Анелированные 9-гидрокси-1,8-диоксо-1,3,4,8-тетрагидро-2н-пиридо[1,2-a]пиразин-7-карбоксамиды - ингибиторы интегразы вич, способы их получения и применения - RU2717101C1
Код документа: RU2717101C1
Чертежи
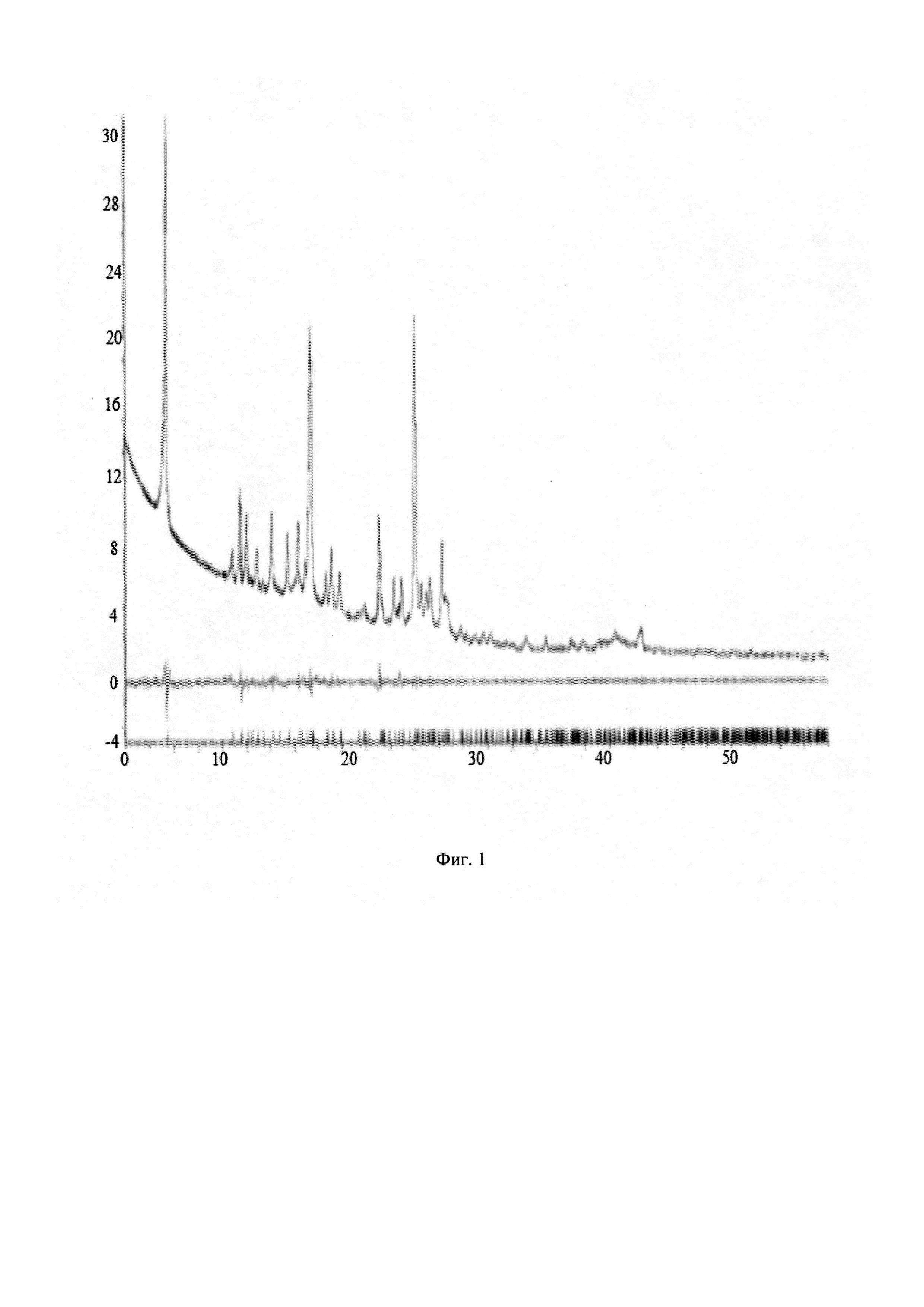

Описание
Настоящее изобретение относится к новому соединению, обладающему противовирусной активностью, в частности ингибиторной активностью в отношении интегразы вируса иммунодефицита человека (ВИЧ).
ВИЧ это ретровирус из рода лентивирусов, вызывающий медленно прогрессирующее заболевание - ВИЧ-инфекцию [Weiss R.A. How does HIV cause AIDS. Science 1993, 260 (5112), 1273-1279. Douek D.C., Roederer M., Koup R.A. Emerging Concepts in the Immunopathogenesis of AID». Annu. Rev. Med. 2009, 60, 471-84]. Вирус иммунодефицита человека независимо открыли в 1983 году в двух лабораториях: в Институте Пастера во Франции под руководством Люка Монтанье и в Национальном институте рака в США под руководством Роберта Галло. Результаты исследований, в которых из тканей пациентов с симптомами СПИДа впервые удалось выделить новый ретровирус, были опубликованы 20 мая 1983 года в журнале Science [

ВИЧ поражает клетки иммунной системы, имеющие на своей поверхности рецепторы CD4: Т-хелперы, моноциты, макрофаги, клетки Лангерганса, дендритные клетки, клетки микроглии. В результате работа иммунной системы угнетается и развивается синдром приобретенного иммунного дефицита (СПИД), организм больного теряет возможность защищаться от инфекций и опухолей, возникают вторичные оппортунистические заболевания, которые не характерны для людей с нормальным иммунным статусом. Без врачебного вмешательства ВИЧ вызывает смерть пациента в среднем через 9-11 лет после заражения (в зависимости от подтипа вируса) [https://ru.wikipedia.org/wiki/Вирус_иммунодефицита_человека].
Согласно глобальной статистики [http://www.lenoblspid.ru/news24/postid/own_news/1166] в 2015 г во всем мире: жили с ВИЧ 36,7 миллиона человек, 2,1 миллиона человек были инфицированы ВИЧ, 1,1 миллиона человек умерли от болезней, обусловленных СПИДом, 78 миллиона человек были инфицированы ВИЧ с момента начала эпидемии из них 35 миллиона человек умерли от болезней, обусловленных СПИДом, с момента начала эпидемии.
ВИЧ передается между людьми посредством обмена жидкостями организма, такими как кровь, сперма, ректальная и вагинальная жидкости и грудное молоко. Он не передается через слюну.
ВИЧ можно ослаблять с помощью комбинированной антиретровирусной терапии (APT), состоящей из нескольких антиретровирусных препаратов с различным механизмами действия. Антиретровирусная терапия (APT) не излечивает эти инфекции, но контролирует репликацию вирусов в организме человека и содействует укреплению иммунной системы и восстановлению ее способностей бороться с инфекциями. При проведении APT, с использованием комбинаций однокомпонентных и/или двухкомпонентных препаратов, продолжительность жизни пациента может быть продлена до 70-80 лет [http://www.who.int/mediacentre/factsheets/fs360/ru/].
Антиретровирусные препараты (АРП) делятся на нуклеозидные ингибиторы обратной транскриптазы (NRTIs), ненуклеозидные ингибиторы обратной транскриптазы (NNRTIs), ингибиторы слияния или входа (fusion inhibitors), капсидные ингибиторы, ингибиторы протеазы (PIs) и ингибиторы интегразы (INIs),.
Примерами однокомпонентных АРП, могут служить Элсульфавирин, VM-1500A [WO 2005/102989, RU 2389719, WO 2010/028968], Рилпивирин [https://pubchem.ncbi.nlm.nih.gov/compound/Rilpivirine#section=Top; https://pubchem.ncbi.nlm.nih.gov/compound/5270790#section=Top] и Эфавиренз [https://pubchem.ncbi.nlm.nih.gov/compound/efavirenz]? которые являются NNRTIs; Ламивудин [https://pubchem.ncbi.nlm.nih.gov/compound/lamivudine#section=2D-Structure], Эмтрицитабин [https://pubchem.ncbi.nlm.nih.gov/compound/Emtricitabine#section=2D-Structure] и их проингибиторы [RU 2659388] - NRTIs; Тенофовир диизопроксил фумарат (Viread®) [https://pubchem.ncbi.nlm.nih.gov/compound/Tenofovir_Disoproxil_Fumarate], Тенофовир алафенамид полуфумарат (TAF, GS-7340, Vemlidy) [https://pubchem.ncbi.nlm.nih.gov/compound/71492247], Тенофовир циклобутилал афенамид (ТЦБА) и ТЦБА фумарат [RU 2647576] - NtRTIs; Элвитегравир [https://pubchem.ncbi.nlm.nih.gov/compound/Elvitegravir] - INI; GS-CA1 (GS-6207) [WO 2018035359; http://www.natap.org/2018/IDWeek/IDWeek_03.htm] - капсидный ингибитор; и Кобицистат [https://pubchem.ncbi.nlm.nih.gov/compound/Cobicistat], который не проявляет противовирусную активность, но является фармакинетическим усиливающим агентом, ингибитором цитохрома Р450 3А (CYP3A).
В последние годы заметное внимание уделяется INIs, в качестве препаратов для комбинированной APT, включающей одновременное использование нескольких АРП с разными механизмами действия. В том числе, для APT длительного подавления ВИЧ (Antiretroviral Therapy as Long Acting Suppression, ATLAS).
В 2007 году администрация США по контролю за продуктами и лекарствами (FDA) одобрила использование INIs ВИЧ в качестве препаратов для APT [https://www.healthline.com/health/hiv-aids/integrase-inhibitors#hiv].
Первым зарегистрированным FDA (Октябрь, 2007) INI препаратом для APT оказался Ралтегравир, разработанный компанией Мэрк [https://aidsinfo.nih.gov/news/803/fda-approves-the-first-integrase-inhibitor-raltegravir-October-12-2007], который был запатентован компанией Шионоги в Европе [ЕР 1422218 (2004]. Позже компания Шионоги запатентовала Каботегравир и Долутегравир [WO 2006/116764 US 8129385 (2012), US 8778943 (2015), ЕР 3260457(2017)].

Позже были запатентованы Биктегравир [WO 2014/100323], INIs A1, A2 [ЕР 3196201] и INIs A3 [WO 2016161382]

где A1, A2: Q - необязательно замещенный карбо- или гетероцикл; А и D - необязательно замещенные гетероциклы; R3 и R8 - независимо водород, галоген, гидрокси, необязательно замещенный низший алкил и другие заместители; R14 и Rx - независимо водород, необязательно замещенный низший алкил и другие заместители.
A3: А - необязательно замещенный 3-7 членный циклоалкил или частично ненасыщенный гетероцикл; А1 представляет собой 5-7 членный насыщенный или необязательно замещенный 4-7 членный моноциклический гетероциклил; R1n представляют собой необязательно одинаковые галоген или С1-С3 алкил; n=1-3; R2 представляет собой необязательно одинаковые Н или С1-С4 алкил.
В настоящее время CAB, DTG и BIC являются наиболее продвинутыми INIs ВИЧ. В зависимости от метода анализа ингибирующая активность CAB имеет значение ЕС50 = 0,25-3,0 нМa, b, DTG - ЕС50 = 1,1-7,4 нМa-c и BIC - EC50 = 1,0-7,5 нМb, c [aHassounah S.A. et al. AAC 2017, 61(12), pii: e01695-17.bYoshinaga T. ey al. AAC 2015, 59(1), 397-406; https://aac.asm.org/content/aac/59/1/397full.pdf; https://aac.asm.org/content/61/12/e01695-17.long.cTsiang M. et al. AAC 2016, 60(12), 7086-7097; https://aac.asm.org/content/60/12/7086].
Долутогравир (торговая марка Тивикай) был одобрен FDA в августе 2013 г. [https://www.drugs.com/history/tivicay.html], а в ноябре 2017 г FDA одобрил препарат Джулука [https://www.drugs.com/history/juluca.html], представляющий собой фиксированную комбинацию 52,6 мг натриевой соли Долутогравира и о 27.5 мг гидрохлорида Рилпивирина [https://www.gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Juluca/pdf/JULUCA-PI-PIL.PDF].
Биктегравир входит в состав препарата Биктарви, представляющего собой фиксированную комбинацию (50 мг биктегравира, 200 мг эмтрицитабина и 25 мг тенофовир алафенамид) [http://www.gilead.com/~/media/files/pdfs/medicines/hiv/biktarvy/biktarvy_pi.pdf]. Биктарви был одобрен FDA в 2018 г. [https://www.google.com/search?q=biktarvy+fda+approval+date&rlz=1C1GGRV_enUS751US751&oq=biktarvy+FDA&aqs=chrome.2.0j69i57j0l2.20836j0j4&sourceid=chrome&ie=UTF-8].
Каботегравир активно изучается в клинике в форме таблетки для перорального приема внутрь и в инъекционной наносуспензии длительного действия, вводимая в мышцу (известная как каботегравир LA или CAB LA (LA означает «Long-acting или долго действующий») [https://aidsinfo.nih.gov/drugs/513/cabotegravir/0/patient. Trezza C. et al. Formulation and pharmacology of long-acting cabotegravir. Curr. Opin. HIV AIDS. 2015, 10(4), 239-245. T.D. McPherson et al. Cabotegravir in the treatment and prevention of Human Immunodeficiency Virus-1. Expert Opin Investig Drugs. 2018, 27(4), 413-420. C.D. Andrews et al. Cabotegravir long acting injection protects macaques against intravenous challenge with SIVmac251. AIDS 2017, 31(4), 461-467.].
Каботегравир в настоящее время активно исследуется в качестве препарата в клинических фазах III ATLAS (NCT02951052), FLAIR (NCT02938520), ATLAS-2M (NCT03299049) и ACTG A5359 (NCT03635788) [https://aidsinfo.nih.gov/drugs/513/cabotegravir/0/patient].
Каботегравир и ингибиторы А1-А3 еще не одобрены FDA.
Несмотря на достигнутые в последние годы результаты по созданию в ряду INIs лекарственных препаратов для комбинированной APT, остается актуальным поиск новых препаратов такого типа с улучшенными характеристиками.
Ниже приведены определения различных терминов, используемых для описания данного изобретения. Эти определения применимы к терминам, как они использованы в данном описании и формуле изобретения, если иным не ограничены в конкретных случаях либо по отдельности, либо как часть большей группы.
Термин «ВИЧ-инфекция» - заболевание, вызываемое ретровирусом, приводящее к прогрессирующему иммунодефициту (СПИДу) и характеризующееся присоединением в терминальной фазе оппортунистических заболеваний.
Термин «СПИД - ассоциированный комплекс (САК) означает раннюю симптомную стадию ВИЧ. Эта стадия связана с риском развития оппортунистических инфекций. Клиническая манифестация САК сопровождается появлением конституциональных симптомов: лихорадка, профузные ночные поты, снижение массы тела на 10% и более, прогрессирующая слабость. Характерны появление дерматологических симптомов, поражение слизистой оболочки полости рта, рецидивирующая герпетическая инфекция, рецидивирующий кожно-слизистый кандидоз. Часто присоединяются заболевания верхних дыхательных путей (синуситы, бронхиты, пневмонии), воспалительные заболевания органов малого таза, дисплазия шейки матки, периферическая нейропатия.
Термин «СПИД» означает синдром приобретенного иммунодефицита или поздняя симптомная стадия. Длительности инфекционного процесса в течение 7-10 лет. В ряде случаев заболевание развивается быстрее и уже через 2-3 года переходит в терминальную стадию. Эту стадию характеризуют тяжелые, угрожаемые жизни инфекции и злокачественные новообразования, которые имеют генерализованную форму. Поражения органов и систем у больных носят необратимое течение.
Термин «галоген» или «гало» означает фтор, бром, хлор и йод.
Термин "необязательно" означает, что впоследствии описанное событие или обстоятельство может произойти или может не произойти и что описание включает случаи, когда указанное событие или обстоятельство происходит, и случаи, в которых не происходит.
Термин «кристаллическая форма» означает структуру вещества, характеризующуюся упаковкой образующих ее молекул в один из видов кристаллической решетки.
Термин «поликристаллическая форма» означает структуру вещества, имеющую поликристаллическое строение, т.е. состоящую из множества мелких монокристаллов, т.е. кристаллитов определенной кристаллической формы.
Термин «сольват» означает продукты присоединения растворителя к растворенным веществам. Частный случай сольватов - гидраты (растворитель - вода). Обычно сольваты образуются в растворе, но нередко (при охлаждении раствора, испарении растворителя и др.). Сольваты могут быть получены в виде кристаллических фаз - кристаллосольватов.
Термин «стериоизомеры» (пространственные изомеры) - химические соединения, имеющие одинаковое строение, но отличающиеся пространственным расположением атомов. Стереоизомеры, представляющие собой зеркальные отражения друг друга, не совмещаемые в пространстве, называются энантиомерами или оптическими изомерами. Оптическая изомерия характерна для соединений, молекулы которых имеют элементы хиральности, например асимметрический (хиральный) атом углерода, связанный с четырьмя разными заместителями. Впервые обнаружена Л. Пастером в 1848 на примере винных кислот и объяснена Я.X. Вант-Гоффом и Ж.А. Ле Белем в 1874 на основе представлений о тетраэдрич. конфигурации углеродных атомов в насыщенных соединениях. Молекулы, содержащие асимметрич. атом углерода, могут быть представлены в виде двух оптич. изомеров, которые не могут быть совмещены в пространстве (т.е. относятся друг к другу как предмет к своему зеркальному изображению). Такие зеркальные изомеры, отличающиеся лишь противоположным расположением одних и тех же заместителей у хирального центра, называются энантиомерами. Энантиомеры, как правило, имеют различную биологическую активность; для них характерна также оптическая активность - способность воздействовать на плоскополяризованный свет (вращать плоскость поляризации). Энантиомеры вращают плоскость поляризации на один и тот же угол, но в противоположном направлении, поэтому их называют оптическим антиподами. Для соединений, имеющих nn хиральных центров в молекуле, количество возможных стереоизомеров составляет 2n2n. Однако при n≥2n≥2 существуют стереоизомеры, которые отличаются друг от друга частью имеющихся в них элементов хиральности. Такие стереоизомеры, не являющиеся энантиомерами, называют диастереомерами.
Термин «фармацевтически приемлемая соль» данного соединения относится к солям, которые сохраняют биологическую эффективность и свойства данного соединения и которые не являются биологически или иным образом нежелательными. Фармацевтически приемлемые основно-аддитивные соли могут быть получены из неорганических и органических оснований. Соли, полученные из неорганических оснований, включают, но не ограничиваются, соли натрия, калия, лития, аммония, кальция и магния. Соли, полученные из органических оснований, включают, но не ограничиваются ими, соли первичных, вторичных и третичных аминов, таких как алкиламины, диалкиламины, триалкиламины, замещенные алкиламины, ди(замещенный алкил)амины, три(замещенный алкил)амины, алкениламины, диалкенила-мины, триалкениламины, замещенные алкениламины и т.п. Также включены амины, где два или три заместителя вместе с атомом азота аминогруппы образуют гетероциклическую или гетероарильную группу.
Фармацевтически приемлемые кислотно-аддитивные соли могут быть получены в данном случае из неорганических кислот. Соли, полученные из неорганических кислот, включают соляную кислоту, бромистоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту и т.п.
Термин «фармацевтически приемлемый носитель» включает любые и все растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые агенты, изотонические и задерживающие всасывание агенты и т.п. Применение таких сред и агентов для фармацевтически активных веществ хорошо известно в данной области техники. Их применяют в терапевтических композициях за исключением случаев, когда любая обычная среда или агент несовместимы с активным ингредиентом. В композиции также могут быть включены дополнительные активные ингредиенты.
Термин «активный компонент» (лекарственное вещество) относится к физиологически активному веществу синтетического или иного (биотехнологического, растительного, животного, бактерицидного и так далее) происхождения, обладающему фармакологической активностью, которое является активным ингредиентом фармацевтической композиции.
Термин «лекарственный препарат» означает вещество (или смесь веществ в виде фармацевтической композиции) в виде таблеток, капсул, инъекций, мазей и др. готовых форм, предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
Термин «наносуспензия» означает, в частности, твердо-жидкостную систему с размером частиц менее одного микрометра. В качестве наночастиц в последние годы используют чистые лекарственные средства. В качестве дисперсионной среды часто используют воду, в которой вещество в значительной степени нерастворимо и вводится субъекту в виде суспензии нано частиц. Нано суспензии имеют большое значение в медицине для веществ, которые слабо растворимы в воде (<5 мг/л), которые, как наносуспензия, улучшают биофармацевтические свойства (например, абсорбция, биодоступность).
Термин «фармацевтическая композиция» обозначает композицию, включающую в себя соединение общей формулы 1 или 2 и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например, сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например, моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, альгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного компонента, одного или в комбинации с другим активным компонентом, может быть введена животным и людям в стандартной форме введения, в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения.
Термин "пролекарство" обозначает соединение, которое при введении in vivo метаболизируется посредством одного или более этапов или процессов, или иным образом превращается в биологически, фармацевтически или терапевтически активную форму соединения. Например, пролекарство означает соединение, которое химически предназначено для эффективного высвобождения исходного лекарственного средства после преодоления биологических барьеров на пути пероральной доставки. Для получения пролекарства фармацевтически активное соединение модифицируют таким образом, что активное соединение будет регенерировано в результате метаболических процессов. Пролекарство может быть предназначено для изменения метаболической стабильности или характеристик доставки лекарственного средства, чтобы замаскировать побочные эффекты или токсичность, для улучшения вкуса лекарственного средства или изменения других характеристик или свойств лекарственного средства. Когда фармацевтически активное соединение является известным, специалисты в данной области техники могут разработать пролекарства такого соединения в соответствии со знаниями фармакодинамических процессов и метаболизма лекарств in vivo (см., например, Nogrady (1985) Medicinal Chemistry A Biochemical Approach, Oxford University Press, New York, pages 388-392).
Термин «инертный наполнитель», используемый в данном описании, относится к соединению, которое используют для получения фармацевтической композиции, и, как правило, безопасному, нетоксичному и ни биологически, ни иным образом нежелательному, и включает в себя вспомогательные вещества, которые являются приемлемыми для применения в ветеринарии, а также фармакологически приемлемыми для человеческого использования. Соединения по данному изобретению могут быть введены отдельно, но обычно их будут вводить в смеси с одним или более фармацевтически приемлемыми эксципиентами, разбавителями или носителями, выбранными с учетом предполагаемого пути введения и стандартно фармацевтической практики.
Термин «терапевтически эффективное количество», используемый здесь, означает количество субстанции, пролекарства или лекарства, необходимое для уменьшения симптомов заболевания у субъекта. Доза субстанции, пролекарства или лекарства будет соответствовать индивидуальным требованиям в каждом конкретном случае. Эта доза может варьироваться в широких пределах в зависимости от многочисленных факторов, таких как тяжесть заболевания, подлежащего лечению, возраста и общего состояния здоровья пациента, других лекарственных средств, с помощью которых пациент проходит лечение, способа и формы введения и опыта лечащего врача. Для перорального введения суточная доза составляет приблизительно от 0,01 до 10 г, включая все значения между ними, в день в монотерапии и/или в комбинированной терапии. Предпочтительная суточная доза составляет примерно от 0,1 до 7 г в день. Как правило, лечение начинают с большой начальной «нагрузочной дозы», чтобы быстро уменьшить или устранить вирус, сопровождающей убывающую дозу до уровня, достаточного для предотвращения всплеска инфекции.
Термин «субъект» означает млекопитающее, которое включает, но не ограничивается ими, крупный рогатый скот, свиней, овец, кур, индеек, буйволов, лам, страусов, собак, кошек и человека, предпочтительно субъектом является человек.
Предметом настоящего изобретения является новый анелированный 9-гидрокси-1,8-диоксо-1,3,4,8-тетрагидро-2Н-пиридо[1,2-а]пиразин-7-карбоксамид, общей формулы 1 и 2, его стереоизомер, его фармацевтически приемлемая соль, его сольват, его кристаллическая или поликристаллическая форма
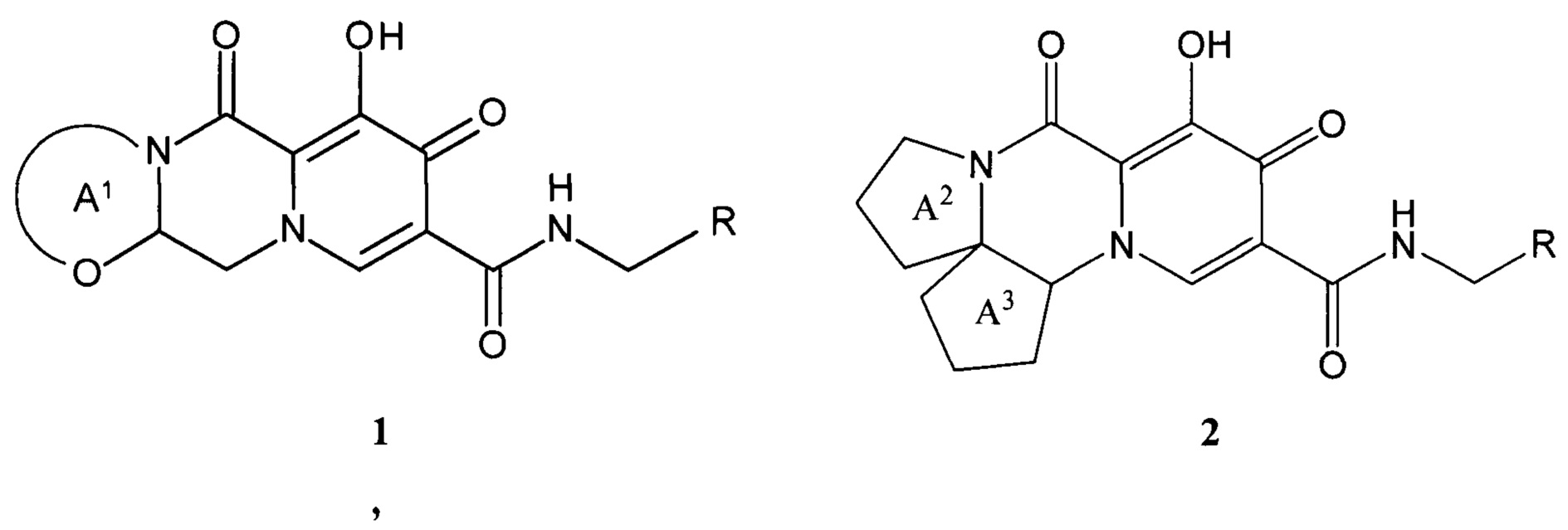
где:
кольцо А1 представляет собой необязательно замещенный метилом 5-7 членный насыщенный гетероцикл или гетеробицикл;
кольцо А2 представляет собой 5-6-членный необязательно замещенный метилом насыщенный или частично насыщенный моноциклический гетероцикл;
кольцо А3 представляет собой 5-6 членный моноциклический насыщенный циклоалкан и тетрагидро-2Н-пиран;
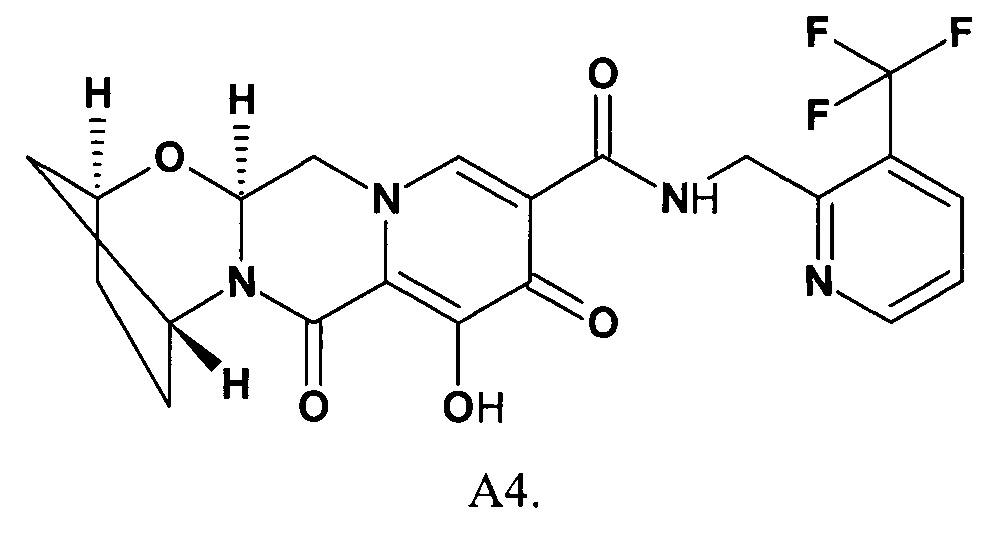
R представляет собой 5-7-членный необязательно замещенный одним, двумя или тремя необязательно одинаковыми заместителями моноциклический или бициклический гетероциклический радикал, включающий 1-4 гетероатома, выбранных из ряда О, S и N, исключая (2S,5R,13aS)-8-гидрокси-7,9-диоксо-N-{[3-(трифторметил)-пиридин-2-ил]метил}-2,3,4,5,7,9,13,13а-октагидро-2,5-метанпиридо[1',2':4,5]пиразино[2,1-b][1,3]оксазепино-10-карбоксамид (формулы А4)
Необязательно замещенный моноциклический гетероарильный радикал R выбирают из ряда, включающего тиенил, фурил, пиразолил, изооксазолил, тиазолил, оксазолил, имидазолил, тиадиазолил, [1,2,5]оксадиазолил, [1,2,4]оксадиазолил, [1,2,4]триазолил, тетразолил, пиридинил, пиридазинил, пиримидинил, пиразинил, 1,2,3-триазинил, 1,2,4-триазинил и 1,3,5-триазинил, имидазо[2,1-b]тиазолил, имидазо[2,1-b][1,3,4]тиадиазолил, бензотиофенил, бензофуранил, индолил, 1,3-бензодиоксол-5-ил, 2,3-дигидро-1,4-бензодиоксин-6-ил, 1,3-бензотиазолил, 1,3-бензооксазолил, бензоимидазолил, 1,3-дигидро-2-оксобензоимидазолил, 2,1,3-бензотиадиазолил, 2,1,3-бензооксадиазолил, хинолинил, изохинолинил, имидазо[1,2-а]пиридинил, 1,2,4-триазоло[4,3-а]пиридинил, имидазо[1,2-а]пиримидинил, имидазо[1,2-а]пиразинил, 1,2,4-триазоло[4,3-b]пиридазинил, 4,5,6,7-тетрагидробензотиофенил, 5,6,7,8-тетрагидро-[1,2,4]триазоло[4,3-а]пиридинил, 1,4,5,6,7,8-гексагидроциклогепта[с]пиразолил, 5,6,7,8-тетрагидро-4Н-циклогепта[d]тиазолил, 5,6,7,8-тетрагидро-4H-циклогепта[d]изотиазол-3-ил, 5,6,7,8-тетрагидро-4H-циклогепта[d]изотиазол-3-ил, [1,2,4]триазоло[4,3-6]пиридазин-3-ил.
Предпочтительно необязательно замещенный моноциклический гетероарильный радикал R выбирают из ряда, включающего 2-тиенил, 2-фурил, 1Н-пиразол-3-ил, 1Н-пиразол-4-ил, 1Н-пиразол-5-ил, изоксазол-4-ил, тиазол-2-ил, 1,3-оксазол-2-ил, имидазол-2-ил, 1,2,3-тиадиазол-5-ил, 1,2,5-оксадиазол-3-ил, 1,2,4-оксадиазол-5-ил, 1Н-1,2,4-триазол-3-ил, 1Н-1,2,3,4-тетразол-5-ил, 2-пиридил, 3-пиридил, 4-пиридил, пиридазин-4-ил, пиримидин-4-ил, пиразин-2-ил, имидазо[2,1-b]тиазол-6-ил, имидазо[2,1-b][1,3,4]тиадиазол-6-ил, бензотиофен-5-ил, бензофуран-2-ил, 1Н-индол-5-ил, 1,3-бензодиоксол-5-ил, 2,3-дигидро-1,4-
бензодиоксин-6-ил, 1,3-бензотиазол-2-ил, 1,3-бензооксазол-2-ил, 1Н-бензимидазол-2-ил, 1,3-дигибро-2-оксобензоимидазол-5-ил, 2,1,3-бензотиадиазол-5-ил, 2,1,3-бензооксадиазол-5-ил, хинолин-2-ил, хинолин-3-ил, хинолин-4-ил, хинолин-5-ил, хинолин-6-ил, хинолин-7-ил, хинолин-8-ил, 1-изохинолинил, имидазо[1,2-а]пиридин-3-ил, 1,2,4-триазоло[4,3-а]пиридин-3-ил, имидазо[1,2-а]пиримидин-2-ил, имидазо[1,2-а]пиразин-3-ил, 1,2,4-триазоло[4,3-b]пиридазин-3-ил, 4,5,6,7-тетрагидробензотиофен-2-ил, 5,6,7,8-тетрагидро-[1,2,4]триазоло[4,3-а]пиридин-3-ил, 1,4,5,6,7,8-гексагидроциклогепта[с]пиразол-3-ил, 5,6,7,8-тетрагидро-4H-циклогепта[d][1,3]тиазол-2-ил, 5,6,7,8-тетрагидро-4H-циклогепта[d]изотиазол-3-ил, 5,6,7,8-тетрагидро-4H-циклогепта[d]изотиазол-3-ил, [1,2,4]триазоло[4,3-b]пиридазин-3-ил.
Предпочтительными заместителями гетероарильного радикала R являются один, два или три независимых заместителя, выбранных из низших С1-С3 алкилов и атомов галогена, предпочтительно F, Cl и Br.
Предметом данного изобретения является соединение общей формулы 1.1, 1.2 и 1.3, в которых R имеет вышеуказанное значение, кольцо А1 представляет собой соответственно 4-метил-1,3-оксазолидин, 4-метил-1,3-оксазинан и (1R,5S)-2-окса-4-азабицикло[3.2.1]октан, их фармацевтически приемлемая соль и их сольват
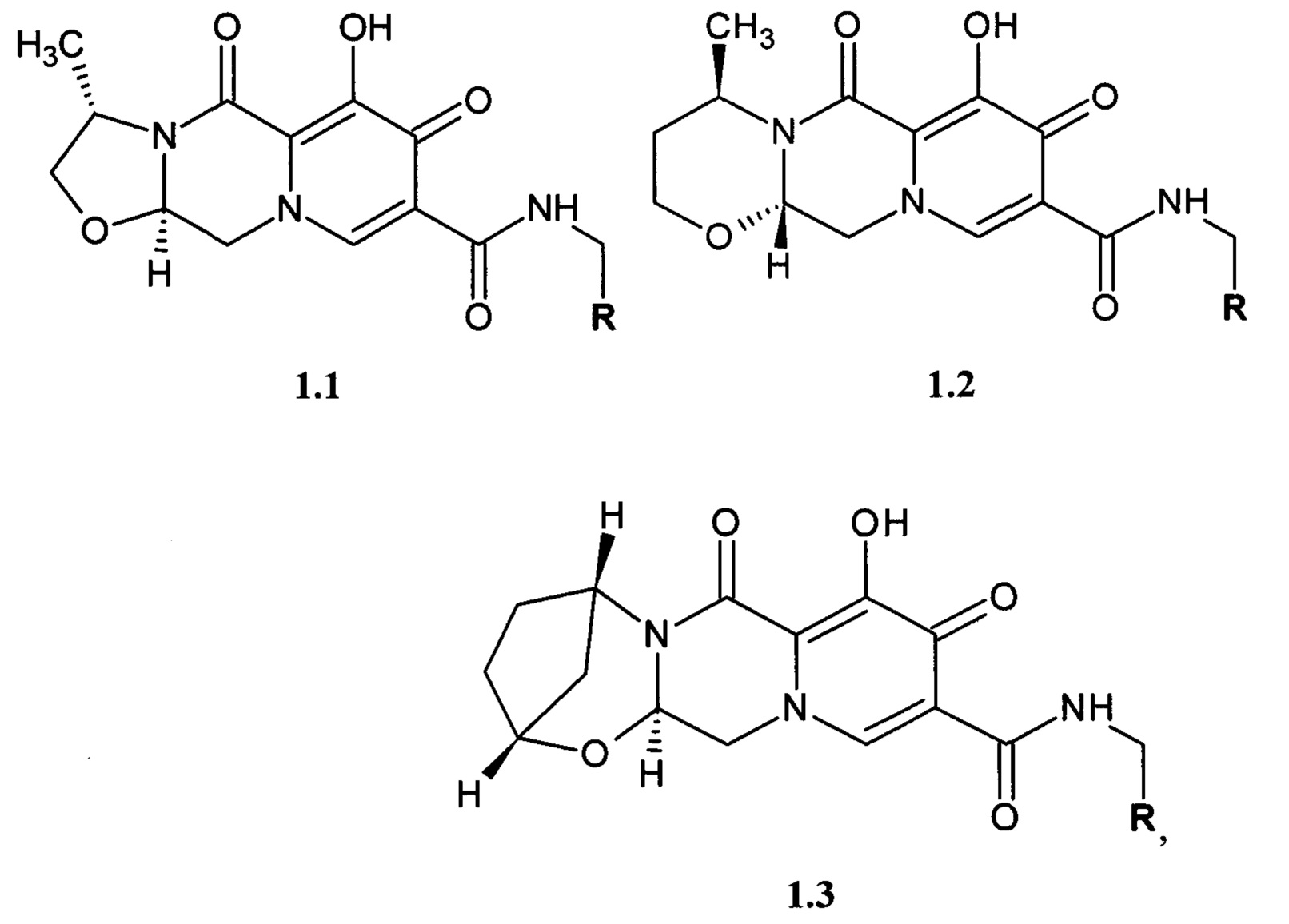
где R имеет вышеуказанное значение.
Предметом данного изобретения является анелированный 9-гидрокси-1,8-диоксо-N-(пиридинилметил)-1,3,4,8-тетрагидро-2Н-пиридо[1,2-а]пиразин-7-карбоксамид общей формулы 2.1 и 2.2, в которых R имеет вышеуказанное значение или их стереоизомер, их фармацевтически приемлемая соль, их сольват, их кристаллическая и поликристаллическая форма или их нанокристаллическая форма

где R имеет вышеуказанное значение.
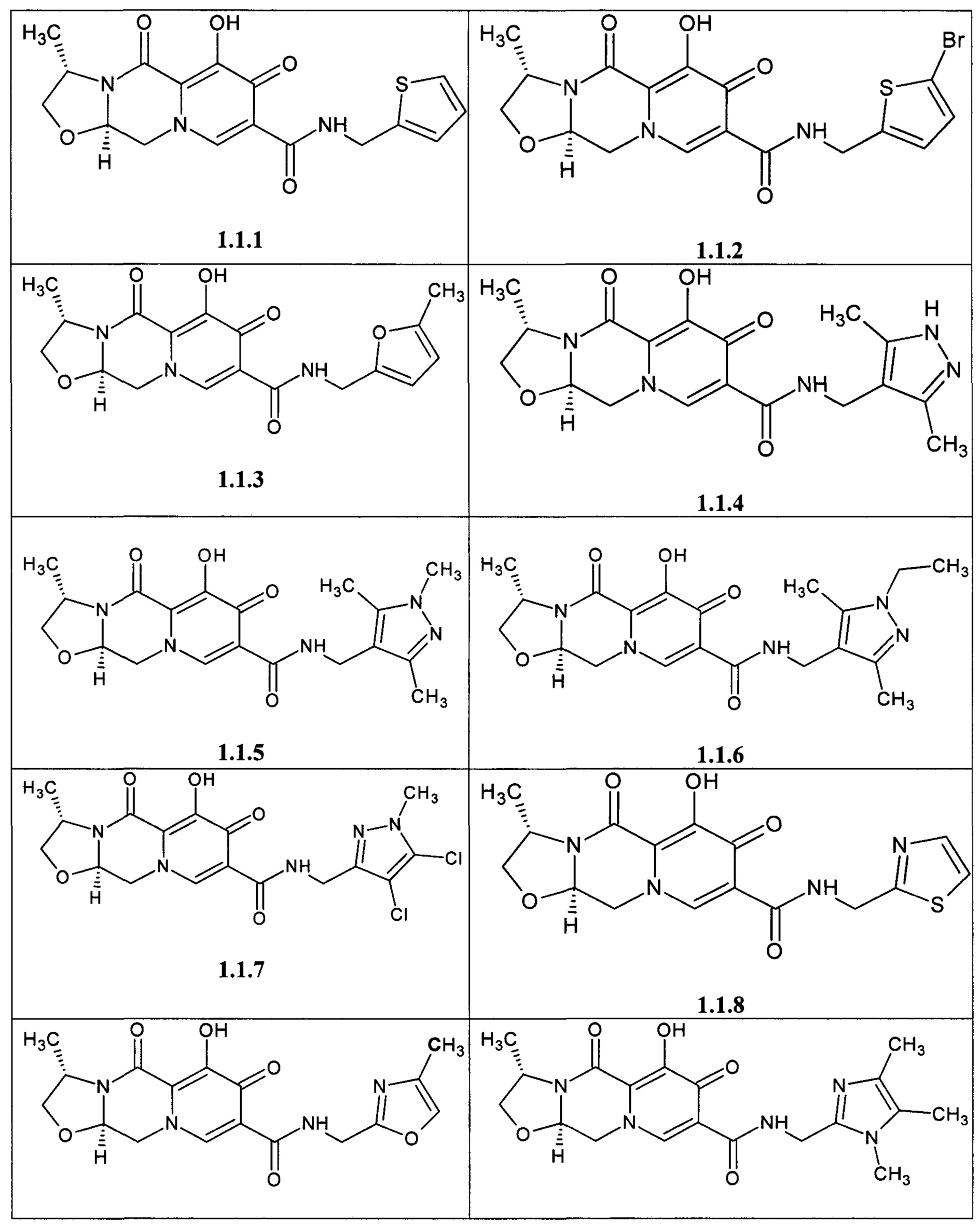
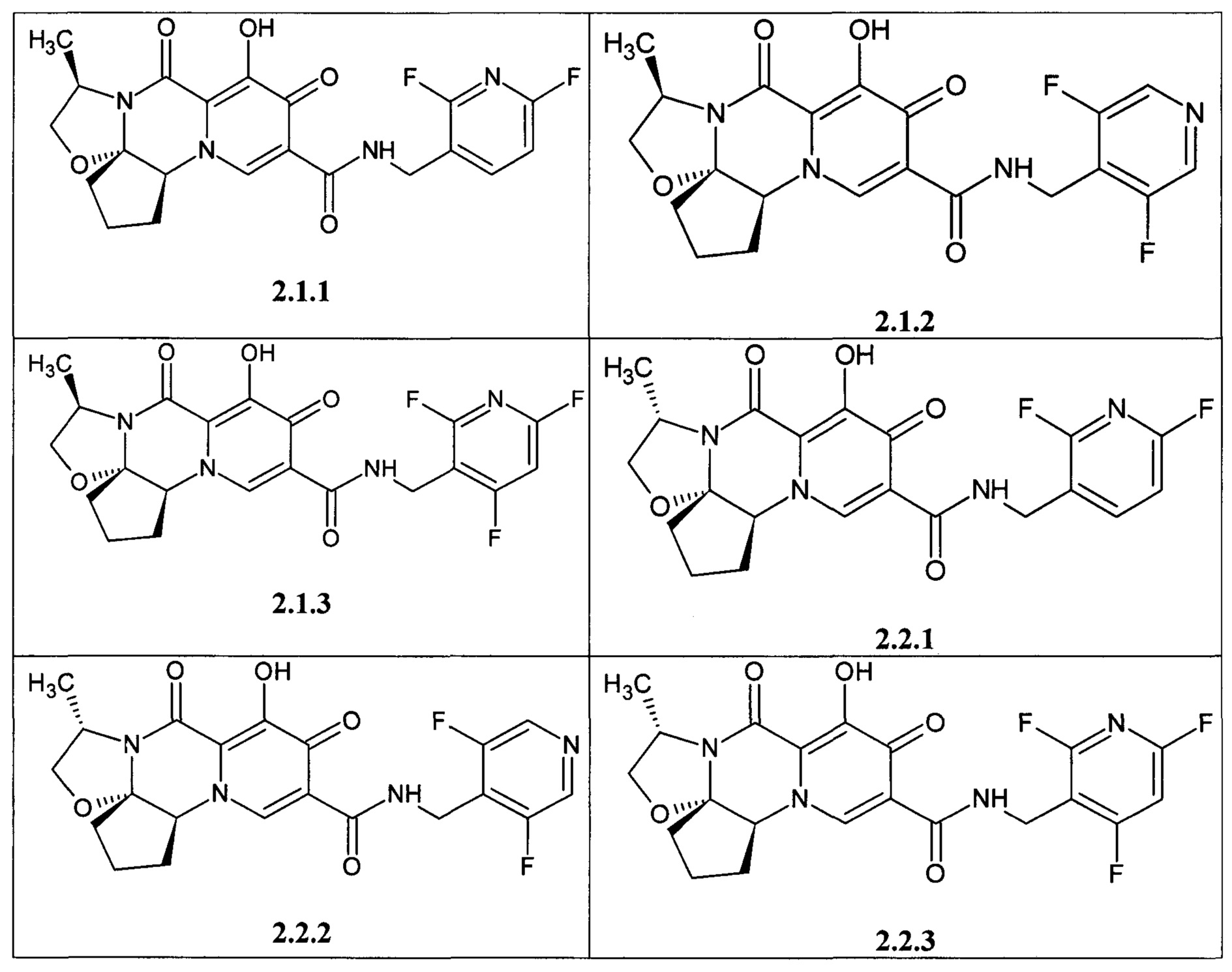
Предметом данного изобретения является анелированный 9-гидрокси-1,8-диоксо-1,3,4,8-тетрагидро-2Н-пиридо[1,2-а]пиразин-7-карбоксамид общей формулы 1 или общей формулы 2, выбранный из ряда включающего:









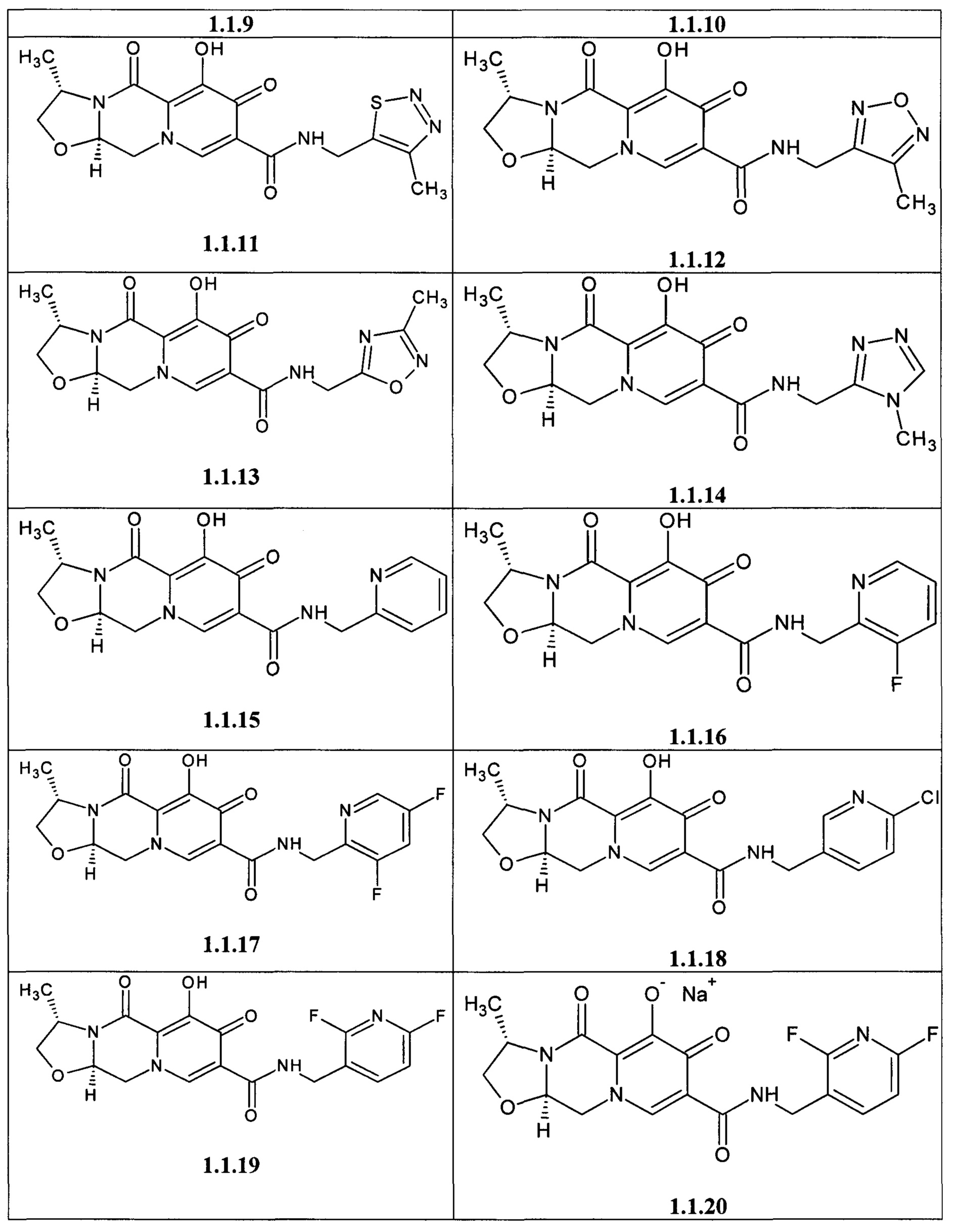
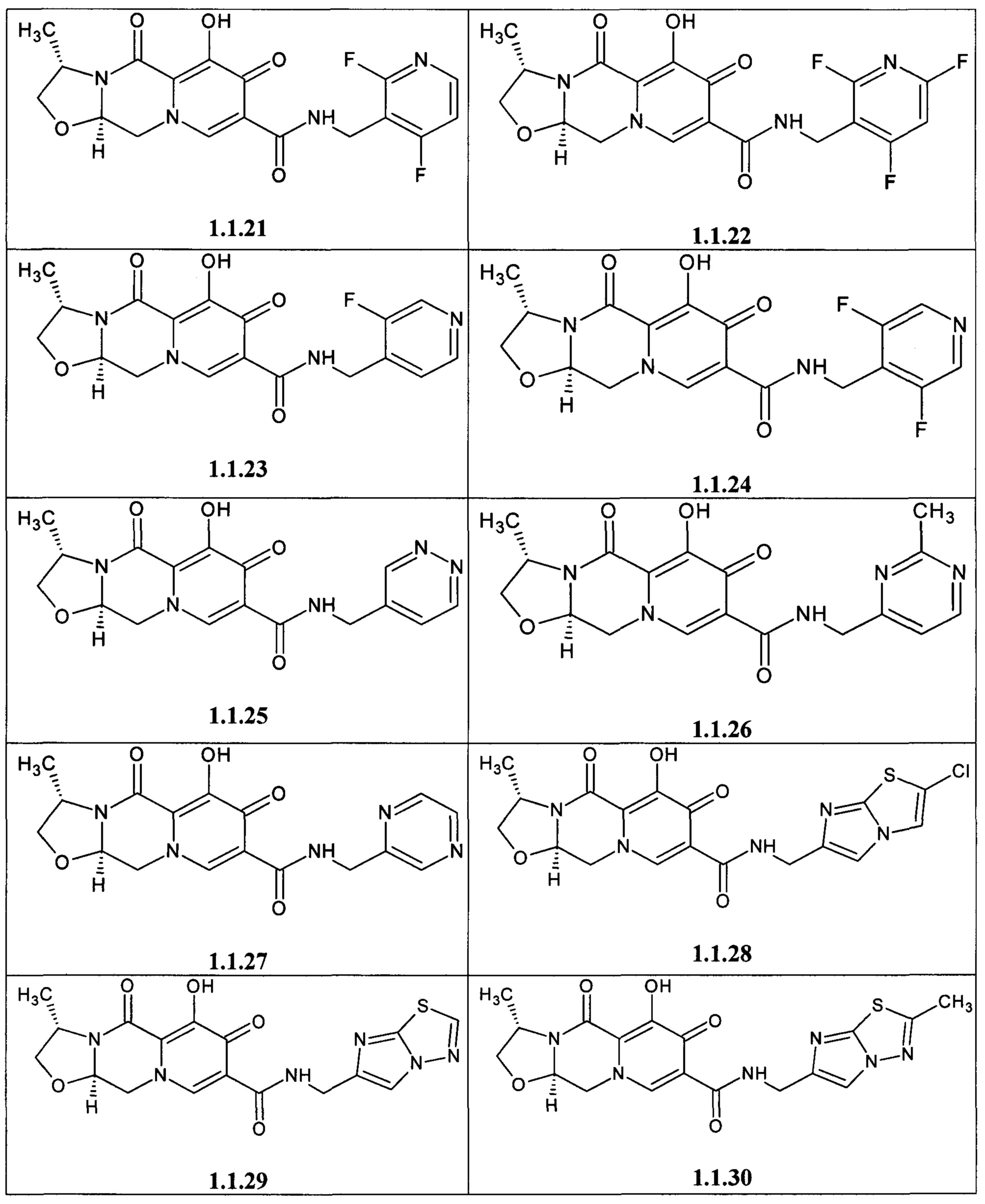
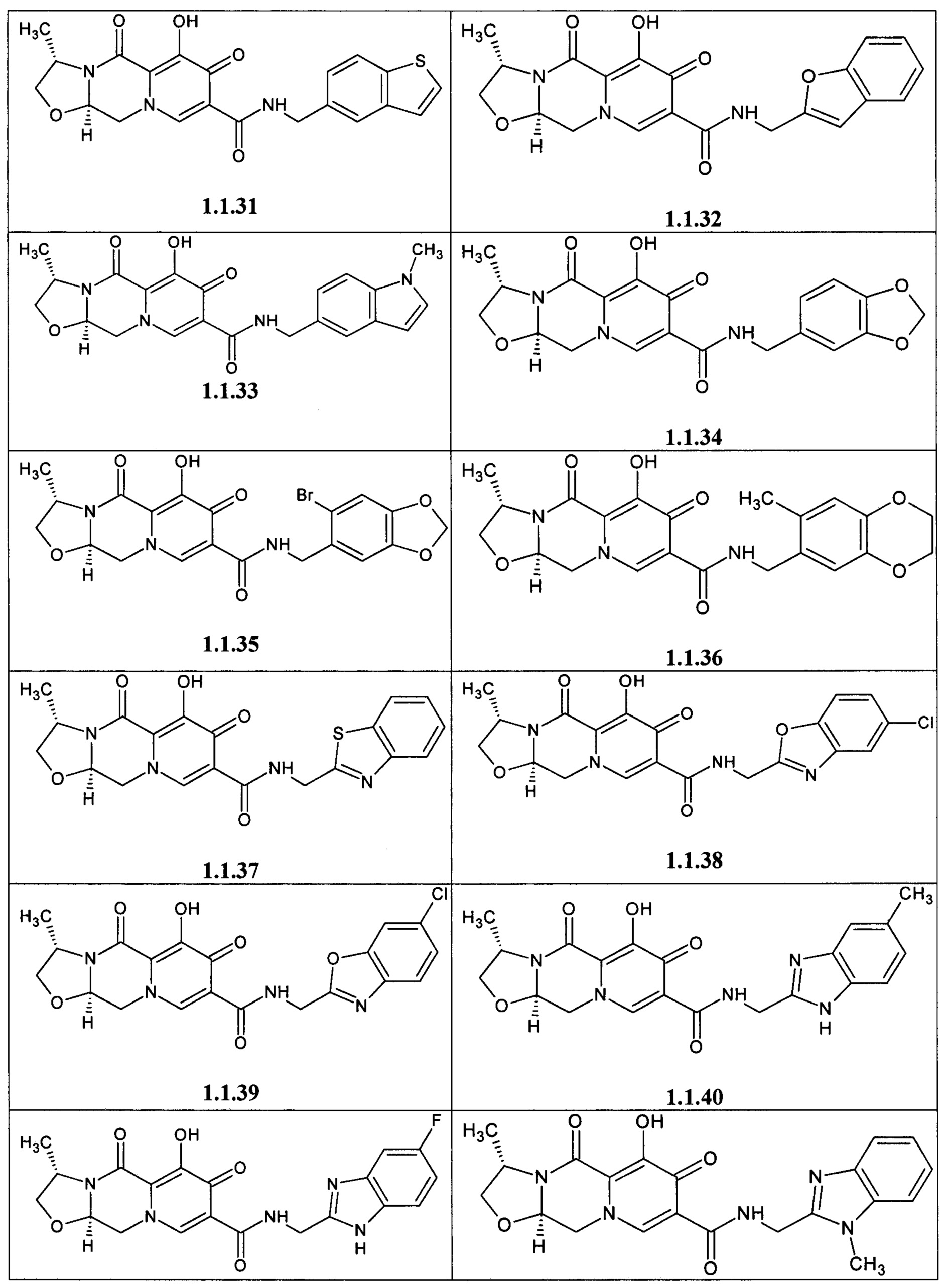

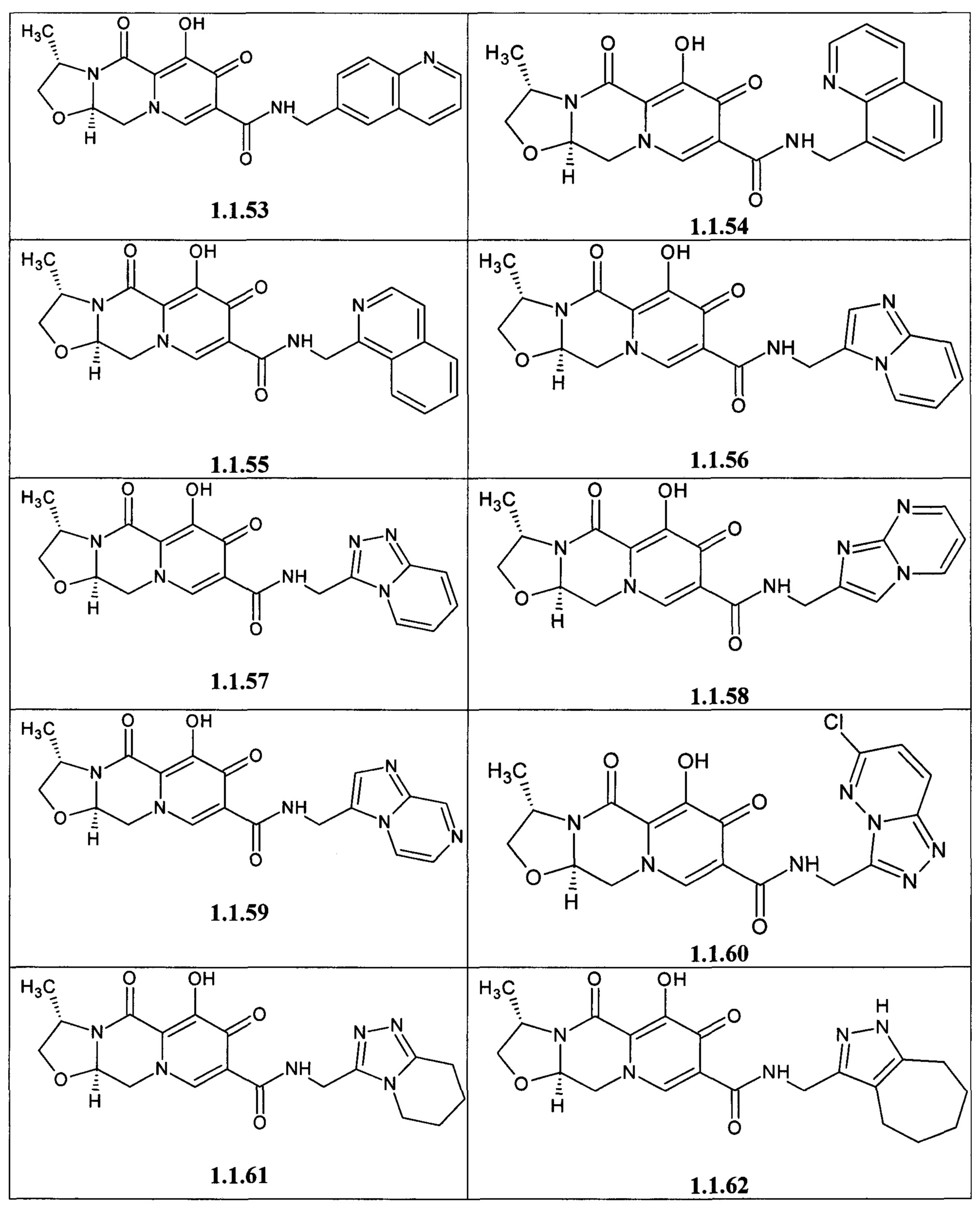

Предметом данного изобретения является способы получения (схема 1) анелированных 9-гидрокси-1,8-диоксо-N-(пиридинилметил)-1,3,4,8-тетрагидро-2Н-пиридо[1,2-а]пиразин-7-карбоксамидов общей формулы (1 и 2) и их стереоизомеров взаимодействием соответствующих бромидов (3, 4) и гетероциклилметиламинов в диметилсульфоксиде в присутствии СО и Pd(PPh3)4 при повышенной температуре с последующим дебензилированием образующихся соединений (5, 6), приводящим к целевым продуктам (1, 2)
Схема 1

где: Bn, R, А1 и А2 имеют вышеуказанное значение.
Предметом данного изобретения является также способ получения (схема 2) анелированных 9-гидрокси-1,8-диоксо-N-(пиридинилметил)-1,3,4,8-тетрагидро-2Н-пиридо[1,2-а]пиразин-7-карбоксамидов общей формулы (1, 2) и их стереоизомеров ацилированием соответствующими кислотами (7 или 8) гетероциклилметиламинов и последующим дебензилированием образующихся продуктов (5, 6), приводящим к целевым продуктам (1, 2)
Схема 2
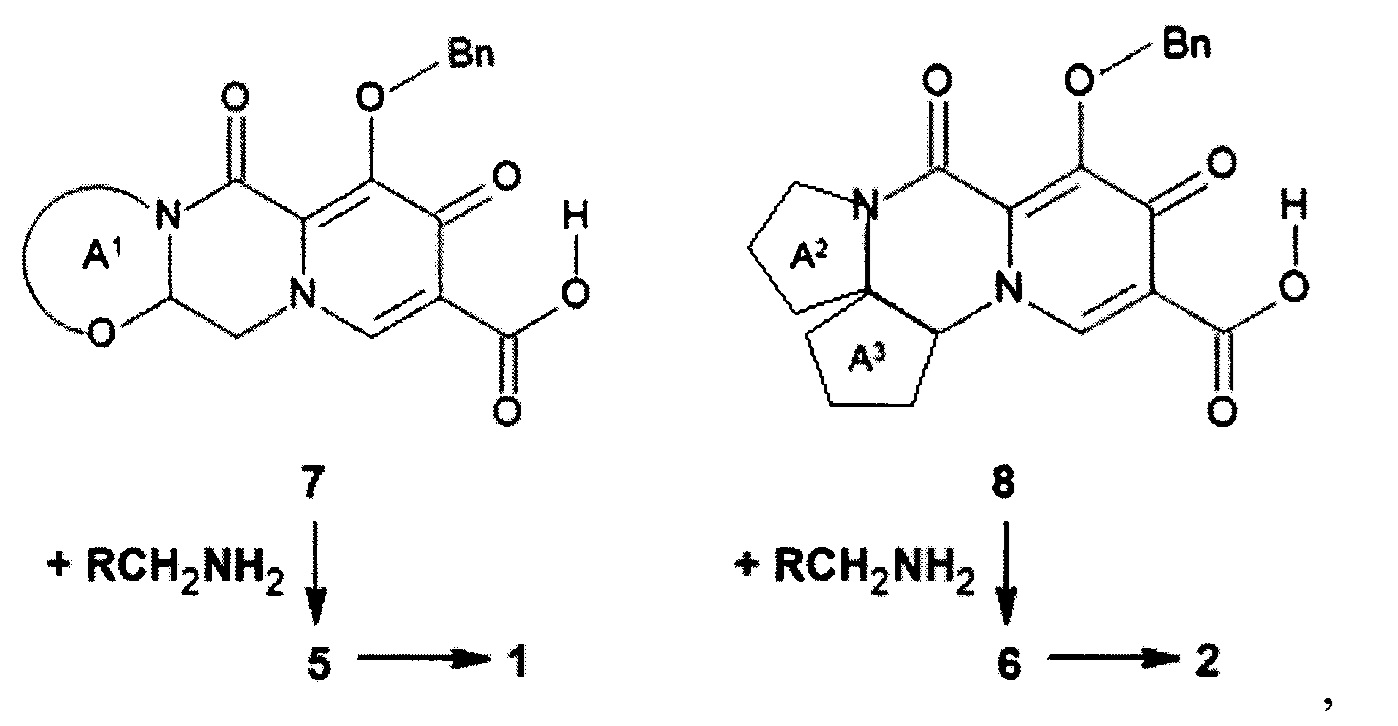
где: Bn имеет вышеуказанное значение.
Наномолярная ингибирующая активность новых соединений общей формулы 1 и 2 (таблица 1) сопоставима с активностью наиболее продвинутых интегразных ингибиторов: CAB, DLG и BIC. Так, например, соединения 1.1.7, 1.1.17, 1.1.19, 1.1.48 имеют ЕС50 соответственно 0,59 нМ, 0,24 нМ, 0.24 нМ и 0.43 нМ. Этот результат оказался неожиданным, так как было известно, что замена в BIC и его аналогах бензильных фрагментов на гетероциклилметильный фрагмент приводит к радикальному снижению активности ингибиторов. В частности известно только 2 ингибитора А4 и А5, включающих вместо бензильного фрагмента пиридин-2-илметильный фрагмент [WO 2014/100323]. Действительно, замена в BIC (соединение 42, ЕС50=2,5 нМ в WO 2014/100323) бензильного фрагмента на пиридин-2-илметильный фрагмент приводит к ингибитору А4 (соединение 49, ЕС50=33,3 нМ в WO 2014/100323), активность которого в 13,3 раза ниже активности BIC. Аналогичная картина наблюдается и у пары ингибиторов А5 (соединение 49, ЕС50=17,8 нМ в WO 2014/100323) и А6 (соединение 66, ЕС50=9,4 нМ в WO 2014/100323). Замена бензильного фрагмента в А6 на пиридин-2-ильный фрагмент приводящая к А5 сопровождается почти двукратным снижением активности.
Новые соединения общей формулы 1 и 2 обладают необходимыми для лекарственных кандидатов ADME свойствами.
В зависимости от их структуры они по разному растворяются в водных растворах (табл. 2). Так, например растворимость в воде (0,00285 мкг/мл) соединения 1.1.48 почти в три раза ниже растворимости CAB (0.008 мкг/мл), что может оказаться существенным преимуществом при использовании 1.1.48 в качестве INI в инъекционной терапии длительного действия ВИЧ. В то же время, например, растворимость натриевой соли 1.1.20 (2,679 мкг/мл) соединения 1.1.19, как и растворимость самого INI 1.1.19 (0.269 мкг/мл) на порядки превышает растворимость CAB, что делает их более перспективными для их использования в качестве INI в пероральной терапии ВИЧ. Это подтверждается, в частности, сравнением фармакокинетических параметров, полученных при пероральном и внутривенном введении крысам соединения 1.1.19 и прототипа CAB в идентичных условиях (Табл. 3). Как следует из таблицы 3 при внутривенном введении крысам фармакокинетические параметры AUCINF, AUClast, Cmax и T1/2 соединения 1.1.4 и значительно выше таковых для CAB. Еще большее различие этих параметров (в два и более раза) наблюдается при переоральном введении крысам этих соединений (табл. 3). Кроме того, время достижения максимальной концентрации в плазме крови (T1/2=16,5 ч) заметно выше такового (T1/2=18,3 ч) чем у CAB, а био доступность соединения 1.1.19 (17,5%), рассчитанная по формуле: F = (AUClastPO/5⋅AUClastIV)⋅100 в два раза выше биодоступности CAB (8,8%).
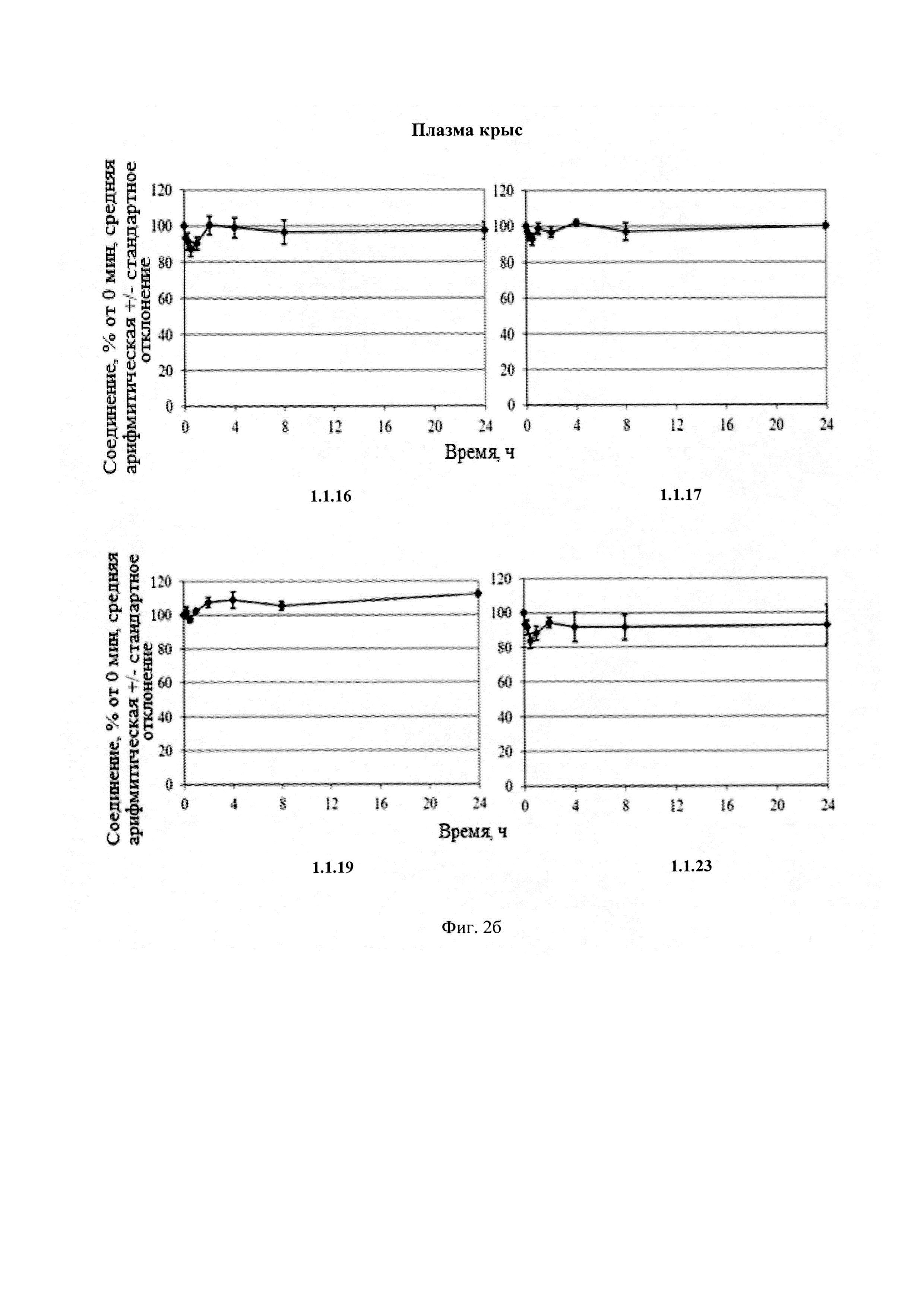
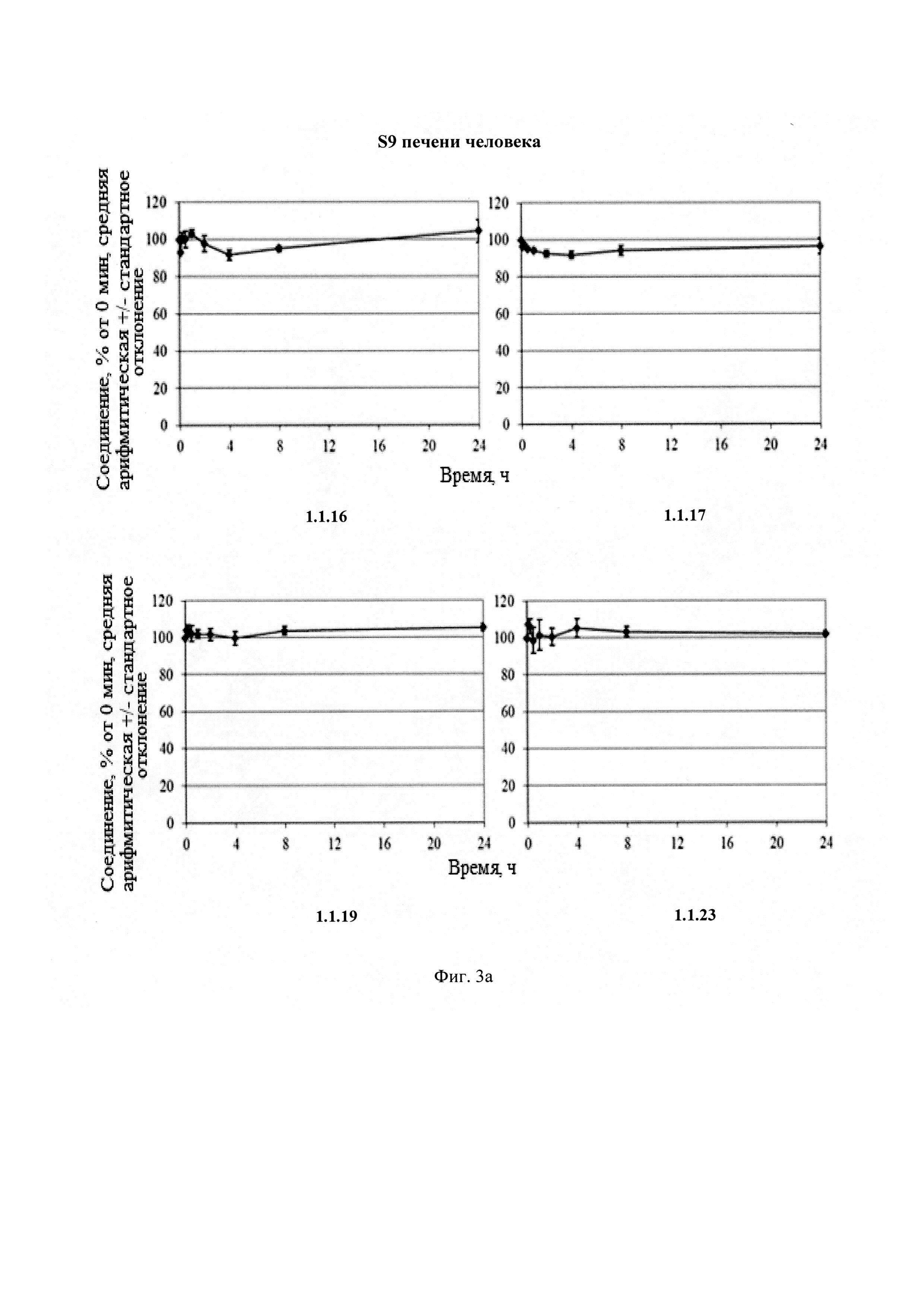
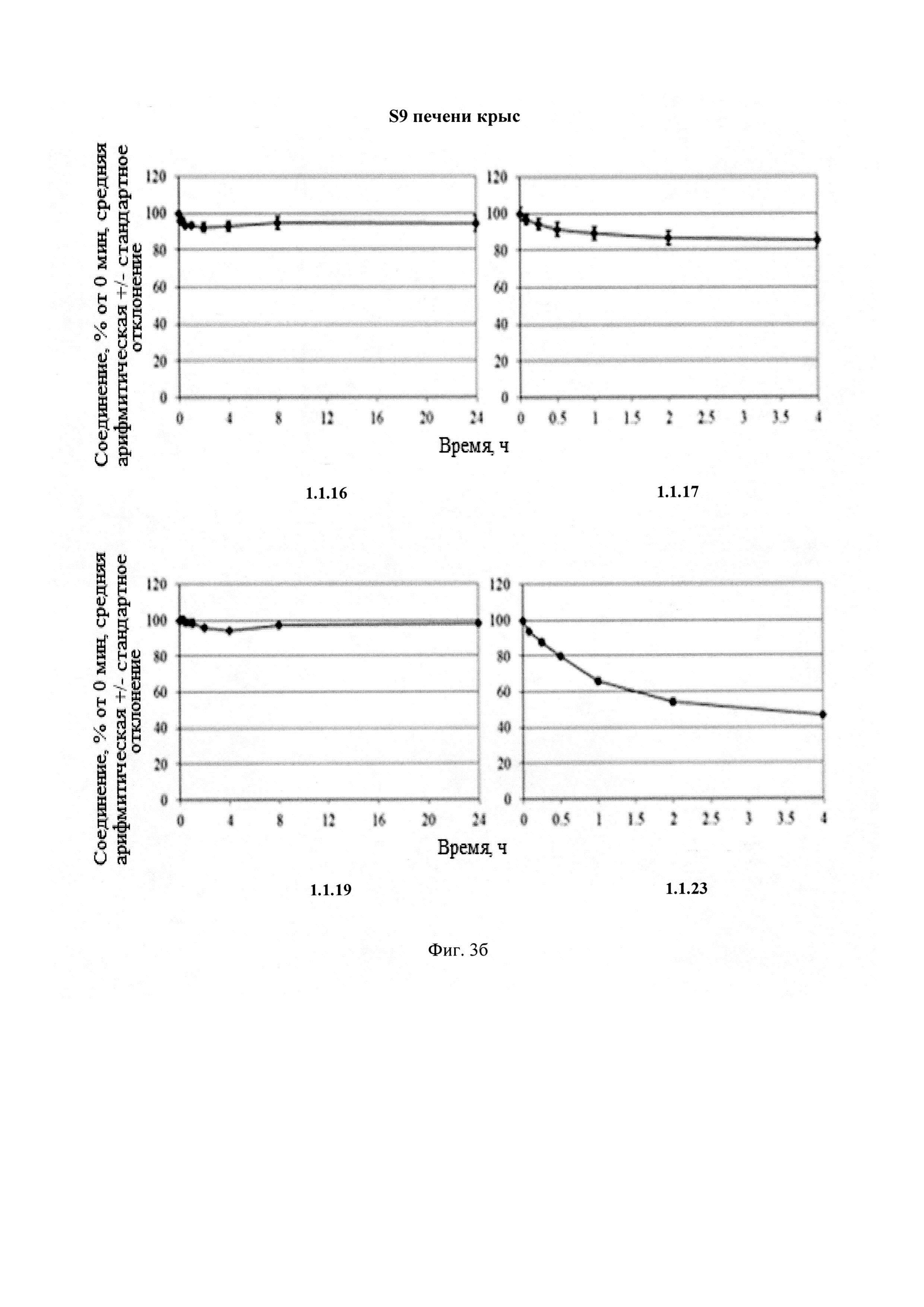
Новые соединения общей формулы 1 и 2 стабильны в плазме человека и крыс (Фиг. 2), S9 фракции печени человека и крыс (Фиг. 3) и обладают высокой степенью связывания с белками плазмы крови человека и крыс (Табл. 4).
Предметом данного изобретения является фармацевтическая композиция, содержащая соединение общей формулы (1) или соединение общей формулы (2) или их стереоизомер, или их фармацевтически приемлемую соль, или их сольват или их кристаллическую или поликристаллическую форму.
Предметом данного изобретения является фармацевтическая композиция, включающая соединение общей формулы 1.1, 1.2, 1.3, 2.1, 2.2 или их стереоизомер, или их фармацевтически приемлемую соль, или их сольват, или их кристаллическую или поликристаллическую форму и фармацевтически приемлемое вспомогательное вещество.
Предметом данного изобретения является фармацевтическая композиция, включающая соединение выбранное из ряда 1.1.1-1.1.65, 1.2.1-1.2.4, 1.3.1-1.3.3, 2.1.1-2.1.3, 2.2.1-2.2.3 или их стереоизомер, или их фармацевтически приемлемую соль, или их сольват, или их кристаллическую или поликристаллическую форму и фармацевтически приемлемое вспомогательное вещество.
Предметом данного изобретения является фармацевтическая композиция, включающая соединение общей формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемую соль, или их сольват, или их кристаллическую или поликристаллическую форму, дополнительно содержит один или более дополнительных терапевтических агентов.
Предметом данного изобретения является фармацевтическая композиция, включающая соединение общей формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемую соль, или их сольват или их кристаллическую или поликристаллическую форму, дополнительно содержит один или более агентов против ВИЧ.
Предметом данного изобретения является фармацевтическая композиция, включающая соединение общей формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемую соль, или их сольват, или их кристаллическую или поликристаллическую форму, дополнительно содержит один или более дополнительных терапевтических агентов, выбранных из группы, состоящей из ингибиторов протеазы ВИЧ, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, нуклеозидных или нуклеотидных ингибиторов обратной транскриптазы ВИЧ, аллостерических ингибиторов интегразы ВИЧ, ингибиторов сборки капсида ВИЧ и их комбинаций.
Предметом данного изобретения является фармацевтическая композиция, включающая соединение общей формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемую соль, или их сольват, или их кристаллическую или поликристаллическую форму, дополнительно содержит первый дополнительный терапевтический агент, выбранный из группы, состоящей из абакавира сульфата, тенофовира, тенофовира дизопроксила, тенофовира дизопроксила фумарата, тенофовира алафенамида, тенофовира алафенамида полуфумарата, тенофовир циклобутилалафенамид, тенофовир циклобутилалафенамид фумарата и тенофовир циклобутилалафенамид полуфумарата, элсульфавирина, VM-1500A, GS-CA1, и второй дополнительный терапевтический агент, выбранный из группы, состоящей из эмтрицитабина и ламивудина.
Предметом данного изобретения является фармацевтическая композиция, включающая соединение общей формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемую соль, или их сольват, или их кристаллическую или поликристаллическую форму, дополнительно содержит первый дополнительный терапевтический агент, выбранный из группы, состоящей из тенофовир циклобутилалафенамида, тенофовир циклобутилалафенамида фумарата или тенофовир циклобутилалафенамид, тенофовир циклобутилалафенамид фумарата и тенофовир циклобутилалафенамид полуфумарата, тенофовир циклобутилалафенамида полуфумарата и эмтрицитабина.
Предметом данного изобретения является фармацевтическая композиция, включающая соединение общей формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемую соль, или их сольват или их кристаллическую или поликристаллическую форму, дополнительно содержит один или более агентов против гепатита С (ВГС) и/или гепатита В (ВГВ).
В качестве агентов против ВГС могут использоваться:
- пролекарства ингибиторов нуклеозида NS5B ВГС, например, Совалди (Sofosbuvir, Sovaldi®, PSI-7977, GS-7977) [https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/204671s004lbl.pdf или циклобутил (S)-2-{(S)-[(2R,3R,4R,5R)-5-(3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-3-гидрокси-4-метил-4-фтор-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноат [Патент RU 2644256 (2018)].
- ингибиторы NS5A HCV, например, Даклинза (Daklinza, Daclatasvir, BMS790052) [https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/206843s006lbl.pdf]; Гепавивир (Hepavivir) (AV-4025) [Ivachtchenko, A.V. et al. Discovery of Novel Highly Potent Hepatitis С Virus NS5A Inhibitor (AV-4025). J. Med. Chem. 2014, 57, 7716-7730. Патент WO 2012/074437 (2012), US 9428491, 2016.]; Омбитасвир (Ombitasvir) (ABT-267) [https://www.drugbank.ca/drugs/DB09296]; Элбасвир (Elbasvir) (MK-8742), [https://www.drugbank.ca/drugs/DB11574]; Велпатасвир (Velpatasvir, VEL, GS-5816), [https://www.drugbank.ca/drugs/DB11613]; Пибрентасвир (Pibrentasvir, ABT-530; ABT530; ABT530, Pibrentasvir) [https://www.drugbank.ca/drugs/DB13878]. Гразопревир (Grazoprevir, MK-5172) [https://www.drugbank.ca/drugs/DB1157].
- ингибиторы NS3 ВГС, например, Нарлапривир (Narlaprevir, SCH 900518) [https://newdrugapprovals.org/tag/narlaprevir/)/].
- ингибиторы NS3/NS4 ВГС, например, Олисио (Simeprevir, Olysio) [https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/205123s001lbl.pdf]; Воксилапревир (Voxilaprevir, GS-9857; GS 9857; GS9857) [https://www.drugbank.ca/drugs/DB12026].
- комбинации указанных ингибиторов ВГС [V. Soriano et al. Treatment of hepatitis С with new fixed dose combinations. Expert Opin Pharmacother. 2017 Aug; 18(12):1235-1242.], в том числе фиксированные комбинации, например, EPCLUSA® (sofosbuvir and velpatasvir) [https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/208341s000lbl.pdf]; Zepatier (Elbasvir / grazoprevir) [https://www.merck.com/product/usa/pi_circulars/z/zepatier/zepatier_pi.pdf]; Viekira XR™ (dasabuvir, ombitasvir, paritaprevir and ritonavir) [https://www.rxabbvie.com/pdf/viekiraxr_pi.pdf]; Mavyret (glecaprevir/pibrentasvir) [https://www.rxabbvie.com/pdf/mavyret_pi.pdf] и др.
В качестве агентов против ВГС мгут использоваться: Бараклюд (Baraclude, entecavir) [https://packageinserts.bms.com/pi/pi_baraclude.pdf]; Эпивир (Epivir, lamivudine) [https://www.ema.europa.eu/documents/product-information/epivir-epar-product-information_en.pdf]; Гепсера (Adefovir, Hepsera, adefovir dipivoxil) [https://www.ema.europa.eu/documents/product-information/hepsera-epar-product-information_en.pdf]; Тизека (Tyzeka, telbivudine) [https://www.accessdata.fda.gov/drugsatfda_docs/label/2006/022011lbl.pdf]; Виреад (Viread, tenofovir) [https://www.ema.europa.eu/documents/variation-report/viread-h-c-419-ii-0120-epar-assessment-report-variation_en.pdf]; Вемлиди (Vemlidy, tenofovir alafenamide fumarate, TAF) [https://www.gilead.com/~/media/files/pdfs/medicines/liver-disease/vemlidy/vemlidy_pi.pdf?la=en]; Циклобутил (S)-2-[[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]-пропаноат фумарат [Патент RU 2674576 (2018)] и др.
Предметом данного изобретения является также фармацевтическая композиция в форме лиофилизата, полученного в результате лиофильной сушки наносуспензии соединения общей формулы 1 или 2, с размером частиц от 200 нм до 900 нм, предпочтительно 200 нм, содержащей фармацевтически приемлемые вспомогательные вещества.
Способ получения фармацевтической композиция в форме лиофилизата заключается в размоле влажных гранул соединения общей формулы 1 или 2, с вспомогательными веществами и водой до размера частиц от 200 нм до 900 нм, предпочтительно 200 нм, и лиофилизации полученной суспензии.
В качестве вспомогательных веществ в фармацевтическая композиция в форме лиофилизата используют вещества, выбранные из маннита, полисорбата, полиэтиленгликоля, полоксамера, маннитола и сахарозы.
Предметом данного изобретения является также инъекционный препарат для долгосрочно поддерживающей терапии ВИЧ-инфекции, включающий фармацевтическую композицию содержащую соединение общей формулы 1 или 2, в форме лиофилизата, фосфатно-буферного солевого раствора и воды для инъекций.
Способ получения нового инъекционного препарата заключается в смешивания фармацевтической композиции содержащую соединение общей формулы 1 или 2, в форме лиофилизата, фосфатно-буферного солевого раствора (PBS) с рН=6.8 и воды для инъекций.
Предметом данного изобретения является способ лечения инфекции ВИЧ у человека, имеющего указанную инфекцию или имеющего риск приобретения указанной инфекции, путем введения указанному человеку терапевтически эффективного количества соединения общей формулы (1) или (2) или их стереоизомера, или их фармацевтически приемлемой соли, или их сольвата, или их кристаллической или поликристаллической формы, или фармацевтической композиции, раскрытой в настоящем описании, или инъекционного препарата.
Предметом данного изобретения является способ профилактики лечения инфекции ВИЧ у человека, имеющего указанную инфекцию или имеющего риск приобретения указанной инфекции, путем введения указанному человеку терапевтически эффективного количества соединения общей формулы (1) или (2) или их стереоизомера, или их фармацевтически приемлемой соли, или их сольвата, или их кристаллической или поликристаллической формы, или фармацевтической композиции или инъекционного препарата, раскрытых в настоящем описании, и дополнительно включает введение указанному человеку терапевтически эффективного количества одного или более дополнительных терапевтических агентов.
Предметом данного изобретения является способ профилактики и лечения инфекции ВИЧ у человека, имеющего указанную инфекцию или имеющего риск приобретения указанной инфекции, путем введения указанному человеку терапевтически эффективного количества соединения общей формулы (1) или (2) или их стереоизомера, или их фармацевтически приемлемой соли, или их сольвата, или их кристаллической или поликристаллической формы, или фармацевтической композиции или инъекционного препарата, раскрытых в настоящем описании, и дополнительно включает введение указанному человеку терапевтически эффективного количества одного или более дополнительных терапевтических агентов, выбранных из группы, состоящей из ингибиторов протеазы ВИЧ, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, нуклеозидных или нуклеотидных ингибиторов обратной транскриптазы ВИЧ, аллостерических ингибиторов интегразы ВИЧ, ингибиторов сборки капсида ВИЧ и их комбинаций.
В конкретном варианте реализации способ профилактики и лечения инфекции ВИЧ у человека, имеющего указанную инфекцию или имеющего риск приобретения указанной инфекции путем введения указанному человеку терапевтически эффективного количества соединения общей формулы (1) или (2) или их стереоизомера, или их фармацевтически приемлемой соли, или их сольвата, или их кристаллической или поликристаллической формы, или фармацевтической композиции или инъекционного препарата, раскрытых в настоящем описании, и дополнительно включает введение указанному человеку терапевтически эффективного количества ненуклеозидных ингибиторов обратной транскриптазы ВИЧ.
Предметом данного изобретения является способ профилактики и лечения инфекции ВИЧ у человека, имеющего указанную инфекцию или имеющего риск приобретения указанной инфекции путем введения указанному человеку терапевтически эффективного количества соединения общей формулы (1) или (2) или их стереоизомера, или их фармацевтически приемлемой соли, или их сольвата, или их кристаллической или поликристаллической формы, или фармацевтической композиции или инъекционного препарата, раскрытых в настоящем описании, и дополнительно включает введение указанному человеку терапевтически эффективного количества первого дополнительного терапевтического агента, выбранного из группы, состоящей из абакавир сульфата, тенофовира, тенофовир дизопроксила, тенофовир дизопроксил фумарата, тенофовир алафенамида, тенофовир алафенамид полуфумарата, тенофовир циклобутилалафенамида, тенофовир циклобутилалафенамида фумарата и тенофовир циклобутилалафенамида полуфумарата, элсульфавирина, VM-1500A, GS-CA1, и второго дополнительного терапевтического агента, выбранного из группы, состоящей из эмтрицитабина и ламивудина.
Предметом данного изобретения является способ профилактики и лечения инфекции ВИЧ у человека, имеющего указанную инфекцию или имеющего риск приобретения указанной инфекции, включает введение указанному человеку единой лекарственной формы для одновременного введения пациенту, например, в виде твердой лекарственной формы для перорального введения, соединения общей формулы (1) или (2) или их стереоизомера, или их фармацевтически приемлемой соли, сольвата совместно или их кристаллической или поликристаллической формы с первым дополнительным терапевтическим агентом, выбранным из группы, состоящей из абакавира сульфата, тенофовира, тенофовира дизопроксила, тенофовира дизопроксила фумарата, тенофовира или их сольвата совместно с первым дополнительным терапевтическим агентом, выбранным из группы, состоящей из абакавир сульфата, тенофовира, тенофовир дизопроксила, тенофовир дизопроксил фумарата, тенофовир алафенамида, тенофовир алафенамид полуфумарата, тенофовир циклобутилалафенамида, тенофовир циклобутилалафенамида фумарата и тенофовир циклобутилалафенамида полуфумарата, элсульфавирина, VM-1500A, GS-CA, и вторым дополнительным терапевтическим агентом, выбранным из группы, состоящей из эмтрицитабина и ламивудина.
Предметом данного изобретения является применение соединения общей формулы (1) или (2) или их стереоизомера, или их фармацевтически приемлемой соли, или их сольвата, или их кристаллической или поликристаллической формы в антиретровирусной терапии (APT), комбинированной антиретровирусной терапии (APT) и антиретровирусной терапии длительного подавления ВИЧ (Antiretroviral Therapy as Long Acting Suppression, ATLAS) для лечения инфекции ВИЧ у человека, имеющего указанную инфекцию или имеющего риск приобретения указанной инфекции.
Предметом данного изобретения является применение фармацевтической композиции или инъекционного препарата, раскрытых в настоящем описании, в антиретровирусной терапии (APT), комбинированной антиретровирусной терапии (APT) и антиретровирусной терапии длительного подавления ВИЧ (Antiretroviral Therapy as Long Acting Suppression, ATLAS) для лечения инфекции ВИЧ у человека, имеющего указанную инфекцию или имеющего риск приобретения указанной инфекции.
Предметом данного изобретения является применение соединения общей формулы (1) или (2) или их стереоизомера, или их фармацевтически приемлемой соли, или их сольвата или фармацевтической композиции или инъекционного препарата, раскрытых в настоящем описании, в доконтактной профилактике до вступления индивидуума в контакт с ВИЧ с целью предотвращения передачи инфекции ВИЧ в случае, если индивидуум вступает в контакт с вирусом, и/или с целью препятствия развитию устойчивой вирусной инфекции и/или с целью предотвращения появления симптомов заболевания и/или предотвращения достижения обнаруживаемых уровней вируса в крови.
Предметом данного изобретения является способ ингибирования репликации ВИЧ действием терапевтически эффективным количеством соединения общей формулы (1) или (2) или их стереоизомера, или их фармацевтически приемлемой соли, или их сольвата, или их кристаллической или поликристаллической формы или фармацевтической композиции или инъекционного препарата, раскрытых в настоящем описании, на ВИЧ в условиях, обеспечивающих ингибирование репликации ВИЧ.
Предметом данного изобретения является применение соединения общей формулы (1) или (2) или их стереоизомера, или их фармацевтически приемлемой соли, или их сольвата или фармацевтической композиции или инъекционного препарата, раскрытых в настоящем описании, для ингибирования активности фермента интегразы ВИЧ.
Предметом данного изобретения является применение соединения общей формулы (1) или (2) или их стереоизомера, или их фармацевтически приемлемой соли, или их сольвата или фармацевтической композиции или инъекционного препарата, раскрытых в настоящем описании, в качестве средства для ингибирования ВИЧ.
Следует понимать, что в любом варианте реализации данного изобретения, изложенного выше, соединение общей формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемая соль, или их сольват, или их кристаллической или поликристаллической формы и любой конкретный заместитель, описанный в настоящей заявке как группа А1, А2, А3 и R в соединениях формул (1) или (2) или в их стереоизомере, или в их фармацевтически приемлемой соли, или в их сольвате, или в их кристаллической или поликристаллической форме, как изложено выше, может независимо быть объединен с другими вариантами реализации и/или заместителями соединения любой из формул (1) или (2) или их стереоизомера, или их фармацевтически приемлемой соли, или их сольвата, или их кристаллической или поликристаллической формы с получением вариантов реализации, конкретно не изложенных выше. Кроме того, в случае, если перечень заместителей приведен для любого конкретного А1, А2, А3 и R в конкретном варианте реализации и/или пункте формулы изобретения, следует понимать, что каждый отдельный заместитель может быть исключен из конкретного варианта реализации и/или пункта формулы изобретения и что оставшийся перечень заместителей будет находиться в рамках вариантов реализации, раскрытых в настоящем описании.
Введение субъекту соединения общей формулы (1) или (2) или их стереоизомера, или их фармацевтически приемлемой соли, или их сольвата, или их кристаллической или поликристаллической формы согласно вариантам реализации, раскрытым в настоящем описании, в чистом виде или в составе соответствующей фармацевтической композиции или инъекционного препарата, раскрытых в настоящем описании, может быть выполнено посредством любого из принятых способов введения средств аналогичного назначения.
Фармацевтические композиции, раскрытых в настоящем описании, согласно вариантам реализации, раскрытым в настоящем описании, могут быть приготовлены посредством объединения соединения согласно вариантам реализации, раскрытым в настоящем описании, с соответствующим фармацевтически приемлемым вспомогательным веществом и могут быть приготовлены в виде препаратов в твердых, полутвердых или жидких газообразных формах, таких как таблетки, капсулы, порошки, гранулы, мази, нано суспензии, растворы, суппозитории, инъекции, ингалянты, гели, микросферы и аэрозоли. Типичные пути введения указанных фармацевтических композиций включают, без ограничения, пероральный, местный, трансдермальный, ингаляционный, парентеральный, сублингвальный, буккальный, ректальный, вагинальный и интраназальный.
Фармацевтические композиции согласно вариантам реализации, раскрытым в настоящем описании, готовят таким образом, чтобы обеспечить биодоступность активных ингредиентов, входящих в их состав, при введении композиции субъекту. Композиции, которые будут введены субъекту или пациенту, представлены в лекарственных формах, содержащих одну или более единиц дозирования, где, например, таблетка может представлять собой лекарственную форму, содержащую одну единицу дозирования, а емкость с соединением согласно вариантам реализации, раскрытым в настоящем описании, в форме аэрозоля может содержать множество единиц дозирования.
Фактические способы получения указанных лекарственных форм известны или будут очевидны специалистам в данной области техники; например, см. Remington.. The Science and Practice of Pharmacy, 20th Edition (Philadelphia College of Pharmacy and Science, 2000). Композиция, подлежащая введению, будет в любом случае содержать терапевтически эффективное количество соединения согласно вариантам реализации, раскрытым в настоящем описании, или его фармацевтически приемлемой соли для лечения представляющего интерес заболевания или состояния в соответствии с рекомендациями настоящего описания.
В одном из вариантов реализации данного изобретения фармацевтическая композиция представляет собой пероральную единичную дозированную форму.
В одном из вариантов реализации фармацевтическая композиция представляет собой твердую пероральную единичную дозированную форму.
В одном из вариантов реализации фармацевтическая композиция представляет собой таблетку.
В одном из вариантов реализации фармацевтическая композиция представляет собой капсулу.
В одном из вариантов реализации фармацевтическая композиция представляет собой лиофилизированную наносуспензию помещенную в предварительно стерилизованный стеклянный флакон, закупоренный предварительно стерилизованной пробкой и загерметизированный.
Фармацевтические композиции, раскрытые в настоящем описании, могут быть получены способами, хорошо известными в фармацевтической области. Например, фармацевтическая композиция, предназначенная для введения посредством инъекции, может быть приготовлена посредством объединения соединения согласно вариантам реализации, раскрытым в настоящем описании, со стерильной дистиллированной водой с образованием раствора или наносуспензии. Для обеспечения образования гомогенного раствора или наносуспензии может быть добавлено поверхностно-активное вещество.
Поверхностно-активные вещества представляют собой соединения, нековалентно взаимодействующие с соединением согласно вариантам реализации, раскрытым в настоящем описании, способствуя растворению или обеспечивая гомогенную наносуспензию соединения в водной системе доставки.
Соединения общей формулы (1) или (2) или их стереоизомера, или их фармацевтически приемлемой соли, или их сольвата, или их кристаллической или поликристаллической формы согласно вариантам реализации, раскрытым в настоящем описании, в чистом виде или в составе соответствующей фармацевтической композиции вводят субъекту или пациенту в терапевтически эффективном количестве, которое будет варьировать в зависимости от множества факторов, включая активность конкретного используемого соединения; метаболическую стабильность и продолжительность действия соединения; возраст, массу тела, общее состояние здоровья, пол и диету пациента; режим и время введения; скорость выведения; сочетание лекарственных средств; тяжесть конкретного расстройства или состояния и субъекта, подвергаемого терапии.
В некоторых вариантах реализации изобретения для лечения или предотвращения инфекции ВИЧ у человека, инфицированного или подверженного риску инфицирования, предложен способ, включающий введение человеку терапевтически эффективного количества соединения, раскрытого в настоящем описании, или его стереоизомера, или их фармацевтически приемлемой соли, или их сольвата, раскрытого в настоящем описании, или фармацевтической композиции, раскрытой в настоящем описании, в комбинации с терапевтически эффективным количеством одного или более (например, одного, двух, трех, одного или двух, или от одного до трех) дополнительных терапевтических агентов (до трех) дополнительных терапевтических агентов.
В некоторых вариантах реализации изобретения приведенный в настоящем описании способ лечения инфекции ВИЧ, включающий введение пациенту в случае необходимости терапевтически эффективного количества соединения или его стереоизомера, или их фармацевтически приемлемой соли, или их сольвата, или их кристаллической или поликристаллической формы, раскрытых в настоящем описании, в комбинации с терапевтически эффективным количеством одного или более (например, одного, двух, трех, одного или двух, или от одного до трех) дополнительных терапевтических агентов, подходящих для лечения инфекции ВИЧ.
Соединение, раскрытое в настоящем описании (например, любое соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемая соль, или их сольват, или их кристаллическая или поликристаллическая форма) может быть объединено с одним или более дополнительными терапевтическими агентами при любой дозе соединения формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемая соль, или их сольват, или их кристаллическая или поликристаллическая форма (например, от 50 мг до 1000 мг соединения).
В одном из вариантов реализации данного изобретения, раскрытом в настоящем описании соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемая соль, или их сольват, или их кристаллическая или поликристаллическая форма в комбинации с одним или более (например, одним, двумя, тремя, одним или двумя, или от одного до трех) дополнительными терапевтическими агентами и фармацевтически приемлемым вспомогательным веществом.
В указанных выше вариантах реализации изобретения дополнительный терапевтический агент может представлять собой агент против ВИЧ. Например, в некоторых вариантах реализации изобретения дополнительный терапевтический агент выбран из группы, состоящей из ингибиторов протеаз ВИЧ, ненуклеозидных или ненуклеотидных ингибиторов обратной транскриптазы ВИЧ, нуклеозидных или нуклеотидных ингибиторов обратной транскриптазы ВИЧ, ингибиторов интегразы ВИЧ, ингибиторов некаталитического (или аллостерического) сайта интегразы ВИЧ, нуклеозидных или нуклеотидных ингибиторов обратной транскриптазы ВИЧ, ингибиторов сборки капсида ВИЧ, ингибиторов проникновения ВИЧ (например, ингибиторов CCR5, ингибиторов gp41 (т+ е+ ингибиторов слияния) и ингибиторов присоединения CD4, ингибиторов CXCR4, ингибиторов gpl20, ингибиторов G6PD и NADH-оксидазы, вакцин против ВИЧ, ингибиторов созревания ВИЧ, агентов, обращающих латентность (например, ингибиторов гистоновой диацетилазы, ингибиторов протеасомы, активаторов протеинкиназы С (РКС) и ингибиторов BRD), соединений, действующих на капсид ВИЧ («ингибиторов капсида», например, ингибиторов полимеризации капсида или соединений, разрушающих капсиды, ингибиторов белка нуклеокапсида ВИЧ р7 (NCp7), ингибиторов белка капсида ВИЧ р24), фармакокинетических усилителей, иммунологических средств лечения (например, модуляторов Pd-1, модуляторов Pd-Ll, модуляторов толл-подобных рецепторов, агонистов ИЛ-15), антител к ВИЧ, биспецифических антител и «антителоподобных» терапевтических белков (например, DARTs®, Duobodies®, Bites®, XmAbs®, TandAbs®, производных Fab), включая лекарственные средства, действующие на gpl20 или gp41 ВИЧ, комбинированных лекарственных средств против ВИЧ, ингибиторов матричного белка ВИЧ р17, антагонистов ИЛ-13, модуляторов пептидил-пролил-цис/транс-изомеразы А, ингибиторов протеиндисульфидизомеразы, антагонистов рецептора комплемента С5а, ингибиторов ДНК-метилтрансферазы, модуляторов vif-гена ВИЧ, ингибиторов фактора вирусной инфекционности ВИЧ-1, ингибиторов ТАТ-белка, модуляторов Nef ВИЧ-1, модуляторов тирозинкиназы Нек, ингибиторов киназы смешанного происхождения-3 (MLK-3), ингибиторов сплайсинга ВИЧ-1, ингибиторов белка Rev, антагонистов интегрина, ингибиторов нуклеопротеина, модуляторов фактора сплайсинга, модуляторов СОММ-домен-содержащего белка 1, ингибиторов рибонуклеазы Н ВИЧ, модуляторов ретроциклина, ингибиторов CDK-9, ингибиторов неинтегринового белка-1 дендритных клеток, связывающего ICAM-3, ингибиторов белка GAG ВИЧ, ингибиторов белка POL ВИЧ, модуляторов фактора Н комплемента, ингибиторов убиквитинлигазы, ингибиторов киназы деоксицитидина, ингибиторов циклинзависимой киназы, стимуляторов пропротеинконвертазы РС9, ингибиторов АТФ-зависимой РНК-хеликазы DDX3X, ингибиторов примирующего комплекса обратной транскриптазы, генной терапии ВИЧ, ингибиторов PI3K, соединений, подобных соединениям, раскрытым в WO 2013/006738, US 2013/0165489, WO 2013/091096 A1, WO 2009/062285, US 20140221380, US 20140221378, WO 2010/130034, WO 2013/159064, WO 2012/145728, WO 2012/003497, WO 2014/100323, WO 2012/145728, WO 2013/159064, WO 2012/003498 и WO 2013/006792 и других лекарственных средств для лечения ВИЧ и их комбинаций.
В некоторых вариантах реализации изобретения соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемая соль, или их сольват, или их кристаллическая или поликристаллическая форма приготовлено в форме таблетки, которая может необязательно содержать одно или более других соединений, применяемых для лечения ВИЧ. В некоторых вариантах реализации таблетка может содержать другой активный ингредиент для лечения ВИЧ, такой как ингибиторы протеаз ВИЧ, ненуклеозидные или ненуклеотидные ингибиторы обратной транскриптазы ВИЧ, нуклеозидные или нуклеотидные ингибиторы обратной транскриптазы ВИЧ, ингибиторы интегразы ВИЧ, ингибитор некаталитического (или аллостерического) сайта интегразы ВИЧ, ингибиторов сборки капсида ВИЧ, фармакокинетические усилители и их комбинации.
В некоторых вариантах реализации изобретения указанные таблетки представляют собой таблетки, подходящие для приема один раз в сутки.
В некоторых вариантах реализации изобретения дополнительный терапевтический агент представляет собой:
- одно или более комбинированное лекарственное средство, выбранных из группы, включающей ATRIPLA® (эфавиренз + тенофовира дизопроксил фумарат + эмтрицитабин), COMPLERA® (EVIPLERA®, рилпивирин + тенофовира дизопроксил фумарат + эмтрицитабин), STRffiILD® (элвитегравир + кобицистат + тенофовира дизопроксил фумарат + эмтрицитабин), долутегравир + абакавира сульфат + ламивудин, TRJUMEQ® (долутегравир + абакавир + ламивудин), ламивудин + невирапин + зидовудин, долутегравир + рилпивирин, атазанавира сульфат + кобицистат, атазанавир + кобицистат, дарунавир + кобицистат, эфавиренз + ламивудин + тенофовира дизопроксил фумарат, тенофовира алафенамид гемифумарат + эмтрицитабин + кобицистат + элвитегравир, тенофовира алафенамид гемифумарат + эмтрицитабин, тенофовира алафенамид + эмтрицитабин, тенофовира алафенамид гемифумарат + эмтрицитабин + рилпивирин, тенофовира алафенамид + эмтрицитабин + рилпивирин, Vacc-4x + ромидепсин, дарунавир + тенофовира алафенамид гемифумарат + эмтрицитабин + кобицистат, АРН-0812, ралтегравир + ламивудин, KALETRA® (ALUVIA®, лопинавир + ритонавир), атазанавира сульфат + ритонавир, COMBIVIR® (зидовудин + ламивудин, AZT+3TC), EPZICOM® (Livexa®, абакавира сульфат + ламивудин, АВС+ЗТС), TRIZIVIR® (абакавира сульфат + зидовудин + ламивудин, ABC+AZT+3TC), TRUYADA® (тенофовира дизопроксил фумарат + эмтрицитабин, TDF+FTC), тенофовир + ламивудин, ламивудин + тенофовира дизопроксил фумарат и ламивудин + тенофовира циклобутилалафенамид или его фумарат или полуфумарат;
- ингибитор протеазы ВИЧ, выбранный из группы, состоящей из ампренавира, атазанавира, фосампренавира, фосампренавира кальция, индинавира, индинавира сульфата, лопинавира, ритонавира, нелфинавира, нелфинавира мезилата, саквинавира, саквинавира мезилата, типранавира, бреканавира, дарунавира, DG-17, TMB-657 (PPL-100) и ТМС-310911;
- ненуклеозидный или ненуклеотидный ингибитор обратной транскриптазы ВИЧ, выбранный из группы, состоящей из делавирдина, делавирдина мезилата, невирапина, этравирина, дапивирина, доравирина, рилпивирина, эфавиренза, КМ- 023, VM-1500, лентинана и AIC-292;
- нуклеозидный или нуклеотидный ингибитор обратной транскриптазы ВИЧ, выбранный из группы, состоящей из VIDEX® и VIDEX® ЕС (диданозин, ddl), зидовудина, эмтрицитабина, диданозина, ставудина, зальцитабина, ламивудина, ценсавудина, абакавира, абакавира сульфата, амдоксовира, элвуцитабина, аловудина, фосфазида, фозивудина тидоксила, априцитабина, амдоксовира, КР-1461, фосалвудина тидоксила, тенофовира, тенофовир дизопроксила, тенофовир дезопроксил фумарата, тенофовир дезопроксил полуфумарата, тенофовир алафенамида, тенофовир алафенамид полуфумарата, тенофовир алафенамид фумарата, тенофовир циклобутилалафенамида, тенофовир циклобутилалафенамид полуфумарата, тенофовир циклобутилалафенамид фумарата, элсульфавирина, VM-1500A, адефовира, адефовира дипивоксила и фестинавира;
- ингибитор интегразы ВИЧ, выбранный из группы, состоящей из куркумина, производных куркумина, цикориевой кислоты, производных цикориевой кислоты, 3,5-дикофеоилхинной кислоты, производных 3,5-дикофеоилхинной кислоты, ауринтрикарбоновой кислоты, производных ауринтрикарбоновой кислоты, фенэтилового эфира кофейной кислоты, производных фенэтилового эфира кофейной кислоты, тирфостина, производных тирфостина, кверцетина, производных кверцетина, ралтегравира, элвитегравира, долутегравира и каботегравира;
- ингибитор некаталитического (или аллостерического) сайта интегразы ВИЧ (NCINI), выбранный из группы, состоящей из СХ-05168, СХ-05045 и СХ-14442;
- ингибиторов сборки капсида ВИЧ, выбранный из группы, состоящей из GS-CA1;
- ингибитор gp41 ВИЧ, выбранный из группы, состоящей из энфувиртида, сифувиртида и альбувиртида;
- ингибитор проникновения ВИЧ, выбранный из группы, состоящей из ценикривирока;
- ингибитор gpl20 ВИЧ, выбранный из группы, состоящей из Radha-108 (рецептола) и BMS-663068;
- ингибитор CCR5, выбранный из группы, состоящей из аплавирока, викривирока, маравирока, ценикривирока, PRO-140, адаптавира (RAP-101), TBR-220 (ТАК-220), нифевирока (TD-0232), TD-0680 и vMIP (Haimipu);
- ингибиторов присоединения CD4, выбранные из группы, состоящей из ибализумаба;
- ингибитор CXCR4, выбранный из группы, состоящей из плериксафора, ALT-1188, vMIP и Haimipu;
- фармакокинетический усилитель, выбранный из группы, состоящей из кобицистата и ритонавира;
- иммунологического лекарственного средства, выбранного из группы, состоящей из dermaVir, интерлейкина-7, плаквенила (гидроксихлорохина), пролейкина (альдеслейкина, ИЛ-2), интерферона альфа, интерферона альфа-2Ь, интерферона альфа-пЗ, пегилированного интерферона альфа, интерферона гамма, гидроксимочевины, микофеноловой кислоты (МРА) и ее эфирного производного микофенолата мофетила (MMF), WF-10, рибавирина, ИЛ-2, ИЛ-12, полимера полиэтиленимина (PEI), гепона, VGV-1, MOR-22, BMS-936559, модуляторов толл-подобных рецепторов (tirl, tlr2, tlr3, tlr4, tlr5, tlr6, tlr7, tlr8, tlr9, tlrlO, tlrll, tlrl2 и tlrl3), ринтатолимода и IR-103;
- вакцины против ВИЧ, выбранной из группы, состоящей из пептидных вакцин, вакцин на основе рекомбинантных субъединиц, живых векторных вакцин, ДНК-вакцин, вакцин на основе вирусо-подобных частиц (вакцин на основе псевдовирионов), пептидных вакцин на основе производных CD4, комбинаций вакцин, rgpl20 (AIDSVAX), ALVAC HIV (vCP1521)/AIDSVAX B/E (gpl20) (RV144), Remune, ITV-1, Contre Vir, Ad5-ENVA-48, DCVax-001 (CDX-2401), PEP-6409, Vacc-4x, Vacc-C5, VAC-3 S, мультивидовой вакцины на основе рекомбинантной ДНК аденовируса серотипа 5 (rAd5), Pennvax-G, VRC-HIV MAB060-00-AB, AVX-101, вакцины Tat Oyi, AVX-201, HIV-LAMP-vax, Ad35, Ad35- GRIN, NAcGM3/VSSP ISA-51, поли-ICLC адъювантных вакцин, Tatlmmune, GTU- multiHIV (FIT-06), AGS-004, gpl40[delta]V2.TVl+ MF-59, rVSVIN HIV-1 gag вакцин, SeV-Gag вакцин, АТ-20, DNK-4, Ad35-GRIN/ENV, TBC-M4, HIVAX, HIV AX-2, NYVAC-HIV-PT1, NYVAC-HIV-PT4, DNA-HIV-PT123, rAAV1-PG9DP, GOVX-B11, GOVX-B21, ThV-01, TUTI-16, VGX-3300, TVI-HIV-1, Ad-4 (Ad4-env Clade С + Ad4-mGag), EN41-UGR7C, EN41-FPA2, PreVaxTat, TL-01, SAV-001, AE-H, MYM-V101, CombiHIVvac, ADVAX, MYM-V201, MVA-CMDR, ETV-01 и DNA-Ad5 gag/pol/nef/nev (HVTN505);
- антитело к ВИЧ, биспецифического антитела и «антителоподобных» терапевтических белков (таких как DARTs®, Duobodies®, Bites®, XmAbs®, TandAbs®, производные Fab), включая BMS-936559, TMB-360 и лекарственное средство, действующее на gpl20 или gp41 ВИЧ, выбранное из группы, состоящей из бавитуксимаба, UB-421, C2F5, C2G12, С4Е10, C2F5+C2G12+C4E10, 3-BNC-117, PGT145, PGT121, MDX010 (ипилимумаба), VRC01, А32, 7 В2, 10Е8 и VRC07;
- агентов, обращающих латентность, выбранных из группы, состоящей из ингибиторов гистоновой диацетилазы, таких как ромидепсин, вориностат, панобиностат; ингибиторов протеасомы, таких как велкейд; активаторов протеинкиназы С (РКС), таких как индолактам, простратин, ингенол В и лактоны ДАТ, иономицин, GSK-343, РМА, SAHA, ингибиторы BRD4, ИЛ-15, JQ1, дисульфирам и амфотерицин В;
- ингибитора белка нуклеокапсида ВИЧ р7 (NCp7), выбранного из группы, состоящей из азодикарбонамида;
- ингибитора созревания ВИЧ, выбранного из группы, состоящей из BMS- 955176 и GSK-2838232;
- ингибитора PI3K, выбранного из группы, состоящей из иделалисиба, AZD- 8186, бупарлисиба, CLR-457, пиктилисиба, нератиниба, ригосертиба, ригосертиба натрия, EN-3342, TGR-1202, алпелисиба, дувелисиба, UCB-5857, таселисиба, XL- 765, гедатолисиба, VS-5584, копанлисиба, КАТ оротата, перифосина, RG-7666, GSK-2636771, DS-7423, панулисиба, GSK-2269557, GSK-2126458, CUDC-907, PQR- 309, INCB-040093, пиларалисиба, BAY-1082439, пуквитиниба мезилата, SAR- 245409, AMG-319, RP-6530, ZSTK-474, MLN-1117, SF-1126, RV-1729, сонолисиба, LY-3023414, SAR-260301
- соединений, раскрытых в WO 2004/096286, WO 2006/110157, WO 2006/015261, WO 2013/006738, US 2013/0165489, US 20140221380, US 20140221378, WO 2013/006792, WO 2009/062285, WO 2010/130034, WO 2013/091096 A1, WO 2013/159064, WO 2012/145728, WO 2012/003497, WO 2014/100323, WO 2012/145728, WO 2013/159064 и WO 2012/003498;
- других лекарственных средств для лечения ВИЧ, выбранных из группы, состоящей из BanLec, МК-8507, AG-1105, TR-452, МК-8591, REP 9, CYT-107, алиспоривира, NOV-205, IND-02, мет-энкефалина, PGN-007, ацеманнана, гамимуна, проластина, 1,5-дикофеоилхинной кислоты, BIT-225, RPI-MN, VSSP, хивирала (Hiviral), 1МО-ЗЮО, SB-728-T, RPI-MN, VIR-576, HGTV-43, МК-1376, rHIV7-shl-TAR-CCR5RZ, MazF-генной терапии, BlockAide, ABX-464, SCY-635, налтрексона, AAV-eCD4-Ig-reHHoii терапии и PA-1050040 (PA-040).
В некоторых вариантах реализации данного изобретения, раскрытое в настоящем описании соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемая соль, или их сольват, или их кристаллическая или поликристаллическая форма объединяют с одним, двумя, тремя, четырьмя или более дополнительными терапевтическими агентами.
Указанные один, два, три, четыре или более дополнительных терапевтических агента могут представлять собой различные терапевтические агенты, выбранные из одного класса терапевтических агентов, и/или они могут быть выбраны из различных классов терапевтических агентов.
В конкретном варианте реализации данного изобретения, раскрытое в настоящем описании соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемая соль, или их сольват, или их кристаллическая или поликристаллическая форма объединяют с ненуклеозидным ингибитором обратной транскриптазы ВИЧ.
В конкретном варианте реализации данного изобретения, раскрытое в настоящем описании соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемая соль, или их сольват объединяют с нуклеозидным или нуклеотидным ингибитором обратной транскриптазы ВИЧ и ненуклеозидным ингибитором обратной транскриптазы ВИЧ.
В другом конкретном варианте реализации данного изобретения, раскрытое в настоящем описании соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемая соль, или их сольват, или их кристаллическая или поликристаллическая форма объединяют с нуклеозидным или нуклеотидным ингибитором обратной транскриптазы ВИЧ и соединением, ингибирующим протеазу ВИЧ.
В другом варианте реализации данного изобретения, раскрытое в настоящем описании соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемая соль, или их сольват, или их кристаллическая или поликристаллическая форма объединяют с нуклеозидным или нуклеотидным ингибитором обратной транскриптазы ВИЧ, ненуклеозидным ингибитором обратной транскриптазы ВИЧ и соединением, ингибирующим протеазу ВИЧ.
В другом варианте реализации данного изобретения, раскрытое в настоящем описании соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемая соль, или их сольват, или их кристаллическая или поликристаллическая форма объединяют с нуклеозидным или нуклеотидным ингибитором обратной транскриптазы ВИЧ, ненуклеозидным ингибитором обратной транскриптазы ВИЧ, ингибиторов сборки капсида ВИЧ, и фармакокинетическим усилителем.
В некоторых вариантах реализации данного изобретения, раскрытое в настоящем описании соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемая соль, или их сольват, или их кристаллическая или поликристаллическая форма объединяют с по меньшей мере одним нуклеозидным ингибитором обратной транскриптазы ВИЧ, ингибитором интегразы и фармакокинетическим усилителем.
В другом конкретном варианте реализации данного изобретения, раскрытое в настоящем описании соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемая соль, или их сольват, или их кристаллическая или поликристаллическая форма объединяют с двумя нуклеозидными или нуклеотидными ингибиторами обратной транскриптазы ВИЧ.
В конкретном варианте реализации данного изобретения, раскрытое в настоящем описании соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемую соль, или их сольват, или их кристаллическую или поликристаллическую форму объединяют с абакавира сульфатом, тенофовиром, тенофовир дизопроксилом, тенофовир дезопроксил фумаратом, тенофовир дезопроксил полуфумаратом, тенофовир алафенамидом, тенофовир алафенамид полуфумаратом, тенофовир алафенамид фумаратом, тенофовир циклобутилалафенамидом, тенофовир циклобутилалафенамид полуфумаратом, тенофовир циклобутилалафенамид фумаратом, элсульфавирином, VM-1500A или GS-CA1.
В конкретном варианте реализации данного изобретения, раскрытое в настоящем описании соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемая соль, или их сольват, или их кристаллическая или поликристаллическая форма объединяют с первым дополнительным терапевтическим агентом, выбранным из группы, состоящей из: абакавир сульфата, тенофовира, тенофовир дизопроксила, тенофовир дезопроксил фумарата, тенофовир дезопроксил полуфумарата, тенофовир алафенамида, тенофовир алафенамид полуфумарата, тенофовир алафенамид фумарата, тенофовир циклобутилалафенамида, тенофовир циклобутилалафенамид полуфумарата, тенофовир циклобутилалафенамид фумарата, элсульфавирина, VM-1500A или GS-CA1, и вторым дополнительным терапевтическим агентом, выбранным из группы, состоящей из эмтрицитабина и ламивудина.
В конкретном варианте реализации данного изобретения, раскрытое в настоящем описании соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемую соль, или их сольват, или их кристаллическую или поликристаллическую форму объединяют с первым дополнительным терапевтическим агентом, выбранным из группы, состоящей из: тенофовира, тенофовир дизопроксила, тенофовир дезопроксил фумарата, тенофовир дезопроксил полуфумарата, тенофовир алафенамида, тенофовир алафенамид полуфумарата, тенофовир алафенамид фумарата, тенофовир циклобутилалафенамида, тенофовир циклобутилалафенамид полуфумарата или тенофовир циклобутилалафенамид фумарата, элсульфавирина, VM-1500A или GS-CA1, и вторым дополнительным терапевтическим агентом, эмтрицитабином.
В конкретном варианте реализации данного изобретения, раскрытое в настоящем описании соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемую соль, или их сольват, или их кристаллическую или поликристаллическую форму объединяют с 5-30 мг тенофовира, тенофовир дизопроксила, тенофовир дезопроксил фумарата, тенофовир дезопроксил полуфумарата, тенофовир алафенамида, тенофовир алафенамид полуфумарата, тенофовир алафенамид фумарата, тенофовир циклобутилалафенамида, тенофовир циклобутилалафенамид полуфумарата, тенофовир циклобутилалафенамид фумарата, элсульфавирина, VM-1500A или GS-CA1, и 200 мг эмтрицитабина.
В некоторых вариантах реализации данного изобретения, раскрытое в настоящем описании соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемую соль, или их сольват, или их кристаллическую или поликристаллическую форму объединяют с 5-10 мг; 5-15 мг; 5-20 мг; 5-25 мг; 25-30 мг; 20-30 мг; 15-30 мг или 10-30 мг тенофовира, тенофовир дизопроксила, тенофовир дезопроксил фумарата, тенофовир дезопроксил полуфумарата, тенофовир алафенамида, тенофовир алафенамид полуфумарата, тенофовир алафенамид фумарата, тенофовир циклобутилалафенамида, тенофовир циклобутилалафенамид полуфумарата, тенофовир циклобутилалафенамид фумарата, элсульфавирина, VM-1500A или GS-CA1, и 200 мг эмтрицитабина.
В конкретном варианте реализации данного изобретения, раскрытое в настоящем описании соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемую соль, или их сольват, или их кристаллическую или поликристаллическую форму объединяют с 10 мг тенофовира, тенофовир дизопроксила, тенофовир дезопроксил фумарата, тенофовир дезопроксил полуфумарата, тенофовир алафенамида, тенофовир алафенамид полуфумарата, тенофовир алафенамид фумарата, тенофовир циклобутилалафенамида, тенофовир циклобутилалафенамид полуфумарата, тенофовир циклобутилалафенамид фумарата, элсульфавирина, VM-1500A или GS-CA1, и 200 мг эмтрицитабина.
В конкретном варианте реализации данного изобретения, раскрытое в настоящем описании соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемую соль, или их сольват, или их кристаллическую или поликристаллическую форму объединяют с 25 мг тенофовира, тенофовир дизопроксила, тенофовир дезопроксил фумарата, тенофовир дезопроксил полуфумарата, тенофовир алафенамида, тенофовир алафенамид полуфумарата, тенофовир алафенамид фумарата, тенофовир циклобутилалафенамида, тенофовир циклобутилалафенамид полуфумарата, тенофовир циклобутилалафенамид фумарата, элсульфавирина, VM-1500A или GS-CA1, и 200 мг эмтрицитабина.
В указанных выше вариантах реализации изобретения дополнительный терапевтический агент может представлять собой указанный выше агент против ВГВ и/или ВГС.
В некоторых вариантах реализации данного изобретения, раскрытое в настоящем описании соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемую соль, или их сольват, или их кристаллическую или поликристаллическую форму или фармацевтическую композицию объединяют с одним или более дополнительными терапевтическими агентами, как описано выше, указанные компоненты композиции вводят одновременно или последовательно. При последовательном введении указанная комбинация может быть введена в виде двух или более введений.
В некоторых вариантах реализации данного изобретения, раскрытое в настоящем описании соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемую соль, или их сольват, или их кристаллическую или поликристаллическую форму объединяют с одним или более дополнительными терапевтическими агентами в единой лекарственной форме для одновременного введения пациенту, например, в виде твердой лекарственной формы для перорального введения с фиксированными дозами компонентов.
В некоторых вариантах реализации данного изобретения, раскрытое в настоящем описании соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемую соль, или их сольват, или их кристаллическую или поликристаллическую форму вводят с одним или более дополнительными терапевтическими агентами.
Совместное введение соединения, раскрытого в настоящем описании, с одним или более дополнительными терапевтическими агентами в целом относится к одновременному или последовательному введению соединения, раскрытого в настоящем описании, и одного или более дополнительных терапевтических агентов с обеспечением присутствия терапевтически эффективных количеств соединения, раскрытого в настоящем описании, и терапевтически эффективных количеств одного или более дополнительных терапевтических агентов в организме пациента.
Совместное введение включает введение единичных доз соединений, раскрытых в настоящем описании, до или после введения единичных доз одного или более дополнительных терапевтических агентов, например, введение соединения, раскрытого в настоящем описании, в течение секунд, минут или часов до или после введения одного или нескольких дополнительных терапевтических агентов. Например, в некоторых вариантах реализации единичную дозу соединения, раскрытого в настоящем описании, вводят первой с последующим введением единичной дозы одного или более дополнительных терапевтических агентов в течение секунд или минут.
В качестве альтернативы, в других вариантах реализации единичную дозу одного или более дополнительных терапевтических агентов вводят первой с последующим введением единичной дозы соединения, раскрытого в настоящем описании, в течение секунд или минут. В некоторых вариантах реализации данного изобретения единичную дозу соединения, раскрытого в настоящем описании, вводят первой с последующим введением единичной дозы одного или более дополнительных терапевтических агентов через несколько часов (например, через 1-12 часов).
В других вариантах реализации единичную дозу одного или более дополнительных терапевтических агентов вводят первой с последующим введением единичной дозы соединения, раскрытого в настоящем описании, через несколько часов (например, через 1-12 часов).
В другом варианте реализации данного изобретения соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемая соль, или их сольват, или их кристаллическая или поликристаллическая форма или фармацевтическая композиция, раскрытая в настоящем описании, предложены для применения в терапии (способе лечения) инфекции ВИЧ у человека, имеющего указанную инфекцию или имеющего риск приобретения указанной инфекции.
В другом варианте реализации данного изобретения соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемая соль, или их сольват, или их кристаллическая или поликристаллическая форма или фармацевтическая композиця, раскрытая в настоящем описании, предложены для применения в терапии инфекции ВИЧ у человека, имеющего указанную инфекцию или имеющего риск приобретения указанной инфекции, причем указанная терапия дополнительно включает введение указанному человеку одного или более дополнительных терапевтических агентов.
В другом варианте реализации данного изобретения соединение формулы (1) или (2) или их стереоизомер, или их новую фармацевтически приемлемая соль, или их сольват, или их кристаллическая или поликристаллическая форма или фармацевтическая композиция, раскрытая в настоящем описании, предложены для применения в терапии инфекции ВИЧ у человека, имеющего указанную инфекцию или имеющего риск приобретения указанной инфекции, причем указанная терапия дополнительно включает введение указанному человеку одного или более дополнительных терапевтических агентов, выбранных из группы, состоящей из ингибиторов протеазы ВИЧ, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, нуклеозидных или нуклеотидных ингибиторов обратной транскриптазы ВИЧ, аллостерических ингибиторов интегразы ВИЧ, ингибиторов сборки капсида ВИЧ и их комбинаций.
В другом варианте реализации данного изобретения соединение формулы (1) или (2) или их стереоизомер, или их фармацевтически приемлемая соль, или их сольват, или их кристаллическая или поликристаллическая форма или фармацевтическая композиция, раскрытая в настоящем описании, предложены для применения в терапии инфекции ВИЧ у человека, имеющего указанную инфекцию или имеющего риск приобретения указанной инфекции, причем указанный способ включает введение указанному человеку единой лекарственной формы для одновременного введения пациенту, например, в виде твердой лекарственной формы для перорального введения, соединения формулы (1) или (2) или их стереоизомера, или их фармацевтически приемлемой соли, или их сольвата, или их кристаллической или поликристаллической формы, или фармацевтическая композиция, раскрытая в настоящем описании, совместно с первым дополнительным терапевтическим агентом, выбранным из группы, состоящей из абакавир сульфата, тенофовира, тенофовир дизопроксила, тенофовир дезопроксил фумарата, тенофовир дезопроксил полуфумарата, тенофовир алафенамида, тенофовир алафенамид полуфумарата, тенофовир циклобутилалафенамида, тенофовир циклобутилалафенамид полуфумарата, тенофовир циклобутилалафенамид фумарата, элсульфавирина, VM-1500A или GS-CA1, и вторым дополнительным терапевтическим агентом, выбранным из группы, состоящей из эмтрицитабина и ламивудина.
Фармацевтические композиции согласно вариантам реализации, раскрытым в настоящем описании, могут быть приготовлены посредством объединения соединения формулы (1) или (2) или их стереоизомера, или их фармацевтически приемлемая соли, или их сольвата с соответствующим фармацевтически приемлемым вспомогательным веществом и могут быть приготовлены в виде препаратов в твердых, полутвердых, жидких или газообразных формах, таких как таблетки, капсулы, порошки, гранулы, мази, растворы, наносуспензии, суппозитории, инъекции, ингалянты, гели, микросферы и аэрозоли. Типичные пути введения указанных фармацевтических композиций включают, без ограничения, пероральный, местный, трансдермальный, ингаляционный, парентеральный, сублингвальный, буккальный, ректальный, вагинальный и интраназальный способы.
Фармацевтические композиции согласно вариантам реализации данного изобретения, раскрытым в настоящем описании, готовят таким образом, чтобы обеспечить биодоступность активных ингредиентов, входящих в их состав, при введении композиции субъекту. Композиции, которые будут введены субъекту или пациенту, представлены в лекарственных формах, содержащих одну или более единиц дозирования, где, например, таблетка может представлять собой лекарственную форму, содержащую одну единицу дозирования, а емкость с соединением согласно вариантам реализации, раскрытым в настоящем описании, в форме аэрозоля может содержать множество единиц дозирования. Фактические способы получения указанных лекарственных форм известны или будут очевидны специалистам в данной области техники; например, см. Remington. The Science and Practice of Pharmacy, 20th Edition (Philadelphia College of Pharmacy and Science, 2000). Композиция, подлежащая введению, будет в любом случае содержать терапевтически эффективное количество соединения согласно вариантам реализации, раскрытым в настоящем описании, для терапии представляющего интерес заболевания или состояния в соответствии с рекомендациями настоящего описания.
Фармацевтические композиции, раскрытые в настоящем описании, могут быть получены способами, хорошо известными в фармацевтической области. Например, фармацевтическая композиция, предназначенная для введения посредством инъекции, может быть приготовлена посредством объединения соединения согласно вариантам реализации, раскрытым в настоящем описании, со стерильной дистиллированной водой с образованием раствора или наносуспензии. Для обеспечения образования гомогенного раствора или суспензии может быть добавлено поверхностно-активное вещество. Поверхностно-активные вещества представляют собой соединения, нековалентно взаимодействующие с соединением согласно вариантам реализации, раскрытым в настоящем описании, способствуя растворению или обеспечивая гомогенную суспензию соединения в водной системе доставки.
Предметом данного изобретения является способ профилактики и лечения инфекции ВИЧ у человека, имеющего указанную инфекцию или имеющего риск приобретения указанной инфекции, путем введения указанному человеку терапевтически эффективного количества соединения общей формулы 1 или 2, или фармацевтической композиции содержащей соединение общей формулы 1 или 2,.
Предметом данного изобретения является способ профилактики и лечения инфекции ВИЧ у человека, имеющего указанную инфекцию или имеющего риск приобретения указанной инфекции, путем перорального введения указанному человеку терапевтически эффективного количества соединения общей формулы 1 или 2, или фармацевтической содержащей соединение общей формулы 1 или 2,.
Предметом данного изобретения является способ лечения инфекции ВИЧ у человека, имеющего указанную инфекцию или имеющего риск приобретения указанной инфекции, путем инъекционного введения указанному человеку терапевтически эффективного количества наносуспензии, содержащей соединение общей формулы (1) или соединение общей формулы (2) или их стереоизомер.
Изобретение иллюстрируется следующими чертежами:
Фиг. 1. Моделирование дифрактограммы образца (3S,11aR)-6-гидрокси-N-[(2,6-дифтор-3-пиридил)метил]-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазол[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамида (1.1.19) методом Паули. Синей линией обозначена экспериментальная дифрактограмма, красной - расчетная, серой - разностная кривая. Вертикальными линиями обозначены пики примесей.
Фиг. 2а. Стабильность соединений общей формулы 1.1 в плазме человека.
Фиг. 2б. Стабильность соединений общей формулы 1.1 в плазме крыс.
Фиг. 3а. Стабильность соединений общей формулы 1.1 в S9 фракции печени человека.
Фиг. 3б. Стабильность соединений общей формулы 1.1 в S9 фракции печени крыс.
Следующие примеры поясняют, но не ограничивают данное изобретение.
Пример 1. Физико-химические исследования соединений общей формулы 1 и 2.
Общие процедуры по химии. Все химические вещества и растворители использовались в том виде, в котором они были получены от поставщиков, без дальнейшей очистки. Неочищенные реакционные смеси концентрировали при пониженном давлении путем удаления органических растворителей на роторном испарителе.
Спектры ядерного магнитного резонанса (NMR) регистрировали с использованием спектрометра Broker DPX-400 при комнатной температуре (к.т.) с тетраметилсиланом в качестве внутреннего стандарта. Химические сдвиги (δ) представлены в частях на миллион (ppm), а сигналы представлены в виде s (синглет), d (дублет), t (триплет), q (квартет), m (мультиплет) или br s. (широкий синглет).
Масс-спектры высокого разрешения (HRMS) получали на масс-спектрометре Orbitrap Elite (Thermo, Бремен, Германия), оборудованном источником ионов HESI.
Высокоэффективная жидкостная хроматография (ВЭЖХ). Чистота конечных соединений была определена с помощью ВЭЖХ и составляла более 98%. Условия ВЭЖХ для оценки чистоты были следующими: ВЭЖХ Shimadzu, XBridge С18, 4,6 мм × 250 мм (3,5 мкм); градиент 0,1% TFA в 5% ацетонитриле / воде (А) и 0,1% TFA ацетонитриле (В); скорость потока 0,5 мл / мин; время сбора 20 мин; длина волны, УФ 214 и 254 нм. Система препаративной ВЭЖХ включала два набора насосов Shimadzu LC-8A, контроллер Shimadzu SCL 10Avp и детектор Shimadzu SPD 10Avp. Использовали колонку Reprosil-Pur C18-AQ 10 мкм, 250 мм × 20 мм. Подвижная фаза имела градиент 0,1% TFA в воде (А) и 0,1% TFA в ацетонитриле (В). ЖХ / МС (LC/MS) проводили на системе РЕ Sciex API 165 с использованием электрораспыления в режиме положительных ионов [M+Н]+ и системы ВЭЖХ Shimadzu, оснащенной колонкой Waters XBridge С18 3,5 мкм (4,6 мм × 150 мм). Диастериоизомеры делили на хиральной ВЭЖХ Phenomenex Lux 5u Cellulose-4, AXIA F, 250×30.00 mm. Скорость потока: 25 мл/мин. Детектор: УФ, 215 нм.
Рентгенофазовый анализ. Дифрактограммы получали на дифрактометре Bruker D8 Advance Vario, оснащенном рентгеновской трубкой с медным анодом и Ge(III)-монохроматором (CuKα1) и позиционно-чувствительным детектором LynxEye, в установках на просвет. Интервал съемки составил 2-60° 1θ, шаг 0.02° 1θ. Анализ проводили в программе Bruker Topas 5 [Bruker TOPAS5 User Manual. - Karlsruhe, Germany: Bruker AXS GmbH, 2015].
Пример 2. Общий способ получения анелированных 9-гидрокси-1,8-диоксо-1,3,4,8-тетрагидро-2Н-пиридо[1,2-а]пиразин-7-карбоксамидов (общей формулы 1 или 2) по схеме 1.
К раствору 0.5 ммоль соответствующего анелированноого 9-бензилокси-7-бром-1,8-диоксо-1,3,4,8-тетрагидро-2Н-пиридо[1,2-a]пиразина формулы 3 или 4 в 2 мл ДМСО добавляли 0.75 ммоль гетероциклилметиламина или его гидрохлорида, 0.131 мл (0.75 ммоль) (плюс 0.75 ммоль на каждый гидрохлорид) диизопропиламина и 29 мг (0.025 ммоль) Pd(PPh)4. Реакционную массу перемешивали 14 ч при 90°С в атмосфере СО. По окончании реакции (контроль LC-MS) реакционную массу упаривали в вакууме. Остаток растворяли в дихлорметане, промывали водой, сушили над сульфатом натрия, упаривали на роторном испарителе и подвергали колоночной хроматографии на силикагеле. Получали соответствующий анелированный 9-бензилокси-1,8-диоксо-1,3,4,8-тетрагидро-2Н-пиридо[1,2-а]пиразин-7-карбоксамид (формулы 6 или 7).
a) Соединение формулы 6 или 7 (0.35 ммоль) растворяли в смеси 18 мл ТГФ и 2 мл метанола, добавляли 0.04 г 10% Pd/C и перемешивали в атмосфере водорода 8 ч. Реакционную массу пропускали через целит, фильтрат упаривали. Остаток обрабатывали эфиром, осадок отфильтровывали, сушили в вакууме и получали соответствующий анелированный 9-гидрокси-1,8-диоксо-1,3,4,8-тетрагидро-2Н-пиридо[1,2-а]пиразин-7-карбоксамид (общей формулы 1 или 2).
b) Соединение формулы 6 или 7 (0.35 ммоль) растворяли в 2 мл трифторуксусной кислоты и перемешивали в 2 ч при комнатной температуре. Раствор упаривали в вакууме, остаток растворяли в хлороформе, промывали насыщенным раствором NaHCO3, сушили над Na2SO4 и упаривали в вакууме. Остаток обрабатывали эфиром, осадок отфильтровывали, сушили в вакууме и получали соответствующий анелированный 9-гидрокси-1,8-диоксо-1,3,4,8-тетрагидро-2Н-пиридо[1,2-a]пиразин-7-карбоксамид (общей формулы 1 или 2).
с) Соединение формулы 6 или 7 (0.35 ммоль) растворяли в 1.5 мл N,N-диметилацетамида, добавляли 0.148 г (3.5 ммоль) LiCl и перемешивали 3 ч при 80°С. По окончании реакции (контроль LC-MS) реакционную массу упаривали в вакууме. Продукт выделяли методом ВЭЖХ и получали соответствующий анелированный 9-гидрокси-1,8-диоксо-1,3,4,8-тетрагидро-2Н-пиридо[1,2-a]пиразин-7-карбоксамид (общей формулы 1 или 2). в том числе:
(3S,11aR)-6-гидрокси-3-метил-5,7-диоксо-N-(2-тиенилметил)-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.1): LC-MS (M+1)=376;1H NMR (DMSO-d6, 400 MHz) δ 11.47 (brs, 1H), 10.33 (t, J=4.8 Hz, 1H), 8.49 (s, 1H), 7.40 (dd, J1=8.8 Hz, J2=4.8 Hz, 1H), 7.03 (d, J=2.8 Hz, 1H), 6.96 (dd, J1=4.8 Hz, J2=3.6 Hz, 1H), 5.39 (dd, J1=10.0 Hz, J2=4.0 Hz, 1H), 4.90 (dd, J1=12.0 Hz, J2=4.0 Hz, 1H), 4.70 (d, J=5.6 Hz, 2H), 4.40 (dd, J1=8.4 Hz, J2=7.2 Hz, 1H), 4.29 (m, 1H), 4.01 (t, J=11.2 Hz, 1H), 3.67 (dd, J1=8.4 Hz, J2=7.2 Hz, 1H), 1.34 (d, J=6.0 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-[(5-метил-2-фурил)метил]-5,7-диоксо-2,3,5,7,11,11a-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.3): LC-MS (ESI) 374 (M+H)+;1Н NMR (DMSO-d6, 400 MHz) δ 11.46 (brs, 1H), 10.20 (t, J=5.2 Hz, 1H), 8.46 (s, 1H), 6.15 (d, J=2.4 Hz, 1H), 5.99 (d, J=2.4 Hz, 1H), 5.39 (dd, J1=9.6 Hz, J2=4.0 Hz, 1H), 4.89 (dd, J1=12.0 Hz, J2=4.0 Hz, 1H), 4.46 (d, J=5.2 Hz, 2H), 4.39 (dd, J1=8.4 Hz, J2=7.2 Hz, 1H), 4.29 (m, 1H), 4.00 (t, J=11.0 Hz, 1H), 3.66 (dd, J1=8.4 Hz, J2=7.2 Hz, 1H), 2.23 (s, 3H), 1.34 (d, J=6.0 Hz, 3H).
(3S,11aR)-6-гидрокси-N-[(3,5-диметил-1Н-пиразол-4-ил)метил]-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.4): LC-MS (ESI) 388 (M+H)+;1Н NMR (DMSO-d6, 400 MHz) δ 12.04 (brs, 1H), 11.42 (brs, 1H), 9.92 (t, J=4.8 Hz, 1H), 8.46 (s, 1H), 5.38 (dd, J1=10.0 Hz, J2=4.0 Hz, 1H), 4.88 (dd, J1=12.0 Hz, J2=4.0 Hz, 1H), 4.38 (dd, J1=8.0 Hz, J2=7.2 Hz, 1H), 4.29 (m, 1H), 4.26 (d, J=5.2 Hz, 2H), 4.00 (t, J=11.2 Hz, 1H), 3.66 (dd, J1=8.0 Hz, J2=7.2 Hz, 1H), 1.33 (d, J=6.4 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-[(1,3,5-триметил-1Н-пиразол-4-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.5): LC-MS (ESI) 402 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.40 (brs, 1H), 9.92 (t, J=4.8 Hz, 1H), 8.46 (s, 1H), 5.38 (dd, J1=9.6 Hz, J2=3.6 Hz, 1H), 4.88 (dd, J1=12.0 Hz, J2=3.6 Hz, 1H), 4.39 (t, J=7.4 Hz, 1H), 4.29 (m, 1H), 4.25 (d, J=4.8 Hz, 2H), 4.00 (t, J=11.0 Hz, 1H), 3.66 (t, J=7.4 Hz, 1H), 3.32 (s, 3H), 2.19 (s, 3H), 2.08 (s, 3H), 1.33 (d, J=6.0 Hz, 3H).
(3S,11aR)-6-гидрокси-N-[(1-этил-3,5-диметил-1Н-пиразол-4-ил)метил]3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.6): LC-MS (ESI) 416 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.40 (brs, 1H), 9.92 (t, J=5.2 Hz, 1H), 8.47 (s, 1H), 5.38 (dd, J1=10.0 Hz, J2=4.0 Hz, 1H), 4.88 (dd, J1=12.0 Hz, J2=4.0 Hz, 1H), 4.39 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 4.28 (m, 1H), 4.25 (d, J=5.6 Hz, 2H), 4.00 (t, J=11.2 Hz, 1H), 3.94 (q, J=7.2 Hz, 2H), 3.66 (dd, J1=8.4 Hz, J2=6.4 Hz, 1H), 2.20 (s, 3H), 2.09 (s, 3H), 1.33 (d, J=6.0 Hz, 3H), 1.24 (t, J=7.2 Hz, 3H).
(3S,11aR)-6-гидрокси-N-[(4,5-дихлор-1-метил-1Н-пиразол-3-ил)метил]3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.7): LC-MS (ESI) 443 (M+H)+;1Н NMR (DMSO-d6, 400 MHz) δ 11.44 (brs, 1H), 10.28 (t, J=5.6 Hz, 1H), 8.46 (s, 1H), 5.39 (dd, J1=10.0 Hz, J2=3.6 Hz, 1H), 4.89 (dd, J1=12.0 Hz, J2=4.0 Hz, 1H), 4.50 (d, J=5.6 Hz, 2H), 4.39 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 4.29 (sxt, J=6.4 Hz, 1H), 4.01 (t, J=11.2 Hz, 1H), 3.80 (s, 3H), 3.66 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 1.34 (d, J=6.0 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-(тиазол-2-илметил)-5,7-диоксо-2,3,5,7,11,11a-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.8): LC-MS (ESI) 377 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.50 (brs, 1H), 10.58 (t, J=6.0 Hz, 1H), 8.49 (s, 1H), 7.74 (d, J=3.2 Hz, 1H), 7.62 (d, J=3.2 Hz, 1H), 5.40 (dd, J1=10.0 Hz, J2=4.0 Hz, 1H), 4.90 (dd, J1=12.0 Hz, J2=4.0 Hz, 1H), 4.84 (d, J=6.0 Hz, 2H), 4.40 (dd, J1=8.4 Hz, J2=7.2 Hz, 1H), 4.30 (m, 1H), 4.01 (dd, J1=12.0 Hz, J2=10.0 Hz, 1H), 3.67 (dd, J1=8.4 Hz, J2=6.4 Hz, 1H), 1.35 (d, J=6.4 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-[(4-метил-1,2,5-оксадиазол-3-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.12): LC-MS (ESI) 376 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.51 (brs, 1H), 10.45 (t, J=5.6 Hz, 1H), 8.47 (s, 1H), 5.39 (dd, J1=10.0 Hz, J2=4.0 Hz, 1H), 4.89 (dd, J1=12.0 Hz, J2=4.0 Hz, 1H), 4.74 (d, J=5.6 Hz, 2H), 4.40 (dd, J1=8.4 Hz, J2=7.2 Hz, 1H), 4.29 (m, 1H), 4.00 (t, J=11.0 Hz, 1H), 3.67 (dd, J1=8.0 Hz, J2=7.2 Hz, 1H), 2.37 (s, 3H), 1.34 (d, J=6.0 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-[(4-метил-4Н-1,2,4-триазол-3-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.14): LC-MS (ESI) 375 (M+H)+;1Н NMR (DMSO-d6, 400 MHz) δ 11.49 (brs, 1H), 10.41 (t, J=5.2 Hz, 1H), 8.48 (s, 1H), 8.40 (s, 1H), 5.39 (dd, J1=10.0 Hz, J2=3.6 Hz, 1H), 4.89 (dd, J1=11.6 Hz, J2=3.6 Hz, 1H), 4.69 (d, J=5.2 Hz, 2H), 4.39 (t, J=7.6 Hz, 1H), 4.29 (m, 1H), 4.01 (t, J=11.2 Hz, 1H), 3.66 (m, 1H), 3.65 (s, 3H), 1.34 (d, J=6.0 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-(пиридил-2-илметил)-5,7-диоксо-2,3,5,7,11,11a-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.15): LC-MS (ESI) 371 (M+H)+;1Н NMR (DMSO-d6, 400 MHz) δ 11.44 (brs, 1H), 10.52 (t, J=5.6 Hz, 1H), 8.53 (d, J=4.0 Hz, 1H), 8.48 (s, 1H), 7.76 (t, J=7.6 Hz, 1H), 7.33 (d, J=8.0 Hz, 1H), 7.28 (m, 1H), 5.39 (dd, J1=10.0 Hz, J2=3.6 Hz, 1H), 4.89 (dd, J1=12.0 Hz, J2=3.6 Hz, 1H), 4.64 (d, J=5.6 Hz, 2H), 4.40 (t, J=7.6 Hz, 1H), 4.30 (m, 1H), 4.01 (t, J=11.2 Hz, 1H), 3.66 (dd, J1=8.0 Hz, J2=6.8 Hz, 1H), 1.34 (d, J=6.0 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-[(2-метилпиримидин-4-ил)метил]-5,7-диоксо-2,3,5,7,11,11a-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.26): LC-MS (M+1)=386;1H NMR (DMSO-d6, 400 MHz) δ 11.50 (brs, 1H), 10.50 (t, J=6.0 Hz, 1H), 8.61 (d, J=5.0 Hz, 1H), 8.46 (s, 1H), 7.16 (d, J=5.0 Hz, 1H), 5.40 (dd, J1=10.0 Hz, J2=3.2 Hz, 1H), 4.89 (dd, J1=12.0 Hz, J2=3.2 Hz, 1H), 4.59 (d, J=6.0 Hz, 2H), 4.40 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 4.30 (m, 1H),4.01 (t, J=11.2 Hz, 1H), 3.67 (dd, J1=8.4 Hz, J2=6.4 Hz, 1H), 2.60 (s, 3H), 1.35 (d, J=6.0 Hz, 3H).
(3S,11aR)-N-(1-бензотиен-5-илметил)-6-гидрокси-3-метил-5,7-диоксо-2,3,5,7,11,11a-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.31): LC-MS (ESI) 426 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.47 (brs, 1H), 10.38 (t, J=6.0 Hz, 1H), 8.50 (s, 1H), 7.96 (d, J=8.4 Hz. 1H), 7.80 (s, 1H), 7.76 (d, J=9.4 Hz, 1H), 7.43 (d, J=9.4 Hz, 1H), 7.33 (dd, J1=8.4 Hz, J2=0.6 Hz, 1H), 5.39 (dd, J1=10.0 Hz, J2=4.0 Hz, 1H), 4.90 (dd, J1=12.0 Hz, J2=4.0 Hz, 1H), 4.66 (d, J=5.6 Hz, 2H), 4.40 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 4.30 (m, 1H), 4.01 (t, J=11.2 Hz, 1H), 3.67 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 1.34 (d, J=6.0 Hz, 3H).
(3S,11aR)-N-(1-бензофуран-2-илметил)-6-гидрокси-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.32): LC-MS (ESI) 410 (М+Н)+;1H NMR (DMSO-d6, 400 MHz) δ 11.50 (brs, 1H), 10.41 (t, J=5.6 Hz, 1H), 8.50 (s, 1H), 7.59 (d, J=7.6 Hz, 1H), 7.54 (d, J=7.6 Hz, 1H), 7.27 (t, J=7.6 Hz, 1H), 7.22 (t, J=7.2 Hz, 1H), 6.75 (s, 1H), 5.40 (dd, J1=10.0 Hz, J2=3.6 Hz, 1H), 4.91 (dd, J1=12.0 Hz, J2=3.6 Hz, 1H), 4.72 (d, J=5.6 Hz, 2H), 4.40 (t, J=7.6 Hz, 1H), 4.28 (sxt, J=6.0 Hz, 1H), 4.02 (t, J=11.0 Hz, 1H), 3.67 (t, J=7.6 Hz, 1H), 1.34 (d, J=6.0 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-[(1-метил-1Н-индол-5-ил)метил]-5,7-диоксо-2,3,5,7,11,11a-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.33): LC-MS (ESI) 423 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.43 (brs, 1H), 10.27 (t, J=6.0 Hz, 1H), 8.50 (s, 1H), 7.48 (s, 1H), 7.39 (d, J=8.8 Hz, 1H), 7.30 (d, J=2.8 Hz, 1H), 7.12 (dd, J1=8.8 Hz, J2=0.8 Hz, 1H), 6.37 (dd, J1=2.8 Hz, J2=0.4 Hz, 1H), 5.39 (dd, J1=10.0 Hz, J2=4.0 Hz, 1H), 4.90 (dd, J1=12.0 Hz, J2=4.0 Hz, 1H), 4.59 (d, J=5.6 Hz, 2H), 4.39 (dd, J1=8.4 Hz, J2=7.2 Hz, 1H), 4.29 (m, 1H), 4.01 (t, J=11.2 Hz, 1H), 3.77 (s, 3H), 3.66 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 1.34 (d, J=6.0 Hz, 3H).
(3S,11aR)-N-(1,3-бензодиоксол-5-илметил)-6-гидрокси-3-метил-5,7-диоксо-2,3,5,7,11,11a-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.34): LC-MS (ESI) 414 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.45 (brs, 1H), 10.24 (t, J=5.8 Hz, 1H), 8.48 (s, 1H), 6.86 (m, 2H), 6.79 (m, 1H), 5.98 (s, 2H), 5.39 (dd, J1=10.0 Hz, J2=4.0 Hz, 1H), 4.89 (dd, J1=12.4 Hz, J2=4.0 Hz, 1H), 4.43 (d, J=6.0 Hz, 2H), 4.40 (dd, J1=8.4 Hz, J2=7.2 Hz, 1H), 4.29 (m, 1H), 4.01 (t, J=11.2 Hz, 1H), 3.67 (dd, J1=8.4 Hz, J2=6.0 Hz, 1H), 1.34 (d, J=6.4 Hz, 3H).
(3S,11aR)-N-[(6-бромо-1,3-бензодиоксол-5-ил)метил]-6-гидрокси-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.35): LC-MS (ESI) 493 (M+H)+;1Н NMR (DMSO-d6, 400 MHz) δ 11.44 (brs, 1H), 10.30 (m, 1H), 8.46 (d, J=2.4 Hz, 1H), 7.23 (d, J=2.4 Hz, 1H), 6.93 (d, J=2.4 Hz, 1H), 6.05 (d, J=2.4 Hz, 2H), 5.38 (m, 1H), 4.88 (m, 1H), 4.46 (m, 2H), 4.39 (m, 1H), 4.29 (m, 1H), 4.00 (m, 1H), 3.66 (m, 1H), 1.34 (dd, J1=6.4 Hz, J2=2.8 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-[(7-метил-2,3-дигидро-1,4-бензодиоксин-6-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.36): LC-MS (ESI) 434 (M+H)+;1Н NMR (DMSO-d6, 400 MHz) δ 11.43 (brs, 1H), 10.14 (t, J=5.6 Hz, 1H), 8.47 (s, 1H), 6.73 (s, 1H), 6.68 (s, 1H), 5.39 (dd, J1=10.0 Hz, J2=4.0 Hz, 1H), 4.89 (dd, J1=12.0 Hz, J2=3.6 Hz, 1H), 4.39 (m, 3H), 4.29 (m, 1H), 4.18 (s, 4H), 4.00 (t, J=11.2 Hz, 1H), 3.66 (dd, J1=8.0 Hz, J2=7.2 Hz, 1H), 2.17 (s, 3H), 1.34 (d, J=6.0 Hz, 3H).
(3S,11aR)-N-(1,3-бензотиазол-2-илметил)-6-гидрокси-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.37): LC-MS (ESI) 427 (M+H)+;1Н NMR (DMSO-d6, 400 MHz) δ 11.54 (brs, 1H), 10.69 (t, J=5.6 Hz, 1H), 8.50 (s, 1H), 8.04 (d, J=8.0 Hz, 1H), 7.95 (d, J=8.0 Hz, 1H), 7.50 (t, J=7.2 Hz, 1H), 7.41 (t, J=7.2 Hz, 1H), 5.40 (dd, J1=9.6 Hz, J2=3.6 Hz, 1H), 4.97 (d, J=5.6 Hz, 2H), 4.90 (dd, J1=12.0 Hz, J2=3.6 Hz, 1H), 4.40 (t, J=7.6 Hz, 1H), 4.30 (m, 1H), 4.02 (t, J=11.2 Hz, 1H), 3.67 (t, J=7.6 Hz, 1H), 1.35 (d, J=6.0 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-[(5-хлоро-1,3-бензооксазол-2-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.38): LC-MS (ESI) 445 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.52 (brs, 1H), 10.54 (t, J=5.6 Hz, 1Н), 8.47 (s, 1H), 7.83 (d, J=2.0 Hz, 1H), 7.75 (d, J=8.8 Hz, 1H), 7.42 (dd, J1=8.8 Hz, J2=2.0 Hz, 1H), 5.39 (dd, J1=10.0 Hz, J2=4.0 Hz, 1H), 4.88 (m, 3H), 4.40 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 4.30 (m, 1H), 4.01 (dd, J1=11.6 Hz, J2=6.4 Hz, 1H), 3.67 (dd, J1=8.4 Hz, J2=6.4 Hz, 1H), 1.35 (d, J=6.4 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-[(6-хлоро-1,3-бензооксазол-2-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.39): LC-MS (ESI) 445 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.52 (brs, 1H), 10.54 (t, J=5.4 Hz, 1H), 8.47 (s, 1H), 7.92 (d, J=1.2 Hz, 1H), 7.72 (d, J=8.4 Hz, 1H), 7.41 (dd, J1=8.4 Hz, J2=1.6 Hz, 1H), 5.40 (dd, J1=10.0 Hz, J2=4.0 Hz, 1H), 4.89 (m, 1H), 4.87 (d, J=6.0 Hz, 2H), 4.40 (dd, J1=8.4 Hz, J2=7.2 Hz, 1H), 4.30 (m, 1H), 4.01 (t, J=11.2 Hz, 1H), 3.67 (dd, J1=8.0 Hz, J2=6.8 Hz, 1H), 1.35 (d, J=6.4 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-[(5-метил-1Н-бензимидазол-2-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.40); LC-MS (ESI) 424 (M+H)+;1Н NMR (DMSO-d6, 400 MHz) δ 12.19 (s, 1H), 11.47 (brs, 1H), 10.48 (s, 1H), 8.47 (s, 1H), 7.32 (m, 2H), 6.96 (m, 1H), 5.40 (m, 1H), 4.90 (m, 1H), 4.72 (m, 2H), 4.40 (m, 1H), 4.30 (m, 1H), 4.02 (m, 1H), 3.67 (m, 1H), 2.39 (s, 3H), 1.35 (d, J=4.4 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-[(5-фторо-1Н-бензимидазол-2-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.41): LC-MS (ESI) 428 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.49 (brs, 1H), 10.51 (brs, 1H), 8.47 (s, 1H), 7.52 (m, 1H), 7.33 (d, J=8.4 Hz, 1H), 7.03 (t, J=8.4 Hz, 1H), 5.40 (m, 1H), 4.90 (m, 1H), 4.77 (brs, 2H), 4.40 (m, 1H), 4.30 (m, 1H), 4.02 (t, J=10.8 Hz, 1H), 3.67 (m, 1H), 1.35 (d, J=5.6 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-[(1-метил-6-хлорбензимидазол-2-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.43): LC-MS (ESI) 458 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.47 (brs, 1H), 10.58 (t, J=5.2 Hz, 1H), 8.50 (s, 1H), 7.65 (d, J=1.2 Hz, 1H), 7.58 (d, J=8.4 Hz, 1H), 7.27 (dd, J1=8.4 Hz, J2=1.2 Hz, 1H), 5.40 (dd, J1=9.6 Hz, J2=3.2 Hz, 1H), 4.90 (m, 1H), 4.85 (d, J=5.2 Hz, 2H), 4.40 (dd, J1=8.4 Hz, J2=7.2 Hz, 1H), 4.30 (m, 1H), 4.02 (t, J=11.0 Hz, 1H), 3.81 (s, 3H), 3.67 (dd, J1=8.4 Hz, J2=7.2 Hz, 1H), 1.34 (d, J=6.0 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-{[1-изопропил-1Н-бензимидазол-2-ил]метил}-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.44): LC-MS (ESI) 452 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.47 (brs, 1H), 10.57 (t, J=5.2 Hz, 1H), 8.52 (s, 1H), 7.70 (d, J=8.0 Hz, 1H), 7.61 (d, J=8.0 Hz, 1H), 7.18 (m, 2H), 5.40 (dd, J1=10.0 Hz, J2=4.0 Hz, 1H), 4.91 (dd, J1=12.0 Hz, J2=4.0 Hz, 1H), 4.86 (d, J=5.2 Hz, 2H), 4.83 (m, 1H), 4.40 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 4.29 (m, 1H), 4.02 (dd, J1=12.0 Hz, J2=10.0 Hz, 1H), 3.66 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 1.56 (d, J=6.8 Hz, 6H), 1.34 (d, J=6.0 Hz, 3H).
(3S,11aR)-6-гидрокси-N-[(2-оксо-2,3-дигидро-1Н-бензоимидазол-5-ил)метил]-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.46): LC-MS (ESI) 426 (M+H)+;1Н NMR (DMSO-d6, 400 MHz) δ 11.46 (brs, 1H), 10.56 (s, 2H), 10.26 (t, J=5.6 Hz, 1H), 8.48 (s, 1H), 6.88 (m, 3H), 5.39 (m, 1H), 4.90 (m, 1H), 4.49 (d, J=5.6 Hz, 2H), 4.40 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 4.29 (m, 1H), 4.01 (t, J=11.0 Hz, 1H), 3.66 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 1.34 (d, J=6.0 Hz, 3H).
(3S,11aR)-N-(2,1,3-бензооксадиазол-5-илметил)-6-гидрокси-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.47): LC-MS (ESI) 412 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.51 (brs, 1H), 10.46 (t, J=6.0 Hz, 1H), 8.49 (s, 1H), 8.03 (d, J=9.4 Hz, 1H), 7.78 (s, 1H), 7.57 (d, J=9.4 Hz, 1H), 5.40 (dd, J1=9.8 Hz, J2=3.4 Hz, 1H), 4.90 (dd, J1=12.0 Hz, J2=3.4 Hz, 1H), 4.68 (d, J=5.6 Hz, 2H), 4.40 (t, J=7.6 Hz, 1H), 4.30 (m, 1H), 4.01 (t, J=11.2 Hz, 1H), 3.67 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 1.35 (d, J=6.0 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-(хинолин-2-илметил)-5,7-диоксо-2,3,5,7,11,11a-гексагидро[1,3]-оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.49): LC-MS (ESI) 421 (M+H)+;1Н NMR (DMSO-d6, 400 MHz) δ 11.44 (brs, 1H), 10.30 (t, J=5.6 Hz, 1H), 8.50 (s, 1H), 7.45 (s, 1H), 7.42 (d, J=8.0 Hz, 1H), 7.16 (dd, J1=8.0 Hz, J2=0.8 Hz, 1H), 5.39 (dd, J1=10.0 Hz, J2=3.6 Hz, 1H), 4.90 (m, 1H), 4.61 (d, J=5.6 Hz, 2H), 4.39 (dd, J1=8.4 Hz, J2=7.2 Hz, 1H), 4.29 (sxt, J=6.4 Hz, 1H), 4.01 (t, J=11.2 Hz, 1H), 3.70 (s, 3H), 3.66 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 2.50 (s, 3H), 1.34 (d, J=6.4 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-(хинолин-3-илметил)-5,7-диоксо-2,3,5,7,11,11a-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.50): LC-MS (ESI) 421 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.50 (brs, 1H), 10.47 (t, J=6.0 Hz, 1H), 8.91 (d, J=2.0 Hz, 1H), 8.50 (s, 1H), 8,24 (s, 1H), 8.02 (d, J=8.0 Hz, 1H), 7.96 (d, J=7.6 Hz, 1H), 7.74 (dt, J1=8.0 Hz, J2=1.2 Hz, 1H), 7.61 (m, 1H), 5.39 (dd, J1=10.0 Hz, J2=4.0 Hz, 1H), 4.89 (dd, J1=12.0 Hz, J2=4.0 Hz, 1H), 4.76 (d, J=6.0 Hz, 2H), 4.40 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 4.30 (m, 1H), 4.01 (t, J=11.2 Hz, 1H), 3.67 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 1.35 (d, J=6.0 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-(2-метилхинолин-4-илметил)-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.51): LC-MS (ESI) 435 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.49 (brs, 1H), 10.47 (t, J=5.6 Hz, 1H), 8.52 (s, 1H), 8.14 (d, J=8.4 Hz, 1H), 7.95 (d, J=8.4 Hz, 1H), 7.73 (t, J=7.6 Hz, 1H), 7.57 (t, J=7.2 Hz, 1H), 7.28 (s, 1H), 5.40 (dd, J1=10.0 Hz, J2=4.0 Hz, 1H), 5.03 (d, J=5.6 Hz, 2H), 4.91 (dd, J1=12.0 Hz, J2=4.0 Hz, 1H), 4.40 (dd, J1=8.4 Hz, J2=7.2 Hz, 1H), 4.30 (m, 1H), 4.02 (t, J=11.2 Hz, 1H), 3.67 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 2.61 (s, 3H), 1.34 (d, J=6.4 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-(хинолин-5-илметил)-5,7-диоксо-2,3,5,7,11,11a-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.52): LC-MS (ESI) 421 (M+H)+;1Н NMR (DMSO-d6, 400 MHz) δ 11.44 (s, 1H), 10.42 (t, J=6.0 Hz, 1H), 8.92 (dd, J1=8.0 Hz, J2=1.2 Hz, 1H),8.61 (d, J=8.0 Hz, 1H), 8.52 (s, 1H), 7.97 (d, J=8.4 Hz, 1H), 7.73 (t, J=8.2 Hz, 1H), 7.58 (m, 2H), 5.39 (dd, J1=10.0 Hz, J2=4.0 Hz, 1H), 5.03 (d, J=6.0 Hz, 2H), 4.91 (dd, J1=12.0 Hz, J2=4.0 Hz, 1H), 4.39 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 4.29 (sxt, J=6.4 Hz, 1H), 4.01 (t, J=11.2 Hz, 1H), 3.66 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 1.33 (d, J=6
(3S,11aR)-6-гидрокси-3-метил-N-(хинолин-8-илметил)-5,7-диоксо-2,3,5,7,11,11a-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.54): LC-MS (ESI) 421 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.41 (brs, 1H), 10.49 (t, J=5.6 Hz, 1H), 8.99 (dd, J1=4.0 Hz, J2=1.6 Hz, 1H), 8.47 (s, 1H), 8.40 (dd, J1=8.0 Hz, J2=1.2 Hz, 1H), 7.91 (d, J=8.0 Hz, 1H), 7.67 (d, J=6.8 Hz, 1H), 7.58 (m, 2H), 5.39 (dd, J1=10.0 Hz, J2=3.6 Hz, 1H), 5.13 (d, J=5.6 Hz, 2H), 4.88 (dd, J1=12.0 Hz, J2=4.0 Hz, 1H), 4.39 (dd, J1=8.0 Hz, J2=7.6 Hz, 1H), 4.29 (m, 1H), 4.00 (t, J=11.2 Hz, 1H), 3.66 (dd. J1=8.0 Hz, J2=6.8 Hz, 1H), 1.34 (d, J=6.0 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-(5,6,7,8-тетрагидро-[1,2,4]триазоло[4,3-а]пиридин-3-илметил)-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.61): LC-MS (ESI) 415 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.48 (brs, 1H), 10.38 (t, J=5.6 Hz, 1H), 8.48 (s, 1H), 5.39 (dd, J1=10.0 Hz, J2=3.6 Hz, 1H), 4.89 (dd, J1=12.0 Hz, J2=3.6 Hz, 1H), 4.64 (d, J=5.6 Hz, 2H), 4.39 (dd, J1=8.0 Hz, J2=7.2 Hz, 1H), 4.29 (sxt, J=6.0 Hz, 1H), 4.01 (t, J=11.2 Hz, 1H), 3.93 (t, J=6.0 Hz, 2H), 3.66 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 2.79 (t, J=6.4 Hz, 2H), 1.88 (m, 2H), 1.79 (m, 2H), 1.34 (d, J=6.0 Hz, 3H).
(3S,11aR)-6-гидрокси-N-(1,4,5,6,7,8-гексагидроииклогепта[d]пиразол-3-илметил)-3-метил-5,7-диоксо-2,3,5,7,11,11а-гвксагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.62): LC-MS (ESI) 428 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 12.13 (brs, 1H), 11.43 (brs, 1H), 10.07 (t, J=5.2 Hz, 1H), 8.47 (s, 1H), 5.40 (dd, J1=10.0 Hz, J2=3.6 Hz, 1H), 4.89 (dd, J1=12.0 Hz, J2=3.6 Hz, 1H), 4.39 (m, 3H), 4.29 (m, 1H), 4.00 (t, J=11.2 Hz, 1H), 3.66 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 2.63 (m, 2H), 2.45 (m, 2H), 1.75 (m, 2H), 1.55 (m, 4H), 1.34 (d, J=6.0 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-5,7-диоксо-N-(5,6,7,8-тетрагидро-4Н-тклогепта[d][1,3]тиазол-2-илметил)-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.63): LC-MS (ESI) 445 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.49 (brs, 1H), 10.47 (m, 1H), 8.48 (s, 1H), 5.39 (m, 1H), 4.90 (m, 1H), 4.67 (m, 2H), 4.40 (m, 1H), 4.30 (m, 1H), 4.01 (m, 1H), 3.67 (m, 1H), 2.82 (m, 2H), 2.73 (m, 2H), 1.78 (m, 2H), 1.60 (m, 4H), 1.34 (dd, J1=6.4 Hz, J2=2.8 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-[(1,2-диметил-1Н-бензимидазол-5-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.64): LC-MS (ESI) 438 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.44 (brs, 1H), 10.30 (t, J=5.6 Hz, 1H), 8.50 (s, 1H), 7.45 (s, 1H), 7.42 (d, J=8.0 Hz, 1H), 7.16 (dd, J1=8.0 Hz, J2=0.8 Hz, 1H), 5.39 (dd, J1=10.0 Hz, J2=3.6 Hz, 1H), 4.90 (m, 1H), 4.61 (d, J=5.6 Hz, 2H), 4.39 (dd, J1=8.4 Hz, J2=7.2 Hz, 1H), 4.29 (sxt, J=6.4 Hz, 1H), 4.01 (t, J=11.2 Hz, 1H), 3.70 (s, 3H), 3.66 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 2.50 (s, 3H), 1.34 (d, J=6.4 Hz, 3H).
(3S,11aR)-6-гидрокси-3-метил-N-(хинолин-4-илметил)-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.65): LC-MS (ESI) 421 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 11.49 (brs, 1H), 10.50 (t, J=6.0 Hz, 1H), 8.85 (d, J=4.0 Hz, 1H), 8.52 (s, 1H), 8.21 (d, J=8.8 Hz, 1H), 8.06 (d, J=8.0 Hz, 1H), 7.79 (t, J=6.8 Hz, 1H), 7.66 (t, J=7.6 Hz, 1H), 7.39 (d, J=4.0 Hz, 1H), 5.40 (dd, J1=10.0 Hz, J2=3.6 Hz, 1H), 5.08 (d, J=6.0 Hz, 2H), 4.91 (dd, J1=12.0 Hz, J2=3.6 Hz, 1H), 4.40 (dd, J1=8.0 Hz, J2=7.2 Hz, 1H), 4.29 (m, 1H), 4.02 (t, J=11.2 Hz, 1H), 3.67 (dd, J1=8.0 Hz, J2=6.8 Hz, 1H), 1.33 (d, J=6.0 Hz, 3H).
(3aS,6R,13aS)-9-гидрокси-N-[(2,6-дифторопиридин-3-ил)метил]-6-метил-8,10-диоксо-1,2,3,5,6,8,10,13а-октагидроциклопента[b][1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-11-карбоксамид (2.1.1): LC-MS (ESI) 447 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 12.28 (s, 1H), 10.39 (t, J=6.0 Hz, 1H), 8.41 (s, 1H), 8.08 (q, J=8.4 Hz, 1H), 7.14 (dd, J1=8.4 Hz, J2=2.0 Hz, 1H), 4.55 (m, 2H), 4.36 (m, 3H), 3.76 (dd, J1=8.0 Hz, J2=6.0 Hz, 1H), 2.33 (m, 1H), 2.14 (m, 1H), 1.77 (m, 3H), 1.38 (d, J=6.0 Hz, 3H).
(3aS,6R,13aS)-9-гидрокси-N-[(3,5-дифторопиридин-4-ил)метил]-6-метил-8,10-диоксо-1,2,3,5,6,8,10,13а-октагидроциклопента[b][1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-11-карбоксамид (2.1.2): LC-MS (ESI) 447 (M+H)+;1Н NMR (DMSO-d6, 400 MHz) δ 12.28 (s, 1H), 10.48 (t, J=6.0 Hz, 1H), 8.50 (s, 2H), 8.38 (s, 1H), 4.66 (m, 2H), 4.34 (m, 3H), 3.75 (dd, J1=8.0 Hz, J2=6.0 Hz, 1H), 2.30 (m, 1H), 2.12 (m, 1H), 1.76 (m, 3H), 1.36 (d, J=6.0 Hz, 3H).
(3aS,6R,13aS)-9-гидрокси-6-метил-N-[(2,4,6-трифторопиридин-3-ил)метил]-8,10-диоксо-1,2,3,5,6,8,10,13а-октагидроциклопента[b][1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-11-карбоксамид (2.1.3): LC-MS (ESI) 465 (M+H)+;1Н NMR (DMSO-d6, 400 MHz) δ 12.28 (s, 1H), 10.42 (t, J=5.6 Hz, 1H), 8.38 (s, 1H), 7.34 (d, J=8.0 Hz, 1H), 4.54 (m, 2H), 4.35 (m, 3H), 3.75 (m, 1H), 2.31 (m, 1H), 2.12 (m, 1H), 1.76 (m, 3H), 1.36 (d, J=6.0 Hz, 3H).
(3aS,6S,13aS)-9-гидрокси-N-[(2,6-дифторопиридин-3-ил)метил]-6-метил-8,10-диоксо-1,2,3,5,6,8,10,13а-октагидроциклопента[b][1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-11-карбоксамид (2.2.1): LC-MS (ESI) 447 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 12.29 (s, 1H), 10.40 (t, J=5.8 Hz, 1H), 8.41 (s, 1H), 8.08 (q, J=8.4 Hz, 1H), 7.15 (dd, J1=8.4 Hz, J2=2.0 Hz, 1H), 4.55 (m, 2H), 4.35 (m, 3H), 3.75 (dd, J1=8.0 Hz, J2=6.4 Hz, 1H), 2.33 (m, 1H), 2.13 (m, 1H), 1.76 (m, 3H), 1.37 (d, J=6.4 Hz, 3H).
(3aS,6S,13aS)-9-гидрокси-N-[(3,5-дифторопиридин-4-ил)метил]-6-метил-8,10-диоксо-1,2,3,5,6,8,10,13а-октагидроциклопента[b][1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-11-карбоксамид (2.2.2): LC-MS (ESI) 447 (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 12.46 (s, 1H), 10.47 (t, J=5.8 Hz, 1H), 8.49 (s, 2H), 8.42 (s, 1H), 5.43 (dd, J1=9.2 Hz, J2=4.0 Hz, 1H), 5.09 (brs, 1H), 4.66 (m, 3H), 4.60 (brs, 1H), 4.01 (dd, J1=12.4 Hz, J2=9.6 Hz, 1H), 1.94 (brs, 4H), 1.84 (d, J=12.0 Hz, 1H), 1.57 (m, 1H).
(3aS,6S,13aS)-9-гидрокси-6-метил-N-[(2,4,6-трифторопиридин-3-ил)метил]-8,10-диоксо-1,2,3,5,6,8,10,13а-октагидроциклопента[b][1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-11-карбоксамид (2.2.3): LC-MS (ESI) 46e (M+H)+;1H NMR (DMSO-d6, 400 MHz) δ 12.28 (s, 1H), 10.42 (t, J=5.8 Hz, 1H), 8.38 (s, 1H), 7.34 (d, J=8.4 Hz, 1H), 4.54 (m, 2H), 4.34 (m, 3H), 3.75 (dd, J1=8.0 Hz, J2=6.0 Hz, 1H), 2.30 (m, 1H), 2.12 (m, 1H), 1.76 (m, 3H), 1.36 (d, J=6.0 Hz, 3H).
Пример 3. Общий способ получения анелированных 9-гидрокси-1,8-диоксо-1,3,4,8-тетрагидро-2Н-пиридо[1,2-a]пиразин-7-карбоксамидов общей формулы 1.1.
(3S,11aR-6-гидрокси-N-[(2,6-дифторопиридин-3-ил)метил]-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.19) получают по схеме 3.
Схема 3. Синтез соединения 1.1.19.

Растворяли 3 г (7,4 ммоль) ((3S,11aR)-6-(бензилокси)-8-бром-3-метил-2,3,11,11а-тетрагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-5,7-диона (3.1) в смеси 250 мл ТГФ и 150 мл метанола. Переносили в автоклав, добавляли триэтиламин (1,3 мл, 8,9 ммоль) и Pd(dppf)Cl2 (0,05 г, 0,07 ммоль), накачивали СО до 15 атм и нагревали до 100°С в течение 3 суток. По окончании реакции (контроль LC-MS) реакционную массу фильтровали через целит, упаривали в вакууме, растворяли в дихлорметане, промывали водой, сушили над сульфатом натрия и упаривали на роторном испарителе. Остаток подвергали колоночной хроматографии на силикагеле, элюент дихлорметан - метанол 60:1. Получали 1.8 г (63%) метил (3S,11aR)-6-(бензилокси)-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]-оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксилагпа (3.2):1Н ЯМР (ДМСО-d6, 400 МГц) δ 8.39 (с, 1H), 7.53 (м, 2Н), 7.34 (м, 3Н), 5.32 (д. д, J1=10.0 Гц, J2=3.6 Гц, 1H), 5.04, 5.17 (ABq, JAB=10.4 Гц, 2Н), 4.67 (д. д, J,=12.0 Гц, J2=3.2 Гц, 1H), 4.29 (м, 2Н), 3.94 (д. д, J1=12.0 Гц, J2=10.0 Гц, 1H), 3.63 (д. д, J1=8.0 Гц, J2=6.4 Гц, 1H), 1.27 (д, J=6.0 Гц).
К раствору метилового эфира 3.2 (1,5 г, 3,9 ммоль) в 15 мл диоксана приливали раствор LiOH × Н2О (0, 33 г, 7,8 ммоль) в 5 мл воды. Перемешивали 3 ч при 50°С. Упаривали диоксан на роторном испарителе. Остаток разбавляли водой и подкисляли 10-% серной кислотой до рН 3. Выпавший осадок отфильтровывали, промывали водой и сушили в вакууме. Получали 1 г (69%) (3S,11aR)-6-(бензилокси)-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоновой кислоты (3.3):1Н ЯМР (ДМСО-d6, 400 МГц) δ 15.45 (уш. с, 1Н), 8.75 (с, 1Н), 7.54 (м, 2Н), 7.36 (м, 3Н), 5.38 (д. д, J1=10.0 Гц, J2=3.2 Гц, 1Н), 5.15, 5.25 (ABq, JAB=10.4 Гц, 2Н), 4.87 (д. д, J1=12.0 Гц, J2=3.2 Гц, 1Н), 4.32 (м, 2Н), 4.13 (т, J=11.0 Гц, 1Н), 3.66 (д. д, J1=7.6 Гц, J2=6.4 Гц, 1Н), 1.28 (д, J=6.0 Гц).
К раствору 1 г (2,7 ммоль) кислоты 3.3 в 20 мл дихлорметана добавляли 1,2 мл (8,1 ммоль) триэтиламин. Перемешивали реакционную массу при комнатной температуре 15 минут и добавляли TBTU (0,82 г, 3,3 ммоль). Смесь перемешивали еще 15 мин и добавляли 0,47 г (3,3 ммоль) [(2,6-дифторпиридин-3-ил)метил]амина, перемешивали 14 ч при комнатной температуре. Промывали реакционную массу 10-% водным раствором поташа и водой. Органический слой сушили над сульфатом натрия и упаривали на роторном испарителе. К остатку прибавляли диэтиловый эфир, полученный осадок отфильтровывали и сушили в вакууме. Использовали дальше без доп. очистки. Получали 0,94 г (70%) (3S,11aR)-6-(бензилокси)-3-метил-5,7-диоксо-N-[(2,6-дифторпиридин-3-ил)метил]-2,3,5,7,11,11a-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамида (3.4.19):1Н ЯМР (ДМСО-d6, 400 МГц) δ 10.45 (t, J=5.9 Гц, 1Н), 8.56 (s, 1Н), 8.13-8.00 (m, 1Н), 7.57-7.52 (m, 2H), 7.40-7.29 (m, 3Н), 7.15 (д. д, J1=8.1 Гц, J2=2.3 Гц, 1Н), 5.35 (д. д, J1=10.0 Гц, J2=3.7 Гц, 1Н), 5.21 (d, J=10.6 Гц, 1Н), 5.07 (d, J=10.6 Гц, 1Н), 4.79 (д. д, J1=12.2 Гц, J2=3.7 Гц, 1H), 4.56 (d, J=5.9 Гц, 2Н), 4.38-4.22 (m, 2Н), 4.08-3.98 (m, 1Н), 3.64 (д. д, J1=8.1 Гц, J2=6.4 Гц, 1Н), 1.27 (d, J=6.1 Гц, 3Н).
Растворяли 0,9 г (1,8 ммоль) соединение 3.4.19 в смеси 18 мл ТГФ и 2 мл метанола, прибавляли 0,09 г 10% Pd/C и перемешивали в атмосфере водорода 8 ч. Реакционную массу пропускали через целит, фильтрат и упаривали. Остаток обрабатывали эфиром, отфильтровывали осадок и сушили в вакууме. Получали 0,6 г (81%) (3S,11aR)-6-гидрокси-N-[(2,6-дифторо-пиридин-3-ил)метил]-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамида (1.1.19): LC-MS (ESI) 07 (M+H)+;1Н ЯМР (ДМСО-d6, 400 МГц) δ 11.49 (уш. с, 1Н), 10.36 (т, J=5.8 Гц, 1Н), 8.45 (с, 1Н), 8.06 (д. д, J1=17.2 Гц, J2=8.4 Гц, 1Н), 7.14 (д. д, J1=8.0 Гц, J2=2.0 Гц, 1Н), 5.38 (д. д, J1=10.0 Гц, J2=3.6 Гц, 1Н), 4.88 (д. д, J1=12.0 Гц, J2=3.6 Гц, 1Н), 4.55 (д, J=5.6 Гц, 2Н), 4.39 (д. д, J1=8.0 Гц, J2=6.8 Гц, 1Н), 4.29 (м, 1Н), 4.00 (т, J=11.0 Гц, 1Н), 3.67 (д. д, J1=8.5 Гц, J2=6.8 Гц, 1Н), 1.34 (д, J=6.4 Гц, 3Н); РФА: Моделирование дифрактограммы образца ингибитора 1.1.19 методом Паули изображено на Фиг. 1. Образец, полученный перекристаллизацией из метанола однофазен. Параметры элементарной ячейки: а=7.1564(5)

Натрий (3S,11aR)-8-({[(2,6-дифторопиридин-3-ил)метил]амино}карбонил)-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-6-олат (1.1.20). К суспензии 0,5 г (1,2 ммоль) (3S,11aR)-6-гидрокси-N-[(2,4-дифтор-3-пиридил)метил]-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазол[3,2-a]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.19) в 80 мл этанола и 20 мл воды добавляют 0,05 г (1,2 ммоль) NaOH при 75°С. Перемешивают 14 ч при комнатной температуре. Выпавший осадок отфильтровывают, промывают абс. этанолом и сушат в вакууме. Получают 0,42 г (80%) натрия (3S,11aR)-8-({[(2,6-дифторопиридин-3-ил)метил]амино}карбонил)-3-метил-5,7-диоксо-2,3,5,7,11,11a-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-6-олат (1.1.20):1Н ЯМР (ДМСО-d6, 400 МГц) δ 10.76 (т, J=5.6 Гц, 1Н), 8.00 (д. д, J1=17.2 Гц, J2=8.0 Гц, 1Н), 7.90 (с, 1Н), 7.11 (д. д, J1=8.0 Гц, J2=1.6 Гц, 1Н), 5.21 (д. д, J1=10.0 Гц, J2=4.0 Гц, 1Н), 4.59 (д. д, J1=12.4 Гц, J2=3.6 Гц, 1Н), 4.52 (т, J=5.6 Гц, 2Н), 4.25 (м, 2Н), 3.74 (т, J=10.8 Гц, 1Н), 3.60 (м, 1Н), 1.27 (д, J=5.2 Гц, 3Н).
Аналогично получают:
(3S,11aR)-6-гидрокси-3-метил-5,7-диоксо-N-(2-тиенилметил)-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.1): LC-MS (ESI) 376 (М+Н)+,1Н NMR (DMSO-d6, 400 MHz) δ 11.47 (brs, 1Н), 10.33 (t, J=4.8 Hz, 1Н), 8.49 (s, 1Н), 7.40 (dd, J1=8.8 Hz, J2=4.8 Hz, 1Н), 7.03 (d, J=2.8 Hz, 1Н), 6.96 (dd, J1=4.8 Hz, J2=3.6 Hz, 1Н), 5.39 (dd, J1=10.0 Hz, J2=4.0 Hz, 1Н), 4.90 (dd, J1=12.0 Hz, J2=4.0 Hz, 1Н), 4.70 (d, J=5.6 Hz, 2Н), 4.40 (dd, J1=8.4 Hz, J2=7.2 Hz, 1Н), 4.29 (m, 1Н), 4.01 (t, J=11.2 Hz, 1Н), 3.67 (dd, J1=8.4 Hz, J2=7.2 Hz, 1Н), 1.34 (d, J=6.0 Hz, 3Н);
(3S,11aR)-6-гидрокси-3-метил-N-[(4-метил-4Н-1,3-оксазол-2-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.14): LC-MS (ESI) 375 (M+H)+,1H NMR (DMSO-d6, 300 MHz, 80°С) δ 10.49 (brs, 1Н), 7.93 (s, 1Н), 5.21 (dd, J1=9.6 Hz, J2=3.6 Hz, 1Н), 4.58-4.85 (m, 3Н), 4.29 (m, 2Н), 3.64 (m, 2Н), 2.30 (s, 3Н), 1.30 (d, J=5.4 Hz, 3H);
(3S,11aR)-6-гидрокси-3-метил-N-[(1,4,5-триметил-1Н-имидазол-2-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.10): LC-MS (ESI) 402 (M+H)+;
(3S,11aR)-6-гидрокси-3-метил-N-[(4-метил-1,2,3-тиадиазол-5-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.11): LC-MS (ESI) 392 (M+H)+,1Н NMR (DMSO-d6, 400 MHz) δ 11.52 (brs, 1H), 10.45 (t, J=5.6 Hz, 1H), 8.49 (s, 1H), 5.39 (dd, J1=9.2 Hz, J2=3.2 Hz, 1H), 4.89 (m, 1H), 4.81 (d, J=5.6 Hz, 2H), 4.40 (dd, J1=7.6 Hz, J2=0.4 Hz, 1H), 4.29 (m, 1H), 4.00 (t, J=11.2 Hz, 1H), 3.66 (t, J=7.6 Hz, 1H), 2.67 (s, 3H), 1.34 (d, J=6.0 Hz, 3H);
(3S,11aR)-6-гидрокси-3-метил-N-[(3-метил-1,2,4-оксадиазол-5-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.13): LC-MS (ESI) 376 (M+H)+;
(3S,11aR)-6-гидрокси-3-метил-N-[(3-фторопиридин-2-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.16): LC-MS (ESI) 389 (M+H)+;1Н ЯМР (ДМСО-d6, 400 МГц) δ 10.61 (t, J=5.4 Гц, 1H), 8.57 (s, 1H), 8.42 (d, J=4.5 Гц, 1H), 7.77-7.67 (m, 1H), 7.57-7.50 (m, 2H), 7.47-7.28 (m, 4H), 5.40-5.31 (m, 1H), 5.25-5.17 (m, 1H), 5.11-5.03 (m, 1H), 4.84-4.70 (m, 3H), 4.37-4.23 (m, 2H), 4.08-3.97 (m, 1H), 3.68-3.60 (m, 1H), 1.28 (d, J=6.1 Гц, 3H);
(2S,11aR)-6-гидрокси-N-[(3,5-дифторопиридин-2-ил)метил]-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.17): LC-MS (ESI) 407 (M+H)+,1Н ЯМР (ДМСО-d6, 400 МГц) δ 11.42 (уш. с, 1H), 10.54 (т, J=5.2 Гц, 1H), 8.49 (д, J=2.4 Гц, 1H), 8.46 (с, 1H), 7.95 (д. т, J1=11.4 Гц, J2=2.4 Гц, 1H), 5.39 (д. д, J1=10.0 Гц, J2=4.0 Гц, 1H), 4.88 (д. д, J1=12.0 Гц, J2=4.0 Гц, 1H), 4.71 (д, J=4.8 Гц, 2H), 4.39 (д. д, J1=8.0 Гц, J2=6.8 Гц, 1H), 4.29 (м, 1H), 4.00 (т, J=11.2 Гц, 1H), 3.66 (д. д, J1=8.0 Гц, J2=6.8 Гц, 1H), 1.34 (д, J=6.4 Гц, 3H);
(3S,11aR)-6-гидрокси-N-[(6-хлоропиридин-3-ил)метил]-3-метил-5,7-диоксо-2,3,5,7,11,11a-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.18): LC-MS (M+1)=405,1H NMR (DMSO-d6, 400 MHz) δ 10.76 (t, J=5.8 Hz, 1H), 8.35 (s, 1H), 7.92 (s, 1H), 7.75 (d, J=8.0 Hz, 1H), 7.45 (d, J=8.0 Hz, 1H), 5.21 (m, 1H), 4.60 (m, 1H), 4.53 (m, 2H), 4.25 (m, 2H), 3.75 (t, J=10.8 Hz, 1H), 3.59 (m, 1H), 1.26 (d, J=4.4 Hz, 3H);
Натрий (3S,11aR)-8-({[(2,6-дифторопиридин-3-ил)метил]амино}карбонил)-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-6-олат (1.1.20): LC-MS (ESI) 407 (M+H)+,1H ЯМР (ДМСО-d6, 400 МГц) δ 11.49 (уш. с, 1H), 10.36 (т, J=5.8 Гц, 1H), 8.45 (с, 1H), 8.06 (д. д, J1=17.2 Гц, J2=8.4 Гц, 1H), 7.14 (д. д, J1=8.0 Гц, J2=2.0 Гц, 1H), 5.38 (д. д, J1=10.0 Гц, J2=3.6 Гц, 1H), 4.88 (д. д, J1=12.0 Гц, J2=3.6 Гц, 1H), 4.55 (д, J=5.6 Гц, 2Н), 4.39 (д. д, J1=8.0 Гц, J2=6.8 Гц, 1Н), 4.29 (м, 1Н), 4.00 (т, J=11.0 Гц, 1Н), 3.67 (д. д, J1=8.5 Гц, J2=6.8 Гц, 1Н), 1.34 (д, J=6.4 Гц, 3Н);
(3S,11aR)-6-гидрокси-3-метил-N-[(2,4,6-трифторопиридин-3-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.22): LC-MS (ESI) 425 (M+H)+,1Н NMR (DMSO-d6, 400 MHz) δ 11.46 (s, 1H), 10.36 (t, J=6.0 Hz, 1Н), 8.43 (s, 1Н), 7.33 (d, J=8.4 Hz, 1Н), 5.37 (dd, J1=10.0 Hz, J2=4.0 Hz, 1Н), 4.85 (dd, J1=12.4 Hz, J2=4.0 Hz, 1Н), 4.55 (d, J=6.0 Hz, 2Н), 4.39 (dd, J1=8.4 Hz, J2=6.8 Hz, 1Н), 4.28 (sxt, J=6.4 Hz, 1Н), 3.99 (dd, J1=11.6 Hz, J2=10.8 Hz, 1Н), 3.66 (dd, J1=8.4 Hz, J2=6.8 Hz, 1Н), 1.33 (d, J=6.4 Hz, 3H);
(3S,11aR)-6-гидрокси-3-метил-N-[(3-фторопиридин-4-ил)метил]-5,7-диоксо-2,3,5,7,11,11a-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.23): LC-MS (ESI) 389 (M+H)+,1Н ЯМР (ДМСО-d6, 400 МГц) δ 10.54-10.44 (m, 1Н), 8.57 (s, 1Н), 8.52 (s, 1Н), 8.38 (d, J=4.4 Гц, 1Н), 7.55 (d, J=7.2 Гц, 2Н), 7.42-7.27 (m, 4H), 5.40-5.30 (m, 1Н), 5.23 (d, J=10.4 Гц, 1Н), 5.08 (d, J=10.4 Гц, 1Н), 4.84-4.74 (m, 1Н), 4.65 (d, J=5.6 Гц, 2Н), 4.38-4.22 (m, 2Н), 4.10-3.96 (m, 1Н), 3.70-3.59 (m, 1Н), 1.28 (d, J=5.7 Гц, 3Н);
(3S,11aR)-6-гидрокси-N-[(3,5-дифторопиридин-4-ил)метил]-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.24): LC-MS (ESI) 407 (M+H)+,1H NMR (DMSO-d6, 400 MHz) δ 11.46 (brs, 1Н), 10.43 (t, J=6.0 Hz, 1Н), 8.50 (s, 2Н), 8.43 (s, 1Н), 5.37 (dd, J1=10.0 Hz, J2=4.0 Hz, 1Н), 4.84 (dd, J1=12.0 Hz, J2=4.0 Hz, 1Н), 4.67 (d, J=6.0 Hz, 2Н), 4.39 (dd, J1=8.4 Hz, J2=6.8 Hz, 1Н), 4.28 (m, 1Н), 3.99 (dd, J1=12.0 Hz, J2=6.4 Hz, 1Н), 3.66 (dd, J1=8.8 Hz, J2=6.8 Hz, 1Н), 1.33 (d, J=6.0 Hz, 3Н);
(3S,11aR)-6-гидрокси-3-метил-N-(пиразин-2-илметил)-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.25): LC-MS (ESI) 372 (M+H)+,1H NMR (DMSO-d6, 400 MHz) δ 11.45 (brs, 1Н), 10.53 (t, J=5.6 Hz, 1Н), 8.63 (s, 1Н), 8.61 (d, J=2.0 Hz, 1Н), 8.54 (d, J=2.0 Hz, 1Н), 8.47 (s, 1Н), 5.39 (dd, J1=10.0 Hz, J2=4.0 Hz, 1Н), 4.89 (dd, J1=12.0 Hz, J2=4.0 Hz, 1Н), 4.71 (d, J=5.6 Hz, 2Н), 4.40 (t, J=7.6 Hz, 1H), 4.30 (m, 1H), 4.01 (t, J=11.0 Hz, 1Н), 3.67 (dd, J1=8.0 Hz, J2=7.2 Hz, 1Н), 1.34 (d, J=6.0 Hz, 3Н);
(3S,11aR)-6-гидрокси-2-метил-N-[(2-хлоримидазо[2,1-b][1,3]тиазол-6-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.28): LC-MS (ESI) 450 (M+H)+,1Н NMR (DMSO-d6, 400 MHz) δ 10.31 (brs, 1Н), 8.06 (s, 1Н), 7.57 (s, 1H), 5.25 (brs, 1H), 4.65 (m, 1H), 4.48 (brs, 2H), 4.28 (brs, 2H), 3.77 (brs, 1H), 3.63 (brs, 1H), 1.29 (brs, 3H);
(3S,11aR)-6-гидрокси-N-(имидазо[2,1-b][1,3,4]тиадиазол-6-илметил)-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.29): LC-MS (ESI) 417 (M+H)+,1H NMR (DMSO-d6, 400 MHz) δ 10.64 (brs, 1H), 9.18 (s, 1H), 8.00 (s, 1H), 7.91 (s, 1H), 5.22 (m, 1H), 4.60 (m, 1H), 4.49 (s, 2H), 4.25 (s, 2H), 3.75 (m, 1H), 3.59 (m, 1H), 1.26 (brs, 3H);
(3S,11aR)-6-гидрокси-3-метил-N-[(2-метилимидазо[2,1-b][1,3,4]тиадиазол-6-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.30): LC-MS (ESI) 431 (M+H)+,1Н NMR (DMSO-d6, 400 MHz) δ 11.43 (brs, 1H), 10.27 (t, J=5.2 Hz, 1H), 8.47 (s, 1H), 7.95 (s, 1H), 5.39 (dd, J1=10.0 Hz, J2=3.6 Hz, 1H), 4.90 (dd, J1=12.0 Hz, J2=3.6 Hz, 1H), 4.49 (d, J=5.2 Hz, 2H), 4.39 (dd, J1=8.4 Hz, J2=7.2 Hz, 1H), 4.29 (m, 1H), 4.01 (t, J=11.2 Hz, 1H), 3.66 (dd, J1=8.4 Hz, J2=7.2 Hz, 1H), 2.70 (s, 3H), 1.34 (d, J=6.0 Hz, 3H);
(3S,11aR)-6-гидрокси-3-метил-N-[(1-метил-бензимидазол-2-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.42): LC-MS (ESI) 424 (M+H)+,1H NMR (DMSO-d6, 400 MHz) δ 11.49 (brs, 1H), 10.60 (t, J=4.8 Hz, 1H), 8.44 (s, 1H), 7.68 (d, J=8.0 Hz, 1H), 7.63 (d, J=7.6 Hz, 1H), 7.37 (t, J=8.0 Hz, 1H), 7.63 (t, J=7.6 Hz, 1H), 5.39 (dd, J1=10.0 Hz, J2=3.6 Hz, 1H), 4.95 (d, J=4.8 Hz, 2H), 4.88 (m, 1H), 4.40 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 4.30 (m, 1H), 3.99 (t, J=11.2 Hz, 1H), 3.89 (s, 3H), 3.67 (dd, J1=8.4 Hz, J2=6.8 Hz, 1H), 1.35 (d, J=6.4 Hz, 3H);
(3S,11aR)-6-гидрокси-3-метил-N-[(6-хлор-1-изопропил-1Н-бензимидазол-2-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[2,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.45): LC-MS (ESI) 486 (M+H)+,1Н NMR (DMSO-d6, 400 MHz) δ 11.47 (brs, 1H), 10.57 (t, J=5.2 Hz, 1H), 8.51 (s, 1H), 7.80 (d, J=2.0 Hz, 1H), 7.61 (d, J=8.8 Hz, 1H), 7.20 (dd, J1=8.8 Hz, J2=2.0 Hz, 1H), 5.40 (dd, J1=10.0 Hz, J2=4.0 Hz, 1H), 4.91 (dd, J1=12.0 Hz, J2=4.0 Hz, 1H), 4.86 (d, J=5.2 Hz, 2H), 4.83 (m, 1H), 4.40 (dd, J1=8.4 Hz, J2=7.2 Hz, 1H), 4.29 (m, 1H), 4.02 (dd, J1=12.0 Hz, J2=10.0 Hz, 1H), 3.66 (dd, J1=8.4 Hz, J2=7.2 Hz, 1H), 1.54 (d, J=6.8 Hz, 6H), 1.34 (d, J=6.0 Hz, 3H);
(3S,11aR)-N-(2,1,3-бензотиадиазол-5-илметил)-6-гидрокси-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.47): LC-MS (ESI) 428 (M+H)+;
(3S,11aR)-6-гидрокси-3-метил-N-(хинолин-6-и)мети)-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.53): LC-MS (ESI) 421 (M+H)+;
(3S,11aR)-6-гидрокси-N-(-изохинолин-1-илметил)-3-метил-5,7-диоксо-2,3,5,7,11,11a-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.55): LC-MS (ESI) 421 (M+H)+,1H NMR (DMSO-d6, 300 MHz, 80°С) δ 11.25 (brs, 1H), 10.64 (t, J=5.2 Hz, 1H), 8.47 (m, 2H), 8.32 (d, J=8.0 Hz, 1H), 7.99 (d, J=8.0 Hz, 1H), 7.75 (m, 3H), 5.42 (dd, J1=10.0 Hz, J2=3.6 Hz, 1H), 5.20 (d, J=5.2 Hz, 2H), 4.85 (dd, J1=12.0 Hz, J2=3.6 Hz, 1H), 4.38 (m, 2H), 4.01 (t, J=10.8 Hz, 1H), 3.70 (m, 1H), 1.38 (d, J=5.6 Hz, 3H);
(3S,11aR)-6-гидрокси-N-(имидазо[1,2-а]пиридин-3-и)метил-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.56): LC-MS (M+1)=410,1Н NMR (DMSO-d6, 400 MHz) δ 10.67 (t, J=5.6 Hz, 1H), 8.48 (d, J=6.4 Hz, 1H), 7.96 (s, 1H), 7.57 (m, 2H), 7.23 (t, J=8.0 Hz, 1H), 6.92 (t, J=1.1 Hz, 1H), 5.20 (dd, J1=9.6 Hz, J2=3.6 Hz, 1H), 4.89 (m, 2H), 4.61 (m, 1H), 4.23 (m, 2H), 3.75 (t, J- 10.8 Hz, 1H), 3.58 (m, 1H), 1.24 (d, J=5.6 Hz, 3H);
(3S,11aR)-6-гидрокси-3-метил-N-(1,2,4-триазоло[4,3-a]пиридин-3-илметил-5,7-диоксо-2,2,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.57): LC-MS (ESI) 411 (М+Н)+;
(3S,11aR)-6-гидрокси-N-(имидазо[1,2-а]пиримидин-2-илметил)-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.58): LC-MS (ESI) 411 (M+H)+,1H NMR (DMSO-d6, 400 MHz) δ 11.46 (brs, 1H), 10.41 (t, J=5.6 Hz, 1H), 8.94 (d, J=5.6 Hz, 1H), 8.50 (m, 2H), 7.79 (s, 1H), 7.03 (m, 1H), 5.40 (m, 1H), 4.91 (m, 1H), 4.67 (d, J=5.6 Hz, 2H), 4.39 (dd, J1=8.0 Hz, J2=7.2 Hz, 1H), 4.30 (m, 1H), 4.02 (m. 1H), 3.67 (dd, J1=8.0 Hz, J2=7.2 Hz, 1H), 1.34 (d, J=6.0 Hz, 3H);
(3S,11aR)-6-гидрокси-N-(имидазо[1,2-а]пиразин-3-илметил)-3-метил-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.59): LC-MS (ESI) 411 (M+H)+,1H NMR (DMSO-d6, 300 MHz, 80°С) δ 10.39 (brs, 1H), 8.98 (s, 1H), 8.59 (d, J=6.0 Hz, 1H), 8.07 (brs, 1H), 7.88 (d, J=6.0 Hz, 1H), 7.76 (s, 1H), 5.23 (m, 1H), 4.89 (m. 2H), 4.65 (m, 1H), 4.26 (s, 2H), 3.72 (brs, 1H), 3.60 (m, 1H), 1.28 (d, J=6.0 Hz, 3H);
(3S,11aR)-6-гидрокси-3-метил-N-[(6-хлоро-1,2,4-триазоло[4,3-b]пиридазин-3-ил)метил]-5,7-диоксо-2,3,5,7,11,11а-гексагидро[1,3]оксазоло[3,2-а]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.60): LC-MS (ESI) 446 (M+H)+,1Н NMR (DMSO-d6, 400 MHz) δ 11.50 (brs, 1H), 10.59 (brs, 1H), 8.47 (m, 2H), 7.52 (d, J=9.6 Hz, 1H), 5.38 (dd, J1=10.0 Hz, J2=4.0 Hz, 1H), 5.04 (d, J=5.6 Hz, 2H), 4.88 (m, 1H), 4.39 (dd, J1=8.2 Hz, J2=7.2 Hz, 1H), 4.29 (sxt, J=6.4 Hz, 1H), 4.00 (t, J=11.0 Hz, 1H), 3.66 (dd, J1=8.2 Hz, J2=7.2 Hz, 1H), 1.34 (d, J=6.4 Hz, 3H);
Пример 4. Общий способ получения анелированных 9-гидрокси-1,8-диоксо-1,3,4,8-тетрагидро-2Н-пиридо[1,2-а]пиразин-7-карбоксамидов общей формулы 1.2 и 1.3.
Ингибиторы 1.2 и 1.3 получают аналогично синтезу соединений общей формулы 1.1 (пример 3), исходя из (4R,12aS)-7-бензилокси-9-бром-4-метил-6,8-диоксо-3,4,6,8,12,12а-гексагидро-2Н-пиридо[1',2':4,5]пиразино-[2,1-b][1,3]оксазин и (2R,5S,13aR)-8-бензилокси-10-бром-7,9-диоксо-2,3,4,5,7,9,13,13а-октагидро-2,5-метанпиридо[1',2':4,5]пиразино[2,1-b][1,3]оксазепин соответственно, в том числе:
(4R,12aS)-7-гидрокси-N-[(2,6-дифторопиридин-3-ил)метил]-4-метил-6,8-диоксо-3,4,6,8,12,12а-гексагидро-2Н-пиридо[1',2':4,5]пиразино[2,1-b][1,3]оксазин-9-карбоксамид (1.2.1); LC-MS (ESI) 421 (М+Н)+,1Н NMR (DMSO-d6, 400 MHz) δ 12.51 (brs, 1H), 10.40 (t, J=5.8 Hz, 1H), 8.48 (s, 1H), 8.05 (q, J=8.8 Hz, 1H), 7.14 (dd, J1=8.0 Hz, J2=2.4 Hz, 1H), 5.44 (m, 1H), 4.79 (m, 1H), 4.55 (m, 3H), 4.34 (dd, J1=13.6 Hz, J2=5.6 Hz, 1H), 4.03 (dt, J1=12.0 Hz, J2=1.6 Hz, 1H), 3.89 (m, 1H), 2.01 (m, 1H), 1.55 (m, 1H), 1.33 (d, J=7.2 Hz, 3H);
(4R,12aS)-7-гидрокси-4-метил-N-[(2,4,6-трифторопиридин-3-ил)метил]-6,8-диоксо-3,4,6,8,12,12а-гексагидро-2Н-пиридо[1',2':4,5]пиразино[2,1-b][1,3]оксазин-9-карбоксамид (1.2.3): LC-MS (ESI) 439 (М+Н)+,1Н NMR (DMSO-d6, 400 MHz) δ 12.49 (brs, 1H), 10.39 (t, J=6.0 Hz, 1H), 8.46 (s, 1H), 7.33 (d, J=8.4 Hz, 1H), 5.43 (dd, J1=9.6 Hz, J2=4.0 Hz, 1H), 4.78 (p, J=6.6 Hz, 1H), 4.53 (m, 3H), 4.32 (dd, J1=13.6 Hz, J2=5.6 Hz, 1H), 4.02 (t, J=12.0 Hz 1H), 3.88 (m, 1H), 2.01 (m, 1H), 1.54 (m, 1H), 1.32 (d, J=7.2 Hz, 3H);
(4R,12aS)-7-гидрокси-N-[(3,5-дифторопиридин-4-ил)метил]-4-метил-6,8-диоксо-3,4,6,8,12,12а-гексагидро-2Н-пиридо[1',2':4,5]пиразино[2,1-b][1,3]оксазин-9-карбоксамид (1.2.4): LC-MS (ESI) 421 (M+H)+,1Н NMR (DMSO-d6, 400 MHz) δ 12.49 (s, 1H), 10.47 (t, J=5.6 Hz, 1H), 8.50 (s, 2H), 8.45 (brs, 1H), 5.43 (m, 1H), 4.78 (m, 1H), 4.66 (d, J=5.6 Hz, 2H), 4.53 (m, 1H), 4.32 (m, 1H), 4.02 (t, J1=12.0 Hz, 1H), 3.89 (m, 1H), 2.00 (m, 1H), 1.54 (d, J=13.6 Hz, 1H), 1.32 (d, J=7.2 Hz, 3H);
(2R,5S,13aR)-8-гидрокси-N-[(2,6-дифторпиридин-3-ил)метил]-7,9-диоксо-2,3,4,5,7,9,13,13а-октагидро-2,5-метанпиридо[1',2':4,5]пиразино[2,1-b][1,3]оксазепин-10-карбоксамид (1.3.1): LC-MS (M+1)=433,1H NMR (DMSO-d6, 400 MHz) δ 12.47 (s, 1H), 10.41 (m, 1H), 8.45 (s, 1H), 8.04 (m, 1H), 7.15 (d, J=7.2 Hz, 1H), 5.44 (m, 1H), 5.10 (brs, 1H), 4.68 (m, 1H), 4.60 (brs, 1H), 4.55 (m, 2H), 4.03 (m, 1H), 1.94 (brs, 4H), 1.84 (m, 1H), 1.58 (m, 1H);
(2R,5S,13aR)-8-гидрокси-N-[(2,4,6-трифторопиридин-3-ил)метил]-7,9-диоксо-2,3,4,5,7,9,13,13а-октагидро-2,5-метанопиридо[1',2':4,5]пиразино[2,1-b][1,3]оксазепин-10-карбоксамид (1.3.2): LC-MS (ESI) 451 (M+H)+,1H NMR (DMSO-d6, 400 MHz) δ 12.46 (s, 1H), 10.40 (t, J=5.6 Hz, 1H), 8.42 (s, 1H), 7.33 (d, J=8.4 Hz, 1H), 5.43 (dd, J1=10.0 Hz, J2=4.0 Hz, 1H), 5.09 (brs, 1H), 4.65 (dd, J1=12.8 Hz, J2=4.0 Hz, 1H), 4.60 (brs, 1H), 4.54 (m, 2H), 4.01 (dd, J1=12.4 Hz, J2=9.6 Hz, 1H), 1.94 (brs, 4H), 1.84 (d, J=12.0 Hz, 1H), 1.57 (m, 1H);
(2R,5S,13aR)-8-гидрокси-N-[(3,5-дифторопиридин-4-ил)метил]-7,9-диоксо-2,3,4,5,7,9,13,13а-октагидро-2,5-метанопиридо[1',2':4,5]пиразино[2,1-b][1,3]оксазепин-10-карбоксамид (1.3.3): LC-MS (ESI) 433 (M+H)+,1Н NMR (DMSO-d6, 400 MHz) δ 12.46 (s, 1H), 10.47 (t, J=5.8 Hz, 1H), 8.49 (s, 2H), 8.42 (s, 1H), 5.43 (dd, J1=9.2 Hz, J2=4.0 Hz, 1H), 5.09 (brs, 1H), 4.66 (m, 3H), 4.60 (brs, 1H), 4.01 (dd, J1=12.4 Hz, J2=9.6 Hz, 1H), 1.94 (brs, 4H), 1.84 (d, J=12.0 Hz, 1H), 1.57 (m, 1H);
Пример 5. Фармацевтическая композиция в виде таблетки.
Крахмал (1600 мг), молотую лактозу (1600 мг), тальк (400 мг), и 900 мг соединения общей формулы 1 или 2 смешивали и прессовали в бар. Полученный брусок измельчали в гранулы и просеивали через сито, чтобы собрать гранулы размером 14-16 меш. Полученные таким образом гранулы были сформированы в таблетки подходящей формы весом 225 или 450 мг каждая.
Пример 6. Фармацевтическая композиция в виде капсул.
Соединения общей формулы 1 или 2 тщательно смешивали с порошком лактозы в соотношении 2:1. Полученную порошкообразную смесь упаковывали в желатиновые капсулы подходящего размера по 30, 60 или 120 мг в каждой капсуле.
Пример 7. Фармацевтическая лиофилизированная нанокомпозиция.
В сосуд (Jar) загружали циркониевый песок (150 мл) (grinding media: 0.5 mm YTZ® Zirconia Grinding and Dispersion Media; Tosoh USA, Inc.) и раствор полоксамера Р338 (10,0 г) и сахарозы или маннитола (11,6 г) в 400 мл фосфатного буферного раствора (PBS, рН=7.4). 150 мл полученного раствора помещали в сосуд (Jar), прибавляли циркониевый песок (150 мл) (grinding media: 0.5 mm YTZ® Zirconia Grinding and Dispersion Media; Tosoh USA, Inc.) и соединение общей формулы 1 или 2 (~15 г). Сосуд (Jar) помещали на мельничный аппарат (U.S. Stoneware 700 Series jar mill) и устанавливали вращение 104 об/мин и перетирали в течение 24 часов. Окончание процесса контролировали анализами распределения частиц на приборе Malvern Zetasizer Nano ZS. По окончании процесса перемалывания вращение останавливали и оставляли на 16-20 часов при температуре 2-8°С для осаждения циркониевого песка. Суспензию фильтровали через стеклянный фильтр пористостью 5-15 микрон и анализировали на установление содержания (концентрации) соединения общей формулы 1 или 2. Получали наносуспензию с размером частиц от 200 нм до 900 нм, предпочтительно 200-250 нм, которую в стерильных условиях разливали по 2 мл в стеклянные стерильные флаконы емкостью 5 мл, замораживали и лиофилизировали. Получали фармацевтическую нанокомпозицию в форме лиофилизата.
Пример 8. Инъекционный препарат.
К полученной в примере 7 нанокомпозиции в форме лиофилизата прибавляют предварительно приготовленный стерильный раствор PBS с рН=6.8 из расчета 2.2 мл на флакон емкостью 5 мл. Флаконы закупоривают предварительно стерилизованными пробками, а затем герметизируют. Получают фармацевтическую наносуспензию, готовую к использованию.
Пример 9. Инъекционный препарат.
Соединение общей формулы 1 или 2 стерилизуют гамма-излучением и подвергают чистому размолу влажных гранул с маннитом, полисорбатом 20, полиэтиленгликолем 3350 и водой для инъекции до целевого среднего размера частиц каботегравира 200 нм. Наносуспензия помещается в предварительно стерилизованные стеклянные флаконы. Флаконы закупоривают предварительно стерилизованными пробками, а затем герметизируют. Получают фармацевтическую наносуспензию, готовую к использованию.
Пример 10. Анти-ВИЧ активность соединений общей формулы 1 и 2 и прототипа (CAB).
а) Ингибирующая активность лимфоцитропный вируса ВИЧ-1 (штамм IIIB) в клеточной линии CEM-SS.
Исследование выполнено компанией ImQuest BioSciences, Inc. (Frederick, MD, USA). Клетки CEM-SS [Foley G.E. et al. Continuous Culture Of Human Lymphoblasts From Peripheral Blood Of A Child With Acute Leukemia. Cancer 1965, 18, 522-529.], полученные из Программы исследований и эталонных реагентов NIH AIDS (Manassas, VA, USA), пассировали в колбах Т-75 перед использованием в среде для культивирования клеток, состоящей из среды RPMI1640 (Lonza; Walkersville, MD, USA), дополненной 10% инактивированной теплом фетальной бычьей сыворотки (Gibco; Grand Island, NY), 2 мкМ L-глутамина (Lonza), 100 U/мкл пенициллина (Lonza) и 100 мкг/ мкл стрептомицина (Lonza) для противовирусного анализа. Клетки CEM-SS содержали в среде RPMI 140, дополненной 10% инактивированной нагреванием фетальной бычьей сывороткой, 2 мМ глутамина, 100 ед/мл пенициллина и 100 мкг/мл стрептомицина.
В качестве вируса для анализа использовали штамм лимфоцитропного вируса ВИЧ-1 IIIB [Popovic, M. et al. Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-III) from patients with AIDS and pre-AIDS. Science. 1984, 224(4648), 497-500.]. Вирус был получен из Программы исследований и эталонных реагентов NIH AIDS и выращивался в клетках CEM-SS для производства исходного пула вирусов. Предварительно титрованную аликвоту вируса удаляли из морозильника (-80°С) и позволяли медленно оттаивать до комнатной температуры в шкафу биологической безопасности. Вирус разводили в среде для тканевых культур таким образом, чтобы количество вируса, добавляемого в каждую лунку в объеме 50 мкл, было определено для того, чтобы убить от 85 до 95% клеток через 6 дней после заражения. ВИЧ-1 IIIB выращивали и титровали в клетках CEM-SS. Ингибирующую активность соединений CEM-SS на ВИЧ-1 соединений оценивали в клетках CEM-SS, как описано ранее [Nara, P.L. at al. Simple, rapid, quantitative, syncytium-forming microassay for the detection of human immunodeficiency virus neutralizing antibody. AIDS Res. Hum. Retroviruses 1987, 3, 283-302.], в микротитровальных анализах против ВИЧ, которые количественно оценивали способность соединения ингибировать индуцированный ВИЧ гибель клеток. Количественное определение проводили с использованием 2,3-бис-(2-метокси-4-нитро-5-сульфенил)-(2Н)-тетразолий-5-карбоксанилида (ХТТ) тетразолия, который метаболизируется в окрашенный формазановый продукт посредством жизнеспособных клетки.
Соединения, предоставленные в таблице 1, были растворены при 10 мкМ в ДМСО и хранились при -20°С. Тестовые материалы были оценены с использованием концентраций 30 и 10 нМ в трех экземплярах для противовирусных анализов. Шестьдесят наномолярных концентраций каждого соединения (в 2 раза больше, чем в тестовой концентрации в лунке) готовили путем разведения 10 μM ДМСО исходного 1:100 в среде для культивирования клеток (10 μL соединения, добавленного к 990 мкл среды) с последующим разведением 1:10 (10 мкл соединения добавляют к 90 мкл среды), затем 6 мкл добавляют к 994 мкл среды. 100 мкл добавляли в 96-луночный микротитровальный планшет в трех экземплярах для обеспечения эффективности и одной колориметрической лунки. Двадцать наномолярных концентраций каждого соединения (в 2 раза больше, чем в тестовой концентрации в лунке) готовили путем разбавления 10 мкМ ДМСО исходного 1:100 в среде для культивирования клеток (10 мкл соединения, добавленного к 990 мкл среды) с последующим разведением 1:100 (10 мкл соединения добавляют к 900 мкл среды), затем 20 мкл добавляют к 980 мкл среды. 100 мкл добавляли в 96-луночный микротитровальный планшет в трех экземплярах для обеспечения эффективности и одной колориметрической лунки.
Azidothymidine (AZT) был приобретен у Sigma Aldrich (США) и оценен в качестве положительного контрольного соединения в противовирусном анализе, используя кривую реакции шести концентраций на каждом планшете для анализа.
Тестируемые соединения первоначально оценивали в отношении штамма IIIB ВИЧ-1 в клетках CEM-SS при тестовых концентрациях 30 и 10 нМ. Данные по противовирусной эффективности обобщены в таблице 1. Соединения, активные при 10 и 30 нМ, были затем оценены в анализе цитопротекции ВИЧ при 2 нМ. Для соединений показавшие активность при 2 нМ была определена Ингибирующая активность (ЕС50). Данные по противовирусной эффективности также приведены в таблице 1.
Контрольное соединение AZT оценивалось параллельно с представленными тестовыми соединениями и давало значения ЕС50 в диапазоне от менее 2 нМ до 7 нМ. При оценке противовирусной активности соединений при 2 нМ, AZT давал значения EC50 в диапазоне от 10 до 20 нМ.

б. Ингибирующая активность ВИЧ-1 (штамм NL4.3), несущий репортерный ген GFP (NL4.3-GFP) в клеточной линии SupT1.
Исследование выполнено компанией RetroVirox Inc. (San Diego, CA, USA). Клетки SupT1 были инфицированы ВИЧ штаммом NL4.3, несущим ген зеленого флуоресцентного белка (NL4.3-GFP). Препарат вируса получали путем трансфекции клеток 293Т провирусной ДНК. Через 48 часов после трансфекции препарат замораживали и хранили до использования. Для повышения эффективности инфекции, суспензию клеток SupT1 осаждали из инфекционной смеси центрифугированием. Тестируемые вещества добавляли к клеткам непосредственно перед добавлением вируса. Через 2 ч инкубации инфекционную смесь заменяли свежей культуральной средой с тестируемыми препаратами. Эффективность инфекции определяли через 45 ч путем подсчета процента флуоресцирующих клеток, по сравнению с неинфицированными клеточными культурами. Цитотоксичность тестируемых соединений определяли параллельно на той же, но не инфицированной клеточной линии SupT1, используя реагент ХТТ. Использовали серийные десятикратные разведения препаратов (начиная от 10 μM для определения антивирусной активности, или от 100 мкМ - для цитотоксичности). В качестве негативного контроля использовали 0,1% ДМСО. Были рассчитаны значения ЕС50. Качество тестов определяли при помощи следующих контролей: отношение сигнала к фону, интегразный ингибитор ралтегравир (1 мкМ), а также воспроизводимость теста. Полученные результаты представлены в Таблице 1.
Пример 11. Определение термодинамической растворимости соединений общей формулы 1 и 2.
Взвешивали в стеклянный флакон по 2 мг испытуемого соединения общей формулы 1 и 2. Добавляли по 0,5 мл универсального буфера (pION) с рН 7,4 либо деионизированной воды в соответствующие флаконы и плотно закрывали крышкой. Помещали флаконы на вращающийся шейкер и инкубировали в течение 24 ч для уравновешивания раствора и твердой фазы в точке насыщения. Затем из флаконов отбирали по 200 мкл полученных растворов (в двух повторах) помещали в 96-луночный фильтровальный планшет MultiScreen Solubility Filter Plate (Millipore) и фильтровали с помощью вакуума (10" Hg) в полипропиленовый планшет с U-образным дном. При необходимости разводили фильтраты с помощью соответствующего буфера (воды). Отбирали 120 мкл фильтрата (либо разбавленного фильтрата) в 96-луночный планшет с UV-прозрачным дном и добавляли 60 мкл/лунку ацетонитрила и 20 мкл/лунку ДМСО, тщательно перемешивали многоканальной пипеткой (ацетонитрил и ДМСО добавляли для соответствия состава раствора в калибровочных стандартах, как описано ниже). Проводили измерение оптической плотности полученных растворов на мультифункциональном ридере Infinite 200 PRO в диапазоне длин волн от 240 нм до 400 нм с шагом 10 нм. Определяли ODλ растворов при длине волны, выбранной для построения калибровочного графика.
Определение концентрации вещества в растворе после теста на растворимость проводили с помощью калибровочного графика. Для этого из отдельной навески готовили исходный раствор в ДМСО с концентрацией 2 мг/мл. При помощи серийных разведении с шагом 2 готовили 6 10-кратных растворов в ДМСО, которые затем разбавляли в 10 раз в соответствующем буфере с 30% содержанием ацетонитрила, чтобы обеспечить полную растворимость тестируемого соединения в смеси. Диапазон финальных концентраций калибровочного графика: 0,2; 0,1; 0,05; 0,025; 0,0125; 0,00625 и 0 мг/мл. Калибровочные стандарты готовили в 96-луночной UV-плашке следующим образом: вносили в лунку 20 мкл 10-кратного сток-раствора в ДМСО, 60 мкл ацетонитрила и 120 мкл соответствующего буфера, перемешивали. Проводили измерение оптической плотности полученных растворов на мультифункциональном ридере Infinite 200 PRO в диапазоне длин волн от 240 нм до 400 нм с шагом 10 нм. Выбирали оптимальную длину волны, обычно соответствующую максимуму поглощения, и по значениям ODλ соединения при данной длине волны строили линейную зависимость ODλ от концентрации соединения.
Расчет концентрации соединения в фильтрате после теста на растворимость проводили по формуле:
Shake-Flask Solubility = (ODλ filtrate - ODλ blank) / (Slope × 1,67 × Filtrate dilution)
где: slope - наклон калибровочного графика; 1,67 - фактор разведения фильтрата ацетонитрилом и ДМСО; Filtrate dilution - дополнительное разведение фильтрата, если применялось. Полученные результаты по термодинамической растворимости соединений общей формулы 1 и 2 представлены в таблице 2.


Пример 12. Фармакокинетика соединений общей формулы 1.1.
Исследования проведены на крысах самцах линии Sprague-Dawley, полученных из питомника Charles River (Германия). Возраст животных к моменту введения составлял 8-10 недель. Животные были предварительно катетеризированы, с тем чтобы все образцы крови в течение исследования каждого вещества можно было брать у одних и тех же животные. Каждое вещество было исследовано на 3х животных при каждом пути введения.
Исследуемые вещества вводили крысам внутривенно в хвостовую вену в виде раствора с концентрацией 0.2 мг/мл в 10% ДМСО/10% Солютол/80% 0.05M N-метилглюкамин в дозе 1 мг/кг. Образцы крови собирали через 5, 15, 30 мин, 1, 2, 4, 8 и 24 часа после введения. Внутрижелудочно вещества вводили животным с помощью гаважа в виде раствора с концентрацией 0,5 мг/мл в 0,5% метилцеллюлозе в дозе 5 мг/кг. Образцы крови собирали через 15, 30 мин, 1, 2, 4, 8 и 24 часа после введения. Кровь собирали в объеме 0,2 мл в полипропиленовые пробирки, содержащие 20 мкл 5% NaЭДТА. Плазму крови отделяли центрифугированием при 1500 g в течение 10 мин и хранили до анализа при температуре -80°С.
К 45 мкл экспериментальных образцов добавляли 5 мкл чистой смеси ацето нитрил: во да 1:1. Пробы тщательно перемешивали на вортексе в течение 10 сек. К образцам добавляли 100 мкл охлажденного до +4°С толбутамида (200 нг/мл в MeCN), перемешивали на вортексе. Образцы осаждали на льду при температуре +4°С в течение 15 минут. Центрифугировали при 2700 g 10 мин при +4°С. По 100 мкл супернатантов переносили в чистую плашку для ВЭЖХ/МС/МС анализа.
Анализ проводили методом ВЭЖХ-МС/МС с помощью ВЭЖХ системы Agilent 1290 Infinity, соединенной с масс-спектрометром АВ Sciex QTrap 6500.
Фармакокинетический анализ данных «концентрация в плазме крови - время» выполнялся модельно-независимым методом с помощью программы Phoenix™ WinNonlin® 6.3 (Pharsight Corp.). Рассчитывались следующие фармакокинетические параметры (Табл. 3): максимальная концентрация в печени (Cmax) и время ее достижения (Tmax), период полувыведения (T1/2), площадь под ФК кривой (AUC0-t, AUC0-inf), клиренс (Cl), константа элиминации (kel).
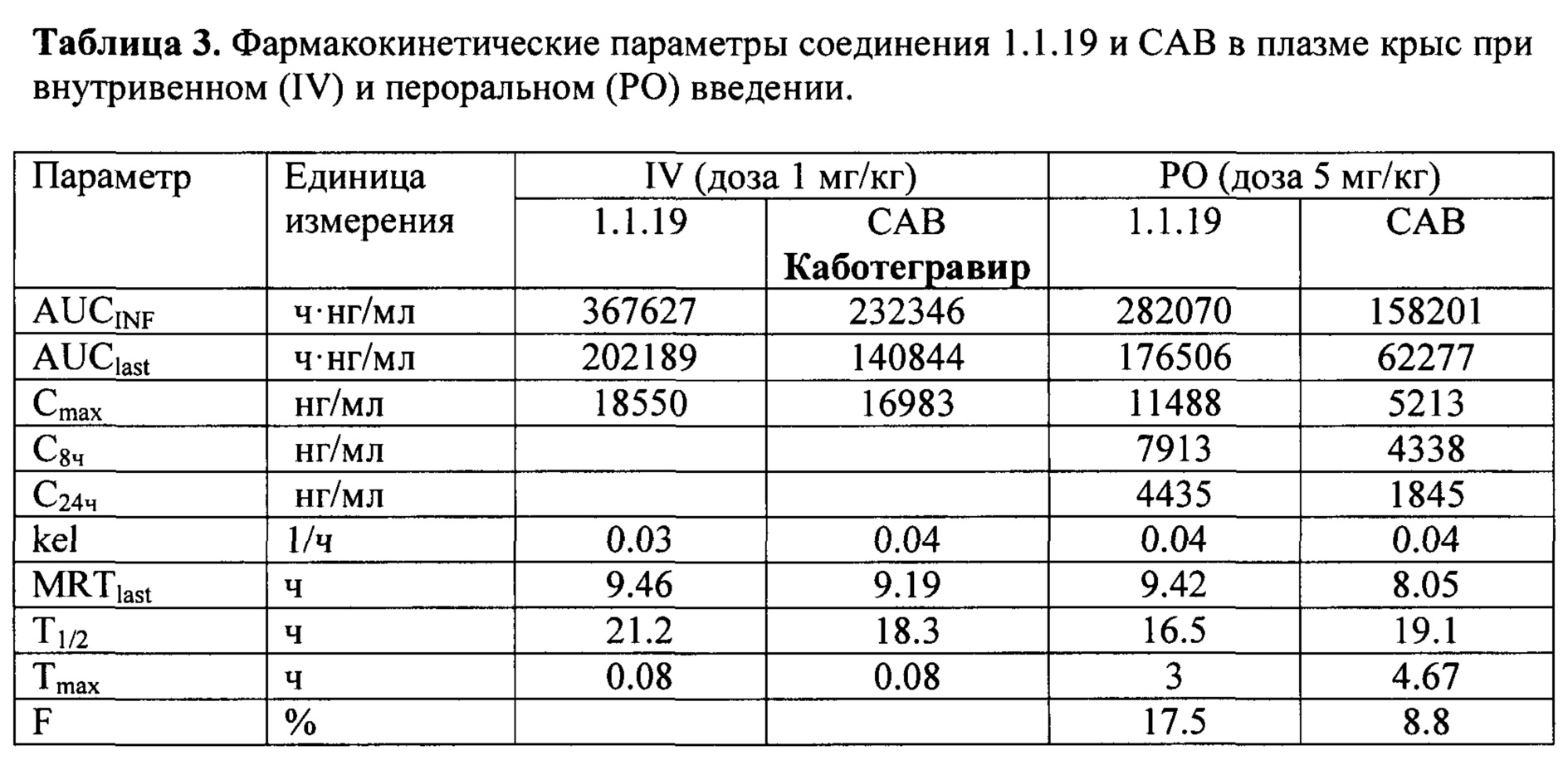
Пример 13. Определение стабильности в биологических средах композиций, включающих соединения общей формулы 1 и 2.
а) Стабильность в плазме крови. Тестируемое соединение в конечной концентрации 1 мкМ инкубировали в пулированной плазме крови человека (Innovative Research). Инкубацию проводили в шейкере-инкубаторе (Vortemp56) при 37°С и перемешивании при 300 об/мин. Через определенные промежутки времени (0, 0.25, 0.5, 1, 2, 4, 6 8, 24 ч) проводился отбор проб в объеме 30 мкл, реакция останавливалась путем добавления к отобранной пробе 180 мкл холодного ацетонитрила, содержащего внутренний стандарт. После чего образцы центрифугировались и в течение 10 мин при 3000 об/мин. 150 мкл супернатанта отбиралось для анализа. Инкубация проводилась в двух повторах, каждая проба измерялась дважды. Анализ проб проводился методом ВЭЖХ-МС/МС, разработанным для каждого тестируемого пролекарства, с использованием хроматографической системы 1290 Infinity II (Agilent Technologies), сопряженной с тандемным масс-спектрометром QTRAP5500 (АВ Sciex). При разработке условий масс-спектрометрического детектирования растворы тестируемых соединений в смеси ацетонитрил-вода 1:1 с концентрацией 100 нг/мл анализировали путем прямого ввода в масс-спектрометр при помощи шприцевого насоса при ионизации электрораспылением в режиме регистрации положительных ионов. При сканировании в режиме полного ионного тока (MS1) определяли молекулярный ион для каждого соединения, основные ионы-продукты регистрировали в режиме MS2. Далее была проведена оптимизация МС/МС метода в режиме MRM для достижения максимальной чувствительности. При количественной обработке хроматограмм использовали наиболее интенсивный MRM-переход для аналита и внутреннего стандарта. Хроматографическое разделение проводили на колонке YMC Triart С18, 50×2.мм, 1,.9 мкм в градиентном режиме элюирования в подвижной фазе состава 0,1% муравьиная кислота в воде - 0.1% муравьиная кислота в ацетонитриле. В качестве внутреннего стандарта использовали толбутамид (Fluka). По кинетике убыли тестируемого пролекарства в противовирусной композиции в процессе инкубации в биологической среде рассчитывали время полураспада (T1/2). Для расчетов использовали нормированные на сигнал внутреннего стандарта значения площадей хроматографических пиков веществ в опытных образцах. По линейной зависимости лог-нормированных площадей хроматографических пиков от времени рассчитывали константу скорости элиминации (k - наклон линейного участка). Далее рассчитывали время полураспада: T1/2=0.693/k. Было установлено (Фиг. 2), что соединения общей формулы 1.1 стабильны в плазме человека и крыс.
б) Стабильность в S9 фракции. Реакционная смесь была приготовлена в 0,1 М калия фосфатном буфере (рН 7,4 BD Gentest) в общем финальном объеме 250 мкл и содержала 1 мМ NADPH-тетранатриевую соль (AppliChem), 7 мМ глюкозо-6-фосфат натриевую соль (Sigma), 1.5 U/мл глюкозо-6-фосфат дегидрогеназу (Sigma), 3.3 мМ MgCl2 (Sigma), 5 мМ уридин-5-дифосфат-глюкуроновой кислоты тринатриевую соль (УДФГК, Sigma) и 1 мкМ тестируемого соединения. Метаболическая реакция была инициирована путем добавления суспензии S9 фракции печени человека (BD Gentest) или крысы, финальная концентрация белка составляла 1 мг/мл. Реакционная смесь инкубировалась при 37°С на шейкере (Vortemp56) с перемешиванием при 400 об/мин. Через определенные промежутки времени (0, 0.25, 0.5, 1, 2, 4, 6 8, 24 ч) проводили отбор проб в объеме 30 мкл, реакцию останавливали путем добавления к отобранной пробе 180 мкл холодного ацетонитрила, содержащего внутренний стандарт. Осаждение белков проводили на льду в течение 15 мин. После чего образцы центрифугировали и в течение 10 мин при 3000 об/мин. 150 мкл супернатанта отбирали для анализа. Инкубацию проводили в двух повторах, каждую пробу измеряли дважды. Установлено (Фиг. 3), что соединения общей формулы 1.1 стабильны в S9 фракции печени человека и крыс.
Пример 14. Связывания с белками плазмы крови соединений 1.1.17 и 1.1.19.
Для определения связывания с белками использовали пулированные образцы плазмы крови, разбавленные фосфатным буфером до 50%. Тест проводили в 48-луночной плашке с тефлоновым покрытием, предназначенной для диализа. Каждая лунка содержит две отдельных камеры, разделенные вертикальной полупроницаемой диализной мембраной с порами 8 кДа. Образец плазмы с 1 мкМ тестируемого соединения вводили в одну из камер, в другую камеру помещали буферный раствор с рН=7,2. С течением времени при 37°С и покачивании происходила пассивная диффузия несвязанного соединения и через 4 ч достигается состояние равновесия между камерами с плазмой и с буферным раствором. Количество свободной фракции оценивали с помощью ВЭЖХ-МС/МС после преципитации белков ацетонитрилом. В ходе исследования также по массовому балансу оценивали стабильность вещества в течение 4 ч и пассивная проницаемость через диализную мембрану в буфере. Контрольное соединение - варфарин. Полученные результаты представлены в таблице 4, из которых следует, что соединения общей формулы 1.1 обладают высокой степенью связывания с белками плазмы крови человека и крыс.

Реферат
Настоящее изобретение относится к новому анелированному 9-гидрокси-1,8-диоксо-1,3,4,8-тетрагидро-2Н-пиридо[1,2-]пиразин-7-карбоксамиду общей формулы 1 или 2 и его стереоизомеру, а также к вариантам способа его получения. Соединения обладают противовирусной активностью в отношении интегразы вируса иммунодефицита человека (ВИЧ). В общей формуле 1 или 2кольцо Апредставляет собой необязательно замещенный метилом 5-7-членный насыщенный гетероцикл или гетеробицикл, кольцо Апредставляет собой необязательно замещенный метилом насыщенный моноциклический гетероцикл, содержащий дополнительно атом кислорода, кольцо Апредставляет собой 5-6-членный моноциклический насыщенный циклоалкан, R представляет собой возможно замещенный одним, двумя или тремя заместителями 5-6-членный моногетероцикл с 1-3 гетероатомами в цикле или 8-10-членный бициклический гетероцикл с 1-4 гетероатомами, где гетероатомы выбраны из О, S, N и где заместители выбраны из атома галогена и С1-С3 алкила. Предпочтительно соединение общей формулы I, представляющее собой (3S,11R)-6-гидрокси-N-[(2,6-дифторопиридин-3-ил)метил]-3-метил-5,7-диоксо-2,3,5,7,11,11-гексагидро[1,3]оксазоло[3,2-]пиридо[1,2-d]пиразин-8-карбоксамид (1.1.19), может быть в виде кристаллической формы, имеющей параметры элементарной ячейки, указанные в формуле изобретения. 9 н. и 10 з.п. ф-лы, 5 ил., 4 табл., 14 пр.
Формула
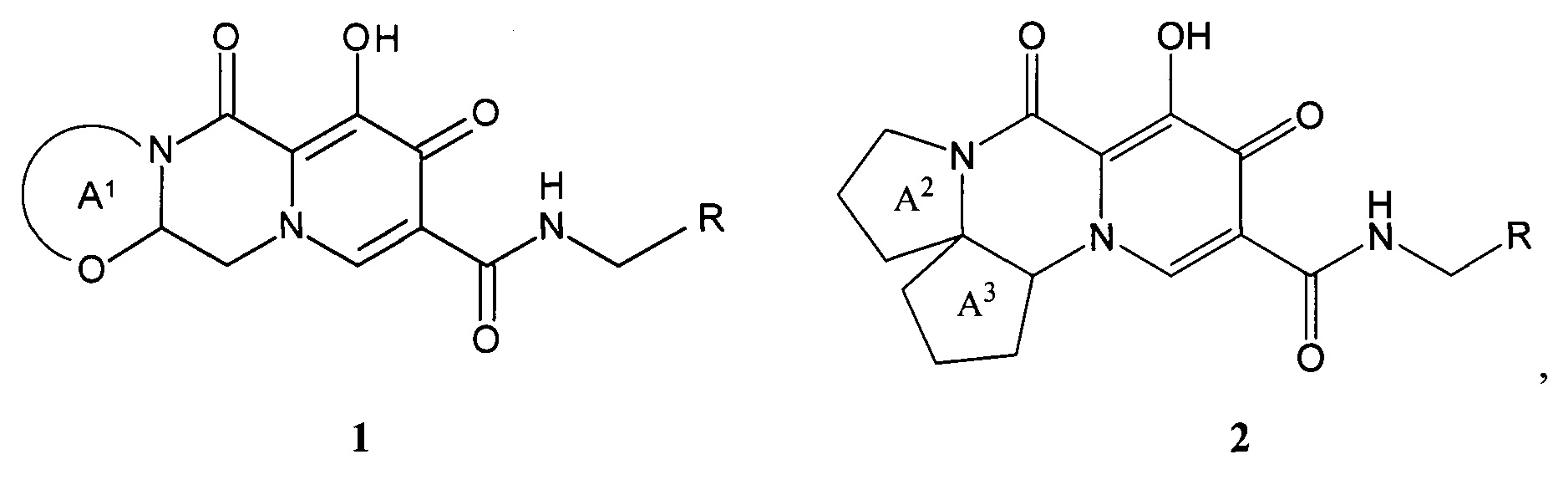

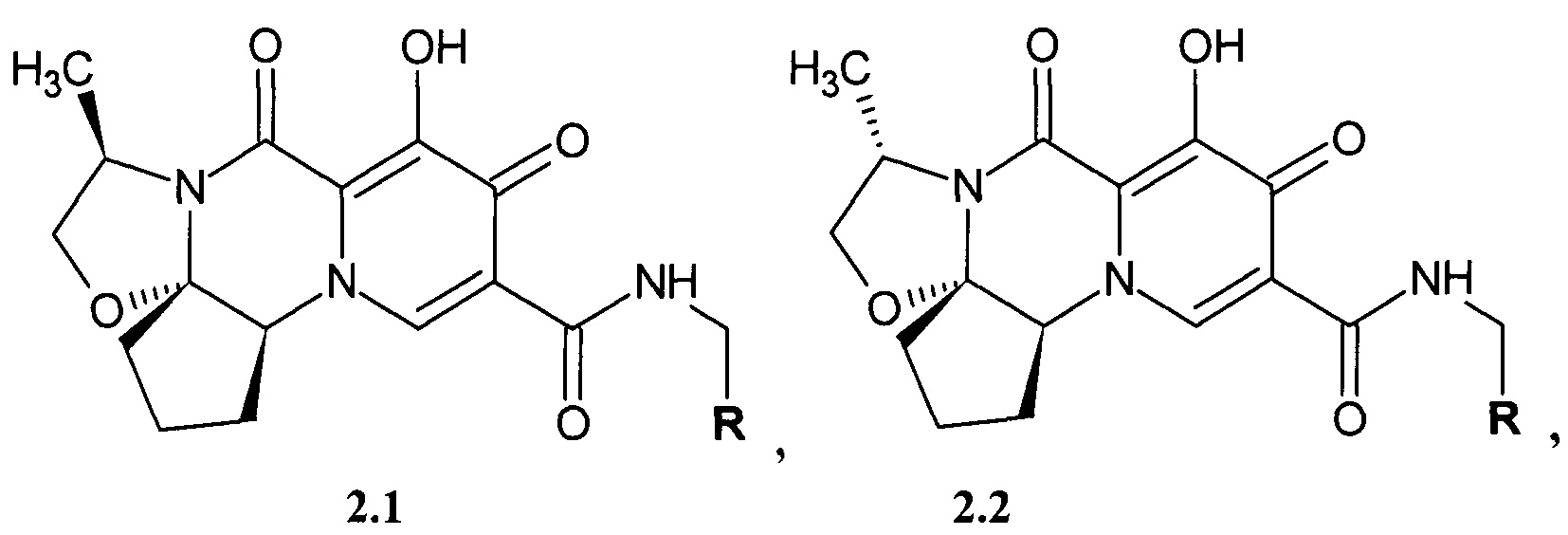







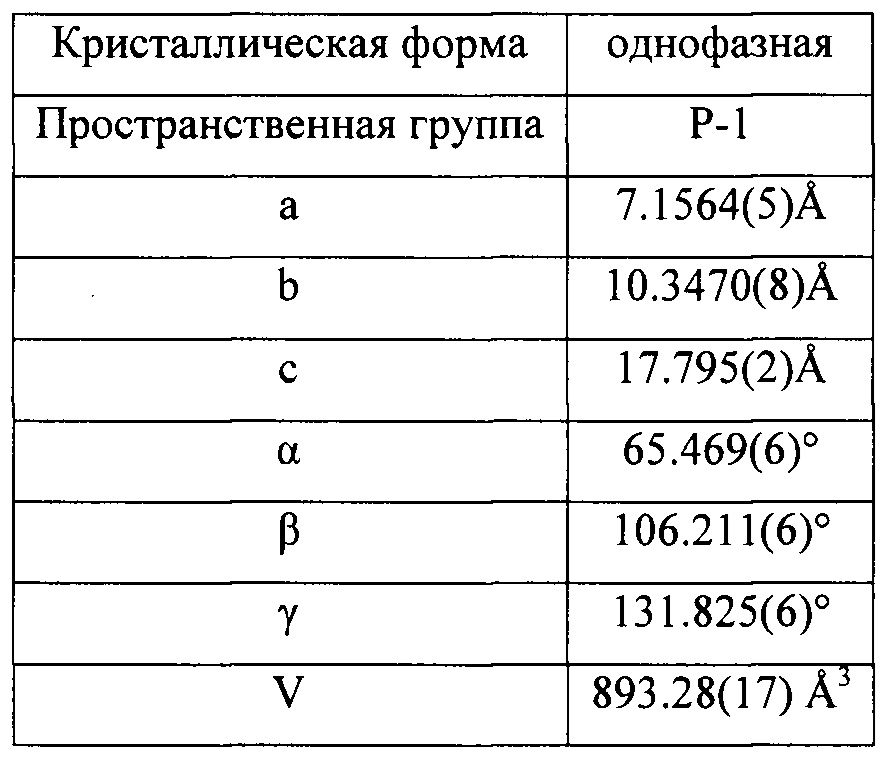
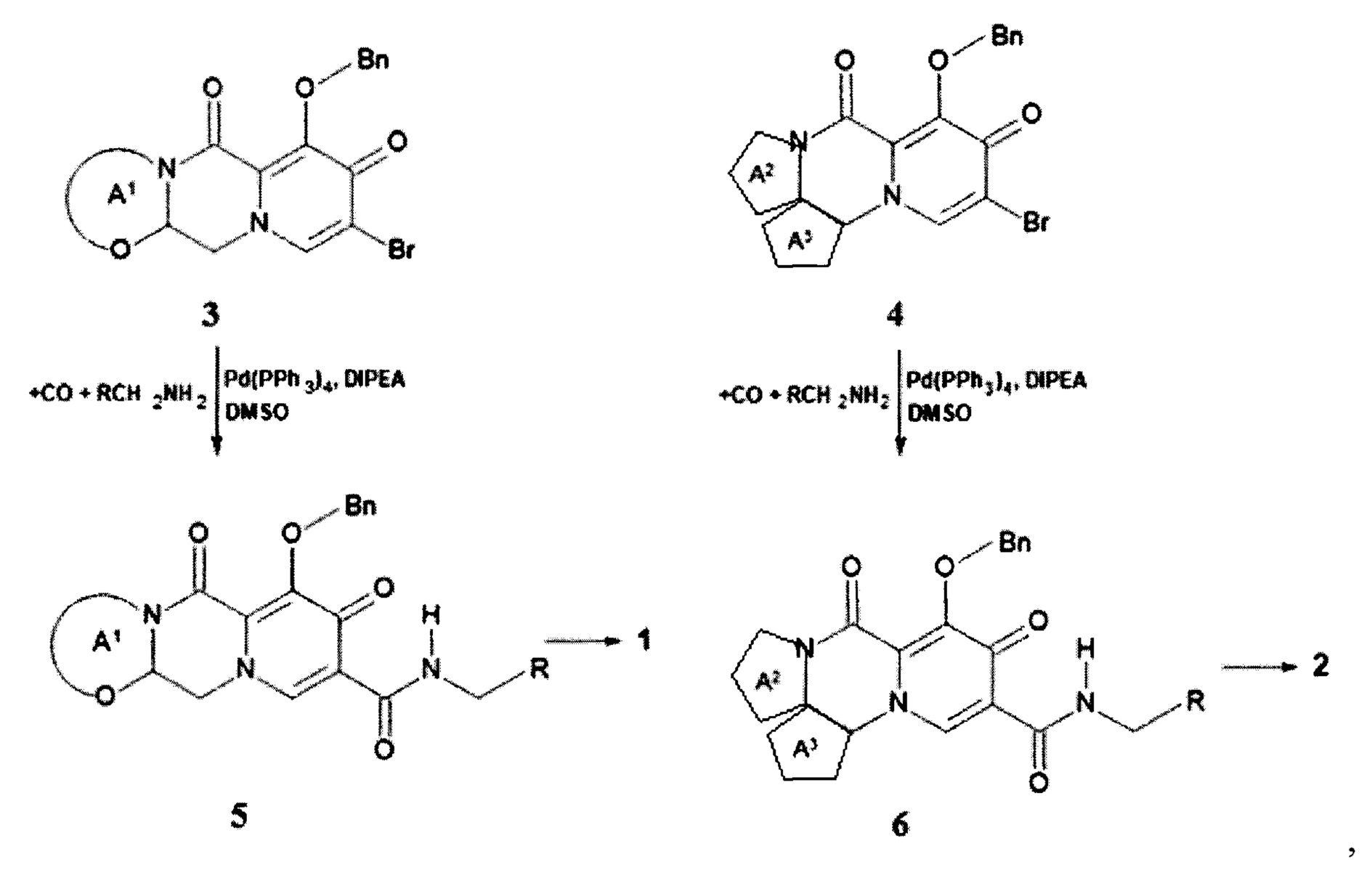
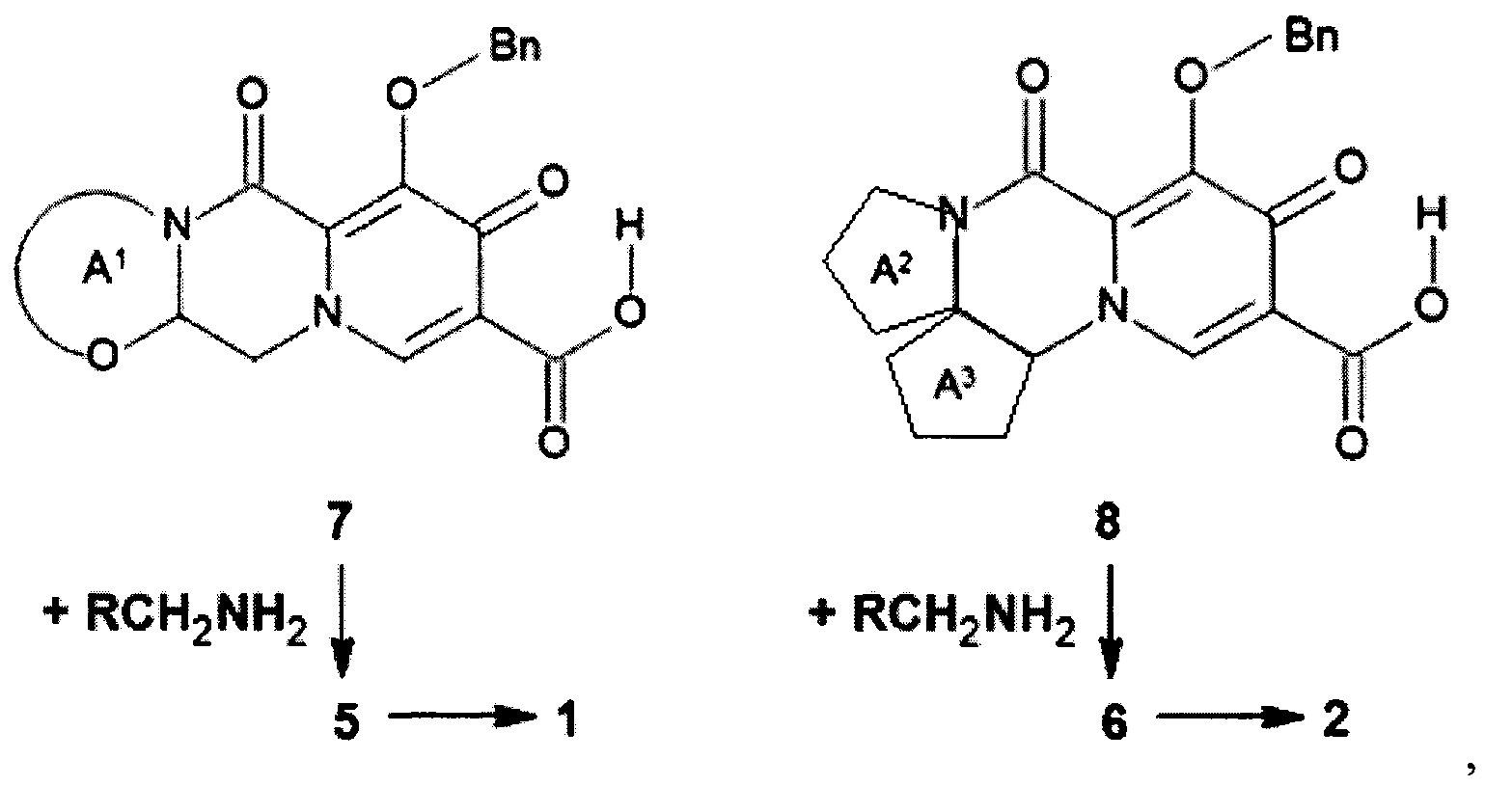
Документы, цитированные в отчёте о поиске
Способ получения амидов из карбонильных соединений
Фармацевтические композиции и соответствующие способы доставки
Комментарии