Композиции для введения лекарственных средств - RU2554814C2
Код документа: RU2554814C2
Чертежи






Описание
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится, в основном, к не вызывающим раздражение, нетоксичным композициям, обеспечивающим увеличенную биодоступность, а конкретнее к содержащим алкилгликозид или алкиловый эфир сахарида композициям для доставки субъекту терапевтических агентов, таких как агонист рецептора 5-HT, в том числе способам и композициям для обеспечения купирования боли при мигрени.
ОПИСАНИЕ ПРЕДШЕСТВУЮЩЕГО УРОВНЯ ТЕХНИКИ
Терапевтические агенты часто объединяют с различными поверхностно-активными веществами. Однако поверхностно-активные вещества часто вызывают раздражение кожи и других тканей, в том числе слизистых оболочек, таких как те, которые встречаются в носовой, ротовой полости, глазу, влагалище, прямой кишке, пищеводе, кишечнике и т.п. Многие поверхностно-активные вещества также вызывают денатурацию белков, уничтожая тем самым их биологическую активность. Другим серьезным ограничивающим разработку и использование таких агентов условием является возможность доставлять их без риска, неинвазивно, эффективно и в неизменяемом состоянии в место действия. Следовательно, идеальное усиливающее поверхностно-активное вещество будет стабилизировать терапевтический агент, будет нетоксичным и не будет вызывать раздражение кожи или поверхностей слизистых оболочек, будет обладать антибактериальной активностью и увеличивать прохождение или абсорбцию терапевтического агента через различные мембранные барьеры без повреждения структурной целостности и биологической функции мембраны и будет увеличивать биодоступность агента.
Ранее был описан ряд подходов к составлению быстро распадающихся или так называемых «быстро диспергируемых» лекарственных форм. Лекарственное вещество проглатывается после распада в ротовой полости, что имеет следствием абсорбцию до попадания в желудок и, в конце концов, желудочную абсорбцию. Термин «абсорбция до попадания в желудок» обычно имеет отношение к абсорбции активного ингредиента в той части пищеварительного тракта, которая предшествует желудку, и включает щечную, подъязычную, ротоглоточную и пищеводную абсорбцию. Термин «желудочная абсорбция» обычно имеет отношение к абсорбции активного ингредиента в желудке или кишечнике. Различные количества лекарственного средства могут абсорбироваться, по мере того как лекарственное средство проходит через предшествующую желудку часть пищеварительного тракта. Однако большая часть лекарственного средства проходит в желудок и абсорбируется обычным способом желудочной абсорбции, которым абсорбируются энтеросолюбильные лекарственные формы, такие как таблетки, капсулы или жидкости. По мере того как лекарственное средство абсорбируется из кишечника, оно доставляется непосредственно в печень, где, в зависимости от конкретной химической структуры, оно может метаболизироваться и элиминироваться ферментами, которые осуществляют обычные процессы детоксификации в клетках печени. Эту элиминацию называют «метаболизмом первого прохождения» или «эффектом первого прохождения» в печени. Результирующие метаболиты, чаще всего являющиеся по существу или полностью неактивными по сравнению с первоначальным лекарственным средством, часто встречаются циркулирующими в кровотоке и впоследствии удаляются в мочу и/или фекалии. Подходы к составлению быстро распадающихся или быстро диспергируемых лекарственных форм предоставлены в заявке на патент США с № 2006/0134194, которая включена сюда посредством ссылки.
Ранее описанные быстро диспергируемые лекарственные формы предусматривают лекарственную форму, которая распадается или растворяется после помещения в ротовую полость для увеличения абсорбции до попадания в желудок или желудочной абсорбции активного ингредиента, однако быстро диспергируемые лекарственные формы настоящего изобретения определяют улучшенные характеристики, такие как ускорение начала действия лекарственного средства и снижение метаболизма лекарственного средства - эффекта первого прохождения.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение основывается, отчасти, на разработке терапевтической композиции, содержащей агент для усиления лекарственного средства, пригодный для увеличения абсорбции и биодоступности лекарственного средства, при избегании в то же самое время различных неблагоприятных токсических эффектов лекарственного средства. В частности, агенты для усиления лекарственного средства настоящего изобретения содержат нетоксичное поверхностно-активное вещество, состоящее из по крайней мере алкилгликозида и/или алкилового эфира сахарида. Одним из преимуществ терапевтических композиций настоящего изобретения является то, что они позволяют вводить и доставлять терапевтические агенты с высокими биодоступностями при концентрациях усиливающих агентов, которые значительно ниже так называемых их «уровней без видимых неблагоприятных эффектов» (их NOAEL). Соответственно, настоящим изобретением обеспечиваются композиции, включающие алкилгликозиды и/или алкиловые эфиры сахаридов и терапевтический агент (например, небольшие органические молекулы лекарственных средств, низкомолекулярные пептиды, такие как Эксенатид, GLP-1 и т.п., белки и не являющиеся пептидами терапевтические полимеры, такие как низкомолекулярный гепарин и ингибирующая РНК), способы введения и применения композиций, например, посредством пероральной, глазной, интраназальной, носослезной, ингаляционной или легочной доставки, доставки в ротовую полость (сублингвальной доставки или доставки к клеткам щеки) или в спинномозговую жидкость (CSF), и способы улучшения болезненного состояния у субъекта посредством ведения таких композиций.
В одном аспекте настоящее изобретение относится к содержащей поверхностно-активное вещество(а) композиции, содержащей по крайней мере один алкилгликозид и/или по крайней мере один алкиловый эфир сахарида, и после примешивания терапевтического агента, лекарственного средства или биологически активного соединения или смешивания с ним поверхностно-активное вещество стабилизирует биологическую активность и увеличивает биодоступность лекарственного средства.
Соответственно, в одном аспекте настоящим изобретением обеспечивается терапевтическая композиция, содержащая по крайней мере одно биологически активное соединение и по крайней мере одно поверхностно-активное вещество, причем поверхностно-активное вещество к тому же состоит из по крайней мере одного алкилгликозида и/или алкилового эфира сахарида или эфира сахарозы, а терапевтическая композиция стабилизирует биологически активное соединение в течение по крайней мере приблизительно 6 месяцев или более при от приблизительно 4°C до приблизительно 25°C.
Настоящим изобретением также обеспечивается способ введения терапевтической композиции, которая содержит поверхностно-активное вещество, включающее по крайней мере один алкилгликозид и/или алкиловый эфир гликозида, смешанное с по крайней мере одним терапевтическим агентом, или лекарственным средством, или биологически активным соединением, и вводится или доставляется субъекту, причем алкил имеет от приблизительно 10 до 24, от 10 до 20, от 10 до 16 или от 10 до 14 атомов углерода, причем поверхностно-активное вещество увеличивает стабильность и биодоступность терапевтического агента.
В еще одном аспекте настоящим изобретением обеспечивается способ увеличения прохождения низкомолекулярного соединения в сердечно-сосудистую систему субъекта посредством введения посредством пероральной, глазной, интраназальной, носослезной, ингаляционной или легочной доставки, доставки в ротовую полость (сублингвальной доставки или доставки к клеткам щеки) или в спинномозговую жидкость (CSF) соединения после его смешивания с увеличивающим абсорбцию количеством подходящего поверхностно-активного вещества, причем поверхностно-активным веществом является нетоксичный и неионный гидрофобный алкил, присоединенный с помощью химической связи к гидрофильному сахариду. Такие низкомолекулярные соединения включают, но без ограничения, никотин, интерферон, PYY, GLP-1, синтетический экзендин-4, гормон паращитовидной железы, гормон роста человека или небольшая органическая молекула. Дополнительные низкомолекулярные соединения включают антисмысловые олигонуклеотиды или молекулы интерферирующих РНК (например, короткую интерферирующую РНК (киРНК) или молекулу, вовлеченную в РНК-интерференцию).
Настоящим изобретением также обеспечивается способ лечения диабета, включающий введение нуждающемуся в этом субъекту посредство пероральной, глазной, интраназальной, носослезной, ингаляционной или легочной доставки или доставки в ротовую полость (сублингвальной доставки или доставки к клеткам щеки) снижающего уровень глюкозы в крови количества терапевтической композиции, например, являющего миметиком инкретина агента или его функционального эквивалента, и увеличивающего абсорбцию количества подходящего нетоксичного, неионного алкилгликозида, имеющего гидрофобную алкильную группу, присоединенную с помощью химической связи к гидрофильному сахариду, увеличивая, тем самым, абсорбцию являющегося миметиком инкретина агента или инсулина и снижая уровень глюкозы в крови, и осуществляя лечение диабета у субъекта.
Настоящим изобретением также обеспечивается способ лечения застойной сердечной недостаточности у субъекта, включающий введение нуждающемуся в этом субъекту посредством пероральной, глазной, интраназальной, носослезной или ингаляционной доставки терапевтически эффективного количества композиции, включающей пептид GLP-1 или его функциональный эквивалент, и увеличивающего абсорбцию количества подходящего нетоксичного, неионного алкилгликозида, имеющего гидрофобный алкил, присоединенный с помощью химической связи к гидрофильному сахариду, осуществляя, тем самым, лечение субъекта.
В другом аспекте настоящим изобретением обеспечивается способ лечения ожирения или связанного с ожирением диабета у субъекта, включающий введение нуждающемуся в этом субъекту посредством пероральной, глазной, интраназальной, носослезной, ингаляционной доставки или доставки в спинномозговую жидкость (CSF) терапевтически эффективного количества композиции, включающей пептид PYY или его функциональный эквивалент, и увеличивающего абсорбцию количества подходящего нетоксичного, неионного алкилгликозида, имеющего гидрофобный алкил, присоединенный с помощью химической связи к гидрофильному сахариду, осуществляя, тем самым, лечение субъекта.
В другом аспекте настоящим изобретением обеспечивается способ увеличения прохождения низкомолекулярного терапевтического соединения в сердечно-сосудистую систему субъекта посредством пероральной, глазной, интраназальной, носослезной, ингаляционной доставки или доставки в спинномозговую жидкость (CSF) соединения и увеличивающего абсорбцию количества подходящего нетоксичного, неионного алкилгликозида, имеющего гидрофобный алкил, присоединенный с помощью химической связи к гидрофильному сахариду, причем соединение имеет молекулярную массу, составляющую приблизительно 1-30 кДа, при условии, что соединением не является инсулин, кальцитонин или глюкагон, когда путем введения является оральный, глазной, назальный или носослезный путь.
Настоящим изобретением также обеспечивается способ увеличения прохождения низкомолекулярного терапевтического соединения в сердечно-сосудистую систему субъекта посредством введения посредством пероральной, глазной, интраназальной, носослезной, ингаляционной или легочной доставки, доставки в ротовую полость (сублингвальной доставки или доставки к клеткам щеки) или в спинномозговую жидкость (CSF) соединения и увеличивающего абсорбцию количества подходящего нетоксичного, неионного алкилгликозида, имеющего гидрофобный алкил, присоединенный с помощью химической связи к гидрофильному сахариду, причем соединение имеет молекулярную массу, составляющую приблизительно 1-30 килодальтон (кДа), при условии, что субъект не страдает диабетом, когда доставка осуществляется посредством орального, глазного, назального или носослезного пути.
В одном аспекте настоящего изобретения обеспечивается фармацевтическая композиция, содержащая подходящий нетоксичный, неионный алкилгликозид, имеющий гидрофобную алкильную группу, присоединенную с помощью химической связи к гидрофильному сахариду, в сочетании с терапевтически эффективным количеством Эксенатида (экзендина-4) в фармацевтически приемлемом носителе.
В одном аспекте настоящим изобретением обеспечивается фармацевтическая композиция, содержащая подходящий нетоксичный, неионный алкилгликозид, имеющий гидрофобную алкильную группу, присоединенную с помощью химической связи к гидрофильному сахариду, в сочетании с терапевтически эффективным количеством GLP-1 в фармацевтически приемлемом носителе.
В одном аспекте настоящим изобретением обеспечивается фармацевтическая композиция, содержащая подходящий нетоксичный, неионный алкилгликозид, имеющий гидрофобную алкильную группу, присоединенную с помощью химической связи к гидрофильному сахариду, в сочетании с терапевтически эффективным количеством никотина в фармацевтически приемлемом носителе.
В одном аспекте настоящим изобретением обеспечивается фармацевтическая композиция, содержащая подходящий нетоксичный, неионный алкилгликозид, имеющий гидрофобную алкильную группу, присоединенную с помощью химической связи к гидрофильному сахариду, в сочетании с терапевтически эффективным количеством интерферона в фармацевтически приемлемом носителе.
В одном аспекте настоящим изобретением обеспечивается фармацевтическая композиция, содержащая подходящий нетоксичный, неионный алкилгликозид, имеющий гидрофобную алкильную группу, присоединенную с помощью химической связи к гидрофильному сахариду, в сочетании с терапевтически эффективным количеством PYY в фармацевтически приемлемом носителе.
В одном аспекте настоящим изобретением обеспечивается фармацевтическая композиция, содержащая подходящий нетоксичный, неионный алкилгликозид, имеющий гидрофобную алкильную группу, присоединенную с помощью химической связи к гидрофильному сахариду, в сочетании с терапевтически эффективным количеством гормона паращитовидной железы в фармацевтически приемлемом носителе.
В одном аспекте настоящим изобретением обеспечивается фармацевтическая композиция, содержащая подходящий нетоксичный, неионный алкилгликозид, имеющий гидрофобную алкильную группу, присоединенную с помощью химической связи к гидрофильному сахариду, в сочетании с терапевтически эффективным количеством пептида, имеющего молекулярную массу, составляющую приблизительно 1-75 кДа, в фармацевтически приемлемом носителе, при условии, что пептидом не является инсулин, кальцитонин и глюкагон.
В одном аспекте настоящим изобретением обеспечивается фармацевтическая композиция, содержащая подходящий нетоксичный, неионный алкилгликозид, имеющий гидрофобную алкильную группу, присоединенную с помощью химической связи к гидрофильному сахариду, в сочетании с терапевтически эффективным количеством эритропоэтина в фармацевтически приемлемом носителе.
В одном аспекте настоящим изобретением обеспечивается фармацевтическая композиция, содержащая терапевтически эффективное количество олигонуклеотида в сочетании с увеличивающим абсорбцию количеством алкилгликозида. Олигонуклеотидом может быть антисмысловой олигонуклеотид или молекулы интерферирующих РНК, такие как киРНК или молекула, вовлеченная в РНК-интерференцию. Олигонуклеотид обычно имеет молекулярную массу, составляющую приблизительно 1-20 кДа, и длину, составляющую приблизительно 1-100, 1-50, 1-30, 1-25 или 15-25 нуклеотидов. В другом аспекте олигонуклеотид имеет молекулярную массу, составляющую приблизительно 5-10 кДа. В одном аспекте алкилгликозидом является тетрадецил-бета-D-мальтозид.
В еще одном аспекте настоящим изобретением обеспечивается способ увеличения биодоступности низкомолекулярного олигонуклеотида у субъекта посредством введения соединения с увеличивающим абсорбцию количеством алкилгликозида, посредством чего увеличивается биодоступность соединения у субъекта. В одном аспекте алкилгликозидом является тетрадецил-бета-D-мальтозид.
В одном аспекте настоящим изобретением обеспечивается способ увеличения прохождения соединения в CSF субъекта, вводя интраназально соединение и увеличивающее абсорбцию количество подходящего нетоксичного, неионного алкилгликозида, имеющего гидрофобную алкильную группу, присоединенную с помощью химической связи к гидрофильному сахариду.
В еще одном аспекте настоящим изобретением обеспечивается фармацевтическая композиция, содержащая подходящий нетоксичный, неионный алкилгликозид, имеющий гидрофобную алкильную группу, присоединенную с помощью химической связи к гидрофильному сахариду, в сочетании с увеличивающим доставку через слизистые оболочки агентом, выбираемым из:
(a) ингибирующего агрегацию агента;
(b) изменяющего заряд агента;
(c) агента для контролирования pH;
(d) ингибирующего деструктивные ферменты агента;
(e) муколитического средства или агента для устранения слизи;
(f) цилиостатического агента;
(g) увеличивающего проникновение через мембрану агента, выбранного из (i) поверхностно-активного вещества; (ii) соли желчной кислоты; (ii) фосфолипидной добавки, смешанной мицеллы, липосомы или носителя; (iii) спирта; (iv) енамина; (v) соединения-донора NO; (vi) длинноцепочечной амфипатической молекулы; (vii) небольшого гидрофобного усилителя проникновения; (viii) натрия или производного салициловой кислоты; (ix) глицеринового эфира ацетоуксусной кислоты; (x) производного циклодекстрина или бета-циклодекстрина; (xi) среднецепочечной жирной кислоты; (xii) хелатообразующего агента; (xiii) аминокислоты или ее соли; (xiv) N-ацетиламинокислоты или ее соли; (xv) фермента, деструктивного в отношении выбранного мембранного компонента; (ix) ингибитора синтеза жирных кислот; (x) ингибитора синтеза холестерина; и (xi) любой комбинации увеличивающих проникновение через мембрану агентов, перечисленных в (i)-(x);
(h) модулирующего физиологию эпителиального контакта агента;
(i) вазодилататора;
(j) избирательного, увеличивающего перенос агента; и
(k) стабилизирующего доставку наполнителя, носителя, мукоадгезива, вспомогательных или комплексообразующих частиц, с которыми соединение эффективно объединяется, ассоциируется, в которых оно содержится, в которые оно включается или с которыми оно связывается, что приводит к стабилизации соединения для увеличения доставки через слизистые оболочки носовой полости, причем составление соединения с увеличивающим интраназальную доставку агентами предусматривает увеличение биодоступности соединения в плазме крови субъекта.
В одном аспекте настоящим изобретением обеспечивается способ увеличения прохождения низкомолекулярного соединения в сердечно-сосудистую систему субъекта посредством пероральной, глазной, интраназальной, носослезной, ингаляционной или легочной доставки, доставки в ротовую полость (сублингвальной доставки или доставки к клеткам щеки) или в спинномозговую жидкость (CSF) (a) соединения; (b) увеличивающего абсорбцию количества подходящего нетоксичного, неионного алкилгликозида, имеющего гидрофобную алкильную группу, присоединенную с помощью химической связи к гидрофильному сахариду; и (c) увеличивающего доставку через слизистые оболочки агента.
В одном аспекте настоящим изобретением обеспечивается способ контролирования потребления калорий посредством введения композиции, содержащей терапевтически эффективное количество экзендина-4, или родственного GLP-1 пептида, с эффективным количеством алкилсахарида Intravail.
В другом аспекте настоящим изобретением обеспечивается способ контролирования уровней глюкозы в крови у субъекта посредством введения композиции, включающей терапевтически эффективное количество экзендина-4, или родственного GLP-1 пептида, с эффективным количеством алкилсахарида Intravail.
Опять же, в другом аспекте, настоящим изобретением обеспечивается лекарственная форма с контролируемым высвобождением, включающая
(a) ядро, включающее
(i) по крайней мере один терапевтический агент или лекарственное средство;
(ii) по крайней мере один алкилгликозид и/или алкиловый эфир гликозида; и
(b) по крайней мере одно оболочечное покрытие, окружающее ядро, причем покрытие является непроницаемым, проницаемым, полупроницаемым или пористым и становится более проницаемым при длительном контакте с содержимым желудочно-кишечного тракта.
В другом варианте осуществления настоящим изобретением обеспечивается способ введения содержащей алкилгликозид композиции посредством введения терапевтически эффективного количества по крайней мере одного алкилгликозида, имеющего алкильную цепь длиной от приблизительно 12 до приблизительно 14 атомов углерода, по крайней мере один сахарид с антибактериальной активностью, и по крайней мере одного терапевтического агента.
Опять же, в другом варианте осуществления, настоящим изобретением обеспечивается композиция, содержащая по крайней мере одно лекарственное средство, выбираемое из группы, состоящей из инсулина, PYY, экзендина-4 или другого родственного GLP-1 пептида, гормона роста человека, кальцитонина, гормона паращитовидной железы (паратгормона), усеченных пептидов паратгормона, таких как PTH 1-34, EPO, интерферона-альфа, интерферона-бета, интерферона-гамма и GCSF, и по крайней мере один алкилсахарид, обладающий антибактериальной активностью.
В одном аспекте настоящим изобретением обеспечивается содержащая антибактериальный алкилсахарид композиция, которая включает н-додецил-4-O-α-D-глюкопиранозил-β-D-глюкопиранозид или н-тетрадецил-4-O-α-D-глюкопиранозил-β-D-глюкопиранозид.
В еще одном аспекте настоящим изобретением обеспечивается водосодержащая композиция лекарственного средства для введения через слизистые оболочки или для чрескожного введения, содержащая по крайней мере одно лекарственное средство и по крайней мере один антибактериальный агент в концентрации, составляющей от приблизительно 0,05% до приблизительно 0,5%.
В другом аспекте настоящим изобретением обеспечивается быстро диспергируемый лекарственный препарат, содержащий матричный материал и алкилсахарид. Препарат может характеризоваться Tmax и эффектом первого прохождения, которые существенно меньше таковых, наблюдаемых в случае эквивалентного препарата, не содержащего алкилсахарид. В одном варианте осуществления препарат может содержать приблизительно от 0,1% до 10% алкилсахарида и демонстрировать Tmax, которое существенно меньше шести часов, и эффект первого прохождения, составляющий менее 40%. Алкилгликозидом может быть любой подходящий алкилгликозид, и в предпочтительном аспекте им является додецилмальтозид, тетрадецилмальтозид, додеканоат сахарозы или моно- и дистеарат сахарозы. Препарат может включать ряд различных терапевтических средств, таких как, но без ограничения, мелатонин, ралоксифен, оланзапен и дифенгидрамин.
В другом аспекте настоящим изобретением обеспечивается способ обеспечения кривой длительной абсорбции посредством уменьшения концентрации алкилсахарида в лекарственном препарате для балансировки доставки через желудок и буккальной доставки. Например, это осуществляют с помощью обеспечения лекарственного препарата, включающего матричный материал и алкилсахарид, который характеризуется Tmax и эффектом первого прохождения, которые существенно меньше таковых, наблюдаемых в случае эквивалентного препарата, не содержащего алкилсахарид.
В одном аспекте настоящим изобретением обеспечивается фармацевтическая композиция, содержащая терапевтически эффективное количество аналога бисфосфоната или аналога триптана в сочетании с увеличивающим абсорбцию количеством алкилгликозида. В различных вариантах осуществления аналогом бисфосфоната может быть этидронат, клодронат, тилудронат, памидронат, неридронат, олпадронат, алендронат, ибандронат, ризедронат, золедронат и/или их фармацевтически приемлемые аналоги. В приводимом в качестве примера варианте осуществления аналогом бисфосфоната является алендронат или его фармацевтически приемлемый аналог. В различных вариантах осуществления аналогом триптана может быть суматриптан, ризатриптан, наратриптан, золмитриптан, элетриптан, алмотриптан, фроватриптан и/или их фармацевтически приемлемые аналоги. В приводимом в качестве примера варианте осуществления аналогом триптана является суматриптан или его фармацевтически приемлемый аналог. В различных вариантах осуществления алкилгликозидом является тетрадецил-бета-D-мальтозид.
В еще одном аспекте настоящим изобретением обеспечивается способ увеличения биодоступности аналога бисфосфоната или аналога триптана у субъекта посредством введения соединения с увеличивающим абсорбцию количеством алкилгликозида, увеличивая, тем самым, биодоступность соединения у субъекта.
В еще одном аспекте настоящим изобретением обеспечивается композиция, включающая терапевтически эффективное количество агониста рецептора 5-HT и алкилсахарид. В различных вариантах осуществления агонистом рецептора 5-HT является суматриптан, наратриптан, ризатриптан, элетриптан, фроватриптан, алмотриптан, золмитриптан, их соли или их комбинации.
В еще одном аспекте настоящим изобретением обеспечивается способ предоставления субъекту уменьшенного, но терапевтически эффективного количества агониста рецептора 5-HT. Способ включает введение композиции для интраназального введения, включающей терапевтически эффективное количество агониста рецептора 5-HT и алкилсахарид, при этом AUC приблизительно равна AUC, обеспечиваемой увеличенным терапевтически эффективным количеством агониста рецептора 5-HT, вводимым в отсутствие алкилсахарида.
В еще одном аспекте настоящим изобретением обеспечивается способ обеспечения быстрого облегчения боли при мигрени у субъекта, включающий введение композиции, включающей терапевтически эффективное количество агониста рецептора 5-HT и алкилсахарид, при этом композиция демонстрирует Tmax, составляющее приблизительно 30 минут или меньше у субъекта, посредством чего обеспечивается быстрое облегчение боли при мигрени.
В еще одном аспекте настоящим изобретением обеспечивается способ обеспечения уменьшенной частоты повторения боли при мигрени у субъекта, включающий введение композиции, включающей терапевтически эффективное количество агониста рецептора 5-HT и алкилсахарид, при этом композиция обеспечивает Tmax, составляющее менее приблизительно 20 минут, посредством чего обеспечивается уменьшенная частота повторения боли при мигрени у субъекта.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг. 1 представляет собой диаграмму, на которой демонстрируется биодоступность в процентах при интраназальном введении по сравнению с внутривенной инъекцией и коэффициенты вариации от субъекта к субъекту для MIACALCIN® (кальцитонина лосося) с алкилгликозидом и без него.
Фиг. 2 представляет собой график, на котором демонстрируется эффект интраназального введения инсулина/0,25% TDM (закрашенные кружки) и внутричерепного введения только инсулина (незакрашенные кружки) на снижение уровней глюкозы в крови.
Фиг. 3 представляет собой график, на котором демонстрируется эффект интраназального введения (закрашенные треугольники) и внутрибрюшинной (IP) инъекции (закрашенные кружки) экзендина-4/0,25% TDM и внутрибрюшинной инъекции только солевого раствора, минус TDM (незакрашенные кружки) на снижение уровней глюкозы в крови после внутрибрюшинной инъекции глюкозы (т.е. в так называемом «тесте на толерантность к глюкозе»).
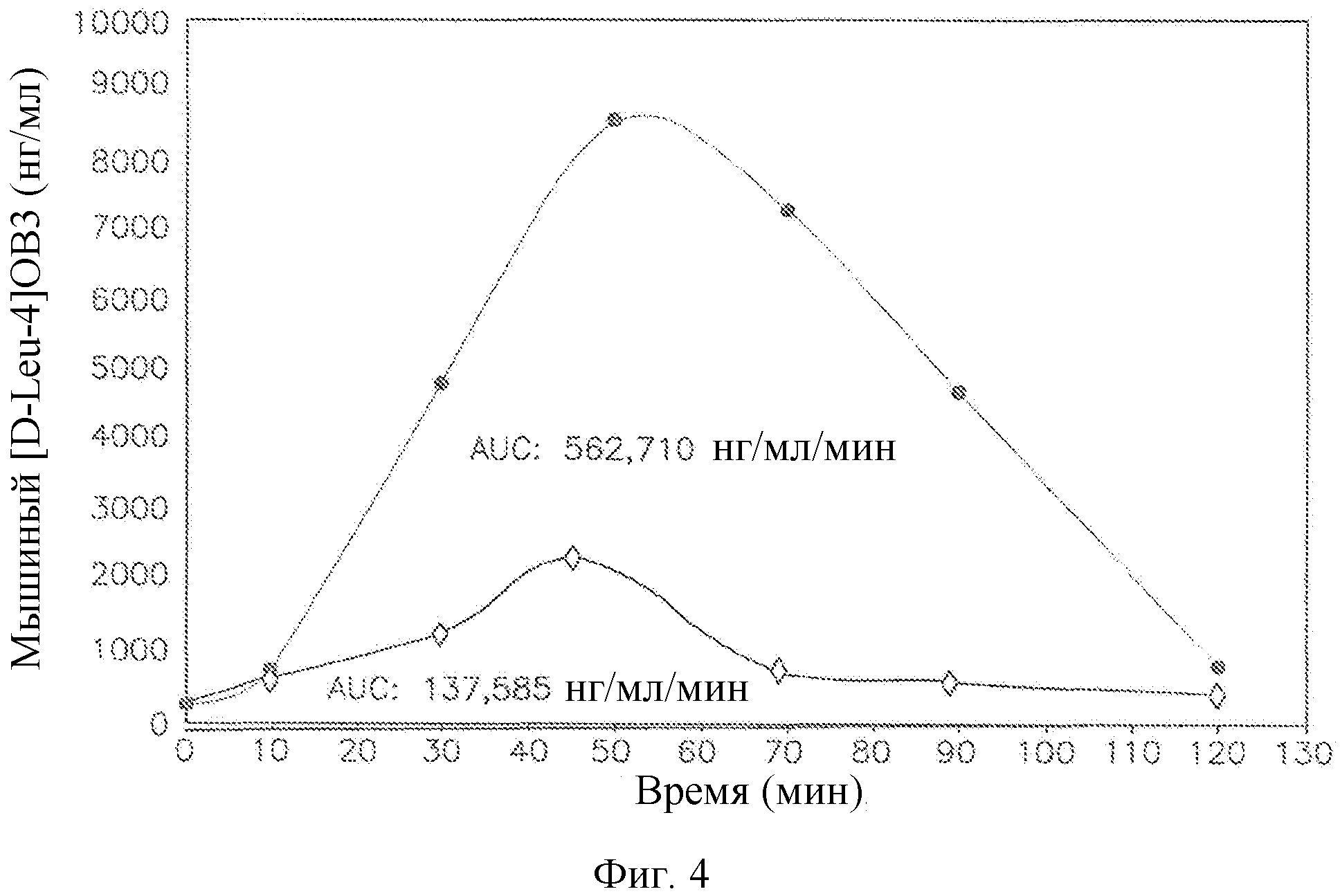
Фиг. 4 представляет собой график, на котором демонстрируется поглощение 1 мг мышиного [D-Leu-4]OB3 в 0,3% алкилгликозиде - тетрадецил-бета-D-мальтозиде (IntravailTM A3) самцами мышей Swiss Webster после введения с помощью кормления через зонд.
Фиг. 5 представляет собой график, на котором демонстрируется поглощение суматриптана в 0,5% алкилгликозиде - тетрадецил-бета-D-мальтозиде (IntravailTM A3) собаками в случае как перорального, так и ректального введения.
Фиг. 6 представляет собой кривую средних уровней в плазме пациентов, которым интраназально введен суматриптан.
Фиг. 7 представляет собой кривую средних уровней в плазме пациентов, которым интраназально введен суматриптан.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение можно легче понять на основе следующего подробного описания конкретных вариантов осуществления и включенных в него примеров.
Настоящее изобретение основывается на обнаружении того, что терапевтические композиции, состоящие из по крайней мере одного лекарственного средства и по крайней мере одного поверхностно-активного вещества, состоящего из по крайней мере одного алкилгликозида и/или по крайней мере одного алкилового эфира сахарида, являются стабильными, не вызывающими раздражение, антибактериальными композициями, которые увеличивают биодоступность лекарственного средства и не имеют видимых неблагоприятных эффектов после введения субъекту.
«Терапевтическая композиция» может состоять из смеси с органическим или неорганическим носителем или наполнителем, и может быть составлена, например, с использованием обычных нетоксичных, фармацевтически приемлемых носителей для таблеток, пилюль, капсул, суппозиториев, растворов, эмульсий, суспензий или другой подходящей для применения формы. Помимо вышеописанных носителей, они могут включать глюкозу, лактозу, маннозу, аравийскую камедь, желатин, маннит, крахмальную пасту, магния трисиликат, тальк, кукурузный крахмал, кератин, коллоидный кремнезем, картофельный крахмал, мочевину, среднецепочечные триглицериды, декстраны и другие носители, подходящие для использовании в производстве препаратов, в твердой, полутвердой или жидкой форме. Кроме того, могут использоваться вспомогательные стабилизаторы, загустители или красители, например, стабилизирующее сухое вещество, такое как дигидроксиацетон, считающийся кетотриозой.
«Лекарственным средством» является любое терапевтическое соединение, или молекула, или терапевтический агент, или биологически активное соединение, включающее, но без ограничения, нуклеиновые кислоты, небольшие молекулы, белки, полипептиды или пептиды и т.п.
Термин «нуклеиновые кислоты» или «олигонуклеотид» также обозначает ДНК, кДНК, РНК, киРНК, молекулу, вовлеченную в РНК-интерференцию, дцРНК и т.п., которые кодируют транслируемые и нетранслируемые области или ингибируют транслируемые или нетранслируемые области структурных генов, кодирующих пептид или белок, или регуляторный район. Например, нуклеиновая кислота настоящего изобретения может включать 5' и 3' нетранслируемые регуляторные нуклеотидные последовательности, а также транслируемые последовательности, связанные со структурным геном. Термин «нуклеиновые кислоты» или «олигонуклеотид» или грамматические эквиваленты, используемые здесь, относится к по крайней мере двум нуклеотидам, ковалентно связанным вместе.
Кроме того, термин «олигонуклеотид» относится к структурам, включающим модифицированные части, такие как модифицированные сахарные составляющие, составляющие в виде модифицированных оснований или составляющие в виде модифицированных связей между сахарами. Эти модифицированные части функционируют схожим с природными основаниями, природными сахарами и природными фосфодиэфирными связями образом. Соответственно, олигонуклеотиды могут иметь составляющие в виде измененных оснований, измененные сахарные составляющие или измененные связи между сахарами. Модифицированные связи могут быть, например, фосфорамидными, фосфоротиоатными, фосфородитиоатными, метилфосфонатными, фосфотриэфирными, фосфорамидатными, O-метилфосфороамидитными связями или пептидонуклеиновыми остовами и связями. Другие аналоги могут включать олигонуклеотиды с положительно заряженными остовами, неионными остовами и нерибозными остовами. Нуклеиновой кислотой может быть ДНК, как геномная, так и кДНК, РНК или гибрид, в котором нуклеиновая кислота содержит любую комбинацию дезоксирибо- и рибонуклеотидов и любую комбинацию природных или модифицированных оснований, включающих урацил, аденин, тимин, цитозин, гуанин, инозин, ксантин, гипоксантин, изоцитозин, изогуанин, галогенизированные основания и т.п. Другие модификации могут включать, например, дезаза- или азапурины и -пиримидины, используемые вместо природных пуриновых и пиримидиновых оснований; пиримидиновые основания, имеющие группы заместителей в 5- или 6-положениях, пуриновые основания, имеющие измененные группы или группы заместителей в 2-, 6- или 8-положениях, или сахара, имеющие группы заместителей в их 2'-положении, замены одного или нескольких атомов водорода сахара, или карбоциклические или ациклические сахара.
Используемый здесь термин «антисмысловая» относится к любой композиции, содержащей последовательность нуклеиновой кислоты, которая комплементарна конкретной последовательности нуклеиновой кислоты. Термин «антисмысловая цепь» используется в отношении цепи нуклеиновой кислоты, которая комплементарна «смысловой» цепи. Антисмысловые молекулы можно создать с помощью любого способа, включающего синтез или транскрипцию. После введения в клетку комплементарные нуклеотиды объединяются с природными последовательностями, созданными клеткой, с образованием дуплексов и с блокированием либо транскрипции, либо трансляции.
Антисмысловые молекулы включают олигонуклеотиды, включающие последовательность одноцепочечной нуклеиновой кислоты (либо РНК, либо ДНК), способные к связыванию с являющимися мишенями, относящимися к рецепторам или лигандам последовательностями мРНК (смысловыми) или ДНК (антисмысловыми). Возможно получение антисмыслового или смыслового олигонуклеотида на основе последовательности кДНК, кодирующей конкретный белок. Антисмысловые или смысловые олигонуклеотиды, кроме того, включают олигонуклеотиды, которые имеют модифицированные сахаро-фосфатные остовы, и в которых связи между сахарами являются устойчивыми к эндогенным нуклеазам. Такие олигонуклеотиды с устойчивыми связями между сахарами являются стабильными in vivo (т.е. способны не поддаваться ферментативной деградации), но сохраняют специфичность последовательности, чтобы быть способными к связыванию с являющимися мишенями нуклеотидными последовательностями.
РНК-интерференция является явлением, в котором введение дцРНК в разнообразный ряд организмов и типов клеток вызывает деградацию комплементарной мРНК. В клетке длинные дцРНК расщепляются на короткие (например, длиной 21-25 нуклеотидов) интерферирующие РНК (киРНК) под действием рибонуклеазы. Впоследствии киРНК собираются, раскручиваясь в процессе, с белковыми компонентами в индуцированный РНК комплекс сайленсинга (RISC). Активированный RISC затем связывается с комплементарными транскриптами с помощью взаимодействий в виде спариваний оснований между антисмысловой цепью киРНК и мРНК. Связанная мРНК затем расщепляется, и специфическая в отношении последовательности деградация мРНК приводит к сайленсингу гена. Как здесь используется, «сайленсинг» относится к механизму, с помощью которого клетки выводят из эксплуатации большие участки хромосомной ДНК, что приводит к подавлению экспрессии конкретного гена. Механизм РНК-интерференции, по-видимому, был развит для защиты генома от эндогенных мобильных генетических элементов и от вирусных инфекций. Таким образом, РНК-интерференцию можно индуцировать введением молекул нуклеиновых кислот, комплементарных мРНК-мишени, подвергаемой деградации.
Другие примеры смысловых или антисмысловых олигонуклеотидов включают такие олигонуклеотиды, которые ковалентно связаны с органическими составляющими и другими составляющими, которые увеличивают сродство олигонуклеотидов к являющейся мишенью последовательности нуклеиновой кислоты, такими как поли-(L-лизин). Опять же дополнительно, интеркалирующие агенты, такие как эллиптицин, и алкилирующие агенты или металлокомплексы могут быть присоединены к смысловым или антисмысловым олигонуклеотидам для модифицирования специфичностей связывания антисмыслового или смыслового олигонуклеотида в отношении являющейся мишенью нуклеотидной последовательности.
Пептид настоящего изобретения может быть полезным для какого-либо лечения или диагностики пептидом или белком маленького - среднего размера (т.е. вплоть до 15 кДа, 30 кДа, 40 кДа, 50 кДа, 60 кДа, 70 кДа, 80 кДа, 90 кДа, 100 кДа, например). Механизмы увеличения абсорбции полипептидов описаны в патенте США № 5661130, который в целом включен, тем самым, посредством ссылки. Композиции настоящего изобретения могут быть смешаны со всеми такими пептидами, хотя степень, в которой увеличиваются эффекты пептидов, может варьировать в соответствии с молекулярной массой и физическими и химическими свойствами пептида и конкретным используемым поверхностно-активным веществом. Примеры полипептидов включают вазопрессин, полипептидные аналоги вазопрессина, десмопрессин, глюкагон, кортикотропин (ACTH), гонадотропин, кальцитонин, C-пептид инсулина, гормон паращитовидной железы (PTH), гормон роста (HG), гормон роста человека (hGH), гормон роста-рилизинг-гормон (GHRH), окситоцин, кортикотропин-рилизинг-гормон (CRH), соматостатин или полипептидные аналоги соматостатина, агонист гонадотропина или полипептидные аналоги агониста гонадотропина, предсердный натрийуретический пептид (ANP) человека, тироксин-рилизинг-гормон (TRH) человека, фолликулостимулирующий гормон (FSH), пролактин, инсулин, инсулиноподобный фактор-I роста (IGF-I) соматомедин-C (SM-C), кальцитонин, лептин и происходящий из лептина короткий пептид OB-3, мелатонин, GLP-1 или глюкагоноподобный пептид-1, GiP, нейропептид гипофизарную аденилатциклазу, ганглиозид GM-1, фактор роста нервов (NGF), нафарелин, (D-tryp6)-LHRH, FGF, антагонисты VEGF, лейпролид, интерферон (например, α, β, γ), низкомолекулярный гепарин, PYY, антагонисты LHRH, фактор роста кератиноцитов (KGF), глиальный нейротрофический фактор (GDNF), грелин и антагонисты грелина. Кроме того, в некоторых аспектах, пептид или белок выбирают из фактора роста, интерлейкина, вакцины на основе полипептида, фермента, эндорфина, гликопротеина, липопротеина или полипептида, вовлеченного в каскад реакций свертывания крови.
Другие лекарственные средства или терапевтические соединения, молекулы и/или агенты включают соединения или молекулы центральной нервной системы, воздействующие на нейромедиаторы или ионные каналы в нейронной мембране, (т.е. антидепрессанты (бупропион)), избирательные агонисты рецептора серотонина 2c, противоэпилептические средства (топирамат, зонисамид), некоторые антагонисты допамина и антагонисты рецептора каннабиноида-1 (римонабант)); лептин/инсулин/молекулы проводящего пути центральной нервной системы (т.е. аналоги лептина, активаторы переноса лептина и/или рецепторов лептина, цилиарный нейротрофический фактор (Аксокин), нейропептид Y и антагонисты связанного с агаути пептида, проопиомеланокортин, регулируемые кокаином и амфетамином транскрипционные активаторы, аналоги альфа-меланоцит-стимулирующего гормона, агонисты рецептора меланокортина-4, ингибиторы протеин-тирозинфосфатазы-1B, антагонисты активируемого пролифератором пероксисом рецептора-гамма, бромокрипин кратковременного действия (эргосет), агонисты соматостатина (октреотид), и адипонектин); молекулы желудочно-кишечного тракта - нервного проводящего пути (т.е. агенты, которые увеличивают активность глюкагоноподобного пептида-1 (экзендин-4, лираглютид, ингибиторы дипептидилпептидазы IV), белок YY3-36, грелин, антагонисты грелина, аналоги амилина (прамлинтид)); и соединения или молекулы, которые могут увеличить основной обмен, «избирательные» бета-3 стимуляторы/агонисты, антагонисты мелалин-концентрирующего гормона, аналоги фитостанола, функциональные масла, P57, ингибиторы амилазы, фрагменты гормона роста, синтетические аналоги дегидроэпиандростерона-сульфата, антагонисты активности 11B-гидроксистероид-дегидрогеназы типа 1 адипоцитов, агонисты кортикотропин-рилизинг-гормона, ингибиторы синтеза жирных кислот, ингибиторы карбоксипептидазы, ингибиторы липаз желудочно-кишечного тракта (ATL962), мелатонин, ралоксифен, оланзарен и дифенгидрамин.
Другие лекарственные средства или терапевтические соединения включают средства против остеопороза, такие как аналоги бисфосфонатов. Аналоги бисфосфонатов, также известные как дифосфонаты, используются клинически для лечения таких состояний, как остеопороз, деформирующий остит (болезни кости Педжета), метастазы в кости (с гиперкальциемией или без нее), множественная миелома, несовершенный остеогенез и другие заболевания, характеризующиеся ломкостью кости. Этот класс лекарственных средств ингибирует функцию остеокластов и резорбцию кости. Примеры бисфосфонатов, смешиваемых с алкилсахаридами для использования в описываемых здесь композициях, включают как не являющиеся N-содержащими, так и N-содержащие аналоги бисфосфонатов. Примеры не являющихся N-содержащими бисфосфонатов включают этидронат (DidronelTM), клодронат (BonefosTM, LoronTM), тилудронат (SkelidTM) и их фармацевтически приемлемые аналоги. Примеры N-содержащих бисфосфонатов включают памидронат (ArediaTM), неридронат, олпадронат, алендронат (FosamaxTM или Fosamax+DTM), ибандронат (BonivaTM), риседронат (ActonelTM) и золедронат (ZometaTM или ReclastTM) и их фармацевтически приемлемые аналоги.
Другие лекарственные средства или терапевтические соединения включают такие лекарственные средства, как аналоги триптанов. В основном аналоги триптанов представляют собой семейство лекарственных средств на основе триптамина, используемых для лечения мигреней и головных болей. Их действие объясняется их связыванием с серотониновыми рецепторами в нервных окончаниях и в кровеносных сосудах головы (вызывая их сужение) и последующим ингибированием выброса провоспалительных нейропептидов. Примеры триптанов, смешиваемых с алкилсахаридами для использования в описываемых здесь композициях, включают суматриптан (ImitrexTM и ImigranTM), ризатриптан (MaxaltTM), наратриптан (AmergeTM и NaramigTM), золмитриптан (ZomigTM), элетриптан (RelpaxTM), алмотриптан (AxertTM и AlmogranTM), фроватриптан (FrovaTM и MigardTM) и их фармацевтически приемлемые соли. Примеры фармацевтически приемлемых солей включают соли соляной, серной или бензойной кислоты, такие как наратриптана-HCl; суматриптана сульфат и ризатриптана бензоат. Солевые формы триптанов демонстрируют увеличенную растворимость в воде по сравнению со «свободным основанием» или незаряженными формами, и поэтому должно быть понятно, что при описании водосодержащих препаратов различных триптанов здесь предполагается, что добавляют или готовят in situ посредством добавления соответствующей кислоты (соляной кислоты, серной кислоты, бензойной кислоты и т.п.) к форме триптана в виде свободного основания растворимую солевую форму триптана.
Терапевтическая композиция настоящего изобретения включает лекарственное средство и увеличивающий абсорбцию лекарственного средства агент, например, поверхностно-активное вещество. Термин «поверхностно-активное вещество» охватывает любое поверхностно-активное вещество, которое меняет межфазное натяжение воды. Обычно поверхностно-активные вещества имеют одну липофильную и одну гидрофильную группу в молекуле. Вообще, группа включает мыла, детергенты, эмульгаторы, диспергаторы и смачивающие агенты, и различные группы антисептиков. Конкретнее, поверхностно-активные вещества включают стеарилтриэтаноламин, натрия лаурилсульфат, лауриламинопропионовую кислоту, лецитин, бензалкония хлорид, бензетония хлорид и глицерина моностеарат; и гидрофильные полимеры, такие как поливиниловый спирт, поливинилпирролидон, натриевая соль карбоксиметилцеллюлозы, метилцеллюлоза, гидроксиметилцеллюлоза, гидроксиэтилцеллюлоза и гидроксипропилцеллюлоза.
Предпочтительно поверхностно-активное вещество настоящего изобретения состоит из по крайней мере одного подходящего алкилгликозида. Как здесь используется, «алкилгликозид» относится к любому сахару, присоединенному с помощью химической связи к какому-либо гидрофобному алкилу, как известно в данной области техники. Под каким-либо «подходящим» алкилгликозидом подразумевается алкилгликозид, который соответствует ограничивающим признакам настоящего изобретения, т.е. тому, что алкилгликозид является нетоксичным и неионным, и тому, что он увеличивает абсорбцию соединения при его введении с соединением посредством пути глазной, интраназальной, носослезной, ингаляционной или легочной доставки, доставки в ротовую полость (сублингвальной доставки или доставки к клеткам щеки) или в спинномозговую жидкость (CSF). Подходящие соединения можно определить с использованием изложенных здесь способов.
Алкилгликозиды настоящего изобретения можно синтезировать с помощью известных процедур, т.е. химически, как описано, например, в Rosevear et al., Biochemistry 19: 4108-4115 (1980) или Koeltzow and Urfer, J Am. Oil Chem. Soc, 61: 1651-1655 (1984), патенте США № 3219656 и патенте США № 3839318, или ферментативно, как описано, например, в Li et al., J Biol. Chem., 266: 10723-10726 (1991) или Gopalan et al., J Biol. Chem. 267: 9629-9638 (1992).
Алкилгликозиды настоящего изобретения могут включать, но без ограничения, такие алкилгликозиды, как октил-, нонил-, децил-, ундецил-, додецил-, тридецил-, тетрадецил-, пентадецил-, гексадецил-, гептадецил- и октадецил-α- или β-D-мальтозид, -глюкозид или -сахарозид, (синтезированные в соответствии с Koeltzow and Urfer; Anatrace Inc., Maumee, Ohio; Calbiochem, San Diego, Calif.; Fluka Chemie, Switzerland); алкилтиомальтозиды, такие как гептил-, октил-, додецил-, тридецил и тетрадецил-β-D-тиомальтозид, (синтезированные в соответствии с Defaye, J. and Pederson, C, "Hydrogen Fluoride, Solvent and Reagent for Carbohydrate Conversion Technology" in Carbohydrates as Organic Raw Materials, 247-265 (F. W. Lichtenthaler, ed.) VCH Publishers, New York (1991); Ferenci, T., J. Bacteriol, 144: 7-11 (1980)); алкилтиоглюкозиды, такие как гептил- или октил-тио-α- или β-D-глюкопиранозид (Anatrace, Inc., Maumee, Ohio; см. Saito, S. and Tsuchiya, T. Chem. Pharm. Bull. 33: 503-508 (1985)); алкилтиосахарозы (синтезированные в соответствии, например, с Binder, T. P. and Robyt, J. F., Carbohydr. Res. 140: 9-20 (1985)); алкилмальтотриозиды (синтезированные в соответствии с Koeltzow and Urfer); амиды длинноцепочечных алифатических карбоновых кислот, образуемые из β-амино-алкиловых эфиров сахарозы; (синтезированные в соответствии с австрийским патентом 382381 (1987); Chem. Abstr., 108: 114719 (1988) иd Gruber and Greber pp. 95-116); производные палатинозы и изомальтамина, связанные амидной связью с алкильной цепью, (синтезированные в соответствии с Kunz, M., "Sucrose-based Hydrophilic Building Blocks as Intermediates for the Synthesis of Surfactants and Polymers" in Carbohydrates as Organic Raw Materials, 127-153); производные изомальтамина, связанные с алкильной цепью посредством мочевины, (синтезированные в соответствии с Kunz); уреиды длинноцепочечных алифатических карбоновых кислот, образуемые из β-амино-алкиловых эфиров сахарозы (синтезированные в соответствии с Gruber and Greber, pp. 95-116); и амиды длинноцепочечных алифатических карбоновых кислот, образуемые из β-амино-алкиловых эфиров сахарозы, (синтезированные в соответствии с австрийским патентом 382381 (1987), Chem. Abstr., 108: 114719 (1988) и Gruber and Greber, pp. 95-116).
Поверхностно-активные вещества настоящего изобретения, состоящие из алкилгликозида и/или эфира сахарозы, имеют характеристические числа гидрофильно-липофильного соотношения (HLB), которые можно рассчитать или определить эмпирически (Schick, M. J. Nonionic Surfactants, p. 607 (New York: Marcel Dekker, Inc. (1967)). Число HLB является прямым отражением гидрофильного характера поверхностно-активного вещества, т.е. чем больше число HLB, тем более гидрофильным является соединение. Числа HLB можно рассчитать по формуле: (20 × М.м. гидрофильного компонента)/(М.м. гидрофобного компонента + М.м. гидрофильного компонента), где М.м. = молекулярная масса (Rosen, M. J., Surfactants and Interfacial Phenomena, pp. 242-245, John Wiley, New York (1978)). Число HLB является непосредственным представлением гидрофильного характера поверхностно-активного вещества, т.е. чем больше число HLB, тем более гидрофильным является соединение. Предпочтительное поверхностно-активное вещество имеет число HLB, составляющее от приблизительно 10 до 20, и даже более предпочтительные пределы от приблизительно 11 до 15.
Как описано выше, может быть выбран, таким образом, гидрофобный алкил любого желательного размера, в зависимости от желаемой гидрофобности и гидрофильности сахаридной составляющей. Например, одними из предпочтительных пределов алкильных цепей являются пределы от приблизительно 9 до приблизительно 24 атомов углерода. Даже более предпочтительными пределами являются пределы от приблизительно 9 до приблизительно 16 или приблизительно 14 атомов углерода. Так же некоторые предпочтительные гликозиды включают мальтозу, сахарозу и глюкозу, связанную гликозидной связью с алкильной цепью, имеющей 9, 10, 12, 13, 14, 16, 18, 20, 22 или 24 атомов углерода, например, нонил-, децил-, додецил- и тетрадецил-сахарозид, -глюкозид и -мальтозид, и т.д. Эти структуры являются нетоксичными, поскольку они распадаются с образованием спирта и олигосахарида, и амфипатическими.
Поверхностно-активные вещества настоящего изобретения могут также включать сахарид. Как здесь используется, «сахаридом» являются, в том числе, и моносахариды, олигосахариды и полисахариды, в форме линейной цепи или циклической форме, или их комбинация, образующая сахаридную цепь. Олигосахаридами являются сахариды, имеющие два или более моносахаридных остатка. Сахарид можно выбрать, например, из любых имеющихся в настоящее время в продаже разновидностей сахаридов, или их можно синтезировать. Некоторые примеры множества возможных для использования сахаридов включают глюкозу, мальтозу, мальтотриозу, мальтотетраозу, сахарозу и трегалозу. Предпочтительные сахариды включают мальтозу, сахарозу и глюкозу.
Поверхностно-активные вещества настоящего изобретения могут также состоять из эфира сахарозы. Как здесь используются, «эфиры сахарозы» являются сахарозными эфирами жирных кислот и представляют собой комплекс сахарозы и жирной кислоты. Эфиры сахарозы могут принимать множество форм из-за восьми гидроксильных групп в сахарозе, пригодных для реакции, и множества групп жирных кислот, от ацетата до более больших, более объемных жирных кислот, которые могут взаимодействовать с сахарозой. Эта податливость означает, что множество продуктов и функциональностей может быть разработано на основе используемой составляющей в виде жирной кислоты. Эфиры сахарозы используются в пищевых и не пищевых продуктах, особенно в качестве поверхностно-активных веществ и эмульгаторов, при этом растет их применение в фармацевтических средствах, косметических средствах, детергентах и пищевых добавках. Они являются биоразрушаемыми, нетоксичными и с мягким действием на кожу.
Поверхностно-активные вещества настоящего изобретения имеют гидрофобную алкильную группу, связанную с гидрофильным сахаридом. Связь между гидрофобной алкильной группой и гидрофильным сахаридом может включать, среди прочих возможностей, гликозидную, тиогликозидную (Horton), амидную (Carbohydrates as Organic Raw Materials, F. W. Lichtenthaler ed., VCH Publishers, New York, 1991), уреидную (австрийский патент 386414 (1988); Chem. Abstr. 110: 137536p (1989); см. Gruber, H. and Greber, G., "Reactive Sucrose Derivatives" in Carbohydrates as Organic Raw Materials, pp. 95-116) или сложноэфирную связь (Sugar Esters: Preparation and Application, J. C. Colbert ed., (Noyes Data Corp., New Jersey), (1974)). Кроме того, предпочтительные гликозиды могут включать мальтозу, сахарозу и глюкозу, присоединенную с помощью гликозидной связи к алкильной цепи, имеющей приблизительно 9-16 атомов углерода, например, нонил-, децил-, додецил- и тетрадецил-сахарозид, -глюкозид и -мальтозид. Опять же эти структуры являются амфипатическими и нетоксичными, поскольку они распадаются с образованием спирта и олигосахарида.
Вышеприведенные примеры иллюстрируют типы гликозидов, которые могут использоваться в заявленных здесь способах, но этот перечень не является исчерпывающим. При выборе гликозида следует также принимать во внимание производные вышеприведенных соединений, которые соответствуют критериям формулы изобретения. Все соединения можно подвергнуть скринингу на эффективность, следуя способам, наставления в отношении которых приведены здесь, и в примерах.
Композиции настоящего изобретения можно вводить в форме, выбираемой из группы, состоящей из таблетки, капсулы, суппозитория, капель, распыляемого раствора, аэрозоля и формы замедленного высвобождения или замедленного удара. Распыляемый раствор и аэрозоль можно с успехом получить благодаря использованию подходящего распылителя. Формой замедленного высвобождения может быть офтальмологическое включение, подверженные эрозии микрочастицы, распухающие мукоадгезивные частицы, pH-чувствительные микрочастицы, наночастицы/латексная система, смолы для ионообменной хроматографии и другие полимерные гели и имплантаты (Ocusert, Alza Corp., California; Joshi, A., S. Ping and K. J. Himmelstein, заявка на патент WO 91/19481). Эти системы поддерживают длительный контакт лекарственного средства с абсорбирующей поверхностью, предотвращая размыв и непродуктивную потерю лекарственного средства. Длительный контакт с лекарственным средством является нетоксичным для кожи и слизистых оболочек.
Содержащие поверхностно-активное вещество композиции настоящего изобретения являются стабильными. Например, Baudys и др. в патенте США № 5726154 демонстрируют, что кальцитонин в водосодержащей жидкой композиции, включающей SDS (натрия додецилсульфат, поверхностно-активное вещество) и органическую кислоту, является стабильным в течение по крайней мере 6 месяцев. Так же содержащие поверхностно-активное вещество композиции настоящего изобретения имеют улучшенные свойства в отношении стабилизации после смешивания с лекарственным средством. В этих композициях органическая кислота не требуется. Например, композиция настоящего изобретения поддерживает стабильность белков и пептидных терапевтических средств в течение приблизительно 6 месяцев или дольше, при хранении при приблизительно от 4°C до 25°C.
Стабильность содержащих поверхностно-активное вещество композиций, отчасти, обусловлена их высоким уровнем без видимых неблагоприятных эффектов (NOAEL). Агентство по охране окружающей среды (EPA) определяет уровень без видимых неблагоприятных эффектов (NOAEL) как уровень воздействия, на котором нет статистически или биологически значимых увеличений частоты или тяжести неблагоприятных эффектов между подвергнутой воздействию популяцией и соответствующим ей контролем. Следовательно, термин «уровень без видимых неблагоприятных эффектов» (или NOAEL) представляет собой наибольшую концентрацию или количество вещества, установленную с помощью эксперимента или наблюдения, которая не вызывает видимого неблагоприятного изменения морфологии, функциональной способности, роста, развития или продолжительности жизни организма-мишени в определенных условиях.
Организация по продовольствию и сельскому хозяйству Объединенных Наций Всемирной Организации Здравоохранения (WHO) установила, что некоторые алкилгликозиды имеют очень высокие NOAEL, что создает возможность для увеличенного поглощения этих алкилгликозидов без какого-либо неблагоприятного эффекта. Это сообщение можно найти во Всемирной паутине на сайте inchem.org/documents/jecfa/jecmono/v10je11.htm. Например, NOAEL для сахарозы додеканоата, эфира сахарозы, используемого в пищевых продуктах, составляет приблизительно 20-30 грамм/килограмм/день, например, человек весом 70 килограмм (приблизительно 154 фунтов) может поглотить приблизительно 1400-2100 грамм (или приблизительно 3-4,6 фунтов) сахарозы додеканоата в день без какого-либо видимого неблагоприятного эффекта. Обычно приемлемое ежедневное поглощение в случае людей составляет приблизительно 1% NOAEL, что означает приблизительно 14-21 грамм, или 14 миллионов микрограмм - 21 миллионов микрограмм в день, неопределенно долго. Определения NOAEL и другие родственные определения можно найти во Всемирной паутине на сайте epa.gov/OCEPAterms. Таким образом, хотя могут быть вызваны некоторые эффекты при использовании уровней алкилгликозида, предполагаемых в настоящем изобретении, эти уровни не считаются неблагоприятными, или предшественниками неблагоприятных эффектов.
Соответственно, субъект, подвергаемый лечению содержащими поверхностно-активное вещество композициями настоящего изобретения, содержащими по крайней мере один алкилгликозид, например, тетрадецилмальтозид (TDM; или Intravail A), в концентрации, составляющей приблизительно 0,125% вес. алкилгликозида, два раза в день, или три раза в день, или более в зависимости от схемы лечения, поглощает приблизительно 200-300 микрограмм в день всего TDM. Значит, эффективная доза TDM в по крайней мере 1000 раз ниже (т.е. составляет 1/1000) NOAEL и опускается гораздо ниже 1% от NOAEL, что является приемлемым ежедневным поглощением; или в этом случае составляет 1/50000 приемлемого ежедневного поглощения. Иначе формулируя, алкилгликозиды настоящего изобретения характеризуются высоким NOAEL, так что количество или концентрация алкилгликозидов, используемых в настоящем изобретении, не вызывает неблагоприятный эффект и может поглощаться без риска, без какого-либо неблагоприятного эффекта.
Содержащие поверхностно-активное вещество композиции настоящего изобретения являются также стабильными, поскольку они являются физиологически нетоксичными и не вызывают раздражение. Термин «нетоксичная» означает, что молекула алкилгликозида обладает достаточно низкой токсичностью, чтобы быть подходящей для введения человеку и поглощения им. Предпочтительные алкилгликозиды не вызывают раздражение тканей, на которые их наносят. Любой используемый алкилгликозид должен обладать минимальной токсичностью в отношении клетки или не обладать ею, так что он не вызывает повреждение клетки. Однако же токсичность в случае любого конкретного алкилгликозида может меняться с изменением концентрации используемого алкилгликозида. Также благотворно, если выбранный алкилгликозид метаболизируется или элиминируется организмом, и если этот метаболизм или элиминация осуществляется таким образом, который не является пагубно токсичным. Термин «не вызывающий раздражение» означает, что агент не вызывает воспаление сразу же, после длительного или повторного контакта с поверхностью кожи или слизистыми оболочками.
Кроме того, один вариант содержащих поверхностно-активное вещество, в частности, эфиры сахарозы, композиций служит в качестве антибактериальных агентов. Агент является «антибактериальным» агентом или веществом, если агент или его эквивалент уничтожает бактерии или подавляет рост или размножение бактерий. Сообщалось об антибактериальной активности эфиров сахарозы и их жирных кислот, см. Tetsuaki et al. (1997) "Lysis of Bacillus subtilis cells by glycerol and sucrose esters of fatty acids," Applied and Environmental Microbiology, 53(3): 505-508. Watanabe и др. (2000) описывают, что галактозы и фруктозы лауреаты являются особенно эффективными моноэфирами углеводов, см. Watanabe et al., (2000) "Antibacterial carbohydrate monoesters suppressing cell growth of Streptococcus mutan in the presence of sucrose," Curr Microbiol 41(3): 210-213. Следовательно, настоящее изобретение не ограничивается описанными здесь эфирами сахарозы, а охватывает эфиры других углеводов, включающие эфиры галактозы и фруктозы, которые подавляют рост и размножение бактерий.
Как правило, все применимые противомикробные агенты являются токсичными веществами. См. Sutton and Porter (2002), "Development of the antimicrobial effectiveness test as USP Chapter <51>," 56(6): 300-311, который в целом включен сюда посредством ссылки. Например, часто используемые противомикробные агенты, такие как бензалкония хлорид, являются в высокой степени токсичными, что продемонстрировано с помощью исследований с использованием электронных микрофотографий, на которых видно значительное разрушение мукоцилиарных поверхностей при концентрациях бензалкония, которые гораздо ниже концентрации, которая обычно используется в препаратах для интраназального введения. См., например, Sebahattin Cureoglu, Murat Akkus, Ustun Osma, Mehmet Yaldiz, Faruk Oktay, Belgin Can, Cengiz Guven, Muhammet Tekin, and Faruk Meric (2002), "The effect of benzalkonium chloride an electron microscopy study," Eur Arch Otorhinolaryngol 259: 362-364.
Структуры поверхностно-активных веществ настоящего изобретения обычно присутствуют на уровне, составляющем от приблизительно 0,01% до 20% вес. Более предпочтительные уровни включения составляют от приблизительно 0,01% до 5% вес., от приблизительно 0,01% до 2% вес., от приблизительно 0,01% до 1%, наиболее предпочтительно от приблизительно 0,01% до 0,125% вес.. Предпочтительно поверхностно-активное вещество составляют в композицию так, чтобы оно было совместимо с другими компонентами, присутствующими в композиции. В композициях в форме жидкости, или геля, или капсулы, или в инъецируемой форме, или в форме распыляемой жидкости наиболее предпочтительно, когда поверхностно-активное вещество составляют в композиции так, чтобы оно увеличивало, или по крайней мере не уменьшало, стабильность любого белка или фермента в этих композициях. Кроме того, настоящее изобретение оптимизирует концентрацию посредством удержания насколько возможно низкой концентрации усилителя абсорбции, при сохранении, однако, желаемого эффекта.
Композиции настоящего изобретения после введения субъекту обеспечивают увеличенную доставку через слизистые оболочки биологически активного соединения(й), или лекарственного средства, с максимальной концентрацией (или Cmax) соединения(й) в ткани, или жидкости, или в плазме крови субъекта, которая составляет приблизительно 15%, 20% или 50% или более Cmax соединения(й) в ткани (например, центральной нервной системе), или жидкости, или плазме крови после внутримышечной инъекции эквивалентной концентрации соединения(й) субъекту.
Показатель того, сколько лекарственного средства или соединения(й) достигает кровотока в заданный период времени, например 24 часа, можно также рассчитать посредством нанесения на график концентраций лекарственного средства в крови в различные моменты времени в течение 24-часового периода или большего периода, а затем определения площади под кривой (AUC) между 0 и 24 часами. Так же показатель эффективности лекарственного средства можно определить на основе времени до достижения максимальной концентрации (tmax) биологически активного соединения(й) в ткани (например, центральной нервной системе), или жидкости, или плазме крови субъекта между приблизительно 0,1 и 1,0 часом. Терапевтические композиции настоящего изобретения ускоряют начало действия лекарственного средства (т.е. уменьшают Tmax) в приблизительно 1,5-2 раза.
Также терапевтические композиции или препараты настоящего изобретения могут вводиться или доставляться нуждающемуся субъекту системно или местно. Подходящие пути могут, например, включать пероральное, глазное, интраназальное, носослезное, ингаляционное или легочное введение, введение в ротовую полость (сублингвальное введение или применение к клеткам щеки), введение через слизистые оболочки, вагинальную, ректальную, парентеральную доставку, в том числе внутримышечную, подкожную, внутривенную, внутрибрюшинную доставку или доставку в спинномозговую жидкость (CSF). Кроме того, вид доставки, например, в виде жидкости, геля, таблетки, распыляемой жидкости и т.д., будет также зависеть от способа доставки субъекту.
Кроме того, терапевтические композиции настоящего изобретения могут состоять из фармацевтически приемлемого носителя. «Фармацевтически приемлемым носителем» является водосодержащий или безводный агент, например, спиртовой или масляный, или их смесь и может содержать поверхностно-активное вещество, смягчающее вещество, смазывающее вещество, стабилизатор, краситель, отдушку, консервант, кислоту или щелочь для доведения pH, растворитель, эмульгатор, гелеобразующий агент, увлажняющий компонент, стабилизатор, смачивающий агент, агент для замедленного высвобождения, гигроскопическое вещество или другой компонент, обычно включаемый в конкретную форму фармацевтической композиции. Фармацевтически приемлемые носители хорошо известны в данной области техники и включают, например, водные растворы, такие как вода или физиологически забуференный солевой раствор, или другие растворители или носители, такие как гликоли, глицерин и масла, такие как оливковое масло, или инъецируемые эфиры органических кислот. Фармацевтически приемлемый носитель может содержать физиологически приемлемые соединения, которые действуют, например, со стабилизацией или увеличением абсорбции специфического ингибитора, например, углеводы, такие как глюкоза, сахароза или декстраны, антиоксиданты, такие как аскорбиновая кислота или глутатион, хелатообразующие агенты, низкомолекулярные белки или другие стабилизаторы или наполнители. Фармацевтически приемлемый носитель может быть также выбран из таких веществ, как дистиллированная вода, бензиловый спирт, лактоза, крахмалы, тальк, магния стеарат, поливинилпирролидон, альгиновая кислота, коллоидный кремнезем, диоксид титана и корригенты.
Кроме того, для уменьшения чувствительности алкилсахаридов или алкиловых эфиров гликозидов к гидролитическому расщеплению лекарственного средства различные атомы кислорода внутри лекарственных средств можно заменить серой (Defaye, J. and Gelas, J. in Studies in Natural Product Chemistry (Atta-ur-Rahman, ed.) Vol. 8, pp. 315-357, Elsevier, Amsterdam, 1991). Например, гетероатомом сахарного цикла может быть либо кислород, либо сера, или связь между моносахаридами в олигосахариде может быть кислородом или серой (Horton, D. and Wander, J. D., "Thio Sugars and Derivatives," The Carbohydrates: Chemistry and Biochemistry, 2d. Ed. Vol. IB, (W. Reyman and D. Horton eds.), pp. 799-842, (Academic Press, New York), (1972)). Олигосахариды могут иметь либо α (альфа), либо β (бета) аномерную конфигурацию (см. Pacsu, E., et al. in Methods in Carbohydrate Chemistry (R. L. Whistler, et al., eds.) Vol. 2, pp. 376-385, Academic Press, New York 1963).
Композицию настоящего изобретения можно приготовить в форме таблетки посредством смешивания терапевтического агента или лекарственного средства и одного алкилгликозида и/или алкилового эфир сахарида в соответствии с настоящим изобретением и подходящего фармацевтического носителя или наполнителя, например, маннита, кукурузного крахмала, поливинилпирролидона или т.п., гранулирования смеси и, наконец, прессования смеси в присутствии фармацевтического носителя, такого как кукурузный крахмал, магния стеарат или т.п. При необходимости приготовляемый таким образом препарат может включать сахаросодержащее или энтеросолюбильное покрытие или быть покрыто таким образом, что активная составляющая высвобождается постепенно, например, в среде с соответствующим pH.
«Энтеросолюбильное покрытие» представляет собой полимер, упаковывающий, окружающий или образующий слой или оболочку вокруг терапевтической композиции или ядра. Также энтеросолюбильное покрытие может содержать лекарственное средство, которое совместимо или несовместимо с покровным слоем. Одна композиция в форме таблетки, которая растворяется или высвобождает лекарственное средство при более высоких уровнях pH (например, при pH, превышающем 4,0, превышающем 4,5, превышающем 5,0 или выше), но не при низких уровнях pH (например, при pH 4 или ниже); или наоборот, может включать энтеросолюбильный покровный полимер с совместимым лекарственным средством.
В предпочтительном варианте осуществления формой дозозависимого высвобождения является таблетка, включающая
(a) ядро, включающее
(i) терапевтический агент или лекарственное средство;
(ii) поверхностно-активное вещество, включающее по крайней мере один алкилгликозид и/или алкиловый эфир сахарида; и
(b) по крайней мере одно оболочечное покрытие, окружающее ядро, причем покрытие является непроницаемым, проницаемым, полупроницаемым или пористым покрытием и становится более проницаемым или пористым при контакте с водосодержащей средой с определенным рН.
Термин «оболочка» является синонимом термина «покрытие» или его эквивалентов. Термины используются для определения зоны лекарственного средства, например, таблетки, которая является непроницаемой, проницаемой, полупроницаемой или пористой для водного раствора(ов) или жидкости(ей) организма, и/или терапевтического агента(ов) или лекарственного средства (средств), заключенных в нее. Если оболочка является проницаемой, полупроницаемой или пористой для лекарственного средства, лекарственное средство может высвобождаться через отверстия или поры оболочки в раствор или in vivo. Пористую оболочку можно изготовить механическим способом (например, «просверливанием» микроскопических отверстий или пор в оболочечном слое, используя лазер), или ее можно обеспечить на основе физико-химических свойств покровного полимера(ов). Оболочечные или покровные полимеры настоящего изобретения хорошо известны в данной области техники и включают сложные эфиры целлюлозы, диэфиры целлюлозы, триэфиры целлюлозы, простые эфиры целлюлозы, простые-сложные эфиры целлюлозы, ацилат целлюлозы, диацилат целлюлозы, триацилат целлюлозы, ацетат целлюлозы, диацетат целлюлозы, триацетат целлюлозы, ацетат-пропионат целлюлозы и ацетат-бутират целлюлозы. Другие подходящие полимеры описаны в патентах США № 3845770, 3916899, 4008719, 4036228 и 411210, которые включены сюда посредством ссылки.
Кроме того, энтеросолюбильное покрытие в соответствии с настоящим изобретением может включать пластификатор и достаточное для воздействия на рН суспензии в растворе или in vivo или его регулирования количество гидроксида натрия (NaOH). Примеры пластификаторов включают триэтилцитрат, триацетин, трибутилсебекат или полиэтиленгликоль. Для воздействия на рН суспензии в растворе или in vivo или его регулирования можно также использовать другие подщелачивающие агенты, включающие гидроксид калия, карбонат кальция, натриевую соль карбоксиметилцеллюлозы, оксид магния и гидроксид магния.
Соответственно, в одном варианте осуществления можно разработать энтеросолюбильное покрытие для высвобождения определенного процента лекарственного средства или лекарственных средств в определенных средах с определенным pH или определенными пределами pH. Например, терапевтическая композиция настоящего изобретения может включать по крайней мере одно энтеросолюбильное покрытие, покрывающее или защищающее по крайней мере одно лекарственное средство, которое является химически нестабильным в кислотной среде (например, желудке). Энтеросолюбильное покрытие защищает лекарственное средство от кислотной среды (например, pH<3), между тем высвобождая лекарственное средство в местах, являющихся менее кислотными, например, районах тонкой и толстой кишки, где pH равняется 3, или 4, или 5, или более. Лекарственное средство такого характера будет перемещаться из одного района желудочно-кишечного тракта в другой район, например, перемещение лекарственного средства из желудка в тонкую кишку (двенадцатиперстную кишку, тощую кишку и подвздошную кишку) занимает от приблизительно 2 до приблизительно 4 часов. Во время этого прохождения или перехода pH меняется с приблизительно 3 (например, в желудке) до 4, или 5, или до приблизительно pH 6 или 7, или более. Следовательно, энтеросолюбильное покрытие позволяет ядру, содержащему лекарственное средство, оставаться по существу интактным и предотвращает преждевременное высвобождение лекарственного средства или препятствует проникновению кислоты и предотвращает дестабилизацию лекарственного средства.
Примеры подходящих энтеросолюбильных полимеров включают, но без ограничения, ацетат-фталат целлюлозы, фталат гидроксипропилметилцеллюлозы, поливинилацетат-фталат, сополимер метакриловой кислоты, шеллак, ацетат-тримеллитат целлюлозы, ацетат-сукцинат гидроксипропилметилцеллюлозы, фталат гидроксипропилметилцеллюлозы, ацетат-фталат целлюлозы, ацетат-сукцинат целлюлозы, ацетат-малат целлюлозы, бензоат-фталат целлюлозы, пропионат-фталат целлюлозы, фталат метилцеллюлозы, карбоксиметилэтилцеллюлозу, фталат этилгидроксиэтилцеллюлозы, шеллак, сополимер стирола и акриловой кислоты, сополимер метилакрилата и акриловой кислоты, сополимер метилакрилата и метакриловой кислоты, сополимер бутилакриалат-стирол-акриловая кислота, сополимер метакриловой кислоты и метилметакрилата, сополимер метакриловой кислоты и этилакрилата, сополимер метилакрилат-метакриловая кислота-октилакрилат, сополимер винилацетата и ангидрида малеиновой кислоты, сополимер стирола и ангидрида малеиновой кислоты, сополимер стирола и моноэфира малеиновой кислоты, сополимер винилметилового эфира и ангидрида малеиновой кислоты, сополимер этилена и ангидрида малеиновой кислоты, сополимер винилбутилового эфира и ангидрида малеиновой кислоты, сополимер акрилонитрил-метилакриалат-ангидрид малеиновой кислоты, сополимер бутилакриалат-стирол-ангидрид малеиновой кислоты, эфир фталевой кислоты и поливинолого спирта, эфир фталевой кислоты и поливинилацеталя, поливинилбутилат фталат и эфир фталевой кислоты и поливинилацетоацеталя, или их комбинации. Квалифицированному в данной области техники специалисту будет понятно, что другие полимеры для гидрофильного, гидрофобного и энтеросолюбильного покрытия могут легко применяться, в одиночку или в любой комбинации, в качестве всего или части покрытия в соответствии с настоящим изобретением.
Терапевтические композиции настоящего изобретения в форме таблетки могут иметь множество покрытий, например, гидрофильное покрытие (например, на основе гидроксипропилметилцеллюлозы), и/или гидрофобное покрытие (например, на основе алкилцеллюлозы), и/или энтеросолюбильное покрытие. Например, ядро таблетки может быть покрыто множеством покрытий одинакового типа или множеством покрытий различных типов, выбираемых из гидрофильного, гидрофобного или энтеросолюбильного покрытия. Следовательно, предполагается возможность разработки таблетки, имеющей по крайней мере один слой (но она может иметь более одного слоя), состоящий из одинаковых или различных покрытий в зависимости от ткани-мишени или назначения лекарственного средства или лекарственных средств. Например, первая композиция может содержаться в слое ядра таблетки, окруженного первым покровным слоем (например, гидрофильным, гидрофобным или энтеросолюбильным покрытием), а вторая такая же или отличная композиция или лекарственное средство в той же самой или отличной дозе может быть включена во второй покровный слой, и т.д. Эта слоистость различных покровов предусматривает первое, второе, третье высвобождение, или более постепенное или дозозависимое высвобождение содержащей одно и то же лекарственное средство или различные лекарственные средства композиции.
В предпочтительном варианте осуществления первая доза первой композиции настоящего изобретения содержится в ядре таблетки с энтеросолюбильным покрытием, так что энтеросолюбильное покрытие защищает содержащуюся в ней композицию и препятствует ее распаду или высвобождению в желудке. В другом примере первая ударная доза терапевтической композиции включена в первый слой и состоит из от приблизительно 10% до приблизительно 40% общего количества композиции в целом, включенной в препарат или таблетку. При второй ударной дозе высвобождается другой процент общей дозы композиции. В настоящем изобретении предусматривается столько доз высвобождения, сколько необходимо в схеме лечения. Таким образом, в определенных аспектах одно покрытие или множество покровных слоев присутствует в количестве, колеблющемся от приблизительно 2% до 6% вес., предпочтительно от приблизительно 2% до приблизительно 5%, даже предпочтительнее от приблизительно 2% до приблизительно 3% вес. от стандартной лекарственной формы с покрытием.
Соответственно, препараты композиций настоящего изобретения делают возможным избирательное высвобождение содержимого твердой капсулы или таблетки в желательном месте, более дистальных частях желудочно-кишечного тракта (например, тонкой и толстой кишке), посредством выбора подходящего pH-растворимого полимера для конкретного района. Механического выталкивания препаратов композиций можно также достичь посредством включения абсорбирующего воду полимера, который увеличивается в объеме после абсорбирования воды внутрь твердой полупроницаемой капсулы, вытесняя, тем самым, композицию через отверстия в твердой капсуле.
Лекарственные средства, особенно подходящие для дозозависимого высвобождения с течением времени включают, но без ограничения, инсулиноподобный фактор-I роста (IGF-I), соматомедин-C (SM-C; диабет, нервная функция, функция почек) инсулин (диабет), кальцитонин (остеопороз), лептин (ожирение; бесплодие), происходящий из лептина короткий пептид (OB-3), hGH (истощение при AID, карликовость), гормон паращитовидной железы (PTH) человека (остеопороз), мелатонин (сон), GLP-1 или глюкагоноподобный пептид-1 (диабет), GiP (диабет), активирующий гипофизарную аденилатциклазу полипептид (PACAP), функция островков (диабет), ганглиозид GM-1 (болезнь Альцгеймера), фактор роста нервов (NGF) (болезнь Альцгеймера), нафарелин (эндометриоз), Synarel® (раствор нафарелина ацетата для интраназального введения), (D-tryp6)-LHRH (бесплодие), FGF (язва двенадцатиперстной кишки, дегенерация желтого пятна, ожоги, раны, повреждения спинного мозга, восстановление кости и хряща после повреждения), антагонисты VEGF (для блокирования рецептора), агонист VEGF (дистресс-синдром новорожденных; ALS), лейпролид (рак предстательной железы и рак молочной железы), интерферон-альфа (хронический гепатит C), низкомолекулярный гепарин (свертывание крови, тромбоз глубоких вен), PYY (ожирение), антагонисты LHRH (бесплодие), LH (лютеинизирующий гормон), антагонисты грелина (ожирение), KGF (болезнь Паркинсона), GDNF (болезнь Паркинсона), G-CSF (эритропоэз при раке), Имитрекс (мигрень), Интегрелин (антикоагуляция), Natrecor® (застойная сердечная недостаточность), натрийуретический пептид В-типа человека (hBNP), SYNAREL® (Searl; раствор нафарелина ацетата для интраназального применения), Сандостатин (заменитель гормона роста), Фортео (остеопороз), назальный аэрозоль DDAVP® (десмопрессина ацетат), Cetrotide® (цетрореликса ацетат для инъекции), AntagonTM(ганиреликса ацетат), Ангиомакс (бивалирудин; ингибитор тромбина), Accolate® (зафирлукаст; инъецируемый препарат), Экзендин-4 (Эксанатид; диабет), SYMLIN® (прамлинтида ацетат; синтетический амилин; диабет), десмопрессин, глюкагон, ACTH (кортикотрофин), C-пептид инсулина, GHRH и аналоги (GnRHa), гормон роста-рилизинг-гормон, окситоцин, кортикотропин-рилизинг-гормон (CRH), предсердный натрийуретический пептид (ANP), тироксин-рилизинг-гормон (TRHrh), фолликулостимулирующий гормон (FSH), пролактин, тобрацин для глазного введения (инфекции роговицы), вазопрессин, десмопрессин, Фузеон (Roche; ингибитор слияния ВИЧ с М.м. 4492) и Эптифибатид.
Кроме того, квалифицированному в данной области техники специалисту будет понятно, что конкретный уровень дозы и частота введения доз в случае любого конкретного субъекта, нуждающегося в лечении, могут варьировать и будут зависеть от ряда факторов, включающих активность конкретного используемого соединения, устойчивости к метаболизму и продолжительности действия этого соединения, возраста, веса тела, общего состояния здоровья, пола, питания, способа и времени введения, скорости выведения, комбинации лекарственных средств, тяжести конкретного состояния и подвергающегося терапии хозяина.
Было установлено, что алкилгликозиды, в частности, алкилмальтозиды, а конкретнее додецилмальтозид (DDM) и тетрадецилмальтозид (TDM), стабилизируют инсулин в растворе и предотвращают агрегацию пептида, см. Hovgaard et al., "Insulin Stabilization and GI absorption," J Control. Rel, 19 (1992) 458-463, приведенный в Hovgaard et al., "Stabilization of insulin by alkylmaltosides: A spectroscopic evaluation," Int. J. Pharmaceutics 132 (1996) 107-113 (ниже «Hovgaard-1»). Кроме того, в Hovgaard-1 показано, что даже спустя 57 дней комплекс DDM-инсулин оставался стабильным и обладал почти полной биологической активностью. Постулируют, что стабильность комплекса обуславливается длиной алкильной группы (числом атомов углерода), и чем выше отношение DDM к инсулину (например, 4:1 и 16:1; см. фиг. 1 в Hovgaard 1), тем лучше. Однако, в соответствии с Hovgaard-1, хотя комплекс DDM-инсулин был стабильным, такая же стабильность не была установлена в случае других мальтозидов. Кроме того, в связанном исследовании Hovgaard и др. (1996) установили, что, когда DDM-инсулин вводился перорально животным in vivo, биодоступность комплекса была низкой (например, 0,5%-1% биодоступность), см. Hovgaard et al., "Stabilization of insulin by alkylmaltoside. B. Oral absorption in vivo in rats," Int. J. Pharmaceutics 132 (1996) 115-121 (Hovgaard-2). Следовательно, усовершенствованным аспектом настоящего изобретения является то, что поверхностно-активное вещество увеличивает биодоступность лекарственного средства для тканей-, органов-, системы-мишени и т.д., а также увеличивает стабильность лекарственного средства.
Соответственно, одним аспектом настоящего изобретения является обеспечение терапевтических композиций, содержащих по крайней мере одно лекарственное средство и одно поверхностно-активное вещество, причем поверхностно-активное вещество к тому же состоит из композиции по крайней мере одного алкилгликозида и/или алкилового эфира сахарида, которая увеличивает биодоступность лекарственного средства. Определение биодоступности лекарственных препаратов описано здесь. Как здесь используется, «биодоступность» представляет собой скорость и степень, с которой (в которой) активное вещество, или составляющая, достигает большого круга кровообращения в виде интактного лекарственного средства. Биодоступность любого лекарственного средства будет зависеть от того, насколько хорошо оно абсорбируется, и какая его доля выходит, покидая печень.
Для определения абсолютной биодоступности исследуемое лекарственное средство и способ введения определяют относительно внутривенной контрольной дозы. Биодоступность внутривенной дозы составляет 100% по определению. Например, животным или добровольцам назначают внутривенные инъекции и соответствующие пероральные дозы лекарственного средства. В течение периода времени отбирают образцы мочи или плазмы, и определяют уровни лекарственного средства в течение этого периода времени.
Определяют площади под кривыми (AUC) зависимости концентрации лекарственного средства от времени, вычерчиваемыми и для внутривенной, и для пероральной дозы, и расчет биодоступности обоих препаратов осуществляют с помощью расчета простой пропорциональной доли. Например, если при введении одинаковых внутривенной и пероральной доз AUC в случае пероральной дозы составляет 50% AUC в случае внутривенной дозы, биодоступность перорального препарата составляет 50%. Заметьте, что биодоступность любого лекарственного средства обуславливается многими факторами, включающими неполную абсорбцию, клиренс при первом прохождении или их комбинацию, (обсуждаемыми подробнее ниже). Кроме того, также определяют максимальную концентрацию (или Cmax) лекарственного средства в плазме относительно максимальной концентрации (Cmax) лекарственного средства в плазме после внутримышечной (IM) инъекции эквивалентной концентрации лекарственного средства. Кроме того, время до достижения максимальной концентрации (или tmax) лекарственного средства в плазме составляет приблизительно 0,1-1,0 час.
Для определения относительной биодоступности более одного препарата лекарственного средства (например, содержащего алкилгликозид или алкиловый эфир сахарида лекарственного препарата), оценивают биодоступность препаратов относительно друг друга, поскольку одно или оба лекарственных средства могли подвергнуться клиренсу при первом прохождении (что обсуждается подробнее ниже) и, следовательно, быть не выявленными. Например, первый пероральный препарат оценивают относительно второго перорального препарата. Второй препарат используют в качестве контроля для оценки биодоступности первого препарата. Этот тип исследования дает показатель относительной эффективности по величине абсорбции лекарственного средства двух препаратов.
Биодоступности лекарственных средств являются неустойчивыми и существенно варьируют от одного лекарственного средства к следующему. Например, назальный аэрозоль MIACALCIN® (кальцитонин лосося от Novartis), рецептурный препарат для лечения остеопороза после менопаузы у женщин, имеет среднюю биодоступность, составляющую приблизительно 3% (в диапазоне 0,3%-30,6%; см. фиг. 1). Информационный в отношении продукта MIACALCIN® листок можно найти во Всемирной паутине на сайтах miacalcin.com/info/howWorks/index.jsp и drugs.com/PDR/Miacalcin_Nasal_Spray.html. Данные о MIACALCIN®, которые были получены различными исследователями с использованием различных способов и являющихся людьми субъектов, демонстрируют большие колебания в биодоступности лекарственного средства, например, у нормальных добровольцев лишь ~3% интраназально введенной дозы являются биодоступными, по сравнению с той же дозой, введенной посредством внутримышечной инъекции (вкладыш в продукт MIACALCIN®). Это представляет колебание в два порядка величины и является нежелательным для потребителя.
Плохую биодоступность лекарственного средства можно также наблюдать в случае NASCOBAL® (Nastech), или цианокобаламина, который используется для лечения и поддержания гематологического статуса у пациентов, находящихся в ремиссии после внутримышечных терапий с использованием витамина B12. Препарат в виде геля вводили интраназально, и биодоступность B12сравнивали с таковой в случае внутримышечных инъекций B12. Максимальные концентрации B12 (или Tmax) были достигнуты через 1-2 часа после интраназального введения, и установлено, что относительно внутримышечной инъекции биодоступность геля для носа с B12составляет приблизительно 8,9% (90% доверительные интервалы, 7,1%-11,2%).
Алкилгликозиды или сложные эфиры сахарозы настоящего изобретения включают любые соединения, известные в настоящее время или открытые позже. Лекарственными средствами, которые являются особенно подходящими для смешивания с алкилгликозидами и/или алкиловыми эфирами сахаридов настоящего изобретения, являются те, которые трудно вводить другими способами, например, лекарственные средства, которые подвергаются деструкции в желудочно-кишечном тракте, или те, которые плохо абсорбируются из желудочно-кишечного тракта, или лекарственные средства, которые могут вводиться самим пациентом посредством глазной, интраназальной, носослезной, ингаляционной доставки или доставки в спинномозговую жидкость (CSF), вместо традиционных способов, таких как инъекция. Некоторые конкретные примеры включают пептиды, полипептиды, белки, нуклеиновые кислоты и другие макромолекулы, например, гормоны в виде пептидов, такие как инсулин и кальцитонин, энкефалины, глюкагон и гипогликемические средства, такие как толбутамид и глибурид, и агенты, которые плохо абсорбируются при использовании энтеральных путей доставки, такие как гризеофульвин, противогрибковое средство. Другие соединения включают, например, никотин, интерферон (например, альфа, бета, гамма), PYY, GLP-1, синтетический экзендин-4 (Эксенатид), гормон паращитовидной железы и гормон роста человека, или другие низкомолекулярные пептиды и белки.
Альтернативно, биодоступность лекарственного средства можно определить посредством измерения уровней клиренса при первом прохождении лекарственного средства печенью. Содержащие алкилгликозиды и/или алкиловые эфиры сахаридов композиции настоящего изобретения, вводимые интраназально или через ротовую полость (сублингвально или посредством доставки к клеткам щеки), не поступают в воротную кровеносную систему печени, избегая, тем самым, клиренса при первом прохождении печенью. Избегание клиренса при первом прохождении этих композиций печенью описывается здесь. Термин «печеночный клиренс при первом прохождении» относится к степени, в которой лекарственное средство удаляется печенью во время его первого прохождения в кровотоке воротной системы через печень в большой круг кровообращения. Его также называют метаболизмом первого прохождения или извлечением при первом прохождении.
Двумя основными путями удаления лекарственного средства из организма являются выведение почками, при этом лекарственное средство является не измененным, и удаление печенью, при этом лекарственное средство метаболизируется. Баланс между этими двумя путями зависит от относительной эффективности двух процессов. В настоящем изобретении описывается удаление печенью или печеночный клиренс. Печеночный клиренс при первом прохождении описывается Birkett и др. (1990 и 1991), которые в целом включены посредством ссылки, см. Birkett et al., Aust Prescr, 13(1990): 88-89; и Birkett et al., Austra Prescr 14: 14-16 (1991).
Кровь, несущая лекарственное средство, из большого круга кровообращения поступает в печень через воротную вену, и печень, в свою очередь, извлекает определенный процент или пропорцию (т.е. 0,5 или 50%) этого лекарственного средства. Оставшаяся часть (т.е. 0,2 или 20%) вновь поступает в большой круг кровообращения через печеночную вену. Этот процент клиренса лекарственного средства называют процентом извлечения печенью. Он представляет собой долю лекарственного средства в крови, которая безвозвратно удаляется (или извлекается) во время первого прохождения крови через печень. Если лекарственное средство не извлекается, процент извлечения печенью равен нулю. Напротив, если лекарственное средство извлекается в высокой степени при первом прохождении через печень, процент извлечения печенью может быть до 100% или 1,0. Как правило, клиренс лекарственного средства печенью зависит в этом случае от скорости доставки этого лекарственного средства в печень (или печеночного кровотока), и от эффективности удаления этого лекарственного средства (или процента извлечения).
Следовательно, общим уравнением, используемым для определения печеночного клиренса, является:
(печеночный клиренс - кровоток)=(несвязанная фракция*собственный клиренс)/кровоток + (несвязанная фракция*собственный клиренс) (1)
«Несвязанная фракция» лекарственного средства зависит от того, настолько сильно лекарственное средство связано с белками и клетками в крови. Как правило, лишь это несвязанное (или свободное) лекарственное средство подходит для диффузии из крови в клетку печени. В отсутствие печеночного кровотока и связывания с белками «собственный клиренс» представляет собой способность печени удалять (или метаболизировать) это лекарственное средство. В биохимических терминах он представляет собой показатель печеночной ферментативной активности в отношении конкретного лекарственное вещества. К тому же, несмотря на то, что собственный клиренс может быть высоким, лекарственные средства не могут выводиться быстрее, чем те, которые направляются в печень. Проще говоря, существует две ситуации, при которых печеночная ферментативная активность является очень высокой или очень низкой (т.е. высокий процент извлечения или низкий процент извлечения).
Когда печеночная ферментативная активность является низкой, уравнение упрощается до:
печеночный клиренс = несвязанная фракция*собственный клиренс (2)
Клиренс к тому же не зависит от кровотока, а вместо этого зависит напрямую от степени связывания с белками в крови и активности метаболизирующих лекарственное средство ферментов относительно этого лекарственного средства.
Напротив, когда печеночная ферментативная активность является высокой, уравнение представляет собой:
печеночный клиренс = печеночный кровоток (3)
При этом сценарии, поскольку ферменты являются столь активными, печень удаляет большую часть лекарственного средства, направленного в нее, и процент извлечения является высоким. Следовательно, единственным фактором, определяющим фактический печеночный клиренс, является скорость поставки лекарственного средства в печень (или печеночный кровоток).
Печеночный клиренс при первом прохождении является важным, поскольку даже небольшие изменения в извлечении лекарственных средств могут привести к большим изменениям в биодоступности. Например, если биодоступность лекарственного средства A при пероральном введении составляет 20% к моменту времени, когда оно достигает большого круга кровообращения, а биодоступность этого же лекарственного средства A при внутривенном введении составляет 100%, в отсутствие других осложняющих факторов, пероральная доза должна, следовательно, превышать в 5 раз внутривенную дозу, чтобы достичь сходных концентраций в плазме.
Во-вторых, в некоторых случаях, когда печеночная ферментативная активность является очень высокой, лекарственные препараты должны быть разработаны так, чтобы лекарственное средство поступало непосредственно в большой круг кровообращения, чтобы избежать печеночного клиренса при первом прохождении в общей сложности. Например, лекарственные средства, вводимые интраназально, сублингвально, буккально, ректально, вагинально и т.д., поступают непосредственно в большой круг кровообращения и не поступают в кровоток воротной системы печени для частичного или полного извлечения печенью. Альтернативно, если лекарственные средства невозможно вводить с использованием вышеуказанных путей, обеспечивается таблетка с по крайней мере одним энтеросолюбильным покровным слоем для предотвращения высвобождения лекарственного средства в желудке (т.е. в высокой степени кислотной среде). Таким образом, целью настоящего изобретения является введение лекарственных средств, используя эти альтернативные пути.
Кроме того, печеночный клиренс при первом прохождении является важным фактором, поскольку многим пациентам назначают более одной схемы приема лекарственных средств, и это может привести к взаимодействиям лекарственных средств, которые увеличивают или уменьшают печеночную ферментативную активность; увеличивая или уменьшая, тем самым, метаболизм представляющего интерес лекарственного средства (увеличивая или уменьшая процент его экстракции печенью).
Следовательно, терапевтические композиции настоящего изобретения могут вводиться непосредственно в большой круг кровообращения и избегать печеночного клиренса при первом прохождении. Избегание клиренса при первом прохождении обеспечивает то, что больше лекарственного средства будет доступно для системы (всего организма). Формулируя иначе, посредством избегания печеночного клиренса при первом прохождении увеличивается биодоступность лекарственного средства.
Настоящее изобретение также относится к способам увеличения прохождения низкомолекулярного соединения в сердечно-сосудистую систему субъекта, включающим введение посредством пероральной, глазной, интраназальной, носослезной, ингаляционной доставки или доставки в спинномозговую жидкость (CSF) соединения и увеличивающего абсорбцию количества подходящего нетоксичного, неионного алкилгликозида, имеющего гидрофобный алкил, присоединенный с помощью химической связи к гидрофильному сахариду.
Состав композиции соответствующим образом выбирают в соответствии с путем введения, таким как оральное введение (пероральный препарат), наружное применение (например, мазь), инъекция (препараты для инъекции) и введение через слизистые оболочки (например, буккальное введение и суппозитории) и т.д. В случае пероральных препаратов и препаратов для введения через слизистые оболочки используют, например, наполнители (например, крахмал, лактозу, кристаллическую целлюлозу, кальция лактат, магния алюминометасиликат и безводный силикат), дезинтеграторы (например, карбоксиметилцеллюлозу и кальциевую соль карбоксиметилцеллюлозы), смазывающие вещества (например, магния стеарат и тальк), агенты для покрытий (например, гидроксиэтилцеллюлозу) и корригенты; в то время как для инъекций используют солюбилизаторы и вспомогательные солюбилизаторы, способные формировать водный инъецируемый раствор, (например, дистиллированную воду для инъекции, физиологический солевой раствор и пропиленгликоль), суспендирующие агенты (например, поверхностно-активное вещество, такое как полисорбат 80), регуляторы pH (например, органическую кислоту и ее соли металлов) и стабилизаторы; а в качестве внешних агентов используют повышающие растворимость в воде или маслах агенты и вспомогательные солюбилизаторы (например, спирты и сложные эфиры жирных кислот), вещества для повышения клейкости (например, карбоксивиниловый полимер и полисахариды) и эмульгаторы (например, поверхностно-активное вещество) используют в качестве внешних агентов. Лекарственное средство и алкилгликозид можно смешать с указанными выше наполнителями, дезинтеграторами, полимерами для покрытий, солюбилизаторами, суспендирующими агентами и т.д. до введения, или они могут вводиться последовательно, в любом порядке. Предпочтительным является их смешивание до введения.
Термин «увеличивающий доставку через слизистые оболочки агент» включает агенты, которые увеличивают высвобождение или растворимость (например, из носителя для доставки препарата), скорость диффузии, способность к проникновению и хронометрирование, поглощение, продолжительность пребывания, стабильность, эффективный полупериод существования, уровни максимальных или устойчивых концентраций, клиренс и другие желательные характеристики доставки через слизистые оболочки (например, определяемые в месте доставки или в выбранном целевом месте активности, таком как кровяное русло или центральная нервная система) соединения(й) (например, биологически активного соединения). Увеличение доставки через слизистые оболочки может происходить посредством любого из множества механизмов, в том числе, например, посредством увеличения диффузии, переноса, устойчивости или стабильности соединения, увеличения текучести мембраны, модулирования доступности или действия кальция и других йонов, которые регулируют внутриклеточное или параклеточное проникновение, солюбилизации компонентов слизистых оболочек (например, липидов), изменения уровней суфгидрильных групп белков и не являющихся белками веществ в слизистых тканях, увеличения потока воды через слизистую поверхность, модулирования физиологии эпителиального контакта, уменьшения вязкости слизи, находящейся сверху слизистого эпителия, уменьшения скорости сукоцилиарного клиренса и других механизмов.
Приводимые в качестве примеров увеличивающие доставку через слизистые оболочки агенты включают следующие агенты и любые их комбинации:
(a) ингибирующий агрегацию агент;
(b) изменяющий заряд агента;
(c) агент для контролирования pH;
(d) ингибирующий деструктивные ферменты агент;
(e) муколитическое средство или агент для устранения слизи;
(f) цилиостатический агент;
(g) увеличивающий проникновение через мембрану агент, выбираемый из (i) поверхностно-активного вещества; (ii) соли желчной кислоты; (ii) фосфолипидной добавки, смешанной мицеллы, липосомы или носителя; (iii) спирта; (iv) енамина; (v) соединения-донора NO; (vi) длинноцепочечной амфипатической молекулы; (vii) небольшого гидрофобного усилителя проникновения; (viii) натрия или производного салициловой кислоты; (ix) глицеринового эфира ацетоуксусной кислоты; (x) производного циклодекстрина или бета-циклодекстрина; (xi) среднецепочечной жирной кислоты; (xii) хелатообразующего агента; (xiii) аминокислоты или ее соли; (xiv) N-ацетиламинокислоты или ее соли; (xv) фермента, деструктивного в отношении выбранного мембранного компонента; (ix) ингибитора синтеза жирных кислот; (x) ингибитора синтеза холестерина; и (xi) любой комбинации увеличивающих проникновение через мембрану агентов, перечисленных в (i)-(x);
(h) модулирующий физиологию эпителиального контакта агент;
(i) вазодилататор;
(j) избирательный, увеличивающий перенос агент; и
(k) стабилизирующий доставку наполнитель, носитель, мукоадгезив, вспомогательные или комплексообразующие частицы, с которыми соединение эффективно объединяется, ассоциируется, в которых оно содержится, в которые оно включается или с которыми оно связывается, что приводит к стабилизации соединения для увеличения доставки через слизистые оболочки носовой полости, причем составление соединения с увеличивающим интраназальную доставку агентами предусматривает увеличение биодоступности соединения в плазме крови субъекта.
Дополнительные увеличивающие доставку через слизистые оболочки агенты включают, например, лимонную кислоту, натрия цитрат, пропиленгликоль, глицерин, аскорбиновую кислоту (например, L-аскорбиновую кислоту), натрия метабисульфит, динатриевую соль этилендиаминтетрауксусной кислоты (EDTA), бензалкония хлорид, гидроксид натрия и их смеси. Например, EDTA или ее соли (например, натриевая или калиевая) используются в количествах, колеблющихся от приблизительно 0,01% до 2% вес. композиции, содержащей консервант алкилсахарид.
Терапевтическими агентами или лекарственными средствами настоящего изобретения могут быть пептиды или белки, применимые в медицине или диагностике, маленького - среднего размера, например, вплоть до приблизительно 15 кДа, 30 кДа, 50 кДа, 75 кДа и т.д., или белок, содержащий приблизительно 1-300 аминокислот или более. В способах настоящего изобретения также предполагается использование небольших молекул, например, органического соединения, которое имеет молекулярную массу, составляющую менее 3 кДа или менее 1,5 кДа.
Механизмы увеличения абсорбции лекарственного средства в соответствии с настоящим изобретением являются в основном применимыми и должны применяться ко всем таким пептидам или белкам, хотя степень, в которой увеличивается их абсорбция, может варьировать в соответствии с молекулярной массой (М.м.) и физико-химическими свойствами пептида или белка и конкретным используемым усилителем. Примеры пептидов или белков включают вазопрессин, полипептидные аналоги вазопрессина, десмопрессин, глюкагон, кортикотропин (ACTH), гонадотропин, кальцитонин, C-пептид инсулина, гормон паращитовидной железы (PTH), гормон роста (HG), гормон роста человека (hGH), гормон роста-рилизинг-гормон (GHRH), окситоцин, кортикотропин-рилизинг-гормон (CRH), соматостатин или полипептидные аналоги соматостатина, агонист гонадотропина или полипептидные аналоги агониста гонадотропина, предсердный натрийуретический пептид (ANP) человека, тироксин-рилизинг-гормон (TRH) человека, фолликулостимулирующий гормон (FSH) и пролактин.
Предпочтительной композицией настоящего изобретения является лекарственное средство в виде пептида - Эксенатид (или экзендин-4) и алкилгликозид. Эксенатид является синтетическим вариантом экзендина-4 и использовался в клинических испытаниях, проводимых AmylinTM Pharmaceuticals. Экзендин-4 является низкомолекулярным пептидом, который представляет собой первое лекарственное средство из нового класса терапевтических средств, известных как являющиеся миметиками инкретинов агенты или гормоны. Гормонами инкретинами являются любые из различных гормонов и факторов желудочно-кишечного тракта, которые функционируют как сильные стимуляторы секреции инсулина, например, желудочный ингибиторный полипептид (GIP), глюкагоноподобный пептид-1 (GLP-1), или Эксенатид, или экзендин-4, или их эквиваленты.
Экзендин-4 представляет собой встречающийся в природе пептид из 39 аминокислот, выделенный из выделений слюнных желез ящерицы-ядозуба, см. Eng et al., "Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas," J. Biol. Chem. 267(15): 7402-7405 (1992). Эксенатид проявляет действия в направлении снижения уровней глюкозы, подобные таковым глюкагоноподобного пептида, или GLP-1. В настоящее время исследуется потенциал Эксенатида в разрешении важных неудовлетворенных терапевтических потребностей многих людей с диабетом типа 2. Клинические испытания говорят о том, что лечение Эксенатидом снижает глюкозу в крови к целевым уровням и сопровождается потерей веса. Наблюдаемые при лечении Эксенатидом эффекты на контролирование глюкозы, скорее всего, обусловлены несколькими действиями, которые схожи с таковыми встречающегося в природе гормона инкретина GLP-1 (см. пример 7). Эти действия включают стимуляцию способности организма продуцировать инсулин в ответ на повышенные уровни глюкозы в крови, ингибирование выброса глюкагона после приемов пищи и уменьшение скорости, с которой питательные вещества проходят в кровяное русло. В исследованиях на животных введение Эксенатида приводило к сохранению и формированию новых бета-клеток, продуцирующих инсулин клеток в поджелудочной железе, которые выходят из строя, по мере прогрессирования диабета типа 2.
Эксенатид, являющиеся миметиками инкретинов агенты или их эквиваленты могут использоваться для лечения различных форм диабета, в том числе, но без ограничения, лабильного диабета, химического диабета или нарушения толерантности к глюкозе, гестационного диабета, несахарного диабета, нейрогенного несахарного диабета, нефрогенного несахарного диабета, гипофизарного несахарного диабета, латентного диабета, липоатрофического диабета, диабета зрелого возраста юноши (MODY), сахарного диабета (DM), сахарного диабета взрослого возраста (сахарного диабета типа 2), инсулинозависимого сахарного диабета (IDDM, или сахарного диабета типа 1), не являющегося инсулинозависимым сахарного диабета (NTDDM), сахарного диабета ювенильного или юношеского возраста, сахарного диабета с подверженностью кетозу, сахарного диабета с устойчивостью к кетозу, сахарного диабета, связанного с нарушением питания (MRDM), тропического или панкреатического сахарного диабета, сахарного диабета, преклинического диабета или диабета, вызванного различными лекарственными средствами, например, тиазидного диабета, стероидного диабета или модели на животном различных диабетов, в том числе, но без ограничения, аллоксанового диабета и экспериментального пункционного диабета.
В другом аспекте терапевтические композиции настоящего изобретения используются для лечения ожирения. Ожирение является общей проблемой как взрослых, так и подростков. Например, PYY3-36 (или AC162352) является гормоном, который играет важную роль в снижении аппетита. Являющийся фрагментом гормона желудочно-кишечного тракта пептид PYY3-36 (PYY) снижает аппетит и потребление пищи после инфузии субъектам нормального веса. Подобно гормону адипоцитов, лептину, PYY снижает потребление пищи посредством модулирования центров аппетита в гипоталамусе. Однако у страдающих ожирением пациентов существуют резистентность к действию лептина, что ограничивает, тем самым, терапевтическую эффективность лептина. В еще одних исследованиях установлено, что PYY снижает потребление пищи. При инъекции PYY выявлено, что они едят в среднем на 30% меньше обычного, что приводит к потере веса. Следовательно, PYY 3-36 обладает потенциалом служить лекарственным средством против ожирения. AmylinTM Pharmaceuticals подало заявку на регистрацию нового экспериментального лекарственно средства - PYY 3-36 в 2003.
Соединения, абсорбцию которых можно увеличить с помощью способа этого изобретения, включают любые соединения, известные в настоящее время или открытые позже, в частности, лекарственные средства, или терапевтические соединения, молекулы или агенты, которые трудно вводить другими способами, например, лекарственные средства, которые подвергаются деструкции в желудочно-кишечном тракте, или те, которые плохо абсорбируются из желудочно-кишечного тракта, или лекарственные средства, которые субъекты могли бы вводить сами более легко посредством глазной, интраназальной, носослезной, ингаляционной или легочной доставки, доставки в ротовую полость (сублингвальной доставки или доставки к клеткам щеки) или в спинномозговую жидкость (CSF), чем с помощью традиционных способов самостоятельного введения, таких как инъекция. Некоторые конкретные примеры включают пептиды, полипептиды, белки и другие макромолекулы, например, гормоны в виде пептидов, такие как инсулин и кальцитонин, энкефалины, глюкагон и гипогликемические средства, такие как толбутамид и глибурид, и агенты, которые плохо абсорбируются при использовании энтеральных путей доставки, такие как гризеофульвин, противогрибковое средство. Другие соединения включают, например, никотин, интерферон (например, альфа, бета, гамма), PYY, GLP-1, синтетический экзендин-4 (Эксенатид), гормон паращитовидной железы и гормон роста человека, или другие низкомолекулярные пептиды и белки.
Как обсуждалось выше, варьирующие количества лекарственного средства могут абсорбироваться, по мере того как лекарственное средство проходит через предшествующие желудку части пищеварительного тракта: щечный карман, подъязычный карман, ротоглотку и пищевод. Однако большая часть лекарственного средства проходит в желудок и абсорбируется обычным способом абсорбции, которым абсорбируются энтеросолюбильные лекарственные формы, такие как таблетки, капсулы, или жидкости. По мере того как лекарственное средство абсорбируется из кишечника, оно доставляется непосредственно в печень, где, в зависимости от конкретной химической структуры, оно может метаболизироваться и элиминироваться ферментами, которые осуществляют обычные процессы детоксификации в клетках печени. Эту элиминацию называют «метаболизмом первого прохождения» или «эффектом первого прохождения» в печени, как обсуждалось ранее. Результирующие метаболиты, чаще всего являющиеся по существу или полностью неактивными по сравнению с первоначальным лекарственным средством, часто встречаются циркулирующими в кровотоке и впоследствии удаляются в мочу и/или фекалии.
Аспекты настоящего изобретения основываются на обнаружении того, что добавление определенных алкилсахаридов, при включении в быстро диспергируемые лекарственные формы, изменяет процент лекарственного средства, который подвергается эффекту первого прохождения, что позволяет, таким образом, фиксированному количеству лекарственного средства вызывать больший клинический результат, или позволяет с помощью меньшего количества лекарственного средства достигать клинический результат, схожий с таковым, достигаемым с помощью в других отношениях большей дозы.
Дополнительные аспекты настоящего изобретения основываются на обнаружении того, что увеличение или уменьшение количества конкретного алкилсахарида, включаемого в быстро диспергируемые лекарственные формы, изменяет или модулирует место абсорбции лекарственного средства, увеличивая или уменьшая, соответственно, процент лекарственного средства, который проходит через ткань щеки по сравнению с другими частями пищеварительного тракта. В тех случаях, когда желательным является ускорение начала действия лекарственного средства, но сохранение обычно большего Tmax, связанного со стандартной пероральной таблеткой, содержание алкилсахарида может быть уменьшено для уменьшения абсорбции тканью щеки, так что часть лекарственного средства сразу же абсорбируется тканью щеки ради быстрого возникновения его действия, а остаток абсорбируется благодаря более медленному процессу желудочной абсорбции. Таким образом было установлено, что посредством выбора концентрации алкилсахарида, меньшей, например, менее 20%, концентрации алкилсахарида, которая, как экспериментально установлено, вызывает максимальную или почти максимальную абсорбцию тканью щеки, можно с успехом получить более широкий пик абсорбции на графике зависимости «системного уровня лекарственного средства» от времени, который считается клинически желательным.
Как далее обсуждается в примерах ниже, добавление определенных алкилсахаридов, имеющих конкретные длины алкильной цепи, к быстро диспергируемым таблеткам изменяет фармакокинетику абсорбции до попадания в желудок лекарственного средства полезным образом. Изменяет фармакокинетику абсорбции до попадания в желудок лекарственного средства полезным образом включение, в частности, приблизительно 0,2%-0,3%, 0,3%-0,4%, 0,4%-0,5%, 0,5%-1,0%, 1,0%-2,0%, 2,0%-3,0%, 3,0%-4,0%, 4,0%-5,0%, 5,0%-6,0%, 6,0%-7,0%, 7,0%-8,0%, 9,0%-10,0% и более 10% алкилгликозида. В приводимых в качестве примеров вариантах осуществления алкилсахаридом является додецилмальтозид, тетрадецилмальтозид и/или сахарозы додеканоат, который при включении в быстро диспергируемую форму таблетки увеличивает количество лекарственного средства, которое поступает в большой круг кровообращения, и уменьшает количество лекарственного средства, которое элиминируется в результате эффекта «первого прохождения» в печени. Кроме того, резко сокращается время до достижения максимальных уровней лекарственного средства, обычно с одного - шести часов до приблизительно 15-45 минут. В случае лечения агрессивных пациентов, подверженных транзиторному психозу, эта более быстрая абсорбция лекарственного средства, приводящая к более быстрому возникновению действия, может принести большую пользу.
Кроме того, другие аспекты настоящего изобретения основываются на обнаружении того, что, что, когда определенные типы быстро растворяющихся или быстро диспергируемых таблеток, помещают между щекой и десной или в тесный контакт с тканью щеки во рту, даже больший процент лекарственного средства непосредственно поступает в большой круг кровообращения, а меньшая часть впоследствии подвергается элиминации первого прохождения в печени. Наконец, было обнаружено, что особенно подходящее место во рту для этого эффекта находится внутри центральной части верхней губы, между внутренней стороной губы и деснами, непосредственно ниже носа. В приводимых в качестве примеров аспектах эти типы быстро растворимых лекарственных форм готовят посредством лиофилизации или вакуумной сушки. В приводимом в качестве примеров аспекте лекарственную форму готовят таким способом, который дает в результате лекарственную форму, которая является в значительной степени пористой.
Предполагается, что термин «быстро диспергируемая лекарственная форма» охватывает все типы лекарственных форм, способных растворяться, полностью или частично, во рту. Однако в приводимых в качестве примеров аспектах быстро диспергируемая лекарственная форма является твердой, быстро диспергируемой системой активного ингредиента и водорастворимой или дисперигируемой в воде матрицы-носителя, которая является инертной по отношению к активному ингредиенту и наполнителям. В различных вариантах осуществления систему можно получить посредством лиофилизации композиции или испарения растворителя из композиции, которая включает активный ингредиент, алкилсахарид и раствор носителя в растворе, в твердое состояние. Хотя ряд растворителей известны в данной области техники как растворители, подходящие для этого применения, одним растворителем, особенно подходящим для применения с настоящим изобретением, является вода. Могут также применяться водно-спиртовые смеси, если растворимость лекарственного средства в смешанном растворителе повышается. В случае плохо растворимых в воде лекарственных средств дисперсии небольших частиц лекарственного средства могут быть суспендированы в водосодержащем геле, который сохраняет равномерное распределение по существу нерастворимого лекарственного средства во время процесса лиофилизации или испарения.
В одном варианте осуществления водосодержащим гелем могут быть самообразующиеся гидрогели, описанные в патенте США № 60/957960, включенном сюда посредством ссылки, образованные с использованием отобранных алкилсахаридов, таких как моно- или дистеарат сахарозы и/или тетрадецилмальтозид. В различных аспектах быстро растворимые композиции настоящего изобретения, помещенные в ротовую полость, распадаются в пределах 20 секунд, предпочтительно менее 10 секунд.
Образующие матрицу агенты, подходящие для использования в быстро растворимых композициях настоящего изобретения описываются по всей этой заявке. Такие агенты включают материалы, происходящие из белков животных или растений, такие как желатины, коллагены, декстрины и белки семян сои, пшеницы и подорожника; камеди, такие как аравийская, гуаровая камедь, агар и ксантановая камедь; полисахариды; альгинаты; каррагенаны; декстраны; карбоксиметилцеллюлозы; пектины; синтетические полимеры, такие как поливинилпирролидон; и комплексы полипептидные/белковые или полисахаридные, такие как комплексы желатин-аравийская камедь. В приводимых в качестве примеров аспектах используется желатин, особенно рыбий желатин или свиной желатин.
Хотя предусматривается, что практически любое лекарственное средство может быть включено в быстро растворимую лекарственную форму, описываемую здесь, особенно подходящие лекарственные средства включают мелатонин, ралоксифен, оланзапен и дифенгидрамин.
Кроме того, в терапевтических композициях настоящего изобретения также предусматриваются не являющиеся пептидами лекарственные средства или терапевтические агенты. Например, в патенте США № 5552534 описываются не являющиеся пептидами соединения, которые имитируют или ингибируют химическую и/или биологическую активность ряда пептидов. Такие соединения можно создать посредством присоединения к определенным видам остовов, таким как тетрагидропиранильный цикл, химических функциональных групп, которые приводят к тому, что соединения становятся по крайней мере частично перекрестно реагирующими с пептидом. Как будет понятно, соединения, которые имитируют или ингибируют пептиды, являются в варьирующих степенях перекрестно реагирующими с ними. Другие методы приготовления пептидомиметиков описаны в патентах США № 5550251 и 5288707. Указанные выше патенты США включены в целом посредством ссылки.
Способ настоящего изобретения также может включать введение, вместе с алкилгликозидом и белком или пептидом, ингибитора протеазы и пептидазы, такого как апротинин, бестатин, ингибитор альфа1-протеиназы, соевый ингибитор трипсина, рекомбинантный ингибитор секреторных лейкоцитарных протеаз, каптоприл и другие ингибиторы преобразующих ангиотензин ферментов (ACE) и тиорфан, чтобы помочь белку или пептиду достичь своего места активности в организме в активном состоянии (т.е. с деградацией, достаточно маленькой, чтобы белок все еще был способен функционировать надлежащим образом). Ингибитор протеазы или пептидазы может быть смешан с алкилгликозидом и лекарственным средством, а затем введен, или он может быть введен отдельно, либо до, либо после введения гликозида или лекарственного средства.
Настоящим изобретением также обеспечивается способ снижения уровня глюкозы в крови у субъекта, включающий введение снижающего уровень глюкозы в крови количества композиции, включающей инсулин, и увеличивающего абсорбцию количества подходящего нетоксичного, неионного алкилгликозида, имеющего гидрофобную алкильную группу, присоединенную с помощью химической связи к гидрофильному сахариду, увеличивая, тем самым, абсорбцию инсулина и снижая уровень глюкозы в крови. «Снижающим уровень глюкозы в крови количеством» такой композиции является такое количество, которое способно вызывать эффект снижения уровней глюкозы в крови, как здесь сообщается. Предпочтительным является количество, которое снижает уровень глюкозы в крови до нормогликемического или почти нормогликемического диапазона. Также предпочтительным является количество, которое приводит к устойчивому снижению уровней глюкозы в крови. Даже более предпочтительным является количество, достаточное для лечения диабета, в том числе сахарного диабета (DM), посредством снижения уровня глюкозы в крови. Таким образом, способ настоящего изобретения может использоваться для лечения сахарного диабета. Предпочтительными алкилгликозидами являются алкилгликозиды, одинаковые с теми, которые описаны выше и проиллюстрированы в примерах.
Также обеспечивается способ повышения уровня глюкозы в крови у субъекта посредством введения повышающего уровень глюкозы в крови количества композиции, включающей глюкагоны и по крайней мере один алкилгликозид и/или алкиловый эфир сахарида. Когда композиция включает инсулин, она может использоваться для вызова известного эффекта инсулина в кровотоке, т.е. снижения уровней глюкозы в крови у субъекта. Такое введение может использоваться для лечения сахарного диабета или родственных заболеваний. «Повышающим уровень глюкозы в крови количеством» глюкагона в такой композиции является такое количество, которое способно вызывать эффект повышения уровней глюкозы в крови. Предпочтительным количеством является количество, которое повышает уровень глюкозы в крови до нормогликемического или почти нормогликемического диапазона. Другим предпочтительным количеством является количество, которое приводит к устойчивому повышению уровней глюкозы в крови. Даже более предпочтительным является количество, достаточное для лечения гипогликемии посредством повышения уровня глюкозы в крови. Таким образом, этот способ может использоваться для лечения гипогликемии. Предпочтительными алкилгликозидами являются алкилгликозиды, одинаковые с теми, которые описаны выше и проиллюстрированы в примерах.
Аналогично, когда эта композиция включает глюкагон, она может использоваться для вызова известного эффекта глюкагона в кровотоке, т.е. повышения уровней глюкозы в крови у субъекта. Следовательно, такое введение может использоваться для лечения гипогликемии, в том числе гипогликемического криза.
Настоящим изобретением также обеспечиваются способы ослабления неврологических нарушений, которые включают введение терапевтического агента в спинномозговую жидкость (CSF). Термин «неврологическое нарушение» обозначает любое нарушение, присутствующее в головном мозге, спинном мозге и связанных тканях, таких как оболочки головного мозга, которое реагирует на соответствующий терапевтический агент. Неожиданная способность терапевтических агентов настоящего изобретения к ослаблению неврологического нарушения обусловлена такой подачей терапевтического агента, что он сохраняется в церебро-вентрикулярном пространстве. Способность способа настоящего изобретения к позволению терапевтическому агенту сохраняться в зоне неврологического нарушения обеспечивает особенно эффективный способ лечения этих нарушений.
Будет, однако, понятно, что конкретный уровень дозы и частота введения доз в случае любого конкретного субъекта, нуждающегося в лечении, могут варьировать и будут зависеть от ряда факторов, включающих активность конкретного используемого соединения, устойчивости к метаболизму и продолжительности действия этого соединения, возраста, веса тела, общего состояния здоровья, пола, питания, способа и времени введения, скорости выведения, комбинации лекарственных средств, тяжести конкретного состояния и подвергающегося терапии хозяина. Однако обычно доза будет приблизительно той, которая является типичной для известных способов введения конкретного соединения. Например, в случае интраназального введения инсулина приблизительная доза могла бы составлять приблизительно 0,5 единиц/кг обычного свиного инсулина (Moses et al.). Оптимально, когда доза соединений, воздействующих на уровни глюкозы в крови, была бы той, которая требуется для достижения соответствующих уровней глюкозы, например, до нормального диапазона, составляющего приблизительно 5-6,7 мМ. Кроме того, подходящее количество может быть определено специалистом со средним уровнем компетентности в данной области техники с использованием лишь стандартного испытания, принимая во внимание приведенные здесь наставления (см. примеры).
Кроме того, композиции настоящего изобретения можно вводить в форме, выбираемой из группы, состоящей из капель, распыляемого раствора, аэрозоля и формы замедленного высвобождения. Распыляемый раствор и аэрозоль можно с успехом получить благодаря использованию подходящего распылителя. Формой замедленного высвобождения может быть офтальмологическое включение, подверженные эрозии микрочастицы, распухающие мукоадгезивные частицы, pH-чувствительные микрочастицы, наночастицы/латексная система, смолы для ионообменной хроматографии и другие полимерные гели и имплантаты (Ocusert, Alza Corp., California; Joshi, A., S. Ping and K. J. Himmelstein, заявка на патент WO 91/19481). Эти системы поддерживают длительный контакт лекарственного средства с абсорбирующей поверхностью, предотвращая размыв и непродуктивную потерю лекарственного средства.
В различных аспектах характеристики композиций настоящего изобретения, включающих агонисты рецепторов 5-HT, сравнивают с растворами IMITREX®. Используемыми здесь растворами являются следующие растворы. IMITREX® (суматриптана сукцинат) для инъекции является избирательным агонистом подтипа рецепторов 5-гидрокситриптамина. Суматриптана сукцинат химически определяется как 3-[2-(диметиламино)этил]-N-метил-индол-5-метансульфонамида сукцинат (1:1). IMITREX® для инъекции представляет собой прозрачный, бесцветный - бледно-желтый, стерильный, апирогенный раствор для подкожной инъекции. Каждые 0,5 мл 8 мг/мл раствора IMITREX® для инъекции содержат 4 мг суматриптана (основания) в виде соли щавелевой кислоты и 3,8 мг натрия хлорида, USP, в воде для инъекции, USP. Каждые 0,5 мл 12 мг/мл раствора IMITREX® для инъекции содержат 6 мг суматриптана (основания) в виде соли щавелевой кислоты и 3,5 мг натрия хлорида, USP, в воде для инъекции, USP. Диапазоном pH обоих растворов является приблизительно 4,2-5,3. Осмоляльность обоих инъецируемых растворов составляет 291 мосмоль. IMITREX® (суматриптана сукцинат) в виде назального аэрозоля в 100-мкл стандартной дозе содержит 5 или 20 мг суматриптана в водном забуференном растворе, содержащем дигидроортокалия фосфат NF, безводный двухосновный фосфат натрия USP, серную кислоту NF, гидроксид натрия NF и очищенную воду USP. pH раствора составляет приблизительно 5,5. Осмоляльность раствора составляет 372 или 742 мосмоль в случае 5- и 20-мг IMITREX® в виде назального аэрозоля, соответственно.
Как здесь используется, рецептор 5-HT включает любой рецептор семейств рецепторов 5-HT1-5-HT7. Такие рецепторы включают 5-HT1A, 5-HT1B, 5-HT1D, 5-HT1E, 5-HT1F, 5-HT2A, 5-HT2B, 5-HT2C, 5-HT3, 5-HT4, 5-HT5A, 5-HT6 и 5-HT7.
Настоящее изобретение конкретнее описывается в следующих примерах, которые, как предполагается, являются лишь иллюстративными, поскольку многочисленные модификации и вариации в них будет очевидны квалифицированным в данной области техники специалистам. Предполагается, что следующие примеры иллюстрируют, а не ограничивают настоящее изобретение.
ПРИМЕР 1
СОДЕРЖАЩИЕ АЛКИЛГЛИКОЗИД И/ИЛИ СЛОЖНЫЙ ЭФИР САХАРОЗЫ ПРЕПАРАТЫ НЕ ВЫЗЫВАЮТ РАЗДРАЖЕНИЕ ИЛИ РАЗРУШЕНИЕ СЛИЗИСТОЙ ОБОЛОЧКИ
Слизистая оболочка носа является в высокой степени васкуляризированной, а, следовательно, оптимальной для высокого проникновения лекарственного средства. Кроме того, при прохождении лекарственного средства (средств) через слизистую оболочку носа оно доступно для центральной нервной системы. Хотя местное применение лекарственных средств является желательным, проблемой для этого способа введения является раздражение слизистой оболочки.
Препарат, состоящий из алкилгликозида (0,125% TDM) в коммерческом безрецептурном (OTC) солевом растворе для носа, применяли in vivo к эпителию носа человека в течение периода времени, составляющего больше одного месяца. Содержащий 0,125% TDM препарат сравнивали с контролем, а именно тем же коммерческим (OTC) солевым раствором для носа, в течение того же периода времени. Результаты свидетельствуют о том, что в продолжение и после 33 дней ежедневного введения TDM (т.е. продолжительности исследования), нет видимого раздражения слизистой оболочки носа (не представленные данные). Таким образом, композиции настоящего изобретения являются нетоксичными и не вызывают раздражение в случае повторного и длительного интраназального введения, что полезно для тех пациентов, которые имеют хроническое и продолжающееся заболевание(я).
Сходное испытание проводили с использованием додеканоата сахарозы, сложного эфира сахарозы. Додеканоат сахарозы применяли in vivo к эпителию носа человека, и в продолжение и после 47 дней (т.е. продолжительности исследования) не выявлено видимое раздражение (не представленные данные). Таким образом, эти результаты свидетельствуют о том, что алкилгликозиды и сложные эфиры сахарозы настоящего изобретения являются нетоксичными и не вызывают раздражение слизистой оболочки при ежедневном введении в течение длительного периода времени.
ПРИМЕР 2
СОДЕРЖАЩИЕ АЛКИЛГЛИКОЗИД И/ИЛИ СЛОЖНЫЙ ЭФИР САХАРОЗЫ КОМПОЗИЦИИ СТАБИЛИЗИРУЮТ ЛЕКАРСТВЕННЫЕ СРЕДСТВА ПОСРЕДСТВОМ УВЕЛИЧЕНИЯ БИОДОСТУПНОСТИ И УМЕНЬШЕНИЯ КОЛЕБАНИЯ БИОДОСТУПНОСТИ ЛЕКАРСТВЕННОГО СРЕДСТВА
Стабильность алкилгликозида зависит, отчасти, от числа атомов углерода или длины алкильной цепи и других длинных алкильных цепей, при этом тетрадецилмальтозид (TDM) оказывает наибольший эффект; но стабилизирующими эффектами также обладают другие высоко разветвленные алкильные цепи, в том числе DDM. В отличие от Hovgaard-1, который описывает предпочтительность высокого отношения алкилгликозида к лекарственному средству, в настоящем изобретении демонстрируется, что это отношение является намного ниже. Например, алкилгликозиды в диапазоне, составляющем от приблизительно 0,01% до приблизительно 6% вес., приводят к подходящей стабилизации лекарственного средства; в то время как Hovgaard-1 показывает, что стабилизация достигается только при намного более высоких отношениях алкилгликозидов к лекарственному средству (10:1 и 16:1). Даже интереснее, алкилгликозиды настоящего изобретения в диапазоне, составляющем от приблизительно 0,01% до приблизительно 6%, увеличивали биодоступность (см. фиг. 1). Это резко отличается от данных Hovgaard-2, который продемонстрировал относительно низкую биодоступность (0,5-1%) при высоких отношениях алкилгликозида к лекарственному средству (10:1 и 16:1).
Фиг. 1 представляет собой диаграмму, на которой сравниваются биодоступности лекарственного средства MIACALCIN® (кальцитонина лосося от Novartis) с алкилгликозидом (TDM) и без него. MIACALCIN® является назальным аэрозолем и распыляется непосредственно на эпителий носа или слизистую оболочку носа. На фиг. 1 демонстрируется, что MIACALCIN® минус алкилгликозид характеризуется очень низкими уровнями биодоступности у людей (вкладыш с описанием продукта MIACALCIN®), по сравнению с MIACALCIN® с алкилгликозидом, вводимым крысам. Конкретнее, интраназальная доставка MIACALCIN® с 0,125% и 0,250% алкилгликозида (TDM) приводила к составляющей от приблизительно 43% до приблизительно 90% биодоступности, соответственно. Биодоступность при интраназальном введении MIACALCIN® без алкилгликозида составляет лишь приблизительно 3% у людей, и не была выявлена у крыс, что говорит о том, что крыса является жесткой моделью для оценки абсорбции лекарственного средства при интраназальном введении у людей. Таким образом, алкилгликозид настоящего изобретения увеличивает абсорбцию и увеличивает биодоступность лекарственного средства.
Кроме того, помимо увеличения биодоступности лекарственного средства, содержащие алкилгликозид композиции настоящего изобретения действенно уменьшают колебание биодоступности лекарственного средства. На фиг. 1 демонстрируется, что интраназальное введение MIACALCIN® с алкилгликозидом (0,125% или 0,25%) характеризуется колебанием биодоступности, составляющим ± 8%, в то время как колебание биодоступности без использования алкилгликозида составляет от 0,3% до 30%, или изменение на два порядка величины. Увеличение биодоступности и уменьшение колебания биодоступности обеспечивает то, что также уменьшается колебание от пациента к пациенту. Представленные на фиг. 1 результаты получены при интраназальном введении, однако схожие результаты ожидаются в случае пероральной, буккальной, вагинальной, ректальной и т.д. доставки и при отличных концентрациях алкилгликозида.
Таким образом, вопреки уровню техники, содержащие алкилгликозид композиции настоящего изобретения, в диапазоне, составляющем от приблизительно 0,01% до приблизительно 6%, приводят к увеличению биодоступности и уменьшению колебания биодоступности. Об этом не сообщалось в других случаях.
ПРИМЕР 3
ГЛАЗНОЕ ВВЕДЕНИЕ АЛКИЛСАХАРИДОВ ПЛЮС ИНСУЛИН ВЫЗЫВАЕТ ГИПОГЛИКЕМИЧЕСКИЕ ЭФФЕКТЫ IN VIVO
Нормальных крыс подвергали анестезии с использованием смеси ксилазин/кетамин для повышения уровней глюкозы в их крови. Повышенные уровни D-глюкозы, которые имеют место в ответ на анестезию, обеспечивают оптимальную систему для определения системного гипогликемического действия лекарственного средства при введении, например, содержащих инсулин глазных капель. Эта модель на животном воспроизводит гипергликемическое состояние, наблюдаемое у страдающих диабетом животных и людей. В экспериментальной группе животных подвергнутым анестезии крысам вводили глазные капли, содержащие инсулин. Уровни глюкозы в крови у животных экспериментальной группы сравнивали с подвергнутыми анестезии животными, которые получили глазные капли без инсулина. Изменение уровней глюкозы в крови и дифференциальные системные ответные реакции отражают эффект инсулина, прошедшего через путь введения, например, глазной путь.
Взрослых самцов крыс Sprague-Dawley (250-350 г) кормили вволю, и эксперименты проводили между 10:00 утра и 3:00 после полудня. Крыс подвергали анестезии с использованием смеси ксилазина (7,5 мг/кг) и кетамина (50 мг/кг), вводимой внутрибрюшинно, и допускали стабилизацию их состояния в течение 50-90 мин до введения глазных капель. Анестезия нормальной крысы с помощью ксилазина/кетамина вызывает повышение значений глюкозы в крови, что обеспечивает оптимальное состояние для определения системного гипогликемического действия содержащих инсулин глазных капель. Значения D-глюкозы в крови определяли посредством забора капли крови из хвостовой вены с 5-10-минутными интервалами на протяжении всего эксперимента и нанесением крови на полоски для глюкометра (Chemstrip bG) в соответствии с инструкциями, предоставленными с прибором (Accu-Chek II, Boehringer Mannheim Diagnostics; Indianapolis, Ind.). Значения D-глюкозы в крови колебались от 200 до 400 мг/дл у подвергнутых анестезии крыс, не страдающих диабетом.
В момент времени 0, после периода стабилизации в течение 50-90 мин, крысам вводили 20 мкл глазных капель, состоящих из забуференного фосфатом солевого раствора (PBS) с 0,2% обычного свиного инсулина или без него и 0,125%-0,5% увеличивающего абсорбцию алкилгликозида (например, TDM) или без него, подвергаемых исследованию. Глазные капли вводили каплями в момент времени 0 с использованием пластикового микродозатора одноразового применения, удерживая глаза открытыми, и крысу удерживали в горизонтальном положении на грелке (37°C) в течение всего протокола. Крыс подвергали дополнительной анестезии, если они демонстрировали признаки пробуждения. Крысам было введено в каждый глаз 20 мкл 0,125-0,5% усилителя абсорбции в забуференном фосфатом солевом растворе, pH 7,4 с 0,2% (50 Е/мл) обычного свиного инсулина (Squibb-Novo, Inc.) всего 2 Е каждому животному (экспериментальному) или без него (контроль). Октил-β-D-мальтозид, децил-β-D-мальтозид, додецил-µ-D-мальтозид, тридецил-β-D-мальтозид и тетрадецил-β-D-мальтозид были получены от Anatrace, Inc. (Maumee, Ohio). Гексилглюкопиранозид, гептилглюкопиранозид, нонилглюкопиранозид, децилсахароза и додецилсахарозы были получены от Calbiochem, Inc. (San Diego, Calif); Сапонин, BL-9 и Brij 78 были получены от Sigma Chemical Co. (St. Louis, Mo.).
Уровни D-глюкозы в крови оставались повышенными, когда животным вводили глазные капли, содержащие: 1) только солевой раствор; 2) только 0,2% обычного свиного инсулина в солевом растворе; или 3) только усилитель абсорбции. Однако, когда крысам вводили глазные капли, содержащие 0,2% обычного свиного инсулина и несколько соединений в виде алкилмальтозидов или алкилсахароз, явное снижение величин D-глюкозы в крови имело место и сохранялось вплоть до двух часов. Инсулин, вводимый в глаз вместе с 0,5% додецил-β-D-мальтозида (см. таблицу I) или 0,5% децил-β-D-мальтозида (см. таблицу III), приводил к быстрому и устойчивому падению уровней глюкозы в крови, которые оставались в нормогликемическом (80-120 мг/дл) или почти нормогликемическом (120-160 мг/дл) диапазоне в продолжение двух часов эксперимента. Следовательно, по крайней мере два алкилмальтозида являются эффективными в достижении достаточной абсорбции инсулина, вводимого глазным путем, с вызовом быстрого и устойчивого падения уровней глюкозы в крови у экспериментально гипергликемических животных. Содержащие поверхностно-активное вещество композиции настоящего изобретения, следовательно, применимы для достижения прохождения в большой круг кровообращения инсулина и других пептидов/белков, например, глюкагона, и являющихся макромолекулами лекарственных средств, и гепарина, вводимых глазным путем в форме глазных капель.
Несколько других алкилмальтозидов, в том числе 0,5% тридецилмальтозида (см. таблицу III) и 0,125% (таблица II) и 0,5% тетрадецилмальтозида, также являются эффективными в качестве усилителей абсорбции в случае глазного введения инсулина. Эти исследования свидетельствуют о том, что алкилмальтозиды с более длинными алкильными цепями (или большим числом атомов углерода), например, додецил-, тридецил- и тетрадецил-β-D-мальтозиды, являются более эффективными. Увеличение числа атомов углерода также вносит вклад в большее значение гидрофобно-гидрофильного структурного соотношения и больший эффект увеличения абсорбции. Более короткие алкильные цепи (с меньшим числом атомов углерода), например, децилмальтозид, или, например, октилмальтозид, вызывают меньший эффект увеличения абсорбции. Отмечено, что наиболее эффективные алкилмальтозиды вызывают эффекты, сравнимые с таковыми, наблюдаемыми при использовании других усилителей абсорбции, таких как сапонин, или превышающие их, и с дополнительным преимуществом, заключающимся в том, что они могут метаболизироваться до нетоксичных продуктов после прохождения в большой круг кровообращения.
Эффекты алкилмальтозидов в качестве усилителей абсорбции являются дозозависимыми, что можно наблюдать при исследовании эффектов различных концентраций, колеблющихся между 0,125-0,5%, в вызове гипогликемического эффекта при объединении с инсулином. Тогда как 0,5% и 0,375% додецилмальтозида выглядят в равной степени эффективными в достижении прохождения в большой круг кровообращения инсулина и снижении уровней глюкозы в крови, 0,25% имеют меньший и более транзиторный эффект, а 0,125% являются неэффективными (таблица I). Аналогично, тридецилмальтозид также демонстрирует дозозависимый эффект снижения концентраций глюкозы в крови при объединении с инсулином, но эффект, достигаемый с использованием даже 0,25% усилителя абсорбции, является устойчивым в течение двухчасового временного курса эксперимента. Дозозависимые эффекты алкилгликозидов наводят, таким образом, на мысль о том, что они добиваются увеличения прохождения белков через глазной путь постепенно, пропорционально концентрации агента.
Связанные с увеличением абсорбции эффекты алкилсахаридов не ограничиваются только алкилмальтозидами, поскольку додецилсахароза (0,125%, 0,25%, 0,375%) также демонстрирует дозозависимый эффект вызова глазной абсорбции инсулина и снижения уровней глюкозы в крови. Этот эффект наблюдается даже при 0,125% алкилсахарида (с 335 мг/дл ± 0,26 мг/дл в момент времени 0 мин до 150 мг/дл ± 0,44 мг/дл в момент времени 120 мин). 0,5% децилсахарозы был также эффективен в снижении уровней глюкозы в крови, но как показано в случае алкилмальтозидов, уменьшение длины алкильной цепи, а, следовательно, гидрофобных свойств молекулы, по-видимому, уменьшает эффективность алкилсахарозных соединений. Однако значительное и устойчивое снижение уровней глюкозы в крови достигается при использовании 0,5% децилсахарозы (с 313 мг/дл ± 0,15 мг/дл в момент времени 0 мин до 164 мг/дл ± 0,51 мг/дл в момент времени 120 мин). Способности к увеличению абсорбции алкилсахаридов с двумя отличными дисахаридными составляющими наводят на мысль о том, что именно физико-химические свойства соединений являются важными для их активности, и что другие алкилсахариды, например, додециллактоза, имеют надлежащее соотношение свойств, чтобы быть в равной степени или более эффективными в качестве усилителей абсорбции при сохранении метаболических и нетоксических свойств являющихся алкилсахаридами усиливающих агентов. Эти алкилсахариды предполагаются настоящим изобретением.
Были также проведены исследования с использованием алкилглюкозидов; 0,5% гексилглюкозид и 0,5% гептилглюкозид были неэффективны в увеличении абсорбции инсулина с поверхности глаза, но 0,5% нонилглюкозида эффективно стимулировал абсорбцию инсулина и снижал уровни глюкозы в крови (с 297 мг/дл до 150 мг/дл). Этот результат еще раз подтверждает, что длина алкильной цепи, а также углеводная составляющая, играет весьма важную роль в эффективном увеличении абсорбции инсулина.
Следует отметить, что эффекты повреждения (т.е. не раздражителей) поверхности глаз не наблюдали при использовании любого из агентов в виде алкилмальтозидов или алкилсахароз в этих исследованиях. Кроме того, быстрые и устойчивые гипогликемические эффекты, вызываемые этими агентами в комбинации с инсулином, говорят о том, что эти усилители абсорбции не оказывают неблагоприятный эффект на биологическую активность гормона, в соответствии с их неденатурирующими, мягкими поверхностно-активными свойствами.
Таким образом, терапевтические композиции настоящего изобретения, состоящие из по крайней мере одного алкилгликозида и лекарственного средства, являются стабильными, а алкилгликозиды увеличивают абсорбцию лекарственного средства.
ПРИМЕР 4
ГЛАЗНОЕ И ИНТРАНАЗАЛЬНОЕ ВВЕДЕНИЕ TDM ПЛЮС ГЛЮКАГОН ВЫЗЫВАЕТ ГИПОГЛИКЕМИЧЕСКИЕ ЭФФЕКТЫ IN VIVO
Поскольку предшествующие примеры продемонстрировали, что введение посредством глазных капель усилителя абсорбции с лекарственных средством, например, инсулином, приводит к значительному прохождению лекарственного средства через носослезную дренажную систему, здесь исследуется терапевтически эффективное введение инсулина с алкилмальтозидами, алкилсахарозой и подобными агентами интраназальным путем.
Тетрадецилмальтозид (TDM) в комбинации с инсулином также вызывал падение уровней D-глюкозы в крови после введения в форме капли интраназального, а также посредством капли в глаза. Глазные капли, содержащие 0,2% обычного свиного инсулина с 0,125% тетрадецилмальтозида, вводили крысам, как описано ранее. Введение композиции вызывает быстрое и значительное падение уровней глюкозы в крови. Падение уровней глюкозы в крови является даже большим при введении капли для носа, содержащей ту же концентрацию инсулина с 0,5% тетрадецилмальтозида (таблица II). Таким образом, интраназальная доставка и введение алкилсахарида вместе с лекарственным средством приводит к снижению уровней глюкозы в крови.
ПРИМЕР 5
ГЛАЗНОЕ ВВЕДЕНИЕ АЛКИЛСАХАРИДОВ ПЛЮС ГЛЮКАГОНА ВЫЗЫВАЕТ ГИПЕРГЛИКЕМИЧЕСКИЕ ЭФФЕКТЫ IN VIVO
Предварительные исследования показали, что сапонин, BL-9 и Brij-78 стимулируют абсорбцию инсулина с поверхности глаза. BL-9 и Brij-78 являются неэффективными в стимулировании абсорбции глюкагона с поверхности глаза, тогда как сапонин является эффективным. Абсорбцию глюкагона с поверхности глаза определяли у крыс, которым вводили глазные капли, содержащие различные поверхностно-активные вещества плюс глюкагон (30 мкг) (Eli Lilly, Indianapolis, Indiana), посредством отслеживания повышения уровней D-глюкозы в крови. В этих экспериментах крыс подвергали анестезии с использованием пентобарбитала натрия, а не ксилазина/кетамина. Эта модификация процедура приводила к базальным уровням глюкозы в крови в нормогликемическом диапазоне и позволяла легко отлеживать гипергликемическое действие любого глюкагона, подвергшегося абсорбции с поверхности глаза.
Подобранные в пары животные, которые получали глазные капли, содержащие только поверхностно-активное вещество или только глюкагон, сравнивали с животными, получающими глазные капли с поверхностно-активным веществом плюс глюкагон. Когда крысам вводят глазные капли, содержащие 0,5% сапонина плюс глюкагон, уровень D-глюкозы в крови значительно повышается, но такой эффект не наблюдается при использовании глазных капель, содержащих 0,5% BL-9 или 0,5% Brij-78 плюс глюкагон. Интересно, что, когда глазные капли, содержащие додецилсахарозу, децилмальтозид или тридецилмальтозид плюс глюкагон, вводят крысам, которые ранее были подвергнуты лечению с использованием глазных капель, содержащих эти поверхностно-активные вещества плюс инсулин, глюкагон абсорбируется, и значения D-глюкозы в крови значительно увеличиваются (таблица III). Этот результат подтверждает, что глазное введение определенных алкилсахаридов может увеличить абсорбцию лекарственных средств, включающих глюкагон и инсулин. Кроме того, теперь возможно лечение гипогликемического криза, используя препарат с по крайней мере алкилсахаридом настоящего изобретения.
ПРИМЕР 6
ИНТРАНАЗАЛЬНОЕ ВВЕДЕНИЕ 0,25% TDM ПЛЮС ИНСУЛИН СНИЖАЕТ УРОВНИ ГЛЮКОЗЫ В КРОВИ IN VIVO
Интраназальное введение лекарственных средств или агентов возможно в моделях на животных, например, мышам и крысам, несмотря на то, что носовое отверстие является очень маленьким. В описываемых здесь экспериментах и результатах использовали модель индуцированной анестезией гипергликемии (описанную в примерах выше). Гипергликемию у животных индуцировали посредством внутрибрюшинной инъекции ксилазина-кетамина, и уровни глюкозы в крови отслеживали в течение периода времени. Сразу же после инъекции ксилазина-кетамина наблюдалось увеличение уровней глюкозы в крови, как продемонстрировано на фиг. 2 (темные незакрашенные кружки), и уровни глюкозы в крови составляли приблизительно 450 мг/дл. Увеличение уровней глюкозы в крови объяснялось ингибированием секреции инсулина поджелудочной железой. Уровни глюкозы в крови достигают максимума, оставляющего приблизительно 482 мг/дл, к 30 минутам после инъекции ксилазина-кетамина (фиг. 2). Затем через приблизительно 33 минут после инъекции ксилазина-кетамина интраназально вводили 6 мкл инсулина (Humalog) в 0,25% тетрадецилмальтозиде (TDM; или Intravail A), используя длинный тонкий микродозатор, и уровни глюкозы в крови контролировали через составляющие приблизительно 15 минут интервалы. После введения композиции 0,25% TDM/инсулин наблюдалось быстрое снижение уровней глюкозы в крови, достигая самого низкого уровня, составляющего приблизительно 80 мг/дл, в момент времени - приблизительно 60 минут, или через приблизительно 30 минут после введения инсулина (фиг. 2). В момент времени - приблизительно 75 минут уровни глюкозы в крови постепенно возвращались к уровню базовой линии у нормогликемической мыши, или приблизительно 80-100 мг/дл.
Вышеприведенные результаты сравнивали с результатами, полученными с использованием животных, подвергнутых лечению только инсулином (в той же дозе), минус 0,25% TDM (фиг. 2, незакрашенные кружки). При лечении только инсулином установлено, что уровни глюкозы в крови не начинают снижаться до достижения вехи - приблизительно 120 минут, или приблизительно 110 минут после введения инсулина. Кроме того, уровни глюкозы в крови, наблюдаемые у животных, подвергнутых лечению только инсулином, никогда не возвращаются к нормогликемическим уровням, как это наблюдалось у животных, получающих инсулин плюс 0,25% TDM (фиг. 2).
Таким образом, эти результаты снова показывают, что композиции настоящего изобретения, состоящие из определенных алкилгликозидов или алкилсахаридов плюс лекарственное средство, например, инсулин, эффективно снижают уровни глюкозы в крови, и что эти эффекты поддаются определению вскоре после введения лекарственного средства.
ПРИМЕР 7
ИНТРАНАЗАЛЬНОЕ ВВЕДЕНИЕ 0,25% TDM (INTRAVAIL A) + ЭКЗЕДИН-4 СНИЖАЕТ УРОВНИ ГЛЮКОЗЫ В КРОВИ IN VIVO
Для описываемых здесь исследованиях использовали модель на мышах ob/ob, см. Friedman, J. M., Nature 404, 632-634 (2000). Все животные получали внутрибрюшинную болюсную инъекцию 2 г/кг глюкозы с целью определения толерантности к глюкозе. В момент времени 0 экспериментальным животным вводили приблизительно 100 микрограмм/кг экзендина-4/0,25% TDM (экзендин-4 от American Peptide) либо в виде 10 мкл капель для носа (фиг. 3; закрашенные треугольники), либо с помощью внутрибрюшинной инъекции (фиг. 3; закрашенные кружки), или животному вводили с помощью внутрибрюшинной инъекции только солевой раствор (без лекарственного средства, без TDM; фиг. 3; незакрашенные кружки). Контрольные животные были заранее представлены и не получали лекарственных средств. Результаты этого исследования представлены на фиг. 3.
На фиг. 3 демонстрируется, что толерантности к глюкозе у животных были различными, поскольку уровни глюкозы в крови варьируют в момент времени 0, когда животные получали болюсную инъекцию глюкозы. Независимо от уровня толерантности к глюкозе в момент времени 0, сразу же после болюсной инъекции глюкозы уровни глюкозы в крови увеличивались у всех трех животных. Уровень глюкозы в крови у животного, получившего только внутрибрюшинную инъекцию солевого раствора, не снижался так быстро, как у экспериментальных животных, получивших лекарственное средство. Кроме того, у животного, получившего только внутрибрюшинную инъекцию солевого раствора, нормогликемический уровень никогда не достигался (фиг. 3, незакрашенные кружки). Напротив, экспериментальные животные, после введения капель для носа, содержащих экзендин-4/TDM, или внутрибрюшинной инъекции экзендина-4/TDM, продемонстрировали быстрое и немедленное снижение уровней глюкозы в крови.
Также экзендин-4, введенный за приблизительно 15-30 минут до болюсной инъекции глюкозы (до момента времени 0 на Фиг. 3; не представленные данные), вызывал даже более выраженный эффект снижения глюкозы в крови, поскольку требуется некоторое количество время, чтобы гормон абсорбировался и был активным. Таким образом, экзендин-4 (или Эксенатид), который в настоящее время находится на стадии клинических испытаний на людях, после объединения с алкилгликозидами настоящего изобретения, эффективно осуществляет лечение гипергликемического состояния посредством снижения уровней глюкозы в крови пациента с гипергликемией.
ПРИМЕР 8
АЛКИЛГЛИКОЗИДЫ ОБЛАДАЮТ АНТИБАКТЕРИАЛЬНОЙ АКТИВНОСТЬЮ ПОСРЕДСТВОМ УМЕНЬШЕНИЯ ЛОГАРИФМИЧЕСКОГО РОСТА БАКТЕРИЙ
Культуры Candida albicans (ATCC № 10231), Aspergillus niger (ATCC № 16404), Escherichia coli (ATCC № 8739), Pseudomonas aeruginosa (ATCC № 9027) и Staphylococcus aureus (ATCC № 6538) были получены из Американской коллекции типовых культур (АТСС), Culture Collection, 10801 University Boulevard, Manassas, VA 20110-2209. Жизнеспособные микроорганизмы, использованные в настоящем изобретении, были микроорганизмами не более пяти пассажей после извлечения из исходной культуры ATCC. Как здесь описано, один пассаж определяют как перенос организмов из стабилизированной культуры в свежую среду, и считаются все переносы.
Полученные из АТСС культуры оживляли в соответствии с инструкциями, предоставленными ATTC. Выращенные в бульоне клетки осаждали центрифугированием, ресуспендировали в 1/20 объема свежего поддерживающего бульона и объединяли с равным объемом 20% (в объемном отношении, в воде) стерильного глицерина. Выращенные на агаре клетки соскабливали с поверхности в поддерживающий бульон, также содержащий 10% глицерина. Небольшие аликвоты суспензии распределяли по стерильным флаконам, и флаконы хранили в жидком азоте или в морозильнике с механическим управлением при температуре, не превышающей приблизительно -50οC. Когда требовался свежий флакон посевного фонда, его извлекали и использовали для засева ряда рабочих исходных культур. Эти рабочие исходные культуры затем использовали периодически (каждый день в случае бактерий и дрожжей) для запуска посевного культивирования.
Все среды, описываемые здесь, должны быть проверены на активацию роста, используя микроорганизмы, указанные выше под названием «исследуемые организмы».
Для определения того, ингибируют ли рост или обладают ли антибактериальной активностью алкилсахариды настоящего изобретения, поверхность подходящего объема твердой агаровой среды засевали из свежей оживленной исходной культуры каждого из указанных микроорганизмов. Условиями для культивирования в случае посевной культуры являются по существу те, которые описаны в таблице IV. Например, подходящие среды могут включать, но без ограничения, казеин-соевую питательную среду или декстрозный агар Сабуро. Культуры бактерий и C. albicans собирали, используя стерильный солевой раствор TS, посредством вымывания поверхностного роста, сбора его в подходящий сосуд и добавления количества стерильного солевого раствора TS, достаточного для получения микробного числа, составляющего приблизительно 1×108 колониеобразующих единиц (cfu) на мл. Для сбора клеток A. niger использовали стерильный солевой раствор TS, содержащий 0,05% полисорбата 80, а затем добавляли количество стерильного солевого раствора TS, достаточное для получения числа, составляющего приблизительно 1×108 cfu на мл.
Альтернативно, организмы исходной культуры можно выращивать в любой подходящей жидкой среде (например, казеин-соевой питательной жидкой среде или декстрозном бульоне Сабуро), а клетки собирают с помощью центрифугирования, и промывают и ресуспендируют в стерильном солевом растворе TS с получением микробного числа, составляющего приблизительно 1×108 cfu на мл. Оценку концентрации посевного материала осуществляли с помощью нефелометрических измерений в случае микроорганизмов, используемых для пробы с заражением. Суспензия должна храниться в холодильнике, если она не используется в пределах 2 часов. Для подтверждения начальной оценки числа cfu на мл определяли число cfu на мл в каждой суспензии, используя средовые условия и периоды инкубации до выделения микробов, которые перечислены в таблице IV (например, от приблизительно 3 до приблизительно 7 дней). Эта величина служит для стандартизации размера посевного материла, используемого в испытании. Суспензии бактерий и дрожжей использовали в пределах 24 часов от момента сбора; в то время как препараты грибов можно хранить в холодильнике в течение вплоть до 7 дней.
Для определения того, какие содержащие алкилгликозиды препараты обладают антибактериальной активностью, препараты готовили в забуференном фосфатом солевом растворе (PBS) при pH 7. В качестве источника питания, либо 1,5 мг/мл бычьего сывороточного альбумина (BSA; см. таблицы V и VI), либо 1 мг/мл PYY добавляли (см. таблицу VII) в среду. BSA (№ CAS: 9048-46-8) был получен от Sigma-Aldrich, St. Louis, MO, США, н-додецил-4-O-α-D-глюкопиранозил-β-D-глюкопиранозид и н-тетрадецил-4-O-α-D-глюкопиранозил-β-D-глюкопиранозид были получены от Anatrace Inc., Maumee, OH, США, а PYY был получен от Bachem California Inc., Torrance, CA, США.
Исследование антибактериальной активности алкилгликозидов проводили в четырех стерильных, бактериологических емкостях с крышками подходящего размера, в которые был перенесен достаточный объем раствора алкилгликозида. Каждую емкость засевали одним из приготовленных и стандартизированных посевных материалов и осуществляли смешивание. Объем суспензионного посевного материала находился между приблизительно 0,5% и приблизительно 1,0% объема раствора алкилгликозида. Концентрация исследуемого микроорганизма, добавляемого к раствору алкилгликозида, была такой, что конечная концентрация исследуемого препарата после засева находилась между приблизительно 1×105 и 1×106 cfu на мл раствора алкилгликозида. Для определения уровня ингибирования роста или уменьшения роста по логарифмической шкале, исходную концентрацию жизнеспособных микроорганизмов в каждом исследуемом препарате определяли на основе концентрации микроорганизмов в каждом стандартизированном посевном материале, определенной чашечным методом подсчета. Засеянные емкости затем инкубировали при приблизительно 22,5°C±2,5. В день 14 и день 28 снова определяли рост или отсутствие роста микроорганизмов в каждой культуре/емкости. Число присутствующих cfu при каждом подсчете определяли чашечным методом подсчета, общепринятым в данной области техники для соответствующих интервалов. Изменение порядков величины бактерий и/или грибов затем определяли вычитанием рассчитанных первыми log10(значений концентраций cfu на мл, присутствующих в начале) (например, день 0) из log10(значений концентраций cfu на мл для каждого микроорганизма через соответствующие исследуемые интервалы) (например, день 14 и день 28; см. таблицы V, VI и VII).
Определение антибактериальной активности других алкилгликозидов могло бы происходить по существу, как здесь описано.
ПРИМЕР 9
ВВЕДЕНИЕ ПРИМАТАМ АЛКИЛГЛИКОЗИДОВ С АНТИСМЫСЛОВЫМИ ОЛИГОНУКЛЕОТИДАМИ
Антисмысловой олигонуклеотид (ASO) с М.м. приблизительно 7000 Дальтон с модифицированным остовом (олигонуклеотид с фосфоротиоатными связями, описанный в патенте США № 7132530), смешанный с алкилгликозидом тетрадецил-бета-D-мальтозидом (IntravailTM), вводили шести яванским макакам, в тощую кишку которых был введен катетер, в дозе, составляющей 10 мг/кг. Животные не ели перед введением. Исследуемые агенты растворяли в буфере PBS и вводили через катетер в тощую кишку каждого животного в объеме 1,5 мл или вводили подкожно, как отмечено в таблице VIII.
Прокол, в котором каждому животному вводили первые 3 в таблице VIII исследуемых агента в три различные даты месяца, включал тройственное пересечение. Между датами введения доз существовал 1-нельный период выведения агента из организма. Двум животным впоследствии вводили четвертый исследуемый агент, содержащий 5% капрата натрия в качестве усилителя абсорбции. Анализ уровней в крови проводили с использованием количественного анализа, включающего твердофазное извлечение с использованием катионных полистироловых наночастиц.
Сначала выполняли твердофазное извлечения из образцов крови. Конъюгаты наночастица-олигонуклеотид образовывали, используя известное количество олигонуклеотида, добавленное к аликвоте каждого образца (200-400 мкл) и разведенное с использованием 800 мкл 50 мМ Трис. HCl (pH 9) в деионизированной воде. Смесь недолго встряхивали перед добавлением 200 мкл суспензии полистироловых наночастиц, приготовленных посредством полимеризации в эмульсии без поверхностно-активного вещества, используя водорастворимые катионные катализаторы для индукции положительного заряда поверхности (сухой остаток: приблизительно 10 мг/мл). Впоследствии смесь снова встряхивали. Через 5-10 мин инкубации суспензию центрифугировали, а супернатант удаляли. Частицы ресуспендировали в 1 мл раствора 0,5 M уксусной кислоты в деионизированной воде/этаноле (1:1) и отделяли от раствора для промывки с помощью центрифугирования. Послу удаления супернатанта частицы ресуспендировали в 1 мл деионизированной воды и отделяли с помощью еще одной стадии центрифугирования. К конъюгатам наночастица-олигонуклеотид добавляли 200 мкл раствора 150 мкМ SDS в нашатырном спирте (25%)/ацетонитриле (60/40), и высвободившиеся олигонуклеотиды отделяли от носителя с помощью центрифугирования. Для исключения загрязнения образцов остаточными частицами супернатант помещали в другую 1,5-мл пробирку и снова центрифугировали. Впоследствии образцы высушивали посредством выпаривания с использованием ротора или лиофилизации и хранили при -20°C до анализа.
Количественный анализ выполняли с помощью капиллярного гель-электрофореза извлеченных образцов. Капиллярный гель-электрофорез (CGE) выполняли с использованием системы для капиллярного электрофореза. Был получен набор для анализа олигонуклеотидов, содержащий покрытые поливиниловым спиртом (PVA) капилляры, раствор полимера B и буфер для олигонуклеотидов. Используя PVA-покрытые капилляры, был выполнен анализ с использованием протокола производителя.
Используя данные, полученные из анализа с использованием CGE, был выполнен количественный анализ олигонуклеотидов с фосфоротиоатными связями. Количество олигонуклеотидов в образцах (nON) рассчитывали, используя формулу:
nON=nStd(εStd/εON)((AON/TON)/(AStd/TStd)),
где nStd представляет собой количество стандартного олигонуклеотида, добавляемого к образцу, εStd и εONпредставляют собой молярные показатели поглощения, и AStd/TStd и AON/TONпредставляют собой коррегированные площади пиков (соотношение площади пика и времени миграции) стандартного и исследуемого соединения, соответственно. Соотношение коррегированных площадей пиков аналита и стандарта называют нормализованной площадью.
AUC рассчитывали на основе кривых зависимостей концентрации от времени на протяжении составляющего 240 минут периода. Относительные биодоступности определяли как отношение каждой AUC к AUC в случае внутривенного введения лекарственного средства. Наполнитель IntravailTM (тетрадецил-бета-D-мальтозид) обеспечивал биодоступность вплоть до 18%. Контроль продемонстрировал не выявляемую абсорбцию без наполнителя - поверхностно-активного вещества. Содержащий капрат натрия препарат продемонстрировал среднюю биодоступность, составляющую 9%.
ПРИМЕР 10
ПРИГОТОВЛЕНИЕ БЫСТРО ДИСПЕРГИРУЕМЫХ ЛЕКАРСТВЕННЫХ ФОРМ ОЛАНЗАПИНА
Быстро диспергируемые лекарственные формы оланзапина готовили следующим образом. Оланзапин, CAS № 132539-06-1, получен от SynFine (Ontario, Канада). Натрийацетатный буфер, 10 мМ, pH 5,0 и pH 6,5 готовят следующим образом. В чистую емкость соответствующего размера с нанесением шкалы для объема поместите 495 мл стерильной воды для инъекции. Добавьте 0,286 мл уксусной кислоты. Добавьте 1 н NaOH для доведения рН до 5,0 (или до pH 6,5). После получения надлежащего pH добавьте добавочное количество воды для доведения общего объема до 500 мл и вновь проверьте pH.
Жидкие препараты, содержащие композиции, проиллюстрированные в таблице IX ниже, готовят посредством медленного добавления рыбного желатина или желатина свиной кожи в ацетатный буфер и предоставления достаточного времени для растворения при перемешивании на всем протяжении процесса. После полного растворения желатина рыбы или свиной кожи добавляют маннит и обеспечивают его растворение. Затем добавляют подсластитель. После его полного диспергирования добавляют активный ингредиент, оланзапин, являющийся одним из примеров соединений настоящего изобретения, для создания конечного раствора. В композицию могут быть также включены дополнительные компоненты, такие как консерванты, антиоксиданты, поверхностно-активные вещества, усилители вязкости, красители, корригенты, подсластители или исправляющие вкус лекарственного средства вещества. Подходящие красители могут включать красный, черный и желтый оксиды железа и красители FD & C, такие как FD & C синий № 2 и FD & C красный № 40, доступные от Ellis & Everard. Подходящие корригенты могут включать мятный, малиновой, солодковый, апельсиновый, лимонный, грейпфрутовый, карамельный, ванильный, вишневый и виноградный ароматизаторы и их комбинации. Подходящие подсластители включают аспартам, ацефульфам калия и таумарин. Подходящие исправляющие вкус лекарственного средства вещества включают натрия бикарбонат. Следует избегать циклодекстринов, поскольку они образуют соединения включения с алкилсахаридами, которые уменьшают эффективность этих наполнителей.
Аликвоты объемом 1 мл каждая вышеуказанных растворов лекарственного средства помещают в лунки 24-луночного пластикового микропланшета одноразового использования. Луночные микропланшеты, содержащие жидкие аликвоты, замораживают при -70°С, и замерший планшет помещают в стеклянную лиофилизационную колбу, присоединяемую к настольной сублимационной установке LabConco Freezone Model 4,5, и подвергают лиофилизации под вакуумом. После лиофилизации быстро диспергируемые таблетки хранят в луночном микропланшете в сухой окружающей среде до исследования. Смешанные моно- и дистеараты сахарозы были предоставлены как подарок Croda Inc. и обозначены CRODESTA F-110. Додецилмальтозид, тетрадецилмальтозид и сахарозы монододеканоат получены от Anatrace Inc., Maumee, OH.
Известно, что лекарственное средство оланзапин, также известный как Зупрекса, хорошо абсорбируется после введения в виде «цельнопроглатываемой» таблетки и достигает максимальных концентраций через приблизительно 6 часов после введения пероральной дозы. Он активно элиминируется посредством метаболизма первого прохождения, при этом метаболизируется приблизительно 40% дозы до достижения большого круга кровообращения. Фармакокинетические исследования показали, что «цельнопроглатываемые» таблетки, содержащие оланзапин, и быстро диспергируемые таблетки, содержащие оланзапин, приготовленные посредством лиофилизации таким образом, как описано выше в этом примере, которые распадаются через приблизительно 3 секунды - 10 секунд после помещения в ротовую полость, являются биоэквивалентными, демонстрируя максимальные концентрации через приблизительно 6 часов после введения. Аналогично, в результате эффекта первого прохождения в печени элиминируется приблизительно 40% дозы до достижения большого круга кровообращения.
В настоящем примере быстро диспергируемые таблетки, содержащие 10 мг оланзапина, готовят посредством лиофилизации, как описано выше в этом примере. После введения быстро диспергируемой таблетки, содержащей оланзапин, посредством приведения ее в контакт с щечной тканью было обнаружено, что добавление определенных алкилсахаридов, имеющих конкретные длины алкильных цепей, в быстро диспергируемые таблетки, содержащие оланзапин, приводит к значительному уменьшению метаболизма (эффекта) первого прохождения оланзапина, наблюдаемому по уменьшению относительной доли метаболитов оланзапина в большом круге кровообращения относительно не подвергнутого метаболизму активного лекарственного средства. Относительные доли оланзапина и метаболитов оланзапина в сыворотке или плазме можно определить, используя хроматограф для HPLC, Perkin Elmer 200, с рефрактометрическим детектором, оснащенным термостатированным элементом. Может использоваться подходящий твердофазный абсорбент, такой как Lichrosorb RP-18 (Merck, Darmstadt, Германия) 250 мм, с использованием подвижной фазы, состоящей из градиента ацетонитрила:воды. Составляющие 20 мкл объемы инъекций с использованием автоматической пипетки Perkin Elmer 200 и скорость потока, составляющая 0,8 мл/минуту, являются приемлемыми для этой цели. В частности, включение от 0,2% вплоть до 10% додецилмальтозида или тетрадецилмальтозида, или сахарозы додеканоата в быстро диспергируемую форму таблетки увеличивает процент лекарственного средства, который поступает в большой круг кровообращения, и уменьшает процент лекарственного средства, который элиминируется в результате эффекта «первого прохождения» в печени. Кроме того, резко сокращается время до достижения максимальных уровней лекарственного средства, обычно с одного-шести часов до всего приблизительно 15-45 минут. В случае применения для лечения агрессивных пациентов, подверженных транзиторному психозу, эта более быстрая абсорбция лекарственного средства, приводящая к более быстрому возникновению действия, может принести большую пользу.
ПРИМЕР 11
ПРИГОТОВЛЕНИЕ БЫСТРО ДИСПЕРГИРУЕМЫХ ЛЕКАРСТВЕННЫХ ФОРМ МЕЛАТОНИНА
Мелатонин или 5-метокси-N-ацетилтриптамин является нейрогормоном, используемым для регуляции циклов сна- бодровстования у пациентов с нарушениями сна. Эндогенный мелатонин секретируется шишковидным телом у всех животных, демонстрирующих циркадианный (суточный) или годичный ритм. Мелатонин играет признанную роль в поддержании ритмов сна- бодровстования, и его пополнение может помочь в регулировании нарушений сна, которые встречаются при смене часовых поясов, скользящем графике работы, депрессии и различных неврологических нарушениях.
Имеющиеся в продаже препараты мелатонина включают пероральные и сублингвальные таблетки, капсулы, настои или отвары, лепешки и системы пероральной аэрозольной доставки. В случае перорального введения мелатонина отслеживается фармакокинетический профиль, отличный от такого эндогенного гормона. После перорального введения мелатонин подвергается значительному метаболизму первого прохождения до 6-сульфаоксимелатонина, результатом чего является биодоступность мелатонина, установленная на уровне 30-50%. DeMuro и др. (2000) сообщили, что абсолютная биодоступность пероральных, содержащих мелатонин таблеток, исследованная на нормальных здоровых добровольцах, пониже на уровне приблизительно 15%. Средний полупериод элиминации мелатонина составляет приблизительно 45 минут.
В соответствии со способом, описанным в примере 10 выше, были приготовлены быстро диспергируемые таблетки, содержащие 1 мг, 5 мг, 10 мг и 20 мг мелатонина с 1%-2% алкилсахарида или без него, как описано в примере 10. Новозеландских белых кроликов помещали в камеру ограниченного объема и подвергали анестезии, используя одно введение ацепромазина/кетамина (0,7 мг/0,03 мг в 0,1 мл) посредством инъекции в краевую вену уха для облегчения введения доз. Это приводило к анестезии в течение периода времени, составляющего приблизительно 10 минут, во время которого животных вводили исследуемый препарат. После этого животные возвращались в сознание. В отдельные моменты времени на протяжении двухчасового периода из центральной ушной артерии забирали 1 мл образцы крови. После забора из каждого образца крови сразу же готовили плазму, используя литий/гепарин в качестве антикоагулянта. Все образцы хранили при -70°C до анализа на мелатонин. Мелатонин измеряли с использованием имеющегося в продаже набора для ELISA, производимого GenWay Biotech Inc., San Diego, CA. Установлено, что после введения посредством контактирования быстро диспергируемых таблеток со щечной тканью в верхней части ротовой полости мелатонин абсорбируется с определяемой по площади под кривой биодоступностью, составляющей по крайней мере 75% в присутствии алкилсахарида и менее 50% в отсутствие алкилсахарида. Мелатонин измеряли с использованием имеющегося в продаже набора для ELISA (№ 40-371-25005), производимого GenWay Biotech Inc., San Diego, CA. Кроме того, в случае таблеток, содержащих алкилсахарид, максимальная концентрация мелатонина, достигается через приблизительно половину периода времени, который требуется в случае таблеток, не содержащих алкилсахариды.
ПРИМЕР 12
ПРИГОТОВЛЕНИЕ БЫСТРО ДИСПЕРГИРУЕМЫХ ЛЕКАРСТВЕННЫХ ФОРМ РАЛОКСИФЕНА
Ралоксифен, также называемый Evista®, используется для лечения и предупреждения остеопороза у женщин после менопаузы, уменьшения риска инвазивного рака молочной железы у женщин после менопаузы, страдающих остеопорозом, и уменьшения риска инвазивного рака молочной железы у женщин после менопаузы, подверженных высокому риску развития инвазивного рака молочной железы. Рекомендуемая доза составляет одна 60-мг таблетка в день. Хотя приблизительно 60% пероральной дозы ралоксифена быстро абсорбируется после перорального введения, значительной является конъюгация с глюкуронидом до попадания в большой круг кровообращения, что приводит к абсолютной биодоступности для ралоксифена, составляющей лишь 2%. Установлено, что быстро диспергируемые таблетки, содержащие 60 мг ралоксифена, приготовленные как описано в патенте США № 5576014, 6696085 B2 или 6024981, имеют очень схожие фармакокинетики с составляющей приблизительно 2% абсолютной биодоступностью. Однако быстро диспергируемая таблетка, содержащая 10 мг или меньше тонкоизмельченного ралоксифена, приготовленного посредством измельчения при сушке распылением (Bend Research Inc., Bend Oregon, or AzoPharma, Miramar, FL) или с помощью чаще используемых стандартных фармацевтических процессов измельчения или дробления, и 0,5%-5% додецилмальтозида, после буккального введения достигает системных уровней лекарственного средства, схожих с таковыми, достигаемыми при использовании 60 мг пероральной таблетки, и в то же самое время приводит к меньшему количеству циркулирующего неактивного конъюгата ралоксифен-глюкуронид.
Хотя клинический результат является, в основном, следствием неконъюгированного лекарственного средства, побочные эффекты могут опосредоваться либо одной из двух форм, либо обеими формами лекарственного средства: активным лекарственным средством и по существу неактивным, конъюгированным с глюкуронидом лекарственным средством. Поэтому уменьшение подвергания воздействию неактивного конъюгата лекарственного средства, в этом случае присутствующего в концентрации, превышающей вплоть до 30 раз концентрацию активного лекарственного средства, дает потенциально значимый клинический результат уменьшения вероятности побочных эффектов. Растворимость в воде ралоксифена составляет приблизительно 0,25 мг/л. В результате, невозможно растворить ралоксифен в воде при приготовлении для лиофилизации для приготовления быстро диспергируемого препарата, как описано в примере 10.
В этом случае можно сформировать самообразующийся гидрогель посредством добавления 1%-30% вес. CRODESTA F-110 в подходящий буфер, который встряхивают и нагревают до 45°С в течение 1 ч. Затем ралоксифен в тонкодисперсной или тонкоизмельченной форме добавляют с получение концентрации в суспензии, составляющей 60 мг/мл, к теплой жидкости, которую снова перемешивают посредством встряхивания до однородного суспендирования и равномерного диспергирования твердого вещества. После охлаждения до комнатной температуры образуется стабильный тиксотропный гидрогель, который можно размельчить, но который поддерживает однородную суспензию. Установлено, что для этой цели особенно подходит ацетатный буфер в диапазоне pH от 2 до 7. Аликвоты объемом 1 мл гелевой суспензии ралоксифена помещают в лунки 24-луночного пластикового микропланшета одноразового применения и подвергают лиофилизации, как описано в примере 1.
Введение этого быстро диспергируемого препарата после подачи к ткани щеки приводит к увеличению (удвоению) абсолютной биодоступности до по крайней мере 4% и соответствующему определяемому уменьшению отношения концентрации циркулирующего конъюгата ралоксифен-глюкуронид к концентрации неконъюгированного ралоксифена.
ПРИМЕР 13
ПРИГОТОВЛЕНИЕ БЫСТРО ДИСПЕРГИРУЕМЫХ ЛЕКАРСТВЕННЫХ ФОРМ ДИФЕНГИДРАМИНА
Дифенгидрамин является седативным антигистаминным препаратом с выраженными свойствами средств с успокаивающими воздействиями на центральную нервную систему и используется в качестве снотворного средства для кратковременного лечения бессонницы, симптоматического ослабления аллергических состояний, в том числе крапивницы и болезни Квинке, ринита и конъюнктивита, расстройств в виде зудящей кожи, тошноты и рвоты, предупреждения и лечения морской болезни, вертиго, непроизвольных движений вследствие побочных эффектов определенных лекарственных средств против психиатрических заболеваний и для контролирования паркинсонизма благодаря антимускариновым свойствам. Особенно желанным свойством дифенгидрамина является наблюдаемое отсутствие каких-либо данных о вызове им зависимости. Благодаря превосходному профилю его безопасности он доступен в виде безрецептурного лекарственного средства, в отличие от некоторых более новых лекарственных средств для сна, таких как Ambien® и Lunesta®, которые могут вызывать аномальное поведение, такое как лунатизм и переедания во сне, наряду с периодическими тяжелыми аллергическими реакциями и опуханием лица, которые явились причиной требования FDA (Управления по контролю качества пищевых продуктов и лекарственных средств) в отношении этикетки, предупреждающей об этих побочных эффектах, в случае этих более новых рецептурных лекарственных средств.
Дифенгидрамина гидрохлорид принимают внутрь в обычных дозах, составляющих 25-50 мг, три или четыре раза в день. Максимальная ежедневная доза для взрослых и детей составляет приблизительно 300 мг. Составляющая 20-50 мг доза может использоваться в качестве снотворного для взрослых и детей старше 12 лет. Лекарственное средство хорошо абсорбируется из желудочно-кишечного тракта; однако оно подвергается значительному метаболизму первого прохождения, который, по-видимому, влияет на системные уровни лекарственного средства. Максимальные концентрации в плазме достигаются через приблизительно 1-4 часа после приема пероральных доз. Дифенгидрамин распределяется по всему организму, в том числе центральной нервной системе, и вследствие его значительного метаболизма в печени лекарственное средство выводится, в основном, с мочой в виде метаболитов с небольшими количествами не измененного лекарственного средства, которое, как установлено, присутствует.
Хотя дифенгидрамин считается безопасным и эффективным для лечения бессонницы и других нарушений, относительно длительный период до начала действия вследствие промедления в достижении максимальных концентраций в плазме, составляющего один - четыре часа, причиняет удобство и уменьшает практическую применимость этого безопасного и эффективного лекарственного средства. Внутривенно введенный дифенгидрамин начинает действовать быстро; однако внутривенное введение не целесообразно в случае амбулаторного использования или несерьезных симптомов. Потребность в препарате дифенгидрамин с быстрым началом действия является очевидной. В случае бессонницы пациенту может быть необходимо принимать современные пероральные формы лекарственно средства задолго до того, как лечь спать, чтобы минимизировать вероятность удлиненной бессонницы, вызывающей беспокойство при ожидании, когда лекарственное средство достигнет системных уровней лекарственного средства, достаточных для проявления своего желательного фармакологического эффекта. В случае применений дифенгидрамина против рвоты быстрое возникновение действия также является в высокой степени желательным для уменьшения тошноты и рвоты как можно скорее. Равно как и в случае лечения морской болезни и вертиго, поскольку эти симптомы могут возникнуть неожиданно, и является как затруднительной, так и нежелательной необходимость ждать один - четыре часа, пока орально введенное лекарственное средство достигнет системных уровней лекарственного средства, достаточных для осуществления своего благотворного эффекта.
Растворимость в воде дифенгидрамина составляет приблизительно 3,06 мг/мл. Поэтому описанный в примере 12 способ может использоваться для приготовления быстро диспергируемой таблетки, содержащей 50 мг лекарственного средства - дифенгидрамина и 1%-2% алкилсахарида. Поскольку дифенгидрамин является слегка горьким, можно добавить исправляющее вкус лекарственного средства количество фармацевтически приемлемого ароматизатора и подсластителя для увеличения аппетитности. Быстро диспергируемые таблетки, приготовленные таким образом, характеризуются более быстрым возникновением действия, чем ««цельнопроглатываемые» таблетки - сироп, жевательные таблетки, лепешки или съедобная пленка, а также характеризуются меньшим метаболизмом первого прохождения.
ПРИМЕР 14
ВВЕДЕНИЕ МЫШАМ АЛКИЛГЛИКОГДИДОВ С МЫШИНЫМ ПЕПТИДОМ ПРОТИВ ОЖИРЕНИЯ [D-Leu-4]OB3
В этом примере демонстрируется поглощение мышиного пептида против ожирения [D-Leu-4]OB3 в 0,3% алкилгликозиде - тетрадецил-бета-D-мальтозиде (IntravailTM A3) самцами мышей Swiss Webster. Синтетический агонист лептина [D-Leu-4]OB3, смешанный с 0,3% алкилгликозида - тетрадецил-бета-D-мальтозида (IntravailTM A3), вводили самцам мышей Swiss Webster возрастом шесть недель в дозе 1 мг с помощью кормления через зонд.
Мышиный [D-Leu-4]OB3 (в концентрации, составляющей 1 мг/200 мкл) растворяли либо в PBS (pH 7,2), либо в 0,3% алкилгликозиде тетрадецил-бета-D-мальтозиде (IntravailTMA3), воссозданном в PBS (pH 7,2), и вводили с помощью кормления через зонд, без анестезии, каждой из 4 мышей на каждый момент времени. Через 10, 30, 50, 70, 90 или 120 минут мышей умерщвляли посредством ингаляции изофлурана (5%) и обескровливали проколом каудальной полой вены. Кровь также брали у четырех мышей, которым не был введен пептид (до введения пептида). Кровь от каждой из четырех мышей, взятую через период времени, объединяли, и готовили образцы сыворотки. Содержание мышиного [D-Leu-4]OB3 в объединенных образцах определяли с помощью конкурентного ELISA.
Эти эксперименты были повторены дважды. Данные, собранные из одного эксперименты, представлены в таблице X и на фиг. 4. Установлено, что данные являются в высокой степени воспроизводимыми. Кривые поглощения были вычерчены с использованием MicrosoftTM Excel, и AUC рассчитывали, используя функцию программы графического построения SigmaPlot 8,0TM (SPSS Science, Chicage, IL). Наименьшее полученное значение для AUC произвольно установлено в 1,0. Относительную биодоступность определяли сравнением всех других значений для AUC с 1,0.
Как показано в таблице X и на фиг. 4, добавление алкилгликозида - тетрадецил-бета-D-мальтозида (IntravailTM A3) в концентрации, составляющей 0,3%, увеличивает относительную абсорбцию пептида OB-3 в 4-раза по сравнению с таковой пептида в только PBS.
ПРИМЕР 15
ВВЕДЕНИЕ СОБАКАМ АЛКИЛГЛИКОЗИДОВ С СУМАТРИПТАНОМ
В этом примере демонстрируется поглощение суматрипана в 0,5% алкилгликозиде - тетрадецил-бета-D-мальтозиде (IntravailTM A3) собаками. Суматриптан, смешанный с 0,5% алкилгликозидом - тетрадецил-бета-D-мальтозидом (IntravailTM A3), вводили собакам в дозе, составляющей 25 мг, с помощью перорального и ректального введения.
Как показано на фиг. 5, добавление алкилгликозида - тетрадецил-бета-D-мальтозида (IntravailTM A3) в концентрации, составляющей 0,3%, увеличивает Cmax суматриптана как в случае перорального, так и в случае ректального введения по сравнению имеющимися в настоящее время 25-мг пероральными таблетками. Установлено, что Cmax для имеющихся в настоящее время таблеток составляет 104 нг/мл в случае собак, которая представлена горизонтальной пунктирной линией на фиг. 5.
ПРИМЕР 16
ВВЕДЕНИЕ АЛКИЛГЛИКОЗИДОВ С ТРИПТАНАМИ УВЕЛИЧИВАЕТ БИОДОСТУПНОСТЬ
Суматриптана сульфат, наратриптана-HCl или ризатриптана бензоат растворяли в 20 мМ натрийацетатном буфере, pH 5,5, содержащем 0%, 0,02%, 0,05%, 0,1%, 0,2%, или 1,0% алкилсахарида. Каждый ряд растворов лекарственных средств вводили шести группам из восьми крыс в каждой посредством 20-мкл инстилляции в одну ноздрю каждого животного. Образцы крови объемом 200 мкл отбирали посредством отбора крови из глаза в течение трехчасового периода времени через 0, 5, 10, 15, 30, 60, 120 и 180 минут. После отбора последнего образца крови каждое животное умерщвляли с помощью CO2. После отбора крови сразу же готовили плазму из каждого образца крови, используя литий/гепарин в качестве антикоагулянта. Образцы плазмы хранили при -70°C до анализа. Уровни лекарственных средств в плазме определяли с помощью HPLC, используя описанный Boulton способ и подобный способ HPLC. Наносили на график данные о концентрации в зависимости от времени для определения Cmax при каждой концентрации алкилсахарида, и соотношение Cmax в случае присутствия алкилсахарида и без него рассчитывали и записывали, как представлено в таблице XI. Устанавливаемые на основе каждого графика значения Tmax определяли посредством исследования графиков зависимости концентрации от времени и записывали, как представлено в таблице XII. Означенные в этой таблице дозы отражают количества триптана в виде свободного основания в каждом случае.
ПРИМЕР 17
ИНТРАНАЗАЛЬНОЕ ВВЕДЕНИЕ СУМАТРИПТАНА С АЛКИЛГЛИКОЗИДОМ УВЕЛИЧИВАЕТ БИОДОСТУПНОСТЬ И CMAX И УСКОРЯЕТ НАЧАЛО ПРОХОЖДЕНИЯ В БОЛЬШОЙ КРУГ КРОВООБРАЩЕНИЯ
Суматриптана сульфат растворяли в фосфатном буфере, приготовленном посредством растворения 0,2 г двухосновного фосфата натрия и 10,0 г дигидроортокалия фосфата в 1 л воды, с доведением pH до 5,5, либо содержащем 0,18% додецилмальтозида («препарат А»), либо не содержащем наполнитель в виде додецилмальтозида («препарат B»), в конечной концентрация суматриптана, составляющей 20 мг в 100 микролитрах аэрозоля. Доведение до конечного pH осуществляли с использованием раствора серной кислоты или гидроксида натрия. Третий препарат, содержащий суматриптан назальный аэрозоль Imitrex®, 20 мг в 100 микролитрах, изготовленный GlaxoSmithKline, был предусмотренным «контролем». В исследовании тройственного пересечения, в котором между введениями доз существовал период выведения агента из организма, составляющий по крайней мере 3 дня, 18 пациентам вводили каждый раствор лекарственного средства в виде назального аэрозоля в дозе 100 микролитров, используя стандартные устройства дозированного интраназального аэрозольного распыления, такие как те, которые производятся Ing. Erich Pfeiffer GmbH, Radolfzell, Германия, Valois Pharma, Le Neubourg, Франция, или Becton Dickinson, New Jersey, США. От каждого пациента брали образцы крови с установленными интервалами, например, через 0,08, 0,17, 0,25, 0,33, 0,42, 0,6, 0,67, 0,83, 1, 2, 3, 4, 6, 8, 12, 24 часов, как представлено на фиг. 6 и 7, для приготовления плазмы из каждого образца крови, используя K2EDTA (дикалия EDTA) в качестве антикоагулянта. Уровни суматриптана в плазме определяли с помощью жидкостной хроматографии высокого разрешения (Ge, Tessier et al. 2004).
На фиг. 6 демонстрируется сравнение средних уровней в плазме всех пациентов в различные указанные моменты времени, наряду со среднеквадратическим отклонением, для контроля в виде назального аэрозоля Imitrex® и препарата B, который не содержит наполнитель в виде алкилсахарида. Контроль и препарат дают приблизительно равную эффективность, при этом Cmax составляет приблизительно 15 нг в мл, а Tmax - 1-2 часов.
На фиг. 7 демонстрируется сравнение средних уровней в плазме всех пациентов в различные указанные моменты времени, наряду со среднеквадратическим отклонением, для контроля в виде назального аэрозоля Imitrex® и препарата А, который содержит 0,18% наполнителя - додецилмальтозида. Cmax в случае препарата A составляет приблизительно 60 нг в мл, или в приблизительно 4 раза превышает Cmax, наблюдаемую в случае контроля в виде назального аэрозоля Imitrex®. T-max снижается с 1-2 часов в случае контроля до приблизительно 8-10 минут в случае препарата A, который содержит алкилсахарид, а предполагаемое терапевтически значимое значение Cmax, составляющее 15 нг/мл, достигаемое контрольным препаратом через 1-2 часа, достигалось через приблизительно 2 минуты в случае препарата A.
На основе полученных данных рассчитывались следующие параметры:
AUC0-t = площадь под кривой зависимости концентрации лекарственного средства в плазме от времени, от момента времени введения до момента времени последней определяемой концентрации.
AUC0-∞ = площадь под кривой зависимости концентрации лекарственного средства в плазме от времени, от момента времени введения до бесконечности.
Cmax = максимальная концентрация лекарственного средства в плазме.
Tmax = время до достижения максимальной концентрации лекарственного средства в плазме.
Полупериод существования = время после введения до тех пор, пока концентрация лекарственного средства в плазме не составит половину от ее максимальной концентрации.
Kel = константа скорости элиминации.
Средние фармакокинетические результаты приведены в виде таблицы ниже:
Средние параметры Cmax в отдельные моменты времени, такие как 0,08, 0,17, 0,25, 0,33, 0,42, 0,6, 0,67, 0,83, 1, 2, 3, 4, 6, 8, 12, 24 часов, для всех субъектов нанесены на график фиг.7.
ПРИМЕР 18
СУБЛИНГВАЛЬНОЕ/БУККАЛЬНОЕ ВВЕДЕНИЕ РИЗАТРИПТАНА (СОЛИ БЕНЗОЙНОЙ КИСЛОТЫ) С ВПЛОТЬ ДО 2% ВЕС. АЛКИЛГЛИКОЗИДА НЕ УВЕЛИЧИВАЕТ ЗНАЧИТЕЛЬНО БИОДОСТУПНОСТЬ ИЛИ НЕ УСКОРЯЕТ ЗНАЧИТЕЛЬНО НАЧАЛО ПРОХОЖДЕНИЯ В БОЛЬШОЙ КРУГ КРОВООБРАЩЕНИЯ
Поскольку биодоступность суматриптама через слизистую оболочку в носу значительно увеличивается в присутствии додецилмальтозида, казалось вероятным, что сопоставимая биодоступность через аналогично образованные слизистые оболочки, такие как подъязычные или щечные оболочки ротовой полости, могла бы быть схожим образом достигнута в случае других триптанов в присутствии алкилсахарида.
Процедура производства: Все наполнители смешивают по законам геометрии и подвергают сжатию в таблетки с использованием подходящего оснащения с круглыми неглубокими выемками. Альтернативно, ризатриптан и алкилгликозид растворяют в воде или водноспиртовых смесях, наполнители гранулируют с использованием раствора, затем гранулы подвергают сушке, измельчению и сжатию в таблетки. Распадающиеся в ротовой полости таблетки Maxalt-MLT (ризатриптана бензоат) 10 мг являются предусмотренным «контролем». Препарат A не содержит алкилгликозиды. Препарат B содержит 0,5% алкилгликозидов. Препарат C содержит 2% алкилгликозидов. Контроль и три препарата распадающихся таблеток, приготовленных, как описано выше, содержащие, соответственно, 0%, 0,5% и 2% додецилмальтозида, вводили 20 пациентам в четыре момента времени, исследование четырехпериодного пересечения, в котором между введениями доз существовал период выведения агента из организма, составляющий по крайней мере 3 дня. Таблетки помещали в ротовую полость, либо под язык, либо между щекой и деснами, и позволяли им распадаться при контактировании со слюной. От каждого пациента брали образцы крови с установленными интервалами, например, через 0,08, 0,17, 0,25, 0,33, 0,42, 0,6, 0,67, 0,83, 1, 2, 3, 4, 6, 8, 12, 24 часов, для приготовления плазмы из каждого образца крови, используя K2EDTA (дикалия EDTA) в качестве антикоагулянта. Уровни ризатриптана в плазме определяли с помощью жидкостной хроматографии высокого разрешения посредством модификации способа, описанного в Ge, Tessier et al. 2004, с включением подходящих стандартов для ризатриптана.
На удивление, в случае сублингвальной или буккальной доставки существует незначительное или не определяемое увеличение биодоступности при добавлении алкилсахарида, как видно в таблице XV ниже. Так же в значениях Cmax и Tmax установлены лишь небольшие изменения.
ПРИМЕР 19
СУБЛИНГВАЛЬНОЕ АЭРОЗОЛЬНОЕ ВВЕДЕНИЕ НАРАТРИПТАНА С ВПЛОТЬ ДО 2% ВЕС. АЛКИЛГЛИКОЗИДА НЕ УВЕЛИЧИВАЕТ ЗНАЧИТЕЛЬНО БИОДОСТУПНОСТЬ ИЛИ НЕ УСКОРЯЕТ ЗНАЧИТЕЛЬНО НАЧАЛО ПРОХОЖДЕНИЯ В БОЛЬШОЙ КРУГ КРОВООБРАЩЕНИЯ
Процесс производства: Наратриптана гидрохлорид растворяют в воде, и добавляют необходимое количество полиэтиленгликоля или этанола. Затем в растворе растворяют алкилгликозид, корригенты, аспартам. С помощью воды осуществляет конечное доведение объема. Препарат с алкилсахаридом - додецилмальтозидом и без него для сублингвальной доставки вводили с использованием насоса для дозированного распыления 20 пациентам в два момента времени, двухпериодное пересечение в течение исследования. Как и в примере 18, на удивление, наблюдается незначительное увеличение прохождения наратриптана через подъязычные слизистые оболочки в присутствии 0,18-0,2% алкилсахарида.
На протяжении этой заявки приводятся ссылки на различные публикации. Описания этих публикаций в целом включены, тем самым, посредством ссылки в эту заявку для более полного описания уровня техники, к которому относится настоящее изобретение.
Ссылки



Хотя настоящее изобретение было описано со ссылкой на специфические детали его конкретных вариантов осуществления в вышеприведенных примерах, будет понятно, что модификации и вариации включены в сущность и объем настоящего изобретения. Соответственно, настоящее изобретение ограничивается только следующей формулой изобретения.
Реферат
Изобретение относится к области фармацевтики, а именно представляет собой композицию и способ увеличения биодоступности терапевтических агентов у субъекта, а также композиции и способы для обеспечения купирования боли при мигрени. Композиции включают по крайней мере один алкилгликозид и по крайней мере один терапевтический агент, такой как агонист рецептора 5-HT, причем длина алкилгликозида составляет от по крайней мере 10 до приблизительно 16 атомов углерода. 2 н. и 14 з.п. ф-лы, 7 ил., 16 табл., 19 пр.
Формула
a) терапевтически эффективное количество аналога триптана, выбираемого из суматриптана, наратриптана, элетриптана, фроватриптана, алмотриптана, золмитриптана, ризатриптана их соли или их комбинации, и
b) алкилсахарид, имеющий алкильную цепь, включающую от 12 до 14 атомов углерода, связанную гликозидной связью с мальтозой, где алкилсахаридом является додецил-β-D-мальтозид, тридецил-β-D-мальтозид, тетрадецил-β-D-мальтозид или их комбинация,
в которой концентрация алкилсахарида составляет 0,02-2% вес. и где композиция сформулирована для интраназального введения.
a) терапевтически эффективное количество аналога триптана, выбираемого из суматриптана, наратриптана, элетриптана, фроватриптана, алмотриптана, золмитриптана, ризатриптана, их соли или их комбинации, и
b) алкилсахарид, выбираемый из додецил-β-D-мальтозида, тридецил-β-D-мальтозида, тетрадецил-β-D-мальтозида или их комбинации, где концентрация алкилсахарида составляет 0,02-2% вес.,
которая предоставляет нуждающемуся в этом субъекту терапевтически эффективное количества аналога триптана, и при этом площадь под кривой (AUC) более чем AUC, обеспечиваемая терапевтически эффективным количеством аналога триптана, вводимым в отсутствие алкилсахарида; или
которая обеспечивает для аналога триптана AUC0-1 ч, превышающую в 1,3 раза, 1,5 раз или более соответствующую AUC0-1 ч, обеспечиваемую в отсутствие алкилсахарида;
которая демонстрирует Tmax, составляющее 30 минут или менее, у субъекта, посредством чего обеспечивается быстрое облегчение боли при мигрени, или
которая обеспечивает Tmax, составляющее менее 20 минут, и где композиция сформулирована для интраназального введения.
Комментарии