Способ получения углеводородных фракций из смесей биологического происхождения - RU2464297C2
Код документа: RU2464297C2
Чертежи

Описание
Данное изобретение относится к способу получения углеводородных фракций, которые можно использовать в качестве дизельного топлива или в качестве компонента дизельного топлива, исходя из смеси биологического происхождения, содержащей эфиры жирных кислот, возможно, с некоторым количеством свободных жирных кислот. Способ включает по меньшей мере одну стадию дезоксигенирования и стадию гидроизомеризации. Применение растительных масел в дизельных двигателях восходит к Рудольфу Дизелю, который в 1900 году продемонстрировал возможность работы дизельных двигателей на кокосовом масле.
Во время Второй мировой войны в качестве топлива для военных автомобилей в Африке применяли как пальмовое масло, так и кокосовое масло. После войны развитие технологий привело к практически исключительному применению топлив, полученных из нефти; к тому же дизельные двигатели были чрезвычайно усовершенствованы, особенно в отношении инжекторов и систем контроля, до такой степени, что они отличались малой гибкостью в отношении применения топлив, отличных от газойля. В то же время от растительных топлив постепенно отошли из-за высокой стоимости производства и непостоянства качества продукта.
Во время нефтяного кризиса семидесятых годов внимание снова было сконцентрировано на использовании растительных масел в качестве дизельных топлив, но это было сложным по ряду причин (образование поверхностного слоя загрязнений в двигателе внутреннего сгорания, забивание инжекторов, растворение в смазочных маслах). Затем исследования были направлены на получение исходя из растительных масел метиловых или этиловых эфиров и их использование в дизельных двигателях. Метиловые и этиловые эфиры жирных кислот получают из растительных масел путем трансэтерификации метанолом или этанолом. Другой подход к превращению растительных масел был предложен в восьмидесятых годах и заключается в их принудительной гидрогенизации с получением углеводородных фракций с температурой кипения, соответствующей дизельным топливам, полученным из нефти. Принудительная гидрогенизация растительных масел вызывает удаление кислорода с одновременным образованием смеси Н2О, СО2 и СО во взаимных соотношениях, изменяющихся в зависимости от рабочих условий. Таким образом, исходный эфир преимущественно превращают в углеводороды, что относится как к жирным кислотам, так и к глицерину. Совместно с углеводородами можно получать небольшое количество свободных спиртов.
Реакцию принудительной гидрогенизации жирных кислот с получением жидких топлив изучали еще в восьмидесятые годы, например, Nunes et al., которые в статье, озаглавленной «Проводимый под давлением гидрокрекинг соевого масла: метод исследования и общий ход протекания реакции» ("Hydrocraquage sous pression d′une huile de soya: procede d'etude et allure generale de la transformation", Rev. Inst. Fr. Pet. 1986, vol.41, page 421 и далее) описывают гидрокрекинг соевого масла с бифункциональным катализатором. При температуре выше 673 К наблюдается декарбонилирование и декарбоксилирование жирных кислот совместно с выраженным гидрогенолизом вследствие присутствия металлического катализатора. Основными продуктами являются углеводороды с линейной цепью.
J.Gusmao et al. («Применение растительных масел в качестве альтернативного источника дизельного топлива: гидрокрекинг на восстановленном Ni/SiO2 и сульфидированном Ni-Mo/Al2O3» ("Utilization of vegetable oils as an alternative source for diesel-type fuel: hydrocracking on reduced Ni/SiO2 and sulphided Ni-Mo/Al2O3"); Catalysis Today 5 (1989); p.533 и далее) показывают, что при гидрогенизации соевого масла полученная углеводородная фракция состоит в основном из линейных парафинов (96 мол.% C15-C16-С17-C18).
Патент США 4992605 описывает способ получения углеводородных фракций в диапазоне С15-C18 путем гидрогенизации таких растительных масел, как подсолнечное масло, рапсовое масло, масло канолы, пальмовое масло, или жирных масел, содержащихся в сосновой пульпе (талловое масло). Эта углеводородная фракция состоит преимущественно из линейных парафинов (С15-C18) и характеризуется высоким цетановым числом, которое имеет такое значение, что эту углеводородную фракцию можно применять для повышения цетанового числа.
В статье «Гидрогенизированные растительные масла для улучшения дизельного топлива» ("Hydroprocessed vegetable oils for diesel fuel improvement", Bioresources Technology, 56 (1996)), приведено краткое описание патентной заявки, описанной в патенте США 4992695, раскрывающем получение гидрогенизованного продукта из масла канолы в лабораторном масштабе. Углеводородная фракция почти полностью состоит из линейных парафинов, а фракция, которая перегоняется в пределах диапазона температур перегонки дизельного топлива, имеет цетановое число в диапазоне от 55 до 90. Другие углеводородные продукты включают легкие C1-С5 углеводороды, воду и СО2. Эта фракция дизельного топлива охарактеризована как «супер-цетановая». Плотность (0,790 г/мл) является сравнимой с дизельным топливом, в то время как вязкость слегка выше. Однако действительное ограничение этой фракции связано с плохими свойствами при низкой температуре (температура помутнения и температура потери текучести, превышающие 20°С), которые связаны с линейностью парафинов. Из этих соображений «супер-цетановую» фракцию можно применять в смеси с обычным дизельным топливом, но не в зимние месяцы.
ЕР 1396531 описывает способ получения углеводородных компонентов из смесей растительного и животного происхождения. Описано получение смеси с содержанием изопарафинов порядка 73%. Способ включает стадию предварительной гидрогенизации, стадию гидродезоксигенирования (ГДО) и стадию изомеризации, которую проводят с использованием принципа противотока. Стадия предварительной гидрогенизации, которую проводят в мягких условиях, необходима для того, чтобы насытить имеющиеся двойные связи и избежать нежелательных побочных реакций на последующих стадиях процесса. На стадии изомеризации является абсолютно необходимым работать в противотоке, чтобы защитить катализатор от дезактивации, вызываемой водой, которая содержится в питающем потоке, поступающем с предшествующей стадии ГДО: при работе в противотоке часть воды, которая содержится в углеводородном питающем потоке, удаляют до того, как указанный питающий поток вступает в контакт со всем количеством катализатора каталитического слоя.
В настоящее время найден способ получения углеводородной смеси, которую можно использовать в качестве дизельного топлива или в качестве компонента газойля, путем гидродезоксигенирования смеси биологического происхождения, содержащей эфиры жирных кислот, возможно, с некоторым количеством свободных жирных кислот, такой как, например, растительные масла (подсолнечное, рапсовое, масло канолы, пальмовое), или же жирные масла, содержащиеся в сосновой пульпе (талловое масло), с последующей гидроизомеризацией, который позволяет получить смеси углеводородов, в которых содержание изопарафинов может превышать 80%, а оставшаяся часть представляет собой н-парафины.
Конкретные составы катализаторов, которые применяют на стадии гидроизомеризации, не только позволяют получить в качестве дизельного топлива продукт высокого качества в сравнении с продуктами из погонов, полученных известными способами, но они также имеют характеристики, которые позволяют применять их в присутствии воды, не подвергая их дезактивированию, или же в любом случае их легко регенерировать непосредственно в ходе гидроизомеризации посредством умеренного подъема температуры.
Таким образом, целью данного изобретения является способ получения углеводородной фракции, которую можно применять в качестве дизельного топлива или в качестве компонента дизельного топлива, из смеси биологического происхождения, содержащей эфиры жирных кислот и, возможно, содержащей также свободные жирные кислоты, который включает следующие стадии:
1) гидродезоксигенирование смеси биологического происхождения;
2) гидроизомеризация смеси, полученной на стадии (1), после возможной обработки с целью очистки, при этом указанную гидроизомеризацию проводят в присутствии каталитической системы, которая включает:
а) носитель кислой природы, включающий полностью аморфный микромезопористый оксид кремния-алюминия, имеющий мольное соотношение SiO2/Al2O3 в диапазоне от 30 до 500, площадь поверхности более 500 м2/г, объем пор в диапазоне от 0,3 до 1,3 мл/г, средний диаметр пор ниже 40 Å,
b) металлический компонент, содержащий один или более металлов группы VIII, возможно, смешанных с одним или более металлов группы VIB.
Смеси биологического происхождения, используемые в способе согласно изобретению, содержат эфиры жирных кислот, возможно, с некоторым количеством свободных жирных кислот, и они могут представлять собой смеси растительного или животного происхождения. Количество жирных кислот может изменяться, например, от 2 до 20 мас.% от общего количества смеси биологического происхождения. Эфиры жирных кислот, которые содержатся в указанных смесях, обычно представляют собой триглицериды жирных кислот, в которых углеводородная цепь жирной кислоты может содержать от 12 до 24 атомов углерода и может быть моно- или полиненасыщенной. Смесь биологического происхождения может быть выбрана из растительных масел, растительных жиров, животных жиров, рыбьих жиров или их смесей. Растительные масла или жиры могут представлять собой подсолнечное, рапсовое масла, масло канолы, пальмовое, соевое, конопляное, оливковое, льняное, арахисовое, касторовое, горчичное, кокосовое масла или жирные масла, которые содержатся в сосновой пульпе (талловое масло), или их смеси. Животные масла или жиры могут быть выбраны из свиного жира, лярда, сала, молочных жиров и их смесей. Также можно использовать жиры или масла как животного, так и растительного происхождения, являющиеся отходами пищевой промышленности. Растительные масла или жиры могут также происходить из растений, выведенных в результате генных манипуляций.
Смеси биологического происхождения, применяемые в способе согласно изобретению, можно также смешивать с другими компонентами перед подачей их в процесс, например их можно смешивать с одним или более углеводородами.
На первой стадии (стадии ГДО) смесь биологического происхождения гидродезоксигенируют водородом в присутствии катализатора гидродезоксигенирования.
На этой стадии происходит гидрогенизация двойной связи, присутствующей в эфирных цепях триглицерида, крекинг структуры триглицерида и дезоксигенирование посредством как декарбоксилирования, так и гидрогенизации, с образованием воды.
Все катализаторы, которые могут быть использованы, представляют собой известные в данной области катализаторы гидрогенизации, содержащие один или более металлов, выбранных из металлов группы VIII и группы VIB, нанесенные на соответствующий носитель. Носители, подходящие для данной цели, состоят из одного или более оксидов металлов, предпочтительно оксида алюминия, оксида кремния, оксида титана, оксида циркония или их смеси. Металл или металлы предпочтительно выбирают из Pd, Pt или из пар металлов Ni-Mo, Ni-W, Co-Mo, причем предпочтительными являются Co-W, Ni-Mo и Со-Мо. Эти катализаторы обычно получают путем пропитки оксидного носителя раствором соответствующей соли металла или металлов. После пропитки проводят термообработку в атмосфере, подходящей для разложения соли-предшественника и получения нанесенного на подложку металла. Можно провести несколько последующих пропиток, чтобы достичь желаемого уровня загрузки по металлу и в случае различных металлов, чтобы дифференцировать их нанесение на подложку. Известны также способы получения указанных катализаторов, применяющие вместо пропитки осаждение предшественника металла из раствора соли этого металла на носитель или же совместное осаждение различных компонентов катализатора, то есть металла и носителя.
Можно также использовать каталитические композиции, такие как Ni-Мо-Р на цеолите, Pd/цеолит, Pt/MSA, где MSA представляет собой оксид кремния-алюминия, имеющий конкретные характеристики, описанные в ЕР 340868, ЕР 659478, ЕР 812804, применяемый также в качестве носителя для каталитических композиций, используемых на последующей стадии гидроизомеризации. Катализаторы, которые с успехом могут быть использованы на стадии ГДО согласно изобретению, описаны, например, в работе J.T.Richardson «Принципы разработки катализаторов» ("Principal of catalyst development"), Plenum Press, New York, 1989, гл.6.
Катализаторы типа Ni-Mo, Ni-W, Co-Mo и Co-W предварительно сульфидируют. Процедуры предварительного сульфидирования проводят в соответствии с известными технологиями.
Для того чтобы поддерживать катализатор в сульфидированной форме, сульфидирующий агент, например диметилдисульфид, подают одновременно с загрузкой биологического происхождения после возможной стадии очистки указанной загрузки в количестве от 0,02 до 0,5 мас.% (140-3400 ppm S).
Альтернативно, возможно осуществлять процесс при совместной подаче газойля прямой перегонки с высоким содержанием S (S>1%), в такой концентрации, которая почти соответствует такому же суммарному содержанию S в загрузке.
Реакцию ГДО проводят в реакционной зоне, содержащей один или более каталитических слоев, в одном или более реакторов. В соответствии с предпочтительным аспектом ее осуществляют в типичном реакторе гидроочистки с неподвижным слоем катализатора. Поток водорода и питающий поток биологического происхождения можно направить прямотоком или противотоком. Реактор может иметь адиабатические каталитические слои в количестве, равном или превышающем 2. Поскольку реакция является экзотермической, с выделением тепла, в каждом слое катализатора происходит подъем температуры. Посредством подачи между одним и другим слоями катализатора потока водорода и/или жидкого питающего потока с определенной температурой можно получить постоянный или возрастающий профиль температуры. Эту рабочую процедуру обычно обозначают как «раздельная подача».
Кроме реактора с адиабатическими слоями можно использовать кожухотрубчатый реактор. Катализатор обычно загружают в трубы, в то время как в межтрубное пространство подают диатермическую жидкость (охлаждающее масло) для отвода тепла реакции.
Независимо от того, применяют ли реактор с адиабатическими слоями или кожухотрубчатый реактор, для лучшего регулирования температурного профиля в реакторе сам реактор может работать с рециркуляцией части потоков в соответствии с типологией, известной как реактор с рециркуляцией. Задачей рециркуляции является разбавление свежего питающего потока в реакторе, таким образом лимитируя температурные максимумы, обусловленные экзотермичностью реакции. Соотношение рециркуляции, то есть количество возвращаемой в реактор фракции по отношению к свежей загрузке, может изменяться от 0,5 до 5 по массе.
Еще одной конфигурацией реактора, которую можно использовать для этого применения, является суспензионный реактор, в котором катализатор гидродезоксигенирования формируют в виде микросфер и диспергируют в реакционной среде. Смешивание газа, жидкости и твердого вещества в этом случае можно проводить механическим перемешиванием или путем принудительной рециркуляции реакционных потоков.
Стадию ГДО предпочтительно проводят при давлении, изменяющемся от 2,5 до 7,0 МПа (от 25 до 70 бар), предпочтительно от 3,0 до 5,0 МПа (от 30 до 50 бар), и при температуре в диапазоне от 240 до 450°С, предпочтительно от 270 до 430°С. Предпочтительно работать при объемной скорости в диапазоне от 0,5 до 2 ч-1, еще более предпочтительно от 0,5 до 1 ч-1. Соотношение Н2/смесь биологического происхождения предпочтительно находится в диапазоне от 400 до 2000 л(н.у.)/л.
Перед стадией ГДО питающий поток биологического происхождения можно соответствующим образом обработать для того, чтобы удалить содержащиеся в нем щелочные металлы (например, Na, K) и щелочноземельные металлы (например, Са), которые могут находиться в загрузке. Эту предварительную обработку можно осуществить путем адсорбции на подходящем материале: например, можно использовать известные способы фильтрации на колонне, заполненной кислой землей или глинами, например монтмориллонитами, бентонитами, смектитами, кислыми сепиолитами. Для этой цели можно использовать имеющиеся в продаже продукты, такие как Filtrol, Tonsil, Bentolites H и L, SAT-1.
Альтернативно можно применять ионообменные смолы или слабокислую промывку, осуществляемую, например, путем контакта с серной кислотой, азотной кислотой или соляной кислотой, предпочтительно при комнатной температуре и атмосферном давлении.
Поток, выходящий со стадии ГДО (1), предпочтительно подвергают обработке с целью очистки перед подачей на последующую стадию гидроизомеризации. Обработка с целью очистки может включать стадию разделения и стадию промывки. В соответствии с этим предпочтительным аспектом потоки, выходящие со стадии (1), направляют в газожидкостный сепаратор высокого давления. Получают газовую фазу, состоящую в основном из водорода, воды, СО и СО2 и легких парафинов (С4-). В небольших количествах могут также присутствовать NH3, PH3 и H2S. После отделения газовую фазу охлаждают и путем конденсации отделяют воду (возможно, содержащую следы спиртов и карбоновых кислот) и способные к конденсации углеводороды. Оставшуюся газовую фазу очищают для обеспечения возможности рециклировать водород на стадию (1) реакции. Для очистки применяют способы, известные в технике - посредством щелочной промывки, например, водными растворами NaOH или Са(ОН)2 или посредством хорошо известных способов очистки аминами (например, МЭА - моноэтаноламином или ДЭА - диэтаноламином). В конце очистки СО2, H2S, PH3 и NH3 удаляют, и полученная таким образом газовая фракция состоит по существу из Н2, возможно, со следами СО. Для того чтобы ограничить накопление СО в рециклируемых газах, его можно удалять медно-аммиачной промывкой или метанированием в соответствии с технологиями, известными специалистам в данной области.
Жидкая фаза, отделенная в сепараторе высокого давления, состоит из углеводородной фракции, состоящей в основном из линейных парафинов с числом атомов углерода от 14 до 21, преимущественно от 15 до 19. В зависимости от рабочих условий в сепараторе жидкая фракция может содержать небольшие количества Н2О и кислородсодержащих соединений, таких как, например, спирты и карбонильные соединения. Содержание остаточной серы может быть ниже 10 ppm. Затем жидкую фракцию можно промыть газообразным углеводородом, например СН4, или азотом или водородом, в десорбере для дополнительного снижения содержания воды.
Полученную смесь углеводородов подают на последующую стадию (2) гидроизомеризации. Стадию гидроизомеризации проводят в присутствии водорода и каталитической композиции, которая включает:
a) носитель кислой природы, включающий полностью аморфный микромезопористый оксид кремния-алюминия, имеющий мольное соотношение SiO2/Al2O3 в диапазоне от 30 до 500, площадь поверхности выше 500 м2/г, объем пор в диапазоне от 0,3 до 1,3 мл/г, средний диаметр пор ниже 40 Å,
b) металлический компонент, содержащий один или более металлов группы VIII, возможно, смешанные с одним или более металлов группы VIB.
Носитель кислой природы (а) каталитической композиции, используемой в данном изобретении, включает оксид кремния-оксид алюминия, предпочтительно имеющий мольное соотношение SiO2/Al2O3 в диапазоне от 50 до 300.
В соответствии с предпочтительным аспектом носитель кислой природы (а) включает оксид кремния-алюминия с пористостью в диапазоне от 0,3 до 0,6 мл/г.
Полностью аморфные микромезопористые оксиды кремния-алюминия, которые можно применять в качестве носителя (а) каталитической композиции стадии гидроизомеризации по настоящему изобретению, описаны в US 5049536, ЕР 659478, ЕР 812804, и они называются MSA. Их порошковые рентгенограммы не имеют кристаллической структуры и не дают никаких пиков. US 5049536, ЕР 659478, ЕР 812804 описывают также различные способы получения оксидов кремния-алюминия, пригодных в качестве носителя (а). В соответствии с ЕР 659478 оксиды кремния-алюминия, которые можно использовать, например, для способа по настоящему изобретению, можно получить исходя из гидроксида тетраалкиламмония, соединения алюминия, которое можно гидролизовать до Al2O3, и соединения кремния, которое можно гидролизовать до SiO2; в этом случае упомянутый гидроксид тетраалкиламмония представляет собой гидроксид тетра(С2-С5) алкиламмония, указанное способное к гидролизу соединение алюминия представляет собой три(С2-С4)алкоксид алюминия, а указанное способное к гидролизу соединение кремния представляет собой тетра(С1-С5)алкилортосиликат; эти реагенты подвергают гидролизу и гелеобразованию, работая при температуре, равной или превышающей температуру кипения, при атмосферном давлении, любого спирта, который получается в качестве побочного продукта указанной реакции гидролиза, без удаления (или существенного удаления) указанных спиртов из реакционной среды. Полученный таким образом гель сушат и прокаливают, предпочтительно в окислительной атмосфере, при температуре в диапазоне от 500 до 700°С, в течение периода порядка 6-10 часов. Предпочтительно проводить процесс, получая водный раствор гидроксида тетраалкиламмония и триалкоксида алюминия и добавляя к указанному водному раствору тетраалкилортосиликат, проводя процесс при температуре ниже, чем температура гидролиза, при таком количестве реагентов, чтобы обеспечить мольное соотношение SiO2/Al2O3 от 30/1 до 500/1, мольное соотношение гидроксид тетраалкиламмония/SiO2 от 0,05/1 до 0,2/1 и мольное соотношение H2O/SiO2 от 5/1 до 40/1, а гидролиз и гелеобразование осуществляют путем нагревания до температуры выше чем примерно от 65°С и до 110°С, работая в автоклаве при аутогенном давлении системы или же при атмосферном давлении в реакторе, снабженном конденсатором.
В соответствии с ЕР 812804 оксид кремния-алюминия, который можно использовать в качестве компонента (а) каталитической композиции для стадии гидроизомеризации, можно получить посредством способа, который включает:
- получение смеси, исходя из тетраалкилортосиликата, спирта на основе С3-С6алкила или двухатомного спирта, гидроксида тетраалкиламмония, имеющего формулу R1(R2)3NOH, где R1 представляет собой С3-С7алкил, a R2представляет собой C1 или С3-С7 алкил, в присутствии способного к гидролизу соединения алюминия; при этом мольные соотношения находятся в следующих диапазонах:
спирт/SiO2≤20
R1(R2)NOH/SiO2=0,05-0,4
H2O/SiO2=1-40
Al2O3/SiO2 - более 0 и менее 0,02;
- проведение гидролиза и последующего гелеобразования указанной смеси при температуре, близкой к температуре кипения присутствующего спирта или смеси спиртов;
- проведение сушки и прокаливания полученного геля.
Носитель кислой природы (а) катализатора, который применяют в способе согласно изобретению, может находиться в форме экструдированного продукта, содержащего традиционные связующие, такие как, например, оксид алюминия, бемит или псевдобемит. Экструдированный продукт можно получить в соответствии со способами, хорошо известными специалистам в данной области. Оксид кремния-алюминия и связующее можно предварительно смешать в массовых соотношениях, находящихся в диапазоне от 30:70 до 90:10, предпочтительно от 50:50 до 70:30. В конце смешивания полученный продукт отверждают в желаемой конечной форме, например в виде экструдированных гранул или таблеток. В соответствии с предпочтительным примером реализации можно использовать способы и связующие, описанные в ЕР 550922 и ЕР 665055, причем предпочтительным является последний и содержание его включено в текст настоящего описания посредством ссылки.
Типичный способ получения компонента с кислой природой (а) в виде экструдированного продукта (ЕР 665055) включает следующие стадии:
(A) получение водного раствора гидроксида тетраалкиламмония (ТАА-ОН), растворимого соединения алюминия, способного гидролизоваться до Al2O3, и соединения кремния, способного гидролизоваться до SiO2, в следующих мольных соотношениях:
SiO2/Al2O3 от 30/1 до 500/1
TAA-OH/SiO2 от 0,05/1 до 0,2/1
H2O/SiO2 от 5/1 до 40/1;
(B) нагревание полученного таким образом раствора для осуществления его гидролиза и гелеобразования и получение смеси А с вязкостью в диапазоне от 0,01 до 100 Па·с;
(C) добавление к смеси А сначала связующего, принадлежащего к группе бемитов или псевдобемитов, в массовом соотношении со смесью А в диапазоне от 0,05 до 0,5, а затем минеральной или органической кислоты в количестве от 0,5 до 8,0 г на 100 г связующего;
(D) нагревание смеси, полученной на стадии (С), до температуры в диапазоне от 40 до 90°С, до получения однородной пасты, которую подвергают экструдированию и гранулированию;
(E) сушка и прокаливание экструдированного продукта в окислительной атмосфере.
Предпочтительно также добавлять на стадии (С) пластификаторы, например метилцеллюлозу, чтобы способствовать образованию однородной и легко поддающейся обработке пасты.
Таким образом получают гранулированный кислый носитель, предпочтительно содержащий количество инертного неорганического связующего от 30 до 70 мас.%, а остальное количество составляет аморфный оксид кремния-алюминия, имеющий по существу такие же характеристики в отношении пористости, площади поверхности и структуры, как и описанные выше для такого же оксида кремния-алюминия без связующего.
Что касается металлов, содержащихся в металлическом компоненте (b) каталитической композиции, применяемой на стадии гидроизомеризации способа согласно изобретению, то их выбирают из металлов группы VIII, возможно, смешанных с одним или более металлов группы VIB. Предпочтительными являются композиции, содержащие только металлы группы VIII. Металл или металлы группы VIII предпочтительно выбирают из Pt, Pd, Ni и Со. В частности, если металлический компонент содержит только металлы группы VIII, то этот металл или металлы предпочтительно выбирают из Pt, Pd и Ni. Если металлический компонент содержит как один или более металлов группы VIII, так и один или более металлов группы VIB, то металл группы VIII предпочтительно выбирают из Ni и Со. Металл группы VIB предпочтительно выбирают из Мо и W.
Металл группы VIII предпочтительно находится в диапазоне от 0,1 до 5 мас.% по отношению к общей массе каталитической композиции. Металл группы VIB, если он присутствует, находится в количестве в диапазоне от 1 до 50, еще более предпочтительно в количестве в диапазоне от 5 до 35 мас.%, по отношению к общей массе каталитической композиции. Массовый процент металла или металлов выражает содержание металлов, рассчитанное на металл в виде элемента; в конечном катализаторе после прокаливания указанный металл находится в форме оксида.
Металлы группы VIII и, возможно, группы VI, которые находятся в каталитической композиции, применяемой на стадии (2) гидроизомеризации, можно нанести на носитель (а) любым способом, известным специалистам в данной области. Каталитические композиции, которые с успехом можно применять на стадии гидроизомеризации по данному изобретению, содержащие один или более металлов группы VIII, а также их получение описаны в ЕР 582347, ЕР 110813 и WO 2005/103207.
В частности, ЕР 582347 описывает каталитические композиции, которые можно использовать при гидроизомеризации н-парафинов, содержащие один или более металлов группы VIII и носитель из алюмосиликагеля, аморфного при анализе с помощью рентгеновских лучей, с мольным соотношением SiO2/Al2O3 в диапазоне от 30 до 500, площадью поверхности в диапазоне от 500 до 1000 м2/г, объемом пор в диапазоне от 0,3 до 0,6 мл/г и диаметром пор преимущественно в диапазоне от 10 до 30 Å. ЕР 110813 описывает каталитические композиции, которые можно использовать для получения средних фракций, содержащие один или более металлов группы VIII и носитель из прокаленного алюмосиликагеля, аморфный при анализе с помощью рентгеновских лучей, с мольным соотношением SiO2/Al2O3 в диапазоне от 30 до 500, площадью поверхности в диапазоне от 500 до 1000 м2/г, объемом пор в диапазоне от 0,2 до 0,8 мл/г и средним диаметром пор в диапазоне от 10 до 40 Å.
WO 2005/103207 описывает каталитические композиции, которые можно использовать для повышения качества дистиллятов, содержащие один или более металлов, выбранных из Pt, Pd, Ir, Ru и Re, и носитель из оксида кремния-алюминия, аморфного при анализе с помощью рентгеновских лучей, с мольным соотношением SiO2/Al2O3 в диапазоне от 30 до 500, площадью поверхности более 500 м2/г, объемом пор в диапазоне от 0,3 до 1,3 мл/г и средним диаметром пор менее 40 Å.
В общем, в композициях, применяемых на стадии (2) гидроизомеризации, содержащих только металл группы VIII, этот металл в соответствии со способами получения, описанными в вышеуказанных патентах, можно ввести посредством пропитки или ионного обмена. В соответствии с первым способом компонент кислой природы (а), также в экструдированной форме, предпочтительно полученный в экструдированной форме в соответствии со способом, описанным в ЕР 665055, смачивают водным раствором соединения метала группы VIII, проводя процесс, например, при комнатной температуре и при рН в диапазоне от 1 до 4. Водный раствор предпочтительно имеет концентрацию металла, выраженную в г/л, в диапазоне от 0,2 до 2,0. Полученный продукт сушат, предпочтительно на воздухе, при комнатной температуре и прокаливают в окислительной атмосфере при температуре в диапазоне от 200 до 600°С.
В случае пропитки спиртовым раствором кислый компонент (а), также в экструдированной форме, предпочтительно полученный в экструдированной форме в соответствии со способом, описанным в ЕР 665055, суспендируют в спиртовом растворе, содержащем металл. После пропитки твердое вещество сушат и прокаливают.
В соответствии со способом ионного обмена кислый компонент (а), также в экструдированной форме, предпочтительно полученный в экструдированной форме в соответствии со способом, описанным в ЕР 665055, суспендируют в водном растворе комплекса или соли металла, проводя процесс при комнатной температуре и рН в диапазоне от 6 до 10. После проведения ионного обмена твердое вещество отделяют, промывают водой, сушат и окончательно термообрабатывают в инертной и окислительной атмосфере. Температуры, которые можно применять для этих целей, находятся в диапазоне от 200 до 600°С.
Соединения металлов, которые с успехом можно использовать в вышеописанных способах получения, представляют собой: H2PtCl6, Pt(NH3)4(OH)2, Pt(NH3)4Cl2, Pd(NH3)4(OH)2, PdCl2, (CH3COO)2Ni, (CH3COO)2Co. Если каталитическая композиция включает более чем один металл группы VIII, то пропитку проводят следующим образом: кислый компонент (а), также в экструдированной форме, предпочтительно полученный в экструдированной форме в соответствии со способом, описанным в ЕР 665055, смачивают раствором соединения первого металла, полученный продукт сушат, возможно, прокаливают и пропитывают раствором соединения второго металла. Продукт сушат, а затем прокаливают в окислительной атмосфере при температуре в диапазоне от 200 до 600°С. Альтернативно можно использовать единый водный раствор, содержащий два или более соединений различных металлов, для одновременного введения указанных металлов.
Перед использованием катализатор активируют известными способами, например посредством восстановительной обработки и предпочтительно посредством сушки и последующего восстановления. Сушку проводят в инертной атмосфере при температурах в диапазоне от 25 до 100°С, в то время как восстановление производят термической обработкой катализатора в восстановительной атмосфере (Н2) в диапазоне температур от 300 до 450°С и давлении предпочтительно в диапазоне от 0,1 до 5 МПа (от 1 до 50 бар). Каталитические композиции, которые с успехом можно использовать на стадии гидроизомеризации настоящего изобретения, содержащие один или более металлов группы VIII, а также их получение описаны в ЕР 908231 и ЕР 105571. В частности, ЕР 908231 описывает каталитические композиции, содержащие смесь металлов, принадлежащих к группам VIB и VIII, и носитель из алюмосиликагеля, аморфного при анализе рентгеновскими лучами, с мольным соотношением SiO2/Al2O3 в диапазоне от 30 до 500, площадью поверхности в диапазоне от 500 до 1000 м2/г, объемом пор в диапазоне от 0,3 до 0,6 мл/г и средним диаметром пор в диапазоне от 10 до 40 Å. Если катализатор гидроизомеризации также содержит в металлической фазе (b) металл группы VIB, то катализатор можно получить посредством пропитки водным или спиртовым раствором. Более конкретно, в соответствии с первым способом оксид кремния-алюминия, также в экструдированной форме, а предпочтительно полученный в экструдированной форме в соответствии со способом, описанным в ЕР 665055, смачивают водным раствором соединения желаемого металла группы VIB, проводя процесс при комнатной температуре или температуре, близкой к комнатной. После пропитки водным раствором твердое вещество сушат, а затем снова пропитывают водным раствором соединения желаемого металла группы VIII. После пропитки водным раствором твердое вещество снова сушат и термообрабатывают в окислительной атмосфере. Подходящие для этой термообработки температуры лежат в диапазоне от 200 до 600°С. Пропитку металлической фазы водным раствором можно также осуществить в одну стадию, когда кислый носитель на основе оксида кремния-алюминия смачивают единым водным раствором, содержащим соединения металлов как группы VIB, так и группы VIII, далее проводя описанные выше процедуры. В способе пропитки спиртовым раствором оксид кремния-алюминия, также в экструдированной форме, а предпочтительно полученный в экструдированной форме в соответствии со способом, описанным в ЕР 665055, суспендируют в спиртовом растворе соединения металла группы VIB и соединения металла группы VIII, проводя процесс при комнатной температуре или при значении температуры, близком к комнатной. После пропитки твердое вещество сушат, предпочтительно на воздухе, при температуре примерно 100°С, и термообрабатывают в окислительной атмосфере, предпочтительно на воздухе.
Конечный катализатор гидроизомеризации можно составить и сформировать в виде экструдированных продуктов, имеющих различные формы (например, цилиндрическую, трехлопастную и т.д.), как это описано, например, в ЕР 1101813.
Каталитические композиции, применяемые на стадии гидроизомеризации настоящего изобретения, характеризуются стойкостью к воде: можно наблюдать замедляющее влияние воды на каталитическую активность, которую, однако, можно восстановить путем повышения температуры, в то время как необратимой дезактивации не наблюдается. Увеличение на несколько (от трех до пяти) градусов Цельсия обычно является достаточным для компенсации снижения активности, которое вызывает содержание воды в загрузке 1000-2000 ppm. Предпочтительно работать с содержанием воды около 1000 ppm, а еще более предпочтительно на уровне ниже 300 ppm.
Реактор для проведения стадии гидроизомеризации представляет собой по конфигурации реактор с неподвижным слоем катализатора. В этом случае контроль температуры не является критическим, поскольку реакция является слабо экзотермической. Из этих соображений пригоден адиабатический реактор с послойным размещением катализатора. В любом случае можно также использовать кожухотрубчатый реактор.
Жидкий питающий поток, выходящий со стадии гидродезоксигенирования, можно направлять в реактор прямотоком или противотоком по отношению к водороду. Если жидкий питающий поток содержит значительный уровень воды и/или кислородсодержащих соединений, которые не участвовали в реакции на первой стадии процесса (>300 ppm кислорода), то предпочтительной является подача противотоком.
Таким образом, воду, присутствующую или полученную из кислородсодержащих соединений в ходе гидроизомеризации, удаляют в газовой фазе в первой части каталитического слоя, снижая таким образом время ее контакта с остальным катализатором. Особенно предпочтительной установкой для этой каталитической стадии является реактор, в котором количество слоев катализатора больше или равно 2, при этом первый слой, покрытый жидким углеводородным потоком, поступающим со стадии гидродезоксигенирования, соответствуя, таким образом, последнему слою, покрытому потоком газообразного водорода, состоит не из катализатора, а из наполнителя из структурированного инертного материала, например керамики или нержавеющей стали, или таблеток или шариков из инертного материала, такого как пемза, альфа-оксид алюминия, стекло. Роль наполнителя состоит в улучшении контакта газа с жидкостью, поскольку загрузка углеводородов, подвергаемая изомеризации, будет вступать в контакт с потоком газообразного водорода до того, как она перетечет на слой катализатора, таким образом подвергаясь дополнительной дегидратации.
Гидроизомеризацию можно осуществить при температуре в диапазоне от 250 до 450°С, предпочтительно от 280 до 380°С, и при давлении в диапазоне от 2,5 до 7,0 МПа (от 25 до 70 бар), предпочтительно от 3 до 5 МПа (от 30 до 50 бар). Предпочтительно проводить процесс при объемной скорости от 0,5 до 2 ч-1. Соотношение Н2/УВ предпочтительно находится в диапазоне от 200 до 1000 л (н.у.)/л.
Смесь, полученную на стадии гидроизомеризации, подвергают перегонке для получения очищенной смеси углеводородов, которую можно использовать в качестве дизельного топлива.
Фиг.1 иллюстрирует схему установки, которую можно применять в способе настоящего изобретения для получения углеводородных фракций, которые можно использовать в качестве дизельного топлива, исходя из смеси биологического происхождения (биологическая смесь), содержащей эфиры жирных кислот и, возможно, некоторое количество свободных жирных кислот. Схема Фиг.1 находится в соответствии с тем, что было описано выше в отношении стадий гидродезоксигенирования (реактор ДЕОКС), очистки с помощью сепаратора высокого давления и промывки (СЕП) и гидроизомеризации (реактор ИЗОМ). На схеме после реактора гидроизомеризации имеются также последующие стадии разделения посредством сепаратора и перегонной установки для выделения полученного газойля. Пунктирной линией показана возможность рециркуляции потока, выходящего с первой стадии.
Для более подробного описания приведены, исключительно для целей иллюстрации конкретных аспектов изобретения, несколько примеров практической реализации способа, являющегося предметом настоящего изобретения, которые, однако, ни в коем случае не следует рассматривать как ограничивающие в целом объем изобретения.
Пример 1 - Получение катализатора Pt/MSA
Реагенты и материалы
Для описанного далее получения применяли следующие имеющиеся в продаже реагенты:
Гидроксид тетрапропиламмония (ТПА-ОН) SACHEM
Триизопропоксид алюминия FLUKA
Тетраэтилсиликат DYNAMIT NOBEL
Оксид алюминия (VERSAL 250, псевдобемит) LAROCHE
Метилцеллюлоза (METHOCEL) FLUKA
Реагенты и/или растворители, которые были использованы и не вошли в вышеприведенный перечень, представляют собой наиболее широко применяемые реагенты и растворители, которые легко можно найти у обычных коммерческих операторов, специализирующихся в данной области.
ПРИМЕР ПОЛУЧЕНИЯ
(i) Получение алюмосиликагеля
100-литровый реактор предварительно промыли 75 литрами раствора, содержащего 1 мас.% гидроксида тетрапропиламмония (ТПА-ОН) в деминерализованной воде, поддерживая жидкость при перемешивании в течение 6 часов при 120°С. Промывной раствор сливают и вводят 23,5 литров деминерализованной воды, 19,6 кг водного раствора 14,4 мас.% ТПА-ОН (13,8 моль) и 600 г (2,94 моль) триизопропоксида алюминия. Смесь нагревают до 60°С и поддерживают при перемешивании при этой температуре в течение 1 часа так, чтобы получить прозрачный раствор. Затем температуру раствора доводят до 90°С и быстро добавляют 31,1 кг (149 моль) тетраэтилсиликата. Реактор закрывают и скорость перемешивания регулируют примерно до 1,2 м/с, поддерживая смесь при перемешивании в течение трех часов, при температуре в диапазоне от 80 до 90°С, под контролем термостата, для отвода тепла, получаемого при реакции гидролиза. Давление в реакторе возрастает примерно до 0,2 МПа (изб.). В конце реакционную смесь выгружают и охлаждают до комнатной температуры с получением однородного и относительно текучего геля (вязкость 0,011 Па·с), имеющего состав со следующими мольными соотношениями:
SiO2/Al2O3=101
ТПА-ОН/SiO2=0,093
H2O/SiO2=21
ii) Получение экструдированного продукта
1150 г оксида алюминия (VERSAL 150), предварительно высушенного в течение 3 часов на воздухе при 150°С, и 190 г метилцеллюлозы загружают в 10-л лопастной смеситель, поддерживаемый при скорости перемешивания 70-80 об/мин. Затем в течение периода времени примерно 15 минут добавляют 5 кг алюмосиликагеля, полученного, как описано выше, и выдержанного в течение примерно 20 часов, и смесь оставляют при перемешивании примерно на 1 час. Добавляют 6 г ледяной уксусной кислоты и температуру в смесителе доводят примерно до 60°С, после чего продолжают перемешивание до получения однородной пасты, имеющей желаемую консистенцию для последующего экструдирования.
Однородную пасту, полученную, как описано выше, загружают в экструдер типа HUTT, экструдируют и нарезают на цилиндрические таблетки, имеющие желаемый размер (примерно 2×4 мм). Продукт выдерживают примерно в течение 6-8 часов, а затем сушат, поддерживая его в потоке воздуха при 100°С в течение 5 часов. Окончательно его прокаливают в муфельной печи при 550°С в течение 3 часов в потоке азота и еще в течение 8 часов - на воздухе.
Таким образом получают пористое твердое вещество с кислыми характеристиками, состоящее по существу из оксида кремния/оксида алюминия (выход 95%, рассчитывая на соответствующие исходные реагенты), имеющее поверхность по БЭТ 608 м2/г.
iii) Пропитка носителя платиной
12,1 мл водного раствора 0,6 М соляной кислоты, содержащего 4,5 г/л гексахлорплатиновой кислоты (H2PtCl6, 0,133 ммоль), добавляют по каплям при медленном перемешивании в стеклянный приемник, содержащий 10 г пористого твердого вещества, полученного, как описано выше. Полученную таким образом смесь оставляют при перемешивании в течение 16 часов при комнатной температуре. Затем воду испаряют при 60°С в токе воздуха в течение примерно 1 часа. Затем полученное твердое вещество сушат, поддерживая его при температуре 150°С в течение двух часов, и прокаливают путем нагревания в муфельной печи в потоке воздуха, изменяя температуру от комнатной до 500°С, в течение трех часов. В результате получают нанесенный на носитель катализатор, который используют на стадии гидроизомеризации, описанной в примере 3 ниже, имеющий следующие характеристики:
59,8 мас.% аморфного алюмосиликагеля (мольное соотношение SiO2/Al2O3=102)
39,9 мас.% оксида алюминия (псевдобемита)
0,3 мас.% платины
Объем пор: 0,6 мл/г
Средний диаметр пор 35 Å
БЭТ: 600 м2/г
Прочность на раздавливание: 10 кг/см2 (радиальная);
90 кг/см2 (аксиальная)
ПРИМЕР 2 - стадия гидродезоксигенирования (ГДО)
Эксперимент проводят в реакторе непрерывного действия, в который подают соевое масло, имеющее характеристики, приведенные в Таблице 1 (рафинированное соевое масло Sipral).
Растительное масло и водород подают на первую стадию прямотоком в присутствии имеющегося в продаже катализатора гидрогенизации UOP UF 210 на основе NiMo/Al2O3 в сульфидированной форме. Сульфидирование катализатора проводят in situ, с использованием газойля, содержащего диметилдисульфид (ДМДС) в концентрации, которую постепенно изменяют от 3 до 9 мас.% при температуре, которую постепенно изменяют в диапазоне от 230 до 370°С, и давлении 7 МПа (70 бар), с соотношением Н2/газойль 1300 л (н.у.)/л и объемной скоростью 0,8 ч-1. Растительное масло подают в реактор в присутствии небольшого количества ДМДС (0,025%), чтобы поддерживать катализатор в сульфидированной форме.
Питающий поток и водород проходят через реактор в направлении сверху вниз.
Используют следующие рабочие условия:
- Средняя температура: 340-350°С
- Объемная скорость: 1 ч-1
- Давление: 3,5 МПа (35 бар)
- Н2/масло: 1500 л (н.у.)/л
Выходящий продукт отделяют в газожидкостном сепараторе от газовой фракции, состоящей из Н2, СО/СО2 и легких углеводородов, почти полностью состоящих из С3Н8. Жидкий продукт после отделения от воды состоит из н-парафинов, характеристики и распределение которых приведены в Таблице 2 и на Фиг.2.
В таблице 2 (строки 2-5) приведен состав получающейся на первой стадии смеси с точки зрения содержания отдельных элементов (С, Н, N, S и О), где сумма составляет почти 100% (с учетом погрешности). Поскольку содержание кислорода рассчитывают по разности, то, пренебрегая содержанием N и S, количество которых определяется в частях на миллион, расчетное разностное содержание кислорода (с точностью до второго знака после запятой) составит 0,53 мас.%.
Для состава смеси с точки зрения типов топлива приведены данные по содержанию газойля как фракции с температурой кипения от 180 до 380°С и фракции более тяжелых продуктов с температурой кипения свыше 380°С; при этом сумма их составляет 96+4=100% (строки 14 и 16); с целью уточнения состава продуктов также приведены данные по содержанию в смеси топлив фракции с температурой кипения выше 340°С (строка 15), откуда следует дополнительная информация, что количество фракции топлива, кипящей при температуре от 340 до 380°С, составляет 5-4=1%.
Общее количество ароматических соединений приведено в таблице в строке 11 с целью доказательства их незначительного количества в смеси с изопарафинами и н-парафинами, это количество ароматики в смеси составляет 3,5%, причем указанные ароматические соединения состоят из измоноароматических соединений 2,9% (строка 8) + диароматических соединений 0,5% (строка 9) + триароматических соединений 0,1% (строка 10).
В последних девяти строках таблицы 2 приведены данные по содержанию н-парафинов и изопарафинов в смеси, что в общем составляет в сумме (90,92+9,08)=100%. При этом следует отметить, что данные распределения парафинов по группам соответственно содержанию атомов углерода получены посредством газохроматографического анализа по расчету области пиков. Специалисту понятно, что такие измерения обладают некоторой погрешностью, и поэтому общая сумма компонентов смеси парафинов составляет величину, очень близкую к 100% (сумма компонентов в строках 1-6 снизу таблицы приблизительно составляет 0,1+0,2+87,7+6,5+2,4+3,1=100).
Пример 3 - Стадия гидроизомеризации
Продукт, полученный на стадии дезоксигенирования, описанной в Примере 2, содержащий 100 ppm остаточной воды, обрабатывают водородом в прямотоке в присутствии катализатора Pt/MSA, полученного в предыдущем примере 1. Используемые рабочие условия приведены в Таблице 3.
Выходящий из реактора гидроизомеризации поток состоит из газовой фазы и жидкой фазы; эти две фазы разделяют в газожидкостном сепараторе, газовая фаза (по результатам анализа методом газовой хроматографии) состоит из легких С3/С4 парафинов (ЛПГ), в то время как отделенную жидкую фазу, содержащую парафины с числом атомов углерода в диапазоне от 5 до 22, анализируют посредством газовой хроматографии для того, чтобы оценить уровень изомеризации, который при данных рабочих условиях составляет 79%, и используют для оценки кривой перегонки.
Затем углеводороды направляют в перегонную колонну, чтобы отделить фракцию бензина (12,7%) от фракции дизельного топлива (87,3%). Эта последняя фракция, содержащая парафины с числом атомов углерода в диапазоне от 12 до 22, была охарактеризована, и основные свойства приведены ниже в Таблице 4:
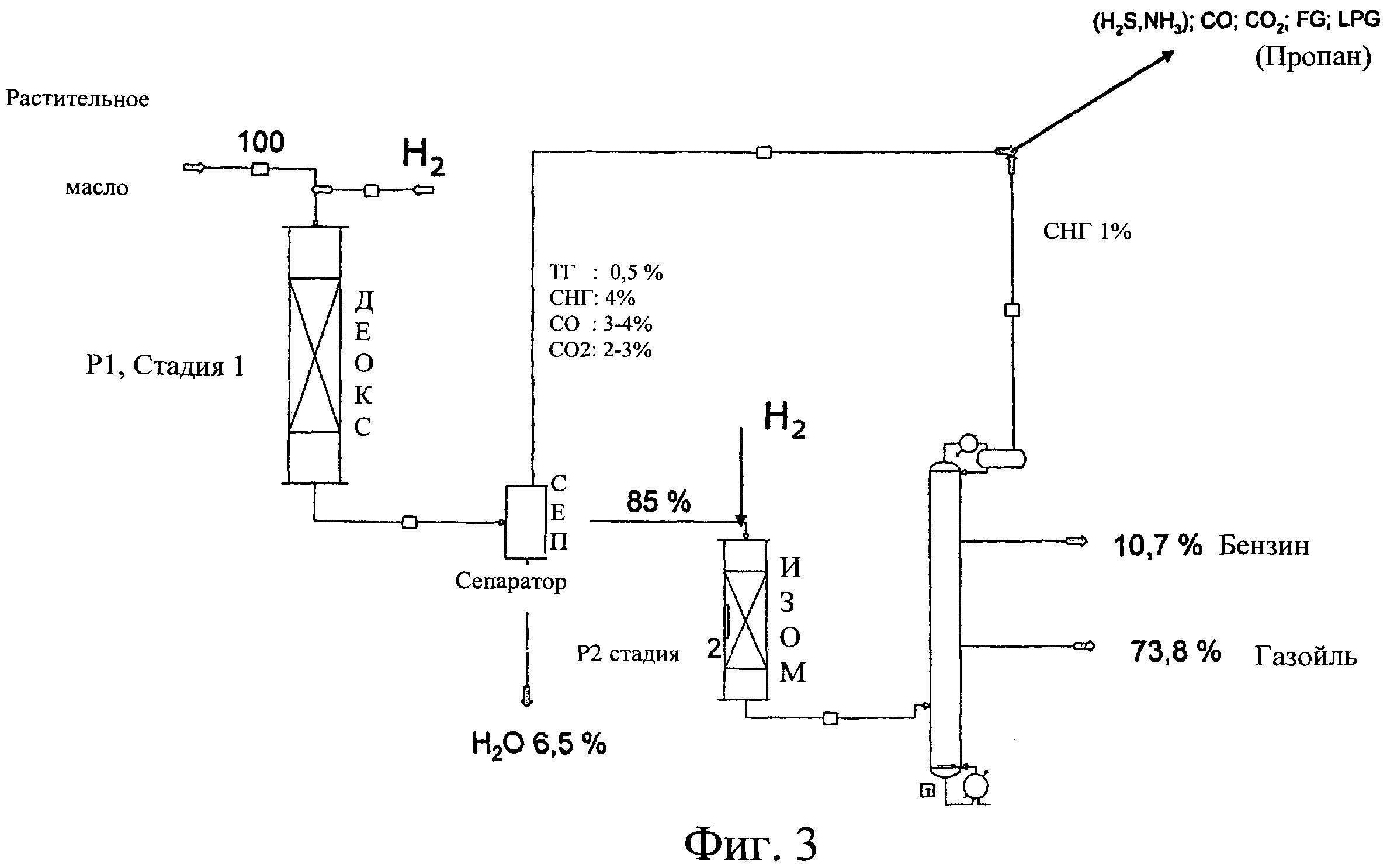
Фиг.3 изображает общий материальный баланс процесса, относящегося к Примерам 2 и 3; стадию 1 гидродезоксигенирования проводят в реакторе 1-ДЕОКС, СЕП представляет собой газожидкостный сепаратор, где отделяют воду, Н2О, от смеси: СО, СО2, FG - топливного газа - ТГ и LPG - сжиженного нефтяного газа - СНГ, а ИЗОМ представляет собой реактор 2, в котором проводят стадию 2 гидроизомеризации. После реактора 2 гидроизомеризации представлена перегонная колонна, из которой получают фракцию дизельного топлива.
Реферат
Описан способ получения углеводородных фракций, которые можно использовать в качестве дизельного топлива или в качестве компонентов дизельного топлива, исходя из смеси биологического происхождения, содержащей эфиры жирных кислот, возможно, с некоторым количеством свободных жирных кислот, который включает следующие стадии: 1) гидродезоксигенирование смеси органического происхождения; 2) гидроизомеризация смеси, полученной на стадии (1), после возможной обработки с целью очистки; причем указанную стадию гидроизомеризации осуществляют в присутствии каталитической системы, которая включает: а) носитель кислой природы, включающий полностью аморфный микромезопористый оксид кремния-алюминия, имеющий мольное соотношение SiO2/Al2O3 в диапазоне от 30 до 500, площадь поверхности более 500 м2/г, объем пор в диапазоне от 0,3 до 1,3 мл/г, средний диаметр пор ниже 40 Å, b) металлический компонент, содержащий один или более металлов группы VIII, возможно, смешанных с одним или более металлов группы VIB. Технический результат - получение углеводородной фракции, которую можно применять в качестве дизельного топлива или в качестве компонента дизельного топлива. 54 з.п. ф-лы, 4 табл., 3 ил., 2 пр.
Формула
1) гидродезоксигенирование смеси органического происхождения;
2) гидроизомеризация смеси, полученной на стадии (1), после возможной обработки с целью очистки, причем указанную гидроизомеризацию осуществляют в присутствии каталитической системы, которая включает:
a) носитель кислой природы, включающий полностью аморфный микро-мезопористый оксид кремния-алюминия, имеющий мольное отношение SiO2/Al2O3 в диапазоне от 30 до 500, площадь поверхности выше 500 м2/г, объем пор в диапазоне от 0,3 до 1,3 мл/г, средний диаметр пор ниже 40 Å,
b) металлический компонент, содержащий один или более металлов группы VIII, возможно смешанных с одним или более металлов группы VIB.
a) носитель кислой природы, включающий полностью аморфный микро-мезопористый оксид кремния-алюминия, имеющий мольное отношение SiO2/Al2O3 в диапазоне от 30 до 500, площадь поверхности выше 500 м2/г, объем пор в диапазоне от 0,3 до 1,3 мл/г, средний диаметр пор ниже 40 Å,
b) металлический компонент, содержащий один или более металлов группы VIII, возможно, смешанных с одним или более металлов группы VIB.
(A) получение водного раствора гидроксида тетраалкиламмония (ТАА-ОН), растворимого соединения алюминия, способного гидролизоваться до Al2O3, и соединения кремния, способного гидролизоваться до SiO2, в следующих мольных отношениях:
SiO2/Al2O3 от 30/1 до 500/1
TAA-OH/SiO2 от 0,05/1 до 0,2/1
H2O/SiO2 от 5/1 до 40/1
(B) нагревание полученного таким образом раствора для осуществления его гидролиза и гелеобразования и получения смеси А с вязкостью в диапазоне от 0,01 до 100 Па·с;
(C) добавление к смеси А сначала связующего, принадлежащего к группе бемитов или псевдобемитов, в массовом отношении со смесью А в диапазоне от 0,05 до 0,5, а затем неорганической или органической кислоты в количестве от 0,5 до 8,0 г на 100 г связующего;
(D) нагревание при перемешивании смеси, полученной на стадии (С), до температуры в диапазоне от 40 до 90°С, до получения однородной пасты, которую подвергают экструзии и гранулированию;
(E) сушка и прокаливание экструдированного продукта в окислительной атмосфере.
Документы, цитированные в отчёте о поиске
Композиция дизельного топлива, включающая компоненты на основе биологического исходного материала, полученные гидрированием и разложением жирных кислот
Комментарии