Фармацевтическая композиция ингибитора папилломавируса - RU2661007C2
Код документа: RU2661007C2
Чертежи

Описание
Настоящее изобретение относится к фармацевтической композиции соединений, применимых в лечении и профилактике связанных с папилломавирусом инфекций, описанных в частности в заявке WO 2007/135106.
Соединения, описанные в заявке WO 2007/135106, имеют следующую структуру:

отличающиеся тем, что:
- А представляет собой необязательно замещенный арил, циклоалкил, циклоалкенил или гетероциклическую группу,
- А представляет собой необязательно замещенный арил или гетероцикл,
- R1 и R2 представляют независимо атом водорода или различные заместители,
- R3 представляет собой кислотную функциональную группу или предшественник или биоизостерный радикал этой функциональной группы,
- G1 представляет собой связь или углеводородную цепь, и
- G2 представляет собой

Однако в WO 2007/135106 не описана лекарственная форма этих соединений.
Эти соединения, применимые в лечении и профилактике связанных с папилломавирусом инфекций, предназначены в частности для местного применения на кожу и слизистые оболочки, в частности в аногенитальной области.
Авторы изобретения тем самым раскрыли очень простые фармацевтические композиции, способные быстро и в высокой степени высвобождать активный ингредиент.
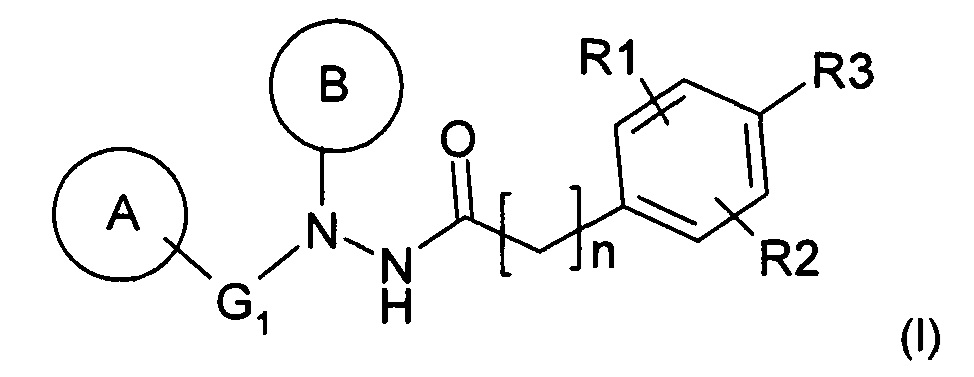
Таким образом, объектом настоящего изобретения является фармацевтическая композиция, включающая соединение общей формулы (I):
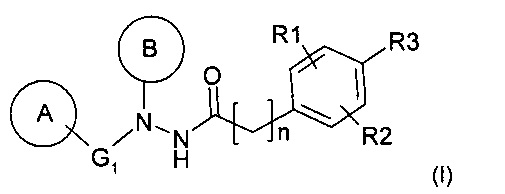
или его фармацевтически приемлемую соль,
отличающаяся тем, что:
- G1 представляет собой связь или линейную или разветвленную, насыщенную или ненасыщенную углеводородную цепь, содержащую от 1 до 4 атомов углерода, необязательно замещенных одной или двумя алкильными группами, предпочтительно идентичными,
- А представляет собой арильную группу, такую как фенил, необязательно замещенную:
- в мета- или пара-положении:
- атомом галогена или цианистой, алкоксильной, галоалкоксильной, ациламиноалкильной или -XR группой, где X представляет собой -О-, -S-, -SO-, -SO2- или -СО- и R представляет собой арилалкильную, циклоалкильную или арильную группу, каждая из которых необязательно замещена одним или двумя заместителями, идентичными или различными, такими как атом галогена, алкоксильная или ацильная группа, или
- циклоалкильной, арильной или арилалкильной группой, каждая из которых необязательно замещена одним или двумя заместителями, идентичными или различными, такими как ацильная или алкоксильная группа,
- и/или в орто или мета-положении алкильной группой,
- В представляет собой арильную группу, предпочтительно фенил, замещенную в орто-положении гетероциклом, предпочтительно N-циклоалкилом, таким как пиперидин-1-ил группа, и необязательно замещенную в орто- положении алкильной группой, такой как метил,
- n - целое число между 1 и 4, предпочтительно между 1 и 2, и более предпочтительно 1,
- R1 представляет собой алкоксильную группу, такую как метоксильную, предпочтительно в орто-положении по отношению к R3,
- R2 представляет собой атом водорода или галогена, такого как хлор или бром, или алкильную группу, такую как метил, предпочтительно в мета-положении по отношению к R3, и
- R3 представляет собой кислотную или эфирную группу, и предпочтительно кислотную,
в комбинации с усиливающим вязкость агентом.
«Фармацевтически приемлемый» в настоящем изобретении означает применяющийся в приготовлении фармацевтической композиции, как правило являющийся безопасным, нетоксичным и не являющийся вредным ни в биологическом, ни в другом отношении, и пригодный как для применения в ветеринарии, так и для применения в фармацевтике для людей.
«Фармацевтически приемлемые соли» соединения в настоящем изобретении означает соли, которые являются фармацевтически приемлемыми, как определено здесь, и которые имеют желаемую фармакологическую активность исходного соединения. Такие соли включают:
(1) гидраты и сольваты,
(2) соли присоединения кислоты, образованные неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобные; или образованные органическими кислотами, такими как уксусная кислота, бензолсульфоновая кислота, бензойная кислота, камфорсульфоновая кислота, лимонная кислота, этансульфоновая кислота, фумаровая кислота, глюкогептоновая кислота, глюконовая кислота, глютаминовая кислота, гликолевая кислота, гидроксинафтоевая кислота, 2-гидроксиэтансульфоновая кислота, молочная кислота, малеиновая кислота, яблочная кислота, миндальная кислота, метансульфоновая кислота, муконовая кислота, 2-нафтален-сульфоновая кислота, пропионовая кислота, салициловая кислота, янтарная кислота, дибензол-L-винная кислота, винная кислота, p-толуолсульфоновая кислота, триметилуксусная кислота, трифторуксусная кислота и тому подобные; и
(3) соли, образованные, когда протон кислоты, присутствующий в исходном соединении, или замещен ионом металла, например ионом щелочного металла (например, Na+, K+ или Li+), ионом щелочноземельного металла (таким как Ca2+ или Mg2+) или ионом алюминия; или связан с органическим или неорганическим основанием. Приемлемые органические основания включают диэтаноламин, этаноламин, N-метилглютамин, триэтаноламин, трометамин и тому подобные. Приемлемые неорганические основания включают алюминия гидроксид, кальция гидроксид, калия гидроксид, натрия карбонат и натрия гидроксид.
В частности это будет соль щелочного метала, и в частности калиевая соль.
«Ненасыщенный» в контексте настоящего изобретения означает группу, включающую одну или более ненасыщенные связи.
«Ненасыщенная связь» в контексте настоящего изобретения означает двойную связь или тройную связь.
«Галоген» в контексте настоящего изобретения означает атом фтора, брома, хлора или йода. Преимущественно является атомом фтора, брома или хлора.
«Алкильная» группа в контексте настоящего изобретения означает линейную или разветвленную насыщенную углеводородную цепь, содержащую от 1 до 6 атомов углерода, в частности следующие группы: метил, этил, n-пропил, изопропил, n-бутил, изобутил, sec-бутил, tert-бутил, n-пентил или n-гексил. Преимущественно является метилом.
«Циклоалкильная» группа в контексте настоящего изобретения означает насыщенную моноцикличную или полицикличную систему, предпочительно моно-, би- или трицикличную, содержащую от 3 до 12 атомов углерода, кольца, способные к объединению или образованию мостиковой связи в парах, такие как следующие группы: циклопропил, циклопентил, циклогексил, циклогептил, циклооктил, адамантил, декалинил или норборнил.
"N-циклоалкильная группа" в контексте настоящего изобретения означает циклоалкильную группу, как определено выше, где атом углерода замещен атомом азота, связь с молекулой осуществляется этим атомом азота. Преимущественно является пипередин-1-ил или пирролидин-1-ил группой.
«Ацильная группа» в контексте настоящего изобретения означает группу формулы -C(O)-Z, где Z представляет собой алкильную группу, как определено выше, или фенил. Преимущественно может являться ацетильной, этилкарбонильной или бензоильной группой.
«Алкоксильная группа» в контексте настоящего изобретения означает алкильную группу, как определено выше, связанную с молекулой посредством атома кислорода. Может являться, в частности, метоксильной, этоксильной, n-пропоксильной, изопропоксильной, n-бутокси, iso-бутоксильной, sec-бутоксильной или tert-бутоксильной группой.
«Галоалкоксильная группа» в контексте настоящего изобретения означает алкоксильную группу, как определено выше, замещенную одним или несколькими атомами галогена, как определено выше. Предпочтительно будет являться фторалкоксильной, то есть алкоксильной группой, замещенной одним или несколькими атомами фтора, такой как -OCF3 или -OCH2CF3 группа.
«Арильная» группа в контексте настоящего изобретения означает ароматическую группу, содержащую предпочтительно от 5 до 10 атомов углерода и включающую одно или несколько соединенных колец, таких как, например, фенильная или нафтильная группа. Преимущественно является фенилом.
«Гетероцикл» в контексте настоящего изобретения означает насыщенную, ненасыщенную или ароматическую моноцикличную или полицикличную систему, и предпочтительно моно- или бицикличную, содержащую от 3 до 12 членов, кольца, способные к объединению, спиро-соединению или образованию мостиковой связи в парах, и включающие от 1 до 4 гетероатомов, идентичных или различных, выбранных из О, S и N, и необязательно включающих одну или две оксо или тиоксогруппы, при этом подразумевается, что в случае полициклической системы одно из колец может быть ароматическим, тогда как другие могут быть ароматическими, насыщенными или ненасыщенными. Преимущественно это относится к следующим группам: пиперидил, пиперазил, фурил, тиенил, пирролил, пиразолил, имидазолил, пиридил, пиримидил, пиразинил, пирадизинил, бензофурил, бензотиенил, индолил, квинолил, изоквинолил, бензодиоксолил, бензодиоксинил, бензо[1,2,5]тиадиазолил, бензо[1,2,5]оксадиазолил, [1,2,3]триазолил и [1,2,4]триазолил.
«Арилалкильная группа» в контексте настоящего изобретения означает арильную группу, как определено выше, связанную с молекулой посредством алкильной группы, как определено выше. Предпочтительно является бензильной группой.
«Ациламиноалкил» в контексте настоящего изобретения означает группу формулы -Alk-NHCO-Alk', где Alk и Alk' представляют, независимо друг от друга, алкильную группу, как определено выше.
«Кислота» в контексте настоящего изобретения означает СООН группу.
«Эфир» в контексте настоящего изобретения означает группу -CO-O-Alk, где Alk представляет собой алкильную группу, как определено ранее.
«Усиливающий вязкость агент» в контексте настоящего изобретения означает соединение, которое увеличивает вязкость текучей среды, такой как жидкость. Таким образом изменяются реологические свойства текучей среды, которая становится более вязкой.
Преимущественно композиция согласно изобретению будет включать от 0,01 до 10 масс. %, в частности от 2 до 8 масс. %, предпочтительно около 5 масс. % соединения формулы (I) по отношению к общей массе композиции.
Усиливающий вязкость агент может представлять собой гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу или карбомер. Усиливающий вязкость агент может также находиться в форме их смеси. Предпочтительно усиливающий вязкость агент будет гидроксипропилцеллюлозой.
Композиция согласно изобретению может включать от 0,01 до 50 масс. %, в частности от 0,05 до 10 масс. %, в особенности от 0,5 до 5 масс. %, предпочтительно от 1 до 5 масс. % этого усиливающего вязкость агента по отношению к общей массе композиции.
Композиция согласно изобретению будет преимущественно также включать растворитель, такой как пропиленгликоль, глицерин или полиэтиленгликоль. Растворитель также может находиться в форме их смеси. Предпочтительно растворитель будет пропиленгликолем.
Композиция согласно изобретению может включать от 40 до 99,9 масс. %, в частности от 80 до 99,5 масс. %, в особенности от 85 до 99 масс. %, предпочтительно от 90 до 95 масс. % этого растворителя по отношению к общей массе композиции.
Таким образом, композиция согласно настоящему изобретению преимущественно будет содержать и предпочтительно будет состоять из соединения формулы (I), усиливающего вязкость агента и растворителя.
Эта фармацевтическая композиция может в частности содержать или состоять из (массовые проценты, выраженные в отношении к общей массе фармацевтической композиции):
- от 0,01 до 10 масс. %, в частности от 2 до 8 масс. % соединения формулы (I),
- от 0,01 до 50 масс. %, в частности от 0,05 до 10 масс. %, в особенности от 0,5 до 5 масс. %, предпочтительно от 1 до 5 масс. % усиливающего вязкость агента, и
- от 40 до 99,9 масс. %, в частности от 80 до 99,5 масс. %, в особенности от 85 до 99 масс. %, предпочтительно от 90 до 95 масс. % растворителя.
Преимущественно эта фармацевтическая композиция будет содержать или будет состоять из (массовые проценты, выраженные в отношении к общей массе фармацевтической композиции):
- от 0,01 до 10 масс. %, в частности от 2 до 8 масс. %, и предпочтительно около 5 масс. % соединения формулы (I),
- от 0,05 до 10 масс. %, в частности от 0,5 до 5 масс. %, в особенности от 1 до 5 масс. %, предпочтительно около 3 масс. % усиливающего вязкость агента, выбранного из гидроксипропилцеллюлозы, гидроксипропилметилцеллюлозы, карбомера и их смесей, и
- от 80 до 99,5 масс. %, в частности от 85 до 99 масс. %, в особенности от 90 до 95 масс. %, предпочтительно около 92 масс. % растворителя, выбранного из пропиленгликоля, глицерина, полиэтиленгликоля и их смесей.
Предпочтительно, композиция согласно настоящему изобретению будет содержать и предпочтительно будет состоять из соединения формулы (I), гидроксипропилцеллюлозы и пропиленгликоля.
Данная композиция согласно изобретению будет таким образом иметь в частности следующую композицию, по отношению к общей массе композиции:
- от 0,01 до 10 масс. %, в частности от 2 до 8 масс. %, предпочтительно около 5 масс. % соединения формулы (I),
- от 0,01 до 50 масс. %, в частности от 0,05 до 10 масс. %, в особенности от 1 до 5 масс. %, предпочтительно около 3 масс. % гидроксипропилцеллюлозы, и
- от 40 до 99,9 масс. %, в частности от 80 до 99,5 масс. %, в особенности от 85 до 99 масс. %, в особенности от от 90 до 95 масс. %, предпочтительно около 92 масс. % пропиленгликоля.
Композицию согласно изобретению будут предоставлять в частности в форме геля.
В этом случае гель будет иметь в частности вязкость от 5000 до 50000 мП, в частности от 15000 до 25000 мП, измеренную в соответствии со стандартами Европейской Фармакопеи 2.2.10. Вязкость измеряют в частности при 25°C с применением вискозиметра модели Брукфильда HDBV+ или эквивалентного, с веретеном №21 на скорости 10 оборотов в минуту. Значения дают после 1 минуты ротации.
Преимущественно G1 представляет собой связь или линейную и насыщенную углеводородную цепь, содержащую от 1 до 4 атомов углерода, и предпочтительно представляет собой связь.
Преимущественным образом радикал А, определенный выше, представляет собой арил, предпочтительно фенил, замещенный в мета или пара-положении, предпочтительно пара, алкоксильной группой, такой как метоксильная, или арильной или арилалкильной группой, такой как фенил или бензил, необязательно замещенной одним или двумя заместителями, идентичными или различными, такими как ацильная или алкоксильная группа.
Преимущественно В представляет собой арильную группу, предпочтительно фенил, замещенный в орто-положении гетероциклом, предпочтительно N-циклоалкилом, таким как пиперидин-1-ил группа, и необязательно замещенным в орто' положении алкильной группой, такой как метил.
Преимущественно R2 представляет собой атом галогена, такой как атом брома, предпочтительно в мета-положении по отношению к R3.
Предпочтительно соединение формулы (I) имеет следующие признаки:
- А представляет собой фенильную группу, замещенную в пара-положении бензильной группой,
- В представляет собой фенильную группу, замещенную в орто-положении пиперидин-1-ил группой, и в орто' положении метильной группой,
- G1 представляет собой связь, и
- R1, R2, R3 и n соответствуют данному выше определению.
В частности, соединение формулы (I) представляет собой 4-[N'-(4-бензил-фенил)-N'-(2-метил-6-пиперидин-1-ил-фенил)-гидразинкарбонилметил]-5-бром-2-метокси-бензойную кислоту или ее фармацевтически приемлемую соль, и в частности ее калиевую соль.
Более того, калиевая соль повышает растворимость этого соединения, в частности по сравнению с его гидрохлоридом, и способствует изготовлению лекарственной формы продукта.
Настоящее изобретение таким образом также имеет объектом калиевую соль 4-[N'-(4-бензил-фенил)-N'-(2-метил-6-пиперидин-1-ил-фенил)-гидразин карбонилметил]-5-бром-2-метокси-бензойной кислоты.
Настоящее изобретение также относится к применению этой конкретной соли в качестве лекарственного средства, в частности для профилактики или лечения папилломавирусной инфекции, и в частности ВПЧ6 или ВПЧ11 инфекции.
Эта калиевая соль дает возможность в частности профилактики или лечения поражений или заболеваний, связанных с папилломавирнусными инфекциями, и в частности с аногенитальными бородавками, такими как кондилома остроконечная и кондилома широкая, ларингеальными, конъюнктивальными или оральными папилломами, рекуррентным респираторным папилломатозом, слабо выраженной или сильно выраженной интраэпителиальной неоплазией, бовеноидным папулезом, обычными, подошвенными, шипига, поверхностными или плоскими бородавками, бородавчатой эпидермодисплазией и карциномами, в частности аногенитальными.
Ее в особенности будут применять для лечения поражений, вызванных в частности ВПЧ6 и ВПЧ11 инфекциями, в частности бородавок и кондилом.
Настоящее изобретение также относится к любой фармацевтической композиции, включающей эту калиевую соль в комбинации с не менее чем одним фармацевтически приемлемым эксципиентом.
Объектом настоящего изобретения также является композиция согласно настоящему изобретению для применения в качестве лекарства, в частности для профилактики или лечения папилломавирусной инфекции, и в частности ВПЧ6 или ВПЧ11 инфекции.
Настоящее изобретение также относится к применению композиции согласно изобретению для приготовления лекарства, применяемого для профилактики или лечения папилломавирусной инфекции, и в частности ВПЧ6 или ВПЧ11 инфекции.
Настоящее изобретение также относится к способу профилактики или лечения папилломавирусной инфекции, и в частности ВПЧ6 или ВПЧ11 инфекции, включающему в себя введение эффективного количества композиции согласно изобретению нуждающемуся в этом лицу.
Композиция будет в частности использоваться местно, в частности в форме геля; оральное введение также может быть предусмотрено.
«Местно» в контексте настоящего изобретения означает местное применение. Такое применение можно осуществлять на кожу или на слизистые оболочки (внешние или внутренние), такие как респираторный тракт, ротовая полость или аногенитальная область. Его также можно осуществлять посредством местной инъекции в поражение или опухоль или рядом с ними. Предпочтительно его осуществляют на кожу и (или) на слизистые оболочки, и в частности в аногенитальной области.
Композиция согласно настоящему изобретению дает возможность в частности профилактики или лечения поражений или заболеваний, связанных с папилломавирнусными инфекциями, и в частности с аногенитальными бородавками, такими как кондилома остроконечная и кондилома широкая, ларингеальными, конъюнктивальными или оральными папилломами, рекуррентным респираторным папилломатозом, слабо выраженной или сильно выраженной интраэпителиальной неоплазией, бовеноидным папулезом, обычными, подошвенными, шипига, поверхностными или плоскими бородавками, бородавчатой эпидермодисплазией и карциномами, в частности аногенитальными.
Композицию согласно настоящему изобретению будут в частности использовать для лечения поражений, вызванных в частности ВПЧ6 и ВПЧ11 инфекциями, в частности бородавок и кондилом.
Композицию согласно настоящему изобретению можно изготавливать посредством смеси различных ингредиентов.
Настоящее изобретение таким образом также имеет объектом способ приготовления композиции согласно настоящему изобретению, включающей соединение формулы (I), усиливающий вязкость агент и растворитель, включающий в себя следующие этапы:
i) смешивание соединения формулы (I) в части растворителя для получения раствора А,
ii) смешивание усиливающего вязкость агента в оставшейся части растворителя для получения раствора В, и
iii) смешивание растворов А и В для получения композиции согласно изобретению.
Предпочтительно, этап (i) будут проводить при комнатной температуре.
Предпочтительно, этап (ii) будут проводить при нагреве для получения гомогенного раствора усиливающего вязкость агента. Этот этап будут проводить в частности при температуре между 60 и 100°C, предпочтительно между 70 и 80°C. Раствор В затем охлаждают перед применением на этапе (iii).
Предпочтительно, этап (iii) будут проводить при температуре между 20 и 40°C. В этом случае содержимое реактора с раствором А будут переносить в реактор, содержащий раствор В, или наоборот. Освобожденный таким образом реактор дополнительно могут промыть небольшим количеством растворителя согласно стандартной практике.
Настоящее изобретение следует лучше понимать на основе следующих не ограничивающих объем изобретения примеров.
Краткое описание графических материалов
На Фигуре 1 представлена диаграмма способа производства композиции согласно изобретению.
Примеры
Активный ингредиент, используемый в примерах, представляет собой калиевую соль 4-[N'-(4-бензил-фенил)-N'-(2-метил-6-пиперидин-1-ил-фенил)-гидразинкарбонилметил]-5-бром-2-метокси-бензойной кислоты (соединение (Ia)).
Один способ получения калиевой соли активного ингредиента основан на трансформации нейтральной формы описываемого соединения, растворенного в этаноле с этанольным гидроксидом калия. Саму нейтральную форму получают при промывке водой соединения в форме гидрохлорида, как заявлено в патенте WO 2007/135106.
Композиции согласно изобретению с 5 масс. % активного ингредиента в форме геля.


Фармацевтическая композиция представляет собой гель, содержащий 5 масс. % компонента (Ia).
Гидроксипропилцеллюлоза:
Технические характеристики соответствуют статье Европейской Фармакопеи №0337.
Источником гидроксипропилцеллюлозы является Klucel® от поставщика ASHLAND по следующим сортам: MF-Pharm, MXF-Pharm и GF-Pharm.
Пропиленгликоль:
Технические характеристики соответствуют статье Европейской Фармакопеи №0430.
Производство фармацевтической композиции от (a) до (g)
Активный ингредиент (Ia) растворяют в фракции пропиленгликоля. Гидроксипропилцеллюлозу диспергируют в «холодном» пропиленгликоле и смесь затем нагревают для получения растворения усиливающего вязкость агента. Когда гель охлаждают до 40°C, добавляют раствор активного ингредиента (Ia) в пропиленгликоле (емкость, содержащую раствор, промывают небольшим количеством пропиленгликоля) и смесь гомогенизируют. Перемешивание продолжают до достижения комнатной температуры.
Способ производства геля представлен на фигуре 1.
Следующую 5000 г партию (в соответствии с композицией (g)) в частности приготовили согласно этому способу:

Активный ингредиент растворяют при комнатной температуре в около 750 г пропиленгликоля для 5000 г партии геля. Гидроксипропилцеллюлозу диспергируют в около 3600 г пропилгликоля. Смесь нагревают при перемешивании при 75±5°C до получения гомогенного геля. Препарат оставляют охлаждаться до 40°C или ниже, продолжая перемешивание.
При перемешивании активный ингредиент в растворе вливают в гидроксипропилцеллюлозу в растворе и емкость с оставшимся пропиленгликолем (около 250 г) промывают. Конечный гель оставляют охлаждаться до 30±5°C при перемешивании до упаковывания.
Полученная фармацевтическая композиция представляет собой вязкий полупрозрачный гель, бесцветный или светло-желтый.
Исследование диффузии через синтетическую мембрану активного ингредиента из фармацевтической композиции (d)
Исследования диффузии через синтетическую диализную мембрану, сделанную из целлюлозы, на основе диффузионной ячейки Франца, проводили с фармацевтической композицией (d).
Делали мониторинг диффузионной кинетики соединения (Ia) через мембрану в течение 6 часов. Когда кинетика прекращалась, более чем 20% активного ингредиента было высвобождено из фармацевтической композиции.
Биологическая активность фармацевтической композиции
Активность активного ингредиента против папилломавируса может быть оценена по различным in vitro и клеточным анализам, таких как описанные Chiang et al. (1992), Proc. Natl. Acad. Sci. USA, 89:5799-5803, White et al. (2003), Journal of Biological Chemistry, 278: 26765-26772, или Fradet-Turcotte et al. (2010), Virology, 399: 65-76.
В настоящем изобретении используются два типа анализов для исследования биологической активности соединений против папилломавируса. Первый анализ ("анализ взаимодействия Е1/Е2") оценивает взаимодействие между Е1 и Е2 белками ВПЧ (папилломавирус человека) в клетках человека. Второй анализ (анализ репликации) измеряет репликацию вирусной геномной ДНК в клетках человека.
- Анализ взаимодействия
Анализ взаимодействия между Е1 и Е2 подобен анализам, часто называемым "двугибридными для млекопитающих" анализами. Он основан на ко-трансфекции репортерного вектора, содержащего ДНК-связывающие сайты для Е2 белка ВПЧ в промоторе, контролирующем экспрессию репортерного гена, и векторов экспрессии, кодирующих Е1 и Е2 белки ВПЧ, Е1 белок объединяется с VP 16 доменом трансактивации. Этот анализ дает возможность проследить взаимодействие между Е1 и Е2 белками, такое взаимодействие является этапом, необходимым для репликации генома ВПЧ.
Для анализов взаимодействия между Е1 и Е2 сконструировали репортерный вектор, содержащий несколько сайтов связывания ДНК для Е2 белка (палиндром 5' ACCGNNNNCGGT-3') выше наименьшего аденовируса большого позднего промотора (major-late promoter, MLP), который контролирует транскрипцию гена, кодирующего люциферазу светляков. Также сконструировали векторы экспрессии ВПЧ Е1 белка, объединенные на N-конце с VP16 доменом трансактивации HSV-1. Ко-трансфекция в клеточных линиях этого репортерного вектора, содержащего Е2 сайты и векторы экспрессии ВПЧ Е2 белка приводит к маргинальному возрастанию активности люциферазы. Ко-трансфекция этого репортерного вектора, содержащего Е2 сайты, векторы экспрессии ВПЧ Е2 белка и векторы экспрессии Е2 белка, объединенного с VP16 доменом, делает возможным формирование в клетках сильной трансактивации E2/E1-VP16 белкового комплекса и приводит к значительному возрастанию активности люциферазы. Это выражает взаимодействие между Е1 и Е2 белками в клетках.
- Анализ репликации
Анализ репликации вирусной геномной ДНК основан на ко-трансфекции репортерного вектора, содержащего ВПЧ начало репликации (ori), и векторов экспрессии, кодирующих Е1 и Е2 белки ВПЧ. Это дает возможность проследить набор биологических функций Е1 and Е2, необходимых для репликации генома HPVs.
Для анализов репликации вирусной геномной ДНК также сконструировали "репликон" репортерного вектора, содержащего ВПЧ11/ВПЧ6 начало репликцации (также называемый LCR, который несет сайты связывания Е1 и Е2 белков ВПЧ) и ген, кодирующий люциферазу светляков под транскрипционным контролем SV40 промотора. Подтвердилось, что присутствие начала репликации ВПЧ не оказывает транскрипционного эффекта на экспрессию гена люциферазы, ни в присутствии ни в отсутствии Е1 или Е2 вирусных белков. Ко-трансфекция этого репликон-вектора и векторов экспрессии белков ВПЧ Е1 и Е2 в линиях клеток человека приводит к возрастанию активности люциферазы, которая зависит от присутствия Е1 и Е2, выражающего возрастание множества репортерных векторов. Это происходит вследствие активности Е1 и Е2 вирусных белков, которые разрешают репликацию в клетках млекопитающих этого репликон-вектора, содержащего вирусное начало репликации.
Для того чтобы показать, что лекарственная форма активного ингредиента в фармацевтической композиции согласно изобретению не изменяет биологическую активность активного ингредиента, не находящегося в лекарственной форме, активность соединения (Ia) измерили посредством исследования биологической активности по отношению к папилломавирусу или фармацевтических композиций (a), (d), (f) и (g) согласно изобретению, или соединения (Ia) в свежеприготовленном Tris-DMSO буферном растворе (25 мМ Tris, рН 8,0, 5% DMSO).
Фармацевтические композиции (a), (d), (f) и (g) растворили в одинаковом буферном растворе для анализа одинаковых концентраций активного ингредиента, а именно в диапазоне от 0,25 до 40 мкМ.
Оценивали ингибиторную активность соединения (Ia), или в растворе, или в фармацевтических композициях (a), (d), (f) и (g), на взаимодействие между Е1 и Е2 белками ВПЧ11/ВПЧ6 в линиях клеток человека, происходящих от почечных эпителиальных клеток или клеток карциномы шейки матки. Различные дозы (0,25-40 мкМ) инкубировали в клеточной среде в течение 2 дней после трансфекции и измеряли активность люциферазы для определения ИК (ингибирующей концентрации)50 соединения на взаимодействие между Е1 и Е2 белками HPVs.
Также оценивали ингибиторную активность соединения (Ia), или в растворе, или в фармацевтических композициях (a), (d), (f) и (g), на Е1- и Е2-зависимую вирусную репликацию ВПЧ11/ВПЧ6 в линиях клеток человека, происходящих от почечных эпителиальных клеток или клеток карциномы шейки матки. Различные дозы (0,25-40 мкМ) инкубировали в клеточной среде в течение от 2 до 6 дней после трансфекции и измеряли активность люциферазы с использованием люминометра для определения ИК50 соединения на репликацию генома HPVs.
Фармацевтические композиции, описанные выше, показывают такую же биологическую активность, как соединение (Ia) в растворе (не в лекарственной форме). Соединение (Ia) в растворе и фармацевтические композиции (a), (d), (f) и (g) ингибируют как взаимодействие между ВПЧ11/ВПЧ6 Е1 и Е2 белками в клетках, так и Е1 и Е2 белок-зависимую вирусную репликацию ВПЧ11 в клетках, с ИК50 около 1 мкМ.
Подкожная абсорбция in vitro и in vivo
Подкожную абсорбцию соединения (Ia) из фармацевтической композиции (g) исследовали после нанесения дозы 10 мг/см2 на кожу человека на основе диффузионной ячейки Франца. Это исследование проводили путем сравнения подкожной абсорбции здоровой кожей и расслоившейся кожей, как модели слабой кератинизации аногенитальной области.
После 8 или 24 часов воздействия фармацевтической композиции (g), количество соединения (Ia), присутствующего в каждом слое кожи (роговой слой, эпидермис, дерма) и прошедшего через кожу (полученная жидкость) определяли посредством ВЭЖХ (высокоэффективная жидкостная хроматография).
Результаты, полученные после 8 часов диффузии, представлены в таблице ниже.

Роговой слой образцов нерасслоившейся кожи удаляют с эпидермиса посредством пилинга адгезивными веществами. Последние наносят на поверхность кожи под постоянным и контролируемым давлением.
Концентрации соединения (Ia), измеренные в коже после 8 часов диффузии, соответствуют местной концентрации от 100 раз (здоровая кожа) до 1000 раз (расслоившаяся кожа) активности активного ингредиента (ИК50), измеренной in cellulo. Такие же результаты получали после 24 часов диффузии. Подобным образом, такие же результаты наблюдали в исследованиях подкожной абсорбции in vitro на коже карликовой свиньи, животной модели, с высокой степенью уверенности прогнозирующей реакцию кожи человека.
Кроме того, при высокой местной концентрации активного ингредиента в коже лишь незначительное количество соединения (Ia), пересекающего кожу карликовой свиньи, попадает в ячейку Франца. Около 0,1% примененной дозы таким образом измеряли в полученной жидкости после 24 часов воздействия дозы 10 мг/см2фармацевтической композиции (g).
Подкожная абсорбция соединения (Ia) из фармацевтической композиции (g) измеряли in vivo во время исследования токсокинетики, проводившегося посредством нанесения дважды в день в течение 42 дней дозы 10 мг/см2 на бока карликовых свиней. Наличие соединения (Ia) измеряли и подсчитывали на первый день и последний день исследования в крови экспериментальных животных с использованием очень чувствительного ЖХ-МС/МС (жидкостная хроматография с тандемной масс-спектрометрией) биоаналитического метода (предел чувствительности = 0,5 нг/мл). Аналогично исследованиям подкожной абсорбции in vitro на человеческой коже и коже карликовых свиней, описанных выше, незначительные количества соединения (Ia) определяли в крови карликовых свиней. Таким образом, измеряли около 0,1% примененной дозы, что показывает низкую подкожную абсорбцию in vivo соединения (Ia) из фармацевтической композиции.
В целом эти результаты показывают высокую местную концентрацию активного ингредиента (соединение (Ia)) в коже после нанесения фармацевтической композиции и его низкое системное воздействие, как желательно для лекарственных препаратов, применяемых местно.
Переносимость
Очень хорошая переносимость фармацевтической композиции (g) была показана как для карликовой свиньи во время доклинического исследования токсичности, так и для здоровых добровольцев во время Фазы Ia клинического исследования. У карликовой свиньи дозу 10 мг/см2 наносили дважды в день на 25, 125 или 250 см2 область на бок животных в течение 42 дней. Высокую переносимость фармацевтической композиции наблюдали вне зависимости от обрабатываемой области. Кроме того, очень хорошая переносимость фармацевтической композиции была показана во время нанесения дважды в день в течение 7 дней в дозе 10 мг/см2 на 25 см2 область поясницы здоровых добровольцев. Не наблюдалось побочных реакций или нежелательных явлений у 8 здоровых обработанных добровольцев.
Микробиологические свойства
Эффективность антибактериальной консервации фармацевтических композиций (а) и (g) анализировали в соответствии с методом Европейской Фармакопеи (метод 5.1.3 с применением стандартов 5.1.3-2).
Результаты представлены в таблице ниже:
Фармацевтическая композиция (а):

Фармацевтическая композиция (g):
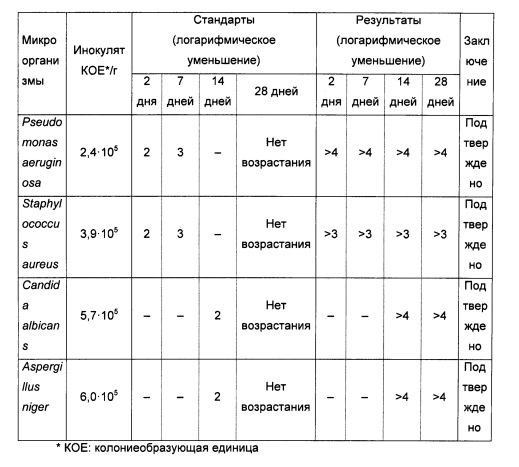
Результаты соответствуют А критерию метода 5.1.3-2 Европейской Фармакопеи.
В результате наличия пропиленгликоля, чьи антибактериальные консервирующие свойства известны специалистам в данной области техники, лекарственная форма защищена от бактериального загрязнения и, следовательно, не требует дополнительных консервирующих средств.
Исследование стабильности
Стабильность фармацевтической композиции (g) исследовали на 5 кг лабораторной партии.
Следующие режимы консервации были проанализированы в соответствии с рекомендациями ICH (Международной конференции по гармонизации):
- Долгосрочный режим: 5°C±3°C,
- Промежуточный режим: 25°C±2°C/60%±5% ОВ (относительная влажность), и
- Ускоренный режим: 40°C±2°C/75%±5% ОВ.
Использовали стандартные методы анализа, включая:
- подтверждение внешнего вида,
- измерение вязкости, и
- анализ активного ингредиента (Ia) посредством ВЭЖХ.
Полученные результаты представлены в следующих таблицах:



Вязкость остается стабильной при всех режимах и продолжительности хранения.
Концентрация активного ингредиента (Ia) и внешний вид геля остается в пределах стандартов на момент истечения срока годности при всех режимах и продолжительности хранения, демонстрируя стабильность геля во время хранения.
Реферат
Изобретение относится к фармацевтической промышленности, а именно к композиции для лечения или профилактики инфекции, связанной с папилломавирусом. Фармацевтическая композиция для лечения или профилактики инфекции, связанной с папилломавирусом, или поражения или заболевания, связанного с папилломавирусной инфекцией, включающая соединение общей формулы (I):или его фармацевтически приемлемую соль, в комбинации с растворителем и усиливающим вязкость агентом, где указанным растворителем является пропиленгликоль и указанным усиливающим вязкость агентом является гидроксипропилцеллюлоза, где указанная композиция имеет форму геля, и где гель имеет вязкость от 5000 до 50000 мП. Способ приготовления фармацевтической композиции, включающий в себя следующие этапы:i) смешивание соединения формулы (I) в части растворителя для получения раствора А,ii) смешивание усиливающего вязкость агента в оставшейся части растворителя для получения раствора В, иiii) смешивание растворов А и В для получения фармацевтической композиции,где указанным растворителем является пропиленгликоль и указанным усиливающим вязкость агентом является гидроксипропилцеллюлоза. Вышеописанная композиция эффективна для лечения или профилактики инфекции, связанной с папилломавирусом, быстро высвобождает активный ингредиент, является стабильной.2 н. и 17 з.п. ф-лы, 3 табл., 1 ил.
Формула
Комментарии