Фармацевтические препаративные формы для ингаляторов сухого порошка, содержащие низкодозовый активный ингредиент - RU2371171C2
Код документа: RU2371171C2
Описание
Настоящее изобретение относится к сухой порошковой препаративной форме для введения в виде сухого порошка для ингаляций, подходящего для эффективного введения низкодозовых лекарственных средств в нижние дыхательные пути пациента. В частности, в изобретении предлагается сухая порошковая препаративная форма для ингаляций, свободнотекучая, физически и химически устойчивая и подходящая для доставки как точных доз, так и пригодных для вдыхания фракций частиц низкодозовых активных ингредиентов, а также способ ее получения.
Предпосылки изобретения
Введение фармакологически активных агентов ингаляцией является широко используемой методикой, особенно для лечения обратимой обструкции дыхательных путей, воспаления и гиперчувствительности. Эта методика также используется для введения определенных активных агентов, имеющих системное действие, которые всасываются через легкие, в кровоток.
Некоторые из наиболее широко используемых систем для введения лекарственных средств в дыхательные пути представляют собой ингаляторы сухого порошка (ИСП).
ИСП могут быть разделены на два основных типа:
i) однодозовые ингаляторы для введения предварительно разделенных разовых доз активного соединения;
ii) многодозовые ингаляторы сухого порошка (МДИСП) либо с предварительно разделенными разовыми дозами, либо с предварительно загруженным количеством активного ингредиента, достаточным для многократного введения; каждая доза создается дозатором внутри ингалятора.
На основе необходимых скоростей потока инспирации (л/мин), которые, в свою очередь, строго зависят от их конструкции и механических особенностей, ИСП также делятся на:
i) устройства с низким сопротивлением (>90 л/мин);
ii) устройства со средним сопротивлением (приблизительно 60 л/мин);
iii) устройства с высоким сопротивлением (приблизительно 30 л/мин).
Сообщаемые скорости потока относятся к падению давления на 4 кПа (килопаскаля) по требованию European Pharmacopoeia (Eur Ph), 4th Ed 2004, страница 3375.
Лекарственные средства, предназначенные для ингаляции в виде сухих порошков, должны использоваться в форме микронизированного порошка, таким образом, они характеризуются частицами с размером частиц в несколько микрон (мкм). В порошках для ингаляции первостепенное значение имеет оценка размера частиц как активного ингредиента, так и возможных наполнителей или носителей. Размер частиц определяют количественно путем измерения характерного диаметра эквивалентной сферы, известного как массовый диаметр (МД) или объемный диаметр (ОД), в зависимости от используемой методики. Распределение размера частиц описывается массовым средним диаметром (МСД) или объемным средним диаметром (ОСД), которые соответствуют диаметру 50 процентов частиц по массе или объему соответственно. ОСД связан с МСД через плотность частиц (принимая независимую от размера плотность частиц). В случае активных ингредиентов для целей ингаляции размер частиц также выражается как массовый аэродинамический диаметр (МАД), а распределение размера частиц - как массовый средний аэродинамический диаметр (МСАД). МАД указывает на способность частиц переноситься взвешенными в воздушном потоке. МСАД соответствует массовому аэродинамическому диаметру 50 процентов частиц по массе. В известном уровне техники термин МСАД также неправильно использовался, чтобы количественно определять диаметр частиц носителя. Частицы активного ингредиента должны иметь такой размер частиц, чтобы достигнуть нижних дыхательных путей. Как правило, пригодными для вдыхания считаются частицы с МАД от 0,5 до 10 микрон, поскольку они в состоянии проникнуть в нижние дыхательные пути, то есть участки бронхиол и альвеол, которые служат местом действия для легочных лекарственных средств и где имеет место всасывание системных лекарственных средств. Большие частицы, главным образом, оседают в полости рта и глотки, таким образом, они не могут достигнуть указанных участков, тогда как меньшие частицы, как полагают, выдыхаются.
Ниже термины «лекарственное средство», «активный ингредиент», «активный» и «активное вещество» используются как синонимы.
Хотя микронизация лекарственного средства существенна для оседания в нижних дыхательных путях во время ингаляции, также известно, что чем мельче частицы, тем сильнее когезия. Сильная когезия препятствует обработке порошка во время производственного процесса (ссыпка, заполнение). Кроме того, она уменьшает текучесть частиц, что в многодозовых ИСП способствует агломерации и прилипанию к стенкам. Указанные явления ухудшают загрузку порошка из резервуара в дозированную камеру. Таким образом, сильная когезия создает проблемы при обработке и точном отмеривании.
Слабая текучесть тоже вредна для вдыхаемой фракции вводимой дозы, поскольку активные частицы не способны покинуть ингалятор и остаются прилипшими к внутренним стенкам ингалятора либо покидают ингалятор в виде крупных агломератов; агломерированные частицы, в свою очередь, не могут достигнуть участков бронхиол и альвеол легких. Неопределенность как в степени агломерации частиц между каждым приведением ингалятора в действие, так и между ингаляторами и различными партиями частиц, тоже приводит к слабой воспроизводимости дозы.
Уровень техники
В известном уровне техники, например в WO 95/09615, один возможный способ улучшения свойств текучести порошков для ингаляции заключается в том, чтобы агломерировать контролируемым способом микронизированные частицы лекарственного средства, чтобы образовались агломераты или шарики относительно высокой плотности и компактности. Способ включает стадии: (i) агломерации микронизированного порошка лекарственного средства при прохождении через шнековый питатель; (ii) сферонизацию агломератов предпочтительно в наклоненном вращающемся контейнере. Способ предпочтительно дополнительно включает (iii) сортировку сферонизированных агломератов по величине.
Альтернативно, мелкие частицы микронизированного активного ингредиента были смешаны со множеством из одного или более наполнителей, таких как лактоза, с получением продукта, который был назван мягким шариком, где частицы микронизированного активного ингредиента и частиц лактозы находятся в агломерированном состоянии.
Например, в WO 95/24889 раскрыта порошковая композиция, содержащая микрочастицы лекарственного средства и по меньшей мере один шарик лактозы с диаметром от 10 до 1500 микрон, причем шарик состоит из множества микрочастиц лактозы.
В EP 441740 заявлен способ и устройство для агломерации и дозирования нетекучих порошков, предпочтительно состоящих из микронизированного формотерола фумарата и мелких частиц лактозы, с тем чтобы сформировать шарики, обладающие улучшенной текучестью. В тексте указано, что соотношение компонентов смеси - формотерола ко всей смеси - находится в пределах от 1:10 до 1:500.
В EP 441740 заявлено, что наличие добавки, такой как смазывающее вещество, является неблагоприятным, так как она формирует конгломераты, и полученные таким образом конгломераты слишком крупные, чтобы обеспечить возможность достаточно точного дозирования очень малых количеств.
В WO 98/31351 и WO 98/31352 заявлена сухая порошковая композиция, содержащая один или более активных ингредиентов и вещество носителя, оба из которых находятся в тонкоизмельченной форме, где композиция имеет насыпную плотность от 0,28 до 0,38 г/мл. Активное вещество и вещество носителя микронизируют, агломерируют и сферонизируют, пока не будет получена желательная насыпная плотность. Размер полученных агломератов предпочтительно находится в пределах от 100 до 2000 микрон, более предпочтительно от 100 до 800 микрон.
Однако мягкие шарики могут обладать столь высокой внутренней когезией, что она может помешать их распаду на мелкие частицы во время ингаляции; такой недостаток можно расценить как специфический критический этап, когда используется многодозовый ингалятор сухого порошка со средним или высоким сопротивлением. В указанных ингаляторах, действительно, меньше энергии доступно для разрушения шариков на мелкие первичные частицы активного ингредиента.
Иным способом порошки для ингаляции были приготовлены путем смешивания микронизированного лекарственного средства с носителем (как правило, физиологически приемлемым веществом, обычно лактозой или маннитом, предпочтительно моногидратом α-лактозы), состоящим из более крупных частиц, чтобы получить "интерактивные упорядоченные смеси".
Однако известно, что силы взаимодействия между частицами, которые возникают между активным ингредиентом и крупными частицами носителя в упорядоченных смесях, могут быть очень мощными, так что они препятствуют высвобождению микронизированных частиц лекарственного средства с поверхности крупных частиц во время ингаляции, что необходимо для того, чтобы частицы лекарственного средства достигли целевого участка в легких.
Поверхность крупных частиц носителя в действительности не гладкая, а имеет шероховатости и трещины, которые являются участками высокой энергии, к которым предпочтительно притягиваются активные частицы и на которых более прочно удерживаются.
Чтобы увеличить высвобождение активных частиц с поверхности крупных частиц носителя во время ингаляции, было предложено несколько подходов.
В EP 0663815 заявлено добавление более мелких частиц наполнителя (<10 микрон) к более крупным частицам наполнителя (>20 микрон) для того, чтобы регулировать количество лекарственного средства, доставленного во время образования аэрозоля, и оптимизировать его.
Подходящие наполнители представляют собой моносахариды, такие как глюкоза и арабиноза, дисахариды, такие как лактоза, сахароза и мальтоза, полисахариды, такие как декстраны, многоатомные спирты, такие как сорбит, маннит и ксилит, и соли, такие как хлорид натрия и карбонат кальция.
Доля активного ингредиента в смеси обычно очень мала, например от 0,01 до 0,1 мг активного вещества на примерно 5 мг смеси наполнителя (соответствует соотношению по массе от 1:50 до 1:500).
Соотношение между активным ингредиентом и более мелкими частицами наполнителя не упомянуто.
В WO 95/11666 описаны способы изменения поверхностных свойств частиц носителя путем устранения любых шероховатостей в форме мелких зерен без существенного изменения размеров частиц, чтобы микронизированные частицы лекарственного средства подвергались более слабому воздействию адгезии между частицами.
Частицы носителя содержат любой приемлемый фармакологически инертный материал, такой как кристаллические сахара, предпочтительно лактозу, и имеют диаметр от 50 до 1000 микрон.
Мелкие зерна имеют диаметр от 1 до 5 микрон. Кроме того, ни о каких соотношениях между активными частицами ингредиента и мелкими зернами или между мелкими зернами и частицами носителя не сообщается.
Активные частицы смешивают с частицами носителя. Авторы заявляют, что полезно прерывать смешивание и просеивать смесь частиц носителя и активных частиц, чтобы уменьшить количество присутствующих крупных агломератов.
'666 умалчивает о наличии и даже увеличении соотношения между активным ингредиентом и мелкими зернами во время просеивания.
В WO 01/78696 ('696) заявлена препаративная форма для применения в устройстве-ингаляторе, которая содержит: i) частицы носителя, имеющие МСАД по меньшей мере 175 микрон; ii) мелкие частицы наполнителя, имеющие МСАД не более 20 микрон; активные частицы.
Частицы носителя могут состоять из любого приемлемого фармакологически инертного вещества, такого как сахара, такие как лактоза.
В заявке на патент '696 не содержится общих сведений о соотношениях между активным ингредиентом и мелкими частицами наполнителя или между активным ингредиентом, мелким наполнителем и/или частицами носителя.
Примеры относятся к сальбутамола сульфату, будезониду и инсулину. Препараты из примеров были приготовлены сначала путем смешивания мелких частиц наполнителя и микронизированного активного ингредиента в соотношении 1:1 по массе в смесителе с высоким усилием сдвига в течение 5 минут, затем путем добавления указанной смеси к крупной фракции.
Ни о каких стадиях просеивания не сообщается.
WO 01/89491 и WO 01/89492 относятся к сухим порошковым композициям, содержащим микронизированный формотерол и его комбинацию с микронизированным глюкокортикостероидом, и способу их приготовления, причем указанный способ включает: i) приготовление смеси микронизированного активного ингредиента и микронизированного носителя/разбавителя, за которым следует ii) добавление дополнительного предварительно микронизированного носителя/разбавителя, который перемешивается с низкой интенсивностью, и iii) либо агломерацию и сферонизацию смеси, либо добавление крупного носителя/разбавителя.
Предпочтительно стадия iii) включает агломерацию и сферонизацию смеси.
Носитель/разбавитель предпочтительно является углеводом, более предпочтительно восстанавливающим углеводом, таким как лактоза, глюкоза.
Микронизированный носитель/разбавитель имеет средний размер частиц менее приблизительно 25 микрон, предпочтительно менее приблизительно 10 микрон, более предпочтительно менее приблизительно 5 микрон, в то время как крупный носитель/разбавитель имеет средний размер частиц более приблизительно 25 микрон. В описании указано, что смесь первого активного ингредиента (формотерола) и носителя/разбавителя по первой стадии может быть приготовлена микронизацией данных двух компонентов вместе или микронизацией каждого компонента по отдельности и затем объединением, чтобы получить микронизированную смесь. В описании нет никакого указания на критичное соотношение между активным ингредиентом и носителем/разбавителем. В примерах микронизированная смесь была затем подвергнута агломерации и сферонизации (то есть в форме мягких шариков), где соотношение между формотерола фумарата дигидратом и лактозой составляло приблизительно 1:190, 1:7696 и 1:170000 масс./масс. Указанные препаративные формы не содержат добавок. Кроме того, цель указанных препаративных форм состоит в том, чтобы устранить проблему химической стабильности формотерола фумарата в присутствии углевода (например, лактозы) и воды, а не улучшать дисперсию активного ингредиента в порошковой композиции.
WO 03/024396 относится к фармацевтическим композициям, включающим: фракцию лекарственного средства, которая содержит частицы лекарственного средства с МСАД не более приблизительно 10 микрон; и по меньшей мере 50% невдыхаемой фракции наполнителя, причем указанная невдыхаемая фракция наполнителя содержит частицы наполнителя низкой плотности, имеющие аэродинамический диаметр, превышающий приблизительно 10 микрон, и геометрический диаметр, превышающий приблизительно 30 микрон.
В дополнительном варианте осуществления изобретения фармацевтические композиции включают вдыхаемую фракцию наполнителя, содержащую частицы наполнителя с аэродинамическим диаметром не более 10 микрон. Предпочтительный наполнитель представляет собой маннит.
Примеры предусматривают стадию предварительного смешивания крупной фракции наполнителя с мелким наполнителем или фракцией лекарственного средства и последующего смешивания гомогенной первой композиции с дополнительным компонентом (то есть лекарственным средством или мелким наполнителем). В заявке на патент не упоминается о критичности соотношения между мелким наполнителем и лекарственным средством. Добавки не упоминаются.
В других документах из уровня техники сообщается о применении частиц носителя и/или наполнителя, содержащих добавку.
WO 96/23485 ('485) относится к порошку, который включает добавку на поверхности частиц носителя, что способствует высвобождению активных частиц из частиц носителя при приведении ингалятора в действие. Добавка представляет собой вещество с антиадгезивными или антифрикционными свойствами, состоящее из одного или более соединений, выбранных из аминокислот (предпочтительно лейцина), фосфолипидов, поверхностно-активных веществ или стеаратов.
Тип и размер носителя такие же, как в WO 95/11666. Фракция мелких зерен тоже может присутствовать. Фракция мелких зерен не охарактеризована, и размер зерен не определен. В заявке на патент '485 никак не упоминается критичность соотношения между активными частицами ингредиента и мелкими зернами.
WO 02/43702 относится к микрочастицам, имеющим МСАД 10 микрон или менее, для применения в фармацевтических композициях для введения в легкие, содержащим частицы активного вещества, содержащие на своей поверхности частицы гидрофобного вещества, подходящего для того, чтобы задержать растворение активного вещества. В качестве гидрофобного вещества могут использоваться различные вещества, такие как гидрофобная аминокислота, предпочтительно лейцин,
C10-C22 карбоновые кислоты и их сложные эфиры, амиды и соли, предпочтительно стеарат магния и поверхностно-активные вещества, такие как лецитин. Указанные композиции могут состоять, по существу, только из микрочастиц, или они могут содержать дополнительные ингредиенты, такие как частицы носителя и ароматизаторы.
WO 00/53157 относится к порошкам для ингаляции, содержащим активный ингредиент и частицы носителя, содержащие в качестве добавки малое количество смазывающего вещества, 0,1-0,5% по массе, предпочтительно стеарата магния.
Преимущественно частицы носителя состоят из одного или более кристаллических сахаров, предпочтительно моногидрата α-лактозы, и имеют размер частиц в пределах 20-1000 микрон, более предпочтительно в пределах 90-150 микрон. Наличие частиц носителя с размером частиц менее 20 микрон не раскрыто.
WO 00/53158 относится к способам получения носителя для порошковых препаративных форм, где мелкая фракция носителя образуется in situ. Также предусмотрены препаративные формы, содержащие частицы активного ингредиента, добавку и порошок носителя, имеющий крупную фракцию и мелкую фракцию со средним аэродинамическим диаметром менее 10 мкм.
WO 00/33789 относится к порошку для лекарственных средств, пригодных для ингаляций, который содержит активный ингредиент и порошок наполнителя, содержащий крупную первую фракции (где по меньшей мере 80% частиц по массе имеют размер частиц по меньшей мере 10 микрон), мелкую вторую фракцию (где по меньшей мере 90% частиц по массе имеют размер частиц не более 10 микрон) и третий агент, который предпочтительно является водорастворимым поверхностно-активным веществом, предпочтительно лейцином. В примерах в способах получения порошков предусматривается конечная стадия смешивания частиц носителя, состоящих из крупной фракции, мелкой фракции и лейцина, с микронизированными частицами кортикостероида.
WO 00/28979 обращается к применению малых количеств стеарата магния в качестве добавки, чтобы улучшить устойчивость сухих порошковых препаративных форм для ингаляции к влажности. Указанная препаративная форма содержит частицы носителя с МСАД 10-500 микрон, предпочтительно 50-200 микрон, и они могут также содержать фракцию частиц пригодного для вдыхания размера, например, 0,1-10% масс./масс. микронизированного носителя. Носитель является фармакологически неактивным веществом.
В тексте сообщается, что препаративные формы можно приготовить путем смешивания носителя, мелко диспергированного лекарственного средства и стеарата магния. Компоненты можно добавлять в любом порядке. В примере 2 крупнозернистый моногидрат лактозы смешивают с микронизированным моногидратом лактозы в барабанном смесителе. После этого просеивают и смешивают формотерола фумарата дигидрат и предварительную смесь просеивают. Полученную таким образом смесь смешивают со стеаратом магния. Об условиях смешивания не сообщается.
WO 01/78695 относится к препаративной форме для применения в устройстве-ингаляторе, которая содержит: i) частицы носителя, имеющие МСАД по меньшей мере 175 микрон; ii) активные частицы; iii) добавку на поверхности частиц носителя, которая способна содействовать высвобождению активных частиц из частиц носителя при приведении устройства-ингалятора в действие. Препаративная форма может дополнительно содержать мелкие частицы наполнителя с МСАД не более 50 микрон, предпочтительно не более 15 микрон.
Частицы носителя получают из любого приемлемого фармакологически инертного вещества, например сахарных спиртов, и предпочтительно они имеют поверхность с относительно большим количеством щелей. В качестве добавок могут использоваться антиадгезивный агент, скользящее вещество, аминокислоты, поверхностно-активные вещества.
Примеры относятся к сальбутамола сульфату и будезониду. Препаративные формы готовят сначала путем совместного перемешивания или совместного размола активного ингредиента и добавки (лейцина, стеарата магния и других) в шаровой мельнице, затем путем перемешивания данной фракции с крупной фракцией носителя и, в случае с мелкими частицами, наполнителя.
В WO 01/78693 заявитель раскрыл порошок для применения в ингаляторе сухого порошка, который содержит i) фракцию мелких частиц, состоящих из смеси, составленной из частиц физиологически приемлемого наполнителя и стеарата магния, причем частицы указанной смеси имеют средний размер частиц менее 35 микрон, предпочтительно менее 15 микрон; ii) фракцию крупных частиц физиологически приемлемого носителя, имеющую размер частиц по меньшей мере 100 микрон, указанная смесь (i) составлена из 90-99 процентов по массе частиц наполнителя и от 10 до 1 процента по массе стеарата магния, и соотношение между мелкими частицами и крупными частицами носителя составляет от 1:99 до 40:60 процентов по массе; указанная смесь далее смешивается с одним или более активными ингредиентами в микронизированной форме.
Частицы носителя получают из любого приемлемого фармакологически инертного вещества, например сахарных спиртов.
Данная заявка на патент также относится к способам получения порошковой препаративной формы: они все предполагают конечную стадию смешивания микронизированных частиц активного ингредиента, а также частиц носителя. В заявке на патент WO 01/78693 показано, что наличие смазывающего вещества, и в особенности стеарата магния, в препаративной форме улучшает характеристики аэрозоля, что позволяет вводить в легкие посредством ингаляции высокую дозу мелких частиц активного ингредиента.
Все вышеупомянутые документы, относящиеся к порошковым препаративным формам для ингаляции, содержащим добавку, не содержат никаких упоминаний о соотношении между мелким наполнителем или носителем и активным ингредиентом и о его значимости для различных вариантов приготовления порошковых препаративных форм.
Заявитель обнаружил, что формирование агломератов может происходить, даже когда микронизированные частицы активного ингредиента смешиваются с крупными частицами наполнителя, то есть во время диспергирования первых на поверхности последних, и что чем мельче частицы активного ингредиента, тем быстрее они стремятся к агломерации. В частности, данное явление наблюдалось, когда использовались низкодозовые активные ингредиенты со значительным количеством частиц, имеющих массовый диаметр менее 1 микрона, даже в присутствии добавки, такой как антиадгезив или смазывающее вещество, которое, как сообщают, способствует улучшению диспергирования активного ингредиента.
Термин «низкодозовые» авторы относят к активным ингредиентам, обладающим особенно высокой активностью, которые присутствуют в порошковых препаративных формах в очень низкой концентрации. Данный фактор, вместе с другими свойствами, такими как высокая адгезионная способность, приводит к проблемам при производстве композиции с хорошей воспроизводимостью доз при введении посредством ИСП.
Формирование агломератов снижает возможность достижения хорошей однородности распределения активного ингредиента в порошковой смеси, а следовательно, высокой точности дозы. Формирование агломератов особенно критично, когда используется низкодозовый активный ингредиент. Фактически, чем ниже концентрация активного ингредиента в процентах по массе к общей массе препаративной формы, тем сильнее негативное влияние агломератов на однородность активного ингредиента в порошковой смеси. Неоднородность порошка из-за формирования агломератов приводит к риску введения избыточной или недостаточной дозы.
Ввиду проблемы, указанной выше, было бы очень выгодно получить порошковую препаративную форму для доставки низкодозового активного ингредиента путем ингаляции с помощью устройства ИСП, которая обладает хорошей однородностью распределения активных частиц ингредиента и, следовательно, адекватной точностью отмеренной дозы, вместе с хорошими характеристиками с точки зрения доставленной дозы и фракции, пригодной для вдыхания.
Сущность изобретения
Техническая проблема, лежащая в основе изобретения, заключается в том, чтобы получить препаративную форму для введения в качестве сухого порошка для ингаляции, подходящую для эффективного введения низкодозовых лекарственных средств в нижние дыхательные пути пациентов. В частности, техническая проблема состоит в том, чтобы получить препаративную форму, которая будет вводиться как сухой порошок для ингаляции, свободнотекучую, физически и химически устойчивую и способную доставлять как точные дозы, так и высокопригодные для вдыхания фракции частиц низкодозовых активных ингредиентов.
Предлагаемое решение представляет собой порошковую препаративную форму для применения в ингаляторе сухого порошка, причем порошок содержит микрочастицы низкодозового активного ингредиента и микрочастицы наполнителя.
По конкретному варианту осуществления изобретения порошковая препаративная форма может дополнительно содержать частицы дополнительного носителя и/или частицы добавки.
Также предложен способ приготовления порошковой препаративной формы для эффективной доставки низкодозовых активных ингредиентов, который обеспечивает лучшую дисперсию активного вещества в порошковой препаративной форме и, следовательно, хорошую однородность, при этом избегает формирования агрегатов, обеспечивая лучшую однородность введенной дозы.
В частности, предложена порошковая препаративная форма для применения в ингаляторе сухого порошка, причем порошок содержит микрочастицы, составленные из микрочастиц низкодозового активного ингредиента и микрочастиц наполнителя, где МСД микрочастиц составляет от 2 до 15 микрон, и по меньшей мере 10% микрочастиц имеют массовый диаметр (МД), превышающий 0,5 микрона. Предпочтительно, чтобы соотношение между микрочастицами активного ингредиента и микрочастицами наполнителя составляло от 1:60 до 1:2000, предпочтительно от 1:100 до 1:1000 по массе.
В случае очень низкодозовых активных ингредиентов соотношение между активным ингредиентом и микрочастицами наполнителя составляет от 1:250 до 1:50.
Определения
Препаративная форма по изобретению содержит терапевтически активное вещество в форме микронизированного порошка (активный ингредиент) и терапевтически неактивные вещества в качестве твердых разбавителей (наполнитель и дополнительный носитель).
В следующем описании термин "наполнитель" определяет терапевтически неактивный твердый разбавитель, присутствующий в препарате в форме микрочастиц, характеризующихся МСД от 2 до 15 микрон, тогда как термины "мелкий носитель" и "крупный носитель" определяют терапевтически неактивные твердые разбавители дополнительного носителя.
Подробное описание изобретения
Характеристики препаративной формы по изобретению и способа ее получения станут более очевидными из следующего подробного описания.
Настоящее изобретение относится к порошковой препаративной форме для применения в ингаляторе сухого порошка, причем порошок содержит микрочастицы низкодозового терапевтически активного ингредиента и микрочастицы терапевтически неактивного вещества или наполнителя, указанные микрочастицы имеют МСД от 2 до 15 микрон, где МСД активного ингредиента составляет менее 10 микрон, предпочтительно менее 6 микрон, более предпочтительно от 1,5 до 4 микрон, и МСД микрочастиц наполнителя составляет от 2 до 15 микрон, и по меньшей мере 10% микрочастиц имеют массовый диаметр (МД), превышающий 0,5 микрона.
Преимущественно микрочастицы наполнителя имеют МСД от 2 до 15 микрон, предпочтительно от 2 до 10, более предпочтительно от 3 до 7 микрон и, в определенных случаях, в особенности, когда используется активный ингредиент с очень низкой дозой, номинальная доза которого равна 4 мкг или ниже, преимущественно МД 90% микрочастиц равен 60 микронам или ниже, предпочтительно равен 50 микронам или ниже, более предпочтительно равен 30 микронам или ниже. В предпочтительном варианте осуществления МД микрочастиц наполнителя составляет от 1 до 20 микрон, предпочтительно от 1 до 15 микрон, и МСД составляет от 3 до 7 микрон.
Соотношение между микрочастицами активного ингредиента и микрочастицами наполнителя составляет от 1:1 до 1:2000 по массе, предпочтительно от 1:15 до 1:1000, более предпочтительно от 1:20 до 1:500. Согласно предпочтительному варианту осуществления соотношение составляет от 1:60 до 1:2000, предпочтительно от 1:100 до 1:1000 по массе.
В случае активных ингредиентов с очень низкой дозой соотношение между активным ингредиентом и микрочастицами наполнителя составляет от 1:250 до 1:500 по массе.
Для целей изобретения низкодозовые активные агенты представляют собой такие активные ингредиенты, номинальная доза которых, введенная после каждого приведения в действие ингалятора, равна или ниже 20 мкг или 12 мкг, или 10 мкг, или 8 мкг, или 6 мкг, или 5 мкг, или 4 мкг, или 3 мкг.
Номинальные дозы активного ингредиента даже от 0,5 до 3,0 мкг можно с успехом ввести в препаративной форме по изобретению.
Частицы наполнителя могут быть составлены из любого аморфного или кристаллического физиологически приемлемого терапевтически неактивного вещества из животного или растительного источника или их комбинации; предпочтительными веществами являются кристаллические сахара и, например, моносахариды, такие как глюкоза или арабиноза, или дисахариды, такие как мальтоза, сахароза, декстроза или лактоза.
Также могут использоваться многоатомные спирты, такие как маннит, сорбит, мальтит, лактит.
Предпочтительным материалом является лактоза и более предпочтительным является моногидрат α-лактозы.
Примерами торгового моногидрата α-лактозы являются Capsulac® и Pharmatose®. Примером торгового маннита является Pearlitol®.
В зависимости от характеристик активного ингредиента и его процентного содержания в препаративной форме и вида ингалятора сухого порошка, используемого для его введения, порошковая препаративная форма по изобретению может содержать по существу только микрочастицы активного ингредиента и наполнителя, или она может содержать частицы дополнительного носителя и/или добавки.
В самом деле было обнаружено, что в определенных случаях, если микронизированные частицы активного ингредиента прежде, чем они будут разбавлены частицами дополнительного носителя, смешиваются с микронизированными частицами наполнителя в предложенном массовом соотношении, можно достичь лучшей дисперсии активного вещества в порошковой препаративной форме и, следовательно, хорошей однородности, избегая формирования агрегатов. Хорошая гомогенность, в свою очередь, приводит к лучшей однородности введенной дозы, особенно когда активный ингредиент является низкодозовым.
Гомогенную дисперсию активного ингредиента в порошке и отсутствие агрегатов активных частиц можно подтвердить, используя спектрофотометор Near Infrared, оснащенный системой получения микроскопических изображений (Near Imaging).
В предпочтительном варианте осуществления изобретения и в особенности когда препаративную форму вводят с помощью многодозового ИСП микрочастицы, состоящие из частиц низкодозового активного ингредиента и микрочастиц наполнителя, разбавляются частицами дополнительного терапевтически неактивного носителя.
Частицы дополнительного носителя могут состоять из мелких частиц, крупных частиц и их смеси.
Преимущественно частицы дополнительного носителя дополнительно содержат одну или более добавок, способствующих высвобождению активных частиц из частиц наполнителя и носителя при приведении ингалятора в действие. Предпочтительная добавка представляет собой антиадгезивное вещество или смазывающее вещество.
Мелкие и крупные частицы дополнительного носителя могут быть составлены из любого аморфного или кристаллического физиологически приемлемого терапевтически неактивного вещества животного или растительного происхождения или их комбинации. Предпочтительные вещества представляют собой кристаллические сахара и, например, моносахариды, такие как глюкоза или арабиноза, или дисахариды, такие как мальтоза, сахароза, декстроза или лактоза. Также можно использовать многоатомные спирты, такие как маннит, сорбит, мальтит, лактит.
Предпочтительным веществом является лактоза и более предпочтительным является моногидрат α-лактозы.
Предпочтительно, чтобы наполнитель и крупные и мелкие частицы носителя были составлены из одного и того же физиологически приемлемого терапевтически неактивного вещества.
В конкретном варианте осуществления изобретения дополнительный носитель состоит из:
i) фракции мелких частиц, составленных из смеси, состоящей из мелких частиц носителя и частиц добавки с антиадгезивными или смазывающими свойствами (мелкая фракция носителя);
ii) фракции крупных частиц носителя (крупная фракция носителя).
Препарат по настоящему изобретению проявляет как отличные реологические свойства, так и физическую и химическую стабильности без воздействия на порошок кондиционирующей обработки, такой как предполагаемая в WO 01/89491 и WO 01/89492. Он также проявляет хорошие характеристики аэрозоля.
Кроме того, в указанном препарате активный ингредиент распределен гомогенно.
Как правило, мелкие частицы носителя имеют МСД менее 50 микрон, предпочтительно менее 35 микрон, более предпочтительно от 3 до 15 микрон с МД, от 1 до 100 микрон, предпочтительно от 1 до 70 микрон.
Преимущественно МСД крупных частиц носителя превышает 90 микрон. Предпочтительно крупные частицы имеют МД от 50 микрон до 500 микрон, более предпочтительно от 150 до 400 микрон, еще более предпочтительно от 210 до 355 микрон.
Крупные частицы носителя могут иметь поверхность с относительно большим количеством трещин, то есть такую, на которой есть щели и впадины, и другие углубленные области, упоминающиеся в данном документе все вместе как трещины.
Крупные частицы носителя "с относительно большим количеством трещин" могут быть определены с точки зрения индекса трещин или коэффициентов складчатости, которые раскрыты в WO 01/78695 и WO 01/78693, и их можно охарактеризовать по изложенному там описанию.
Преимущественно индекс трещин составляет не менее 1,5, в то время как коэффициент складчатости - по меньшей мере 1,25.
Указанные крупные частицы носителя могут также быть охарактеризованы с точки зрения насыпной плотности после уплотнения или полного объема интрузии, измеренных, как сообщено в WO 01/78695.
Преимущественно насыпная плотность после уплотнения крупных частиц носителя составляет менее 0,8 г/см3, предпочтительно от 0,8 до 0,5 г/см3.
Полный объем интрузии составляет по меньшей мере 0,8 см3, предпочтительно по меньшей мере 0,9 см3.
Добавка может содержать комбинацию одного или более веществ. Преимущественно добавка является материалом с антиадгезивными свойствами, таким как аминокислоты, лейцин и изолейцин. Добавка также может состоять из одного или более водорастворимых поверхностно-активных веществ, например лецитина, в частности соевого лецитина.
Предпочтительно добавка является нерастворимым в воде смазывающим веществом, таким как стеарат магния; стеарилфумарат натрия; лаурилсульфат натрия, стеариловый спирт, стеариновая кислота и монопальмитат сахарозы. В более предпочтительном варианте осуществления изобретения добавка является стеаратом магния, и его частицы по меньшей мере частично формируют непрерывную оболочку вокруг поверхности частиц наполнителя.
Преимущественно МСД добавки составляет менее 50 микрон, предпочтительно менее 35 микрон, более предпочтительно менее 15 микрон.
Преимущественно количество добавки в окончательной препаративной форме составляет от 0,02 до 4,0 процента по массе (что приравнивается к 4 г на 100 г окончательной препаративной формы), предпочтительно от 0,05 до 2,0 процента по массе от суммарной массы препаративной формы.
В случае стеарата магния его количество составляет от 0,02 до 1,0 процента по массе, предпочтительно от 0,05 до 0,5 процента по массе, более предпочтительно от 0,1 до 0,4 процента по массе от суммарной массы препаративной формы.
Изобретение также предлагает способ получения препаративной формы.
В конкретном варианте осуществления способ включает приготовление микрочастиц, состоящих из микрочастиц активного ингредиента и микрочастиц физиологически приемлемого наполнителя.
Указанные микрочастицы могут быть приготовлены путем смешивания и последующей микронизации двух компонентов вместе посредством размалывания. Альтернативно, каждый компонент может быть подвергнут микронизации по отдельности, а затем их объединяют путем смешивания.
В первом варианте осуществления микрочастицы получают путем смешивания и последующей микронизации двух компонентов вместе в ступке или мельнице.
В качестве первой стадии частицы активного ингредиента и частицы наполнителя смешивают вместе в обычном смесителе, таком как смеситель Turbula, работающего на скорости от 8 до 72 об/мин, предпочтительно от 16 до 32 об/мин в течение по меньшей мере одного часа, предпочтительно по меньшей мере двух часов, более предпочтительно до пяти часов, затем их вместе микронизируют в мельнице.
Для получения микрочастиц по изобретению подходит широкий диапазон мельничных устройств, таких как шаровая мельница, молотковая мельница или ножевая мельница, и их рабочих режимов.
Предпочтительно частицы микронизируют совместно при использовании струйной мельницы путем соответствующего модулирования релевантных параметров.
В действительности было обнаружено, что при соответствующем модулировании давления и других параметров, таких как скорость подачи, при которой работает струйная мельница, можно управлять процессом микронизации таким образом, чтобы увеличить выход процесса, а также чтобы достичь желательного распределения размера частиц. Указанный размер частиц оптимален для того, чтобы избежать формирования устойчивых агломератов, когда микрочастицы смешивают с частицами дополнительного носителя.
Например, когда используется струйная мельница Glove Box 100 W, микрочастицы, соответствующие требованиям изобретения, получают при использовании давления измельчения 7 бар и скорости подачи 1,5-2,0 кг/ч.
Преимущественно начальный МД частиц наполнителя перед микронизацией составляет от 20 до 1000 микрон, предпочтительно от 50 до 400 микрон, более предпочтительно от 212 до 355 микрон.
Частицы активного ингредиента могут также находиться в микронизированной форме до того, как их будут измельчать совместно с частицами наполнителя.
Альтернативно, микрочастицы, состоящие из микронизированных частиц активного ингредиента и микронизированных частиц физиологически приемлемого наполнителя, можно получить путем микронизации каждого компонента по отдельности и последующего их объединения путем смешивания обычным способом и, например, как описано ранее на странице 27, строки 5-10.
В варианте осуществления способа по изобретению микрочастицы имеют начальный МСД от 2 до 15 микрон, предпочтительно от 2 до 10 микрон, более предпочтительно от 3 до 7 микрон. МД микрочастиц составляет от 1 до 20 микрон, предпочтительно от 1 до 15 микрон, и по меньшей мере 10% микрочастиц имеют МД, превышающий 0,5 микрона.
В вышеописанных вариантах осуществления для получения микрочастиц соотношение между микрочастицами активного ингредиента и микрочастицами наполнителя составляет от 1:5 до 1:100 по массе, предпочтительно от 1:9 до 1:90, более предпочтительно от 1:15 до 1:80, более предпочтительно от 1:20 до 1:75, еще более предпочтительно от 1:20 до 1:60.
Способ может дополнительно включать стадию добавления путем смешивания микрочастиц с частицами дополнительного носителя, как определено выше.
Частицы дополнительного носителя могут содержать мелкие частицы, крупные частицы и их смесь. Преимущественно частицы дополнительного носителя дополнительно содержат одну или более добавок, способствующих высвобождению активных частиц из частиц наполнителя и носителя при приведении ингалятора в действие. Предпочтительная добавка представляет собой антиадгезивное вещество или смазывающее вещество.
Процесс смешивания может быть выполнен согласно способам, раскрытым в уровне техники и известным специалисту в данной области техники, и, например, как было сообщено прежде на странице 27, строки 5-10.
Частицы дополнительного носителя, содержащие мелкие частицы носителя, крупные частицы носителя, необязательно частицы добавки и их смеси, могут быть приготовлены путем смешивания вместе мелкого носителя, крупного носителя и необязательно частиц добавки в любом порядке и сочетании, в течение по меньшей мере двух часов.
Если частицы дополнительного носителя содержат мелкие и крупные частицы носителя, соотношение между мелкими частицами носителя и крупными частицами носителя составляет от 1:99 до 40:60 процентам по массе, предпочтительно от 5:95 до 30:70 процентам по массе, еще более предпочтительно от 10:90 до 20:80 процентам по массе.
Преимущественно мелкие и/или крупные частицы дополнительного носителя содержат от 0,02 до 10 процентов по массе частиц добавки от массы окончательного препарата, предпочтительно от 0,05 до 5% масс./масс., более предпочтительно от 0,1 до 1 процента по массе от суммарной массы окончательного препарата.
В дополнительном варианте осуществления дополнительный носитель содержит мелкую фракцию носителя и крупную фракцию носителя, как определено со страницы 23, строка 27, до страницы 24, строка 4. Мелкую фракцию носителя можно получить по способам, описанным в заявке на патент WO 01/78693, содержание которого полностью включено в настоящую заявку.
Мелкие частицы носителя и частицы добавки совместно микронизируют, чтобы уменьшить размер их частиц до МСАД менее 35 микрон и необязательно создать частицы добавки полностью или частично, непрерывно или с перерывами покрывающие поверхность частиц наполнителя. Полученную в результате смесь затем смешивают с крупными частицами носителя так, что мелкие частицы фракции носителя прилипают к поверхности крупных частиц носителя.
Альтернативно, мелкую фракцию носителя можно приготовить путем смешивания в смесителе с высокой интенсивностью мелких частиц носителя и частиц добавки, имеющих МСД менее 35 микрон.
В предпочтительном варианте осуществления изобретения частицы дополнительного носителя состоят из мелкой фракции носителя и крупной фракции носителя, как определено выше, где мелкая фракция носителя составлена из от 90 до 99 процентов по массе частиц моногидрата α-лактозы и от 10 до 1 процента по массе частиц стеарата магния, предпочтительно от 98 до 2, и соотношение между мелкой фракцией носителя и крупной фракцией носителя, созданной из частиц моногидрата α-лактозы, составляет от 15 до 85 до 5:95, предпочтительно 10-90 процентов по массе.
Преимущественно частицы стеарата магния покрывают, по меньшей мере частично, поверхность либо мелких, либо крупных частиц носителя.
Преимущественно препаративная форма по изобретению имеет кажущуюся плотность перед уплотнением по меньшей мере 0,5 г/мл, предпочтительно от 0,6 до 0,7 г/мл, и индекс Carr менее 25, предпочтительно менее 15.
Препарат по изобретению может находиться в форме "твердых шариков", полученных путем воздействия на смесь процесса сферонизации.
Под термином "твердые шарики" авторы подразумевают сферические или полусферические единицы, ядро которых создано из крупных частиц носителя, которые после введения легко подвергаются деагрегации.
Под термином "сферонизация" авторы подразумевают процесс "округления" частиц, который происходит во время обработки.
Стадия сферонизации будет выполняться путем смешивания крупной фракции носителя и мелкой фракции носителя в подходящем смесителе, например барабанном смесителе, таком как Turbula, ротационных смесителях или смесителях высокой интенсивности, таких как Diosna, в течение по меньшей мере 5 минут, предпочтительно в течение по меньшей мере 30 минут, более предпочтительно в течение по меньшей мере двух часов, еще более предпочтительно в течение четырех часов. В общем случае специалист в данной области техники отрегулирует время смешивания и скорость вращения смесителя, чтобы получить гомогенную смесь.
Отношение между микрочастицами и частицами дополнительного носителя будет зависеть от типа используемого ингаляторного устройства и необходимой дозы активного ингредиента. Преимущественно соотношение между микрочастицами и частицами дополнительного носителя составляет от 5:95 до 0,1:99, предпочтительно от 10:90 до 0,25:99,75 по массе, более предпочтительно от 2:98 до 0,5:99,5 по массе.
Микрочастицы смешивают с частицами носителя в подходящем смесителе, предпочтительно в смесителе Turbula, в течение по меньшей мере 30 минут, предпочтительно в течение одного часа, предпочтительно в течение двух часов, более предпочтительно в течение по меньшей мере трех часов, работая при скорости от 8 до 72 об/мин, предпочтительно при 16 или 32 об/мин.
После смешивания однородность содержимого активного ингредиента, выраженная в виде относительного стандартного отклонения (RSD), составляет менее 6%, предпочтительно менее 5%, более предпочтительно равна/составляет менее 2,5%, еще более предпочтительно равна или составляет менее 1,5%.
В определенных случаях, и в особенности в присутствии активного ингредиента с очень низкой дозой, когда гомогенность порошка и отсутствие агрегатов особенно критичны, чтобы гарантировать однородность доз, препаративную форму по изобретению получают следующим способом.
Частицы активного ингредиента в микронизированной форме и терапевтически неактивный твердый разбавитель, состоящий из микронизированных частиц физиологически приемлемого наполнителя, желательно часть дополнительного носителя, содержащего мелкие частицы носителя и/или крупные частицы носителя и необязательно частицы добавки, продавливают через сито, чтобы облегчить дисперсию активного ингредиента и избежать формирования агломератов.
Преимущественно в данном конкретном варианте осуществления соотношение между активными частицами ингредиента и частицами наполнителя составляет от 1:1 до 1:3 по массе, предпочтительно от 1:1,5 до 1:2, и соотношение между активными частицами ингредиента и терапевтически неактивным твердым разбавителем, состоящим из частиц наполнителя и дополнительных частиц носителя, составляло от 1:10 до 1:100 по массе, предпочтительно от 1:10 до 1:50, более предпочтительно от 1:20 до 1:30 по массе.
Предпочтительной добавкой является смазывающее вещество, более предпочтительной - стеарат магния.
Преимущественно размер ячейки сита составляет от 100 до 400 микрон, предпочтительно от 200 до 300 микрон и более предпочтительно составляет 250 микрон.
В предпочтительном варианте осуществления указанный процесс выполняют путем использовании микронизированных активных частиц ингредиента, микронизированных частиц наполнителя, крупных частиц носителя и стеарата магния в качестве добавки, таким образом, чтобы соотношение между активным ингредиентом и частицами наполнителя составляло от 1:1,5 до 1:2 по массе и соотношение между активным ингредиентом и наполнителем, крупными частицами носителя и стеаратом магния составляло от 1:10 до 1:20 по массе.
В более предпочтительном варианте осуществления микронизированные частицы наполнителя, стеарат магния и крупные частицы носителя предварительно перемешивают в соотношении от 14,7:0,3:85 до 5:0,2:94,8 по массе, предпочтительно 9,8:0,2:90 по массе, перед добавлением активных микрочастиц ингредиента и продавливанием смеси через сито.
В указанном варианте осуществления наполнитель и крупный носитель состоят из моногидрата α-лактозы, и МСД наполнителя составляет от 2 мкм до 15 мкм.
Способ дополнительно включает стадию добавления путем смешивания смеси, полученной после пропускания через сито, с дополнительным носителем таким образом, чтобы процентное содержание активного ингредиента в окончательной препаративной форме составляло от 0,005 до 0,05% от суммарной массы окончательной препаративной формы. Процесс смешивания может быть выполнен, как сообщалось на странице 27, строки 5-10.
Частицы активного ингредиента, упомянутые по всему описанию изобретения, будут содержать эффективное количество по меньшей мере одного низкодозового активного вещества, которое можно ввести в легкие в форме порошка для ингаляции посредством ИСП. Вещество может действовать или локально, на легочном уровне, или, после проникновения в кровоток, на системном уровне.
Частицы активного ингредиента преимущественно состоят по существу из одного или более терапевтически активных агентов.
Подразумевается, что ссылки данного документа, относящиеся к любому активному агенту, включают любое физиологически приемлемое производное. В случае β2-агонистов физиологически приемлемые производные включают соли, сольваты и сольваты солей.
Подходящие терапевтически активные агенты включают лекарственные средства, которые обычно вводятся перорально путем ингаляции для лечения респираторных заболеваний. Примерами высокоактивных респираторных лекарственных средств служат β2-агонисты длительного действия, такие как 8-гидрокси-5-[(1R)-1-гидрокси-2-[[(1R)-2-(4-метоксифенил)-1-метилэтил]амино]этил-2(1H)-хинолинон и его соли.
Подходящие для целей изобретения соли включают гидрохлорид, фосфат, салицилат и соли миндальной кислоты.
Предпочтительной солью является гидрохлорид, иногда также называемый ΤΑ 2005 и описываемый в дальнейшем под экспериментальным кодом CHF 4226.
В других случаях активный ингредиент можно выбрать из низкодозового активного вещества для системного применения, например пептидов или полипептидов, таких как циклоспорин, инсулин, гормон роста человека, кальцитонин и эритропоэтин, или ложных или антисмысловых олигонуклеотидов.
Активные частицы предпочтительно содержат CHF 4226.
Преимущественно номинальная доза CHF 4226 составляет от 0,5 до 8 мкг, предпочтительно от 1 до 4 мкг, более предпочтительно от 1 до 2 мкг или от 2 до 4 мкг.
Конкретная реализация изобретения относится к 8-гидрокси-5-[(1R)-1-гидрокси-2-[[(1R)-2-(4-метоксифенил)-1-метилэтил]амино]этил-2(1H)-хинолинона гидрохлориду или CHF 4226 в порошковой препаративной форме, содержащей микрочастицы наполнителя и, необязательно, мелкие и/или крупные частицы дополнительного носителя и/или добавки согласно определениям настоящей заявки на патент, где CHF 4226 присутствует в количестве от 0,005 до 0,05% суммарной массы окончательной препаративной формы.
В более конкретной реализации изобретения препаративная форма содержит микрочастицы CHF 4226 в количестве от 0,005 до 0,05%, микрочастицы наполнителя, состоящие из моногидрата α-лактозы с МСД от 2 до 15 микрон, крупные частицы носителя, состоящие из моногидрата α-лактозы с МД от 212 до 355 микрон, и стеарат магния, и ее получают способом, раскрытым со страницы 34, строка 6, до страницы 35, строка 1.
Если желательно, активные частицы могут содержать CHF 4226 в комбинации с дополнительными активными ингредиентами, выбранными из группы кортикостероидов, таких как будезонид и его эпимеры, беклометазон дипропионат, триамцинолона ацетонид, флутиказона пропионат, флунизолид, мометазона фуроат, рофлепонид и циклезонид; группы антихолинергических или антимускариновых агентов, таких как ипратропиум бромид, окситропиум бромид, тиотропиум бромид, гликопирролата бромид или реватропат и их энантиомеры; группы ингибиторов фосфодиэстеразы-4 (PDE-4), таких как циломиласт и рофлумиласт, и их комбинаций, при условии, что они совместимы друг с другом в условиях хранения и применения.
Дополнительные активные ингредиенты, если присутствуют, добавляют к первому активному ингредиенту и/или к частицам наполнителя, необязательно в присутствии частиц дополнительного носителя, затем вместе смешиваются и необязательно вместе просеиваются или вместе смешиваются и вместе подвергаются размолу согласно сообщенной выше идее, чтобы сформировать микрочастицы.
Препаративная форма по изобретению особенно подходит для введения низкодозовых активных ингредиентов путем использования ИСП устройства с высоким или средним сопротивлением.
Препаративная форма также может содержать дополнительные компоненты, такие как ароматические средства.
Изобретение проиллюстрировано следующими примерами.
На основе раскрытия в настоящей заявке специалист в данной области техники будет в состоянии воспроизвести предложенную здесь идею путем использования других видов наполнителей или дополнительного носителя и их смеси.
Пример 1 - Получение микрочастиц, содержащих CHF 4226 гидрохлорид в качестве активного ингредиента и различные типы моногидрата α-лактозы, путем cовместного размола при различном давлении
Микронизированный гидрохлорид CHF 4226 и различные типы моногидрата α-лактозы в различных соотношениях по массе смешивали в смесителе Turbula в течение подходящего времени смешивания при 32 об/мин, затем совместно размалывали в аппарате струйной мельницы в различных рабочих режимах, чтобы получить различное распределение размеров частиц.
Микрочастицы характеризовали с точки зрения однородности распределения активного ингредиента, размера частиц и насыпной плотности.
Размер частиц полученных микрочастиц определяли лазерным дифракционным анализом. Учитывали следующие параметры: i) ОМД в микронах 10%, 50% и 90% частиц, выраженный как d (v, 0,1), d (v, 0,5) и d (v, 0,9) соответственно, которые соответствуют массовому диаметру, принимая для частиц плотность, независимую от размера, ii) полидисперсность порошка, то есть ширину распределения размера частиц, которая выражается промежутком (промежуток = [d (v, 0,9) - d (v, 0,1)]/d (v, 0,5) в соответствии с Chew NY et al. J Pharm Pharmaceut Sci, 2002, 5, 162-168).
Насыпную плотность вычисляли способом, изложенным далее.
Порошковые смеси (100 г) насыпали в градуированный стеклянный цилиндр и измеряли насыпной объем перед уплотнением, V0; кажущуюся плотность до уплотнения (насыпная плотность, dv) вычисляли, деля массу пробы на объем V0. После 1250 легких ударов описанного аппарата измеряли кажущийся объем после уплотнения (V1250), и подсчитывали кажущуюся плотность после уплотнения (насыпная плотность после уплотнения, ds).
Релевантные данные изложены в таблице 1.
Все препараты показали хорошую однородность распределения активного ингредиента, поскольку они продемонстрировали RSD ниже 6%.
Как можно оценить по таблице 1, было получено различное распределение размера микрочастиц за счет изменения скорости подачи, с которой работал аппарат струйной мельницы. Во всех фракциях микрочастиц имеется по меньшей мере 10% частиц с МД выше 0,5 микрона и МСД выше 2 микрон, за исключением партии 2. Различные партии микрочастиц затем добавляли к носителю, полученному из более крупных частиц. Партии 1, 3 и 4 однородно рассеивали в носителе, и после подходящего времени смешивания никаких агломератов не наблюдалось. В препаративной форме, приготовленной, начиная с партии 2, состоящей из микрочастиц, имеющих МСД менее 2 микрон, агломераты все еще присутствовали после более длительного периода, то есть 10 часов смешивания.
Агломераты отделяли просеиванием, и распределение их размера частиц определяли лазерным диффракционным анализом.
Результаты приведены в таблице 2 для сравнения с распределением размера частиц у микрочастиц партии 2.
Из анализа следует, что агломераты сформированы из более мелких частиц фракции микрочастиц, состоящей из наполнителя и активного ингредиента.
Из этого следует, что фракции, имеющие d (v, 0,1) и d (v, 0,5) частиц, сместившиеся к более мелкому размеру, то есть равному 0,5 микрона или менее и менее 2 микрон соответственно, дают начало устойчивым агломератам, которые невозможно диспергировать даже после долгого времени смешивания (более 10 часов). Это вредно для однородности распределения активного ингредиента в окончательной препаративной форме.
Пример 2 - Препаративная форма микрочастиц, полученая из CHF 4226 гидрохлорида и различных типов моногидрата α-лактозы путем совместного перемешивания
Микронизированный CHF 4226 гидрохлорид, имеющий МСД 1,8 микрона, и микронизированный моногидрат α-лактозы, имеющий МСД приблизительно 12,5 микрона, в соотношениях 1:2, 1:9, 1:24 и 1:99 процентам по массе смешивали в ступке, чтобы получить различные партии микрочастиц.
Микрочастицы были охарактеризованы с точки зрения размера частиц (лазерный дифракционный анализ) и однородности распределения активного ингредиента.
Результаты приведены в таблице 3.
Все партии микрочастиц показали хорошую однородность распределения активного ингредиента, поскольку они продемонстрировали RSD ниже 6%, и во всех случаях по меньшей мере 10% частиц имели МД выше 0,5 микрона с МСД выше 2 микрон и равным 15 микрон или ниже.
Различные партии микрочастиц затем добавляли к носителю, состоящему из крупных частиц. Партии 2, 3 и 4 однородно распределяли в окончательной препаративной форме. В данном виде препаративной формы можно заметить, что партия 1, где соотношение между активным ингредиентом и наполнителем было 1:2, не показала хорошей однородности распределения активного ингредиента, и RSD был равен 13,27%.
Пример 3 - Приготовление препаративной формы, состоящей из микрочастиц, состоящих из совместно микронизированного CHF 4226 гидрохлорида и моногидрата α-лактозы в соотношении 1:49 масс./масс., и носителя, содержащего мелкую фракцию носителя и крупную фракцию носителя
a) Приготовление мелкой фракции носителя.
SpheroLac® 100, моногидрат α-лактозы с начальным МД 50-400 микрон (МСД приблизительно 170 микрон) и стеарат магния с начальным МД 3-35 микрон (МСД приблизительно 10 микрон) в соотношении 98:2 процента по массе совместно размалывали в аппарате струйной мельницы.
b) Добавление мелкой фракции носителя к крупной фракции носителя. 89,5 процентов по массе моногидрата α-лактозы CapsuLac® (212-355 микрон) помещали в контейнер из нержавеющей стали на 240 мл, затем добавляли 10 процентов по массе мелкой фракции носителя. Смесь перемешивали в лабораторном смесителе Turbula в течение 4 часов при 32 об/мин, чтобы получить носитель.
c) Добавление фракции микрочастиц к носителю. Микронизированный CHF 4226 гидрохлорид и моногидрат α-лактозы Capsulac® (212-355 микрон) в соотношении 1:49 процентов из партии 3 примера 1 добавляли к носителю в подходящем количестве, чтобы получить соотношение 1 мкг активного ингредиента на 10 мг окончательной препаративной формы и перемешивали в смесителе Turbula в течение трех часов при 32 об/мин. Количество стеарата магния в окончательной препаративной форме составляло 0,2 процента по массе.
Пример 4 - Технологическая характеризация препаративной формы из примера 3
Препаративную формуа из примера 3 охарактеризовывали по ее параметрам плотности/текучести и однородности распределения активного ингредиента.
Насыпная плотность была вычислена, как сообщалось в примере 1.
Свойства текучести проверяли по методу, приведенному ниже.
Порошковые смеси (приблизительно 110 г) засыпали в сухую воронку, оборудованную отверстием подходящего диаметра, которое блокировалось подходящим устройством. Открытие нижней части воронки деблокировали и регистрировали время, необходимое для того, чтобы вся проба высыпалась из воронки. Текучесть выражали в секундах и десятых долях секунд, отнесенных к 100 г пробы.
Текучесть также оценивали по индексу Carr, вычисленному по следующей формуле:
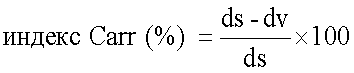
Индекс Carr менее 25 обычно считают показателем хороших характеристик текучести.
Однородность распределения активного ингредиента оценивали путем взятия в различных частях смеси 10 проб, каждая из которых была приблизительно эквивалентна разовой дозе.
Результаты приведены в таблице 4.
Препаративная форма по изобретению показывает хорошую однородность распределения активного ингредиента, что подтверждается низким RSD. Она также показывает очень хорошие свойства потока, что подтверждается индексом Carr; данный параметр очень важен, чтобы получить постоянство отмеренной дозы, когда используется многодозовый ингалятор сухого порошка с резервуаром для порошка.
Пример 5 - Определение характеристик аэрозоля препарата из примера 3
Количество порошка для ингаляции загружали в многодозовый ингалятор сухого порошка.
Оценку характеристик аэрозоля производили, используя аппарат (аппарат C) Multi Stage Liquid Impinger (MSLI), в соответствии с условиями, сообщенными в Eur Ph 4th Ed, 2004, par 2.9.18, страницы 213-219. После распыления 10 доз аппарат MSLI демонтировали, и количество лекарственного средства, осевшее на уровнях, извлекали путем промывания смесью растворителей, а затем количественно определяли высокоэффективной жидкостной хроматографией (ВЭЖХ). Вычисляли следующие параметры: i) введенная доза, которая представляет собой количество лекарственного средства, введенное устройством и извлеченное в импакторе; ii) доза мелких частиц (FPD), которая представляет собой количество введенной дозы, выделенной ниже 5 микрон; iii) фракция мелких частиц (FPF), которая представляет собой процентное содержание дозы мелких частиц от введенной дозы, достигнувшей уровня 2 MSLI; iv) МСАД.
Результаты с точки зрения характеристик аэрозоля приведены в таблице 5.
Характеристики аэрозоля препарата очень хороши с FPF, превышающей 50%.
Пример 6 - Препаративная форма, состоящая из микрочастиц, состоящих из CHF 4226 гидрохлорида и моногидрата α-лактозы в различных масс./масс. соотношениях, полученных путем совместного перемешивания предварительно микронизированных частиц, и носителя, содержащего мелкую фракцию носителя и крупную фракцию носителя
a) Приготовление препаративных форм.
Носитель получали, как описано в примере 3.
Микрочастицы получали, как описано в примере 2.
Микрочастицы добавляли к носителю в подходящем количестве, чтобы получить соотношение 1 мкг активного ингредиента на 10 мг окончательного препарата, и смешивали в смесителе Turbula в течение одного часа при 32 об/мин.
b) Технологическая характеризация окончательных препаративных форм.
Окончательные препаративные формы были охарактеризованы по их параметрам плотности/текучести и однородности распределения активного ингредиента, как изложено в примере 4. Результаты изложены в таблице 6.
c) Определение характеристик аэрозоля окончательных препаративных форм. Характеристики аэрозоля определяли, как изложено в примере 5. Результаты приведены в таблице 7.
Все препаративные формы показывают превосходные характеристики с точки зрения как однородности распределения активного ингредиента (RSD ниже 5%), так и характеристик аэрозоля.
В случае соотношения 1:24 и 1:99 была достигнута FPF, превышающая 50%.
Пример 7 - Приготовление препаративной формы, содержащей микрочастицы CHF 4226, микрочастицы наполнителя, крупные частицы носителя и стеарат магния
a) Приготовление терапевтически неактивного твердого разбавителя, состоящего из смеси микрочастиц наполнителя, крупных частиц носителя и стеарата магния.
Частицы моногидрата α-лактозы с МД от 1 до 15 микрон и МСД от 3 до 7 микрон смешивали со стеаратом магния и крупными частицами носителя - моногидратом α-лактозы с МД от 212 до 355 микрон - в соотношении 9,8:0,2:90 по массе соответственно.
b) Приблизительно к 60 г смеси a) добавляли 3,2 г микронизированного CHF 4226 таким образом, что соотношение между активным ингредиентом и терапевтически неактивным твердым разбавителем составило приблизительно 1:20 по массе, и полученную в результате смесь просеивали через 250-мкм сито.
c) Смесь, полученную на стадии b), затем добавляли к оставшейся смеси твердых разбавителей a), чтобы получить количество 4 мкг активного ингредиента на 10 мг окончательной препаративной формы (0,04%), и смешивали в промышленном смесителе Turbula в течение 1 часа при 16 об/мин. Количество стеарата магния в окончательной препаративной форме составило 0,2 процента по массе.
Препаративную форму охарактеризовали по ее параметрам плотности/текучести, однородности распределения активного ингредиента и характеристикам аэрозоля.
Насыпную плотность вычисляли, как сообщено в примере 1. Свойства текучести и однородность распределения определяли, как сообщено в примере 4.
Оценку характеристик аэрозоля выполняли с помощью каскадного импактора Андерсена в соответствии с условиями, сообщенными в Eur Ph 3rd Ed Suppl. 2001, par 2.9.18, страницы 123-124, определяя те же самые параметры, о которых сообщалось в примере 5. Результаты приведены в таблицах 8 и 9.
Насколько можно оценить, препаративная форма по изобретению показывает хорошую однородность распределения активного ингредиента, что демонстрируется низким RSD, а также очень хорошие свойства потока, что демонстрируется скоростью потока. Характеристики аэрозоля препарата тоже очень хороши с FPF, превышающим 60%.
Кроме того, оказалось, что активный ингредиент в препаративной форме физически и химически устойчив после хранения в течение трех месяцев на складе при контролируемых условиях окружающей среды, что демонстрируется анализом на CHF 4226, который оставался по существу неизменным, и характеристиками аэрозоля после хранения, где вводимая доза составляла 3,0 мкг и оставалась неизменной.
Кроме того, не было обнаружено никаких продуктов деградации.
Реферат
Настоящее изобретение относится к области лекарственных средств, в частности к порошковой препаративной форме для применения в ингаляторе сухого порошка, причем порошок содержит микрочастицы, состоящие из микрочастиц активного ингредиента, представляющего собой 8-гидрокси-5-[(1R)-1-гидрокси-2-[[(1R)-2-(4-метоксифенил)-1-метилэтил]амино]этил-2(1Н)-хинолинон или его соль, и микрочастиц физиологически приемлемого наполнителя, представляющего собой моногидрат α-лактозы, где МСД микрочастиц составляет от 2 до 15 мкм и по меньшей мере 10% микрочастиц имеют массовый диаметр (МД), превышающий 0,5 мкм; а также дополнительный носитель, содержащий мелкую фракцию носителя с МСД менее 35 мкм, состоящую из моногидрата α-лактозы и стеарата магния, и крупную фракцию носителя с МСД более 90 мкм, состоящего из моногидрата α-лактозы, с МД, составляющим от 212 до 355 мкм, и стеарат магния. Кроме того, изобретение относится к способам получения указанной порошковой препаративной формы. Полученная порошковая форма имеет улучшенные характеристики. 3 н. и 19 з.п. ф-лы, 9 табл.
Формула
МСД микрочастиц составляет от 2 до 15 мк; и
по меньшей мере 10% микрочастиц имеют массовый диаметр (МД), превышающий 0,5 мк;
а также дополнительный носитель, содержащий мелкую фракцию носителя с МСД менее 35 мк, состоящую из моногидрата α-лактозы и стеарата магния, и крупную фракцию носителя с МСД более 90 мк, состоящего из моногидрата α-лактозы, с МД, составляющим от 212 до 355 мк, и стеарат магния,
причем указанная порошковая препаративная форма доставляет номинальную дозу активного ингредиента, составляющую 20 мкг или менее, при каждом приведении указанного ингалятора порошка в действие.
Комментарии