Железосодержащая композиция очистителя - RU2729485C1
Код документа: RU2729485C1
Описание
Перекрестная ссылка на родственные заявки
Для данной заявки испрашивается приоритет по предварительной заявке США № 62/378 751, поданной 24 августа 2016 г. и озаглавленной “Iron-Containing Cleaning Composition” («Железосодержащая чистящая композиция»).
Область техники
Настоящее изобретение касается композиции очистителя.
Уровень техники
Известны способы предварительной обработки металлических поверхностей фосфатом цинка в целях улучшения их коррозионной устойчивости и адгезии лакокрасочных покрытий. Химическая обработка фосфатом цинка выполняется на большом разнообразии металлов с некоторыми известными и широко используемыми примерами, представленными черными металлами, металлическим цинком и алюминием и многими сплавами. При их применении эти способы предварительной обработки приводят к образованию неорганического кристаллического фосфатного слоя, который может инкорпорировать некоторые элементы из поверхности металлической подложки. Для улучшения надежности цинк-фосфатного покрытия в условиях жестких сред к растворам для обработки фосфатом цинка обычно добавляются такие элементы, как никель и марганец. Эти элементы модифицируют поверхность подвергаемой воздействию подложки и встраиваются в осаждающиеся кристаллы фосфата цинка, снижая их растворимость и измельчая размер кристаллов и форму, что приводит к более плотному покрытию. Вышеупомянутые трехкатионные цинк-фосфатные составы особенно эффективны в отношении содействия адгезии лако-красочных составов и обеспечения коррозионной устойчивости подложек, имеющих оцинкованные поверхности, таких как стальные, подвергнутые цинкованию или электролитическому цинкованию, подложки.
Кроме того, в качестве улучшающих производительность известны герметизирующие композиции, содержащие хром в комбинации с цинк-фосфатными композициями для предварительной обработки. Эти герметизирующие композиции снижают пористость покрытия и образуют защитный слой на открытой для воздействий подложке. При всей его эффективности, широкое применение хрома вызвало обеспокоенность связанными с экологией и здравоохранением проблемами, что привело к отказу лакокрасочной промышленности от использования хрома везде, где это возможно. В последнее время пристальное внимание в связи с заботой о здоровье стал привлекать и никель. Это внимание к никелю вызвало интерес к развитию свободной от никеля цинк-фосфатной технологии при сохранении эффективности цинкования и содействовало быстрому развитию технологий предварительной обработки, основанных на цирконии. Не содержащий никеля фосфат цинка исследовался и ранее, но покрытия, наносимые в соответствии с такой технологией, всегда страдали от недостаточной адгезии на цинковых поверхностях. Существует необходимость в обеспечении дополнительной эффективности не содержащих никеля фосфатных систем в случаях, когда краска наносится на цинковые поверхности.
Композиции для предварительной обработки с оксидом циркония обеспечивают экологичную альтернативу цинк-фосфатным композициям для предварительной обработки. Некоторые из эксплуатационных преимуществ включают уменьшение водопотребления, возможность управления способом при температурах окружающей среды и значительно сниженное шламообразование. Рецептуры композиций для предварительной обработки на циркониевой основе не включают никеля и не содержат хрома. Кроме того, в областях мира, где в целях препятствования эвтрофикации введены нормативы в отношении фосфоросодержащих соединений, композиции для предварительной обработки на основе оксида циркония являются превосходным выбором. При том, что применение оксида циркония представляет подтвержденную технологию, существует необходимость в дальнейшем улучшении адгезии и защиты от коррозии лакокрасочных систем, подвергаемых циклическим воздействиям определенных типов условий на цинковых поверхностях.
Был предложен ряд подходов для улучшения эффективности применения композиций для предварительной обработки на цинковых поверхностях. В одном примере предлагается щелочной раствор, содержащий ионы железа (III) в комбинации с ортофосфатными или фосфатными (PO43-) комплексообразующими реагентами в количестве от 100 ч./млн. до 4000 ч./млн. и имеющий pH по меньшей мере 10,5, для целей осаждения на цинковых поверхностях тонкого пассивирующего слоя фосфата железа до нанесения композиции для предварительной обработки. Условие применения в композиции ортофосфата для получения фосфата железа является нежелательным, поскольку регулирующие органы в различных странах мира надеются сдерживать эвтрофикацию, ассоциируемую с использованием ортофосфатов. Это особенно нежелательно для технологий с оксидом циркония, поскольку они представляют единственную технологию предварительной обработки, способную отвечать самым строгим нормативам по фосфатам, когда используются совместно с не содержащим фосфатов очистителем. Было бы желательным предоставление способа максимально возможного улучшения эффективности фосфатной или циркониево-оксидной композиции для предварительной обработки на цинковых поверхностях в системе, которая ограничивает применение ортофосфатов. Другой подход к улучшению эффективности композиции для предварительной обработки на цинковых поверхностях состоит в применении в течение контактного времени от 2 до 60 секунд щелочной композиции для предварительного ополаскивания, содержащей от 5 до 400 ч./млн. ионов железа (III), минимум 0,5 г/л гидроксидных ионов (pH приблизительно 12,5), от 0,0 до 4,0 г/л ионов кобальта, комплексообразующие реагенты и, не обязательно, источник силиката. Во всех случаях предварительное ополаскивание выполняется перед этапом предварительной обработки и следует за этапом щелочной очистки, выполняемым с помощью очистителя Parco Cleaner 1533, представляющего собой, согласно описанию изготовителя, средство с высоким содержанием фосфатов, предназначенное для условий работы от средних до тяжелых. Эта последовательность действий типична для способа предварительной обработки, использующего известное в данной области предварительное ополаскивание. Обычно при этом подходе наилучшие результаты достигались при добавлении кобальта к высокощелочному, железосодержащему средству предварительного ополаскивания цинковых поверхностей. Кобальт, переходный металл, с точки зрения REACH (Registration, Evaluation and Authorisation of Chemicals - технический регламент ЕС «Порядок государственной регистрации, экспертизы и лицензирования химических веществ») оказывается перед неопределенным будущим и, таким образом, может представляться не самым предпочтительным решением для промышленности. Авторы заявляют, что травление алюминия из-за щелочных свойств раствора делает такое предварительное ополаскивание неподходящим для алюминиевых изделий и подложек. Было бы желательным разработать систему для улучшения адгезии и коррозионных характеристик циркониево-оксидных композиций для предварительной обработки на цинке, которая не содержала бы ортофосфата или кобальта и могла применяться в процессах, использующих алюминиевые изделия и подложки, предлагая тем самым надежное решение, обеспечивающее возможность обработки многих металлов.
Краткое описание существа изобретения
Настоящее изобретение касается водной композиции щелочного очистителя, содержащей катион железа, катион молибдена, катион кобальта или их комбинации; и щелочной компонент; причем pH водной щелочной композиции равен по меньшей мере 10 и данная водная щелочная композиция включает не более 50 ч./млн. фосфата по отношению к общей массе композиции.
Настоящее изобретение также касается обрабатывающей системы, предназначенной для обработки металлических подложек, содержащей (a) водную щелочную композицию, содержащую катион железа, катион молибдена, катион кобальта или их комбинации; и щелочной компонент; причем pH водной щелочной композиции равен по меньшей мере 10 и данная водная щелочная композиция включает не более 50 ч./млн. фосфата по отношению к общей массе композиции; и (b) композицию предварительной обработки, предназначенную для обработки по меньшей мере одного участка подложки и содержащую катион металла Группы IVB.
Настоящее изобретение также касается обрабатывающей системы, предназначенной для обработки металлических подложек, содержащей (a) водную щелочную композицию, содержащую катион железа, катион молибдена, катион кобальта или их комбинации; и щелочной компонент; причем pH водной щелочной композиции равен по меньшей мере 10 и данная водная щелочная композиция включает не более 50 ч./млн. фосфата по отношению к общей массе композиции; и (b) композицию предварительной обработки, предназначенную для обработки по меньшей мере одного участка подложки и содержащую фосфат металла.
Также раскрываются подложки, подвергаемые обработке раскрываемой здесь системой.
Подробное описание существа изобретения
Композиция щелочного очистителя
Как указывалось выше, настоящее изобретение касается водной щелочной композиции очистителя, содержащей или в некоторых случаях состоящий по существу из, или в некоторых случаях состоящий из катионов железа, катионов молибдена и/или катионов кобальта, и щелочного компонента, причем pH данной водной щелочной композиции равен по меньшей мере 10 и данная водная щелочная композиция содержит не более 50 ч./млн. фосфата по отношению к общей массе водной щелочной композиции. В данном контексте термин «фосфат» относится к аниону PO43 -и включает ортофосфаты, но исключает фосфонаты (определены ниже). В данном контексте термин «композиция очистителя» относится к композиции, содержащей источник щелочности, структурообразователи, хелаторы и поверхностно-активные вещества и, не обязательно, противовспенивающие компоненты, антикоррозийные добавки и органические растворители/носители помимо воды, и она применяется для обработки металлической подложки с целью удаления масел и других загрязнений с по меньшей мере одного участка поверхности металлической подложки до выполнения любых последующих этапов обработки, таких как введение очищенной поверхности подложки в контакт с композицией(-ми) предварительного ополаскивания, композицией(-ми) предварительной обработки, композицией(-ми) для ополаскивания после обработки и/или композициями для нанесения покрытий электроосаждением, порошковых покрытий, или жидкими композициями. В отличие от очистителя, композиция «предварительного ополаскивания» не включает хелаторов или поверхностно-активных веществ и применяется для обработки очищенной металлической поверхности, то есть поверхности подложки, которая является по существу, фактически или полностью не содержащей масел и других загрязнителей.
Согласно настоящему изобретению, катион железа композиции очистителя может содержать железо (II) и/или железо (III) и может находиться в форме соли. Неограничивающие примеры анионов, подходящих для образования солей с катионом железа, включают нитрат, сульфат, ацетат, хлорид, цитрат, глюконат, сульфамат или их комбинации.
Согласно настоящему изобретению, катион железа может быть представлен в водной композиции щелочного очистителя, если он присутствует, в количестве по меньшей мере 50 ч./млн. по отношению к общей массе композиции щелочного очистителя, таком как по меньшей мере 100 ч./млн., таком как по меньшей мере 250 ч./млн., и в некоторых случаях может быть представлен в количестве, не превышающем 500 ч./млн. по отношению к общей массе композиции щелочного очистителя, таком как не более 400 ч./млн., таком как не более 300 ч./млн. Катион железа может быть представлен в водной композиции щелочного очистителя, если он присутствует, в количестве от 50 ч./млн. до 500 ч./млн. по отношению к общей массе композиции щелочного очистителя, таком как от 100 ч./млн. до 400 ч./млн., таком как от 250 ч./млн. до 300 ч./млн.
Как упоминалось выше, согласно настоящему изобретению, водная композиция щелочного очистителя может содержать катион молибдена. Катион молибдена может быть представлен в композиции очистителя в форме соли. Неограничивающие примеры анионов, подходящих для образования солей с катионом молибдена, включают молибденовую кислоту, молибдат натрия, молибдат аммония или их комбинации.
Молибдаты могут подвергаться реакциям конденсации при изменениях величины pH раствора. Было точно установлено, что в щелочном растворе (pH 7 - 12) молибдат существует в виде дискретных единиц MoO42 -. В диапазоне pH 6 - 7 соединения молибдена главным образом присутствуют в форме протонированных молибдатов (то есть HMoO41-). При более кислых величинах pH (например, при pH 3 - 5) молибдат образует смесь HMoO4 и HMoO41-, которая подвергается реакциям многократной конденсации при высоких концентрациях (то есть более 10-3 M Mo6+). При значениях рН менее 3 образуются соединения, являющиеся в концентрированных растворах октамолибдатами (то есть Mo8O264-). В отличие от разбавленных растворов молибдата (менее 10-5 M Mo6+) при pH менее 3 образуются мономерные соединения протонированного молибдата. Они включают H2MoO4 или H3MoO41+. Учитывая различия в степени конденсации MoO42- в зависимости от концентрации Мо (VI) и pH раствора, не удивительно, что восстановительный потенциал молибдата может быть вариабельным. Таким образом, соединения, которые осаждаются на металлической подложке, могут находиться в некотором диапазоне степеней окисления. Однако молибден будет осаждаться в восстановленной относительно MoO42- форме, где Мо присутствует в степени окисления 6+.
Согласно настоящему изобретению, катион молибдена может быть представлен в композиции очистителя, если он присутствует, в количестве по меньшей мере 10 ч./млн. по отношению к общей массе щелочной композиции, таком как по меньшей мере 50 ч./млн., таком как по меньшей мере 100 ч./млн., и в некоторых случаях может быть представлен в количестве, не превышающем 400 ч./млн. по отношению к общей массе щелочной композиции, таком как не более 300 ч./млн., таком как не более 200 ч./млн. Согласно настоящему изобретению, катион молибдена может быть представлен в щелочной композиции, если он присутствует, в количестве от 10 ч./млн. до 400 по отношению к общей массе щелочной композиции, таком как от 50 ч./млн. до 300 ч./млн., таком как от 100 ч./млн. до 200 ч./млн.
Как упоминалось выше, согласно настоящему изобретению, водная композиция щелочного очистителя может содержать катион кобальта. Катион кобальта может быть представлен в композиции очистителя в форме соли. Неограничивающие примеры анионов, подходящих для образования солей с катионом кобальта, включают нитрат, сульфат, ацетат, хлорид, цитрат, глюконат, сульфамат или их комбинации.
Согласно настоящему изобретению, катион кобальта может быть представлен в водной композиции щелочного очистителя, если он присутствует, в количестве по меньшей мере 50 ч./млн. по отношению к общей массе композиции очистителя, таком как по меньшей мере 100 ч./млн., таком как по меньшей мере 250 ч./млн., и в некоторых случаях может быть представлен в количестве, не превышающем 500 ч./млн. по отношению к общей массе композиции очистителя, таком как не более 400 ч./млн., таком как не более 300 ч./млн. Согласно настоящему изобретению, катион кобальта может быть представлен в водной композиции щелочного очистителя, если он присутствует, в количестве от 50 ч./млн. до 500 ч./млн. по отношению к общей массе композиции очистителя, таком как от 100 ч./млн. до 400 ч./млн., таком как от 250 ч./млн. до 300 ч./млн.
Согласно настоящему изобретению, водная композиция щелочного очистителя может содержать щелочной компонент. Щелочной компонент не ограничен при условии, что он предоставляет для водной композиции щелочного очистителя источник гидроксидных ионов. Например, щелочной компонент может быть гидроксидом натрия, гидроксидом калия и подобными соединениями, или же их комбинациями.
Согласно настоящему изобретению, щелочной компонент может быть представлен в водной композиции щелочного очистителя в количестве, достаточном для доведения pH композиции очистителя до величины по меньшей мере 10. Согласно настоящему изобретению, pH водной композиции щелочного очистителя может равняться по меньшей мере 10, такой как по меньшей мере 10,5, например, по меньшей мере 11, например, по меньшей мере 12, и в некоторых случаях может быть не выше 14, например, не выше 12,5, например, не выше 12. Согласно настоящему изобретению, pH водной композиции щелочного очистителя может составлять от 10 до 14, например, от 10,5 до 12,5, например, от 11 до 12.
Согласно настоящему изобретению, композиция очистителя может содержать фосфонаты или фосфоновую кислоту, которые здесь определяются как химические соединения, которые имеют по меньшей мере одну углерод-фосфорную связь и три связи кислород-фосфор. Эти соединения могут быть представлены в общей форме как R - PO3X2, где R отвечает группе с по меньшей мере одним атомом углерода, связанным с атомом фосфора, и X соответствует водороду или катиону металла. Неограничивающие примеры фосфоновой кислоты или фосфонатов, где R – углеродная цепь, включают метилфосфоновую кислоту, этилфосфоновую кислоту, бутилфосфоновую кислоту, винилфосфоновую кислоту и докозилфосфоновую кислоту. Другие неограничивающие примеры могут включать фосфоновую кислоту или фосфонаты, которые содержат связанные с углеродом гетероатомы помимо фосфора, например, этридроновую кислоту (присутствует в Dequest 2010), иминоди(метилфосфоновую кислоту), N,N-бис(фосфонометил)глицин, N-(фосфонометил)иминодиуксусную кислоту, нитрилотри(метилфосфоновую кислоту) (присутствует в Dequest 2000) и диэтилентриаминпентакис(метилфосфоновую кислоту). Примеры фосфоновой кислоты или фосфонатов, которые являются полидентатными и не включают других связанных с углеродом гетероатомов помимо фосфора, представляют метиленбис(фосфоновая кислота), метантриилтрис(фосфоновая кислота) и метантетраилтетракис(фосфоновая кислота). Свойства фосфоновой кислоты или фосфонатов в высокой степени зависят от их молекулярной структуры. Неограничивающие свойства включают, но не ограничиваются гидрофобностью, гидрофильностью и прочностью связи с металлическими катионам или поверхностями подложки. Безотносительно к какой-либо конкретной теории, когда фосфоновая кислота или фосфонаты добавляются к щелочной чистящей композиции настоящего изобретения, можно было ожидать, что не все фосфонаты приведут к идентичным результатам. Например, трудно предполагать, что монодентатные фосфонаты (такие как CH3CH2CH2CH2PO32-) будут демонстрировать столь же сильное связывание с поверхностью подложки или металлическими катионами в растворе, как полидентатные фосфонаты (например, этридонат, который является бидентатным). Более высокая прочность связывания полидентатных фосфонатов может лучше стабилизировать металлические ионы в растворе или модифицировать скорость травления подложки. Кроме того, нельзя ожидать, что все полидентатные фосфонаты способны обеспечивать одинаковую эффективность, так как предполагается, что размер кольца образующегося хелата способен влиять на стабильность образующихся в растворе комплексных соединений фосфонатов и металлических ионов. Например, этридонат образует шестичленное кольцо с представленным металлом центром в отличие от имино(метилфосфоната), который образует восьмичленное кольцо с металлическими ионами в растворе. С учетом разницы в размере кольца, вероятно, первые соединения более устойчивы, чем последние, поскольку 6-членные кольца термодинамически более выгодны, чем восьмичленные кольца. Эти описанные выше свойства не предполагаются для рассмотрения в качестве исчерпывающих, а скорее являются иллюстрирующими причины того, что специалисты в данной области не стали бы ожидать идентичной эффективности от любой фосфоновой кислоты или фосфоната.
Фосфонаты или фосфоновая кислота могут отличаться соотношением фосфора и углерода, которое здесь определяется как «отношение P-C» и представляет собой общий атомный процент содержания фосфора в фосфонате, деленный на общий атомный процент углерода в данной молекуле. Например, докозилфосфоновая кислота имеет отношение P-C, равное 0,12. Отношение P-C этидроната составляет 2,58. Одним из свойств, которое описывает отношение P-C, является гидрофильность при том, что более низкая величина отношения P-C указывает на более выраженную гидрофобность фосфоновой кислоты. Согласно настоящему изобретению, отношение P-C для фосфоната в щелочной чистящей композиции может составлять по меньшей мере 0,10, являясь таким, как по меньшей мере 0,20, таким, как по меньшей мере 0,30, таким, как по меньшей мере 0,40, и в некоторых случаях может быть не больше, чем 3,20, таким, как не более 5,25, таким, как не более 7,75, таким, как не более 10,3. Согласно настоящему изобретению, величина отношения P-C может составлять от 0,10 до 10,3, являясь такой, как от 0,20 до 7,75, такой, как от 0,30 до 5,25, такой, как от 0,40 до 3,20.
Как указывалось выше, согласно настоящему изобретению, композиция очистителя содержит не более 50 ч./млн. фосфата по отношению к общей массе водной щелочной композиции. В некоторых случаях, согласно настоящему изобретению, композиция очистителя и/или осаждаемые из нее слои могут быть по существу не содержащими, или в некоторых случаях могут быть фактически не содержащими, или в некоторых случаях могут быть полностью не содержащими одного или нескольких фосфатов. Выражение «композиция очистителя и/или осаждаемые из нее слои, которые являются по существу не содержащими фосфата» означает, что фосфат преднамеренно не добавляется, но может быть представлен в ничтожно малых количествах, например, из-за примесей или неизбежных загрязнений из окружающей среды, коммунальных источников водоснабжения и т.п. Другими словами, количество материала является настолько малым, что не влияет на свойства композиции; это может, кроме того, подразумевать, что фосфат не присутствует в композициях очистителя и/или осажденных из них слоях в таких уровнях содержания, которые оказывают нагрузку на окружающую среду. Понятие «по существу не содержит» означает, что композиции очистителя и/или осажденные из них слои содержат менее 25 ч./млн. любого фосфата по отношению к общей массе, соответственно, композиции очистителя или слоя, если фосфат он присутствует. Понятие «фактически не содержит» означает, что композиции очистителя и/или содержащие их слои содержат менее 10 ч./млн. любого фосфата. Понятие «полностью не содержит» означает, что композиции очистителя и/или содержащие их слои содержат менее 1 ч./млрд. любого фосфата.
Также, согласно настоящему изобретению, водная композиция щелочного очистителя может, кроме того, содержать хелатор. Хелатор может содержать, например, карбоксилаты, такие как тартраты, цитраты или глюконаты, комплексные соединения на основе ацетатов, такие как метилглицинацетат, этилендиаминтетраацетат или нитрилотриацетат, фосфаты, такие как пятизамещенный трифосфат натрия или четырехзамещенный пирофосфат калия, фосфонаты, поликарбоксилаты, кислоты, сложные эфиры, соли любого из вышеупомянутых или их комбинации. Хелатор может быть представлен в водной композиции щелочного очистителя в количестве по меньшей мере 10 ч./млн., таком как по меньшей мере 50 ч./млн., таком как по меньшей мере 100 ч./млн., и в некоторых случаях может быть представлен в количестве не больше, чем 10 000 ч./млн., таком как не более 5 000 ч./млн., таком как не более 2 500 ч./млн. по отношению к общей массе композиции очистителя. Хелатор может присутствовать в водной композиции щелочного очистителя в количестве от 10 ч./млн. до 10 000 ч./млн., таком как от 50 ч./млн. до 5 000 ч./млн., таком как от 100 ч./млн. до 2 500 ч./млн. по отношению к общей массе композиции.
Согласно настоящему изобретению, водная композиция щелочного очистителя может также содержать окисляющий компонент. Такой окисляющий компонент может содержать, например, пероксиды, персульфаты, перхлораты, гипохлорит, нитрит, барботируемый кислород, броматы, пероксибензоаты, озон, нитробензолсульфонат натрия или их комбинации. Окисляющий компонент может быть представлен в водной композиции щелочного очистителя в количестве по меньшей мере 10 ч./млн., таком как по меньшей мере 50 ч./млн., таком как по меньшей мере 100 ч./млн., и в некоторых случаях может быть представлен в количестве не больше, чем 5 000 ч./млн., таком как не более 2 500 ч./млн., таком как не более 1 000 ч./млн. по отношению к общей массе композиции очистителя. Окисляющий компонент может присутствовать в водной композиции щелочного очистителя в количестве от 10 ч./млн. до 5 000 ч./млн., таком как от 50 ч./млн. до 2 500 ч./млн., таком как от 100 ч./млн. до 1 000 ч./млн. по отношению к общей массе композиции.
Согласно настоящему изобретению, водная композиция щелочного очистителя может, кроме того, содержать поверхностно-активное вещество. Такое поверхностно-активное вещество может быть анионным, неионогенным, катионным или амфотерным. Поверхностно-активное вещество может содержать, например, этоксилаты спиртов (такие как Tomadol-1-n или Tomadol 91-6, предлагаемые на рынке Evonik Industries, или SEACO 9AE, представленный Sea-Land Chemical Company), алкилфенолэтоксилаты (такие как Makon NF-12, доступный в Surfachem), алкилдифенилсульфонаты (такие как Dowfax 2A1, предлагаемый The Dow Chemical Company), сульфаты (такие как Niaproof 08, доступный в Niacet), фосфатные эфиры (такие как Triton H-66, предлагаемый The Dow Chemical Company), простые эфиры (такие как Triton DF20, предлагаемый на рынке The Dow Chemical Company), сополимеры стирола и малеинового ангидрида (SMA), алкилсултаины (такие как Mirataine ASC и Mirataine CBS, предлагаемые в продаже Rhodia) или их комбинации. Поверхностно-активное вещество может быть представлено в водной щелочной композиции в количестве по меньшей мере 25 ч./млн. по отношению к общей массе щелочной композиции, таком как по меньшей мере 100 ч./млн., таком как по меньшей мере 200 ч./млн., таком как по меньшей мере 500 ч./млн., и в некоторых случаях может быть представлено в количестве не больше, чем 10 000 ч./млн. по отношению к общей массе щелочной композиции, таком как не более 5 000 ч./млн., таком как не более 3 000 ч./млн., таком как не более 2 000 ч./млн. Поверхностно-активное вещество может быть представлено в водной щелочной композиции в количестве от 25 ч./млн. до 10 000 ч./млн. по отношению к общей массе щелочной композиции, таком как от 100 ч./млн. до 5 000 ч./млн., таком как от 200 ч./млн. до 3 000 ч./млн., таком как от 500 ч./млн. до 2 000 ч./млн.
Согласно настоящему изобретению, композиция щелочного очистителя также может необязательно содержать ингибитор коррозии, такой как антикоррозийная добавка, препятствующая мгновенной коррозии стальных подложек, прошедших обработку на обрабатывающей линии. Ингибитор коррозии может содержать, например, нитрит натрия, Hostacor 2098, Halox 515, амины или их комбинации. Согласно настоящему изобретению, ингибитор коррозии может быть представлен в композиции очистителя, если он присутствует, в количестве по меньшей мере 10 ч./млн. по отношению к общей массе композиции очистителя, таком как по меньшей мере 25 ч./млн., таком как по меньшей мере 75 ч./млн., и в некоторых случаях может быть представлен в количестве, не превышающем 10 000 ч./млн. по отношению к общей массе композиции очистителя, таком как не более 5 000 ч./млн., таком как не более 1 500 ч./млн., таком как не более 1 000 ч./млн. Согласно настоящему изобретению, ингибитор коррозии может быть представлен в композиции очистителя, если он присутствует, в количестве от 10 ч./млн. до 10 000 ч./млн. по отношению к общей массе композиции очистителя, таком как от 25 ч./млн. до 5 000 ч./млн., таком как от 75 ч./млн. до 1 500 ч./млн., таком как от 100 ч./млн. до 1 000 ч./млн.
Согласно настоящему изобретению, водная композиция щелочного очистителя может также содержать осаждаемые соединения. Осаждаемые соединения могут содержать, например, силикат, силаны, фосфоновую кислоту, ангидриды или их комбинации. Осаждаемые соединения могут быть представлены в водной композиции щелочного очистителя в количестве по меньшей мере 25 ч./млн., таком как по меньшей мере 50 ч./млн., таком как по меньшей мере 100 ч./млн., и в некоторых случаях могут быть представлены в количестве не большем, чем 5 000 ч./млн., таком как не более 2 500 ч./млн., таком как не более 1 000 ч./млн. по отношению к общей массе композиции. Осаждаемые соединения могут присутствовать в водной щелочной композиции в количестве от 25 ч./млн. до 5 000 ч./млн., таком как от 50 ч./млн. до 2 500 ч./млн., таком как от 100 ч./млн. до 1 000 ч./млн. по отношению к общей массе композиции.
Согласно настоящему изобретению, водная композиция щелочного очистителя может, кроме того, содержать противовспенивающий компонент. Подходящие противовспенивающие агенты включают, например, BYK-011, BYK-20, BYK-32 и BYK 34, коммерчески доступные в BYK-Chemie GmbH, Drewplus L-419, предлагаемый Ashland, и FOAM BAN HV-820G, представленный на рынке Munzing Chemie GmbH. Противовспенивающий компонент может быть представлен в водной щелочной композиции в количестве по меньшей мере 100 ч./млн., таком как по меньшей мере 250 ч./млн., таком как по меньшей мере 500 ч./млн., и в некоторых случаях может быть представлен в количестве не большем, чем 10 000 ч./млн., таком как не более 5 000 ч./млн., таком как не более 2 500 ч./млн. по отношению к общей массе композиции очистителя. Противовспенивающий компонент может быть представлен в водной щелочной композиции в количестве от 100 ч./млн. до 10 000 ч./млн., таком как от 250 ч./млн. до 5 000 ч./млн., таком как от 500 ч./млн. до 2 500 ч./млн. по отношению к общей массе композиции.
Согласно настоящему изобретению, водная щелочная композиция может содержать водную среду и может необязательно содержать другие материалы, такие как вспомогательные средства, традиционно применяемые в области композиций очистителей. В водной среде могут присутствовать диспергируемые в воде органические растворители, например, спирты с вплоть до около 8 атомов углерода, такие как метанол, изопропанол и другие подобные; или гликолевые эфиры, такие как моноалкильные эфиры этиленгликоля, диэтиленгликоля или пропиленгликоля и другие подобные. В случае их присутствия диспергируемые в воде органические растворители в типичном случае используются в количествах вплоть до около 2 об.% по отношению к общему объему водной среды.
Системы обработки
Согласно настоящему изобретению, описанная выше водная композиция щелочного очистителя может быть частью системы обработки, предназначенной для обработки металлической подложки. Такая система обработки может содержать, или в некоторых случаях состоять по существу из, или в некоторых случаях состоять из описанной выше водной щелочной композиции для обработки участка подложки и композиции для предварительной обработки, предназначенной для обработки по меньшей мере одного участка подложки, прошедшего обработку водной щелочной композицией.
Например, система обработки может содержать, или в некоторых случаях состоять из, или в некоторых случаях состоять по существу из: a) водной композиции щелочного очистителя, содержащей, или в некоторых случаях состоящей по существу из, или в некоторых случаях состоящей из катионов железа, катионов молибдена и/или катионов кобальта и щелочного компонента, при том, что pH водной композиции щелочного очистителя равен по меньшей мере 10 и водная композиция щелочного очистителя содержит не больше, чем 50 ч./млн. фосфата по отношению к общей массе водной композиции щелочного очистителя; и b) композиции для предварительной обработки, содержащей металл Группы IVB, предназначенной для обработки по меньшей мере одного участка подложки и содержащей, или в некоторых случаях состоящей по существу из, или в некоторых случаях состоящей из катионов металла Группы IVB, более подробно описанной ниже.
Например, в качестве варианта, система обработки, предназначенная для обработки металлических подложек, может содержать, или в некоторых случаях состоять из, или в некоторых случаях состоять по существу из: a) водной композиции щелочного очистителя, содержащей, или в некоторых случаях состоящей по существу из, или в некоторых случаях состоящей из катионов железа, катионов молибдена и/или катионов кобальта и щелочного компонента, причем pH водной щелочной композиции равен по меньшей мере 10 и водная щелочная композиция содержит не больше, чем 50 ч./млн. фосфата по отношению к общей массе водной щелочной композиции; b) не обязательно, активирующего ополаскивателя, предназначенного для обработки по меньшей мере одного участка подложки; и c) металло-фосфатной композиции для предварительной обработки, предназначенной для обработки по меньшей мере одного участка подложки. Согласно настоящему изобретению, металло-фосфатная композиция для предварительной обработки может содержать, например, ион цинка, и/или ион железа, и фосфатный ион.
Согласно настоящему изобретению, водная композиция щелочного очистителя системы обработки может быть такой, как описано выше, и может быть приведена в контакт с подложкой любым из множества различных способов, таким как окунание или погружение, напыление, импульсное распыление, окунание, сопровождаемое напылением, распыление, сопровождаемое окунанием, нанесение кистью или с помощью валиков. Согласно настоящему изобретению, водная композиция щелочного очистителя при нанесении на металлическую подложку может находиться при температуре в пределах от 10°C до 90°C, такой как от 25°C до 75°C. Например, контакт с подложкой может осуществляться при температуре окружающей среды или при комнатной температуре. Контактное время часто составляет по меньшей мере 60 секунд, такое как по меньшей мере 90 секунд, такое как по меньшей мере 120 секунд. Согласно настоящему изобретению, контактное время часто находится в диапазоне от 60 секунд до 120 секунд, таком как от 75 секунд до 100 секунд.
Как упоминалось выше, согласно настоящему изобретению, система обработки также может содержать композицию для предварительной обработки. Для целей настоящего изобретения термин «композиция для предварительной обработки» относится к композиции, которая при контакте с подложкой вступает с ней в реакцию, химически изменяет поверхность подложки и связывается с ней с образованием защитного слоя. Например, композиция для предварительной обработки системы обработки может быть металло-фосфатной композицией для предварительной обработки или композицией для предварительной обработки, содержащей металл Группы IVB, такими, как описанные ниже.
Металло-фосфатная композиция для предварительной обработки
Согласно настоящему изобретению, композиция для предварительной обработки может быть металло-фосфатной композицией для предварительной обработки, содержащей ион металла и фосфатный ион. В данном контексте термин «металло-фосфатная композиция для предварительной обработки» относится к композиции, содержащей фосфаты цинка, железа и/или других двухвалентных металлов, известных в данной области, которые при контакте с подложкой вступают с ней в реакцию, химически изменяют поверхность подложки и связываются с ней с образованием защитного слоя.
Согласно настоящему изобретению, металлический ион в металло-фосфатной композиции для предварительной обработки может быть цинком и содержание ионов цинка в композиции для предварительной обработки может составлять, если они присутствуют, по меньшей мере 500 ч./млн., например, по меньшей мере 800 ч./млн., и в некоторых случаях может быть не больше, чем 1 500 ч./млн., например, не больше, чем 1 200 ч./млн. по отношению к общей массе композиции для предварительной обработки. Согласно настоящему изобретению, содержание ионов цинка в композиции для предварительной обработки может составлять, если они присутствуют, от 500 ч./млн. до 1 500 ч./млн., например, по меньшей мере от 800 ч./млн. до 1 200 ч./млн. по отношению к общей массе композиции для предварительной обработки. Источником ионов цинка могут быть стандартные источники цинковых ионов, такие как нитрат цинка, оксид цинка, карбонат цинка, металлический цинк и другие подобные.
Согласно настоящему изобретению, металлический ион в металло-фосфатной композиции для предварительной обработки может быть железом и содержание ионов железа в композиции для предварительной обработки может составлять, если они присутствуют, по меньшей мере 5 ч./млн., например, по меньшей мере 8 ч./млн., например, по меньшей мере 10 ч./млн., и в некоторых случаях может быть не больше, чем 550 ч./млн., например, не больше, чем 250 ч./млн., например, не больше, чем 100 ч./млн. по отношению к общей массе композиции для предварительной обработки. Согласно настоящему изобретению, содержание ионов цинка в композиции для предварительной обработки может составлять, если они присутствуют, от 5 ч./млн. до 550 ч./млн., например, по меньшей мере от 8 ч./млн. до 250 ч./млн., например, по меньшей мере от 10 ч./млн. до 100 ч./млн. по отношению к общей массе композиции для предварительной обработки.
Согласно настоящему изобретению, содержание фосфата в композиции для предварительной обработки может составлять по меньшей мере 8 000 ч./млн., насчитывая по меньшей мере 12 000 ч./млн., и в некоторых случаях может быть не больше, чем 20 000 ч./млн., таким как не больше, чем 14 000 ч./млн. по отношению к общей массе композиции для предварительной обработки. Согласно настоящему изобретению, содержание фосфата в композиции для предварительной обработки может составлять от 8 000 ч./млн. до 20 000 ч./млн., например, от 12 000 ч./млн. до 14 000 ч./млн. по отношению к общей массе композиции для предварительной обработки. Источником фосфатного иона может быть фосфорная кислота, монофосфат натрия, двузамещенный фосфат натрия и другие подобные.
Согласно настоящему изобретению, в дополнение к описанным выше катионам металло-фосфатная композиция для предварительной обработки также может содержать ионы натрия, калия и/или аммония для целей регулирования содержания свободной кислоты и/или общей кислотности. Свободная кислотность и общая кислотность могут быть определены так, как описано в следующих ниже примерах. Согласно настоящему изобретению, металло-фосфатная композиция для предварительной обработки может иметь показатель свободной кислотности от 0,1 пунктов до 2 пунктов, такой как от 0,5 пунктов до 1,5 пунктов, такой как от 0,7 пунктов до 1,1 пунктов. Согласно настоящему изобретению, металло-фосфатная композиция для предварительной обработки может иметь показатель общей кислотности от 5 пунктов до 40 пунктов, такой как от 7,5 пунктов до 10,5 пунктов, такой как от 10 пунктов до 30 пунктов, такой как от 15 пунктов до 24 пунктов.
Согласно настоящему изобретению, металло-фосфатная композиция для предварительной обработки может иметь pH от 3,0 до 6,5, такой как от 3,0 до 4,0, такой как от 4,5 до 6,0.
Согласно настоящему изобретению, металло-фосфатная композиция для предварительной обработки может также содержать ускоряющую добавку. Ускоряющая добавка может быть представлена в количестве, достаточном для ускорения образования металло-фосфатного покрытия, и может присутствовать в композиции для предварительной обработки в количестве по меньшей мере 500 ч./млн., таком как по меньшей мере 1 000 ч./млн., таком как по меньшей мере 2 500 ч./млн., и в некоторых случаях может быть представлена в количестве не большем, чем 20 000 ч./млн., таком как не больше, чем 10 000 ч./млн., такой как не больше, чем 5 000 ч./млн., по отношению к общей массе композиции для предварительной обработки. Согласно настоящему изобретению, ускоряющая добавка может быть представлена в композиции для предварительной обработки в количестве от 500 ч./млн. до 20 000 ч./млн., таком как от 1 000 ч./млн. до 10 000 ч./млн., таком как от 2 500 ч./млн. до 5 000 ч./млн. по отношению к общей массе композиции для предварительной обработки. Подходящие для применения ускоряющие добавки могут включать оксимы, такие как оксим ацетальдегида и ацетоксим, нитриты, такие как нитрит натрия и нитрит аммония, пероксиды, такие как пероксид водорода, хлораты, такие как хлорат натрия, или сульфонаты, такие как нитробензолсульфонат натрия, или их комбинации.
Согласно настоящему изобретению, металло-фосфатная композиция для предварительной обработки может также содержать (свободный) фторидный ион, ион нитрата и различные металлические ионы, такие как ионы никеля, ионы кобальта, ионы кальция, ионы магния, ионы марганца, ионы железа, ионы меди и другие подобные.
Фторид, присутствующий в металло-фосфатной композиции для предварительной обработки, может вводиться в виде фторидов аммония и щелочных металлов, фторангидридов, фтороборной кислоты, фторкремниевой кислоты и/или других неорганических фторидов. Неисключительные примеры фторидов включают фторид цинка, фторид цинка-алюминия, фторид никеля, фторид аммония, фторид натрия, фторид калия и фтористоводородную кислоту, а также другие подобные материалы, известные специалистам в данной области.
Фториды, присутствующие в металло-фосфатной композиции для предварительной обработки и не связанные с ионами металлов или водородным ионом, определяются здесь как «свободный фторид»; их содержание может быть измерено в качестве технологического параметра в ванне с металло-фосфатной композицией с помощью, например, двухканального настольного измерителя Orion Dual Star, оснащенного фторидным ион-селективным электродом (“ISE”), предлагаемым компанией Thermoscientific, фторидным ион-селективным комбинированным электродом Symphony®, выпускаемым компанией VWR International, или другими подобными электродами. См., например, Light and Cappuccino, "Determination of fluoride in toothpaste using an ion-selective electrode” («Определение фторида в зубной пасте с помощь ион-селективного электрода»), J. Chem. Educ., 52:4, 247 - 250, апрель 1975. Фторидный ISE может быть стандартизован посредством погружения электрода в растворы с известными концентрациями фторидов и фиксацией показаний в милливольтах, а затем нанесением этих выраженных в милливольтах данных на график с логарифмическими координатами. Показания в милливольтах, снятые с неизвестного образца, затем сравниваются с этим калибровочным графиком, в результате чего определяется концентрация фторида. В качестве варианта, фторидный ISE может применяться с измерительным устройством, которое выполняет калибровочные вычисления внутренним образом и, таким образом, данные по концентрации неизвестного образца могут считываться непосредственно.
Фторидный ион является небольшим, отрицательно заряженным ионом с высокой плотностью заряда, вследствие чего в водном растворе он часто образует комплексы с металлическими ионами, имеющими высокую плотность положительного заряда, или с водородным ионом. Находящиеся в растворе фторидные анионы, которые являются связанными ионной или ковалентной связью с металлическими катионами или водородным ионом, определяются здесь как «связанные фториды». Закомплексованные таким образом фторидные ионы не поддаются измерениям с помощью фторидных ISE, если раствор, в котором они присутствуют, не смешан с регулирующим ионную силу буфером (например, цитратным анионом или ЭДТА), который высвобождает фторидные ионы из таких комплексных соединений. В таком случае (все) фторидные ионы оказываются пригодными для измерений фторидным ISE, а результаты таких измерений известны как «общий фторид». В качестве варианта, общий фторид может вычисляться путем сравнения массы фторида, вводимого в металло-фосфатную композицию предварительной обработки, с общей массой композиции.
Свободный фторид может быть представлен в металло-фосфатной композиции для предварительной обработки в количестве по меньшей мере 100 ч./млн., таком как по меньшей мере 150 ч./млн., по меньшей мере 200 ч./млн., и в некоторых случаях может быть представлен в количестве, не превышающем 2000 ч./млн., таком как не более 1000 ч./млн., таком как не более 500 ч./млн., и в некоторых случаях может присутствовать в количестве от 100 ч./млн. до 3000 ч./млн., таком как от 150 ч./млн. до 1 000 ч./млн., таком как от 200 ч./млн. до 500 ч./млн. свободного фторида по отношению к общей массе композиции для предварительной обработки.
Общий фторид может быть представлен в металло-фосфатной композиции для предварительной обработки в количестве по меньшей мере 200 ч./млн., таком как по меньшей мере 300 ч./млн., таком как по меньшей мере 400 ч./млн., и в некоторых случаях может быть представлен в количестве, не превышающем 2 500 ч./млн., таком как не более 1750 ч./млн., таком как не более 1250 ч./млн., и в некоторых случаях может присутствовать в количестве от 200 ч./млн. до 2 500 ч./млн., таком как от 300 ч./млн. до 1 750 ч./млн., таком как от 400 ч./млн. до 1250 ч./млн. свободного фторида по отношению к общей массе композиции для предварительной обработки.
Согласно настоящему изобретению, ион нитрата может быть представлен в металло-фосфатной композиции для предварительной обработки в количестве по меньшей мере 1 000 ч./млн., таком как по меньшей мере 2 000 ч./млн., и в некоторых случаях может быть представлен в количестве, не превышающем 10 000 ч./млн., таком как не более 5 000 ч./млн., и в некоторых случаях может присутствовать в количестве от 1 000 ч./млн. до 10 000 ч./млн., таком как от 2 000 ч./млн. до 5 000 ч./млн. по отношению к общей массе композиции для предварительной обработки.
Согласно настоящему изобретению, ион кальция может быть представлен в металло-фосфатной композиции для предварительной обработки в количестве по меньшей мере 100 ч./млн., таком как по меньшей мере 500 ч./млн., и в некоторых случаях может быть представлен в количестве, не превышающем 4 000 ч./млн., таком как не более 2 500 ч./млн., и в некоторых случаях может присутствовать в количестве от 100 ч./млн. до 4 000 ч./млн., таком как от 500 ч./млн. до 2 500 ч./млн. по отношению к общей массе композиции для предварительной обработки.
Согласно настоящему изобретению, ион марганца может быть представлен в металло-фосфатной композиции для предварительной обработки в количестве по меньшей мере 100 ч./млн., таком как по меньшей мере 200 ч./млн., таком как по меньшей мере 500 ч./млн., и в некоторых случаях не более 1 500 ч./млн., таком как не более 1 000 ч./млн., таком как не более 800 ч./млн., и в некоторых случаях в количестве от 100 ч./млн. до 1 500 ч./млн., таком как от 200 ч./млн. до 1 000 ч./млн., таком как от 500 ч./млн. до 800 ч./млн. по отношению к общей массе композиции для предварительной обработки.
Согласно настоящему изобретению, ион железа может быть представлен в металло-фосфатной композиции для предварительной обработки в количестве по меньшей мере 5 ч./млн., таком как по меньшей мере 10 ч./млн., таком как по меньшей мере 50 ч./млн., и в некоторых случаях не превышающем 500 ч./млн., таком как не более 300 ч./млн., и в некоторых случаях может присутствовать в композиции для предварительной обработки в количестве от 5 ч./млн. до 500 ч./млн., таком как от 5 ч./млн. до 20 ч./млн., таком как от 50 ч./млн. до 300 ч./млн. по отношению к общей массе композиции для предварительной обработки.
Согласно настоящему изобретению, ион меди может быть представлен в металло-фосфатной композиции для предварительной обработки в количестве по меньшей мере 1 ч./млн., таком как по меньшей мере 3 ч./млн., и в некоторых случаях может быть представлен в количестве, не превышающем 30 ч./млн., таком как не более 15 ч./млн., и в некоторых случаях может присутствовать в количестве от 1 ч./млн. до 30 ч./млн., таком как от 3 ч./млн. до 15 ч./млн. по отношению к общей массе композиции для предварительной обработки.
Согласно настоящему изобретению, ион никеля может быть представлен в металло-фосфатной композиции для предварительной обработки в количестве по меньшей мере 100 ч./млн., таком как по меньшей мере 200 ч./млн., таком как по меньшей мере 300 ч./млн., и в некоторых случаях может быть представлен в композиции для предварительной обработки в количестве, не превышающем 1 800 ч./млн., таком как не более 1 200 ч./млн., таком как не более 800 ч./млн., и в некоторых случаях может присутствовать в композиции для предварительной обработки в количестве от 100 ч./млн. до 1 800 ч./млн., таком как от 200 ч./млн. до 1 200 ч./млн., таком как от 300 ч./млн. до 800 ч./млн. по отношению к общей массе композиции для предварительной обработки.
Согласно настоящему изобретению, металло-фосфатная композиция для предварительной обработки может быть по существу не содержащей, или в некоторых случаях фактически не содержащей, или в некоторых случаях полностью не содержащей никеля. В данном контексте выражение «по существу не содержащая», когда оно используется в отношении отсутствия никеля, означает, что никель, если он присутствует в ванне, содержащей композицию для предварительной обработки, в композиции для предварительной обработки и/или в слоях, образованных из нее и ее содержащих, если он присутствует, то присутствует лишь в ничтожно малом количестве 5 ч./млн. или менее по отношению к общей массе композиции или слоя(-ев), исключая, в зависимости от обстоятельств, никель, полученный из занесенных в ванну примесей, из подложки (подложек) и/или в результате растворения оборудования. В данном контексте выражение «фактически не содержащая», когда оно используется в отношении отсутствия никеля, означает, что никель, если он присутствует в ванне, содержащей композицию для предварительной обработки, в композиции для предварительной обработки и/или в слоях, образованных из нее и ее содержащих, если он присутствует, то присутствует лишь в ничтожно малом количестве 1 ч./млн. или менее по отношению к общей массе композиции или слоя(-ев), исключая, в зависимости от обстоятельств, никель, полученный из занесенных в ванну примесей, из подложки (подложек) и/или в результате растворения оборудования. В данном контексте выражение «полностью не содержащая», когда оно используется в отношении отсутствия никеля, означает, что никель отсутствует в ванне, содержащей композицию для предварительной обработки, в композиции для предварительной обработки и/или в слое, образованном из нее и ее содержащем (то есть ванна, содержащая композицию для предварительной обработки, композиция для предварительной обработки и/или слой, образованный из нее и ее содержащий, содержат 0 ч./млн. никеля, исключая никель, полученный из занесенных в ванну примесей, из подложки (подложек) и/или в результате растворения оборудования.
Металло-фосфатная композиция для предварительной обработки может быть нанесена на подложку распылением или погружением подложки в кислую фосфатную ванну, содержащую указанную композицию для предварительной обработки при температуре, в типичном случае находящейся в пределах от 25°C до 75°C, в типичном случае на время от 1 до 3 минут, такое как от 1 минуты до 2 минут, такое как от 1 минуты до 90 секунд. Покрытие, которое образуется на подложке в результате введения подложки в контакт с металло-фосфатной композицией для предварительной обработки, может иметь толщину от 0,25 мкм до 8 мкм и массу покрытия от 70 мг/фут2 до 800 мг/фут2.
Неожиданно было обнаружено, что очистка подложки композицией щелочного очистителя настоящего изобретения, сопровождаемая предварительной обработкой металло-фосфатной композицией для предварительной обработки, приводит к подложке, которая имеет значительно увеличенную энергию разрушения по сравнению с подложкой, очищенной с применением чистящей композицией, не включающей железа и/или кобальта, за которой следовала предварительная обработка металло-фосфатной композицией для предварительной обработки; к такой как, например, подложка, которая имеет энергию разрушения по меньшей мере 1000 Дж/м2, такую как по меньшей мере 1500 Дж/м2, такую как по меньшей мере 2000 Дж/м2 по результатам испытаний согласно протоколу, описанному в примерах. Согласно настоящему изобретению, очистка подложки композицией щелочного очистителя настоящего изобретения, сопровождаемая предварительной обработкой металло-фосфатной композицией для предварительной обработки, приводит к получению подложки, которая имеет по меньшей мере 2X увеличение энергии разрушения относительно подложки, очищенной композицией очистителя, не включающей железа и/или кобальта, сопровождаемой предварительной обработкой металло-фосфатной композицией для предварительной обработки; такое как по меньшей мере 5X увеличение энергии разрушения, такое как по меньшей мере 10X увеличение энергии разрушения по результатам испытаний согласно протоколу, описанному в примерах.
Кроме того, неожиданно было обнаружено, что при отсутствии фосфатных ионов железо из композиции щелочного очистителя настоящего изобретения осаждается на поверхности металлов в виде металлического железа. Ранее в данной области предполагалось, что осаждение фосфата железа из чистящей композиции является критически важным для улучшения эффективности композиции для предварительной обработки. В системе, которая не содержит ортофосфатов или PO43-, фосфат железа образовываться не может. Осаждение металлического железа влечет за собой улучшение защиты от коррозии, признаком чего оказывается уменьшение распространения коррозии при скрайбировании в испытании “scribe creep”, а также оказывает положительное влияние на адгезию в том, что касается снижения потерь краски при оценке сухой и влажной адгезии методом перекрестной штриховки или увеличенной энергии разрушения в ходе оценки адгезии при отслаивании под углом 180° (t-peel test), результаты которых были получены согласно описанному в примерах протоколу.
Кроме того, неожиданно было обнаружено, что по данным испытаний, проводившихся согласно описанному в примерах протоколу, очистка подложки с помощью композиции щелочного очистителя настоящего изобретения, сопровождаемая предварительной обработкой не содержащей никеля металло-фосфатной композицией для предварительной обработки, позволяет получить подложку, которая имеет коррозионную характеристику, сопоставимую с характеристикой подложки, обрабатывавшейся содержащей никель металло-фосфатной композицией для предварительной обработки и стандартной композицией очистителя (то есть композицией очистителя, которая не включает железо и/или кобальт).
Кроме того, неожиданно было обнаружено, что очистка подложки с помощью композиции щелочного очистителя настоящего изобретения, сопровождаемая предварительной обработкой композицией для предварительной обработки, содержащей фосфат железа, позволяет получить подложку, обладающую коррозионной характеристикой, которая значительно улучшена по сравнению с подложкой, очищенной чистящей композицией, не включающей железа и/или кобальта, с последующей предварительной обработкой композицией для предварительной обработки, содержащей фосфат железа. Согласно настоящему изобретению, подложка с композицией щелочного очистителя настоящего изобретения, сопровождаемой предварительной обработкой композицией для предварительной обработки, содержащей фосфат железа, позволяет получить подложку, которая в испытаниях, выполнявшихся согласно приведенному в примерах протоколу, демонстрирует средний показатель распространения коррозии при скрайбировании не более 4,5 мм, такой как не более 4 мм. Согласно настоящему изобретению, очистка подложки композицией щелочного очистителя настоящего изобретения, сопровождаемая предварительной обработкой с помощью металло-фосфатной композиции для предварительной обработки, приводит к получению подложки, которая по результатам испытаний, выполненных согласно приведенному в примерах протоколу, демонстрирует по меньшей мере 50%-ое снижение показателя распространения коррозии при скрайбировании относительно подложки, очищенной композицией очистителя, не включающей железа и/или кобальта, с последующей предварительной обработкой металло-фосфатной композицией для предварительной обработки, такое как по меньшей мере 55%-ое снижение, такое как по меньшей мере 60%-ое снижение, такое как по меньшей мере 65%-ое снижение, такое как по меньшей мере 70%-ое снижение.
Кроме того, было неожиданно обнаружено, что энергия разрушения может значительно изменяться в зависимости от вида фосфонатов, бифосфонатов, полифосфонатов и/или фосфоновой кислоты, используемых в композиции очистителя. Некоторые из очистителей, полученных с использованием комбинаций вышеупомянутых молекул, по результатам испытаний, выполненных согласно приведенному в примерах протоколу, показали существенные улучшения в том, что касается энергии разрушения в композициях очистителя, содержащих железо и кобальт. Было найдено, что молекулы, имеющие соотношение P-C в описанных здесь диапазонах, оказались особенно эффективными в обеспечении увеличения энергии разрушения.
Композиция предварительной обработки с металлами Группы IVB
Согласно настоящему изобретению, композиция для предварительной обработки настоящего изобретения может быть композицией для предварительной обработки металлом Группы IVB, содержащей катион металла Группы IVB. Например, катион металла Группы IVB, применяемый в композиции для предварительной обработки металлом Группы IVB, может быть соединением циркония, титана, гафния, скандия или их смесью. Подходящие соединения циркония включают, но не ограничиваются гексафторциркониевой кислотой, ее солями с щелочными металлами и аммонием, карбонатом аммония-циркония, нитратом цирконила, сульфатом цирконила, карбоксилатами циркония и гидроксикарбоксилатами циркония, такими как ацетат циркония, оксалат циркония, гликолят аммония-циркония, лактат аммония-циркония, цитрат аммония-циркония и их смеси. Подходящие соединения титана включают, но не ограничиваются фтортитановой кислотой и ее солями. Подходящее соединение гафния включает, но не ограничивается нитратом гафния.
Согласно настоящему изобретению, катион металла Группы IVB может быть представлен в композиции для предварительной обработки, содержащей металл Группы IVB, в общем количестве по меньшей мере 20 ч./млн. металла по отношению к общей массе композиции для предварительной обработки, таком как по меньшей мере 50 ч./млн. или в некоторых случаях по меньшей мере 70 ч./млн., и в некоторых случаях может быть представлен в композиции для предварительной обработки, содержащей металл Группы IVB, в общем количестве, не превышающем 1 000 ч./млн. металла по отношению к общей массе композиции для предварительной обработки, таком как не более 600 ч./млн. металл, таком как не более 300 ч./млн. металла. Согласно настоящему изобретению, катион металла Группы IVB может быть представляет в композиции для предварительной обработки, содержащей металл Группы IVB, в общем количестве от 20 ч./млн. металла до 1 000 ч./млн. металла по отношению к общей массе композиции для предварительной обработки, таком как от 50 ч./млн. металла до 600 ч./млн. металла, таком как от 70 ч./млн. металла до 300 ч./млн. металла. В данном контексте понятие «общее количество», когда оно применяется в отношении количества металла Группы IVB, означает сумму всех металлов Группы IV, присутствующих в композиции для предварительной обработки, содержащей металл Группы IVB,.
Согласно настоящему изобретению, композиция для предварительной обработки, содержащая металл Группы IVB, также может содержать ион электроположительного металла. В данном контексте термин «ион электроположительного металла» относится к металлическим ионам, которые будут восстанавливаться подвергаемой обработке металлической подложкой при контактировании раствора композиции для предварительной обработки с поверхностью металлической подложки. Специалистам в данной области известно, что склонность химических соединений к переходу в восстановленное состояние называется восстановительным потенциалом, выражается в вольтах и измеряется относительно стандартного водородного электрода, восстановительный потенциал которого произвольно задается равным нулю. Ниже в Таблице 1 приводятся данные по восстановительному потенциалу нескольких элементов (согласно CRC, выпуск 82, 2001 - 2002). Элемент или ион является более легко восстанавливаемым, чем другой элемент или ион, если в следующей далее таблице имеет величину разности потенциалов E* более положительную, чем у элемента или иона, с которым он сравнивается.
Таблица 1
Таким образом, очевидно, что когда металлическая подложка содержит один из перечисленных выше материалов, таких как холоднокатаная сталь, горячекатаная сталь, сталь с покрытием из металлического цинка, соединений цинка или цинковых сплавов, сталь горячего цинкования, сталь с покрытием из гальванила, сталь, плакированная цинковым сплавом, алюминиевые сплавы, сталь с покрытием из алюминия, сталь с покрытием из алюминиевого сплава, магний и магниевые сплавы, подходящие для осаждения на ней ионы электроположительных металлов включают, например, никель, медь, серебро и золото, а также их смеси.
Согласно настоящему изобретению, когда данный ион электроположительного металла включает медь, в качестве источника медных ионов в композициях для предварительной обработки могут выступать как растворимые, так и нерастворимые соединения. Например, источником снабжения медными ионами композиции для предварительной обработки может являться водорастворимое соединение меди. Конкретные примеры таких соединений включают, но не ограничиваются сульфатом меди, нитратом меди, тиоционатом меди, тетрагидратом четырехзамещенного этилендиаминтетраацетата натрия и меди, бромидом меди, оксидом меди, гидроксидом меди, хлоридом меди, фторидом меди, глюконатом меди, цитратом меди, лауроилсаркозинатом меди, лактатом меди, оксалатом меди, тартратом меди, малатом меди, сукцинатом меди, малонатом меди, малеатом меди, бензоатом меди, салицилатом меди, медными комплексами аминокислот, фумаратом меди, глицерофосфатом меди, натрий-медь-хлорофиллином, фторсиликатом меди, фторборатом меди и йодатом меди, а также медными солями карбоновых кислот в гомологическом ряду от муравьиной кислоты до декановой кислоты и медными солями многоосновных кислот ряда от щавелевой кислоты до пробковой кислоты.
Когда ионы меди, поступающие из такого водорастворимого медного соединения, осаждаются в качестве примеси в форме сульфата меди, оксида меди и т.д., предпочтительным может оказаться добавление комплексообразующего реагента, который подавляет осаждение ионов меди, тем самым стабилизируя их в композиции в виде комплексного соединения меди.
Согласно настоящему изобретению, соединение меди может быть добавлено в виде комплексной соли меди, такой как Cu-EDTA, которая может устойчиво существовать в композиции для предварительной обработки как таковая, а также возможно получение комплексного соединения меди, способного стабильно находиться в композиции для предварительной обработки в результате соединения комплексообразующего реагента с веществом, которое само по себе является трудно растворимым. Его пример включает комплексное соединение Cu-EDTA, образованное комбинацией CuSO4 и EDTA•2Na.
Согласно настоящему изобретению, ион электроположительного металла может быть представлен в композиции для предварительной обработки в количестве по меньшей мере 2 ч./млн. (в расчете на металлический ион) по отношению к общей массе композиции для предварительной обработки, таком как по меньшей мере 4 ч./млн., таком как по меньшей мере 6 ч./млн., таком как по меньшей мере 8 ч./млн., таком как по меньшей мере 10 ч./млн. Согласно настоящему изобретению, ион электроположительного металла может быть представлен в композиции для предварительной обработки в количестве, не превышающем 100 ч./млн. (в расчете на металлический ион) по отношению к общей массе композиции для предварительной обработки, таком как не превышающее 80 ч./млн., таком как не превышающее 60 ч./млн., таком как не превышающее 40 ч./млн., таком как не превышающее 20 ч./млн. Согласно настоящему изобретению, ион электроположительного металла может быть представлен в композиции для предварительной обработки в количестве от 2 ч./млн. до 100 ч./млн. (в расчете на металлический ион) по отношению к общей массе композиции для предварительной обработки, таком как мере от 4 ч./млн. до 80 ч./млн., таком как от 6 ч./млн. до 60 ч./млн., таком как от 8 ч./млн. до 40 ч./млн. Количество ионов электроположительного металла в композиции для предварительной обработки может находиться в диапазоне между указанными величинами, включая и сами указанные величины. Согласно настоящему изобретению, в композиции для предварительной обработки, содержащей металл Группы IVB, может быть представлен источник фторида. В данном контексте количество раскрываемого или указываемого в композиции для предварительной обработки фторида упоминается «как свободный фторид» и измеряется в единицах частей фторида на миллион. Свободный фторид здесь ограничивается как поддающийся измерению с помощью фтор-селективного ISE. Помимо свободного фторида композиция для предварительной обработки может также содержать «связанный фторид», который описывается выше. Сумма концентраций связанного и свободного фторида соответствует общему фториду, который может быть определен, как здесь описано. Общий фторид может вводиться в композицию для предварительной обработки с помощью фтористоводородной кислоты, а также фторидов щелочных металлов и аммония или фторидов водорода. Помимо этого, общий фторид может поступать в композицию для предварительной обработки из присутствующих в композиции для предварительной обработки соединений металлов Группы IVB, включая, например, гексафторциркониевую кислоту или гексафтортитановую кислоту. Другие комплексные фториды, такие как H2SiF6 или HBF4, могут быть добавлены к композиции для предварительной обработки для обеспечения наличия общего фторида. Специалистам в данной области понятно, что присутствие свободного фторида в ванне с композицией для предварительной обработки может воздействовать на осаждение композиции для предварительной обработки и на травление подложки, поэтому измерение этого параметра ванны имеет критически важное значение. Уровни содержания свободного фторида будут зависеть от pH и добавления хелаторов в ванну с композицией для предварительной обработки и указывают на степень ассоциации фторида с металлическими ионами/протонами, присутствующими в ванне с композицией для предварительной обработки. Например, композиции для предварительной обработки с идентичными уровнями общих фторидов могут иметь различные уровни свободных фторидов, которые будут находиться под влиянием pH и присутствия хелаторов в растворе композиции для предварительной обработки.
Согласно настоящему изобретению, свободный фторид в композиции для предварительной обработки может быть представлен в количестве по меньшей мере 15 ч./млн. по отношению к общей массе композиции для предварительной обработки, таком как по меньшей мере 50 ч./млн. свободного фторида, таком как по меньшей мере 100 ч./млн. свободного фторида, таком как по меньшей мере 200 ч./млн. свободного фторида. Согласно настоящему изобретению, свободный фторид в композиции для предварительной обработки может быть представлен в количестве, не превышающем 2500 ч./млн. по отношению к общей массе композиции для предварительной обработки, таком как не превышающее 1000 ч./млн. свободного фторида, таком как не превышающее 500 ч./млн. свободного фторида, таком как не превышающее 250 ч./млн. свободного фторида. Согласно настоящему изобретению, свободный фторид в композиции для предварительной обработки может быть представлен в количестве от 15 ч./млн. свободного фторида до 2500 ч./млн. свободного фторида по отношению к общей массе композиции для предварительной обработки, таком как от 50 ч./млн. фторида до 1000 ч./млн., таком как от не превышающего 200 ч./млн. свободного фторида до 500 ч./млн. свободного фторида, таком как от не превышающего 100 ч./млн. свободного фторида до 250 ч./млн. свободного фторида.
Согласно настоящему изобретению, композиция для предварительной обработки, содержащая металл Группы IVB, может содержать источник молибдена. Согласно настоящему изобретению, источник молибдена, используемого в композиция для предварительной обработки, содержащей металл Группы IVB, может находиться в форме соли. Подходящие соли молибдена могут включать молибдат натрия, молибдат кальция, молибдат калия, молибдат аммония, хлорид молибдена, ацетат молибдена, сульфамат молибдена, формиат молибдена или лактат молибдена.
Согласно настоящему изобретению, молибден может быть представлен в композиции для предварительной обработки, содержащей металл Группы IVB, в количестве по меньшей мере 5 ч./млн. (в расчете на элементарный металл), таком как по меньшей мере 20 ч./млн., таком как по меньшей мере 50 ч./млн., и может быть представлен в количестве, не превышающем 500 ч./млн., таком как не превышающее 300 ч./млн., таком как не превышающее 150 ч./млн. по отношению к общей массе композиции для предварительной обработки, содержащей металл Группы IVB,. Молибден может быть представлен в композиции для предварительной обработки, содержащей металл Группы IVB, в количестве от 5 ч./млн. до 500 ч./млн., таком как от 5 ч./млн. до 150 ч./млн. по отношению к общей массе композиции для предварительной обработки с металлом Группы IVB. Согласно настоящему изобретению, мольное отношение металла Группы IVB к молибдену может находиться между 100 : 1 и 1 : 10, например, между 30 : 1 и 1 : 1.
Согласно настоящему изобретению, композиции для предварительной обработки, содержащие металл Группы IVB, также могут содержать литий. Согласно настоящему изобретению, источник лития, используемого в композиции для предварительной обработки, может быть в форме соли. Подходящие соли лития могут включать нитрат лития, сульфат лития, фторид лития, хлорид лития, гидроксид лития, карбонат лития и йодид лития.
Согласно настоящему изобретению, литий может быть представлен в композиции для предварительной обработки, содержащей металл Группы IVB, в количестве от 5 до 500 ч./млн., таком как от 25 до 125 ч./млн. по отношению к общей массе композиции для предварительной обработки. Согласно настоящему изобретению, литий может быть представлен в композиции для предварительной обработки в количестве менее 200 ч./млн. Количество лития в композиции для предварительной обработки может находиться в диапазоне между указанными величинами, включая и сами указанные величины. Согласно настоящему изобретению, мольное отношение металла Группы IVB к литию может находиться между 100 : 1 и 1 : 100, например, между 12 : 1 и 1 : 50.
Согласно настоящему изобретению, композиция для предварительной обработки, содержащая металл Группы IVB, также может содержать смолистое связующее вещество. Подходящие смолы включают продукты реакции одного или нескольких алканоламинов и эпоксифункционального материала, содержащего по меньшей мере две эпоксигруппы, такие, как раскрываемые в патенте США № 5 653 823. В некоторых случаях такие смолы содержат бета-гидроксиэфирную, имидную или сульфидную функциональную группу, включенную с использованием диметилпропионовой кислоты, имида фталевой кислоты, или меркаптоглицерина в качестве дополнительного реагента при приготовлении смолы. В качестве варианта, продукт реакции может являться продуктом реакции диглицидилового эфира бисфенола A (коммерчески доступен в Shell Chemical Company под наименованием EPON 880), диметилолпропионовой кислоты и диэтаноламина при мольном соотношении от 0,6 до 5,0 : 0,05 до 5,5 : 1. Как раскрывается в патентах США № 3 912 548 и № 5 328 525, другие подходящие смолистые связующие вещества включают растворимые в воде и диспергируемые в воде полиакриловые кислоты; фенолформальдегидные смолы, как описывается в патенте США № 5 662 746; растворимые в воде полиамиды, такие, как раскрываются в WO 95/33869; сополимеры малеиновой или акриловой кислоты с аллиловым эфиром, как описывается в канадской патентной заявке 2 087 352; и растворимые в воде и диспергируемые смолы, включая эпоксидные смолы, аминопласты, фенолформальдегидные смолы, таннины и поливинилфенолы, как уже указывалось в патенте США № 5 449 415.
Согласно настоящему изобретению, смолистое связующее вещество может быть представлено в композиции для предварительной обработки, содержащей металл Группы IVB, в количестве от 0,005 до 30 масс.%, таком как от 0,5 до 3 масс.% по отношению к общей массе композиции для предварительной обработки, содержащей металл Группы IVB.
Согласно настоящему изобретению, композиция для предварительной обработки, содержащая металл Группы IVB, может быть по существу не содержащей или в некоторых случаях полностью не содержащий какого-либо смолистого связующего вещества. В данном контексте термин «по существу не содержащий» при его использовании в отношении отсутствия смолистого связующего вещества в композиции для предварительной обработки, содержащей металл Группы IVB, означает, что любое смолистое связующее вещество присутствует в композиции для предварительной обработки, содержащей металл Группы IVB, в ничтожно малом количестве менее 0,005 масс.% по отношению к общей массе композиции для предварительной обработки. В данном контексте понятие «полностью не содержащий» означает, что в композиции для предварительной обработки вообще нет никаких смолистых связующих веществ.
Согласно настоящему изобретению, композиция для предварительной обработки, содержащая металл Группы IVB, также может необязательно содержать источник фосфатных ионов. Например, в некоторых случаях фосфатные ионы могут быть представлены в количестве более 5 ч./млн. по отношению к общей массе композиции для предварительной обработки, содержащей металл Группы IVB, таком как 10 ч./млн., таком как 20 ч./млн. В некоторых случаях фосфатные ионы могут быть представлены в количестве, не превышающем 60 ч./млн. по отношению к общей массе композиции для предварительной обработки, содержащей металл Группы IVB, таком как не превышающее 40 ч./млн., таком как не превышающее 30 ч./млн. В некоторых случаях фосфатные ионы могут быть представлены в количестве от 5 ч./млн. до 60 ч./млн. по отношению к общей массе композиции для предварительной обработки, содержащей металл Группы IVB, таком как от 10 ч./млн. до 40 ч./млн., таком как от 20 ч./млн. до 30 ч./млн.
В качестве варианта, согласно настоящему изобретению, композиция для предварительной обработки, содержащая металл Группы IVB, в некоторых случаях может исключать фосфатные ионы, или содержащие фосфаты соединения, и/или образование отложений, таких как орто-фосфат алюминия, фосфат железа, и/или фосфат цинка, образующиеся в случае использования обрабатывающего агента, основанного на фосфате цинка. Когда композиция, и/или слой, или покрытие, содержащие такой материал, являются по существу не содержащими, фактически не содержащими или полностью не содержащими фосфата, при этом подразумеваются фосфатные ионы или соединения, содержащие фосфат в любой форме.
Таким образом, согласно настоящему изобретению, композиция для предварительной обработки, содержащая металл Группы IVB, и/или осажденные из нее слои могут быть по существу не содержащими, или в некоторых случаях могут быть фактически не содержащими, или в некоторых случаях могут быть полностью не содержащими фосфатов. Понятие «по существу не содержащие» означает, что композиции для предварительной обработки, содержащие металл Группы IVB, и/или осажденные из них слои содержат менее 25 ч./млн. фосфата по отношению к общей массе, соответственно, композиции или слоя, если он присутствует. Понятие «фактически не содержащие» означает, что композиции для предварительной обработки, содержащие металл Группы IVB, и/или содержащие их слои содержат менее 10 ч./млн. фосфатов. Понятие «полностью не содержащие» означает, что композиции для предварительной обработки, содержащие металл Группы IVB, и/или содержащие их слои содержат менее 1 ч./млрд. фосфатов, если они присутствуют.
Согласно настоящему изобретению, композиция для предварительной обработки, содержащая металл Группы IVB, может исключать хром или содержащие хром соединения. В данном контексте термин «содержащее хром соединение» относится к материалам, которые включают шестивалентный хром. Неограничивающие примеры таких материалов включают хромовую кислоту, триоксид хрома, ангидрид хромовой кислоты, бихроматные соли, такие как бихромат аммония, бихромат натрия, бихромат калия и кальция, бария, магния, цинка, кадмия и бихромат стронция. Соответственно, когда композиция для предварительной обработки, содержащая металл Группы IVB, и/или покрытие, или слой, образованные из нее, являются по существу не содержащими, фактически не содержащими или полностью не содержащими хрома, это ограничение включает хром в любой форме, такой как, но не ограничиваясь упомянутыми выше соединениями, содержащими шестивалентный хром.
Таким образом, согласно настоящему изобретению, настоящие композиции для предварительной обработки, содержащие металл Группы IVB, и/или покрытия, или слои, соответственно, осаждаемые из них, могут быть по существу не содержащими, могут быть фактически не содержащими и/или могут быть полностью не содержащими одного или нескольких любых элементов или соединений, перечисленных в предыдущем абзаце. Если композиция для предварительной обработки, содержащая металл Группы IVB, и/или покрытие, или слой, соответственно, образованные из нее, являются по существу не содержащими хрома или его производных, это означает, что хром или его производные преднамеренно не добавляются, но могут быть представлены в ничтожно малых количествах, например, в качестве примесей или из-за неизбежного загрязнения из окружающей среды. Другими словами, количество материала является настолько малым, что не влияет на свойства композиции предварительной обработки, содержащей металл Группы IVB; в случае хрома, это может, кроме того, означать, что данный элемент или его соединения не присутствуют в композиции предварительной обработки, содержащей металл Группы IVB, и/или покрытиях, или слоях, соответственно, образованных из нее, в таких уровнях содержания, которые оказывают нагрузку на окружающую среду. Термин «по существу не содержащие» означает, что композиции для предварительной обработки, содержащие металл Группы IVB, и/или покрытие, или слои, образованные из них, содержат по отношению к общей массе, соответственно, композиции или слоя менее 10 ч./млн. любых или всех из элементов или соединений, перечисленных в предыдущем абзаце, если они присутствуют. Термин «фактически не содержащие» означает, что композиции для предварительной обработки, содержащие металл Группы IVB, и/или покрытия, или слои, соответственно, из них образованные, содержат менее 1 ч./млн. любых или всех из элементов или соединений, перечисленных в предыдущем абзаце, если они присутствуют. Термин «полностью не содержащие» означает, что композиции для предварительной обработки, содержащие металл Группы IVB, и/или покрытия, или слои, соответственно, из них образованные, содержат менее 1 ч./млрд. любых или всех из элементов или соединений, перечисленных в предыдущем абзаце, если они присутствуют.
Согласно настоящему изобретению, pH композиции для предварительной обработки, содержащей металл Группы IVB, может в некоторых случаях равняться 6,5 или быть меньше, таким как 5,5 или менее, таким как 4,5 или менее, таким как 3,5 или менее. Согласно настоящему изобретению, pH композиции для предварительной обработки, содержащей металл Группы IVB, в некоторых случаях может располагаться в диапазоне от 2,5 до 6,5, таком как от 3,0 до 5,5, и может быть, в зависимости от необходимого, отрегулирован и/или поддержан с помощью, например, любой кислоты и/или основания. Например, согласно настоящему изобретению, pH композиции может поддерживаться посредством введения кислого материала, включая растворимые в воде и/или диспергируемые в воде кислоты, такие как азотная кислота, серная кислота и/или фосфорная кислота. Согласно настоящему изобретению, pH композиции может поддерживаться введением основного материала, включая растворимые в воде и/или диспергируемые в воде основания, такие как гидроксид натрия, карбонат натрия, гидроксид калия, гидроксид аммония, аммиак и/или амины, такие как триэтиламин, метилэтиламин или их смеси.
Композиция для предварительной обработки, содержащая металл Группы IVB, может содержать носитель, часто водную среду, так, чтобы композиция была представлена в форме раствора или дисперсии металла Группы IVB в носителе. Согласно настоящему изобретению, раствор или дисперсия могут быть приведены в контакт с подложкой любым из множества различных способов, таким как окунание или погружение, напыление, импульсное распыление, окунание, сопровождаемое напылением, распыление, сопровождаемое окунанием, нанесение кистью или с помощью валиков. Согласно изобретению, раствор или дисперсия при их нанесении на металлическую подложку находятся при температуре в пределах от 60 до 185°F (от 15 до 85°C). Например, способ предварительной обработки может выполняться при температуре окружающей среды или при комнатной температуре. Продолжительность контакта часто составляет от 10 секунд до 5 минут, например, от 30 секунд до 2 минут. Покрытие, которое образуется на подложке после введения подложки в контакт с композицией для предварительной обработки, содержащей металл Группы IVB, может иметь толщину от 20 нм до 400 нм и массу покрытия от 10 мг/фут2 до 250 мг/фут2 в выражении элементарного металла Группы IVB. Масса покрытия может быть определена посредством снятия пленки с подложки и определения химического состава композиции с помощью различных аналитических методик (таких как XRF (рентгеновская флуоресценция), ICP (спектрометрия с индуктивно связанной плазмой) и т.д.). Толщина полученного в результате предварительной обработки слоя может быть определена, используя ряд аналитических методик, включая, но не ограничиваясь профилированием по глубине с помощью XPS (рентгеновская фотоэмиссионная спектроскопия) или TEM (просвечивающая электронная микроскопия).
После контактирования с композицией для предварительной обработки, содержащей металл Группы IVB, подложка может быть промыта водопроводной водой, деионизированной водой и/или водным раствором ополаскивающих агентов для удаления любых остатков. Подложка необязательно может быть высушена, например, воздушной сушкой, или сушкой горячим воздухом, например, при использовании воздушного ножа, испарением воды в результате кратковременного подвергания воздействию высокой температуры, например, при сушке подложки в сушильном шкафу с температурой от 15°C до 200°C или в нагревательной установке, применяющей, например, инфракрасный нагрев, такой как в течение 10 минут при 70°C, или посредством пропускания подложки между отжимными гуммированными валиками. Неожиданно было обнаружено, что очистка подложки чистящей композицией настоящего изобретения, сопровождаемая предварительной обработкой композицией для предварительной обработки, содержащей металл Группы IVB, приводит к подложке, которая имеет значительно увеличенную энергию разрушения по сравнению с подложкой, очищенной с применением чистящей композицией, не включающей железа и/или кобальта, с последующей предварительной обработкой композицией для предварительной обработки, содержащей металл Группы IVB, к такой, как, например, подложка, которая имеет энергию разрушения по меньшей мере 1000 Дж/м2, такую как по меньшей мере 1500 Дж/м2, такую как по меньшей мере 2000 Дж/м2 по результатам испытаний согласно протоколу, описанному в примерах. Например, очистка подложки чистящей композицией настоящего изобретения, сопровождаемая предварительной обработкой композицией для предварительной обработки, содержащей металл Группы IVB, позволяет получить подложку, которая имеет по меньшей мере 1X увеличение энергии разрушения относительно подложки, очищенной чистящей композицией, которая не включает железа и/или кобальта, с последующей предварительной обработкой композицией для предварительной обработки, содержащей металл Группы IVB, такое как по меньшей мере 1,5X увеличение энергии разрушения, такое как по меньшей мере 2X увеличение энергии разрушения по данным испытаний согласно протоколу, описанному в примерах.
Кроме того, неожиданно было обнаружено, что очистка подложки композицией очистителя настоящего изобретения, сопровождаемая предварительной обработкой композицией для предварительной обработки, содержащей металл Группы IVB, позволяет получить подложку, которая имеет значительно улучшенную коррозионную устойчивость, заключающуюся в снижении распространения коррозии при скрайбировании по сравнению с подложкой, очищенной чистящей композицией, которая не включает железо и/или кобальт, с последующей предварительной обработкой композицией для предварительной обработки, содержащей металл Группы IVB, а именно, подложка демонстрирует, например, распространение коррозии при скрайбировании до величины менее 10 мм, такое как менее 9 мм, такое как менее 8 мм, такое как менее 7 мм, такое как менее 6 мм по результатам испытаний, выполненных согласно описанному в примерах протоколу. Например, очистка подложки чистящей композицией настоящего изобретения, сопровождаемая предварительной обработкой композицией для предварительной обработки, содержащей металл Группы IVB, позволяет получить подложку, которая демонстрирует по меньшей мере 10% снижение распространения коррозии при скрайбировании по сравнению с подложкой, очищенной чистящей композицией, которая не включает железа и/или кобальта, с последующей предварительной обработкой композицией для предварительной обработки, содержащей металл Группы IVB, такое как по меньшей мере на 25% меньшее распространение коррозии при скрайбировании, такое как по меньшей мере на 50% меньшее распространение коррозии при скрайбировании, такое как по меньшей мере на 70% меньшее распространение коррозии при скрайбировании, такое как по меньшей мере на 80% меньшее распространение коррозии при скрайбировании по результатам испытаний согласно протоколу, описанному в примерах. Также неожиданно было обнаружено, что эти результаты улучшаются при включении молибдена и лития в композицию для предварительной обработки, содержащую металл Группы IVB, что демонстрируется сниженным распространением коррозии при скрайбировании по сравнению с подложкой, очищенной чистящей композицией, которая не включает железо и/или кобальт, с последующей предварительной обработкой содержащей металл Группы IVB композицией для предварительной обработки, которая содержит молибден и литий, так, например, подложка показывает распространение коррозии при скрайбировании менее 8 мм, такое как менее 5 мм, такое как менее 4, такое как менее 3 мм, определенные по результатам испытаний, проведенных согласно протоколу, описанному в примерах.
Кроме того, неожиданно было обнаружено, что очистка подложки чистящей композицией настоящего изобретения, включающей молибден и/или железо, сопровождаемая предварительной обработкой композицией для предварительной обработки, содержащей металл Группы IVB, приводит к подложке, показывающей результаты испытаний по распространению коррозии при скрайбировании, проведенных согласно описанному в примерах протоколу, менее 6 мм, такие как менее 5 мм, такие как менее 4 мм, которые являются по меньшей мере столь же как хорошими или улучшенными по сравнению с подложкой, очищенной чистящей композицией, которая не включает молибден и/или железо, с последующей предварительной обработкой композицией для предварительной обработки, содержащей металл Группы IVB. В частности, включение одного молибдена или в комбинации с железом дает подложку, которая показывает результаты испытаний по распространению коррозии при скрайбировании по меньшей мере столь же хорошие или улучшенные по сравнению с подложкой, очищенной очистителем настоящего изобретения, содержащим кобальт и/или железо, что является значимым результатом в отношении проблем, вызываемых содержащими кобальт композициями, в связи с заботой о здоровье и окружающей среде.
Кроме того, было неожиданно обнаружено, что энергия разрушения может значительно изменяться в зависимости от вида фосфонатов, бифосфонатов, полифосфонатов и/или фосфоновой кислоты, используемых в композиции очистителя. Некоторые из очистителей, полученных с использованием комбинаций вышеупомянутых молекул, показали значительные улучшения показателя энергии разрушения для композиций очистителя, содержащих железо и кобальт. Химическая природа фосфоната, полифосфоната, бисфосфоната или фосфоновой кислоты может быть приспособлена для получения наибольшего выигрыша в адгезии грунтового покрытия. Было найдено, что молекулы, имеющие соотношение P-C в описанных здесь диапазонах, оказались особенно эффективными в обеспечении увеличения энергии разрушения.
Согласно настоящему изобретению, после контактирования подложки с композицией для предварительной обработки подложка может быть введена в контакт со второй композицией для предварительной обработки. Вторая композиция для предварительной обработки может быть композицией для предварительной обработки, содержащей металл Группы IIIB (описана ниже) и/или вышеописанной композицией для предварительной обработки, содержащей металл Группы IVB. Например, после контактирования подложки с металло-фосфатной композицией для предварительной обработки далее эта подложка может быть введена в контакт со второй композицией для предварительной обработки, содержащей композицию для тонкопленочной предварительной обработки.
Активирующий ополаскиватель
Согласно настоящему изобретению, система обработки может необязательно включать активирующий ополаскиватель, предназначенный для обработки по меньшей мере одного участка подложки. В данном контексте понятие «активирующий ополаскиватель» относится к непрерывной водной среде, содержащей диспергированные и/или суспендированные в ней частицы фосфата металла, которая наносится на по меньшей мере один участок подложки и/или в которую по меньшей мере один участок подложки погружается для «активирования» или «кондиционирования» подложки с тем, чтобы содействовать образованию металло-фосфатного покрытия на по меньшей мере одном участке подложки, прошедшем обработку активирующим ополаскивателем. В данном контексте «активирование» или «кондиционирование» поверхности подложки означает создание на поверхности подложки участков зародышеобразования. Безотносительно к какой-либо конкретной теории предполагается, что такие участки зародышеобразования поддерживают образование на поверхности подложки кристаллов фосфатов металлов, когда впоследствии такая поверхность подложки обрабатывается металло-фосфатной композицией для предварительной обработки. Полагают, например, что активация поверхности подложки создает участки зародышеобразования, которые поддерживают образование на поверхности подложки кристаллов фосфатов цинка и цинка/железа, когда поверхность подложки подвергается предварительной обработке цинк-фосфатной композицией для предварительной обработки.
Согласно настоящему изобретению, частицы фосфата металла из дисперсии частиц фосфатов двухвалентных или трехвалентных металлов, или их комбинации могут иметь размер D90, не превышающий 10 мкм, такой как не более 8 мкм, такой как не более 5 мкм, такой как не более 2 мкм, такой как не более 1 мкм, и в некоторых случаях может составлять по меньшей мере 0,06 мкм, например, по меньшей мере 0,1 мкм, например, по меньшей мере 0,2 мкм. Согласно настоящему изобретению, частицы фосфатов металлов из дисперсии фосфатных частиц двухвалентных или трехвалентных металлов, или их комбинации могут иметь размер D90 от 0,06 мкм до 8 мкм, такой как от 0,1 мкм до 5 мкм, такой как от 0,2 мкм до 2 мкм.
В данном контексте термин «размер частиц D90» относится к объемно-массовому распределению частиц, при котором 90% частиц имеют диаметр, меньший, чем величина D90. Согласно настоящему изобретению, размер частиц может быть измерен с помощью таких инструментальных средств, как Mastersizer 2000 производства Malvern Instruments, Ltd., Malvern, Worcestershire, Великобритания, или подобных приборов. Mastersizer 2000 направляет луч лазера (диаметр 0,633 мм, длина волны 633 нм) через дисперсию частиц (в дистиллированной, деионизированной или фильтрованной воде со степенью помутнения 2 - 3%) и оценивает рассеяние света дисперсии (параметры измерения 25°C, 2200 об/мин, задержка перед измерением 30 с, 10 с фоновых измерений, 10 с измерений на образце). Количество света, рассеиваемого дисперсией, обратно пропорционально размеру частиц. Рассеянный свет измеряется последовательностью детекторов, и данные затем анализируются программным обеспечением (программное обеспечение Malvern Mastersizer 2000, версия 5.60) для построения распределения частиц по размерам, из которого стандартным образом может определяться размер частиц. Согласно настоящему изобретению, образец дисперсии частиц перед анализом может быть необязательно подвергнут обработке ультразвуком.
Согласно настоящему изобретению, частицы фосфатов металлов могут быть по существу порошкообразными, таким образом, что более 50% частиц фосфатов металлов в композиции для активирующего ополаскивателя оказываются порошкообразными, например, более 60%, например, более 70%, например, более 80%, например, более 90%. Согласно настоящему изобретению, частицы фосфатов металлов могут быть полностью порошкообразными, таким образом, что порошкообразными оказываются 100% частиц. В данном контексте термин «порошкообразный» относится к частицам, имеющим неоднородную форму.
Согласно настоящему изобретению, фосфаты металлов (как общее металлическое соединение) могут быть представлены в композиции активирующего ополаскивателя в количестве по меньшей мере 50 ч./млн. по отношению к общей массе активирующего ополаскивателя, таком как по меньшей мере 150 ч./млн., и в некоторых случаях могут быть представлены в композиции активирующего ополаскивателя в количестве, не превышающем 5 000 ч./млн. по отношению к общей массе композиции активирующего ополаскивателя, таком как не превышающее 1 500 ч./млн. Согласно настоящему изобретению, фосфаты металлов (как общее металлическое соединение) могут быть представлены в композиции активирующего ополаскивателя в количестве от 50 ч./млн. до 5 000 ч./млн. всего фосфата металла по отношению к общей массе композиции активирующего ополаскивателя, таком как от 150 ч./млн. до 1 500 ч./млн.
Согласно настоящему изобретению, двухвалентный или трехвалентный металл фосфата металла может быть представлен цинком, железом, кальцием, марганцем, алюминием или их комбинациями. Если применяются комбинации различных фосфатов металлов, они могут содержать одинаковые или различные металлы и могут быть выбраны, в частности, из упоминаемых в последующем фосфатов цинка, железа, кальция, марганца и алюминия.
Подходящие фосфаты цинка, пригодные для применения в ванне для активирующего ополаскивания, включают без ограничения Zn3(PO4)2, Zn2Fe(PO4)2, Zn2Ca(PO4)2, Zn2Mn(PO4)2 или их комбинации.
Подходящие фосфаты железа, пригодные для применения в ванне для активирующего ополаскивания, включают без ограничения FePO4, Fe3(PO4)2 или их комбинации.
Подходящие фосфаты кальция, пригодные для применения в ванне для активирующего ополаскивания, включают без ограничения CaHPO4, Ca3(PO4)2 или их комбинации.
Подходящие фосфаты марганца, пригодные для применения в ванне для активирующего ополаскивания, включают без ограничения Mn3(PO4)2, MnPO4 или их комбинации.
Подходящие фосфаты алюминия, пригодные для применения в ванне для активирующего ополаскивания, включают без ограничения AlPO4.
Согласно настоящему изобретению, активирующий ополаскиватель может, кроме того, содержать диспергирующий агент. Диспергирующий агент может быть ионным или неионогенным. Подходящие ионные диспергирующие агенты, пригодные для применения в активирующем ополаскивателе, могут содержать ароматическую органическую кислоту, производное фенола, фенольную смолу или их комбинации. Подходящие неионогенные диспергирующие агенты, пригодные для применения в активирующем ополаскивателе, могут включать неионогенные полимеры, в частности, состоящие из мономеров (или их остатков), включающих пропиленоксид, этиленоксид, стирол, одноосновную кислоту, такую как (мет)акриловая кислота, двухосновную кислоту, такую как малеиновая кислота или итаконовая кислота, кислотный ангидрид, такой как акриловой ангидрид или малеиновый ангидрид, или их комбинации. Примеры подходящих коммерчески доступных неионогенных диспергирующих агентов включают DISPERBYK®-190, предлагаемый на рынке BYK-Chemie GmbH, и ZetaSperse® 3100, предлагаемый Air Products Chemicals Inc.
Согласно настоящему изобретению, активирующий ополаскиватель может, кроме того, включать сульфатную соль металла. Металл в сульфате металла может быть тем же самым или отличным от металла в частицах фосфата металла. Согласно настоящему изобретению, металл в сульфатной соли металла может содержать двухвалентный металл, трехвалентный металл или их комбинации, такой как, например, никель, медь, цинк, железо, магний, кобальт, алюминий или их комбинации.
Согласно настоящему изобретению, сульфатный ион сульфатной соли металла может быть представлен в активирующем ополаскивателе в количестве по меньшей мере 10 ч./млн. по отношению к общей массе активирующего ополаскивателя, таком как по меньшей мере 25 ч./млн., таком как по меньшей мере 50 ч./млн., таком как по меньшей мере 100 ч./млн., таком как по меньшей мере 200 ч./млн., таком как по меньшей мере 500 ч./млн., и в некоторых случаях в количестве, не превышающем предел растворимости сульфатной соли металла в активирующем ополаскивателе, таком как не более 5 000 ч./млн., таком как не более 1 000 ч./млн., таком как не более 500 ч./млн., таком как не более 200 ч./млн., таком как не более 100 ч./млн. Согласно настоящему изобретению, сульфатный ион из сульфатной соли металла может быть представлен в количестве от 10 ч./млн. до 5 000 ч./млн. по отношению к общей массе активирующего ополаскивателя, таком как от 25 ч./млн. до 5 000 ч./млн., таком как от 50 ч./млн. до 1 000 ч./млн., таком как от 200 ч./млн. до 500 ч./млн.
Согласно настоящему изобретению, активирующий ополаскиватель может включать смачивающее вещество. Согласно настоящему изобретению, смачивающие вещества могут быть представлены в количествах вплоть до 2 масс.%, таких как вплоть до 0,5 масс.% по отношению к общей массе активирующего ополаскивателя. В некоторых случаях смачивающие вещества могут быть представлены в количествах от 0,1 масс.% до 2 масс.% по отношению к общей массе активирующего ополаскивателя, таких как от 0,3 масс.% до 0,5 масс.%. В данном контексте «смачивающее вещество» уменьшает поверхностное натяжение на границе раздела фаз между поверхностью частиц дисперсной фазы и водной средой с тем, чтобы позволить водной среде более равномерно входить в контакт или «смачивать» поверхность частиц дисперсной фазы.
Согласно настоящему изобретению, активирующий ополаскиватель может иметь pH от 6 до 12, такой как от 6,5 до 9, такой как от 7,5 до 8,5, такой как от 7 до 8. Щелочной компонент может быть представлен в активирующем ополаскивателе в количестве, достаточном для регулирования показателя pH активирующего ополаскивателя. Подходящие щелочные компоненты могут включать, например, гидроксид натрия, карбонат натрия, триполифосфат натрия, ортофосфат калия или их комбинации.
Согласно настоящему изобретению, активирующий ополаскиватель может также включать биоцид. Подходящие биоциды включают, например, метилхлоризотиазолинон, метилизотиазолинон или их комбинации. В случае его применения биоцид может быть представлен в количестве по меньшей мере 10 ч./млн. по отношению к общей массе активирующего ополаскивателя, таком как по меньшей мере 20 ч./млн., таком как по меньшей мере 80 ч./млн., таком как по меньшей мере 100 ч./млн., и в некоторых случаях в количестве, не превышающем 140 ч./млн., таком как не превышающее 120 ч./млн., таком как не превышающее 40 ч./млн., таком как не превышающее 30 ч./млн. Согласно настоящему изобретению, биоцид может быть представлен в количестве от 10 ч./млн. до 140 ч./млн. по отношению к общей массе активирующего ополаскивателя, таком как от 10 ч./млн. до 40 ч./млн., таком как от 20 ч./млн. до 30 ч./млн., таком как от 80 ч./млн. до 140 ч./млн., таком как от 100 ч./млн. до 120 ч./млн. Специалистам в данной области очевидно, что биоциды могу быть включены в активирующий ополаскиватель в количествах, основывающихся на указаниях изготовителя.
Согласно настоящему изобретению, активирующий ополаскиватель может, кроме того, содержать диоксид кремния. Согласно настоящему изобретению, диоксид кремния может быть осажденным диоксидом кремния, таким как синтетический аморфный осажденный диоксид кремния. Согласно настоящему изобретению, диоксид кремния может быть хрупким под сдвиговым усилием. В данном контексте «хрупкий под сдвиговым усилием» означает, что размер частиц может быть уменьшен под действием сдвига. Согласно настоящему изобретению, диоксид кремния может содержать, например, диоксид кремния Hi-SilTM EZ 160G (коммерчески доступен в PPG Industries, Inc). Согласно настоящему изобретению, в случае его присутствия диоксид кремния может быть представлен в количестве по меньшей мере 50 ч./млн. по отношению к общей массе активирующего ополаскивателя, таком как по меньшей мере 100 ч./млн., таком как по меньшей мере 150 ч./млн., и в некоторых случаях в количестве, не превышающем 5000 ч./млн. по отношению к общей массе активирующего ополаскивателя, таком как не превышающее 1000 ч./млн., таком как не превышающее 500 ч./млн. Согласно настоящему изобретению, диоксид кремния может быть представлен в активирующем ополаскивателе в количестве от 50 ч./млн. до 5 000 ч./млн. по отношению к общей массе активирующего ополаскивателя, таком как от 100 ч./млн. до 1 000 ч./млн., таком как от 150 ч./млн. до 500 ч./млн.
Активирующий ополаскиватель может, кроме того, необязательно содержать дополнительные компоненты в дополнение к диспергирующему агенту (то есть компоненты, отличные от диспергирующего агента), такие как неионогенные поверхностно-активные вещества и вспомогательные средства, традиционно используемые в данной области. Такие дополнительные необязательные компоненты включают поверхностно-активные вещества, которые выступают в качестве противовспенивающих компонентов. Могут использоваться амфотерные и/или неионогенные поверхностно-активные вещества. Противовспенивающие поверхностно-активные вещества могут быть представлены, если они присутствуют, в количествах по меньшей мере 0,1 масс.% по отношению к общей массе активирующего ополаскивателя, таком как по меньшей мере 0,5 масс.%, и в некоторых случаях могут быть представлены в количествах, не превышающих 1 масс.%, таких как не превышающие 0,7 масс.% по отношению к общей массе активирующего ополаскивателя. В некоторых случаях противовспенивающие поверхностно-активные вещества могут быть представлены, если они присутствуют, в количествах от 0,1 масс.% до 1 масс.%, таких как от 0,5 масс.% до 0,7 масс.% по отношению к общей массе активирующего ополаскивателя.
Согласно настоящему изобретению, активирующий ополаскиватель может, кроме того, содержать модификатор реологии в дополнение к диспергирующему агенту (а именно, вещество, отличное от диспергирующего агента). Модификатор реологии может содержать, например, полиуретаны, акриловые полимеры, латексы, стирол/бутадиен, поливиниловые спирты, глины, такие как аттапульгит, бентонит, монтмориллонит и другие, материалы на основе целлюлозы, такие как карбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, или желатин, камеди, такие как гуар и ксантан, или их комбинации.
Согласно настоящему изобретению, активирующий ополаскиватель в некоторых случаях может по существу или в некоторых случаях полностью не содержать частиц фосфата титана. В данном контексте термин «по существу не содержащий» при его использовании в отношении отсутствия частиц фосфата титана в активирующем ополаскивателе означает, что никакие присутствующие в активирующем ополаскивателе частицы фосфата титана умышленным образом в него не добавляются и присутствуют в ничтожно малом количестве менее 5 ч./млн. по отношению к общей массе активирующего ополаскивателя. В данном контексте термин «полностью не содержащий» при его использовании в отношении отсутствия частиц фосфата титана означает, что никаких частиц фосфата титана не присутствует вообще.
Согласно настоящему изобретению, активирующий ополаскиватель в некоторых случаях может необязательно содержать коллоидные частицы фосфата титана. Согласно настоящему изобретению, титан может быть представлен (если он присутствует) в композиции активирующего ополаскивателя в количестве по меньшей мере 1 ч./млн. по отношению к общей массе активирующего ополаскивателя, таком как по меньшей мере 2 ч./млн., и в некоторых случаях может быть представлен в активирующем ополаскивателе в количестве, не превышающем 6 ч./млн. по отношению к общей массе композиции активирующего ополаскивателя, таком как не превышающее 3,5 ч./млн. Согласно настоящему изобретению, титан может быть представлен в активирующем ополаскивателе, если он присутствует, в количестве от 1 ч./млн. до 6 ч./млн., таком как от 2 ч./млн. до 3,5 ч./млн., а показатель pH может находиться в диапазоне от 7,5 до 10, таком как от 8 до 9,5.
Ванна для активирующего ополаскивания может быть приготовлена посредством смешивания композиции активирующего ополаскивателя, взятой в виде концентрата, с водной средой, такой как вода. Согласно настоящему изобретению, активирующий ополаскиватель может, кроме того, содержать хелатирующий агент. Хелатор может содержать, например, карбоксилаты, такие как тартраты, цитраты или глюконаты, комплексные соединения на основе ацетатов, такие как этилендиаминтетраацетат или нитрилотриацетат, фосфаты, такие как пятизамещенный трифосфат натрия или четырехзамещенный пирофосфат калия, фосфонаты, поликарбоксилаты, кислоты, сложные эфиры, соли любого из вышеупомянутых или их комбинации.
Согласно настоящему изобретению, активирующий ополаскиватель может быть нанесен на поверхность подложки с применением методик распыления, нанесения валиком или погружения. Активирующий ополаскиватель может быть нанесен на подложку при температуре, например, от 15°C до 50°C, такой как от 25°C до 35°C в течение любого подходящего промежутка времени, такого как по меньшей мере 1 секунда, такого как по меньшей мере 10 секунд, такого как по меньшей мере 2 минуты, такого как по меньшей мере 5 минут.
Композиция предварительного ополаскивания
Согласно настоящему изобретению, система обработки может, кроме того, необязательно содержать композицию предварительного ополаскивания, предназначенную для обработки подложки. Согласно настоящему изобретению, композиция для предварительного ополаскивания может содержать источник фторида. В данном контексте количество фторида, указываемого или раскрываемого в композиции для предварительного ополаскивания, упоминается как «общий фторид» и измеряется в единицах частей фторида на миллион.
Часто композиция для предварительного ополаскивания может содержать носитель, часто водную среду, так, чтобы композиция для предварительного ополаскивания могла находиться в форме раствора или дисперсии композиции для предварительного ополаскивания в носителе. В этих случаях раствор или дисперсия могут быть приведены в контакт с подложкой любым из множества различных способов, таким как окунание или погружение, напыление, импульсное распыление, окунание, сопровождаемое напылением, распыление, сопровождаемое окунанием, нанесение кистью или с помощью валиков. Согласно настоящему изобретению, раствор или дисперсия при их нанесении на металлическую подложку находятся при температуре в пределах от 50 до 200°F, такой как от 75 до 125°F. Например, способ предварительного ополаскивания может выполняться при температуре окружающей среды или при комнатной температуре. Продолжительность контакта часто составляет от 15 секунд до 10 минут, например, от 30 секунд до 2 минут.
Фторид, присутствующий в композиции для предварительного ополаскивания, может быть представлен в виде общего фторида в выражении ч./млн. Общий фторид может быть измерен или рассчитан, как описано выше. Общий фторид может вводиться в композицию для предварительного ополаскивания с помощью фтористоводородной кислоты, а также фторидов щелочных металлов и аммония или фторидов водорода. Помимо этого, общий фторид может поступать в композицию для предварительного ополаскивания из присутствующих в композиции для предварительной обработки соединений металлов Группы IVB, включая, например, гексафторциркониевую кислоту или гексафтортитановую кислоту. Для обеспечения наличия общего фторида к композиции для предварительного ополаскивания могут быть добавлены и другие комплексные фториды, такие как H2SiF6 или HBF4.
Источник общего фторида может быть представлен в композиции для предварительного ополаскивания в количестве по меньшей мере 10 ч./млн. по отношению к общей массе композиции для предварительного ополаскивания, таком как по меньшей мере 100 ч./млн., в соответствии с измерениями, выполненными, как описано выше. Источник общего фторида может быть представлен в композиции для предварительного ополаскивания в количестве, не превышающем 5 000 ч./млн. по отношению к общей массе композиции для предварительного ополаскивания, таком как по меньшей мере 2 000 ч./млн. Источник общего фторида может быть представлен в композиции для предварительного ополаскивания в количестве от 10 ч./млн. до 5 000 ч./млн. по отношению к общей массе композиции для предварительного ополаскивания, таком как от 100 ч./млн. до 2 000 ч./млн.
Показатель pH композиции для предварительного ополаскивания может быть ниже 7, таким как от 2,5 до 5, и может быть отрегулирован при варьировании количества растворенного комплексного металло-фторидного иона, присутствующего в композиции, или же, в зависимости от необходимого, может быть отрегулирован с помощью, например, любой кислоты или основания. Например, pH композиции для предварительного ополаскивания может поддерживаться введением основного материала, включая растворимые в воде и/или диспергируемые в воде основания, такие как гидроксид натрия, карбонат натрия, гидроксид калия, гидроксид аммония, аммиак и/или амины, такие как триэтиламин, метилэтиламин или их комбинации.
Общий фторид может вводиться в композицию для предварительного ополаскивания с помощью фтористоводородной кислоты, а также фторидов щелочных металлов и аммония или фторидов водорода. Помимо этого, общий фторид может поступать в композицию для предварительного ополаскивания из присутствующих в композиции для предварительной обработки соединений металлов Группы IVB, включая, например, гексафторциркониевую кислоту или гексафтортитановую кислоту. Для обеспечения наличия общего фторида к композиции для предварительного ополаскивания могут быть добавлены и другие комплексные фториды, такие как H2SiF6 или HBF4.
Источник свободного фторида может быть представлен в композиции для предварительного ополаскивания в количестве по меньшей мере 10 ч./млн. по отношению к общей массе композиции для предварительного ополаскивания, таком как по меньшей мере 100 ч./млн., в соответствии с измерениями, выполненными, как описано выше. Источник свободного фторида может быть представлен в композиции для предварительного ополаскивания в количестве, не превышающем 5 000 ч./млн. по отношению к общей массе композиции для предварительного ополаскивания, таком как по меньшей мере 2 000 ч./млн. Источник свободного фторида может быть представлен в композиции для предварительного ополаскивания в количестве от 10 ч./млн. до 5 000 ч./млн. по отношению к общей массе композиции для предварительного ополаскивания, таком как от 100 ч./млн. до 2 000 ч./млн.
Раствор для нанесения покрытия
Согласно настоящему изобретению, система обработки может необязательно содержать раствор для нанесения покрытия, предназначенный для обработки подложки до ее обработки композицией для предварительной обработки, содержащей металл Группы IVB. Согласно настоящему изобретению, раствор для нанесения покрытия может осаждать на поверхности подложки электроположительный металл посредством введения подложки в контакт с раствором для нанесения покрытия, содержащим растворимую соль металла, такую как растворимая соль меди, при том, что металл подложки растворяется, в то время как находящийся в растворе металл, такой как медь, осаждается на поверхности подложки.
Упомянутый выше раствор для нанесения покрытия может являться водным раствором растворимой в воде соли металла. Согласно настоящему изобретению, растворимая в воде соль металла может быть растворимым в воде соединением меди. Конкретные примеры растворимых в воде соединений меди, являющихся подходящими для применения в настоящем изобретении, включают, но не ограничиваются цианидом меди, двойным цианидом меди и калия, сульфатом меди, нитратом меди, пирофосфатом меди, роданидом меди, тетрагидратом четырехзамещенного этилендиаминтетраацетата меди и натрия, бромидом меди, оксидом меди, гидроксидом меди, хлоридом меди, фторидом меди, глюконатом меди, цитратом меди, лауроилсаркозинатом меди, формиатом меди, ацетатом меди, пропионатом меди, бутиратом меди, лактатом меди, оксалатом меди, фитатом меди, тартратом меди, малатом меди, сукцинатом меди, малонатом меди, малеатом меди, бензоатом меди, салицилатом меди, аспартатом меди, глутаматом меди, фумаратом меди, глицерофосфатом меди, натрий-медь-хлорофиллином, фторсиликатом меди, фторборатом меди и йодатом меди, а также медными солями карбоновых кислот в гомологическом ряду от муравьиной кислоты до декановой кислоты, медными солями многоосновных кислот ряда от щавелевой кислоты до пробковой кислоты и медными солями оксикарбоновых кислот, включая гликолевую, молочную, виннокаменную, яблочную и лимонную кислоты.
Когда ионы меди, поступающие из такого водорастворимого медного соединения, осаждаются в качестве примеси в форме сульфата меди, оксида меди и т.д., предпочтительным может оказаться добавление комплексообразующего реагента, который подавляет осаждение ионов меди, тем самым стабилизируя их в растворе в виде комплексного соединения меди.
Согласно настоящему изобретению, соединение меди может быть добавлено в виде комплексной соли меди, такой как K3Cu(CN)4 или Cu-EDTA, которая сама по себе может устойчиво существовать в растворе для нанесения покрытия, а также возможно получение комплексного соединения меди, способного устойчиво присутствовать в растворе для нанесения покрытия, посредством соединения комплексообразующего реагента с веществом, которое само по себе является труднорастворимым. Их примеры включают медно-цианидный комплекс, образованный соединением CuCN и KCN или соединением CuSCN и KSCN или KCN, и комплексное вещество Cu-EDTA, образованное соединением CuSO4 и EDTA•Na2.
Что касается комплексообразующего реагента, то использоваться может вещество, которое способно образовывать комплексное соединение с ионами меди; их примеры включают такие неорганические соединения, как цианидные соединения и роданидные соединения, а также многоосновные карбоновые кислоты, их конкретные примеры включают этилендиаминтетрауксусную кислоту, соли этилендиаминтетрауксусной кислоты, такие как двузамещенная натриевая соль этилендиаминтетрауксусной кислоты, аминокарбоновые кислоты, такие как нитрилотриуксусная кислота и иминодиацетилуксусная кислота, оксикарбоновые кислоты, такие как лимонная кислота, винная кислота, янтарная кислота, щавелевая кислота, этилендиаминтетраметиленфосфоновая кислота и аминоуксусная кислота.
Согласно настоящему изобретению, электроположительный металл, такой как медь, вносится в раствор для нанесения покрытия в количестве по меньшей мере 1 ч./млн., таком как по меньшей мере 50 ч./млн., или в некоторых случаях по меньшей мере 100 ч./млн. общего металла (в расчете на элементарный металл) и может быть внесен в раствор для нанесения покрытия в количестве, не превышающем 5 000 ч./млн., таком как не превышающее 1 000 ч./млн., или в некоторых случаях не превышающее 500 ч./млн. общего металла (в расчете на элементарный металл). Количество электроположительного металла в растворе для нанесения покрытия может составлять от 1 ч./млн. до 5 000 ч./млн., например, от 50 ч./млн. до 1 000 ч./млн., например, от 100 ч./млн. до 500 ч./млн.
В дополнение к растворимой в воде соли металла и необязательному комплексообразующему реагенту раствор для нанесения покрытия может также включать и другие добавки. Например, может применяться стабилизатор, такой как 2-меркаптобензотиазол. Другие возможные материалы включают поверхностно-активные вещества, которые выступают в качестве увлажнителей подложки или противовспенивающих добавок. Могут использоваться анионные, катионные, амфотерные или неионогенные поверхностно-активные вещества. Подходящими также являются совместимые смеси таких материалов. По отношению к общему объему раствора пеноудаляющие сурфактанты часто присутствуют при уровнях содержания до 1 процента, например, вплоть до 0,1 об.%, а увлажнители часто присутствуют при уровнях вплоть до 2 процентов, например, вплоть до 0,5 об.%.
Согласно настоящему изобретению, водный раствор для нанесения покрытия в ходе применения может иметь pH менее 7 и в некоторых случаях pH может находиться в пределах от 1 до 4, например, от 1,5 до 3,5. Согласно настоящему изобретению, pH раствора поддерживается посредством внесения кислоты. Показатель pH раствора может быть отрегулирован с помощью неорганических кислот, таких как фтористоводородная кислота, фтороборная кислота и фосфорная кислота, включая их смеси; органических кислот, таких как молочная кислота, уксусная кислота, лимонная кислота, сульфаминовая кислота или их смеси; и растворимых в воде или диспергируемых в воде оснований, таких как гидроксид натрия, гидроксид аммония, аммиак или амины, такие как триэтиламин, метилэтиламин или их смеси.
Согласно настоящему изобретению, раствор для нанесения покрытия может быть приведен в контакт с подложкой любым из множества различных способов, включая, например, окунание или погружение, напыление, импульсное распыление, окунание, сопровождаемое напылением, распыление, сопровождаемое окунанием, нанесение кистью или с помощью валиков. В некоторых воплощениях может применяться технология окунания или погружения, и раствор при его нанесении на металлическую подложку находится при температуре в пределах от 60 до 185°F (от 15 до 85°C). Продолжительность контакта может составлять от 10 секунд до пяти минут, например, от 30 секунд до 2 минут. После извлечения подложки из раствора для нанесения покрытия подложка, если желательно, может быть промыта водой и высушена.
Согласно настоящему изобретению, осадок из раствора для нанесения покрытия, то есть электроположительный металл, может присутствовать на подложке в количестве в пределах от 1 до 1 000 миллиграммов на квадратный метр (мг/м2), например, от 10 до 400 мг/м2. Толщина получаемого из раствора для нанесения покрытия осадка может варьировать, но вообще он является очень тонким, часто имеющим толщину менее 1 микрометра, такую как от 1 до 500 нанометров, такую как от 10 до 300 нанометров.
Композиция для ополаскивания после обработки.
Согласно настоящему изобретению, система обработки может необязательно включать композицию для ополаскивания после обработки, предназначенную для обработки по меньшей мере одного участка подложки после того, как подложка была подвергнута обработке композицией для предварительной обработки. Общеизвестно в данной области, что композиция для ополаскивания после обработки может содержать органический или неорганический состав для ополаскивания после обработки или герметизирующий состав, такой как хроматный или бесхроматный герметизирующий состав, или ополаскивание эпоксидной смолой. Примеры композиций для ополаскивания после обработки включают Chemseal 19, Chemseal 59 и Chemseal 100, предлагаемые на рынке компанией PPG. В качестве варианта, композиция для ополаскивания после обработки может содержать композицию для ополаскивания после обработки на основе циркония, такую как композиция, содержащая цирконий, цирконий и триэтаноламин или цирконий и смолу.
Система обработки необязательно может быть по существу не содержащей, фактически не содержащей или полностью не содержащей фосфата. Термин «по существу не содержащая» в отношении системы обработки означает, что каждый компонент системы обработки, такой как описанные выше водная щелочная композиция и композиция для предварительной обработки, содержащая металл Группы IVB, содержит менее 25 ч./млн. фосфата (при его наличии вообще) по отношению к общей массе, соответственно, каждого компонента системы обработки. Термин «фактически не содержащая» в отношении системы обработки означает, что каждый компонент системы обработки содержит, соответственно, менее 10 ч./млн. фосфата, если содержит вообще. Термин «полностью не содержащая» в отношении системы обработки означает, что каждый компонент системы обработки содержит, соответственно, менее 1 ч./млрд. фосфата, если содержит вообще.
Композиция для нанесения электроосаждаемых покрытий
Согласно настоящему изобретению, система обработки может необязательно содержать композицию для нанесения электроосаждаемых покрытий, предназначенную для нанесения покрытия на обработанную подложку.
Ванны для электроосаждения в типичном случае снабжаются как двухкомпонентные: (i) смоляной смесью и (ii) пастой. Согласно настоящему изобретению, смоляная смесь может содержать (a) основной пленкообразующий полимер (например, смола, содержащая группу катионной, содержащей активный водород соли), имеющий реакционноспособные функциональные группы, (b) отвердитель, который является реакционноспособным по отношению к функциональным группам пленкообразующего полимера, и (c) любые дополнительные диспергируемые в воде непигментированные компоненты.
Известно большое разнообразие основных пленкообразующих полимеров, которые могут применяться в ваннах для электроосаждения изобретения при условии, что эти полимеры являются «диспергируемыми в воде». В данном контексте «диспергируемый в воде» означает, что материал приспособлен для того, чтобы растворяться, диспергироваться и/или эмульгироваться в воде. Основные пленкообразующие полимеры, используемые в данном изобретении, имеют катионную природу. Другими словами, основной пленкообразующий полимер содержит катионные солевые группы, обычно приготавливаемые нейтрализацией функциональной группы на пленкообразующем полимере кислотой, что позволяет данному основному пленкообразующему полимеру электролитически осаждаться на катоде.
Примеры основных пленкообразующих полимеров, подходящих для применения в катионных композициях для нанесения покрытия электроосаждением, включают без ограничений катионные полимеры, полученные из полиэпоксида, акрила, полиуретана и/или полиэфира, полимеры, содержащие гидроксильную группу, полимеры, содержащие группу аминной соли, или их комбинации. Согласно настоящему изобретению, основной пленкообразующий полимер может быть сополимером полимеров, перечисленных в предыдущем предложении.
Согласно настоящему изобретению, основной пленкообразующий полимер может быть катионным полимером (катионной смолой), получаемым из полиэпоксида. Например, основной пленкообразующий полимер может быть приготовлен посредством взаимодействия материала, содержащего полиэпоксидные и полигидроксильные группы, выбираемого из спиртовых, содержащих гидроксильные группы материалов, и фенольных, содержащих гидроксильные группы материалов, для продолжения цепи или увеличения молекулярной массы полиэпоксида. Как более подробно обсуждается ниже, продукт данной реакции может быть затем приведен во взаимодействие с образующим катионные солевые группы агентом для создания катионного полимера.
Согласно настоящему изобретению, продолжающий цепь полиэпоксид в типичном случае готовится следующим образом: содержащие полиэпоксидные и полигидроксильные группы материалы реагируют друг с другом «беспримесным» образом или в присутствии инертного органического растворителя, такого как кетон, включая метилизобутилкетон и метиламилкетон, ароматические соединения, такие как толуол и ксилол, и гликолевые эфиры, такие как диметиловый эфир диэтиленгликоля. Такая реакция обычно проводится при температуре от около 80°C до 160°C в течение времени от около 30 до 180 минут, до тех пор, пока не будет получен смолоподобный продукт реакции, содержащий эпоксигруппы.
Согласно настоящему изобретению, эквивалентное соотношение реагентов (то есть материалов, содержащих эпокси- и полигидроксильные группы) располагается в диапазоне от 1,00 : 0,50 до 1,00 : 2,00.
Согласно настоящему изобретению, полиэпоксид в типичном случае имеет по меньшей мере две 1,2-эпоксигруппы. Эпоксисоединения могут быть насыщенными или ненасыщенными, циклическими или ациклическими, алифатическими, алициклическими, ароматическими или гетероциклическими. Более того, эпоксисоединения могут содержать заместители, такие как галогеновые, гидроксильные и простые эфирные группы.
Примеры полиэпоксидов представлены обладающими 1,2-эпоксиэквивалентностью, превышающей единицу и/или равной двум; то есть полиэпоксидами, которые в среднем имеют две эпоксидные группы на молекулу. Подходящие полиэпоксиды включают полиглицидиловые эфиры многоатомных спиртов, такие как циклические многоатомные спирты и полиглицидиловые эфиры многоатомных фенолов, такие как бисфенол A. Эти полиэпоксиды могут быть получены этерификацией многоатомных фенолов с эпигалогидрином или дигалогидрином, такими как эпихлоргидрин или дихлоргидрин, в присутствии щелочи. Помимо многоатомных фенолов для приготовления полиглицидиловых эфиров циклических многоатомных спиртов могут применяться и другие циклические многоатомные спирты. Примеры других циклических многоатомных спиртов включают алициклические многоатомные спирты, в частности, циклоалифатические многоатомные спирты, такие как гидрогенизированный бисфенол A, 1,2-циклогександиол и 1,2-бис(гидроксиметил)циклогексан.
Согласно настоящему изобретению, полиэпоксиды имеют эквивалентную массу эпоксида ≥ 180. Согласно настоящему изобретению, полиэпоксиды могут иметь эквивалентную массу эпоксида ≤ 2000. Согласно настоящему изобретению, полиэпоксиды могут иметь эквивалентную массу эпоксида, которая располагается между любой комбинацией величин, указанных в предшествующих предложениях, включая и сами упомянутые величины. Например, полиэпоксиды могут иметь диапазон эквивалентных масс эпоксида от 186 до 1200.
Также в настоящем изобретении могут применяться содержащие эпоксигруппы акриловые полимеры. Согласно настоящему изобретению, акриловые полимеры, содержащие эпоксигруппы, имеет эквивалентную массу эпоксида ≥750, такую как эквивалентная масса эпоксида ≤2000. Согласно настоящему изобретению, содержащий эпоксигруппы акриловый полимер имеет эпоксиэквивалентную массу, которая располагается между любой комбинацией величин, указанных в предшествующих предложениях, включая и сами упомянутые величины.
Примеры содержащих полигидроксильные группы материалов, используемых для продолжения цепи или увеличения молекулярной массы полиэпоксида (а именно, через реакцию между гидрокси- и эпоксигруппами) включают спиртовые, содержащие гидроксильные группы материалы и содержащие гидроксильные группы фенольные материалы. Примеры содержащих гидроксильные группы спиртовых материалов представляют простые многоатомные спирты, такие как неопентилгликоль; полиэфирполиолы, такие как описываются в патенте США № 4 148 772; простые полиэфиры многоатомных спиртов, такие как описываются в патенте США № 4 468 307; и уретандиолы, такие как описываются в патенте США № 4 931 157. Примеры фенольных, содержащих гидроксильные группы материалов представляют многоатомные фенолы, такие как бисфенол A, флороглюцин, катехин и резорцин. Также могут применяться смеси спиртовых, содержащих гидроксильные группы материалов и фенольных, содержащих гидроксильные группы материалов.
Основной пленкообразующий полимер может содержать группы катионной соли, которые могут быть введены в молекулу смолы следующим образом. Смолистый продукт реакции, приготовленный как описано выше, далее вводится в реакцию с образующим катионные солевые группы агентом. В данном контексте под «агентом образования катионных солевых групп» подразумевается материал, который является реакционноспособным по отношению к эпоксигруппам и который может быть подкислен до, в течение или после протекания реакции по эпоксигруппам для образования катионных солевых групп. Примеры подходящих материалов включают амины, такие как первичные или вторичные амины, которые могут быть подкислены после реакции с эпоксигруппами для образования групп аминных солей, или третичные амины, которые могут быть подкислены до реакции с эпоксигруппами и которые после реакции с эпоксигруппами образуют группы четвертичных аммонийных солей. Примерами других образующих катионные солевые группы агентов являются сульфиды, которые могут быть смешаны с кислотой до реакции с эпоксигруппами и образовывать группы тройных сульфониевых солей после последующей реакции с эпоксигруппами.
Когда в качестве агентов, образующих катонные солевые группы, применяются амины, использоваться могут моноамины, гидроксилсодержащие амины, полиамины или их комбинации.
Третичные и вторичные амины применяются чаще, чем первичные амины, поскольку первичные амины являются полифункциональными относительно эпоксигрупп и проявляют большую склонность к превращению реакционной смеси в гель. Если применяются полиамины или первичные амины, они могут применяться в существенном стехиометрическом избытке относительно количества эпоксигрупп в полиэпоксиде с тем, чтобы препятствовать гелеобразованию, а избыточный амин может быть удален из реакционной смеси вакуумной отгонкой или с помощью другой методики в конце реакции. Чтобы гарантировать наличие избытка амина, к амину может добавляться эпоксид.
Примеры гидроксилсодержащих аминов включают, но не ограничиваются алифатическими аминоспиртами, диалканоламинами, алкилалканоламинами и аралкилалканоламинами, содержащими от 1 до 18 атомов углерода, например, от 1 до 6 атомов углерода в каждой алканольной, алкильной и арильной группе. Конкретные примеры включают этаноламин, N-метилэтаноламин, диэтаноламин, N-фенилэтаноламин, N,N-диметилэтаноламин, N-метилдиэтаноламин, 3-аминопропилдиэтаноламин и N-(2-гидроксиэтил)пиперазин.
Также могут применяться амины, такие как моно-, ди- и триалкиламины, и смешанные арилалкиламины, которые не содержат гидроксильных групп, или амины, замещенные иными, помимо гидроксила, группами, которые не оказывают отрицательного воздействия на реакцию между амином и эпоксидом. Конкретные примеры включают этиламин, метилэтиламин, триэтиламин, N-бензилдиметиламин, дикокоамин, 3-диметиламинопропиламин и N,N-диметилциклогексиламин.
Также в настоящем изобретении могут применяться смеси указанных выше аминов.
Реакция первичного и/или вторичного амина с полиэпоксидом происходит после смешивания амина и полиэпоксида. Амин может быть добавлен к полиэпоксиду, или наоборот. Реакция может проводиться беспримесным образом или в присутствии подходящего растворителя, такого как метилизобутилкетон, ксилол, или 1-метокси-2-пропанол. Реакция в целом является экзотермической и может быть желательным охлаждение. Однако для ускорения реакции возможно нагревание до умеренной температуры в пределах от 50°C до 150°C.
Продукту реакции первичного и/или вторичного амина и полиэпоксида придается катионная форма и способность к диспергированию в воде посредством по меньшей мере частичной нейтрализации кислотой. Подходящие кислоты включают органические и неорганические кислоты. Неограничивающие примеры подходящих органических кислот включают муравьиную кислоту, уксусную кислоту, метансульфоновую кислоту и молочную кислоту. Неограничивающие примеры подходящих неорганических кислот включают фосфорную кислоту и сульфаминовую кислоту. Под «сульфаминовой кислотой» подразумевается сама сульфаминовая кислота или ее производные, такие, как имеющие формулу

где R – водород или алкильная группа, имеющая от 1 до 4 атомов углерода.
Указывается, что смеси вышеупомянутых кислот также могут применяться в настоящем изобретении.
Степень нейтрализации катионной композиции для нанесения электроосаждаемых покрытий варьирует в зависимости от конкретного продукта реакции. При этом кислота должна вноситься в таком количестве, которое является достаточным для диспергирования в воде композиции для нанесения электроосаждаемых покрытий. Как правило, количество используемой кислоты отвечает по меньшей мере 20 процентам от полной нейтрализации. Также возможно применение избытков кислоты относительно количества, требующегося для полной 100-процентной нейтрализации. Например, количество кислоты, используемой для нейтрализации композиции для нанесения электроосаждаемых покрытий, может составлять ≥1% относительно общего содержания аминов в композиции для нанесения электроосаждаемых покрытий, и количество кислоты, используемой для нейтрализации композиции для нанесения электроосаждаемых покрытий, может быть ≤100% относительно общего содержания аминов в композиции для нанесения электроосаждаемых покрытий. Согласно настоящему изобретению, суммарное количество кислоты, используемой для нейтрализации композиции для нанесения электроосаждаемых покрытий, располагается между любой комбинацией величин, указанных в предшествующих предложениях, включая и сами упомянутые величины. Например, общее количество кислоты, используемой для нейтрализации композиции для нанесения электроосаждаемых покрытий, может составлять 20%, 35%, 50%, 60% или 80% относительно общего содержания аминов в композиции для нанесения электроосаждаемых покрытий.
При реакции третичного амина с полиэпоксидом такой третичный амин может быть предварительно подвергнут нейтрализации кислотой для образования соли амина, и затем соль амина реагирует с полиэпоксидом для получения смолы, содержащей группу соли четвертичного основания. Реакция проводится посредством смешивания соли амина с полиэпоксидом в воде. Как правило, вода присутствует в количествах, находящихся в диапазоне от 1,75 до 20 масс.% по отношению к общему сухому веществу реакционной смеси.
При образовании смолы, содержащей группу соли четвертичного аммониевого основания, температура реакции может варьировать от самой низкой температуры, при которой проходит реакция, обычно это комнатная температура или несколько ее превышающая, и до максимальной температуры 100°C (при атмосферном давлении). При более высоких давлениях возможно использование более высоких температур реакции. Температура реакции может находиться в пределах от 60°C до 100°C. Могут использоваться такие растворители, как пространственно затрудненный сложный эфир, простой эфир или пространственно затрудненный кетон, но их применение необходимым не является.
В дополнение к первичному, вторичному и третичному аминам, раскрываемым выше, часть амина, которая реагирует с полиэпоксидом, может быть представлена кетимином полиамина, таким, как описывается в патент США № 4 104 147, с колонки 6, строка 23 по колонку 7, строка 23. Группы кетимина распадаются при диспергировании продукта реакции амина и эпоксидной смолы в воде. По меньшей мере одна часть присутствующего в смоле активного водорода, (a) содержит группы первичного амина, полученные в результате реакции содержащего кетимин соединения и содержащего эпоксигруппы материала, такого, как описан выше.
В дополнение к смолам, содержащим группы аминных солей и солей четвертичного аммония, в композиции настоящего изобретения могут применяться катионные полимеры, содержащие группы третичного сульфония. Примеры этих смол и способ их приготовления описаны в патентах США №№ 3 793 278 и 3 959 106.
Подходящие содержащие активный водород, содержащие катионную солевую группу смолы могут включать сополимеры одного или нескольких алкильных эфиров акриловой кислоты или (мет)акриловой кислоты, необязательно вместе с одним или несколькими другими полимеризуемыми этиленненасыщенными мономерами. Подходящие алкиловые эфиры акриловой кислоты или (мет)акриловой кислоты включают метил(мет)акрилат, этил(мет)акрилат, бутил(мет)акрилат, этилакрилат, бутилакрилат и 2-этилгексилакрилат. Другие подходящие сополимеризуемые этиленненасыщенные мономеры включают нитрилы, такие акрилонитрил и (мет)акрилонитрил, винил- и винилиденгалогениды, такие как винилхлорид и винилиденфторид, и виниловые эфиры, такие как винилацетат. Могут быть применены обладающие кислотной функциональностью и функциональностью ангидридов этиленненасыщенные мономеры, такие как акриловая кислота, (мет)акриловая кислота или ангидрид, итаконовая кислота, малеиновая кислота или ангидрид, либо фумаровая кислота. Также подходящими являются амид-функциональные мономеры, включая акриламид, (мет)акриламид и N-алкилзамещенные (мет)акриламиды. При условии отсутствия требований к высокому уровню сопротивления фоторазложению полимера возможно применение виниловых ароматических соединений, таких как стирол и винилтолуол.
Функциональные группы, такие как гидроксильные и аминогруппы, могут быть включены в акриловый полимер посредством использования функциональных мономеров, таких как гидроксиалкилакрилаты и метакрилаты или аминоалкилакрилаты и метакрилаты. Эпоксидные функциональные группы (для преобразования в катионные солевые группы) могут быть включены в акриловый полимер при использовании функциональных мономеров, таких как глицидилакрилат и метакрилат, 3,4-эпоксициклогексилметил(мет)акрилат, 2-(3,4-эпоксициклогексил)этил(мет)акрилат или аллилглицидиловый эфир. В качестве варианта, эпоксидные функциональные группы могут быть введены в акриловый полимер при реакции карбоксильных групп на акриловом полимере с эпигалогидрином или дигалогидрином, такими как эпихлоргидрин или дихлоргидрин.
Акриловый полимер может быть приготовлен с помощью стандартных методик инициируемой свободными радикалами полимеризации, например, известной в данной области полимеризацией в растворе или эмульсии, при использовании подходящих катализаторов, которые включают органические пероксиды и азосоединения, и, не обязательно, агентов передачи цепи, таких как димер альфа-метилстирола и третичный додецилмеркаптан. Дополнительные акриловые полимеры, которые являются подходящими для образования содержащих активный водород катионных полимеров и которые могут применяться в композициях для нанесения электроосаждаемых покрытий настоящего изобретения, включают смолы, описанные в патентах США №№ 3 455 806 и 3 928 157.
Как указывалось выше, основной пленкообразующий полимер также может быть получен из полиуретана. Среди пригодных для применения полиуретанов представлены полимерные многоатомные спирты, которые синтезируются реакцией полиэфирполиолов или акриловых полиолов, таких как упоминаемые выше, с полиизоцианатом, таким, для которого эквивалентное соотношение OH/NCO составляет более 1:1, с тем, чтобы в продукте присутствовали свободные гидроксильные группы. Многоатомные спирты с меньшей молекулярной массой, такие как раскрываются выше в связи с их применением в приготовлении полиэфира, могут также применяться вместо или в комбинации с полимерными многоатомными спиртами.
Дополнительные примеры полиуретановых полимеров, подходящих для получения содержащего активный водород катионного полимера, включают полиуретан, полимочевину и поли(уретан-мочевинные)полимеры, приготовленные в результате реакции полиэфирполиолов и/или полиэфирполиаминов с полиизоцианатами. Такие полимеры полиуретана описаны в патенте США № 6 248 225.
Эпоксидные функциональные группы могут быть введены в полиуретан способами, хорошо известными в данной области. Например, эпоксидные группы могут вводиться при реакции глицидилового спирта со свободными изоцианатными группами.
Также могут быть приготовлены полиуретаны, содержащие группу сульфония, – по меньшей мере неполной реакцией гидроксифункциональных сульфидных соединений, таких как тиодигликоль и тиодипропанол, которая приводит к включению серы в главную цепь полимера. Серосодержащий полимер затем в присутствии кислоты реагирует с полифункциональным эпоксисоединением с образованием группы сульфония. Подходящие полифункциональные эпоксисоединения включают этиленоксид, пропиленоксид, глицидиловый спирт, фенилглицидиловый эфир и CARDURA E, предлагаемый компанией Resolution Performance Products.
В дополнение к возможности его получения из полиэпоксида или полиуретана, основной пленкообразующий полимер также может быть получен из полиэфира. Такие полимеры могут быть приготовлены известным способом посредством конденсации многоатомных спиртов и поликарбоновых кислот. Подходящие многоатомные спирты включают, например, этиленгликоль, пропиленгликоль, бутиленгликоль, 1,6-гексиленгликоль, неопентилгликоль, диэтиленгликоль, глицерин, триметилолпропан и пентаэритрит. Примеры многоосновных карбоновых кислот, подходящих для приготовления полиэфира, включают янтарную кислоту, адипиновую кислоту, азелаиновую кислоту, себациновую кислоту, малеиновую кислоту, фумаровую кислоту, фталевую кислоту, тетрагидрофталевую кислоту, гексагидрофталевую кислоту и тримеллитовую кислоту. Помимо упомянутых выше многоосновных карбоновых кислот возможно применение функциональных аналогов кислот, таких как ангидриды в случае их существования или низшие алкильные эфиры кислот, такие как сложные метиловые эфиры. Кроме того, также в качестве компонентов полиэфира могу применяться оксикислоты и/или лактоны, такие как капролактон и/или 12-гидроксистеариновая кислота.
Полиэфиры содержат долю свободных гидроксильных групп (образующуюся в результате применения избытков многоатомного спирта и/или высших многоатомных спиртов в ходе приготовления полиэфира), которые являются доступными для реакций отверждения.
Эпоксидные функциональные группы могут быть введены в полиэфир при реакции карбоксильных групп на полиэфире с эпигалогидрином или дигалогидрином, такими как эпихлоргидрин или дихлоргидрин. В качестве варианта, кислотно-функциональный полиэфир может быть введен в эпоксидный полимер реакцией карбоксильных групп с избытком полиэпоксида.
При реакции содержащего эпоксигруппы полимера описанного выше типа с сульфидом в присутствии кислоты могут вводиться группы солей сульфония, как описано в патентах США №№ 3 959 106 и 4 715 898. Сульфониевые группы могут присоединяться к главным цепям описанного полиэфира при использовании подобных условий реакции.
Согласно настоящему изобретению, основной пленкообразующий полимер может быть представлен в композиции для нанесения электроосаждаемых покрытий в количестве ≥40 масс.% по отношению к общей массе сухого вещества смеси смолы, присутствующей в композиции для нанесения электроосаждаемых покрытий. Согласно настоящему изобретению, основной пленкообразующий полимер может быть представлен в композиции для нанесения электроосаждаемых покрытий в количестве ≤95 масс.% по отношению к общей массе сухого вещества смеси смолы, присутствующей в композиции для нанесения электроосаждаемых покрытий. Согласно настоящему изобретению, массовая процентная доля содержания основного пленкообразующего полимера в композиции для нанесения электроосаждаемых покрытий может располагаться между любой комбинацией величин, указанных в предшествующих предложениях, включая и сами упомянутые величины. Например, основной пленкообразующий полимер может быть представлен в композиции для нанесения электроосаждаемых покрытий в количестве от 50 до 75 масс.% по отношению к общей массе сухого вещества смеси смолы, присутствующей в композиции для нанесения электроосаждаемых покрытий.
Как указывалось выше, в дополнение к (a) основному пленкообразующему полимеру смесь смолы, кроме того, содержит (b) отвердитель (сшивающий агент), который является реакционноспособным по отношению к реакционноспособным функциональным группам пленкообразующего полимера, таким как группы активного водорода. Очевидно, что специалисты в данной области смогут определить подходящий отвердитель для любого конкретного основного пленкообразующего полимера, исходя из функциональности такого основного пленкообразующего полимера. Например, отвердители, которые могут быть применены в настоящем изобретении, включают, но не ограничиваются уретаном, эфиром изоциановой кислоты или их комбинациями.
Следует понимать, что неограничивающие примеры уретановых отвердителей включают продукты (i) реакции между амином и карбонатом и/или (ii) реакции между спиртом и изоцианатом.
Неограничивающие примеры подходящих циклических карбонатов, которые могут быть применены для получения уретанового отвердителя, включают, но не ограничиваются пропиленкарбонатом, этиленкарбонатом, бутиленкарбонатом или их комбинациями. Неограничивающие примеры подходящих ациклических карбонатов, которые могут быть применены для образования уретана, включают, но не ограничиваются диметилкарбонатом, диэтилкарбонатом, метилэтилкарбонатом, дипропилкарбонатом, метилпропилкарбонатом, дибутилкарбонатом или их комбинациями. Согласно настоящему изобретению, ациклический карбонат может содержать диметилкарбонат. Неограничивающие примеры подходящих аминов, которые могут быть применены для образования уретана, включают, но не ограничиваются диэтилентриамином, дипропилентриамином, бис-гексаметилентриамином, изофорондиамином, 4’-бис-аминоциклогексиламином, ксилилендиамином, N-гидроксиэтилэтилендиамином, гексаметилентриамином, трис-аминоэтиламином или их комбинациями. Согласно настоящему изобретению, отвердитель может быть продуктом реакции полиамина и циклического карбоната, и первичные амины полиамина могут вступать в реакцию с циклическим карбонатом. Согласно настоящему изобретению, продукт реакции полиамина и циклического карбоната затем может быть введен во взаимодействие с эпокси-функциональным полимером, таким, как используемые для приготовления основного наполнителя и/или измельченного наполнителя. Более конкретно, вторичный амин из продукта реакции может быть введен во взаимодействие с эпокси-функциональной группой эпокси-функционального полимера.
Неограничивающие примеры подходящих эфиров изоциановой кислоты, которые могут применяться для образования уретанового отвердителя, включают, но не ограничиваются толуилендиизоцианатом, метилендифенил-4,4'-диизоцианатом, изофорондиизоцианатом, гексаметилендиизоцианатом, ксилилендиизоцианатом, тетраметилксилилендиизоцианатом, алифатическими диизоцианатами с прямой цепью, такими как 1,4-тетраметилендиизоцианат, норборнандиизоцианат и 1,6-гексаметилендиизоцианат, изофорондиизоцианат и 4,4'-метилен-бис(циклогексилизоцианат), ароматическими диизоцианатами, такими как п-фенилендиизоцианат, дифенилметан-4,4'-диизоцианат и 2,4- или 2,6-толуолдиизоцианат, высшими полиизоцианатами, такими как трифенилметан-4,4',4"-триизоцианат, 1,2,4-бензолтриизоцианат и полиметиленполифениленизоцианат, и тримерами 1,6-гексаметилендиизоцианатов или их комбинациями. Следует заметить, что димеры, тримеры и производные эфиров изоциановой кислоты с более высокой функциональностью также могут применяться в настоящем изобретении. Неограничивающие примеры подходящих спиртов, которые могут применяться для образования уретана, включают, но не ограничиваются метанолом, этанолом, пропанолом, изопропанолом, бутанолом, гликолевыми эфирами и другими спиртами.
Как указывается выше, подходящие отвердители для полимеров, содержащих группы солей амина, катионных акриловых полимеров и/или содержащих гидроксильные группы полимеров включают эфиры изоциановой кислоты, а также блокированные изоцианаты. Следует заметить, что для целей настоящего изобретения «изоцианаты» также включают полиизоцианаты, и наоборот. Полиизоцианатный отверждающий агент может быть полностью блокированным полиизоцианатом с по существу отсутствующими свободными изоцианатными группами или же он может быть частично блокированным и способным реагировать с главной цепью смолы, как описано в патенте США № 3 984 299. Полиизоцианат может быть алифатическим, ароматическим полиизоцианатом или их комбинацией. Согласно настоящему изобретению, возможно применение диизоцианатов при том, что могут применяться и другие высшие полиизоцианаты вместо или в комбинации с диизоцианатами.
Также могут применяться изоцианатные преполимеры, например, продукты реакции полиизоцианатов с многоатомными спиртами, такими как неопентилгликоль и триметилолпропан, и с полимерными многоатомными спиртами, такими как поликапролактондиолы и поликапролактонтриолы (соотношение эквивалентов NCO/OH выше единицы). Возможно применение смеси дифенилметан-4,4'-диизоцианата и полиметиленполифениленизоцианата.
В качестве блокирующего агента для полиизоцианата в композиции для нанесения электроосаждаемых покрытий настоящего изобретения может использоваться любой подходящий спирт или многоатомный спирт при условии, что такой агент снимает блокировку при температуре отверждения и если не вызывается образования гелеобразного продукта. Например, подходящие спирты включают, без ограничения, метанол, этанол, пропанол, изопропанол, бутанол, 2-этилгексанол, бутоксиэтанол, гексилоксиэтанол, 2-этилгексилоксиэтанол, н-бутанол, циклогексанолфенилкарбинол, метилфенилкарбинол, монобутиловый эфир этиленгликоля, монобутиловый эфир диэтиленгликоля, монометиловый эфир этиленгликоля, монометиловый эфир пропиленгликоля или их комбинации.
Согласно настоящему изобретению, блокирующий агент содержит один или несколько 1,3-гликолей и/или 1,2-гликолей. Согласно настоящему изобретению, блокирующий агент может содержать один или несколько 1,2-гликолей, в типичном случае один или несколько C3 - C6 1,2-гликолей. Например, блокирующий агент может быть выбран по меньшей мере из одного из 1,2-пропандиола, 1,3-бутандиола, 1,2-бутандиола, 1,2-пентандиола, триметилпентендиола и/или 1,2-гександиола.
Согласно настоящему изобретению, полиизоцианатные отвердители могут быть применены во взаимодействии с основными катионными пленкообразующими полимерами в количествах ≥5 масс.% по отношению к общей массе сухого вещества смеси смолы в ванне для электроосаждения. Согласно настоящему изобретению, полиизоцианатные отвердители могут быть применены во взаимодействии с основными катионными пленкообразующими полимерами в количествах ≤60 масс.% по отношению к общей массе сухого вещества смеси смолы в ванне для электроосаждения. Согласно настоящему изобретению, количество основного пленкообразующего полимера может располагаться между любой комбинацией величин, указанных в предшествующих предложениях, включая и сами упомянутые величины. Например, полиизоцианатные отвердители могут быть применены во взаимодействии с основными катионными пленкообразующими полимерами в количествах, находящихся в диапазоне от 20 до 50 масс.% по отношению к общей массе сухого вещества смеси смолы в ванне для электроосаждения.
Другие подходящие блокирующие агенты включают оксимы, таким как метилэтилкетоксим, ацетоксим и циклогексаноноксим, а также лактамы, такие как эпсилон-капролактам.
Как указывалось выше, отверждающий агент, который применяется в настоящем изобретении, может быть эфирным отвердителем. Следует заметить, что для целей настоящего изобретения понятие «эфир» также включает и полиэфиры. Соответственно, эфирный отвердитель может быть полиэфирным отверждающим агентом. Подходящие полиэфирные отверждающие агенты включают материалы, имеющие более одной сложноэфирной группы на молекулу. Сложноэфирные группы присутствуют в количестве, достаточном для обеспечения сшивающего эффекта, например, при температурах вплоть до 250°C и времени отверждения вплоть до 90 минут. Следует понимать, что приемлемые температуры отверждения и продолжительность времени отверждения будут зависеть от предназначенных для нанесения покрытия подложек и их конечного применения.
Соединения, в целом подходящие для применения в качестве полиэфирного отвердителя, могут быть полиэфирами многоосновных карбоновых кислот. Их неограничивающие примеры включают бис(2-гидроксиалкил)эфиры дикарбоновых кислот, такие как бис(2-гидроксибутил)эфир азелаиновой кислоты и бис(2-гидроксиэтил)терефталат; трис(2-этилгексаноил)тримеллитат; и поли(2-гидроксиалкил)эфиры полуэфиров кислот, получаемые из ангидрида дикарбоновой кислоты и спирта, включая многоатомные спирты. Последний тип является подходящим для придания полиэфиру конечной функциональности, превышающей 2. Один подходящий пример включает полиэфир, получаемый реакцией эквивалентных количеств ангидрида дикарбоновой кислоты (например, янтарного ангидрида или фталевого ангидрида) с трехатомным или четырехатомным спиртом, таким как глицерин, триметилолпропан или пентаэритрит, при температурах ниже 150°C с последующим взаимодействием кислого полиэфира с по меньшей мере эквивалентным количеством эпоксиалкана, такого как 1,2-эпоксибутан, этиленоксид или пропиленоксид. Полиэфирный отвердитель (ii) может содержать ангидрид. Другой подходящий полиэфир содержит более низкомолекулярный полиалкиленгликольтерефталат с концевыми 2-гидроксиалкильными группами.
Согласно настоящему изобретению, полиэфирный отвердитель может содержать по меньшей мере по одной сложноэфирной группе на молекулу, в которой соседний с этерифицированным гидроксилом атом углерода имеет свободную гидроксильную группу.
Также подходящим является тетрафункциональный полиэфир, получаемый из полуэфирного интермедиата, приготавливаемого при реакции тримеллитового ангидрида с пропиленгликолем (мольное отношение 2:1) с последующим взаимодействием такого интермедиата с 1,2-эпоксибутаном и глицидиловым эфиром разветвленной одноосновной карбоновой кислоты.
Согласно настоящему изобретению, когда содержащая активный водород смола содержит солевые катионные группы, полиэфирный отвердитель может быть по существу не содержащим кислоты. Для целей настоящего изобретения выражение «по существу не содержащий кислоты» означает наличие кислоты в количестве менее 0,2 мЭкв/г. В случае водных систем, предназначенных, например, для катодного нанесения покрытий электроосаждением, подходящие полиэфирные отвердители могут включать не являющиеся кислотными полиэфиры, приготавливаемые из ангидрида многоосновной карбоновой кислоты, один или нескольких гликолей, спирты, гликоль-моноэфиры, многоатомные спирты и/или моноэпоксиды.
Подходящие ангидриды многоосновных кислот могут включать ангидриды дикарбоновых кислот, такие как янтарный ангидрид, фталевый ангидрид, тетрагидрофталевый ангидрид, тримеллитовый ангидрид, гексагидрофталевый ангидрид, метилгексагидрофталевый ангидрид, диангидрид 3,3’,4,4’-бензофенонтетракарбоновой кислоты и пиромеллитовый диангидрид. Могут применяться смеси ангидридов.
Подходящие спирты могут включать линейные, циклические или разветвленные спирты. Спирты могут быть алифатическими, ароматическими или аралифатическими по своей природе. В данном контексте термины «гликоли» и «моноэпоксиды» предназначаются для включения соединений, содержащих не более двух спиртовых групп на молекулу, которые способны реагировать с функциональными группами карбоновых кислот или ангидридов при температуре ниже 150°C.
Подходящие моноэпоксиды могут включать глицидиловые эфиры разветвленных одноосновных карбоновых кислот. Кроме того, могут применяться алкиленоксиды, такие как этиленоксид или пропиленоксид. Подходящие гликоли могут включать, например, этиленгликоль и полиэтиленгликоли, пропиленгликоль и полипропиленгликоли, и 1,6-гександиол. Возможно применение смесей гликолей.
Не обладающие кислотными свойствами полиэфиры могут быть приготовлены, например, при взаимодействии в одну или несколько стадий тримеллитового ангидрида (TMA) с глицидиловыми эфирами разветвленных одноосновных карбоновых кислот в мольном соотношении от 1:1,5 до 1:3, при необходимости с применением катализатора этерификации, такого как октоат олова или бензилдиметиламин, при температурах 50 - 150°C. Помимо этого, тримеллитовый ангидрид может вступать в реакцию с 3 эквивалентами моноспирта, такого как 2-этилгексанол.
В качестве варианта, тримеллитовый ангидрид (1 моль) может быть вначале приведен в реакцию с гликолем или моноалкильным эфиром гликоля, таким как монобутиловый эфир этиленгликоля, взятыми в мольном соотношении от 1:0,5 до 1:1, после чего полученному продукту дают возможность реагировать с 2 молями глицидилового эфира разветвленной одноосновной карбоновой кислоты. Кроме того, ангидрид многоосновной карбоновой кислоты (то есть содержащий по две или три карбоксильные функциональные группы на молекулу) или смесь ангидридов многоосновных карбоновых кислот может одновременно вводиться в реакцию с гликолем, таким как 1,6-гександиол и/или моноэфир гликоля, и с моноэпоксидом, после чего, если это необходимо, продукт может прореагировать с моноэпоксидами. Для водных композиций эти не являющиеся кислотными полиэфиры также могут быть подвергнуты модифицированию полиаминами, такими как диэтилентриамин, для образования полиэфирамидов. Такие «аминомодифицированные» полиэфиры могут быть введены в описанные выше линейные или разветвленные аминовые аддукты для образования самоотверждающихся эфиров аминовых аддуктов.
Некислотные полиэфиры описанных выше типов в типичном случае являются растворимыми в органических растворителях и, как правило, могут быть легко смешаны с основной пленкообразующей смолой, описанной выше.
Полиэфиры, подходящие для применения в водной системе или смесях таких материалов, диспергируются в воде, обычно в присутствии смол, содержащих катионные солевые группы.
Согласно настоящему изобретению, отверждающий агент, используемый в композиции для нанесения покрытия электроосаждением, может быть карбамат-функциональным отвердителем, таким как описывается в патенте США № 5 902 473, который включен здесь посредством ссылки.
Согласно настоящему изобретению, по меньшей мере одна доля отверждающего агента может быть химически связана с основным пленкообразующим полимером. Согласно настоящему изобретению, отверждающий агент может не являться химически связанным с основным пленкообразующим полимером и добавляется в виде добавки к композиции для нанесения электроосаждаемых покрытий.
Пигментная паста может содержать один или несколько пигментов, диспергируемый в воде полимер и, не обязательно, добавки, такие как поверхностно-активные вещества, смачивающие вещества, катализаторы, диспергирующие агенты или их комбинации. Следует заметить, что диспергируемый в воде полимер из пигментной пасты может быть как тем же самым, так и отличным от основного пленкообразующего полимера в смеси смолы. Пигментная композиция, используемая в пигментной пасте, может принадлежать к стандартному типу, содержащему такие пигменты, как, например, оксиды железа, хромат стронция, сажу, угольную пыль, диоксид титана, тальк, барит, а также цветные пигменты, такие как желтый кадмий, красный кадмий, желтый хром и т.п. Согласно настоящему изобретению, пигментная композиция может содержать функциональные пигменты, такие как, но не ограничиваясь электропроводящими и/или фотохромными пигментами. Содержание пигмента в дисперсии обычно выражается в виде массового соотношения пигмента и смолы. В практике изобретения, в случае применения пигмента отношение содержания пигмента к содержанию связующего обычно находится внутри диапазона от около 0,02:1 до 1:1. Упомянутые выше другие добавки обычно находятся в дисперсии в количестве от 0,01 до 3 масс.% по отношению к общей массе сухих веществ смеси смолы.
Первый и второй компоненты ванны для электроосаждения совместно диспергируются в водной среде, которая содержит воду и обычно коалесцирующие растворители, с тем, чтобы образовать ванну для электроосаждения. Подходящие для применения коалесцирующие растворители, которые могут использоваться в ванне для электроосаждения, включают, но не ограничиваются углеводородами, спиртами, сложными эфирами, простыми эфирами и/или кетонами. Согласно настоящему изобретению, коалесцирующие растворители включают спирты, многоатомные спирты и кетоны. Конкретные коалесцирующие растворители включают изопропанол, бутанол, 2-этилгексанол, изофорон, 2-метоксипентанон, этилен- и пропиленгликоль и моноэтиловые, монобутиловые и моногексиловые эфиры этиленгликоля. Согласно настоящему изобретению, количество коалесцирующего растворителя, используемого в ванне для электроосаждения, может составлять ≥ 0,01 масс.% по отношению к общей массе водной среды, используемой для создания ванны для электроосаждения. Согласно настоящему изобретению, количество коалесцирующего растворителя, используемого в ванне для электроосаждения, может составлять ≤ 25 масс.% по отношению к общей массе водной среды, используемой для создания ванны для электроосаждения. Согласно настоящему изобретению, количество коалесцирующего растворителя, используемого в ванне для электроосаждения, может располагаться между любой комбинацией величин, указанных в предшествующих предложениях, включая и сами упомянутые величины. Например, количество коалесцирующего растворителя, используемого в ванне для электроосаждения, может находиться в диапазоне между 0,05 и 5 масс.% по отношению к общей массе водной среды, используемой для создания ванны для электроосаждения.
Как упоминалось ранее, настоящее раскрытие также касается систем и способов для обработки различных подложек. Подходящие подложки, которые могут использоваться с системами и способами обработки настоящего изобретения, включают металлические подложки, подложки из металлических сплавов и/или подложки, которые были металлизированы, такие как никелированная пластмасса. Согласно настоящему изобретению, такой металл или металлический сплав могут содержать или являться холоднокатаной сталью, горячекатаной сталью, сталью с покрытием из металлического цинка, соединений цинка или цинковых сплавов, такой как сталь с гальваническим цинковым покрытием, сталь горячего цинкования, сталь с покрытием из гальванила и сталь, плакированная цинковым сплавом. Также в качестве подложки могут быть использованы алюминиевые сплавы серий 2XXX, 5XXX, 6XXX или 7XXX, а также плакированные алюминиевые сплавы и алюминиевые литейные сплавы серии A356. В качестве подложки также могут быть использованы магниевые сплавы серий AZ31B, AZ91C, AM60B или EV31A. Подложка, применяемая в настоящем изобретении, может также содержать титан и/или титановые сплавы. Другие подходящие цветные металлы включают медь и магний, а также сплавы этих материалов. Подходящие для применения в настоящем изобретении металлические подложки включают часто используемые при сборке корпусов транспортных средств (например, без ограничения, дверей, панелей кузова, люков тронковых судов, панелей крыши, капотов, потолков или стрингеров, заклепок, компонентов шасси и/или обшивки, используемых в самолетах), рам транспортных средств, деталей транспортных средств, мотоциклов, колес, мелких металлические деталей, включая крепежные элементы, то есть гайки, болты, винты, штифты, гвозди, скобы, кнопки и другие подобные, промышленных конструкций и компонентов, таких как бытовые приборы, включая стиральные машины, сушилки, холодильные устройства, печи, посудомоечные машины и другие подобные, сельскохозяйственное оборудование, средства ухода за садом и газоном, установки кондиционирования воздуха, тепловые насосы, садовую мебель и другие изделия. В данном контексте «транспортное средство» или его вариации включает, но не ограничивается гражданскими, коммерческими и военными воздушными судами и/или наземными транспортными средствами, такими как автомобили, мотоциклы и грузовики. Кроме того, предназначенная для обработки с помощью способов настоящего изобретения подложка может представлять обрезанную кромку подложки притом, что остальная часть ее поверхности обработана и/или покрыта каким-либо иным образом. Металлическая подложка, подвергнутая обработке в соответствии со способами настоящего изобретения, может быть в форме, например, металлического листа или заготовки.
Композиция для нанесения электроосаждаемых покрытий настоящего изобретения может быть нанесена на множество подложек, таких как описанные выше. Соответственно, настоящее изобретение, кроме того, касается подложки, покрытой, по меньшей мере частично, описанной здесь композицией для нанесения электроосаждаемых покрытий. Понимается, что композиция для нанесения покрытия электроосаждением может быть нанесена на подложку в виде монопокрытия или как слой в многослойном композиционном покрытии. Согласно настоящему изобретению, по меньшей мере один участок поверхности металлической поверхности, на которую наносится покрытие, может быть предварительно обработан фосфатной композицией для предварительной обработки, такой как цинк-фосфатная композиция для предварительной обработки или композиция для предварительной обработки металлом Группы IVB.
Кроме того, композиция для нанесения электроосаждаемых покрытий настоящего изобретения может быть нанесена на подложку для придания той широкого разнообразия свойств, таких как, но не ограничиваясь коррозионной устойчивостью, стойкостью к расщеплению, пригодностью к заполнению (то есть способностью скрывать шероховатость нижележащей подложки), сопротивлением истиранию, стойкостью к повреждениям при ударной нагрузке, огне- и/или теплостойкостью, стойкостью к воздействию химикатов, стойкостью к действию ультрафиолетового света и/или структурной целостностью.
Композиция для нанесения электроосаждаемых покрытий может быть нанесена погружением подложки в ванну для электроосаждения, при этом данная подложка выступает в качестве электрода в электрической цепи с противоэлектродом, а к системе прикладывается электрический потенциал с тем, чтобы обеспечить осаждение на поверхности подложки композиции для нанесения электроосаждаемых покрытий. В зависимости от подложки, композиция для нанесения электроосаждаемых покрытий может быть нанесена (то есть выполнено электроосаждение) на подложку при использовании напряжения, величина которого может располагаться в пределах от 1 вольта до нескольких тысяч вольт. Согласно настоящему изобретению, прикладываемое напряжение находится в диапазоне от 50 вольт до 500 вольт. При этом плотность тока может составлять между 0,5 амперами и 5 амперами на квадратный фут. Однако понятно, что плотность тока имеет тенденцию к снижению в ходе электроосаждения, что является свидетельством образования изолирующей пленки.
После того, как покрытие оказывается нанесено на подложку посредством электроосаждения, такое покрытие может быть отверждено путем подвергания подложки термической обработке при повышенной температуре в пределах от 90°C до 260°C в течение интервала времени от 1 минуты до 40 минут.
Композиция для нанесения электроосаждаемых покрытий настоящего изобретения может быть использована в слое наносимого электроосаждением покрытия, являющегося частью многослойного композиционного покрытия, содержащего подложку с различными слоями покрытия. Слои покрытия могут включать слой для предварительной обработки, такой как фосфатный слой (например, слой фосфата цинка) или описанный выше слой металла Группы IVB для предварительной обработки, слой наносимого электроосаждением покрытия, которое образуется из композиции для нанесения электроосаждаемых покрытий настоящего изобретения, и слои подходящего наружного покрытия (например, первый слой двухслойного покрытия, слой прозрачного покрытия, окрашенное монопокрытие и композиции композита типа пигментированная грунтовка/прозрачный наружный слой). Понимается, что подходящие слои поверхностного покрытия включают любые из известных в данной области, при этом каждый может независимым образом быть водоразбавляемым, органорастворимым, находиться в форме твердого дисперсного материала (то есть порошковой композиции для нанесения покрытий) или в форме суспензии порошка. Поверхностный слой в типичном случае включает пленкообразующий полимер, сшивающий материал и, если имеется цветной первый слой двухслойного покрытия или монопокрытие, один или несколько пигментов. Согласно настоящему изобретению, между слоем наносимого электроосаждением покрытия и первым слоем двухслойного покрытия располагается грунтовочный слой. Согласно настоящему изобретению, один или несколько слоев поверхностного покрытия наносятся на по существу неотвержденный нижележащий слой. Например, слой прозрачного покрытия может быть нанесен на по меньшей мере один участок по существу неотвержденного первого слоя двухслойного покрытия (влажный по влажному) и оба слоя могут быть подвергнуты одновременному отверждению в ходе последующего этапа способа.
Кроме того, слои поверхностного покрытия могут быть нанесены непосредственно на слой электроосаждаемого покрытия. Другими словами, в этом случае на подложке отсутствует грунтовочный слой. Например, первый слой двухслойного покрытия наносится непосредственно на по меньшей мере один участок слоя электроосаждаемого покрытия.
Также следует понимать, что слои поверхностного покрытия могут наноситься на нижележащий слой, невзирая на то, что такой нижележащий слой не был полностью отвержден. Например, слой покровного лака может быть нанесен на первый слой двухслойного покрытия даже в том случае, когда такой первый слой не был подвергнут этапу отверждения. Далее оба слоя могут быть отверждены в течение последующего этапа отверждения, что устраняет, таким образом, необходимость в отдельном отверждении первого слоя двухслойного покрытия и слоя покровного лака.
Согласно настоящему изобретению, дополнительные ингредиенты, такие как красители и наполнители, могут присутствовать в различных композициях для нанесения покрытий, из которых образуется слой поверхностного покрытия. Использоваться могут любые подходящие красители и наполнители. Например, к покрытию может добавляться окрашивающее вещество в любой подходящей форме, такой как дискретные частицы, дисперсия, растворы и/или хлопья. В покрытиях настоящего изобретения может использоваться индивидуальное окрашивающее вещество или смесь из двух или более окрашивающих веществ. Следует заметить, что в целом краситель может присутствовать в слое многослойного композита в любом количестве, достаточном для придания желаемого свойства, визуального и/или цветового эффекта.
Примеры окрашивающих веществ включают пигменты, красители и колерующие средства, такие как применяемые в лакокрасочной промышленности и/или перечисленные в списке Международной ассоциации производителей пигментов (DCMA), а также композиции, сообщающие особые эффекты. Окрашивающее вещество может включать, например, тонко измельченный твердый порошок, который является нерастворимым, но смачиваемым в условиях применения. Окрашивающее вещество может быть органическим или неорганическим и может быть агломерированным или неагломерированным. Окрашивающие вещества могут вноситься в покрытия при измельчении или простым смешиванием. Окрашивающие вещества могут вноситься в покрытия посредством истирания с применением измельченного наполнителя, такого как акриловый измельченный наполнитель, использование которого известно специалистам в данной области.
Примеры пигментов и/или композиций пигментов включают, но не ограничиваются карбазолдиоксазиновым необработанным красителем, азо-, моноазо-, дисазокрасителями, нафтолом AS, солевого типа (баканы), бензимидазолоновыми, конденсационными, металлокомплексными, изоиндолиноновыми, изоиндолиновыми и полициклическими фталоцианиновыми, хинакридоновыми, периленовыми, периноновыми, дикетопирролопирроловыми, тиоиндиговыми, антрахиноновыми, индатроновыми, антрапиримидиновыми, флавантроновыми, пирантроновыми, антантроновыми, диоксазиновыми, триарилкарбониевыми, хинофталоновыми пигментами, дикетопирролопирролом красным ("DPP red BO"), диоксидом титана, газовой сажей, оксидом цинка, оксидом сурьмы и т.п., а также органическими или неорганическими препятствующими прохождению ультрафиолетового света пигментами, такими как оксид железа, прозрачный красный или желтый оксид железа, фталоцианиновый синий, и их смесями. Термины «краситель» и «окрашенный наполнитель» могут использоваться равнозначно.
Примеры красителей включают, но не ограничиваются такими, которые являются красителями на водной основе и/или на основе органических растворителей, такими как кислотные красители, азокрасители, основные красители, прямые красители, дисперсные красители, активные красители, органорастворимые красители, сернистые красители, протравные красители, например, ванадат висмута, антрахиноновые, периленовые, алюминиевые красители, хинакридоновые, тиазоловые, тиазиновые, азокрасители, индигоидные, нитрокрасители, нитрозокрасители, оксазиновые, фталоцианиновые, хинолиновые, стильбеновые красители и трифенилметановые красители.
Примеры колерующих средств включают, но не ограничиваются пигментами, диспергируемыми в носителях на водной основе или растворимыми в воде, такими как AQUA-CHEM 896, предлагаемый компанией Degussa, Inc, CHARISMA COLORANTS и MAXITONER INDUSTRIAL COLORANTS, предлагаемые в продаже отделением Accurate Dispersions компании Eastman Chemical, Inc.
Как отмечалось ранее, окрашивающее вещество может находиться в форме дисперсии, включая, но не ограничиваясь дисперсией наночастиц. Дисперсия наночастиц может включать одно или несколько тонко диспергированных, состоящих из наночастиц окрашивающих веществ и/или частицы окрашивающего вещества, которые обеспечивают желаемый видимый цвет, и/или непрозрачность, и/или визуальный эффект. Дисперсия наночастиц может включать окрашивающие вещества, такие как пигмент или краситель, имеющие размер частиц менее 150 нм, например, менее 70 нм или менее 30 нм. Наночастицы могут быть получены размалыванием исходных органических или неорганических пигментов с использование абразивного материала с размером частиц менее 0,5 мм. Пример дисперсий наночастиц и способы их изготовления раскрываются в патенте США № 6 875 800 B2, который является включенным в данное описание посредством ссылки. Дисперсии наночастиц могут быть также получены кристаллизацией, осаждением, конденсацией из газовой фазы и химическим истиранием (то есть частичным растворением). Для минимизации повторной агломерации наночастиц в покрытии может быть применена дисперсия наночастиц, покрытых смолой. Для целей настоящего изобретения «дисперсия покрытых смолой наночастиц» относится к непрерывной фазе, в которой диспергированы дискретные «композитные микрочастицы», содержащие наночастицы и покрытие из смолы на этих наночастицах. Пример дисперсии покрытых смолой наночастиц и способов их получения представлен в заявке США № 10/876 031, поданной 24 июня 2004 г., которая включена здесь посредством ссылки, и в предварительной заявке США № 60/482 167, поданной 24 июня 2003 г., которая также включена здесь посредством ссылки.
Согласно настоящему изобретению, пример композиций для создания специальных эффектов, которые могут быть применены в одном или нескольких слоях многослойного композиционного покрытия, включает пигменты и/или композиции, которые обеспечивают один или более визуальных эффектов, таких как отражение, перламутровый эффект, эффект металлического блеска, фосфоресценция, флуоресценция, фотохромизм, фоточувствительность, термохромизм, гониохромизм и/или изменения цвета. Дополнительные композиции для создания специальных эффектов могут обеспечивать другие видимые свойства, такие как отражательная способность, непрозрачность или текстура. Например, композиции для создания специальных эффектов могут приводить к цветовому сдвигу, то есть к изменению цвета покрытия при рассмотрении покрытия под различными углами. Пример композиции для создания цветовых эффектов раскрывается в патенте США № 6 894 086, включенном здесь посредством ссылки. Дополнительные композиции с цветовым эффектом могут включать слюду с прозрачным покрытием и/или синтетическую слюду, диоксид кремния с покрытием, оксид алюминия с покрытием, прозрачный жидкокристаллический пигмент, жидкокристаллическое покрытие и/или любую композицию, в которой интерференция вызывается наличием различий в показателе преломления в материале, а не различием в показателях преломления между поверхностью материала и воздухом.
Согласно настоящему изобретению, в некотором количестве слоев многослойного композита может применяться светочувствительная композиция и/или фотохромная композиция, которая обратимо изменяет свой цвет, когда подвергается воздействию одного или нескольких источников света. Фотохромные и/или светочувствительные композиции могут быть активированы в результате воздействия излучения с определенной длиной волны. При возбуждении композиции изменяется ее молекулярная структура, а измененная структура демонстрирует новый цвет, который отличается от исходного цвета композиции. При прекращении действия облучения фотохромные и/или светочувствительные композиции возвращаются в состояние покоя, в котором исходный цвет композиции восстанавливается. Например, фотохромная и/или светочувствительная композиция может быть бесцветной в невозбужденном состоянии и демонстрировать окраску в возбужденном состоянии. Полное изменение цвета может проявиться в течение времени от миллисекунд до нескольких минут, например, от 20 секунд до 60 секунд. Пример фотохромных и/или светочувствительных композиций включает фотохромные красители.
Согласно настоящему изобретению, светочувствительная композиция и/или фотохромная композиция может быть связана и/или по меньшей мере частично соединена, например, ковалентными связями с полимером и/или полимерными материалами полимеризующегося компонента. В отличие от некоторых покрытий, в которых светочувствительная композиция может мигрировать из покрытия и кристаллизоваться в подложке, светочувствительная композиция и/или фотохромная композиция, связанная и/или по меньшей мере частично присоединенная к полимерному и/или полимеризующемуся компоненту, в соответствии с настоящим изобретением способна лишь к минимальной миграции из покрытия. Пример светочувствительных композиций и/или фотохромных композиций и способов их изготовления раскрывается в зарегистрированной 16 июля 2004 г. заявке США № 10/892919, включенный здесь посредством ссылки.
Согласно настоящему изобретению, неожиданно было обнаружено, что обработка подложки водной щелочной композицией, содержащей соль железа и щелочной компонент, приводит к значительному улучшению адгезии наносимого впоследствии покрытия, что было установлено по результатам испытаний, выполненных согласно представленному в примерах протоколу. Этот результат был неожиданным.
Для целей подробного описания следует понимать, что изобретение может допускать различные альтернативные варианты и порядок следования этапов за исключением случаев, если явно указано иное. Более того, за исключением любых рабочих примеров или иных случаев, где это специально обозначается, все числовые значения, такие как выражающие величины, количества, процентные доли, диапазоны, поддиапазоны и фракции, могут быть прочитаны как предваряемые словом «около», даже если в явном виде оно отсутствует. Соответственно, если не указывается иного, представленные в нижеследующем описании и прилагаемой формуле изобретения числовые параметры являются приближенными и могут изменяться в зависимости от желаемых для достижения с помощью настоящего изобретения свойств. По меньшей мере, и не как попытка ограничения применимости доктрины эквивалентов к объему формулы изобретения, каждый числовой параметр должен истолковываться по меньшей мере с учетом количества представленных значимых цифр и с применением обычных методов округления. В случаях, когда здесь описывается закрытый или открытый числовой диапазон, все числа, величины, количества, процентные доли, поддиапазоны и фракции внутри или охватываемые данным числовым диапазоном должны рассматриваться как определенно в него включаемые и принадлежащие оригинальному раскрытию этой заявки, как если бы эти числа, величины, количества, процентные доли, поддиапазоны и фракции были явно отображены в их полноте.
Несмотря на то, что числовые диапазоны и параметры, определяющие объем изобретения в целом, являются приближенными, представленные в конкретных примерах численные величины устанавливаются настолько точно, насколько это возможно. Однако любая численная величина по своему существу содержит некоторые погрешности, заведомо следующие из стандартного разброса, обнаруживаемого при соответствующих контрольных измерениях.
В данном контексте «сухое вещество смеси смолы» включает отверждающий агент, смолу, используемую для приготовления основного пленкообразующего полимера, и/или пигментную пасту и любой дополнительный диспергируемый в воде непигментированный компонент(-ы).
В данном контексте, если не указывается иного, термин во множественном числе может охватывать и его единственное число, и наоборот, если не указывается иного. Например, хотя в тех случаях, когда дается ссылка на соль железа, щелочной компонент, соль кобальта, композицию для предварительной обработки, она может означать единственное число, возможно также применение и комбинации (то есть множества) этих компонентов. Помимо этого, использование в данной заявке союза «или», если специальным образом не заявляется иного, означает «и/или», даже при том, что «и/или» может в некоторых случаях использоваться явно.
В данном контексте «включающий», «содержащий» и другие подобные термины понимаются в контексте этой заявки как являющиеся синонимичными «содержащему» и поэтому являются открытыми и не исключают присутствия дополнительных неописанных и/или неперечисленных элементов, материалов, ингредиентов или этапов способа. В контексте данной заявки «состоящий из» понимается как исключающий присутствие любого неопределенного элемента, ингредиента или этапа способа. В контексте данной заявки «состоящий по существу из» понимается как включающий указанные элементы, материалы, ингредиенты или этапы способа, а также «те, которые существенно не воздействуют на основной и новый признак(-и)» того, что описывается.
В данном контексте термины «на», «нанесенный на», «образованный на», «осажденный на» обозначают образованный, наложенный, осажденный или обеспеченный на, но не обязательно в контакте с поверхностью. Например, композиция для нанесения электроосаждаемых покрытий, «осажденная на» подложку, не препятствует присутствию одного или нескольких других промежуточных слоев покрытия из той же самой или другой композиции, располагающихся между композицией для нанесения электроосаждаемых покрытий и подложкой.
Если здесь не указывается иного, термин «по существу не содержит» при его использовании в отношении отсутствия какого-либо конкретного материала, означает, что такой материал, если он присутствует в композиции, в ванне, содержащей композицию и/или в образованных из и содержащих композицию слоях, наличествует лишь в ничтожно малом количестве 5 ч./млн. или менее по отношению к общей массе композиции, ванны и/или слоя (слоев), в зависимости от обстоятельств. Если здесь не указывается иного, термин «фактически не содержит» при его использовании в отношении отсутствия какого-либо конкретного материала означает, что такой материал, если он присутствует в композиции, в ванне, содержащей композицию и/или в образованных из и содержащих композицию слоях, наличествует лишь в ничтожно малом количестве 1 ч./млн. или менее по отношению к общей массе композиции, ванны и/или слоя (слоев), в зависимости от обстоятельств. Если здесь не указывается иного, термин «полностью не содержит», когда он используется в отношении отсутствия какого-либо конкретного материала, означает, что такой материал, если он присутствует в композиции, в ванне, содержащей композицию и/или в образованных из и содержащих композицию слоях, отсутствует в композиции, в содержащей композицию ванне и/или в слоях, образованных из и содержащих то же самое (то есть композиция, ванна, содержащая композицию, и/или слой, образованный из и содержащий композицию, содержат 0 ч./млн. такого материала). Когда композиция, ванна, содержащая композицию, и/или слой (слои), образованный из и содержащий то же самое, является по существу не содержащим, фактически не содержащим или полностью не содержащим конкретного материала, это означает, что такой материал исключен оттуда, кроме тех случаев, когда материал может быть представлен в результате, например, переноса из предшествующих ванн для обработки в поточной линии, из муниципальных источников водоснабжения, на подложке (подложках) и/или вследствие растворения оборудования.
В данном контексте термин «металл Группы IVB » относится к элементу, который находится в Группе IVB CAS-версии Периодической таблицы элементов, представленной, например, в Handbook of Chemistry and Physics, 63-е издание (1983), и соответствующей Группе 4 в действующей нумерации IUPAC (Международный союз теоретической и прикладной химии).
В данном контексте термин «соединение металла Группы IVB » относится к соединениям, которые включают по меньшей мере один элемент, который находится в Группе IVB CAS-версии Периодической таблицы элементов.
В данном контексте «соль» относится к ионному соединению, составленному из металлических катионов и неметаллических анионов и имеющему общий электрический заряд равный нулю. Соли могут быть гидратированными или безводными.
В данном контексте «водная композиция» относится к раствору или дисперсии в среде, которая содержит преимущественно воду. Например, такая водная среда может содержать воду в количестве более 50 масс.%, или более 70 масс.%, или более 80 масс.%, или более 90 масс.%, или более 95 масс.% по отношению к общей массе среды. Например, водная среда может состоять по существу из воды.
При том, что были подробно описаны конкретные аспекты данного изобретения, специалистам в данной области очевидно, что в свете общей идеи данного раскрытия могут быть разработаны различные модификации и альтернативные варианты. Соответственно, подразумевается, что описанные конкретные конфигурации являются лишь иллюстративными и не ограничивающими объем данного изобретения, широта которого определяется прилагаемой формулой изобретения и всеми и любыми ее эквивалентами. Принимая во внимание предшествующее описание, настоящее изобретение, таким образом, относится, в частности, но не ограничиваясь только этим, к следующим объектам 1 - 26.
Объекты
1. Водная композиция щелочного очистителя, содержащая:
катион железа, катион молибдена, катион кобальта или их комбинации; и
щелочной компонент;
причем показатель pH водной щелочной композиции равен по меньшей мере 10 и водная щелочная композиция включает не более 50 ч./млн. фосфата по отношению к общей массе композиции.
2. Композиция очистителя из объекта 1, в которой катион железа присутствует в количестве от 50 ч./млн. до 500 ч./млн. по отношению к общей массе композиции.
3. Композиция очистителя из объекта 1 или объекта 2, в которой катион молибдена присутствует в количестве от 10 ч./млн. до 400 ч./млн. по отношению к общей массе композиции.
4. Композиция очистителя из любого из предшествующих объектов, в которой катион кобальта присутствует в количестве от 50 ч./млн. до 5800 ч./млн. по отношению к общей массе композиции.
5. Композиция очистителя из любого из предшествующих объектов, дополнительно содержащая, хелатор, окислитель, поверхностно-активное вещество, осаждаемые соединения или их комбинации.
6. Композиция очистителя из объекта 5, в которой поверхностно-активное вещество является анионным, неионогенным, катионным или амфотерным соединением.
7. Композиция очистителя по любому из предшествующих объектов, дополнительно содержащая фосфонат.
8. Композиция очистителя любого из предшествующих объектов, в которой фосфонат имеет отношение P-C, отвечающее по меньшей мере 0,10.
9. Композиция очистителя объекта 7 или объекта 8, в которой фосфонат содержит полидентатный фосфонат.
10. Композиция очистителя любого из предшествующих объектов, в которой композиция по существу не содержит, фактически не содержит или полностью не содержит фосфата.
11. Система обработки, предназначенная для обработки металлических подложек, содержащая:
a) водную композицию щелочного очистителя любого из предшествующих объектов; и:
b) композицию для предварительной обработки, предназначенную для обработки по меньшей мере одного участка подложки и содержащую катион металла Группы IVB.
12. Система объекта 11, в которой композиция для предварительной обработки дополнительно содержит электроположительный металл, катион лития, катион молибдена или их комбинации.
13. Система объекта 11 или объекта 12, в которой система по существу не содержит, фактически не содержит или полностью не содержит фосфата.
14. Система любого из объектов 11 - 13, дополнительно содержащая по меньшей мере одно из композиции для предварительного ополаскивания, композиции для ополаскивания после обработки, раствора для нанесения покрытия, композиции для нанесения электроосаждаемых покрытий, композиции для нанесения порошкового покрытия и жидкой композиции.
15. Способ обработки металлической подложки, содержащий введение по меньшей мере одного участка подложки в контакт с композицией очистителя согласно любому из объектов 1 - 10.
16. Способ объекта 15, в котором контактирование осуществляют в течение от 60 секунд до 120 секунд.
17. Подложка, пригодная для получения с помощью системы любого из объектов 10 - 14.
18. Подложка, пригодная для получения способом объекта 15 или 16.
19. Система обработки, предназначенная для обработки металлических подложек, содержащая:
a) композицию очистителя любого из объектов 1 - 10;
b) необязательно активирующий ополаскиватель, предназначенный для обработки по меньшей мере одного участка подложки; и
c) композицию для предварительной обработки, предназначенную для обработки по меньшей мере одного участка подложки и содержащую фосфат металла.
20. Система объекта 19, в которой активирующий ополаскиватель содержит дисперсию частиц фосфата металла, имеющих размер частиц D90 не более 10 мкм, при том, что фосфат металла содержит двухвалентные или трехвалентные металлы, или их комбинации.
21. Система объекта 19, в которой активирующий ополаскиватель содержит дисперсию частиц фосфата металла, имеющих размер частиц D90 не более 1 мкм, при том, что фосфат металла содержит двухвалентные или трехвалентные металлы, или их комбинации.
22. Система объекта 19, в которой активирующий ополаскиватель содержит коллоидные частицы фосфата титана.
23. Система любого из объектов 19 - 22, в которой активирующий ополаскиватель дополнительно содержит сульфатную соль металла, причем металл сульфатной соли металла содержит никель, медь, цинк, железо, магний, кобальт, алюминий или их комбинации.
24. Система любого из объектов 19 - 23, в которой композиция для предварительной обработки по существу не содержит никеля.
25. Система любого из объектов 19 - 24, дополнительно содержащая по меньшей мере одну композицию из:
второй композиции для предварительной обработки, предназначенной для обработки по меньшей мере одного участка подложки и содержащей катион металла Группы IIIB и/или Группы IVB;
композиции для ополаскивания после обработки; и
композиции для нанесения электроосаждаемых покрытий, предназначенной для нанесения покрытия на по меньшей мере один участок подложки.
26. Подложка, подвергнутая обработке системой обработки любого из объектов 19 - 25.
Примеры
Пример 1. Цинк-фосфатная композиция для предварительной обработки
Ванны с щелочным очистителем
Не содержащие фосфатов ванны с щелочным очистителем были приготовлены следующим образом.
Стандартная ванна с щелочным очистителем была приготовлена при 1,25 об.% концентрации Chemkleen 2010LP (бесфосфатный щелочной очиститель, выпускаемый PPG) и 0,125% Chemkleen 181 ALP (бесфосфатная смешанная поверхностно-активная добавка, предлагаемая PPG). Была приготовлена предназначенная для струйной очистки ванна объемом 10 галлонов. Для очистки погружением была приготовлена ванна объемом 5 галлонов. Ванны готовились на деионизированной воде. Показатель pH каждой ванны равнялся 12.
Модифицированный очиститель №1. К пяти галлонам указанной выше стандартной ванны щелочного очистителя было добавлено 28,5 г натриевой соли D-глюконовой кислоты (производства Sigma Aldrich Corporation), 17,7 г гексагидрата нитрата кобальта (предлагается Fisher Scientific, Inc) и 23,5 г нитрата железа (выпускается Sigma Aldrich Corporation). Расчетные концентрации кобальта и железа в этой ванне составляли 190 ч./млн. и 172 ч./млн., соответственно. Показатель pH полученной ванны равнялся 11,8.
Модифицированный очиститель №2. К пяти галлонам указанной выше стандартной ванны щелочного очистителя было добавлено 28,5 г натриевой соли D-глюконовой кислоты (производства Sigma Aldrich Corporation) и 47,0 г нитрата железа (выпускается Sigma Aldrich Corporation). Расчетная концентрация железа в этой ванне отвечала 344 ч./млн. Показатель pH полученной ванны равнялся 11,8.
Приготовление активатора
Активирующий ополаскиватель на основе фосфата цинка был приготовлен следующим образом: 4717,6 г состоящего из фосфата цинка пигмента было просеяно в предварительно смешанную смесь из 1700,18 г деионизированной воды, 1735,92 г диспергирующего вещества (Disperbyk-190, предлагается на рынке BYK-Chemie GmbH) и 56,36 г противовспенивающего компонента (BYK-011, предлагается BYK-Chemie GmbH) и смешивалось в течение 30 минут с помощью смесительного устройства Fawcett Air Mixer, модель LS - 103A, с наклонным зубом/стальной лопастью Коулса типа 1. Были добавлены и перемешивались еще в течение 20 минут дополнительные 680,47 г деионизированной воды, а также 2590,25 г DisperbyK-190 и 55,5 г BYK-011. Затем эта смесь была подвергнута измельчению при пропускании в режиме рециркуляции через горизонтальную мельницу HM 1.5 L (производство Premier Mill Corp.), содержащую 0,5 мм наполнитель из оксида циркония, со временем пребывания 60 минут. На протяжении процесса измельчения были добавлены дополнительные 1180 г деионизированной воды. В ходе измельчения было отобрано несколько промежуточных технологических проб, так что конечный выход составил 10 455,62 г. Этот материал имел концентрацию фосфата цинка 36,78 масс.%. Конечный продукт имел средневзвешенный размер частиц D90, равный 0,26 мкм (ультразвуковая обработка). В данном контексте термин «размер частиц D90» относится к объемно-массовому распределению частиц, при котором 90% частиц имеют диаметр, меньший, чем величина D90. Согласно настоящему изобретению, размер частиц измерялся с помощью устройства Mastersizer 2000 производства Malvern Instruments, Ltd., Malvern, Worcestershire, Великобритания. Mastersizer 2000 направляет луч лазера (диаметр 0,633 мм, длина волны 633 нм) через дисперсию частиц (в дистиллированной, деионизированной или фильтрованной воде со степенью помутнения 2 - 3%) и оценивает рассеяние света дисперсии (параметры измерения 25°C, 2200 об/мин, задержка перед измерением 30 с, 10 с фоновых измерений, 10 с измерений на образце). Количество света, рассеянного дисперсией, обратно пропорционально размеру частиц. Рассеянный свет измеряется последовательностью детекторов и данные затем анализируются программным обеспечением (программное обеспечение Malvern Mastersizer 2000, версия 5.60) для построения распределения частиц по размерам, из которого стандартным образом может определяться размер частиц.
Ванна для активирующего ополаскивания была приготовлена посредством добавления по 1,36 г указанной выше дисперсии фосфата цинка на 1 литр деионизированной воды с тем, чтобы получить ванну для активации с концентрацией фосфата цинка 0,5 г на литр.
Приготовление ванны с фосфатом цинка Chemfos 700LT
Емкость объемом в 5 галлонов была заполнена приблизительно на три четверти деионизированной водой. К ней было добавлено 760 мл Chemfos 700A, 1,5 мл Chemfos FE и 42 мл Chemfos AFL (все – производства PPG). К этому было добавлено 29,5 г нитрата цинка (выпускаемого Fischer Scientific), 9,5 мл Chemfos F (предлагаемого на рынке компанией PPG), 136,8 мл Zetaphos N (предлагаемого PPG) и 130 мл раствора NaOH (5% (отношение массы к объему) NaOH производства Fisher Scientific, растворенного в деионизированной воде). Свободная кислота ванны использовалась при содержании, соответствовавшем 0,7 - 0,8 пунктам свободной кислоты, 15,8 - 16,0 пунктам общей кислоты и 2,6 - 2,7 газовым пунктам нитрита. Количество нитрита в растворе измерялось с помощью бродильной трубки, используя протокол, описанный в листе технической информации для Chemfos Liquid Additive (PPG Industries, Inc, Cleveland, OH). Бродильная трубка была заполнена на 70 мл образцом, взятым из ванны с композицией для предварительной обработки, до уровня чуть ниже устья трубки. В трубку было добавлено приблизительно 2,0 г сульфаминовой кислоты и трубка была перевернута для смешивания сульфаминовой кислоты и раствора композиции для предварительной обработки. Произошло выделение газа, который вытеснял жидкость к верхней части бродильной трубки, уровень подъема жидкости отслеживался и регистрировался. Данный уровень соответствовал газовым пунктам, измеренным в растворе в миллилитрах.
Приготовление испытательной пластины
Для каждой пробы прежде всего были очищены следующим образом по две пластины из стали горячего цинкования (4” x 6”, производство ACT Test Panels, LLC): контрольные пластины были подвергнуты струйной очистке в окрасочной камере из нержавеющей стали с помощью дающих V-образную струю форсунок при давлении 10 - 15 фунт/кв.дюйм, используя подробно описанный выше раствор стандартного щелочного очистителя в течение двух минут при 49°C, что сопровождалось ополаскиванием при погружении в неионизированную (DI) воду на 15 секунд и струйной промывкой DI-водой в течение 15 секунд. Комплекты испытательных пластин №1 и №2 были очищены в подробно описанной выше ванне стандартного щелочного очистителя в течение 30 секунд при 49°C, затем немедленно погружались в модифицированный очиститель №1 или №2, соответственно, на две минуты при 49°C, что сопровождалось ополаскиванием при погружении на 15 секунд в DI-воду и струйной промывкой DI-водой в течение 15 секунд. Затем пластины были погружены на одну минуту в вышеописанную активирующую ванну (20°C - 25°C). Далее пластины погружались на две минуты в ванну с Chemfos 700 LT при 30°C или 35°C, с перемешиванием. Все пластины затем были подвергнуты струйной промывке DI-водой в течение 20 - 30 секунд. Пластины были высушены теплым воздухом с помощью ручного высокоскоростного фена производства Oster® (модель номер 078302-300-000) в режиме высокой фиксации при температуре около 50 - 55°C до тех пор, пока пластина не становилась сухой (около 1 - 5 минут).
После высыхания пластины были подвергнуты электроосаждению EPIC 200, катодного электропокрытия, предлагаемого PPG Industries. Электропокрытие наносилось до достижения целевой толщины в 0,69 мил. Выпрямитель (Модель Xantrex XFR600-2) был установлен в режим “Coulomb Controlled” (контроль по заряду). Задавались условия, отвечавшие 27 кулон, отсутствию ограничений по силе тока, номинальному напряжения 220 В и времени нарастания тока 30 с. Ванна для электроосаждения поддерживалась при 90°F с перемешиванием со скоростью 340 об/мин. После нанесения электропокрытия пластины были подвергнуты термической обработке в сушильном шкафу (Despatch Model LFD-1-42) при 177°C в течение 25 минут. Толщина покрытия измерялась с помощью измерителя толщины лакокрасочной пленки (Fischer Technology Inc., модель FMP40C).
Испытания пластин
Пластины были проверены на адгезию с помощью способа испытаний "T- peel” для измерения энергии разрушения, требующейся для отрыва покрытия от подложки.
Покрытия на пластинах сначала были приведены в напряженное состояние посредством подвергания их в течение 24 часов вымачиванию в DI-воде при 60°C. После извлечения пластин из водяной бани они оставлялись для восстановления на срок от двух до трех часов. Пластины с электроосажденным покрытием затем разрезались вдоль на четыре полосы с размерами 1” x 6”. Были приготовлены образцы для испытаний на сопротивление отслаиванию посредством прежде всего отгибания одного конца каждой полосы на 90°; далее испытываемые поверхности были очищены изопропиловым спиртом и подвергнуты плазменной обработке (Diener Electronic модель ATTO B с насосом Duo 2.5) в течение 5 минут (после откачки давления до 0,17 мбар при подаче газообразного N2 в течение 1 минуты до плазменной обработки). Затем пары пластин из каждого комплекта были соединены друг с другом с помощью быстроотверждающегося клея (3M Scotch-Weld DP 460) так, чтобы получить Т-образное соединение. Клеевое соединение перед испытанием оставлялось для отверждения под условиями окружающей среды на 24 часа. Испытание проводилось посредством разделения соединения с помощью разрывной машины Instron 5567 при номинальной скорости движения траверсы 250 мм/мин. Энергия разрушения GC вычислялась по формуле
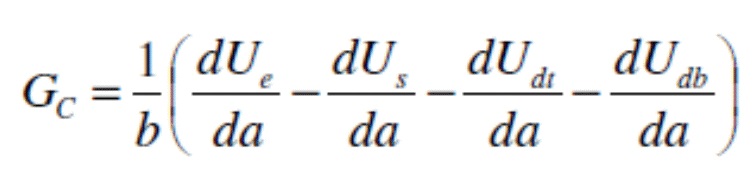
где a – длина трещины, b - ширина, dUe – потенциальная энергия внешней нагрузки, dUs – энергия деформации, запасенная в подложке, dUdt – энергия, рассеянная в виде деформации растяжения, и dUdb – энергия, рассеянная при пластическом изгибе подложки. Результаты представлены в нижеследующей Таблице 2.
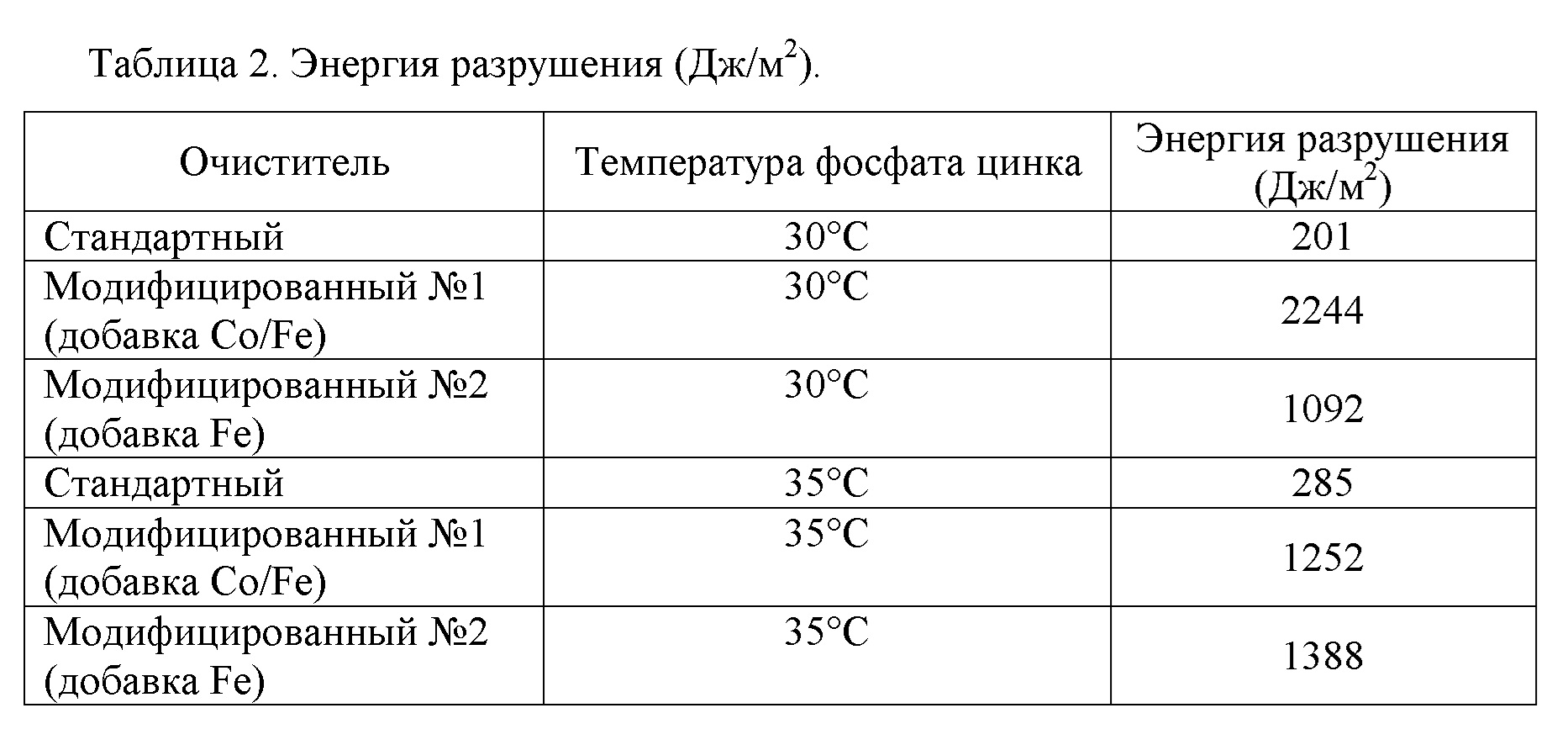
Пластины, обработанные железосодержащими очистителями, продемонстрировали значительно более высокие величины энергии разрушения, что указывает на значительно лучшую адгезию.
Пример 2. Предварительная обработка цирконием
Ванны с щелочным очистителем
Ванны с щелочным очистителем были идентичны применявшимся в Примере 1.
Композиция для предварительной обработки цирконием (Zircobond 1.5)
Было приготовлено согласно инструкциям изготовителя пять галлонов раствора ZircoBond 1.5 ((“ZB 1.5”) – содержащей цирконий композиции для предварительной обработки, коммерчески доступной в PPG Industries, Inc). Раствор имел pH 4,72 и содержал 175 ч./млн. циркония, 30 ч./млн. меди и 101 ч./млн. свободного фторида.
Приготовление испытательной пластины
Для каждой пробы прежде всего были очищены следующим образом по две пластины из стали горячего цинкования (4” x 6”, производство ACT Test Panels, LLC). Пластины, предназначенные для проведения испытаний со стандартным щелочным очистителем, а также модифицированными очистителями №1 и №2, были подвергнуты струйной очистке с использованием подробного описанного выше раствора стандартного щелочного очистителя в течение 30 секунд при 49°C, затем немедленно погружались в стандартный щелочной очиститель, модифицированный очиститель №1, или модифицированный очиститель №2, соответственно, на две минуты при 49°C, что сопровождалось ополаскиванием при погружении на 15 секунд в DI-воду и струйной промывкой DI-водой в течение 15 секунд. Пластины были затем погружены на две минуты в ванну композиции для предварительной обработки цирконием, с перемешиванием. Все пластины затем были подвергнуты струйной промывке DI-водой в течение 20 - 30 секунд. Пластины были высушены теплым воздухом с помощью ручного высокоскоростного фена производства Oster® (модель номер 078302-300-000) в режиме высокой фиксации при температуре около 50 - 55°C до тех пор, пока пластина не становилась сухой (около 1 - 5 минут).
После высыхания пластины были подвергнуты электроосаждению катодного электропокрытия EPIC 200. Электропокрытие наносилось до достижения целевой толщины в 0,66 - 0,72 мил. Выпрямитель (Sorensen производства Ametek, модель XG300-5.6) был установлен в режим “Coulomb Controlled”. Задавались условия, отвечавшие 24 кулонам, отсутствию ограничений по силе тока, номинальному напряжению 200 В и времени нарастания тока 30 с. Ванна для электроосаждения поддерживалась при 90°F с перемешиванием со скоростью 340 об/мин. После нанесения электропокрытия пластины были подвергнуты термической обработке в сушильном шкафу (Despatch Model LFD-1-42) при 177°C в течение 25 минут. Толщина покрытия измерялась с помощью измерителя толщины лакокрасочной пленки (Fischer Technology Inc., модель FMP40C).
Испытания пластин
Покрытые пластины были протестированы, используя ту же самую процедуру, которая описана в Примере 1. Результаты представлены в нижеследующей Таблице 3.
Таблица 3. Энергия разрушения (Дж/м2)
Пример 3. Предварительная обработка цирконием (Zb 1.5) с модифицированными железом очистителями, улучшающими коррозионную характеристику.
Ванны с щелочным очистителем
Ванны с щелочным очистителем были идентичны применявшимся в Примере 1.
Контрольная предварительная обработка фосфатом цинка
Был приготовлен активатор добавлением 1,1 г/л Versabond RC (также известный как RC30, предлагаемый на рынке PPG Industries, Inc) к емкости с залитыми в нее пятью галлонами (18,79 литров) деионизированной воды, предназначенный для применения непосредственно перед применением описанной ниже цинк-фосфатной ванны.
Объединением перечисленных ниже ингредиентов было приготовлено 1 500 г раствора Chemfos 700A/AL/M – концентрата цинк-фосфатной композиции.

Была приготовлена цинк-фосфатная композиция для предварительной обработки Chemfos 700A/AL/M заполнением пятигаллоной емкости деионизированной водой приблизительно на три четверти. К ней было добавлено 756 г упомянутого выше контрольного концентрированного раствора CF700A/AL/M, 56,7 г CF-F (PPG), 122,85 г CF-F/F (PPG) 15,4 г CF-AZN (PPG), 321,3 г Buffer M (PPG) и 8,5 г AAO (Sigma - Aldrich). Концентрации свободной и общей кислоты были отрегулированы с помощью Buffer M (предлагаемого на рынке PPG Industries, Inc) так, чтобы достигнуть показателей содержания свободной кислоты в 0,85 пунктов и общей кислоты в 17,2 пунктов. Было добавлено 35 ч./млн. раствора пероксида водорода (35% Alfa Aesar), применяемого в качестве ускорителя.
Предварительная обработка цирконием (ZircoBond 1.5).
Было приготовлено согласно инструкциям изготовителя пять галлонов раствора ZircoBond 1.5 ((“ZB 1.5”) – содержащей цирконий композиции для предварительной обработки, коммерчески доступной в PPG Industries, Inc). Раствор имел pH 4,73 и содержал 175 ч./млн. циркония, 30 ч./млн. меди и 107 ч./млн. свободного фторида.
Приготовление испытательной пластины для предварительной обработки на основе циркония.
Испытательные пластины были приготовлены с помощью того же способа, который был описан в отношении предварительно обработанных цирконием пластин из Примера 2.
Приготовление испытательных пластин для контрольной предварительной цинк-фосфатной обработки.
Для каждой пробы прежде всего были очищены следующим образом по две пластины из стали горячего цинкования (4” x 6”, производство ACT Test Panels, LLC). Испытательные пластины подвергались струйной очистке с использованием раствора стандартного щелочного очистителя в течение 30 секунд при 49°C, затем немедленно погружались в стандартный щелочной очиститель на две минуты при 49°C, что сопровождалось ополаскиванием при погружении на 15 секунд в DI-воду и струйной промывкой DI-водой в течение 15 секунд. Пластины затем вносились в Versabond RC (активирующая ванна) на одну минуту при температуре ванны 80°F. Далее пластины непосредственно помещались на три минуты в подробно описанный выше раствор цинк-фосфатной ванны, находящейся при 50°C при перемешивании. В завершение пластины были подвергнуты струйной промывке DI-водой в течение 20 - 30 секунд. Пластины были высушены теплым воздухом с помощью ручного высокоскоростного фена производства Oster® (модель номер 078302-300-000) в режиме высокой фиксации при температуре около 50 - 55°C до тех пор, пока пластина не становилась сухой (около 1 - 5 минут).
Электропокрытие.
После высушивания на все подвергнутые предварительной обработке цирконием и фосфатом цинка пластины способом электроосаждения было нанесено катодное электропокрытие EnviroPrime 2010. Электропокрытие наносилось до достижения целевой толщины в 0,71 - 0,86 мил. После нанесения электропокрытия пластины были подвергнуты термической обработке в сушильном шкафу (Despatch Model LFD-1-42) при 177°C в течение 25 минут. Толщина покрытия измерялась с помощью измерителя толщины лакокрасочной пленки (Fischer Technology Inc., модель FMP40C).
Испытания пластин
Пластины были проверены на образование вздутий вследствие распространения коррозии после скрайбирования ("scribe creep”) с помощью циклического коррозионного испытания GMW 14872. Было измерено распространение коррозии после скрайбирования от повреждения краски в месте нанесения риски до повреждения краски с левой и правой стороны от царапины. Риска наносилась на пластину перед ее помещением в климатическую камеру на срок длительностью в 30 циклов. Показатели распространения коррозии при скрайбировании для каждого испытания показаны в нижеследующей Таблице 4.
Измерения распространения коррозии при скрайбировании – использовалось среднее по результатам испытаний двух пластин. Длина риски равнялась 4 дюймам (10,16 см). Данные по ширине повреждения краски снимались в 8 точках вдоль по риске. Из этих усредненных по двум пластинам результатам были получены данные по распространению коррозии при скрайбировании, отмеченные ниже в Таблице 4. Кроме того, было измерено максимальное среднее распространения коррозии при скрайбировании. Это означает, что была измерена максимальная величина распространения коррозии при скрайбировании по всей длине риски – на расстоянии слева от риски и справа от риски. Максимальные величины с левой стороны и максимальные величины с правой стороны были добавлены, чтобы получить показатель максимального распространения коррозии при скрайбировании. Средний показатель по этим двум пластинам показан в таблице ниже в виде максимального среднего распространения коррозии при скрайбировании. Измерения были сделаны с помощью штангенциркуля с цифровой индикацией Fowler Sylvac, модель S 235.

Пример 4. Предварительная обработка на основе циркония
Ванны с щелочным очистителем
Ванны с щелочным очистителем были идентичны применявшимся в Примере 1.
Контрольная предварительная обработка фосфатом цинка
Был приготовлен активатор добавлением 1,1 г/л Versabond RC (также известный как RC30, предлагаемый на рынке PPG Industries, Inc) к емкости с залитыми в нее пятью галлонами (18,79 литров) деионизированной воды, предназначенный для применения непосредственно перед применением описанной ниже цинк-фосфатной ванны.
Согласно инструкциям изготовителя была приготовлена ванна для предварительной обработки фосфатом цинка Chemfos 700AL (CF 700AL) посредством заполнения пятигаллонной емкости деионизированной водой приблизительно на три четверти. К ней было добавлено 700 мл Chemfos 700A, 1,5 мл Chemfos FE, 51 мл Chemfos AFL и 350 мл Chemfos 700B (все производства PPG). К этому было добавлено 28,6 г гексагидрата нитрата цинка и 2,5 г нитрита натрия (оба производства Fischer Scientific), а концентрация свободной кислоты ванны регулировалась так, чтобы соответствовать 0,7 - 0,8 пунктам свободной кислоты, 15 - 19 пунктам общей кислоты и 2,2 - 2,7 газовым пунктам нитрита натрия. В ходе регулирования в целях получения надлежащих количеств свободной и общей кислоты использовался Chemfos 700B способом, соответствовавшим информации в листе технической информации .
Температура ванны равнялась 125°F и пластины погружались в ванну на 2 минуты.
Предварительная обработка цирконием (ZircoBond 2.0)
Было приготовлено согласно инструкциям изготовителя пять галлонов раствора ZircoBond 2.0 (содержащей цирконий композиции для предварительной обработки, коммерчески доступной в PPG Industries, Inc). Раствор имел pH 4,6 и содержал 175 ч./млн. циркония, 30 ч./млн. меди, 5 ч./млн. лития, 85 ч./млн. молибдена и 85 ч./млн. свободного фторида.
Приготовление испытательной пластины для предварительной обработки цирконием.
Для каждого испытания прежде всего были следующим образом очищены по две горячеоцинкованные, не имеющие покрытия пластины производства ACT Test Panels Code (HDG60G60E).
Пластины помещались на две минуты в описанные выше щелочные чистящие растворы при температуре 120°F. Затем пластины были ополоснуты деионизированной водой, вначале ополаскиванием при погружении на 10 - 15 секунд, а затем струйным ополаскиванием в течение 15 - 30 секунд с использованием деионизированной воды.
После очистки в соответствующих очищающих растворах пластины помещались для выполнения испытаний 1 - 3 в циркониевые ванны на срок в две минуты при слабом перемешивании. Температура поддерживалась на уровне 80°F. Сразу после завершения обработки в ваннах с ZircoBond 2.0 пластины в течение 20 - 30 секунд обмывались струйным ополаскиванием DI-водой. Пластины были высушены теплым воздухом с помощью ручного высокоскоростного фена производства Oster® (модель номер 078302-300-000) в режиме высокой фиксации при температуре около 50 - 55°C до тех пор, пока пластина не становилась сухой (около 1 - 5 минут). Для испытания №4 пластины были очищены и ополоснуты таким же образом, как и перед этапом предварительной обработки. Затем пластины на одну минуту при температуре окружающей среды помещались в раствор Versabond Rinse Conditioner. Из него пластины напрямую перемещались в ванну CF700AL, где выдерживались в течение двух минут при температуре 125°F. После этого пластины были ополоснуты и высушены, как описано выше.
Приготовление испытательных пластин для контрольной предварительной обработки фосфатом цинка.
Для каждой пробы прежде всего были очищены следующим образом по две пластины из стали горячего цинкования (4” x 6”, производство ACT Test Panels, LLC). Испытательные пластины подвергались струйной очистке с использованием раствора стандартного щелочного очистителя в течение двух минут при 49°C, затем немедленно погружались в стандартный щелочной очиститель на две минуты при 49°C, что сопровождалось ополаскиванием при погружении на 15 секунд в DI-воду и струйной промывкой DI-водой в течение 15 секунд. Пластины затем вносились в Versabond RC (активирующая ванна) на одну минуту при температуре ванны 80°F. Далее пластины непосредственно помещались на три минуты в подробно описанный выше раствор цинк-фосфатной ванны, находящейся при 50°C при перемешивании. В завершение пластины были подвергнуты струйной промывке DI-водой в течение 20 - 30 секунд. Пластины были высушены теплым воздухом с помощью ручного высокоскоростного фена производства Oster® (модель номер 078302-300-000) в режиме высокой фиксации при температуре около 50 - 55°C до тех пор, пока пластина не становилась сухой (около 1 - 5 минут).
Электропокрытие
После высыхания панели были подвергнуты электроосаждению предлагаемого PPG Industries электропокрытия ED7200. Электропокрытие наносилось до достижения целевой толщины в 0,60 мил. Выпрямитель (Модель Xantrex XFR600-2) был установлен в режим “Coulomb Controlled”. Заданные параметры были следующими: 20 кулон, ограничение в 0,5 ампер, номинальное напряжение 220 В в случае композиции для предварительной обработки с фосфатом цинка и 180 В в случае композиции на основе циркония, время нарастания тока 30 с. В ходе электроосаждения поддерживалась температура 90°F и скорость перемешивания 360 об/мин. После нанесения электропокрытия пластины были подвергнуты термической обработке в сушильном шкафу (Despatch Model LFD-1-42) при 177°C в течение 25 минут. Толщина покрытия измерялась с помощью измерителя толщины лакокрасочной пленки (Fischer Technology Inc., модель FMP40C).
Испытания пластин
Пластины были проверены на образование вздутий вследствие распространения коррозии после скрайбирования с помощью Honda Salt Dip для измерения распространения коррозии при скрайбировании. Было измерено распространение коррозии при скрайбировании от повреждения краски в месте нанесения риски до повреждения краски с левой и правой стороны от риски. Х-образная риска наносилась на пластину перед ее помещением во влажную камеру на срок длительностью в 15 дней. Иммерсионный раствор представлял собой 5% раствор соли NaCl при температуре 55°C.
После испытания и сушки при комнатной температуре на риску была наклеена клейкая лента Scotch Brand 898 Fiber Tape и оторвана для снятия продуктов коррозии и всей отслоившейся краски вдоль царапины с тем, чтобы сделать возможным выполнение оценки повреждения краски с левой и правой стороны от риски.
Измерение распространения коррозии при скрайбировании – длина риски составляла по 8 см для каждой из ножек литеры X. Данные по расстоянию между участками повреждения краски снимались через каждый сантиметр за исключением области, отстоящей на один сантиметр от точки пересечения ножек литеры Х. Тем самым это позволило получить в общей сложности 14 точек измерения для обеих (правой и левой) ножек риски X. Из этих усредненных данных по обеим ножкам риски X были получены средние показатели по распространению коррозии при скрайбировании для трех пластин, показанные ниже в Таблице 5. Измерения были сделаны с помощью штангенциркуля с цифровой индикацией Fowler Sylvac, модель S 235.
Таблица 5
Пример 5. Предварительная обработка фосфатом цинка
Ванны с щелочным очистителем
Ванны с щелочным очистителем были идентичны применявшимся в Примере 1.
Активатор №1
Versabond Rinse Conditioner (коммерческий продукт PPG). Ванна для активирующего ополаскивания была приготовлена посредством добавления 1,1 г Versabond RC на 1 литр деионизированной воды с тем, чтобы получить ванну для активации с концентрацией фосфата цинка 0,5 г на литр.
Активатор №2
Активирующий ополаскиватель на основе фосфата цинка был приготовлен следующим образом: 4717,6 г состоящего из фосфата цинка пигмента было просеяно в предварительно смешанную смесь из 1700,18 г деионизированной воды, 1735,92 г диспергирующего вещества (DisperbyK-190, предлагается на рынке BYK-Chemie GmbH) и 56,36 г противовспенивающего компонента (BYK-011, предлагается BYK-Chemie GmbH) и смешивалось в течение 30 минут с помощью смесительного устройства Fawcett Air Mixer, модель LS-103A, с наклонным зубом/стальной лопастью Коулса типа 1. Были добавлены и перемешивались еще в течение 20 минут дополнительные 680,47 г деионизированной воды, а также 2590,25 г DisperbyK-190 и 55,5 г BYK-011. Затем эта смесь была подвергнута измельчению при пропускании в режиме рециркуляции через горизонтальную мельницу HM 1.5 L (производство Premier Mill Corp.), содержащую 0,5 мм наполнитель из оксида циркония, со временем пребывания 60 минут. На протяжении процесса измельчения были добавлены дополнительные 1180 г деионизированной воды. В ходе измельчения было отобрано несколько промежуточных технологических проб, так что конечный выход составил 10 455,62 г. Этот материал имел концентрацию фосфата цинка 36,78 масс.%. Конечный продукт имел средневзвешенный размер частиц D90, равный 0,26 мкм (ультразвуковая обработка).
Ванна для активирующего ополаскивания была приготовлена посредством добавления 1,36 г указанной выше дисперсии фосфата цинка на 1 литр деионизированной воды с тем, чтобы получить ванну для активации с концентрацией фосфата цинка 0,5 г на литр.
Приготовление концентрата фосфата цинка Chemfos 700A/AL/M (контроль)
Было приготовлено 1 500 г концентрированного раствора объединением следующих ингредиентов:

Цинк-фосфатная ванна №1: приготовление фосфата цинка Chemfos 700A/AL/M (контроль).
Цинк-фосфатная Chemfos 700A/AL/M ванна была приготовлена следующим образом. Емкость объемом в 5 галлонов была заполнена приблизительно на три четверти деионизированной водой. К ней было добавлено 756 г упомянутого выше контрольного концентрированного раствора CF700A/AL/M, 56,7 г CF-F (PPG), 122,85 г CF-F/F (PPG) 15,4 г CF-AZN (PPG), 321,3 г Buffer M (PPG) и 8,5 г AAO (Sigma - Aldrich). Свободная кислота и общая кислота были отрегулированы с помощью Buffer М таким образом, чтобы достичь содержания свободной кислоты, соответствующего 0,85 газовым пунктам, и общей кислоты в 17,2 газовых пунктов. Было добавлено 35 ч./млн. пероксида водорода (35% раствор Alfa Aesar), применяемого в качестве ускорителя.
Приготовление концентрата фосфата цинка Chemfos 700A/AL/M (отсутствие никеля).
Был приготовлен не содержащий никеля концентрат фосфата цинка Chemfos 700A/AL/M объединением тех же самых, упомянутых выше ингредиентов за исключением того, что раствор нитрата никеля был заменен равным количеством деионизированной воды.
Цинк-фосфатная ванна №2: приготовление фосфата цинка Chemfos 700A/AL/M (отсутствие никеля).
Цинк-фосфатная Chemfos 700A/AL/M (отсутствие никеля) ванна была приготовлена следующим образом. Емкость объемом в 5 галлонов была заполнена приблизительно на три четверти деионизированной водой. К ней было добавлено 756 г упомянутого выше, не содержащего никеля концентрированного раствора CF700A/AL/M, 56,7 г CF-F (PPG), 122,85 г CF-F/F (PPG) 15,4 г CF-AZN (PPG), 321,3 г Buffer M (PPG) и 8,5 г AAO (Sigma - Aldrich). Свободная кислота и общая кислота были отрегулированы с помощью Buffer М таким образом, чтобы достичь содержания свободной кислоты, соответствующего 0,75 газовым пунктам, и общей кислоты в 17,3 газовых пункта. Было добавлено 35 ч./млн. пероксида водорода (35% раствор Alfa Aesar), применяемого в качестве ускорителя.
Матрица испытания
Приготовление испытательной пластины
Для каждой пробы прежде всего были очищены следующим образом по две пластины с нанесенным на них электролитическим цинкованием покрытием MBZE 8 Steel (предлагается Chemetall): контрольные пластины были подвергнуты струйной очистке в окрасочной камере из нержавеющей стали с помощью дающих V-образную струю форсунок при давлении 10 - 15 фунт/кв.дюйм, используя подробно описанный выше стандартный раствор Chemkleen 2010LP в течение двух минут при 49°C, что сопровождалось ополаскиванием при погружении в DI-воду на 15 секунд и струйной промывкой DI-водой в течение 15 секунд. Комплекты испытательных пластин №3 и №4 были очищены в подробно описанной стандартной ванне Chemkleen 2010LP в течение 30 секунд при 49°C, затем немедленно погружались в модифицированный очиститель №1 или №2, соответственно, на две минуты при 49°C, что сопровождалось ополаскиванием при погружении на 15 секунд в DI-воду и струйной промывкой DI-водой в течение 15 секунд. Затем пластины погружались на одну минуту в вышеописанную активирующую ванну №1 или №2 (20°C - 25°C). Далее пластины были погружены на три минуты в контрольную или в не содержащую никеля фосфатно-цинковую ванну, находящуюся при 50°C с перемешиванием. Все пластины затем были подвергнуты струйной промывке DI-водой в течение 20 - 30 секунд. Пластины были высушены теплым воздухом с помощью ручного высокоскоростного фена производства Oster® (модель номер 078302-300-000) в режиме высокой фиксации при температуре около 50 - 55°C до тех пор, пока пластина не становилась сухой (около 1 - 5 минут).
После высыхания панели были подвергнуты электроосаждению EnviroPrime 2010, катодного электропокрытия, предлагаемого PPG Industries. Электропокрытие наносилось до достижения целевой толщины в 0,785 мил. Выпрямитель (Модель Xantrex XFR600-2) был установлен в режим “Coulomb Controlled”. Условия задавались так, чтобы отрегулировать количество кулонов (25 - 35°C) с заданием ограничения в 0,75 А при номинальном напряжении 210 В и времени нарастания тока 30 с. В ходе электроосаждения поддерживалась температура 32,78°C и скорость перемешивания 340 об/мин. После нанесения электропокрытия пластины были подвергнуты термической обработке в сушильном шкафу (Despatch Model LFD-1-42) при 175°C в течение 25 минут. Толщина покрытия измерялась с помощью измерителя толщины лакокрасочной пленки (Fischer Technology Inc., модель FMP40C).
После получения электропокрытия с помощью Spraymation были нанесены B1:B2 порошковая окрасочная система и прозрачный слой. Коды продуктов, диапазоны толщины сухой пленки и условия выполнения термической обработки показаны в таблице ниже. Все краски коммерчески доступны в PPG.
Как только на пластины были полностью нанесены поверхностное покрытие и прозрачный слой, пластины были процарапаны на разметочном столе Corrocutter 639 (производство Erichsen) с помощью 1 мм чертилки Sikkens в двух местах по поверхности пластины и помещены на 12 недель в климатическую камеру для циклических испытаний VDA 233 - 102. Результаты представлены ниже в Таблице 6. Результаты отображены в виде U/2 (среднее распространения коррозии при скрайбировании) = (d - 1)/2, где d – среднее общего расслаивания на царапине (после отрыва пленки).
Таблица 6.
Пример 6. Добавление Мо к щелочному очистителю с металлом Группы IVb
Приготовление щелочного очистителя I. Чистая пятигаллоная емкость была наполнена 18,93 литрами деионизированной воды. К ней был добавлен 250 мл Chemkleen 2010LP (бесфосфатный щелочной очиститель, поставляемый PPG Industries, Inc) и 25 мл Chemkleen 181ALP (бесфосфатная смешанная поверхностно-активная добавка, предлагаемая в продаже PPG Industries, Inc). Образец щелочного очистителя объемом 10 мл титровался 0,100 Н серной кислотой, чтобы измерить свободную и общую щелочность. Свободная щелочность была измерена по конечной точке фенолфталеина (изменение от розового к бесцветному), а общая щелочность была измерена по конечной точке бромкрезолового зеленого (цветовой переход от голубого к желтому). Измеренная величина pH щелочного очистителя равнялась 12,0.
Приготовление щелочного очистителя II. Этот очиститель был приготовлен таким же способом, как и щелочной очиститель I за исключением гексагидрата нитрата кобальта (17,70 г), приобретенного у Thermofisher Acros Organics (Gel, Бельгия) и D-глюконата натрия (28,50 г), приобретенного у Thermofisher Acros Organics (Geel, Бельгия), которые были добавлены к смеси CK2010LP и CK181ALP. Показатель pH раствора с помощью гидроксида натрия был доведен до 12,0. Была вычислена концентрация Co, которая составила 189 ч./млн. по отношению к общей массе композиции.
Приготовление щелочного очистителя III. Этот очиститель был приготовлен из щелочного очистителя II добавлением нонагидрата нитрата железа (23,53 г), предлагаемого на рынке Fisher Scientific (Hampton, NH). Показатель pH раствора с помощью гидроксида натрия был доведен до 12,0. Вычисленная концентрация Co составила 189 ч./млн., а вычисленная концентрация железа равнялась 172 ч./млн. по отношению к общей массе композиции.
Приготовление щелочного очистителя IV. Этот очиститель был приготовлен таким же способом, как и щелочной очиститель I за исключением 9,50 г дигидрата молибдата натрия, поставляемого Thermofisher Acros Organics (Geel, Бельгия), и 28,50 г d-глюконата натрия, которые были добавлены к смеси CK2010LP и CK181ALP. Показатель pH раствора с помощью гидроксида натрия был доведен до 12,0. Рассчитанная концентрация Мо составила 199 ч./млн. по отношению к общей массе композиции.
Приготовление щелочного очистителя V. Этот очиститель был приготовлен из щелочного очистителя IV добавлением нонагидрата нитрата железа (23,53 г), предлагаемого на рынке Fisher Scientific (Hampton, NH). Показатель pH раствора с помощью гидроксида натрия был доведен до 12,0. Вычисленная концентрация Mo составила 199 ч./млн., а вычисленная концентрация железа равнялась 172 ч./млн. по отношению к общей массе композиции.
Приготовление щелочного очистителя VI. Этот очиститель был приготовлен таким же способом, как и щелочной очиститель I за исключением 3,50 г дигидрата молибдата натрия, поставляемого Thermofisher Acros Organics (Geel, Бельгия), и 28,50 г d-глюконата натрия, которые были добавлены к смеси CK2010LP и CK181ALP. Показатель pH раствора с помощью гидроксида натрия был доведен до 12,0. Рассчитанная концентрация Мо составила 73 ч./млн. по отношению к общей массе композиции.
Общий мониторинг ванн для предварительной обработки. После добавления к ванне композиции для предварительной обработки всех ингредиентов был измерен показатель pH ванны с помощью pH-метра (интерфейсный, DualStar pH/ISE двухканальный настольный измеритель производства Therm°Fisher Scientific, Waltham, Massachusetts, США с pH-электродом Fisher Scientific Accumet (Ag/AgCl электрод сравнения) посредством погружения pH-электрода в раствор композиции для предварительной обработки. Содержание свободного фторида измерялось с помощью DualStar pH/ISE двухканального настольного измерителя (Therm°Fisher Scientific), оснащенного фторид-селективным электродом (Orion ISE Fluoride Electrode, твердотельный, выпускаемый Therm°Fisher Scientific), посредством погружения ISE в раствор для предварительной обработки и позволяя датчику прийти в равновесное состояние. Далее pH при необходимости был доведен до указанного диапазона pH с помощью буфера Chemfil (щелочной буферирующий раствор, коммерчески доступный в PPG Industries, Inc, или гексафторциркониевой кислоты (45 масс.% в воде, доступна в Honeywell International, Inc, Morristown, NJ). Содержание свободного фторида при необходимости приводилось в диапазон 25 - 150 ч./млн. с помощью Chemfos AFL (частично нейтрализованный водный раствор гидродифторида аммония, коммерчески доступный в PPG Industries, Inc и приготавливаемый согласно инструкциям поставщика). Количество меди в каждой ванне измерялось с помощью колориметра DR/890 (выпускается HACH, Loveland, Colorado, США) и при использовании индикатора (CuVer1 Copper Reagent Powder Pillows, предлагается HACH).
Приготовление ванны A. Ванна A была приготовлена согласно инструкциям изготовителя для Zircobond 1.5. Цирконий вводился в ванны для предварительной обработки посредством добавления гексафторциркониевой кислоты (45 масс.% в воде), предлагаемой Honeywell International, Inc (Morristown, NJ), а медь вносилась добавлением 2 масс.% раствора Cu, который был приготовлен разбавлением раствора нитрата меди (18 масс.% Cu в воде), производимого Shepherd Chemical Company (Cincinnati, OH). Ванна для предварительной обработки была приготовлена в пятигаллоной емкости (объем 18,93 литров). Величины pH и содержания свободного фторида были отрегулированы с помощью 39,0 г буфера Chemfil и 21,0 г Chemfos AFL, соответственно. Конечные параметры ванны были: pH 5,1, Cu 34 ч./млн., Zr 200 ч./млн. и свободные фториды 92 ч./млн.
Приготовление ванны B. Ванна B была приготовлена согласно инструкциям изготовителя для Zircobond 1.5 с использованием материалов, описанных в ходе приготовления ванны A. Ванна для предварительной обработки была приготовлена в пятигаллоной емкости (объем 18,93 литров). Величины pH и содержания свободного фторида были отрегулированы с помощью 47,0 г буфера Chemfil и 15,0 г Chemfos AFL, соответственно. Конечные параметры ванны были: pH 4,8, Cu 34 ч./млн., Zr 200 ч./млн. и свободные фториды 93 ч./млн.
Оцинкованные горячим способом стальные пластины (Gardobond MBZ1/EA, 105 мм x 190 мм x 0,75 мм, смазанные, без обработки, производство Chemetall) перед применением щелочного очистителя были разрезаны пополам с получением пластин размерами 5,25 см x 9,5 см.
Пластины были обработаны с применением способов обработки A либо B, представленных в Таблицах 7 и 8 ниже. В случае пластин, обработанных согласно способу обработки A, такие пластины были очищены погружением, обезжирены в течение 120 секунд в пятигаллонной емкости 125°F) и ополоснуты деионизированной водой при погружении на 30 секунд в ванну с деионизированной водой (75°F), что сопровождалось струйным ополаскиванием деионизированной водой с помощью описанной выше форсунки (75°F) в течение 30 секунд. Все пластины на 120 секунд (80°F) погружались в композицию для предварительной обработки с металлом Группы IVB, ополаскивались струйным ополаскиванием деионизированной водой (75°F) в течение 30 секунд и высушивались в течение 120 секунд горячим воздухом (140°F) с помощью высокоскоростного ручного фена производства Oster® (номер модели 078302-300-000) в режиме высокой фиксации.
В случае пластин, обработанных согласно способу обработки B, пластины были очищены, предварительно обработаны и ополоснуты, как при Способе A, за исключением того, что щелочной очиститель был модифицирован добавлением Fe, Co, и/или Мо.
Таблица 7. Способ обработки A
Таблица 8. Способ обработки B
После окончания выполнения способов обработки A или B все пластины были подвергнуты электроосаждению покрытия ED7000Z (катодное электропокрытие с компонентами, коммерчески доступными в PPG), приготовленного смешиванием смолы E6433Z (51,0 масс.%), пасты E6434Z (8,9 масс.%) и деионизированной воды (40,1 масс.%). Краска была подвергнута ультрафильтрации с удалением 25% материала, который был восполнен свежей деионизированной водой. В качестве источника постоянного тока применялся выпрямитель (Xantrax, модель XFR600-2, Elkhart, Indiana или Sorensen XG 300-5.6, Ameteck, Berwyn, Pennsylvania). Условия нанесения электропокрытия были следующими: номинальное напряжение 180 - 200 В, время нарастания тока 30 с и плотность тока 1,6 мА/см2. Температура электропокрытия поддерживалась на уровне 90°F. Толщина пленки была регулируемой по времени с целевой толщиной осаждаемой пленки 0,6 +0,1 мил. Показатель DFT контролировался изменением величины заряда (кулоны), проходящего через пластины. После осаждения электропокрытия пластины были подвергнуты термической обработке в сушильном шкафу (Despatch Model LFD-1-42) при 177°C в течение 25 минут. Затем пластины были подвергнуты испытаниям на стойкость к коррозии или же на них наносилось поверхностное покрытие перед выполнением испытаний на силу адгезии.
На пластины с электропокрытием были нанесены риски в виде вертикальной линии в середине пластины, углубляющейся к металлической (стальной) подложке. Пластины с нанесенными рисками были подвергнуты GM циклическим коррозионным испытаниям GMW14872. Пластины были подвергнуты струйной обработке со средой-наполнителем (MB-2, пластмассовые частицы неправильной формы с твердостью по шкале Мооса 3,5 и размерами в диапазоне 0,58 - 0,84 мм, выпускаемые Maxi-Blast, Inc, South Bend, Indiana) при помощи линейной конвейерной системы пескоструйной обработки IL-885 (входящее давление воздуха 85 фунт/кв.дюйм, Empire Abrasive Equipment Company, модель IL885 - M9655) после выполнения испытания на стойкость к коррозии с тем, чтобы удалить слабо держащиеся продукты коррозии и краску. Испытания пластин выполнялись троекратно для каждого набора условий. Усредненные по трем пластинам данные по распространению коррозии при скрайбировании показаны в Таблице 10 ниже. Распространение коррозии при скрайбировании относится к площади потери краски вокруг риски, возникающей или вследствие коррозии, либо из-за отслаивания (например, от поврежденной краски до поврежденной краски).
Также на пластины с электропокрытием (не подвергавшиеся испытаниям на коррозионную устойчивость) был нанесен белый поверхностный слой. Такой поверхностный слой доступен в PPG Industries, Inc в виде трехкомпонентной системы, составленной из грунтовочного покрытия, первого слоя двухслойного покрытия и покровного лака. Коды продуктов, диапазоны толщины сухой пленки и условия выполнения термической обработки показаны в Таблице 9 ниже.
Таблица 9. Трехкомпонентная система покрытия
Далее была исследована адгезия краски к пластинам, обработанным согласно каждому из способов обработки A и B, в сухих (защищенных) и влажных (незащищенных) условиях. Были проверены три пластины и в Таблице 10 представлена средняя величина адгезии для защищенных и незащищенных условий. Для испытаний адгезии в сухих условиях использовалось лезвие, которым наносилось одиннадцать линий, параллельных и перпендикулярных длине одной из пластин с электропокрытием. Образовавшаяся сетка из процарапанных линий представляла собой квадрат от 0,5” x 0,5” до 0,75” х 0,75”. Сухая адгезия оценивалась с помощью ленты 3M Fiber 898, которая плотно прижималась к процарапанной сетке пальцем с последующим многократным прижимающим разглаживанием ленты по пластине перед выполнением отрыва от ее поверхности. Площадь штриховки была оценена в отношении потери краски по шкале от 0 до 10 баллов, где 0 соответствовал полной потере краски, а 10-ю баллами оценивалось полное отсутствие какой-либо потери краски (см. ниже). В автомобильной промышленности приемлемой считается величина адгезии 8. Для испытания адгезии в условиях воздействия после нанесения поверхностного покрытия пластина была на десять дней погружена в деионизированную воду (40°C), после чего пластины были извлечены, вытерты полотенцем для осушения и оставлены на сорок пять минут при температуре окружающей среды перед нанесением на нее штриховки и отрыванием ленты для оценки адгезии краски, как описано выше.
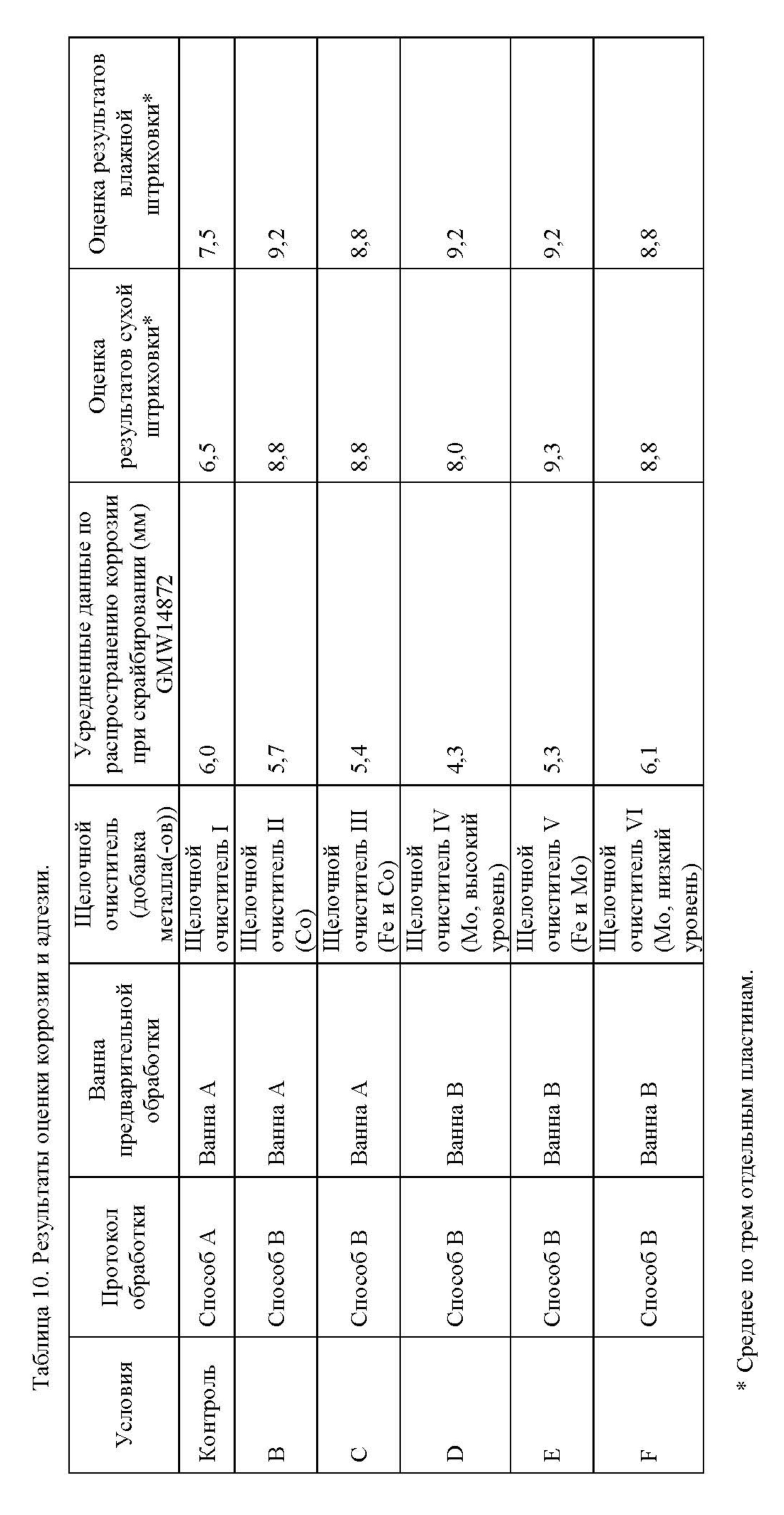
Шкала оценок, используемая в Примере 6, следующим образом выглядела в Таблице 11 и ограничивалась высокой оценкой, соответствовавшей сильной адгезии между поверхностью подложки, пленкой композиции для предварительной обработки и слоем органического покрытия (например, электропокрытия, поверхностного покрытия или порошкового покрытия).
Таблица 11. Штриховка, описание оценки.
Испытание на незащищенной штриховке представляет собой важный критерий, поскольку слабая адгезия на штриховке указывает на наличие слабости внутри пакета автомобильного покрытия. Это особенно важно в случае подложек HDG (оцинкованные горячим способом), где адгезия краски является проблемным вопросом. Связанные с адгезией сложности дополнительно усиливаются, поскольку внешняя обшивка автомобильных конструкций часто бывает именно HDG, так как она обеспечивает превосходную коррозионную устойчивость.
Эти данные демонстрируют, что применение композиции очистителя настоящего изобретения значительно улучшило противокоррозионные свойства и адгезию по сравнению с контролем. Очиститель, содержащий молибден в высоких концентрациях или молибден + железо, продемонстрировал более высокую эффективность при его использовании с композицией для предварительной обработки, содержащей металл Группы IVB, наглядно показывая, что молибден способен заменить кобальт в щелочном очистителе в отношении повышения эффективности, а также исключить потенциальные проблемы соблюдения нормативно-правового регулирования, связанные с применением кобальта.
Пример 7
Ванны с щелочным очистителем
Ванны с щелочным очистителем были идентичны применявшимся в Примере 1.
Предварительная обработка цирконием. Была приготовлена согласно инструкциям изготовителя содержащая Zircobond Zb4200 Dm (доступен в PPG Industries) ванна для предварительной обработки цирконием. Раствор имел pH 4,5 и содержал 190 ч./млн. циркония, 20 ч./млн. меди и 75 ч./млн. свободного фторида. Температура ванны составляла 80°F и, когда пластины пропускались через ванну, это производилось в течение 2 минут при слабом перемешивании погружным смесительным устройством (Poly Science Sous Vide).
Предварительная обработка фосфатом железа. Chemfos 158 был приготовлен следующим образом. К 10-галлоной ванне с водопроводной водой было добавлено 4 об.% (1,5 литра) концентрата Chemfos 158 (предлагается на рынке PPG). Ванна находилась в распылительном резервуаре из нержавеющей стали, предназначенном для применений, связанных с предварительной обработкой и очисткой. Показатель pH ванны с помощью буфера ChemFil был доведен до величины 5,06. В режиме эксплуатации температура ванны равнялась 140°F, а продолжительность использования составляла 90 секунд. Помимо этого, данный резервуар имел ряд V-образных форсунок, предназначенных для целей нанесения растворов, а уровень прикладываемого давления отвечал 15 - 20 фунт/кв.дюйм.
Приготовление пластины
Каждый набор из 3 оцинкованных горячим способом пластин ACT (категория подвергаемых воздействиям) 60G60U был очищен в одной из указанных выше композиций очистителя в течение 2 минут при температуре 120°F, пластины ополаскивались в ванне с деионизированной водой в течение 10 - 15 секунд, что сопровождалось струйным ополаскиванием с помощью 7 переключаемых форсунок, установленных в режим «ливень». Затем пластины поступали в описанные выше ванны предварительной обработки одного из двух типов. После этапа предварительной обработки пластины были вновь подвергнуты струйному ополаскиванию в течение 10 - 15 секунд, а в завершение пластины были высушены теплым воздухом с помощью высокоскоростного ручного фена производства Oster® (модель номер 078302-300-000) в режиме высокой фиксации при температуре около 50 - 55°C до тех пор, пока пластина не становилась сухой (около 1 - 5 минут). Данные наборы были следующими:
набор 1 – стандартный Chemkleen 2010 – ZB4200;
набор 2 – стандартный Chemkleen 2010 – Chemfos 158;
набор 3 – модифицированный очиститель 1 – Chemfos 158;
набор 4 – модифицированный очиститель 2 – Chemfos 158.
После высыхания пластины были подвергнуты электроосаждению катодного электропокрытия 1K AdvantEdge Industrial, предлагаемого PPG Industries. Электропокрытие наносилось до достижения целевой толщины в 1,00 мил. Выпрямитель (Xantrex модель XFR600-2) был установлен в режим “Time Controlled” (контроль по времени). Условия задавались так, чтобы отрегулировать время (90 секунд) с заданием ограничения в 0,5 А при номинальном напряжении 200 В и времени нарастания тока 30 секунд. В ходе электроосаждения поддерживалась температура 90°F и скорость перемешивания 340 об/мин. После нанесения электропокрытия пластины были подвергнуты термической обработке в сушильном шкафу (Despatch Model LFD-1-42) при 160°C в течение 30 минут. Толщина покрытия измерялась с помощью измерителя толщины лакокрасочной пленки (Fischer Technology Inc., модель FMP40C).
На пластины с электропокрытием были нанесены риски в виде 10,2 см вертикальной линии в середине пластины, углубляющейся к металлической подложке. Пластины с нанесенными рисками были подвергнуты GM циклическим коррозионным испытаниям GMW14872 длительностью 30 дней. Пластины были подвергнуты струйной обработке со средой-наполнителем (MB-2, пластмассовые частицы неправильной формы с твердостью по шкале Мооса 3,5 и размерами в диапазоне 0,58 - 0,84 мм, выпускаются Maxi-Blast, Inc, South Bend, Indiana) при помощи линейной конвейерной системы пескоструйной обработки IL-885 (входящее давление воздуха в 85 фунт/кв.дюйм регулировалось до 40 фунт/кв.дюйм для применения Empire Abrasive Equipment Company, данные модели: IL885 - M9655) после выполнения испытания на стойкость к коррозии с тем, чтобы удалить слабо удерживаемые продукты коррозии и краску. Были сняты показания по расстоянию между поврежденными участками краски, измерявшиеся через каждый сантиметр царапины и обеспечившие в общей сложности 10 точек измерения. Из этих данных брались средние значения по трем пластинам с тем, чтобы вычислить средний показатель распространения коррозии при скрайбировании, величины которого представлены в Таблице 12 ниже. Измерения были сделаны с помощью штангенциркуля с цифровой индикацией Fowler Sylvac, модель S 235.
Таблица 12
Пример 8
Приготовление ванны щелочного очистителя
Не содержащие фосфатов ванны с щелочным очистителем были приготовлены следующим образом.
Была приготовлена стандартная ванна с Chemkleen 2010LP при 1,25 об.% концентрации Chemkleen 2010LP (бесфосфатный щелочной очиститель, выпускаемый PPG) и 0,125% Chemkleen 181 ALP (бесфосфатная смешанная поверхностно-активная добавка, предлагаемая PPG). Была приготовлена пятигаллонная ванна с деионизированной водой.
Модифицированные очистители. Для каждого модифицированного очистителя была приготовлена пятигаллонная ванна Chemkleen 2010LP так же, как описывается выше. К этим ваннам было добавлено 28,5 г натриевой соли D-глюконовой кислоты (производства Sigma Aldrich Corporation), 17,7 г гексагидрата нитрата кобальта (предлагается Fisher Scientific, Inc) и 23,5 г нитрата железа (выпускается Sigma - Aldrich Corporation). Расчетные концентрации кобальта и железа в этих ваннах составляли 190 ч./млн. и 172 ч./млн., соответственно. Затем добавлены различные фосфонатные материалы, подробно представленные в нижеследующей таблице.
Приготовление активатора
Ванна для активирующего ополаскивания была приготовлена посредством добавления по 1,36 г описанной выше дисперсии фосфата цинка на 1 литр деионизированной воды с тем, чтобы получить ванну для активации с концентрацией фосфата цинка 0,5 г на литр.
Приготовление цинк-фосфатной ванны
Был приготовлен не содержащий никеля концентрат фосфата цинка посредством смешивания следующих ингредиентов в представленном порядке и при тщательном перемешивании до прозрачности раствора:
Этот концентрат фосфата цинка использовался для приготовления цинк-фосфатной ванны посредством разбавления до концентрации 3,7 об.% деионизированной водой и регулирования кислотности до показателя свободной кислотности в 0,7 - 0,9 газовых пунктов с помощью состава Chemfos Make-up B, предлагаемого PPG Industries. Был добавлен нитрит натрия для поддержания величины 2,3 – 2,7 газовых пункта.
Предварительная обработка цирконием
К 5 галлонам деионизированной воды было добавлено 16,75 граммов 45% гексафторциркониевой кислоты (предлагается на рынке Honeywell International Inc), 30 граммов 2 масс.% раствора меди (приготовлен растворением в деионизированной воде гемипентагидрата нитрата меди, предлагаемого Fisher Scientific) и 15,4 граммов Chemfos AFL (предлагается PPG). С помощью Chemfil Buffer (доступен в PPG) ванна была отрегулирована так, чтобы эксплуатироваться в диапазоне pH от 4,5 до 5,0.
Приготовление испытательной пластины
Для каждой пробы прежде всего были очищены следующим образом по две пластины из стали горячего цинкования (4” x 6”, производство ACT Test Panels, LLC). Испытательные пластины погружались в ванну с выбранным очистителем на две минуты при 49°C, что сопровождалось ополаскиванием при погружении в DI-воде в течение 15 секунд и струйным ополаскиванием DI-водой еще в течение 15 секунд. На этой стадии пластины для предварительной обработки цинк-фосфатной композицией были на 60 секунд погружены в раствор активатора при температуре окружающей среды, а затем немедленно погружены на 120 секунд в фосфатную ванну при 52°C. Для выполнения предварительной обработки цирконием пластины на две минуты при 27°C погружались в ванну для предварительной обработки цирконием. После предварительной обработки (фосфатом цинка или цирконием) все пластины затем подвергались струйному ополаскиванию DI-водой в течение 20 - 30 секунд. Пластины были высушены теплым воздухом с помощью высокоскоростного ручного фена производства Oster® (модель номер 078302-300-000) в режиме высокой фиксации при температуре около 50 - 55°C до тех пор, пока пластина не становилась сухой (около 1 - 5 минут).
После высыхания пластины были подвергнуты электроосаждению катодного электропокрытия EPIC 200. Электропокрытие наносилось до достижения целевой толщины в 0,66 - 0,72 мил. Выпрямитель (Sorensen производства Ametek, модель XG300-5.6) был установлен в режим “Coulomb Controlled”. Задавались условия, отвечавшие 24 кулонам при отсутствии ограничений по силе тока с номинальным напряжения 220 В при OVP (Over Voltage Protection – защита от перенапряжения) 300 В и времени нарастания тока 30 с. Раствор для электроосаждения нагревался до 90°F при скорости перемешивания 340 об/мин. После нанесения электропокрытия пластины были подвергнуты термической обработке в сушильном шкафу (Despatch Model LFD-1-42) при 177°C в течение 25 минут. Толщина покрытия измерялась с помощью измерителя толщины лакокрасочной пленки (Fischer Technology Inc., модель FMP40C).
Испытания пластин
Пластины с нанесенным на них электропокрытием были разрезаны вдоль на полосы размерами 15 мм x 110 мм. Были приготовлены образцы для испытаний на сопротивление отслаиванию посредством прежде всего отгибания одного конца каждой полосы на 90°; далее испытываемые поверхности были очищены изопропиловым спиртом и подвергнуты плазменной обработке (Diener Electronic модель ATTO B с насосом Duo 2.5) в течение 5 минут (после откачки давления до 0,17 мбар при подаче газообразного N2 в течение 1 минуты до плазменной обработки). Затем пары пластин из каждого комплекта были соединены друг с другом с помощью быстроотверждающегося клея (3M Scotch-Weld DP 460) так, чтобы получить Т-образное соединение. Клеевое соединение перед испытанием оставлялось для отверждения под условиями окружающей среды на 24 часа. Испытание проводилось посредством разделения соединения с помощью разрывной машины Instron 5567 при номинальной скорости движения траверсы 250 мм/мин. Энергия разрушения GC вычислялась по формуле
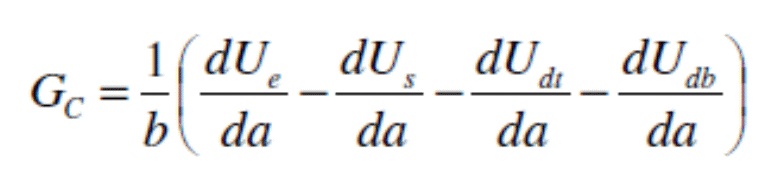
где a – длина трещины, b – ширина, dUe – потенциальная энергия внешней нагрузки, dUs – энергия деформации, запасенная в подложке, dUdt – энергия, рассеянная в деформации растяжения, и dUdb – энергия, рассеянная в пластическом изгибе подложки. Результаты представлены в следующей далее таблице.
Специалистам в данной области очевидно, что без отступления от общей идеи изобретения, описанной и иллюстрируемой здесь, в свете приведенного выше раскрытия возможно внесение многочисленных модификаций и изменений. Поэтому, соответственно, следует понимать, что предшествующее раскрытие является лишь иллюстративным в отношении различных представленных в качестве примеров объектов этой заявки и что специалистами в данной области могут быть легко сделаны многочисленные модификации и изменения, которые находятся в пределах сущности и объема этой заявки и сопутствующей формулы изобретения. Специалистам в данной области очевидно, что без отступления от общей идеи изобретения, описанной и иллюстрируемой здесь, в свете приведенного выше раскрытия возможно внесение многочисленных модификаций и изменений. Поэтому, соответственно, следует понимать, что предшествующее раскрытие является лишь иллюстративным в отношении различных представленных в качестве примеров объектов этой заявки и что специалистами в данной области могут быть легко сделаны многочисленные модификации и изменения, которые находятся в пределах сущности и объема этой заявки и сопутствующей формулы изобретения.
Реферат
Изобретение относится к водной композиции щелочного очистителя для обработки металлической подложки. Водная щелочная композиция содержит катион железа и катион молибдена в количестве от 10 до 400 ч./млн. по отношению к общей массе композиции и щелочной компонент, причем показатель pH композиции равен по меньшей мере 10 и она включает не более 50 ч./млн. фосфата по отношению к общей массе композиции. Также предложены системы обработки, содержащие водную щелочную композицию, предназначенную для обработки по меньшей мере одного участка подложки, и композиция для предварительной обработки, предназначенная для обработки по меньшей мере одного участка подложки. Предложены подложки, обработанные в соответствии с раскрываемыми системами обработки. Железо из водной щелочной композиции изобретения осаждается на поверхности металлов в виде металлического железа и обеспечивает повышение эффективности композиции для предварительной обработки, улучшение защиты от коррозии и положительно влияет на адгезию. 6 н. и 13 з.п. ф-лы, 14 табл., 8 пр.
Формула
Документы, цитированные в отчёте о поиске
Композиция для подготовки поверхности, способ приготовления указанной композиции и способ подготовки поверхности
Комментарии