Частицы резината фенилэфрина - RU2675788C2
Код документа: RU2675788C2
Чертежи









Описание
ОБЛАСТЬ ПРИМЕНЕНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к частицам фенилэфрина, подходящим для твердых, полутвердых или жидких дозированных форм. Частицы фенилэфрина, на которые можно нанести покрытие, высвобождают фенилэфрин со скоростью, обеспечивающей фармацевтически подходящие концентрации в плазме в течение продолжительного периода времени. Настоящее изобретение также относится к способу производства дозированных форм, содержащих частицы фенилэфрина, и к способам ослабления заложенности носа и дыхательных путей у субъектов-людей при пероральном введении дозированных форм. Дозированные формы могут дополнительно содержать один или более дополнительных терапевтически активных агентов, которые выбирают из одного или более из группы, состоящей из антигистаминных, противозастойных, обезболивающих, противовоспалительных, жаропонижающих, противокашлевых и отхаркивающих средств.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Фенилэфрин представляет собой эффективный вазоконстриктор, обладающий как прямым, так и непрямым симпатомиметическим эффектом [Hoffman 2001]. Преобладающим и прямым эффектом является агонизм в отношении α1-адренергических рецепторов. Стимуляция α1-адренергических рецепторов, расположенных на емкостных кровеносных сосудах слизистой оболочки носа (посткапиллярных венулах), приводит в результате к вазоконстрикции, уменьшению объема крови и уменьшению объема слизистой оболочки носа (снятию отека полости носа) [Johnson 1993]. Суженные кровеносные сосуды позволяют меньшему количеству жидкости поступать в слизистые оболочки носа, горла и синусов, что приводит к уменьшению воспаления носовых перегородок, а также к уменьшению образования слизи [Johnson 1993]. Таким образом, благодаря сужению кровеносных сосудов, преимущественно расположенных в носовых ходах, фенилэфрин вызывает уменьшение заложенности носа [Hoffman 2001, Empey 1981].
Фенилэфрин представляет собой безрецептурное (OTC), пероральное противозастойное средство для носа категории I (считающееся безопасным и эффективным (GRASE)). В мире фенилэфрин имеется в продаже с 1960-х годов и, начиная с 1996 г., фенилэфрин широко применяется в США. Гидрохлорид фенилэфрина, который интенсивно применяется у взрослых и детей как безрецептурное лекарственное средство от кашля и простуды, показан для применения взрослыми и детьми с целью временного облегчения заложенности носа, вызванной острым вирусным ринофарингитом, поллинозом или другими дыхательными аллергиями (аллергическим ринитом). Он имеется в продаже в виде таблеток 10 мг для перорального введения взрослым. Схема дозирования представляет собой однократную дозу 10 мг фенилэфрина каждые четыре часа, но не более 60 мг (шесть доз) за 24 часа. Полная информация содержится в монографии по маркировке OTC для утвержденных лекарственных средств.
Фенилэфрин, химическое название (R)-1-(3-гидроксифенил)-2-метиламиноэтанол, имеется в продаже в виде соли гидрохлорида. Эмпирическая формула представляет собой C9H13NO2⋅HCl; молекулярная масса составляет 203,67. Композиция, которая представляет собой кристаллический порошок от белого до беловатого цвета, имеет следующую химическую структуру:

Основными путями метаболизма фенилэфрина являются конъюгация с сульфатами (преимущественно в кишечной стенке) и окислительное дезаминирование обеими формами A и B моноаминоксидазы [Suzuki 1979]. Также происходит глюкуронирование, но в меньшей степени. В одном исследовании после перорального введения дозы 30 мг в течение восьми часов [Ibrahim 1983] фенилэфрин претерпевал метаболизм до фенилэфринсульфата, мета-гидроксиминдальной кислоты, фенилэфринглюкуронида и мета-гидроксифенилгликольсульфата в количестве 47%, 30%, 12% и 6% от дозы соответственно. После внутривенной инъекции фенилэфрина преобладающим путем метаболизма является дезаминирование [Hengstmann 1982], тогда как после перорального введения преобладающим путем является конъюгация с сульфатами. Метаболиты фенилэфрина фазы I и фазы II у людей представлены ниже. Процентные показатели на схеме означают процент от пероральной дозы (в соответствии с данными Ibrahim).

Данные об эффективности, полученные в клинических исследованиях применения фенилэфрина быстрого высвобождения у взрослых, указывают на то, что фенилэфрин является эффективным назальным противозастойным средством.
Ацетаминофен представляет собой производное парааминофенола, обладающее обезболивающей и жаропонижающей активностью. Его применяют для временного облегчения небольшой ломоты и боли, связанных с острым вирусным ринофарингитом, боли в спине, головной боли, зубной боли, менструальных спазмов и мышечных болей; а также для временного облегчения небольших болей при артрите и для уменьшения лихорадки. Доза ацетаминофена для взрослых в США составляет 1000 мг каждые четыре–шесть часов, при этом максимальная доза составляет 4000 мг за 24 часа. Доза ацетаминофена продленного высвобождения для взрослых составляет 1300 мг каждые восемь часов, при этом максимальная доза составляет 3900 мг за 24 часа.
Ацетаминофен преимущественно претерпевает метаболизм в печени посредством трех основных параллельных путей: глюкуронирования, сульфатирования и окисления [Miners 1983; Slattery 1989; Lee 1992; Miners 1992]. Оба пути метаболизма, как глюкуронирование, так и окисление, следуют процессу, имеющему константу скорости первого порядка, что означает, что концентрация метаболизированного ацетаминофена возрастает по мере возрастания концентрации в печени. Сульфатный путь метаболизма следует кинетике Михаэлиса - Ментен, что означает, что концентрация метаболизированного ацетаминофена остается постоянной, как только концентрация в печени возрастает выше уровня насыщения.
Схематическое изображение метаболизма ацетаминофена представлено ниже. Менее 9% терапевтической дозы выводится с мочой в неизмененном виде [Miners 1992]. Основным путем метаболизма является глюкуронирование, при котором 47–62% дозы ацетаминофена образует конъюгаты с глюкуронидом. Такие глюкуронидные конъюгаты неактивны и нетоксичны [Koch-Weser 1976], секретируются в желчь и выводятся с мочой. Конъюгация с глюкуронидами катализируется преимущественно одной изоформой глюкуронилтрансферазы (UGT1A6) [Court 2001] с уридин-5’-дифосфоглюкуроновой кислотой в качестве незаменимого кофактора.
Вторым основным путем метаболизма ацетаминофена является сульфатирование, при котором 25-36% дозы образует конъюгаты с сульфатом. Такие конъюгаты сульфатных эфиров также являются неактивными и нетоксичными [Koch-Weser 1976] и быстро выводятся с мочой. Сульфатирование опосредуется сульфотрансферазами, которые представляют собой гетерогенные цитозольные ферменты, и 3'-фосфоаденозин-5'-фосфат является их кофактором. Фактором, регулирующим скорость сульфатирования ацетаминофена, является скорее активность сульфотрансферазы, чем истощение запасов сульфата [Blackledge 1991].
Третьим путем метаболизма является окисление, при котором 5-8% дозы ацетаминофена претерпевает метаболизм посредством ферментной системы цитохрома P-450. Изоферментом цитохрома P-450, который преимущественно отвечает за метаболизм ацетаминофена, является CYP2E1 [Manyike 2000]. Когда ацетаминофен претерпевает метаболизм посредством CYP2E1, он образует высокоактивное промежуточное соединение N-ацетил-пара-бензохинонимин (NAPQI). Поскольку NAPQI является высокоактивным, его уровень невозможно измерить вне тканей печени; а также он не может накапливаться. Это промежуточное соединение быстро инактивируется гепатоцеллюлярными запасами глутатиона с образованием цистеина и конъюгатов меркаптурата, которые оба являются неактивными и нетоксичными [Koch-Weser 1976]. Такие конъюгаты выводятся с мочой [Mitchell 1974].

25–36%47–62%5–8%
Существует необходимость в менее частой доставке фенилэфрина. Менее частое введение приводит в результате к лучшему соблюдению режима лечения пациентами. Кроме того, постоянные терапевтические уровни активных компонентов в плазме могут быть более эффективны и в равной степени эффективны по сравнению с колебаниями, наблюдаемыми при введении многократных доз традиционной композиции быстрого высвобождения. Устойчивые эффективные уровни могли бы уменьшить тяжесть и частоту побочных эффектов, наблюдаемых при высоких пиковых уровнях в плазме. Таким образом, необходимы композиции фенилэфрина, которые можно вводить менее часто, например, один раз каждые 6, 8, 12, 16, 20 часов или 24 часа.
Также существует необходимость в согласовании длительности действия фенилэфрина с активными веществами, которые обеспечивают более продолжительное действие, чем фенилэфрин быстрого высвобождения.
В опубликованной заявке на патент США № 20070281020, выданной Schering-Plough Corporation, описано введение субъекту-человеку таблетки устойчивого высвобождения, содержащей 30 мг фенилэфрина, гидроксипропилметилцеллюлозу, натриевую соль карбоксиметилцеллюлозы, Коллидон CL-M, коллоидный диоксид кремния и стеарат магния, и сравнение таблетки устойчивого высвобождения с тремя дозами по 10 мг фенилэфрина быстрого высвобождения.
В патенте США № 8,282,957, выданном McNeil-PPC, Inc., описаны частицы фенилэфрина с покрытием, содержащие гидрохлорид фенилэфрина, модифицированный крахмал и Эудрагит NE30D™, покрытые первым слоем покрытия, содержащим Эудрагит RS PO, ацетилтрибутилцитрат и стеарат магния, и вторым слоем покрытия, содержащим Эудрагит NE30D™, Эудрагит FS30D™, стеарат магния, лаурилсульфат натрия и симетикон, а также их применение в фармацевтических дозированных формах, включая дозированные формы, содержащие ацетаминофен.
В патенте США № 6,001,392, выданном Warner Lambert Company, описан комплекс лекарственного средства и смолы, который содержит смесь покрытого и непокрытого Амберлита™ IR69, сшитого с дивинилбензолом.
В опубликованной заявке на патент США № 20100068280, выданной Schering-Plough Corporation, описаны фармацевтические дозированные формы, содержащие фенилэфрин в форме устойчивого высвобождения. В соответствии с вариантом осуществления однократную дозу фенилэфрина в таблетке, содержащей 30 мг фенилэфрина, моногидрат лактозы, Метоцел K100M CR, Клуцел EXF и стеарат магния, сравнивали с двумя таблетками фенилэфрина быстрого высвобождения по 10 мг, вводимыми с интервалом 4 часа, в исследовании биоэквивалентности.
В опубликованных заявках на патенты США №№ 20050266032 и 20060057205, выданных Sovereign Pharmaceuticals, описаны фармацевтические дозированные формы, содержащие фенилэфрин. В соответствии с вариантом осуществления фенилэфрин включен в комплекс с ионообменной смолой с помощью, например, полистиролсульфоната натрия, и покрыт полимером отсроченного высвобождения, например, Эудрагитом® L 100, Коллидоном® MAE и Аквакоутом® cPD. В этом варианте осуществления формула содержит 45 мг фенилэфрина устойчивого высвобождения и 15 мг фенилэфрина быстрого высвобождения.
В патенте США № 8,062,667, выданном Tris Pharma, Inc., описаны комплексы лекарственного средства с ионообменной смолой, имеющие покрытие. В соответствии с вариантом осуществления фенилэфрин включен в комплекс с ионообменной смолой с помощью, например, полистиролсульфоната натрия, и покрыт препаратом КОЛЛИКОУТ™ SR-30D, триацетином и водой.
В патенте США № 8,394,415, выданном McNeil-PPC, Inc., описана жидкая композиция, содержащая ибупрофен быстрого высвобождения и комплекс фенилэфрина продленного высвобождения с определенной ионообменной смолой, покрытый первым и вторым слоями покрытия, содержащими определенные компоненты.
В заявке на патент США № 11/761,698, выданный McNeil-PPC, Inc., описана твердая композиция, содержащая ибупрофен быстрого высвобождения (НВ) и фенилэфрин, покрытый первым слоем покрытия, содержащим этилцеллюлозу, и вторым слоем покрытия, содержащим защитное покрытие.
В патенте США № 20100068280, выданном Schering-Plough Healthcare Products, Inc., описано исследование биодоступности, в котором сравнивали 10 мг гидрохлорида фенилэфрина, вводимого в капсулах Энтерион™, 10 мг Судафеда ФЭ™ и 30 мг гидрохлорида фенилэфрина, вводимого в капсулах Энтерион™.
В заявке на патент США № 2007014239, выданной Coating Place, Inc., описаны способ и композиция для нанесения одного или более лекарственных средств на одну или более частиц ионообменной смолы с образованием несущих лекарственное средство частиц смолы.
Продолжает существовать необходимость в препаратах фенилэфрина, обладающих обсуждаемыми выше свойствами.
ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к частицам фенилэфрина, которые доставляют фенилэфрин или его фармацевтически приемлемую соль субъекту, нуждающемуся в этом, в результате чего обеспечивают максимальную концентрацию фенилэфрина в плазме в период от приблизительно 0,1 до приблизительно 16 часов, предпочтительно от приблизительно 0,5 до приблизительно 5 часов, более предпочтительно от приблизительно 1 до приблизительно 4,5 часа после приема внутрь, и при этом фенилэфрин поддерживается на уровне, превышающем приблизительно 20, приблизительно 40, приблизительно 60, приблизительно 80, приблизительно 100, приблизительно 120, приблизительно 140, приблизительно 160, приблизительно 180 или приблизительно 200 пг/мл, в течение по меньшей мере приблизительно 6, приблизительно 8, приблизительно 12, приблизительно 16, приблизительно 20 и/или приблизительно 24 часов после приема внутрь.
В соответствии с предпочтительным вариантом осуществления изобретение относится к частицам резината фенилэфрина с покрытием, которые доставляют фенилэфрин или его фармацевтически приемлемую соль субъекту, нуждающемуся в этом, в результате чего обеспечивают максимальную концентрацию фенилэфрина в плазме в период от приблизительно 0,1 до приблизительно 16 часов, предпочтительно от приблизительно 0,5 до приблизительно 5 часов, более предпочтительно от приблизительно 1 до приблизительно 4,5 часа после приема внутрь, и при этом фенилэфрин поддерживается на уровне, превышающем приблизительно 20, приблизительно 40, приблизительно 60, приблизительно 80, приблизительно 100, приблизительно 120, приблизительно 140, приблизительно 160, приблизительно 180 или приблизительно 200 пг/мл, в течение по меньшей мере приблизительно 6, приблизительно 8, приблизительно 12, приблизительно 16, приблизительно 20 и/или приблизительно 24 часов после приема внутрь.
Настоящее изобретение также относится к фармацевтическим дозированным формам, содержащим частицы фенилэфрина, которые доставляют фенилэфрин или его фармацевтически приемлемую соль субъекту, нуждающемуся в этом, в результате чего обеспечивают максимальную концентрацию фенилэфрина в плазме в период от приблизительно 0,1 до приблизительно 16 часов, предпочтительно от приблизительно 0,5 до приблизительно 5 часов, более предпочтительно от приблизительно 1 до приблизительно 4,5 часа после приема внутрь, и при этом фенилэфрин поддерживается на уровне, превышающем приблизительно 20, приблизительно 40, приблизительно 60, приблизительно 80, приблизительно 100, приблизительно 120, приблизительно 140, приблизительно 160, приблизительно 180 или приблизительно 200 пг/мл, в течение по меньшей мере приблизительно 6, приблизительно 8, приблизительно 12, приблизительно 16, приблизительно 20 и/или приблизительно 24 часов после приема внутрь.
В другом варианте осуществления частицы фенилэфрина, которые обеспечивают продленное высвобождение фенилэфрина, комбинируют с фенилэфрином быстрого высвобождения.
В другом варианте осуществления частицы фенилэфрина комбинируют с одним или более дополнительных терапевтических агентов быстрого или устойчивого высвобождения. Такой агент или агенты можно включать в композицию для быстрого высвобождения после приема внутрь, для устойчивого высвобождения, для высвобождения в толстом кишечнике одновременно по меньшей мере с некоторым количеством фенилэфрина или любую их комбинацию. В одном варианте осуществления дополнительный терапевтический агент не имеет покрытия. В другом варианте осуществления дополнительный терапевтический агент имеет покрытие.
Дополнительное терапевтическое средство может представлять собой антигистаминное, противозастойное, обезболивающее, противовоспалительное, жаропонижающее, противокашлевое, отхаркивающее или любое другое терапевтическое средство или комбинацию таких средств, полезных для облегчения симптомов таких заболеваний, как острый вирусный ринофарингит, сезонная и другие формы аллергии, поллиноз или заболевания пазух носа, любое из которых может привести к увеличению выделений из носа. Предпочтительно одним или более из дополнительных терапевтических агентов является ацетаминофен.
Примеры антигистаминных средств и деконгестантов включают, помимо прочего, бромфенирамин, хлорциклизин, дексбромфенирамин, бромгексан, фениндамин, фенирамин, пириламин, тонзиламин, приполидин, эфедрин, псевдоэфедрин, фенилпропаноламин, хлорфенирамин, декстрометорфан, дифенгидрамин, доксиламин, астемизол, терфенадин, фексофенадин, нафазолин, оксиметазолин, монтелукаст, пропилгекседрин, трипролидин, клемастин, акривастин, прометазин, оксомемазин, меквитазин, буклизин, бромгексин, кетотифен, эбастин, оксатамид, ксилометазолин, лоратадин, дезлоратидин и цетиризин, их изомеры, фармацевтически приемлемые соли и эфиры.
Примеры подходящих обезболивающих, противовоспалительных и жаропонижающих средств включают, без ограничений, нестероидные противовоспалительные препараты (НПВП), такие как производные пропионовой кислоты (например, ибупрофен, напроксен, кетопрофен, флурбипрофен, фенбуфен, фенопрофен, индопрофен, флупрофен, пирпрофен, карпрофен, оксапрозин, пранопрофен и супрофен) и ингибиторы циклооксигеназы, такие как целекоксиб; ацетаминофен; ацетилсалициловая кислота; производные уксусной кислоты, такие как индометацин, диклофенак, сулиндак и толметин; производные фенамовой кислоты, такие как мефенамовая кислота, меклофенамовая кислота и флуфенамовая кислота; производные бифенилкарбодиловой кислоты, такие как дифлунизал и флуфенизал; оксикамы, такие как пироксикам, судоксикам, изоксикам и мелоксикам; их изомеры, фармацевтически приемлемые соли и пролекарственные формы.
Примеры средств от кашля и отхаркивающих средств включают, помимо прочего, дифенгидрамин, декстрометорфан, носкапин, клофедианол, ментол, бензонатат, этилморфин, кодеин, ацетилцистеин, карбоцистеин, амброксол, алкалоиды красавки обыкновенной, собренол, гваякол и гвайфенезин, их изомеры, фармацевтически приемлемые соли и пролекарственные формы.
Другой аспект изобретения представляет собой способ лечения симптомов острого вирусного ринофарингита, гриппа, аллергий или неаллергического ринита у субъекта, нуждающегося в этом, включающий введение частиц фенилэфрина по изобретению. В некоторых вариантах осуществления частицы фенилэфрина вводят приблизительно каждые 6, 8, 12, 16, 20 часов или 24 часа. В одном предпочтительном варианте осуществления частицы фенилэфрина вводят приблизительно каждые 12 часов. В другом предпочтительном варианте осуществления частицы резината фенилэфрина вводят приблизительно каждые 8 часов.
Некоторые варианты осуществления изобретения представляют собой способы поддержания устойчивой биодоступности фенилэфрина у субъекта, включающие пероральное введение субъекту частиц фенилэфрина, при этом по меньшей мере часть фенилэфрина всасывается из толстого кишечника, и при этом концентрация фенилэфрина в плазме субъекта составляет по меньшей мере приблизительно 20, приблизительно 40, приблизительно 60, приблизительно 80, приблизительно 100, приблизительно 120, приблизительно 140, приблизительно 160, приблизительно 180 или приблизительно 200 пг/мл приблизительно через 6 часов после введения композиции. В конкретных вариантах осуществления концентрация фенилэфрина в плазме субъекта составляет по меньшей мере приблизительно 20, приблизительно 40, приблизительно 60, приблизительно 80, приблизительно 100, приблизительно 120, приблизительно 140, приблизительно 160, приблизительно 180 или приблизительно 200 пг/мл приблизительно через 8 часов после введения композиции. В конкретных вариантах осуществления концентрация фенилэфрина в плазме субъекта составляет по меньшей мере приблизительно 20, приблизительно 40, приблизительно 60, приблизительно 80, приблизительно 100, приблизительно 120, приблизительно 140, приблизительно 160, приблизительно 180 или приблизительно 200 пг/мл приблизительно через 12 часов после введения композиции. В конкретных вариантах осуществления концентрация фенилэфрина в плазме субъекта составляет по меньшей мере приблизительно 20, приблизительно 40, приблизительно 60, приблизительно 80, приблизительно 100, приблизительно 120, приблизительно 140, приблизительно 160, приблизительно 180 или приблизительно 200 пг/мл приблизительно через 20 часов после введения композиции. В конкретных вариантах осуществления концентрация фенилэфрина в плазме субъекта составляет по меньшей мере приблизительно 20, приблизительно 40, приблизительно 60, приблизительно 80, приблизительно 100, приблизительно 120, приблизительно 140, приблизительно 160, приблизительно 180 или приблизительно 200 пг/мл приблизительно через 24 часа после введения композиции. Некоторые другие варианты осуществления изобретения представляют собой способы введения фенилэфрина субъекту, включающие пероральное введение частиц фенилэфрина, при этом указанная композиция доставляет по меньшей мере некоторое количество фенилэфрина в толстый кишечник, где фенилэфрин высвобождается в толстом кишечнике и всасывается из толстого кишечника.
Настоящее изобретение может быть понято более полно при рассмотрении фигур, подробного описания и примеров, которые представлены далее.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На Фиг. 1 представлен профиль средней концентрации фенилэфрина в плазме после введения частиц резината фенилэфрина продленного высвобождения (ПВ) с покрытием, содержащих 20 мг фенилэфрина. Как показано на Фиг. 1, на оси y представлена концентрация свободного фенилэфрина в плазме в пикограммах (пг) на миллилитр (мл). На оси x представлено время в часах. На Фиг. 1 показано, что средняя концентрация фенилэфрина достигает пика (Cmax) приблизительно через 2 часа. На Фиг. 1 также показан вторичный пик приблизительно через 12 часов.
На Фиг. 2 представлены отдельные профили концентрации фенилэфрина в плазме после введения частиц резината фенилэфрина ПВ с покрытием, содержащих 20 мг фенилэфрина. Как показано на Фиг. 2, межсубъектная вариабельность для фенилэфрина модифицированного высвобождения является удовлетворительной. Диапазон Cmax наблюдался в период от приблизительно 1 часа до приблизительно 4,5 часа. Вторичный пик приблизительно через 12 часов отмечали у всех субъектов.
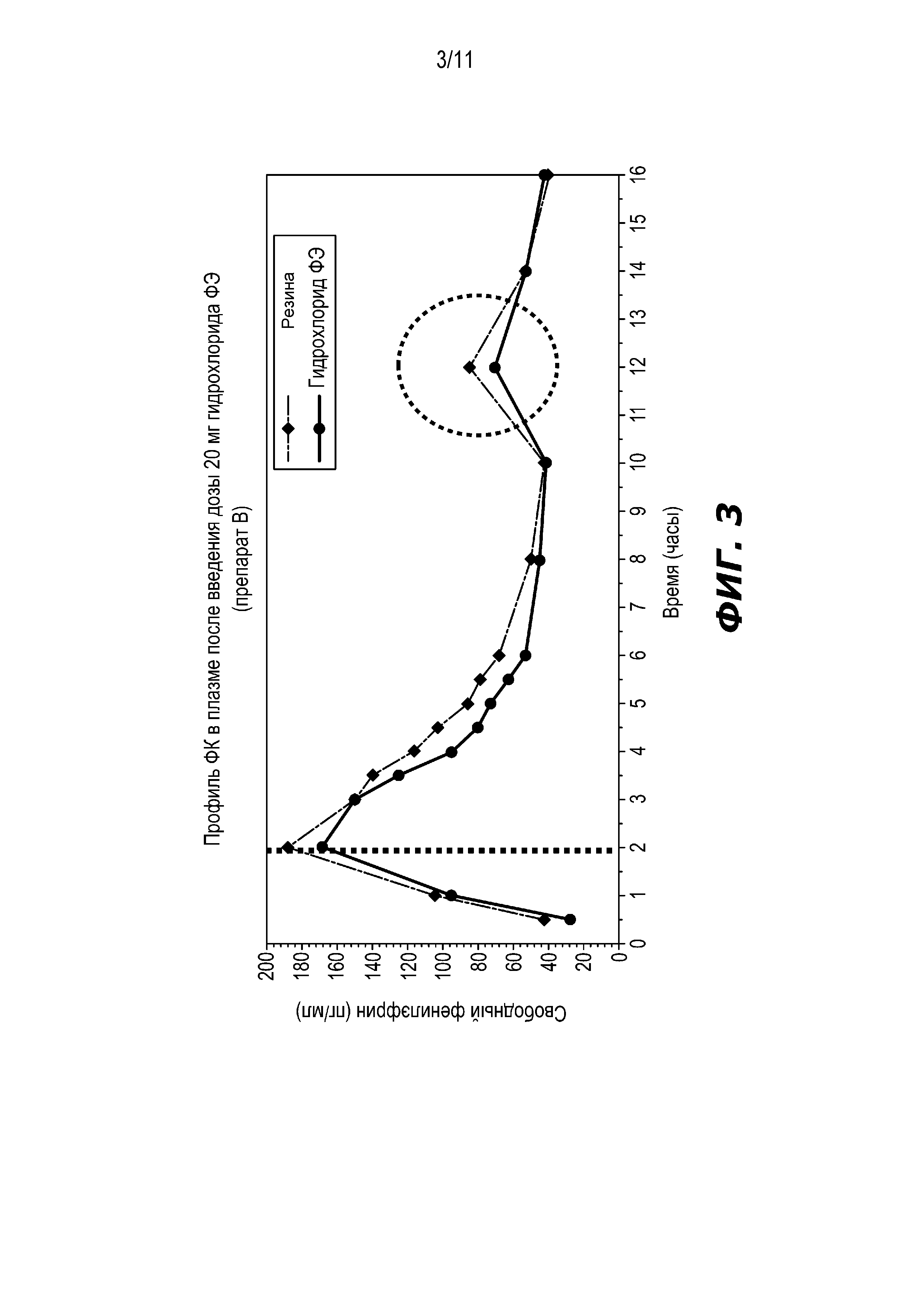
На Фиг. 3 представлен профиль средней концентрации фенилэфрина в плазме после введения частиц гидрохлорида фенилэфрина ПВ с покрытием, содержащих 20 мг фенилэфрина. На Фиг. 3 пунктирная линия представляет собой профиль из Фиг. 1, показанный для сравнения. Для частиц резината фенилэфрина наблюдали немного более высокую Cmax. Вторичный пик приблизительно через 12 часов виден на обоих профилях. Это может быть результатом меньшего количества фенилэфрина, претерпевающего пресистемный метаболизм в стенке кишечника в результате быстрого перемещения частиц вниз по желудочно-кишечному тракту (ЖКТ). Высвобождение фенилэфрина в толстом кишечнике приведет в результате к усилению всасывания в более позднем периоде.
На Фиг. 4 представлены отдельные профили концентрации фенилэфрина в плазме после введения частиц гидрохлорида фенилэфрина ПВ с покрытием, содержащих 20 мг фенилэфрина.
На Фиг. 5 представлен профиль средней концентрации фенилэфрина в плазме после введения частиц резината фенилэфрина ПВ с покрытием, содержащих 15 мг фенилэфрина, и жидкого гидрохлорида фенилэфрина быстрого высвобождения (НВ), содержащего 5 мг фенилэфрина («смесь ПВ-НВ»). На Фиг. 5 сплошная линия представляет смесь ПВ-НВ. Опять же, кривая для данного препарата согласуется с тем, что наблюдали для композиций резината и гидрохлорида. В случае смеси ПВ-НВ существует два пика в течение первых 2 часов; один обусловлен преимущественно дозой НВ, а другой аккумуляцией доз НВ и ПВ. Достижение Cmax происходило быстрее и поддерживалось в течение более длительного периода времени. Таким образом, смесь ПВ-НВ оказывается полезной в отношении начала эффективности.
На Фиг. 6 представлены отдельные профили концентрации фенилэфрина в плазме после введения частиц резината фенилэфрина ПВ с покрытием, содержащих 15 мг фенилэфрина, и жидкого гидрохлорида фенилэфрина НВ, содержащего 5 мг фенилэфрина.
На Фиг. 7 представлен профиль средней концентрации фенилэфрина в плазме после введения жидкого гидрохлорида фенилэфрина НВ, содержащего 20 мг фенилэфрина. На Фиг. 7 сплошная линия представляет профиль имеющегося в продаже в настоящее время жидкого препарата НВ, а пунктирная линия представляет собой профиль из Фиг. 5, показанный для сравнения. Cmax смеси НВ-ПВ ниже, чем Cmax формы НВ.
На Фиг. 8 представлены отдельные профили концентрации фенилэфрина в плазме после введения жидкого гидрохлорида фенилэфрина НВ, содержащего 20 мг фенилэфрина.
На Фиг. 9 представлен профиль средней концентрации фенилэфрина в плазме после введения частиц резината фенилэфрина ПВ с покрытием, содержащих 22,5 мг фенилэфрина, и жидкого гидрохлорида фенилэфрина НВ, содержащего 7,5 мг фенилэфрина («смесь ПВ-НВ»), а также сравнение с жидким гидрохлоридом фенилэфрина НВ, содержащим 20 мг фенилэфрина.
На Фиг. 10A и 10B представлено сравнение профиля средней концентрации фенилэфрина в плазме: (1) после введения частиц резината фенилэфрина ПВ с покрытием, содержащих 15 мг фенилэфрина, и жидкого гидрохлорида фенилэфрина НВ, содержащего 5 мг фенилэфрина (Фиг. 10A); а также (2) после введения частиц резината фенилэфрина ПВ с покрытием, содержащих 22,5 мг фенилэфрина, и жидкого гидрохлорида фенилэфрина НВ, содержащего 7,5 мг фенилэфрина (Фиг. 10B); с (3) жидким гидрохлоридом фенилэфрина НВ, содержащим 20 мг фенилэфрина.
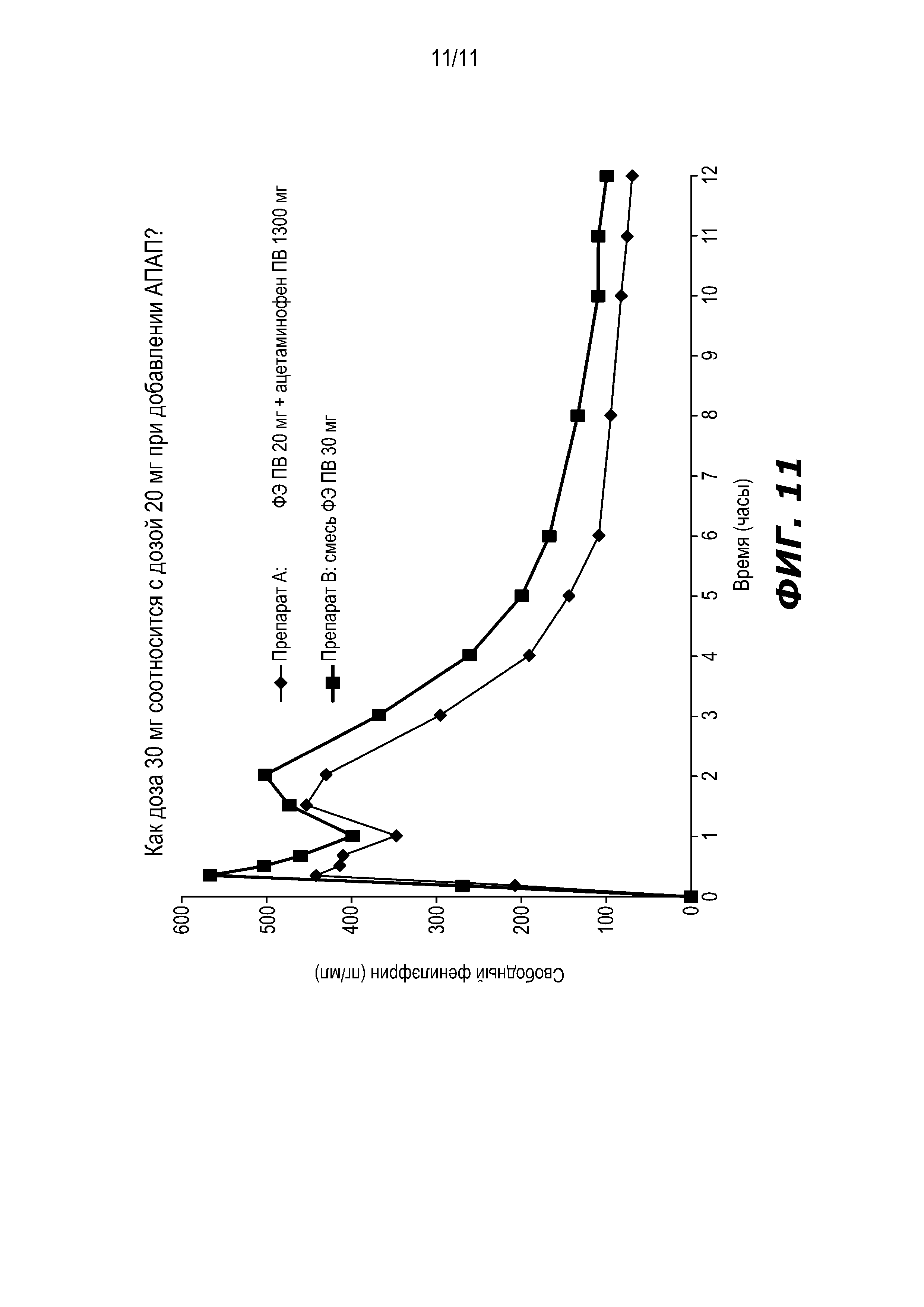
На Фиг. 11 представлено сравнение профилей средней концентрации фенилэфрина в плазме после (1) введения частиц резината фенилэфрина ПВ с покрытием, содержащих 15 мг фенилэфрина, и жидкого гидрохлорида фенилэфрина НВ, содержащего 5 мг фенилэфрина, с (2) комбинацией: (a) частиц резината фенилэфрина ПВ с покрытием, содержащих 15 мг фенилэфрина; (b) жидкого гидрохлорида фенилэфрина НВ, содержащего 5 мг фенилэфрина; и (с) 1300 мг ацетаминофена ПВ.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Считается, что специалист в данной области, основываясь на представленном в настоящем документе описании, сможет использовать настоящее изобретение в самом полном объеме. Представленные ниже конкретные варианты осуществления следует рассматривать исключительно как иллюстративные, которые ни в коей мере не ограничивают остальную часть описания.
Если не определено иное, все технические и научные термины, применяемые в настоящем документе, имеют общепринятое значение, понятное среднему специалисту в области, к которой относится настоящее изобретение. Кроме того, все публикации, заявки на патенты, патенты и другие материалы, упомянутые в настоящем документе, включены в настоящий документ путем ссылки. В настоящем документе все процентные значения, если не указано иное, даны по массе. Кроме того, все указанные в настоящем документе диапазоны предполагают включение любых комбинаций значений между двумя конечными значениями включительно.
ОПРЕДЕЛЕНИЯ
При использовании в настоящем документе фармацевтически приемлемая соль фенилэфрина включает, без ограничений, гидрохлорид фенилэфрина, битартрат фенилэфрина, таннат фенилэфрина и т. д. В одном предпочтительном варианте осуществления фармацевтически приемлемая соль фенилэфрина представляет собой гидрохлорид фенилэфрина.
Используемый в настоящем документе термин «AUC» для любого упоминаемого лекарственного средства означает «площадь под кривой зависимости концентрация-время» от введения дозы или активации лекарственного средства до момента, рассчитанного по формуле трапеций. AUC представляет собой параметр, отражающий кумулятивную концентрацию лекарственного средства в плазме по времени и являющийся показателем общего количества и доступности лекарственного средства в плазме.
Используемый в настоящем документе термин «Cmax» означает максимальную (или пиковую) концентрацию лекарственного средства, которая достигается в исследуемом диапазоне после введения лекарственного средства и до введения второй дозы.
Используемый в настоящем документе термин «кристаллическая форма» означает такую неаморфную форму активного компонента, которая проявляет кристаллоподобные свойства, включающие, без ограничений, способность к преломлению видимого света. Термин «кристаллическая» также можно использовать для описания активного компонента в его чистой форме, т. е., например, без добавления к нему других эксципиентов.
Термин «отсроченное высвобождение» подразумевает наличие по меньшей мере одного периода времени после введения, в течение которого активный компонент не высвобождается из дозированной формы, т. е. высвобождение активного компонента (-ов) происходит в момент времени, отличающийся от момента, следующего сразу за пероральным введением.
Используемый в настоящем документе термин «растворяющая среда» означает любую подходящую жидкую среду, в которой может растворяться дозированная форма суспензии настоящего изобретения, такую как, например, растворяющая среда in vitro, применяемая для тестирования препарата, или желудочно-кишечные среды. Подходящая растворяющая среда in vitro, применяемая для тестирования растворимости активного компонента или компонентов из дозированной формы суспензии настоящего изобретения, включает среды, описанные в Фармакопее США.
Используемые в настоящем документе термины «дозировка», «дозированная форма» или «доза» означают количество фармацевтической композиции, содержащей терапевтически активный агент (-ы), вводимый за один раз. «Дозировка», «дозированная форма» или «доза» включает введение одной или более единиц фармацевтической композиции, вводимых в одно и то же время. В одном варианте осуществления дозированная форма представляет собой таблетку. В одном варианте осуществления дозированная форма представляет собой многослойную таблетку. В варианте осуществления, содержащем многослойную таблетку, один слой может содержать часть быстрого высвобождения, а другой слой может содержать часть продленного высвобождения. В варианте осуществления, содержащем многослойную таблетку, один слой может содержать частицы резината фенилэфрина, а другой слой может содержать форму фенилэфрина быстрого высвобождения и/или второй активный компонент. В одном варианте осуществления дозированная форма, содержащая частицы резината фенилэфрина, представляет собой заполненную жидкостью гелевую капсулу.
Используемый в настоящем документе термин «комплекс лекарственное средство - смола» означает связанную форму активного компонента, включающую, без ограничений, фармацевтически активные компоненты и ионообменную смолу. Комплекс лекарственное средство - смола специалисты в данной области техники также называют «резинатом». В соответствии с изобретением может применяться ионообменная смола Амберлит™ IRP 69 производства The Dow Chemical Company, представляющая собой нерастворимую сильнокислую натриевую форму катионообменной смолы, полученную из сульфированного сополимера стирола и дивинилбензола. Подвижным или обменным катионом является натрий, который может обмениваться со многими катионными (основными) соединениями, включая, например, медь, цинк, железо, кальций, стронций, магний и литий, или замещаться ими. Адсорбция лекарственного средства на частицах ионообменной смолы с образованием комплекса лекарственное средство - смола представляет собой хорошо известную методику, описанную в патентах США №№ 2,990,332 и 4,221,778. Лекарственное средство по существу смешивают с водной суспензией смолы, после чего комплекс промывают и высушивают. Адсорбцию лекарственного средства на смоле можно обнаружить путем измерения изменения pH реакционной среды или путем измерения изменения концентрации натрия или лекарственного средства. Образованный комплекс лекарственное средство - смола можно собрать и промыть этанолом и/или водой для обеспечения удаления какого-либо несвязанного лекарственного средства. Обычно комплексы высушивают воздухом в лотках при комнатной или повышенной температуре. Комплекс лекарственное средство - смола имеет соотношение фенилэфрина и смолы, равное от приблизительно 0,25: 1 до приблизительно 0,65: 1, предпочтительно от приблизительно 0,30: 1 до приблизительно 0,55: 1, предпочтительно от приблизительно 0,35: 1 до приблизительно 0,45: 1.
«Энтеросолюбильный» означает способный растворяться при pH выше приблизительно 5,0, или выше приблизительно 5,5, или выше приблизительно 6,0, или таком значении, которое наблюдается в кишечнике.
Под «продленным высвобождением» подразумевают по существу непрерывное контролируемое высвобождение активного компонента из дозированной формы после ее введения, при этом время для полного высвобождения, т. е. истощения, активного компонента из дозированной формы является более длительным, чем обусловленное формой быстрого высвобождения того же активного компонента. Типы продленного высвобождения включают контролируемое, устойчивое, пролонгированное, нулевого порядка, первого порядка, импульсное и т. п.
Используемый в настоящем документе термин «быстрое высвобождение» означает, что характеристики растворения по меньшей мере одного активного компонента соответствуют спецификациям Фармакопеи США для таблеток быстрого высвобождения, содержащих данный активный компонент. Активный компонент, имеющий свойство быстрого высвобождения, может растворяться в содержимом желудочно-кишечного тракта без тенденции к отсроченному или пролонгированному растворению активного компонента.
«Жидкие дозированные формы» могут, без ограничений, включать суспензии или настойки, где один или более активных компонентов находится в растворенном, частично растворенном либо в нерастворенном или суспендированном состоянии.
В настоящем документе термин «модифицированное высвобождение» применяют по отношению к измененному высвобождению или растворению активного компонента в растворяющей среде, такой как жидкости желудочно-кишечного тракта. Типы модифицированного высвобождения включают: 1) продленное высвобождение; или 2) отсроченное высвобождение. По существу, композиции дозированных форм модифицированного высвобождения получают таким образом, чтобы сделать активный (-е) компонент (-ы) доступным (-и) в течение продленного периода времени после приема внутрь, что, таким образом, позволяет снизить частоту дозирования по сравнению с дозированием того же (тех же) активного (-ых) компонента (-ов) в традиционной дозированной форме. Дозированные формы модифицированного высвобождения также позволяют применять комбинации активных компонентов, в которых продолжительность действия одного активного компонента может отличаться от продолжительности действия другого активного компонента.
При использовании в настоящем документе термин «фармакодинамика» или «ФД» представляет собой исследование взаимосвязи между концентрацией лекарственного средства в месте действия и полученным в результате эффектом.
При использовании в настоящем документе термин «фармакокинетика» или «ФК» означает исследование динамики всасывания, распределения, метаболизма и выведения лекарственного средства.
При использовании в настоящем документе термин «фенилэфрин» означает дигидробензолметанол, 3-гидрокси-α-[(метиламино)метил] и включает, без ограничений, его фармацевтически приемлемые соли, сложные эфиры, изомеры или производные.
При использовании в настоящем документе термин «скорость высвобождения лекарственного средства» относится к количеству лекарственного средства, высвобождаемого из дозированной формы за единицу времени, например, миллиграммы лекарственного средства, высвобождаемые за час (мг/ч). Скорости высвобождения лекарственных средств рассчитывают в условиях тестирования растворения дозированной формы in vitro, известных в данной области техники. При использовании в настоящем документе скорость высвобождения, полученная в определенное время «после введения», относится к скорости высвобождения лекарственного средства in vitro, полученной в определенное время после начала соответствующего теста на растворение, например, тестов, описанных в издании 24 Фармакопеи США (United States Pharmacopeia 24, United States Pharmacopeia Convention, Inc., Rockville, MD).
При использовании в настоящем документе термин «полупроницаемый» означает, что вода может проходить через мембрану, и другие молекулы, включая соли и активные компоненты, описанные в настоящем документе, могут медленно диффундировать через такую мембрану, когда мембрана находится в контакте с соответствующей растворяющей средой, например, желудочно-кишечными средами или растворяющей средой in vitro.
«Полутвердые дозированные формы» означают дозированные формы, которые обладают высокой вязкостью и некоторыми общими свойствами с жидкостями, включая, без ограничений: (1) способность по существу соответствовать чему-либо, что оказывает давление на них и вызывает деформацию их формы; и (2) отсутствие способности течь так же свободно, как жидкость. Полутвердые дозированные формы также обладают некоторыми общими свойствами с твердыми веществами, включая, без ограничений, более высокую плотность и определенную форму. Полутвердые дозированные формы могут, без ограничений, включать гели, жевательные дозированные формы, жевательные формы на основе пектина, кондитерские жевательные формы, формы типа формующегося желатина.
«Твердые дозированные формы» означают дозированные формы, которые являются по существу твердыми при комнатной температуре и имеют плотность по меньшей мере приблизительно 0,5 г/см3. Твердые дозированные формы могут, без ограничений, включать агломерированные таблетки, лекарственные средства типа капсул, капсулы, заполненные порошком или гранулами, пакеты-саше, заполненные порошком или гранулами, прессованные таблетки, таблетки с покрытием, жевательные дозированные формы и быстрорастворимые дозированные формы.
При использовании в настоящем документе термин «по существу покрытый» применительно к частицам означает, что менее чем приблизительно 20%, например, менее чем приблизительно 15% или менее чем приблизительно 1,0% площади поверхности частицы открыто, то есть не имеет желаемого покрытия. Используемый в настоящем документе термин «по существу покрывает» или «по существу непрерывный» при использовании для описания покрытия означает, что покрытие является по существу непрерывным и по существу покрывает всю поверхность сердцевины или нижележащего слоя таким образом, что открытым остается лишь небольшое количество активного компонента или нижележащего слоя или не остается совсем. Покрытия, наносимые на частицы, могут наслаиваться, при этом каждый слой готовят в системе водных (на основе воды) или органических растворителей и последовательно добавляют до тех пор, пока не будет достигнут желаемый уровень покрытия.
При использовании в настоящем документе термин «терапевтический эффект» означает любой эффект или действие активного компонента, предназначенные для диагностики, лечения, излечения, облегчения или профилактики заболевания, либо воздействие на структуру или любую функцию организма.
Конкретные варианты осуществления настоящего изобретения проиллюстрированы представленными ниже примерами. Настоящее изобретение не ограничено конкретными ограничениями, установленными в данных примерах.
ПРИМЕРЫ
Частицы фенилэфрина продленного высвобождения были разработаны с целью включения в жидкие и твердые дозированные формы. Частицы фенилэфрина продленного высвобождения могут применяться для согласования продолжительности действия с другими активными веществами (в частности, обезболивающими), которые могут обеспечивать более длительное действие, чем фенилэфрин. Такие активные вещества включают, без ограничений, ацетаминофен, ибупрофен и напроксен, а также их соли и производные.
Пример 1. Получение композиции, содержащей частицы фенилэфрина продленного высвобождения с покрытием
Получили композицию, содержащую частицы фенилэфрина, покрытые полимерным покрытием. Композиция, обеспечивающая высвобождение фенилэфрина в течение продленного периода времени, была стабильна при 25°C/ОВ 60% в течение 24 месяцев и при 40°C/ОВ 75% в течение 3 месяцев. Многие гранулированные композиции фенилэфрина нестабильны во времени и подвергаются значительному разложению.
Серию 3,203 кг частиц фенилэфрина с покрытием получили по формуле, указанной в таблице 1. Количественная формула и состав партии, соответственно, представлены в таблице 1.
Наслоение частиц
1. Очищенную воду по Фарм. США добавляли в контейнер подходящего размера из нержавеющей стали.
2. Аккуратно встряхивая, добавляли дисперсию сополимера этилакрилата по NF и метилметакрилата по NF (Эудрагит® NE 30D).
3. Помешивая и встряхивая, добавляли фенилэфрин HCl по Фарм. США и перемешивали.
4. Смесь, полученную на стадии 3, использовали для покрытия (наслоения) прежелатинизированного модифицированного крахмала по NF.
Высушивание и просеивание
5. Наслоенный гидрохлорид фенилэфрина/прежелатинизированный модифицированный крахмал, полученный на стадии 4, высушивали и просеивали через сито № 20.
Нанесение раствора этилцеллюлозного покрытия на наслоенные частицы
6. Аккуратно встряхивая, в контейнер подходящего размера в указанном порядке добавляли следующее: изопропиловый спирт по Фарм. США, затем ацетон по NF, затем ацетилтрибутилцитрат по NF.
7. Встряхивая, добавляли этилцеллюлозу по NF (Этоцел® Стандарт Премиум 10) и перемешивали до образования прозрачного раствора.
8. К раствору при встряхивании добавляли стеарат магния.
9. Просеянные наслоенные частицы фенилэфрина/прежелатинизированного модифицированного крахмала, полученные на стадии 5, покрывали раствором, полученным на стадии 8, используя подходящее устройство для нанесения покрытия в псевдоожиженном слое, оборудованное перегородкой Вюрстера.
Отверждение
10. Частицы, полученные на стадии 9, выдерживали в печи.
Нанесение покрытия из Эудрагита® NE30D и Аквакоута ECD® на частицы, покрытые Этоцелем®
11. В контейнер подходящего размера добавляли Эудрагит® NE30D, затем очищенную воду по Фарм. США и водную дисперсию этилцеллюлозы по NF (Аквакоут ECD®) и перемешивали, аккуратно встряхивая.
12. На покрытые Этоцелем® наслоенные частицы, полученные на стадии 10, наносили раствор покрытия, используя подходящее устройство для нанесения покрытия в псевдоожиженном слое, оборудованное перегородкой Вюрстера.
13. Покрытые частицы, полученные на стадии 12, смешивали с коллоидным диоксидом кремния по NF.
Отверждение
14. Частицы, полученные на стадии 13, выдерживали в печи.
Анализ растворения
Частицы фенилэфрина с покрытием, полученные на стадии 14, анализировали на растворение в период от 0 до 14 часов, используя аппарат, описанный в Фармакопее США <Общая глава <711> Растворение>, аппарат II, вращающиеся лопасти с УФ-детектированием при 274 нм. Растворяющая среда в течение первого часа представляла собой 750 мл 0,1 н. HCl, а затем со второго по четырнадцатый часы 1000 мл 0,05 M натрий-фосфатного буфера, pH 6,8. Температура составляла 37°C, и скорость вращения составляла 50 об/мин. Исследование растворения показало, что процент высвободившегося фенилэфрина по сравнению со стандартом, полученным при 100%-ном содержании фенилэфрина в композиции, был меньшим или равным 50% за 1 час, большим или равным 30% за 3 часа и большим или равным 50% за 8 часов. Используемый способ описан ниже, и результаты представлены в таблице 2 ниже.
Аппарат для осуществления способа растворения по Фарм. США (2 лопасти, 50 об/мин)
1. Убеждаются в том, что температура растворяющей среды достигла целевого значения (37°C).
2. Взвешивают образцы, эквивалентные 45 мг гидрохлорида фенилэфрина. Добавляют образцы (на поверхность раствора среды) в каждый сосуд, содержащий 750 мл 0,1 н. соляной кислоты, и начинают тест на растворение при скорости вращения лопастей 50 об/мин. Через 1 час обработки в 0,1 н. соляной кислоте выполняют измерение, соответствующее временной отметке 1 час. Сразу переходят к стадии растворения в буфере путем добавления 250 мл 0,20 М трехосновного фосфата натрия. pH буферной среды составляет 6,8±0,05.
3. Измеряют УФ поглощение гидрохлорида фенилэфрина, высвободившегося в среду, используя оптоволоконную систему LEAP со встроенными зондами для измерения УФ при 274 нм.
4. Количество растворенного гидрохлорида фенилэфрина можно определить, используя УФ поглощение раствора испытуемого образца по сравнению с тем же показателем раствора стандарта при длине волны 274 нм. Количество растворенного гидрохлорида фенилэфрина также можно определить, используя способ анализа, представленный ниже.
Способ анализа
Приготовление образца
1. Точно взвешивают приблизительно 1600 мг частиц гидрохлорида фенилэфрина и переносят их в мерную колбу на 200 мл. (Рекомендуется добавлять 1 мл 1%-ного водного раствора уксусной кислоты для смачивания частиц во избежание образования твердых комков).
2. Добавляют 70 мл 1%-ного раствора уксусной кислоты/ацетонитрила; встряхивают колбу на шейкере с платформой при низкой скорости в течение 1 часа. Примечание: периодически поворачивают колбу для удаления частиц, собирающихся над уровнем растворителя.
3. Добавляют приблизительно 50 мл 1%-ного раствора уксусной кислоты/водного раствора в колбу и непрерывно встряхивают ее при низкой скорости в течение 1 часа.
4. Доводят объем 1%-ным водным раствором уксусной кислоты и тщательно перемешивают.
5. Фильтруют аликвоту, используя фильтр Millipore Millex PVDF с размером пор 0,45 мкм. Перед сбором фильтрата для дальнейшего разведения отбрасывают первые 1–2 мл фильтрата.
6. Пипеткой переносят 6 мл фильтрата в мерную колбу на 50 мл, разводят до объема 1%-ным водным раствором уксусной кислоты и тщательно перемешивают.
Анализ фенилэфрина
Вводят стандарты (0,05 мг/мл гидрохлорида фенилэфрина в 1%-ном водном растворе уксусной кислоты) и образцы в подходящую систему высокоэффективной жидкостной хроматографии (ВЭЖХ) в условиях, аналогичных предложенным ниже. Параметры могут быть изменены для оптимизации хроматографии. Определяют содержание фенилэфрина HCl с помощью площадей пика растворов испытуемых образцов в условиях проведения теста по сравнению с площадями пика стандартного раствора.
Условия хроматографии ВЭЖХ
Способ определения продуктов разложения
Приготовление образца
2. Точно взвешивают приблизительно 1600 мг частиц гидрохлорида фенилэфрина и переносят их в мерную колбу на 200 мл. (Рекомендуется добавлять 1 мл 1%-ного водного раствора уксусной кислоты для смачивания частиц во избежание образования твердых комков).
2. Добавляют 70 мл 1%-ного раствора уксусной кислоты/ацетонитрила; встряхивают колбу на шейкере с платформой при низкой скорости в течение 1 часа. Примечание: периодически поворачивают колбу для удаления частиц, собирающихся над уровнем растворителя.
3. Добавляют приблизительно 50 мл 1%-ной уксусной кислоты/водного раствора в колбу и непрерывно встряхивают ее при низкой скорости в течение 1 часа.
4. Доводят объем 1%-ным водным раствором уксусной кислоты и тщательно перемешивают.
5. Фильтруют аликвоту, используя фильтр Millipore Millex PVDF с размером пор 0,45 мкм. Перед сбором фильтрата для дальнейшего разведения отбрасывают первые 1–2 мл фильтрата.
6. Пипеткой переносят 6 мл фильтрата в мерную колбу на 50 мл, разводят до объема 1%-ным водным раствором уксусной кислоты и тщательно перемешивают.
Анализ фенилэфрина
Вводят стандарты (0,00025 мг/мл гидрохлорида фенилэфрина в 1%-ном водном растворе уксусной кислоты) и образцы в подходящую систему высокоэффективной жидкостной хроматографии (ВЭЖХ) в условиях, аналогичных предложенным ниже. В целях оптимизации хроматографии параметры могут быть изменены. Определяют количество продуктов разложения фенилэфрина HCl с помощью площадей пиков растворов испытуемых образцов по сравнению с площадями пиков раствора стандарта.
Условия хроматографии ВЭЖХ
Пример 2. Получение частиц резината фенилэфрина продленного высвобождения с покрытием
Получали частицы, содержащие фенилэфрин и катионообменную смолу, и дополнительно покрывали их полупроницаемой мембраной. Соотношение количеств компонентов покрытия, которое в некоторой степени может варьировать, может составлять, например, ацетат целлюлозы:гидроксипропилцеллюлоза 2:1, 3:1, 4:1 или 5:1. Уровень покрытия, который в некоторой степени может варьировать, может составлять, например, 50%, 45%, 40%, 35%, 30%, 25% или 20% от массы частицы с покрытием. Большинство частиц в исходной катионообменной смоле имело размер частицы от приблизительно 74 мкм до приблизительно 177 мкм (микрон).
Частицы резината фенилэфрина, которые обеспечивают высвобождение фенилэфрина в течение продленного периода времени, были стабильны при 25°C/ОВ 60% в течение 24 месяцев и при 40°C/ОВ 75% в течение 3 месяцев. Многие гранулированные композиции фенилэфрина нестабильны во времени и подвергаются значительному разложению.
Серию 3,846 кг частиц резината фенилэфрина с покрытием получили по формуле, указанной в таблице 3. Количественная формула и состав партии представлены в таблице 3 и таблице 4 соответственно.
Частицы резината фенилэфрина с покрытием получали, используя следующие стадии обработки.
Просеивание
1. Полистиролсульфонат натрия по Фарм. США, имеющий желаемый размер частиц, пропускали через сито 170 меш, и затем собирали фракцию, оставшуюся на сите.
Промывание
2. Полистиролсульфонат натрия по Фарм. США, полученный на стадии 1, диспергировали в очищенной воде и перемешивали.
3. При перемешивании часть суспензии, полученной на стадии 2, фильтровали и промывали очищенной водой по Фарм. США. Фильтрование продолжали до удаления большей части воды.
4. Смолу переносили в контейнер.
5. Стадии 3 и 4 повторяли до полного удаления суспензии.
Ввод лекарственного средства
6. Очищенную воду по Фарм. США добавляли в контейнер подходящего размера из нержавеющей стали.
7. Помешивая, гидрохлорид фенилэфрина добавляли в контейнер и перемешивали до полного растворения.
8. Непрерывно помешивая, добавляли промытую смолу, полученную на стадии 5, и смешивали с суспензией.
9. При перемешивании часть суспензии, полученной на стадии 8, отбирали и промывали очищенной водой по Фарм. США. Фильтрование продолжали до удаления большей части воды.
10. Промытый и профильтрованный резинат фенилэфрина, полученный на стадии 9, переносили в контейнер.
11. Стадии 9 и 10 повторяли до тех пор, пока не была профильтрована вся суспензия.
Высушивание
12. Резинат фенилэфрина высушивали.
Получение раствора покрытия
13. Очищенную воду по Фарм. США и ацетон по NF добавляли в контейнер из нержавеющей стали подходящего размера.
14. Гидроксипропилцеллюлозу по NF медленно добавляли в контейнер и перемешивали до растворения. Ацетат целлюлозы по NF медленно добавляли и перемешивали до растворения.
15. Ацетон по NF добавляли до достижения желаемой массы раствора.
Нанесение покрытия
16. На резинат фенилэфрина, полученный на стадии 12, наносили раствор покрытия, полученный на стадии 15, в подходящем оборудовании для нанесения покрытия в псевдоожиженном слое, оборудованном колонной Вюрстера.
17. Резинат фенилэфрина с покрытием выгружали в контейнер.
Высушивание
18. Резинат фенилэфрина с покрытием высушивали.
Просеивание
19. Высушенный резинат фенилэфрина с покрытием просеивали через стандартное сито США № 40 меш и собирали фракцию, прошедшую через сито.
Анализ растворения
Частицы резината фенилэфрина с покрытием, полученные на стадии 19, анализировали на растворение в период от 0 до 14 часов, используя аппарат, описанный в Фармакопее США <Общая глава <711> Растворение>, аппарат II, вращающиеся лопасти с УФ-детектированием при 274 нм. Растворяющая среда в течение первого часа представляла собой 750 мл 0,1 н. HCl, а со второго по четырнадцатый часы 1000 мл 0,05 M натрий-фосфатного буфера, pH 6,8. Температура составляла 37°C, и скорость вращения составляла 50 об/мин. Исследование растворения показало, что процент высвободившегося фенилэфрина по сравнению со стандартом, полученным при 100%-ном содержании фенилэфрина в композиции, был меньшим или равным 50% за 1 час, большим или равным 30% за 3 часа и большим или равным 50% за 8 часов. Используемый способ описан ниже, и результаты представлены в таблице 5 ниже.
Аппарат 2 для осуществления способа растворения по Фарм. США (лопасти), 50 об/мин
1. Убеждаются в том, что температура растворяющей среды достигла целевого значения.
2. Добавляют образец (на поверхность раствора среды) в каждый сосуд, содержащий 750 мл 0,1 н. соляную кислоту и начинают тест на растворение при скорости вращения лопастей 50 об/мин. Через 1 час обработки в 0,1 н. соляной кислоте отбирают образец, соответствующий временной отметке 1 час, и сразу переходят к стадии растворения в буфере путем добавления 250 мл 0,20 М трехосновного фосфата натрия. pH среды должен составлять 6,8±0,05.
3. Отбирают 10 мл растворов образца для исследования растворения из каждого сосуда через 1 час, 3 часа, 6 часов (необязательно) и 8 часов. Фильтруют растворы образца через полнопоточные фильтры Varian (10 мкм).
4. Количество растворенного фенилэфрина можно определить при помощи УФ поглощения по сравнению с поглощением раствора стандарта при длине волны 274 нм. Количество растворенного фенилэфрина также можно определить способом количественного анализа фенилэфрина.
5. Корректируют количество, растворенное через 3, 6 и 8 часов, путем добавления количества, отобранного в более ранние моменты времени. Используют программу DISSL (или эквивалентную) или вручную корректируют по промежуточным образцам.
Пример 3. Анализ распределения размера частиц
Несколько партий смолы и частиц на основе смолы анализировали на распределение размера частиц. Образцы включали: (1) смолу Амберлит™ IRP69, имеющуюся в продаже от компании The DOW Chemical Company; (2) смолу, не содержащую лекарственное средство, имеющую выбранные размеры частиц (полученную способом A или способом B соответственно); и (3) частицы резината, содержащие лекарственное средство (т. е. содержащие фенилэфрин). При анализе распределения размера частиц использовали приблизительно 75 грамм на образец в анализаторе FMC Syntron Sieve (FMC Technologies, г. Хьюстон, штат Техас, США) в условиях напряжения 90 вольт в течение 11 минут. Сита обрабатывали способом легкого припудривания стеаратом магния, чтобы предотвратить слипание во время работы. Полученные результаты показаны в таблицах 6 и 7.
Распределение размера частиц можно анализировать в меньшем масштабе, используя, например, ультразвуковой просеиватель ATM L3P Sonic Sifter (Advantech Manufacturing, г. Нью Берлин, штат Висконсин, США), работа которого основана на использовании ультразвуковых импульсов в сочетании с механическим перемешиванием для обеспечения эффективного разделения частиц.
Изучали влияние соотношения лекарственного средства и смолы на эффективность ввода лекарственного средства. Результаты представлены ниже в таблицах 8 и 9.
Способ количественного анализа фенилэфрина - измерения для таблиц 8 и 9
Приготовление образца
1. Точно взвешивают подходящее количество образца резината фенилэфрина с покрытием (содержащее эквивалент 25 мг гидрохлорида фенилэфрина) и переносят взвешенный образец в мерную колбу объемом 500 мл.
2. Добавляют 400 мл разбавителя (1 н. HCl); встряхивают колбу на шейкере с платформой при низкой скорости в течение не менее 2 часов.
3. Чтобы исключить собирание частиц над уровнем растворителя, периодически смывают частицы в раствор разбавителем.
4. Доводят до объема разбавителем и тщательно перемешивают.
5. Фильтруют аликвоту, используя шприцевой фильтр Millipore Millex PVDF с размером пор 0,45 мкм или эквивалент. Перед сбором остатка во флакон для ВЭЖХ для анализа отбрасывают первые приблизительно 5 мл фильтрата.
Анализ фенилэфрина
Вводят стандарты (0,05 мг/мл гидрохлорида фенилэфрина в 1 н. HCl) и образцы в подходящую систему ВЭЖХ в условиях, аналогичных предложенным ниже. Параметры могут быть изменены для оптимизации хроматографии. Аналитические результаты действительны, если соблюдены критерии пригодности системы.
Условия хроматографии ВЭЖХ
Пример 4. Анализ растворения материала для исследования ФК
Частицы резината фенилэфрина с покрытием, используемые в первом исследовании ФК, втором исследовании ФК и в исследовании ФД примера 5, анализировали на растворение в период от нуля до 8 часов, используя способ, описанный в примере 2. Результаты показаны ниже в таблице 10A.
Частицы резината фенилэфрина с покрытием, используемые в первом исследовании ФК, втором исследовании ФК и в исследования ФД примера 5, также анализировали на растворение в период от нуля до 8 часов, используя способ, описанный ниже. Результаты показаны ниже в таблице 10B.
Аппарат 2 для осуществления способа растворения по Фарм. США (лопасти), 75 об/мин
1. Убеждаются в том, что температура растворяющей среды достигла целевого значения.
2. Добавляют образцы (непосредственно в раствор среды, используя подходящую пробирку) в каждый сосуд, содержащий 750 мл 0,1 н. соляной кислоты, и начинают тестирование растворения при скорости вращения лопастей 75 об/мин. Через 1 час обработки в 0,1 н. соляной кислоте отбирают образец, соответствующий временной отметке 1 час, и сразу переходят к стадии растворения в буфере путем добавления 250 мл 0,20 М трехосновного фосфата натрия. pH среды должен составлять 6,8±0,05.
3. Отбирают 10 мл растворов образца для исследования растворения из каждого сосуда через 1 час, 3 часа, 6 часов (необязательно) и 8 часов. Фильтруют растворы образца через полнопоточные фильтры Varian (10 мкм).
4. Определяют количество растворенного фенилэфрина при помощи УФ поглощения по сравнению с поглощением раствора стандарта при длине волны 274 нм.
Количество растворенного фенилэфрина также можно определить способом количественного анализа фенилэфрина.
5. Корректируют количество, растворенное через 3, 6 и 8 часов, путем добавления количества, отобранного в более ранние моменты времени. Используют программу DISSL (или эквивалентную) или вручную корректируют по промежуточным образцам.
Анализ стабильности
Частицы резината фенилэфрина с покрытием, используемые в первом исследовании ФК и во втором исследовании ФК примера 5, анализировали на стабильность после хранения в течение 1 месяца при 25°C и относительной влажности 60% и в течение 1 месяца при 40°C и относительной влажности 75%. Уровни 3-гидроксибензальдегида во всех образцах были меньше или равны 0,5%; уровни 4,6 изомера фенилэфрина (гидрохлорида N-метил-4,6-дигидрокси-1,2,3,4-тетрагидроксиизохинолона) и 4,8 изомера фенилэфрина (гидрохлорида N-метил-4,8-дигидрокси-1,2,3,4-тетрагидроксиизохинолона) были меньше или равны 2,0%. Через 1 месяц хранения в каждой среде общее количественное содержание продуктов разложения относительно фенилэфрина было менее или равно 2,0%.
Способ определения продуктов разложения
Приготовление образца для способа определения продуктов разложения
1. Точно взвешивают подходящее количество образца резината фенилэфрина с покрытием (содержащее эквивалент 25 мг гидрохлорида фенилэфрина) и переносят взвешенный образец в мерную колбу объемом 500 мл.
2. Добавляют 400 мл разбавителя (1 н. HCl); встряхивают колбу на шейкере с платформой при низкой скорости в течение не менее 2 часов.
3. Чтобы исключить собирание частиц над уровнем растворителя, периодически смывают частицы в раствор разбавителем.
4. Доводят до объема разбавителем и тщательно перемешивают.
5. Фильтруют аликвоту, используя шприцевой фильтр Millipore Millex PVDF с размером пор 0,45 мкм или эквивалент. Перед сбором остатка во флакон для ВЭЖХ для анализа отбрасывают первые приблизительно 5 мл фильтрата.
Способ анализа фенилэфрина для определения продуктов разложения
Вводят стандарты (0,00025 мг/мл гидрохлорида фенилэфрина в 1 н. HCl) и образцы в подходящую систему ВЭЖХ в условиях, аналогичных предложенным ниже. Параметры могут быть изменены для оптимизации хроматографии. Аналитические результаты действительны, если соблюдены критерии пригодности системы.
Условия хроматографии ВЭЖХ
Пример 5. Клинические исследования
Провели два фармакокинетических (ФК) исследования и фармакодинамическое (ФД) исследование.
A. Первое исследование ФК
Поисковое исследование провели на шестнадцати субъектах с целью определения фармакокинетического профиля, биодоступности и метаболизма частиц фенилэфрина продленного высвобождения с покрытием, полученных в примере 1, и частиц резината фенилэфрина продленного высвобождения с покрытием, полученных в примере 2. Субъекты должны были получать четыре препарата после ночного голодания. Период вымывания между четырьмя периодами составлял семь дней. В обоих случаях частицы с покрытием, эквивалентные дозе 20 мг гидрохлорида фенилэфрина, вводили здоровым субъектам в яблочном пюре. Кроме того, оценивали комбинацию частиц резината фенилэфрина продленного высвобождения, полученных в примере 2, и имеющегося в продаже жидкого препарата быстрого высвобождения. При комбинированной терапии частицы резината фенилэфрина продленного высвобождения с покрытием, эквивалентные 15 мг гидрохлорида фенилэфрина, вводили в яблочном пюре, и 10 мл жидкости, эквивалентные 5 мг гидрохлорида фенилэфрина, вводили пероральным шприцем.
Частицы фенилэфрина продленного высвобождения с покрытием, полученные в примере 1, и частицы резината фенилэфрина продленного высвобождения с покрытием, полученные в примере 2, сравнивали с назальной противозастойной жидкостью Судафед ФЭ® для детей без снотворного эффекта компании McNeil-PPC, Inc. (гидрохлорид фенилэфрина 2,5 мг/5 мл). В таблице 11 суммированы препараты, применявшиеся в первом исследовании ФК.
Частицы резината фенилэфрина ПВ с покрытием и частицы гидрохлорида фенилэфрина ПВ вводили перорально после перемешивания отмеренного количества в кружке 113 г (4 унции) яблочного пюре непосредственно перед введением. Указанные однократные дозы проглатывали без пережевывания с последующим приемом 240 мл воды. Жидкий гидрохлорид фенилэфрина вводили перорально, используя пероральный шприц. В целях стандартизации условий дозирования сравнительного препарата после приема первой из двух пероральных доз жидкости по 10 мг вводили кружку 113 г (4 унции) яблочного пюре и 240 мл воды.
Серийные образцы крови отбирали в пробирки K3-ЭДТА в определенные моменты времени в течение 8 или 16 часов после введения дозы.
B. Второе исследование ФК
Второе поисковое исследование провели: (i) для определения того, могут ли 30 мг фенилэфрина обеспечить достижение подобных максимальных концентраций лекарственного средства по сравнению с двумя дозами фенилэфрина быстрого высвобождения по 10 мг, вводимыми с интервалом 4 часа, и (ii) для оценки ФК профиля ПВ и биодоступности 20 мг фенилэфрина и 1300 мг ацетаминофена.
Второе поисковое исследование провели на двадцати субъектах с целью определения фармакокинетических профилей, биодоступности и метаболизма: (1) комбинации (а) частиц резината фенилэфрина продленного высвобождения с покрытием, полученных в примере 2 и эквивалентных 15 мг гидрохлорида фенилэфрина, (b) 10 мл жидкого фенилэфрина, эквивалентного 5 мг гидрохлорида фенилэфрина, и (с) 1300 мг ацетаминофена продленного высвобождения; (2) комбинации (а) частиц резината фенилэфрина продленного высвобождения с покрытием, полученных в примере 2 и эквивалентных 22,55 мг гидрохлорида фенилэфрина, и (b) жидкого фенилэфрина, эквивалентного 7,5 мг гидрохлорида фенилэфрина; (3) комбинации (а) жидкого фенилэфрина, эквивалентного 20 мг гидрохлорида фенилэфрина, и (b) 1300 мг ацетаминофена продленного высвобождения; и (4) жидкого фенилэфрина, эквивалентного 20 мг гидрохлорида фенилэфрина. В таблице 12 суммированы препараты, применявшиеся во втором исследовании ФК.
Серийные образцы крови отбирали в пробирки K3-ЭДТА в определенные моменты времени в течение 12 или 20 часов.
Результаты
Результаты исследований ФК представлены на Фиг. 1–11 и в таблице 13 ниже.
Цифры округляли.
В итоге результаты показали следующее.
Смесь ПВ-НВ, содержащая 20 мг фенилэфрина, имела Cmax, составлявшую 50% по сравнению с дозой 10 мг НВ, и AUCinf, которая была на 15% больше, чем для двух доз по 10 мг НВ (20 мг).
Смесь ПВ-НВ, содержащая 30 мг фенилэфрина, имела Cmax, составлявшую 85% по сравнению с дозой 10 мг НВ, и AUCinf, которая была на 61% больше, чем для двух доз по 10 мг НВ (20 мг).
Смесь ПВ-НВ, содержащая 20 мг фенилэфрина и 1300 мг ацетаминофена, имела Cmax, составлявшую 80% от дозы 10 мг НВ, и AUCinf, которая была на 22% больше, чем для двух доз по 10 мг НВ (20 мг).
Эти результаты демонстрируют, что композиция настоящего изобретения обеспечивает эффективность в течение продленного периода времени.
Эти результаты также демонстрируют, что композиция настоящего изобретения способна соответствовать продолжительности действия ацетаминофена продленного высвобождения.
Эти результаты также демонстрируют, что воздействие фенилэфрина увеличивается, а профиль ФК фенилэфрина улучшается по сравнению с дозой 10 мг фенилэфрина быстрого высвобождения при комбинировании фенилэфрина с ацетаминофеном. Это может быть обусловлено конкуренцией за метаболизм в кишечной стенке, приводящей к увеличенному всасыванию фенилэфрина и отсутствию влияния на ацетаминофен, а также композицией продленного высвобождения, обеспечивающей увеличенное всасывание фенилэфрина за счет избегания метаболизма в кишечной стенке в нижних отделах ЖКТ.
C. Исследование фармакодинамики
Рандомизированное двойное слепое плацебо-контролируемое исследование провели с целью определения эффективности композиций фенилэфрина и фенилэфрина-ацетаминофена продленного высвобождения у субъектов с заложенностью и болевыми симптомами вследствие инфекций верхних дыхательных путей. Дозу 30 мг ПВ, дозу 45 мг ПВ и дозу 30 мг ПВ, вводимую совместно с 1300 мг ацетаминофена, оценивали и сравнивали с плацебо. В каждом примере использовали частицы резината фенилэфрина ПВ с покрытием настоящего изобретения. Каждая из доз (30 мг ПВ, 45 мг ПВ и 30 мг ПВ, вводимая совместно с 1300 мг ацетаминофена) была эффективна по сравнению с плацебо по баллу оценки тяжести: (1) отечности/заложенности носа; (2) давления/болезненности синуса; и (3) застоя крови в голове в период от 0 до 12 часов в день 1.
Приведенные выше примеры не предназначены для ограничения объема настоящего изобретения, который может быть определен в формуле изобретения. В частности, специалистам в данной области известны различные эквиваленты и заменители, применимые в вышеизложенном описании, и они рассматриваются как входящие в объем изобретения.
Литература
Blackledge HM, O’Farrell J, Minton NA, et al. The effect of therapeutic doses of paracetamol on sulphur metabolism in man. Hum Exp Toxicol 1991 May; 10(3): 159–65.
Court MH, Duan SX, Von Moltke LL, et al. Interindividual variability in acetaminophen glucuronidation by human liver microsomes: Identification of relevant acetaminophen UDP-glucuronosyltransferase isoforms. J Pharmacol Exp Ther 2001; 299(3):998–1006.
Empey DW and Medder KT. Nasal Decongestants. Drugs 1981; 21:438–443.
Hengstmann JH, Goronzy J. Pharmacokinetics of 3H-phenylephrine in man. Eur J Clin
Pharmacol 1982; 21:335–341.
Hoffman BB. Chapter 10: Catecholamines, Sympathomimetic Drugs, and Adrenergic
Receptor Antagonists. В: Goodman & Gilman’s The Pharmacologic Basis of Therapeutics
– 10th Ed. Hardman JG and Limbird LE, eds. McGraw-Hill, Medical Publishing Division, USA, 2001.
Ibrahim KE, Midgley JM, Crowley IR, and Willaims CM. The mammalian metabolism of R-(-)-m-synephrine. J Pharm Pharmacol. 1983; 35:144–147.
Johnson DA, Hricik JG. The pharmacology of a-adrenergic decongestants. Pharmacother
1993; 13:110S–115S.
Koch-Weser J. Medical Intelligence: Drug Therapy. N Engl J Med 1976 Dec 2; 295(23):1297–1300.
Manyike PT, Kharasch ED, Kalhorn TF, et al. Contribution of CYP2E1 and CYP3A to acetaminophen reactive metabolite formation. Clin Pharmacol Ther 2000 Mar; 67(3):275–282.
Miners JO, Atwood J, Birkett DJ. Influence of sex and oral contraceptive steroids on paracetamol metabolism. Br J Clin Pharmacol 1983; 16:503–509.
Miners JO, Osborne NJ, Tonkin AL, et al. Perturbation of paracetamol urinary metabolic ratios by urine flow rate. Br J Clin Pharmacol 1992; 34:359–362.
Mitchell JR, Thorgeirsson SS, Potter WZ, et al. Acetaminophen-induced injury: Protective role of glutathione in man and rationale for therapy. Clin Pharmacol Ther 1974; 16:676–684.
Slattery JT, McRorie TI, Reynolds R, et al. Lack of effect of cimetidine on acetaminophen disposition in humans. Clin Pharmacol Ther 1989 Nov; 46(5):591–597.
Suzuki O. Matsumoto T. Oya M, Katsumata Y. Oxidation of synephrine by type A and type B monoamine oxidase. Experientia 1979; 35:1283–1284.
Реферат
Настоящая группа изобретений относится к медицине, а именно к терапии и оториноларингологии, и касается лечения заложенности носа. Для этого вводят фармацевтическую композицию, содержащую комплекс лекарственное средство-смола, включающий фенилэфрин и полистиролсульфонат натрия, и однослойное покрытие. При этом указанный полистиролсульфонат натрия содержит частицы с размером от 74 до 177 мкм до комбинирования с фенилэфрином, где по меньшей мере 90% указанных частиц имеет размер частиц от 74 до 177 мкм. Однослойное покрытие на указанном комплексе составляет от 30 до 45% масс. по сравнению с массой фармацевтической композиции, и включает от 65 до 85% ацетата целлюлозы. Такой состав композиции обеспечивает облегчение заложенности носа в течение не менее 8 часов за счет длительного поддержания высокой концентрации фенилэфрина в плазме. 2 н. и 5 з.п. ф-лы, 11ил., 13 табл., 5 пр.
Формула
Документы, цитированные в отчёте о поиске
Композиции с модифицированным высвобождением, содержащие комплексы лекарственное вещество - ионообменная смола
Комментарии