Способ биологического производства н-бутанола - RU2461627C2
Код документа: RU2461627C2
Чертежи
Описание
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Изобретение включает способ биоконверсии ферментируемого источника углерода до н-бутанола с высоким выходом под действием микроорганизма, созданного технологией метаболической инженерии.
ПРЕДШЕСТВУЮЩИЙ ИЗОБРЕТЕНИЮ УРОВЕНЬ ТЕХНИКИ
н-Бутанол представляет собой бесцветную нейтральную жидкость средней летучести с ограниченной смешиваемостью (примерно 7-8%) с водой, но легко смешиваемую со всеми обычными растворителями, такими как гликоли, кетоны, спирт, альдегиды, простые эфиры и ароматические и алифатические углеводороды. н-Бутанол используется 1) для получения других химических веществ, 2) в качестве растворителя и 3) в качестве ингредиента в таких препаратах, как косметические средства. Основные применения н-бутанола в качестве сырья заключаются в синтезе акрилат/метакрилатных сложных эфиров, простых эфиров гликолей, н-бутилацетата, аминосодержащих смол и н-бутиламинов. В настоящее время в мире расходуется более 9 миллионов тонн н-бутанола ежегодно.
Совсем недавно было показано, что н-бутанол является более хорошим биотопливом, чем этанол, вследствие более низкой упругости паров, более высокого энергосодержания (близкого к бензину) и меньшей склонности к разделению в присутствии воды. Кроме того, н-бутанол можно смешивать в более высоких концентрациях, чем этанол, для использования в двигателях обычных транспортных средств, и его применение не заставляет автомобилестроительные предприятия идти на компромисс по вопросу соответствия природоохранительному законодательству; он также подходит для транспортировки по трубопроводам и, как результат, имеется потенциальная возможность для быстрого внедрения его вместо бензина и для того, чтобы избежать необходимости в создании дополнительных крупномасштабных поставляющих инфрастуктур.
н-Бутанол может быть получен в виде смеси ацетон/н-бутанол/этанол (ABE) в результате ферментации углевода под действием сольвентогенных клостридий. АВЕ-ферментации проходят в две фазы. В процессе первой ацидогенной фазы высокая скорость роста сопровождается продуцированием уксусной и масляной кислот. На второй сольвентогенной фазе скорость роста уменьшается, и продуцируются растворители (ABE) с сопутствующим расходованием органических кислот, полученных на первой фазе. В течение всей ферментации продуцируются двуокись углерода и водород.
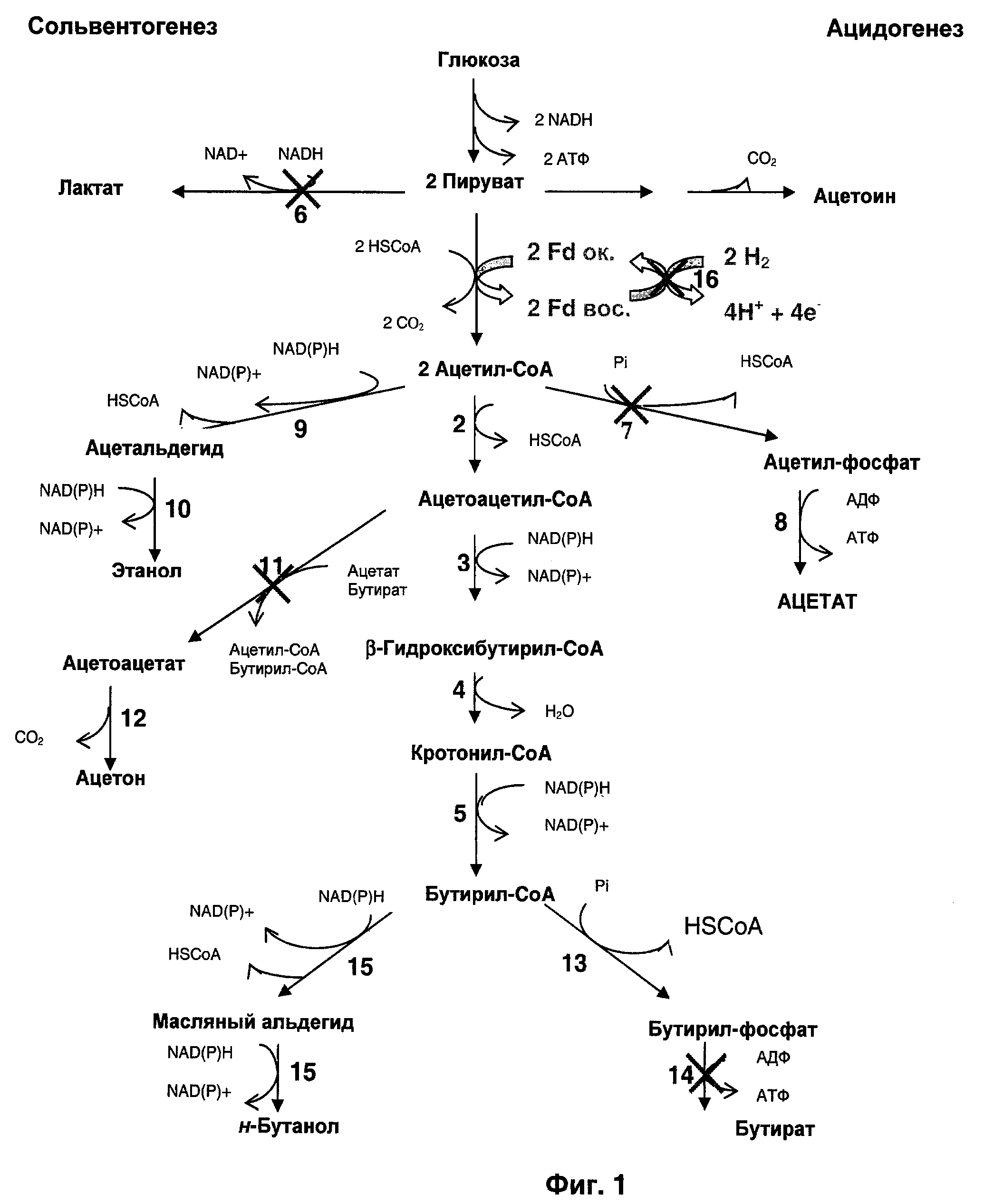
Для биологического получения н-бутанола, представленного на Фиг.1, необходимо образование бутирил-CoA в качестве промежуточного соединения, который может быть восстановлен, в зависимости от физиологических условий, под действием двух разных бифункциональных альдегид-алкогольдегидрогеназ, кодируемых генами adhE1 и adhE2. Бутирил-CoA также может быть превращен в масляную кислоту под действием фосфотрансбутирилазы и бутираткиназы, кодируемых соответственно генами ptb и buk. Ацетон получается из ацетоацетил-CoA (промежуточного соединения при получении бутирил-CoA) под действием CoA-трансферазы и ацетоацетатдекарбоксилазы, кодируемых соответственно генами ctfAB и adc. Водород продуцируется под действием содержащей только один атом железа гидрогеназы, кодируемой геном hydA. Если культивирование проводят в присутствии монооксида углерода, гидрогеназного ингибитора, основными продуктами ферментации являются н-бутанол, этанол и лактат. Лактат получается из пирувата под действием лактатдегидрогеназы, кодируемой геном Idh.
Штаммы Clostridium acetobutylicum с инактивированным геном buk (полученные единичным кроссинговером с использованием нерепликативной плазмиды) уже описаны в статье (Green et al., 1996). Нерепликативный вектор pJC4BK, содержащий внутренний buk-фрагмент длиной 0,8 т.п.н., был интегрирован в хромосомный ген buk, что приводило к инактивации эндогенного гена. Полученный штамм обозначали как "мутантный PJC4BK", исходя из названия плазмиды. Как установлено в этой статье, такая генная интеграция полностью не элиминировала ни ферментативную активность, ни образование бутирата вследствие нестабильности этого типа генной инактивации, которая могла вызывать возврат к дикому типу в результате вырезания плазмиды. Этот мутантный штамм затем был использован в нескольких исследованиях (Green и Bennett, 1998; Desai и Harris, 1999; Harris и др., 2000).
Традиционно промышленную АВЕ-ферментацию проводили только в периодическом режиме вследствие нестабильности непрерывных культур, продуцирующих Clostridia. Описано несколько способов ферментации с получением растворителей. С применением этих способов получают н-бутанол, ацетон и этанол в соотношении 6:3:1. В литературе сообщается о выходах растворителей, составляющих 29-34% (18-25% только для н-бутанола) от ферментируемого источника углерода. Суммарная концентрация растворителей 16-24 г/л и концентрация н-бутанола 10-14 г/л представляют собой, как правило, предельную величину вследствие токсичности получаемого н-бутанола. Однако, по-видимому, такие низкие титры растворителя более не являются экономическим ограничением данного способа, поскольку недавно продемонстрировано, что растворители можно извлекать в процессе ферментации посредством применения "недорогой" технологии "извлечения из газа" (gas striping).
Проблема, которая должна быть решена настоящим изобретением, заключается в получении стабильного мутантного штамма, не имеющего никакой бутираткиназной активности, который можно было бы культивировать в течение нескольких генераций без какой-либо возможности возврата к генотипу дикого типа. Этот штамм будет полезен для биологического получения н-бутанола с высоким выходом из недорогого углеродного субстрата, такого как глюкоза или другие сахара, под действием генетически стабильных культур Clostridia. Количество биохимических стадий, необходимых для инактивации, и сложность регулирования метаболизма неизбежно влекут за собой, что касается промышленно осуществимого способа продуцирования н-бутанола, необходимость применения катализатора на основе цельной клетки, созданного технологией метаболической инженерии.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Заявителем решена сформулированная проблема и согласно настоящему изобретению предложен способ биоконверсии ферментируемого источника углерода до н-бутанола в качестве основного продукта под действием генетически стабильных культур Clostridia. В качестве модельного субстрата используют глюкозу, а рекомбинантный Clostridium acetobutylicum используют в качестве модели хозяина. В одном из аспектов этого изобретения стабильный рекомбинантный С.acetobutylicum, не способный метаболизировать бутирил-CoA до бутирата, конструируют посредством делетирования гена, кодирующего бутираткиназу (buk). В другом аспекте этого изобретения рекомбинантный С.acetobutylicum, не способный продуцировать ацетон, конструируют последством делетирования генов, кодирующих CoA-трансферазу {ctfAB). В следующем аспекте этого изобретения рекомбинантный штамм, не способный продуцировать лактат, конструируют посредством делетирования гена, кодирующего лактатдегидрогеназу (Idh). Кроме того, рекомбинантный С.acetobutylicum, не способный продуцировать ацетат, конструируют посредством делетирования генов, кодирующих фосфотрансацетилазу и/или ацетаткиназу (pta и ack). В конечном аспекте этого изобретения поток продуцирования водорода уменьшают и затем поток восстановительного эквивалента (reducing equivalent) переориентируют на продуцирование н-бутанола посредством ослабления гена, кодирующего гидрогеназу (hydA).
В общем случае настоящее изобретение может включать использование любого углеродного субстрата, который легко конвертируется до ацетил-CoA.
Соответственно, задачей настоящего изобретения является разработка рекомбинантного микроорганизма, полезного для продуцирования н-бутанола, содержащего: (a) по меньшей мере делецию одного из двух генов, вовлеченных в конверсию бутирил-CoA в бутират, и (b) по меньшей мере делецию одного из двух генов, кодирующих CoA-трансферазную активность. Возможно, что рекомбинантный микроорганизм может 1) содержать инактивирующие мутации в эндогенных генах, выбранных из группы, состоящей из: (a) гена, кодирующего полипептид с лактатдегидрогеназной активностью, (b) гена, кодирующего полипептид с фосфотрансацетилазной или лактаткиназной активностью, и 2) иметь ослабление в гене, кодирующем полипептид с гидрогеназной активностью.
В другом воплощении изобретения предложен стабильный способ продуцирования н-бутанола с высоким выходом из рекомбинантного микроорганизма, включающий: (a) приведение в контакт рекомбинантного микроорганизма по настоящему изобретению по меньшей мере с одним источником углерода, выбранным из группы, состоящей из моносахаридов, олигосахаридов, полисахаридов и одноуглеродных субстратов, посредством чего продуцируется н-бутанол; возможны (b) извлечение н-бутанола в процессе продуцирования посредством стадии "извлечения из газа" и (c) очистка н-бутанола из конденсата путем дистилляции.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Сопровождающий графический материал, включенный в это описание и составляющий часть этого описания, представляет собой типичные примеры изобретения и вместе с описанием служит для разъяснения принципов этого изобретения.
На Фиг.1 изображена генетическая инженерия основного пути метаболизма при разработке системы продуцирования бутанола из углеводов.
1: пируват-ферредоксин-оксидоредуктаза; 2: тиолаза; 3: β-гидроксибутирил-CoA-дегидрогеназа; 4: кротоназа; 5: бутирил-CoA-дегидрогеназа; 6: лактатдегидрогеназа; 7: фосфотрансацетилаза; 8: ацетаткиназа; 9: ацетальдегиддегидрогеназа; 10: этанолдегидрогеназа; 11: CoA-транфераза (ацетоацетил-CoA:ацетат/бутират:CoA-трансфераза); 12: ацетоацетат-декарбоксилаза; 13: фосфотрансбутирилаза; 14: бутираткиназа; 15: (масляный альдегид)-бутанолдегидрогеназа; 16: гидрогеназа.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Применяемые в данном изобретении следующие термины можно использовать для интерпретации формулы изобретения и описания.
Термин "микроорганизм" относится ко всем видам одноклеточных организмов, включая прокариотические организмы типа бактерий и эукариотические организмы типа дрожжей.
Выражение "соответствующая культуральная среда" относится к культуральной среде, адаптированной для используемого микроорганизма, как хорошо известно специалисту в данной области техники.
Термин "углеродный субстрат" или "источник углерода" обозначает любой источник углерода, который может быть метаболизирован микроорганизмом, причем данный субстрат содержит по меньшей мере один атом углерода. Авторы изобретения упоминают, в частности, такие возобновляемые, недорогие и ферментируемые источники углерода, как моносахариды, олигосахариды, полисахариды, одноуглеродные субстраты и полиолы, такие как глицерин. "Одноуглеродный субстрат" определяют как углеродсодержащие молекулы, имеющие только один атом углерода, как например метанол.
Моносахариды формулы (CH2O)n также называются озы или "простые сахара"; моносахариды включают сахарозу, фруктозу, глюкозу, галактозу и маннозу.
Другие источники углерода, содержащие более одного моносахарида, называются дисахаридами, трисахаридами, олигосахаридами и полисахаридами. Дисахариды включают сахарозу, лактозу и мальтозу. Крахмал и гемицеллюлоза представляют собой полисахариды, также известные как "сложные сахара".
Следовательно, термин "источник углерода" обозначает любой продукт, перечисленный выше, и их смеси.
Термин "ослабление" относится к уменьшению экспрессии гена или уменьшению активности белка, продукта этого гена. Специалисту в данной области техники известны многочисленные способы достижения этого результата, например:
- введение мутации в ген, уменьшающей уровень экспрессии этого гена или уровень активности кодируемого белка;
- замена природного промотора гена на менее сильный промотор, приводящая к снижению экспрессии;
- применение элементов, дестабилизирующих соответствующую информационную РНК или соответствующий белок;
- делетирование гена, если никакой экспрессии не требуется.
Термин "делетированный ген" означает, что существенная часть кодирующих последовательностей указанного гена была удалена. Предпочтительно, чтобы было удалено по меньшей мере 50% кодирующей последовательности и более предпочтительно по меньшей мере 80%.
В описании настоящего изобретения ферменты идентифицируют по их специфическим активностям. Таким образом, это определение включает все полипептиды, которые имеют определенную специфическую активность, также присутствующую в других организмах, более конкретно в других микроорганизмах. Зачастую ферменты со схожими активностями могут быть идентифицированы путем отнесения их к некоторым семействам, определенным как PFAM или COG.
PFAM (база данных по семействам белков, полученная из выравниваний и скрытых моделей Маркова; http://www.sanger.ac.uk/Software/Pfam/) предоставляет большую коллекцию выравниваний белковых последовательностей. Каждая PFAM дает возможность визуализировать многочисленные выравнивания, посмотреть белковые домены, оценить распределение среди организмов, получить доступ к другим базам данных и визуализировать известные белковые структуры.
COGs (кластеры ортологических групп белков; http://www.ncbi.nlm.nih.gov/COG/) получают путем сравнения белковых последовательностей из 43 полностью секвенированных геномов, представляющих 30 основных филогенетических линий. Каждый COG определяется по меньшей мере из трех линий, что позволяет проводить идентификацию предковых консервативных доменов.
Способы идентификации гомологичных последовательностей и процента их гомологии общеизвестны среди специалистов в данной области техники и включают, в частности, программы BLAST, которыми можно воспользовать из вебсайта http://www.ncbi.nlm.nih.gov/BLAST/, используя параметры по умолчанию, указанные на этом вебсайте. Полученные последовательности далее можно использовать (например для выравнивания), применяя, например, программы CLUSTALW (http://www.ebi.ac.uk/clustalw/) или MULTALIN (http://prodes.toulouse.inra.fr/multalin/cqi-bin/multalin.pl) с параметрами по умолчанию, указанными на этих вебсайтах.
Используя ссылки, приведенные в GenBank для известных генов, специалисты в данной области техники способны определить эквивалентные гены в других организмах, бактериальных штаммах, дрожжах, грибах, млекопитающих, растениях и т.д. Эту рутинную работу предпочтительно выполняют, используя консенсусные последовательности, которые можно определить путем проведения выравниваний последовательностей с генами, происходящими из других микроорганизмов, и конструирования вырожденных зондов с целью клонирования соответствующего гена в другой организм. Эти рутинные методы молекулярной биологии общеизвестны среди специалистов в данной области техники и описаны, например, в Sambrook et al. (Molecular Cloning: a Laboratory Manual. 2nd ed. Cold Spring Harbor Lab., Cold Spring Harbor, New York, 1989).
Согласно настоящему изобретению предложен способ ферментативного периодического или непрерывного продуцирования н-бутанола путем культивирования микроорганизма в соответствующей культуральной среде, содержащей источник углерода, и одновременного извлечения н-бутанола из культуральной среды, причем в данном микроорганизме делетирован по меньшей мере один ген, вовлеченный в образование бутирата.
Согласно конкретному воплощению изобретения предложен способ, где микроорганизм модифицирован с потерей способности к превращению бутирил-CoA в бутират вследствие делетирования по меньшей мере одного гена, кодирующего фосфотрансбутирилазу (ptb) или бутираткиназу (buk). Делетирование генов в Clostridia может быть выполнено с использованием способа, недавно описанного в заявке на патент PCT/EP2006/066997, позволяющего проводить 1) замену гена с целью делетирования гена устойчивости к эритромицину и 2) удаление гена устойчивости к эритромицину с использованием рекомбиназы.
В другом воплощении изобретения микроорганизм не способен продуцировать ацетон вследствие ослабления или делетирования по меньшей мере одного гена, кодирующего CoA-трансферазу (ctfAB) или ацетоацетат-декарбоксилазу (adc). Делетирование одного из этих генов может быть проведено с использованием способа, недавно описанного в заявке на патент PCT/EP2006/066997.
В следующем воплощении изобретения микроорганизм, используемый в способе по изобретению, не способен продуцировать лактат. В частности, это может происходить вследствие делетирования гена Idh, кодирующего лактатдегидрогеназу. Делетирование Idh может быть проведено с использованием способа, недавно описанного в заявке на патент PCT/EP2006/066997.
В другом воплощении микроорганизм модифицирован таким образом, чтобы он был не способен продуцировать ацетат. Этого результата можно достичь путем делетирования по меньшей мере одного из генов, кодирующих фосфотрансацетилазу (pta) или ацетаткиназу (ack). Делетирование одного из этих генов может быть проведено с использованием способа, недавно описанного в заявке на патент PCT/EP2006/066997.
Согласно одному из воплощений изобретения также предложен микроорганизм с уменьшенным потоком продуцирования водорода и затем с переориентированием потока восстановительного эквивалента на продуцирование н-бутанола; это может быть выполнено посредством ослабления гена, кодирующего гидрогеназу (hydA), фермента, обеспечивающего снижение восстановительного эквивалента в форме продуцирования водорода. Ослабление hydA может быть выполнено посредством замены природного промотора на менее сильный промотор или на элемент, дестабилизирующий соответствующую информационную РНК или соответствующий белок. При необходимости, полного ослабления гена также можно достичь путем делетирования соответствующей последовательности ДНК.
Предпочтительно, что используемый микроорганизм выбран среди группы, состоящей из С.acetobutylicum, С.beijerinckii, С.saccharoperbutylacetonicum или С.saccharobutylicum.
В другом воплощении изобретения культура является непрерывной и стабильной.
В другом воплощении способ по изобретению включает следующие стадии:
(a) приведение в контакт микроорганизма по меньшей мере с одним источником углерода, выбранным из группы, состоящей из глюкозы, ксилозы, арабинозы, сахарозы, моносахаридов, олигосахаридов, полисахаридов, целлюлозы, ксилана, крахмала или его производных и глицерина, посредством чего продуцируется н-бутанол;
(b) извлечение н-бутанола в процессе ферментации посредством "извлечения из газа" и
(c) выделение н-бутанола из конденсата путем дистилляции.
Специалисты в данной области техники способны определить условия культивирования для микроорганизмов по изобретению. В частности, клостридии ферментируют при температуре от 20°C до 55°C, предпочтительно от 25°C до 40°C и более конкретно при примерно 35°С для С.acetobutylicum.
Как правило, ферментацию проводят в ферментерах с неорганической культуральной средой известного определенного состава, адаптированного к используемым бактериям, содержащей по меньшей мере один простой источник углерода и, если необходимо, косубстрат, необходимый для продуцирования метаболита.
Изобретение также относится к микроорганизму, который определен ранее. Предпочтительно, этот микроорганизм выбран из группы, состоящей из С.acetobutylicum, С.beijerinckii, С.saccharoperbutylacetonicum или С.saccharobutylicum.
ПРИМЕР 1
Конструирование штаммов, не способных продуцировать бутират: Clostridium acetobutylicum Δсас1515 Δuрр Δbuk
Для делетирования гена buk используют стратегию гомологичной рекомбинации, описанную Croux & Soucaille (2006) в заявке на патент PCT/EP2006/066997. Эта стратегия позволяет осуществлять вставку кассеты устойчивости к эритромицину, одновременно делетируя большую часть рассматриваемого гена. buk-делеционную кассету в pCons::upp конструировали, как изложено ниже.

Два фрагмента ДНК, окружающие buk, подвергали ПЦР-амплификации с использованием полимеразы Pwo, применяя суммарную ДНК из С.acetobutylicum в качестве матрицы и две специфические пары олигонуклеотидов. Используя пары праймеров BUK 1-BUK 2 и BUK 3-BUK 4, получали соответственно два ДНК-фрагмента. С использованием обоих праймеров BUK 1 и BUK 4 вводят BamHI-сайт, тогда как праймеры BUK 2 и BUK 3 имеют комплементарный участок, с помощью которого вводят Nrul-сайт. ДНК-фрагменты BUK 1-BUK 2 и BUK 3-BUK 4 соединяли в эксперименте по ПЦР-слиянию, используя праймеры BUK 1 и BUK 4, и полученный фрагмент клонировали в pCR4-TOPO-Blunt, получая pTOPO:buk. По уникальному Stul-сайту pTOPO:buk вводили ген устойчивости к антибиотикам MLS (макролид-линкозамид-стрептограмин) с FRT (Flippase Recognition Target)-последовательностями (распознаваемыми флиппазой последовательностями-мишенями) по обеим сторонам из Stul-фрагмента от pUC18-FRT-MLS2. BUK-делеционную кассету, полученную после переваривания полученной плазмиды с помощью BamHI, клонировали в pCons::upp по BamHI-сайту, получая плазмиду pREPΔBUK::upp.
Плазмиду pREPΔBUK::upp использовали для трансформации путем электропорации штамма С.acetobutylicum MGCΔcac15Δupp. После селекции на чашке Петри на предмет клонов, устойчивых к эритромицину (40 мкг/мл), одну колонию культивировали в течение 24 часов в жидкой синтетической среде с эритромицином в концентрации 40 мкг/мл и 100 мкл неразбавленной культуры наносили на RCA (обогащенный агар для клостридий) с эритромицином в концентрации 40 мкг/мл и 5-FU (5-5-фторурацил) в концентрации 400 мкМ. Колонии, устойчивые как к эритромицину, так и к 5-FU, наносили методом реплик как на RCA с эритромицином в концентрации 40 мкг/мл, так и на RCA с тиамфениколом в концентрации 50 мкг/мл для селекции клонов, у которых устойчивость к 5-FU ассоциирована также с чувствительностью к тиамфениколу. Генотип клонов, устойчивых к эритромицину и чувствительных к тиамфениколу, проверяли ПЦР-анализом (используя праймеры BUK 0 и BUK 5, локализованные вне buk-делеционной кассеты). Штамм Δcac15ΔuppΔbuk::mlsR, утративший pREPΔbuk::upp, выделяли.
Штамм Δcac15ΔuppΔbuk::mlsR трансформировали вектором pCLF1.1, экспрессирующим ген Flp1, кодирующий рекомбиназу Flp (флиппазу) из S.cerevisiae. После трансформации и селекции на чашке Петри на предмет устойчивости к тиамфениколу (50 мкг/мл) одну колонию культивировали на синтетической жидкой среде с тиамфениколом в концентрации 50 мкг/мл и соответствующие разведения наносили на RCA с тиамфениколом в концентрации 50 мкг/мл. Тиамфениколустойчивые клоны наносили методом реплик как на RCA с эритромицином в концентрации 40 мкг/мл, так и на RCA с тиамфениколом в концентрации 50 мкг/мл. Генотип клонов с чувствительностью к эритромицину и устойчивостью к тиамфениколу проверяли ПЦР-анализом, используя праймеры BUK 0 и BUK 5. Проводили два последовательных 24-часовых культивирования штамма Δcac15ΔuppΔbuk с чувствительностью к эритромицину и устойчивостью к тиамфениколу с целью удаления pCLF1.1. Штамм Δcac15ΔuppΔbuk, утративший pCLF1.1, выделяли в соответствии с его чувствительностью как к эритромицину, так и к тиамфениколу.
ПРИМЕР 2
Конструирование штаммов, не способных продуцировать бутират и ацетон: С.acetobutylicum Δcac1515 Δupp Δbuk ΔctfAB
Для делетирования генов ctfAB используют стратегию гомологичной рекомбинации, описанную Croux & Soucaille (2006) в заявке на патент PCT/EP2006/066997. Эта стратегия позволяет осуществлять вставку кассеты устойчивости к эритромицину, одновременно делетируя большую часть рассматриваемых генов. ctfAB-делеционную кассету в pCons::upp конструировали, как изложено ниже.

Два фрагмента ДНК, окружающие ctfAB, подвергали ПЦР-амплификации с использованием полимеразы Pwo, применяя суммарную ДНК из С.acetobutylicum в качестве матрицы и две специфические пары олигонуклеотидов. Используя пары праймеров CTF 1-CTF 2 и CTF 3-CTF 4, получали соответственно два ДНК-фрагмента. С использованием обоих праймеров CTF 1 и CTF 4 вводят BamHI-сайт, тогда как праймеры CTF 2 и CTF 3 имеют комплементарный участок, с помощью которого вводят Stul-сайт. ДНК-фрагменты CTF 1-CTF 2 и CTF 3-CTF 4 соединяли в эксперименте по ПЦР-слиянию, используя праймеры CTF 1 и CTF 4, и полученный фрагмент клонировали в pCR4-TOPO-Blunt, получая pTOPO:CTF. По уникальному Stul-сайту pTOPO:CTF ген устойчивости к антибиотикам MLS с FRT-последовательностями по обеим сторонам вводили из Stul-фрагмента от pUC18-FRT-MLS2. UPP-делеционную кассету, полученную после переваривания полученной плазмиды с помощью BamHI, клонировали в pCons::upp по BamHI-сайту, получая плазмиду pREPΔCTF::upp.
Плазмиду pREPΔCTF::upp использовали для трансформации путем электропорации штамма С.acetobutylicum MGCΔcac15ΔuppΔbuk. После селекции на чашке Петри на предмет клонов, устойчивых к эритромицину (40 мкг/мл), одну колонию культивировали в течение 24 часов в жидкой синтетической среде с эритромицином в концентрации 40 мкг/мл и 100 мкл неразбавленной культуры наносили на RCA с эритромицином в концентрации 40 мкг/мл и 5-FU в концентрации 400 мкМ. Колонии, устойчивые как к эритромицину, так и к 5-FU, наносили методом реплик как на RCA с эритромицином в концентрации 40 мкг/мл, так и на RCA с тиамфениколом в концентрации 50 мкг/мл для селекции клонов, у которых устойчивость к 5-FU ассоциирована также с чувствительностью к тиамфениколу. Генотип клонов, устойчивых к эритромицину и чувствительных к тиамфениколу, проверяли ПЦР-анализом (используя праймеры CTF 0 и CTF 5, локализованные вне ctfAB-делеционной кассеты). Штамм Δcac15ΔuppΔbuk ΔctfAB::mlsR, утративший pREPΔCTF::upp, выделяли.
Штамм Δcac15ΔuppΔbukΔctfAB::mlsR трансформировали вектором pCLF1.1, экспрессирующим ген Flp1, кодирующий рекомбиназу Flp из S. cerevisiae. После трансформации и селекции на чашке Петри на предмет устойчивости к тиамфениколу (50 мкг/мл) одну колонию культивировали на синтетической жидкой среде с тиамфениколом в концентрации 50 мкг/мл и соответствующие разведения наносили на RCA с тиамфениколом в концентрации 50 мкг/мл. Тиамфениколустойчивые клоны наносили методом реплик как на RCA с эритромицином в концентрации 40 мкг/мл, так и на RCA с тиамфениколом в концентрации 50 мкг/мл. Генотип клонов с чувствительностью к эритромицину и устойчивостью к тиамфениколу проверяли ПЦР-анализом, используя праймеры CTF 0 и CTF 5. Проводили два последовательных 24-часовых культивирования штамма Δcac15ΔuppΔbukΔctfAB с чувствительностью к эритромицину и устойчивостью к тиамфениколу с целью удаления pCLF1.1 Штамм Δcac15ΔuppΔbukΔctfAB, утративший pCLF1.1, выделяли в соответствии с его чувствительностью как к эритромицину, так и к тиамфениколу.
ПРИМЕР 3
Конструирование штаммов, не способных продуцировать бутират, ацетон и лактат: С.acetobutylicum Δcac1515 Δupp Δbuk ΔctfAB Δldh
Для делетирования гена ldh используют стратегию гомологичной рекомбинации, описанную Croux & Soucaille (2006) в заявке на патент PCT/EP2006/066997. Эта стратегия позволяет осуществлять вставку кассеты устойчивости к эритромицину, одновременно делетируя большую часть рассматриваемых генов. Idh-делеционную кассету в pCons::upp конструировали, как изложено ниже.
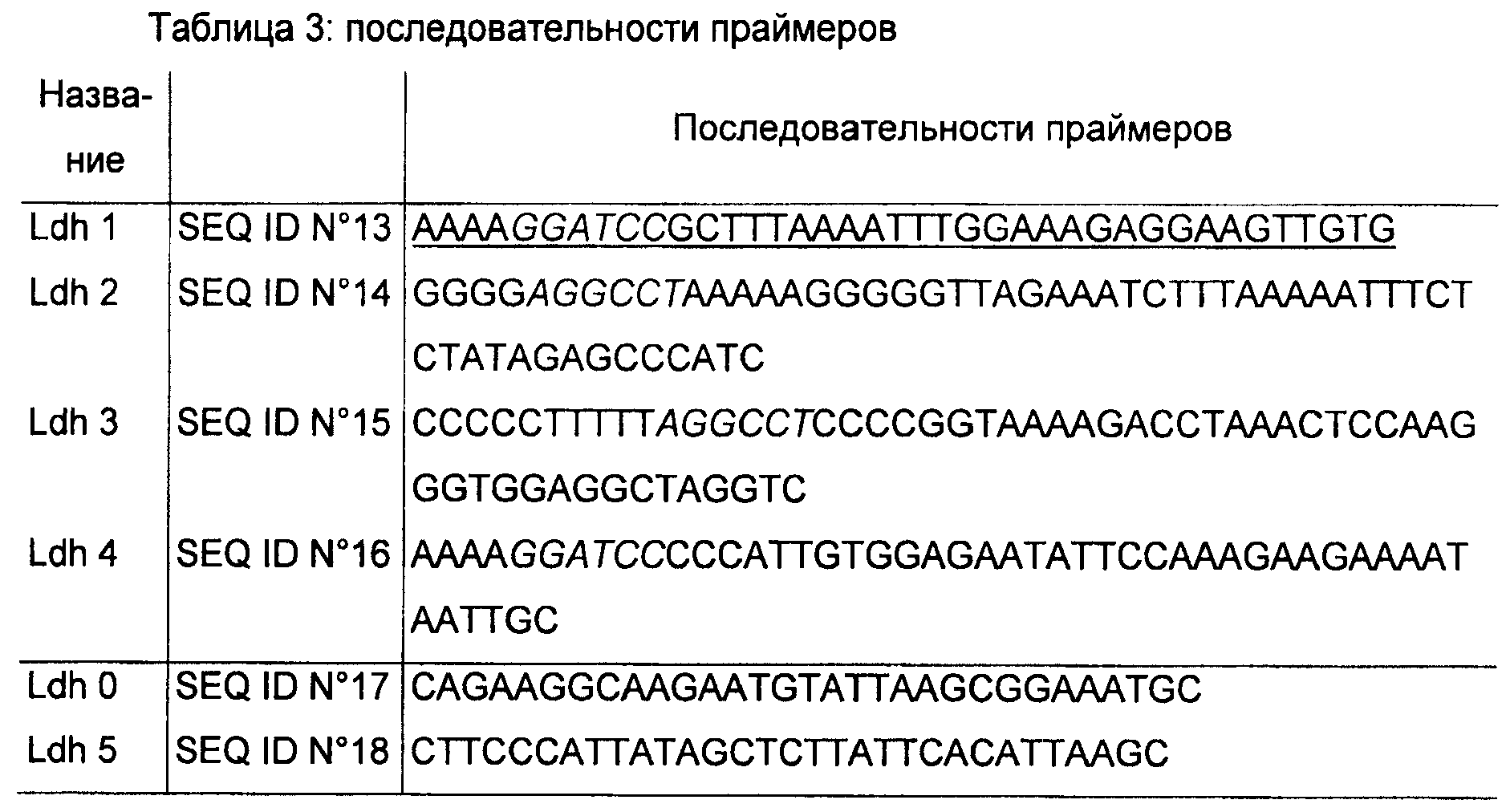
Два фрагмента ДНК, окружающие Idh (САС267), подвергали ПЦР-амплификации с использованием полимеразы Pwo, применяя суммарную ДНК из С.acetobutylicum в качестве матрицы и две специфические пары олигонуклеотидов. Используя пары праймеров LDH 1-LDH 2 и LDH 3-LDH 4, получали ДНК-фрагменты 1135 п.о. и 1177 п.о. соответственно. С использованием обоих праймеров LDH 1 и LDH 4 вводят BamHI-сайт, тогда как праймеры LDH 2 и LDH 3 имеют комплементарный участок, с помощью которого вводят Stul-сайт. ДНК-фрагменты LDH 1-LDH 2 и LDH 3-LDH 4 соединяли в эксперименте по ПЦР-слиянию, используя праймеры LDH 1 и LDH 4, и полученный фрагмент клонировали в pCR4-TOPO-Blunt, получая pTOPO:LDH. По уникальному Stul-сайту pTOPO:LDH вводили ген устойчивости к антибиотикам MLS с FRT-последовательностями по обеим сторонам из Stul-фрагмента в 1372 п.о. от pUC18-FRT-MLS2. UPP-делеционную кассету, полученную после переваривания полученной плазмиды с помощью BamHI, клонировали в pCons::upp по BamHl-сайту, получая плазмиду pREPΔLDH::upp.
Плазмиду pREPΔLDH::upp использовали для трансформации путем электропорации штамма С.acetobutylicum MGCΔcac15ΔuppΔbukΔctfAB. После селекции на чашке Петри на предмет клонов, устойчивых к эритромицину (40 мкг/мл), одну колонию культивировали в течение 24 часов в жидкой синтетической среде с эритромицином в концентрации 40 мкг/мл и 100 мкл неразбавленной культуры наносили на RCA с эритромицином в концентрации 40 мкг/мл и 5-FU в концентрации 400 мкМ. Колонии, устойчивые как к эритромицину, так и к 5-FU, наносили методом реплик как на RCA с эритромицином в концентрации 40 мкг/мл, так и на RCA с тиамфениколом в концентрации 50 мкг/мл для селекции клонов, у которых устойчивость к 5-FU ассоциирована также с чувствительностью к тиамфениколу. Генотип клонов, устойчивых к эритромицину и чувствительных к тиамфениколу, проверяли ПЦР-анализом (используя праймеры LDH 0 и LDH 5, локализованные вне ldh-делеционной кассеты). Штамм Δcac15ΔuppΔbukΔctfAB ΔIdh::mIsR, утративший pREPΔLDH::upp, выделяли.
Штамм Δcac15ΔuppΔbukΔctfABΔldh::mlsR трансформировали вектором pCLF1.1, экспрессирующим ген Flp1, кодирующий рекомбиназу Flp из S. cerevisiae. После трансформации и селекции на чашке Петри на предмет устойчивости к тиамфениколу (50 мкг/мл) одну колонию культивировали на синтетической жидкой среде с тиамфениколом в концентрации 50 мкг/мл и соответствующие разведения наносили на RCA с тиамфениколом в концентрации 50 мкг/мл. Тиамфениколустойчивые клоны наносили методом реплик как на RCA с эритромицином в концентрации 40 мкг/мл, так и на RCA с тиамфениколом в концентрации 50 мкг/мл. Генотип клонов с чувствительностью к эритромицину и устойчивостью к тиамфениколу проверяли ПЦР-анализом, используя праймеры LDH 0 и LDH 5. Проводили два последовательных 24-часовых культивирования штамма Δcac15ΔuppΔbukΔctfABΔldh с чувствительностью к эритромицину и устойчивостью к тиамфениколу с целью удаления pCLF1.1. Штамм Δcac15ΔuppΔbukΔctfABΔldh, утративший pCLF1.1, выделяли в соответствии с его чувствительностью как к эритромицину, так и к тиамфениколу.
ПРИМЕР 4
Конструирование штаммов, не способных продуцировать бутират, ацетон, лактат и ацетат: С.acetobutylicum Δcac1515 Δupp Δbuk ΔctfAB Δldh Δpta-ack
Для делетирования генов pta и ack используют стратегию гомологичной рекомбинации, описанную Croux & Soucaille (2006) в заявке на патент PCT/EP2006/066997. Эта стратегия позволяет осуществлять вставку кассеты устойчивости к эритромицину, одновременно делетируя большую часть рассматриваемых генов; pta-ack-делеционную кассету в pCons::upp конструировали, как изложено ниже.

Два фрагмента ДНК, окружающие pta-ack, подвергали ПЦР-амплификации с использованием полимеразы Pwo, применяя суммарную ДНК из С.acetobutylicum в качестве матрицы и две специфические пары олигонуклеотидов. Используя пары праймеров РА 1-РА 2 и РА 3-РА 4, получали соответственно два ДНК-фрагмента. С использованием обоих праймеров РА 1 и РА 4 вводят BamHI-сайт, тогда как праймеры РА 2 и РА 3 имеют комплементарный участок, с помощью которого вводят StuI-сайт.ДНК-фрагменты РА 1-РА 2 и РА 3-РА 4 соединяли в эксперименте по ПЦР-слиянию, используя праймеры РА 1 и РА 4, и полученный фрагмент клонировали в pCR4-TOPO-Blunt, получая рТОРО:РА. По уникальному StuI-сайту рТОРО:РА вводили ген устойчивости к антибиотикам MLS с FRT-последовательностями по обеим сторонам из StuI-фрагмента от pUC18-FRT-MLS2. UPP-делеционную кассету, полученную после переваривания полученной плазмиды с помощью BamHI, клонировали в pCons::upp по BamHI-сайту, получая плазмиду pREPΔPA::upp.
Плазмиду pREPΔPA::upp использовали для трансформации путем электропорации штамма С.acetobutylicum MGCΔcac15ΔuppΔbukΔctfABΔldh. После селекции на чашке Петри на предмет клонов, устойчивых к эритромицину (40 мкг/мл), одну колонию культивировали в течение 24 часов в жидкой синтетической среде с эритромицином в концентрации 40 мкг/мл и 100 мкл неразбавленной культуры наносили на RCA с эритромицином в концентрации 40 мкг/мл и 5-FU в концентрации 400 мкМ. Колонии, устойчивые как к эритромицину, так и к 5-FU, наносили методом реплик как на RCA с эритромицином в концентрации 40 мкг/мл, так и на RCA с тиамфениколом в концентрации 50 мкг/мл для селекции клонов, у которых устойчивость к 5-FU ассоциирована также с чувствительностью к тиамфениколу. Генотип клонов, устойчивых к эритромицину и чувствительных к тиамфениколу, проверяли ПЦР-анализом (используя праймеры РА 0 и РА 5, локализованные вне pta-ack-делеционной кассеты). Штамм Δcac15ΔuppΔbuk ΔctfABΔldhΔpta-ack::mlsR, утративший pREPΔPA::upp, выделяли.
Штамм Δcac15ΔuppΔbukΔctfABΔIdhΔpta-ack::mlsR трансформировали вектором pCLF1.1, экспрессирующим ген Flp1, кодирующий рекомбиназу Flp из S. cerevisiae. После трансформации и селекции на чашке Петри на предмет устойчивости к тиамфениколу (50 мкг/мл) одну колонию культивировали на синтетической жидкой среде с тиамфениколом в концентрации 50 мкг/мл и соответствующие разведения наносили на RCA с тиамфениколом в концентрации 50 мкг/мл. Тиамфениколустойчивые клоны наносили методом реплик как на RCA с эритромицином в концентрации 40 мкг/мл, так и на RCA с тиамфениколом в концентрации 50 мкг/мл. Генотип клонов с чувствительностью к эритромицину и устойчивостью к тиамфениколу проверяли ПЦР-анализом, используя праймеры РА 0 и РА 5. Проводили два последовательных 24-часовых культивирования штамма Δcac15ΔuppΔbukΔctfABΔldhΔpta-ack с чувствительностью к эритромицину и устойчивостью к тиамфениколу с целью удаления pCLF1.1. Штамм Δcac15ΔuppΔbukΔctfABΔldhΔpta-ack, утративший pCLF1.1, выделяли в соответствии с его чувствительностью как к эритромицину, так и к тиамфениколу.
ПРИМЕР 5
Конструирование штаммов с низким продуцированием водорода: С.acetobutylicum Δcac1515 Δupp Δbuk ΔctfAB Δldh ΔhydA
Для делетирования гена hydA используют стратегию гомологичной рекомбинации, описанную Croux & Soucaille (2006) в заявке на патент PCT/EP2006/066997. Эта стратегия позволяет осуществлять вставку кассеты устойчивости к эритромицину, одновременно делетируя большую часть рассматриваемых генов; hydA-делеционную кассету в pCons::upp конструировали, как изложено ниже.

Два фрагмента ДНК, окружающие hydA (САС028), подвергали ПЦР-амплификации с использованием полимеразы Pwo, применяя суммарную ДНК из С.acetobutylicum в качестве матрицы и две специфические пары олигонуклеотидов. Используя пары праймеров HYD 1-HYD 2 и HYD 3-HYD 4, получали ДНК-фрагменты 1269 п.о. и 1317 п.о. соответственно. С использованием обоих праймеров HYD 1 и HYD 4 вводят BamHI-сайт, тогда как праймеры HYD 2 и HYD 3 имеют комплементарный участок, с помощью которого вводят Stul-сайт. ДНК-фрагменты HYD 1-HYD 2 и HYD 3-HYD 4 соединяли в эксперименте по ПЦР-слиянию, используя праймеры HYD 1 и HYD 4, и полученный фрагмент клонировали в pCR4-TOPO-Blunt, получая pTOPO:HYD. По уникальному StuI-сайту pTOPO:HYD вводили ген устойчивости к антибиотикам MLS с FRT-последовательностями по обеим сторонам из Stul-фрагмента в 1372 п.о. от pUC18-FRT-MLS2. UPP-делеционную кассету, полученную после переваривания полученной плазмиды с помощью BamHI, клонировали в pCons::upp по BamHI-сайту, получая плазмиду pREPΔHYD::upp.
Плазмиду pREPΔHYD::upp использовали для трансформации путем электропорации штамма С.acetobutylicum MGCΔcac15ΔuppΔbukΔctfABΔldh. После селекции на чашке Петри на предмет клонов, устойчивых к эритромицину (40 мкг/мл), одну колонию культивировали в течение 24 часов в жидкой синтетической среде с эритромицином в концентрации 40 мкг/мл и 100 мкл неразбавленной культуры наносили на RCA с эритромицином в концентрации 40 мкг/мл и 5-FU в концентрации 400 мкМ. Колонии, устойчивые как к эритромицину, так и к 5-FU, наносили методом реплик как на RCA с эритромицином в концентрации 40 мкг/мл, так и на RCA с тиамфениколом в концентрации 50 мкг/мл для селекции клонов, у которых устойчивость к 5-FU ассоциирована также с чувствительностью к тиамфениколу. Генотип клонов, устойчивых к эритромицину и чувствительных к тиамфениколу, проверяли ПЦР-анализом (используя праймеры HYD 0 и HYD 5, локализованные вне hudA-делеционной кассеты). Штамм Δcac15ΔuppΔbukΔctfABΔldhΔhydA::mlsR, утративший pREPΔHYD::upp, выделяли.
Штамм Δcac15ΔuppΔbukΔctfABΔldhΔhydA::mlsR трансформировали вектором pCLF1.1, экспрессирующим ген Flp1, кодирующий рекомбиназу Flp из S. cerevisiae. После трансформации и селекции на чашке Петри на предмет устойчивости к тиамфениколу (50 мкг/мл) одну колонию культивировали на синтетической жидкой среде с тиамфениколом в концентрации 50 мкг/мл и соответствующие разведения наносили на RCA с тиамфениколом в концентрации 50 мкг/мл. Тиамфениколустойчивые клоны наносили методом реплик как на RCA с эритромицином в концентрации 40 мкг/мл, так и на RCA с тиамфениколом в концентрации 50 мкг/мл. Генотип клонов с чувствительностью к эритромицину и устойчивостью к тиамфениколу проверяли ПЦР-анализом, используя праймеры HYD 0 и HYD 5. Проводили два последовательных 24-часовых культивирования штамма Δcac15ΔuppΔbukΔctfABΔldhΔhydA с чувствительностью к эритромицину и устойчивостью к тиамфениколу с целью удаления pCLF1.1. Штамм Δcac15ΔuppΔbukΔctfABΔldhΔhydA, утративший pCLF1.1, выделяли в соответствии с его чувствительностью как к эритромицину, так и к тиамфениколу.
ПРИМЕР 6
Периодическая ферментация н-бутанолпродуцирующих штаммов
Первоначально штаммы анализировали в колбах для анаэробного культивирования в синтетической среде, описанной Soni и др. (Soni et al., 1987, Appl. Microbiol. Biotechnol. 27: 1-5), дополненной 2,5 г/л ацетата аммония. Ночную культуру при 35°C использовали для инокуляции культуры объемом 30 мл до получения OD600, равной 0,05. После инкубации культуры в течение 3 суток при 35°C проводили анализ глюкозы, органических кислот и растворителей посредством HPLC, используя колонку Biorad НРХ 97Н для разделения и рефрактометр для детекции.
Штаммы с надлежащим фенотипом потом тестировали в условиях продуцирования в 300 мл ферментерах (DASGIP), используя протокол анаэробного периодического культивирования.
Для этой цели ферментер заполняли 250 мл синтетической среды, барботировали азотом в течение 30 мин и инокулировали 25 мл предкультуры до оптической плотности (OD600 нм) от 0,05 до 0,1.
Температуру культуры поддерживали постоянной при 35°C и pH постоянно подводили до 5,5, используя раствор NH4OH. В процессе ферментации скорость перемешивания поддерживали 300 оборотов в минуту.
ПРИМЕР 7
Непрерывная ферментация н-бутанолпродуцирующих штаммов
Наилучший н-бутанолпродуцирующий штамм анализировали при культивировании в хемостате в синтетической среде, описанной Soni и др. (Soni et al., 1987, Appl. Microbiol.Biotechnol. 27: 1-5). Ночную культуру при 35°C использовали для инокуляции 300 мл ферментеров (DASGIP), применяя протокол анаэробного культивирования в хемостате.
Для этой цели ферментер заполняли 250 мл синтетической среды, барботировали азотом в течение 30 мин и инокулировали 25 мл предкультуры до оптической плотности (OD600 нм) от 0,05 до 0,1. Через 12 часов периодического культивирования при 35°C, pH 5,5 (регулировали с использованием раствора NH4OH) и скорости перемешивания 300 оборотов в минуту, ферментер непрерывно подпитывали не содержащей кислорода синтетической средой при скорости разведения 0,05 ч-1, в то время как объем поддерживали постоянным путем последовательного удаления ферментационной среды. За стабильностью следили, используя анализ продуктов с применением протокола HPLC, описанного ранее.
ПРИМЕР 8
Оценка н-бутанолпродуцирующих штаммов в периодических культурах.
Продуцирующие штаммы оценивали в небольших колбах. 10% размороженных культур (обычно 3 мл) использовали для инокуляции 30 мл синтетической среды (MSL4). Для киллинга любых вегетативных клеток, присутствующих до инициации роста, применяли тепловой шок при 80°C в течение 15 минут. Затем культуры выращивали при 37°C в течение 6-7 суток. Внеклеточные соединения определяли количественно посредством HPLC, используя следующие параметры: концентрация элюента (H2SO4): 0,25 мМ; поток: 0,5 мл/мин; температура: 25°C, время: 50 минут.
Как можно видеть в Таблице 2, после делетирования гена, кодирующего бутираткиназу (buk), максимальная концентрация бутирата, детектируемая в среде, уменьшается от 3,13 г/л через 2 суток культивирования штамма С.acetobutylicum Δcac15 Δupp до 0,43 г/л через 5 суток культивирования штамма С.acetobutylicum Δсас15 Δupp Δbuk::MSLr. Отметим, что выход спирт/глюкоза (Ybu+Yet) значительно увеличивается, в то время как выход ацетон/глюкоза (Yac) значительно уменьшается.

Реферат
Изобретение относится к области биотехнологии и касается способа биологического получения н-бутанола и микроорганизма Clostridia acetobutylicum, используемого в таком способе. Представленный способ заключается в культивировании микроорганизма Clostridia acetobutylicum, у которого делетирован кодирующий бутираткиназу ген buk, в соответствующей культуральной среде, содержащей источник углерода, и извлечения н-бутанола из культуральной среды. Используемый в способе микроорганизм может также содержать инактивирующие мутации в эндогенных генах, кодирующих полипептид с лактатдегидрогеназной активностью, полипептид с фосфотрансацетилазной или лактаткиназной активностью и иметь ослабление в гене, кодирующем полипептид с гидрогеназной активностью. Представленное изобретение позволяет получать н-бутанол с высоким выходом из недорогого углеродного субстрата, такого как глюкоза или другие сахара, под действием генетически стабильных культур Clostridia acetobutylicum. 2 н. и 8 з.п. ф-лы, 1 ил., 7 табл., 8 пр.
Формула
- ctfAB, кодирующий СоА-трансферазу,
- adc, кодирующий ацетоацетат-декарбоксилазу.
- pta, кодирующего фосфотрансацетилазу,
- ack, кодирующего ацетаткиназу.
a) ферментацию микроорганизма, продуцирующего н-бутанол,
b) удаление н-бутанола в процессе ферментации посредством "извлечения из газа",
c) выделение н-бутанола из конденсата путем дистилляции.
Комментарии