Замещенные 2,6-диоксопиперидины, фармацевтическая композиция на их основе и способы снижения уровней tnf-α - RU2177944C2
Код документа: RU2177944C2
Чертежи
Описание
Настоящее изобретение относится к замещенным 2,6-диоксопиперидинам, к фармацевтическим композициям на их основе и к способам снижения уровней фактора некроза опухолей α у млекопитающего посредством их введения.
Предпосылки изобретения
Фактор некроза опухолей α, или TNF-α,является цитокином,
который выделяется главным образом мононуклеарными фагоцитами в ответ на ряд иммуностимуляторов. При введении животным или людям он вызывает воспаление, лихорадку, сердечно-сосудистые эффекты,
кровотечение, коагуляцию и острые фазовые реакции, схожие с наблюдаемыми во время острых инфекций и шоковых состояний. Таким образом, избыточное или нерегулируемое продуцирование TNF-α
вовлечено в ряд болезненных состояний. Они включают в себя наличие в крови эндотоксинов и/или синдром токсического шока (Tracey еt al. , Nature 330, 662-664 (1987) и Hinshaw et al. , Circ. Shock 3-0,
279-292 (1990)); кахексию (Dezube et al. , Lancet, 335 (8690), 662 (1990)) и респираторный дистресс-синдром взрослых (РДСВ), когда концентрация TNF-α свыше 12000 пг/мл была обнаружена в
полученном путем аспирации легочном материале пациентов с РДСВ (Millar et al. , Lancet 2 (8665), 712-714 (1989)). Системная инфузия рекомбинантного TNF-α также приводила к изменениям, обычно
наблюдаемым при РДСВ (Ferrai-Baliviera et al. , Arch. Surg. 124 (12), 1400-1405 (1989)).
TNF-α, по-видимому, вовлечен в заболевания, связанные с костной резорбцией, включая артрит. Будучи активированными, лейкоциты вызывают костную резорбцию, активность, вклад в которую TNF-α подтверждается экспериментальными данными (Bertolini et al. , Nature 319, 516-518 (1986) и Johnson et al. , Endocrinology 124 (3), 1424-1427 (1989)). Было также показано, что TNF-α стимулирует костную резорбцию и ингибирует костеобразование in vitro и in vivo посредством стимуляции образования и активации остеокластов в сочетании с ингибированием функции остеобластов. Несмотря на то, что TNF-α может быть вовлечен во многие заболевания, связанные с костной резорбцией, включая артрит, наиболее очевидной связью с заболеванием является ассоциация между продуцированием TNF-α опухолью или тканями хозяина и злокачественной гиперкальциемией (Calci Tissue Int. (US) 46 (Suppl. ), S3-10 (1990)). В реакции "трансплантат против хозяина" повышенные уровни сывороточного TNF-α ассоциировались с основным осложнением после срочных аллогенных трансплантаций костного мозга (Holler et al. , Blood, 75 (4), 1011-1016 (1990)).
Церебральная малярия является летальным сверхострым неврологическим синдромом, ассоциированным с высокими уровнями TNF-α в крови, и наиболее серьезным осложнением, встречающимся у пациентов с малярией. Уровни сывороточного TNF-α непосредственно коррелировали с тяжестью заболевания и прогнозом у пациентов с острыми приступами малярии (Grau et al. , N. Engl. J. Med. 320 (24), 1586-1591 (1989)).
Известно, что индуцируемое макрофагами развитие кровеносных сосудов опосредовано TNF-α. Показано (Leibovich et al. , Nature, 329, 630-632 (1987)), что TNF-α в очень низких дозах индуцирует in vivo образование капиллярных кровеносных сосудов в роговице крыс и развитие хориаллантоисных мембран у цыплят, и высказано предположение, что TNF-α является кандидатом на роль агента, индуцирующего развитие кровеносных сосудов при воспалении, заживлении ран и опухолевом росте. Кроме этого, продуцирование TNF-α ассоциировалось с раковыми состояниями, в частности, с индуцированными опухолями (Ching et al. , Brit. J. Cancer (1955) 72, 339-343 и Koch, Progress in Medical Chemistry, 22, 166-242 (1985)).
TNF-α также играет определенную роль в сфере хронических воспалительных заболеваний легких. Отложение частиц диоксида кремния приводит к силикозу, заболеванию с прогрессирующей дыхательной недостаточностью, вызванной фиброзной реакцией. Антитело к TNF-α полностью блокировало индуцированный диоксидом кремния фиброз легких у мышей (Pignet et al. , Nature, 344, 245-247 (1990)). Высокие уровни продуцирования TNF-α (в сыворотке и изолированных макрофагах) продемонстрированы в животных моделях фиброза, индуцированного диоксидом кремния и асбестом (Bissonnette et al. , Inflammation 13 (3), 329-339 (1989)). Кроме этого, обнаружено, что альвеолярные макрофаги от пациентов с легочным саркоидозом спонтанно выделяют огромные количества TNF-α в сравнении с макрофагами от нормальных доноров (Baughman et al. , J. Lab. Clin. Med. 115 (1), 36-42 (1990)).
TNF-α также вовлечен в воспалительную реакцию, сопровождающую реперфузию, называемую реперфузионным повреждением, и является главной причиной поражения ткани после прекращения кровоснабжения (Vedder et al. , PNAS 87, 2643-2646 (1990)). Кроме этого, TNF-α изменяет свойства эндотелиальных клеток и обладает различными про-коагулянтными активностями, такими как способность вызывать увеличение про-коагулянтной активности тканевого фактора и подавление пути антикоагулянтного белка С, равно как и регуляция, ведущая к уменьшению экспрессии тромбомодулина (Sherry et al. , J. Cell Biol. 107, 1269-1277 (1988)). TNF-α обладает про-воспалительными активностями, которые совместно с его ранним продуцированием (во время начальной стадии воспаления) делают его возможным медиатором тканевого повреждения при некоторых важных нарушениях, включающих в себя инфаркт миокарда, удар и циркуляторный шок, но не ограничивающихся ими. Особенно важным может быть идуцируемая TNF-α экспрессия факторов адгезии, таких как фактор межклеточной адгезии (ICAM) или фактор эндотелиальной лейкоцитарной адгезии (ELAM) на эндотелиальных клетках (Munro et al. , Am. J. Path. 135 (1), 121-132 (1989)).
Показано, что блокирование TNF-α моноклональными анти-TNF-α антителами является благотворным при ревматоидном артрите (Elliot et al. , Int. J. Pharmac. , 1995, 17 (2), 141-145) и болезни Крона (Croh'n) (von Dullemen et al. , Gastroenterology, 1995 109 (1), 129-135).
Более того, теперь известно, что TNF-α является сильнодействующим активатором ретровирусной репликации, включая активацию ВИЧ-1 (Duh et al. , Proc. Nat. Acad. Sci. 86, 5974-5978 (1989); Poll et al. , Proc. Nat. Acad. Sci. 87, 782-785 (1990); Monto et al. , Blood 79, 2670 (1990); Clouse et al. , J. Immunol. 142, 431-438 (1989); Poll et al. , AIDS Res. Hum. Retrovirus, 191-197 (1992)). СПИД является результатом заражения Т-лимфоцитов вирусом иммунодефицита человека (ВИЧ). Идентифицированы по меньшей мере три типа штаммов ВИЧ, а именно ВИЧ-1, ВИЧ-2 и ВИЧ-3. Как следствие ВИЧ-инфекции, происходит ослабление иммунитета, опосредованного Т-клетками, и у инфицированных индивидуумов обнаруживаются тяжелые заболевания, вызываемые условно-патогенными микроорганизмами, и/или необычные новообразования. Для внедрения ВИЧ в Т-лимфоциты требуется активация Т-лимфоцитов. Другие вирусы, такие как ВИЧ-1, ВИЧ-2, инфицируют Т-лимфоциты после активации Т-клеток, и экспрессия белка и/или репликация таких вирусов опосредована либо поддерживается такой активацией Т-клеток. После того как Т-лимфоцит инфицирован ВИЧ, этот Т-лимфоцит должен продолжать поддерживаться в активированном состоянии для обеспечения возможности экспрессии генов ВИЧ и/или репликации ВИЧ. Цитокины, в особенности TNF-α, вовлечены в опосредованную активированными Т-клетками экспрессию белка ВИЧ и/или вирусную репликацию путем их участия в поддержании активации Т-лимфоцитов. Следовательно, воздействие на активность цитокинов, такое как предотвращение или ингибирование продуцирования цитокинов, особенно TNF-α, у ВИЧ-инфицированного индивидуума, помогает ограничить поддержание Т-лимфоцитов, вызванное ВИЧ-инфекцией.
Моноциты, макрофаги и родственные клетки, такие как клетки Купфера (Kupffer) и глиальные клетки, также вовлечены в поддержание ВИЧ-инфекции. Эти клетки, подобно Т-клеткам, являются мишенями для вирусной репликации, и уровень вирусной репликации зависит от состояния активации данных клеток (Rosenberg et al. , The Immunopathogenesis of HIV Infection, Advances in Immunology, 57 (1989)). Показано, что цитокины, такие как TNF-α, активируют репликацию ВИЧ в моноцитах и/или макрофагах (Poli et al. , Proc. Natl. Acad. Sci. 87, 782-784 (1990)), следовательно, предотвращение или ингибирование продуцирования цитокинов или активности цитокинов помогает в ограничении прогрессирования ВИЧ для Т-клеток. Дополнительные исследования идентифицировали TNF-α в качестве общего фактора в активации ВИЧ in vivo и раскрыли ясный механизм действия через ядерный регуляторный белок, обнаруженный в цитоплазме клеток (Osborn et al. , PNAS 86 2336-2340). Эти данные подтверждают тот факт, что уменьшение синтеза TNF-α может оказывать антивирусное действие при ВИЧ-инфекциях путем уменьшения транскрипции и, таким образом, продуцирования вируса.
Вирусная репликация латентного ВИЧ при СПИДе в линиях Т-клеток и макрофагов может быть индуцирована TNF-α (Folks et al. , PNAS 86 2365-2368 (1989)). Способность TNF-α активировать ген-регуляторный белок (NFkB), обнаруженный в цитоплазме клеток, который способствует репликации ВИЧ посредством связывания с вирусной регуляторной генной последовательностью (LTR) (Osborn et al. , PNAS 86 2336-2340 (1989)), дает возможность предположить молекулярный механизм для вирус-индуцирующей активности. Повышенный уровень сывороточного TNF-α и высокие уровни спонтанного продуцирования TNF-α в моноцитах периферической крови пациентов (Wright et al. , J. Immunol. 141 (1), 99-104 (1988)) дает возможность предположить участие TNF-α в СПИД ассоциированной кахексии. По причинам, аналогичным указанным выше, TNF-α задействован в различных ролях при других вирусных инфекциях, таких как вирус цитомегалии (CMV), вирус гриппа, аденовирус и семейство вирусов герпеса.
Ядерный фактор kB (NFkB) является плеотропным транскрипционным активатором (Lenardo et al. , Cell 1989, 58, 227-29). NFkB как транскрипционный активатор вовлечен в разнообразные заболевания и воспалительные состояния; полагают, что он регулирует уровень цитокинов, включая TNF-α, но не ограничиваясь им, а также является активатором транскрипции ВИЧ (Dbaibo et al. , J. Biol. Chem. 1993, 17762-66; Duh et al. , Proc. Natl. Acad. Sci. 1989, 86, 5974-78; Bachelerie et al. , Nature 1991, 350, 709-12; Boswas et al. , J. Acquired Immune Deficiency Syndrome 1993, 6, 778-786; Suzuki et al. , Biochem. and Biophys. Res. Comm. 1993, 193, 277-83; Suzuki et al. , Biochem. and Biophys. Res. Comm. 1992, 189, 1709-15; Suzuki et al. , Biochem. Mol. Bio. Int. 1993, 31 (4), 693-700; Shakhov et al. , Proc. Natl. Acad. Sci. USA 1990, 171, 35-47 и Staal et al. , Proc. Natl. Acad. Sci. USA 1990, 87, 9943-47). Таким образом, ингибирование связывания NFkB может регулировать транскрипцию гена(ов) цитокинов, и посредством этой модуляции и других механизмов может быть полезным для ингибирования большого числа болезненных состояний. Описанные здесь соединения могут ингибировать действие NFkB в ядре и, таким образом, являются полезными при лечении разнообразных заболеваний, включающих в себя ревматоидный артрит, ревматоидный спондилит, остеоартрит, другие артритные состояния, септический шок, сепсис, эндотоксический шок, болезнь "трансплантат против хозяина", истощение, болезнь Крона, неспецифический язвенный колит, рассеянный склероз, системную красную волчанку, узловую эритему при лепре, ВИЧ, СПИД и условно-патогенные инфекции при СПИДе, но не ограничивающихся ими. На уровни TNF-α и NFkB воздействуют по механизму обратной связи. Как отмечено выше, соединения по настоящему изобретению влияют на уровни и TNF-α, и NFkB.
Многие клеточные функции опосредованы уровнями аденозин 3', 5'-циклического монофосфата (цАМФ). Такие клеточные функции могут содействовать воспалительным состояниям и заболеваниям, включая астму, воспаление и другие состояния (Lowe and Cheng Drugs of the Future, 17 (9), 799-807, 1992). Показано, что повышение цАМФ в воспалительных лейкоцитах ингибирует их активацию и последующее выделение медиаторов воспаления, включая TNF-α и NFkB. Кроме этого, повышенные уровни цАМФ приводят к расслаблению дыхательной гладкой мышцы.
Таким образом, снижение уровней TNF-α, и/или повышение уровней цАМФ составляет полезную терапевтическую стратегию лечения многих воспалительных, инфекционных, иммунологических и злокачественных заболеваний. Они включают в себя септический шок, сепсис, эндотоксический шок, гемодинамический шок и септический синдром, повреждение в результате ишемической реперфузии, малярию, микобактериальную инфекцию, менингит, псориаз, застойную сердечную недостаточность, фиброзное заболевание, кахексию, отторжение трансплантата, онкогенные и злокачественные состояния, астму, аутоиммунное заболевание, условно-патогенные инфекции при СПИДе, ревматоидный артрит, ревматоидный спондилит, остеоартрит, другие артритные состояния, болезнь Крона, неспецифический язвенный колит, рассеянный склероз, системную красную волчанку, узловую эритему при лепре, лучевое поражение, онкогенные состояния и гипероксическое альвеолярное повреждение, но не ограничиваются ими. Предыдущие попытки, направленные на подавление влияния TNF-α, простираются от использования стероидов, таких как дексаметазон и преднизолон, до применения как поликлональных, так и моноклональных антител (Beutler et al. , Science 234, 470-474 (1985); WO 92/11383).
Подробное описание
Настоящее изобретение основано на открытии того, что определенные классы соединений неполипептидной природы, описанные здесь более полно, снижают уровни TNF-α.
В частности,
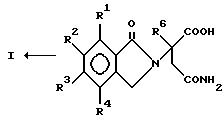
данное изобретение относится к (а) соединениям формулы:
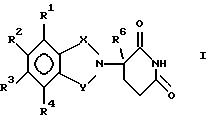
в которой:
один из Х и Y представляет собой С= О, а другой из Х и Y представляет собой С= О или CH2;
(1) каждый из R1, R2, R3 и R4 независимо от других представляет собой галогено, алкил из 1-4 атомов углерода или алкокси из 1-4 атомов углерода, или
(2) один из R1, R2, R3 и R4 представляет собой -NHR5, а оставшиеся из R1, R2, R3 и R4 представляют собой водород;
R5 представляет собой водород или алкил из 1-8 атомов углерода;
R6 представляет собой водород, алкил из 1-8 атомов углерода, бензил или галогено;
при условии, что R6 другой, чем водород, если Х и Y представляют собой С= О, и
(1) каждый из R1, R2, R3 и R4 представляет собой фтор, или
(2) один из R1, R2, R3 и R4 представляет собой аминогруппу; и
(б) полученным присоединением кислоты солям указанных соединений, которые содержат способный к протонированию атом азота.
Предпочтительную группу соединений формулы I представляют те из них, в которых каждый из R1, R2, R3 и R4 независимо от других представляет собой галогено, алкил из 1-4 атомов углерода или алкокси из 1-4 атомов углерода, и R6 представляет собой водород, метил, этил или пропил. Вторую предпочтительную группу соединений формулы I представляют те из них, в которых один из R1, R2, R3 и R4 представляет собой -NH2, оставшиеся из R1, R2, R3 и R4 представляют собой водород, и R6 представляет собой водород, метил, этил или пропил.
Если не определено особо, термин "алкил" означает одновалентную насыщенную разветвленную или прямую углеводородную цепь, содержащую от 1 до 8 атомов углерода. Представителями таких алкильных групп являются метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил. "Алкокси" относится к алкильной группе, связанной с остальной частью молекулы через эфирный атом кислорода. Представителями таких алкоксигрупп являются метокси, этокси, пропокси, изо-пропокси, бутокси, изо-бутокси, втор-бутокси и трет-бутокси. R1, R2, R3 и R4 предпочтительно представляют собой хлор, фтор, метил или метокси.
Соединения формулы I применяют под наблюдением квалифицированных специалистов для ингибирования нежелательных эффектов TNF-α.Соединения могут быть введены перорально, ректально или парентерально, отдельно или в сочетании с другими терапевтическими агентами, включая антибиотики, стероиды и так далее, нуждающемуся в лечении млекопитающему.
Соединения по настоящему изобретению также могут применяться местно при лечении или профилактике локальных болезненных состояний, опосредованых либо усиленных избыточным продуцированием TNF-α, соответственно таких, как вирусные инфекции, например, вызываемые вирусами герпеса, или вирусный конъюнктивит, псориаз, атопический дерматит и так далее.
Данные соединения также могут быть использованы в ветеринарии для лечения млекопитающих, отличных от человека, нуждающихся в предотвращении или ингибировании продуцирования TNF-α. Опосредованные TNF-α заболевания животных для терапевтического или профилактического лечения включают в себя болезненные состояния, указанные выше, но в особенности вирусные инфекции. Примеры включают в себя вирус иммунодефицита кошек, вирус инфекционной анемии лошадей, вирус артрита коз, visna вирус и maedi вирус, равно как и другие лентивирусы.
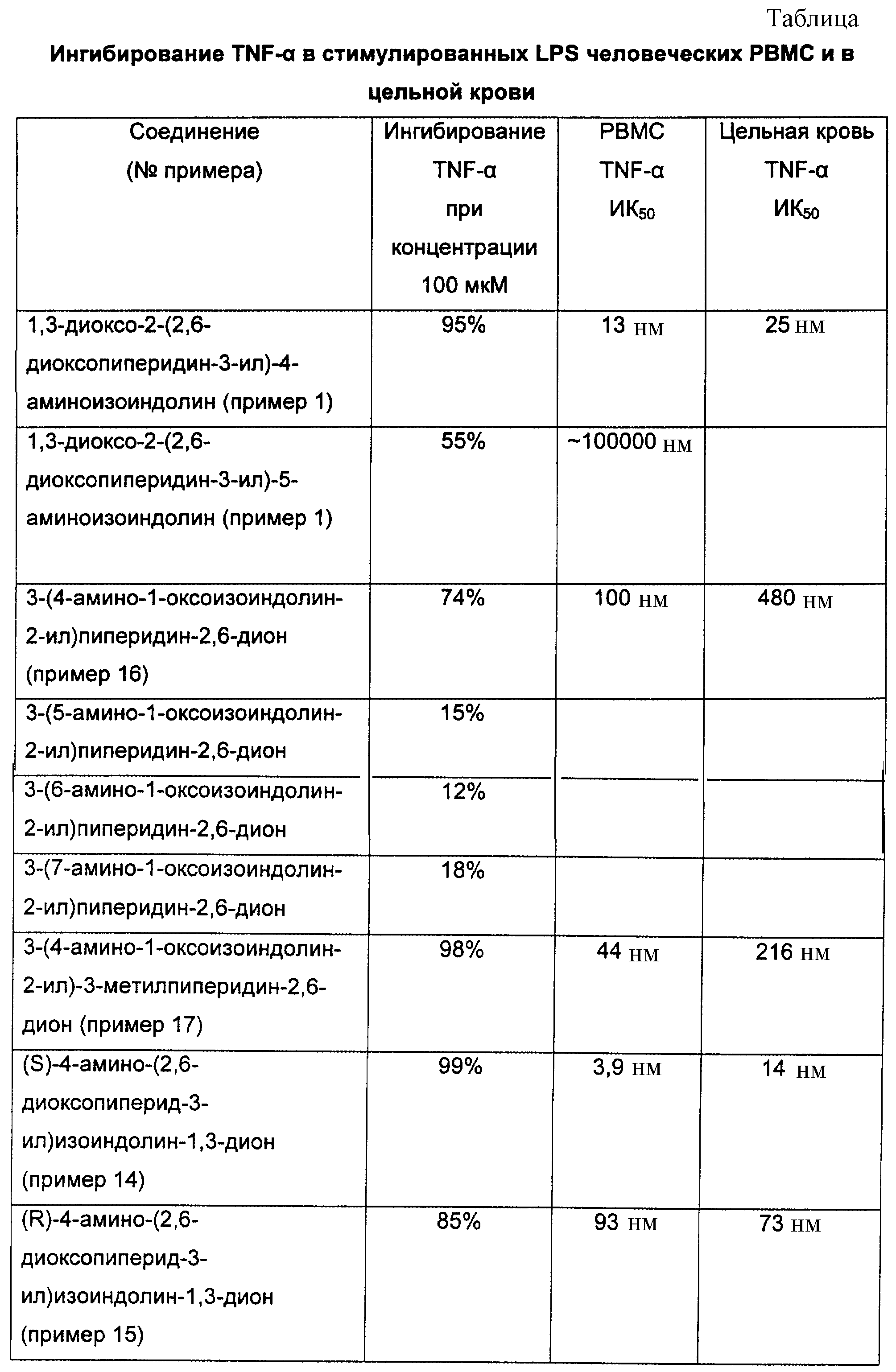
Известны (Jonsson, Acta Pharma. Succica 9, 521-542 (1972)) соединения, в которых один из R1, R2, R3, R4 представляет собой аминогруппу, а R5 и R6, равно как и оставшиеся из R1, R2, R3, R4 представляют собой водород, как, например, 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-4-аминоизоиндолин или 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-5-аминоизоиндолин.
Соединения могут быть получены общеизвестными способами. В частности, соединения могут быть получены путем взаимодействия 2,6-диоксопиперидин-3-аммонийхлорида и (низший алкил)-2-бромметилбензоата в присутствии акцептора кислоты, такого как диметиламинопиридин или триэтиламин.
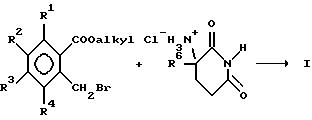
Промежуточные соединения - замещенные бензоаты известны либо могут быть получены общепринятыми способами. Например, (низший алкил)-орто-толуилат бромируют с помощью N-бромсукцинимида под действием света с получением (низший алкил)-2-бром-метилбензоата.
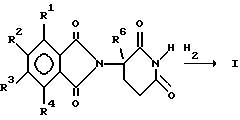
С другой стороны, диальдегид приводят во взаимодействие с 2,6-диоксопиперидин-3-аммонийхлоридом:
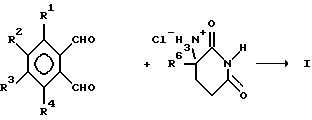
В следующем способе диальдегид приводят во взаимодействие с глутамином и образовавшуюся 2-(1-оксоизоиндолин-2-ил)глутаровую кислоту затем циклизуют, получая 1-оксо-2-(2,6-диоксопиперидин-3-ил)-изоиндолин формулы I:
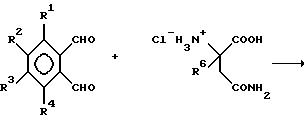
И наконец, селективно восстанавливают соответствующим образом замещенное фталимидное промежуточное соединение
Аминосоединения могут быть получены путем каталитического гидрирования соответствующего нитросоединения:
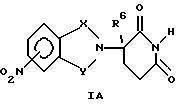
Нитро-промежуточные соединения формулы IA известны либо могут быть получены общепринятыми способами. Например, нитрофталевый ангидрид приводят во взаимодействие с гидрохлоридом α-аминоглутаримида (называемого по другому 2, 6-диоксопиперидин-3-иламмонийхлоридом) в присутствии ацетата натрия и ледяной уксусной кислоты, получая промежуточное соединение формулы IA, в котором Х и Y оба являются С= О.
По второму пути синтеза (низший алкил)-нитро-орто-толуилат бромируют с помощью N-бромсукцинимида под действием света с получением (низший алкил)-2-(бромметил)нитробензоата. Его приводят во взаимодействие с 2,6-диоксопиперидин-3-аммонийхлоридом, например в диметилформамиде, в присутствии триэтиламина, получая промежуточное соединение формулы II, в котором один из Х представляет собой С= О, а другой представляет собой CH2.
С другой стороны, если один из R1, R2, R3 и R4 представляет собой защищенную аминогруппу, то защитная группа может быть отщеплена с получением соответствующего соединения, в котором один из R1, R2, R3 и R4 представляет собой аминогруппу. Используемые здесь защитные группы означают такие группы, которые вообще не обнаруживаются в конечных терапевтических соединениях, но которые намеренно вводятся на какой-либо стадии синтеза для защиты групп, которые без такой защиты могут быть изменены в ходе химических манипуляций. Такие защитные группы удаляют на более поздней стадии синтеза, и соединения, несущие подобные защитные группы, важны, таким образом, в основном в качестве химических промежуточных соединений (несмотря на то, что некоторые из производных также демонстрируют биологическую активность). В соответствии с этим точная структура защитных групп не является критической. Многочисленные реакции для введения и удаления таких защитных групп описаны в ряде стандартных работ ("Protective Groups in Organic Chemistry", Plenum Press, London and New York, 1973; Greene Th. W. "Protective Groups in Organic Synthesis", Wiley, New York, 1981; "The Peptides", Vol. I, Schroder and Lubke, Academic Press, London and New York, 1965; "Methoden der organischen Chemie", Houber-Weyl, 4th Edition, Vol. 15/I, Georg Thieme Verlag, Stuttgart 1974), описания которых включены здесь посредством ссылок. Аминогруппа может быть защищена в виде амида с использованием ацильной группы, которую можно селективно удалить в мягких условиях, в особенности бензилоксикарбонильной, формильной или низшей алканоильной группы, разветвленной в 1-ом или α -положении относительно карбонильной группы, в частности, третичного алканоила, такого как пивалоил, низшей алканоильной группы, которая замещена в положении α относительно карбонильной группы, например трифторацетила.
Соединения по настоящему изобретению содержат центр хиральности и могут существовать в виде оптических изомеров. Как рацематы этих изомеров, так и сами индивидуальные изомеры, равно как и диастереомеры, когда имеются два хиральных центра, входят в объем настоящего изобретения. Рацематы могут быть использованы сами по себе или могут быть разделены на их индивидуальные изомеры механически, как, например, с помощью хроматографии с использованием хирального адсорбента. С другой стороны, индивидуальные изомеры могут быть получены в хиральной форме либо выделены химически из смеси путем образования солей с хиральной кислотой, такой как индивидуальные энантиомеры 10-камфорсульфоновой кислоты, камфорной кислоты, α-бромкамфорной кислоты, метоксиуксусной кислоты, винной кислоты, диацетилвинной кислоты, яблочной кислоты, пирролидон-5-карбоновой кислоты и им подобных, с последующим высвобождением одного или обоих разделенных оснований, возможным повторением процесса для того, чтобы получить одно или оба в существенно чистой от другого форме, то есть в форме с оптической чистотой > 95%.
Кроме этого, настоящее изобретение относится к физиологически приемлемым нетоксичным солям соединений формулы I, полученным присоединением кислот. Такие соли включают в себя соли, полученные с помощью органических и неорганических кислот, таких как, без ограничения, соляная кислота, бромисто-водородная кислота, фосфорная кислота, серная кислота, метансульфоновая кислота, уксусная кислота, винная кислота, молочная кислота, янтарная кислота, лимонная кислота, яблочная кислота, малеиновая кислота, сорбиновая кислота, аконитовая кислота, салицилловая кислота, фталевая кислота, эмбоновая кислота, энантовая кислота и им подобные.
Лекарственные формы для перорального введения включают в себя таблетки, капсулы, драже и похоже оформленные, прессованные фармацевтические формы, содержащие от 1 до 100 мг лекарственного средства на стандартную дозу. Для парентерального введения, которое включает в себя внутримышечный, внутриоболочечный, внутривенный и внутриартериальный способы введения, могут быть использованы изотонические солевые растворы, содержащие от 20 до 100 мг/мл. Ректальное введение может осуществляться посредством использования суппозиториев, изготовленных на основе традиционных носителей, таких как масло какао.
Таким образом, фармацевтические композиции содержат одно или более чем одно соединение по настоящему изобретению, объединенное по меньшей мере с одним фармацевтически приемлемым носителем, разбавителем или эксципиентом. При получении таких композиций активные ингредиенты обычно смешивают с эксципиентом или разбавляют им либо заключают внутрь такого носителя, который может быть в форме капсулы или пакетика. В том случае, когда эксципиент служит в качестве разбавителя, он может представлять собой твердый, полутвердый или жидкий материал, который действует как наполнитель, носитель или среда для активного ингредиента. Таким образом, данные композиции могут быть в форме таблеток, драже, порошков, эликсиров, суспензий, эмульсий, растворов, сиропов, мягких и твердых желатиновых капсул, суппозиториев, стерильных растворов для инъекций и стерильных упакованных порошков. Примеры подходящих эксципиентов включают в себя лактозу, декстрозу, сахарозу, сорбит, маннит, крахмал, аравийскую камедь, кальция силикат, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду, сироп и метилцеллюлозу; препараты могут дополнительно содержать смазывающие вещества, такие как тальк, магния стеарат и минеральное масло, увлажняющие агенты, эмульгирующие и суспендирующие агенты, консерванты, такие как метил- или пропилгидроксибензоаты, подсластители и корригенты.
Композиции предпочтительно изготавливают в виде стандартной лекарственной формы, означающей физически дискретные единицы, пригодные в качестве единичной дозы, или предварительно определенную часть единичной дозы для введения в режиме разовой дозы или множественных доз субъекту-человеку или другим млекопитающим, причем каждая единица содержит предварительно определенное количество активного вещества, вычисленное с целью получения желаемого терапевтического эффекта, вместе с подходящим фармацевтическим эксципиентом. Данные композиции могут быть изготовлены таким образом, чтобы обеспечить немедленное, непрерывное или задержанное высвобождение активного ингредиента после введения пациенту посредством использования методик, хорошо известных специалистам.
Следующие примеры служат для дальнейшего раскрытия природы этого изобретения, но не должны истолковываться как ограничивающие его объем, который определен исключительно в приложенной формуле изобретения.
ПРИМЕР 1
1,3-Диоксо-2-(2,6-диоксопиперидин-3-ил)-5-аминоизоиндолин
Смесь 1,3-диоксо-2-(2,
6-диоксопиперидин-3-ил)-5-нитроизоиндолина (по другому называемого как N-(2,6-диоксопиперидин-3-ил)4-нитрофталимид) (1 г; 3,3 ммоль) и 10% Pd/C (0,13 г) в 1,4-диоксане (200 мл) гидрируют при 50
фунт-сила/кв. дюйм (344,75 кПа) в течение 6,5 часов. Катализатор отфильтровывают на целите (Celite) и фильтрат концентрируют in vacuо. Остаток кристаллизуют из этилацетата (20 мл), получая 0,62 г
(69%) 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-5-аминоизоиндолина (иначе называемого как N-(2,6-диоксопиперидин-3-ил)-4-аминофталимид) в виде твердого вещества оранжевого цвета. Перекристаллизация из
смеси диоксан/этилацетат дает 0,32 г твердого вещества желтого цвета: т. пл. 318,5-320,5oС; ВЭЖХ (Nova-Pak С18, 15/85 ацетонитрил/0,1%-ная Н3РО4) 3,97 мин
(98,22%);1H ЯМР (ДMCO-d6 ) δ 11.08 (s, 1H), 7.53-7.50 (d, J= 8.3 Гц, 1Н), 6.94 (s, 1H), 6.84-6.81 (d, 3= 8.3 Гц, 1H), 6.55 (s, 2H), 5.05-4.98 (m, 1H), 2.87-1.99 (m, 4H);13C ЯМР (ДMCO-d6) δ 172.79, 170.16, 167.65, 167.14, 155.23, 134.21, 125.22, 116.92, 116.17, 107.05, 48.58, 30.97, 22.22. Аналитич. рассчитано для C13H11N3O4: С, 57.14; Н, 4.06; N, 15.38. Обнаружено: С, 56.52; Н, 4.17; N, 14.60.
Подобным же образом из 1-оксо-2-(2,6-диоксопиперидин-3-ил)5-нитроизоиндолина, 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4-нитроизоиндолина, 1-оксо-2-(2,6-диоксопиперидин-3-ил)-6-нитроизоиндолина, 1-оксо-2-(2,6-диоксопиперидин-3-ил)-7-нитроизоиндолина и 1,3-диоксо-2-(2, 6-диоксопиперидин-3-ил)-4-нитроизоиндолина соответственно получают 1-оксо-2-(2,6-диоксопиперидин-3-ил)-5-аминоизоиндолин, 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4-аминоизоиндолин, 1-оксо-2-(2, 6-диоксопиперидин-3-ил)-6-аминоизоиндолин, 1-оксо-2-(2,6-диоксопиперидин-3-ил)-7-аминоизоиндолин и 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-4-аминоизоиндолин, соответственно после гидрирования.
ПРИМЕР 2
1,3-Диоксо-2-(2,6-диоксопиперидин-3-ил)-5-нитроизоиндолин
Смесь 4-нитрофталевого ангидрида (1,7 г; 8,5 ммоль), α-аминоглутаримида гидрохлорида (1,4 г; 8,5
ммоль) и ацетата натрия (0,7 г; 8,6 ммоль) в ледяной уксусной кислоте (30 мл) кипятят с обратным холодильником в течение 17 часов. Смесь концентрируют in vacuo и остаток перемешивают с метиленхлоридом
(40 мл) и водой (30 мл). Водный слой отделяют, экстрагируют метиленхлоридом (2•40 мл). Объединенные растворы в метиленхлориде высушивают над сульфатом магния и концентрируют in vacuo, получая 1,
4 г (54%) 1,3-диоксо-2-(2,6-диоксопиперидин-3-ил)-5-нитроизоиндолина в виде твердого вещества светло-коричневой окраски. Аналитический образец получают перекристаллизацией из метанола: т. пл. 228,
5-229,5oС;1Н ЯМР (ДMCO-d6) δ 11.18 (s, 1H), 8.69-8.65 (dd, J= 1.9 и 8.0 Гц, 1Н), 8.56 (d, J= 1.9 Гц, 1H), 8.21 (d, J= 8.2 Гц, 1H), 5.28 (dd, J= 5.3 и 12.8 Гц,
1H), 2.93-2.07 (m, 4H);13C ЯМР (ДMCO-d6) δ 172.66, 169.47, 165.50, 165.23, 151.69, 135.70, 132.50, 130.05, 124.97, 118.34, 49.46, 30.85, 21.79. Аналитич. рассчитано для
C13H9N3O6: С, 51.49; Н, 2.99; N, 13.86. Обнаружено: С, 51.59; Н, 3.07; N, 13.73.
1-Оксо-2-(2,6-диоксопиперидин-3-ил)-5-нитроизоиндолин, 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4-нитроизоиндолин, l-оксо-2-(2,6-диоксопиперидин-3-ил)-6-нитроизоиндолин и 1-оксо-2-(2,6-диоксопиперидин-3-ил)-7-нитроизоиндолин могут быть получены путем взаимодействия 2,6-диоксопиперидин-3-аммонийхлорида с метиловым эфиром 2-бромметил-5-нитробензойной кислоты, метиловым эфиром 2-бромметил-4-нитробензойной кислоты, метиловым эфиром 2-бромметил-6-нитробензойной кислоты и метиловым эфиром 2-бромметил-7-нитробензойной кислоты, соответственно, в диметилформамиде в присутствии триэтиламина. В свою очередь, метиловые эфиры 2-(бромметил)нитробензойной кислоты получают из соответствующих метиловых эфиров нитро-орто-толуиловых кислот обычным бромированием с помощью N-бромсукцинимида под действием света.
ПРИМЕР 3
1-Оксо-2-(2,6-диоксопиперидин-3-ил)-4,5,6,7-тетрафторизоиндолин
Смесь 16,25 г 2,6-диоксопиперидин-3-аммонийхлорида, 30,1 г метилового эфира 2-бромметил-3,4,5,
6-тетрафторбензойной кислоты и 12,5 г триэтиламина в 100 мл диметилформамида перемешивают при комнатной температуре в течение 15 часов. Далее смесь концентрируют in vacuо и остаток смешивают с
метиленхлоридом и водой. Водный слой отделяют и повторно экстрагируют метиленхлоридом. Объединенные растворы в метиленхлориде высушивают над сульфатом магния и концентрируют in vacuo, получая
1-оксо-2-(2,6-диоксопиперидин-3-ил)-4,5,6,7-тетрафторизоиндолин.
Подобным же образом получают 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4,5,6,7-тетрахлоризоиндолин, 1-оксо-2-(2, 6-диоксопиперидин-3-ил)-4,5,6,7-тетраметилизоиндолин и 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4,5,6,7-тетраметоксиизоиндолин, заменяя 2-бромметил-3,4,5,6-тетрафторбензоат эквивалентными количествами 2-бромметил-3,4,5,6-тетрахлорбензоата, 2-бромметил-3,4,5,6-тетраметилбензоата и 2-бромметил-3,4,5,6-тетраметоксибензоата, соответственно.
ПРИМЕР 4
N-бензилоксикарбонил-α
-метил-глутаминовая кислота
К перемешиваемому раствору α-метил-D, L-глутаминовой кислоты (10 г; 62 ммоль) в 2 н. гидроксиде натрия (62 мл) при 0-5oС добавляют в течение 30
мин бензилхлорформиат (12,7 г; 74,4 ммоль). По окончании добавления реакционную смесь перемешивают при комнатной температуре в течение 3 часов. В течение этого времени значение рН поддерживают равным
11 путем добавления 2 н. гидроксида натрия (33 мл). Реакционную смесь затем экстрагируют эфиром (60 мл). Водный слой охлаждают в ледяной бане и далее подкисляют с помощью 4 н. соляной кислоты (34 мл)
до рН 1. Полученную смесь экстрагируют этилацетатом (3•100 мл). Объединенные этилацетатные экстракты промывают рассолом (60 мл) и высушивают (MgSО4). Растворитель удаляют in vacuо,
получая 15,2 г (83%) N-бензилоксикарбонил-α-метил-глутаминовой кислоты в виде масла:1H ЯМР (CDC13) δ 8.73 (m, 5H), 5.77 (b, 1H), 5.09 (s, 2H), 2.45-2.27 (m, 4H),
2.0 (s, 3Н).
Подобным же образом из α-этил-D, L-глутаминовой кислоты и α-пропил-D, L-глутаминовой кислоты получают N-бензилоксикарбонил-α-этилглутаминовую кислоту и N-бензилоксикарбонил-α-пропилглутаминовую кислоту, соответственно.
ПРИМЕР 5
N-бензилоксикарбонил-α-метил-глутаминовый ангидрид
Перемешиваемую смесь
N-бензилоксикарбонил-α-метил-глутаминовой кислоты (15 г, 51 ммоль) и уксусного ангидрида (65 мл) кипятят с обратным холодильником в атмосфере азота в течение 30 мин. Реакционную смесь охлаждают
до комнатной температуры и далее концентрируют in vacuo, что позволяет получить N-бензилкарбонил-α-метилглутаминовый ангидрид в виде масла (15,7 г), который можно использовать в последующей
реакции без дальнейшей очистки:1H ЯМР (CDC13) δ 7.44-7.26 (m, 5H), 5.32-5.30 (m, 2H), 5.11 (s, 1H), 2.69-2.61 (m, 2H), 2.40-2.30 (m, 2H), 1.68 (s, 3H).
Подобным же образом из N-бензилоксикарбонил-α-этилглутаминовой кислоты и N-бензилоксикарбонил-α-пропилглутаминовой кислоты получают N-бензилкарбонил-α-этилглутаминовый ангидрид и N-бензилкарбонил-α-пропилглутаминовый ангидрид, соответственно.
ПРИМЕР 6
N-бензилоксикарбонил-α-метилизоглутанин
Перемешиваемый раствор
N-бензилкарбонил-α-метилглутаминового ангидрида (14,2 г; 51,5 ммоль) в метиленхлориде (100 мл) охлаждают в ледяной бане. Газообразный аммиак барботируют в охлажденный раствор в течение 2 часов.
Реакционную смесь перемешивают при комнатной температуре в течение 17 часов и далее экстрагируют водой (2•50 мл). Объединенные водные экстракты охлаждают в ледяной бане и подкисляют 4 н.
соляной кислотой (32 мл) до рН 1. Полученную смесь экстрагируют этилацетатом (3•80 мл). Объединенные этилацетатные экстракты промывают рассолом (60 мл) и далее высушивают (MgSО4).
Растворитель удаляют in vacuo, получая 11,5 г N-бензилоксикарбонил-α-амино-α-метилизоглутамина:1H ЯМР (СDС13/ДМСО) δ 7.35 (m, 5H), 7.01 (s, 1H), 6.87 (s,
1H), 6.29 (s, 1H), 5.04 (s, 2H), 2.24-1.88 (m, 4Н), 1.53 (s, 3H).
Подобным же образом из N-бензилкарбонил-α-этилглутаминового ангидрида и N-бензилкарбонил-α -пропилглутаминового ангидрида получают N-бензилоксикарбонил-α-амино-α-этилизоглутамин и N-бензилоксикарбонил-α-амино-α-пропилизоглутамин, соответственно.
ПРИМЕР 7
N-бензилоксикарбонил-α-амино-α-метилглутаримид
Перемешиваемую смесь N-бензилоксикарбонил-α-метилизоглутамина (4,60 г; 15,6 ммоль), 1,
1'-карбонилдиимидазола (2,80 г; 17,1 ммоль) и 4-диметиламинопиридина (0,05 г) в тетрагидрофуране (50 мл) нагревают в атмосфере азота до температуры дефлегмации в течение 17 часов. Реакционную смесь
затем концентрируют in vacuо до масла. Масло суспендируют в воде (50 мл) в течение 1 часа. Полученную суспензию фильтруют и твердое вещество промывают водой и высушивают на воздухе, что позволяет
получить 3,8 г неочищенного продукта в виде твердого вещества белого цвета. Неочищенный продукт очищают флэш-хроматографией (метиленхлорид: этилацетат 8: 2), что позволяет получить 2,3 г (50%)
N-бензилоксикарбонил-α-амино-α-метилглутаримида в виде твердого вещества белого цвета: т. пл. 150,5-152,5oС;1H ЯМР (СDС13) δ 8.21 (s, 1H), 7.34
(s, 5H), 5.59 (s, 1H), 5.08 (s, 2H), 2.74-2.57 (m, 3Н), 2.28-2.25 (m, 1H), 1.54 (s, 3Н);13C ЯМР (СDС13) δ 174.06, 171.56, 154.68, 135.88, 128.06, 127.69, 127.65, 66.15,
54.79, 29.14, 28.70, 21.98; ВЭЖХ: Waters Nova-Pak C18-колонка, 4 микрона; 3,9•150 мм, 1 мл/мин, 240 нм, 20/80 СН3СN/0,1%-ная Н3РО4 (водн. ), 7,56 мин
(100%). Аналитич. рассчитано для C14H16N2O4: С, 60,86; Н, 5,84; N, 10,14. Обнаружено: С, 60,88; Н, 5,72; N, 10,07.
Подобным же образом из N-бензилоксикарбонил-α-амино-α-этилизоглутамина и N-бензилоксикарбонил-α-амино-α-пропилизоглутамина получают N-бензилоксикарбонил-α-амино-α-этилглутаримид и N-бензилоксикарбонил-α-амино-α-пропилглутаримид, соответственно.
ПРИМЕР 8
-α-Амино-α-метилглутаримида гидрохлорид
N-Бензилоксикарбонил-α-амино-α-метилглутаримид (2,3 г; 8,3 ммоль) растворяют при слабом нагревании в этаноле (200 мл) и полученному раствору позволяют охладиться до комнатной
температуры. К этому раствору добавляют 4 н. соляную кислоту (3 мл) с последующим добавлением 10% Pd/C (0,4 г). Смесь гидрируют в аппарате Парра (Parr) при давлении водорода 50 фунт-сила/кв. дюйм (344,
75 кПа) в течение 3 часов. Для растворения продукта к смеси добавляют воду (50 мл). Смесь фильтруют через слой целита (Celite), который промывают водой (50 мл). Фильтрат концентрируют in vacuo, что
позволяет получить твердый остаток. Его суспендируют в этаноле (20 мл) в течение 30 мин. Суспензию фильтруют, что позволяет получить 1,38 г (93%) α-амино-α-метилглутаримида гидрохлорида
в виде твердого вещества белого цвета:lН ЯМР (ДМСО-d6) δ 11.25 (s, 1H), 8.92 (s, 3Н), 2.84-2.51 (m, 2Н), 2.35-2.09 (m, 2H), 1.53 (s, 3Н); ВЭЖХ, Waters Nova-Pak C18-колонка, 4 микрона, 1 мл/мин, 240 нм, 20/80 СН3СN/0,1%-ная Н3РО4 (водн. ), 1,03 мин (94,6%).
Подобным же образом из N-бензилоксикарбонил-α-амино-α-этилглутаримида и N-α-амино-α-пропилглутаримида получают α-амино-α-этилглутаримида гидрохлорид и α-амино-α -пропилглутаримида гидрохлорид, соответственно.
ПРИМЕР 9
3-(3-Нитрофталимидо)-3-нетилпиперидин-2,6-дион
Перемешиваемую смесь α-амино-α-метилглутаримида
гидрохлорида (1,2 г; 6,7 ммоль), 3-нитрофталевого ангидрида (1,3 г; 6,7 ммоль) и ацетата натрия (0,6 г; 7,4 ммоль) в уксусной кислоте (30 мл) нагревают до температуры дефлегмации в атмосфере азота в
течение 6 часов. Затем смесь охлаждают и концентрируют in vacuo. Полученное твердое вещество суспендируют в воде (30 мл) и метиленхлориде (30 мл) в течение 30 мин. Суспензию фильтруют, твердое
вещество промывают метиленхлоридом и высушивают in vасuо (60oC, < 1 мм), что позволяет получить 1,44 г (68%) 3-(3-нитрофталимидо)-3-метилпиперидин-2,6-диона в виде твердого вещества
беловатой окраски: т. пл. 265-266,5oС;1H ЯМР (ДМСО-d6) δ 11.05 (s, 1H), 8.31 (dd, J= 1.1 и 7.9 Гц, 1Н), 8.16-8.03 (m, 2H), 2.67-2.49 (m, 3Н), 2.08-2.02 (m, 1H),
1.88 (s, 3Н);13С ЯМР (ДМСО-d6) δ 172.20, 171.71, 165.89, 163.30, 144.19, 136.43, 133.04, 128.49, 126.77, 122.25, 59.22. 28.87, 28.49, 21.04; ВЭЖХ, Water Nova-Pak/C18-колонка, 4 микрона, 1 мл/мин, 240 нм, 20/80 CH3CN/0,1%-ная Н3РО4 (водн. ), 7,38 мин (98%). Аналитич. рассчитано для С14Н11N3
О4: С, 53,00; Н, 3,49; N, 13,24. Обнаружено: С, 52,77; Н, 3,29; N, 13,00.
Подобным же образом из α-амино-α-этилглутаримида гидрохлорида и α -амино-α-пропилглутаримида гидрохлорида получают 3-(3-нитрофталимидо)-3-этилпиперидин-2,6-дион и 3-(3-нитрофталимидо)-3-пропилпиперидин-2,6-дион, соответственно.
ПРИМЕР 10
3-(3-Аминофталимидо)-3-метилпиперидин-2,6-дион
3-(3-Нитрофталимидо)-3-метилпиперидин-2,6-дион (0,5 г; 1,57 ммоль) растворяют при слабом нагревании в ацетоне (250 мл) и затем охлаждают
до комнатной температуры. К этому раствору в атмосфере азота добавляют 10% Pd/C (0,1 г). Смесь гидрируют в аппарате Парра при давлении водорода 50 фунт-сила/кв. дюйм (344,75 кПа) в течение 4 часов.
Далее смесь фильтруют через целит (Celite) и слой промывают ацетоном (50 мл). Фильтрат концентрируют in vacuo, получая твердое вещество желтого цвета. Его суспендируют в этилацетате (10 мл) в течение
30 минут. Суспензию затем фильтруют и высушивают (60oС, < 1 мм), что позволяет получить 0,37 г (82%) 3-(3-аминофталимидо)-3-метилпиперидин-2,6-диона в виде твердого вещества желтого
цвета: т. пл. 268-269oС;1Н ЯМР (ДMCO-d6)) δ 10.98 (s, 1H), 7.44 (dd, J= 7.1 и 7.3 Гц, 1H), 6.99 (d, J= 8.4 Гц, 1H), 6.94 (d, J= 6.9 Гц, 1H), 6.52 (s, 2H),
2.71-2.47 (m, 3Н), 2.08-1.99 (m, 1H), 1.87 (s, 3Н);13С ЯМР (ДМСО-d6) δ 172.48, 172.18, 169.51, 168.06, 146.55, 135.38, 131.80, 121.51, 110.56, 108.30, 58.29, 29.25, 28.63,
21.00; ВЭЖХ, Waters Nova-Pak/C18-колонка, 4 микрона, 1 мл/мин, 240 нм, 20/80 СН3NН/0,1%-ная Н3РО4 (водн. ), 5,62 мин (99,18%). Аналитич. рассчитано для
C14H13N3О4: С, 58,53; Н, 4,56; N, 14,63. Обнаружено: С, 58,60; Н, 4,41; N, 14,36.
Подобным же образом из 3-(3-нитрофталимидо)-3-этилпиперидин-2,6-диона и 3-(3-нитрофталимидо)-3-пропилпиперидин-2,6-диона получают 3-(3-аминофталимидо)-3-этилпиперидин-2,6-дион и 3-(3-аминофталимидо)-3-пропилпиперидин-2, 6-дион, соответственно.
ПРИМЕР 11
Метиловый эфир 2-бромметил-3-нитробензойной кислоты
Перемешиваемую смесь метилового эфира 2-метил-3-нитробензойной кислоты (17,6 г;
87,1 ммоль) и N-бромсукцинимида (18,9 г; 105 ммоль) в четыреххлористом углероде (243 мл) осторожно кипятят с обратным холодильником, освещая электрической лампочкой мощностью 100 ватт, расположенной
на расстоянии 2 см от реакционной смеси, в течение ночи. Через 18 часов реакционную смесь охлаждают до комнатной температуры и фильтруют. Фильтрат промывают водой (2•120 мл), рассолом (120 мл)
и высушивают (MgSО4). Растворитель удаляют in vacuo, получая твердое вещество желтого цвета. Продукт очищают флэш-хроматографией (гексан: этилацетат, 8: 2), получая 22 г (93%) метилового
эфира 2-бромметил-3-нитробензойной кислоты в виде твердого вещества желтого цвета: т. пл. 69-72oС;1Н ЯМР (СDС13) δ 8.13-8.09 (dd, J= 1.36 и 7.86 Гц, 1H),
7.98-7.93 (dd, J= 1.32 и 8.13 Гц, 1H), 7.57-7,51 (t, J= 7.97 Гц, 1Н), 5.16 (s, 2H), 4.0 (s, 3H);13С ЯМР (СDС13) δ/ 65.84, 150.56, 134.68, 132.64, 132.36, 129.09, 53.05,
22.70; ВЭЖХ: Waters Nova-Pak C18-колонка, 4 микрона, 1 мл/мин, 240 нм, 40/60 СН3СN/0,1%-ная Н3РО4 (водн. ), 8,2 мин (99%). Аналитич. рассчитано для С9Н8NO4Br: С, 39,44; Н, 2,94; N, 5,11; Br, 29,15. Обнаружено: С, 39,51; Н, 2,79; N, 5,02; Br, 29,32.
ПРИМЕР 12
3-(1-Оксо-4-нитроизоиндолин-1-ил)-3-метилпиперидин-2,6-дион
К перемешиваемой смеси α-амино-α-метилглутаримида гидрохлорида (2,5 г; 14,0 ммоль) и метилового эфира
2-бромметил-3-нитробензойной кислоты (3,87 г; 14,0 ммоль) в диметилформамиде (40 мл) добавляют триэтиламин (3,14 г; 30,8 ммоль). Полученную смесь нагревают в атмосфере азота до температуры дефлегмации
в течение 6 часов. Смесь охлаждают и затем концентрируют in vacuo. Полученное твердое вещество суспендируют в воде (50 мл) и CH2Cl2 в течение 30 мин. Суспензию фильтруют, твердое
вещество промывают метиленхлоридом и высушивают in vacuo (60oС, < 1 мм), что позволяет получить 2,68 г (63%) 3-(1-оксо-4-нитроизоиндолин-1-ил)-3-метилпиперидин-2,6-диона в виде
твердого вещества беловатой окраски: т. пл. 233-235oС;1Н ЯМР (ДМСО-d6) δ 10.95 (s, 1H), 8.49-8.46 (d, J= 8.15 Гц, 1Н), 8.13-8.09 (d, J= 7.43 Гц, 1H), 7.86-7.79
(t, J= 7.83 Гц, 1Н), 5.22-5.0 (dd, J= 19.35 и 34.6 Гц, 2H), 2.77-2.49 (m, 3H), 2.0-1.94 (m, 1H), 1.74 (s, 3H);13С ЯМР (ДМСО-d6) δ 173.07, 172.27, 164.95, 143.15, 137.36,
135.19, 130.11, 129.32, 126.93, 57.57, 48.69, 28.9, 27.66, 20.6; ВЭЖХ, Waters Nova-Pak С18-колонка, 4 микрона, 1 мл/мин, 240 нм, 20/80 СН3СN/0,1%-ная Н3РО4
(водн. ), 4,54 мин (99,6%). Аналитич. рассчитано для C14H13N3CO5: С, 55,45; Н, 4,32; N, 13,86. Обнаружено: С, 52,16; Н, 4,59; N, 12,47.
Заменяя α-амино-α-метилглутаримида гидрохлорид эквивалентными количествами α-амино-α-этилглутаримида гидрохлорида и α-амино-α-пропилглутаримида гидрохлорида, получают соответственно 3-(1-оксо-4-нитроизоиндолин-1-ил)-3-этилпиперидин-2,6-дион и 3-(1-оксо-4-нитроизоиндолин-1-ил)-3-пропилпиперидин-2,6-дион.
ПРИМЕР 13
3-(1-Оксо-4-аниноизоиндолин-1-ил)-3-метилпиперидин-2,6-дион
3-(1-Оксо-4-нитроизоиндолин-1-ил)-3-метилпиперидин-2,6-дион (1,0 г; 3,3 ммоль) растворяют при слабом нагревании в метаноле (500 мл)
и позволяют раствору охладиться до комнатной температуры. К этому раствору в атмосфере азота добавляют 10% Pd/C (0,3 г). Смесь гидрируют в аппарате Парра при давлении водорода 50 фунт-сила/кв. дюйм
(344,75 кПа) в течение 4 часов. Смесь фильтруют через целит (Celite) и целит промывают метанолом (50 мл). Фильтрат концентрируют in vacuo до твердого вещества беловатого цвета. Его суспендируют в
метиленхлориде (20 мл) в течение 30 мин. Суспензию далее фильтруют и твердое вещество высушивают (60oС, <1 мм), что позволяет получить 0,54 г (60%)
3-(1-оксо-4-аминоизоиндолин-1-ил)-3-метилпиперидин-2,6-диона в виде твердого вещества белого цвета: т. пл. 268-270oС;1H ЯМР (ДMCO-d6) δ 10.85 (s, 1H),
7.19-7.13 (t, J= 7.63 Гц, 1H), 6.83-6.76 (m, 2Н), 5.44 (s, 2Н), 4.41 (s, 2H), 2.71-2.49 (m, 3H), 1.9-1.8 (m, 1H), 1.67 (s, 3Н);13С ЯМР (ДMCO-d6) δ 173.7, 172.49, 168.0,
143.5, 132.88, 128.78, 125.62, 116.12, 109.92, 56.98, 46.22, 29.04, 27.77, 20.82; ВЭЖХ, Waters Nova-Pak/C18-колонка, 4 микрона, 1 мл/мин, 240 нм, 20/80 СН3СN/0,1%-ная Н3РО4 (водн. ), 1,5 мин (99,6%). Аналитич. рассчитано для C14H15N3О3: С, 61,53; Н, 5,53; N, 15,38. Обнаружено: С, 58,99; Н, 5,48; N, 14,
29.
Из 3-(1-оксо-4-нитроизоиндолин-1-ил)-3-этилпиперидин-2,6-диона и 3-(1-оксо-4-нитроизоиндолин-1-ил)-3-пропилпиперидин-2,6-диона аналогичным способом получают 3-(1-оксо-4-аминоизоиндолин-1-ил)-3-этилпиперидин-2,6-дион и 3-(1-оксо-4-аминоизоиндолин-1-ил)-3-пропилпиперидин-2,6-дион, соответственно.
ПРИМЕР 14
S-4-амино-2-(2,
6-диоксопиперид-3-ил)изоиндолин-1,3-дион
А. 4-Нитро-N-этоксикарбонилфталимид
Этилхлорформиат (1,89 г; 19,7 ммоль) в атмосфере азота добавляют по каплям в течение 10 мин к
перемешиваемому раствору 3-нитрофталимида (3,0 г; 15,6 ммоль) и триэтиламина (1,78 г; 17,6 ммоль) в диметилформамиде (20 мл) при 0-5oС. Реакционной смеси позволяют нагреться до комнатной
температуры и перемешивают в течение 4 часов. Далее смесь медленно добавляют к перемешиваемой смеси льда и воды (60 мл). Полученную суспензию фильтруют и твердое вещество кристаллизуют из хлороформа
(15 мл) и петролейного эфира (15 мл), что позволяет получить 3,1 г (75%) продукта в виде твердого вещества беловатой окраски: т. пл. 100-100,5oС;1H ЯМР (CDC13)
δ 8.25 (d, J= 7.5 Гц, 1Н), 8.20 (d, J= 8.0 Гц, 1Н), 8.03 (t, J= 7.9 Гц, 1Н), 4.49 (q, J= 7.1 Гц, 2Н), 1.44 (t, J= 7.2 Гц, 3Н);13С ЯМР (СDС13) δ 161.45, 158.40,
147.52, 145.65, 136.60, 132.93, 129.65, 128.01, 122.54, 64.64, 13.92; ВЭЖХ, Waters Nova-Pak/C18, 3,9•150 мм, 4 микрона, 1 мл/мин, 240 нм, 30/70 CH3CN/0,1%-ная Н3
РO4 (водн. ), 5,17 мин (98,11%). Аналитич. рассчитано для C11H8N2O6: С, 50,00; Н, 3,05; N, 10,60. Обнаружено: С, 50,13; Н, 2,96; N, 10,54.
Б. Трет-бутил N-(4-нитрофталоил)-L-глутамин
Перемешиваемую смесь 4-нитро-N-этоксикарбонилфталимида (1,0 г; 3,8 ммоль), трет-бутилового эфира L-глутамина гидрохлорида (0,90 г; 3,8
ммоль) и триэтиламина (0,54 г; 5,3 ммоль) в тетрагидрофуране (30 мл) нагревают до температуры дефлегмации в течение 24 часов. Тетрагидрофуран удаляют in vacuo и остаток растворяют в метиленхлориде (50
мл). Раствор метиленхлорида промывают водой (2•15 мл), рассолом (15 мл) и затем высушивают (сульфат натрия). Растворитель удаляют in vacuo и остаток очищают флэш-хроматографией (7: 3,
метиленхлорид: этилацетат), получая 0,9 г (63%) стеклообразного материала:1Н ЯМР (СDС13) δ 8.15 (d, J= 7.9 Гц, 2Н), 7.94 (t, J= 7.8 Гц, 1Н), 5.57 (b, 2H), 4.84 (dd, J=
5.1 и 9.7 Гц, 1H), 2.53-2.30 (m, 4H), 1.43 (s, 9H); ВЭЖХ, Waters Nova-Pak/C18, 3,9•150 мм, 4 микрона, 1 мл/мин, 240 нм, 30/70 СН3СN/0,1%-ная Н3РО4
(водн. ), 6,48 мин (99,68%). Хиральный анализ, Daicel Chiral Pak AD, 0,4•25 см, 1 мл/мин, 240 нм, 5,32 мин (99,39%). Аналитич. рассчитано для C17H19N3O7: С, 54,11; Н, 5,08; N, 11,14. Обнаружено: С, 54,21; Н, 5,08; N, 10,85.
В. N-(4-нитрофталоил)-L-глутамин
Газообразный хлористый водород барботируют в перемешиваемый при
5oС раствор трет-бутил N-(4-нитрофталоил-L-глутамина (5,7 г; 15,1 ммоль) в метиленхлориде (100 мл) в течение 25 мин. Далее смесь перемешивают при комнатной температуре в течение 16 часов.
Добавляют эфир (50 мл) и полученную смесь перемешивают в течение 30 мин. Полученную суспензию фильтруют, получая 4,5 г неочищенного продукта в виде твердого вещества, которое непосредственно
используют на следующей стадии:1Н ЯМР (ДМСО-d6) δ 8.36 (dd, J= 0.8 и 8.0 Гц, 1H), 8.24 (dd, J= 0.8 и 7.5 Гц, 1H), 8.11 (t, J= 7.9 Гц, 1H), 7.19 (b, 1H), 6.72 (b, 1H),
4.80 (dd, J= 3.5 и 8.8 Гц, 1H), 2.30-2.10 (m, 4H).
Г. (S)-2-(2,6-диоксо(3-пиперидил))-4-нитроизоиндолин-1,3-дион
Перемешиваемую суспензию N-(4-нитрофталоил)-L-глутамина (4,3 г;
13,4 ммоль) в безводном метиленхлориде (170 мл) охлаждают до -40oС (баня IPA (изофталевая кислота)/сухой лед). К смеси по каплям добавляют тионилхлорид (1,03 мл; 14,5 ммоль) с последующим
добавлением пиридина (1,17 мл; 14,5 ммоль). Через 30 минут добавляют триэтиламин (2,06 мл; 14,8 ммоль) и смесь перемешивают при температуре от -30 до -40oС в течение 3 часов. Смеси
позволяют нагреться до комнатной температуры, ее фильтруют и промывают метиленхлоридом, что позволяет получить 2,3 г (57%) неочищенного продукта. Перекристаллизация из ацетона (300 мл) позволяет
получить 2 г продукта в виде твердого вещества белого цвета: т. пл. 259,0-284,0oС (разл. );1H ЯМР (ДМСО-d6) δ 11.19 (s, 1H), 8.34 (d, J= 7.8 Гц, 1Н), 8.23 (d,
J= 7.1 Гц, 1H), 8.12 (t, J= 7.8 Гц, 1H), 5.25-5.17 (dd, J= 5.2 и 12.7 Гц, 1H), 2.97-2.82 (m, 1H), 2.64-2.44 (m, 2H), 2.08-2.05 (m, 1H);13С ЯМР (ДМСО-d6) δ 172.67, 169.46,
165.15, 162.50, 144.42, 136.78, 132.99, 128.84, 127.27, 122.53, 49.41, 30.84, 21.71; ВЭЖХ, Waters Nova-Pak/C18; 3,9•150 мм, 4 микрона, 1 мл/мин, 240 нм, 10/90 CH3CN/0,
1%-ная Н3РO4 (водн. ), 4,27 мин (99,63%). Аналитич. рассчитано для С13Н9N3O6: С, 51,49; Н, 2,99; N, 13,86. Обнаружено: С, 51,67; Н, 2,
93; N, 13,57.
Д. S-4-амино-2-(2,6-диоксопиперид-3-ил)изоиндолин-1,3-дион
Смесь (S)-3-(4'-нитрофталимидо)-пиперидин-2,6-диона (0,76 г; 2,5 ммоль) и 10% Pd/C (0,3 г) в ацетоне
(200 мл) гидрируют в аппарате Парра-Шакера (Parr-Shaker) при давлении водорода 50 фунт-сила/кв. дюйм (344,75 кПа) в течение 24 часов. Смесь фильтруют через целит и фильтрат концентрируют in vacuo.
Твердый остаток суспендируют в горячем этилацетате в течение 30 мин и фильтруют, получая 0,47 г (69%) продукта в виде твердого вещества желтого цвета: т. пл. 309-310oС;1H ЯМР
(ДMCO-d6) δ 11.10 (s, 1H), 7.47 (dd, J= 7.2 и 8.3 Гц, 1H), 7.04-6.99 (dd, J= 6.9 и 8.3 Гц, 2H), 6.53 (s, 2H), 5.09-5.02 (dd, J= 5.3 и 12.4 Гц, 1H), 2.96-2.82 (m, 1H), 2.62-2.46 (m,
2H), 2.09-1.99 (m, 1H);13C ЯМР (ДМСО-d6) δ 172.80, 170.10, 168.57, 167.36, 146.71, 135.44, 131.98, 121.69, 110.98, 108.54, 48.48, 30.97,22.15; ВЭЖХ, Waters Nova-Pak/C18; 3,9•150 мм, 4 микрона, 1 мл/мин, 240 нм, 15/85 СН3СN/0,1%-ная Н3РО4 (водн. ), 4,99 мин (98,77%). Хиральный анализ, Daicel Chiral Pak AD, 0,46•
25 см, 1 мл/мин, 240 нм, 30/70 гексан/IPA, 9,55 мин (1,32%), 12,55 мин (97,66%). Аналитич. рассчитано для C13H11N3О4: С, 57,14; Н, 4,06; N, 15,38.
Обнаружено: С, 57,15; Н, 4,15; N, 14,99.
ПРИМЕР 15
R-4-амино-2-(2,6-диоксопиперид-3-ил)изоиндолин-1,3-дион
А. Трет-бутиловый эфир N-(4-нитрофталоил)-D-глутамина
Перемешиваемую смесь 4-нитро-N-этоксикарбонилфталимида (5,9 г; 22,3 ммоль), трет-бутилового эфира D-глутамина (4,5 г; 22,3 ммоль) и триэтиламина (0,9 г; 8,9 ммоль) в тетрагидрофуране (100 мл)
кипятят с обратным холодильником в течение 24 часов. Смесь разбавляют метиленхлоридом (100 мл) и промывают водой (2•50 мл), рассолом (50 мл) и затем высушивают. Растворитель удаляют in vacuо и
остаток очищают флэш-хроматографией (2%-ный СН3ОН в метиленхлориде), что позволяет получить 6,26 г (75%) продукта в виде стеклообразного материала:1Н ЯМР (CDC13)
δ 8.12 (d, J= 7.5 Гц, 2Н), 7.94 (dd, J= 7.9 и 9.1 Гц, 1H), 5.50 (b, 1H), 5.41 (b, 1H), 4.85 (dd, J= 5.1 и 9.8 Гц, 1Н), 2.61-2.50 (m, 2Н), 2.35-2.27 (m, 2H), 1.44 (s, 9Н);13С ЯМР
(CDC13) δ 173.77, 167.06, 165.25, 162.51, 145.07, 135.56, 133.78, 128.72, 127.27, 123.45, 83.23, 53.18, 32.27, 27.79, 24.42; ВЭЖХ, Waters Nova-Pak/C18; 3,9•150 мм,
4 микрона, 1 мл/мин, 240 нм, 25/75 СН3СN/0,1%-ная Н3РO4 (водн. ), 4,32 мин. (99,74%). Хиральный анализ, Daicel Chiral Pak AD, 0,46•25 см, 1 мл/мин, 240 нм,
55/45 гексан/IPA 5,88 мин (99,68%). Аналитич. рассчитано для C17H19N3О7: С, 54,11; Н, 5,08; N, 11,14. Обнаружено: С, 54,25; Н, 5,12; N, 10,85.
Б. N-(4-нитрофталоил)-D-глутамин
Газообразный хлористый водород барботируют в перемешиваемый при 5oС раствор трет-бутилового эфира N-(4-нитрофталоил)-D-глутамина (5,9 г;
15,6 ммоль) в метиленхлориде (100 мл) в течение 1 часа, после чего перемешивание продолжают еще час при комнатной температуре. Добавляют эфир (100 мл) и перемешивают в течение еще 30 минут. Смесь
фильтруют, твердое вещество промывают эфиром (60 мл) и высушивают (40oС, <1 мм рт. ст. (133,322 Па)), что позволяет получить 4,7 г (94%) продукта:1H ЯМР (ДМСО-d6) δ 8.33 (d, J= 7.8 Гц, 1Н), 8.22 (d, J= 7.2 Гц, 1Н), 8.11 (t, J= 7.8 Гц, 1Н), 7.19 (b, 1H), 6.72 (b, 1H). 4.81 (dd, J= 4.6 и 9.7 Гц, 1H), 2.39-2.12 (m, 4H);13С ЯМР
(ДMCO-d6) δ 173.21, 169.99, 165.41, 162.73, 144.45, 136.68, 132.98, 128.80, 127.23, 122.52, 51.87, 31.31, 23.87.
В. (R)-2-(2,
6-диоксо(3-пиперидил))-4-нитроизоиндолин-1,3-дион
Перемешиваемую суспензию N-(4'-нитрофталоил)-D-глутамина (4,3 г; 13,4 ммоль) в безводном метиленхлориде (170 мл) охлаждают до -40o
С с помощью бани со смесью изопропанол/сухой лед. По каплям добавляют тионилхлорид (1,7 г; 14,5 ммоль) с последующим добавлением пиридина (1,2 г; 14,5 ммоль). Через 30 мин добавляют триэтиламин (1,5 г;
14,8 ммоль) и смесь перемешивают при температуре от -30 до -40oС в течение 3 часов. Смесь фильтруют, твердое вещество промывают метиленхлоридом (50 мл) и высушивают (60oС,
<1 мм рт. ст. (133,322 Па)), получая 2,93 г продукта. Другие 0,6 г продукта получают из метиленхлоридного фильтрата. Обе фракции объединяют (3,53 г) и перекристаллизовывают из ацетона (450 мл),
что позволяет получить 2,89 г (71%) продукта в виде твердого вещества белого цвета: т. пл. 256,5-257,5oС;1H ЯМР (ДМСО-d6) δ 11.18 (s, 1H), 8.34 (dd, J= 0.8 и
7.9 Гц, 1H), 8.23 (dd, J= 0.8 и 7.5 Гц, 1H), 8.12 (t, J= 7.8 Гц, 1Н), 5.22 (dd, J= 5.3 и 12.8 Гц, 1Н), 2.97-2.82 (m, 1Н), 2.64-2.47 (m, 2H), 2.13-2.04 (m, 1H);13С ЯМР (ДМСО-d6)
δ 172.66, 169.44, 165.14, 162.48, 144.41, 136.76, 132.98, 128.83, 127.25, 122.52, 49.41, 30.83, 21.70; ВЭЖХ, Waters Nova-Pak/C18, 3,9•150 мм, 4 микрона, 1 мл/мин, 240 нм,
10/90 СН3СN/0,1%-ная Н3РO4 (водн. ), 3,35 мин (100%). Аналитич. рассчитано для C13H9N3O6: С, 51,49; Н, 2,99; N, 13,86.
Обнаружено: С, 51,55; Н, 2,82; N, 13,48.
Г. (R)-4-амино-2-(2,6-диоксопиперид-3-ил)изоиндолин-1,3-дион
Смесь R-3-(4'-нитрофталимидо)-пиперидин-2,6-диона (1,0 г; 3,3 ммоль) и
10% Pd/C (0,2 г) в ацетоне (250 мл) гидрируют в аппарате Парра-Шакера при давлении водорода 50 фунт-сила/кв. дюйм (344,75 кПа) в течение 4 часов. Смесь фильтруют через целит и фильтрат концентрируют
in vacuo. Полученное твердое вещество желтого цвета суспендируют в горячем этилацетате (20 мл) в течение 30 мин, получая после фильтрования и высушивания 0,53 г (59%) продукта в виде твердого вещества
желтого цвета: т. пл. 307,5-309,5oС;1Н ЯМР (ДМСО-d6) δ 11.06 (s, 1H), 7.47 (dd, J= 7.0 и 8.4 Гц, 1H), 7.02 (dd, J= 4.6 и 8.4 Гц, 2H), 6.53 (s, 2H), 5.07 (dd,
J= 5.4 и 12.5 Гц, 1H), 2.95-2.84 (m, 1H), 2.62-2.46 (m, 2H), 2.09-1.99 (m, 1H);13C ЯМР (ДМСО-d6) δ 172.78, 170.08, 168.56, 167.35, 146.70, 135.43, 131.98, 121.68, 110.95,
108.53, 48.47, 30.96, 22.14; ВЭЖХ, Waters Novа-Pak/C18, 3,9•150 мм, 4 микрона, 1 мл/мин, 240 нм, 10/90 СН3СN/0,1%-ная Н3РО4 (водн. ), 3,67 мин (99,
68%). Хиральный анализ, Daicel Chiral Pak AD, 0,46•25 см, 1 мл/мин, 240 нм, 30/70 гексан/IPA 7,88 мин (97,48%). Аналитич. рассчитано для C13H11N3О4:
С, 57,14; Н, 4,06; N, 15,38. Обнаружено: С, 57,34; Н, 3,91; N, 15,14.
ПРИМЕР 16
3-(4-Амино-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион
А. Метиловый эфир
2-бромметил-3-нитробензойной кислоты
Перемешиваемую смесь метилового эфира 2-метил-3-нитробензойной кислоты (14,0 г; 71,7 ммоль) и N-бромсукцинимида (15,3 г; 86,1 ммоль) в четыреххлористом
углероде (200 мл) мягко нагревают в течение 15 часов при температуре дефлегмации, освещая колбу электрической лампочкой мощностью 100 ватт, расположенной на расстоянии 2 см. Смесь фильтруют и твердое
вещество промывают метиленхлоридом (50 мл). Фильтрат промывают водой (2•100 мл), рассолом (100 мл) и высушивают. Растворитель удаляют in vacuo и остаток очищают флэш-хроматографией
(гексан/этилацетат, 8/2), что позволяет получить 19 г (96%) продукта в виде твердого вещества желтого цвета: т. пл. 70,0-71,5oС;1Н ЯМР (CDC13) δ 8.12-8.09 (dd.
J= 1.3 и 7.8 Гц, 1Н), 7.97-7.94 (dd, J= 1.3 и 8.2 Гц, 1H), 7.54 (t, J= 8.0 Гц, 1Н), 5.15 (s, 2Н), 4.00 (s, 3Н);13C ЯМР (CDC13) δ 165.85, 150.58, 134.68, 132.38, 129.08,
127.80, 53.06, 22.69; ВЭЖХ, Waters Nova-Pak/C18, 3,9•150 мм, 4 микрона, 1 мл/мин, 240 нм, 40/60 СН3СN/0,1%-ная Н3РO4 (водн. ), 7.27 мин (98,92%)
Аналитич. рассчитано для C9H8NO4Br: С, 39,44; Н, 2,94; N, 5,11; Br, 29,15. Обнаружено: С, 39,46; Н, 3,00; N, 5,00; Br, 29,11.
Б. Трет-бутиловый эфир
N-(1-оксо-4-нитроизоиндолин-2-ил)-L-глутамина
Триэтиламин (2,9 г; 28,6 ммоль) добавляют по каплям к перемешиваемой смеси метилового эфира 2-бромметил-3-нитробензойной кислоты (3,5 г; 13,0
ммоль) и трет-бутилового эфира L-глутамина гидрохлорида (3,1 г; 13,0 ммоль) в тетрагидрофуране (90 мл). Смесь нагревают до температуры дефлегмации в течение 24 часов. К охлажденной смеси добавляют
метиленхлорид (150 мл) и смесь промывают водой (2•40 мл), рассолом (40 мл) и высушивают. Растворитель удаляют in vacuо и остаток очищают флэш-хроматографией (3% СН3ОН в
метиленхлориде), что позволяет получить 2,84 г (60%) неочищенного продукта, который непосредственно используют в следующей реакции:1Н ЯМР (СDС13) δ 8.40 (d, J= 8.1 Гц,
1Н), 8.15 (d, J= 7.5 Гц, 1Н), 7.71 (t, J= 7.8 Гц, 1Н), 5.83 (s, 1H), 5.61 (s, 1H), 5.12 (d, J= 19.4 Гц, 1Н), 5.04-4.98 (m, 1H), 4.92 (d, J= 19.4 Гц, 1H), 2.49-2.22 (m, 4H), 1.46 (s, 9H); ВЭЖХ, Waters
Nova-Pak/C18, 3,9•150 мм, 4 микрона, 1 мл/мин, 240 нм, 25/75 СН3СN/0,1%-ная Н3РО4 (водн. ), 6,75 мин (99,94%).
В.
N-(1-оксо-4-нитроизоиндолин-2-ил)-L-глутамин
Газообразный хлористый водород барботируют в перемешиваемый при 5oС раствор трет-бутилового эфира
N-(1-оксо-4-нитроизоиндолин-2-ил)-L-глутамина (3,6 г; 9,9 ммоль) в метиленхлориде (60 мл) в течение 1 часа. Затем смесь перемешивают при комнатной температуре в течение еще одного часа. Добавляют эфир
(40 мл) и полученную смесь перемешивают в течение 30 минут. Суспензию фильтруют, промывают эфиром и высушивают, что позволяет получить 3,3 г продукта:1Н ЯМР (ДМСО-d6) δ
8.45 (d, J= 8.1 Гц, 1H), 8.15 (d, J= 7.5 Гц, 1H), 7.83 (t, J= 7.9 Гц, 1Н), 7.24 (s, 1H), 6.76 (s, 1H), 4.93 (s, 2H), 4.84-4.78 (dd, J= 34.8 и 10.4 Гц, 1H), 2.34-2.10 (m, 4H);13С ЯМР
(ДМСО-d6) δ 173.03, 171.88, 165.96, 143.35, 137.49, 134.77, 130.10, 129.61, 126.95, 53.65, 48.13, 31.50, 24.69. Аналитич. рассчитано для C13H13N3
O6: С, 50,82; Н, 4,26; N, 13,68. Обнаружено: С, 50,53; Н, 4,37; N, 13,22.
Г. (S)-3-(1-оксо-4-нитроизоиндолин-2-ил)пиперидин-2,6-дион
Перемешиваемую суспендированную
смесь N-(1-оксо-4-нитроизоиндолин-2-ил)-L-глутамина (3,2 г; 10,5 ммоль) в безводном метиленхлориде (150 мл) охлаждают до -40oС с помощью бани со смесью изопропанол/сухой лед. К охлажденной
смеси по каплям добавляют тионилхлорид (0,82 мл; 11,3 ммоль) с последующим добавлением пиридина (0,9 г; 11,3 ммоль). Через 30 мин добавляют триэтиламин (1,2 г; 11,5 ммоль) и смесь перемешивают при
температуре от -30 до -40oС в течение 3 часов. Смесь выливают в ледяную воду (200 мл) и водный слой экстрагируют метиленхлоридом (40 мл). Метиленхлоридный раствор промывают водой (2•
60 мл), рассолом (60 мл) и высушивают. Растворитель удаляют in vacuo и твердый остаток суспендируют с помощью этилацетата (20 мл), получая 2,2 г (75%) продукта в виде твердого вещества белого цвета:
т. пл. 285oС;1Н ЯМР (ДМСО-d6) δ 11.04 (s, 1H), 8.49-8.45 (dd, J= 0.8 и 8.2 Гц, 1H), 8.21-8.17 (dd, J= 7.3 Гц, 1H), 7.84 (t, J= 7.6 Гц, 1H), 5.23-5.15 (dd, J=
4.9 и 13.0 Гц, 1H), 4.96 (dd, J= 19.3 и 32.4 Гц, 2Н), 3.00-2.85 (m, 1H), 2.64-2.49 (m, 2H), 2.08-1.98 (m, 1H);13С ЯМР (ДMCO-d6) δ 172.79, 170.69, 165.93, 143.33, 137.40,
134.68, 130.15, 129.60, 127.02, 51.82, 48.43, 31.16, 22.23; ВЭЖХ, Waters Novа-Pak/C18, 3,9•150 мм, 4 микрона, 1 мл/мин, 240 нм, 20/80 СН3СN/0,1%-ная Н3РО4 (водн. ), 3,67 мин (100%). Аналитич. рассчитано для С13Н11N3О5: С, 53,98; Н, 3,83; N, 14,53. Обнаружено: С, 53,92; Н, 3,70; N, 14,10.
Д. (S)-3-(1-оксо-4-аминоизоиндолин-2-ил)пиперидин-2,6-дион
Смесь (S)-3-(1-оксо-4-нитроизоиндолин-2-ил)пиперидин-2,6-диона (1,0 г; 3,5 ммоль) и 10% Pd/C (0,3 г) в метаноле (600 мл) гидрируют
в аппарате Парра-Шакера при давлении водорода 50 фунт-сила/кв. дюйм (344,75 кПа) в течение 5 часов. Смесь фильтруют через целит (Celite) и фильтрат концентрируют in vacuo. Твердое вещество
суспендируют в горячем этилацетате в течение 30 мин, фильтруют и высушивают, что позволяет получить 0,46 г (51%) продукта в виде твердого вещества белого цвета: т. пл. 235,5-239oС;1H ЯМР (ДМСО-d6) δ 11.01 (s, 1H), 7.19 (t, J= 7.6 Гц, 1H), 6.90 (d, J= 7.3 Гц, 1H), 6.78 (d, J= 7.8 Гц, 1Н), 5.42 (s, 2Н), 5.12 (dd, J= 5.1 и 13.1 Гц, 1Н), 4.17 (dd, J= 17.0 и
28.8 Гц, 2Н), 2.92-2.85 (m, 1H), 2.64-2.49 (m, 1H), 2.34-2.27 (m, 1H), 2.06-1.99 (m, 1H);13C ЯМР (ДМСО-d6) δ 172.85, 171.19, 168.84, 143.58, 132.22, 128.79, 125.56,
116.37, 110.39, 51.48, 45.49, 31.20, 22.74; ВЭЖХ, Waters Nova-Pak/C18, 3,9•150 мм, 4 микрона, 1 мл/мин, 240 нм, 10/90 СН3СN/0,1%-ная Н3РО4 (водн. ),
0,96 мин (100%). Хиральный анализ: Daicel Chiral Pak AD, 40/60 гексан/IPA, 6,60 мин (99,42%); аналитич. рассчитано для С13Н13N3O3: С, 60,23; Н, 5,05; N, 16,
21. Обнаружено: С 59,96; Н, 4,98; N, 15,84.
ПРИМЕР 17
3-(4-Амино-1-оксоизоиндолин-2-ил)-3-метилпиперидин-2,6-дион
А. N-бензилоксикарбонил-3-амино-3-метилпиперидин-2,
6-дион
Перемешиваемую смесь N-бензилоксикарбонил-α-метил-изоглутамина (11,3 г; 38,5 ммоль), 1,1'-карбонилдиимидазола (6,84 г; 42,2 ммоль) и 4-диметиламинопиридина (0,05 г) в
тетрагидрофуране (125 мл) нагревают в атмосфере азота до температуры дефлегмации в течение 19 часов. Реакционную смесь концентрируют in vacuо до масла. Масло суспендируют в воде (50 мл) в течение 1
часа, затем фильтруют, промывают водой, высушивают на воздухе, что позволяет получить 7,15 г твердого вещества белого цвета. Неочищенный продукт очищают флэш-хроматографией (2: 8, этилацетат:
метиленхлорид), что позволяет получить 6,7 г (63%) продукта в виде твердого вещества белого цвета: т. пл. 151-152oС;1H ЯМР (СDС13) δ 8.24 (s, 1H), 7.35 (s, 5Н),
5.6 (s, 1H), 5.09 (s, 2Н), 2.82-2.53 (m, 3H), 2.33-2.26 (m, 1H), 1.56 (s, 3Н);13C ЯМР (СDС13) δ 174.4, 172.4, 154.8, 136.9, 128.3, 127.8, 127.7, 65.3, 54.6, 29.2, 29.0,
22.18; ВЭЖХ: Waters Nova-Pak/C18-колонка, 4 микрона, 3,9•150 мм, 1 мл/мин, 240 нм, 20/80 СН3СN/Н3РО4 (водн. ), 6,6 мин (100%). Аналитич. рассчитано
для Cl4Hl6N2O4: C 60,86; Н, 5,84; N, 10,14. Обнаружено: С, 60,94; Н, 5,76; N, 10,10.
Б. 3-Амино-3-метилпиперидин-2,6-дион
N-Бензилоксикарбонил-3-амино-3-метилпиперидин-2,6-дион (3,0 г; 10,9 ммоль) растворяют при мягком нагревании в этаноле (270 мл) и затем охлаждают до комнатной температуры. К этому раствору добавляют 4
н НС1 (7 мл) с последующим добавлением 10% Pd/C (0,52 г). Смесь гидрируют при давлении водорода 50 фунт-сила/кв. дюйм (344,75 кПа) в течение 3 часов. Далее для растворения продукта к смеси добавляют
воду (65 мл). Смесь фильтруют через набивку целита и целитную набивку промывают водой (100 мл). Фильтрат концентрируют in vacuo до твердого остатка. Это твердое вещество суспендируют в этаноле (50 мл)
в течение 30 мин. Суспензию фильтруют, что позволяет получить 3,65 г (94%) продукта в виде твердого вещества белого цвета:1Н ЯМР (ДМСО-d6) δ 11.25 (s, 1H), 8.9 (s, 3Н),
2.87-2.57 (m, 2Н), 2.35-2.08 (m, 2H), 1.54 (s, 3Н); ВЭЖХ (Waters Nova-Pak/C18-колонка, 4 микрона, 1 мл/мин, 240 нм, 15/85 СН3СN/Н3РO4 (водн. ), 1,07 мин,
100%).
В. 3-Метил-3-(4-нитро-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион
К перемешиваемой смеси α-амино-α-метил-глутаримида гидрохлорида (2,5 г; 14,0 ммоль) и
метилового эфира 2-бромметил-3-нитробензойной кислоты (3,87 г; 14 ммоль) в диметилформамиде (40 мл) добавляют в атмосфере азота триэтиламин (3,14 г; 30,8 ммоль). Смесь нагревают до температуры
дефлегмации в течение 6 часов. Смесь охлаждают и далее концентрируют in vacuo. Твердый остаток суспендируют в воде (50 мл) и метиленхлориде в течение 30 мин. Суспензию фильтруют, твердое вещество
промывают метиленхлоридом и высушивают (60oС, < 1 мм). Перекристаллизация из метанола (80 мл) дает 0,63 г (15%) продукта в виде твердого вещества беловатой окраски: т. пл.
195-197oС;1Н ЯМР (ДМСО-d6) δ 10.95 (s, 1H), 8.49-8.46 (d, J= 8.2 Гц, 1Н), 8.13-8.09 (d, J= 7.4 Гц, 1Н), 7.86-7.79 (t, J= 7.8 Гц, 1Н), 5.22-5.0 (dd, J= 19.4 и
34.6 Гц, 2Н), 2.77-2.49 (m, 3Н), 2.0-1.94 (m, 1H), 1.74 (s, 3Н);13С ЯМР (ДМСО-d6) δ 173.1, 172.3, 165.0, 143.2, 137.4, 135.2, 130.1, 129.3, 126.9, 57.6, 48.7, 28.9, 27.7,
20.6; ВЭЖХ (Waters Nova-Pak/C18-колонка, 4 микрона, 1 мл/мин, 240 нм, 20/80 СН3СN/Н3РO4 (водн. ), 4,54 мин, 99,6%). Аналитич. рассчитано для C14
H13N3О5: С, 55,45; Н, 4,32; N, 13,86. Обнаружено: С, 55,30; Н, 4,48; N, 13,54.
Г. 3-Метил-3-(4-амино-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион
3-Метил-3-(4-нитро-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион (1,0 г; 3,3 ммоль) растворяют при мягком нагревании в метаноле (500 мл) и затем охлаждают до комнатной температуры. К этому раствору в
атмосфере азота добавляют 10% Pd/C (0,3 г). Смесь гидрируют в аппарате Парра-Шакера при давлении водорода 50 фунт-сила/кв. дюйм (344,75 кПа) в течение 4 часов. Смесь фильтруют через набивку целита и
набивку целита промывают метанолом (50 мл). Фильтрат концентрируют in vacuo до твердого вещества беловатой окраски. Твердое вещество суспендируют в метиленхлориде (20 мл) в течение 30 мин. Суспензию
фильтруют и твердое вещество высушивают (60oС, < 1 мм). Твердое вещество перекристаллизовывают из метанола (3 раза, 100 мл за раз), получая 0,12 г (13,3%) продукта в виде твердого
вещества белого цвета: т. пл. 289-292oС;1H ЯМР (ДMCO-d6) δ 10.85 (s, 1H), 7.19-7.13 (t, J= 7.6 Гц, 1H), 6.83-6.76 (m, 2H), 5.44 (s, 2H), 4.41 (s, 2H),
2.71-2.49 (m, 3Н), 1.9-1.8 (m, 1H), 1.67 (s, 3Н);13C ЯМР (ДМСО-d6) δ 173.7, 172.5. 168.0, 143.5, 132.9, 128.8, 125.6, 116.1, 109.9, 57.0, 46.2, 29.0, 27.8, 20.8; ВЭЖХ
(Waters Nova-Pak/C18-колонка, 4 микрона, 1 мл/мин, 240 нм, 20/80 СН3СN/Н3РО4 (водн. ), 1,5 мин, 99,6%). Аналитич. рассчитано для C14H15
N3O3: С, 61,53; Н, 5,53; N, 15,38. Обнаружено: С, 61,22; Н, 5,63; N, 15,25.
ПРИМЕР 18
Таблетки, содержащие по 50 мг 1,3-диоксо-2-(2,
6-диоксопиперидин-3-ил)-5-аминоизоиндолина каждая, могут быть приготовлены следующим образом:
Составляющие (на 1000 таблеток):
1,3-диоксо-2-(2,
6-диоксо- пиперидин-3-ил)-5-амино- изоиндолин - 50,0 г
лактоза - 50,7 г
пшеничный крахмал - 7,5 г
полиэтиленгликоль 6000 - 5,0 г
тальк - 5,0 г
стеарат
магния - 1,8 г
деминерализованная вода - сколько требуется
Твердые ингредиенты первоначально продавливают через сито с размером ячейки 0,6 мм. После этого смешивают активный
ингредиент, лактозу, тальк, стеарат магния и половину крахмала. Вторую половину крахмала суспендируют в 40 мл воды и эту суспензию добавляют к кипящему раствору полиэтиленгликоля в 100 мл воды.
Полученную пасту добавляют к порошкообразным материалам и смесь гранулируют, при необходимости добавляя воду. Гранулят высушивают в течение ночи при 35oС, продавливают через сито с размером
ячейки 1,2 мм и прессуют для формирования таблеток диаметром приблизительно 6 мм, которые вогнуты с обеих сторон.
ПРИМЕР 19
Таблетки, содержащие по 100 мг 1,3-диоксо-2-(2,
6-диоксопиперидин-3-ил)-5-аминоизоиндолина каждая, могут быть приготовлены следующим образом:
Составляющие (на 1000 таблеток):
1,3-диоксо-2-(2,
6-диоксо- пиперидин-3-ил)-5-амино- изоиндолин - 100,0 г
лактоза - 100,0 г
пшеничный крахмал - 47,0 г
стеарат магния - 3,0 г
Твердые ингредиенты первоначально
продавливают через сито с размером ячейки 0,6 мм. После этого смешивают активный ингредиент, лактозу, стеарат магния и половину крахмала. Вторую половину крахмала суспендируют в 40 мл воды и эту
суспензию добавляют к 100 мл кипящей воды. Полученную пасту добавляют к порошкообразным материалам и смесь гранулируют, при необходимости добавляя воду. Гранулят высушивают в течение ночи при 35oС, продавливают через сито с размером ячейки 1,2 мм и прессуют для формирования таблеток диаметром приблизительно 6 мм, которые вогнуты с обеих сторон.
ПРИМЕР 20
Таблетки для жевания, содержащие по 75 мг 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4-аминоизоиндолина каждая, могут быть приготовлены следующим образом:
Состав (на 1000 таблеток):
1-оксо-2-(2,6-диоксо- пиперидин-3-ил)-4-амино- изоиндолин - 75,0 г
маннит - 230,0 г
лактоза - 150,0 г
тальк - 21,0 г
глицин - 12,5 г
стеариновая кислота
- 10,0 г
сахарин - 1,5 г
5%-ный раствор желатина - сколько требуется
Все твердые ингредиенты первоначально продавливают через сито с размером ячейки 0,25 мм. Маннит и
лактозу смешивают, гранулируют с добавлением раствора желатина, продавливают через сито с размером ячейки 2 мм, высушивают при 50oС и еще раз продавливают через сито с размером ячейки 1,7
мм. 1-Оксо-2-(2,6-диоксопиперидин-3-ил)-4-аминоизоиндолин, глицин и сахарин аккуратно смешивают, добавляют маннит, лактозный гранулят, стеариновую кислоту и тальк, все тщательно перемешивают и
прессуют для формирования таблеток диаметром приблизительно 10 мм, которые вогнуты с обеих сторон и имеют желобок для разламывания на верхней стороне.
ПРИМЕР 21
Таблетки,
содержащие по 10 мг 1-оксо-2-(2,6-диоксопиперидин-3-ил)-5-аминоизоиндолина каждая, могут быть приготовлены следующим образом:
Состав (на 1000 таблеток):
1-оксо-2-(2,
6-диоксо- пиперидин-3-ил)-5-амино- изоиндолин - 10,0 г
лактоза - 328,5 г
кукурузный крахмал - 17,5 г
полиэтиленгликоль 6000 - 5,0 г
тальк - 25,0 г
стеарат
магния - 4,0 г
деминерализованная вода - сколько требуется
Твердые ингредиенты первоначально продавливают через сито с размером ячейки 0,6 мм. После этого смешивают активный имидный
ингредиент, лактозу, тальк, стеарат магния и половину крахмала до образования однородной смеси. Вторую половину крахмала суспендируют в 65 мл воды и эту суспензию добавляют к кипящему раствору
полиэтиленгликоля в 260 мл воды. Полученную пасту добавляют к порошкообразным материалам, все перемешивают и гранулируют, при необходимости добавляя воду. Гранулят высушивают в течение ночи при 35oС, продавливают через сито с размером ячейки 1,2 мм и прессуют для формирования таблеток диаметром приблизительно 10 мм, которые вогнуты с обеих сторон и имеют бороздку для разламывания на
верхней стороне.
ПРИМЕР 22
Желатиновые капсулы сухого заполнения, содержащие по 100 мг l-oкco-2-(2,6-диоксопиперидин-3-ил)-6-аминоизоиндолина каждая, могут быть приготовлены
следующим образом:
Состав (на 1000 капсул)
1-оксо-2-(2,6-диоксо- пиперидин-3-ил)-6-амино- изоиндолин - 100,0 г
микрокристаллическая целлюлоза - 30,0 г
лаурилсульфат
натрия - 2,0 г
стеарат магния - 8,0 г
Лаурилсульфат натрия просеивают в 1-оксо-2-(2,6-диоксопиперидин-3-ил)-6-аминоизоиндолин через сито с размером ячейки 0,2 мм и два компонента
смешивают в течение 10 минут до образования однородной смеси. Далее через сито с размером ячейки 0,9 мм добавляют микрокристаллическую целлюлозу и все еще раз смешивают в течение 10 минут до
образования однородной смеси. И наконец, через сито с размером ячейки 0,8 мм добавляют стеарат магния и, после перемешивания в течение еще 3 минут, данной смесью, порциями по 140 мг каждая, заполняют
желатиновые капсулы сухого заполнения размером 0 (удлиненные).
ПРИМЕР 23
0,2%-ный раствор для инъекции или инфузии может быть приготовлен, например, следующим образом:
1-оксо-2-(2,6-диоксо- пиперидин-3-ил)-7-амино- изоиндолин - 5,0 г
хлорид натрия - 22,5 г
фосфатный буфер рН 7,4 - 300,0 г
деминерализованная вода - до 2500,0 мл
1-Оксо-2-(2,6-диоксопиперидин-3-ил)-7-аминоизоиндолин растворяют в 1000 мл воды и фильтруют через микрофильтр. Добавляют буферный раствор и общий объем доводят водой до 2500 мл. Для получения
стандартных лекарственных форм порции по 1,0 или 2,5 мл каждая вводят в стеклянные ампулы (содержащие соответственно 2,0 или 5,0 мг имида каждая).
ПРИМЕР 24
Таблетки,
содержащие по 50 мг 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4,5,6,7-тетрафторизоиндолина каждая, могут быть приготовлены следующим образом:
Составляющие (на 1000 таблеток)
1-оксо-2-(2,
6-диоксо- пиперидин-3-ил)-4,5,6,7- тетрафторизоиндолин - 50,0 г
лактоза - 50,7 г
пшеничный крахмал - 7,5 г
полиэтиленгликоль 6000 - 5,0 г
тальк - 5,0 г
стеарат магния - 1,8 г
деминерализованная вода - сколько требуется
Твердые ингредиенты первоначально продавливают через сито с размером ячейки 0,6 мм. После этого смешивают активный
ингредиент, лактозу, тальк, стеарат магния и половину крахмала. Вторую половину крахмала суспендируют в 40 мл воды и эту суспензию добавляют к кипящему раствору полиэтиленгликоля в 100 мл воды.
Полученную пасту добавляют к порошкообразным материалам и смесь гранулируют, при необходимости добавляя воду. Гранулят высушивают в течение ночи при 35oС, продавливают через сито с размером
ячейки 1,2 мм и прессуют для формирования таблеток диаметром приблизительно 6 мм, которые вогнуты с обеих сторон.
ПРИМЕР 25
Таблетки, содержащие по 100 мг 1-оксо-2-(2,
6-диоксопиперидин-3-ил)-4,5,6,7-тетрахлоризоиндолина каждая, могут быть приготовлены следующим образом:
Составляющие (на 1000 таблеток)
1-оксо-2-(2,6-диоксо- пиперидин-3-ил)-4,5,6,
7- тетрахлоризоиндолин - 100,0 г
лактоза - 100,0 г
пшеничный крахмал - 47,0 г
стеарат магния - 3,0 г
Все твердые ингредиенты первоначально продавливают через сито с
размером ячейки 0,6 мм. После этого смешивают активный ингредиент, лактозу, стеарат магния и половину крахмала. Вторую половину крахмала суспендируют в 40 мл воды и эту суспензию добавляют к 100 мл
кипящей воды. Полученную пасту добавляют к порошкообразным материалам и смесь гранулируют, при необходимости добавляя воду. Гранулят высушивают в течение ночи при 35oС, продавливают через
сито с размером ячейки 1,2 мм и прессуют для формирования таблеток диаметром приблизительно 6 мм, которые вогнуты с обеих сторон.
ПРИМЕР 26
Таблетки для жевания, содержащие по
75 мг 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4,5,6,7-тетрафторизоиндолина каждая, могут быть приготовлены следующим образом:
Состав (на 1000 таблеток)
1-оксо-2-(2,
6-диоксо- пиперидин-3-ил)-4,5,6,7- тетрафторизоиндолин - 75,0 г
маннит - 230,0 г
лактоза - 150,0 г
тальк - 21,0 г
глицин - 12,5 г
стеариновая кислота - 10,
0 г
сахарин - 1,5 г
5%-ный раствор желатина - сколько требуется
Все твердые ингредиенты первоначально продавливают через сито с размером ячейки 0,25 мм. Маннит и лактозу
смешивают, гранулируют с добавлением раствора желатина, продавливают через сито с размером ячейки 2 мм, высушивают при 50oС и еще раз продавливают через сито с размером ячейки 1,7 мм.
1-Оксо-2-(2,6-диоксопиперидин-3-ил)-4,5,6,7-тетрафторизоиндолин, глицин и сахарин аккуратно смешивают, добавляют маннит, лактозный гранулят, стеариновую кислоту и тальк, все тщательно перемешивают и
прессуют для формирования таблеток диаметром приблизительно 10 мм, которые вогнуты с обеих сторон и имеют желобок для разламывания на верхней стороне.
ПРИМЕР 27
Таблетки,
содержащие по 10 мг 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4,5,6,7-тетраметилизоиндолина каждая, могут быть приготовлены следующим образом:
Состав (на 1000 таблеток)
1-оксо-2-(2,
6-диоксо- пиперидин-3-ил)-4,5,6,7- тетраметилизоиндолин - 10,0 г
лактоза - 328,5 г
кукурузный крахмал - 17,5 г
полиэтиленгликоль 6000 - 5,0 г
тальк - 25,0 г
стеарат магния - 4,0 г
деминерализованная вода - сколько требуется
Твердые ингредиенты первоначально продавливают через сито с размером ячейки 0,6 мм. После этого смешивают активный
имидный ингредиент, лактозу, тальк, стеарат магния и половину крахмала до образования однородной смеси. Вторую половину крахмала суспендируют в 65 мл воды и эту суспензию добавляют к кипящему раствору
полиэтиленгликоля в 260 мл воды. Полученную пасту добавляют к порошкообразным материалам, все смешивают и гранулируют, при необходимости добавляя воду. Гранулят высушивают в течение ночи при 35oС, продавливают через сито с размером ячейки 1,2 мм и прессуют для формирования таблеток диаметром приблизительно 10 мм, которые вогнуты с обеих сторон и имеют бороздку для разламывания на
верхней стороне.
ПРИМЕР 28
Желатиновые капсулы сухого заполнения, содержащие по 100 мг 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4,5,6,7-тетраметоксиизоиндолина каждая, могут быть
приготовлены следующим образом:
Состав (на 1000 капсул)
1-оксо-2-(2,6-диоксо- пиперидин-3-ил)-4,5,6,7- тетраметокси- изоиндолин - 100,0 г
микрокристаллическая целлюлоза - 30,
0 г
лаурилсульфат натрия - 2,0 г
стеарат магния - 8,0 г
Лаурилсульфат натрия просеивают в 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4,5,6,7-тетраметоксиизоиндолин через сито с
размером ячейки 0,2 мм и два компонента смешивают в течение 10 минут до образования однородной смеси. Далее через сито с размером ячейки 0,9 мм добавляют микрокристаллическую целлюлозу и все еще раз
перемешивают в течение 10 минут до образования однородной смеси. И наконец, через сито с размером ячейки 0,8 мм добавляют стеарат магния и, после перемешивания в течение еще 3 минут данной смесью
порциями по 140 мг каждая заполняют желатиновые капсулы сухого заполнения размером 0 (удлиненные).
ПРИМЕР 30
0,2%-ный раствор для инъекции или инфузии может быть приготовлен,
например, следующим образом:
1-оксо-2-(2,6-диоксопиперидин-3-ил)- 4,5,6,7-тетрафторизоиндолин - 5,0 г
хлорид натрия - 22,5 г
фосфатный буфер рН 7,4 - 300,0 г
деминерализованная вода - до 2500,0 мл
1-Оксо-2-(2,6-диоксопиперидин-3-ил)-4,5,6,7-тетрафторизоиндолин растворяют в 1000 мл воды и фильтруют через микрофильтр. Добавляют буферный раствор и
общий объем доводят водой до 2500 мл. Для получения стандартных лекарственных форм порции по 1,0 или 2,5 мл каждая вводят в стеклянные ампулы (содержащие соответственно 2,0 или 5,0 мг имида
каждая).
ПРИМЕР 31
Таблетки, содержащие по 50 мг 1-оксо-2-(2,6-диоксо-3-метилпиперидин-3-ил)-4,5,6,7-тетрафторизоиндолина каждая, могут быть приготовлены следующим образом:
Составляющие (на 1000 таблеток)
1-оксо-2-(2,6-диоксо-3-метил- пиперидин-3-ил)-4,5,6,7- тетрафторизоиндолин - 50,0 г
лактоза - 50,7 г
пшеничный крахмал - 7,5 г
полиэтиленгликоль 6000 - 5,0 г
тальк - 5,0 г
стеарат магния - 1,8 г
деминерализованная вода - сколько требуется
Твердые ингредиенты первоначально продавливают через
сито с размером ячеек 0,6 мм. После этого смешивают активный ингредиент, лактозу, тальк, стеарат магния и половину крахмала. Вторую половину крахмала суспендируют в 40 мл воды и эту суспензию
добавляют к кипящему раствору полиэтиленгликоля в 100 мл воды. Полученную пасту добавляют к порошкообразным материалам и смесь гранулируют, при необходимости добавляя воду. Гранулят высушивают в
течение ночи при 35oС, продавливают через сито с размером ячейки 1,2 мм и прессуют для формирования таблеток диаметром приблизительно 6 мм, которые вогнуты с обеих сторон.
ПРИМЕР 32
Таблетки, содержащие по 100 мг 1-оксо-2-(2,6-диоксопиперидин-3-ил)-4-аминоизоиндолина каждая, могут быть приготовлены следующим образом:
Составляющие (на 1000 таблеток)
1-оксо-2-(2,6-диоксо- пиперидин-3-ил)-4-амино- изоиндолин - 100,0 г
лактоза - 100,0 г
пшеничный крахмал - 47,0 г
стеарат магния - 3,0 г
Все твердые
ингредиенты первоначально продавливают через сито с размером ячейки 0,6 мм. После этого смешивают активный ингредиент, лактозу, стеарат магния и половину крахмала. Вторую половину крахмала
суспендируют в 40 мл воды и эту суспензию добавляют к 100 мл кипящей воды. Полученную пасту добавляют к порошкообразным материалам и смесь гранулируют, при необходимости добавляя воду. Гранулят
высушивают в течение ночи при 35oС, продавливают через сито с размером ячеек 1,2 мм и прессуют для формирования таблеток диаметром приблизительно 6 мм, которые вогнуты с обеих сторон.
ПРИМЕР 33
Таблетки для жевания, содержащие по 75 мг 2-(2,6-диоксо-3-метилпиперидин-3-ил)-4-аминофталимида каждая, могут быть приготовлены следующим образом:
Состав (на 1000
таблеток)
2-(2,6-диоксо-3-метилпиперидин- 3-ил)-4-аминофталимид - 75,0 г
маннит - 230,0 г
лактоза - 150,0 г
тальк - 21,0 г
глицин - 12,5 г
стеариновая кислота - 10,0 г
сахарин - 1,5 г
5%-ный раствор желатина - сколько требуется
Все твердые ингредиенты первоначально продавливают через сито с размером ячейки 0,25
мм. Маннит и лактозу смешивают, гранулируют с добавлением раствора желатина, продавливают через сито с размером ячейки 2 мм, высушивают при 50oС и еще раз продавливают через сито с размером
ячейки 1,7 мм. 2-(2,6-диоксо-3-метилпиперидин-3-ил)-4-аминофталимид, глицин и сахарин аккуратно смешивают, добавляют маннит, лактозный гранулят, стеариновую кислоту и тальк, все тщательно перемешивают
и прессуют для формирования таблеток диаметром приблизительно 10 мм, которые вогнуты с обеих сторон и имеют желобок для разламывания на верхней стороне.
ПРИМЕР 34
Таблетки,
содержащие по 10 мг 2-(2,6-диоксоэтилпиперидин-3-ил)-4-аминофталимида каждая, могут быть приготовлены следующим образом:
Состав (на 1000 таблеток)
2-(2,
6-диоксоэтилпиперидин-3-ил)- 4-аминофталимид - 10,0 г
лактоза - 328,5 г
кукурузный крахмал - 17,5 г
полиэтиленгликоль 6000 - 5,0 г
тальк - 25,0 г
стеарат
магния - 4,0 г
деминерализованная вода - сколько требуется
Твердые ингредиенты первоначально продавливают через сито с размером ячейки 0,6 мм. После этого смешивают активный имидный
ингредиент, лактозу, тальк, стеарат магния и половину крахмала до образования однородной смеси. Вторую половину крахмала суспендируют в 65 мл воды и эту суспензию добавляют к кипящему раствору
полиэтиленгликоля в 260 мл воды. Полученную пасту добавляют к порошкообразным материалам, все смешивают и гранулируют, при необходимости добавляя воду. Гранулят высушивают в течение ночи при 35oС, продавливают через сито с размером ячейки 1,2 мм и прессуют для формирования таблеток диаметром приблизительно 10 мм, которые вогнуты с обеих сторон и имеют бороздку для разламывания на
верхней стороне.
ПРИМЕР 35
Желатиновые капсулы сухого заполнения, содержащие по 100 мг 1-оксо-2-(2,6-диоксо-3-метилпиперидин-3-ил)-4,5,6,7-тетрафторизоиндолина каждая, могут
быть приготовлены следующим образом:
Состав (на 1000 капсул)
1-оксо-2-(2,6-диоксо-3- метилпиперидин-3-ил)-4,5,6,7- тетрафторизоиндолин - 100,0 г
микрокристаллическая
целлюлоза - 30,0 г
лаурилсульфат натрия - 2,0 г
стеарат магния - 8,0 г
Лаурилсульфат натрия просеивают в 1-оксо-2-(2,6-диоксо-3-метилпиперидин-3-ил)-4,5,6,
7-тетрафторизоиндолин через сито с размером ячейки 0,2 мм и два компонента смешивают в течение 10 минут до образования однородной смеси. Далее через сито с размером ячейки 0,9 мм добавляют
микрокристаллическую целлюлозу и все еще раз перемешивают в течение 10 минут до образования однородной смеси. И наконец, через сито с размером ячейки 0,8 мм добавляют стеарат магния и после
перемешивания в течение еще 3 минут смесью порциями по 140 мг каждая заполняют желатиновые капсулы сухого заполнения размером 0 (удлиненные).
ПРИМЕР 36
0,2%-ный раствор для
инъекции или инфузии может быть приготовлен, например, следующим образом:
1-оксо-2-(2,6-диоксо-3-метил- пиперидин-3-ил)-4,5,6,7-тетрафтор- изоиндолин - 5,0 г
хлорид натрия - 22,5
г
фосфатный буфер рН 7,4 - 300,0 г
деминерализованная вода - до 2500,0 мл
1-Оксо-2-(2,6-диоксо-3-метилпиперидин-3-ил)-4,5,6,7-тетрафторизоиндолин растворяют в 1000 мл воды и
фильтруют через микрофильтр. Добавляют буферный раствор и общий объем
доводят водой до 2500 мл. Для получения стандартных лекарственных форм порции по 1,0 или 2,5 мл каждая вводят в стеклянные
ампулы (содержащие соответственно 2,0 или 5,0 мг имида каждая).
Пример 37
2-(2,6-Диоксо-пиперидин-3-ил)-4-пентиламино-изоиндол-1,3-дион
Диметиловый эфир
3-пентиламино-фталевой кислоты
К перемешиваемому раствору диметилового эфира 3-амино-фталевой кислоты (3,14 г, 15 ммоль) в метиленхлориде (50 мл) в атмосфере азота добавляли валеральдегид (2,0 мл, 18,75 ммоль) и уксусную кислоту (5,18 мл, 90 ммоль). Смесь перемешивали в течение 5 минут, после чего добавляли триацетоксиборгидрид натрия (6,36 г, 30 ммоль). Реакционную смесь перемешивали в течение 30 минут, разбавляли метиленхлоридом (50 мл), промывали водой (2•100 мл), насыщенным водным бикарбонатом натрия (2•100 мл), рассолом (100 мл) и сушили (МgSO4). Растворитель выпаривали в вакууме с получением 4,19 г продукта (100%), который использовали без последующей очистки.1H ЯМР (ДМСО-d6) δ 7.31 (t, J= 7.9 Гц, 1Н), 6.81-6.75 (m, 2H), 3.85 (s, 3Н), 3.82 (s, 3Н), 3.15 (bs, 2Н), 1.68-1.60 (m, 2Н), 1.45-1.32 (m, 4H), 0.89 (t, J= 6.9 Гц, 3Н).
3-Пентиламино-фталевая кислота
Диметиловый эфир 3-пентиламино-фталевой кислоты (4,19 г, 15 ммоль) обрабатывали таким же образом, как описано выше для синтеза 3-(2-метоксиэтиламино)-фталевой кислоты. Продукт реакции, который содержал смесь дикислоты и монометиловых эфиров, использовали без последующей очистки.
2-(2,6-Диоксо-пиперидин-3-ил)-4-пентиламино-изоиндол-1,3-дион
К перемешиваемому раствору 3-пентиламино-фталевой кислоты (2,51 г, 10 ммоль) в уксусной кислоте (50 мл) добавляли 3-амино-пиперидин-2,6-диона гидрохлорид (1,81 г, 11 ммоль). Реакционную смесь нагревали с обратным холодильником в течение ночи. Растворитель выпаривали в вакууме, и остаток растворяли в этилацетате (100 мл). Этилацетатную смесь промывали водой (2•100 мл), насыщенным водным бикарбонатом натрия (2•100 мл), рассолом (1•100 мл) и сушили (MgSО4). Растворитель выпаривали в вакууме, и твердый остаток очищали хроматографией (25% этилацетата/гексан) с получением 1,82 г (53%) продукта в виде желтого твердого вещества: т. пл. 141-143oС;1Н ЯМР (ДМСО-d6) δ 11.09 (s, 1H), 7.58 (t, J= 8.3 Гц, 1Н), 7.08 (d, J= 8.6 Гц, 1Н), 7.02 (d, J= 7.0 Гц, 1H), 6.519 (t, J= 5.7 Гц, 1H), 5.06 (dd, J= 5.3 и 12.4 Гц, 1H), 3.32-3.25 (m, 2Н), 2.97-2.82 (m, 1H), 2.62-2.46 (m, 2H), 2.06-2.01 (m, 1H), 1.61-1.55 (m, 2H), 1,35-1,32 (m, 4H), 0.88 (t, J= 6.7 Гц, 3Н);13С ЯМР (ДМСО-d6) δ 172.72, 170.02, 168.92, 167.25, 146.39, 136.21, 132.14, 117.08, 110.31, 108.99, 48.52, 41.77, 30.94, 28.46, 28.33, 22.12, 13.85; Анализ: Вычислено для C18H21N3О4: С, 62.96; Н, 6.17; N, 12.15.
Биологические анализы
Ингибиторную в отношении TNF-α активность заявленных соединений измеряли в стимулированных липополисахаридами (LPS) человеческих мононуклерных клетках периферической крови (РВМС), как
описано в Muller, G. W. , et аl. , J. Med. Chem. 1996, 39, 3238. Анализ на ингибирование TNF-α цельной крови человека проводили аналогичным образом, за исключением того, что гепаризированную
свежую цельную кровь человека вносили непосредственно в микротитрационные планшеты. Анализ затем продолжали так же, как анализ РВМС.
Результаты анализов соединений по изобретению на их способность ингибировать продукцию TNF-α приведены в таблице.
TNF-α ИК50 известного незамещенного структурного аналога заявленных соединений 2-(2, 6-диоксо-3-пиперидил)изоиндолин-1,3-диона (талидомида) в анализе РВМС, стимулированных LPS, составляет ~ 200 мкм (~ 200000 нм) (Muller, G. W. , et al. , Bioorg. Med. Chem. Lett. , 1998, 8, 2669). Таким образом, из данных приведенной выше таблицы следует, что ингибиторная в отношении TNF-α активность заявленных соединений во много раз выше, чем активность талидомида.
Реферат
Настоящее изобретение относится к соединениям общей формулы (I), где один из Х и Y представляет собой C= O, а другой С= O или СН2; R1, R2, R3, R4 - один из них представляет собой -NHR5, а оставшиеся представляют собой водород; R5, R6 представляют собой водород или алкил при условии, что R6 другой, чем водород, если Х и Y представляет собой С= O, и один из R1, R2 , R3, R4 представляют собой аминогруппу. Новые соединения формулы (I) обладают свойством ингибировать TNF-α и поэтому могут быть использованы в фармкомпозиции в качестве активного ингредиента. Также соединения формулы (I) могут найти применение в способе снижения нежелательных уровней TNF-α у млекопитающих. 4 с. и 4 з. п. ф-лы, 1 табл.
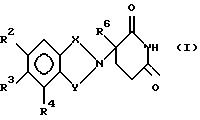
Формула
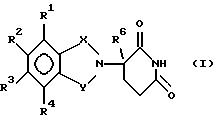
в которой один из Х и Y представляет собой С= O, а другой из Х и Y представляет собой С= O или СН2;
один из R1, R2, R3 и R4 представляет собой -NHR5, а оставшиеся из R1, R2, R3 и R4 представляют собой водород;
R5 представляет собой водород или алкил из 1-8 атомов углерода;
R6 представляет собой водород или алкил из 1-8 атомов углерода; при условии, что R6 другой, чем водород, если Х и Y представляют собой С= O, и один из R1, R2, R3 и R4 представляет собой аминогруппу.
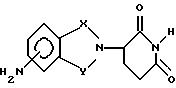
в котором один из Х и Y представляет собой С= O, а другой из Х и Y представляет собой С= O или CH2.
Комментарии