Фармацевтические композиции пиримидин-2,4,6-трионов - RU2411043C2
Код документа: RU2411043C2
Чертежи











Описание
Настоящее изобретение относится к фармацевтической композиции пиримидин-2,4,6-трионов (триоксопиримидинов), способам ее получения и применения.
Матриксные металлопротеазы (ММП) являются семейством цинк- и кальцийзависимых протеаз, которые способны разрушать внеклеточный матрикс (ВКМ) и базальную мембрану (Egeblad, M. and Werb, Z., Nat. Rev. Cancer 2 (2002) 161-174; Overall, C.M. and Lopez-Otin, С., Nat. Rev. Cancer 2 (2002) 657-672). Предполагается, что они играют главную роль в эмбриональном развитии и росте (Holmbeck, К., et al., Cell 99 (1999) 81-92; Vu, Т.Н., et al., Cell 93 (1998) 411-422), а также в ремоделировании и восстановлении ткани (Shapiro, S.D., Curr. Opin. Cell Biol. 10 (1998) 602-608; Lund, L.R., et al., EMBO J. 18 (1999) 4645-4656). Поэтому чрезмерное или неадекватное экспрессирование ММП может способствовать патогенезу многих процессов ремоделирования ткани, включая прогрессирование опухоли (Egeblad, M., and Werb, Z., Nat. Rev. Cancer 2 (2002) 161-174; Overall, C.M. and Lopez-Otin, С., Nat. Rev. Cancer 2 (2002) 657-672) и образование аневризмы (Carmeliet, P., et al., Nat. Genet. 17 (1997) 439-444). Воздействие ММП не ограничивается только разложением ВКМ (Chang, С. and Werb, D., Trends Cell Biol. 11 (2001) S.37-43). После разложения посредством ММП-9 становятся доступными пептидные факторы роста, которые секвестированы белками ВКМ (Manes, S., et al., J. Biol. Chem. 274 (1999) 6935-6945). ММП может увеличивать биологическую доступность VEGF (эндотелиального фактора роста сосудов) (Bergers, G., et al., Nat. Cell Biol. 2 (2000) 737-744), но также генерируют ингибиторы ангиогенеза, такие как ангиостатин, путем расщепления плазминогена (Dong, Z., et al., Cell 88 (1997) 801-810). Предполагается, что ММП участвуют в активации стволовых клеток костного мозга (Janowska-Wieczorek A., et al., Blood 93 (1999) 3379-3390). Высокая концентрация ММП-9 наблюдалась при индуцированной G-CSF активации НРС (Carstanjen, D., et al., Transfusion 42 (2002) 588-596).
Триоксопиримидины являются соединениями хорошо известного структурного класса. Такие соединения описаны, например, в патентах US №№ 6242455 и 6110924; WO 97/23465; WO 98/58915; WO 01/25217, которые включены в настоящее изобретение в качестве ссылки, и в публикации Grams, F., et al., Biol. Chem. 382 (2001) 1277-1285, и являются эффективными и высокоселективными по отношению к ММП-2, ММП-9 и ММП-14.
Циклодекстрины являются циклическими углеводами, образованными из крахмала. Они отличаются друг от друга количеством глюкопиранозных звеньев в структуре. Исходные циклодекстрины содержат 6, 7 и 9 глюкопиранозных звеньев и называются альфа-, бета- и гамма-циклодекстринами соответственно. α-, β- и γ-Циклодекстрины, полученные ферментативным превращением крахмала, различаются диаметром своей гидрофобной полости и обычно пригодны для включения многочисленных липофильных веществ.
Триоксопиримидины, которые являются высокоактивными ингибиторами ММП, плохо растворимы в воде и водных растворителях. Поэтому объектом настоящего изобретения является водная композиция, в которой растворим такой триоксопиримидин, причем такую водную композицию такого триоксопиримидина можно использовать в качестве фармацевтической композиции.
Краткое содержание изобретения
Согласно изобретению неожиданно было установлено, что комплекс триоксопиримидин-циклодекстрин, образованный производным триоксопиримидина, характеризующимся описанной ниже формулой (I), и растворимым в воде циклодекстрином (далее обозначаемым, как ЦД), обладает повышенной растворимостью в воде, превосходной стабильностью и оказывает слабое местное раздражающее воздействие и применим в качестве терапевтического средства.
Также установлено, что такой комплекс триоксопиримидина с циклодекстрином и вспомогательным веществом, таким как L-лизин или L-аргинин, обладает улучшенной растворимостью в воде и биологической доступностью и оказывает слабое местное раздражающее воздействие и применим в качестве терапевтического средства. Соответственно настоящее изобретение относится к комплексу триоксопиримидин-циклодекстрин, образованному производным триоксопиримидина или его солью и циклодекстрином, предпочтительно - α-, β- или γ-циклодекстрином или растворимым в воде производным циклодекстрина (растворимый в воде определяется, как обладающий растворимостью, составляющей не менее 0,5 г/100 мл воды при 25°С), в котором производное триоксопиримидина описывается формулой (I).
Кроме того, настоящее изобретение относится к комплексу триоксопиримидин-циклодекстрин, образованному производным триоксопиримидина, описываемым формулой (I), или его солью и циклодекстрином, предпочтительно - α-, β- или γ-циклодекстрином или растворимым в воде производным циклодекстрина (растворимый в воде определяется, как обладающий растворимостью, составляющей не менее 0,5 г/100 мл воды при 25°С), в присутствии вспомогательного вещества, такого как L-лизин или L-аргинин, предпочтительно - L-лизин.
Такой комплекс, предлагаемый в настоящем изобретении, является комплексом включения триоксопиримидин-циклодекстрин и находится в жидкой или твердой форме.
В комплексе, предлагаемом в настоящем изобретении, предпочтительно, если 1 моль триоксопиримидина образует комплекс с количеством циклодекстрина, предпочтительно - β- или γ-циклодекстрина или его производного, составляющим примерно от 1 до 2 моль, и включается в него.
Настоящее изобретение также относится к фармацевтическому средству, предназначенному для лечения нуждающегося в нем пациента, предпочтительно для лечения воспалительных заболеваний бронхов, содержащему в качестве активного компонента в фармацевтически эффективном количестве комплекс триоксопиримидин-циклодекстрин, предлагаемый в настоящем изобретении.
Фармацевтическое средство, предлагаемое в настоящем изобретении, применимо для лечения, профилактики или предупреждения патологий, обусловленных очень значительным или неприемлемым экспрессированием ММП. Предпочтительно, если такое лечение является терапевтическим, профилактическим или предупредительным лечением ревматоидного артрита, опухолей, метастатической инвазии, остеопороза, дегенерации желтого пятна, диабетической ретинопатии, изъязвления роговицы, атеросклероза, воспалительных заболеваний бронхов, таких как астма, хроническое обструктивное заболевание легких или эмфизема.
Настоящее изобретение также относится к композиции для инъекции, содержащей комплекс триоксопиримидин-циклодекстрин, предлагаемый в настоящем изобретении, в фармацевтически эффективном количестве.
Другим объектом настоящего изобретения является жидкая водная композиция комплекса, предлагаемого в настоящем изобретении, в которой фармацевтически приемлемым носителем является вода, и композиция для введения представляет собой водный раствор. Активное вещество, предлагаемое в настоящем изобретении, находится в виде комплекса, образованного включением в циклодекстрин в водном растворе.
Другим объектом настоящего изобретения является жидкая водная композиция комплекса, предлагаемого в настоящем изобретении, в присутствии L-лизина (концентрация L-лизина равна от 10 до 1000 мМ, предпочтительно - от 10 до 500 мМ и более предпочтительно - от 10 до 100 мМ), в которой фармацевтически приемлемым носителем является вода, и композиция для введения представляет собой водный раствор. Активное вещество, предлагаемое в настоящем изобретении, находится в виде комплекса, образованного включением в циклодекстрин в водном растворе в присутствии L-лизина.
Другим объектом настоящего изобретения является комплекс, предлагаемый в настоящем изобретении, в твердом состоянии, комплекс находится в форме порошка, растворимого в воде, предназначенного для растворения перед введением или введения в таком виде, в котором он находится.
Другим объектом настоящего изобретения является комплекс, включенный в различные галеновы формы, соответствующие необходимой форме введения, которыми могут быть таблетки, капсулы, гранулированные системы, растворы для перорального введения, суспензии для перорального введения, растворы, суспензии и имплантаты для парентерального введения, растворы или порошки для ингаляции, кремы и мази гидрофильного или липофильного типа, водные или водно-спиртовые гели, лосьоны для местного, чрескожного или вагинального применения, внутриматочные устройства, растворы, суспензии, имплантаты для применения в офтальмологии, суппозитории, суспензии, аэрозольные препараты, растворы и вспененные препараты для ректального применения.
Настоящее изобретение также относится к применению такого фармацевтического средства в фармацевтически эффективном количестве для лечения таких заболеваний у пациента, страдающего от такого заболевания, предпочтительно - воспалительных заболеваний бронхов. Комплекс, предлагаемый в настоящем изобретении, предпочтительно вводить местно, подкожно, чрескожно, перорально или патентерально.
Настоящее изобретение также относится к способу получения фармацевтического средства, предпочтительно - предназначенного для лечения таких заболеваний, предпочтительно - воспалительных заболеваний бронхов, представляющего собой комплекс триоксопиримидина с циклодекстрином в фармацевтически эффективном количестве в воде или забуференном водном растворе, предпочтительно - дополнительно содержащего вспомогательное вещество, буферное вещество, консервант, растворитель и/или изменяющий вязкость агент.
Предпочтительными циклодекстринами являются
- альфа-циклодекстрин и его синтетические производные, такие как ГПαЦД, метилированный αЦД, гидроксибутил-αЦД, мальтозил-αЦД, глюкозил-αЦД.
- бета-циклодекстрин и его синтетические производные, такие как ГПβЦД, СБОβЦД, СМβЦД, ДИМЕβЦД, ТРИМЕβЦД, гидроксибутил-βЦД, глюкозил-РЦД, мальтозил-βЦД
- гамма-циклодекстрин и его синтетические производные, такие как ГПγЦД, СМγЦД и ДИМЕγЦД, гидроксибутил-γЦД, глюкозил-γЦД, мальтозил-γЦД.
Настоящее изобретение также относится к применению фармацевтической композиции, включающей терапевтически эффективное количество пиримидин-2,4,6-триона и по меньшей мере одного циклодекстрина, а также, возможно, фармацевтически приемлемый носитель для приготовления лекарственного средства, предназначенного для терапевтического, профилактического или предупредительного лечения указанных выше заболеваний.
Настоящее изобретение также относится к применению фармацевтической композиции, включающей терапевтически эффективное количество, а) пиримидин-2,4,6-триона, b) по меньшей мере одного циклодекстрина, с) L-лизина или L-аргинина, предпочтительно - L-лизина, а также, d) возможно, фармацевтически приемлемый носитель для приготовления лекарственного средства, предназначенного для терапевтического, профилактического или предупредительного лечения указанных выше заболеваний.
Подробное описание изобретения
Пиримидин-2,4,6-трионами (триоксопиримидинами), предлагаемыми в настоящем изобретении, являются описываемые формулой (I)

в которой
R1 обозначает С3-С20алкил, которые необязательно может включать одну или несколько групп -S-, -О- или -NH-; или
группу W-V, в которой
W обозначает химическую связь или фенил; и
V обозначает фенил, фенилоксигруппу, фенилтиогруппу, фенилсульфинил, фенилсульфонил или фениламиногруппу, и эти фенильные фрагменты могут быть незамещенными или один или несколько раз замещенными галогеном, гидроксигруппой, С1-С6алкилом, С1-С6алкоксигруппой, С1-С6-алкилтиогруппой, С1-С6алкилсульфинилом, С1-С6-алкиламиногруппой, цианогруппой, нитрогруппой или С1-С6-алкилсульфонилом; и
R2 обозначает С1-С10алкил, и эта алкильная группа является незамещенной или один или два раза замещенной гидроксигруппой или аминогруппой и необязательно может включать одну или несколько групп -S-, -О- или -NH-; бензоильную группу, которая может быть незамещенной или один или несколько раз замещенной галогеном, гидроксигруппой, нитрогруппой, С1-С6-алкоксигруппой, С1-С6-алкиламиногруппой, С1-С6-алкилтиогруппой, С1-С6-алкилсульфинилом, С1-С6-алкилсульфонилом, амидосульфонилом, С1-С6-алкиламидосульфонилом, бис-С1-С6-алкиламидосульфонилом; гетероароматическую ацильную группу; или
фенильную или гетероарильную группу, которые являются незамещенными или один или несколько раз замещенными галогеном, гидроксигруппой, С1-С6-алкоксигруппой, С1-С6-алкиламиногруппой, С1-С6-диалкиламиногруппой, цианогруппой, С1-С6-алкилом, С1-С6-алкенилом, С1-С6-алкинилом, С1-С6-ацилом, С1-С6-алкилтиогруппой, С1-С6-алкилсульфонилом, С1-С6-алкилсульфинилом, С1-С6-алкиламинокарбонилом, аминокарбонилом, С1-С6-алкиламидосульфонилом, амидосульфонилом, бис-С1-С6-алкиламидосульфонилом, нитрогруппой, С1-С6-алкоксикарбонилом, карбоксигруппой.
Объектом настоящего изобретения является применение соединений формулы (I), а также их фармацевтически приемлемых солей, энантиомерных форм, диастереоизомеров и рацематов для приготовления новых фармацевтических препаратов.
При использовании в настоящем изобретении для R1 термин "С3-С20-алкил" означает линейный или разветвленный насыщенный углеводородный радикал, содержащий от 3 до 20, предпочтительно - от 4 до 12 и более предпочтительно - от 8 до 12 атомов углерода. Примерами являются бутил, гексил, октил, децил, 2-этилгексил, 2-этилоктил. Предпочтительными С3-С20алкильными остатками являются н-октил и н-децил. С3-С20Алкильная группа может включать одну или несколько групп -S-, -О- или -NH-, предпочтительно - -O-. Примерами таких С3-С20алкильных групп являются 5-этокси-н-пентил, 9-метокси-н-октил.
Заместители в фенильных фрагментах в "V" предпочтительно расположены в п- или м-положении.
Предпочтительной группой "W-V" являются п-бутоксифенил, бифенил, феноксифенил, п-хлорфеноксифенил, п-бромфеноксифенил, 3,4-дихлорфеноксифенил.
Термин "C1-С10-алкил" при использовании для R2 означает линейный или разветвленный насыщенный углеводородный радикал, содержащий от 1 до 10, предпочтительно - от 1 до 6 и более предпочтительно - от 1 до 4 атомов углерода. Указанный C1-С10-алкил может включать одну или несколько групп -S-, -О- или -NH-, предпочтительно - -О- и более предпочтительно - таким образом, чтобы образовывалась группа, состоящая из этиленоксильных фрагментов. Предпочтительными примерами C1-С10-алкильных групп являются гидроксиэтил; гидроксипропил; этоксиэтил; 1,2-бисэтоксиэтил; 1,2-бисгидроксиэтил.
Термин "гетероароматическая" при использовании в выражении "гетероароматическая ацильная группа" для R2 означает 5- или 6-членное ароматическое кольцо, в котором 1, 2 или 3 атома кольца представляют собой кислород, азот или серу, а остальными атомами кольца являются атомы углерода. Указанная гетероароматическая группа может быть сконденсирована с другим фенильным кольцом. Примерами таких гетероароматических ацильных групп являются фуранкарбоксильная, тиофенкарбоксильная, имидазолилкарбоксильная, 3-бензтиофенкарбоксильная, пиридилкарбоксильная. Предпочтительными примерами являются фуранкарбоксильная и тиофенкарбоксильная.
Термин "гетероарил" при использовании в настоящем изобретении означает гетероароматическую группу, определенную выше. Примерами гетероарильных групп являются электронно-дефицитные остатки, такие как азотсодержащие 6-членные кольца, такие как пиридин, пиримидин, пиразин и 1,3,5-триазин. Особенно предпочтительными гетероарильными группами являются пиримидинильная и пиразинильная.
Заместители, которые могут содержаться в фенильных или гетероарильных группах в R2, в основном расположены в любом положении, пригодном для проведения соответствующей реакции замещения. Предпочтительно, чтобы 1 или 2 заместителя находились в пара- и/или мета-положении.
Термин "С1-С6-алкил" при использовании в настоящем изобретении по отдельности или в комбинации с С1-С6-алкоксигруппой, С1-С6-алкиламиногруппой, С1-С6-диалкиламиногруппой, С1-С6-ацилом, С1-С6-алкилтиогруппой, С1-С6-алкилсульфонилом, С1-С6-алкилсульфинилом С1-С6-алкиламинокарбонилом, С1-С6-алкиламидосульфонилом, бис-С1-С6-алкиламидосульфонилом или С1-С6-алкоксикарбонилом означает линейный или разветвленный насыщенный углеводородный радикал, содержащий от 1 до 6, предпочтительно - от 1 до 4 атомов углерода. Предпочтительными примерами являются метил, этил, пропил, изопропил и трет-бутил.
Термин "С1-С6-алкенил" при использовании в настоящем изобретении означает линейный или разветвленный ненасыщенный углеводородный радикал, содержащий от 2 до 6, предпочтительно - от 2 до 5 атомов углерода и 1 или 2 двойных связи. Если содержатся 2 двойных связи, то они могут быть изолированными или сопряженными двойными связями, предпочтительно - сопряженными двойными связями. Предпочтительными примерами являются аллил и пентадиенил.
Термин "С2-С6-алкинил" при использовании в настоящем изобретении означает линейный или разветвленный углеводородный радикал, содержащий от 2 до 6, предпочтительно - от 2 до 4 атомов углерода. Предпочтительным примером является пропаргил.
Термин "галоген" означает фтор, хлор, бром, йод, предпочтительно - хлор или бром.
Выражение "несколько раз" при использовании в настоящем изобретении означает 1, 2, 3 или 4 раза, предпочтительно - 1 или 2 раза.
Термин "фармацевтически приемлемая соль" при использовании выше в настоящем изобретении означает обычные молекулярные соли с кислотами или молекулярные соли с основаниями, которые сохраняют биологическую эффективность и характеристики соединений формулы (I) и образуются из подходящих нетоксичных органических или неорганических кислот или органических или неорганических оснований. Примеры молекулярных солей с кислотами включают соли, полученные из неорганических кислот, таких как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, сульфаминовая кислота, фосфорная кислота и азотная кислота, и соли, полученные из органических кислот, таких как п-толуолсульфоновая кислота, салициловая кислота, метансульфоновая кислота, щавелевая кислота, янтарная кислота, лимонная кислота, яблочная кислота, молочная кислота, фумаровая кислота и т.п. Примеры молекулярных солей с основаниями включают соли, полученные из гидроксидов аммония, калия, натрия и четвертичного аммония, такого как, например, тетраметиламмонийгидроксид. Химическое превращение фармацевтического соединения (т.е. лекарственного средства) в соль является методикой, хорошо известной химикам-фармацевтам и использующейся для обеспечения лучшей физической и химической стабильности, гигроскопичности, сыпучести и растворимости соединений (см., например, Ansel, H., et. al., Pharmaceutical Dosage Forms and Drug Delivery Systems, 6th ed., (1995), pp.196 and 1456-1457).
Соединения, предлагаемые в настоящем изобретении, можно получить так, как это описано в ЕР 0869947 и WO 01/25217.
В контексте настоящего изобретения особенно предпочтительными являются следующие соединения:
5-бифенил-4-ил-5-[4-(4-нитрофенил)-пиперазин-1-ил]пиримидин-2,4,6-трион
(соединение I)
5-(4-феноксифенил)-5-(4-пиримидин-2-илпиперазин-1-ил)-пиримидин-2,4,6-трион
(соединение II)
5-[4-(4-хлорфенокси)-фенил]-5-(4-пиримидин-2-илпиперазин-1-ил)-пиримидин-2,4,6-трион
(соединение III)
5-[4-(3,4-дихлорфенокси)-фенил]-5-(4-пиримидин-2-илпиперазин-1-ил)-пиримидин-2,4,6-трион
(соединение IV)
5-[4-(4-бромфенокси)-фенил]-5-(4-пиримидин-2-илпиперазин-1-ил)-пиримидин-2,4,6-трион
(соединение V).
Также очевидно, что, если производное триоксопиримидина (I) содержит кислотный фрагмент, такой как карбоксильную группу или сульфонильную группу, то с помощью кислотного фрагмента производное может образовать соль с основанием.
В дополнение к описанным выше молекулярным солям триоксопиримидин может образовать гидратированную или сольватированную форму. Гидраты и сольваты включают и свободное основание соединения формулы (I), и соль соединения формулы (I). Они также включают таутомеры соединения формулы (I).
Циклодекстрины (ЦД), предлагаемые в настоящем изобретении, являются циклическими олигосахаридами, полученными ферментативным разложением крахмала, которые содержат разное количество глюкопиранозных звеньев, чаще всего 6, 7 или 8: эти циклодекстрины соответственно называются α-, β- и γ-циклодекстринами (αЦД, βЦД и γЦД). Циклодекстрины, предлагаемые в настоящем изобретении, представляют собой сами циклодекстрины или производные циклодекстрина, которые растворимы в воде по меньшей мере в количестве, равном 0,5 г/100 мл при 25°С.
Растворимый в воде циклодекстрин, предпочтительно применяющийся в настоящем изобретении, означает циклодекстрин, обладающий растворимостью в воде, по меньшей мере такой же, как β-циклодекстрин. Примерами таких растворимых в воде циклодекстринов являются сульфобутилциклодекстрин, гидроксипропилциклодекстрин, мальтозилциклодекстрин и их соли. Предпочтительными являются сульфобутил-β-циклодекстрин, гидроксипропил-β-циклодекстрин, мальтозил-β-циклодекстрин и их соли.
Предпочтительными циклодекстринами, предлагаемыми в настоящем изобретении, также являются метилциклодекстрины (продукты метилирования циклодекстринов), диметилциклодекстрины (ДИМЕЦД) (предпочтительно - замещенные в положениях 2 и 6), триметилциклодекстрины (предпочтительно - замещенные в положениях 2, 3 и 6), "статистически метилированные" циклодекстрины (предпочтительно - статистически замещенные в положениях 2, 3 и 6, но содержащие от 1,7 до 1,9 метальных групп в пересчете на глюкопиранозное звено, СМβЦД), гидроксипропилциклодекстрины (ГПЦД, гидроксипропилированные циклодекстрины предпочтительно - статистически замещенные в основном в положениях 2 и 3 (ГП-βЦД, ГП-γЦД)), сульфобутоксициклодекстрины (СБОЦД), гидроксиэтилциклодекстрины, карбоксиметилэтилциклодекстрины, этилциклодекстрины, амфифильные циклодекстрины, полученные прививкой углеводородных цепей к гидроксигруппам и способные образовывать наночастицы, холестеринциклодекстрины и триглицеридциклодекстрины, полученные прививкой моноаминированных циклодекстринов (со спейсерной группой).
Вспомогательными веществами, предлагаемыми в настоящем изобретении, являются L-лизин или L-аргинин, предпочтительно - L-лизин. Такие вспомогательные вещества можно использовать для увеличения растворимости кислотных компонентов за счет образования тройного комплекса. Комплекс триоксопиримидин-циклодекстрин, предлагаемый в настоящем изобретении, можно получить путем приготовления водного раствора, содержащего триоксопиримидин или его соль и растворимый в воде циклодекстрин. Растворимый в воде циклодекстрин используют в количестве, предпочтительно составляющем 1 моль или более в пересчете на 1 моль триоксопиримидина или его соли, более предпочтительно - 1-10 моль и особенно предпочтительно - 1-2 моль циклодекстрина на 1 моль триоксопиримидина.
Чем выше концентрация растворимого в воде циклодекстрина, тем сильнее увеличивается растворимость триоксопиримидина. На методику приготовления водного раствора не налагается каких-либо специальных ограничений и, например, его готовят путем использования воды или буферного раствора в температурном диапазоне, составляющем примерно от -5 до 35°С.
Когда водный раствор циклодекстрина перемешивают с избытком триоксопиримидина формулы I, то происходит образование комплекса этих двух молекул. Однако установление равновесия требует по меньшей мере примерно нескольких дней, так что через несколько часов или даже через один день улучшенная растворимость триоксопиримидинов, предлагаемых в настоящем изобретении, не обнаруживается. Фильтрование раствора позволяет извлечь комплекс, растворенный в фильтрате, поскольку комплекс растворим в воде. Комплекс также можно получить путем смешивания известного количества солюбилизованного триоксопиримидина формулы I в водном растворе с известным количеством солюбилизованного ЦД, соотношение которых рассчитано заранее.
Другая методика получения комплекса заключается в прибавлении раствора триоксопиримидина формулы I в растворителе (например, спирте, ацетоне и т.п.) к водному раствору циклодекстрина. Комплекс может образоваться после достаточно длительного перемешивания, или после выпаривания растворителя, или даже в присутствии растворителя.
Во всех этих методиках получения комплекса триоксопиримидин-ЦД в качестве вспомогательного вещества можно использовать раствор L-лизина или L-аргинина (при концентрации аминокислоты, составляющей от 10 до 1000 мМ, предпочтительно - от 10 до 500 мМ и более предпочтительно - от 10 до 100 мМ). В качестве вспомогательного вещества предпочтительно использовать раствор L-лизина.
Лиофилизация или распыление растворов комплекса, предлагаемого в настоящем изобретении, позволяет получить комплекс в твердой форме. Таким образом можно получить комплекс в форме аморфного порошка. Комплекс в твердом состоянии также можно получить после растворения ЦД и триоксопиримидина формулы I в подходящем органическом растворителе с последующим выпариванием растворителя.
Для получения твердых комплексов можно использовать другие методики, например, энергичное перемешивание суспензии триоксопиримидина формулы I и ЦД в очень небольшом количестве воды, последующий сбор комплекса после сушки, или использование СО2 в надкритическом состоянии для смешивания триоксопиримидина формулы I и ЦД в присутствии СО2 в надкритическом состоянии.
Комплекс, предлагаемый в настоящем изобретении, можно получить, например, по методике, которая сама по себе известна, из раствора или с использованием пасты, при которой отношение массы циклодекстрина к массе триоксопиримидина составляет от 2 (2:1) до 540 (540:1) и предпочтительно - от 2 до 25, особенно предпочтительно - в диапазоне от 2,6 до 3,5 (для комплекса с циклодекстрином состава 1:1) или от 5,2 до 6,2 (для комплекса с циклодекстрином состава 1:2) при молекулярной массе циклодекстрина, равной примерно 1300.
Предпочтительно готовить комплекс из концентрированного водного препарата циклодекстрина. Концентрация циклодекстрина в препарате предпочтительно равна от 50 до 400 мМ. Предпочтение отдается концентрации циклодекстрина, равной от 100 до 250 мМ. В зависимости от консистенции смеси интенсивно перемешивают или замешивают. Содержание циклодекстрина в мас.% приводится в пересчете на полную массу водного препарата циклодекстрина.
Также предпочтительно готовить комплекс из концентрированного водного препарата циклодекстрина в присутствии раствора L-лизина (концентрация L-лизина равна от 10 до 1000 мМ, предпочтительно - от 10 до 500 мМ и более предпочтительно - от 10 до 100 мМ). Концентрация циклодекстрина в препарате предпочтительно равна от 50 до 400 мМ. Предпочтение отдается концентрации циклодекстрина, равной от 100 до 250 мМ. В зависимости от консистенции смеси интенсивно перемешивают или замешивают. Содержание циклодекстрина в мас.% приводится в пересчете на полную массу водного препарата циклодекстрина.
Температура проведения реакции обычно равна от 20 до 80°С, предпочтительно - от 20 до 60°С, особенно предпочтительно - от 25 до 45°С. Длительность проведения реакции зависит от температуры и составляет по меньшей мере несколько дней. Предпочтение отдается длительности проведения реакции, равной не менее 7 дней, чтобы установилось равновесие комплексообразования. Затем реакционную смесь фильтруют, если еще имеется нерастворившееся вещество, или используют непосредственно, если растворение прошло до конца. При необходимости комплекс может выделить, например, с помощью хроматографических методик. Предпочтительно, чтобы концентрации и соотношения количеств триоксопиримидина и циклодекстрина были такими, чтобы образование комплекса прошло до конца (было достигнуто равновесие) и не обнаруживался нерастворенный и не образовавший комплекс триоксопиримидин.
Согласно изобретению было установлено, что комплексы триоксопиримидина формулы I и циклодекстрина резко повышают растворимость триоксопиримидина в воде. Также установлено, что образование комплекса не оказывает неблагоприятного влияния на фармакологические характеристики триоксопиримидина.
Согласно изобретению было установлено, что комплексы триоксопиримидина формулы I, циклодекстрина и вспомогательного вещества, такого как L-лизин или L-аргинин, резко повышают растворимость триоксопиримидина в воде. Также установлено, что образование комплекса не оказывает неблагоприятного влияния на фармакологические характеристики триоксопиримидина.
Все эти характеристики позволяют приготовить жидкие препараты в виде растворов для инъекции или для распыления и позволяют улучшить биологическую доступность, особенно пероральную. Комплекс триоксопиримидин-циклодекстрин, предлагаемый в настоящем изобретении, можно применять в том виде, в котором он получен, или в порошкообразной форме, которую получают путем удаления содержащейся в нем воды. Примеры методик удаления воды включают лиофилизацию и сушку при пониженном давлении. Особенно предпочтительным является препарат, полученный лиофилизацией.
Комплекс триоксопиримидин-циклодекстрин, предлагаемый в настоящем изобретении, оказывает свое воздействие при пероральном или парентеральном введении и его предпочтительно готовить в виде препарата для парентерального введения, предпочтительно - в виде композиции для инъекции, или препарата для местного применения, предпочтительно - в виде аэрозольного препарата.
Дозу комплекса, предлагаемого в настоящем изобретении, можно менять в соответствии с возрастом, массой тела и тяжестью симптомов, проявляющихся у пациента, и комплекс можно вводить в виде одной дозы или разделенных доз. Примеры форм препаратов включают таблетки, капсулы, порошки и гранулы. Их можно приготовить по известным технологиям с использованием типичных добавок, таких как инертные наполнители, смазывающие вещества и связующие.
Настоящее изобретение относится к способу, применяющемуся для лечения воспалительных заболеваний бронхов у страдающего ими млекопитающего, нуждающегося в таком лечении, например, предпочтительно - астмы и хронического обструктивного заболевания легких (ХОЗЛ), путем введения пациенту комплекса, предлагаемого в настоящем изобретении, в фармацевтически эффективном количестве. Астма является воспалительным заболеванием бронхиального дерева, связанным или не связанным с воздействием аллергена. Это воспаление провоцирует у пациентов симптомы, стимулируя сокращения гладких мышц бронхов, усиливая секрецию слизи и вызывая морфологические изменения бронхов, что считается факторами, осложняющими течение этого заболевания. Гиперчувствительность дыхательных путей является отличительным признаком этого заболевания и она приводит к большинству симптомов. Бронхиальное дерево является очень сложной тканью, содержащей множество типов клеток (эпителиальные клетки, гладкомышечные клетки, воспалительные клетки, нервные клетки, клетки, вырабатывающие слизь, фибробласты и т.п.), и ремоделирование бронхов, которое включает множество аспектов, в основном заключается в осаждении компонентов внеклеточного матрикса на стенках бронхов и гиперплазию клеток, вырабатывающих слизь. Применение комплексов, предлагаемых в настоящем изобретении, подавляет приток воспалительных клеток в области смывания альвеол бронхов и перибронхиальную ткань и подавляет гиперчувствительность, которая определяется, как аномальная реакция на стимулирующие агенты, такие как метахолин. Обзор по этому заболеванию и современным методикам его лечения приведен, например, в публикации: GINA Workshop Report, Global Strategy for Asthma Management and Prevention (NIH Publication No. 02-3659).
Поэтому настоящее изобретение также относится к способу лечения или предупреждения хронических обструктивных заболеваний легких у страдающего ими млекопитающего, нуждающегося в таком лечении, путем применения комплексов, предлагаемых в настоящем изобретении. При таком заболевании бронхи воспалены и слизистые железы гиперплазированы и продуцируют большое количество слизи. Стенки бронхов являются аномальными и осаждение аномальных компонентов внеклеточного матрикса увеличивает сопротивление потоку воздуха. Это заболевание и современные методики его лечения описаны, например, в публикации Fabbri, L.M., and Hurd, S.S., Eur. Respir. J. 22 (2003) 1-2.
Поэтому настоящее изобретение также относится к способу лечения или предупреждения эмфиземы у страдающего ею млекопитающего, нуждающегося в таком лечении, путем применения комплексов, предлагаемых в настоящем изобретении. При таком заболевании стенки альвеол разрушаются вследствие протекания протеолитических процессов и это разрушение нарушает перенос кислорода в кровь. Патофизиологические нарушения также возникают вследствие чрезмерного расширения, которое приводит к аномалиям газообмена вследствие нарушения функции дыхательных мышц и вследствие гипертензии в легочных артериях, что в поздних стадиях заболевания приводит к сердечной недостаточности.
В контексте настоящего изобретения комплексы триоксопиримидин-циклодекстрин предпочтительно вводить пациенту, нуждающемуся в таком лечении, в течение нескольких месяцев или лет. Комплексы предпочтительно вводить в виде аэрозолей жидкостей или порошков в нетоксичных дозах, составляющих микро- или макромоли в пересчете на килограмм массы тела в сутки.
Точная дозировка комплексов, предлагаемых в настоящем изобретении, будет меняться, но ее легко определить. Обычно суточная доза комплексов будет находиться в диапазоне от 1 мкмоль/кг в сутки до 100 нмоль/кг в сутки (концентрация триоксопиримидина в комплексе).
Предпочтительно, если фармацевтические композиции представляют собой водные композиции, являющиеся физиологически совместимыми. Предпочтительно, если композиции дополнительно включают фармацевтически приемлемую добавку, такую как буферную систему, консервант и/или вспомогательное вещество. Подходящие буферные системы основаны на фосфате натрия, ацетате натрия или борате натрия. Консерванты необходимы для предотвращения микробного загрязнения фармацевтической композиции при ее применении. Подходящими консервантами являются, например, бензалконийхлорид, хлорбутанол, метилпарабен, пропилпарабен, фенилэтиловый спирт, сорбиновая кислота. Такие консерванты обычно используют в количестве, составляющем от 0,01 до 1% мас./об.
Подходящие вспомогательные вещества и фармацевтические препараты описаны в публикации Remington's Pharmaceutical Sciences, 16th ed., 1980, Mack Publishing Co., edited by Oslo et al. Обычно в препарате используют количество фармацевтически приемлемой соли, необходимое для того, чтобы препарат стал изотоническим. Примеры фармацевтически приемлемых веществ включают физиологический раствор, раствор Рингера и раствор декстрозы. Значение рН раствора предпочтительно составляет от примерно 5 до примерно 8 и более предпочтительно - от примерно 7 до примерно 7,5.
Если в качестве вспомогательных веществ для образования комплекса используют L-лизин или L-аргинин, то значение рН раствора предпочтительно составляет от примерно 6 до примерно 8,5 и более предпочтительно - от примерно 7,5 до примерно 8,5.
Предпочтительный препарат, предлагаемый в настоящем изобретении, представляет собой препарат для инъекции или распыления, предпочтительно приготовленный из ЦД и триоксопиримидина в молярном отношении, составляющем от 1 до 500.
Комплекс получают путем растворения ЦД в воде, прибавления триоксопиримидина формулы I и нагревания на водяной бане, пока последний полностью не растворится. Раствор предпочтительно стерилизовать путем фильтрования. Предпочтительно, если раствор обладает осмоляльностью, равной 200-400, предпочтительно - примерно 300 мОс/кг. Значение рН равно примерно 7,2. В зависимости от требований концентрацию триоксопиримидина и/или ЦД можно менять. Предпочтительно регулировать тоничность путем прибавления NaCl.
Предпочтительный препарат для распыления содержит триоксопиримидин, ЦД, NaCl и воду. Особенно предпочтительной является комбинация, содержащая (в 200 мл раствора): триоксопиримидина 0,05-0,2 г, предпочтительно - 0,1 г; 10-50 г ЦД, предпочтительно - 20 г ЦД, предпочтительно - ГПβЦД; хлорида натрия 1,2-1,5 г, предпочтительно - 1,42 г (изотоничность) и воду, предпочтительно - апирогенную, стерильную, очищенную воду, прибавляемую для доведения объема до 200 мл.
Раствор готовят путем растворения ЦД в 100 мл очищенной воды, прибавления триоксопиримидина и NaCl при перемешивании, так чтобы они растворились, и дополнительного прибавления воды, так чтобы получить 200 мл раствора. Раствор предпочтительно стерилизовать путем фильтрования через полипропиленовую мембрану с отверстиями размером 0,22 мкм или с помощью способа стерилизации паром.
Другими предпочтительными препаратами являются препараты для применения в офтальмологии, препараты для перорального применения, внутриматочные устройства. Также можно рассмотреть сочетание с другими системами, такими как, например, нано- или микрочастицы или липосомы.
Приведенные ниже примеры, литературные ссылки и чертежи предоставлены для облегчения понимания настоящего изобретения, полный объем которого указан в прилагаемой формуле изобретения. Следует понимать, что в описанные методики можно внести изменения без отклонения от сущности настоящего изобретения.
Описание чертежей
На фиг.1 приведены данные по растворимости соединения I, полученные при использовании СМβЦД и ГП-β-ЦД. Обе фазовые диаграммы растворимости относятся к типу Ар, что означает, что ЦД образуют комплексы состава 1:1 и 1:2. Затем рассчитаны константы устойчивости и их значения приведены в таблице 6.
Фиг.2: Спектры ЯМР комплекса соединения I с ДИМЕβЦД (верхняя часть) и самого ДИМЕβЦД (нижняя часть).
Фиг.3: Спектры ЯМР соединения I (сверху), ДИМЕβЦД (справа) и Т-ROESY (в середине).
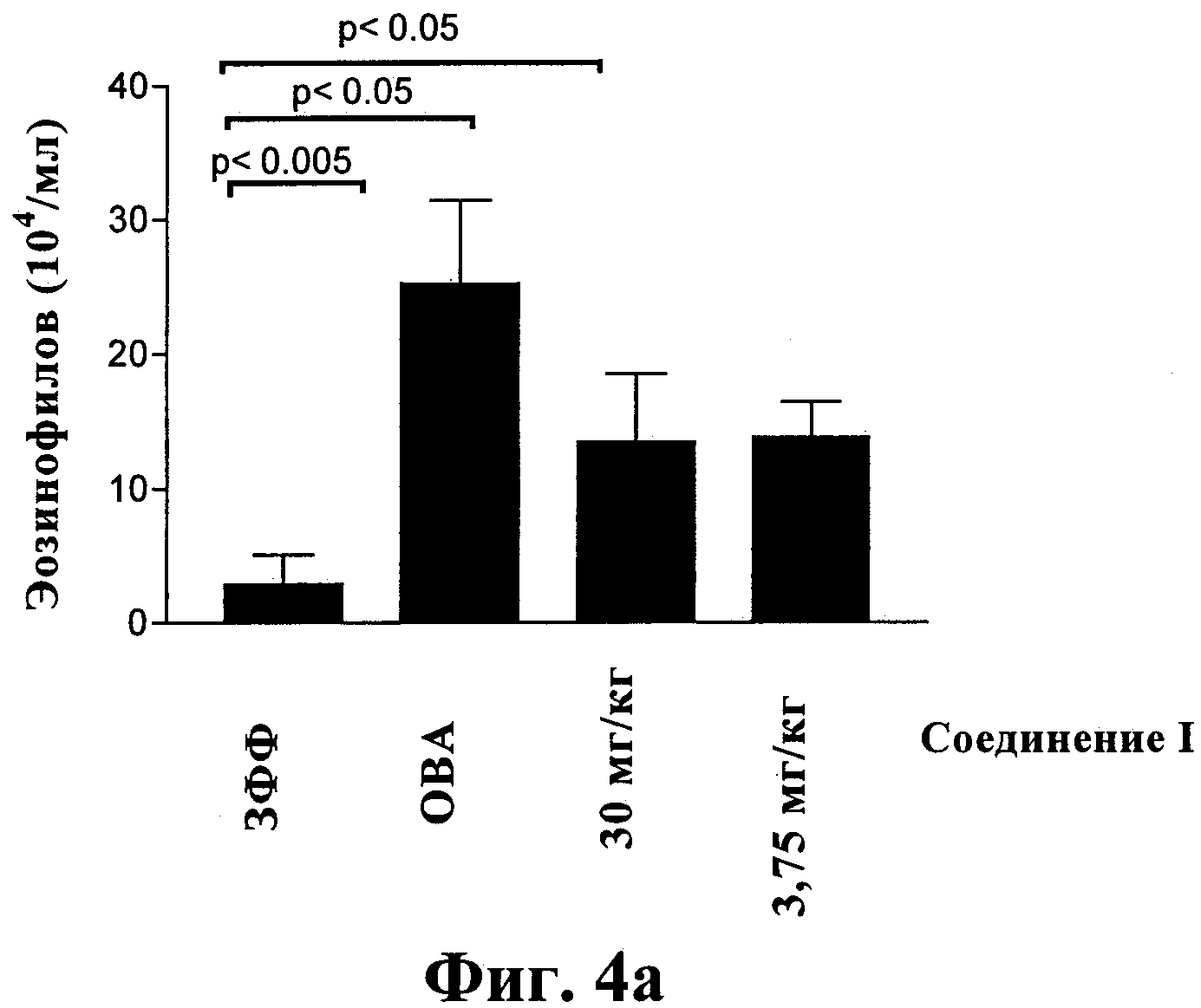
Фиг.4: Влияние внутрибрюшинной инъекции суспензии соединения I на количество эозинофилов по данным БАЛ (фиг.2а) и показатель перибронхиального воспаления (фиг.2b). Контрольными являлись мыши, на которых воздействовали только с помощью ЗФФ и на которых не воздействовали аллергеном (ЗФФ), и мыши, на которых воздействовали с помощью ОВА путем ингаляции и с помощью плацебо путем внутрибрюшинной инъекции (ОВА).
Фиг.5: Терапевтическое воздействие комплекса соединение I-ГП-β-ЦД, флутиказона и плацебо, введенных в виде аэрозолей, на исследованную с помощью БАЛ эозинофилию (5а), показатель перибронхиального воспаления (5b) и показатель инфильтрации эозинофилов в ткань (5с) в модели кратковременного (5 дней) воздействия аллергена.
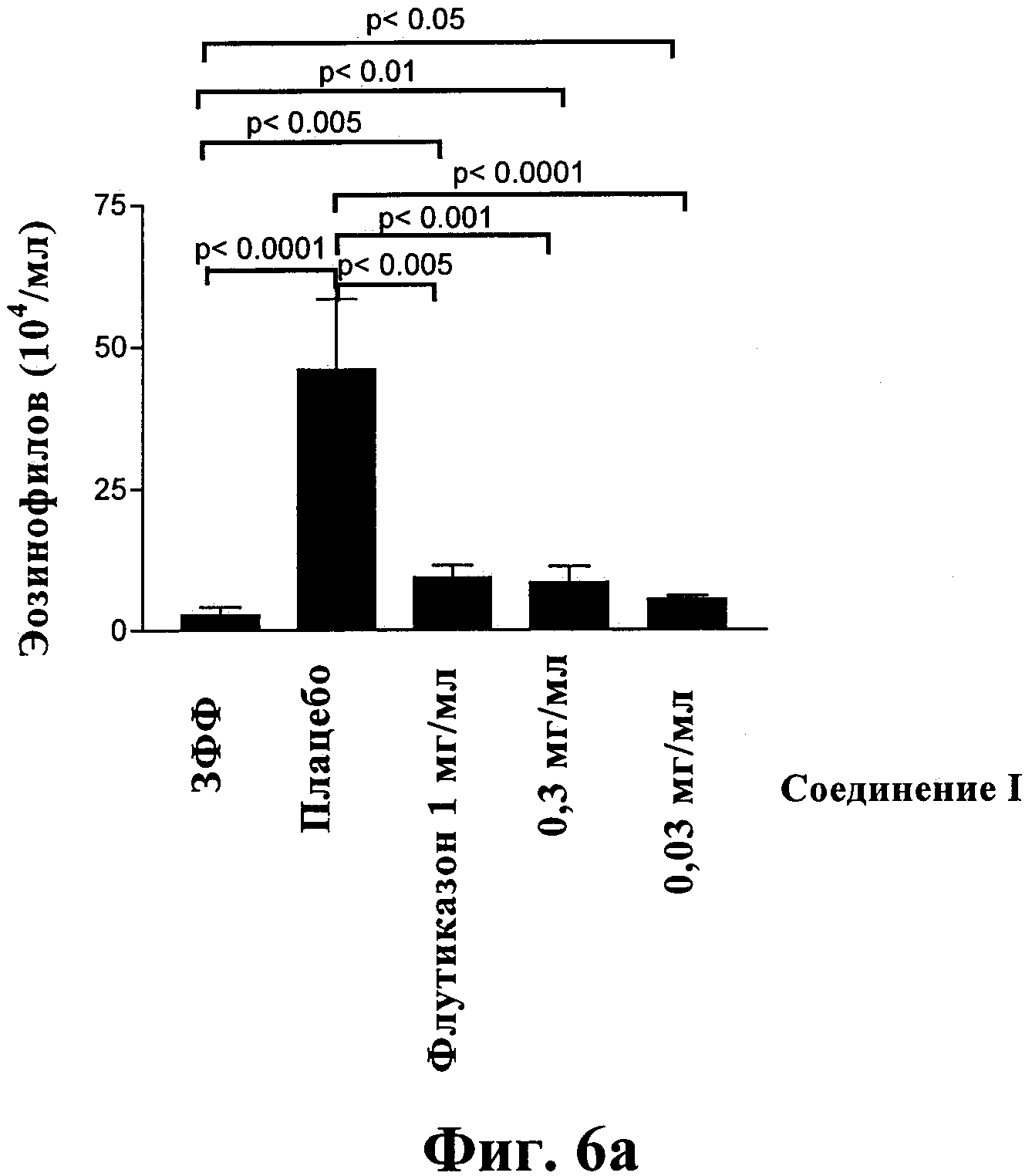
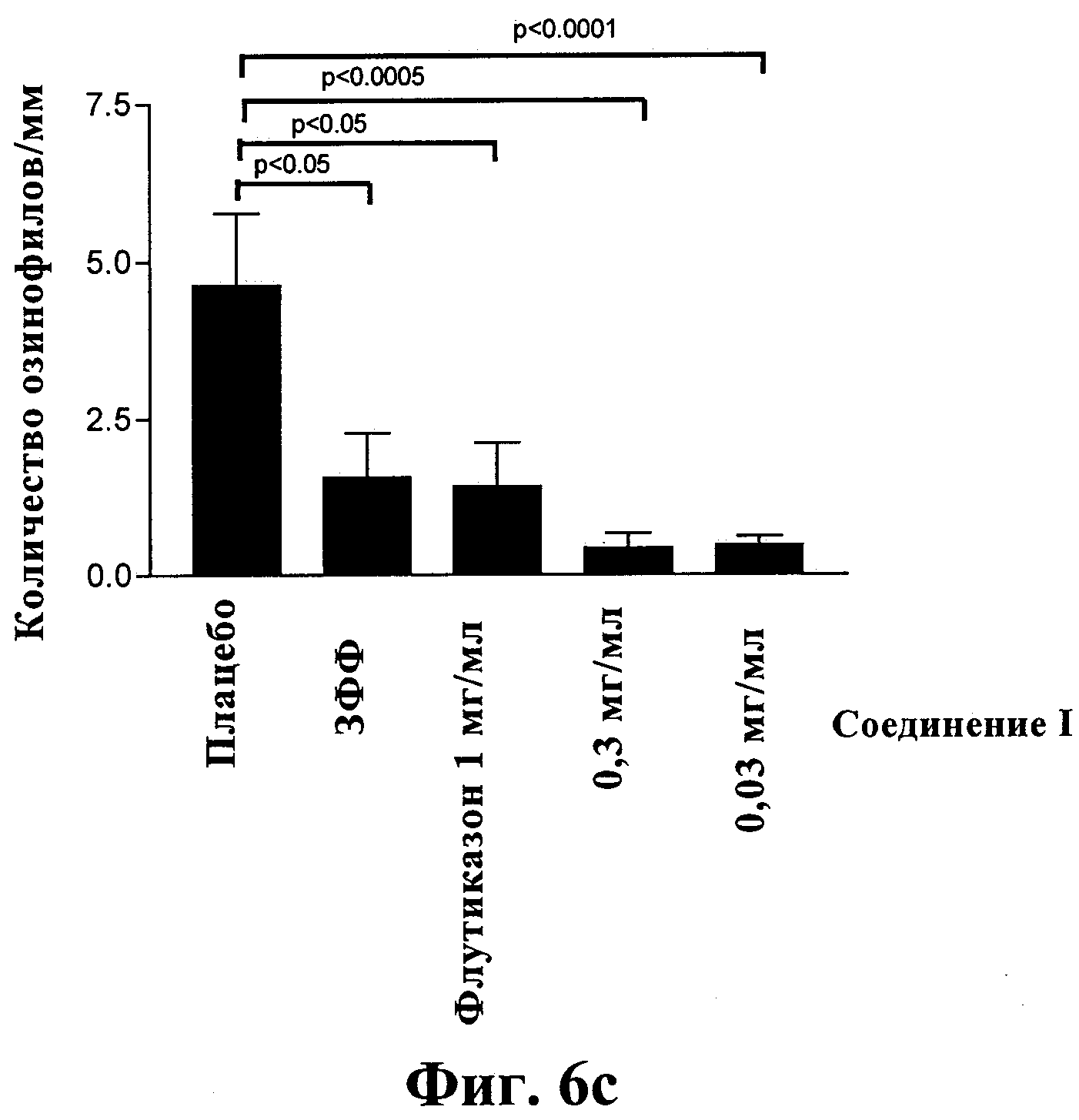
Фиг.6: Терапевтическое воздействие комплекса соединение I-ГП-β-ЦД, флутиказона и плацебо (ЗФФ), введенных в виде аэрозолей, на исследованную с помощью БАЛ эозинофилию (6а), показатель перибронхиального воспаления (6b) и показатель инфильтрации эозинофилов в ткань (6с) в модели длительного (11 недель) воздействия аллергена. Мышей, сенсибилизированных, но не подвергнутых воздействию аллергена (ЗФФ), и мышей, сенсибилизированных и подвергнутых воздействию ОВА (плацебо), лечили путем ингаляции ЗФФ.
Фиг.7: Фазовая диаграмма растворимости соединения I с ГП-β-ЦД в очищенной воде (●), в присутствии L-лизина 50 мМ (х) или L-лизина 500 мМ (▲).
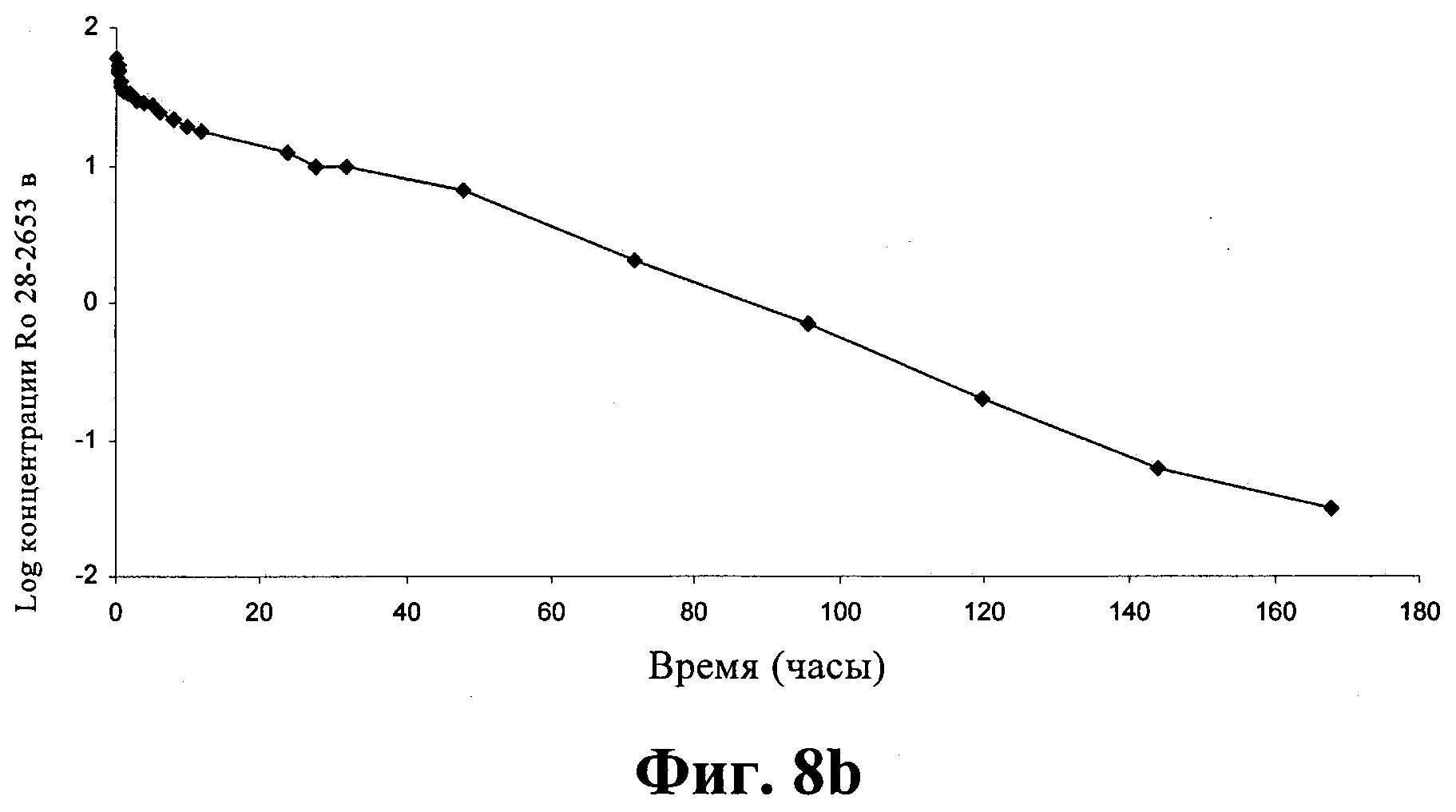
Фиг.8: Зависимость средней (±СП = стандартное отклонение) концентрации соединения I в сыворотке (а) и логарифма средней концентрации соединения I в сыворотке (b) от времени после внутривенного введения (5 мг/кг) овцам (n=6).
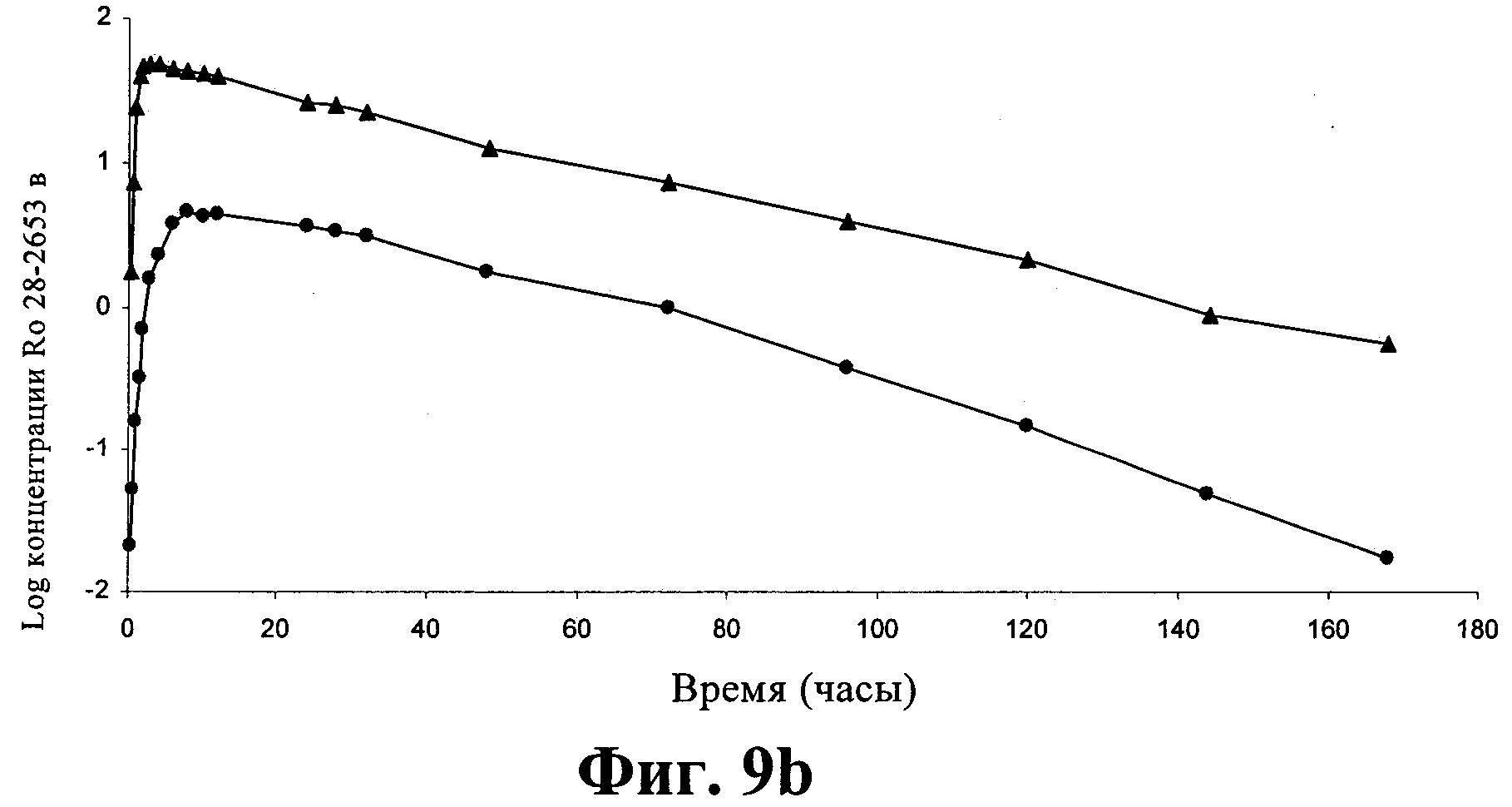
Фиг.9: Зависимость средней (±СП) концентрации соединения I в сыворотке (а) и логарифма средней концентрации соединения I в сыворотке (b) от времени после перорального введения (15 мг/кг) раствора (▲) и суспензии (●) овцам (n=5 для введения раствора и n=6 для введения суспензии).
Аббревиатуры
ЦД циклодекстрин
βЦД β-циклодекстрин
γЦД γ-циклодекстрин
ДИМЕβЦД диметил-β-циклодекстрин
ТРИМЕβЦД триметил-β-циклодекстрин
ГПβЦД гидроксипропил-β-циклодекстрин
СМβЦД статистически метилированный β-циклодекстрин
ВВ внутривенное
Пример 1
Приготовление растворимого комплекса соединения I с циклодекстрином (ЦД)
1.1 Отвешивают 20 мг соединения I. Прибавляют 2 мл раствора ГПβЦД 200 мМ. Перемешивают в течение 24 ч при 37°С. Фильтруют через миллипористый фильтр Millex HV с отверстиями размером 0,45 мкм. Полученный после фильтрования раствор содержит комплекс соединение I-ЦД.
1.2. Отвешивают 2,5 мг соединения I. Прибавляют 2 мл раствора ГПβЦД 200 мМ. Перемешивают при 37°С в течение 24 ч или до полного растворения соединения I. Полученный таким образом раствор содержит комплекс соединение I-ЦД.
Пример 2
Исследование фазовой диаграммы растворимости
При образовании комплекса соединение I, практически нерастворимое в воде (<0,6 мкг/мл, ММ (молекулярная масса): 485), значительно солюбилизируется. Увеличение растворимости соединения I является доказательством образования комплекса соединения I с ЦД. Обнаружено, что комплексообразование и установление равновесия протекает на 20% через 1 день, на 40% через 4 дня и на 100% через 7 дней. Диаграммы растворимости (фиг.1) получают путем прибавления избытка соединения I к растворам ЦД увеличивающихся концентраций. После 7 дней перемешивания в термостатируемых банях при 37°С эти растворы фильтруют и количество солюбилизированного соединения I определяют с помощью ВЭЖХ (высокоэффективная жидкостная хроматография). Дают возможность образоваться комплексам βЦД и γЦД и их синтетических аналогов с соединением I. Используют β-ЦД и ГПβЦД, выпускающиеся фирмой ROQUETTE (France), СМβЦД и γЦД, выпускающиеся фирмой Wacker (Germany).
Приготовление водных растворов ЦД:
- РЦД: Растворы концентрации 2, 4, 8, 10, 12, 16 мМ.
- ГПβЦД: Растворы концентрации 10, 25, 50, 75, 100, 150, 200 мМ.
- СМβЦД: Растворы концентрации 10, 25, 50, 75, 100, 150, 200 мМ.
- γЦД: Растворы концентрации 10, 25, 50, 100, 75, 150 мМ.
Образование комплекса:
Колбы, содержащие соединение I и циклодекстрины, помещают в термостатируемые бани при 37°С на 7 дней и содержимое перемешивают, чтобы установилось равновесие комплексообразования. Затем суспензии фильтруют через миллипористый фильтр с отверстиями размером 0,22 мкм, изготовленный из поливинилиденфторида (ПВДФ), и фильтрат растворяют в ДМСО (диметилсульфоксид) и готовят образцы разной концентрации для построения калибровочного графика. Затем концентрации определяют с помощью аттестованной методики ВЭЖХ, описанной ниже.
Определение концентрации соединения I: Методика ВЭЖХ
Аппаратура:
Насос: Merck-Hitachi модель L-7100, пробоотборник: Merck-Hitachi L-7200, печь: Merck-Hitachi L-7350, детектор: детектор с диодной матрицей Merck-Hitachi L-7455, интерфейс: D-7000, обработка данных с помощью программного обеспечения "Chromatography Data Station Software", выпускающегося фирмой Merck-Hitachi.
Стационарная фаза:
Колонка: Lichrocart (125×4 мм внутренний диаметр), заполненная стационарной фазой октилсилан С8 LiChorspher® 60RP-Select В (5 мкм) Merck.
Условия хроматографирования:
Подвижная фаза: смесь фосфатного буфера 0,05 М до рН 3 с метанолом (30/70, об./об.). Газ удаляют с помощью ультразвуковой обработки в течение 15 мин, выходной поток: 1 мл/мин, детектирование в УФ-области при длине волны 265 нм, рабочая температура: 30°С, инжектируемый объем: 20 мкл.
Результаты определения содержания соединения I с помощью ВЭЖХ приведены в представленных ниже таблицах (таблицы 1-4) для каждого циклодекстрина. При отсутствии циклодекстрина растворимость найдена равной 0,56 мкг/мл.
Соединение I образует комплексы со всеми исследованными циклодекстринами, поскольку наблюдается увеличение растворимости. Также можно непосредственно обнаружить, что образование комплекса соединения I с СМβЦД значительно и совершенно неожиданно увеличивает растворимость соединения I. То же наблюдается и для ГП-β-ЦД. В таблице 5 приведены данные по растворимости для каждого циклодекстрина при максимальной использованной концентрации. Увеличение растворимости рассчитано по сравнению с растворимостью соединения I в воде (при отсутствии циклодекстрина), которая найдена равной 0,56 мкг/мл.
На основании этих результатов фазовые диаграммы растворимости построены по методике, описанной в публикации Higuchi, Т. and Connors, K.A., Advances in Analytical Chemistry and Instrumentation 4 (1965) 117-212.
Большие значения K1:1 показывают, что в воде полости производных β-ЦД очень хорошо вмещают молекулярный фрагмент соединения I, участвующий во включении. При концентрации РЦД, равной от 0 до 4 мМ, растворимость соединения I увеличивается и достигает плато при концентрации βЦД, равной 8 мМ. При концентрация βЦД, превышающей 8 мМ, образуется дополнительный комплекс состава 1:2 (соединение I: βЦД), обладающий меньшей растворимостью (1,5 мкг/мл). Поэтому полученная фазовая диаграмма является диаграммой типа AL. Для γ-ЦД, ГП-βЦД и СМβЦД получается диаграмма типа Ар. Рассчитанная константа устойчивости, равная 346 М-1, показывает, что полость в ЦД является слишком большой, чтобы обеспечить достаточные взаимодействия.
Растворимости соединения I в закомплексованной и незакомплексованной формах являются разными. Например, соединение I обладает хорошей растворимостью в ацетонитриле (±700 мкг/мл), а ГП-β-ЦД и комплекс соединение I-ЦД нерастворимы в этом растворителе. При этих условиях лекарственное средство остается включенным и становится нерастворимым в этом растворителе. Это различие растворимостей соединения I в свободной и закомплексованной форме можно использовать для определения степени комплексообразования.
Пример 3
Растворимость разных триоксопиримидинов в присутствии ГПβЦД
Растворимость исследуют в соответствии с примерами 1 и 2. Результаты приведены в таблице 7.
Пример 4
Исследования фазовых диаграмм растворимости при использовании раствора L-лизина в качестве вспомогательного вещества
Исследования растворимости проводили по методике, описанной в публикации Higuchi, Т. and Connors, K.A., Advances in Analytical Chemistry and Instrumentation 4 (1965) 117-212. Избыточные количества соединения I прибавляли к растворам ГП-β-ЦД увеличивающихся концентраций (0-200 мМ) в 5 мл растворяющей среды - очищенной воды или растворов L-лизина (50 или 500 мМ). Стеклянные емкости закрывали и суспензии встряхивали на водяной бане при 25°С до установления равновесия комплексообразования (7 дней). Аликвоты фильтровали через мембранный фильтр с отверстиями размером 0,45 мкм, изготовленный из ПВДФ, и содержание соединения I определяли с помощью аттестованной методики жидкостной хроматографии (ЖХ).
На фиг.7 приведена фазовая диаграмма растворимости для соединения I, полученная при 25°С в присутствии ГП-β-ЦД в очищенной воде, в 50 мМ растворе L-лизина и в 500 мМ растворе L-лизина. В этих трех случаях растворимость соединения I в воде увеличивается при увеличении концентрации ЦД. Диаграмма растворимости, полученная при отсутствии L-лизина, подтверждает отмеченный ранее результат: растворимость соединения I в 200 мМ растворе ГП-β-ЦД составляет примерно 5,5 мг/мл (11 мМ), что соответствует примерно 10000-кратному увеличению растворимости соединения I в воде.
В присутствии L-лизина растворимость соединения I в растворах ГП-β-ЦД является еще более высокой. Растворимость в 200 мМ растворе ГП-β-ЦД увеличивается примерно в 2 и 7 раз в присутствии 50 мМ и 500 мМ раствора L-лизина соответственно. В таблице 8 приведены данные по растворимости соединения I в разных средах. Результаты свидетельствуют о синергетическом эффекте при одновременном присутствии L-лизина и ГП-β-ЦД. Растворимость в присутствии и 500 мМ L-лизина, и 200 мМ ГП-β-ЦД (38,14 мг/мл) является большей, чем ожидаемая при суммировании влияния ГП-β-ЦД и L-лизина по отдельности (5,53 мг/мл и 0,09 мг/мл). Этот синергетический эффект L-лизина и ГП-β-ЦД обеспечивает значительное увеличение растворимости соединения I в воде (70000-кратное при присутствии 500 мМ L-лизина и 200 мМ ГП-β-ЦД).
Пример 5
Исследования с помощью ЯМР
Готовили растворы ДИМЕ-β-ЦД в D2O концентрации 10 мМ. Поскольку растворимость соединения I в воде является слишком низкой, снять спектры только соединения I в D2O невозможно. Для отнесения сигналов протонов снимали спектры ЯМР соединения I в ДМСО. Все спектры ЯМР протонов снимали на спектрометре Bruker DRX500 на частоте 500 МГц. Температура установлена равной 298 К. Калибровку проводили по сигналу остатка растворителя и в качестве вторичного стандарта использовали HDO. При экспериментах T-ROESY использовали время смешивания, равное 300 мс. Результаты обрабатывали на компьютерной станции Silicon Graphics INDY с использованием программы WINNMR, выпускающейся фирмой Broker. Сопоставление спектра ЯМР чистого соединения I и в присутствии избытка ДИМЕβЦД показало, что сигналы протонов Н-3 и Н-5 испытывают сильнопольный сдвиг. Этот сдвиг является доказательством образования включения. Анализ спектров T-ROESY показал, что соединение I включено в полость ЦД. В полость ЦД могут входить две разные части молекулы.
Пример 6
Исследования с помощью молекулярного моделирования
Расчеты молекулярных моделей проводили с помощью программы Gaussian 94 с использованием для β-ЦД кристаллографической структуры POBRON, взятой из базы данных Cambridge Data Base. Расчеты проведены для двух предельных пространственных конформаций соединения I. Полученные результаты показали, что включение энергетически возможно и что структура с включением обладает высокой стабильностью. Стабильность можно объяснить образованием водородных связей между атомом кислорода и протоном атома азота барбитуратных ядер и молекулами спирта, расположенными снаружи от ЦД. Все фармацевтические композиции, включающие соединение I и циклодекстрин (предпочтительно - βЦД, γЦД и их синтетические производные), как в виде комплексов, так и в виде ассоциатов, включены в объем настоящего изобретения независимо от формы и их терапевтического применения. В действительности, если соединение I и циклодекстрины в препарате не находятся в виде комплекса, то он образуется in situ.
Пример 7
Фармацевтические композиции
Ниже в качестве неограничивающих примеров описаны составы препаратов.
Предпочтительным примером препарата для инъекции является следующий:
- ГП-βЦД - 200 мМ; соединение 1-1 мг/мл; стерильная вода для инъекции - сколько требуется.
Для 25 мл раствора:
a) Приготовление раствора:
Отвешивают 6,77 г ГПβЦД (4,2% H2O) и растворяют в 25 мл воды для инъекции. Прибавляют 25 мг соединения I и нагревают на водяной бане до полного растворения последнего. Раствор стерилизуют фильтрованием.
b) Характеристики раствора:
Осмолярность раствора равна 308 мОс/кг. Значение рН равно 7,2. Концентрации соединения I и/или ЦД при необходимости можно изменить. Предпочтительно регулировать тоничность путем прибавления NaCl. Предпочтительным препаратом для распыления является:
Для 200 мл раствора:
- Соединение I - 0,1 г (ММ: 485)
- Апирогенный ГПβЦД - 20,15 г (ММ: 1300)
- Хлорид натрия -1,42 г (для обеспечения изотоничности)
- Апирогенная, стерильная, очищенная вода - сколько требуется до 200 мл.
a) Отвешивают 20,15 г апирогенного ГПβЦД (3,2% Н2О, ROQUETTE) и растворяют в 100 мл очищенной воды.
b) Отвешивают 0,1 г соединения I и 1,42 г хлорида натрия и прибавляют к раствору (а) при энергичном перемешивании для его растворения.
c) Прибавляют количество воды, необходимое для получения 200 мл раствора.
d) Стерилизуют путем фильтрования через полипропиленовую мембрану с отверстиями размером 0,22 мкм.
Пример 8
Фармакокинетические исследования биологической доступности
Растворы для фармакокинетических исследований готовили с использованием комбинации ГП-β-ЦД и L-лизина, что позволяло обеспечить высокую концентрацию соединения I, при биологически совместимом значении рН.
Приготовление дозированных форм
Раствор для внутривенного введения соединение I/ГП-β-ЦД готовили растворением соединения I (10 мг/мл) в растворе, содержащем ГП-β-ЦД (200 мМ), L-лизин (20 мМ) и воду для инъекции. Значения осмоляльности (примерно 325 мОсм/кг) и рН (примерно 8,2) этого раствора соответствуют требованиям, предъявляемым к растворам для инъекции. Раствор стерилизовали путем пропускания через стерильный фильтр из ацетилцеллюлозы с отверстиями размером 0,20 мкм при асептических условиях.
Раствор для перорального введения соединение I/ГП-β-ЦД готовили растворением соединения I (15 мг/мл) в растворе, содержащем ГП-β-ЦД (200 мМ), L-лизин (50 мМ) и воду.
Суспензия соединения I состояла из соединения I (15 мг/мл), полисорбата 80 (0,1 мг/мл) в качестве смачивающего агента, симальдрата (VEEGUM HV®, 1% мас./об.) и метилцеллюлозы (METHOCEL A400®, 0,4% мас./об.) в качестве загущающих агентов.
Схема эксперимента на животных и введения лекарственного средства
В качестве подопытных животных использовали 6 здоровых овец (2 самцов и 4 самок), обладающих массой тела, равной от 45 до 82 кг. Во время исследования корм и воду животным предоставляли без ограничения.
Исследование, проведенное по схеме, приведенной в таблице 9, являлось рандомизированным двухпараметрическим перекрестным с пероральным введением с последующим внутривенным введением. Перед каждым введением проводилась фаза выведения длительностью в 3 недели.
В случае пероральных дозированных форм каждое животное получало соединение I в дозе, равной 15 мг/(кг массы тела), в виде обоих препаратов. Для подбора доз овец взвешивали в день введения лекарственного препарата. Пробы крови брали из яремной вены перед пероральным введением и через 0,25, 0,5, 1, 1, 5, 2, 3, 4, 6, 8, 10, 12, 24, 28, 32, 48, 72, 96, 120, 144, 168 ч после него.
В случае внутривенной дозированной формы все 6 овец получали 5 мг соединения 1/(кг массы тела). Растворы вводили в левую яремную вену, а пробы крови брали из правой яремной вены перед внутривенным введением и через 5, 10, 15, 20, 30, 45 мин, 1, 1, 5, 2, 3, 4, 5, 6, 8, 10, 12, 24, 28, 32, 48, 72, 96, 120, 144, 168 ч после его начала.
Все пробы крови центрифугировали с до проведения анализа сыворотку хранили при - 80°С.
Методика биологического анализа
Для определения содержания этого соединения в сыворотке с помощью ЖХ разработана полностью автоматическая методика. Очистку образца проводили путем присоединения форколонки, заполненной материалом с ограниченной доступностью (МОД), а именно LiChrospher RP-8 ADS (диоксид кремния с привитыми алкильными и диольными цепями), к аналитической колонке и с использованием схемы переключения колонок. Сорбенты ADS относятся к группе сорбентов с обращенной фазой на внутренней поверхности и успешно применялись для очистки биологических образцов перед проведением анализа с помощью ЖХ (Yu, Z., and Westerlund, D., Chromatographia 44 (1997) 589-594; Hubert, Ph., et al., S. T. P. Pharma Pratiques 9 (1999) 160-180; Souverain, S., et al., Journal of Chromatography В 801 (2004) 141-156). Условия проведения эксперимента описаны в предыдущей публикации (Chiap P., et al., Journal of Chromatography В 817 (2005), 109-117). Проведена полная проверка достоверности методики в соответствии с новым подходом, основанным на профилях точности с учетом полной погрешности измерения (Hubert, P., et al., Analytica Chimica Acta 391 (1999) 135-148; Hubert, Ph., et al., S. T. P. Pharma Pratiques 13 (2003) 27-64; Hubert, Ph., et al., J. Phann. Biomed. Anal. 36 (2004) 579-586.
Ввиду необходимости определения высоких концентраций для проведения биологического анализа диапазон концентраций методики расширен до 50 мкг/мл. Проведена частичная повторная проверка достоверности и получены хорошие результаты для функции отклика, истинности, точности, правильности и линейности.
Фармакокинетика и статистический анализ
При исследовании внутривенного введения фармакокинетические параметры определяли для каждого животного с использованием линейной двухкамерной модели с распределением и выведением первого порядка (Boroujerdi, M., Pharmacokinetics, Principles and Applications. McGrow-Hill Companies, USA, 2002). Значения площади под кривой (ППК0-168) рассчитывали по линейной формуле трапеций для периода отбора проб. Значения ППК, экстраполированные на бесконечность (ППК0-∞), общего клиренса (C1t), периода биологического полувыведения (T1/2β) и общего объема распределения (Vdt) рассчитывали по обычным уравнениям камерного анализа (Boroujerdi, M., Pharmacokinetics, Principles and Applications. McGrow-Hill Companies, USA, 2002).
При исследовании перорального введения фармакокинетические параметры определяли для каждого животного при использовании и суспензии, и раствора с помощью линейной однокамерной модели с распределением и выведением первого порядка (Boroujerdi, M., Pharmacokinetics, Principles and Applications. McGrow-Hill Companies, USA, 2002). Значения ППК0-168 рассчитывали, как это описано выше, суммированием по формуле трапеций. Значения ППК0-∞ определяли по следующему уравнению (уравнение I):

где К и ka соответственно обозначают общую константу скорости выведения и константу всасывания и Со обозначает концентрацию, экстраполированную на начальное значение.
Максимальные концентрации лекарственного средства в плазме (Cmax) и соответствующие времена (Tmax) для каждого животного определяли двумя разными способами: непосредственно по зависимостям концентрация-время (Cmax эксперим. и Tmax эксперим.) и расчетом по следующим уравнениям (уравнения 2 и 3) (Cmax рассчитанное и Tmax рассчитанное):


Абсолютную биологическую доступность (Fабсолютное) рассчитывали по следующему соотношению (уравнение 4):

где Dпероральн и DBB обозначают количества лекарственных средств, введенных перорально и ВВ соответственно.
Все фармакокинетические параметры приведены в виде средних значений ± стандартные отклонения за исключением абсолютной биологической доступности, рассчитанной по средней ППК0-∞.
Данные считали ошибочными, если отдельное значение ППК было больше или меньше, чем среднее значение ±2 стандартных отклонения. На основании этого одyа овца была исключена из схемы определения фармакокинетических параметров после перорального введения и из статистического анализа.
Сопоставление фармакокинетических параметров для этих двух пероральных дозированных форм проводили с помощью двухфакторного дисперсионного анализа (двухфакторного ANOVA). После логарифмического преобразования, проводимого для нормировки распределения, сопоставляли средние значения всех рассчитанных параметров. Результаты считали статистически значимыми при 5% критическом уровне (р<0,05).
Фармакокинетика соединения I после внутривенного введения
Зависимость средней концентрации соединения I в сыворотке от времени, полученная после однократного внутривенного введения овце раствора (5 мг/кг), приведена на фиг.9а. Фиг.9b (зависимость логарифма средней концентрации соединения I в сыворотке от времени) показывает, что фармакокинетика соединения I описывается двухкамерной моделью. Различные фармакокинетические параметры, рассчитанные после этого внутривенного введения, приведены в таблице 10.
Фаза распределения является непродолжительной (примерно 30 мин) и показывает, что соединение I быстро распределяется в организме. Общий объем распределения невелик (примерно 8 л), что показывает, что распределение соединения I происходит только во внеклеточных жидкостях и что диффузия соединения I в ткани должна быть не очень существенной. С другой стороны, период биологического полувыведения соединения I является длительным (примерно 15,5 ч) и поэтому выведение лекарственного средства является очень медленным. С учетом этого небольшого объема распределения накопление к организме обусловлено не сохранением, например, в жире, а может быть обусловлено сильным связыванием белками или другими компонентами плазмы. Также рассчитано значение общего клиренса, найденное равным примерно 358,5 мл/ч.
Фармакокинетика соединения I после перорального введения суспензии и раствора
Зависимости средней концентрации соединения I в сыворотке от времени, полученные после перорального введения одной дозы (15 мг/кг) раствора и суспензии соединения I, приведены на фиг.10а. После логарифмического преобразования средней концентрации в сыворотке представляется, что фармакокинетика после перорального введения описывается однокамерной моделью (фиг.10b). Фармакокинетические параметры приведены в таблице 11.
Концентрация соединения I в сыворотке после введения раствора явно выше, чем концентрация, полученная при такой же дозе, введенной в виде суспензии. Фаза всасывания, наблюдающаяся для раствора (примерно 4 ч), короче, чем после введения суспензии (примерно 10 ч). Также можно видеть, что фармакокинетические параметры раствора и суспензии значимо различаются (р<0,05) (таблица 11). Средние пиковые концентрации соединения I в сыворотке равны примерно 54 и 5 мкг/мл после введения раствора и суспензии соответственно. Значение Cmax для раствора примерно в 10 раз больше значения для суспензии. Для раствора получено в три раза меньшее значение Tmax(примерно 3,8 ч), чем для суспензии (примерно 11 ч). Значения ППК подчиняются такой же зависимости, как и значения Cmax: значения ППК после введения раствора являются примерно в 10 раз большими, чем значения после введения суспензии. Вследствие этого после сопоставления с ВВ раствором обнаруживается, что абсолютная биологическая доступность в случае раствора (80%) больше, чем в случае суспензии (8%).
Пример 9
Исследования in vivo (подавление ангиогенеза)
Для изучения возможного воздействия комплекса соединение I-циклодекстрин использовали модель реваскуляризации. Кольцо аорты разрезали и помещали в культуральную среду. Эта культуральная среда содержала одно из следующих:
- не содержала активное действующее вещество
- содержала комплекс соединение 1-циклодекстрины (конечная концентрация равна 10-6 М, 10-7 М)
- содержала соединение I, растворенное в ДМСО (конечная концентрация равна 10-6, 10-7 М).
При отсутствии ингибитора матриксной металлопротеиназы, соединения I, наблюдается образование новых сосудов (ангиогенез). В присутствии только соединения I, растворенного в ДМСО, или в форме комплекса включения в циклодекстрин ангиогенез значительно подавляется.
Пример 10
Применение композиций, содержащих соединение I и ГПβЦД, для лечения вызванного аллергеном воспаления дыхательных путей и гиперчувствительности бронхов с помощью экспериментальной модели астмы на мышах
Материалы
Использовали ГП-β-ЦД (степень замещения = 0,64), выпускающийся фирмой Roquette (France). Использовали апирогенный забуференный фосфатом физиологический раствор (ЗФФ), выпускающийся фирмой Bio-Wittaker (Verviers, Belgium), и метахолин, выпускающийся фирмой Sigma-Aldrich (Germany). Все остальные материалы являлись чистыми для анализа. Во всем исследовании использовали стерильную воду для инъекций. Готовили стерильные, апирогенные и изотонические растворы ЦД концентрации 20, 50 и 75 мМ. Имеющийся в продаже раствор флутиказона для ингаляций (Flixotide® 1 мг/мл) приобретали у фирмы Glaxo-Smithkline (Genval, Belgium)
Сенсибилизация, воздействие аллергена и схемы лечения
Для изучения изменения воспаления дыхательных путей после внутрибрюшинной инъекции соединения I мышей сенсибилизировали с помощью 10 мкг овальбумина, адсорбированного на оксиде алюминия (aluminject, perbio, Erembodegem, Belgium), вводимого внутрибрюшинно в дни 0 и 7, и затем на них воздействовали аэрозолями овальбумина (ОВА) 1% или ЗФФ в течение 30 мин в дни от 21 до 24. Внутрибрюшинные инъекции выполняли за 30 мин до ингаляций ОВА. Использовали следующие препараты для инъекций: кремофор 10%-ДМСО 10%-ЗФФ 80%-соединение I 30 мг/кг (суспензия); кремофор 10%-ДМСО 10%-ЗФФ 80%-соединение I 3,75 мг/кг (раствор); ГПβЦД 200 мМ - соединение I 7,5 мг/кг (раствор); ГПβЦД 200 мМ. Все результаты сопоставляли с результатами, полученными для мышей, сенсибилизированных с помощью ОВА и подвергнутых воздействию ЗФФ и ОВА, которых лечили с помощью ЗФФ путем внутрибрюшинных инъекций. Для изучения изменения воспаления дыхательных путей после ингаляции соединения I мышей сенсибилизировали так, как описано выше. Использовали две схемы, называемые пробой с кратковременным воздействием и пробой с длительным воздействием. В случае пробы с кратковременным воздействием на мышей в дни от 21 до 27 в течение 30 мин воздействовали аэрозолями комплекса соединения I при концентрациях активного соединения в водном растворе, равных 0,03 и 0,3 мг/мл; мыши находились в камере для воздействия, изготовленной из плексигласа (30×20×15 см). В дни от 23 до 27 через 30 мин после ингаляции соединения I на мышей воздействовали аэрозолями ОВА. В случае так называемой пробы с длительным воздействием на мышей в течение 30 мин воздействовали аэрозолями соединения I при концентрациях в водном растворе, равных 0,03 и 0,3 мг/мл, в комплексе с ГПβЦД в течение 5 дней в нечетные недели и аэрозолями ОВА в течение 3 дней в нечетные недели в течение 11 недель. В четные недели ингаляции не проводили.
Аэрозоли вырабатывали с помощью ультразвукового распылителя SYSTAM (Système Assistance Medical, Le Ledat, France) при частоте колебаний, равной 2,4 МГц при различных интенсивностях колебаний и объемах вентиляции. Интенсивность колебаний была установлена постоянной в позиции 6 и объем вентиляции составлял 25(ν1/2) л/мин.
Исследование чувствительности дыхательных путей
Через 24 ч после последнего воздействия аллергена гиперчувствительность бронхов определяли путем измерения значения параметра Penh с помощью барометрического плетизмографа по методике, предложенной в публикации Hamelmann, E., et al., Am. J Respir. Crit. Care Med. 156 (1997) 766-775). Значение параметра Penh измеряли в исходном состоянии и через 5 мин после ингаляции увеличивающихся доз (25, 50, 75 и 100 мМ) метахолина (Mch).
Бронхоальвеолярный лаваж (БАЛ) и гистологическое исследование
Сразу же после исследования чувствительности дыхательных путей мышей умерщвляли и 1 мл ЗФФ, не содержащего ионов кальция и магния, но с прибавлением 0,05 мМ раствора натриевой соли ЭДТК (этилендиаминтетрауксусная кислота) по каплям 4 раза вливали через трахеальную канюлю и извлекали путем проводимого вручную осторожного отсасывания. Извлеченную после бронхоальвеолярного лаважа жидкость (БАЛ) центрифугировали (1800 оборотов/мин в течение 10 мин при 4°С). Таблетку клеток дважды промывали и затем повторно суспендировали в 1 мл ЗФФ. Полное количество клеток подсчитывали в камере Thoma и количества разных клеток в образцах, содержащих не менее 400 клеток, определяли для цитоцентрифугированных препаратов (Cytospin 2; Cytospin, Shandon td., Runcorn, Cheshire, U.K.) с использованием стандартных морфологических критериев после окрашивания с помощью Diff-Quick (Dade, Germany). После БАЛ вскрывали грудную клетку и пережимали левый главный бронх. Левое легкое вырезали и сразу же замораживали в жидком N2 для химического анализа белка и экстракции мРНК, а правое легкое обрабатывали для проведения гистологического исследования. По описанной ранее методике (Cataldo, D.D., et al. Am. J. Pathol. 161 (2002) 491-498) правое легкое заполняли 4% параформальдегидом и помещали в парафин. Срезы толщиной 5 мкм для всех долей окрашивали гематоксилином и эозином. Степень перибронхиальной инфильтрации оценивали с помощью показателя воспаления. Срезы кодировали и перибронхиальное воспаление оценивали по слепой схеме с использованием воспроизводимой системы показателей, описанной ранее (Cataldo, D.D., et al., Am. J. Pathol. 161 (2002) 491-498). Каждый исследованный срез ткани оценивали с помощью показателей, равных от 0 до 3. Значение 0 присваивали в случае, когда воспаление не обнаруживалось, значение 1 - в случае, когда вокруг бронхов иногда скапливались воспалительные клетки, значение 2 - в случае, когда большинство бронхов было окружено тонким слоем (1-5 клеток) воспалительных клеток, и значение 3 - в случае, когда большинство бронхов было окружено толстым слоем (>5 клеток) воспалительных клеток. После того как для каждой мыши исследовали 5-7 случайным образом выбранных срезов тканей, показатели воспаления можно было представить в виде средних значений для каждого животного и можно было сопоставить группы. Другой показатель, показатель инфильтрации эозинофилов в ткань, который характеризует количество эозинофилов, инфильтрованных в стенки бронхов, определяли следующим образом: после окрашивания конго красным исследовали по 7 бронхов для каждой мыши. Подсчитывали количество эозинофилов вокруг бронхов вплоть до стенок дыхательных путей, измеряли периметр эпителиальной базальной мембраны и результаты представляли в виде количества эозинофилов в пересчете на 1 мм периметра базальной мембраны. Левое легкое мгновенно замораживали в жидком азоте и измельчали с помощью прибора Mikro-Dismembrator S (Braun Biotech International, Melsungen, Germany) и до исследования экстракты хранили при -80°С. Почки вырезали, помещали в парафин, срезы толщиной 5 мкм окрашивали гематоксилином и эозином. Пробы крови отбирали путем пункции сердца и до проведения анализов сыворотку хранили при -80°С.
Все исследования in vivo были утверждены местным Ветеринарным комитетом по этике.
Внутрибрюшинная инъекция соединения I
По данным БАЛ внутрибрюшинная инъекция соединения I (раствора или осадка) при дозах, равных от 3,75 до 30 мг/кг, по сравнению с плацебо приводила к уменьшению вызванного аллергеном эозинофильного воспаления дыхательных путей (фиг.4а). При таких же дозах соединения I и с одинаковой эффективностью для всех исследованных препаратов также значительно уменьшались показатели перибронхиального воспаления (фиг.4b). Показатель инфильтрации эозинофилов в ткань значительно уменьшался после внутрибрюшинной инъекции соединения I при дозах, равных 7,5 и 25 мг/кг.
Ингаляционное воздействие соединения I и комплексов соединение I-ГПβЦД
Внутреннюю активность соединения I сначала оценивали, как активность противовоспалительного средства местного действия путем исследования раствора соединения I концентрации 40 мг/мл в чистом ДМСО при кратковременном воздействии. При сопоставлении с ингаляцией чистого ДМСО ингаляция этого препарата приводила к значительному уменьшению количества эозинофилов, определенного с помощью БАЛ (р<0,005), показателей перибронхиального воспаления (р<0,01), а также гиперчувствительности бронхов (р<0,05).
При исследовании с помощью кратковременного воздействия мы оценивали воздействие препаратов, содержащих комплексы соединения I с ГП-β-ЦД, на воспаление дыхательных путей и гиперчувствительность. Воздействие ингаляции препаратов, содержащих комплексы соединение 1-ГП-β-ЦД, сопоставляли с воздействием плацебо (ЗФФ) или флутиказон (1 мг/мл), применявшегося в качестве эталонного средства. Ингаляция таких препаратов, содержащих соединение I в дозах, равных 0,03 и 0,3 мг/мл, по данным БАЛ по сравнению с плацебо приводила к значительному уменьшению эозинофильного воспаления в степени, сравнимой со степенью для флутиказона (р<0,0001) (фиг.5а). По сравнению с плацебо также уменьшались показатели перибронхиального воспаления (р<0,0001) (фиг.5b), а также показатель инфильтрации эозинофилов в ткань (р<0,01) (фиг.5с).
После длительного воздействия эозинофилия, вызванная воздействием аллергена, по данным БАЛ значительно уменьшилась после лечения путем ингаляции препаратов, содержащих комплексы соединение 1-ГП-β-ЦД (р<0,001) и в такой же степени, как после лечения флутиказоном (фиг.6а). Показатель перибронхиального воспаления также значительно уменьшился после ингаляции препаратов, содержащих комплексы соединение 1-ГП-β-ЦД, а также флутиказона (р<0,0001) (фиг.6b). Показатель инфильтрации эозинофилов в ткань также уменьшился после лечения путем ингаляции соединения I в степени, сравнимой со степенью для случая лечения мышей флутиказоном (р<0,01) (фиг.6с).
Список литературы
Bergers, G., et al., Nat. Cell Biol. 2 (2000) 737-744
Boroujerdi, M., Pharmacokinetics, Principles and Applications. McGrow-Hill Companies, USA, 2002
Carmeliet, P., et al., Nat. Genet. 17 (1997) 439-444 Carstanjen, D., et al., Transfusion 42 (2002) 588-596
Cataldo, D.D., et al., Am. J. Pathol. 161 (2002) 491-498
Chang, С., and Werb, D., Trends Cell Biol. 11 (2001) S37-43
Chiap, P., et al., Journal of Chromatography В 817 (2005), 109-117
Dong, Z., et al., Cell 88 (1997) 801-810
Egeblad, M., and Werb, Z., Nat. Rev. Cancer 2 (2002) 161-174
EP 0869947
Fabbri, L.M., and Hurd, S.S., Eur. Respir. J. 22 (2003) 1-2
GINA Workshop Report, Global Strategy for Asthma Management and Prevention (NIH Publication No. 02-3659)
Grams, F., et al., Biol. Chem. 382 (2001) 1277-1285
Hamelmann, E., et al., Am. J Respir. Crit. Care Med. 156 (1997) 766-775
Higuchi, Т., and Connors, K.A., Advances in Analytical Chemistry and Instrumentation 4 (1965) 117-212
Holmbeck, К., et al., Cell 99 (1999) 81-92
Hubert, P., et al., Analytica Chimica Acta 391 (1999) 135-148
Hubert, Ph., et al., J. Pharm. Biomed. Anal. 36 (2004) 579-586
Hubert, Ph., et al., S. Т. P. Pharma Pratiques 9 (1999) 160-180
Hubert, Ph., et al., S. T. P. Pharma Pratiques 13 (2003) 27-64
Lund, L.R., et al., EMBO J. 18 (1999) 4645-4656
Manes, S., et al., J. Biol. Chem. 274 (1999) 6935-6945
Overall, C.M., and Lopez-Otin, С., Nat. Rev. Cancer 2 (2002) 657-672
Remington's Pharmaceutical Sciences, 16th ed., 1980, Mack Publishing Co., edited by Oslo et al.
Shapiro, S.D., Curr. Opin. Cell Biol. 10 (1998) 602-608
Souverain, S., et al., Journal of Chromatography В 801 (2004) 141-156
US 6110924
US 6242455
Vu, Т.Н., et al., Cell 93 (1998) 411-422
WO 01/25217
WO 97/23465
WO 98/58915
Yu, Z., and Westerlund, D., Chromatographia 44 (1997) 589-594
Реферат
Изобретение относится к области фармакологии и медицины и касается комплекса триоксопиримидин-циклодекстрин, образованного из производного триоксопиримидина или его соли и растворимого в воде производного циклодекстрина, обладающего улучшенной растворимостью, и фармацевтической композиции на его основе, являющейся ингибитором матриксных металлопротеаз. 2 н.з. и 8 з.п. ф-лы, 9 ил., 11 табл.
Формула

в которой R1 обозначает группу W-V, в которой
W обозначает фенил; и
V обозначает фенил, фенилоксигруппу, фенилтиогруппу, фенилсульфинил, фенилсульфонил или фениламиногруппу, и эти фенильные фрагменты могут быть незамещенными или один или несколько раз замещенными галогеном; и
R2 обозначает фенильную или гетероарильную группу, которые являются незамещенными или один или несколько раз замещенными нитрогруппой.
5-(4-феноксифенил)-5-(4-пиримидин-2-илпиперазин-1-ил)-пиримидин-2,4,6-трион;
5-[4-(4-хлорфенокси)-фенил]-5-(4-пиримидин-2-илпиперазин-1-ил)-пиримидин-2,4,6-трион;
5-[4-(3,4-дихлорфенокси)-фенил]-5-(4-пиримидин-2-илпиперазин-1-ил)-пиримидин-2,4,6-трион;
5-[4-(4-бромфенокси)-фенил]-5-(4-пиримидин-2-илпиперазин-1-ил)-пиримидин-2,4,6-трион
или его соль.
Комментарии