Рекомбинантный аденовирусный вектор и способы его применения - RU2162342C2
Код документа: RU2162342C2
Чертежи











Описание
Для образования рекомбинантных аденовирусов, применимых в генотерапии, необходимо использовать клеточную линию, в которой по "транс" типу синтезируются продукты вирусных генов E1 области, делетированных у исходных вирусов. В настоящее время доступна единственная клеточная линия 293, первоначально описанная в 1977 Graham с соавт. Клетки линии 293 содержат приблизительно 12% (4,3 кб) левой части генома аденовируса типа 5 (Aiello, 1979; Spector, 1983).
Аденовирусные векторы, исследованные на настоящий момент для целей генной терапии, обычно имеют делеции генов Ad2 или Ad5, расположенные от точки, отстоящей на 400 кб от 5'-конца вирусного генома до точки, отстоящей приблизительно на 63,3 кб от 5'-конца, с общей делецией области E1 (2,9 кб). Таким образом, существует ограниченная область гомологии приблизительно в 1 кб между последовательностями ДНК рекомбинантного вируса и ДНК Ad5 в клеточной линии. Данная гомология определяет область потенциальной рекомбинации между вирусными и клеточными аденовирусными последовательностями. Такая рекомбинация приводит к образованию вируса фенотипически дикого типа, несущего область Ad5 E1 из клеток 293. По-видимому, именно такое рекомбинационное событие обусловливает частое обнаружение аденовируса дикого типа в препаратах рекомбинантного вируса. Кроме того, было прямо показано, что такая рекомбинация является причиной контаминации вирусом дикого типа рекомбинантного вируса Ad2/CFTR-1, созданного на основе Ad2 (Rich et al., 1993).
В силу высокой степени гомологии последовательностей в подгруппе аденовирусов типа C, подобная рекомбинация более вероятна, если для создания вектора использован любой аденовирус группы C (типы 1,2,5,6).
При мелкомасштабном производстве рекомбинантных аденовирусов проблема контаминации вирусом дикого типа может быть решена процедурой отбора, при которой контаминированные партии вируса просто отбрасываются. При увеличении масштабов культивирования для нужд генотерапии повышается вероятность загрязнения каждой отдельной партии вируса вирусом дикого типа и возрастают трудности получения неконтаминированных препаратов рекомбинантного вируса.
В текущем году будет диагностировано более миллиона случаев первичного рака, а количество смертей, обусловленных онкологическими заболеваниями, достигнет полумиллиона (Американское Противораковое Общество, 1993). Мутации гена p53 являются наиболее частым генетическим повреждением, ассоциированным с опухолями человека, они встречаются в 50-60% опухолей человека (Hollstein et al.,1991: Bartek et al., 1991: Levine, 1993). Целью генотерапии p53-дефицитных опухолей является, например, введение нормальной функциональной копии гена p53 дикого типа для восстановления контроля клеточной пролиферации. P53 играет ключевую роль в клеточном цикле, останавливая рост с тем, чтобы могли произойти репарация или апоптоз в ответ на повреждение ДНК. Недавно было показано, что p53 дикого типа является необходимым компонентом системы апоптоза, индуцируемого облучением или лечением некоторыми химиотерапевтическими препаратами (Lowe et al. , 1993, A и B). Поскольку мутации p53 с высокой частотой обнаруживаются в опухолях человека, вероятно, что эти опухоли стали устойчивыми к химио- и радиотерапии в силу утраты p53 дикого типа. "Доставка" функционального p53 в эти опухоли с высокой вероятностью сделает их чувствительными к апоптозу, обычно связанному с повреждением ДНК, индуцированным облучением или химиотерапией.
Одним из критических моментов в успешной терапии заболеваний человека при помощи генов-супрессоров опухолей является возможность воздействия на значительную долю опухолевых клеток. С этой целью на различных опухолевых моделях широко применяли ретровирусные векторы. Например, для лечения опухолей печени применение ретровирусных векторов оказалось малоэффективным, поскольку с их помощью не удавалось достигнуть высокого уровня переноса генов, необходимого для генотерапии in vivo (Huber, B.E. et al., 1991; Caruso M. etal., 1993).
С целью создания более длительного источника продукции вируса исследователи попытались преодолеть проблему низкой частоты переноса генов с помощью прямой инъекции в солидные опухоли упаковочных клеток, продуцирующих ретровирусные векторы (Caruso, M. et al., 1993; Ezzidine, Z.D. et al., 1991; Culver, K. W. et al., 1992). Однако данный подход оказался неприемлемым для лечения больных, поскольку в ответ на введение упаковочных клеток развивалась воспалительная реакция.
Другим недостатком ретровирусных векторов является их потребность в делящихся клетках для эффективной интеграции и экспрессии перенесенного рекомбинантного гена (Hubner, В.Е., 1991). Стабильная интеграция ретровирусного генома в существенный ген клетки-хозяина может привести к возникновению и наследованию различных патологий.
Рекомбинантные аденовирусы имеют существенные преимущества по сравнению с ретровирусными векторами и с другими методами переноса генов (см. обзор Siegfried, 1993). Никогда не было показано, что аденовирусы способны вызывать опухоли у человека и их вполне безопасно использовали в качестве живых вакцин (Straus, 1984). Дефектные по репликации рекомбинантные аденовирусы могут быть получены заменой области E1, необходимой для репликации, на ген, предназначенный для переноса. Аденовирус не интегрирует с геномом клеток человека, и тем самым значительно снижается риск инсерционного мутагенеза, возможного при использовании ретровирусных и аденоассоциированных (AAV) векторов. Отсутствие стабильной интеграции повышает безопасность еще и за счет того, что эффект перенесенного гена является временным, поскольку экстрахромосомная ДНК будет утрачиваться по мере деления нормальных клеток. Стабильный рекомбинантный аденовирус с высоким титром может быть получен в количествах, не достижимых в случае ретровирусных или AAV векторов, что создает возможность для лечения большого числа больных. Кроме того, аденовирусные векторы обеспечивают высокоэффективный перенос генов in vivo в разнообразные опухолевые клетки и ткани. Так, например, было показано, что перенос генов при помощи аденовирусов имеет существенное значение при генотерапии таких заболеваний как цистофиброз (Rosenfeld et al., 1992; Rich et al., 1993) и дефицит альфа-1-антитрипсина (Lemarchand et al.,1992). Хотя в настоящее время используются и альтернативные методы переноса генов, например катионные комплексы липосомы/ДНК, ни один из этих методов пока не является столь же эффективным, как перенос генов с помощью аденовирусов.
Как и в случае дефектных по p53 опухолей, целью генотерапии других опухолей является восстановление контроля над пролиферацией клеток. При дефиците p53 введение функционального гена восстанавливает контроль над клеточным циклом и делает возможной гибель клеток путем апоптоза под действием терапевтических препаратов. Аналогично, гемотерапия в равной степени может быть основана на манипуляциях с другими генами-супрессорами опухолей, которые могут применяться независимо или в сочетании с терапевтическими препаратами для контроля клеточного цикла опухолевых клеток и/или индукции гибели клеток. Кроме того, гены, которые не кодируют белки - регуляторы клеточного цикла, но непосредственно вызывают гибель клеток, например "суицидные" гены или гены, кодирующие токсичные для клетки белки, также могут быть использованы в генотерапевтических процедурах для остановки клеточного цикла в опухолевых клетках.
Независимо от того, какой ген использован для восстановления контроля над клеточным циклом, теоретические предпосылки и практическая значимость такого подхода остаются неизменными. А именно: необходимо получить высокую эффективность переноса генов для экспрессии терапевтических количеств рекомбинантного продукта. Для успеха генотерапевтического подхода важен правильный выбор вектора, обеспечивающего высокую эффективность переноса генов с минимальным риском для пациента.
Таким образом, существует потребность в векторах и методах, обеспечивающих высокую эффективность переноса генов и высокий уровень экспрессии белков, которые были бы достаточно безопасны для генотерапевтических процедур. Настоящее изобретение отвечает указанным целям и, кроме того, предоставляет ряд дополнительных преимуществ.
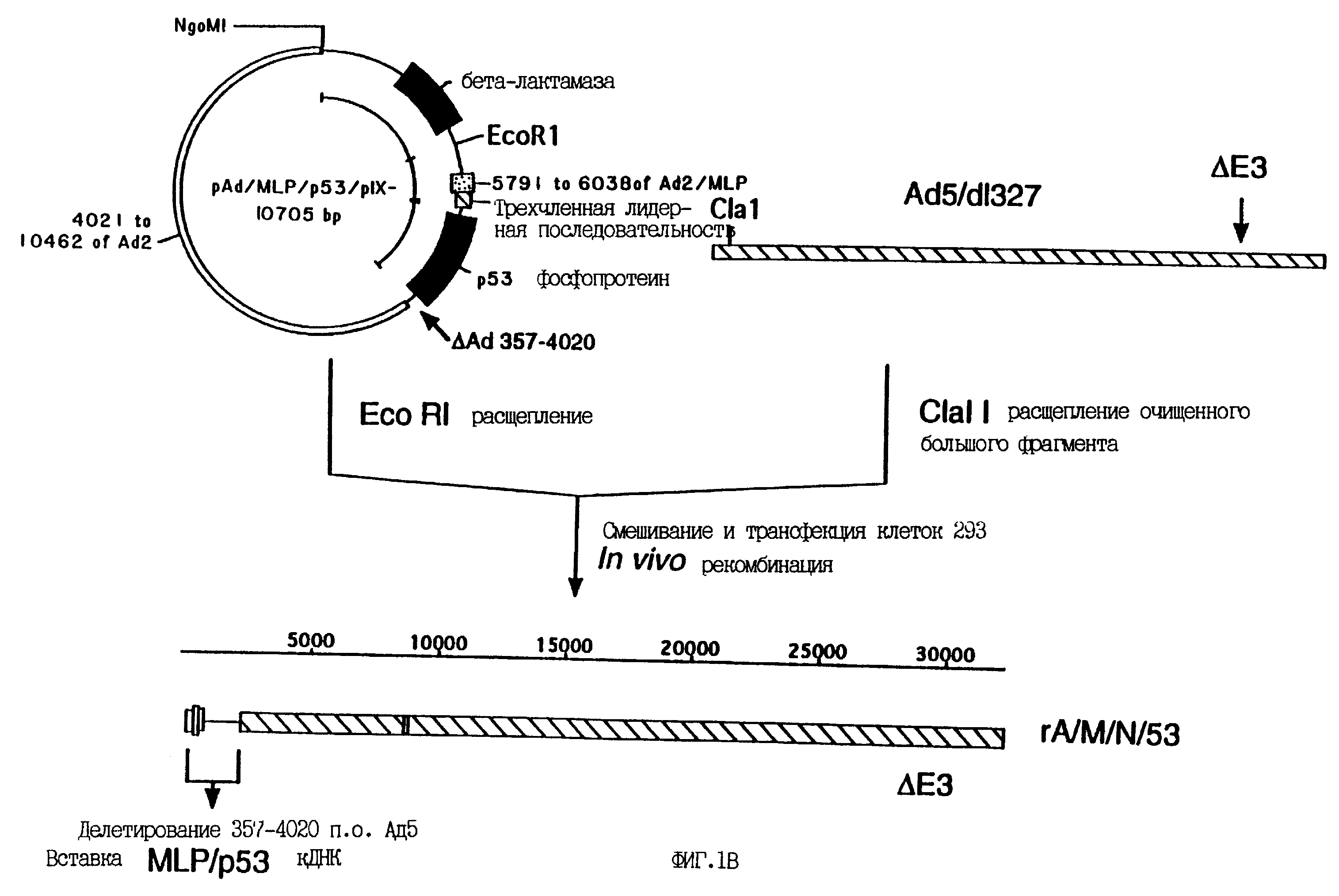
На фиг. 1 представлен заявленный в настоящем изобретении аденовирусный вектор. Сборку конструкта производили, как показано на фиг. 1. Полученный вирус имеет 5'-концевую делецию аденовирусных последовательностей, простирающуюся от нуклеотида 356 до нуклеотида 4020 и устраняющую гены E1a и E1b, а также все кодирующие последовательности белка IX, оставляя интактным общий сайт полиаденилирования генов Eld и P1X, что позволяет использовать его для терминации транскрипции любого желаемого гена.
На фиг. 2 приведена аминокислотная последовательность p110RB.
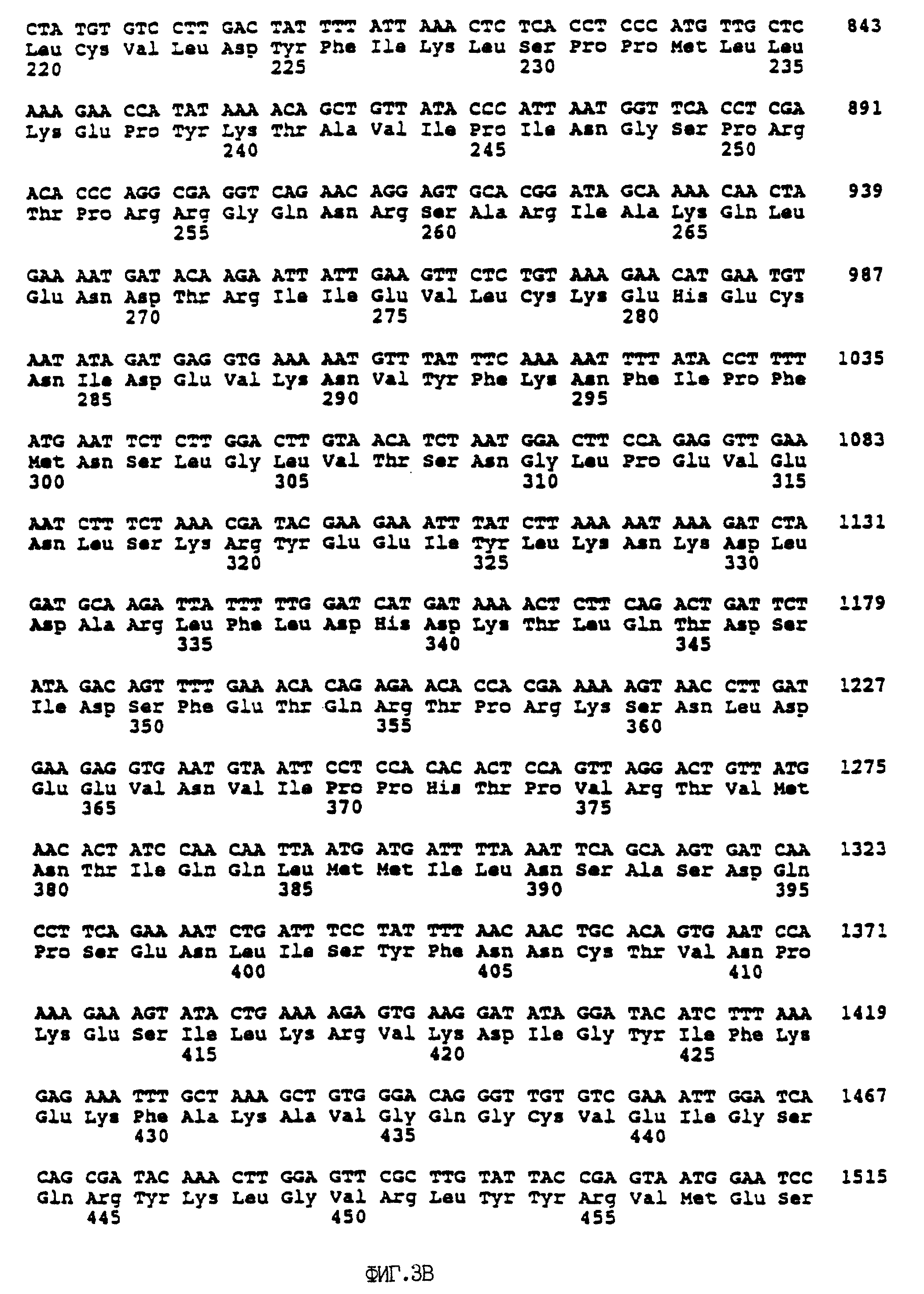
На фиг. 3 представлена последовательность ДНК, кодирующей белок-супрессор ретинобластомы.
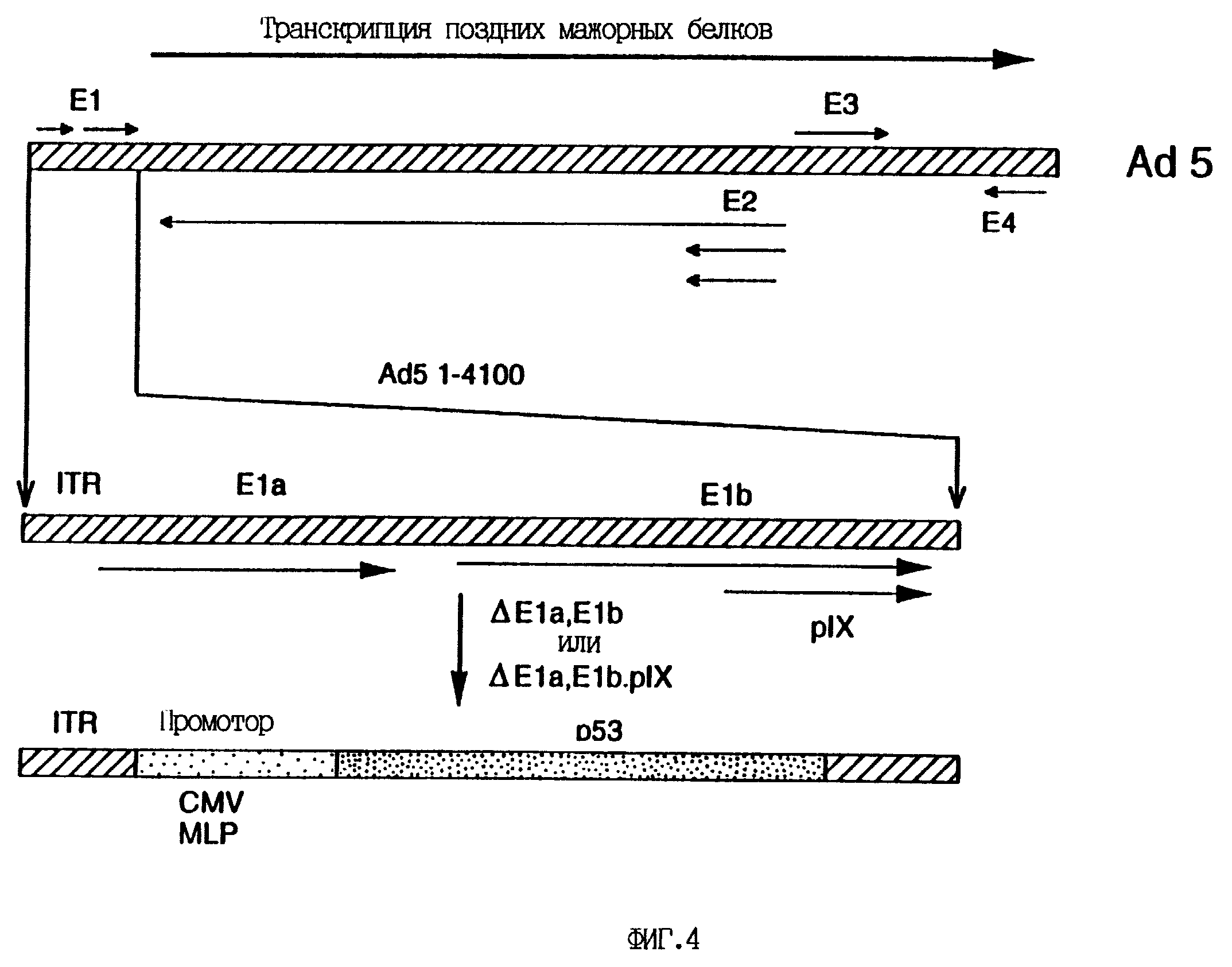
На фиг. 4 схематически представлены рекомбинантные конструкты P53/аденовирус, заявленные в настоящем изобретении. P53-рекомбинанты основаны на Ad 5, у которого область E1 (нуклеотиды 360-3325) была заменена на полноразмерную (1.4 кб) кДНК p53. При этом экспрессия p53 направлялась промотором Ad 2 MLP (A/М/53) или промотором цитомегаловируса человека (CMV) (A/C/53), за которыми следовала трехчленная лидерная кДНК Ad 2. Контрольный вирус A/М имел те же делеции генома Ad 5, что и вирус A/М/53, но не имел 1.4 кб-вставки кДНК p53. Оставшиеся последовательности E1b (705 нуклеотидов) делетировали для получения конструктов A/M/N/53 и A/C/N/53 с делецией по белку IX. Данные конструкты также имели 1.9 кб Xba 1-делецию в области E3 аденовируса типа 5.
Фиг. 5A и 5B иллюстрируют экспрессию белка p53 в опухолевых клетках, инфицированных вирусами A/М/53 и A/C/53. Фиг. 5A: клетки Saos-2 (остеосаркома) были заражены с указанной множественностью инфекции вирусом A/М/53 или вирусом A/C/53 и подвергнуты анализу спустя 24 часа после заражения. Антитела pAb 1801 к p53 использовали для окраски иммуноблотов образцов, уравненных по общей концентрации белка. В качестве маркера использовали эквивалентные по белку образцы экстракта клеток SW480, которые экспрессируют мутантный p53 в больших количествах. "О" под заголовком "A/C/53" означает псевдоинфекцию, и данный трек содержит лизат необработанных клеток Saos-2. Фиг. 5B: клетки гепатоцеллюлярной карциномы Hep B3 заражали вирусом A/М/53 или вирусом A/C/53 с указанной множественностью инфекции и анализировали, как описано в разделе "A". Стрелка указывает положение белка p53.
На фиг. 6A-6C показано зависимое от p53 изменение морфологии клеток Saos-2. Субконфлюэнтные клетки Saos-2 (1 · 105 клеток/10 см чашку) оставляли неинфицированными (A), инфицировали с множественностью 50 контрольным вирусом A/М (B) или вирусом A/C/53 (C). Клетки фотографировали спустя 72 часа после заражения.
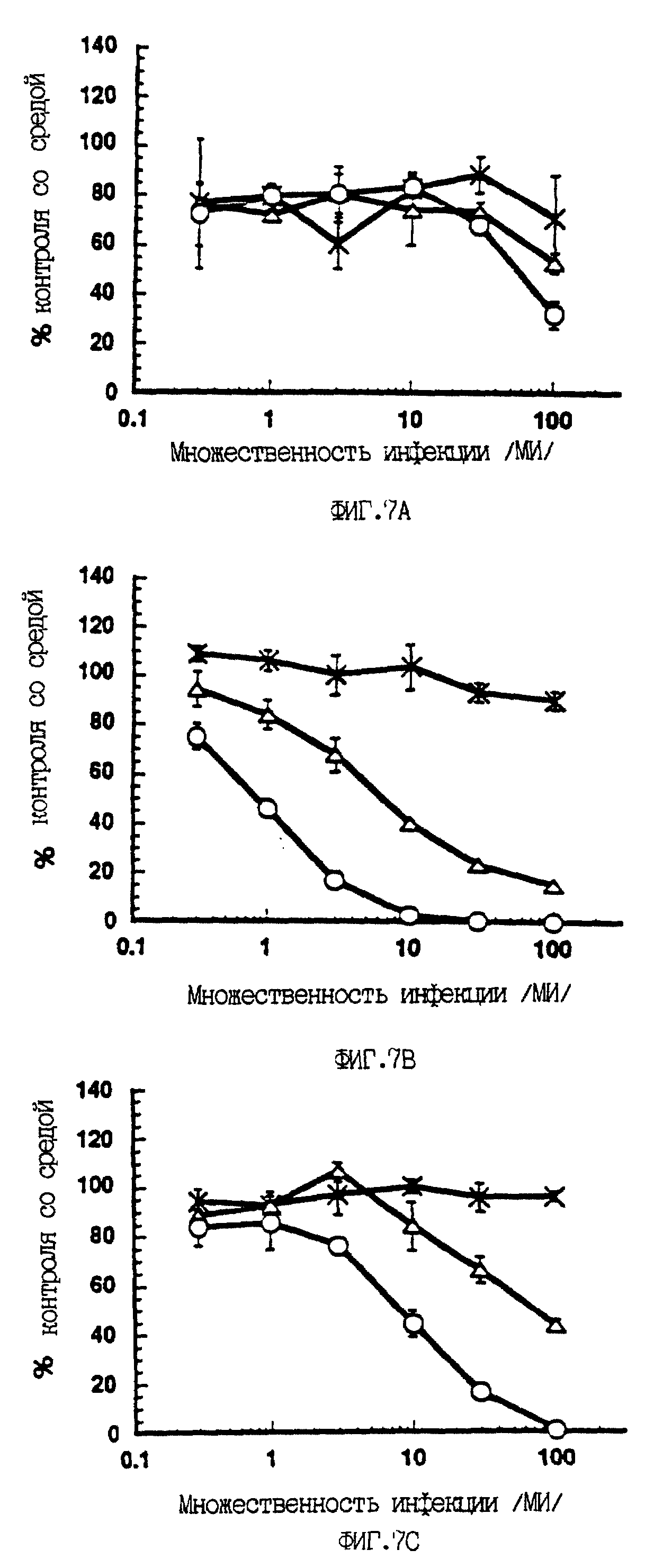
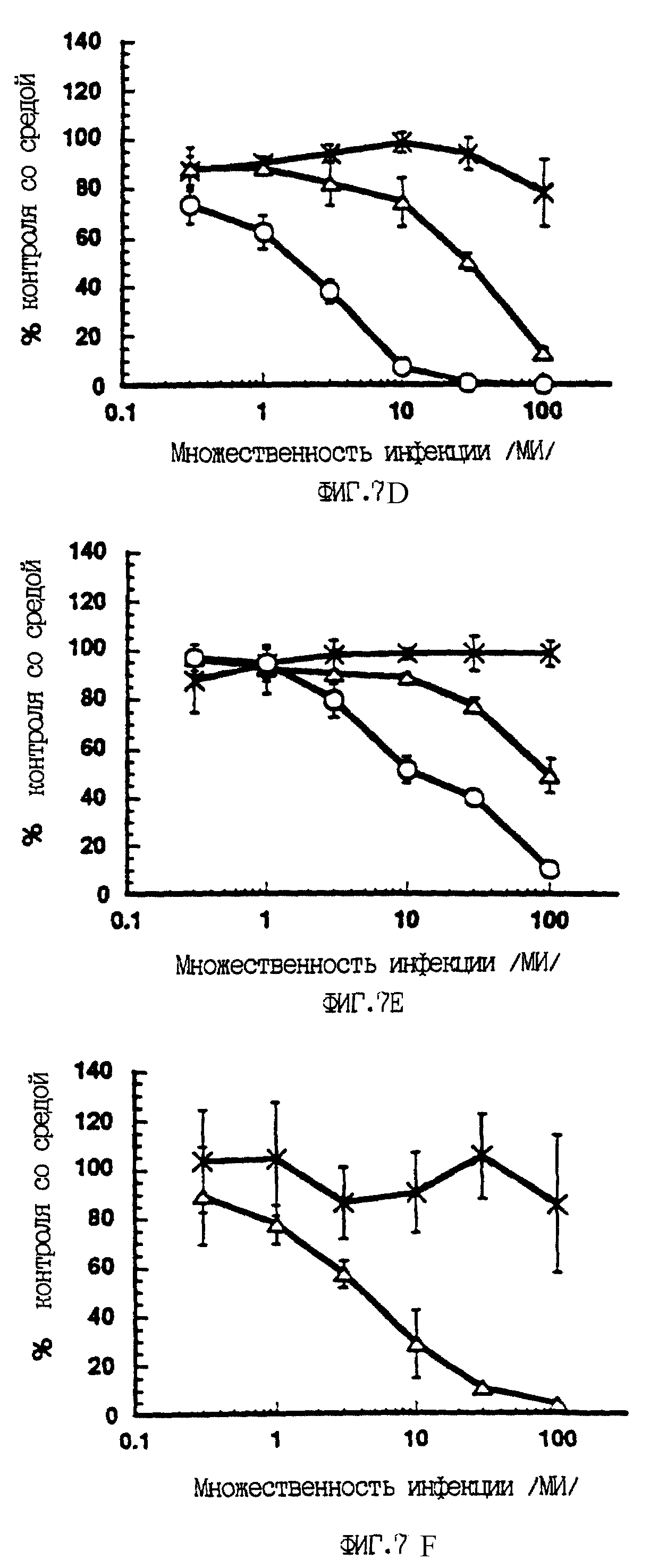
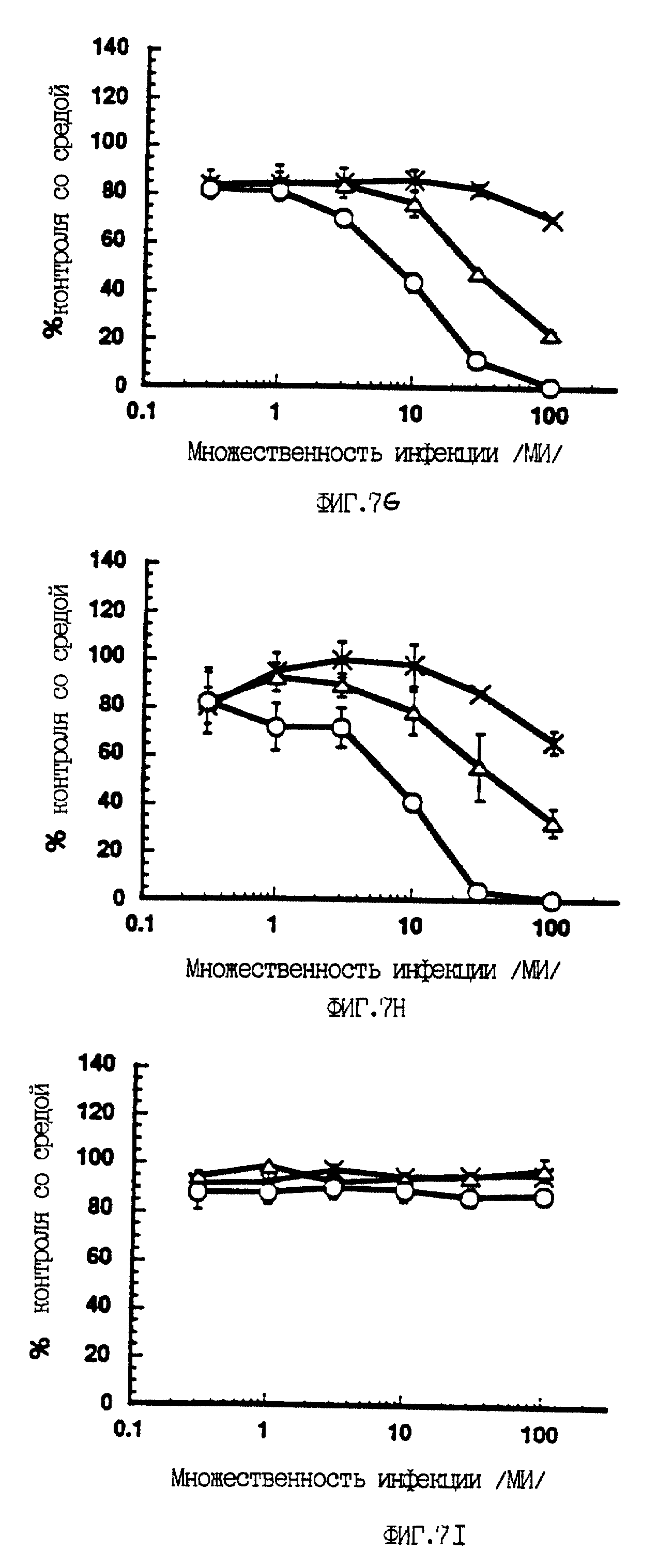
На фиг. 7 показано зависимое от p53 ингибирование синтеза ДНК в опухолевых клеточных линиях человека, инфицированных вирусами A/M/N/53 и A/C/N/53. Клетки девяти различных опухолевых линий инфицировали или контрольным аденовирусом A/М (-x-x-), или экспрессирующими p53 A/M/N/53 (- - -) или A/C/N/53 (-о-о-) с возрастающей множественностью инфекции, как показано на фигуре. Для каждой клеточной линии приведен тип опухоли и статус p53 (wt - дикий тип; null - белок не экспрессируется; mut - экспрессируется мутантный белок). Спустя 72 часа после заражения определяли синтез ДНК, как это описано ниже в Эксперименте N ll. Результаты приведены для трех измерений для каждой дозы (среднее +/- стандартное отклонение) и представлены как процент контроля со средой в зависимости от множественности инфекции. Клетки H69 тестировали только с вирусами A/М и A/M/N/53.
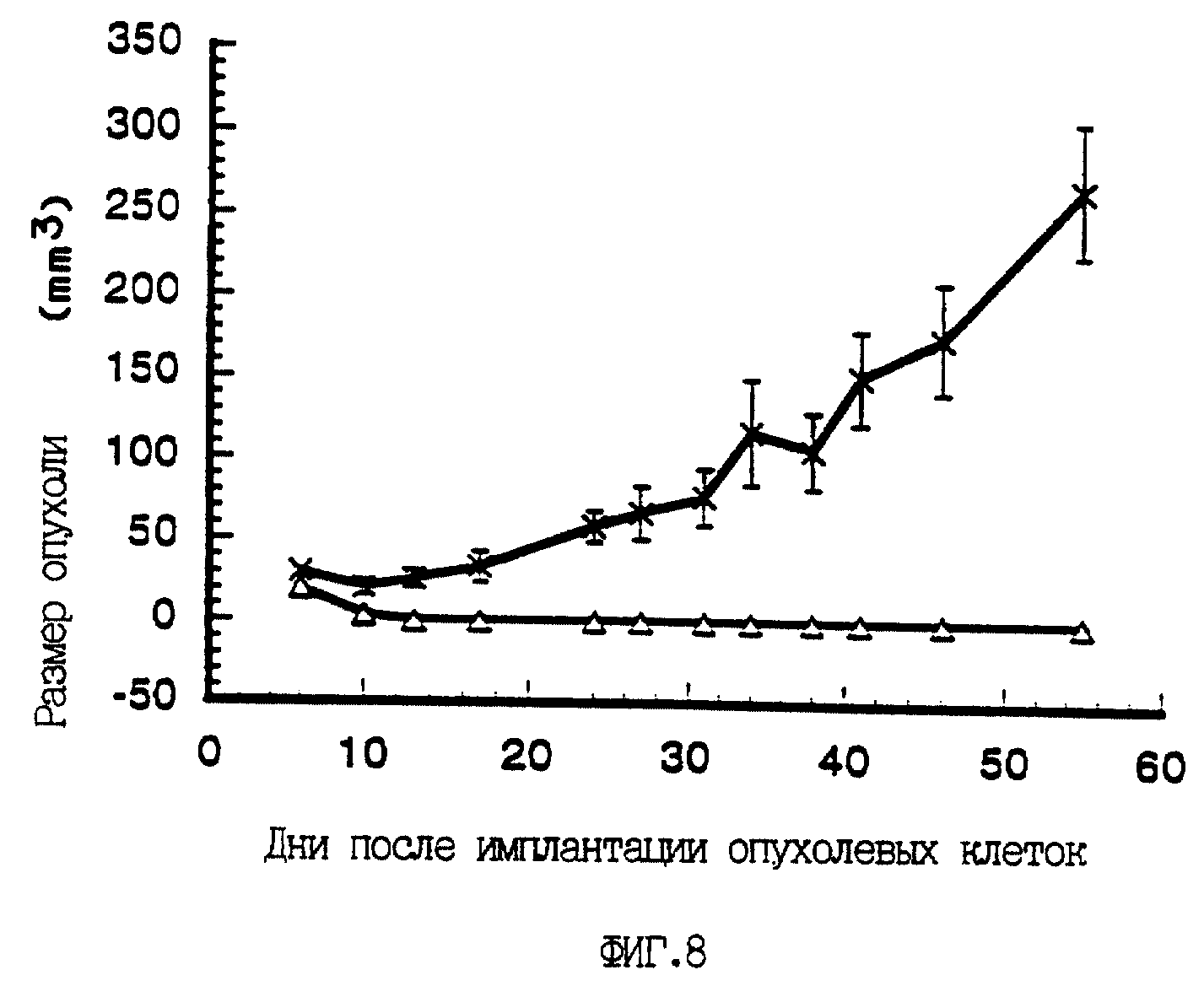
На фиг. 8 представлена туморогенность клеток Saos-2, инфицированных p53, для голых мышей. Клетки Saos-2 инфицировали или контрольным вирусом A/М или p53-рекомбинантом A/M/N/53 с множественностью инфекции 30. Обработанные клетки вводили подкожно в бок голым мышам и дважды в неделю в течение 8 недель определяли размеры опухолей (как описано в Эксперименте II). Результаты выражали как зависимость размера опухоли от дней, прошедших с момента имплантации опухолевых клеток как для контрольных A/М- инфицированных (-x-x-) клеток, так и для A/M/M/53- инфицированных клеток (- - -). Границы ошибок отражают средний размер опухолей (+/- стандартное отклонение) для каждой группы из 4-х животных для каждой временной точки.
Фиг. 9 иллюстрирует экспрессию rAd/p53 РНК в опухолях. Голым мышам подкожно вводили клетки H69 (SCLC) и в течение 32 дней позволяли развиться опухолям, которые к этому времени достигали размеров около 25-50 мм3. Затем случайно отобранным мышам вводили перитуморально по 2·109 бляшкообразующих единиц или контрольного вируса A/C/бетагал или вируса A/C/53. На 2-й и 7-й дни после инъекции опухоли вырезали и из каждого образца опухоли выделяли полиА РНК. Проводили РТ-ПЦР с равными количествами РНК и с праймерами, специфичными к мРНК рекомбинантного p53. ПЦР-амплификацию проводили в течение 30 циклов при 94oC 1 мин, 55oC 1.5 мин, 72oC 2 мин, и окончательное наращивание занимало 10 мин при 72oC. Использовали термосайклер Omnigen (Hybaid). Для ПЦР использовали следующие праймеры: 5'трехчленная лидерная кДНК (5'- CGCCACCGAGGGACCTGAGCGAGTC-3') и 3'p53 праймер (5'- TTCTGGGAAGGGACAGAAGA-3'). На линиях 1, 2, 4 и 5 представлены образцы из обработанных p53 опухолей, полученные, как показано, на 2-й или 7-й дни. На линии 3 и 8 были нанесены образцы из опухолей, обработанных бета-гал. Линии 7, 8 и 9 являются соответственными повторами линий 4, 5 и 6, но амплификацию проводили с праймерами актина, чтобы удостовериться в одинаковой загрузке. На линии 10 приведен положительный контроль с плазмидой, содержащей трехчленник/p53.
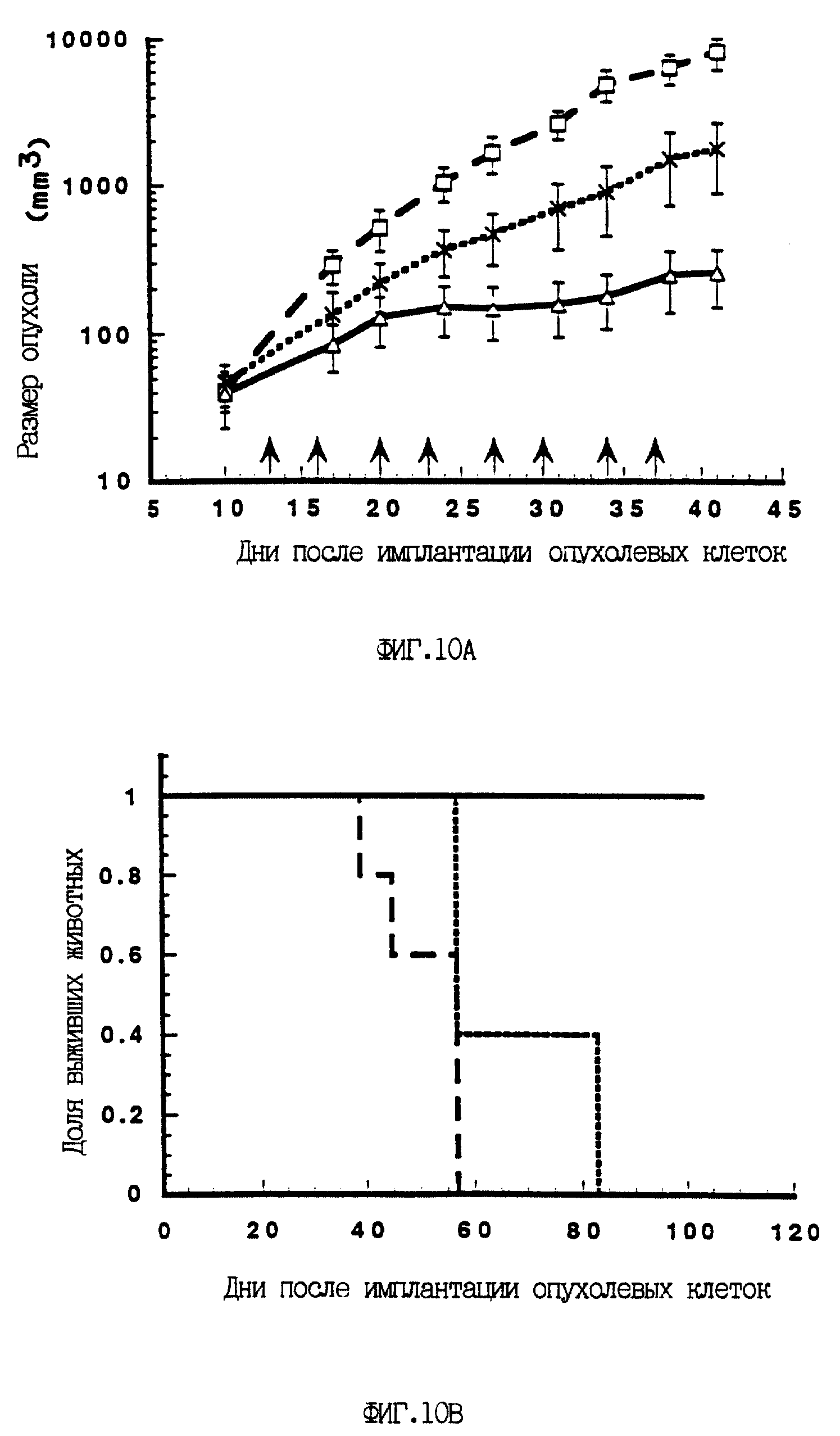
На фиг. 10A и 10B показаны подавление опухолей in vivo и увеличение времени выживания под действием A/M/N/53. Голым мышам подкожно вводили клетки H69 (SCLC) и в течение 2-х недель давали возможность развиться опухолям. Дважды в неделю мышам перитуморально вводили или просто буфер (---), или контрольный аденовирус A/М (-x-x-), или вирус A/M/N/53 (- - -) (доза обоих вирусов составляла 2·109 бое на инъекцию, всего 8 доз). Размеры опухолей определяли дважды в неделю и объем опухолей оценивали, как это описано в Эксперименте N II. A) Размер опухолей в случае введения каждого вируса ставили в зависимость от времени (число дней), прошедшего с введения клеток H69. Границы ошибок отражают средний размер опухолей (+/- стандартное отклонение) для каждой группы из 5-ти животных. Стрелки указывают дни введения вируса. B) Приведена доля выживших мышей для каждой группы в зависимости от времени, прошедшего после введения клеток H69, обработанных только буфером (---), контрольным вирусом A/М (... ... ...) или вирусом A/M/N/53 (_).
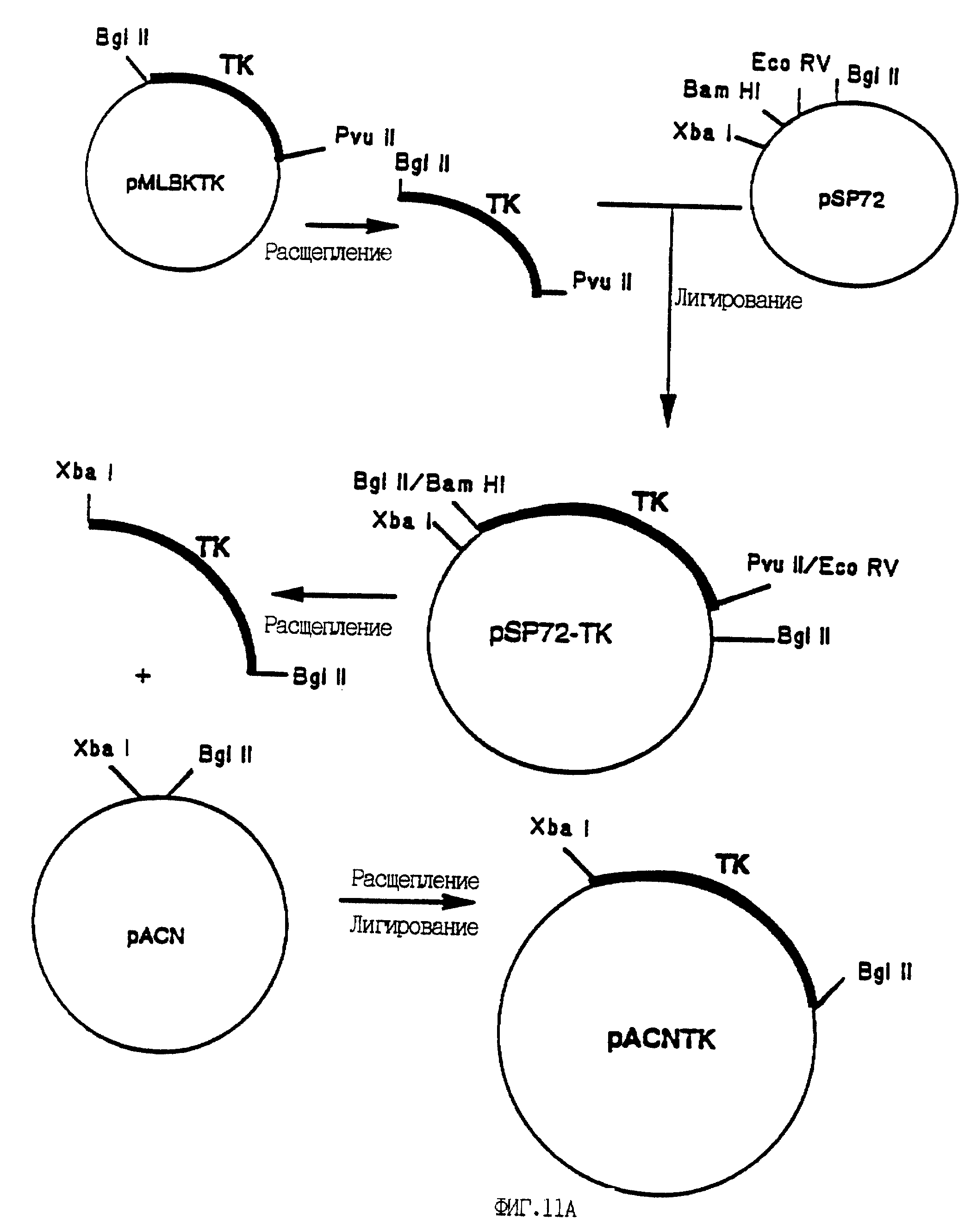
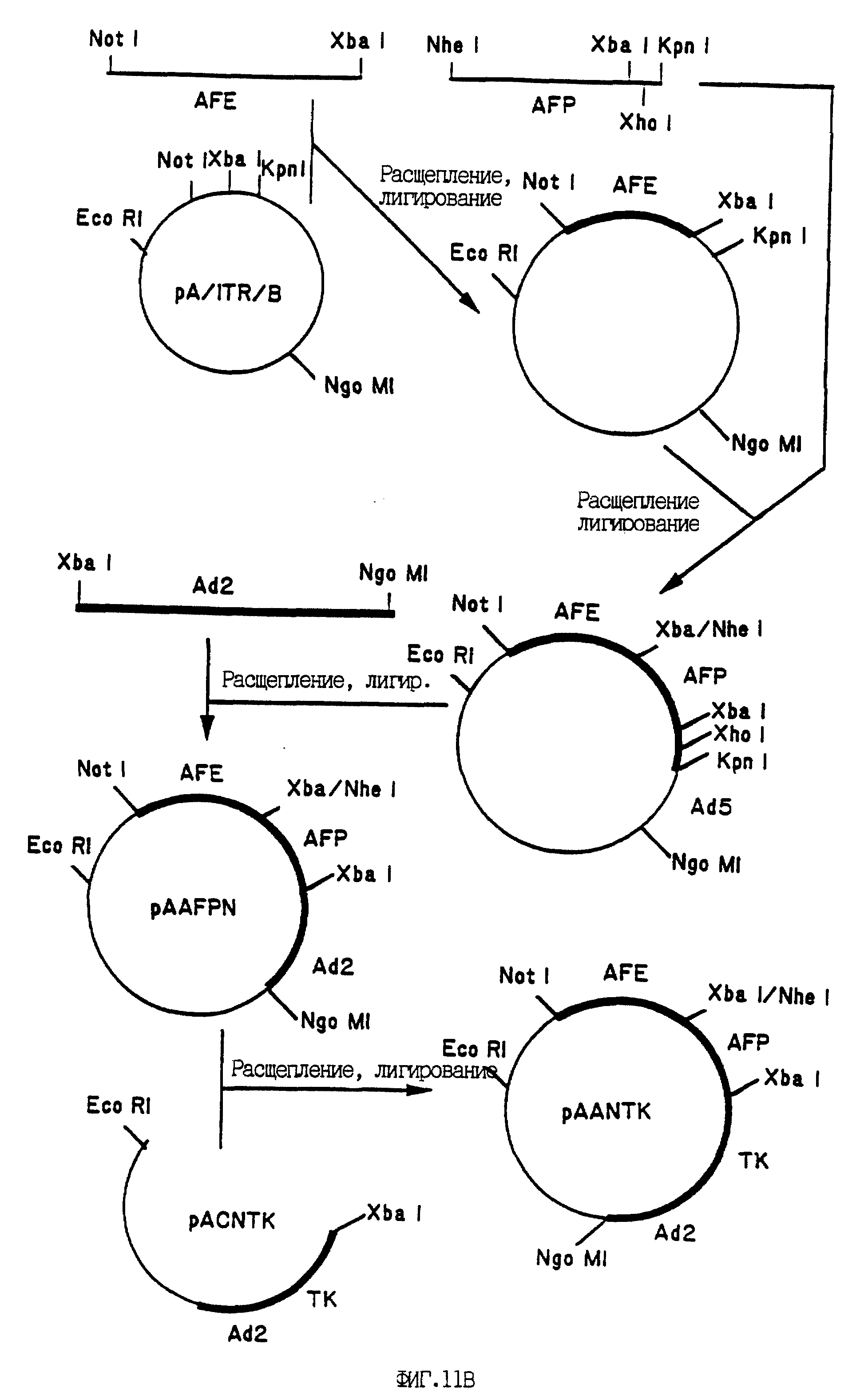
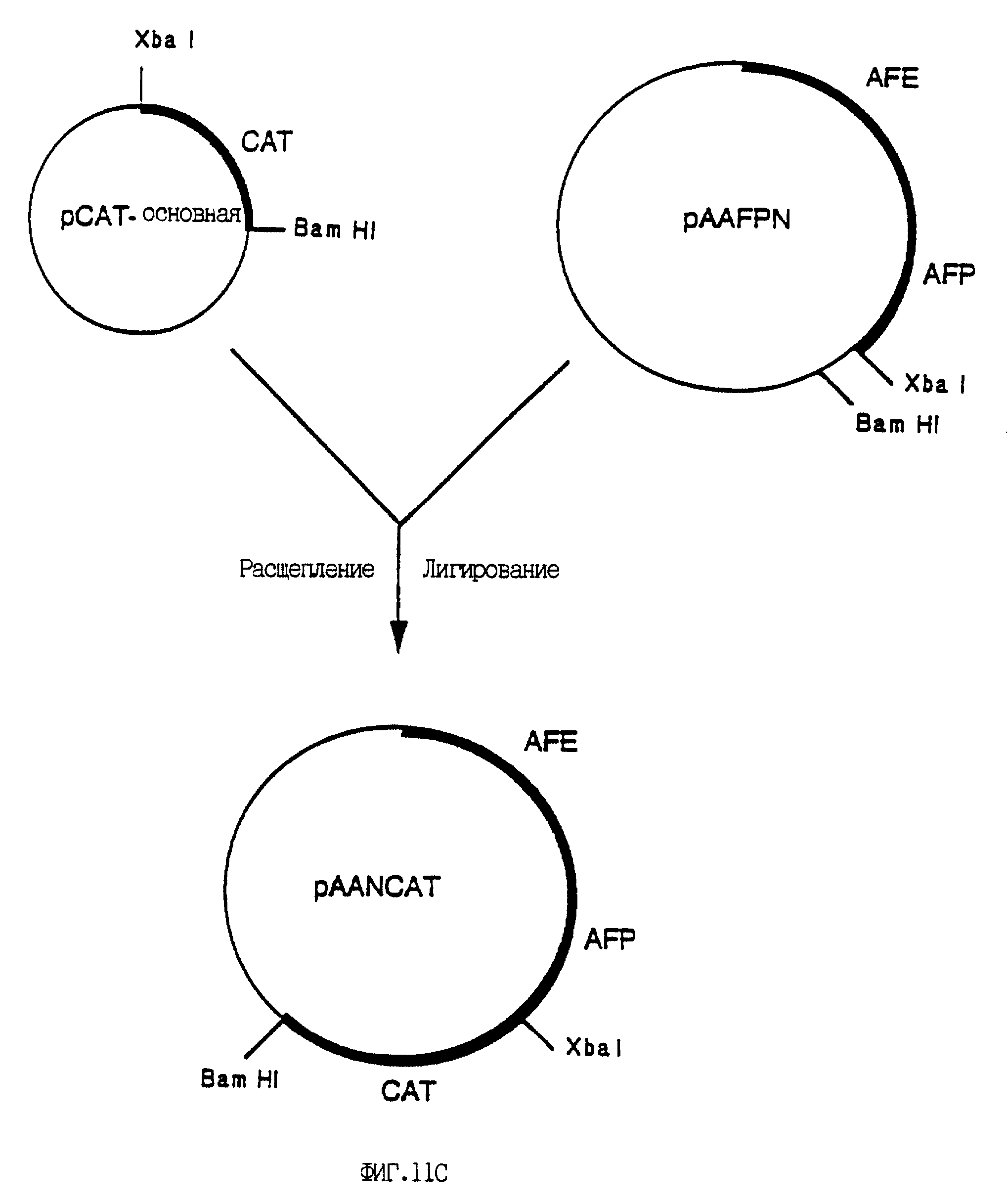
На фиг. 11A - 11C приведены карты рекомбинантных плазмидных конструктов. Конструирование плазмид проводили, как описано ниже. Жирными линиями в конструктах обозначены представляющие интерес гены, а жирным шрифтом обозначены сайты рестрикции, которые использованы для соединения фрагментов с получением плазмид, как это указано стрелками. На фиг. 11A показано конструирование плазмиды pACNTK путем субклонирования гена HSV-TK в полилинкер вектора клонирования, с последующим выделением гена TK с желаемыми концами для клонирования в вектор pACM. Вектор pACN содержит аденовирусные последовательности, необходимые для рекомбинации in vivo, приводящей к образованию рекомбинантного аденовируса (см. фиг. 12). На фиг. 11B приведено конструирование плазмиды pAANTK, начинающееся с ПЦР-амплификации фрагментов, кодирующих энхансер (AFP-E) и промотор (AFP-P) гена альфа-фетопротеина (АФП), с последующим субклонированием их в несколько этапов в конечную плазмиду, где промотор и энхансер АФП находятся "выше" гена HSV-TK, за которым следуют аденовирусные (Ad2) последовательности, необходимые для рекомбинации in vivo, приводящей к образованию рекомбинантного аденовируса. На фиг. 11C показано конструирование плазмиды pAANCAT, начинающееся с выделения из коммерчески доступной плазмиды гена хлорамфениколацетилтрансферазы (CAT) с последующим субклонированием его в плазмиду pAAN (см.выше) с получением конечной плазмиды pAANCAT, в которой транскрипция гена CAT, находящегося в окружении аденовирусных последовательностей, направляется промотором/энхансером АФП.
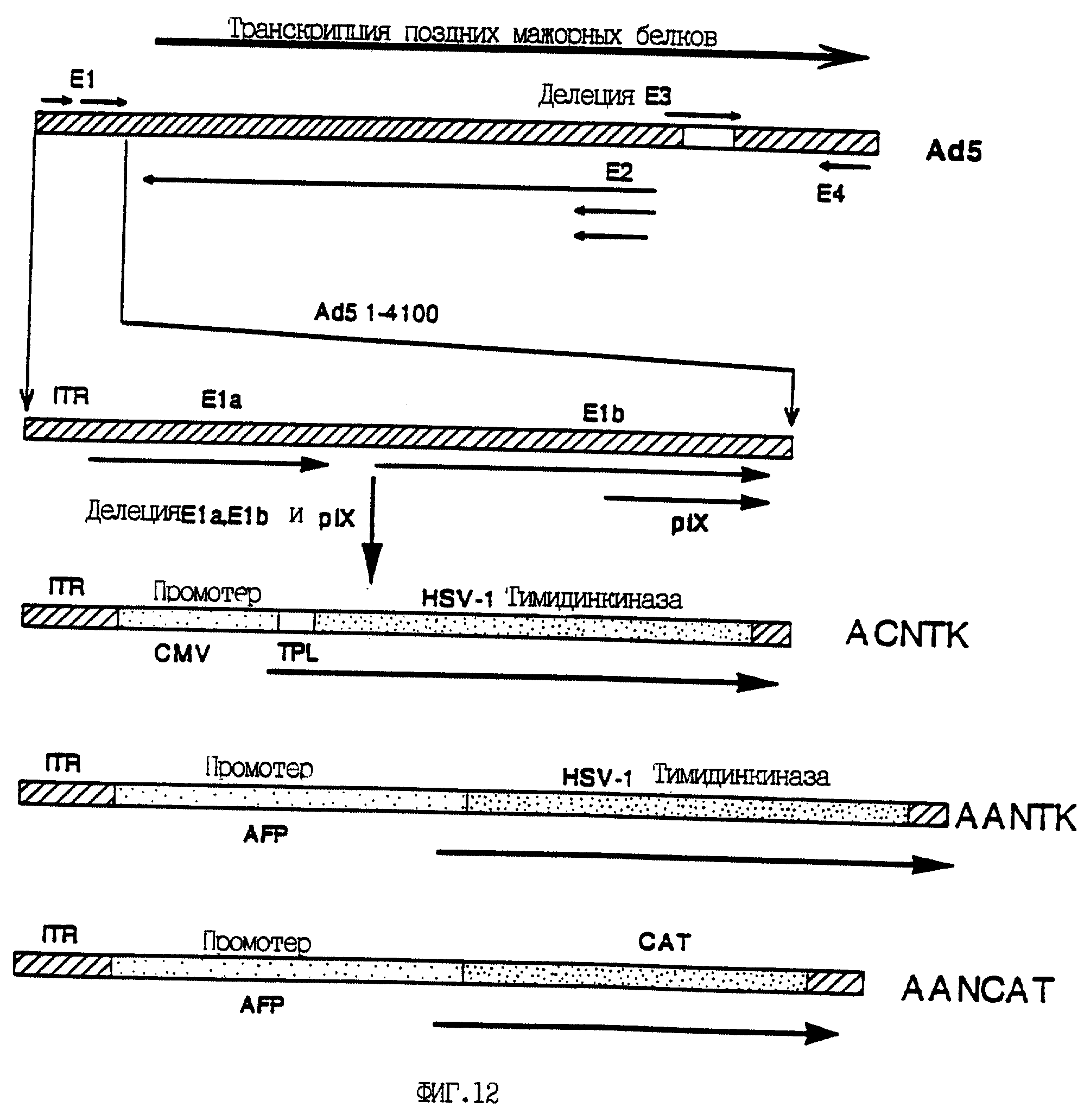
На фиг. 12 приведены схематические карты рекомбинантных аденовирусов ACNTK, AANTK и AANCAT. Для получения рекомбинантных аденовирусов из плазмид, приведенных на фиг. 11, 4 части (20 мкг) каждой из плазмид pACNTK, pAANTK или pAANCAT были линеаризованы рестриктазой EcoRI и котрансфицированы с 1-й частью (5 мкг) большого фрагмента рестрицированного Clal рекомбинантного аденовируса (rACbeta-gal), содержащего делецию области E3 (Wills et al., 1994). В полученных вирусах нуклеотиды 360-4021 вируса Ad5 заменены или промотором CMV и трехчленной лидерной кДНК (TPL) или промотором/энхансером АФП, направляющими экспрессию гена HSV-1 TK или гена CAT. Полученные рекомбинантные аденовирусы обозначены соответственно ACNTK, AANTK и AANCAT.
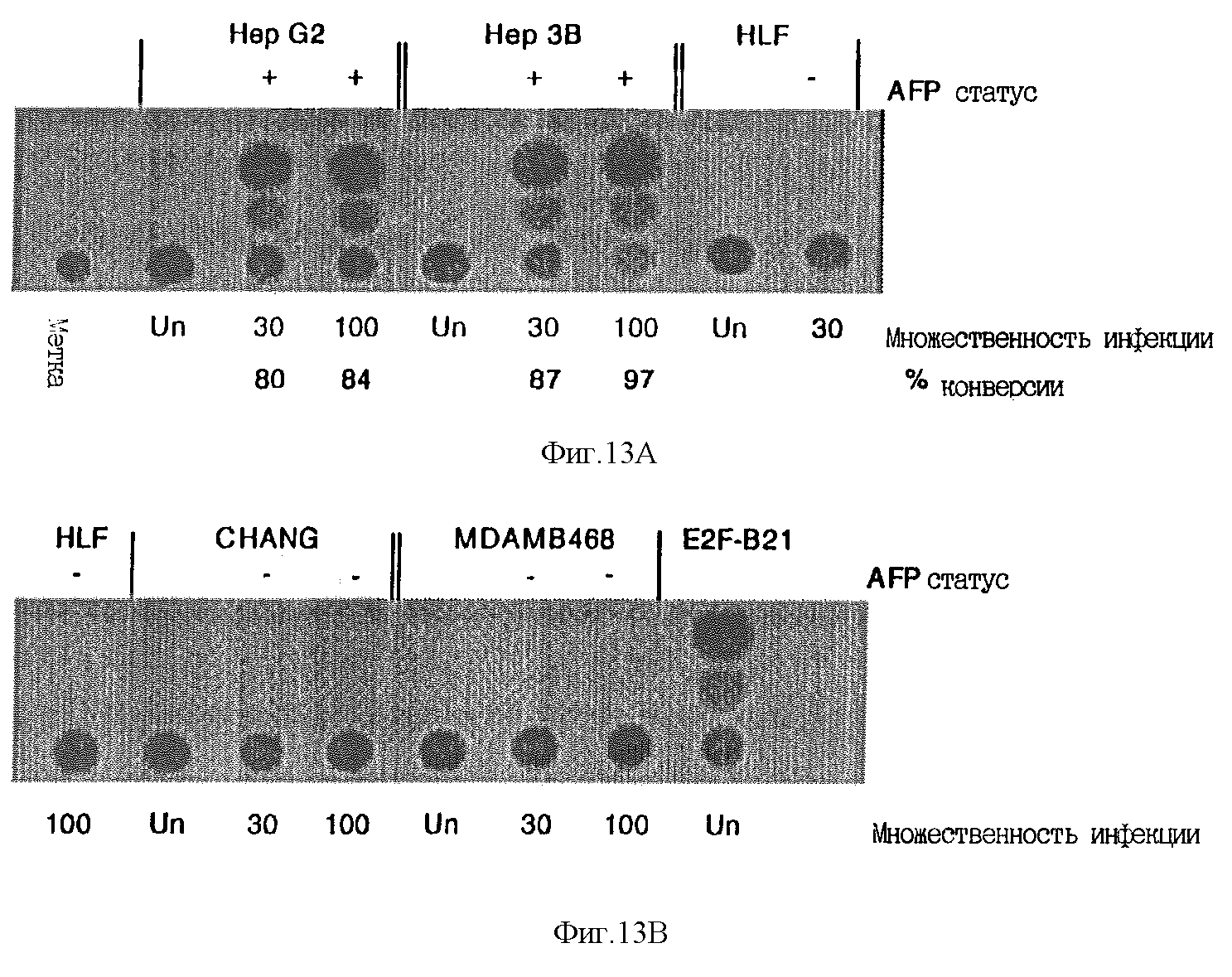
Фиг. 13 демонстрирует специфичность промотора при экспрессии CAT в составе рекомбинантных аденовирусных векторов. Два миллиона клеток (2 · 106) указанной линии инфицировали с множественностью инфекции 30 или 100 рекомбинантным аденовирусом AANCAT или оставляли неинфицированными (UN). Клетки Hep G2 и Hep 3B экспрессировали АФП, тогда как остальные клетки не экспрессировали. Через три дня клетки собирали, концентрацию белка в клеточных лизатах выравнивали и определяли активность CAT, как это описано ниже в разделе "Методы". Равное число неинфицированных клеток служило контролем фоновой активности CAT, при том что14C-хлорамфеникол (только14C) и экстракт стабильной клеточной линии B21, экспрессирующей CAT, служили отрицательным и положительным контролями соответственно. Указан процент конверсии ацетил-CoA, при этом видно, что экспрессия CAT ограничена клетками, экспрессирующими АФП.
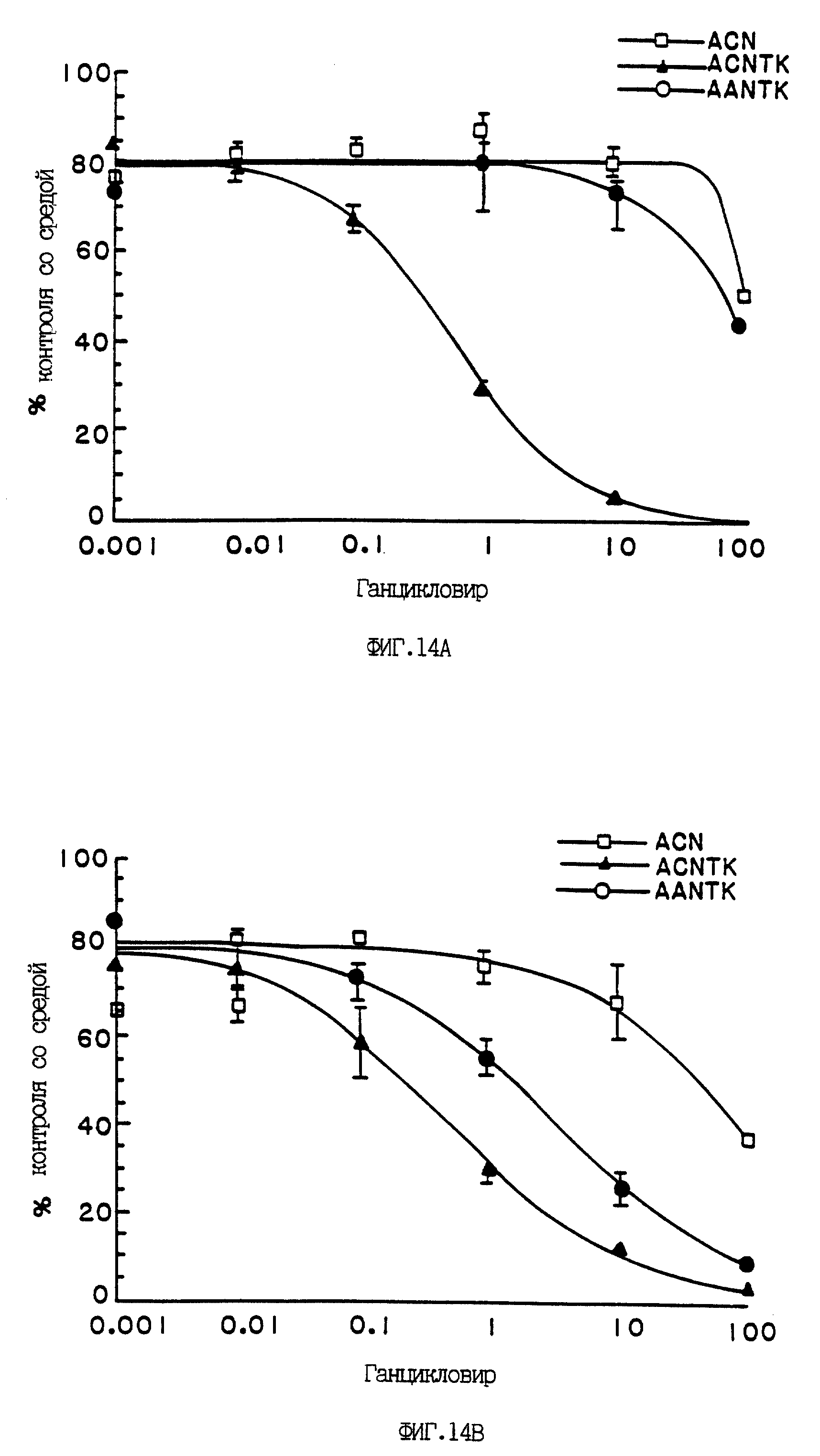
На фиг. 14 показан эффект обработки TK/GCV клеток гепатоцеллюлярных линий и зависимость данного эффекта от специфичности промотора. Клетки линий Hep-G2 (АФП-положительная) и HLF (АФП-отрицательная) инфицировали в течение ночи одним из следующих вирусов: ACNTK [- -], AANTK [- -] или контрольным ACN [-□-] с множественностью инфекции, равной 30, а затем обрабатывали единичной дозой ганцикловира в указанной концентрации. Пролиферацию клеток контролировали, добавляя3H-тимидин приблизительно за 18 часов до сбора клеток. Включение3H-тимидина в клеточные нуклеиновые кислоты измеряли через 72 часа после заражения (TopCount, Packard) и выражали в процентах (среднее +/- среднее отклонение) по отношению к необработанному контролю. Результаты указывают на неизбирательное дозозависимое подавление пролиферации под действием конструкта с CMV-промотором, при том что ген TK под контролем АФП-промотора избирательно подавляет клетки Hep-G2.
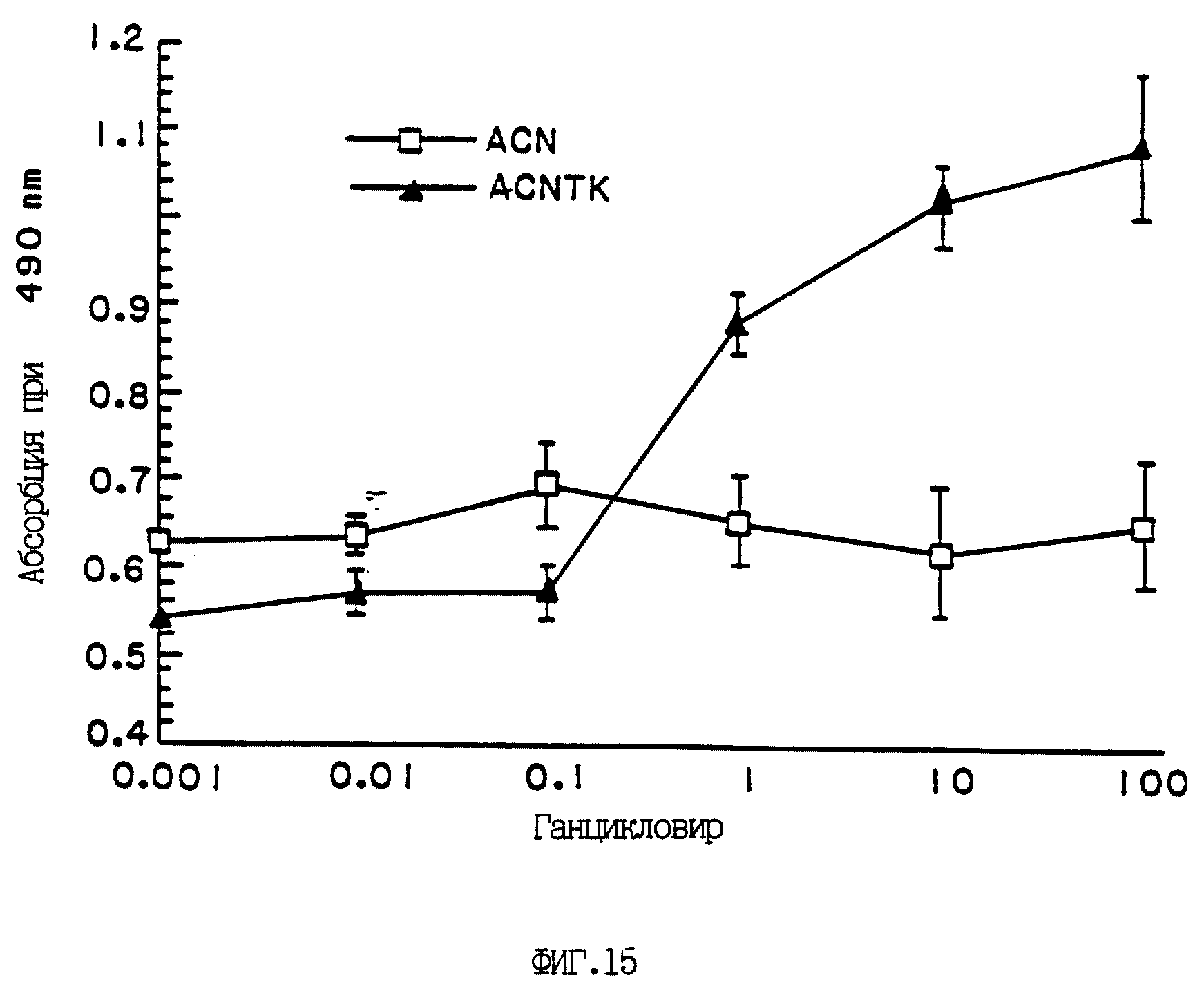
Фиг. 15 иллюстрирует цитотоксичность ACNTK в комплексе с ганцикловиром для клеток гепатоцеллюлярной карциномы (HCC). Клетки HLF заражали с множественностью инфекции 30 вирусом ACNTK [-·-] или контрольным вирусом ACN [-□-], а затем обрабатывали ганцикловиром в указанных дозах. Спустя 72 часа после обработки анцикловиром колориметрически определяли количество лактатдегидрогеназы (LDH), высвобожденной в клеточный супернатант. Приведен график зависимости количества LDH (среднее +/- стандартная ошибка) в зависимости от концентрации ганцикловира для двух обработанных вирусом групп.
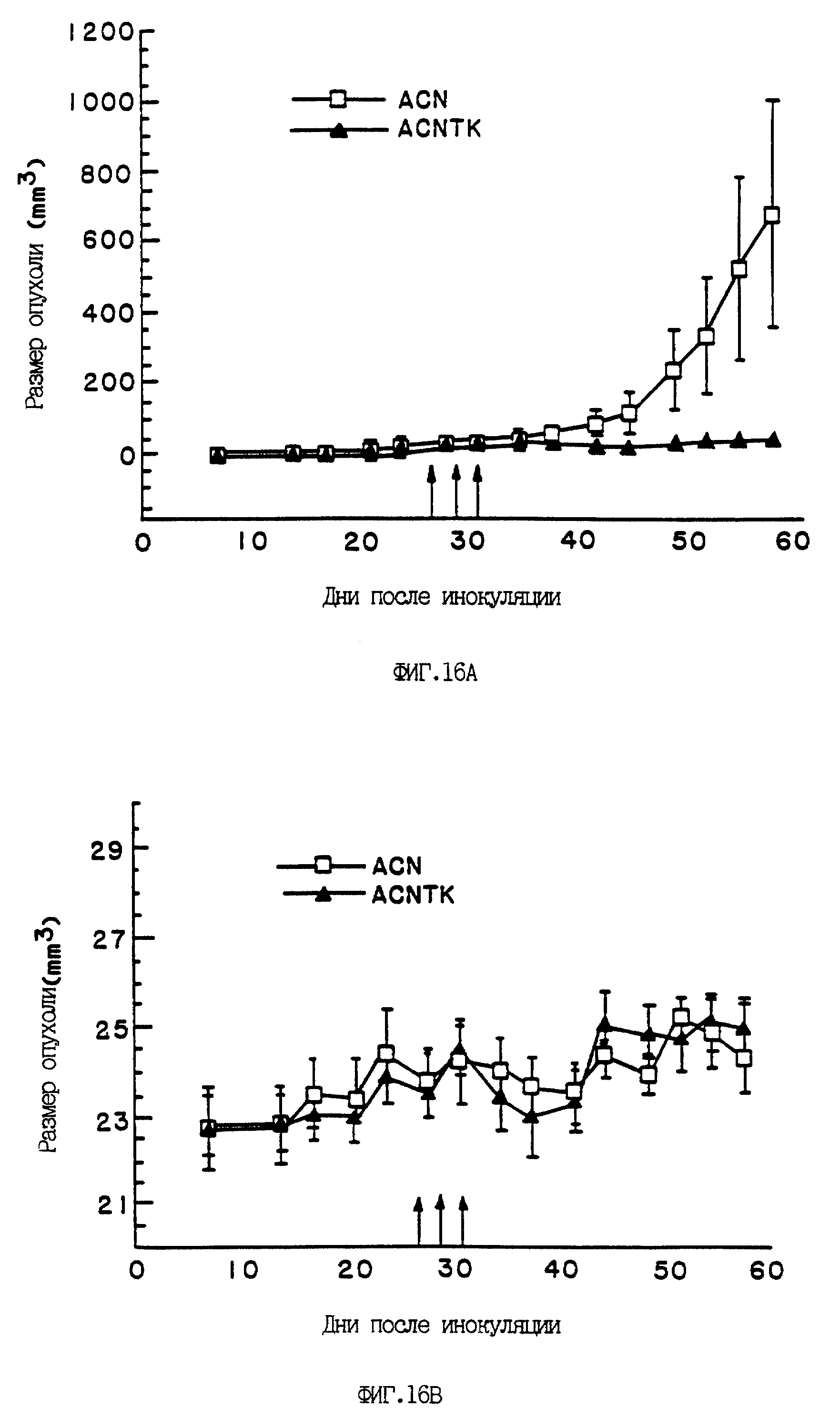
На фиг. 16A и 16B показан эффект ACNTK в комплексе с ганцикловиром на сформировавшиеся гепатоцеллюлярные опухоли (HCC) у голых мышей. Самкам голых мышей подкожно в бок вводили десять миллионов (1·107) клеток Hep 3B и в течение 27 дней давали возможность сформироваться опухолям. Затем мышам интратуморально или перитуморально вводили вирус ACNTK [-·-] или контрольный вирус ACN [-□-] (1·109 инфекционных единиц в объеме 100 мкл) через день, всего три дозы (указаны стрелками). Инъекции ганцикловира (100 мг/кг, интраперитонеально) начинались 24 часа спустя после первого введения вируса и продолжались в течение 10 дней.
На фиг. 6A приведен график зависимости размера опухолей в случае каждого вируса от количества дней, прошедших после заражения (среднее +/- средняя ошибка). На фиг. 6B показан график зависимости среднего веса тела для каждой группы животных, обработанных вирусом, в зависимости от числа дней, прошедших с момента заражения.
Для уменьшения частоты контаминации аденовирусом дикого типа желательно понизить способность аденовируса или клеточной линии к рекомбинации. Например, аденовирус из группы, обладающей низкой гомологией с вирусами группы C, может быть использован для конструирования рекомбинантных вирусов с незначительной предрасположенностью к рекомбинации с Ad5-последовательностями в клетках 293. Однако, альтернативно, снижение частоты рекомбинации между вирусными и клеточными последовательностями может быть достигнуто за счет увеличения размера делеции в рекомбинантном вирусе и, следовательно, уменьшения протяженности общей последовательности между ним и AdS-генами клеток 293.
Делеции, лежащие на расстоянии 3,5 кб от 5'-конца аденовирусного генома, могут затрагивать ген аденовирусного белка IX? и их присутствие в аденовирусном векторе не может считаться желательным.
У аденовирусов ген белка IX кодирует минорный компонент наружного аденовирусного капсида, который стабилизирует девятичленные гексоны, составляющие в основном вирусный капсид (Stewart, 1993). Исследования делеционных мутантов аденовирусов первоначально дали основания считать белок IX не необходимым компонентом аденовируса, хотя его отсутствие было ассоциировано с увеличенной по сравнению с вирусом дикого типа термолабильностью (Colby and Shenk, 1981). Недавно было обнаружено, что белок IX необходим для упаковки полноразмерной вирусной ДНК в капсиды и что в отсутствие этого белка в качестве рекомбинантных вирусов могут размножаться только те, которые содержат геном по меньшей мере на 1 кб меньше, чем у вируса дикого типа (Ghosh-Chooudhury et al., 1987). С учетом указанных ограничений делеции белка IX не рассматривались при конструировании аденовирусных векторов.
В данной заявке приведены ссылки на стандартные учебники молекулярной биологии, которые содержат определения и способы выполнения основных методик, используемых в настоящем изобретении. См., например, Sambrook et al., (1989), а также приведенные в этой книге ссылки. Эта книга и другие цитированные публикации включены в текст настоящего описания в качестве ссылок.
В противоположность известному из уровня техники в настоящем изобретении заявлено использование рекомбинантных аденовирусов, имеющих делеции в гене белка IX, что приводит к снижению риска контаминации вирусом дикого типа при получении вирусных препаратов для диагностических и терапевтических целей, таких как генотерапия. Термин "рекомбинант" означает вирусное потомство, сформированное в результате генно-инженерных манипуляций. Данные делеции могут удалять дополнительно 500-700 пар оснований из последовательностей ДНК, в норме присутствующих в обычных E1-делетированных вирусах (возможны меньшие, не столь желательные делеции частей гена p1X, которые также включены в объем настоящего изобретения) и доступны для рекомбинации с последовательностями Ad 5, интегрированными в геном клеток 293. Рекомбинантные аденовирусы, основанные на любом представителе группы C, серотипа 1, 2, 5 и 6, включены в объем настоящего изобретения. Данным изобретением также защищен рекомбинантный вирус, основанный на гибриде Ad2/Ad5, который обеспечивает экспрессию кДНК p53 человека под контролем главного позднего промотора аденовируса типа 2. Этот конструкт был собран, как показано на фиг. 1. Полученный вирус несет 5'-делецию аденовирусных последовательностей с 357-го по 4020-й нуклеотиды, и в нем удалены гены E1a и E1b, так же как и все кодирующие последовательности белка IX. При этом оставлен интактным сайт полиаденилирования, общий для генов E1b и белка IX, что позволяет терминировать транскрипцию любого желаемого гена. Альтернативно, делеция может быть увеличена на 30-40 пар оснований, что не влияет на соседний ген белка IVa2, хотя в данном случае требуется экзогенный сигнал полиаденилирования для терминации транскрипции генов, введенных в рекомбинантный вирус. Исходный вирус с данной делецией легко размножается в клетках 293 без признаков контаминации вирусом дикого типа и направляет сильную экспрессию p53 с транкрипционного блока, введенного в сайт делеции.
Емкость рекомбинантных вирусов, имеющих описанную выше делецию гена белка IX, составляет около 2,6 килобаз. Это достаточно для большинства генов, включая кДНК p53. Емкость векторов может быть повышена путем введения в аденовирусный "скелет" других делеции, например делеции в ранних областях 3 или 4 (см. Graham and Prevec, 1991). Например, может быть использован аденовирусный "скелет" с делецией 1,9 кб не необходимых последовательностей в ранней области 3. С такой дополнительной делецией емкость вектора возрастает приблизительно до 4,5 килобаз, что достаточно для большинства крупных кДНК, включая кДНК гена-супрессора ретинобластомы.
В настоящем изобретении описан рекомбинантный аденовирусный вектор, характеризующийся полной или частичной делецией ДНК аденовирусного белка IX и имеющий ген, кодирующий чужеродный белок, или его функциональный фрагмент, или мутант. Данные векторы применимы для безопасного рекомбинантного получения диагностических и терапевтических полипептидов и белков, и, что более важно, для введения генов при генотерапии. Так, например, представленный в настоящем изобретении аденовирусный вектор может содержать чужеродный ген, направляющий экспрессию белка, который участвует в регуляции клеточного цикла, такого как p53, Rb, или митозин, или белка, индуцирующего гибель клеток, такого как кодируемый обычным "суицидным" геном тимидинкиназы. (Последний должен использоваться в сочетании с метаболитом тимидинкиназы для достижения эффекта). В представленных в настоящем изобретении векторах могут быть использованы любые кассеты экспрессии. Под "кассетой экспрессии" подразумевается молекула ДНК, включающая промотор/энхансер транскрипции, например, такой как промотор/энхансер цитомегаловируса (CMV), чужеродный ген, и, как в некоторых описанных ниже случаях, сигнал полиаденилирования. Термином "чужеродный ген" обозначена молекула ДНК, не представленная в нужной ориентации и в нужном положении в геномной ДНК аденовируса дикого типа. Чужеродный ген может представлять собой молекулу ДНК размером до 4,5 килобаз. "Вектор экспрессии" означает вектор, обеспечивающий экспрессию вставленных последовательностей ДНК при наращивании в подходящих клетках-хозяевах, другими словами, белок или полипептид, кодируемый данной ДНК, синтезируется в данных клетках. Рекомбинантный аденовирусный вектор экспрессии может содержать часть гена, кодирующего аденовирусный белок IX, при том что биологически активный белок IX или его фрагмент не продуцируются. Примером такого вектора является вектор экспрессии, рестрикционная карта которого приведена на фиг. 1 или 4.
В заявленном в настоящем изобретении векторе могут быть также использованы индуцибельные промоторы. Данные промоторы инициируют транскрипцию только в присутствии дополнительной молекулы. Примерами индуцибельных промоторов служат промоторы генов бета-интерферона, генов теплового шока, гена металлотионеина, а также промоторы генов, экспрессирующихся под действием стероидных гормонов. Тканеспецифическая экспрессия достаточно изучена, и тканеспецифические и индуцибельные промоторы хорошо известны из уровня техники. Указанные гены используются для регулирования экспрессии чужеродного гена после его переноса в клетку-мишень.
В объем настоящего изобретения включен также рекомбинантный аденовирусный вектор экспрессии, аналогичный описанным выше, но имеющий менее протяженные делеции последовательностей гена белка IX, в частности в одном из воплощений изобретения делеции расположены от точки, отстоящей на 3500 пар оснований от 5' конца вирусного генома до точки, отстоящей примерно на 4000 пар оснований от 5'-конца. В другом, отдельном воплощении изобретения рекомбинантный аденовирусный вектор экспрессии может иметь также дополнительную делецию не необходимых последовательностей ДНК аденовируса в ранней области 3 и/или 4 и/или делецию последовательностей ДНК аденовируса, обозначаемых как E1a и E1b.
В данном случае чужеродный ген может представлять молекулу ДНК размером до 4.6 килобаз. В другом воплощении изобретения вектор имеет делецию размером до сорока нуклеотидов, расположенную к 3'-концу по отношению к делеции E1a и E1b и p1X, а также в состав вектора включена чужеродная молекула ДНК, кодирующая сигнал полиадефилирования, расположенная таким образом по отношению к чужеродному гену, чтобы регулировать его экспрессию.
Согласно целям настоящего изобретения рекомбинантный аденовирусный вектор может быть получен из группы аденовирусов дикого типа, серотипов 1, 2, 5 или 6.
В одном из воплощений изобретения рекомбинантный аденовирусный вектор экспрессии имеет в качестве чужеродного гена ген, кодирующий функциональный белок-супрессор опухолей или его биологически активный фрагмент. Термин "функциональный" по отношению к генам-супрессорам опухолей означает гены-супрессоры опухолей, которые кодируют белки-супрессоры опухолей, которые в свою очередь эффективно подавляют превращение нормальных клеток в опухолевые. Функциональные гены могут включать, например, обычные гены дикого типа и модифицированные нормальные гены, которые сохраняют свою способность кодировать эффективные белки-супрессоры опухолей, а также другие противоопухолевые гены, такие как кодирующие "суицидный" белок или токсин.
Аналогично, термин "нефункциональный" используется в данном контексте как синоним термина "инактивированный". Нефункциональные или дефектные гены могут возникнуть в результате различных событий, включая, например, точечные мутации, делеции, метилирование и другие явления, хорошо известные из уровня техники.
В контексте данного изобретения под "активным фрагментом" гена подразумеваются меньшие участки гена, которые сохраняют способность кодировать белки с антиопухолевой активностью. P56 RB, более подробно описанный ниже, является одним из примеров активного фрагмента функционального гена-супрессора опухолей. Модификации генов-супрессоров опухолей, такие как добавления, делеции или замены, также применимы по отношению к их активным фрагментам при том, чтобы последние сохраняли функциональную активность немодифицированного гена.
Другим примером гена-супрессора опухолей является ген ретинобластомы (RB). Полная нуклеотидная последовательность кДНК RB и предсказанная аминокислотная последовательность белка RB (обозначенного p110RB) были опубликованы Lee et al., (1987) и приведены на фиг. 3. Также полезной для экспрессии ретинобластомного белка-супрессора опухолей является молекула ДНК, кодирующая аминокислотную последовательность, приведенную на фиг. 2, или имеющая последовательность ДНК, приведенную на фиг. 3. Усеченный вариант p110RB, p56RB, также может оказаться полезным. Последовательность p56RB опубликована Huang et al., (1991). В представленных в настоящем изобретении векторах могут быть использованы дополнительные гены-супрессоры опухолей, кодирующие соответствующие белки. Для иллюстрации можно назвать некоторые из них: белок p16 (Kamb et al.,1994), белок p21, белок WT1 опухоли Вилма, митозин, h-NUC или белок DCC карциномы прямой кишки. Митозин описан X.Zhu и W-H Lee в заявке на изобретение США N 08/141,239, поданной 22 октября 1993 г, и в дальнейшем частичном продолжении указанной заявки тех же авторов, юридической выписке N P-CJ 1191, выданной 24 октября 1994 г. Оба документа приведены в настоящей заявке в качестве ссылок. Аналогично, h-NUC описан W-H Lee и P.L. Chen в заявке на изобретение США N 08/170,586, поданной 20 декабря 1993 г и приведенной здесь в качестве ссылки.
Как известно из уровня техники, термин "протеин" ("белок") означает линейный полимер аминокислот, соединенных пептидными связями в специфическую последовательность. Термин "аминокислота" относится к D или L стереоизомерным формам аминокислоты, если другое не оговорено специально. В объем настоящего изобретения включены также эквивалентные белки или эквивалентные пептиды, имеющие биологическую активность очищенного белка-супрессора опухолей дикого типа. "Эквивалентные белки" и "эквивалентные полипептиды" означают соединения, отличающиеся от линейной последовательности природных белков или полипептидов, но которые имеют аминокислотные замены, не изменяющие их биологической активности. Данные эквиваленты могут отличаться от исходных последовательностей заменой одной или более аминокислот сходными аминокислотами, например, сходно заряженными аминокислотами, или же заменой или модификацией боковых цепей или функциональных групп.
Определение функционального белка-супрессора опухолей распространяется на любой белок, чье присутствие снижает туморогенность, злокачественность или гиперпролиферативный фенотип клетки-хозяина. Согласно данному определению примерами генов- супрессоров опухолей являются (но не ограничиваются указанными) p110RB, p56RB, митозин, h-NUC и p53. "Туморогенность" означает способность образовывать опухоли или способность вызывать образование опухолей и является синонимом неопластического роста. "Злокачественность" характеризует способность туморогенной клетки к метастазированию и созданию угрозы для жизни хозяйского организма. "Гиперпролиферативный фенотип" характеризует рост и деление клеток, которые происходят со скоростью, превышающей нормальную для клеток данного типа. Понятие "неопластический" относится к клеткам, утратившим эндогенный функциональный белок-супрессор опухолей, или означает неспособность клетки экспрессировать эндогенную нуклеиновую кислоту, кодирующую функциональный белок-супрессор опухолей.
Примером заявленного в настоящем изобретении вектора является рекомбинантный аденовирусный вектор экспрессии, содержащий в качестве чужеродного гена ген, кодирующий белок p53 или его активный фрагмент. Кодирующая последовательность гена p53 приведена в Таблице 1.
Любой из описанных здесь векторов экспрессии является применимым в качестве средства диагностики или терапии. Векторы могут быть использованы для скрининга многочисленных генов-супрессоров опухолей в отношении их применимости для генотерапии. Например, подозреваемые в неопластичности клетки могут быть взяты у пациента или у животного. Затем в соответствующих условиях данные клетки могут быть обработаны эффективным количеством рекомбинантного вектора, заявленного в настоящем изобретении и несущего один из функциональных генов-супрессоров. Вызывает ли введение данного гена реверсию злокачественного фенотипа, можно определить путем колониеобразования в мягком агаре или введением обработанных клеток голым мышам.
Если злокачественный фенотип ревертирован, то данный ген может рассматриваться как положительный кандидат для успешной генотерапии данного пациента или животного. При фармацевтическом применении такой ген может использоваться в сочетании с одним или несколькими фармацевтически приемлемыми носителями. Данные носители хорошо известны из уровня техники и включают такие водные растворы, как забуференный физиологический раствор, или другие растворители - гликолы, глицерин, растительные масла (например, оливковое масло) или органические эфиры, пригодные для инъекций.
Фармацевтически приемлемые носители могут использоваться для доставки растворимых композиций в клетку in vitro или для введения субъекту in vivo. Фармацевтически приемлемые носители могут содержать физиологически приемлемое вещество, которое, например, стабилизирует композицию или же снижает или повышает абсорбцию агента. Такие физиологически приемлемые вещества могут включать, например, углеводы, такие как глюкоза, сахароза и декстраны, антиоксиданты, такие как аскорбиновая кислота и глютатион, хелатирующие агенты, низкомолекулярные белки или другие стабилизаторы. Иные физиологически приемлемые вещества могут представлять собой увлажняющие агенты, эмульгаторы, диспергирующие агенты или консерванты, которые необходимы для предотвращения роста микроорганизмов. Известно множество консервантов, например фенол и аскорбиновая кислота. Опытный исследователь хорошо понимает, что выбор фармацевтически приемлемого носителя, включая физиологически приемлемые соединения, зависит, например, от способа введения полипептида и конкретных физико-химических характеристик отдельного полипептида. Например, такие физиологически приемлемые вещества, как моностерат алюминия или желатин, особенно полезны как замедляющие агенты, которые пролонгируют абсорбцию введенной субъекту фармацевтической композиции. Другие примеры носителей, стабилизаторов и адъювантов можно найти в книге Martin, Remington's Pharm. Sci. , 15th Ed. (Mack Publ.Co., Easton, 1975). Фармацевтические композиции могут быть при желании инкорпорированы в липосомы, микросферы или другой полимерный матрикс (Gregoriages, Liposome Technology, Vol.l (CRC Press, Boca Raton, Florida, 1984). Липосомы, состоящие из фосфолипидов или других липидов, являются нетоксичными, физиологически приемлемыми и метаболизируемыми носителями, которые сравнительно просто приготовить и ввести. В контексте данного изобретения термин "фармацевтическая композиция" относится к любой композиции, упоминаемоей в данном изобретении, в сочетании с одним или более приведенных выше фармацевтически приемлемых носителей. Данные композиции могут применяться в терапевтических или профилактических целях. Они могут быть введены в контакт с клетками-хозяевами in vivo, ex vivo или in vitro в эффективных количествах. In vitro и ex vivo означает приведение в контакт с клетками-хозяевами, как это описано ниже. В случае in vivo способы введения фармацевтического препарата, содержащего описанный в настоящем изобретении вектор, хорошо известны из уровня техники и включают (но не ограничиваются указанными) оральное введение, интратуморальное, внутривенное, внутримышечное или интраперитонеальное. Введение может быть постоянным или прерывистым и изменяться в зависимости от состояния больного, подвергаемого лечению, и характера заболевания, точно так же, как это происходит в случае применения других терапевтических композиций (Landmann et al., 1992; Aulitzky et al. , 1991; Lantz et al., 1990; Supersaxo et al.,1988; Demetri et al., 1989; LeMaistre et al., 1991).
Кроме того, в объем настоящего изобретения включены трансформированные прокариотические и эукариотические клетки- хозяева, например клетки млекопитающих или клетки других животных, в которые введен описанный выше рекомбинантный аденовирусный вектор экспрессии. К подходящим прокариотическим клеткам относятся (но не ограничиваются указанными) такие бактериальные клетки, как клетки E.coli. Методы трансформации клеток-хозяев ретровирусными векторами хорошо известны (см. Sambrook et al., 1989) и включают (но не ограничиваются указанными) трансфекцию, электропорацию и микроинъекцию.
В контексте настоящей заявки термин "животное" считается синонимом понятия "млекопитающее" и означает (но не ограничивается указанными животными) корову, свинью, кошку, обезьяну, собаку, лошадь, мышь, крысу или человека. Кроме того, клетки-хозяева могут включать (но не ограничиваться указанными) любые неопластические или опухолевые клетки, такие как остеосаркома, карцинома яичника, рак молочной железы, меланома, гепатокарцинома, рак легких, рак мозга, рак толстой кишки, гематопоэтические клетки, рак простаты, цервикальная карцинома, ретинобластома, карцинома пищевода, рак мочевого пузыря, нейробластома или рак почек.
Кроме того, любая клеточная линия эукариот, способная экспрессировать E1a и E1b или E1a, E1b и p1X, является подходящим хозяином для данного вектора. В одном из воплощений настоящего изобретения в качестве клеток-хозяев использованы эукариотические клетки 293, доступные из Амириканской коллекции клеточных культур (12301 Parklawn Drive, Rockville, Maryland, U.S. A. 20231).
Любые из описанных здесь трансформированных клеток-хозяев могут быть использованы для целей диагностики или терапии. При фармацевтическом использовании они могут быть комбинированы с различными фармацевтически приемлемыми носителями. Удобные фармацевтически приемлемые носители хорошо известны из уровня техники и, например, некоторые из них описаны выше. Композиции в эффективных количествах могут быть применены терапевтически или профилактически, как более подробно описано ниже.
Настоящее изобретение также относится к способу трансформации клетки-хозяина. Указанный способ состоит в обеспечении контакта клетки-хозяина в соответствующих условиях, т. е. прокариотической или эукариотической клетки-хозяина, с одним из векторов экспрессии, описанных в данной заявке. Трансформированные согласно данному способу клетки-хозяева также включены в объем настоящего изобретения. Контактирование может быть достигнуто in vitro, in vivo или ex vivo с использованием методов, известных из уровня техники (Sambrook et al., 1989), при том, что векторы экспрессии применяются в эффективных количествах. В настоящую заявку также включен способ получения рекомбинантного белка или полипептида при выращивании трансформированной клетки-хозяина в соответствующих условиях, благоприятствующих транскрипции и трансляции введенного чужеродного гена. Методы экспрессии рекомбинантов в различных клетках-хозяевах, таких как клетки млекопитающих, дрожжей, насекомых или в бактериальных клетках, широко известны и включают, например, описанные Sambrook et al., supra. Транслированный чужеродный белок может быть затем выделен обычными методами, такими как очистка на колонках или очистка с помощью антител к данному белку. Выделенный белок или полипептид также включен в объем настоящего изобретения. Применительно к белку термины "очищенный" или "выделенный" означают, что данный белок в значительной степени свободен от исходных белков или нуклеиновых кислот, обычно связанных с данным белком или полипептидом в его естественном окружении (в клетке-хозяине).
Настоящее изобретение также относится к животным, несущим введенные векторы экспрессии или трансформированные клетки-хозяева. Таких "трансгенных" животных получают при помощи способов, хорошо известных из уровня техники, например, как описано в Патенте США N 5,175,384, или обычными методами терапии ex vivo, как описано Culver et al., 1991.
Как более подробно описано ниже, рекомбинантные аденовирусы, экспрессирующие супрессор опухолей - p53 дикого типа, могут эффективно подавлять синтез ДНК и супрессировать рост широкого спектра опухолевых клеток человека, включая и те, которые имеют клиническое значение. Кроме того, рекомбинантные аденовирусы могут экспрессировать гены-супрессоры опухолей, такие как p53, в развившихся in vivo опухолях без необходимости прямого введения в опухоль или предварительной обработки раковых клеток ex vivo. Экспрессируемый p53 является функциональным и эффективно подавляет рост опухоли in vivo и значительно увеличивает время выживания, как это показано на модели голых мышей, инокулированных клетками рака легкого человека.
Таким образом, заявленные в настоящем изобретении векторы особенно подходят для генотерапии. Соответственно, в объем настоящего изобретения включены и способы генотерапии с использованием указанных векторов. Вектор очищают и затем эффективное количество данного вектора вводят субъекту in vivo или ex vivo. Методы генотерапии хорошо известны из уровня техники. См., например, Larrik, J.W. и Burck, K.L. (1991) и Kreigler, M. (1990). "Субъект" означает любое животное, млекопитающее (корову, свинью, кошку, собаку, лошадь, мышь, крысу) или же человека. При том, что введенный в состав вектора чужеродый ген кодирует белок-супрессор опухолей или другой противоопухолевый белок, данный вектор применим для лечения субъекта и снижения у последнего числа гиперпролиферативных клеток, для подавления пролиферации опухолевых клеток или облегчения конкретной специфической патологии. Патологические гиперпролиферативные клетки являются характерными для следующих заболеваний: гиперплазия щитовидной железы - болезнь Грэйва, псориаз, доброкачественная гипертрофия простаты, синдром Ли-Фраумени, включая рак молочной железы, саркомы и другие неоплазии, рак мочевого пузыря, рак толстого кишечника, рак легких, различные лейкозы и лимфомы. Примерами непатологических гиперпролиферативных клеток служат эпителиальные клетки протоков молочной железы при лактации и клетки, ассоциированные с заживлением ран. Патологические гиперпролиферативные клетки характеризуются потерей контактного ингибирования и сниженной способностью к избирательному прикреплению, что отражает изменения поверхностных свойств клетки и последующее нарушение межклеточных взаимодействий. Данные нарушения включают стимуляцию деления и способность секретировать протеолитические ферменты.
Кроме того, в настоящее изобретение включен способ истощения соответствующего образца потологических гиперпролиферативных клеток млекопитающих, которые контаминируют гематопоэтические предшественники в процессе пересадки костного мозга. Это достигается путем введения гена-супрессора опухолей дикого типа в клетки соответствующего образца (полученные из аутологичной периферической крови или костного мозга) посредством заявленного в настоящем изобретении вектора. Термин "соответствующий образец" подразумевает гетерогенный клеточный препарат, полученный от пациента, т.е. смешанную популяцию клеток, содержащую как фенотипически нормальные, так и патологические клетки. Термин "введение" включает (но не ограничивается указанным ниже) введение в клетку или внутривенную инъекцию, прямое введение в опухоль, интратуморальное введение, интраперитонеальное введение, аэрозольное введение в легкие или местное применение. Подобные процедуры могут сочетаться с применением фармацевтически приемлемого носителя из числа описанных выше.
Термин "сниженная туморогенность" введен для характеристики клеток, которые были превращены в менее туморогенные или нетуморогенные клетки. Клетки со сниженной туморогенностью или не образуют опухолей при введении in vivo, или же лагпериод до проявления опухолевого роста in vivo составляет недели или месяцы и/или рост опухолевой массы, определяемый в трех измерениях, происходит медленнее по сравнению с опухолями, в которых ген-супрессор опухолей инактивирован или нефункционален.
Термин "эффективное количество" означает такое количество вектора или антиракового белка, которое позволяет достигнуть положительного результата при контроле пролиферации клеток. Например, одна доза содержит от 108 до 1013 инфекционных единиц вируса. Типичный курс лечения предусматривает введение одной такой дозы ежедневно в течение пяти дней. Эффективное количество может варьировать в зависимости от характера заболевания, состояния больного и других факторов, хорошо известных из уровня техники. Эффективные количества легко определяются опытным исследователем.
В объем настоящего изобретения включен также способ облегчения заболевания, для которого характерно наличие гиперпролиферативных клеток или генетического дефекта, путем введения субъекту эффективного количества вектора, описанного выше и содержащего чужеродный ген, продукт которого способен снижать тяжесть заболевания при соответствующих условиях. Термин "генетический дефект" подразумевает любое заболевание или аномалию, возникшие в результате наследуемых факторов, например, серповидно-клеточную анемию или болезнь Тэй-Сакса.
Настоящее изобретение относится также к способу снижения пролиферации опухолевых клеток у субъекта путем введения в опухолевую массу эффективного количества аденовирусного вектора экспрессии, содержащего антиопухолевый ген, отличающийся от гена-супрессора опухолей. Антиопухолевый ген может кодировать, например, тимидинкиназу (ТК). Затем субъекту вводят эффективное количество терапевтического агента, который в присутствии антиопухолевого гена является токсичным для клетки. В конкретном случае тимидинкиназы такими терапевтическими агентами являются метаболиты тимидинкиназы, например, ганцикловир (GCV), 6-метоксипурин арабинонуклеозид (araM) или их функциональные эквиваленты. Для проявления токсического эффекта по отношению к клетке-хозяину ген тимидинкиназы и метаболит тимидинкиназы должны использоваться конкурентно. Однако в клетке GCV фосфорилируется и становится мощным ингибитором синтеза ДНК, а araM превращается в токсический анаболит araATP. Для снижения пролиферации опухолевых клеток могут быть использованы и другие антиопухолевые гены в сочетании с соответствующими терапевтическими агентами. Подобные сочетания генов и терапевтических агентов хорошо известны из уровня техники. Другим примером может служить заявленный в настоящем изобретении вектор, экспрессирующий фермент цитозиндезаминазу. Подобный вектор может быть использован в сочетании с введением препарата 5-фторурацил (Austin and Huber, 1993). Еще один пример - сочетание недавно открытого гена Deo-дельта E.coli с 6-метил-пурин-2'-дезоксинуклеозидом (Sorscher et al., 1994).
Использование описанных выше генов-супрессоров опухолей, а также использование других антиопухолевых генов как самих по себе, так и в сочетании с соответствующим терапевтическим агентом, позволяет оказывать влияние на неконтролируемый рост клеток и пролиферативные характеристики опухолей. Таким образом, в настоящем изобретении заявлен способ терапии для остановки неконтролируемого клеточного роста у пациента, что приводит к облегчению симптомов заболевания и смягчению имеющейся у больного кахексии. Результаты подобной терапии выражаются (но не ограничиваются указанным) в увеличенном сроке жизни пациента, уменьшении опухолевой массы, апоптозе опухолевых клеток или снижении числа циркулирующих опухолевых клеток. Способы оценки положительных эффектов при таком терапевтическом подходе хорошо известны из уровня техники.
Объектом настоящего изобретения является рекомбинантный аденовирусный вектор экспрессии, характеризующийся частичной или полной делецией ДНК аденовирусного белка IX, и несущий чужеродный ген, кодирующий чужеродный белок, при том, что чужеродный белок является продуктом суицидного гена или его функциональным агентом. Описанный выше антиопухолевый ген ТК служит примером суицидного гена, поскольку продукт его экспрессии является или может являться летальным для клетки. Для ТК летальность индуцируется наличием GCV. Ген ТК получен из вируса простого герпеса при помощи методов, хорошо известных из уровня техники. Для целей настоящего изобретения источником гена тимидинкиназы (ТК) вируса простого repneca (HSV-1) служит плазмидар/BKTK в клетках E.coli HB 101 (из ATCC # 39369). Однако имеются и многие другие источники.
Ген ТК может быть введен в опухолевую массу посредством аденовирусного вектора экспрессии в сочетании с соответствующим фармацевтически приемлемым носителем. Введение может быть проведено, например, путем прямой инъекции в опухолевую массу. При таких специфических неоплазиях, как гепатоцеллюлярная карцинома (HCC), можно использовать введение в печеночную артерию, поскольку кровоснабжение большинства HCC осуществляется именно через эту артерию. Для контроля пролиферации клеток опухоли и снижения массы опухоли индуцируется гибель клеток путем введения пациентам метаболита ТК, такого как ганцикловир (GCV). Метаболит ТК может быть введен системно, локально в опухоль или, в случае HCC, инъецирован в печеночную артерию. Метаболит ТК предпочтительно вводить ежедневно, однако в зависимости от обстоятельств частота введений может быть повышена или снижена. Метаболит ТК может вводиться одновременно или последовательно с введением ТК- содержащего вектора. Опытные исследователи могут определить дозы и продолжительность введения, которые являются терапевтически эффективными.
Способ опухолеспецифической доставки гена-супрессора опухолей может быть осуществлен при обеспечении контакта ткани- мишени данного животного с эффективным количеством рекомбинантного аденовирусного вектора экспрессии, заявленного в настоящем изобретении. Ген должен кодировать антиопухолевый агент, такой как функциональный ген-супрессор опухолей или суицидный ген. "Обеспечение контакта" означает любой способ эффективной доставки вектора, например, внутригуморальное введение.
Использование заявленного в настоящем изобретении аденовирусного вектора для приготовления медицинских препаратов, предназначенных для терапии определенных заболеваний, также является предметом настоящего изобретения.
Ниже приведены примеры, чтобы проиллюстрировать, но не ограничить, объем настоящего изобретения.
ЭКСПЕРИМЕНТ N I
Для указанных исследований в качестве исходного материала была выбрана плазмида pAd/MLP/p53/E1b-. Данная плазмида основана на pML2, производной от pBR322 (pBR322 с делецией
оснований 1140-2490), и содержит последовательности аденовируса типа 5 с 1-й по 5788-ю пару оснований с делецией оснований 4357-3327. Вместо делеции Ad5 357-3327 введена транскрипционная кассета,
состоящая из позднего промотора Ad2, трехчленной лидерной кДНК Ad2 и кДНК p53 человека. Таким образом, получен типичный вектор с заменой последовательностей E1, делецией генов E1a и E1b Ad5, но
содержащий ген белка IX Ad5 (см. обзор, посвященный аденовирусным векторам: Graham and Prevec (1992)). ДНК Ad2 была получена от Gibco BRL. Эндонуклеазы рестрикции и Т4 ДНК-лигаза были получены от New
England Biolabs. Компетентные клетки E.coli DH5-альфа были куплены у Gibco BRL, а клетки 293 получены из Американской Коллекции Клеточных Культур (ATCC). Смола для очистки ДНК Prep-A-Gene была куплена
у фирмы BioRad. LB-бульон для выращивания бактерий был куплен у фирмы Difco. Колонки Quagen для выделения ДНК были куплены у фирмы Quagen, Inc. Вирус Ad5 dl327 был получен от R.J.Schneider, NYU. Набор
для трансфекции ДНК MBS был куплен у фирмы Stratagene.
Один (1) мкг pAd/MLP/p53/E1b- рестрицировали 20 единицами каждого из ферментов Ecl 136II и NgoMI в соответствии с рекомендациями производителя. Пять (5) мкг ДНК Ad2 рестрицировали 20 единицами каждого из ферментов DraI и NgoMI в соответствии с рекомендациями производителя. Рестрикционные смеси были нанесены в раздельные лунки 0.8%-ного агарозного геля и подвергнуты электрофорезу при 100 вольтах в течение 2 часов. Рестрикционный фрагмент pAd/MLP/p53/E1b- 4268 кб и рестрикционный фрагмент 6437 кб Ad2 были выделены из геля при помощи смолы для экстракции ДНК Prep-A-Gene в соответствии с рекомендациями производителя. Рестрикционные фрагменты смешали и обработали Т4 ДНК-лигазой в общем объеме 50 мкл при 16oC в течение 16 часов согласно рекомендациям производителя. Вслед за лигированием 5 мкл реакционной смеси использовали для трансформации клеток E.coli OH5-альфа (отбор вели по устойчивости к ампициллину). Шесть бактериальных колоний, полученных в результате данной процедуры, использовали для инокуляции 2-мл культур в ростовой среде LB, которые подращивали в течение ночи при 37oC при постоянном встряхивании. По стандартной методике (Sambrook et al., 1989) из каждой бактериальной культуры была выделена плазмидная ДНК. Четвертая часть каждого образца плазмидной ДНК была подвергнута рестрикции 20-ю единицами эндонулкеазы Xhol для отбора "правильного" рекомбинанта, содержащего рестрикционные фрагменты Xhol в 3627, 3167 и 1445 пар оснований. Пять из шести проверенных образцов содержали искомый рекомбинант. Один из данных клонов был использован для инокуляции 1-литровой культуры в среде LB и последующего выделения больших количеств плазмидной ДНК. После инкубации в течение ночи из 1-литровой культуры была выделена ДНК с помощью колонок Qiagen согласно рекомендациям производителя. Полученная плазмида была обозначена Pad/MLP/p53/PIX-. Образцы данной плазмиды были депонированы в Американской Коллекции Культур Клеток (ATCC), 12301 Parklawn Drive, Rockville, Maryland, USA, 12301, 22 октября 1993. Депонирование произведено согласно условиям Будапештского соглашения о международном депонировании микроорганизмов для целей патентования. Данный депозит получил номер ATCC 75576.
Для конструирования рекомбинантного аденовируса 10 мкг плазмиды Pad/MLP/p53/PIX-линеаризовали 40 единицами рестриктазы EcoRI. ДНК аденовируса 5-го типа dl327 (Thimmappaya, 1982) рестрицировали эндонуклеазой Clal и большой фрагмент (приблизительно 33 т.п.о.) очищали центрифугированием в градиенте сахарозы. Десять (10) мкг рестрицированной EcoRI плазмиды Pad/MLP/p53/PIX- и 2.5 мкг рестрицированной Clal ДНК Ad5 dl327 смешивали и использовали для трансфекции приблизительно 106 клеток 293 с помощью набора MBS для трансфекции клеток млекопитающих согласно рекомендациям производителя. Через восемь (8) дней после трансфекции клетки 293 рассевали в соотношении 1: 3 в свежей среде и еще через 2 дня на трансфицированных клетках становился виден цитопатический эффект, индуцированный аденовирусом. На 13-й день после трансфекции из клеток по стандартной методике (Graham and Prevec, 1991) выделяли ДНК и проводили рестрикционный анализ при помощи рестриктазы Xhol. Экспрессию p53, обусловленную вирусом, верифицировали последующей инфекцией клеток остеосаркомы Saos-2 содержащим вирус лизатом и иммуноблоттингом с анти-p53 моноклональными антителами 1801 (Novocasta Lab.Ltd., UK).
ЭКСПЕРИМЕНТ N II
Материалы и методы
Линии клеток
Рекомбинантные аденовирусы выращивали и накапливали на клетках 293 (эмбриональная почка человека) ATCC CRL 1573, выращиваемых на среде DME с 10%
телячьей сыворотки (Hyclone). Клетки Saos-2 выращивали на среде Кайна с добавлением 15% эмбриональной телячьей сыворотки. Клетки HeLa и Hep 3B выращивали на среде DME с добавлением 10% эмбриональной
телячьей сыворотки. Все остальные клетки выращивали на среде Кайна с добавлением 10% эмбриональной телячьей сыворотки. Клетки Saos-2 были любезно предоставлены доктором Эриком Станбриджем. Все
остальные клеточные линии были получены из ATCC.
Конструирование рекомбинантных аденовирусов
Для конструирования Ad5/p53 вирусов 1.4 килобазный Hindlll-Smal фрагмент,
содержащий полноразмерную кДНК p53 (Таблица 1), был выделен из плазмиды pGEM1-p53-B-T (любезно предоставлена Dr.Wen Hwa Lee) и вставлен в сайт клонирования вектора экспрессии pSP72 (Promega) с
использованием стандартных методов клонирования (Sambrook et al., 1989). Вставка p53 была выделена из данного вектора после рестрикции Xhol-Bglll и последующего гель-электрофореза. Затем кодирующие
последовательности p53 встраивали в аденовирусные векторы для переноса генов pNL3C или pNL3CMV (любезно предоставлены Dr.Robert Schneider), которые содержат инвертированный концевой повтор Ad5 5',
вирусные сигналы упаковки, энхансер E1a и главный промотор поздних белков Ad2 (MPL) или промотор раннего гена цитомегаловируса человека (CMV), за которыми следуют трехчленная лидерная кДНК и
последовательности 3325-5525 Ad5 в основной части PML2. В этих новых конструктах область E1 Ad5 (360-3325 пары оснований) заменена p53 под контролем Ad2 MPL (A/М/53) или промотором CMV человека
(A/C/53), причем в обоих случаях за p53 следует трехчленная лидерная кДНК (см. фиг. 4). Вставки p53 используют остающийся "ниже" сайт полиаденилирования E1b.
Были получены дополнительные рекомбинанты с p53 под контролем MPL или CMV (A/M/N/53, A/C/N/53), которые имели делецию 705 нуклеотидов последовательностей Ad5 для удаления кодирующей области белка IX (PIX). В качестве контроля из исходной плазмиды pNL3C был получен рекомбинантный аденовирус без вставки p53 (A/М). Другим контролем служил рекомбинантный аденовирус, кодирующий ген бета-галактозидазы под контролем промотора CMV (A/C/бета-гал). Плазмиды линеаризовали рестрикцией Nrul или EcoRI и котрансфецировали с большим фрагментом Clal-рестрицированного мутантного Ad5 dl309 или dl327 с использованием набора для Ca/PO4-трансфекции (Stratagene). Выделяли вирусные бляшки и рекомбинанты идентифицировали рестрикционным анализом и ПЦР с использованием рекомбинантных специфических праймеров к последовательностям трехчленной лидерной ДНК, расположенным "ниже" последовательностей кДНК p53. Затем рекомбинантный вирус очищали методом конечных разведений, выделяли вирусные частицы и титровали стандартными методами (Graham and van der Eb, 1973; Graham and Prevec, 1991).
Обнаружение белка p53
Клетки Saos-2 или Hep3B (5·105) инфицировали указанными рекомбинантными аденовирусами в течение 24 часов с возрастающей множественностью инфекции (в
бляшкообразующих единицах вируса на клетку). Затем клетки однократно
ополаскивали PBS и лизировали в буфере следующего состава: 50 мМ Tris-HCl, pH 7.5, 250 мМ NaCl, 0.1% NP4O, 50 мМ NaF, 5 мМ ЕДТА, 10 мкг/мл апротинина, 10 мкг/мл лейпептина и 1 мМ PMSF.
Клеточные белки (приблизительно 30 мкг) разделяли в 10% SDS-PAGE и переносили на нитроцеллюлозу. Мембраны инкубировали с анти-альфа-p53 антителами PAb 1801 (Novocastro), а затем с конъюгатом овечьих
антимышиных lgG с пероксидазой хрена. Белок p53 визуализировали хемилюминесценцией (набор ECL, Amersham) с пленкой Kodak XAR-5.
Определение скорости синтеза ДНК
Клетки
(5·103/лунку) засевали в ячейки 96-луночных планшетов (Costar) и позволяли им прикрепиться в течение ночи (37oC, 7% CO2). Затем клетки в течение 24 часов
инфицировали частицами очищенного рекомбинантного вируса с множественностью инфекции от 0,3 до 100. Спустя 24 часа после заражения производили замену среды и инкубацию продолжали до 72 часов. За 18
часов до сбора клеток добавляли 3H-тимидин (Amersham, 1 мкКи/лунку). Клетки осаждали на стекловолоконные фильтры и определяли уровень связавшейся радиоактивности при помощи сцинтилляционного
бета-счетчика. Включение 3H-тимидина выражали как процент (+/- среднее отклонение) от контроля среды и ставили в зависимость от множественности инфекции.
Туморогенность для голых
мышей
Приблизительно 2.4·108 клеток Saos-2, растущих на флаконах Т225, обрабатывали буфером (1% сахарозы в PBS), содержащим очищенный вирус A/M/N/53 или A/М с
множественностью инфекции от 3 до 30. После инкубации с вирусом в течение ночи клетки вводили подкожно в левый и правый бок атимическим голым мышам BALB/c (по 4 мыши в группе). В один бок вводили
клетки, обработанные вирусом A/M/N/53, а в другой - вирусом A/М, таким образом, каждая мышь служила и для контроля и для опыта. Животные получали также двусторонние инъекции клеток, обработанных
буфером, что служило дополнительным контролем. Размеры опухолей (длина, ширина и высота), а также вес тела определяли дважды в неделю в течение 8 недель. Объем опухолей определяли для каждого
животного исходя из сферической геометрии опухоли, при том что за радиус принимали одну вторую от среднего значения всех трех измерений опухоли.
Анализ опухолевых РНК
Атимическим голым мышам BALB/c (приблизительно 5-недельного возраста) вводили подкожно в правый бок 1·107 клеток H69 (мелкоклеточная карцинома легкого). Опухолям позволяли
сформироваться в течение 32-х дней, к этому времени они достигали объема 25-50 мм3. Мышам проводили перитуморальные инъекции рекомбинантных аденовирусов A/C/53 или A/C/бета-гал (2·
109 бляшкообразующих единиц, бое) в подкожное пространство под опухолевой массой. Опухоли вырезали на 2-й и 7-й дни после введения аденовирусов и ополаскивали в PBS. Опухоли
гомогенизировали и выделяли из них тотальную РНК с помощью набора TriReagent (Molecular Research Center, Inc.,). ПолиА РНК выделяли с помощью системы выделения мРНК PolyATract (Promega) и
приблизительно 10 нг каждого образца РНК использовали для определения экспрессии мРНК p53 с помощью РТ-ПЦР (Wang et al., 1989). Праймеры были сконструированы таким образом, чтобы амлифицировать
последовательности между аденовирусной трехчленной лидерной кДНК и расположенной "ниже" кДНК p53. Тем самым достигалась амплификация только рекомбинантных, но не эндогенных последовательностей p53.
Гемотерапия опухолей у голых мышей посредством p53
Приблизительно 1·107 опухолевых клеток H69 (SCLC) в объеме 200 мкл вводили подкожно самкам атимических голых
мышей BALB/c. Опухоли формировались в течение 2-х недель и затем животных рандомизировали по размеру опухолей (N= 5/группа). Проводили перитуморальные инъекции аденовируса A/M/N/53 или контрольного
аденовируса A/М (2·109 бое/инъекция) или только буфера (1% сахароза в PBS) дважды в неделю, всего 8 доз на группу. В течение 7 недель дважды в неделю определяли размеры опухолей и
вес тела; объем опухолей оценивали, как описано ранее. Кроме того, определяли действие терапии на выживание мышей.
Результаты
Конструирование рекомбинантного
p5З-аденовируса
Содержащие p53 аденовирусы конструировали путем замены части области E1a и E1b аденовируса типа 5 на кДНК p53 под контролем промотора Ad2 MPL (A/М/53) или промотора CMV
(A/C/53) (схематически представлено на фиг. 4). Такая замена области E1 резко нарушает способность рекомбинантных аденовирусов к репликации, позволяя им размножаться лишь в клетках 293, которые служат
источником продуктов гена El Ad5 по "транс" типу (Graham et al., 1977). После идентификации рекомбинантных аденовирусов как рестрикционным анализом, так и ПЦР, полная кДНК p53 одного из рекомбинантных
аденовирусов (A/М/53) была просеквенирована для того, чтобы убедиться в отсутствии мутаций. Затем очищенные препараты p53-рекомбинантов были использованы для инфекции клеток HeLa с целью обнаружения
аденовируса дикого типа. Клетки HeLa, которые являются непермиссивными для репликации E1-делетированного аденовируса, были инфицированы 1-4·109 инфекционных единиц рекомбинантного
аденовируса, затем их культивировали в течение 3-х недель и наблюдали появление цитопатического действия (ЦПД). При этом репликации рекомбинантного аденовируса или контаминации вирусом дикого типа
обнаружено не было. В культурах контрольных клеток, инфицированных аденовирусом дикого типа, ЦПД было очевидным, и чувствительность данного метода составляет приблизительно 1 из 109.
Экспрессия белка p53 с помощью рекомбинантного аденовируса
Для того чтобы определить, экспрессируют ли рекомбинантные p53-аденовирусы белок p53, данными вирусами были инфицированы
линии клеток, которые не экспрессируют эндогенного p53. Опухолевые клеточные линии человеческого происхождения Saos-2 (остеосаркома) и Hep3B (гепатоцеллюлярная карцинома) инфицировали в течение 24
часов p53-рекомбинантными аденовирусами A/М/53 или A/C/53 с множественностью инфекции в пределах от 0.1 до 200 бое/клетку. Вестерн-анализ лизатов, приготовленных из инфицированных клеток, показал
дозозависимую экспрессию p53 в клетках обоих типов (фиг. 3). В неинфицированных клетках p53 обнаружен не был. Уровни эндогенного p53 дикого типа в норме весьма низки и практически не обнаруживаются
Вестерн-анализом клеточных лизатов (Bartek et al., 1991). Однако очевидно, что p53 дикого типа легко выявляется после заражения вирусом A/М/53 или A/C/53 с низкой множественностью инфекции (фиг. 5),
что свидетельствует о том, что даже небольшие дозы p53-рекомбинантных аденовирусов способны продуцировать потенциально эффективные количества p53.
p53-зависимые изменения морфологии
Реинтродукция p53 дикого типа в p53-негативные клетки остеосаркомы линии Saos-2 приводила к характерному увеличению и уплощению этих обычно веретеновидных клеток (Chen et al., 1990).
Субконфлюэнтные клетки Saos-2 (1·105 / 10 см чашку) инфицировали с множественностью инфекции 50 вирусом A/C/53 или контрольным вирусом A/М и инкубировали при 37oC в
течение 72 часов. К этому времени контрольные неинфицированные клетки образовывали монослой. В это время ожидаемые морфологические изменения становились хорошо заметными в чашках с клетками,
обработанными A/C/53 (фиг. 6, панель C), но не в чашках с неинфицированными клетками (фиг. 6, панель A) или в контрольных инфицированных чашках (фиг. 6, панель B). Данный эффект не является функцией
плотности клеток, поскольку в контрольной чашке клетки, засеянные с меньшей плотностью, сохраняли нормальную морфологию к 72-м часам инкубации, когда их плотность была одинаковой с таковой для
A/C/53-инфицированных клеток. Более ранние результаты указывают на высокие уровни экспрессии белка p53 в клетках Saos-2 при множественности инфекции 50 (фиг. 5A), а также подтверждают тот факт, что
белок p53, экспрессируемый при помощи рекомбинантных аденовирусов, является биологически активным.
Ингибирование синтеза клеточной ДНК посредством p53
Для дальнейшего изучения
p5З-рекомбинантных аденовирусов проверяли путем включения меченого 3H-тимидина их способность подавлять пролиферацию опухолевых клеток человека. Ранее было показано, что введение p53 дикого типа в
клетки, которые не экспрессируют эндогенного p53 дикого типа, может останавливать клетки при переходе из фазы G1 в фазу S клеточного цикла, что приводит к подавлению включения меченого тимидина во
вновь синтезируемую ДНК (Baker et al. , 1990; Mercer et al., 1990; Diller et al., 1990). Различные опухолевые клеточные линии, дефицитные по p53, инфицировали аденовирусными векторами A/M/N/53,
A/C/N/53 или не экспрессирующим p53 контрольным рекомбинантным аденовирусом (A/М). Наблюдали сильное дозозависимое подавление синтеза ДНК при заражении рекомбинатными вирусами A/M/N/53 и A/C/N/53 в
случае семи из девяти проверенных опухолевых клеточных линий (фиг. 7). Оба конструкта были способны подавлять синтез ДНК в этих опухолевых клетках человека независимо от того, экспрессировали ли они
мутантный p53 или вовсе не экспрессировали p53. В данном эксперименте было также показано, что конструкт A/C/N/53 является значительно более эффективным, нежели A/M/N/53. В клетках Saos-2
(остеосаркома) и MDA-MB468 (рак молочной железы) при инфекции конструктом A/C/N/53 с множественностью не выше 10 удавалось достигнуть почти 100%-ного подавления синтеза ДНК. При тех дозах, когда
подавление синтеза ДНК контрольным аденовирусом достигало только 10-30%, наблюдалось 50-100%-ное подавление синтеза ДНК при использовании любого p53-рекомбинантного аденовируса. Напротив, ни с одним
конструктом не наблюдалось какого-либо p53-специфического эффекта в сравнении с контрольным вирусом в случае клеток Hep G2 (клеточная линия гепатокарциномы, экспрессирующая эндогенный p53 дикого типа,
Bressac et al., 1990) и лейкозной клеточной линии K562 (p53-ноль).
Туморогенность для голых мышей
B более строгом тесте на функционирование рекомбинантных аденовирусов,
содержащих p53, опухолевые клетки инфицировали ex vivo и затем вводили голым мышам для проверки способности рекомбинантов подавлять рост опухолей in vivo. Клетки Saos-2, инфицированные вирусом
A/M/N/53 или контрольным вирусом A/М с множественностью инфекции от 3 до 30, вводили в разные бока голым мышам. Затем в течение 8 недель дважды в неделю определяли размеры опухолей. При
множественности инфекции, равной 30, ни у одного из животных рост p53-обработанных опухолей не наблюдался, при том что рост контрольных опухолей происходил нормально (фиг. 8). Прогрессирующее
увеличение опухолей, обработанных контрольным вирусом, было сходным с таковым, наблюдавшимся у животных, обработанных только буфером. Наблюдались явные различия в росте опухолей в случае контрольного
аденовируса и p53-рекомбинантного аденовируса при множественности инфекции 3, хотя у 2-х из 4-х p53-обработанных мышей рост опухолей начался приблизительно через 6 недель. Таким образом,
рекомбинантный аденовирус A/M/N/53 способен обусловливать p53-специфическую супрессию опухолей in vivo.
Экспрессия Ad/p53 in vivo
При том что обработка опухолевых клеток ех
vivo с последующим введением их животным является важным тестом на подавление опухоли, более важными в клиническом плане представляются эксперименты по введению p53-рекомбинантных аденовирусов и
экспрессии p53 в опухолях, развившихся in vivo. С этой целью клетки H69 (SCLC, p53- null) вводили подкожно голым мышам и позволяли образовываться опухолям в течение 32-х дней. В это время однократно
вводили 2·109 бое вируса A/C/53 или A/C/бета-гал в перитуморальное пространство. На второй или седьмой день после введения вируса опухоли вырезали и из каждой опухоли выделяли полиА
РНК. Для обнаружения мРНК p53 в p53-обработанных опухолях использовали РТ- ПЦР с праймерами, специфичными относительно рекомбинантного p53. Сигнала p53 не было обнаружено в опухолях, вырезанных у
животных, обработанных вирусом с бета-галактозидазой (фиг. 9, линии 3 и 6). Амплификация с актиновыми праймерами служила контролем в реакции РТ-ПЦР (фиг. 9, линии 7-9), а плазмида, содержащая
последовательности рекомбинантного p53, служила в качестве положительного контроля для p53-специфической полосы (фиг. 9, линия 10). Данный эксперимент показывает, что p53-рекомбинантный аденовирус
может специфически направлять экспрессию p53 мРНК в развившихся опухолях после однократного введения в перитуморальное пространство. Он также демонстрирует персистенцию вируса in vivo по меньшей мере
в течение недели после инфекции p53-рекомбинантным аденовирусом.
Эффективность in vivo
Для определения достоверности генотерапии развившихся опухолей была использована модель
голых мышей - носителей опухоли. В правый бок голым мышам вводили подкожно клетки H69 и позволяли опухолям развиваться в течение 2-х недель. Затем мышам дважды в неделю перитуморально вводили буфер
или рекомбинантный вирус (всего 8 инъекций). На протяжении всей обработки опухоли у мышей, получавших буфер или контрольный вирус A/М, продолжали быстро расти, тогда как опухоли у мышей, обработанных
вирусом A/M/N/53, росли гораздо медленнее (фиг. 10A). После прекращения инъекций контрольные опухоли продолжали расти, а опухоли, обработанные p53, росли незначительно или не росли совсем по меньшей
мере в течение недели в отсутствие дополнительного экзогенного p53 (фиг. 10A). Несмотря на то, что у контрольных животных, получавших только буфер, наблюдался ускоренный рост опухолей в сравнении с
любой группой животных, обработанных вирусом, значительной разницы в весе тела между тремя группами за все время обработки обнаружено не было. Изъязвление опухолей у отдельных животных снижало
достоверность определений размеров опухолей после 42-го дня. Однако продолжение наблюдений за животными для определения времени выживания показало преимущество в выживании p53-обработанных животных
(фиг. 10B). Последнее из обработанных контрольным аденовирусом животное умерло на 83-й день, тогда как все контрольные животные, получавшие только буфер, умерли к 56-му дню. Напротив, все пять
животных, обработанных вирусом A/M/N/53, выжили (130-й день после введения клеток) (фиг. 10B). Совокупность этих данных подтверждает специфическое влияние p53 на рост опухолей и время выживания у
животных с развившимися p53-дефицитными опухолями.
Аденовирусные векторы, экспрессирующие p53
Были сконструированы рекомбинантные аденовирусные векторы, способные к экспрессии
высоких уровней белка p53 дикого типа дозозависимым образом. Каждый вектор содержал делеции областей E1a и E1b, что делало вирус дефектным по репликации (Challberg and Kelly, 1979; Horowotz, 1991).
Важно отметить, что данные делеции захватывали последовательности, кодирующие белки 19 кд и 5 кд E1b. Показано, что белок 19 кд участвует в ингибировании апоптоза (White et al., 1992: Rao et al.,
1992), а белок 55 кд способен связываться с белком p53 дикого типа (Samow et al., 1982; Heuvel et al., 1990). При делеции данных аденовирусных последовательностей удаляются потенциальные ингибиторы
функции p53, способные действовать путем прямого связывания с p53 или же посредством ингибирования p53-опосредованного апоптоза. Были получены дополнительные конструкты, в которых были делетированы
остающиеся 3' E1b последовательности, включая все последовательности, кодирующие белок IX. Хотя, как показано, делеции в области E3 приводят к снижению емкости вектора приблизительно на 3 кб по
сравнению с аденовирусом дикого типа (Ghosh-Choudhury et al. , 1987), подобные делеции были проведены в конструктах A/M/N/53 и A/C/N/53, так что емкость указанных векторов укладывается в указанные
рамки. Делетированием области pIX содержание аденовирусных последовательностей, гомологичных таковым, содержащимся в клетках 293, было снижено до 300 пар оснований, что уменьшило вероятность
возникновения путем рекомбинации компетентного по репликации аденовируса дикого типа. Конструкты, утратившие кодирующую последовательность pIX, имели одинаковую эффективность по сравнению с
конструктами, имеющими данную последовательность.
Эффективность p53-аденовируса in vitro
В соответствии со строгой дозозависимостью экспрессии p53 в инфицированных клетках
было показано дозозависимое p53- специфическое подавление роста опухолевых клеток. В опухолевых клетках различных типов, в которых не экспрессировался p53 дикого типа, подавлялось клеточное деление,
что демонстрировалось снижением синтеза ДНК. Недавно Bacchetti и Graham (1993) в аналогичных экспериментах показали p53-специфическое подавление синтеза ДНК в клеточной линии карциномы яичника SKOV-3
посредством рекомбинантного p53-аденовируса. Ингибирование роста с помощью заявленных в настоящем изобретении p53-рекомбинантов было показано не только на карциноме яичника, но и на ряде других
опухолевых клеточных линий человека, представляющих клинически важные неоплазии человека, включая линии, в которых происходит повышенная экспрессия мутантного белка p53. При той множественности
инфекции, когда рекомбинант A/C/N/53 проявлял 90-100% эффективность подавления синтеза ДНК в опухолях данных типов, активность контрольного аденовируса составляла менее 20%.
Хотя Feinstein et al., (1992) показал, что реинтродукция p53 дикого типа может вызывать дифференцировку и увеличение доли клеток в G1 по отношению к S+G2 для лейкозных клеток K562, авторами изобретения p53-специфического эффекта на данных клетках не наблюдалось. Horvath и Weber (1988) сообщали, что лимфоциты периферической крови человека в высокой степени непермиссивны для аденовирусной инфекции. В отдельных экспериментах рекомбинантный аденовирус A/C/бета-гал с трудом инфицировал неотвечающие клетки K562, в то время как другие клеточные линии, в том числе контрольные клетки линии Hep G2 и клетки с ярко выраженным эффектом p53, легко инфицировались. Таким образом, по меньшей мере частично вариации в эффективности обусловлены различиями в инфекционности вируса, хотя и другие факторы также могут иметь значение.
Результаты, полученные с вирусом A/M/N/53 и представленные на фиг. 8, показывают, что и в условиях in vivo возможно полное подавление роста опухолей. Возобновление роста опухолей у двух животных из четырех, обработанных p53 с низкой множественностью инфекции, объясняется низким процентом клеток, инфицированных p53 рекомбинантом при данной дозе. Полное подавление роста опухолей в случае вируса A/M/N/53 в высокой дозе, однако, показывает, что способность к возобновлению роста опухоли может быть преодолена.
Эффективность
p53-аденовируса in vivo
Приведенные здесь эксперименты, а также работы других групп (Chen et al. , 1990, Takahashi et al., 1992) показывают, что опухолевые клетки человека, в которых не
происходит экспрессии p53 дикого типа, могут быть обработаны p53 ex vivo, что приводит к супрессии роста опухолей при последующем введении данных клеток животным. Авторы предоставили первое
доказательство возможности генотерапии опухолей посредством переноса гена-супрессора в опухоли, развившиеся in vivo. Данный метод приводит к супрессии (подавлению) роста опухолей и увеличению времени
выживания. В системе, описанной авторами, доставка агента в опухолевые клетки не основывается на прямой инъекции в опухолевую массу. Напротив, p53-рекомбинантный аденовирус вводили в перитуморальное
пространство и в опухоли обнаруживали экспрессию мРНК p53. Экспрессируемый рекомбинантами p53 был функционален и в значительной степени подавлял рост опухоли в сравнении с контрольными опухолями,
обработанными аденовирусами, не экспрессирующими p53. Однако подавление роста опухолей наблюдалось в обоих группах - как обработанных p53-содержащими, так и p53-несодержащими вирусами, в отличие от
контроля (обработка только буфером). Было показано, что у голых мышей местная экспрессия фактора некроза опухолей (TNF), гамма-интерферона, интерлейкина-2 (IL-2), интерлейкинов 4 или 7 (IL-4, IL-7)
может приводить к временной супрессии опухолей, зависимой от Т-клеток (Hoch et al. , 1992). Экспозиция моноцитов с аденовирусом также является слабым индуктором интерферонов альфа и бета (см. обзор
Gooding and Wold, 1990). Таким образом, не удивительно, что некоторое подавление опухолей у голых мышей наблюдалось в случае контрольного аденовируса. Подобное вирус-обусловленное подавление опухолей
не наблюдалось в случае опухолевых клеток Saos-2, обработанных ex vivo контрольным вирусом. P53-специфическое подавление опухолей in vivo становится особенно очевидно при продолжительном мониторинге
животных (фиг. 10). Время выживания p53-обработанных мышей резко возрастало, так 5 из пяти опытных животных были живы спустя 130 дней после введения клеток в сравнении с 0 из пяти, обработанных
контрольным аденовирусом. У выживших животных рост опухолей продолжался, что, по-видимому, обусловлено тем фактом, что не все клетки были первоначально инфицированы p53-рекомбинантным аденовирусом.
Это обстоятельство может быть преодолено более высокими дозами вируса или более частыми его введениями. Кроме того, выключение промотора (Palmer et al.,1991) или дополнительные мутации могут
обусловливать устойчивость данных клеток к обработке p53-рекомбинантными аденовирусами. Например, мутации в недавно описанном гене WAF1, который индуцируется p53 дикого типа и затем подавляет переход
клетки в S-фазу клеточного цикла (El-Deiry et al., 1993; Hunter, 1993), могут приводить к образованию p53-устойчивых опухолей.
Эксперимент N III
В настоящем примере показано
применение "суицидных" генов и тканеспецифической экспрессии таких генов в генотерапевтических подходах, описываемых в данном изобретении. В качестве мишени была выбрана гепатоцеллюлярная карцинома,
поскольку она является одной из самых распространенных неоплазий человека и приводит к гибели около 1250000 людей во всем мире ежегодно. Частота этого вида рака особенно высока в Юго-Восточной Азии и
Африке, где он ассоциирован с инфекцией гепатитом B и C и воздействием афлатоксина. Единственным методом лечения гепатоцеллюлярной карциномы на сегодня является хирургический, хотя менее 20% больных
рассматриваются как кандидаты на операцию (Ravoet С. et al. , 1993). Однако представленные здесь способы снижения пролиферативного потенциала опухолей применимы и к другим неоплазиям, отличным от
гепатоцеллюлярной карциномы.
Линии клеток
Все клеточные линии за исключением линии HFL были получены из Американской Коллекции Клеточных Культур (АТСС), 12301 Parklawn Drive,
Rockville, Maryland. Кодовые номера АТСС приведены в скобках. Клеточная линия эмбриональной печени человека 293 (CRL 1573) была использована для получения и наращивания рекомбинантных аденовирусов.
Клетки выращивали на среде DМЕ с добавлением 10% телячьей сыворотки (Hyclone). Клеточные линии гепатоцеллюлярной карциномы Hep 3B (HB 8064), Hep G2 (HB 8065) и HFL поддерживали на среде DME/F12 с
добавлением 10% эмбриональной телячьей сыворотки так же, как и линии рака молочных желез MDA-MB468 (HTB 132) и BT-549 (HTB 122). Печеночные клетки линии Chang (CCL 13) выращивали на среде MEM с
добавлением 10% эмбриональной телячьей сыворотки. Клеточная линия HLF была получена от д-ров T.Morsaki и H.Kitsuki (медицинский факультет Университета Киушу, Япония).
Конструирование
рекомбинантного вируса
Два аденовирусных вектора экспрессии, обозначенные здесь как ACNTK и AANTK, лишенные функции белка IX (приведены на фиг. 11), способны обеспечивать экспрессию
"суицидного" гена тимидинкиназы (ТК) в опухолевых клетках. Третий аденовирусный вектор экспрессии, обозначенный AANCAT, был сконструирован для дальнейшей демонстрации осуществимости специфической
целенаправленной экспрессии генов в особых типах клеток при помощи аденовирусных векторов. Данные аденовирусные конструкции "собирали", как это показано на фиг. 11 и 12, и они являются производными
тех конструкций, которые были ранее описаны для экспрессии генов-супрессоров опухолей.
Для экспрессии перенесенного гена была использована экспрессионная кассета, в которой для транскрипции гена ТК или гена хлорамфениколацетилтрансферазы (CAT) применялись ранний промотор/энхансер цитомегаловируса человека (Boshart, М. et al., 1985) или энхансер/промотор альфа-фетопротеина человека (Watanabe, K. et al., 1987; Nakabayashi, H. et al., 1989). Промотор CMV способен направлять устойчивую экспрессию генов в клетках различных типов, а конструкты с промотором/энхансером АФП экспрессируются специфически в клетках гепатоцеллюлярной карциномы (HCC), в которых экспрессия АФП наблюдается у 70-80% больных с данным типом опухолей. В конструкте с промотором/энхансером CMV для усиления трансляции транскрипта ТК использована трехчленная лидерная последовательность аденовируса типа 2 (Berkner, K.L and Sharp, 1985). В дополнение к делеции E1 у обоих аденовирусных векторов делетировано по 1.9 кб ДНК в области E3. ДНК, делетированная в E3 области, не является необходимой для репликации вируса и подобная делеция увеличивает на эквивалентную величину (1.9 кб) емкость рекомбинантного вируса в отношении чужеродной ДНК (Graham and Prevec, 1991).
Для демонстрации специфичности промотора/энхансера АФП был сконструирован вирус AANCAT, в котором маркерный ген хлорамфениколацетилтрансферазы (CAT) поставлен под контроль промотора/энхансера АФП. В вирусном конструкте ACNTK трехчленная лидерная последовательность Ad2 помещена между геном ТК и промотором/энхансером CMV. Показано, что такой трехчленный лидер усиливает трансляцию соединенных с ним генов. Замены в области E1 нарушают способность рекомбинантных вирусов к репликации, ограничивая их размножение только клетками 293, которые предоставляют продукты гена E1 Ad5 по "транс" типу (Graham et al., 1977).
Аденовирусный вектор ACNTK: плазмида pMLBKTK в E.coli HB 101 (из АТСС, # 39369) была использована как источник гена тимидинкиназы (ТК) вируса простого герпеса (HSV-1). Ген ТК был вырезан из плазмиды в виде фрагмента 1.7 кб при рестрикции ферментами Bgl II и Pvu II, а затем субклонирован по сайтам BamHI и EcoRV в плазмиду pSP72 (Promega) с использованием стандартных методов клонирования (Sambrook et al., 1989). Вставку ТК-гена затем вырезали из данного вектора в виде 1.7 кб фрагмента рестриктазами Xba 1 и Bgl II и клонировали в плазмиду pACN (Wills et al., 1994). Двадцать (20) микрограммов данной плазмиды, обозначенной pACNTK, линеаризовали при помощи рестриктазы EcoRI и котрансфицировали в клетки 293 (ATCC CRL 1573) с 5 мкг переваренной Cla I плазмиды ACBGL (Wills et al., 1994 supra) и использованием набора для кальций-фосфатной трансфекции (Stratagene, San Diego, California). Изолировали вирусные бляшки и идентифицировали рекомбинанты ACNTK при помощи рестрикционного анализа выделенной ДНК. При этом использовали рестриктазы Xhol и BsiWI. Положительные рекомбинанты затем очищали методом конечных разведений, наращивали и титровали стандартными методами (Graham and Prevec, 1991).
Аденовирусный вектор AANTK: промотор гена альфа-фетопротеина (AFP-P) и энхансер того же гена (AFP-E) были клонированы из геномной ДНК человека (Clonetech) путем ПЦР-амплификации с использованием праймеров с сайтами рестрикции на концах. Праймеры, использованные для выделения фрагмента AFP-P (210 пар оснований), содержали сайт рестрикции Nhel в 5'-праймере и линкер Xbal, Xhol, Kpnl в 3'-праймере. Последовательность 5'- праймера была следующей: 5'-CGC GCT AGC TCT GCC CCA AAG AGC T-З'. Последовательность 3'-праймера была следующей: 5'-CGC GGT ACC CTC GAG TCT AGA TAT TGC CAG TGG TGG AAG-3'. Праймеры, использованные для выделения фрагмента AFE (1763 пар оснований), имели сайт рестрикции Notl в 5'-праймере и сайт рестрикции Xbal в 3'-праймере. Последовательность 5'-праймера была следующей: 5'-CGT GCG GCC GCT GGA GGA CTT TGA GGA TGT CTG TC-3'. Последовательность 3'-праймера была следующей: 5'-CGC TCT AGA GAG ACC AGT TAG GAA GTT TTC GCA-3'. Для ПЦР-амплификации ДНК денатурировали 7 минут при 97oC, затем следовали 5 циклов амплификации при 97oC 1 минута, 53oC 1 минута, 72oC 2 минуты и, наконец, наращивание при 72oC 10 минут. Амплифицированный фрагмент AFE рестрицировали ферментами Notl и Xbal и встраивали по сайтам Notl и Xbal в плазмидный вектор pA/1TR/B, содержащий последовательности аденовируса типа 5 (1-350 и 3330-5790), разделенные полилинкером, содержащим сайты рестрикции Notl, Xhol, Xbal, Hindlll, Kpnl, BamHI, Ncol, Smal и Bgl II. Амплифицированный фрагмент AFP-E переваривали ферментами Nhel и Kpnl и встраивали в содержащий AFP-E конструкт, описанный выше по сайтам рестрикции указанных ферментов. Этот новый конструкт затем рестрицировали ферментами Xbal и NgoMI для удаления аденовирусных последовательностей 3330-5780, которые заменяли рестрикционным фрагментом Xbal-NgoMl плазмиды pACN, содержащим нуклеотиды 4021-10457 аденовируса типа 2, с получением плазмиды pANN, содержащей как промотор, так и энхансер альфа-фетопротеина. Данный конструкт затем рестрицировали ферментами EcoRI и Xbal для выделения фрагмента размером 2.3 кб, содержащего обращенный концевой повтор вируса Ad5, AFP-E и AFP-P, который затем лигировали с EcoRI-Xbal 8.5 - кб фрагментом описанной выше плазмиды pACNTK с получением плазмиды pAANTK, в которой ген TK в аденовирусном окружении поставлен под контроль промотора и энхансера гена альфа- фетопротеина. Данную плазмиду затем линеаризовали при помощи EcoRI и котрансфицировали вместе с большим фрагментом Clal рестрицированной плазмиды ALBGL, как описано выше, и рекомбинанты, обозначенные AANTK, выделяли и очищали, как описано выше.
Аденовирусный вектор AANCAT: ген хлорамфениколацетилтрансферазы (CAT) был выделен из Basic Vector pCAT (Promega Corporation) при рестрикции последнего ферментами Xbal и BamHI. Полученный 1.64 - кб фрагмент лигировали с рестрицированной Xbal и BamHI плазмидой pAAN (описана ранее) с получением плазмиды pAANCAT. Данную плазмиду затем линеаризовали при помощи EcoRI и котрансфицировали с большим фрагментом рестрицированной Clal rA/C/7бета-gal с полученим AANCAT.
Экспрессия репортерного гена:
экспрессия бета-галактозидазы
Клетки засевали по 1·105 клеток на лунку в 24-луночные культуральные планшеты (Costar) и оставляли на ночь для прикрепления (37oC,
7% CO2). Заражение вирусом ACBGL проводили в течение ночи при множественности инфекции, равной 30. Спустя 24 часа клетки фиксировали 3.7%-ным формальдегидом, отмывали PBS и окрашивали
реагентом X-gal (1 мг/мл, USB). Результаты выражали в принятых символах (+, ++, +++), определяя процент окрашенных клеток при каждой множественности инфекции. [+=1-33%, ++=33-67%, и +++≥
67%).
Экспрессия репортерного гена: экспрессия CAT
Клетки HepG2, Hep B3, HLF, Chang и MDA-MB468 засевали по 2·106 на 10-см чашки (по три чашки каждой
культуры) и инкубировали ночь при 37oC, 7% CO2. Затем клетки на каждой чашке заражали или AANCAT с множественностью инфекции 30 или 100, или же оставляли незараженными и
инкубировали в течение 3 дней. Затем клетки снимали трипсином, отмывали PBS и суспендировали в 100 мкл 0.25 М Трис-HCl pH 7.8. Образцы трижды замораживали и оттаивали, супернатант переносили в новые
пробирки и инкубировали при 60oC 10 минут. Затем образцы центрифугировали при 4oC 5 минут и в супернатантах определяли концентрацию белка по Брэдфорду (Bio-Rad Protein Assay
Kit). Концентрацию белка в пробах выравнивали, доводя конечный объем до 75 мкл с использованием 0.25 М Трис, 25 мкл 4 мМ ацетил-КоА и 1 мкл14C-хлорамфеникола. Пробы инкубировали ночь при
37oC. Затем к каждой пробе добавляли 500 мкл этилацетата, перемешивали на встряхивателе и центрифугировали 5 минут при комнатной температуре. Верхнюю фазу переносили в новые пробирки и
испаряли этилацетат центрифугированием в вакууме. Продукты реакции растворяли в 25 мкл этилацетата и наносили на пластину для тонкослойной хроматографии (TLC). Пластину помещали в уравновешенную (95%
хлороформа, 5% метанола) камеру для TLC. Растворителю давали возможность мигрировать в верхнюю часть камеры, затем пластину высушивали и экспонировали с рентгеновской пленкой.
Пролиферация клеток: включение3H-тимидина
Клетки засевали по 5·103
клеток/ячейка в ячейки 96-луночных планшетов (Costar) и инкубировали в течение ночи (37oC, 7% CO2). Серийно разведенные вирусы ACN, ACNTK или AATK в среде DMЕМ с 15% телячьей эмбриональной сыворотки и 1% глутамина использовали для заражения клеток с множественностью
инфекции, равной 30. Инкубация с вирусом продолжалась в течение ночи, после чего к клеткам добавляли ганцикловир в логарифмических разведениях от 0.001 до 100 мкМ (микроМоль). За 12-18 часов до сбора
клеток в каждую ячейку добавляли 1 мкКи3H-тимидина (Amersham). Спустя 72 часа после заражения клетки осаждали на стекловолоконные фильтры и определяли количество включенного1
H-тимидина методом жидкостной сцинтилляции (TopCount, Packard). Результаты представлены как процент от контрольной пролиферации без обработки и приведены в виде эффективной дозы (ED50 +/- стандартное
отклонение) для 50%-ного снижения пролиферации над контролем со средой. Значения ED50 оценивали по совпадению с логическим уравнением "доза-ответ".
Цитоксичность: высвобождение
лактатдегидрогеназы (LDH)
Клетки (HFL, гепатоцеллюлярная карцинома человека, HCC) после заражения ACN или ACTK обрабатывали ганцикловиром, как это описано в разделе "Пролиферация клеток".
Спустя 72 часа после добавления ганцикловира клетки центрифугировали и удаляли супернатант. Уровни лактатдегидрогеназы определяли колориметрически (Promega, Cytotox 96TM). Среднее высвобождение LDH
(+/- среднее отклонение) ставили в зависимость от множественности инфекции.
Терапия in vivo
Клетки гепатоцеллюлярной карциномы человека (Hep 3B) вводили подкожно десяти
самкам атимических мышей nu/nu (Simonsen Laboratories, Gilroy, CA). Каждое животное получало в левый бок около 1·107 клеток. Опухолям давали сформироваться в течение 27 дней, а затем
мышей рандомизировали по размеру опухоли. Мышам вводили через день интратуморально или перитуморально вирус ACNTH или контрольный вирус ACN (1·109 инфекционных единиц в 100 мкл).
Всего каждая мышь получила три инъекции. Спустя 24 часа после первой инъекции аденовируса мышам интраперитонеально вводили ганцикловир (Cytovene, 100 мг/кг). Эти инъекции проводили ежедневно в течение
10 дней. Дважды в неделю проверяли размер опухолей и общий вес животного. Замеры опухолей проводили в трех измерениях при помощи градуированных штангенциркулей и объем опухолей определяли по формуле:
4/3π3, где r - одна вторая среднего размера опухоли.
Результаты
Рекомбинантные аденовирусы были использованы для заражения трех клеточных линий
гепатоцеллюлярной карциномы (HLF, Hep3B и Hep-G2). В качестве контролей были использованы линия клеток печени человека (Chang) и две линии рака молочной железы (MDAMB468 и BT549). Для демонстрации
специфичности промотора/энхансера АФП был сконструирован вирус AANCAT. Данный вирус был использован для заражения клеток, которые экспрессируют (Hep 3B, Hep-G2) или не экспрессируют (HLE, Chang,
MDAMB468) маркер гепатоцеллюлярных опухолей альфа-фетопротеин (АФП). Как показано на фиг. 13, AANCAT направляет экспрессию маркерного гена CAT только в тех HCC клетках, которые способны к экспрессии
АФП (фиг. 13). Эффективность ACNTH и AANTK для лечения HCC проверяли с использованием включения 3H-тимидина с целью определения эффекта экспрессии HSV-TK в сочетании с обработкой ганцикловиром на
пролиферацию клеток. Клетки указанных линий инфицировали ACNTK или AANTK или же контрольным вирусом ACN (Wills et al.,1994 supra), который не направлял экспрессии HSV-TK, а затем обрабатывали
возрастающими дозами ганцикловира. Эффективность данной обработки являлась функцией концентрации ганцикловира. Определяли такую концентрацию ганцикловира, которая требовалась для 50%-ного подавления
включения3H-тимидина (ED50). Для каждой клеточной линии определяли также относительное число клеток, в которых экспрессировались перенесенные аденовирусом гены, по отношению к клеткам, в
которых экспрессировался маркерный ген бата-галактозидазы, перенесенный контрольным вирусом. Данные, представленные на фиг. 14 и в таблице 2, показывают, что комбинированная обработка вирусом ACNTK и
ганцикловиром способна ингибировать пролиферацию клеток всех проверенных линий (контролем в данном случае служила обработка контрольным аденовирусом ACN в сочетании с ганцикловиром). Наоборот,
вирусный вектор AANTK был эффективен в тех гепатоцеллюлярных линиях, которые экспрессировали АФП. Кроме того, комбинация AANTK/ганцикловир была более эффективна при засеве клеток с высокой
плотностью.
"Голые" мыши с опухолями, вызванными введением клеток Hep3B (группа из пяти животных), получали интратуморально или перитуморально эквивалентные дозы ACNTK или контрольного вируса ACN. Спустя 24 часа после первого введения рекомбинантного аденовируса начиналось ежедневное введение ганцикловира каждой мыши. Дважды в неделю определяли с помощью штангенциркуля размеры опухоли у каждого животного и средние размеры опухолей представлены на фиг. 16. На 58-й день средний размер опухолей был ниже у обработанных ACNTK мышей, однако разница не была статистически значимой (p<0.09, t-test). Приведенные данные свидетельствуют о специфическом эффекте ACNTK на рост опухолей in vivo. Значимых различий в среднем весе тела между группами животных обнаружено не было.
Несмотря на то, что изобретение изложено в отношении приведенных воплощений, необходимо понимать, что возможные различные модификации не противоречат идее изобретения. Соответственно, объем притязаний данного изобретения ограничен нижеприведенной формулой.
Литература
AIELLO, L. et al. (1979) Virology 94:460-469.
AMERICAN CANCER SOCIETY. (1993) Cancer Facts and Figures.
AULITZKY et al. (1991) Eur. J. Cancer 27(4):462-467.
AUSTIN, E.A. and HUBER, B.E. (1993) Mol. Pharmaceutical 43:380-387.
BACCHETTI, S. AND GRAHAM, F. (1993) International Journal of Oncology 3: 781-788.
BAKER S.J., MARKOWITZ, S., FEARON E.R., WILLSON, J.K.V., AND VOGELSTEIN, B. (1990) Science 249:912-915.
BARTER, J. , BARTKOVA, J., VOJTESEK, B., STASKOVA, Z., LUKAS, J., REJTHAR, A. , KOVARIK, J., MIDGLEY, C.A., GANNON, J.V., AND LANE, D.P. (1991) Oncogene 6:1699-1703.
BERKNER, K.L. and SHARP (1985) Nucleic Acids Res 13:841-857.
BOSHART, M. et al. (1985) Cell 41:521-530.
BRESSAC, B., GALVIN, К.M., LIANG, Т.J., ISSELBACHER, К.J., WANDS, J.R., AND OZTURK, M. (1990) Proc. Natl. Acad. Sci. USA 87:1973-1977.
CARUSO M. et al. (1993) Proc. Natl. Acad. Sci. USA 90:7024-7028.
CHALLBERG, M.D., KELLY, T.J. (1979) Biochemistry 76:655-659.
CHEN P. L. , CHEN Y. , BOOKSTEIN R., AND LEE W.H. (1990) Science 250: 1576-1580.
CHEN, Y. , CHEN, P.L., ARNAIZ, N., GOODRICH, D., AND LEE, W.H. (1991) Oncogene 6:1799-1805.
CHENG, JL, YEE, J.K., YEARGIN, J., FRIEDMANN, Т., AND HAA3, M. (1992) Cancer Research 52:222-226.
COLBY, W.W. AND SHENK, T. J. (1981) Virology 39:977-980.
CULVER et al. (1991) P.N.A.S. (U.S.A.) 88:3155-3159.
CULVER, K.W. et al. (1992) Science 256:1550-1552.
DEMETRI et al. (1989) J. Clin. Oncol. 7(10):1545-1553.
DILLER, L., et al. (1990) Mol. Cell. Biology 10:5772-5781.
EL-DEIRY, W.S., et al. (1993) Cell 75:817-825.
EZZIDINE, Z.D. et al. (1991) The New Biologist 3:608-614.
FEINSTEIN, E., GALE, R.P., REED, J., AND CANAANI, E. (1992) Oncogene 7: 1853-1857.
GHOSH-CHOUDHURY, G., HAJ-AHMAD, Y., AND GRAHAM, F.L. (1987) EMBO Journal 6:1733-1739.
GOODING, L.R., AND WOLD, W.S.M. (1990) Crit. Rev. Immunol. 10:53-71.
GRAHAM F.L., AND VAN DER ERB A.J. (1973) Virology 52:456-467.
GRAHAM, F. L. AND PREVEC, L. (1992) Vaccines: New Approaches to Immunological Problems. R.W. Ellis (ed), Butterworth-Heinemann, Boston, pp. 363-390.
GRAHAM, F. L. , SMILEY, J., RUSSELL, W.C. AND NAIRN, R. (1977) J. Gen. Virol. 36:59-74.
GRAHAM F.L. AND PREVEC L. (1991) Manipulation of adenovirus vectors. In: Methods in Molecular Biology. Vol 7 - Gene Transfer and Expression Protocols. Murray E.J. (ed.) The Humana Press Inc., Clifton N.J., Vol 7:109-128.
HEUVEL, S. J.L., LAAR, T., KAST, W.M., MELIEF, C.J.M., ZANTEMA, A., AND VAN DER EB, A.J. (1990) EMBO Journal 9:2621-2629.
HOCK, H., DORSCH, M., KUZENDORF, U., QIN, Z., DIAMANTSTEIN, T., AND BLANKENSTEIN, T. (1992) Proc. Natl. Acad. Sci. USA 90:2774-2778.
HOLLSTEIN, M., SIDRANSKY, D., VOGELSTEIN, B., AND HARRIS, C. (1991) Science 253:49-53.
HOROWITZ, M.S. (1991) Adenoviridae and their replication. In Fields Virology. B.N. Fields, ed. (Raven Press, New York) pp. 1679-1721.
HORVATH, J., AND WEBER, J.M. (1988) J. Virol. 62:341-345.
HUANG et al. (1991) Nature 350:160-162.
HUBER, B.E. et al. (1991) Proc. Natl. Acad. Sci. USA 88:8039-8043.
HUNTER, T. (1993) Cell 75:839-841.
JONES, N. AND SHENK, T. (1979) Cell 17:683-689.
KAMB et al. (1994) Science 264:436-440.
KEURBITZ, S. J. , PLUNKETT, B.S., WALSH, W.V., AND KASTAN, M.B. (1992) Proc. Natl. Acad. Sci. USA 89: 7491-7495.
KREIGLER, M. Gene Transfer and Expression: A Laboratory Manual, W.H. Freeman and Company, New York (1990).
LANDMANN et al. (1992) J. Interferon Res. 12(2):103-111.
LANE, D.P. (1992) Nature 358:15-16.
LANTZ et al. (1990) Cytokine 2(6):402-406.
LARRICK, J. W. and BURCK, K.L. Gene Therapy: Application of Molecular Biolocry. Elsevier Science Publishing Co., Inc. New York, New York (1991).
LEE et al. (1987) Science 235:1394-1399.
LEMAISTRE et al. (1991) Lancet 337:1124-1125.
LEMARCHAND, P., et al. (1992) Proc. Natl. Acad. Sci. USA 89:6482-6486.
LEVINE, A. J. (1993) The Tumor Suppressor Genes. Annu. Rev. Biochem. 1993. 62:623-651.
LOWE S. W. , SCHMITT, E.M., SMITH, S.W., OSBORNE, B.A., AND JACKS, J. (1993) Nature 362:847-852.
LOWE, S. W. , RULEY, H.E., JACKS, T., AND HOUSMAN, D.E. (1993) Cell 74: 957-967.
MARTIN (1975) In: Remington's Pharm. Sci. 15th Ed. (Mack Publ. Co., Easton).
MERCER, W.E., et al. (1990) Proc. Natl. Acad. Sci. USA 87:6166-6170.
NAKABAYASHI, H. et al. (1989) The Journal of Biological Chemistry 264: 266-271.
PALMER, T.D., ROSMAN, G.J., OSBORNE, W.R., AND MILLER, A.D. (1991) Proc. Natl. Acad. Sci USA 88:1330-1334.
RAO, L. , DEBBAS, M., SABBATINI, P., HOCKENBERY, D., KORSMEYER, S., AND WHITE, E. (1992) Proc. Natl. Acad. Sci. USA 89:7742-7746.
RAVOET C. et al. (1993) Journal of Surgical Oncology Supplement 3:104-111.
RICH, D.P., et al. (1993) Human Gene Therapy 4:460-476.
ROSENFELD, M.A., et al. (1992) Cell 68:143-155.
SAMBROOK J., FRITSCH E.F., AND MANIATIS T. (1989). Molecular Cloning: A Laboratory Manual. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor).
SARNOW, P. , HO, Y.S., WILLIAMS, J., AND LEVINE, A.J. (1982) Cell 28: 387-394.
SHAW, P. , BOVEY, R., TARDY, S., SAHLI, R., SORDAT, B., AND COSTA, J. (1992) Proc. Natl. Acad. Sci. USA 89:4495-4499.
SIEGFRIED, W. (1993) Exp. Clin. Endocrinol. 101:7-11.
SORSCHER, E.J. et al. (1994) Gene Therapy 1:233-238.
SPECTOR, D.J. (1983) Virology 130:533-538.
STEWART, P.L. et al. (1993) EMBO Journal 12:2589-2599.
STRAUS. S. E. (1984) Adenovirus infections in humans. In: The Adenoviruses. Ginsberg HS, ed. New York: Plenum Press, 451-496.
SUPERSAXO et al. (1988) Pharm. Res. 5(8):472-476.
TAKAHASHI, T., et al. (1989) Science 246:491-494.
TAKAHASHI, T., et al. (1992) Cancer Research 52:2340-2343.
THIMMAPPAYA, B. et al. (1982) Cell 31:543-551.
WANG, A.M., DOYLE, M.V., AND MARK, D.F. (1989) Proc. Natl. Acad. Sci USA 86:9717-9721.
WATANABLE, K. et al. (1987) The Journal of Biological Chemistry 262: 4812-4818.
WHITE, E., et al. (1992) Mol. Cell. Biol. 12:2570-2580.
WILLS, K.N. et al. (1994) Hum. Gen. Ther. 5:1079-1088.
YONISH-ROUACH, E., et al. (1991) Nature 352:345-347.
Реферат
Изобретение относится к рекомбинантным аденовирусным векторам экспрессии, характеризующимся частичной или полной делецией фрагмента ДНК аденовируса, кодирующего белок IX, и содержащим ген чужеродного белка, или его функциональный фрагмент, или мутантную форму. Предложена фармацевтическая композиция, включающая рекомбинантный аденовирусный вектор экспрессии, который содержит вставку экзогенной ДНК, содержащей ген, кодирующий чужеродный белок, и аденовирусную ДНК, имеющую делецию, начинающуюся в положении от 357 до 360 и заканчивающуюся в положении от 4020 до 4050. Указанная фармацевтическая композиция может быть использована в генной терапии, для трансформации гиперпролиферативных клеток млекопитающих, терапии рака, ингибировании пролиферации опухоли у животных, для снижения пролиферации опухолевых клеток. 7 с. и 13 з.п. ф-лы, 2 табл., 16 ил.
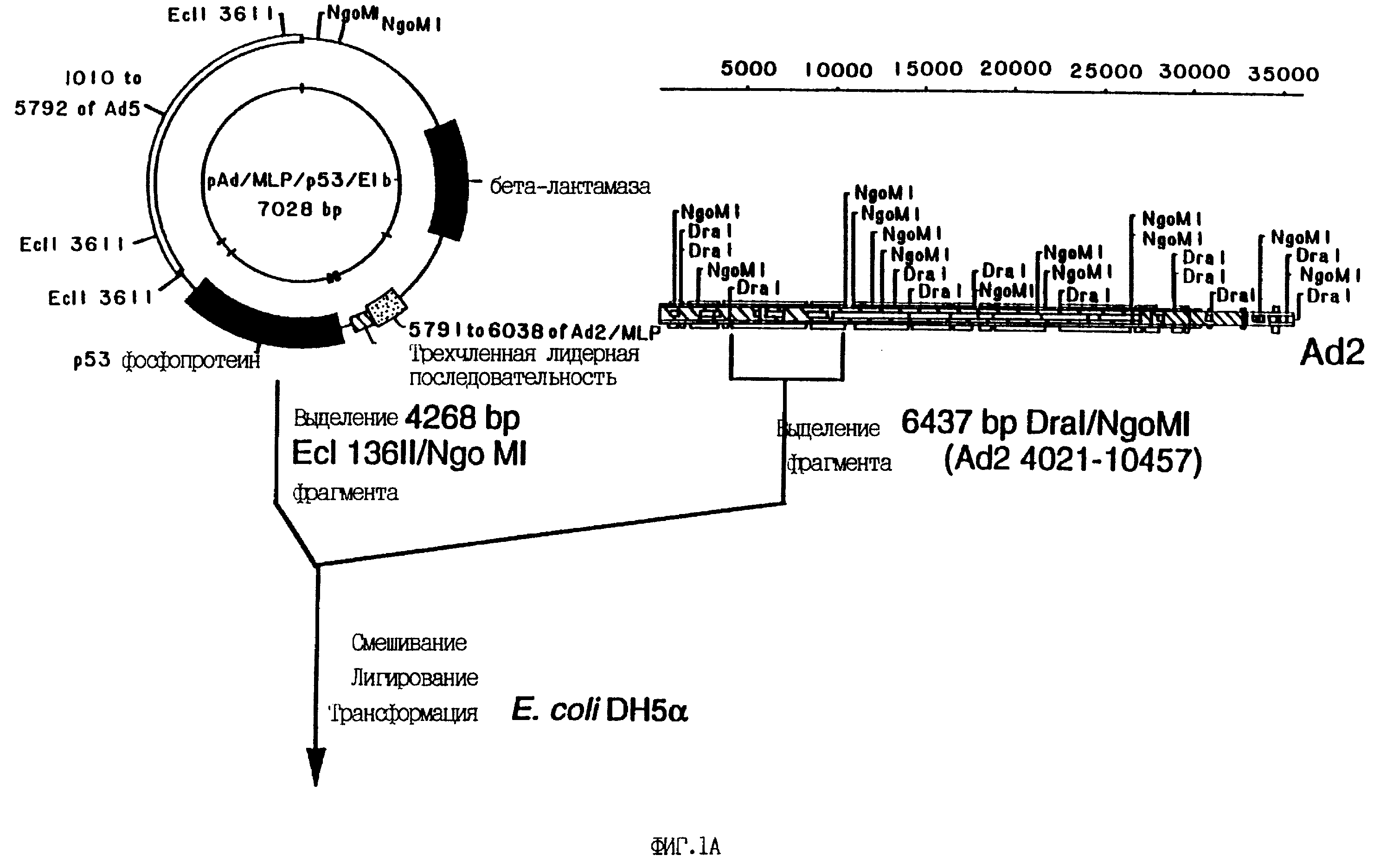
Формула
25.10.93 - пп.1 - 9, 11 - 15;
19.05.94 - пп.10, 16 - 20.
Комментарии