Составы и соединения для доставки активных агентов - RU2326110C2
Код документа: RU2326110C2
Описание
Изобретение относится к соединениям для доставки к мишени активных агентов, в частности биологически или химически активных агентов. Эти соединения хорошо подходят для получения нековалентных смесей с активными агентами для введения в организм через рот, толстую кишку, легкие или другими путями. Описаны также способы получения и введения таких составов.
Обычные средства доставки активных агентов часто имеют жесткие ограничения в виде биологических, химических и физических барьеров. Обычно эти барьеры налагаются средой, по которой происходит доставка, окружающей средой мишени, в которую производится доставка, и/или самой мишенью.
Биологически и химически активные агенты особенно уязвимы для таких барьеров.
При введении животным биологически и химически активных фармацевтических и терапевтических агентов барьеры налагаются организмом. Примерами физических барьеров являются кожа, липидные двойные слои и мембраны различных органов, которые являются относительно непроницаемыми для определенных активных агентов, однако должны быть пройдены перед достижением мишени, например сердечно-сосудистой системы. Химические барьеры включают без ограничения изменения рН в желудочно-кишечном (GI) тракте и ферменты разложения.
Эти барьеры имеют особое значение для создания систем пероральной доставки. Пероральная доставка многих биологически или химически активных агентов является предпочтительным способом для введения в организм при условии отсутствия биологических, химических и физических барьеров. Среди множества агентов, которые обычно не предназначены для перорального введения, можно назвать биологически или химически активные пептиды, в частности кальцитонин и инсулин, полисахариды, и в особенности - мукополисахариды, включая без ограничения гепарин, гепариноиды, антибиотики и другие органические вещества. Эти агенты могут быстро утрачивать эффективность или разрушаться в желудочно-кишечном тракте под действием гидролиза в кислой среде, ферментов и т.п. Кроме того, размер и структура макромолекулярных лекарственных препаратов могут препятствовать поглощению.
Первые способы перорального введения уязвимых фармакологических агентов были основаны на совместном введении адъювантов (например, резорцинов и неионных поверхностно-активных веществ, в частности полиоксиэтиленолеилового эфира и н-гексадецилполиэтиленового эфира) для искусственного увеличения проницаемости стенок кишечника, а также на совместном введении ферментных ингибиторов (например, ингибиторы панкреатического трипсина, диизопропилфторфосфат (DFF) и трасил) для подавления ферментного разложения. Липосомы также описаны в качестве систем доставки лекарств, в частности инсулина и гепарина. Однако широкий спектр применений таких систем доставки лекарственных препаратов ограничен вследствие того, что (1) системы требуют токсичного количества адъювантов или ингибиторов, (2) отсутствуют пригодные транспортируемые грузы, т.е. активные агенты с низкими молекулярными массами, (3) системы обладают низкой стабильностью и недостаточным сроком хранения, (4) системы сложны для изготовления, (5) системы не обеспечивают защиту активного агента (груза), (6) системы неблагоприятно изменяют активный агент или (7) системы не допускают или не способствуют поглощению активного агента.
Позднее для доставки фармацевтических средств стали использовать белковые микросферы. Так, например, см. патенты США 5401516, 5443841 и RE 35862. Кроме того, для доставки фармацевтических средств использовали определенные модифицированные аминокислоты. См., например, патенты США 5629020, 5643957, 5766633, 5776888 и 5866536.
Тем не менее, сохраняется потребность в простых и экономичных системах доставки, которые являются несложными в получении и могут доставлять широкий спектр активных агентов различными путями.
Обеспечены соединения и составы, полезные для доставки активных агентов. Настоящее изобретение включает соединения, имеющие следующую формулу, а также их соли или их смеси.
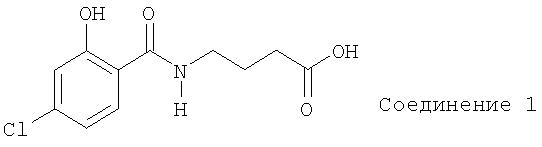
Составы согласно настоящему изобретению содержат по меньшей мере один активный агент, предпочтительно - биологически или химически активный агент, и по меньшей мере одно из соединений или их солей согласно настоящему изобретению. Разработали также способы получения и введения таких соединений.
Кроме того, получили дозированные формы, содержащие указанные соединения. Дозированная форма может быть твердой (в частности, таблетки, капсулы или частицы, например, порошок или саше) или жидкой.
Также разработали способы введения биологически активного агента животному, которому требуется такой агент, в особенности через рот, толстую кишку или легкие, вместе с составами согласно настоящему изобретению, и способы лечения с использованием указанных составов. Кроме того, разработали способ лечения заболевания у животного, включающий введение состава согласно настоящему изобретению животному, которое нуждается в этом.
Соединения
Соединения могут иметь форму карбоновой кислоты и/или ее солей. Соли включают без ограничения органические и неорганические соли, например соли щелочных металлов, в частности натрия, калия и лития, соли щелочноземельных металлов, в частности магния, кальция или бария, соли аммония, основные аминокислоты, в частности лизин или аргинин, и органические амины, в частности диметиламин или пиридин. Предпочтительными солями являются соли натрия. Соли могут быть одно- и многовалентными солями, в частности однонатриевыми солями и динатриевыми солями. Соли могут быть также сольватами, в том числе сольватами этанола.
Кроме того, можно использовать полиаминокислоты и пептиды, содержащие одно или несколько указанных соединений.
Аминокислота представляет собой любую карбоновую кислоту, содержащую по меньшей мере одну свободную аминовую группу, и включает природные и синтетические аминокислоты. Полиаминокислоты являются пептидами (которые представляют собой две или более аминокислот, соединенных пептидной связью) или двумя или несколькими аминокислотами, соединенными связью, образованной другими группами, которые могут быть соединены, например, эфирной или ангидридной связью. Пептиды могут изменять длину от дипептидов с двумя аминокислотами до полипептидов, содержащих несколько сотен аминокислот. Одна или несколько аминокислот или пептидных единиц может быть ацилирована или сульфонирована.
Описанные здесь соединения могут быть получены из аминокислот и могут быть легко приготовлены из аминокислот способами, которые могут реализовать специалисты на основании настоящего описания и способов, описанных в WO 96/30036, WO 97/36480, US 5643957 и US 5650386. Так, например, указанные соединения можно получить в результате реакции одинарной аминокислоты с соответствующим ацилирующим или амино-модифицирующим агентом, который реагирует со свободной аминогруппой, присутствующей в аминокислоте с образованием амидов. С тем, чтобы избежать нежелательных побочных реакций, можно использовать защитные группы, как это известно специалистам. Относительно защитных групп можно указать работу Т.W.Greene, Protecting Groups in Organic Synthesis, Wiley, New York (1981), содержание которой приводится здесь в качестве ссылки.
Соли соединений согласно настоящему изобретению можно получить способами, известными специалистами. Так, например, соли натрия можно получить путем растворения соединения в этаноле и добавления водного раствора гидроксида натрия.
Соединение можно очистить путем перекристаллизации или фракционирования на одном или нескольких твердых хроматографических носителях, отдельных или связанных в тандем. Пригодные системы растворителей для перекристаллизации включают без ограничения ацетонитрил, метанол и тетрагидрофуран. Фракционирование можно выполнить на пригодном хроматографическом носителе, в частности на глиноземе, используя смеси метанола и н-пропанола в качестве подвижной фазы, а также способом обращено-фазной хроматографии, используя смеси трифторуксусной кислоты и ацетонитрила в качестве мобильной фазы, и способом ионообменной хроматографии, используя воду или соответствующий буфер в качестве подвижной фазы. В случае применения анионообменной хроматографии предпочтительно использовать градиент хлорида натрия 0-500 мМ.
Согласно одному из вариантов реализации изобретения состав используют в его безводной форме.
Активные агенты
Активные агенты, пригодные для применения согласно настоящему изобретению, включают биологически активные агенты и химически активные агенты, в том числе, но без ограничения: пестициды, фармакологические агенты и терапевтические агенты.
Так, например, биологически или химически активные агенты включают без ограничения белки, полипептиды, пептиды, гормоны, полисахариды, в особенности, смеси мукосахаридов, углеводы, липиды, другие органические соединения и в особенности соединения, которые не проходят (или проходят лишь частью введенной дозы) через слизистую оболочку желудочно-кишечного тракта и/или являются чувствительными к химическому расщеплению кислотами и ферментами в желудочно-кишечном тракте, или комбинацию указанных агентов.
Другие примеры включают без ограничения следующие агенты, имеющие синтетические, природные или рекомбинатные источники:
гормоны роста, в том числе гормоны роста человека (hGH), рекомбинантные гормоны роста человека (rhGH), бычьи гормоны роста и свиные гормоны роста, гормоны, выделяющие гормоны роста, интерфероны, включая α, β и γ, интерлейкин-1, интрелейкин-2, инсулин, включая свиной, бычий, человека и рекомбинантный инсулин человека, возможно, содержащие противоионы, включая ионы натрия, цинка, кальция и аммония, инсулиноподобный фактор роста, включая IGF-1, гепарин, включая нефракционированный гепарин, гепариноиды, дерматаны, хондроитины, гепарин с низкой молекулярной массой, гепарин с очень низкой молекулярной массой и гепарин со сверхнизкой молекулярной массой, кальцитонин, включая кальцитонин лосося, угря, свиньи и человека, эритропоэтин, предсердный натрийуретический фактор, антигены, моноклональные антитела, соматостатин, ингибиторы протеазы, адренокортикотропин, гормон, выделяющий гонадотропин, окситоцин, гормон, выделяющий лейтинизирующий гормон, гормон, стимулирующий фолликулы, глюкоцереброзидазу, тромбопоэтин, филграстим, простагландины, циклоспорин, вазопрессин, кромолин натрия (натриевый или динатриевый хромогликат), ванкомицин, десферриоксамин (DFO), паратироидный гормон (РТН), включая его фрагменты, противомикробные, в том числе противогрибковые агенты, витамины, аналоги, фрагменты, миметики или производные этих соединений, модифицированные полиэтиленгликолем (PEG), а также их любые сочетания. Другие пригодные формы инсулина, включая без ограничения синтетические формы инсулина, описаны в патентах США №№4421685, 5474978 и 5534488, каждый из которых полностью включен в данное описание в качестве ссылки.
Системы доставки
Составы согласно настоящему изобретению содержат носитель и один или несколько активных агентов. В одном из вариантов реализации одно или несколько соединений носителя или соли этих соединений или полиаминокислоты или пептиды, из которых эти соединения или соли образуют один или несколько компонентов, можно использовать в качестве носителя путем смешивания с активным агентом перед введением.
Вводимые составы могут иметь форму жидкости. В качестве растворителя дозированного раствора можно использовать воду (например, для лососевого кальцитонина, паратироидного гормона и эритропоэтина), 25% водный раствор пропиленгликоля (например, для гепарина) и фосфатный буфер (например, для rhGH). Другие растворители включают полиэтиленгликоли, сорбит, мальтитол и сахарозу. Дозированные растворы можно получить путем смешивания раствора соединения носителя с раствором активного агента непосредственно перед введением. В альтернативном варианте реализации раствор носителя (или активного агента) можно смешивать с твердой формой активного агента (или носителя). Соединение носителя и активный агент можно смешивать также в форме сухих порошков. Кроме того, соединение носителя и активный агент можно смешивать в процессе изготовления.
Дозированные растворы могут содержать добавки, в частности, буферные фосфатные соли, лимонную кислоту, гликоли или иные диспергирующие агенты. В раствор можно вводить стабилизирующие добавки, предпочтительно - с концентрацией в пределах примерно от 0,1 до 20% (масса/объем).
В альтернативном варианте реализации составы можно вводить в твердой форме, в частности в форме таблетки, капсулы или частиц, например порошка или пакетика-саше. Твердые дозированные формы можно получить путем смешивания твердой формы соединения с твердой формой активного агента. Альтернативно твердую форму можно получить из раствора соединения и активного агента известными способами, в частности лиофилизацией, осаждением, кристаллизацией и получением твердой дисперсии.
Составы для введения согласно настоящему изобретению могут включать также один или несколько ингибиторов ферментов. Такие ингибиторы ферментов включают без ограничения такие соединения как актинонин или эпиактинонин и их производные. Другие ингибиторы ферментов включают без ограничения апротинин (трасилол) и ингибитор Боумэна-Бирка.
Доза активного агента, которую применяют в соединении, вводимом согласно настоящему изобретению, является достаточно эффективной для выполнения задачи конкретного активного агента для указания цели. Доза активного агента в соединениях обычно является фармакологически, биологически, терапевтически или химически эффективной дозой. Однако эта доза может быть меньшей в том случае, когда соединение используют в стандартной дозированной форме, поскольку стандартная дозированная форма может содержать несколько составов/соединений активного агента или может содержать разделенную фармакологически, биологически, терапевтически или химически эффективную дозу. В этом случае суммарную, эффективную дозу можно ввести кумулятивными дозами, которые в сумме содержат эффективную дозу активного агента.
Необходимую суммарную дозу активного агента можно определить способами, известными специалистам. Однако соединения согласно изобретению могут доставлять активные агенты более эффективно, чем известные ранее соединения, поэтому пациенту можно вводить более низкие дозы биологически или химически активных агентов по сравнению с использовавшимися ранее дозированными стандартными формами, обеспечивая такие же концентрации в крови и/или такие же терапевтические эффекты.
Составы и соединения согласно настоящему изобретению доставляют биологически и химически активные агенты, в особенности посредством оральной, интраназальной, сублингвальной, интрадуоденальной, подкожной, буккальной, ободочно-кишечной, ректальной, вагинальной, мукозальной, пульмонарной, трансдермальной, интрадермальной, парентеральной, внутривенной, внутримышечной и окулярной систем, а также путем пересечения гематоэнцефалического барьера.
Стандартные дозированные формы могут включать также один или несколько наполнителей, разбавителей, дезинтеграторов, лубрикантов, пластификаторов, красителей, ароматизаторов, агентов, маскирующих вкус, сахаров, подсластителей, солей и растворителей, включая без ограничения воду, 1,2-пропандиол, этанол, оливковое масло или их сочетание.
Соединения и составы согласно изобретению применимы для введения биологически или химически активных агентов животным, включая без ограничения птиц, в частности кур, млекопитающих, в частности грызунов, коров, свиней, собак, кошек, приматов и, в особенности, человека, а также насекомых.
Система особенно полезна для доставки химически или биологически активных агентов, которые в противном случае разрушились бы или оказались бы менее эффективными при условиях, возникающих до того, как активный агент достигнет зоны мишени (т.е. зоны, в которой активный агент должен быть выделен из соединения носителя) в организме животного, куда он был введен. Составы и соединения согласно настоящему изобретению полезны, в частности, для перорального введения активных агентов, в особенности тех, которые не могут быть доставлены перорально обычным способом или для которых желательно улучшение процесса доставки.
Составы, содержащие соединения и активные агенты, полезны для доставки активных агентов в выбранные биологические системы в увеличенной или улучшенной биодоступности активного агента по сравнению с введением активного агента без носителя. Доставку можно усовершенствовать путем доставки более активного агента через определенный период времени или путем доставки активного агента в определенный период времени (т.е. эффект ускоренной или задержанной доставки) или в течение определенного периода времени (т.е. продолжительная доставка).
После введения активный агент, присутствующий в составе или в стандартной дозированной форме, поглощается системой кровообращения. Биодоступность агента легко определить путем измерения известной фармакологической активности в крови, например увеличения времени свертывания крови, вызываемого гепарином, или уменьшения концентрации кальция в крови, вызываемого кальцитонином. В альтернативном случае можно измерить концентрацию в крови непосредственно самого активного агента.
ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ПРИМЕРОВ РЕАЛИЗАЦИИ
Приведенные ниже примеры иллюстрируют изобретение без ограничения. Все части даны по массе, если не указано иного.
Пример 1 - Получение соединений
1а. Получение соединения 1
4-хлорсалициловую кислоту (10,0 г, 0,0579 моль) добавили в 250 мл одногорлую круглодонную колбу, содержащую около 50 мл метиленхлорида. Начали перемешивание, которое продолжали до окончания реакции. Добавили в колбу отдельными порциями связывающий агент 1,1-карбонилдиимидазол (9,39 г, 0,0579 моль) в твердой фазе. Реакционную смесь перемешивали при комнатной температуре около 20 минут, затем добавили остаток связующего агента, а после этого в колбу при перемешивании добавили гидрохлорид этил-4-аминобутаноата (9,7 г, 9,0579 моль). Из дополнительной воронки добавили по каплям триэтиламин (10,49 мл, 0,0752 моль). Промыли дополнительную воронку метиленхлоридом. Реакционную смесь оставили на ночь при перемешивании при комнатной температуре.
Реакционную смесь перелили в делительную воронку и промыли 2N HCl, получив эмульсию. Выдержали эмульсию в течение двух дней. Затем профильтровали эмульсию через целит на воронке Бюхнера. Фильтрат снова залили в делительную воронку для разделения слоев. Органический слой высушили над сульфатом натрия, который затем отфильтровали, и сконцентрировали фильтрат в роторном испарителе. Полученный осадок гидролизовали 2N NaOH, выдержали в течение ночи при замораживании, а затем снова гидролизовали. Раствор подкислили 3N HCl и полученную твердую фазу отделили, высушили в вакууме и дважды перекристаллизовали с помощью смеси метанола и воды. Твердую фазу осаждали в течение ночи, отделили и высушили. Осадок растворили в 2N NaOH и довели рН образца до 5 с помощью 2N HCl. Собрали осадок и провели анализ способом ВЭЖХ, получив один пик. Затем этот осадок перекристаллизовали в смеси этанола и воды, отделили и высушили в вакууме, получив 4,96 г (33%) 4-(4-хлор-2-гидроксибензоил)аминомасляной кислоты. (C11H12ClNO4, молекулярная масса 257,67). Температура плавления: 131-133°С. Анализ сжиганием: %С: 51,27 (рассчитано), 51,27 (найдено), %Н: 4,69 (рассчитано), 4,55 (найдено), %N: 5,44 (рассчитано), 5,30 (найдено). Анализ1H ЯМР: (d6 - ДМСО): δ 13,0, s, 1H (СООН); δ 12,1, s, 1H (ОН); δ 8,9, t, 1H (NH); δ 7,86, d, 1H (H орто к амиду); δ 6,98, d, 1H (орто к фенолу ОН); δ 6,96, d, 1H (H мета к амиду); δ 3,33, m, 2H (CH2 смежный с NH); δ 2,28, t, 2H (СН2 смежный с СООН); δ 1,80, m, 2H (алифатический СН2 бета к NH и СН2 бета к СООН).
1b. Дополнительный способ получения соединения 1
4-хлорсалициловую кислоту (25,0 г, 0,1448 моль) добавили в 250 мл одногорлую круглодонную колбу, содержащую около 75-100 мл метиленхлорида. Начали перемешивание, которое продолжали до окончания реакции. Добавили в колбу отдельными порциями связывающий агент 1,1-карбонилдиимидазол (23,5 г, 0,1448 моль) в твердой фазе. Реакционную смесь перемешивали при комнатной температуре около 20 минут, затем добавили остаток связывающего агента, а после этого в колбу при перемешивании добавили гидрохлорид этил-4-аминобутаноата (24,3 г, 0,1448 моль). Из дополнительной воронки добавили по каплям триэтиламин (26,0 мл, 0,1448 моль). Промыли дополнительную воронку метиленхлоридом. Реакционную смесь оставили на ночь при перемешивании при комнатной температуре.
Перелили реакционную смесь в делительную воронку и промыли 2N HCl, получив эмульсию. Профильтровали эмульсию через целит в воронке с оплавленным стеклом. Фильтрат снова залили в делительную воронку для разделения слоев. Органический слой промыли водой и соляным раствором и высушили над сульфатом натрия, который затем отфильтровали и сконцентрировали фильтрат в роторном испарителе. Полученный осадок гидролизовали 2N NaOH в течение ночи. Раствор подкислили 2N HCl и полученный коричневый осадок перекристаллизовали с помощью смеси метанола и воды, а затем отфильтровали в горячем состоянии нерастворимый материал черного цвета. Белые частицы осадили, отделили и высушили, получив 11,68 г (37,0%) 4-(4-хлор-2-гидроксибензоил)аминомасляной кислоты. (C11H12ClNO4, молекулярная масса 257,67). Температура плавления: 129-133° С. Анализ сжиганием: %С: 51,27 (рассчитано), 51,26 (найдено), %Н: 4,69 (рассчитано), 4,75 (найдено), %N: 5,44 (рассчитано), 5,32 (найдено). Анализ1H ЯМР: (d6 - ДМСО): δ 13,0, s, 1Н (СООН); δ 12,1, s, 1H (ОН); δ 8,9, t, 1H (NH); δ 7,86, d, 1H (Н орто к амиду); δ 6,98, d, 1H (орто к фенолу ОН); δ 6,96, d, 1H (Н мета к амиду); δ 3,33, m, 2Н (CH2 смежный с NH); δ 2,28, t, 2H (CH2 смежный с СООН); δ 1,80, m, 2H (алифиатический СН2 бета к NH и СН2 бета к СООН).
1с. Дополнительный способ получения соединения 1
(4-[(4-хлор-2-гидроксибензоил)амино]масляная кислота)
Пятигорлую круглодонную колбу объемом 22 л оснастили подвесным смесителем, 1-литровой ловушкой Дина-Старка с обратным холодильником, термопарой для измерения температуры и нагревательным кожухом. Следующую реакцию провели в атмосфере сухого азота. Реагент н-бутанол (5000 мл) и 4-хлорсалициловую кислоту (2000 г, 11,59 моль) загрузили в реакционную колбу. Ловушку Дина-Старка заполнили н-бутанолом (1000 мл). Добавили концентрированную серную кислоту (50 г). Реакционную смесь нагревали для кипячения с обратным холодильником в течение примерно 120 часов. За это время в ловушке собрали около 206 мл воды. Сняли нагревательный кожух и выдержали реакционную смесь для охлаждения до комнатной температуры. Опустошили и сняли ловушку Дина-Старка. Залили деионизованную воду (1000 мл). Двухфазную смесь перемешивали в течение 10 минут. Прекратили перемешивание и дали фазам разделиться. Нижнюю водную фазу слили сифоном и удалили. В реакционную смесь добавили 10% (по массе) водный раствор бикарбоната натрия (1000 мл). Смесь перемешивали в течение 10 минут. Реакционную смесь проверили индикаторной бумагой для измерения рН, чтобы убедиться в том, что рН раствора больше 7. Добавили в реакционную смесь воду (500 мл). Прекратили перемешивание и дали фазам разделиться. Нижний водный слой слили сифоном и удалили. Реакционную смесь промыли еще одной порцией 500 мл деионизованной воды. Реактор включили на дистилляцию при атмосферном давлении в тарированный ресивер объемом 5 л. Смесь дистиллировали до тех пор, пока температура реактора поднималась до 140-150°С. Затем переключили дистилляцию с дистилляции при атмосферном давлении на вакуумную дистилляцию. Давление в дистилляционной установке медленно понижали до 100 мм рт.ст. Снизили температуру реактора и удалили дистилляцией остаточный н-бутанол и н-бутиловый эфир (побочный продукт реакции). Прекратили нагревание и дали реакционной смеси охладиться до комнатной температуры. Вакуум заменили осушенным азотом. Неочищенный бутиловый эфир перенесли в 5-литровый реактор вакуумной дистилляционной установки. Произвели дистилляцию неочищенного бутилового эфира при давлении от 0,2 до 0,5 мм рт.ст. Головной погон, собранный при температуре верхней части колонны <40°С, удалили. Фракцию бутил-4-хлор-2-гидроксибензоата собрали при температуре верхней части колонны в пределах от 104 до 112°С. Эта фракция имела массу 2559 г. Выход составил 96%.
Пятигорлую круглодонную колбу объемом 22 л оснастили подвесным смесителем, обратным холодильником, термопарой для измерения температуры и нагревательным кожухом. Реактор продули азотом. Бутил-4-хлор-2-гидроксибензоат (2559 г, 11,2 моль) и реагент метанол (10000 мл) загрузили в реакционную колбу и перемешали содержимое до получения раствора. Реакционную смесь профильтровали через воронку Бюхнера и возвратили в реактор. Увеличили скорость перемешивания и быстро ввели в свободное пространство реактора газообразный аммиак. Добавление газообразного аммиака производили до тех пор, пока температура реактора не достигла 45°С. Прекратили добавление аммиака и снизили скорость перемешивания. Выдержали реакционную смесь для охлаждения до комнатной температуры. Повторили добавление газообразного аммиака, как описано выше, до окончания протекания реакции, что указывала жидкостная хроматография. Для завершения реакции потребовалось семь загрузок аммиака в течение пяти дней. Примерно половину растворителя удалили путем дистилляции при атмосферном давлении. Реакционную смесь охладили до комнатной температуры и добавили 5 л деионизованной воды. Концентрированную соляную кислоту (примерно 500 мл) медленно добавили в реактор до получения рН реакционной смеси между 4 и 5. Полученный осадок собрали путем вакуумной фильтрации через большую воронку с фильтром из пористого стекла. Осадок на фильтре промыли 2000 мл деионизованной воды и высушили при 50°С в течение 32 часов, получив 1797 г 4-хлор-2-гидроксибензамид. Выход составил 94%.
Пятигорлую круглодонную колбу объемом 22 л оснастили подвесным смесителем, обратным холодильником, дополнительной воронкой, термопарой для измерения температуры и нагревательным кожухом. Реактор продули азотом. Ацетонитрил (4700 мл) и 4-хлор-2-гидроксибензамид (1782 г, 10,4 моль) загрузили в реакционную колбу и начали перемешивание. Пиридин (1133 мл, 14,0 моль) загрузили в реактор. Полученную реакционную суспензию охладили до температуры менее 10°С на ледяной бане. Этилхлороформиат (1091 мл, 1237 г, 11,4 моль) поместили в дополнительную воронку и медленно добавили к перемешиваемой реакционной смеси таким образом, что при добавлении температура реакционной смеси не превысила 15°С. После окончания добавления этилхлороформиата температуру реакционной смеси поддерживали в пределах от 10 до 15°С в течение 30 минут. Удалили ледяную баню, и реакционная смесь нагрелась до комнатной температуры. Затем медленно нагрели реакционную смесь до получения флегмы и выдержали при этой температуры в течение 18 часов. Анализ реакционной смеси способом жидкостной хроматографии показал, что реакция завершилась только на 80%. Примерно половину растворителя удалили путем дистилляции при атмосферном давлении. Реакционную смесь охладили вначале до комнатной температуры, а затем - до температуры <10°С с помощью ледяной бани. В реакционную смесь добавили пиридин (215 мл, 2,65 моль). Этилхлороформиат (235 г, 2,17 моль) медленно добавили через дополнительную воронку в холодную реакционную смесь. После окончания добавления этилхлороформиата температуру реакционной смеси поддерживали в пределах от 10 до 15°С в течение 30 минут. Удалили ледяную баню, и реакционная смесь нагрелась до комнатной температуры. Затем медленно нагрели реакционную смесь для кипячения с обратным холодильником и выдержали при этой температуре в течение 18 часов, после чего анализ реакционной смеси способом жидкостной хроматографии показал, что реакция закончилась. Реакционную смесь охладили вначале до комнатной температуры, а затем - до температуры <10°С с помощью ледяной бани. Через дополнительную воронку медленно добавили воду (1600 мл) и полученную суспензию выдержали при температуре <10°С в течение 90 минут. Полученный осадок собрали путем вакуумной фильтрации через большую воронку с фильтром из пористого стекла. Осадок на фильтре промыли деионизованной водой и высушили в вакууме при 50°С в течение 18 часов, получив 1914 г 7-хлор-2Н-1,3-бензоксазин-2, 4(3Н)-диона в форме порошка желто-коричневого цвета. Выход составил 83%.
Пятигорлую круглодонную колбу объемом 22 л оснастили подвесным смесителем, обратным холодильником, термопарой для измерения температуры и нагревательным кожухом. Следующую реакцию провели в атмосфере сухого азота. При продувке азотом загрузили 7-хлор-2Н-1,3-бензоксазин-2,4(3Н)-дион (1904 г, 9,64 моль), этил-4-бромбутаноат (1313 мл, 9,18 моль) и N,N-диметилацетамид (4700 мл). Реакционную смесь нагрели до 70°С. Карбонат натрия (1119 г, 10,55 моль) ввели в прозрачный раствор пятью равными частями с интервалом около 40 минут. Реакционную смесь выдержали в течение ночи при 70°С. Неорганический осадок удалили с помощью вакуумной фильтрации через воронку с фильтром из пористого стекла. Реакционную колбу промыли этанолом 2В (2000 мл) и использовали эту жидкость для промывки для промывания осадка на фильтре. Промыли реакционную колбу деионизованной водой. Возвратили фильтрат в промытую реакционную колбу. Охладили фильтрат на ледяной бане. Через дополнительную воронку медленно добавили деионизованную воду (9400 мл). Охлажденную смесь выдержали при перемешивании в течение ночи. Полученный осадок отделили с помощью вакуумной фильтрации через воронку с фильтром из пористого стекла. Готовый осадок промыли деионизованной водой. Этил-3-(4-бутаноат)-7-хлор-2Н-1, 3-бензоксазин-2,4-(3Н)-дион имел массу 2476,0 г. Выход составил 82,2%.
Реактор из нержавеющей стали объемом 12 л оснастили подвесным смесителем, обратным холодильником, дополнительной воронкой, термопарой для измерения температуры и нагревательным кожухом. Следующую реакцию провели в атмосфере сухого азота. Воду (3 л) и этил-3-(4-бутаноат)-7-хлор-2Н-1,3-бензоксазин-2,4-(3Н)-дион (1118 г, 3,58 моль) загрузили в реактор и начали перемешивание. Раствор гидроксида натрия (574 г, 14,34 моль) в воде (2 л) медленно добавили в реакционную суспензию. Реакционную смесь нагрели до 70°С в течение 6 часов, а затем выдержали для медленного охлаждения до комнатной температуры. Профильтровали реакционную смесь через воронку Бюхнера.
Пятигорлую круглодоннкю колбу объемом 22 л оснастили подвесным смесителем, обратным холодильником, термопарой для измерения температуры и дополнительной воронкой. В реактор залили деионизованную воду (1880 мл) и концентрированную соляную кислоту (1197 г, 12,04 моль). К раствору кислоты через дополнительную воронку медленно добавили вышеуказанный гидролизат. С помощью добавления соляной кислоты (160 мл, 1,61 моль) довели рН полученной суспензии до 3. Образовавшийся осадок отделили фильтрацией через воронку с фильтром из пористого стекла и высушили в вакуумной печи при 50°С в течение 24 часов, получив 1109,3 г 4-[(4-хлор-2-гидроксибензоил)амино]масляной кислоты в форме светлого порошка. Выход - количественный.
ПРИМЕР 1d: Получение безводного (4-[(4-хлор-2-гидроксибензоил)амино]бутаноата натрия
Пятигорлую круглодонную колбу объемом 22 л оснастили подвесным смесителем, обратным холодильником, термопарой для измерения температуры и нагревательным кожухом. Следующую реакцию провели в атмосфере сухого азота. Реагент ацетон (13000 мл) и 4-[(4-хлор-2-гидроксибензоил)амино]масляную кислоту (500,0 г, 1,94 моль) загрузили в реактор и начали перемешивание. Реакционную суспензию нагрели до 50°С, получив мутный бурый раствор. Теплый раствор перекачали через фильтр, работающий под давлением, с бумагой Ватман №1 в чистый реактор объемом 22 л. Прозрачный фильтрат желтого цвета нагрели до 50°С при перемешивании. Раствор гидроксида натрия (50%, водный, 155 г, 1,94 моль) загрузили в реактор при интенсивном перемешивании. После окончания добавления основания реактор кипятили с обратным холодильником (60°С) в течение 2,5 часов, а затем выдержали для медленного охлаждения до комнатной температуры. Продукт отделили фильтрацией через воронку с фильтром из пористого стекла и высушили в вакуумной печи при 50°С в течение 24 ч, получив 527,3 г (4-[(4-хлор-2-гидроксибензоил)амино]бутаноата натрия в форме светлого порошка. Выход составил 97,2%.
ПРИМЕР 1е: Получение 4-[4-хлоро-(2-гидроксибензоил)амино]бутаноата натрия моногидрата
Колбу объемом 22 л оснастили подвесным смесителем. Деионизованную воду (2000 мл) и 4-[(4-хлор-2-гидроксибензоил)амино]масляную кислоту (380,0 г, 1,47 моль) добавили и начали перемешивание. Добавили в реактор раствор гидроксида натрия (59,0 г, 1,48 моль) в воде (500 мл). Воду (1500 мл) добавили в реактор и полученную суспензию нагревали до образования полного раствора. Реакционную смесь охладили до температуры окружающей среды, а затем сконцентрировали до сухого состояния при пониженном давлении. Полученный осадок соскребли из колбы и высушили в вакууме при 50°С, получив 401,2 г (4-[(4-хлоро-2-гидроксибензоил)амино]бутаноата натрия в форме моногидрата в виде светлого порошка. Выход составил 96,9%.
ПРИМЕР 1f: Получение натрий-4-[4-хлор-(2-гидроксибензоил)амино]бутаноата посредством сольвата изопропанола
Четырехгорлую круглодонную колбу объемом один литр оснастили подвесным смесителем, обратным холодильником, термопарой для измерения температуры и нагревательным кожухом. Следующую реакцию провели в атмосфере сухого азота. Изопропанол (400 мл) и (4-[(4-хлор-2-гидроксибензоил)амино]масляную кислоту (25,0 г, 0,09 моль) загрузили в реактор и начали перемешивание. Реакционную суспенизию нагрели до 50°С, получив мутный бурый раствор. Теплый раствор перекачали через фильтр, работающий под давлением, с бумагой Ватман №1 в чистый реактор объемом 1 л. Прозрачный фильтрат желтого цвета нагрели до 62°С при перемешивании. Раствор гидроксида натрия (50%, водный, 7,2 г, 0,09 моль) загрузили в реактор при интенсивном перемешивании. После окончания добавления основания реактор нагрели до кипячения с обратным холодильником (72°С), а затем выдержали для медленного охлаждения до комнатной температуры. Продукт отделили вакуумной фильтрацией через воронку с фильтром из пористого стекла и высушили в вакуумной печи при 50°С в течение 24 часов, получив 23,16 г (4-[4-хлор-(2-гидроксибензоил)амино]бутаноата натрия в виде бежевого порошка. Выход составил 92%.
ПРИМЕР 1g: Изготовление капсул
Капсулы для введения приматам, содержащие мононатриевую соль соединения 1 (полученного согласно примеру 1d) и инсулин, изготовили следующим образом. Мононатриевая соль соединения 1 и кристаллы цинкового инсулина человека QA307X: проинсулин (полученный из рекомбинантной ДНК) (производства Eli-Lilly & Со, Индианополис, Индиана) вначале просеяли через стандартное сито Тайлера, 35 меш, и взвесили требуемое количество. Просеянную мононатриевую соль соединения 1 и инсулин смешали способом геометрического просеивания в стеклянной ступке соответствующего размера. Материалы в ступке хорошо перемешали стеклянным пестиком. Для удаления материала со стенок ступки использовали шпатель. Полученный состав перенесли в пластмассовую весовую лодочку для заполнения капсул. Вручную упаковали состав в твердые желатиновые капсулы Torpac, размер №0 (производства Torpac, Inc., Фэирфилд, Нью-Джерси). Масса наполнения каждой капсулы зависела от массы каждого отдельного животного. Дозы соединения 1 в капсулах составляли 100 мг/кг, 75 мг/кг и 50 мг/кг (как мононатриевая соль). Дозы инсулина в капсулах составляли от 0,25 до 0,5 мг на кг.
Пример 2 - Инсулин: пероральная доставка
А. Опыты на крысах
Составы для перорального дозирования (РО) соединения носителя (полученного в примере 1а или 1b, как указано ниже) и цинковый рекомбинантный инсулин человека (поставляемый Calbiochem - Novabiochem Corp., Ла Джолла, Калифорния (каталог №407694)) растворили в деионизованной воде. Обычно 500 мг соединения носителя добавляли к 1,5 мл воды. Свободную кислоту соединения носителя превратили в соль натрия путем перемешивания полученного раствора и добавления одного эквивалента гидроксида натрия. Раствор перемешали с помощью вихревого смесителя, затем нагрели (примерно до 37°С) и обработали ультразвуком. С помощью NaOH или HCl довели рН примерно до 7-8,5. В случае необходимости добавляли дополнительное количество NaOH, чтобы обеспечить равномерную растворимость и откорректировать рН. (Так, например, для соединения 1а к 501 мг соединения в 1,5 мл воды добавили в сумме 258,5 мкл 10N NaOH, получив конечное значение рН 7,73). Затем добавили воду, чтобы довести общий объем примерно до 2,4 мл, и перемешали с помощью вихревого смесителя. Около 1,25 мг инсулина из основного инсулинового раствора (15 мг/мл, получили из 0,5409 г инсулина и 18 мл деионизованной воды, откорректировали HCl и NaOH до рН 8,15 и получили прозрачный раствор с помощью 40 мл концентрированной HCl, 25 мкл 10N NaOH и 50 мкл 1N NaOH) добавили к раствору и смешали путем переворачивания. Величины конечной дозы соединения носителя, дозы инсулина и объемной дозы представлены ниже в таблице 1.
Протоколы введения доз и отбора проб описаны ниже. Самцов крыс Sprague-Dawley весом около 200-250 г выдержали без пищи 24 часа и ввели им кетамин (44 мг/кг) и хлорпромазин (1,5 мг/кг) за 15 минут до введения дозированных растворов и повторно, как это требуется для поддержания анестезии. Испытуемой группе из пяти животных ввели один из дозированных растворов. Для перорального введения доз French катетер Rusch 8 длиной 11 см присоединили к шприцу с пипеткой на конце. Шприц заполнили дозированным раствором, пропустив раствор через катетер, который затем вытерли досуха. Катетер поместили в пищевод, оставив 1 см трубки выступающим после резцов. Раствор вводили путем нажатия на поршень шприца.
Пробы крови брали последовательно из хвостовой артерии обычно через 15, 30, 60, 120 и 180 минут после введения раствора. Содержание сывороточного инсулина определяли с помощью инсулинового тест-набора ELISA (набор №DSL-10-1600, поставляемый Diagnostic System Laboratories, Inc., Уэбстер, Техас), модифицируя стандартный протокол, чтобы оптимизировать чувствительность и линейный диапазон стандартной кривой для объемов и концентраций образцов, используемых в настоящем протоколе. Концентрации сывороточного инсулина человека (мкЕд/мл) измеряли в каждой указанной временной точке у каждого из пяти животных в каждой группе, получающей дозированные препараты. Пять значений для каждой временной точки усредняли и результаты наносили на график зависимости концентрации сывороточного инсулина от времени. Значения максимума (пика) и площади под кривой (AUC) указаны в таблице 1. Предшествующие эксперименты не выявили измеримого содержания инсулина человека после перорального введения одного только инсулина человека.
Б. Опыты на обезьянах
Все протоколы опытов на животных соответствуют "Принципам ухода за лабораторными животными" и утверждены Институционным комитетом по уходу за животными и их использованию (IACUC).
Протокол дозирования для введения капсул каждому животному был следующим. Перед введением доз препаратов у животных были взяты базовые пробы плазмы. Группы из четырех обезьян cynomolgus, двух самцов и двух самок, весом 2-3 кг выдерживали без пищи 4 часа перед введением препаратов и до 2 часов после введения препаратов. Животных анестезировали путем внутримышечного введения 10 мг/кг кетамина гидрохлорида непосредственно перед введением препаратов. Каждому животному вводили переменные дозы состава 1 (25-100 мг/кг) в сочетании с переменными дозами инсулина 0,25-0,5 мг/кг инсулина в 1 капсуле. Животным в течение всего периода введения препаратов давали воду, а также 400 мл сока - в ночь перед введением препаратов и в период введения. Животное связали ремнями. Капсулу поместили в пистолет для введения пилюль, который представляет собой пластмассовый инструмент, содержащий взводимый поршень и разрезной резиновый наконечник для вкладывания капсулы. Пистолет ввели в пищевод животного. Поршень пистолета взвели, чтобы вытолкнуть капсулу из наконечника в пищевод. Затем убрали пистолет. Рот животного держали закрытым и примерно 5 мл воды с помощью обратного осмоса ввели в рот сбоку, чтобы вызвать глотательный эффект. Далее продолжали массировать глотку животного, чтобы вызывать глотательный эффект.
Цитратные пробы крови (1 мл каждая) получали с помощью венипункции из соответствующей вены за 1 час до введения препаратов, а также через 10, 20, 30, 40 и 50 минут и через 1, 1,5, 2, 3, 4 и 6 часов после введения препаратов. Каждый полученный образец плазмы делили на две части. Одну часть замораживали при -80°С и отправляли в другую лабораторию для определения содержания инсулина. Другую часть использовали для определения содержания глюкозы крови. Четырем обезьянам инсулин вводили также подкожно (0,02 мг/кг). Пробы крови получали и анализировали, как описано выше.
Определение содержания инсулина. Содержание сывороточного инсулина определяли с помощью инсулинового тест-набора ELISA (DSL, Уэбстер, Техас).
Анализ содержания глюкозы. Содержание глюкозы крови определяли с помощью системы мониторинга глюкозы ONETOUCH® производства Live Scan Inc., Ньютаун, Пенсильвания.
Результаты представлены ниже в таблице 1А.
Пример 3 - Кромолин - пероральная доставка
Дозированные растворы, содержащие соединение носителя (полученное в примере 1b) и кромолин, динатриевую соль (кромолин) (производства Sigma, Милуоки, Висконсин) приготовили в деионизованной воде. Свободную кислоту соединения носителя превратили в натриевую соль с помощью одного эквивалента гидроксида натрия. Эту смесь перемешали в вихревом смесителе и поместили в устройство ультразвуковой обработки (при температуре около 37°). рН откорректировали примерно до 7-7,5 с помощью водного раствора NaOH. В случае необходимости добавляли дополнительное количество NaOH, чтобы получить равномерное растворение и повторно корректировали рН. Смесь перемешали в вихревом смесителе для получения однородного раствора, также используя в случае необходимости обработку ультразвуком и нагревание. Раствор соединения носителя смешали с кромолином из основного раствора (175 мг кромолина/мл в деионизованной воде, в случае необходимости корректировали рН примерно до 7,0 с помощью NaOH или HCl, основной раствор хранили в замороженном состоянии завернутым в фольге, затем оттаивали и нагревали примерно до 30°С перед применением). Смесь перемешали в вихревом смесителе для получения однородного раствора, также используя в случае необходимости обработку ультразвуком и нагревание. рН корректировали примерно до 7-7,5 с помощью водного раствора NaOH. Затем раствор разбавили водой до желаемого объема (обычно 2,0 мл) и концентрации и хранили перед использованием завернутым в фольге. Конечные дозы соединения носителя и кромолина и объемы доз указаны ниже в таблице 2.
Типичные протоколы введения препаратов и отбора проб были следующими. Самцов крыс Sprague-Dawley весом 200-250 г выдерживали без пищи в течение 24 часов и анестезировали кетамином (44 мг/кг) и хлорпромазином (1,5 мг/кг) за 15 минут до введения препаратов и повторно по мере необходимости поддержания анестезии. Испытуемой группе из пяти животных ввели один из дозирующих растворов. French катетер Rusch 8 длиной 11 см присоединили к шприцу с пипеткой на конце. Шприц заполнили дозирующим раствором, пропустив раствор через катетер, который затем вытерли досуха. Катетер поместили в пищевод, оставив 1 см трубки, выступающим после резцов. Раствор вводили путем нажатия на поршень шприца.
Пробы крови брали из хвостовой артерии обычно через 0,25, 0,5, 1,0 и 1,5 часа после введения раствора. Содержание сывороточного кромолина определяли способом ВЭЖХ. Образцы приготовили следующим образом: 100 мкл сыворотки смешали со 100 мкм 3N HCl и 300 мкл этилацетата в пробирке Эппендорфа. Пробирку установили в вихревой смеситель на 10 минут, а затем центрифугировали в течение 10 минут при 10000 оборотов в минуту. 200 мкл этила цетатного слоя перенесли в пробирку Эппердорфа, содержащую 67 мкл 0,1 М фосфатного буфера. Пробирку установили в вихревой смеситель на 10 минут, а затем центрифугировали в течение 10 минут при 10000 оборотов в минуту. Слой фосфатного буфера перенесли в ампулу для ВЭЖХ и ввели в колонку ВЭЖХ = Keystone Exsil Amino 150×2, т.е. 5 мкм, 100 ангстрем (производства Keystone Scientific Products, Inc.); подвижная фаза = 35% буфера (68 мМ КН2PO4 с рН, откорректированным до 3,0 с помощью 85% Н3PO4)/65% ацетонитрила, объем впрыска = 10 мкл, скорость потока=0,30 мл/минута, время удерживания кромолина = 5,5 минут, спектральное поглощение обнаружили при 240 нм). Предшествующие опыты давали базовые значения, близкие к нулю.
Результаты, полученные на животных в каждой группе, усреднили для каждого момента времени. Максимальные из этих усредненных значений (т.е. средняя пиковая концентрация сывороточного кромолина) представлены ниже в таблице 2.
Пример 4: Рекомбинантный гормон роста человека (rhGH) - пероральная доставка
Предназначенные для перорального введения через желудочный зонд дозированные растворы соединения носителя (полученные в примерах 1а и 1b и указанные ниже в таблице 3) и rhGH приготовили в фосфатном буфере. Свободную кислоту состава носителя превратили в натриевую соль с помощью одного эквивалента гидроксида натрия. Обычно раствор соединения получали в фосфатном буфере и перемешивали, добавив один эквивалент гидроксида натрия (1,0 Н) и получая натриевую соль. В случае необходимости добавляли дополнительное количество NaOH, чтобы получить равномерное растворение, и повторно корректировали рН. Готовые дозированные растворы получили путем смешивания раствора соединения носителя с основным раствором rhGH (15 мг rhGH/мл, полученного смешиванием порошкообразных продуктов: 15 мг rhGH, 75 мг D-маннитола, 15 мг глицина и 3,39 мг двухосновного фосфата натрия с последующим разбавлением 2% глицерином). Дозы состава и rhGH, a также объемы дозы указаны ниже в таблице 3.
Типичные протоколы введения препаратов и отбора проб были следующими. Самцов крыс Sprague-Dawley весом 200-250 г выдерживали без пищи в течение 24 часов и анестезировали кетамином (44 мг/кг) и хлорпромазином (1,5 мг/кг) за 15 минут до введения растворов и повторно по мере необходимости поддержания анестезии. Испытуемой группе из пяти животных ввели один из дозированных растворов. French катетер Rusch 8 длиной 11 см присоединили к шприцу с пипеткой на конце. Шприц заполнили дозированным раствором, пропустив раствор через катетер, который затем вытерли досуха. Катетер поместили в пищевод, оставив 1 см трубки, выступающим после резцов. Раствор вводили путем нажатия на поршень шприца.
Пробы крови брали последовательно из хвостовой артерии обычно через 15, 30, 45 и 60 минут после введения раствора. Концентрации сывороточного rhGH определяли с помощью тест-набора для иммунологического анализа rhGH (набор №K1F4015 производства Genzyme Corporation Inc., Кембридж, Массачусетс). Предшествующие опыты давали базовые значения, близкие к нулю.
Результаты, полученные на животных в каждой группе, усреднили для каждого момента времени. Максимальные из этих усредненных значений (т.е. средняя пиковая концентрация сывороточного rhGH) представлены ниже в таблице 3. (В тех случаях, когда не указаны стандартное отклонение (SD) или среднеквадратичная ошибка (SE), пять образцов, полученных в соответствующий момент времени, объединяли перед проведением анализа).
Пример 5 - Интерферон - пероральная доставка
Дозированные растворы, содержащие соединение носителя (полученный в примере 1b) и интерферон человека (IFN), приготовили в деионизованной воде. Свободную кислоту состава носителя превратили в натриевую соль с помощью одного эквивалента гидроксида натрия. Обычно раствор состава получали в фосфатном буфере и перемешивали, добавив один эквивалент гидроксида натрия (1,0 Н) и получая натриевую соль. Эту смесь перемешали в вихревом смесителе и поместили в устройство ультразвуковой обработки (при температуре около 37°). рН откорректировали примерно до 7-8,5 с помощью водного раствора NaOH. Смесь перемешали в вихревом смесителе для получения однородной суспензии или раствора, также используя в случае необходимости обработку ультразвуком и нагревание. В случае необходимости добавляли также дополнительное количество NaOH для обеспечения равномерной растворимости и повторно корректировали рН. Раствор соединения носителя смешали с основным раствором IFN (примерно 22,0-27,5 мг/мл в фосфатном буферном солевом растворе) и разбавили до желаемого объема (обычно 3,0 мл). Конечные дозы соединения носителя и IFN и объемы доз указаны ниже в таблице 4.
Типичные протоколы введения препаратов и отбора проб были следующими. Самцов крыс Sprague-Dawley весом 200-250 г выдерживали без пищи в течение 24 часов и анестезировали кетамином (44 мг/кг) и хлорпромазином (1,5 мг/кг) за 15 минут до введения растворов и повторно по мере необходимости поддержания анестезии. Испытуемой группе из пяти животных ввели один из дозированных растворов. French катетер Rusch 8 длиной 11 см присоединили к шприцу с пипеткой на конце. Шприц заполнили дозированным раствором, пропустив раствор через катетер, который затем вытерли досуха. Катетер поместили в пищевод, оставив 1 см трубки, выступающим после резцов. Раствор вводили путем нажатия на поршень шприца.
Пробы крови брали последовательно из хвостовой артерии обычно через 0, 15, 30, 45 60 и 90 минут после введения раствора. Концентрации сывороточного IFN определяли с помощью тест-набора для цито-иммунологического анализа альфа-интерферона человека (каталог №КНС4012, Biosource International, Камарильо, Калифорния). Предшествующие опыты давали базовые значения, близкие к нулю. Результаты, полученные на животных в каждой группе, усреднили для каждого момента времени. Максимальные из этих усредненных значений (т.е. средняя пиковая концентрация сывороточного IFN) представлены ниже в таблице 4.
Вышеупомянутые патенты, патентные заявки, методики анализов и публикации полностью приведены здесь в качестве ссылки.
Множество вариантов реализации настоящего изобретения очевидно для специалистов в свете вышеприведенного подробного описания. Все эти очевидные варианты включены в прилагаемую формулу изобретения.
Реферат
Изобретение относится к 4-[(4-хлоро-2-гидроксибензоил)амино]масляной кислоте, ее соли и сольватам (формула I), предназначенным для использования в составах и стандартных дозированных формах для доставки активных агентов. Состав для доставки активных агентов получают путем смешивание (А) по меньшей мере одного активного агента, (Б) соединения формулы I и дополнительно добавляют (В) растворитель. Активный агент представляет собой инсулин, гормон роста человека, рекомбинантный гормон роста человека, кромолин натрия, гепарин, кальцитонин, паратироидный гормон, а также выбран из группы, включающей биологически активный агент, химически активный агент и их сочетание. Активный агент вводят животному, которое нуждается в этом агенте, посредством перорального введения состава или твердой дозированной формы для перорального введения, содержащих соединение формулы (I). Биологически активный агент вводят животному, которое нуждается в этом агенте, также посредством перорального введения указанному животному состава, содержащего соединение формулы I и биологически активный агент. Технический результат - соединения для доставки к мишени биологически или химически активных агентов. 7 н. и 38 з.п. ф-лы, 5 табл.

Формула

Комментарии