Способы депарафинизации жидкого нефтепродукта и смазочных масел - RU2214441C2
Код документа: RU2214441C2
Чертежи






Описание
Изобретение относится к способу каталитической депарафинизации смазочных масел. Более конкретно настоящее изобретение относится к способу депарафинизации исходного смазочного нефтяного сырья, где по крайней мере часть недогона фракционирующей колонны возвращают на рециркуляцию в исходное сырье.
Некоторые способы депарафинизации нефтяного сырья хорошо известны. Депарафинизация требуется, когда парафиновые масла необходимо использовать в продуктах, которые должны быть подвижными при низких температурах, например в смазочных маслах, в маслах для подогрева и топливе для реактивных двигателей. Высокомолекулярные нормальные с прямой цепью, замещенные и слегка разветвленные парафины, присутствующие в таких нефтепродуктах, представляют собой парафины, которые приводят к высоким температурам текучести и температурам помутнения нефти. Если должны быть получены достаточно низкие температуры текучести, то парафины должны быть полностью или частично удалены. В прошлом для удаления таких парафинов применяли различные методы удаления растворителем, например депарафинизация МЕК; однако эти способы имеют высокие эксплуатационные расходы, значительное воздействие на окружающую среду и дают масла, которые хуже по качеству по сравнению с маслами, полученными каталитической депарафинизацией. Процессы каталитической депарафинизации являются более экономичными и удаляют парафины селективной изомеризацией и крекингом парафиновых компонентов для получения продуктов с более низкой молекулярной массой, некоторые из которых можно удалить перегонкой.
Благодаря своей селективности известные катализаторы депарафинизации, как правило, включают в себя алюмосиликатный цеолит, имеющий размер пор, который соответствует размеру либо только одних н-парафинов с линейной цепью, либо их смеси с парафинами со слегка разветвленной цепью, но который не позволяет проникнуть более сильно разветвленным материалам, циклопарафинам большего размера и ароматике. Для этой цели в способах депарафинизации были предложены цеолиты такие, как ZSM-5, ZSM-11, ZSM-12, ZSM-23, ZSM-35 и ZSM-38. Их использование описывается в патентах США 3700585; 3894938; 4176050; 4181598; 4222855; 4229282 и 4247388, приведенных в качестве ссылки.
Поскольку работа многих процессов депарафинизации этого основаны на реакциях крекинга, ряд полезных продуктов разрушается до соединений с более низкой молекулярной массой. Например, твердые парафины могут быть подвергнуты крекингу до бутана, пропана, этана и метана, и тоже самое может произойти с более легкими н-парафинами, которые не вносят вклад в воскообразную природу нефти. Поскольку эти более легкие продукты, как правило, представляют меньшую ценность, чем материалы с более высокой молекулярной массой, является желательным ограничить степень крекинга, который имеет место во время осуществления каталитической депарафинизации.
Заявка на патент Европы 225053 описывает способ производства смазочных масел с помощью частичной депарафинизации сырья, содержащего в своей основе смазочное масло, посредством изомеризационной депарафинизации, после чего следует стадия селективной депарафинизации. Стадию изомеризационной депарафинизации проводят, используя катализатор депарафинизации, представляющий собой цеолит с высоким содержанием кремния и большими размерами пор, например кремнезем γ или цеолит бета, которые изомеризуют парафиновые компоненты исходного сырья до менее воскообразных изопарафинов с разветвленной цепью. Стадия селективной депарафинизации может быть проведена либо в растворителе, например, депарафинизации МЕК, либо каталитическую депарафинизацию, предпочтительно использующую цеолит с хорошо развитой внутренней поверхностью, например ZSM-22 или ZSM-23.
Патент США 4437976 описывает двустадийный процесс депарафинизации и гидрообработки углеводородов, где температуру текучести углеводородного исходного сырья, кипящего от 400oF (204/4oС) до 1050oF (565,6oС), уменьшают каталитической депарафинизацией исходного сырья в присутствии цеолитного катализатора и затем подвергают по крайней мере его жидкую часть гидрированию в присутствии гидрирующего катализатора, включающего в себя гидрирующий компонент и кремниевый пористый кристаллический материал из класса цеолитов ZSM-5, ZSM-11, ZSM-23 и ZSM-35.
Патент США 4575416, Chester et al., описывает способ гидродепарафинизации с первым цеолитным катализатором, имеющим индекс проницаемости не менее чем 1, со вторым каталитическим компонентом со специфическими характеристиками и с гидрирующим компонентом.
Патент США 5149421 описывает катализатор депарафинизации, который проявляет превосходную селективность в отношении природы продуктов, получаемых в процессе депарафинизации. Используя в способе депарафинизации катализатор на основе силикоалюмофосфатных молекулярных сит, жидкие нефтепродукты подвергают эффективной депарафинизации и получаемые продукты имеют более высокую молекулярную массу, чем продукты, полученные с использованием других алюмосиликатных цеолитов. Полученные по этому способу депарафинизации продукты имеют лучшие вязкости и коэффициенты вязкости при данной температуре текучести по сравнению с вышеописанным способом прототипа, использующего алюмосиликатные цеолиты.
Тем не менее было бы выгодно иметь способ, который дает увеличенный выход по сравнению с выходами, получаемыми в известных процессах, или более сильное снижение температуры текучести при том же выходе. Настоящее изобретение обеспечивает такой процесс.
Настоящее изобретение преодолевает проблемы и недостатки разработанных к настоящему времени процессов, предоставляя способ каталитической депарафинизации сырья, представляющего собой жидкие нефтепродукты, который дает превосходный выход смазочного масла.
Способ депарафинизации жидких нефтепродуктов по настоящему изобретению включает следующие стадии:
(a) взаимодействие в условиях депарафинизации исходного
сырья для смазочного масла в присутствии добавленного газообразного водорода с каталитической системой, включающей силикоалюмофосфатные молекулярные сита с промежуточным размером пор и гидрирующий
компонент, где гидрирующий компонент присутствует в количестве примерно от 0,01 до 10%, основываясь на массе силикоалюмофосфатного молекулярного сита, и дополнительно включающей катализатор, выбранный
из группы, состоящей из алюмосиликатного цеолита, аморфного катализатора и их смесей, где подвергают депарафинизации по крайней мере часть указанного перерабатываемого сырья;
(b) подачу по
крайней мере части подвергнутого депарафинизации исходного сырья в ректификационную колонну, где по крайней мере часть указанного подвергнутого депарафинизации исходного сырья разделяют на фракции с
получением по крайней мере одной головной фракции и одной фракции недогона, и
(с) смешивание от около 1 до около 80 мас.% недогона ректификационной колонны с исходным углеводородным нефтяным
сырьем для увеличения выхода или уменьшения температуры текучести подвергнутого депарафинизации исходного сырья из фракции недогона с исходным углеводородным нефтяным сырьем стадии (а).
На чертеже изображена упрощенная блок-схема одного варианта реализации способа по настоящему изобретению.
Способ депарафинизации жидкого нефтепродукта по настоящему изобретению
включает следующие стадии:
(a) взаимодействие в условиях депарафинизации исходного сырья для смазочного масла в присутствии добавленного газообразного водорода с каталитической системой,
включающей силикоалюмофосфатные молекулярные сита с промежуточным размером пор и гидрирующий компонент, где гидрирующий компонент присутствует в количестве примерно от 0,01 до 10%, основываясь на
массе силикоалюмофосфатного молекулярного сита, и дополнительно включающей катализатор, выбранный из группы, состоящей из алюмосиликатного цеолита, аморфного катализатора и их смесей, где подвергают
депарафинизации по крайней мере часть указанного перерабатываемого сырья;
(b) подачу по крайней мере части подвергнутого депарафинизации исходного сырья в ректификационную колонну, где по
крайней мере часть указанного подвергнутого депарафинизации исходного сырья разделяют на фракции, с получением по крайней мере одной головной фракции и одной фракции недогона, и
(с)
смешивание от около 1 до около 80 мас.% недогона ректификационной колонны с исходным углеводородным нефтяным сырьем для увеличения выхода или уменьшения температуры текучести подвергнутого
депарафинизации исходного сырья из фракции недогона с исходным углеводородным нефтяным сырьем стадии (а).
Каталитическая система включает в себя катализатор, выбранный из группы, состоящей из алюмосиликатного цеолитного катализатора с промежуточным размером пор, аморфного катализатора и их смесей. Для предварительной обработки сырье может быть подвергнуто гидрокрекингу или жидкостной экстракции и гидроочистке. Этот тип процесса и типичные условия гидрокрекинга описываются в патенте США 4921594 от 1 мая 1990 (Miller), включенный в качестве ссылки. Последующие обработки могут включать гидроочистку, обсуждаемую ниже.
Не ограничиваясь какой-либо теорией, в одном из вариантов воплощения изобретения механизм депарафинизации представляет собой изомеризацию и/или крекинг парафиновых компонентов. Типично каталитическую депарафинизацию, например каталитический процесс депарафинизации ISODEWAXING компании Шеврон, осуществляют для улучшения температуры текучести и коэффициента вязкости, по сравнению с депарафинизацией в растворителе.
Способ по настоящему изобретению может быть использован для депарафинизации прежде всего смазочных масел. Перерабатываемое сырье включает фракции дистиллята, например продукты гидрокрекинга, вплоть до сырья с высокой температурой кипения такого, как деасфальтированная нефть и нефть, подвергнутая жидкостной экстракции. Перерабатываемое сырье будет обычно представлять собой С10+ сырье, как правило, кипящее выше примерно 350oF (177oC), поскольку более легкие фракции нефти обычно будут свободны от значительных количеств воскообразных компонентов. Однако настоящий способ особенно подходит для сырья для смазочного масла, масел для нагрева и других фракций, температура текучести и вязкость которых необходимо поддерживать внутри определенных пределов спецификации. Масляное сырье обычно будет кипеть при температуре выше 230oС (450oF), в более обычном случае - выше 315oС (600oF).
Сырье, подвергаемое гидрообработке, представляет собой традиционный источник сырья этого вида, поскольку оно имеет более высокое содержание водорода в противоположность сырью, перерабатываемому с помощью растворителя, и является относительно свободным от гетероатомов (например, соединений серы и азота), которые могут ухудшить эксплуатационные характеристики катализаторов депарафинизации и гидроочистки. Исходное сырье по настоящему изобретению будет обычно представлять собой С10+ сырье, содержащее парафины, олефины, циклопарафины, ароматику и гетероциклические соединения и значительную часть н-парафинов с высокой молекулярной массой и слаборазветвленных и замещенных парафинов, которые придают воскообразную природу перерабатываемому сырью. В течение переработки молекулы сырья подвергаются крекингу или гидрокрекингу с образованием жидких материалов, которые придают низкую вязкость продукту. Однако степень происходящего крекинга ограничивают для сохранения выхода ценных жидкостей.
Типичное сырье включает легкий газойль, тяжелый газойль и слабо крекированную нефть, кипящую выше 350oF (177oС). В одном варианте осуществления изобретения перерабатываемое сырье содержит основную часть исходного сырья, представляющего собой жидкие нефтепродукты, кипящего выше примерно 350oF (177oС), и содержит углеводороды с нормальной цепью и слаборазветвленной цепью. Термин "основная часть" означает более чем 50 мас.%.
В то время как способ по настоящему изобретению может быть успешно реализован на практике, когда сырье содержит органический азот (азотсодержащие примеси), предпочтительным является, чтобы содержание органического азота в сырье было бы менее чем 50 млн.д. (мас.), более предпочтительно менее чем 10 млн. д. (мас. ). Особенно хорошие результаты, с точки зрения активности и продолжительности цикла катализатора (период между последующими регенерациями или пуском и первой регенерацией), получаются, когда сырье содержит менее чем 10 млн.д. (мас.) органического азота.
Обычно катализаторами, используемыми в способе по настоящему изобретению, являются силикоалюмофосфатные сита с промежуточным размером пор (SAPOs). Подходящие SAPOs представляют собой любые обычные SAPO с промежуточным размером пор. SAPOs используются отдельно или в сочетании с цеолитами и/или аморфными катализаторами. Примеры силикоалюмофосфатных молекулярных сит, которые можно использовать в настоящем изобретении, описаны в патентах США 4440871 и 5149421, приведенных здесь в качестве ссылки.
В способе по настоящему изобретению применяют катализатор, представляющий собой силикоалюмофосфатные молекулярные сита с промежуточным размером пор, чтобы конвертировать твердые парафиновые компоненты в невоскообразные компоненты и уменьшить их температуру текучести примерно на температуру от 30oF (16,7oС) до 60oF (33,3oС). Количество используемого катализатора зависит от условий реакции.
В предпочтительном варианте осуществления изобретения, конечный катализатор может быть смесью и включать в себя силикоалюмофосфатные молекулярные сита с промежуточным размером пор, металлический компонент, представляющий собой платину или палладий для гидрирования, и неорганическую оксидную матрицу. Предпочтительные силикоалюмофосфатные молекулярные сита с промежуточным размером пор, подходящие для использования в способе по настоящему изобретению, включают SAPO-11, SAPO-31 и SAPO-41. Наиболее предпочтительным силикоалюмофосфатом является SAPO-11, наиболее предпочтительным металлическим компонентом является платина и наиболее предпочтительным связующим веществом оксид алюминия. Описания SAPO-11, SAPO-31 и SAPO-41 и способов их получения даны в цитируемых ранее патентах и в R. Szostak, Handbook of Molecular Sieves (Van Norstrand Reinhold, 1992), pp. 410-413, 415-416, 419-420, приведенных здесь в качестве ссылки.
Молекулярные сита могут быть смешаны с другими материалами, которые устойчивы к температурам и другим условиям, используемым в процессе депарафинизации. Такие материалы матрицы включают активные и неактивные материалы и синтетические или природные цеолиты, а также неорганические материалы такие, как глины, кремнезем и оксиды металлов. Последние могут находиться либо в природном виде, либо в форме гелеобразных осадков, золей или гелей, включая смеси кремнезема и оксидов металлов. Неактивные материалы соответственно служат в качестве связующих веществ или в качестве разбавителей, чтобы контролировать степень конверсии в способе депарафинизации так, что продуты могут быть получены экономично без применения других средств контроля за скоростью реакции.
Силикоалюмофосфаты могут быть смешаны с природными глинами, например бентонитом и каолином. Эти материалы, т.е. глины, оксиды и т.д., частично функционируют в качестве связующего вещества для катализатора. Желательным является получение катализатора, имеющего хорошую устойчивость к истиранию потому, что при переработке нефти катализатор часто подвергается грубому воздействию и огромным нагрузкам в реакторе. Это приводит к разрушению катализатора на фрагменты, которые могут закупорить реактор.
Встречающиеся в природе глины, которые можно смешивать с силикоалюмофосфатом, включают семейства монтмориллонита и каолина, которые включают суббентониты, и каолины, обычно известные как Dixie, McNamee, Georgia и Florida глины или другие, в которых главный составляющий минеральный компонент представляет собой галлуазит, каолинит, диккит, накрит или анауксит. Также в качестве носителя могут использоваться волокнистые глины такие, как галлуазит, сепиолит и аттапульгит. Такие глины можно использовать в виде сырья как первоначально добытые или вначале подвергнутыми кальцинированию, кислотной обработке или химическому модифицированию.
В дополнение к вышеуказанным материалам, силикоалюмофосфаты могут быть смешаны с пористыми материалами матрицы, например, с неорганическими матричными материалами и смесями матричных материалов таких, как кремнезем, глинозем, оксид титана, оксид магния, смешанные оксиды кремния и алюминия, смешанные оксиды кремния и магния, смешанные оксиды кремния и циркония, смешанные оксиды кремния и тория, смешанные оксиды кремния и бериллия, смешанные оксиды кремния и титана, смешанные оксиды титана и циркония, а также трехкомпонентные составы такие, как смешанные оксиды кремния, алюминия и тория, смешанные оксиды кремния, алюминия и титана, смешанные оксиды кремния, алюминия и магния и смешанные оксиды кремния, магния и циркония. Матрица может быть в форме совместного геля или однородной физической смеси.
Силикоалюмофосфатные катализаторы, используемые по настоящему изобретению, также могут быть смешаны с другими цеолитами такими, как синтетические и природные фоязиты, (например, Х и Y) эриониты и мордениты. Также они могут быть смешаны с чисто синтетическими цеолитами такими, как цеолиты ZSM серий. Также в пористую неорганическую матрицу может быть вставлена комбинация цеолитов.
Примеры подходящих для использования в способе по настоящему изобретению алюмосиликатных цеолитных катализаторов, включают ZSM-22, ZSM-23 и ZSM-35. Они описаны в R. Szostak, Handbook of Molecular Sieves (Van Norstrand Reinhold, 1992), на страницах 538-542 и 545-546, которые включаются здесь ссылкой, и в патентах США 4481177; 4076842 и 4016245, описания которых включаются здесь ссылкой.
Катализатор, содержащий силикоалюмофосфатные молекулярные сита, и алюмосиликатный цеолитный катализатор применяют в способе по настоящему изобретению в эффективном массовом соотношении силикоалюмофосфатного молекулярного сита с промежуточным размером пор к алюмосиликатному цеолитному молекулярному ситу с промежуточным размером пор, чтобы увеличить выход конвертированного исходного сырья. Предпочтительные соотношения равны примерно от 1:5 до 20:1. Цеолит, используемый в способе, предпочтительно имеет индекс проницаемости, измеренный примерно от 400 до 454oС, равный примерно от 4 до 12.
В другом варианте воплощения способа по настоящему изобретению в процессе депарафинизации по настоящему изобретению можно использовать SSZ-48, предпочтительно главным образом в водородной форме. Не ограничиваясь теорией, предполагают, что SSZ-48 депарафинизирует посредством селективного удаления парафинов с прямой цепью. Обычно коэффициент вязкости продукта, подвергнутого депарафинизации, улучшают (по сравнению с депарафинизацией сырья в растворителе), когда содержащее твердые парафины сырье контактирует с SSZ-48 при условиях изомеризационной депарафинизации (также упоминаемой как гидродепарафинизация).
При получении
цеолитов SSZ-48 в качестве матрицы кристаллизации используют катион декагидрохинолиния. Катион декагидрохинолиния может иметь следующую структуру:

Связанный с катионом анион (X-) может представлять собой любой анион, который не препятствует образованию цеолита. Типичными анионами являются галоген, например фторид, хлорид, бромид и йодид, гидроксид, ацетат, сульфат, тетрафторборат, карбоксилат и подобные. Гидроксид является наиболее предпочтительным анионом.
В общем, SSZ-48 готовят контактом активного источника одного или более оксидов, выбранных из группы, состоящей из оксидов одновалентных элементов, оксидов двухвалентных элементов, оксидов трехвалентных элементов и оксидов четырехвалентных элементов, с матричным реагентом, содержащим катион декагидрохинолиния.
SSZ-48 готовят из реакционной смеси, имеющей, состав, показанный в приведенной ниже таблице 1.
На практике SSZ-48 готовят посредством способа предусматривающего
(а) приготовление водного раствора, содержащего источник по крайней
мере одного оксида, который может образовывать кристаллические молекулярные сита, и катион декагидрохинолиния, имеющего анионный противоион, который не препятствует образованию SSZ-48;
(b)
выдерживание водного раствора при условиях, которые достаточны для образования кристаллов SSZ-48, и
(с) извлечение кристаллов SSZ-48.
Соответственно SSZ-48 может содержать кристаллический материал и матричный реагент в сочетании с металлическими и неметаллическими оксидами, связанными в тетраэдрическую координацию через разделяющие атомы кислорода с образованием сшитой трехмерной кристаллической структуры. Металлические и неметаллические оксиды включают в себя один или комбинацию оксидов первого(ых) четырехвалентного(ых) элемента(ов) и один или комбинацию второго(ых) четырехвалентного(ых) элемента(ов), отличающихся от первого(ых) четырехвалентного(ых) элемента(ов), трехвалентного(ых) элемента(ов), пятивалентного(ых) элемента(ов) или их смесь. Первый(ые) четырехвалентный(ые) элемент(ы) предпочтительно выбирают из группы, состоящей из кремния, германия и их комбинации. Более предпочтительно первый четырехвалентный элемент представляет собой кремний. Второй четырехвалентный элемент (который отличается от первого четырехвалентного элемента), трехвалентный элемент и пятивалентный элемент предпочтительно выбирают из группы, состоящей из алюминия, галлия, железа, бора, титана, индия, ванадия и их комбинаций. Более предпочтительно второй трехвалентный или четырехвалентный элемент представляет собой алюминий или бор.
Типичные источники оксида алюминия для реакционной смеси включают алюминаты, оксид алюминия, коллоиды алюминия, оксид алюминия, нанесенный на золь диоксида кремния, гидратированные алюмогели, например Аl(ОН)3, и соединения алюминия такие, как АlСl3 и Аl2(SO4)3. Типичные источники оксида кремния включают силикаты, гидрогели кремния, кремниевую кислоту, дымящийся кремнезем, коллоидный кремнезем, тетраалкилортосиликаты и гидроксиды кремния. Бор, а также галлий, германий, титан, индий, ванадий и железо можно добавлять в форме, которая соответствует их алюминиевым и кремниевым составляющим.
Реагент, являющийся источником цеолита, может обеспечить источник алюминия или бора. В большинстве случаев источник цеолита также обеспечивает источник кремнезема. Источник цеолита в своей деалюминированной или деборированной форме можно также использовать как источник кремнезема с дополнительным кремнием, добавленным с использованием, например, обычных источников перечисленных выше. Использование реагента, являющегося источником цеолита, в качестве источника оксида алюминия для настоящего способа наиболее полно описывается в патенте США 5187132 от 16 февраля 1993 (Zones et al.), который озаглавлен "Приготовление боросиликатных цеолитов", включенном здесь в качестве ссылки.
Обычно в реакционной смеси используют гидроксид щелочного металла и/или гидроксид щелочноземельного металла, например гидроксид натрия, калия, лития, цезия, рубидия, кальция и магния; однако, этот компонент может быть не включен при условии, что эквивалентная основность сохраняется. Для получения гидроксид-иона может быть использован матричный реагент. Таким образом, это может быть выгодным для ионного обмена, например, галогена на гидроксид-ион, при этом уменьшая или исключая требуемое количество гидроксида щелочного металла. Катион щелочного металла или катион щелочноземельного металла может являться частью кристаллического оксидного материала в том виде, как он синтезирован для сбалансировки в нем валентных зарядов электронов.
Повышенную температуру реакционной смеси поддерживают до тех пор, пока не сформируются кристаллы цеолита SSZ-48. Гидротермическую кристаллизацию обычно проводят при автогенном давлении, при температуре между 100 и 200oС, предпочтительно между 135 и 160oС. Период кристаллизации обычно больше чем 1 день и предпочтительно равен примерно от 3 дней до 20 дней.
Предпочтительно цеолит получают, используя мягкое перемешивание или размешивание.
В течение стадии гидротермической кристаллизации кристаллам SSZ-48 дают возможность самопроизвольно образовывать зародыши из реакционной смеси. Для уменьшения времени, необходимого для того, чтобы имела место полная кристаллизация, может быть благоприятным использование кристаллов SSZ-48 в качестве материала зародышей кристаллизации. Кроме того, введение затравки может вести к увеличенной чистоте полученного продукта путем промотирования ядрообразования и/или образования SSZ-48 вместо образования каких-либо нежелательных фаз. При использовании в качестве зародышей кристаллизации кристаллы SSZ-48 добавляют в количестве между 0,1 и 10% по массе диоксида кремния, используемого в реакционной смеси.
Как только образовались кристаллы цеолита, твердый продукт отделяют от реакционной смеси стандартными методами механического разделения, например фильтрацией. Кристаллы промывают водой и затем сушат, например, при температуре от 90 до 150oС в течение от 8 до 24 часов, чтобы получить кристаллы цеолита SSZ-48, как они синтезированы. Стадию сушки можно провести при атмосферном давлении или под вакуумом.
SSZ-48, как получен, имеет мольное отношение оксида, выбранного из оксида кремния, оксида германия и их смесей, к оксиду, выбранному из оксида алюминия, оксида галлия, оксида железа, оксида бора, оксида титана, оксида индия, оксида ванадия и их смесей, больше чем примерно 40, и имеет рентгенографические характеристики, приведенные в таблице 2 ниже.
SSZ-48 далее имеет состав, непосредственно после синтеза и в безводном состоянии, в терминах соотношений молей, показанный в таблице 3 ниже.
Существует метод увеличения мольного отношения кремнезема к бору посредством использования стандартных обработок кислотным выщелачиванием или хелатами. Более низкие отношения кремнезема к оксиду алюминия можно также получить, используя методы, приводящие к внедрению алюминия в кристаллический каркас. Например, внедрение алюминия может происходить при термической обработке цеолита в смеси со связующим веществом, представляющим собой оксид алюминия, или растворенным источником оксида алюминия. Такие процедуры описываются в патенте США 4559315, Chang et al., от 17 декабря 1985, приведенном здесь в качестве ссылки.
Цеолиты SSZ-48 после синтеза имеют кристаллическую структуру, порошковая рентгенограмма которой показывает характеристические линии, показанные в вышеприведенной таблице 2, и тем самым отличаются от других известных цеолитов.
После прокаливания цеолиты SSZ-48 имеют кристаллическую структуру, порошковая рентгенограмма которой включает характеристические линии, показанные в таблице 4.
Рентгенограммы определяли стандартными методами. Излучение представляло собой К-альфа/дублет меди. Высоту пиков и положение как функцию 2θ, где θ представляет собой угол Брегга, рассчитывали из относительных интенсивностей пиков и рассчитывали d, межплоскостное расстояние, в ангстремах, соответствующее записанным линиям.
Изменение в измерениях угла рассеивания (2θ), обусловленное ошибкой прибора и различиями между индивидуальными образцами, оценивают при ±0,30 градусах.
Вышеприведенная рентгенограмма из таблицы 2 представляет собой образец цеолитов непосредственно после синтеза или после приготовления. Меньшие изменения рентгенограммы могут быть результатом изменений в мольном соотношении кремнезема и оксида алюминия или кремнезема к бору в конкретном образце из-за изменений в постоянной решетки. Кроме того, достаточно маленькие кристаллы будут влиять на форму и интенсивность пиков, приводя к значительному уширению пиков.
Характерные пики рентгенограммы кальцинированного SSZ-48 показаны в таблице 4. Прокаливание также может привести к изменениям в интенсивностях пиков по сравнению с образцами материала непосредственно после приготовления, а также меньшим сдвигам в рентгенограмме. Цеолит, который получили обменом катионов металлов или других катионов, присутствующих в цеолите, на различные другие катионы (такие как H+ или NH4+), дает по существу такие же рентгенограммы, хотя снова могут быть незначительные отклонения в величинах межплоскостных расстояний и различия в относительных интенсивностях пиков. Несмотря на эти незначительные изменения, основная кристаллическая решетка остается неизменной после таких обработок.
Кристаллический SSZ-48 можно использовать в том состоянии, в котором он находится непосредственно после синтеза, но предпочтительно его надо термически обработать (прокалить). Обычно является желательным удалить катион щелочного металла ионным обменом и заместить его водородом, аммонием или любым желательным катионом металла. Цеолит можно подвергнуть выщелачиванию хелатообразующими реагентами, например ЭДТУ или растворами разбавленной кислоты, чтобы увеличить мольное отношение оксида кремния к оксиду алюминия. Также цеолит можно обработать водяным паром; обработка водяным паром помогает стабилизировать кристаллическую решетку, чтобы противостоять воздействию кислоты.
SSZ-48 и любой другой цеолит, используемый в настоящем способе, можно использовать в однородной смеси с гидрирующими компонентами такими, как вольфрам, ванадий, молибден, рений, никель, кобальт, хром, марганец или благородными металлами такими, как палладий или платина, для таких целей, в которых операция гидрирования-дегидрирования является желательной. Платина и палладий являются предпочтительными.
Металлы в цеолиты можно также ввести путем замены некоторых катионов в цеолите на катионы металлов посредством стандартной методики ионного обмена (смотри, например, патенты США 3140249 от 7 июля 1964 (Plank et al.); 3140251 от 7 июля 1964 (Plank et al.) и 3140253 от 7 июля 1964 (Plank et al. ), приведенные здесь в качестве ссылки. Типичные замещающие катионы могут включать катионы металлов, например редкоземельных металлов, металлов группы IA, группы IIА и группы VIII, а также их смеси. Среди замещающих катионов металлов особенно предпочтительными являются катионы металлов таких, как редкие земли, Mn, Ca, Mg, Zn, Cd, Pt, Pd, Ni, Co, Ti, Al, Sn и Fe.
Методы введения каталитически активных металлов в молекулярные сита описываются в литературе, и подходящими для использования в настоящем способе являются существовавшие ранее методы введения металла и обработки молекулярных сит для получения активного катализатора такие, как ионный обмен, импрегнирование или окклюзия в течение приготовления сита. Такие методики описываются в патентах США 3236761; 3226339; 3236762; 3620960; 3373109; 4202996; 4440781 и 4710485, приведенных здесь в качестве ссылки. Количество металла находится в диапазоне примерно от 0,01 до 10% от веса цеолита, предпочтительно примерно от 0,2 до 5%.
Водород, аммоний и металлические компоненты могут быть введены в цеолиты посредством ионного обмена. Они могут быть также импрегнированны металлами или металлы можно физически и однородно добавить в смесь с цеолитом, используя известные стандартные методики.
Типичные методы ионного обмена включают контакт синтетического цеолита с раствором, содержащим соль желаемого замещающего катиона или катионов. Хотя может применяться широкое разнообразие солей, особенно предпочтительными являются хлориды и другие галогениды, ацетаты, нитраты и сульфаты. Обычно цеолиты прокаливают перед процедурой ионного обмена для удаления органических веществ, присутствующих в каналах и на поверхности, поскольку это приводит к более эффективному ионному обмену. Характерные методы ионного обмена описываются во многих патентах, включая патенты США 3140249 от 7 июля 1964 (Plank et al.); 3140251 от 7 июля 1964 (Plank et al.) и 3140253 от 7 июля 1964 (Plank et al.), приведенных здесь в качестве ссылки.
Вслед за взаимодействием с раствором соли желаемого замещающего катиона цеолит обычно промывают водой и сушат при температурах в диапазоне примерно от 65 до 200oС. После промывки цеолит можно прокалить на воздухе или в атмосфере инертного газа при температурах в диапазоне примерно от 200 до 800oС в течение периода времени от 1 до 48 часов или более, чтобы получить каталитически активный продукт, необходимый в процессах конверсии углеводородов.
Независимо от катионов, присутствующих в синтезированной форме SSZ-48, пространственное окружение атомов, которые образуют основную кристаллическую решетку цеолита, остается в значительной степени неизменным.
Гидрирующий компонент присутствует в количестве, необходимом для получения эффективного катализатора гидродепарафинизации и гидроизомеризации, предпочтительно в диапазоне примерно от 0,05 до 5% по массе. Катализатор может работать в таком режиме, чтобы увеличить изомеризационную депарафинизацию при издержках из-за реакций крекинга.
Любые два или более из цеолитов, применяемых в настоящем способе, можно использовать в качестве катализатора депарафинизации в форме слоистого катализатора. То есть катализатор включает в себя первый слой, включающий, например, цеолит SSZ-48 и по крайней мере один металл VIII группы, и второй слой, включающий другой алюмосиликатный цеолит, например цеолит, который необязательно является более селективным, чем цеолит SSZ-48. Использование слоистых катализаторов описывается в патенте США 5149421 от 22 сентября 1992 (Miller), который приведен здесь в качестве ссылки. Слоистость также может включать в себя слой цеолита, например SSZ-48, покрытого слоем нецеолитного компонента, предназначенного либо для гидрокрекинга, либо для гидроочистки. Вместо слоистого строения другой полезный вариант этой концепции представляет собой однородно перемешанную каталитическую систему.
Пригодные в настоящем изобретении аморфные катализаторы представляют собой любые аморфные катализаторы, имеющие гидрирующее и/или изомеризационное действие на перерабатываемое сырье. Такие аморфные катализаторы описаны, например, в патенте США 4383913, приведенном здесь в качестве ссылки.
Они включают в себя, например, аморфные каталитические неорганические оксиды, например каталитически активные смешанные оксиды кремния и алюминия, глины, синтетические или активированные кислотой глины, кремнеземы, глиноземы, смешанные оксиды кремния и алюминия, смешанные оксиды кремния и циркония, смешанные оксиды кремния и магния, смешанные оксиды алюминия и бора, смешанные оксиды алюминия и титана, столбовидные или сшитые глины и подобные и их смеси.
Процесс проводят в условиях каталитической депарафинизации. Такие условия известны и описываются, например, в патентах США 5591322; 5149421 и 4181598, приведенных в качестве ссылки. Условия каталитической депарафинизации в огромной степени зависят от используемого сырья и от желательной температуры текучести. Предпочтительно, чтобы в течение процесса каталитической депарафинизации в реакционной зоне присутствовал водород. Отношение водорода к сырью, т. е. скорость циркуляции водорода, обычно лежит между примерно 500 и 30000 SCF/bbl (стандартный кубический фут на баррель) (89-5343 л (н.у.)/л), предпочтительно примерно от 1000 до 20000 SCF/bbl (от 178 до 3562 л (н.у.)/л). Как правило, водород подлежит отделению от продукта и повторному возвращению в реакционную зону.
Процент недогона фракционирующей колонны, повторно возвращаемого в сырье, представляет собой эффективное количество для увеличения общего выхода. Предпочтительно процент повторного возвращения в цикл равен примерно от 1 до 100 или более предпочтительно примерно от 10 до 50. Соотношение недогона фракционирующей колонны и исходного сырья является эффективным либо для уменьшения температуры текучести без какой-либо потери в выходе, либо для увеличения полного выхода при сохранении температуры текучести. Предпочтительно отношение равно примерно от 1:100 до 60:100 или, более предпочтительно, примерно от 1:100 до 40:100.
Возможно использование алюмосиликатного цеолитного катализатора с промежуточным размером пор и/или аморфного катализатора в таком же реакторе как и катализатор, представляющий собой силикоалюмофосфатные молекулярные сита, или их можно использовать в отдельном реакторе. Когда в одном реакторе используют два или более катализаторов, они могут образовывать последовательные слои или быть смешаны. Когда катализаторы размещены в последовательные слои, SAPO необязательно представляет собой первый или второй слой. Когда в одном реакторе используют два или более катализаторов, они могут быть однородно перемешанными. В способе по настоящему изобретению можно использовать любой обычный катализатор слоевой конфигурации.
Стадию каталитической изомеризации по настоящему изобретению можно проводить путем взаимодействия сырья, подлежащего депарафинизации, со стационарным неподвижным слоем катализатора, со стационарным псевдоожиженным слоем или с подвижным слоем по желанию. Простым и, следовательно, предпочтительным является режим работы в слое со струйным течением жидкости, в котором сырью позволяют течь струей через стационарный неподвижный слой, предпочтительно в присутствии водорода.
Используемые условия каталитической депарафинизации
зависят от используемого сырья и желаемой температуры текучести.
В способе по настоящему изобретению температура, как правило, равна примерно от 200 до 475oС, предпочтительно между примерно 250 и примерно 450oС. Давление, как правило, равно примерно от 15 psig (103, 41 кПа) до 3000 psig (20682 кПа), предпочтительно между примерно 200 psig (1378,8 кПа) и 3000 psig (20682 кПа). Часовая объемная скорость жидкости (LHSV) предпочтительно будет от 0,1 до 20, более предпочтительно между примерно 0,2 и 10.
Часто является желательным использовать мягкое гидрирование (иногда упоминаемое как гидроочистка). Стадия гидроочистки является выгодной при приготовлении продукта с приемлемой стабильностью (например, смазочного масла), поскольку ненасыщенные продукты имеют тенденцию являться нестабильными и распадаются при воздействии воздуха и света. Стадию гидроочистки можно выполнить после стадии изомеризации. Гидроочистку типично проводят при температурах, находящихся в диапазоне примерно от 190 до 340oС, при давлениях примерно от 400 psig (2757,6 кПа) до 3000 psig (20682 кПа), при объемной скорости жидкого продукта (LHSV) примерно от 0,1 до 20 и скоростями рециркулирования водорода примерно от 400 (71 л (н.у.)/л) до 1500 SCF/bbl (267 л (н. у.)/л).
Применяемый гидрирующий катализатор должен быть достаточно активным не только для того, чтобы гидрировать олефины и диены во фракциях смазочного масла, но также уменьшать содержание присутствующей ароматики.
Подходящие гидрирующие катализаторы включают традиционные металлические гидрирующие катализаторы, в частности металлы VIII группы, такие, как кобальт, никель, палладий и платина. Эти металлы типично соединены с носителями такими, как боксит, глинозем, силикагель, композиты на основе оксидов кремния и алюминия и кристаллические алюмосиликатные цеолиты и другие молекулярные сита. Палладий представляет собой особенно предпочтительный гидрирующий металл. Если это желательно, могут быть использованы неблагородные металлы VIII группы с молибдатами. Можно использовать оксиды или сульфиды металлов. Подходящие катализаторы описываются в патентах США 3852207; 4157294; 4921594; 3904513 и 4673487, приведенных здесь в качестве ссылки.
На чертеже показана упрощенная блок-схема одного варианта воплощения способа по настоящему изобретению. Поток исходного сырья для смазочного масла 5 и поток водорода 10 подают в зону каталитической депарафинизации 15, например аппарат каталитической депарафинизации ISODEWAXING. Поток 20 из зоны каталитической депарафинизации 15 подают в зону гидроочистки 25. Поток 30 из зоны гидроочистки 25 подают в ректификационную колонну, работающую при атмосферном давлении, 35 для начальной перегонки. Различные потоки продуктов, например поток газойля 36, поток лигроина 37, поток топлива для реактивных двигателей 38 и поток недогона 40 удаляют из ректификационной колонны, работающей под атмосферном давлением, 35. Поток недогона 40 из ректификационной колонны, работающей под атмосферном давлением, 35 подают в ректификационную колонну, работающую под вакуумом, 45 для дальнейшего разделения на фракции. Различные потоки продуктов, например поток дизельного топлива 50, поток 55 дистилляционных масел средней вязкости 60, поток 60 дистилляционных масел средней вязкости 100 и поток недогона (или поток дистилляционных масел средней вязкости 300) 65 удаляют из ректификационной колонны, работающей под вакуумом, 45. Часть потока недогона 65 удаляют в виде потока 70 дистилляционных масел средней вязкости 300, а часть возвращают на повторную переработку как поток 75 для смешения с потоком свежего сырья 5.
Далее настоящее изобретение будет разъяснено следующими примерами, которые предназначены быть чисто иллюстративными для настоящего изобретения.
Преимущества операции рецикла недогона фракционирующей колонны были показаны при крупномасштабном испытании на пилотной установке. Для демонстрационного цикла реактор изодепарафинизации содержал примерно 5000 см3 катализатора SAPO-11, импрегнированного благородным металлом, и реактор гидроочистки содержал примерно 5000 см3 катализатора гидроочистки от компании Chevron. Перегонка в оперативном режиме давала 3 фракции смазочного масла и фракцию среднего дистиллята. Пилотная установка устроена таким образом, чтобы смоделировать блок-схему, показанную на чертеже.
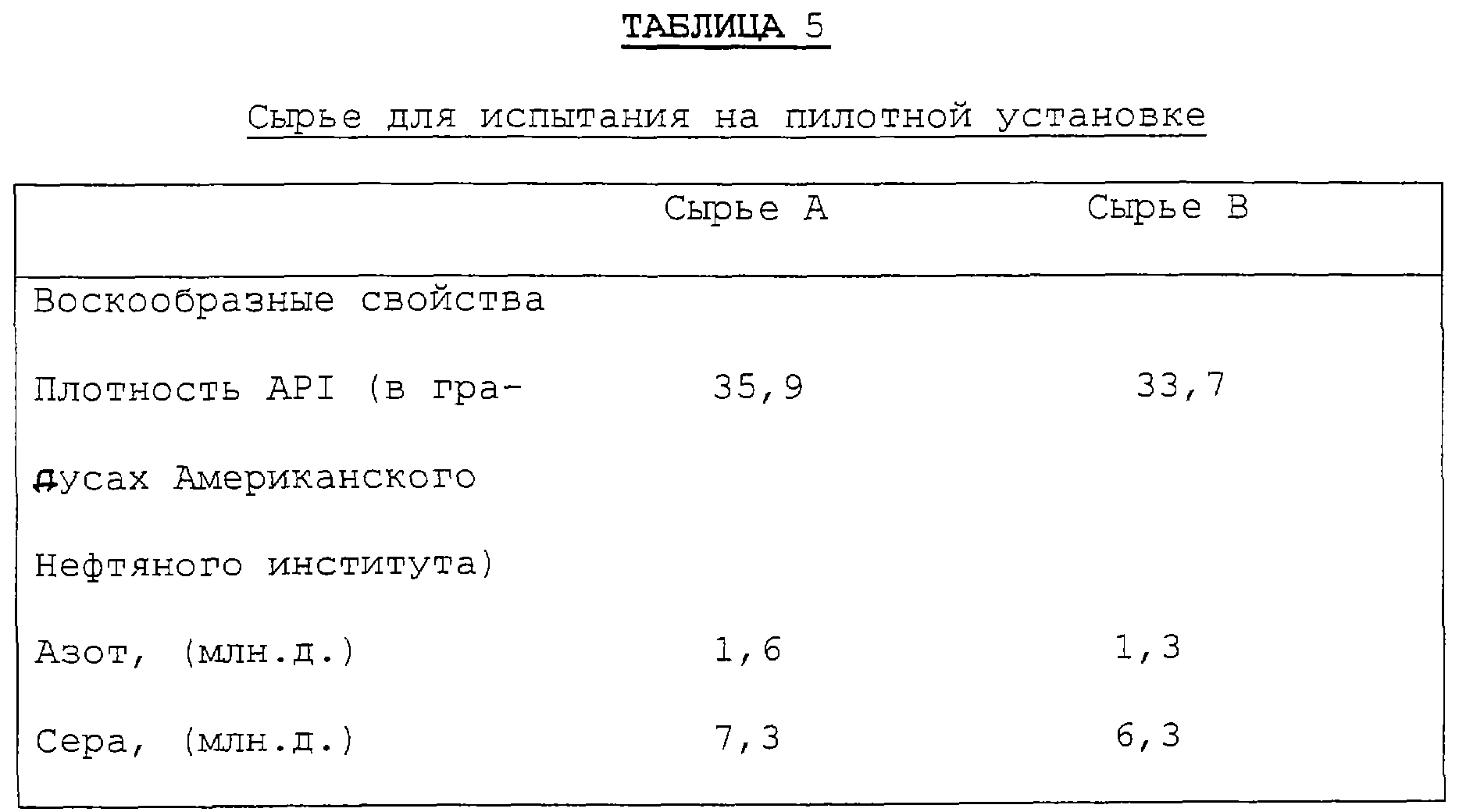
Были протестированы два типа перерабатываемого сырья с широкими интервалами температур кипения. Данные контроля этого перерабатываемого сырья представлена в таблице 5. После изодепарафинизации и гидроочистки весь жидкий продукт фракционировали на 3 окончательные маслосодержащие погоны - дистиллятное масло 60, дистиллятное масло 100 и дистиллятное масло 300. Таблица 6 суммирует улучшения эксплуатационных характеристик, которые получены возвращением на повторную переработку недогона фракционирующей колонны.
Для испытаний 1-4 скорость подачи свежего сырья поддерживали примерно постоянной в то время, как изменяли процент недогона фракционирующей колонны, направляемого на повторную переработку, и средневесовую температуру слоя изодепарафинизатора (WAT). Аппарат для гидроочистки в течение этих циклов работал при примерно постоянной температуре.
А. Сравнение испытания 1 и испытания 2 показывает, что при повторном возвращении на переработку значительной части недогона фракционирующей колонны является возможным уменьшить WAT изодепарафинизатора и, хотя степень депарафинизации не вполне одинакова в обоих случаях, резко увеличить общий выход масла - от 70 до 80%. Повторное возвращение на переработку также сдвигает температуры текучести 100N и 300N намного ближе друг к другу - разница между температурами текучести равна 18oС в испытании 2, но только 9oС в испытании 1. Это значит, что при возвращении на повторную переработку 100N не должна быть избыточно депарафинизирована так, чтобы сделать приемлемой температуру текучести по 300N.
В. Испытание 3 провели до такой же температуры текучести 300N, как в испытании 2. При возвращении на повторную переработку (испытание 3) оказалось возможным увеличить общий выход смазочного масла на 3% и увеличить выход ценной фракции 100N с низкой температурой текучести на 4%. В данном случае снова при возвращении на повторную переработку степень избыточной депарафинизации 100N уменьшается.
С. Испытание 4 провели при той же средневесовой температуре слоя изодепарафинизатора, как в испытании 2, но в испытании 4 перерабатываемое сырье изодепарафинизатора содержало 13% недогона фракционирующий колонны (направленный на повторную переработку). Хотя общий выход смазочного масла остается таким же, в испытании 4 300N было на 2% меньше, а 60N на 2% больше. Более важно, что в испытании 4 температуры текучести конечных фракций масла значительно ниже. Таким образом, возвращение на повторную переработку может улучшить свойства продукта конечных масел без изменения общего выхода.
Для испытаний 5-7 меняли скорость подачи свежего сырья также, как процент недогона из фракционирующей колонны. Здесь снова аппарат для гидроочистки работал при приблизительно постоянной температуре.
А. Для испытания 6 скорость подачи свежего сырья увеличили на 23% без повторного возвращения на переработку. Чтобы сохранить примерно такую же температуру текучести по 300N, WAT изодепарафинизатора должна была быть увеличена на 5oF (2,8oС), но в этом случае (без повторного возвращения на переработку), общий выход смазочного масла оставался таким же, в то время как температура текучести фракций смазочного масла незначительно увеличилась (стала хуже).
В. Для испытания 7 скорость подачи свежего сырья по существу поддерживали постоянной и исходное сырье для изодепарафинизатора содержало 14% недогона из фракционирующей колоны. В этом случае оказалось возможным слегка понизить температуру катализатора изодепарафинизатора, в тоже время близко придерживаясь тех же самых температур текучести продукта, и оказалось, что общий выход смазочного масла увеличился на 2%. Возможно более важным является то, что выход 100N увеличился на 4% в то время, как температура текучести 100N слегка упала.
Указанные выше примеры сравнения показывают неожиданное улучшение эксплуатационных характеристик при повторном возвращении на переработку части недогона фракционирующей колонны. Поскольку увеличение количества недогона, возвращаемого на повторную переработку, в конечном счете будет ограничивать количество свежего сырья, которое может быть переработано, экономические ограничения обычно будут диктовать максимальное количество, возвращаемое на повторную переработку.
Для обоих типов сырья существует положительный эффект по выходу и температуре текучести в случае возврата на повторную переработку.
НАДПИСИ К ЧЕРТЕЖУ
5 - Исходное сырье
10 - Водород
15 - Аппарат каталитической депарафинизации
20 - Поток сырья подвергнутого каталитической
депарафинизации
25 - Аппарат гидроочистки
30 - Поток на выходе из аппарата гидроочистки
35 - Ректификационная колонна, работающая при атмосферном давлении
36
- Газойль
37 - Лигроин
38 - Топливо для реактивных двигателей
40 - Недогон
45 - Ректификационая колонна, работающая под вакуумом
50 - Дизельное топливо
55 - Дистиляционное масло средней вязкости 60
60 - Дистиляционное масло средней вязкости 100
65 - Недогон
70 - Дистиляционное масло средней вязкости 300
75
- Поток на повторную переработкуе
Реферат
Использование: нефтехимия. Сущность: проводят (1) взаимодействие исходного углеводородного нефтяного сырья в присутствии добавленного газообразного водорода с катализатором, выбранным из группы, состоящей из SAPO-11, SAPO-31 или SAPO-41 силикоалюмофосфатных молекулярных сит, имеющих промежуточный размер пор, и гидрирующим компонентом и их смесями, где по крайней мере часть исходного сырья конвертируют, (2) подачу по крайней мере части конвертированного исходного сырья в фракционирующую колонну, где по крайней мере часть конвертированного исходного сырья разделяют на фракции, с получением по крайней мере одной головной фракции и одной фракции недогона и (3) смешивание по крайней мере части фракции недогона с углеводородным нефтяным сырьем со стадии (1). Технический результат - повышение выхода целевого продукта или снижение температуры текучести при неизмененном выходе. 2 с. и 8 з.п. ф-лы, 1 ил., 6 табл.

Комментарии