С в течение 48 часов со встряхиванием при 200 об/мин. Результаты суммированы в таблице 4 ниже.
Как показано в таблице 4, в штаммах Corynebacterium glutamicum, трансформированных FBR-serA*, берущим начало от Corynebacterium, наблюдалось накопление O-фосфосерина от 0,1 до 0,3 г/л.
Получение продуцирующей O-фосфосерин E. coli и получение с ее помощью O-фосфосерина
Пример 5: Получение штамма E. coli со сниженной активностью фосфосеринфосфатазы (SerB)
E. coli модифицировали путем делеции гена serB (SEQ ID NO: 16), кодирующего фосфосеринфосфатазу, которая катализирует синтез L-серина из O-фосфосерина. Делеционный мутант E. coli K12 получали с помощью одностадийного метода инактивации (Datsenko KA and Wanner BL, Proc. Natl. Acad. Sci., 97:6640-6645, 2000) для делеции маркерного гена устойчивости к антибиотику. Для получения штамма с делецией serB, во-первых, была проведена ПЦР на плазмиде pKD3 (Datsenko KA and Wanner BL, Proc. Natl. Acad. Sci., 97:6640-6645, 2000; GenBank № AY048742) с использованием пары праймеров SEQ ID NO: 33 и 34. Продукт ПЦР был введен в компетентные клетки E. coli K12, содержащие pKD46 (Datsenko KA and Wanner BL, Proc. Natl. Acad. Sci., 97:6640-6645, 2000; GenBank №AY048746), путем электропорации. После этого штаммы, проявлявшие устойчивость к хлорамфениколу, были подвергнуты ПЦР для подтверждения делеции serB и затем трансформированы pCP20 (Datsenko KA and Wanner BL, Proc. Natl. Acad. Sci., 97:6640-6645, 2000) для удаления маркера устойчивости к антибиотику. Полученный мутантный штамм был назван CA07-0012.
Кроме того, для снижения фосфосеринфосфатазной активности был модифицирован кодон инициации serB следующим образом. Ген serB дикого типа с ATG в качестве кодона инициации был получен с помощью ПЦР на геномной ДНК E. coli W3110, служившей в качестве матрицы. Мутантный serB с CTG в качестве кодона инициации был сконструирован с помощью сшивающей ПЦР. Пару праймеров SEQ ID NO: 35 и 36 использовали в ПЦР для амплификации serB дикого типа, а пару праймеров SEQ ID NO: 37 и 38 использовали в ПЦР для амплификации мутантного serB. Продукты ПЦР обрабатывали HindIII и клонировали в pccRAC1 (Epicentre) в сайт рестрикции HindIII для конструирования pccBACl-Pself-ATG-serB и pccBAC1-Pself-CTG-serB соответственно. Вектор serB дикого и мутантного типа вводили в CA07-0012 для сравнения фосфосеринфосфатазной активности.
Пример 6: Анализ продукции O-фосфосерина штаммом со сниженной активностью SerB
Мутантный штамм CA07-0012 с недостаточностью фосфосеринфосфатазы с ожидаемой аккумуляцией O-фосфосерина наносили на чашки LB и инкубировали в течение ночи в инкубаторе при 33°С. Затем появившиеся на чашках LB колонии инокулировали с помощью платиновой петли в 25 мл среды для титрования, показанной в таблице 5, и инкубировали при 33°С в течение 48 часов со встряхиванием при 200 об/мин. Результаты суммированы в таблице 6 ниже.
Для увеличения роста и продукции O-фосфосерина CCA07-0012 культивировали в течение 48 часов в среде для титрования таблицы 5, дополнительно содержащей 1 г/л L-глицина. Результаты суммированы в таблице 7 ниже.
Как показано в таблице 7, добавление L-глицина в культуральную среду обеспечило увеличение скорости роста штамма и продукции O-фосфосерина.
Пример 7: Конструирование вектора, несущего мутантный ген фосфоглицератдегидрогеназы (SerA*), берущий начало от E. coli
Были сконструированы берущие начало от E. coli гены serA*(G335V) (SEQ ID NO: 18), serA*(G336V, G337V) (SEQ ID NO: 19) и serA* (G336V, R338G) (SEQ ID NO: 20), кодирующие соответствующие мутанты 3-фосфоглицератдегидрогеназы, фермента, катализирующего синтез 3-фосфогидроксипирувата из 3-фосфоглицерата. Сообщалось, что эти мутанты резистентны к действию серина по механизму обратной связи (FBR) (Grant GA, Xu XL and Hu Z, Biochem., 39: 7316-7319, 2000; Grant GA, Hu Z and Xu XL, J. Biol. Chem., 276: 17844-17850, 2001). Введение мутантных генов в хромосому E. coli проводили с помощью метода сшивающий ПЦР. Фрагменты ДНК, содержащие мутации, получали с использованием следующих праймеров.
Праймеры SEQ ID NO: 39 и 41 обычно использовали с геном SerA*. Для введения мутаций в ген serA проводили ПЦР с парой праймеров SEQ ID NO: 42 и 43 для serA*(G336V), с парой праймеров SEQ ID NO: 44 и 45 для serA*(G336V, G337V) и с парой праймеров SEQ ID NO: 46 и 47 для serA*(G336V, R338G). Праймеры синтезировали на основании информации, касающейся гена K12 W3110 (GenBank номер регистрации AP 003471) и прилегающих к нему нуклеотидных последовательностей, зарегистрированных в NIH GenBank.
Пример 8: Клонирование берущего начало от E. coli гена serA, гена serA* и гена 3-фосфосеринаминотрансферазы (serC)
serA (SEQ ID NO: 17, EC 1.1.1.95), serC (SEQ ID NO: 21, EC 2.6.1.52), serA*(G335V), serA*(G336V, G337V) и serA*(G336V, R338G) клонировали следующим образом. serA и serC были получены проведением ПЦР на геномной ДНК E. coli W3110, а serA*(G336V), serA*(G336V, G337V) и serA*(G336V, R338G) конструировали с помощью ПЦР с использованием фрагментов ДНК примера 7 в качестве матриц. В ПЦР использовали праймеры SEQ ID NO: 48 и 49 для serA и SEQ ID NO: 50 и 51 для serC. После обработки EcoRV и HindII продукты ПЦР клонировали в рекомбинантный вектор pCL-Prmf, сконструированный путем вставки промотора rmf E. coli в вектор pCL1920 (GenBank №AB236930), с получением соответствующих рекомбинантных векторов, названных pCL-Prmf-serA, pCL-Prmf-serC, pCL-Prmf-serA*(G336V), pCL-Prmf-serA*(G335V, G337V) и pCL-Prmf-serA*(G336V, R338V) соответственно.
Кроме того, были сконструированы плазмиды, в которых serA, один из трех мутантов serA и/или serC образуют оперон, т.е. pCL-Prmf-serA-(RBS)serC, pCL-Prmf-serA*(G336V)-(RBS)serC, pCL-Prmf-serA*(G336V, G337V)-(RBS)serC и pCL-Prmf-serA*(G336V, R338V)-(RB5)serC. Для этого был получен фрагмент (RBS)serC с использованием праймеров SEQ ID NO: 51 и 52 и клонирован по сайту HindIII в pCL-Prmf-serA, pCL-Prmf-serA*(G336V), pCL-Prmf-serA*(G336V, G337V) и pCL-Prmf-serA* (G336V, R338V).
Пример 9: Получение берущих начало от E. coli штаммов, усиленных serA, serA* и serC, и анализ продукции O-фосфосерина
Восемью плазмидами, сконструированными в примере 8, трансформировали CA07-0012, и полученные рекомбинантные штаммы оценивали на предмет продукции O-фосфосерина. Каждый штамм наносили на чашки LB и инкубировали в течение ночи при 33°С. Затем появившиеся на чашках LB колонии инокулировали в 25 мл среды для титрования, показанной в таблице 8, и культивировали при 33°С в течение 48 часов со встряхиванием при 200 об/мин. Результаты суммированы в таблице 9 ниже.
Как видно из таблицы 9, продукция O-фосфосерина штаммом CA07-0012 возрастала после его трансформации serA и продукция O-фосфосерина увеличивалась в большей степени после введения одного из трех мутантных serA*. Штаммы, в которых serA или один из трех мутантных serA* и serC активировались одновременно, показали более высокую продукцию O-фосфосерина по сравнению со штаммами, в которых происходила изолированная активация serA или serA*. Наибольшая продукция O-фосфосерина была обнаружена в штамме, в котором одновременно активировались мутантный serA* и serC.
Пример 10: Получение штамма E. coli с недостаточностью PhnC/PhnD/PhnE алкилфосфонатного ABC-транспортера (phnCDE оперона)
Сообщалось, что в E. coli PhnC/PhnD/PhnE алкилфосфонатный ABC-транспортер переносит O-фосфосерин в цитоплазму (Wanner BL and Metcalf WW. FEMS Microbiol. Lett., 15:133-139, 1992). Оперон phnCDE, кодирующий белок PhnC/PhnD/PhnE алкилфосфонатного ABC-транспортера, удаляли из штамма с делецией serB с получением штамма CA07-0016. Для делеции phnCDE использовали пару праймеров SEQ ID NO: 53 и 54. Делецию производили способом, сходным со способом примера 5.
Кроме того, в CA07-0016 была введена pCL-Prmf-serA*(G336V)-(RBS)serC, сконструированная в примере 8.
Пример 11: Анализ продукции O-фосфосерина штаммом E. coli с недостаточностью phnCDE оперона
Штаммы CA07-0016 и CA07-0016/pCL-Prmf-serA*(G336V)-(RBS)serC, полученные в примере 10, оценивали на предмет продукции O-фосфосерина. Каждый штамм наносили на чашки LB или чашки LB(спектиномицин) и инкубировали в течение ночи при 33°С. Затем появившиеся на чашках LB или чашках LB(спектиномицин) колонии инокулировали с помощью платиновой петли в 25 мл среды для титрования, показанной в таблице 8, и культивировали при 33°С в течение 48 часов со встряхиванием при 200 об/мин. Результаты суммированы в таблице 10 ниже.
Как видно из таблицы 10, штамм с делецией оперона phnCDE проявил лишь слабое повышение продукции O-фосфосерина.
Пример 12: Получение штамма E. coli с недостаточностью щелочной фосфатазы (phoA), кислой фосфатазы (aphA)
В штамме E. coli с делецией фосфосеринфосфатазы были дополнительно удалены ген phoA, кодирующий щелочную фосфатазу, и ген aphA, кодирующий кислую фосфатазу. Фрагмент ДНК, использованный для делеции phoA, был получен путем проведения ПЦР на плазмиде pkD3 с парой праймеров SEQ ID NO: 55 и 56. С другой стороны, фрагмент ДНК, использованный для делеции aphA, был получен сходным образом с использованием пары праймеров SEQ ID NO: 57 и 58. Каждый штамм с делецией получали способом, сходным со способом примера 5. Штамм с делецией и phoA, и aphA был получен путем электропорации фрагмента ДНК для делеции aphA в компетентные клетки штамма с делецией phoA, который был вновь трансформирован pKD46. После этого трансформанты, которые были устойчивы к хлорамфениколу, подвергали ПЦР для подтверждения делеции aphA и затем трансформировали pVP20 для удаления маркера устойчивости к антибиотику. Полученные мутантные штаммы и их генотипы суммированы в таблице 11 ниже.
В каждый штамм с делецией была введена pCL-Prmf-serA*(G336V)-(RBS)serC, сконструированная в примере 8, способом, сходным со способом примера 10.
Пример 13: Анализ способности штамма E. coli с недостаточностью щелочной фосфатазы (phoA), кислой фосфатазы (aphA) разрушать O-фосфосерин
Полученные в примере 12 штаммы проверяли на предмет продукции OPS и неспособность к разрушению OPS. Каждый штамм наносили на чашки LB или чашки LB(спектиномицин) и инкубировали в течение ночи при 33°С. Затем появившиеся на чашках LB или чашках LB(спектиномицин) колонии инокулировали с помощью платиновой петли в 25 мл среды для титрования, показанной в таблице 8, и культивировали при 33°С в течение 72 часов со встряхиванием при 200 об/мин. Результаты суммированы в таблице 12 ниже. Неспособность к разрушению OPS оценивали по изменению уровня иона фосфата, определяемого с помощью анализа иона фосфата.
Как видно из таблицы 12, штамм с делецией aphA проявлял феномен аномального роста, тогда как штаммы, лишенные phoA или обоих phoA и aphA, проявляли в некоторой мере повышенную продукцию O-фосфосерина и пониженную способность разрушать O-фосфосерин. С другой стороны, штамм, в котором ни phoA, ни aphA не были удалены, разрушал O-фосфосерин, накопленный за 72 часа, при одновременном росте уровня PO4.
Пример 14: Получение штаммов с недостаточностью оперона phnCDE, phoA и aphA
Штамм с недостаточностью serB (CA07-0012) модифицировали путем дополнительной делеции phnCDE, кодирующего PhnC/PhnD/PhnE алкилфосфонатный ABC-транспортер, кодирующего щелочную фосфатазу phoA и кодирующего кислую фосфатазу aphA. Полученные таким образом штаммы представлены в таблице 13 ниже. Для получения мутантов с делециями использовали одностадийный метод инактивации, описанный в примере 5.
В каждый штамм с делецией была введена pCL-Prmf-serA*(G336V)-(RBS)serC, сконструированная в примере 8, способом, сходным со способом примера 10.
Пример 15: Анализ продукции O-фосфосерина штаммами E. coli с недостаточностью phnCDE оперона, phoA и aphA
Штаммы, полученные в примере 14, оценивали на предмет продукции OPS. Каждый штамм наносили на чашки LB или чашки LB(спектиномицин) и инкубировали в течение ночи при 33°С. Затем появившиеся на чашках LB или чашках LB(спектиномицин) колонии инокулировали с помощью платиновой петли в 25 мл среды для титрования, показанной в таблице 8, и культивировали при 33°С в течение 72 часов со встряхиванием при 200 об/мин. Результаты суммированы в таблице 14 ниже.
Было обнаружено, что CA07-0020 и CA07-0022 обладают повышенной продукцией OPS и пониженной способностью к разрушению O-фосфосерина по сравнению с CA07-0012. Это свойство также было обнаружено у штаммов, дополнительно трансформированных pCL-Prmf-serA*(G336V)-(RBS)serC.
Пример 16: Получение мутантов E. coli с недостаточностью оперона phnCDE и генов phoA и aphA и содержащих замену фосфоглицератдегидрогеназы (serA*)
В CA07-0022 кодирующий 3-фосфоглицератдегидрогеназу serA был заменен на хромосоме serA*(G336V), serA*(G336V, G337V) или serA*(G336V, R338G), которые все, как сообщалось, резистентны к действию серина по механизму обратной связи, следующим образом.
Для введения мутаций в ген serA на хромосоме были сконструированы следующие праймеры. ПЦР проводили с парой праймеров SEQ ID NO: 40 и 41 на serA*(G336V), serA*(G336V, G337V) и serA*(G336V, R338G), полученных в примере 7. После совместной обработки SacI и BamHI полученные таким образом продукты ПЦР клонировали в pSG76C по сайту SacI и BamHI. Полученным рекомбинантным вектором трансформировали E. coli BW, которую затем наносили на чашки LB. Появившиеся на чашках колонии подвергали секвенированию оснований и отбирали трансформанты, в которые были введены мутации. Из них были выделены плазмиды с помощью обычного минипрепаративного метода. В соответствии с введенными мутациями плазмиды были названы pSG76C-serA*(G336V), pSG76C-serA*(G336V, G337V) и pSG76C-serA*(G336V, R338G).
Каждый из мутантов E. coli получали, как описано ранее (Posfai G, Kolisnychenko V, Bereczki Z and Blattner FR, Nucleic Acids Res. 27: 4409-4415, 1999), и из них удаляли маркерный ген устойчивости к антибиотику. Для получения мутанта serA*(G335V) в компетентные клетки CA07-0022 с помощью электропорации вводили pSG76C-serA*(G336V). Штаммы, устойчивые к хлорамфениколу, были подвергнуты ПЦР для подтверждения введения serA*(G336V). Штамм трансформировали pST76-ASceP (Posfai G, Kolisnychenko V, Bereczki Z and Blattner FR, Nucleic Acids Res. 27: 4409-4415, 1999) для удаления маркерного гена устойчивости к антибиотику. Полученный штамм был назван CA07-0022 serA*(G336V). Штамм CA07-0022 serA*(G336V) трансформировали pSG76C-serA*(G336V, G337V) и pSG76C-serA*(G336V, R338G) сходным образом с получением мутантов serA*(G336V, G337V) и serA*(G336V, R338G), названных CA07-0022 serA*(G336V, G337V) и serA*(G336V, R338G) соответственно.
Пример 17: Анализ продукции O-фосфосерина мутантами E. coli с недостаточностью phnCDE оперона, генов aphA и aphA и содержащими замену фосфоглицератдегидрогеназы (serA*)
Штаммы, полученные в примере 16, оценивали на предмет продукции O-фосфосерина. Каждый штамм наносили на чашки LB или чашки LB(спектиномицин) и инкубировали в течение ночи при 33°С. Затем появившиеся на чашках LB или чашках LB(спектиномицин) колонии инокулировали с помощью платиновой петли в 25 мл среды для титрования, показанной в таблице 8, и культивировали при 33°С в течение 48 часов со встряхиванием при 200 об/мин. Результаты суммированы в таблице 15 ниже.
Штаммы, в которых serA был заменен генами, резистентными к действию серина по механизму обратной связи, проявляли несколько пониженную скорость роста, но повышенную продукцию O-фосфосерина.
Пример 18: Получение мутантных штаммов E. Coli, с недостаточностью оперона phnCDE, phoA и aphA и содержащих замененную фосфоглицератдегидрогеназу (serA*) и повышенную 3-фосфосеринаминотрансферазу, и анализ продукции O-фосфосерина
В штаммы, полученные в примере 16, то есть CA07-0022 serA*(G336V), CA07-0022 serA*(G336V, G337V) и CA07-0022 serA* (G336V, R338G), была введена плазмида, полученная в примере 8, то есть pCL-Prmf-serC. Полученные мутанты оценивали на предмет продукции O-фосфосерина таким же образом, что и в примере 9. Результаты суммированы в таблице 16 ниже.
Как видно из таблицы 16, активированные serC штаммы проявляли улучшенную продукцию O-фосфосерина. Этот феномен был даже более выражен в случае штамма, в котором serA был модифицирован в ген, резистентный к действию серина по механизму обратной связи.
Пример 19: Штамм с повышенной трансгидрогеназой пиридиновых нуклеотидов (PntAB) и конструирование вектора, содержащего глутаматдегидрогеназу (GdhA)
Для получения штамма, в котором повышен pntAB, кодирующий трансгидрогеназу пиридиновых нуклеотидов, промотор pntAB был заменен промотором trc с помощью мутантной системы loxP (Arakawa H et al., BMC Biotechnol. 1: 7, 2001). Для этого проводили ПЦР на плазмиде pmlox-trc(ref) с использованием пары праймеров SEQ ID NO: 59 и 60, и полученный таким образом продукт ПЦР вводили с помощью электропорации в компетентные клетки CA07-0022 serA*(G336V), содержащие pKD46. Трансформанты, проявившие устойчивость к хлорамфениколу, были подвергнуты ПЦР для подтверждения замены промотора и затем трансформированы pJW168 (Le Borgne S et al., Methods Mol Biol. 267: 135-43, 2004) для удаления маркерного гена устойчивости к антибиотику. Полученный штамм был назван CA07-0022 serA*(G336V) P(trc)-pntAB. Праймеры, использованные для ПЦР, были сконструированы на основании информации о гене K12 W3110 (GenBank номер регистрации AP002223, AP002224) и прилегающих нуклеотидных последовательностях, зарегистрированных в NHI GenBank.
Ген gdhA, кодирующий глутаматдегидрогеназу, амплифицировали с помощью ПЦР с парой праймеров SEQ ID NO: 61 и 62 с получением единственного полинуклеотида. Оба праймера SEQ ID NO: 61 и 62 содержат сайт рестрикции ферментом HindIII. Праймеры были сконструированы на основании информации о гене K12 W3110 (GenBank номер регистрации AP 002380) и прилегающих нуклеотидных последовательностях, зарегистрированных в NHI GenBank.
ПЦР начинали с денатурации при 94°С в течение 3 мин и продолжали 25 циклами денатурации при 94°С в течение 30 сек, отжига при 56°С в течение 30 сек и элонгации при 72°С в течение 2 мин с последующей элонгацией при 72°С в течение 7 мин. В результате был получен полинуклеотид длиной 1714 п.н. После обработки HindIII продукт ПЦР клонировали в сайт HindIII pCC1BAC и вводили в E. coli DH5σ, которую затем наносили на чашки LB. Секвенирование оснований позволило провести отбор развившихся колоний, которые не содержали мутаций в их гене gdhA. Была выделена плазмида с помощью обычного минипрепаративного метода и названа pCC1BAC-P(native)-gdhA.
Пример 20: Введение pntAB и gdhA в продуцирующий OPS штамм и анализ продукции OPS
Для получения продуцирующего OPS штамма, в котором pntAB и gdhA были повышены, штамм CA07-0022 serA*(G336V) или штамм CA07-0022 serA*(G336V) P(trc)-pntAB трансформировали pCL-P(trc)-serA*(G336V)-serC и pCCIBAC-P(native)-gdhA по отдельности или в сочетании, как показано в следующей таблице. Каждый трансформант инкубировали в течение ночи при 33°С на чашках LB. Колонии инокулировали с помощью платиновой петли в 25 мл среды для титрования, показанной в таблице 8, и культивировали при 33°С в течение 48 часов со встряхиванием при 200 об/мин.
Как видно из таблицы 17, штамм был улучшен в отношении продукции O-фосфосерина, когда в нем была повышена pntAB. Повышение и pntAB, и gdhA вызывало большее увеличение продукции O-фосфосерина по сравнению с контролем. Из этого понятно, что pntAB и gdhA играют важную роль в продукции OPS.
Пример 21: конструирование векторов, несущих гены E. coli, кодирующие белок оттока O-ацетилсерина/цистеина (ydeD), пермеазу оттока O-ацетилсерина/цистеина (yfiK), белок оттока гомосерина/гомосеринлактона (rhtB), белок оттока треонина/гомосерина (rhtC), транспортер арсенита/антимонита (asrB) и субъединицу транспортера лейцина/изолейцина/валина (livHM)
Для выделения продуцированного O-фосфосерина необходим подходящий фактор экспорта, ни об одном из которых ранее не сообщалось. В этом контексте из множества ранее описанных генов транспортеров было отобрано шесть генов, то есть кодирующий белок оттока O-ацетилсерина/цистеина ydeD, кодирующий пермеазу оттока O-ацетилсерина/цистеина yfiK (Franke I, Resch A, Dassler T, Maier T and Bock A, J. Bacteriology, 185: 1161-166, 2003), кодирующий белок оттока гомосерина/гомосеринлактона rhtB, кодирующий белок оттока треонина/гомосерина RhtC, кодирующий транспортер арсенита/антимонита asrB и кодирующий субъединицу транспортера лейцина/изолейцина/валина (livHM), которые были клонированы и оценены.
Каждый ген был получен путем проведения ПЦР на геномной ДНК E. coli W3110 с парой праймеров SEQ ID NO: 63 и 64 для ydeD, с парой праймеров SEQ ID NO: 65 и 66 для yfiK, с парой праймеров SEQ ID NO: 67 и 68 для rhtB, с парой праймеров SEQ ID NO: 69 и 70 для rhtC, с парой праймеров SEQ ID NO: 71 и 72 для asrB и с парой праймеров SEQ ID NO: 73 и 74 для livHM. После обработки EcoRV и HindIII каждый из полученных таким образом продуктов ПЦР клонировали по сайту EcoRV и HindIII в pCL-Prmf-GFP с получением рекомбинантных векторов, названных pCL-Prmf-ydeD, pCL-Prmf-yfiK, pCL-Prmf-rhtB, pCL-Prmf-rhtC, pCL-Prmf-arsB и pCL-Prmf-livHM.
Пример 22: Введение векторов, несущих гены, кодирующие YdeD, YfiK, RhtB, RhtC, AsrB, livHM E. coli, в продуцирующий O-фосфосерин штамм и анализ продукции O-фосфосерина
Штамм CA07-0022 serA*(G336V) трансформировали шестью плазмидами, сконструированными в примере 21 и оценивали продукцию O-фосфосерина таким же образом, как и в примере 9. Результаты представлены в таблице 18 ниже.
Как показано в таблице 18, штаммы, трансформированные ydeD, mdtG или livHM, проявляли пониженную скорость роста и сниженную продукцию OPS, тогда как трансформация yfiK, rhtB или rhtC повышала скорость роста и продукцию OPS (таблица 18).
Пример 23: Получение штамма с недостаточностью фосфоглицератмутазы (gpmI, gpmA и gpmB)
gpmI, gpmA и gpmB, каждый из которых кодирует фосфоглицератмутазу, были удалены из CA07-0022 serA*(G336V) по отдельности или в сочетании с получением мутантных штаммов, названных CA07-0022 serA*(G336V)ДgpmI, CA07-0022 serA*(G336V)ДgpmA, CA07-0022 serA*(G336V)ДgpmB, CA07-0022 serA*(G336V)ДgpmIДgpmA, CA07-0022 serA*(G336V)ДgpmAДgpmB и CA07-0022 serA*(G336V)ДgpmIДgpmAДgpmB соответственно. Штаммы с делециями gpmA и gpmB были получены способом, сходным со способом примера 5, с использованием пары праймеров SEQ ID NO: 75 и 76 для gpmA и пары праймеров SEQ ID NO: 81 и 82 для gpmB. Для конструирования штамма с делецией gpmI, как описано в примере 16, мутация gpmI, содержащая стоп-кодон, была введена с помощью pSG76C. Мутант gpmI, содержащий стоп-кодон, был амплифицирован с помощью сшивающей ПЦР с использованием праймеров SEQ ID NO: с 77 по 81 на геномной ДНК K12 W3110, служившей в качестве матрицы, и клонирован в pSG76 по сайту SacI/BamHI.
Пример 24: Анализ продукции OPS штаммами с недостаточностью gpmI, gpmA и gpmB
Штаммы, полученные в примере 23, оценивали на предмет продукции OPS таким же образом, что и в примере 9. Результаты суммированы в таблице 19 ниже.
Как можно видеть из таблицы 19, когда каждый из gpmI, gpmA и gpmB был удален, а другие нет, потребление сахара мутантными штаммами снижалось, но продукция ими OPS возрастала по сравнению с исходным штаммом. В частности, штамм, лишенный и gpmA, и gpmB, обладал сходным потреблением сахара, но повышенной продукцией OPS по сравнению со штаммами, лишенными либо gpmA, либо gpmB. Таким образом, понятно, что делеция gpmI, gpmA и gpmB обеспечивает продукцию большего количества 3-фосфоглицерата, предшественника OPS, что тем самым ведет к повышенной продукции OPS.
Пример 25: Получение штаммов с недостаточностью 2-амино-3-кетобутират-CoA-лигазы (kbl), L-сериндезаминазы I (sdaA)
Ген kbl, кодирующий 2-амино-3-кетобутират-CoA-лигазу, и ген sdaA, кодирующий L-сериндезаминазу I, были удалены из CA07-0022 serA*(G336V) с получением CA07-0022 serA*(G336V)Дkbl и CA07-0022 serA*(G336V)ДsdaA соответственно. Мутантные штаммы с делециями kbl и sdaA были получены способом, сходным со способом примера 5, с использованием пары праймеров SEQ ID NO: 83 и 84 для kbl и пары праймеров SEQ ID NO: 85 и 86 для sdaA.
Пример 26: Анализ ОП и потребления сахара штаммами с недостаточностью kbl/sdaA в зависимости от концентрации глицина
Штаммы, полученные в примере 25, оценивали на предмет ОП, потребления сахара и продукции O-фосфосерина при их инкубации в условиях той же среды, которая описана в таблице 8 примера 9, за исключением того, что глицин использовали в количестве от 0 до 2,5 г/л.
Как можно видеть из таблицы 20, ОП и скорость потребления сахара во всех трех штаммах возрастала при повышении уровня глицина в среде. В частности, штамм с делецией sdaA проявлял значительное повышение ОП и скорости потребления сахара при концентрации глицина 1 г/л. Продукция OPS штаммом с делецией kbl улучшалась в присутствии 2,5 г/л глицина.
Пример 27: Получение штамма с недостаточностью iclR
Транскрипционный фактор iclR был удален из CA07-0022 serA*(G336V) с получением CA07-0022 serA*(G336V)ДiclR. Мутантный штамм с делецией был получен с помощью одностадийного метода инактивации, как в примере 5, и маркерный ген устойчивости к антибиотику был удален. Для получения штамма с делецией iclR проводили ПЦР с парой праймеров SEQ ID NO: 87 и 88.
Пример 28: Анализ продукции OPS штаммом с недостаточностью iclR
Штамм, полученный в примере 27, оценивали на предмет продукции OPS тем же способом, что и в примере 9.
Как видно из данных таблицы 21, было обнаружено, что продукция OPS штаммом с делецией iclR увеличена.
Пример 29: Конструирование векторов, несущих ацетил-CoA-синтазу (asc), мономер пируватоксидазы (poxB), ацетаткиназу (ackA) и фосфатацетилтрансферазу (pta) E. coli
Для повышения продукции и повторного использования ацетата в продуцирующем O-фосфосерин штамме были сконструированы экспрессионные плазмиды, несущие кодирующий ацетил-CoA-синтазу asc, кодирующий мономер пируватоксидазы poxB, кодирующий ацетаткиназу ackA и кодирующий фосфатацетилтрансферазу pta соответственно.
Каждый ген был получен путем проведения ПЦР на геномной ДНК E. coli W3110 с парой праймеров SEQ ID NO: 89 и 90 для acs, с парой праймеров SEQ ID NO: 91 и 92 для poxB и с парой праймеров SEQ ID NO: 93 и 94 для ackA и pta. После обработки HindIII каждый из продуктов ПЦР клонировали по сайту EcoRV и HindIII в вектор pCL-Prmf-GFP, сконструированный путем вставки промотора rmf E. coli в pCL1920, с получением в результате pCL-Prmf-acs, pCL-Prmf-poxB и pCL-Prmf-ackA-pta. Затем эти плазмиды обрабатывали EcoRI с получением вставок ДНК, то есть Prmf-acs, Prmf-poxB и Prmf-ackA-pta, которые затем вставляли в вектор pCC1BAC (EcoRI) (CopyControlTM pcc1BACTM, Epicentre, кат. № CBAC311) для конструирования pCC1BAC-Prmf-acs, pCC1BAC-Prmf-poxB и pCC1BAC-Prmf-ackA-pta соответственно.
Пример 30: Получение продуцирующего OPS штамма E.coli с повышенными acs, poxB, ackA, pta и анализ продукции OPS
Штамм The CA07-0022 serA*(G336V) трансформировали тремя векторами, полученными в примере 29, и анализировали на предмет продукции OPS тем же способом, что и способ примера 9.
Как можно видеть из таблицы 22, скорость роста штамма, трансформированного poxB, снижалась, тогда как введение acs или ackA-pta повышало скорость роста и продукцию OPS.
Пример 31: Конструирование векторов, несущих малатсинтазу A (aceB), мономер изоцитратлиазы (aceA), фосфоенолпируваткарбоксикиназу (pckA), малатсинтазу G (glcB) и малатдегидрогеназу (maeB) E. coli
Были сконструированы плазмиды, которые обеспечивают экспрессию в E. coli и aceB, кодирующего малатсинтазу A, и aceA, кодирующего мономер изоцитратлиазы, pckA, кодирующего фосфоенолпируваткарбоксикиназу, glcB, кодирующего малатсинтазу G, и maeB, кодирующего малатдегидрогеназу соответственно.
Гены получали путем проведения ПЦР на геномной ДНК E. coli W3110 с парой праймеров SEQ ID NO: 95 и 96 для aceBA, с парой праймеров SEQ ID NO: 97 и 98 для pckA, с парой праймеров SEQ ID NO: 99 и 100 для glcB и с парой праймеров SEQ ID NO: 101 и 102 для maeB. После обработки HindIII каждый из полученных таким образом продуктов ПЦР клонировали по сайту EcoRV и HindIII в вектор pCL-Prmf-GFP, сконструированный путем вставки промотора rmf E. coli в pCL1920, с получением в результате pCL-Prmf-aceBA, pCL-Prmf-pckA, pCL-Prmf-glcB и pCL-Prmf-maeB.
Пример 32: Получение продуцирующего OPS штамма E. coli с повышенными aceB, aceA, pckA, glcB и maeB и анализ продукции O-фосфосерина
Штамм CA07-0022 serA*(G336V) трансформировали четырьмя векторами, полученными в примере 31, и оценивали на предмет продукции O-фосфосерина тем же способом, что и способ примера 9.
Как можно видеть из таблицы 23, скорость потребления сахара и продукция OPS штаммом несколько снижалась после трансформации aceBA и скорость роста значительно снижалась после трансформации pckA, тогда как введение glcB или maeB повышало продукцию OPS.
Пример 33: Конструирование векторов, несущих глиоксалаткарболигазу (glc), тартронатсемиальдегидредуктазу 2 (glxR) и глицераткиназу II (glxK)
Кодирующий глиоксалаткарболигазу glc, кодирующий тартронатсемиальдегидредуктазу 2 glxR и кодирующий глицераткиназу II glxK, все из которых участвуют в превращении глиоксилата в 3-фосфоглицерат, клонировали следующим образом. Гены были получены путем проведения ПЦР на геномной ДНК E. coli W3110 с парой праймеров SEQ ID NO: 103 и 104 для gcl и с парами праймеров SEQ ID NO: с 105 по 108 для glxR-glxK. После гидролиза EcoRV и HindIII каждый из продуктов ПЦР клонировали по сайтам EcoRV и HindIII в вектор pCL-Prmf-GFP, сконструированный путем вставки промотора rmf E. coli в pCL1920, с получением рекомбинантных плазмид, названных pCL-Prmf-gcl, pCL-Prmf-glxR-glxK и pCL-Prmf-glxR-glxK-Prmf-gcl соответственно.
Пример 34: Введение векторов, несущих glc, glxR, glxK, в продуцирующий O-фосфосерин штамм и анализ продукции O-фосфосерина
Три плазмиды, сконструированные в примере 33, были введены в CA07-0022 serA*(G336V), который затем оценивали на предмет продукции O-фосфосерина таким же образом, что и в примере 9. Результаты суммированы в таблице 24 ниже.
Как можно видеть из таблицы 24, конечная продукция O-фосфосерина штаммами, трансформированными соответственно gcl, glxR-glxK и glxR-glxK-gcl, была снижена, но скорость роста и потребление сахара были повышены по сравнению с самим штаммом CA07-0022 serA*(G336V). В частности, было обнаружено, что введение glxR-glxK вызывает наибольшее повышение скорости роста и скорости потребления сахара.
Пример 35: Оценка продуцирующего O-фосфосерин штамма в ферментере
Штаммы CA07-0022 serA*(G336V)/pCL-Prmf-serA*(G336V)-serC инкубировали при 33°С в течение 24 часов на агаровых чашках MMYE (2 г/л глюкозы, 2 мМ сульфата магния, 0,1 мМ хлорида кальция, 6 г/л пирофосфата натрия, 0,5 г/л хлорида натрия, 3 г/л дигидрофосфата калия, 10 г/л дрожжевого экстракта, 18 г/л агара), содержащих 50 мкг/мл спектиномицина. Полученные колонии соскабливали с 1/10 площади каждой агаровой чашки, инокулировали в среду для посева, содержащую 50 мкг/мл спектиномицина, 10 г/л L-глюкозы, 0,5 г/л сульфата магния, 3 г/л дигидрофосфата калия, 10 г/л дрожжевого экстракта, 0,5 г/л хлорида натрия, 1,5 г/л хлорида аммония, 12,8 г/л пирофосфата натрия, 1 г/л глицина), в сосуде с перегородкой и инкубировали при 30°С в течение шести часов со встряхиванием при 200 об/мин. Полученную посевную культуру добавляли к 300 мл основной среды в 1-л ферментере в количестве, составляющем 16% от объема основной среды и затем инкубировали при 33°С и pH 7,0. Состав основной среды представлен в таблице 25 ниже.
Во время инкубации pH культуральной среды доводили до 7,0 аммиачной водой. После истощения глюкозы в культуральной среде проводили ферментацию с подпиткой партии путем добавления раствора глюкозы 520 г/л. После ферментации в течение 80 часов O-фосфосерин был продуцирован в концентрации 19,5 г/л по данным измерения с помощью ВЭЖХ.
Усовершенствование и характеристика сульфгидрилазы O-фосфосерина (OPS) (OPSS)
Пример 36: Усовершенствование сульфгидрилазы OPS (OPSS)
Сообщалось, что Aeropyrum pernix, Mycobacterium tuberculosis и Trichomonas vaginalis содержат сульфгидрилазу O-фосфосерина (OPSS), фермент, который использует O-фосфо-L-серин (OPS) вместо O-ацетилсерина (OAS) в E. coli в качестве субстрата для синтеза цистеина (Mino K and Ishikawa K, FEBS letters, 551: 133-138, 2003; Burns KE, Baumgart S, Dorrestein PC, Zhai H, McLafferty FW and Begley TP, J. Am. Chem. Soc., 127: 11602-11603, 2005; Westrop GD, Goodall G, Mottram JC and Coombs GH, J. Biol. Chem., 281: 25062-25075, 2006). На основании этих сообщений авторы настоящего изобретения обнаружили два типа сульфгидрилазы OPS, которые превращают OPS в цистеин, в Aeropyrum pernix и Mycobacterium tuberculosis H37Rv. Из них фермент OPSS из Mycobacterium tuberculosis H37Rv был использован для скрининга аминокислотной гомологии. В результате было получено три типа OPSS из Mycobacterium smegmatis str. MC2 155, Rhodococcus jostii RHA1 и Nocardia farcinica IFM 10152.
Для получения OPSS из каждого штамма была сконструирована векторная система pET28a (Novagen), которая обычно используется для экспрессии ферментов. Все матрицы и праймеры, использованные для клонирования пяти разных генов сульфгидрилазы OPS, и полученные рекомбинантные плазмиды суммированы в таблице 26 ниже. Подходящие сочетания матриц и праймеров, как представлено в таблице 26, были использованы для ПЦР для амплификации соответствующих генов OPSS. Продукты ПЦР и вектор pET28a гидролизовали NdeI и HindIII (37°С в течение 3 часов). Каждый из фрагментов генов лигировали с гидролизованным вектором pET28a (Novagen). Секвенирование оснований подтвердило конструирование экспрессионных векторов, несущих каждый из генов OPSS. Экспрессионные векторы фермента были введены в E. coli (DE3) с получением штаммов, способных экспрессировать ферменты OPSS. Наименования ферментов представлены в таблице 26 ниже.
Экспрессию ферментов осуществляли в соответствии с инструкциями производителя системы pET (Novagen). Одиночные колонии каждого штамма с чашек LB инокулировали в 5 мл бульона LB и инкубировали при 37°С в течение 16 часов со встряхиванием при 200 об/мин. Культуры переносили в 25 мл свежего бульона LB (в 250-мл флаконах) и инкубировали до ОП600, равной 0,5-0,6 (в течение 2-3 часов), в тех же условиях, сразу после чего к среде добавляли 1 мМ IPTG для индукции подлежащих экспрессии ферментов во время инкубации при 18°С в течение 18 часов со встряхиванием при 200 об/мин. Ферменты очищали с использованием колонок Ni-NTA для His-tag с помощью His SpinTrap (GE Healthcare). Из пяти выделенных таким образом ферментов OPSS четыре были обнаружены в растворимой форме, а один (Rjo-OPSS) находился в тельцах включений по данным анализа с помощью 14% SDS-ПААГ-электрофореза.
Пример 37: Анализ сульфгидрилазы OPS (OPSS) на предмет активности в синтезе цистеина
Ферменты сульфгидрилазы OPS, полученные из четырех штаммов микроорганизмов, анализировали на способность катализировать превращение O-фосфосерина (OPS) в цистеин. В плане условий и методов анализа (ферментативного анализа cysM) дается ссылка на предшествующие сообщения (Mino K and Ishikawa K, FEBS letters, 551: 133-138, 2003; Burns KE, Baumgart S, Dorrestein PC, Zhai H, McLafferty FW and Begley TP, J. Am. Chem. Soc., 127: 11602-11603, 2005; Westrop GD, Goodall G, Mottram JC and Coombs GH, J. Biol. Chem., 281: 25062-25075, 2006). Количество использованного субстрата представлено в единицах на мл. Условия анализа ферментативной активности суммированы в таблице 27 ниже.
Растворы для реакции, исключая ферменты, инкубировали при 37°С в течение 5 мин, после чего к раствору для реакции добавляли 50 мг очищенной сульфгидрилазы OPS. В заранее определенные моменты времени инкубации при 37°С отбирали 100 мл раствора ферментативной реакции и смешивали с 100 мл 33,2% ТХУ для остановки ферментативной реакции. Концентрации цистеина в растворах ферментативной реакции количественно анализировали путем измерения поглощения ОП560 согласно методу Гейтонд. Активность в отношении синтеза цистеина четырех разных ферментов сульфгидрилазы OPS суммирована в таблице 28 ниже. Титры синтеза цистеина ферментов OPSS выражены в виде степени превращения в цистеин за время реакции.
Было подтверждено, что OPS-сульфгидрилазные ферменты из Aeropyrum pernix и Mycobacterium tuberculosis H37Rv, о которых сообщалось ранее (Mino K and Ishikawa K, FEBS letters, 551: 133-138, 2003; Westrop GD, Goodall G, Mottram JC and Coombs GH, J. Biol. Chem., 281: 25062-25075, 2006), обладают способностью использовать OPS в качестве субстрата для синтеза цистеина. Впервые была обнаружена способность к синтезу цистеина новой сульфгидрилазы OPS из Mycobacterium smegmatis str. MC2 155, которая была получена путем скрининга аминокислотной гомологии с ферментом Mtb-OPSS. Как видно из данных таблицы 28, степень превращения OPS в цистеин Ape-OPSS достигает почти 100% за один час. Конечная степень превращения ферментом Msm-OPSS, который был впервые отобран посредством скрининга фермента на основе OPSS из Mycobacterium tuberculosis H37Rv, о которой сообщалось ранее, составляла 43,7%, что в 4,3 раза превышало степень превращения Mtb-OPSS. С другой стороны, новая сульфгидрилаза OPS из Nocardia farcinica IFM 10152, полученная скринингом гомологии, проявляла несущественную активность в отношении превращения O-фосфосерина в цистеин.
Пример 38: Получение Mtb-T и Msm-T, которые кодируют укороченные на 5 C-концевых аминокислотных остатков Mtb-OPSS и Msm-OPSS
Сообщалось, что OPSS из Mycobacterium tuberculosis H37Rv (Mtb-OPSS), которая катализирует превращение OPS в цистеин с помощью дополнительных ферментов mec+ и cysO, способна использовать источник серы, содержащий S2-, для превращения OPS в цистеин даже в отсутствие дополнительных ферментов, когда из нее удалены пять C-концевых аминокислотных остатков (Agren D, Schnell R and Schneider G, FEBS letters, 583: 330-336, 2009). На основании этого сообщения была получена Mtb-T (SEQ ID NO: 11), которая может быстро превращать OPS в присутствии S2- в качестве источника серы. Была также получена Msm-T из Msm-OPSS (SEQ ID NO: 9), которая имеет аминокислотную гомологию с Mtb-OPSS. Были сконструированы экспрессионные векторы, несущие два мутантных фермента. Для этого проводили ПЦР с pfu на геномной ДНК Mycobacterium tuberculosis H37Rv или Mycobacterium smegmatis в присутствии пары праймеров SEQ ID NO: 119, 120, 121 и 122. Полученные таким образом фрагменты генов OPSS обрабатывали NdeI и HindIII и клонировали в вектор pET28a, гидролизованный теми же ферментами рестрикции, для конструирования рекомбинантных экспрессионных векторов, названных pET28a-Mtb-T и pET28a-Msm-T соответственно. Рекомбинантные экспрессионные векторы были введены в E. coli (DE3). Экспрессия двух мутантных ферментов OPSS была подтверждена с помощью 14% SDS-ПАГЭ. Два мутантных фермента OPSS были очищены и экспрессированы в тех же условиях, что и в примере 36. В результате были получены Mtb-T (SEQ ID NO: 11) и Msm-T (SEQ ID NO: 10).
Пример 39: Анализ Mtb-T и Msm-T на предмет способности к образованию цистеина
На основании сообщения о том, что мутанты OPSS из Mycobacterium tuberculosis H37Rv, лишенные пяти C-концевых аминокислотных остатков, проявляют повышенное сродство к содержащему группу S2- источнику серы в отсутствие вспомогательных ферментов (Agren D, Schnell R and Schneider G, FEBS letters, 583: 330-336, 2009), были получены Mtb-T и Msm-T. Их ферментативную активность оценивали путем измерения конечной степени образования цистеина. Ферментативную активность анализировали в тех же условиях и таким же образом, что и в примере 17. Продуцированный цистеин количественно определяли методом Гейтонд.
Как видно из таблицы 29, Msm-T, лишенный пяти C-концевых аминокислотных остатков OPSS из Mycobacterium smegmatis str. MC2 155, обеспечивал степень образования цистеина из субстрата, равную 100% за один час.
При модификации аминокислотной последовательности сульфгидрилаза O-фосфосерина (OPSS) может более эффективно катализировать биосинтез L-цистеина.
Пример 40: Потребность в кофакторе для активности сульфгидрилазы OPS
Для проверки влияния кофакторов на образование цистеина под действием OPSS степень превращения в цистеин под действием Msm-T измеряли в отсутствие и в присутствии PLP (пиридоксаль-5'-фосфата) и DTT (дитиотреитола). Для этого субстраты в бульоне 50 мМ OPS и 100 мМ Na2S вводили во взаимодействие при 37°С в течение 30 мин в присутствии 25 мМ DTT или 0,2 мМ PLP. Продуцированный таким образом цистеин количественно определяли методом Гейтонд. Как видно из таблицы 30, степень превращения в цистеин в присутствии и PLP, и DTT была в 2,3 раза выше, чем в отсутствие и PLP, и DTT. Таким образом, наблюдалось положительное действие и PLP, и DTT на превращение.
Пример 41: Влияние температуры на активность сульфгидрилазы OPS
Была оценена степень превращения в цистеин под действием Ape-OPSS и Msm-T в зависимости от температуры. Ферментативную активность при 37°С и 60°С измеряли через 2, 5, 10, 30 и 60 мин прохождения реакции. Реакцию проводили в условиях 100 мМ HEPES (pH 7,4), 5 мМ OPS, 10 мМ Na2S, 0,2 мМ PLP и 50 мкг/мл CysM. Количество продуцированного цистеина определяли методом Гейтонд. В условиях буфера, как показано на фиг.2, Ape-OPSS проявляла более высокую начальную скорость реакции при 37°С, а также большую активность при 60°С по сравнению с Msm-T.
Пример 42: Температурная стабильность сульфгидрилазы OPS
Была произведена оценка термостабильности Ape-OPSS и Msm-T. Каждый из ферментов разбавляли до концентрации 2 мг/мл в бульоне OPS и обрабатывали нагреванием при 37°С и 60°С в течение 10, 30, 60, 120 и 240 мин с последующим проведением реакции при 37°С в течение 30 мин в условиях 5 мМ OPS, 10 мМ Na2S, 0,2 мМ PLP и 100 мМ HEPES (pH 7,4). Для этой реакции использовали 10 мкг/мл Ape-OPSS и 50 мкг/мл Msm-T. Количество продуцированного цистеина определяли методом Гейтонд. Было обнаружено, что Ape-OPSS сохраняет свою исходную активность, несмотря на термообработку при 60°С в течение 4 часов, в то время как активность Msm-T сохранялась при 37°С, но снижалась до 50% после термообработки при 60°С в течение 30 мин. Результаты представлены в таблице 31 ниже.
Была произведена оценка сохранения ферментативной активности при 37°С, когда Msm-T использовали в количестве 50 мкг/мл, представляющем собой реальную концентрацию в бульоне OPS. Обработку 50 мкг/мл Msm-T производили в отсутствие Na2S, но в присутствии 50 мМ бульона OPS и 0,2 мМ PLP при 37°C в течение 0,5, 1, 2, 4 и 6 часов, после чего добавляли Na2S для индукции ферментативной реакции. После проведения реакции в течение 30 мин измеряли активность Msm-T. Количество продуцированного цистеина определяли методом Гейтонд. В результате активность Msm-T снижалась ниже 50% через 2 часа реакции при 37°С в бульоне OPS (Таблица 32).
Пример 43: Влияние pH на сульфгидрилазу OPS
Измеряли степень превращения в цистеин под действием Ape-OPSS и Msm-T в зависимости от pH. Реакцию проводили при 37°С в течение 10 мин в 100 мМ буфере с Ape-OPSS и Msm-T, каждая в концентрации 50 мкг/мл. Для этого использовали K-фосфатный буфер с pH 6,4/7,0/7,4/8,0, трис-HCl буфер с pH 7,0/7,4/8,0/8,5/8,8 и Na-карбонатный буфер с pH 8,0/8,5/9,0/10,0. Количественный анализ продуцированного цистеина проводили методом Гейтонд. Как видно из фиг.3, Msm-T проявляла наибольшую активность при pH от 8,0 до 9,0 независимо от буфера. Что касается Ape-OPSS, то наибольшая активность была обнаружена в K-фосфате (pH 7,4) с оптимумом pH, различающемся в разных буферах.
Пример 44: Влияние ионов на активность сульфгидрилазы OPS
Влияние ионов на активность ферментов OPSS оценивали следующим образом. Ферменты вводили в реакцию при 37°С на 30 мин в реакционной смеси, содержащей 5 мМ OPS, 10 мМ Na2S, 0,2 мМ PLP и 100 мМ HEPES (pH 7,4), в присутствии (NH4)2SO4 (1, 3, 5, 10, 20 г/л), KH2PO4 (0,5, 1, 2, 4, 8 г/л) или NH4Cl (0,2, 0,5, 1, 2 г/л). Ape-OPSS и Msm-T использовали в концентрации 10 мкг/мл и 50 мкг/мл, соответственно. Количество продуцированного цистеина определяли методом Гейтонд.
Не было обнаружено изменений в степени превращения в цистеин при добавлении к реакционной смеси (NH4)2SO4 или KH2PO4. С другой стороны, как видно из таблицы 33, степень превращения в цистеин снижалась при увеличении концентрации NH4Cl. В частности, максимальная ферментативная активность снижалась до менее 70% при добавлении 2 г/л NH4Cl. Таким образом, наблюдалось негативное действие NH4Cl на превращающую активность сульфгидрилазы OPS.
Пример 45: Влияние источника серы на активность сульфгидрилазы OPS в отношении синтеза цистеина
Был проведен эксперимент для оценки влияния источников серы на активность каждого фермента в отношении синтеза цистеина. Каждый фермент (50 мкг/мл Ape-OPSS, 50 мкг/мл Msm-T) вводили в реакцию при 37°С в течение 1 часа в реакционной смеси, содержащей 5 мМ OPS, 0,2 мМ PLP и 100 мМ HEPES, в присутствии 10 мМ Na2S, NaSH или Na2S2O3. Количество продуцированного цистеина определяли методом Гейтонд. Наблюдалось, что Ape-OPSS предпочитает Na2S2O3 в качестве источника серы, а Msm-T предпочитает Na2S. Результаты суммированы в таблице 34 ниже.
Пример 46: Конструирование экспрессионного вектора, несущего сульфгидрилазу OPS (система pCL-Pcj1), и экспрессия в E. coli
ПЦР проводили с использованием праймеров SEQ ID NO: 123 и 124 на векторе pET28a-Msm-T, служившем в качестве матрицы. Полученный таким образом продукт ПЦР обрабатывали EcoRV и HindIII и клонировали в pCL-P(CJ1) для конструирования рекомбинантного вектора, названного pCL-P(CJ1)-Msm-T. Для оценки разницы в степени экспрессии Msm-T между системой pET и системой pCL-Pcj1 были получены штаммы, экспрессирующие фермент. Система pET была введена в Rosetta (DE3), тогда как для системы pCL-Pcj1 был использован штамм K12G. Одиночные колонии, собранные с чашек LB, инокулировали в 5 мл бульона LB и культивировали при 37°С в течение 16 часов со встряхиванием при 200 об/мин. Эти культуры переносили в 25 мл свежего бульона LB, содержащего канамицин или спектиномицин и 0,2% глюкозы (в 250-мл сосудах), и инкубировали до ОП600, равной 0,5-0,6, после чего к среде сразу добавляли 1 мМ IPTG для индукции подлежащих экспрессии ферментов. Во время инкубации при 37°С со встряхиванием при 200 об/мин измеряли уровни экспрессии фермента через разные промежутки времени культивирования (8, 16, 24 часа). Уровни экспрессии фермента в двух системах анализировали на 14% SDS-ПАГЭ (фиг.4).
Пример 47: Синтез цистеина сульфгидрилазой OPS с очищенным бульоном для ферментации OPS
Были определены скорости превращения очищенного OPS в цистеин под действием Msm-T и Ape-OPSS. В присутствии 75 мкг/мл каждого из ферментов и 0,2 мМ PLP, 60 мМ OPS, очищенного из бульона ферментации OPS, вводили во взаимодействие с 120 мМ Na2S при 37°С или 70°С в течение 30, 60, 90 и 120 мин. Реакцию проводили только при 37°С для Msm-T, но и при 37°С, и при 70°С для Ape-OPSS. Количество продуцированного цистеина определяли методом Гейтонд. Как видно на фиг.5, очищенный из бульона ферментации OPS служил хорошим субстратом для ферментативного превращения в цистеин. В частности, скорость превращения под действием Ape-OPSS повышалась при 70°С даже при использовании очищенного из бульона ферментации OPS.
Пример 48: Синтез цистеина сульфгидрилазой OPS при использовании бульона ферментации OPS.
При использовании в качестве субстрата бульона ферментации OPS измеряли скорости превращения в цистеин под действием Msm-T и Ape-OPSS в зависимости от концентраций ферментов. В присутствии 100 мМ Na2S и 0,2 мМ PLP 50 мМ OPS бульона ферментации вводили во взаимодействие с 5 мкг/мл или 50 мкг/мл каждой из Msm-T и Ape-OPSS при 37°С. Количество продуцированного цистеина определяли методом Гейтонд. Как видно на фиг.6, наибольшая скорость превращения обнаруживалась при 50 мкг/мл Msm-T. Кроме того, при использовании бульона ферментации OPS в качестве субстрата активность Msm-T превышала активность Ape-OPSS.
Пример 49: Скорость превращения в цистеин в зависимости от концентрации OPS
Для оценки влияния концентрации OPS на скорость превращения под действием Msm-T к бульону ферментации OPS добавляли заранее определенные количества очищенного OPS для индукции реакции превращения. Фермент использовали в количестве 50 мкг. Количество цистеина в реакционном растворе определяли методом Гейтонд. Степень превращения под действием Msm-T достигала 100% при концентрации OPS около 30 г/л.
При концентрации OPS выше 50 г/л и скорость превращения, и процент превращения снижались. Из этих результатов понятно, что когда в качестве субстрата используется бульон ферментации OPS, имеется оптимальное соотношение концентраций между OPS и ферментом.
Реферат
Настоящее изобретение относится к биохимии и представляет собой способ получения цистеина, включающий культивирование рекомбинантного микроорганизма, в котором снижена активность эндогенной фосфосеринфосфатазы (SerB), для продукции О-фосфосерина (OPS) и введение во взаимодействие полученной культуры, содержащей OPS, или OPS, выделенной из культуры, с сульфидом в присутствии О-фосфосеринсульфгидрилазы (OPSS) или микроорганизма, экспрессирующего OPSS, для получения цистеина, причем рекомбинантный микроорганизм представляет собой рекомбинантную бактерию. Изобретение относится также к получению производного цистеина, согласно которому получают цистеин, как указано выше, который затем превращают в производное цистеина. Изобретение позволяет получать цистеин и его производное с высокой степенью эффективности. 3 н. и 28 з.п. ф-лы, 6 ил., 35 табл., 49 пр.
Формула
1) культивирование рекомбинантного микроорганизма, в котором снижена активность эндогенной фосфосеринфосфатазы (SerB), для продукции О-фосфосерина (OPS); и
2) введение во взаимодействие культуры со стадии 1), содержащей OPS, или OPS, выделенной из культуры со стадии 1), с сульфидом в присутствии О-фосфосеринсульфгидрилазы (OPSS) или микроорганизма, экспрессирующего OPSS, для получения цистеина,
причем рекомбинантный микроорганизм представляет собой рекомбинантную бактерию.
i) SerA представляет собой фермент, выбранный из группы, состоящей из аминокислотных последовательностей SEQ ID NO: с 3 по 7; и
ii) SerC имеет аминокислотную последовательность SEQ ID NO:
1) культивирование рекомбинантного микроорганизма, в котором снижена активность эндогенной фосфосеринфосфатазы (SerB), для продукции О-фосфосерина (OPS); и
2) введение во взаимодействие культуры со стадии 1), содержащей OPS, или OPS, выделенной из культуры со стадии 1), с сульфидом в присутствии О-фосфосеринсульфгидрилазы (OPSS) или микроорганизма, экспрессирующего OPSS, для получения цистеина; и
3) превращение цистеина, полученного на стадии 2), в производное цистеина,
где рекомбинантный микроорганизм представляет собой рекомбинантную бактерию.
Комментарии