Новые способы - RU2682658C1
Код документа: RU2682658C1
Чертежи


Описание
По настоящей заявке испрашивается приоритет на основании предварительной заявке на патент США No. 61/911416, поданной 3 декабря, 2013; 61/925607, поданной 9 января, 2014; 61/975702, поданной 4 апреля, 2014; и 62/032326, поданной 1 августа, 2014, содержание каждой из которых включено в качестве ссылки в полном объеме.
Область техники
Настоящее изобретение относится к применению конкретных замещенных гетероциклических конденсированных гамма-карболинов, согласно настоящему описанию, в свободной форме или в форме фармацевтически приемлемой соли, в качестве фармацевтических средств и фармацевтических композиций в качестве первичной или дополнительной терапии при лечении острых и остаточных фаз шизофрении, в частности, включая лечение остаточных симптомов, таких как активная социальная устраненность, пассивная социальная отгороженность, эмоциональное отчуждение, тревога, напряженность, стереотипное мышление, и соматическая озабоченность. Соединения, раскрытые в настоящем описании, можно использовать для лечения симптомов острой фазы и симптомов остаточной фазы, которые появляются во время обострений, но определяют остаточную фазу заболевания, например, шизофрении, после того, как острые симптомы ослабевают. Таким образом, соединения, описанные в настоящем документе, могут быть использованы отдельно или в комбинации с другими антипсихотическими лекарственными средствами, а также другими активными средствами, которые лечат сопутствующие расстройства, такие как депрессия и/или расстройства сна. Так как профиль ответа для всех симптомов, связанных с шизофренией, может быт особенно полезным в улучшении социальной функции, соединения, описанные в настоящем документе, могут быть использованы для улучшения социальной интеграции и социальной функции. Такой профиль ответа может быть полезным для продромальной, острой и остаточной фаз шизофрении.
Предпосылки создания изобретения
Психозами, особенно шизофренией, страдает 1,1% населения во всем мире. Это заболевание включает три фазы: продромальную фазу, активную фазу и остаточную фазу. Продромальная фаза представляет собой раннюю фазу, при которой наблюдаются субклинические признаки и симптомы. Эти симптомы могут включать потерю интереса к обычным занятиям, отдаление от друзей и членов семьи, спутанность сознания, проблемы с концентрацией, чувство вялости и апатию. Активная фаза характеризуется обострением позитивных симптомов, таких как бред, галлюцинации и подозрительность. Остаточная фаза характеризуется негативными симптомами, такими как эмоциональное отчуждение, пассивная социальная отгороженность, и стереотипное мышление, и симптомами общей психопатологии, включая активную социальную устраненность, тревогу, напряженность, и соматическую озабоченность. Симптомы остаточной фазы часто сопровождаются депрессией, когнитивной дисфункцией и бессонницей. В совокупности эти симптомы остаточной фазы не очень хорошо поддаются лечению многими антипсихотическими лекарственными средствами, в настоящее время доступными на рынке, и поэтому, как правило, наблюдаются после того, как симптомы активной фазы прошли после лечения. Эта фаза заболевания, когда пациенты хотели бы вернуться к более продуктивной и полноценной жизни, но так как остаточные негативные симптомы и когнитивные нарушения не вылечили должным образом, эта задача не состоялась. Сохраняется острая необходимость в антипсихотических препаратах, которые могут лечить не только симптомы активной или острой фазы, а также симптомы остаточной фазы психозов, например, шизофрении.
Замещенные конденсированные гетероциклические гамма-карболины известны как агонисты или антагонисты рецепторов 5-HT2, в частности, рецепторов 5-HT2A и 5-HT2C, в лечении расстройства центральной нервной системы. Эти соединения раскрыты в патенте США No. 6548493; 7238690; 6552017; 6713471; U.S. RE39680, U.S. RE39679, U.S. No. 7183282 и 7071186, в качестве новых соединений и применения в медицине при лечении расстройств, связанных с модуляцией рецептора 5-HT2A, таких как ожирение, тревога, депрессия, психоз, шизофрения, расстройства сна, сексуальные расстройства, мигрень, аутизм, состояний, связанных с головной болью, социальных фобий, и нарушений со стороны желудочно-кишечного тракта, таких как нарушение моторики желудочно-кишечного тракта. В PCT/US08/03340 и заявке США No. 10/786935 также описаны способы получения замещенных гетероциклических конденсированных гамма-карболинов и использование этих гамма-карболинов в качестве агонистов и антагонистов серотонина, пригодных для контроля и профилактики расстройств центральной нервной системы, таких как аддиктивное поведение и расстройства сна.
В дополнение, в WO 2009/145900, WO 2011/133224, WO 2013/155505, WO 2013/155504 и WO 2013/155506 предложены кроме того замещенные гетероциклические конденсированные гамма-карболиновые соединения и/или их использование для лечения одного или нескольких нарушений, связанных с 5-HT2A, транспортером серотонина (SERT) и/или путями дофамина D1/D2.
Хотя, приведенные выше ссылки, относящиеся к замещенным гетероциклическим конденсированным гамма-карболиновым соединениям, предлагают лечение некоторых расстройств, связанных с психозом и/или депрессией, ни одна из этих ссылок не раскрывает лечение остаточных симптомов психоза, в частности, остаточных симптомов шизофрении.
Сущность изобретения
Было обнаружено, что особым образом замещенные гетероциклические конденсированные гамма-карболиновые соединения (соединения, описанные в настоящем документе ниже) обладают механизмами действия, которые, как полагают, обладают потенциалом для получения первого в своей группе антипсихотической терапии. Соединения формулы I объединяют активный антагонизм серотонинового 5-HT2A-рецептора, модуляцию фосфопротеина рецептора дофамина или DPPM, глутаматергическую модуляцию и ингибирование обратного захвата серотонина в одно лекарственное средство-кандидат для лечения острой и резидуальной шизофрении. При дофаминовых D2-рецепторах, соединения формулы I имеют двойные свойства и действуют как постсинаптические антагонисты и пресинаптические частичные агонисты. Соединения формулы I также стимулируют фосфорилирование глутаматергических NMDA-рецепторов, NR2B или GluN2B, в мезолимбической системе, определенным образом. Считается, что эта региональная избирательность в областях головного мозга опосредует эффективность антипсихотических лекарственных средств, вместе с серотонинергическими, глутаматергическими и дофаминергическими взаимодействиями, может привести к антипсихотической эффективности для позитивных, негативных, аффективных и когнитивных симптомов, связанных с шизофренией. Ингибирование обратного захвата серотонина может привести к антидепрессивной активности при лечении шизоаффективного расстройства, сопутствующей депрессии и/или в качестве отдельного лечения большого депрессивного расстройства. Считается, что соединения формулы I могут быть также полезны для лечения биполярного расстройства и других психических и нейродегенеративных расстройств, в частности, поведенческих нарушений, связанных с деменцией, аутизма и других заболеваний ЦНС. Эти особенности соединений формулы I могут быть в состоянии улучшить качество жизни пациентов с шизофренией и повысить социальную функцию, чтобы дать им возможность более полно интегрироваться в свои семьи и свое место работы. Кроме того, соединения формулы I могут быть показаны для лечения расстройств, в низких дозах (например, сон, агрессивность и ажитация) или в высоких дозах (например, острая и резидуальная шизофрения, биполярные расстройства и расстройства настроения).
Поскольку соединения, описанные ниже, являются эффективными при лечении не только острых симптомов, но и остаточных симптомов психоза, настоящее изобретение, следовательно, обеспечивает, в одном из аспектов, способы использования конкретных гетероциклических конденсированных гамма-карболиновых соединений (соединения, описанные в настоящем документе ниже), или отдельно, или в качестве вспомогательной терапии, при лечении остаточных симптомов психоза, в частности, шизофрении. Это новое и неожиданное свойство.
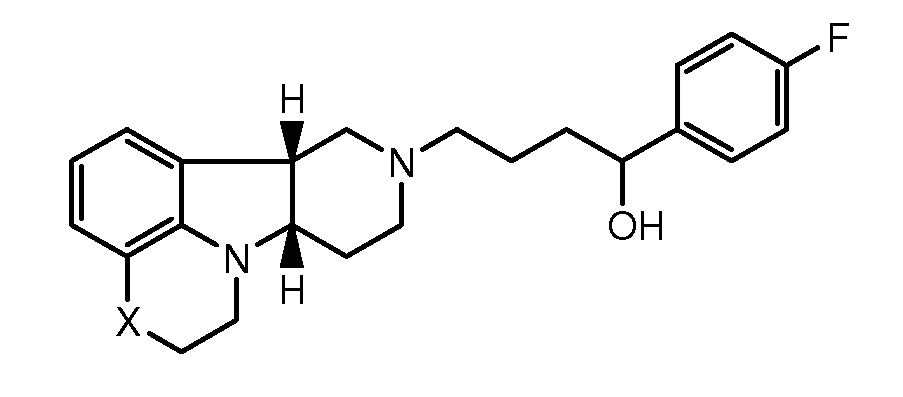
Таким образом, настоящее изобретение относится к способу (Способ А) для лечения остаточных симптомов, включающему введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы I:

где:
X представляет собой -O-, -NH- или -N(CH3)-;
Y представляет собой -O-, -C(R2)(OH)-, -C(R3)(OR1) или -C(O)-;
R1 представляет собой C1-6алкил (например, метил) или физиологически гидролизуемый и приемлемый ацил, например, выбранный из -C(O)-C1-21алкила (например, -C(O)-C1-5алкил, -C(O)-C6-15алкил или -C(O)-C16-21алкил), например, где R1 представляет собой -C(O)-C6алкил, -C(O)-C7алкил, -C(O)-C9алкил, -C(O)-C11алкил, -C(O)-C13алкил или -C(O)-C15алкил, например, где такое соединение гидролизуется с образованием природной или неприродной, насыщенной или ненасыщенной жирной кислоты формулы R1-OH и соединения формулы 1, где Y представляет собой -C(R2)(OH)-, например, где соединение гидролизуется с образованием гидроксисоединения формулы I, где Y представляет собой -C(R2)(OH)-, с одной стороны, и октановой кислоты, декановой кислоты, додекановой кислоты, тетрадекановой кислоты или гексадекановой кислоты, с другой стороны);
R2 представляет собой H или -C1-6алкил (например, метил); и
R3 представляет собой H или -C1-6алкил (например, метил);
например, где термин "алкил" относится к линейной цепи углеводородной группы, необязательно, насыщенной или ненасыщенной и, необязательно замещенной одной или несколькими гидрокси или C1-22алкокси (например, этокси) группами;
в свободной форме или в форме фармацевтически приемлемой соли.
В другом варианте осуществления соединение формулы I способа А представляет собой соединение, где:
X представляет собой -O-, -NH- или -N(CH3)-;
Y представляет собой -O-, -C(H)(OH)-, -C(H)(OR1) или -C(O)-; и
R1 представляет собой физиологически гидролизуемый и приемлемый ацил, например, выбранный из -C(O)-C1-21алкила (например, -C(O)-C1-5алкила, -C(O)-C6-15алкила или -C(O)-C16-21алкила), например, где R1 представляет собой -C(O)-C6алкил, -C(O)-C7алкил, -C(O)-C9алкил, -C(O)-C11алкил, -C(O)-C13алкил или -C(O)-C15алкил, например, где такое соединение гидролизуется с образованием природной или неприродной, насыщенной или ненасыщенной жирной кислоты формулы R1-OH и соединения формулы I, где Y представляет собой -C(R2)(OH)-, например, где соединение гидролизуется с образованием гидроксисоединения формулы I, где Y представляет собой -C(R2)(OH)-, с одной стороны, и октановой кислоты, декановой кислоты, додекановой кислоты, тетрадекановой кислоты или гексадекановой кислоты, с другой стороны);
например, где ʺалкилʺ относится к линейной цепи углеводородной группы, необязательно, насыщенной или ненасыщенной и, необязательно замещенной одной или несколькими гидрокси или C1-22алкокси (например, этокси) группами;
в свободной форме или в форме фармацевтически приемлемой соли.
В еще одном варианте осуществления пациент способа А страдает от остаточных симптомов психоза, например, шизофрения (например, остаточный подтип), бредовое расстройство (например, соматического типа), большая депрессия с психозом, биполярное расстройство с психотическими симптомами, кратковременное психотическое расстройство, шизофреноформное расстройство, шизоаффективное расстройство или психоз, вызванный заболеванием или употреблением наркотиков. Предпочтительно, пациент страдает от остаточных симптомов шизофрении.
В другом дополнительном варианте осуществления, симптомы остаточной фазы включают: негативные симптомы, такие как притупленный аффект, эмоциональное отчуждение, малоконтактность, пассивная или апатическая социальная отгороженность, затруднение в абстрактном мышлении, нарушение спонтанности и плавности речи и стереотипного мышления; общие психопатологические симптомы, такие как соматическая озабоченность, тревога, чувство вины, напряженность, манерность и позирование, депрессия, моторная заторможенность, малоконтактность, необычное содержание мыслей, дезориентированность, нарушение внимания, снижение оценки и критичности к себе, расстройство воли, ослабление контроля импульсивности, поглощенность и активная социальная устраненность; когнитивные нарушения и расстройства сна (например, бессонница). В конкретном варианте осуществления, способ А лечит остаточные симптомы (негативные, эффективные и когнитивные симптомы), связанные с шизофренией.
В другом дополнительном варианте осуществления эффективное количество соединения формулы I составляет от приблизительно 1 мг до приблизительно 140 мг на дозу в день, в другом варианте осуществления от приблизительно 2,5 мг до приблизительно 120 мг, в другом варианте осуществления от приблизительно 10 мг до приблизительно 120 мг на дозу в день, в другом варианте осуществления приблизительно 60 мг до приблизительно 120 мг на дозу в день, в еще одном варианте осуществления от приблизительно 10 мг до приблизительно 60 мг на дозу в день, в другом варианте осуществления от приблизительно 20 мг до приблизительно 60 мг на дозу в день, в еще одном варианте осуществления приблизительно 20 мг, приблизительно 40 мг или приблизительно 60 мг на дозу в день. Соединения по настоящему изобретению являются эффективными при лечении как позитивных симптомов, которые имеют место во время активной или острой фазы психоза (например, эффективны при лечении позитивных симптомов, таких как бред и галлюцинации), так и негативных и других остаточных симптомов, как правило, наблюдаемых в остаточной фазе. Предпочтительно, эффективное количество для лечения острых и остаточных симптомов шизофрении составляет приблизительно 60 мг в день. В конкретном варианте осуществления дозировки, раскрытые в настоящем документе, для перорального введения основаны на количестве соединений формулы I в форме соли присоединения кислоты, в частности, в форме соли присоединения толуолсульфоновой кислоты.
Таким образом, настоящее изобретение относится к способам, как изложено ниже:
1.1 Способ А, содержащий соединение формулы I, где X представляет собой -N(CH3);
1.2 Способ А, содержащий соединение формулы I, где X представляет собой -NH;
1.3 Способ А, содержащий соединение формулы I, где X представляет собой O;
1.4 Способ A или любой из 1.1-1.3, содержащий соединение формулы I, где Y представляет собой -C(O)-;
1.5 Способ A или любой из 1.1-1.3, содержащий соединение формулы I, где Y представляет собой -O-;
1.6 Способ A или любой из 1.1-1.3, содержащий соединение формулы I, где Y представляет собой -C(R2)(OH)-, например, -C(H)(OH)-;
1.7 Способ A или любой из 1.1-1.3, содержащий соединение формулы I, где Y представляет собой -C(R3)(OR1), например, -C(H)(OR1);
1.8 Способ А или формула 1.7, где R1 представляет собой -C(O)-C1-21алкил (например, -C(O)-C1-5алкил, -C(O)-C6-15алкил или -C(O)-C16-21алкил), предпочтительно, указанный алкил является прямоцепочечным, необязательно насыщенным или ненасыщенным и необязательно замещенным одной или несколькими гидрокси или C1-22алкокси (например, этокси) группами, например, R1 представляет собой -C(O)-C6алкил, -C(O)-C7алкил, -C(O)-C9алкил, -C(O)-C11алкил, -C(O)-C13алкил или -C(O)-C15алкил, где такое соединение гидролизуется с образованием остатка природной или неприродной, насыщенной или ненасыщенной жирной кислоты, например, соединение гидролизуется с образованием гидроксисоединения, с одной стороны, и октановой кислоты, декановой кислоты, додекановой кислоты, тетрадекановой кислоты или гексадекановой кислоты, с другой стороны);
1.9 Способ А или формула 1.7, где R1 представляет собой - C(O)-C6-15алкил, например, -C(O)-C9алкил;
1.10 Способ А или формула 1.7, где R1 представляет собой -C(O)-C1-5алкил, например, -C(O)-C3алкил;
1.11 Способ А или формула 1.7, где R1 представляет собой -C1-6алкил (например, метил);
1.12 Способ А или любая из формулы 1.1-1.11, где R2 представляет собой H или -C1-6алкил (например, метил);
1.13 Способ А или любая из формулы 1.1-1.11, где R2 представляет собой H;
1.14 Способ А или любая из формулы 1.1-1.11, где R2 представляет собой -C1-6алкил (например, метил);
1.15 Способ А или любая из формулы 1.1-1.11, где R3 представляет собой H или -C1-6алкил (например, метил);
1.16. Способ А или любая из формулы 1.1-1.11, где R3 представляет собой H;
1.17 Способ А или любая из формулы 1.1-1.11, где R3 представляет собой -C1-6алкил (например, метил);
1.18 любой из предшествующих способов, где соединение формулы I выбрано из группы, состоящей из соединения формулы I, где:
X представляет собой -O- и Y представляет собой -C(H)(OH)-,
X представляет собой -NH- и Y представляет собой -C(H)(OH)-,
X представляет собой -N(CH3)- и Y представляет собой -C(H)(OH)-,
X представляет собой -O- и Y представляет собой -C(O)-,
X представляет собой -O- и Y представляет собой -O-,
X представляет собой -N(CH3)- и Y представляет собой -C(O)-,
X представляет собой -N(CH3)- и Y представляет собой -O-,
X представляет собой -NH- и Y представляет собой -C(O)-,
X представляет собой -NH- и Y представляет собой -O-,
X представляет собой -N(CH3)- и Y представляет собой -C(H)(OR1),
X представляет собой -NH- и Y представляет собой -C(H)(OR1), или
X представляет собой -O- и Y представляет собой -C(H)(OR1);
X представляет собой -O- и Y представляет собой -C(CH3)(OH)-,
X представляет собой -NH- и Y представляет собой C(CH3)(OH)-,
X представляет собой -N(CH3)- и Y представляет собой C(CH3)(OH)-,
1.19 любой из предшествующих способов, где X представляет собой -O- и Y представляет собой -C(O)- в соединении формулы I;
1.20 любой из предшествующих способов, где X представляет собой -NH- и Y представляет собой -C(H)(OH)- в соединении формулы I;
1.21 любой из предшествующих способов, где X представляет собой -N(CH3)- и Y представляет собой -C(H)(OH)- в соединении формулы I;
1.22 любой из предшествующих способов, где X представляет собой -O- и Y представляет собой -C(O)- в соединении формулы I;
1.23 любой из предшествующих способов, где X представляет собой -O- и Y представляет собой -O- в соединении формулы I;
1.24 любой из предшествующих способов, где X представляет собой -N(CH3)- и Y представляет собой -C(O)- в соединении формулы I;
1.25 любой из предшествующих способов, где X представляет собой -O- и Y представляет собой -C(H)(OH)- в соединении формулы I
1.26 любой из предшествующих способов, где X представляет собой -NH- и Y представляет собой -C(H)(OH)- в соединении формулы I;
1.27 любой из предшествующих способов, где X представляет собой -N(CH3)- и Y представляет собой -C(H)(OH)- в соединении формулы I;
1.28 любой из предшествующих способов, где X представляет собой -N(CH3)- и Y представляет собой -C(H)(OR1) и R1 представляет собой -C(O)-C1-21алкил в соединении формулы I;
1.29 любой из предшествующих способов, где X представляет собой -N(H)- и Y представляет собой -C(H)(OR1) и R1 представляет собой -C(O)-C1-21алкил в соединении формулы I;;
1.30 любой из предшествующих способов, где X представляет собой -O- и Y представляет собой -C(H)(OR1) и R1 представляет собой -C(O)-C1-21алкил в соединении формулы I;
1.31 Любой из предшествующих способов, в котором соединение формулы I представляет собой соединение Формулы IA:
1.32 Любой из предшествующих способов, в котором соединение формулы I представляет собой соединение Формулы IB:

1.33 Любой из предшествующих способов, в котором соединение формулы I представляет собой соединение Формулы IC:

1.34 Любой из предшествующих способов, в котором соединение формулы I представляет собой соединение Формулы ID (иногда упоминается в настоящем описании как соединение B):

1.35 Любой из предшествующих способов, в котором соединение формулы I представляет собой соединение Формулы IE (иногда упоминается в настоящем описании как соединение A):

1.36 Любой из предшествующих способов, в котором соединение формулы I представляет собой соединение Формулы IF:

1.37 Любой из предшествующих способов, в котором соединение формулы I представляет собой соединение Формулы IG:

1.38 Любой из предшествующих способов, в котором соединение формулы I вводят перорально, например, один раз в день в виде одной суточной дозы или два раза в день в разделенной дозе, например, в виде таблетки или капсулы.
1.39 Любой из предшествующих способов, в котором эффективным количеством соединения формулы I является ежедневная пероральная доза 10-120 мг/день, например, приблизительно 60 мг соли присоединения п-толуолсульфоновой кислоты соединения формулы IE:

1.40 Любой из предшествующих способов, где соединение формулы I вводят в виде инъецируемой формы с замедленным высвобождением, например, инъецируемой лекарственной формы депо, например, вводят один или два раза в месяц, например, в форме биоразлагаемой микрочастицы, например, соединение формулы IE:

например, в форме свободного основания.
1.41 Любой из предшествующих способов, включающий введение инъецируемого состава длительного действия соединения формулы I, например, композиции 2, например, любой из композиций 2.1, и след. как изложено ниже.
1.42 Любой из предшествующих способов, дополнительно включающий введение одного или нескольких других терапевтических средств, таких как дополнительные антипсихотические средства и/или антидепрессивные средства и/или снотворные средства;
1.43 Способ 1.380, где одно или несколько других терапевтических средств выбраны из антидепрессивных средств, таких как соединения, которые модулируют активность ГАМК (например, усиливают активность и облегчает передачу ГАМК), агонист рецептора ГАМК-B, модулятор 5-HT (например, агонист рецептора 5-HT1A, антагонист рецептора 5-HT2A, обратный агонист рецептора 5-HT2A, и тому подобное), агонист рецептора мелатонина, модулятор ионного канала (например, блокатор), антагонист рецептора серотонина 2/ингибитор обратного захвата серотонина (SARI), антагонист орексиновых рецепторов, агонист рецептора H3, антагонист норадренергического рецептора, агонист галанинового рецептора, антагонист рецептора CRH, гормон роста человека, агонист рецептора гормона роста, эстроген, агонист рецептора эстрогена, препарат нейрокинина-1; и антипсихотические средства, например, атипичные антипсихотические средства, в свободной форме или в форме фармацевтически приемлемой соли;
1.44 Способ 1.420 или 1.430, где одно или несколько других терапевтических средств представляют собой антипсихотические средства, например, хлорпромазин, галоперидол, дроперидол, флуфеназин, локсапин, мезоридазин молиндон, перфеназин, пимозид, прохлорперазин промазин, тиоридазин, тиотиксен, трифтороперазин, клозапин, арипипразол, оланзапин, кветиапин, рисперидон, зипрасидон, палиперидон, азенапин, луразидон, илоперидон, карипразин, амисульприд, зотепин, сертиндол, где одно или несколько других терапевтических средств вводят как вспомогательное средство к соединению формулы I или соединение формулы I представляет собой вспомогательное средство к одному или нескольким другим терапевтическим средствам;
1.45 Любой из предшествующих способов, где эффективное количество составляет от 1 мг до 120 мг в день или 10 мг до 120 мг в день, или 10 мг до 60 мг в день, или 10 мг до 40 мг в день, или 1 мг до 10 мг в день, или 10 мг в день, 20 мг в день, 40 мг в день или 60 мг в день; В конкретном варианте осуществления изобретения эффективное количество соединения формулы I, раскрытого в этой формуле, основано на количестве соединения формулы I в форме соли присоединения кислоты, непролекарственная форма, например, в форме соли присоединения толуолсульфоновой кислоты, непролекарственная форма.
1.46 Способ А или любой из 1.38-1.45, где одно или несколько других терапевтических средств представляют собой антидепрессивные средства, например, один или несколько антидепрессантов, выбранных из селективных ингибиторов обратного захвата серотонина (SSRI) (например, выбранных из циталопрама, эсциталопрама оксалата, флуоксетина, флувоксамина малеата, пароксетина, сертралина, дапоксетина), ингибиторов обратного захвата серотонина-норэпинефрина (SNRI) (например, выбранных из венлафаксина, десвенлафаксина, дулоксетина, милнаципран, левомилнаципрана, сибутрамина) и трициклических антидепрессантов; тройные ингибиторы обратного захвата, анксиолитики, буспирон, и тразодон;
1.47 Способы 1.45, где соединение формулы I вводят как вспомогательное средство к одному или нескольким другим терапевтическим средствам, таким как SSRI-антидепрессанты или SSRI-антидепрессанты вводят как вспомогательное средство к соединению формулы I;
1.48 Способ 1.47, где указанное одно или несколько антидепрессивных средств выбраны из SSRI, таких как циталопрам (целекса, ципрамил, эмокаль, сепрам, серопрам), эсциталопрама оксалат (лексапро, ципралекс, Esertia), флуоксетин (прозак, фонтекс, серомекс, серонил, сарафем, флуктин (Fluctin) (EUR)), флувоксамина малеат (лювокс, фаверин), пароксетин (паксил, сероксат, аропакс, дероксат, пароксат), сертралин (золофт, люстрал, Serlain), дапоксетин;
в свободной форме или в форме фармацевтически приемлемой соли;
1.49 Любой из вышеуказанных способов, где соединение формулы I вводят как часть инъекционной композиции микросфер длительного действия;
1.50 Способ 1.49, где инъекционная композиция микросфер длительного действия представляет собой композицию по любому из 2.1-2.22 в настоящем документе ниже.
В конкретном варианте осуществления способа А и сл., пациент является пациентом, который не реагирует или не реагирует должным образом на лечение другим антипсихотическим средством, например, хлорпромазином, галоперидолом, дроперидолом, флуфеназином, локсапином, мезоридазин молиндоном, перфеназином, пимозидом, прохлорперазин промазином, тиоридазином, тиотиксеном, трифтороперазином, клозапином, арипипразолом, оланзапином, кветиапином, рисперидоном, зипрасидоном, палиперидоном, азенапином, луразидоном, илоперидоном, карипразином, амисульпридом, зотепином, сертиндолом. Таким образом, соединение формулы I можно вводить в качестве первичной терапии или вспомогательной терапии, например, дополнительно к другому антипсихотическому средству.
В другом аспекте настоящее изобретение относится к способу (Способ В) лечения любого из следующих нарушений: шизоаффективное расстройство, сопутствующая депрессия, большое депрессивное расстройство, биполярное расстройство (например, биполярное расстройство I типа и/или биполярное расстройство II типа), расстройства аутистического спектра (например, аутическое расстройство, синдром Аспергера, первазивное расстройство развития неуточненное (PDD-NOS), расстройство Ретта (синдром Ретта), дезинтегративное расстройство детского возраста), включающему введение пациенту, нуждающемуся в этом, эффективного количества соединения Формула I, в свободной форме или в форме фармацевтически приемлемой соли. В конкретном варианте осуществления нарушение способа В представляет собой биполярное расстройство (например, биполярное расстройство I типа и/или биполярное расстройство II типа). В другом конкретном варианте осуществления нарушение способа В представляет собой расстройства аутистического спектра. В еще одном конкретном варианте осуществления нарушение способа В представляет собой большое депрессивное расстройство.
Таким образом, в зависимости от комбинации нарушений, подлежащих лечению, соединения формулы I могут быть использованы стратегически. Например, при низких дозах (например, пероральная суточная доза 1-10 мг, например, 1 мг, 5 мг и 10 мг соединения формулы I в форме соли присоединения толуолсульфоновой кислоты), соединения формулы I являются полезными для лечения расстройства сна, агрессивности и ажитации, болезни Альцгеймера и других деменций, расстройства аутистического спектра, болезни Паркинсона и интермиттирующего эксплозивного расстройства (IED). При более высокой дозе (например, пероральная суточная доза 60 мг соединения формулы I в форме соли присоединения толуолсульфоновой кислоты), соединения формулы I являются полезными для лечения острой обострившейся и резидуальная шизофрении, биполярной депрессии, большого депрессивного расстройства, генерализованного тревожного расстройства. При очень высоких дозах (например, пероральная суточная доза 120 мг соединения формулы I в форме соли присоединения толуолсульфоновой кислоты), введение утром может вызывать сонливость/успокоение. Поэтому при таких высоких суточных дозах, введение в вечернее время является предпочтительным.
Соединения формулы I могут существовать в свободной форме или в форме соли, например, в форме соли присоединения кислоты. В настоящем описании, если не указано иное, выражения, такие как "соединения формулы I", "антипсихотические средства", "антидепрессивные средства", "другими терапевтические средства", и тому подобное не следует понимать как охватывающее соединения в любой форме, например, в свободной форме или в форме соли присоединения кислоты, или когда соединения содержат кислые заместители, в форме соли присоединения основания. Соединения формулы I предназначены для использования в качестве фармацевтических средств, поэтому их фармацевтически приемлемые соли являются предпочтительными. Соли, которые являются неподходящими для фармацевтического использования, могут быть полезными, например, для выделения или очистки свободных соединений формулы I или их фармацевтически приемлемых солей, которые, поэтому также включены. Фармацевтически приемлемые соли включают, например, гидрохлорид, мезилат и тозилат. Предпочтительно, соединения формулы I, в частности, где X представляет собой -N(CH3)- и Y представляет собой -C(O)-, находятся в форме тозилатной соли (соль присоединения толуолсульфоновой кислоты). Где размеры дозировки солей приведены по массе, например, миллиграммы в день или миллиграммы на единицу дозы, размер дозировки соли может быть основан на массе соответствующего свободного основания или как указано иное. В конкретном варианте осуществления, доза соединений формулы I в форме соли присоединения кислоты для перорального введения на основе массы соли присоединения толуолсульфоновой кислоты, но не формы свободного основания. Например, в конкретном варианте осуществления, размер дозировки 10 мг, 60 мг, 120 мг соединения формулы I основан, соответственно, на 10 мг, 60 мг и 120 мг соединения формулы I в форме соли присоединения толуолсульфоновой кислоты, а не в расчете на количество свободного основания. Например, доза 60 мг соединения для перорального введения соединений формулы I (например, где X представляет собой -N(CH3)- и Y представляет собой -C(O)-) в форме соли присоединения толуолсульфоновой кислоты относится к соединению в форме тозилатной соли, что эквивалентно приблизительно 41,7 мг указанного соединения в форме свободного основания.
Настоящее изобретение также обеспечивает вышеуказанные способы, например, способ А, например, любой из 1.1-1.50 и способ B, где соединение формулы I, в свободной форме или в форме фармацевтически приемлемой соли вводят в композиции, где указанное соединение формулы I в свободной форме или в форме фармацевтически приемлемой соли находится в смеси или в сочетании с фармацевтически приемлемым разбавителем или носителем.
В конкретном варианте осуществления настоящее изобретение также обеспечивает вышеуказанные способы, например, способ А, например, любой из 1.1-1.50 и способ B, где соединение формулы I в свободной форме или в форме фармацевтически приемлемой соли вводят в составе с немедленным освобождением или замедленным или отсроченным высвобождением, например, лекарственная форма депо.
В одном из вариантов осуществления композиция с замедленным или отсроченным высвобождением содержит соединения формулы I, описанные в настоящем документе (например, соединение формулы I или любое из тех, которые описаны в любой из формул 1.1-1.50) в полимерной матрице. В другом варианте осуществления соединения формулы I диспергируют или растворяют в полимерной матрице. В еще одном варианте осуществления полимерная матрица содержит стандартные полимеры, используемые в лекарственных формах депо, такие как полимеры, выбранные из полиэфира жирной гидроксикислоты и его производных, или полимера алкил-альфа-цианоакрилата, полиалкиленоксалата, поли(орто)эфира, поликарбоната, полиорто-карбоната, полиаминокислоты, сложного эфира гиалуроновой кислоты, и их смесей. В другом варианте осуществления полимер выбран из группы, состоящей из полилактида, поли-d,l-лактида, полигликолида, PLGA 50:50, PLGA 75:25, PLGA 85:15 и PLGA 90:10 полимера. В другом варианте осуществления полимер выбран из поли(гликолевой кислоты), поли-D,L-молочной кислоты, поли-L-молочной кислоты, сополимеров вышеизложенных, поли(алифатических карбоновых кислот), сополиоксалатов, поликапролактона, полидиоксонона, поли(ортокарбонатов), поли(ацеталей), поли(молочной кислоты-капролактона), полиортоэфиров, поли(гликолевой кислоты капролактона), полиангидридов и природных полимеров, включающих альбумин, казеин и воски, такие как, глицерола моно- и дистеарат и тому подобное. В конкретном варианте осуществления, полимерная матрица содержит поли(d,l-лактид-со-гликолид). Соединение формулы I в полимерной матрице, может быть, в смеси или в сочетании с фармацевтически приемлемым разбавителем или носителем.
Составы с замедленным или отсроченным высвобождением, как описано выше, особенно полезны для замедленного или отсроченного высвобождения, где соединения формулы I, высвобождаются при разрушении полимерной матрицы. Эти составы могут высвобождать соединения формулы I в течение периода до 180 дней, например, приблизительно от 14 до приблизительно 30 до приблизительно 180 дней. Например, полимерная матрица может разрушаться и высвобождать соединения формулы I в течение периода приблизительно 30, приблизительно 60 или приблизительно 90 дней. В другом примере полимерная матрица может разрушаться и высвобождать соединения формулы I в течение периода приблизительно 120 или приблизительно 180 дней.
В еще одном дополнительном варианте осуществления композиция с замедленным или отсроченным высвобождением получают для введения путем инъекции.
Например, настоящее изобретение обеспечивает инъекционные составы длительного действия (LAI) соединений формулы I. Такие LAI могут быть оптимизированы, относительно композиции носителя, размера частицы, молекулярной массы носителя, загрузки активного ингредиента, и дозы, например, как описано в примерах. В дополнение для удобства введения и для обеспечения соблюдения больным режима и схемы лечения, составы LAI соединений формулы I неожиданно обеспечивают преимущества в отношении фармакокинетики и побочных эффектов. Когда соединение формулы I вводят с использованием состава LAI, в противоположность пероральной лекарственной форме, метаболизм первого прохода в печень избегается, а это означает, что меньшая часть соединение формулы I метаболизируется до достижения мозга. Составы с замедленным или отсроченным высвобождением, как описано в настоящем документе, обычно дают меньшее количество экстрапирамидных побочных эффектов, а также обеспечивают лучшую переносимость препарата и снижение общей дозы, чем соответствующие составы с немедленным высвобождением. Составы с замедленным или отсроченным высвобождением, и особенно, инъекционные составы длительного действия, позволяют пациенту достигать и поддерживать терапевтически эффективные уровни лекарственного средства в ЦНС при получении гораздо более низкой общей дозы, чем было бы необходимо для достижения такого же уровня лекарственного средства в орагнизме с использованием составов для перорального введения с немедленным высвобождением. Для инъекционных составов длительного действия, в частности, этот эффект происходит частично из-за предотвращения метаболизма первого прохода, который происходит с пероральными препаратами, включая пероральные лекарственные средства с замедленным и отсроченным высвобождением.
Эффективное количество соединения формулы I при введении в виде инъекционного состава длительного действия, таким образом, оказывается значительно ниже, чем эффективное количество при пероральном введении, например, от приблизительно 100 мг в месяц до приблизительно 600 мг в месяц, и, предпочтительно, от 150 мг в месяц до 300 мг в месяц.
В конкретном варианте осуществления, таким образом, изобретение относится к инъекционному составу длительного действия (композиция 2), содержащему полимерные микросферы, где микросферы содержат:
поли(D,L-лактид-со-гликолид) (PLGA) полимерную матрицу и
эффективное количество соединения формулы I, как описано выше, в свободной форме или в форме фармацевтически приемлемой соли, соединение формулы I диспергируют, растворяют или инкапсулируют в полимерную матрицу.
Например, изобретение относится к:
2.1 Композиции 2, где полимер PLGA представляет собой полимер с соотношением приблизительно 75:25 PLA/PLG с концевыми группами карбоновой кислоты или карбонового эфира.
2.2 Композиции 2 или 2.1, где полимер PLGA представляет собой полимер с приблизительно 75:25 PLA/PLG концевыми группами карбоновой кислоты.
2.3 Композиции 2, 2.1 или 2.2, где соединение формулы I находится в форме свободного основания.
2.4 Любой предшествующей композиции, где соединение формулы I выбрано из соединений формулы IA, IB, IC, ID, IE.
2.5 Любой предшествующей композиции, где соединение формулы I представляет собой соединение формулы IE:

например, в форме свободного основания.
2.6 Любой предшествующей композиции, где соединение формулы I представляет собой соединение формулы ID:

например, в форме свободного основания.
2.7 Любой предшествующей композиции, где средний диапазон молекулярной массы для полимера PLGA составляет, например, 20 кД-200 кД, например, 24000-38000 дальтон, или приблизительно 113000 дальтон или приблиительно 159000 дальтон.
2.8 Любой предшествующей композиции, где временные рамки для полной деградации микросфер и высвобождения инкапсулированных лекарственных соединений составляют, например, менее 6 месяцев, менее 4 месяцев, менее 3 месяцев, менее 2 месяцев или менее 1 месяца.
2.9 Любой предшествующей композиции, где диаметр микросфер, например, средний диаметр (или D50), 10-процентильный диаметр (D10), 25-процентильный диаметр (D25), 75-процентильный диаметр (D75), или 90-процентильный диаметр (D90), составляет от приблизительно 10 мкм до приблизительно 200 мкм, например, от приблизительно 20 мкм до приблизительно 160 мкм, или от приблизительно 20 мкм до приблизительно 120 мкм, или от приблизительно 20 мкм до приблизительно 100 мкм, или от приблизительно 20 мкм до приблизительно 80 мкм, или от приблизительно 20 мкм до приблизительно 70 мкм, или от приблизительно 20 мкм до приблизительно 60 мкм, или от приблизительно 20 мкм до приблизительно 50 мкм, или от приблизительно 20 мкм до приблизительно 40 мкм, или от приблизительно 20 мкм до приблизительно 30 мкм, или от приблизительно 25 мкм до приблизительно 70 мкм, или от приблизительно 25 мкм до приблизительно 60 мкм, или от приблизительно 25 мкм до приблизительно 50 мкм, или от приблизительно 25 мкм до приблизительно 40 мкм, или от приблизительно 30 мкм до приблизительно 60 мкм, или от приблизительно 30 до 50 мкм, или от приблизительно 30 мкм до приблизительно 40 мкм, или от приблизительно 30 мкм до приблизительно 120 мкм, или от приблизительно 40 мкм до приблизительно 120 мкм, или от приблизительно 40 мкм до приблизительно 100 мкм, или от приблизительно 40 мкм до приблизительно 80 мкм, или от приблизительно 40 мкм до приблизительно 70 мкм, или от приблизительно 40 мкм до приблизительно 60 мкм, или от приблизительно 40 мкм до приблизительно 50 мкм, или от приблизительно 50 мкм до приблизительно 100 мкм, или от приблизительно 50 мкм до приблизительно 80 мкм, или от приблизительно 50 мкм до приблизительно 70 мкм, или от приблизительно 50 мкм до приблизительно 60 мкм, или от приблизительно 60 мкм до приблизительно 100 мкм, или от приблизительно 60 мкм до приблизительно 90 мкм, или от приблизительно 60 мкм до приблизительно 80 мкм, или от приблизительно 60 мкм до приблизительно 70 мкм, или от приблизительно 70 мкм до приблизительно 100 мкм, или от приблизительно 70 мкм до приблизительно 90 мкм, или от приблизительно 70 мкм до приблизительно 80 мкм, или от приблизительно 75 мкм до приблизительно 110 мкм, или от приблизительно 40 мкм до приблизительно 160 мкм, или от приблизительно 50 мкм до приблизительно 160 мкм, или от приблизительно 50 мкм до приблизительно 120 мкм, или приблизительно 20 мкм, приблизительно 30 мкм, приблизительно 40 мкм, приблизительно 50 мкм, приблизительно 60 мкм, приблизительно 70 мкм, приблизительно 80 мкм, приблизительно 90 мкм или приблизительно 100 мкм.
2.10 Любой предшествующей композиции, где диаметр микросфер, например, средний диаметр (или D50), 10-процентильный диаметр (D10), 25-процентильный диаметр (D25), 75-процентильный диаметр (D75) или 90-процентильный диаметр (D90), составляет от 10 мкм до 160 мкм, например, 20 мкм-70 мкм, 25 мкм-70 мкм, 40-120 мкм, или 20 мкм-60 мкм, или 20 мкм-50 мкм, 30 мкм-60 мкм, 30 мкм-50 мкм, 40 мкм - 50 мкм, или приблизительно 30 мкм, или приблизительно 40 мкм, или приблизительно 50 мкм.
2.11 Любой предшествующей композиции, где количество соединения формулы I, диспергированное, растворенное или инкапсулированное в каждой микросфере, в среднем, составляет от приблизительно 5% по массе до приблизительно 50% по массе, например, от приблизительно 10% по массе до приблизительно 50% по массе, или от приблизительно 20% по массе до приблизительно 40% по массе, или от приблизительно 30% по массе до приблизительно 40% по массе, или, например, приблизительно 8,5% по массе, или приблизительно 16% по массе, или приблизительно 30% по массе, или приблизительно 35% по массе, или приблизительно 40% по массе.
2.12 Любой предшествующей композиции, где характеристическая вязкость составляет приблизительно от 0,1 до приблизительно 1, например, приблизительно 0,3 до приблизительно 0,4, приблизительно 0,7, приблизительно 0,8, приблизительно 0,9 дл/г.
2.13 Любой из предшествующих композиций для использования в способе А, например, любом из способов 1.1 и сл. или в способе B, как описано выше.
2.14 Любой из предшествующих композиций для применения у пациентов, испытывающих трудности при обычном режиме лечения, умышленно или неумышленно.
2.15 Любой из предшествующих композиций для введения пациентам на еженедельной, раз в две недели или месячной основе или один раз в 2, 3 4, 5 или 6 месяцев.
2.16 Любой из предшествующих композиций для внутримышечной, внутрибрюшинной, интратекальной, эпидуральной или подкожной инъекции, например, подкожной или внутримышечной инъекции, например, внутримышечной инъекции.
2.17 Любой из предшествующих композиций для внутримышечной инъекции.
2.18 Любой из предшествующих композиций, дополнительно содержащей антиоксидант, например, в количестве, эффективном для ингибирования или уменьшения окисления соединения формулы 1.
2.19 Любой из предшествующих композиций, дополнительно содержащей антиоксидант, где антиоксидант представляет собой водорастворимый антиоксидант (например, аскорбиновую кислоту, липоевую кислоту), или жирорастворимый антиоксидант (например, липоевую кислоту, витамин Е, токоферолы, каротины или фенольные антиоксиданты), или нейтральный или слабоосновной антиоксидант или каталитический антиоксидант (например, эбселен), или металлсодержащий антиоксидант.
2.20 Любой из предшествующих композиций, дополнительно содержащей антиоксидант, где антиоксидант представляет собой липидные или нейтральные или слабоосновные антиоксиданты, например, где полимер содержит концевые карбоксильные группы.
2.21 Любой из предшествующих композиций, дополнительно содержащей антиоксидант, где антиоксидант представляет собой фенольный антиоксидант (например, бутилированный гидроксианизол (ВНА) или бутилированный гидрокситолуол (ВНТ)).
2.22 Любой из предшествующих композиций, дополнительно содержащей антиоксидант, где антиоксидант представляет собой ВНТ.
В еще одном варианте осуществления настоящее изобретение относится к способам А или В, как описано выше в настоящем документе, где соединение формулы I формулируется в пероральной осмотической системе доставки с контролируемым высвобождением (OROS) для доставки соединений формулы I, например, аналогично системам, описанным в WO 2000/35419 и EP 1539115 (опубликованная заявка США No. 2009/0202631), содержание каждой из заявок включено в качестве ссылки, в полном объеме. Поэтому в одном из вариантов осуществления настоящее изобретение относится к способам А или В, как описано выше в настоящем документе, где соединение формулы I формулируется в лекарственное средство, содержащее (а) желатиновую капсулу, содержащую соединение формулы I, в свободной форме или в форме фармацевтически приемлемой соли, как описано выше; (b) многослойную стенку, наложенную на желатиновую капсулу, содержащую, в порядке снаружи от капсулы: (i) барьерный слой, (ii) расширяемый слой, и (iii) полупроницаемый слой; и (c) отверстие, образованное или образуемое через стенку. (Композиция P.1)
В еще одном варианте осуществления настоящее изобретение относится к способам А или В, как описано выше в настоящем документе, где соединение формулы I формулируется в композицию, включающую желатиновую капсулу, содержащую жидкость, соединения формулы I, в свободной форме или в форме фармацевтически приемлемой соли, как описано выше, желатиновая капсула окружена многослойной стенкой, содержащей барьерный слой, контактирующий с внешней поверхностью желатиновой капсулы, расширяемый слой, контактирующий с барьерным слоем, полупроницаемый слой, охватывающий расширяемый слой, и выходное отверстие, образованное или образуемое через стенку. (Композиция P.2)
В еще одном варианте осуществления настоящее изобретение относится к способам А или В, как описано выше в настоящем документе, где соединение формулы I формулируется в композицию, включающую желатиновую капсулу, содержащую жидкость, соединение формулы I, в свободной форме или в форме фармацевтически приемлемой соли, как описано выше, желатиновая капсула окружена многослойной стенкой, содержащей барьерный слой, контактирующий с внешней поверхностью желатиновой капсулы, расширяемый слой, контактирующий с барьерным слоем, полупроницаемый слой, охватывающий расширяемый слой, и выходное отверстие, образованное или образуемое в стенке, где барьерный слой образует уплотнение между расширяемым слоем и окружающей средой у выходного отверстия. (Композиция P.3)
В еще одном варианте осуществления настоящее изобретение относится к композиции, включающей желатиновую капсулу, содержащую жидкость, соединение формулы I, в свободной форме или в форме фармацевтически приемлемой соли, как описано выше в настоящем документе, желатиновая капсула окружена барьерным слоем, контактирующим с внешней поверхностью желатиновой капсулы, расширяемым слоем, контактирующим с частью барьерного слоя, полупроницаемым слоем, охватывающим по меньшей мере расширяемый слой, и выходное отверстие, образованное или образуемое в лекарственной форме, проходящее от наружной поверхности желатиновые капсулы к среде применения. (Композиция P.4). Расширяемый слой может быть образован в одном или нескольких дискретных участках, таких как, например, два участка, расположенные на противоположных сторонах или концах желатиновой капсулы.
В конкретном варианте осуществления соединения формулы I в пероральной осмотической системе доставки с контролируемым высвобождением (то есть, в композиции P.1-С.4), как описано выше, находятся в жидкой лекарственной форме, лекарственная форма может быть чистой, жидким активным агентом, жидким активным агентом в растворе, суспензией, эмульсией или самоэмульгирующейся композицией или тому подобное.
Более подробную информацию о композициях пероральной осмотической системы доставки с контролируемым высвобождением, включая характеристики желатиновой капсулы, барьерного слоя, расширяемого слоя, полупроницаемого слоя и отверстия могут быть найдены в WO 2000/35419, содержание которой включено в качестве ссылки в полном объеме. Другие пероральные осмотические системы доставки с контролируемым высвобождением можно найти в EP 1539115 (опубликованная заявка США No. 2009/0202631), содержание которой включено в качестве ссылки в полном объеме.
В еще одном варианте осуществления настоящее изобретение относится к способам А или В, как описано выше, где соединение формулы I, формулируется в устройство, содержащее (а) два или несколько слоев, указанных двух или нескольких слоев, содержащих первый слой и второй слой, указанный первый слой, содержащий соединение формулы I, в свободной форме или в форме фармацевтически приемлемой соли, как описано выше, указанный второй слой, содержащий полимер; (b) наружную стенку, окружающую указанные два или несколько слоев; и (c) отверстие в указанной наружной стене. (Композиция P.5)
Композиция Р.5 предпочтительно использует полупроницаемую мембрану, окружающую трехслойное ядро, описанное здесь: в этих вариантах осуществления первый слой называется первым лекарственным слоем и содержит небольшое количество лекарственного средства (например, соединения формулы I) и осмотический агент, такой как соль, средний слой, называемый вторым лекарственным слоем, содержит более высокие количества лекарственного средства, эксципиенты и не содержит соли; и третий слой, называемый выталкивающим слоем, содержит осмотические агенты и не содержит лекарственное средство. По меньшей мере, одно отверстие проделано сквозь мембрану на краю первого лекарственного слоя таблетки, имеющей форму капсулы. (Композиция P.6)
Композиция P.5 или P.6 может содержать мембрану, ограничивающую отсек, мембрану, окружающую внутренний защитный подслой, по меньшей мере одно выходное отверстие, образованное или образуемое в нем, и по меньшей мере часть мембраны является полупроницаемой; расширяющийся слой, расположенный в внутри отсека, удаленного от выходного отверстия, и выполненный в подвижной связи с полупроницаемой частью мембраны; первый лекарственный слой, примыкающий к выходному отверстию; и второй лекарственный слой, расположенный внутри отсека между первым лекарственным слоем и расширяющимся слоем, лекарственные слои, содержащие соединение формулы I в свободной форме или в форме его фармацевтически приемлемой соли. В зависимости от относительной вязкости первого лекарственного слоя и второго лекарственного слоя, получают различные профили высвобождения. Это является крайне важным для установления оптимальной вязкости для каждого слоя. В настоящем изобретении вязкость модулируют путем добавления соли, например, хлорида натрия. Профиль доставки из ядра зависит от массы, состава и толщины каждого из лекарственных слоев. (Композиция P.7)
В конкретном варианте осуществления композиция P.7 содержит первый лекарственный слой, содержащий соль, и второй лекарственный слой, не содержащий соль. Композиция P.5-P.7 может необязательно содержать слой, облегчающий выход, между мембраной и лекарственными слоями. Композиции P.1-P.7 обычно упоминаются как композиция пероральной осмотической системы доставки с контролируемым высвобождением.
Кроме того, изобретение относится к фармацевтической композиции, как описано выше, содержащей соединение формулы I в свободной форме или в форме фармацевтически приемлемой соли, например, как описано в любом из способов A или 0-1.50, или способе B в смеси с фармацевтически приемлемым разбавителем или носителем, например, в составе с немедленным или замедленным или отсроченным высвобождением, включая инъекционный состав длительного действия, для использования при лечении остаточных симптомов, как описано в любом из способов A, или 1.1-1.50, или для использования в лечении расстройств, как описано в способе B.
В другом аспекте настоящее изобретение относится к применению соединения формулы I или фармацевтической композиции, содержащей соединение формулы I, в свободной форме или в форме фармацевтически приемлемой соли, как описано в способах A или 1.1-1.50, сформулированному в состав с немедленным или замедленным или отсроченным высвобождением, включая инъекционный состав длительного действия, как описано выше, (в производстве лекарственного средства) для лечения остаточных симптомов, как описано в любом из способов А или 1.1-1.50, или для лечения расстройств, как описано в способе B.
В другом аспекте настоящее изобретение относится к применению соединения формулы I или любой фармацевтической композиции, как описано выше (например, композиции P.1-P.7 или композиция 2), содержащей соединение формулы I в свободной форме или в форме фармацевтически приемлемой соли, как описано в способах A или 1.1-1.50, где соединение находится в смеси или сочетании с антиоксидантом. Не будучи связанными какой-либо конкретной теорией, полагают, что присутствие антиоксиданта в композиции стабилизируют соединения формулы I. В предпочтительном варианте осуществления инъецируемые микросферы длительного действия содержат антиоксидант, при этом считается, что антиоксидант стабилизирует соединение формулы I в процессе высвобождения из микросферы.
Подробное описание изобретения
Настоящее изобретение обеспечивает неудовлетворенную потребность в лечении не только острых симптомов, а также остаточных симптомов психозов, в частности, шизофрении. Пациентов, страдающих шизофренией, в настоящее время лечат типичными или атипичными антипсихотическими средствами. Эти средства, которые могут быть эффективными для лечения позитивных симптомов психоза, как правило, являются недостаточными для лечения остаточных симптомов. Таким образом, другое или дополнительное лечение необходимо для улучшения результатов. В одном из вариантов осуществления способы по настоящему изобретению используют соединение формулы I, отдельно, как описано выше, или используют в комбинации соединения формулы I и одного или нескольких различных антипсихотических средств для лечения остаточных симптомов и острых симптомов психоза.
Как используется в настоящем документе ʺостаточные симптомыʺ включают негативные симптомы и общие психопатологические симптомы, как описано в Шкале позитивных и негативных симптомов (PANSS) для шизофрении, описанных Kay et al., Schizophr. Bull. (1987) 13(2):261-276, содержание которых включено в качестве ссылки в полном объеме. Негативные симптомы включают: притупленный аффект, эмоциональное отчуждение, малоконтактность, пассивно/апатическая социальная отгороженность, затруднение в абстрактном мышлении, нарушение спонтанности и плавности речи и стереотипного мышления. Общие психопатологические симптомы включают: соматическую озабоченность, тревога, чувство вины, напряженность, манерность и позирование, депрессия, моторная заторможенность, малоконтактность, необычное содержание мыслей, дезориентированность, нарушение внимания, снижение оценки и критичности к себе, расстройство воли, ослабление контроля импульсивности, поглощенность и активная социальная устраненность. Остаточные симптомы могут также включать депрессию, когнитивные нарушения и расстройства сна (например, бессонница). Из этих остаточных симптомов, соединения формулы I являются особенно полезными для лечения пассивной социальной отгороженности, стереотипного мышления, соматической озабоченности, тревоги, напряженности, активной социальной устраненности и депрессии. Большинство пациентов с шизофренией показывают дефициты в социальной функции, препятствующие успешной реинтеграции в общество. Дефициты социальной функции могут быть определены с помощью шкалы просоциальных факторов PANSS. Просоциальный фактор состоит из синдромов из позитивной субшкалы, негативной субшкалы и субшкалы общей психопатологии, таких как активная социальная устраненность, эмоциональное отчуждение, пассивная социальная отгороженность, стереотипное мышление, галлюцинаторное поведение и подозрительность. Этот фактор показано, чувствителен к изменению в условиях клинического исследования. Поскольку соединения формулы I, в частности, соединение А, как определено в примере 1, уменьшают негативные симптомы и также лечат ряд других симптомов проявлений, полагают, что соединения формулы I могут быть полезны при лечении дефицитов социальной функции. Таким образом, соединения по настоящему изобретению являются особенно полезными в улучшении социальной интеграции и социальной функции у пациентов, страдающих шизофренией. Социальная функция представляет собой способность распознавать, понимать, обрабатывать и использовать внешние сигналы, чтобы решать проблемы, сохранять работоспособность и вести межличностные отношения.
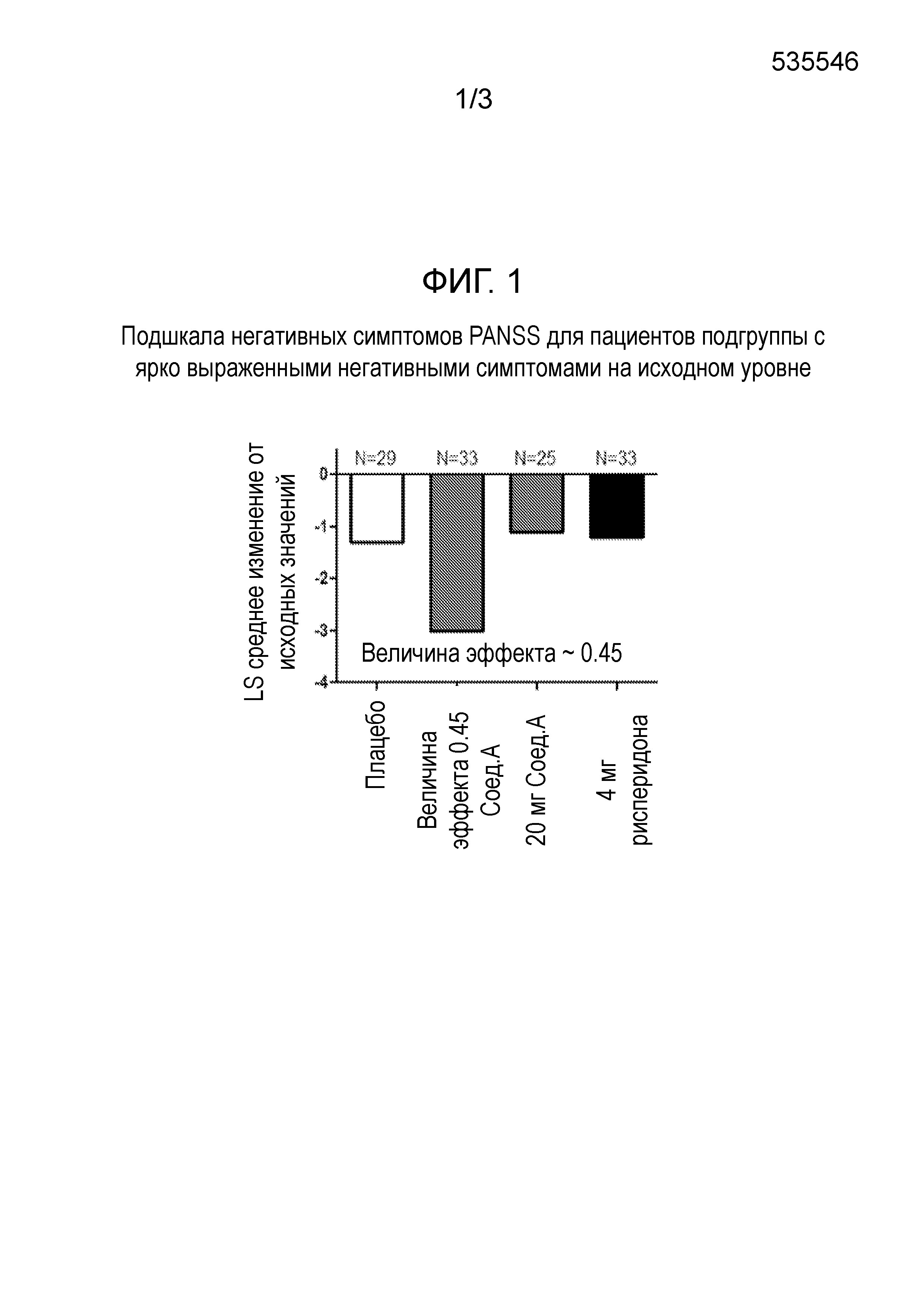
Лечение этих остаточных симптомов также особенно эффективно у пациентов с шизофренией, также страдающих от депрессии. 60 мг Соединения формулы I, в частности, соединения А, как определено в примере 1, в форме соли присоединения толуолсульфоновой кислоты, вводимого один раз в день утром, улучшает симптомы, связанные с шизофренией, измеренные статистически достоверным и клинически значимым снижением общего балла по шкале PANSS после 28 дней лечения. Это соединение А также не вызывает гиперпролактинемию, ЭПС/акатизию, увеличение массы тела или проблемы сердечно-сосудистой безопасности. В той же дозировке, это соединение А также статистически значимо улучшает субшкалу позитивной симптоматики и субшкалу общей психопатологии шкалы PANSS. Кроме того, соединение формулы I, в частности, соединение А, как определено в примере 1, в форме соли присоединения толуолсульфоновой кислоты, при 60 мг улучшает негативные симптомы в подгруппе пациентов с выраженными негативными симптомами в начале исследования. Важно отметить, что оно также улучшает некоторые пункты в субшкале негативных симптомов и субшкале общей психопатологии, указывающие на улучшение социальной функции.
Термин ʺострые симптомыʺ психоза или шизофрении относится к позитивным симптомам шкалы PANSS, таким как бред, концептуальная дезорганизация, галлюцинаторное поведение и подозрительность.
Термин "психоз" относится к заболеваниям, таким как шизофрения, бредовое расстройство, большая депрессия с психозом, биполярное расстройство с психотическими симптомами, кратковременное психотическое расстройство, шизофреноформное расстройство, шизоаффективное расстройство или психоз, вызванный заболеванием или употреблением наркотиков. Предпочтительно, пациент, страдающий от остаточных симптомов психоза, представляет собой пациента, страдающего от остаточных симптомов шизофрении.
Термин ʺбиполярное расстройствоʺ относится к расстройству, характеризуемому резкими сменами настроения. Индивиды с биполярным расстройством могут испытывать сильные чувства повышенного возбуждения, раздражительность и импульсивность с грандиозными убеждениями и скачками мыслей, называемыми маниакальным эпизодом. Симптомы депрессии могут включать ощущение усталости, безнадежности и отчаяние, трудности сосредоточения и суицидальные мысли. Некоторые люди испытывают оба типа симптомов в одном «смешанном» эпизоде. Тяжелые симптомы биполярного расстройства могут быть связаны с галлюцинациями или бредом, иначе называемыми психозом.
Термин ʺлечениеʺ следует соответственно понимать как охватывающее профилактику и лечение или уменьшение симптомов заболевания и/или лечение причины заболевания.
Термин "пациент" может включать человека или пациента, не являющегося человеком.
Если не указано иное или ясно из контекста, следующие термины в данном описании, имеют следующие значения:
(i) ʺАлкилʺ, как используется в настоящем описании, представляет собой насыщенный или ненасыщенный углеводородный радикал, например, от одного до двадцати одного атома углерода в длину, который может быть линейным или разветвленным (например, н-бутил или трет-бутил), предпочтительно, линейным, если не указано иное. Например, "C1-21 алкил" обозначает алкил, имеющий 1-21 атомов углерода. В одном варианте осуществления алкил необязательно замещен одной или несколькими гидрокси или C1-22алкокси (например, этокси) группами. В другом варианте осуществления алкил содержит 1-21 атомов углерода, предпочтительно, с прямой цепью, и, необязательно, насыщенный или ненасыщенный, например, R1 представляет собой алкильную цепь, содержащую от 1 до 21 атомов углерода, предпочтительно, 6-15 атомов углерода, 16-21 атомов углерода, например, так что вместе с -С(O)-, к которой он присоединяется, например, при расщеплении из соединения формулы I, образует остаток природной или неприродной, насыщенной или ненасыщенной жирной кислоты.
Способы получения соединений формулы I
Соединения формулы I и их фармацевтически приемлемые соли могут быть получены с использованием способов, как это описано и проиллюстрировано в любом из следующих патентов или заявок: патент США No. 6548493; 7238690; 6552017; 6713471; U.S. RE39680; U.S. RE39679; PCT/US08/03340; заявка США No. 10/786935; WO 2011/133224 A1, WO 2009/114181 и WO 2013/155505. Исходные материалы для этих процессов, если не являются коммерчески доступными, могут быть получены в соответствии с процедурами, выбранными из области химии, с использованием методик, подобных или аналогичных синтезу известных соединений. Все ссылки, приведенные в настоящем описании, включены в качестве ссылки в полном объеме.
Соединения формулы I относятся к соединениям формулы I или любому из соединений, описанных в способах 1.1-1.50, которые включают:

в свободной форме или в форме фармацевтически приемлемой соли. Соединения формулы I также включают другие конкретные соединения, где Y представляет собой -C(H)(OH)- или -C(H)(OR1), где R1 определен ранее и X представляет собой -O-, -N(H)- или -N(CH3)-. Они включают, например:

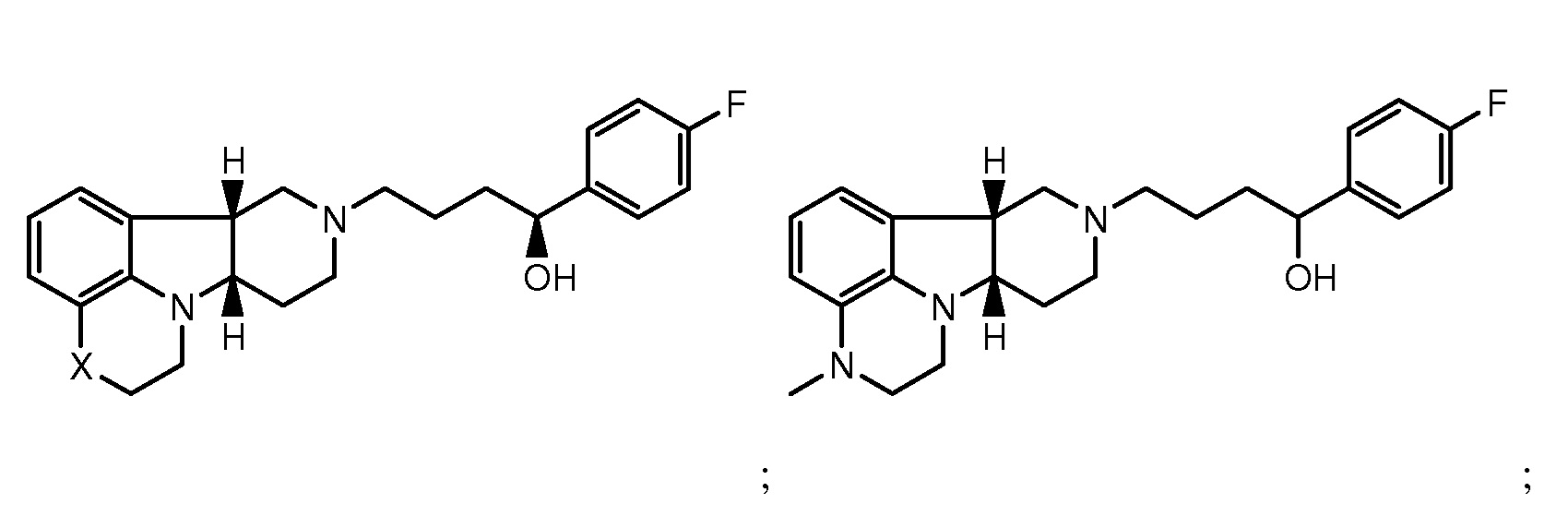
в свободной форме или в форме фармацевтически приемлемой соли. Конкретное соединение формулы I также включает соединение А, которое представляет собой соединение формулы I, где X представляет собой -N(CH3)- и Y представляет собой -C(O)-. Термины "соединения формулы I" и "соединения формулы I" могут быть использованы взаимозаменяемо, и могут быть использованы в качестве единственного терапевтического средства, или они также могут быть использованы в комбинации или для совместного введения с другими активными средствами. Кроме того, в способах по настоящему изобретению выражение "соединение формулы I" может включать более чем одно из соединений формулы I.
Соединения формулы I могут в некоторых случаях также существовать в форме пролекарства. Форма пролекарства представляет собой соединение, которое преобразуется в организме в активное соединение формулы I. Термин ʺпролекарствоʺ является термином, принятым в данной области, и относится к предшественнику лекарственного средства перед введением, который образует или высвобождает активный метаболит in vivo после введения, в результате некоторых химических или физиологических процессов. Например, когда соединения формулы I содержат гидрокси- или карбокси-заместители, эти заместители могут образовывать физиологически гидролизуемые и приемлемые сложные эфиры. Как используется в настоящем документе, ʺфизиологически гидролизуемые и приемлемые сложные эфирыʺ означают сложные эфиры соединений формулы I, которые гидролизуются в физиологических условиях с получением кислот (в случае соединений формулы I, которые имеют гидроксизаместители) или спиртов (в случае соединений формулы I, которые имеют карбоксизаместители), которые сами по себе являются физиологически приемлемыми в дозах, предназначенных для введения. Например, где Y соединения формулы I представляет собой -C(H)(OR1), и R1 представляет собой физиологически гидролизуемый и приемлемый ацил, например, -C(O)-C1-21алкил, например, -C(O)-C3алкил или -C(O)-C9алкил, эти соединения могут гидролизоваться в физиологических условиях с получением соединения формулы I, где Y представляет собой -C(H)(OH) с одной стороны и C1-21алкил-C(O)OH, например, C3алкил-C(O)OH или C9алкил-C(O)OH с другой стороны. В частности, соединения формулы I, где Y представляет собой -O-, -C(R2)(OH)-, -C(R3)(OR1) или -C(O)-; R1 представляет собой C1-6 алкил (например, метил) и R2 и R3 представляют собой H или C1-6 алкил (например, метил) являются активными фрагментами. Напротив, соединения формулы I, где R1 представляет собой -C(O)-C1-21алкил является физиологически активным фрагментом, будут иметь слабую активность или отсутствие активности, но в физиологических условиях будут подвергаться гидролизу для получения соединений формулы I, R1 отщепляется оставляя -C(R2)(OH) или -C(R3)(OH), и другой продукт гидролиза является не токсичным при релевантных концентрациях, например, при концентрациях, которые будут предоставлены гидролизу in vivo дозы пролекарственного соединения. В некоторых физиологических условиях, соединения формулы I, где R1 представляет собой C1-6 алкил (например, метил), могут также проходить превращение in vivo в более активное соединение, где R1 представляет собой H, и, следовательно, эти соединения могут считаться и активными фрагментами и пролекарствами.
Как будет понятно, темин пролекарство охватывает обычные фармацевтические пролекарственные формы. Где используют пролекарство (например, соединение формулы (I), где R1 представляет собой -C(O)-C1-21алкил), величина дозы рассчитывается на основании количества активного соединения формулы I, например, Y представляет собой -O-, -C(R2)(OH)-, -C(R3)(OR1) или -C(O)-; R1 представляет собой C1-6 алкил (например, метил), R2 и R3 представляют собой H или C1-6 алкил (например, метил), в частности, соединение формулы (I), где Y представляет собой C(=O) или Y представляет собой C(H)(OH) в форме свободного основания или в форме соли, например, в форме соли присоединения толуолсульфоновой кислоты.
Соединения формулы I могут содержать один или несколько хиральных атомов углерода. Соединения, таким образом, существуют в отдельных изомерных формах, например, энантиомерных или диастереомерных формах или в виде смесей отдельных форм, например, рацемических/диастереомерных смесей. Любой изомер может присутствовать, в котором асимметричный центр находится в (R) -, (S)- или (R,S)- конфигурации. Изобретение следует понимать как охватывающее как отдельные оптически активные изомеры, так и их смеси (например, рацемические/диастереомерные смеси). Соответственно, соединения формулы I могут быть рацемической смесью, или они могут быть преимущественно, например, в чистой или по существу чистой, изомерной форме, например, более 70% энантиомерный/диастереомерный избыток ("эи"), предпочтительно более 80% эи, более предпочтительно, более 90% ее, наиболее предпочтительно, более 95% эи. Очистка указанных изомеров и разделение указанных смесей изомеров может быть осуществлено с помощью стандартных методов, известных в данной области (например, колоночной хроматографией, препаративной ТСХ, препаративной ВЭЖХ, псевдодвижущимся слоем и тому подобное).
Геометрические изомеры по природе заместителей около двойной связи или кольца могут присутствовать в цис- (Z) или транс- (Е) форме, и обе изомерные формы включены в объем настоящего изобретения.
Соединения формулы I могут быть включены в виде состава с замедленным или отсроченным высвобождением, например, депо-препарата, например, путем диспергирования, растворения или инкапсулирования соединений формулы I в полимерную матрицу, как описано выше в P.1-P.7, так, что соединение постоянно высвыбождается по мере разрушения полимера со временем. Высвобождение соединений формулы I из полимерной матрицы обеспечивает контролируемое и/или отсроченное и/или замедленное высвобождение этих соединений, например, из фармацевтического депо, индивиду, например, теплокровному животногму, такому как человек, которому вводят фармацевтическое депо. Таким образом, фармацевтическое депо доставляет соединения формулы I субъекту в концентрациях, эффективных для лечения конкретного заболевания или медицинского состояния в течение продолжительного периода времени, например, 14-180 дней, предпочтительно, приблизительно 30, приблизительно 60 или приблизительно 90 дней.
Полимеры, пригодные для полимерной матрицы в композиции по настоящему изобретению (например, депо композиция по настоящему изобретению), могут включать полиэфир жирной гидроксикислоты и их производные или другие агенты, такие как полимолочная кислота, полигликолевая кислота, полилимонная кислота, полияблочная кислота, поли-бета.-гидроксимасляная кислота, полимер с раскрытым кольцом эпсилон.-капро-лактон, сополимер молочной кислоты и гликолевой кислоты, сополимер 2-гидроксимасляной кислоты-гликолевой кислоты, сополимер полимолочной кислоты-полиэтиленгликоля или сополимер полигликолевой кислоты-полиэтиленгликоля), полимер алкил альфа-цианакрилата (например, поли(бутил-2-цианакрилат)), полиалкиленоксалат (например, политриметиленоксалат или политетраметиленоксалат), полиортоэфир, поликарбонат (например, полиэтиленкарбонат или полиэтиленпропиленкарбонат), полиортокарбонат, полиаминокислоты (например, поли-гамма-L-аланин, поли-.гамма.-бензил-L-глутаминовая кислота или поли-y-метил-L-глутаминовая кислота), сложный эфир гиалуроновой кислоты, и тому подобное, и один или несколько из этих полимеров, могут быть использованы.
Если полимеры представляют собой сополимеры, они могут быть любыми из случайных, блочных и/или привитых сополимеров. Когда вышеуказанные альфа-гидроксикарбоновые кислоты, гидроксидикарбоновые кислоты и гидрокситрикарбоновые кислоты обладают оптической активностью в их молекулах, может быть использован любой из D-изомеров, L-изомеров и/или DL-изомеров. Среди других, полимер альфа-гидроксикарбоновой кислоты (предпочтительно, полимер молочной кислоты-гликолевой кислоты), его сложный эфир, сложные эфиры поли-альфа-цианоакриловой кислоты, и тому подобное могут быть использованы, и сополимер молочной кислоты-гликолевой кислоты (также называемый поли(лактид-альфа-гликолид) или поли(молочная-со-гликолевая)кислота), и именуемый далее PLGA) является предпочтительным. Таким образом, в одном аспекте полимер, используемый для полимерной матрицы представляет собой PLGA. Как используется в настоящем описании термин PLGA включает полимеры молочной кислоты (также называемый как полилактид, поли(молочная кислота), или PLA). Наиболее предпочтительно, полимер представляет собой биоразлагаемый полимер поли(d,l-лактид-со-гликолид).
В предпочтительном варианте осуществления изобретения полимерная матрица по настоящему изобретению представляет собой биосовместимый и биоразлагаемый полимерный материал. Термин "биосовместимый" определяется как полимерный материал, который не токсичен, не является канцерогенным, и не вызывает значительного воспаления тканей организма. Материал матрицы должен быть биоразлагающимся, где полимерный материал должен разлагаться процессами в организме на продукты, которые легко выводятся и не накапливаются в организме. Продукты биоразложения должны быть также биосовместимыми с организмом в том смысле, что полимерная матрица должна быть биосовместима с организмом. Конкретные примеры подходящих полимерных материалов матрицы включают поли(гликолевую кислоту), поли-D,L-молочную кислоту, поли-L-молочную кислоту, сополимеры вышеуказанных соединений, поли(алифатические карбоновые кислоты), сополиоксалаты, поликапролактон, полидиоксонон, поли(ортокарбонаты), поли(ацетали), поли(молочную кислоту-капролактон), полиортоэстеры, поли(гликолевая кислота-капролактон), полиангидриды и природные полимеры, включающие альбумин, казеин и воски, так ие, как глицерола моно- и дистеарат и подобные. Предпочтительным полимером для использования в практике настоящего изобретения является dl-(полилактид-со-гликолид). Предпочтительно, чтобы молярное соотношение лактида к гликолиду в таком сополимере было в пределах от приблизительно 75:25 до 50:50.
Подходящие полимеры PLGA могут иметь средневесовую молекулярную массу от приблизительно 5000 до 500000 дальтонов, например, приблизительно 150000 дальтонов, или 20000-200000 дальтонов, например, 24000-38000 дальтонов, или приблизительно 113000 дальтонов или приблизительно 159000 дальтонов. Например, полимер PLGA имеет средневесовую молекулярную массу 24000-38000 дальтонов. В зависимости от скорости разложения, которые должны быть достигнуты, могут быть использованы различные молекулярные массы полимеров. Для диффузионного механизма высвобождения лекарственного средства полимер должен оставаться целым до тех пор, пока все лекарственное средство не выйдет из полимерной матрицы, и затем распадаться. Лекарственное средство может также высвобождаться из полимерной матрицы по мере биоэрозирования полимерного наполнителя.
PLGA могут быть получены обычными способами, или могут быть коммерчески доступны. Например, PLGA может быть получен полимеризацией с раскрытием кольца с подходящим катализатором из циклического лактида, гликолида и т.д. (см. EP-0058481B2; Effects of polymerization variables on PLGA properties: molecular weight, composition and chain structure).
Считают, что PLGA является биоразлагаемым благодаря разложению всей твердой полимерной композиции в результате разрыва гидролизуемых и ферментативно расщепляемых сложноэфирных связей в биологических условиях (например, в присутствии воды и биологических ферментов, находящихся в тканях теплокровных животных, таких как человек) с образованием молочной кислоты и гликолевой кислоты. И молочная кислота, и гликолевая кислота являются водорастворимыми, нетоксичными продуктами нормального метаболизма, которые могут далее подвергаться биоразложению с образованием диоксида углерода и воды. Другими словами, считается, что PLGA разлагается посредством гидролиза его сложноэфирных групп в присутствии воды, например, в организме теплокровного животного, такого как человек, с образованием молочной кислоты и гликолевой кислоты и созданием кислого микроклимата. Молочная и гликолевая кислоты представляют собой побочные продукты различных метаболических путей в организме теплокровного животного, такого как человек, в нормальных физиологических условиях и поэтому являются хорошо переносимыми и вызывают минимальную системную токсичность.
В другом варианте осуществления изобретения полимерная матрица, подходящая для осуществления настоящего изобретения, может включать звездчатый полимер, где структура полиэфира является звездчатой. Эти полиэфиры имеют один остаток полиола, как центральная часть, окруженный цепями кислотного остатка. Фрагментом полиола может быть, например, глюкоза или, например, маннитол. Эти сложные эфиры известны и описаны в GB 2145422 и в патенте США No. 5538739, содержание которых включено в качестве ссылки.
Звездообразные полимеры могут быть получены с использованием полигидроксисоединений, например, полиола, например, глюкозы и маннитола в качестве инициатора. Полиол содержит по меньшей мере 3 гидроксигруппы и имеет молекулярную массу до приблизительно 20000 дальтон, с по меньшей мере 1, предпочтительно по меньшей мере 2, например, как среднее 3 гидроксигруппы полиола, находящиеся в форме эфирных групп, которые содержат полилактидные или со-полилактидные цепи. Разветвленные сложные полиэфиры, например, поли (d,l-лактид-со-гликолид) имеют центральный глюкозный фрагмент, включающий отходящие от него линейные полилактидные цепи.
Состав с замедленным или отсроченным высвобождением, как описано выше, может содержать полимер в форме микрочастиц (например, микросферы) или наночастиц, или в жидкой форме, с соединениями формулы I, диспергированными или инкапсулированными в нем. Под "микрочастицами" понимаются твердые частицы, которые содержат соединения формулы I, либо в растворе, либо в твердой форме, где такое соединение диспергируется или растворяется внутри полимера, который служит в качестве матрицы частицы. Соответствующим подбором полимерных материалов можно получить препарат микрочастиц, в котором полученные микрочастицы демонстрируют как свойства диффузионного выхода, так и свойства биораспада. Это находит применение в многофазных схемах выхода.
Когда полимер представлен в форме микрочастиц, микрочастицы можно получать, используя любой подходящий способ, такой как выпаривание растворителя или способ экстракции растворителем. Например, в способе выпаривания растворителя, соединения формулы I и полимер могут быть растворены в подходящем летучем органическом растворителе (например, кетоне, таком как ацетон, галогенированном углеводороде, таком как хлороформ или метиленхлорид, галогенированном ароматическом углеводороде, циклическом простом эфире, таком как диоксан, сложном эфире, таком как этилацетат, нитриле, таком как ацетонитрил, или спирте, таком как этанол) и диспергированы в водной фазе, содержащей подходящий стабилизатор эмульсии (например, поливиниловый спирт, PVA). Органический растворитель затем выпаривают с образованием микрочастиц с соединениями формулы I, инкапсулированными в них. В способе экстракции растворителем соединения формулы I и полимер могут быть растворены в полярном растворителе (таком как ацетонитрил, дихлорметан, метанол, этилацетат или метилформиат) и затем диспергированы в водной фазе (такой как раствор вода/PVA). Получают эмульсию с образованием микрочастиц с соединениями формулы I, инкапсулированными в них. Распылительная сушка является альтернативным способом изготовления для получения микрочастиц.
Другой способ получения микрочастиц по настоящему изобретению также описан в патенте США No. 4389330 и патенте США No. 4530840, содержания которых включены в качестве ссылки.
Микрочастицу по настоящему изобретению можно получить любым способом, способным производить микрочастицы в пределах размера, приемлемого для применения в качестве инъекционной композиции. Один предпочтительный способ получения тот, который описан в патенте США No. 4389330. В этом способе активный агент растворяют или диспергируют в соответствующем растворителе. К среде, содержащей агент, добавляют полимерный материал матрицы в таком количестве относительно активного ингредиента, которое дает продукт с нужной нагрузкой активным агентом. В другом случае все ингредиенты продукта микрочастиц можно смешивать вместе в среде растворителя.
Растворители для соединений формулы I и полимерного материала матрицы, которые можно использовать в практике настоящего изобретения, включают органические растворители, такие как ацетон; галогенированные углеводороды, такие как хлороформ, метиленхлорид и прочие; ароматические углеводородные соединения; галогенированные ароматические углеводородные соединения; циклические эфиры, спирты, такие как бензиловый спирт, этилацетат и другие. В одном из вариантов осуществления растворителем для использования в практике настоящего изобретения может быть смесь бензилового спирта и этилацетата. Дополнительная информация для получения микрочастиц, пригодных для настоящего изобретения, можно найти в публикации патента США номер 2008/0069885, содержание которого включено в настоящее описание в качестве ссылки в полном объеме.
Количество соединений формулы I включено в микрочастицы, обычно в диапазоне от приблизительно 1% масс. до приблизительно 90% масс., предпочтительно 30-50% масс., более предпочтительно 35-40% масс. Под мас. % подразумеваются части соединений формулы I на общую массу микрочастицы.
Состав с замедленным или отсроченным высвобождением может включать фармацевтически приемлемый разбавитель или носитель, такие как смешивающиеся с водой разбавитель или носитель.
В одном варианте осуществления состав с замедленным или отсроченным высвобождением представляет собой инъецируемый состав длительного действия. В еще одном варианте осуществления инъецируемый состав длительного действия формулируют с использованием полимерных микросфер, например, микросфер, содержащих матрицу PLGA с соединениями формулы I в свободной форме или форме фармацевтически приемлемой соли, диспергированными, растворенными или инкапсулированными в них. В предпочтительном варианте осуществления полимер PLGA представляет собой полимер с соотношением 75:25 PLA/PLG (лактид:гликолид) с концевыми группами карбоновой кислоты или карбонового эфира. В предпочтительном варианте осуществления соединение формулы I присутствует в микросфере в форме свободного основания. Микросферы могут быть получены с использованием способов, известных в данной области, например, с помощью способа эмульгирования-выпаривания растворителя без микро-просеивания, или способом эмульгирования-выпаривания растворителя с сухим микро-просеиванием, или способом эмульгирования-выпаривания растворителя с мокрым микро-просеиванием.
Скорость, при которой микросферы PLGA разлагаются, в значительной степени зависит от выбранного диапазона молекулярной массы молекул полимера и от размера микросфер. В одном из вариантов осуществления, диапазон средней молекулярной массы для полимера PLGA составляет 24000-38000 дальтон. В другом варианте осуществления полимер PLGA имеет среднюю молекулярную массу приблизительно 113000 дальтон. В еще одном варианте осуществления полимер PLGA имеет среднюю молекулярную массу приблизительно 159000 дальтон. В некоторых вариантах осуществления, временные рамки для полной деградации микросфер и высвобождения инкапсулированных лекарственных соединений составляют, например, менее 6 месяцев, менее 4 месяцев, менее 3-х месяцев, менее 2 месяцев или менее 1 месяца.
Диаметр микросфер, например, средний диаметр (или D50), 10-процентильный диаметр (D10), 25-процентильный диаметр (D25), 75-процентильный диаметр (D75), или 90-процентильный диаметр (D90), может быть от приблизительно 1 мкм до приблизительно 100 мкм, или от приблизительно 2 мкм до приблизительно 80 мкм, или от приблизительно 2 мкм до приблизительно 60 мкм, или приблизительно 2 мкм до приблизительно 50 мкм, или приблизительно 2 мкм до приблизительно 40 мкм, или приблизительно 2 мкм до приблизительно 30 мкм, или приблизительно 5 мкм до приблизительно 35 мкм, или от приблизительно 5 мкм до приблизительно 25 мкм, или от приблизительно 5 мкм до приблизительно 20 мкм, или от приблизительно 10 мкм до приблизительно 20 мкм, или приблизительно 10 мкм до приблизительно 200 мкм, от приблизительно 20 мкм до приблизительно 160 мкм, или от приблизительно 20 мкм до приблизительно 120 мкм, или от приблизительно 20 мкм до приблизительно 100 мкм, или от приблизительно 20 мкм до приблизительно 80 мкм, или от приблизительно 20 мкм до приблизительно 70 мкм, или от приблизительно 20 мкм до приблизительно 60 мкм, или от приблизительно 20 мкм до приблизительно 50 мкм, или от приблизительно 20 мкм до приблизительно 40 мкм, или от приблизительно 20 мкм до приблизительно 30 мкм, или от приблизительно 25 мкм до приблизительно 70 мкм, или от приблизительно 25 мкм до приблизительно 60 мкм, или от приблизительно 25 мкм до приблизительно 50 мкм, или от приблизительно 25 мкм до приблизительно 40 мкм, или от приблизительно 30 мкм до приблизительно 60 мкм, или от приблизительно 30 до 50 мкм, или от приблизительно 30 мкм до приблизительно 40 мкм, или от приблизительно 30 мкм до приблизительно 120 мкм, или от приблизительно 40 мкм до приблизительно 120 мкм, или от приблизительно 40 мкм до приблизительно 100 мкм, или от приблизительно 40 мкм до приблизительно 80 мкм, или от приблизительно 40 мкм до приблизительно 70 мкм, или от приблизительно 40 мкм до приблизительно 60 мкм, или от приблизительно 40 мкм до приблизительно 50 мкм, или от приблизительно 50 мкм до приблизительно 100 мкм, или от приблизительно 50 мкм до приблизительно 80 мкм, или от приблизительно 50 мкм до приблизительно 70 мкм, или от приблизительно 50 мкм до приблизительно 60 мкм, или от приблизительно 60 мкм до приблизительно 100 мкм, или от приблизительно 60 мкм до приблизительно 90 мкм, или от приблизительно 60 мкм до приблизительно 80 мкм, или от приблизительно 60 мкм до приблизительно 70 мкм, или от приблизительно 70 мкм до приблизительно 100 мкм, или от приблизительно 70 мкм до приблизительно 90 мкм, или от приблизительно 70 мкм до приблизительно 80 мкм, или от приблизительно 75 мкм до приблизительно 110 мкм, или от приблизительно 40 мкм до приблизительно 160 мкм, или от приблизительно 50 мкм до приблизительно 160 мкм, или от приблизительно 50 мкм до приблизительно 120 мкм, или приблизительно 20 мкм, приблизительно 30 мкм, приблизительно 40 мкм, приблизительно 50 мкм, приблизительно 60 мкм, приблизительно 70 мкм, приблизительно 80 мкм, приблизительно 90 мкм, или приблизительно 100 мкм. Такие измерения размера частиц могут быть сделаны, например, с помощью фотомикроскопии, сканирующей электронной микроскопии, лазерной дифракции, рассеяния света, и другими способами, известными специалистам в данной области.
Количество лекарственного средства, инкапсулированного в каждую микросферу, в среднем, может составлять от приблизительно 5% масс. до приблизительно 50% масс., например, 10% масс. до приблизительно 50% масс., или от приблизительно 20% масс. до приблизительно 40% масс., или от приблизительно 30% масс. до приблизительно 35% масс., или приблизительно 8,5% масс., или приблизительно 16% масс., или приблизительно 30% масс., или приблизительно 35% масс., или приблизительно 40% масс. В предпочтительном варианте осуществления количество лекарственного средства, инкапсулированное в каждую микросферу составляет приблизительно 8,5% по массе или приблизительно 16% по массе.
Детали композиции пероральной осмотической системы доставки с контролируемым высвобождением могут быть найдены в EP 1539115 (публикация США No. 2009/0202631) и WO 2000/35419, содержание каждой из которых включено в качестве ссылки в полном объеме.
Термин "терапевтически эффективное количество" означает любое количество соединений формулы I, которое при введении индивиду, страдающему заболеванием или расстройством, является эффективным, чтобы вызвать уменьшение, ремиссию или регрессию заболевания или расстройства, в течение периода времени, предназначенного для лечения.
Депрессия, как оценивается, появляется у 50% пациентов, страдающих шизофренией. Пациенты, страдающие шизофренией с сопутствующей депрессией, по сравнению с пациентами только с шизофренией, как правило, имеют худшее общее психическое и физическое здоровье, более низкое качество жизни, большее ухудшение социальных отношений, меньшую удовлетворенность сном, способность выполнять ежедневную деятельность, работоспособность, перемещение, социальную поддержку и чувство собственного достоинства. Таким образом, симптомы депрессии усугубляют недостатки психосоциального функционирования и повышают риск психотического рецидива. В отличие от антагонистов дофаминовых рецептор, соединения формулы I, в частности, формулы А, как определено в примере 1, нормализует активность дофамина в мозге, в частностия, в префронтальной коре. Соединения формулы I, в частности, формулы А, как описано в примере 1, связываются с 5-HT2A и дофаминовыми D2 рецепторами. Кроме того, соединения формулы I также модулируют глутаматергический фосфопротеин (D1/GluN2B) и дофамин фосфопротеин (DPPM). Соединения формулы I также демонстрируют наномолярную аффинность связывания к транспортеру серотонина по сравнению с известными антидепрессантами. Таким образом, в дополнение к лечению острых симптомов (например, галлюцинации и бред) и остаточных симптомов, соединения формулы I могут быть также использованы для лечения депрессии и/или расстройств сна. Для пациентов с шизофренией, которые также страдают от депрессии, соединения формулы I особенно полезны для улучшения общих значений PANSS и негативных симптомов. В то время как одно из атипичных антипсихотических лекарственных средств, рисперидон, является также полезным для лечения некоторых негативных симптомов, это соединение является менее эффективным по сравнению с подгруппой пациентов, страдающих депрессией, на момент начала исследования получающих соединение формулы I (см. Фиг. 2 и 3 и Пример 1). Таким образом, в некоторых вариантах осуществления способы по настоящему изобретению особенно полезны для лечения депрессии и/или расстройств сна у пациентов, страдающих острыми и/или остаточными симптомами психоза, а также для лечения острых и/или остаточных симптомов психоза у пациентов, страдающих от депрессии и/или расстройств сна.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения остаточных симптомов путем введения соединения формулы I в комбинации с одним или несколькими другими терапевтическими средствами, такими как антипсихотическое средство(а) и/или анти-депрессивное средство(а) и/или снотворное средство. В таких способах, антипсихотические и/или анти-депрессивные и/или снотворные средства могут быть вспомогательным средством к соединению формулы I или соединение формулы I может быть вспомогательным средством к антипсихотическому средству и/или анти-депрессивному средству и/или снотворному средству. Как используется в настоящем описании термин "дополнительное" или "вспомогательное средство" относится к любому лечению, которое используется в сочетании с другим, для увеличения вероятности излечения, или для повышения эффективности первого лечения. Другими словами, дополнительная терапия действует как вспомогательное средство для первичного лечения. В другом варианте осуществления соединение формулы I используют в качестве монотерапии для лечения острых симптомов и/или остаточных симптомов шизофрении, а также депрессии и/или расстройства сна (например, инсомния) у пациентов, страдающих шизофренией.
Другие терапевтические средства, которые дополнительно могут быть введены пациенту, нуждающемуся в этом, включают соединения, которые модулируют активность ГАМК (например, усиливают активность и облегчают передачу ГАМК), агонист рецептора ГАМК-B, модулятор 5-HT (например, агонист рецептора 5-HT1A, антагонист рецептора 5-HT2A, обратный агонист рецептора 5-HT2A, и тому подобное), агонист мелатонинового рецептора, модулятор ионного канала (например, блокатор), антагонист рецептора серотонина 2/ингибитор обратного захвата серотонина (SARI), антагонист орексиновых рецепторов, агонист рецептора H3, антагонист норадренергического рецептора, агонист галанинового рецептора, антагонист рецептора CRH, гормон роста человека, агонист рецептора гормона роста, эстроген, агонист рецептора эстрогена, препарат нейрокинина-1; и антипсихотические средства, например, атипичные антипсихотические средства, в свободной форме или в форме фармацевтически приемлемой соли.
Термин "ГАМК" относится к гамма-аминомасляной кислоте. Соединения ГАМК представляют собой соединения, которые связываются с ГАМК-рецептором, и включают, но ими не ограничиваются, одно или несколько из доксепина, алпразолама, бромазепама, клобазама, клоназепама, клоразепата, диазепама, флунитразепама, флуразепама, лоразепама, мидазолама, нитразепама, оксазепама, темазапама, триазолама, индиплона, зопиклона, эсзопиклона, залеплона, золпидема, габаксадола, вигабатрина, тиагабина, EVT 201 (Evotec Pharmaceuticals) или эстазолама.
Другие дополнительные терапевтические средства представляют собой антагонисты 5HT2A-рецептора, такие как кетансерин, рисперидон, эпливансерин, волинансерин (Sanofi-Aventis, Франция), прувансерин, пимавансерин (ACP-103), MDL 100907 (Sanofi-Aventis, Франция), HY10275 (Eli Lilly), APD125 (Arena Pharmaceuticals, Сан-Диего, Калифорния) или AVE8488 (Sanofi-Aventis, Франция).
Еще другие дополнительные терапевтические средства включают пизотифен.
Другие дополнительные терапевтические средства представляют собой агонисты 5HT1A-рецептора, такие как репинотан, саризотан, эптапирон, буспирон или MN-305 (MediciNova, Сан-Диего, Калифорния).
Другие дополнительные соединения представляют собой агонисты мелатонинового рецептора, такие как мелатонин, рамелтеон (Розерем®, Takeda Pharmaceuticals, Япония), VEC-162 (Vanda Pharmaceuticals, Роквилл, штат Мэриленд), PD-6735 (исследования II фазы) или агомелатин.
Другие дополнительные терапевтические средства представляют собой блокаторы ионного канала, такие как ламотриджин, габапентин или прегабалин.
Другие дополнительные терапевтические средства представляют собой антагонисты орексиновых рецепторов, такие как орексин, 1,3-биарилмочевина, SB-334867-a (GlaxoSmithKline, Великобритания), GW649868 (GlaxoSmithKline) или производное бензамида, например.
Другие дополнительные терапевтические средства представляют собой антагонист рецептора серотонина 2/ингибиторы обратного захвата серотонина (SARI), такие как Org 50081 (Organon - Нидерланды), ритансерин, нефазодон, серзон или тразодон.
Другие дополнительные терапевтические средства представляют собой препараты нейрокинина-1, такие как касопитант (GlaxoSmithKline).
Конкретные примеры дополнительных терапевтических средств включают модафинил, армодафинил, доксепин, алпразолам, бромазепам, клобазам, клоназепам, клоразепат, диазепам, флунитразепам, флуразепам, лоразепам, мидазолам, нитразепам, оксазепам, темазапам, триазолам, индиплон, зопиклон, эсзопиклон, залеплон, золпидем, габаксадол, вигабатрин, тиагабин, EVT 201 (Evotec Pharmaceuticals), эстазолам, кетансерин, рисперидон, эпливансерин, волинансерин (Sanofi-Aventis, Франция), прувансерин, MDL 100907 (Sanofi-Aventis, Франция), HY10275 (Eli Lilly), APD125 (Arena Pharmaceuticals, Сан-Диего, Калифорния), AVE8488 (Sanofi-Aventis, Франция), репинотан, саризотан, эптапирон, буспирон, MN-305 (MediciNova, Сан-Диего, Калифорния), мелатонин, рамелтеон (Розерем®, Takeda Pharmaceuticals, Япония), VEC-162 (Vanda Pharmaceuticals, Роквилл, штат Мэриленд), PD-6735 (исследования II фазы), агомелатин, ламотриджин, габапентин, прегабалин, орексин, 1,3-биарилмочевину, SB-334867-a (GlaxoSmithKline, Великобритания), GW649868 (GlaxoSmithKline), производное бензамида, Org 50081 (Organon - Нидерланды), ритансерин, нефазодон, серзон, тразодон, касопитант (GlaxoSmithKline), амитриптилин, амоксапин, бупропион, циталопрам, кломипрамин, дезипрамин, доксепин, дулоксетин, эсциталопрам, флуоксетин, флувоксамин, имипрамин, изокарбоксазид, мапротилин, миртазапин, нефазодон, нортриптилин, пароксетин, фенелзин сульфат, протриптилин, сертралин, транилципромин, тразодон, тримипрамин, венлафаксин, хлорпромазин, галоперидол, дроперидол, флуфеназин, локсапин, мезоридазин молиндон, перфеназин, пимозид, прохлорперазин промазин, тиоридазин, тиотиксен, трифтороперазин, клозапин, арипипразол, оланзапин, кветиапин, рисперидон, зипрасидон, палиперидон, азенапин, луразидон, илоперидон, карипразин, амисульприд, зотепин, сертиндол, в свободной форме или в форме фармацевтически приемлемой соли.
Комбинированные композиции по настоящему изобретению могут включать смеси комбинированных лекарственных средств, а также две или несколько отдельных композиций лекарственных средств, что отдельные композиции могут быть, например, совместно вводимыми вместе пациенту в одно и тоже различное время.
В другом аспекте настоящее изобретение относится к применению соединения формулы I или любой из фармацевтических композиций, описанных выше (Композиции P.1-P.7 и композиции 2, и 2.1-2.17), содержащей соединение формулы I в свободной форме или в форме фармацевтически приемлемой соли, как описано в способах А или 1.1-1.50, где соединение находится в смеси с антиоксидантом. В предпочтительном варианте осуществления инъецируемые составы микросфер длительного действия по настоящему изобретению содержат антиоксидант. В некоторых вариантах осуществления настоящего изобретения антиоксидант представляет собой водорастворимый антиоксидант (например, аскорбиновую кислоту, липоевую кислоту), в то время как в других вариантах осуществления настоящего изобретения антиоксидант представляет собой жирорастворимый антиоксидант (например, липоевая кислота, витамин Е, токоферолы, каротины и фенольные антиоксиданты). В некоторых вариантах осуществления настоящего изобретения антиоксидант представляет собой нейтральный или слабоосновной антиоксидант. Другие возможные антиоксиданты включают каталитические антиоксиданты (например, эбселен) и металлсодержащие антиоксиданты. В предпочтительном варианте осуществления антиоксидант представляет собой фенольный антиоксидант, такой как бутилированный гидрокситолуол (ВНТ) или бутилированный гидроксианизол (ВНА). В более предпочтительном варианте осуществления антиоксидант представляет собой ВНТ.
Дозы, используемые в практике настоящего изобретения, будет, конечно, варьировать в зависимости, например, от конкретного заболевания или патологического состояния, подлежащего лечению, от конкретного используемого соединения формулы I, способа введения и требуемой терапии. Если не указано иное, количество соединения формулы I для введения (которое вводят в форме свободного основания, пролекарства или в форме соли), относится к или основано на количестве соединения формулы I в форме свободного основания (то есть, вычисление количества основывается на количестве свободного основания). В конкретном варианте осуществления, однако, дозу соединения формулы I вводят в расчете на количество в форме соли, например, форме соли присоединения толуолсульфоновой кислоты. Соединения формулы I можно вводить любым подходящим способом, в том числе перорально, парентерально или чрескожно, но предпочтительно вводят перорально. В качестве монотерапии, соединение формулы I можно вводить приблизительно от 1 мг до 120 мг в день или от 10 мг до 120 мг в день, или от 10 мг до 60 мг в день, или от 10 мг до 40 мг в день, или от 1 мг до 10 мг в день, или 10 мг в день, 20 мг в день, 40 мг в день или 60 мг в день, предпочтительно, приблизительно 60 мг соединения в форме соли присоединения толуолсульфоновой кислоты в день. Когда вводят 120 мг, предпочтительно, вводить в ночное время.
В качестве инъецируемого состава микросферы длительного действия, дозировка соединения формулы I, зависит от среднего содержания лекарственного средства в микросферах (выраженного в % масс./масс.) и от дозы вводимого микросферами (мг/кг). В качестве монотерапии, соединение формулы I можно вводить в виде микросфер для обеспечения дозировки 1-50 мг/кг соединения формулы I, например, 5-25 мг/кг, предпочтительно, 5-10 мг/кг, например, приблизительно 5 мг/кг. Доза приблизительно 5 мг/кг соединения формулы I может быть обеспечена, например, использованием дозы 60 мг/кг микросфер, где каждая микросфера содержит в среднем содержание приблизительно 8,5% масс./масс. соединения формулы I.
Дозы соединения формулы I и/или другого антипсихотического и/или антидепрессивного средства способа А могут быть такими же, как или ниже чем разрешенные дозы для лекарственного средства, клинические или литературные тест-дозы или дозы, используемые для лекарственного средства в качестве монотерапии. Например, суточная доза соединения формулы I для введения в комбинации с другим антипсихотическим средством и/или антидепрессивным средством составляет от приблизительно 1 мг до приблизительно 140 мг, в другом варианте осуществления от приблизительно 1 мг до приблизительно 120 мг, в другом варианте осуществления от приблизительно 10 мг до приблизительно 120 мг, в другом варианте осуществления от приблизительно 10 мг до приблизительно 60 мг, в другом варианте осуществления от приблизительно 10 мг до приблизительно 40 мг, в другом варианте осуществления от приблизительно 20 мг до приблизительно 40 мг, в другом варианте осуществления от приблизительно 1 мг до приблизительно 10 мг, и в еще одном варианте осуществления, приблизительно 60 мг соединения в форме свободного основания или в форме соли присоединения толуолсульфоновой кислоты. Количество антипсихотического средства для введения в комбинации с соединением формулы I составляет от приблизительно 0,01 мг до приблизительно 1000 мг, в другом варианте осуществления от приблизительно 0,1 мг до приблизительно 600 мг, например, от приблизительно 1 мг до приблизительно 200 мг, например, от приблизительно 1 мг до приблизительно 50 мг, например, от приблизительно 1 мг до приблизительно 15 мг, например, приблизительно 4 мг. Количество антидепрессивного средства для введения в комбинации с соединением формулы I составляет от приблизительно 0,01 мг до приблизительно 2000 мг, в другом варианте осуществления от приблизительно 0,1 мг до приблизительно 200 мг, в другом варианте осуществления от приблизительно 10 мг до приблизительно 200 мг. В конкретном варианте осуществления второе терапевтическое средство представляет собой антипсихотическое средство, рисперидон, в суточной дозе от приблизительно 2 мг до приблизительно 4 мг и антидепрессант представляет собой сертралин и суточная доза сертралина составляет от приблизительно 20 мг до приблизительно 100 мг.
В конкретном варианте осуществления дозы соединения формулы I и/или второго (или третьего) терапевтического средства способа А ниже, чем при использовании в виде монотерапии. Таким образом, в конкретном варианте осуществления, суточная доза соединения формулы I ниже, чем 100 мг один раз в день, или менее 60 мг, или менее 40 мг, или менее 30 мг, или менее 20 мг, или менее 10 мг. В другом предпочтительном варианте осуществления дозы как соединения формулы I, так и антипсихотического средства и/или антидепрессивного средства способа А, ниже, чем дозы, используемые для отдельных лекарственных средств в качестве монотерапии. Таким образом, в конкретном варианте осуществления, например, способ А включает введение (1) соединения формулы I в дозе ниже 100 мг один раз в день, например, менее 60 мг или менее 40 мг; и/или (2) антидепрессанта, например, SSRI, такого как сертралин, при суточной дозе менее 50 мг, более предпочтительно, менее 20 мг, еще более предпочтительно, менее 10 мг, наиболее предпочтительно, менее 6 мг; и/или (3) антипсихотического средства, например, рисперидона, при суточной дозе менее 4 мг, в свободной форме или в форме фармацевтически приемлемой соли.
Для лечения расстройств, описанных в настоящем документе, где используется состав с замедленным или отсроченным высвобождением для достижения более длительного срока действия, дозы будут выше по сравнению с композицией короткого действия, например, выше чем 1-100 мг, например, например, 25 мг, 50 мг, 100 мг, 500 мг, 1000 мг, или более 1000 мг. В конкретном варианте осуществления схема приема для состава с замедленным или отсроченным высвобождением включает пероральную начальную дозу с немедленным высвобождением вместе с депо высвобождением для того, чтобы обеспечить устойчивый уровень лекарственного средства в крови. Длительно действия соединений формулы I можно контролировать с помощью манипуляцией композиции полимера, т.е. соотношением полимер:лекарственное средство и размером микрочастицы. Где состав по настоящему изобретению представляет собой депо-лекарственную форму, введение путем инъекции, является предпочтительным. В предпочтительном варианте осуществления состав представляет собой инъецируемый состав микросфер длительного действия, как описано выше.
Соединения, которые будут вводиться в способах по настоящему изобретению, могут быть в форме свободной кислоты или свободного основания или в виде фармацевтически приемлемых солей. Выражение "фармацевтически приемлемые соли" относится к производным раскрытых соединений, где исходное соединение модифицируют созданием его кислотных или основных солей. Примеры фармацевтически приемлемых солей включают, но ими не ограничиваясь, соли минеральной или органической кислоты основных остатков, таких как амины; щелочные или органические соли кислотных остатков, таких как карбоновые кислоты; и подобные. Фармацевтически приемлемые соли включают стандартные нетоксичные соли или четвертичные аммониевые соли исходного соединения, образованного, например, из нетоксичных неорганических или органических кислот. Например, такие стандартные нетоксичные соли включают производные неорганических кислот, таких как, но ими не ограничиваясь, хлористоводородная, бромистоводородная, серная, сульфаминовая, фосфорная, азотная и тому подобные; и соли, полученные из органических кислот, таких как, но ими не ограничиваясь, уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, памовая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изетионовая и тому подобные. Предпочтительно, соединения формулы I в форме соли присоединения толуолсульфоновой кислоты.
Фармацевтически приемлемые соли соединений для использования в способах по настоящему изобретению, можно синтезировать из исходного соединения, которое содержит основную или кислотную часть, обычными химическими способами. Обычно, такие соли можно получить реакцией свободных основных форм этих соединений со стехиометрическим количеством соответствующей кислоты в воде или в органическом растворителе, или в смеси двух; как правило, неводная среда, такая как диэтиловый эфир, этилацетат, этанол, изопропанол и ацетонитрил являются предпочтительными. Дополнительные детали получения этих солей, например, солей толуолсульфокислоты в аморфной или кристаллической форме, могут быть найдены в PCT/US08/03340 (WO 2008/112280) и/или WO 2009/114181 и WO 2011/133224.
Фармацевтические композиции, содержащие соединения формулы I, могут быть получены с использованием традиционных разбавителей или эксципиентов и способов, известных в галеновой области. Например, соединения могут быть введены в широком разнообразии различных лекарственных форм, то есть, они могут быть объединены с различными фармацевтически приемлемыми инертными носителями в форме таблеток, капсул, лепешек, пастилок, твердых леденцов, порошков, спреев, водной суспензии, растворов для инъекций, эликсиров, сиропов и тому подобное.
Для перорального введения фармацевтические композиции могут принимать форму, например, таблеток или капсул, полученных обычными способами с фармацевтически приемлемыми эксципиентами, такими как связующие средства (например, предварительно желатинизированный кукурузный крахмал, поливинилпирролидон или гидроксипропилметилцеллюлоза); наполнители (например, лактоза, микрокристаллическая целлюлоза или фосфат кальция); лубриканты (например, стеарат магния, тальк или диоксид кремния); дезинтегранты (например, картофельный крахмал или натрий крахмалгликолят); или смачивающие вещества (например, лаурилсульфат натрия). Таблетки могут быть покрыты способами, хорошо известными в данной области. Жидкие препараты для перорального введения могут принимать форму, например, растворов, сиропов или суспензий, или они могут быть представлены в виде сухого продукта для восстановления водой или другим подходящим носителем перед использованием. Такие жидкие препараты могут быть получены обычными способами с фармацевтически приемлемыми добавками, такими как суспендирующие средства (например, сорбитовый сироп, метилцеллюлоза или гидрогенизированные пищевые жиры); эмульгирующее вещество (например, лецитин или аравийская камедь); неводные носители (например, миндальное масло, жирные сложные эфиры или этиловый спирт); и консерванты (например, метил или пропил п-гидроксибензоаты или сорбиновая кислота).
Для состава с немедленным высвобождением, соединение формулы I, в свободной форме или в форме фармацевтически приемлемой соли, как описано выше, может быть сформулировано с фармацевтически приемлемым разбавителем или носителем. Для состава с пролонгированым или замедленным высвобождением, соединение соединения формулы I, в свободной форме или форме фармацевтически приемлемой соли, как описано выше в настоящем описании, может быть сформулировано, как описано выше в настоящем описании в любой из композиций P.1-P.7.
В конкретном варианте осуществления изобретения соединение формулы I формулируется в капсуле следующим образом:
Для трансбуккального введения композиция может иметь форму таблеток или пастилок, полученных обычным способом.
Соединения формулы I могут быть получены для парентерального введения путем инъекции, в том числе с использованием обычных методик катетеризации или инфузии. Составы для инъекции могут быть представлены в стандартной лекарственной форме, например, в ампулах, или в многодозовых контейнерах, с добавлением консерванта. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных носителях, а также могут содержать препаратообразующие средства, такие как суспендирующие, стабилизирующие и/или диспергирующие средства. В качестве альтернативы, активный ингредиент может быть в форме порошка для восстановления подходящим носителем, например, стерильной апирогенной водой, перед использованием.
Соединения формулы I также могут быть приготовлены в виде ректальных композиций, таких как суппозитории или удерживающие клизмы, например, содержащие обычные основы для суппозиториев, такие как масло какао или другие глицериды.
Для интраназального введения или введения путем ингаляции, соединения формулы I обычно доставляются в виде раствора или суспензии из контейнера пульверизатора, который сжимается или накачивается пациентом, или в качестве аэрозольного спрея из контейнера под давлением или небулайзера, с использованием подходящего пропеллента, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа. В случае аэрозоля под давлением, разовая доза может определяться за счет клапана для доставки отмеренного количества. Контейнер, находящийся под давлением, или распылитель может содержать раствор или суспензию активного соединения. Капсулы и картриджи (изготовленные, например, из желатина) для использования в ингаляторе или инсуффляторе могут изготовляться содержащими порошковую смесь активного соединения и подходящего порошкового основания, такого как лактоза или крахмал. Когда водные суспензии и/или эликсиры желательны для перорального введения, основной активный ингредиент(ы) в них, может быть объединен с различными подслащивающими или ароматизирующими средствами, окрашивающими веществами или красителями и, при желании, эмульгирующими и/или суспендирующими средствами, а также, вместе с такими разбавителями, как вода, этанол, пропиленгликоль, глицерин, и различными их комбинациями.
Все ссылки, приведенные выше, включены в настоящее описание в качестве ссылки в полном объеме.
Нижеследующие примеры приведены для иллюстрации настоящего изобретения, но не должно быть истолкованы как ограничивающие настоящее изобретение.
Пример 1: Лечение острых, а также остаточных симптомов шизофрении
Для целей этого примера, соединение А относится к соединению формулы I, где X представляет собой -N(CH3)- и Y представляет собой -C(O)-; соль тозилата, если не указано иное. Выполняли рандомизированное двойное слепое плацебо-контролируемое клиническое исследование. Четыре группы лечения, продолжительность лечения 4 недели, QAM дозирование выполняли следующим образом: 60 мг соединения А; 120 мг соединения А; положительный контроль (4 мг рисперидона); и плацебо. Первичный результат измеряли изменением относительно исходных значений общего балла по шкале позитивных и негативных синдромов (PANSS) в 28-й день. Вторичные показатели исследовали ключевые дифференцирующие признаки.
Группа пациентов, получавшая 60 мг соединения А, получала 60 мг соединения А один раз в день в течение 28 дней. Группа пациентов, получавшая 120 мг соединения А, получала 60 мг соединения А в 1 день исследования, затем 120 мг соединения А один раз в день в течение 27 дней. Группа пациентов, получавшая рисперидон, получала 2 мг рисперидон в 1 день исследования, а затем 4 мг рисперидона один раз в день в течение 27 дней. Группа пациентов, получавшая плацебо, получала плацебо один раз в день в течение 28 дней. Все дозы вводили утром с завтраком.
Основные критерии включения: Клинический диагноз шизофрении согласно DSM-IV-TR (Руководство по диагностике и статистике психических расстройств) подтверждали модифицированной SCID-CT. Балл по краткой психиатрической оценочной шкале (BPRS) 40 или выше (18-ти пунктам набрали 1-7 каждый). Минимальный балл 4 или выше по меньшей мере двух из следующих позитивных пунктов: подозрительность, концептуальная дезорганизация, галлюцинаторное поведение, необычное содержание мыслей. Текущий обострившийся эпизод длится не дольше, чем 4 недели. Соответствующая история болезни и/или независимый репортер должен проверить, что текущее состояние является обострившимся состоянием для человека. Предшествующий ответ на антипсихотическую терапию в течение последних 5 лет, определенный как клинически значимый и зарегистрированное уменьшение в бреде и/или галлюцинациях во время зарегистрированного обострившегося эпизода (и по меньшей мере 3 месяца до воздействия антипсихотической терапии).
Исследование имело высокий процент завершения у индивидов (74%) по сравнению со средним показателем процента завершения 62% в других 4-недельных антипсихотических исследованиях. 19% завершили в течение периода исследования лечения (в течение дня 1-28). 7% завершили лечение в рамках исследования через 28 дней, но пропали для отслеживания. При 60 мг, исследование показало, что соединение А демонстрирует антипсихотическую эффективность на первичной конечной точке изменения общего балла по шкале PANSS от исходных значений в день 28:
Для пациентов с выраженными негативными симптомами на исходном уровне (например, у пациентов с баллом 4 или выше по меньшей мере 3 пункта негативного симптома на исходном уровне), 60 мг соединения A улучшили негативные симптомы качественно (см. Фиг. 1).
В подгруппе пациентов с симптомами депрессии, которые являются вторичными по отношению к шизофрении, измеряли минимально допустимый уровень показателей для депрессии (Шкала депрессии при шизофрении Калгари, CDSS, балл > 6) на исходном уровне (на долю которого приходится около 16% пациентов в исследовании), 60 мг соединения А значительно уменьшило депрессию, как измерено с помощью CDSS (p=0,044). Соединение А при дозе 60 мг также улучшило общий балл по шкале PANSS (см. Фиг. 2) и негативные симптомы (см. фиг. 3).
При дозе 60 мг, соединение А также показало значительное улучшение в изменении просоциального фактора PANSS от исходного уровня по сравнению с плацебо (см. Фиг. 4).
ПРИМЕРЫ 2-10: Получение инъецируемых составов микросфер длительного действия
Пример 2: Получение микросфер PLGA (PLA/PLG=75/25, 0,32-0,44 дл/г) соединения A в форме свободного основания (партия A)
Для целей примеров 2-10, Соединение А относится к соединению формулы I, где X представляет собой -N(CH3)- и Y представляет собой -C(O)- в форме свободного основания.
2 г Поли(винилового спирта) (ПВС, гидролизованный на 87-89%, типичная молекулярная масса 13000-23000) в 400 мл деионизированной воды обрабатывали ультразвуком в ультразвуковой ванне в течение 5-10 минут, а затем фильтровали с получением 0,5% водного раствора ПВС. Фильтрат переносили в 500-мл колбу с плоским дном. 0,80 г соединения A и 1,2 г полимера PLGA (PLA/PLG=75/25, 0,32-0,44 дл/г, кислотные концевые группы) растворяли в 15 мл дихлорметана. Этот раствор добавляли в 0,5% водный раствор ПВС по каплям при интенсивном перемешивании. Использовали механическую верхнеприводную мешалку и скорость перемешивания составляла приблизительно 700 ppm в течение добавления. После завершения добавления смесь перемешивали при этой скорости в течение часа, а затем перемешивали при приблизительно 550 ppm в течение 4 часов. Поток аргона применяли в течение всего процесса для способствования испарению дихлорметана из водного раствора. Микросита с размером 75 мкм укладывали поверх микросита 20 мкм. Суспензию медленно вливали на расположенные друг над другом микросита, и затем промывали водой по меньшей мере пять раз. Микросферы, собранные на микросите 20 мкм, переносили в 50-мл пробирку фирмы Falcon с деионизированной водой и затем лиофилизировали с получением 1,43 г микросфер PLGA соединения А с распределением по размеру 20-60 мкм. Содержание лекарственного средства в полученных микросферах составляло 37,5%, как определяли с помощью ВЭЖХ. Начальное содержание для этого препарата составляло 40% (рассчитывали на основании 0,8 г соединения А в форме свободного основания в 1,2 г полимера PLGA). Эффективность включения лекарственного средства составила 93,8%.
Пример 3: Получение микросфер PLGA (PLA/PLG=75/25, 0,32-0,44 дл/г) соединения A в форме свободного основания (партия B)
Эту партию PLGA микросфер соединения А получали с использованием методики, аналогичной той, которая описана в примере 2, за исключением того, что использовали стерилизованную воду в течение всего процесса вместо деионизированной воды и все лабораторное оборудование в контакте с микросферами стерилизовали. Получали 1,49 г микросфер PLGA соединения А с распределением по размеру 20-50 мкм. Содержание лекарственного средства в полученных микросферах составляло 38%, как определяли с помощью ВЭЖХ. Начальное содержание лекарственного средства составляло 40% и эффективность включения лекарственного средства составила 85,8%.
Пример 4: Получение микросфер PLGA (PLA/PLG=75/25, 0,89 дл/г) соединения A в форме свободного основания (партия C)
2 г Поли(винилового спирта) (ПВС, гидролизованный на 87-89%, типичная молекулярная масса 13000-23000) в 400 мл деионизированной воды обрабатывали ультразвуком в ультразвуковой ванне в течение 5-10 минут, а затем фильтровали с получением 0,5% водного раствора ПВС. Фильтрат переносили в 500-мл колбу с плоским дном. 0,80 г Соединения A и 1,2 г полимера PLGA (PLA/PLG=75/25; 0,89 дл/г; молекулярная масса: 159000 Д; с концевыми эфирными группами) растворяли в 16 мл дихлорметана с обработкой ультразвуком. Этот раствор добавляли в 0,5% водный раствор ПВС по каплям при интенсивном перемешивании. Использовали механическую верхнеприводную мешалку и скорость перемешивания составляла приблизительно 700 ppm в течение добавления. После завершения добавления смесь перемешивали при этой скорости в течение часа, а затем перемешивали при приблизительно 550 ppm в течение 4 часов. Поток аргона применяли в течение всего процесса для способствования испарению дихлорметана из водного раствора. Микросита с размером 75 мкм укладывали поверх микросита 20 мкм. Суспензию медленно вливали на расположенные друг над другом микросита, и затем промывали водой по меньшей мере пять раз. Микросферы, собранные на микросите 20 мкм, переносили в 50-мл пробирку фирмы Falcon с деионизированной водой и затем лиофилизировали с получением 0,78 г микросфер PLGA соединения А с распределением по размеру 20-70 мкм. Содержание лекарственного средства в полученных микросферах составляло 36%, как определяли с помощью ВЭЖХ. Начальное содержание для этого препарата составляло 40% (рассчитывали на основании 0,8 г соединения А в форме свободного основания в 1,2 г полимера PLGA). Эффективность включения лекарственного средства составила 90%.
Пример 5: Получение микросфер PLGA (PLA/PLG=75/25, 0,68 дл/г) соединения A в форме свободного основания (партия D)
Эту партию PLGA микросфер соединения А получали с использованием методики, аналогичной той, которая описана в примере 4, за исключением того, что использовали новый тип полимера PLGA (PLA/PLG=75/25; 0,68 дл/г; молекулярная масса: 113000 Д; кислотные концевые группы) в получении микросферы. Получали 1,11 г микросфер PLGA соединения А с распределение размера 25-75 мкм. Содержание лекарственного средства в полученных микросферах составляло 36%, как определяли с помощью ВЭЖХ. Начальное содержание лекарственного средства составляло 40% и эффективность включения лекарственного средства составила 90%.
Пример 6: Получение микросфер PLGA (PLA/PLG=75/25, 0,68 дл/г) соединения A в форме свободного основания (партия E)
Эту партию PLGA микросфер получали с использованием методики, аналогичной той, которая описана в примере 4, за исключением того, что использовали другой тип полимера PLGA (PLA/PLG=75/25; 0,68 дл/г; молекулярная масса: 113000 Д; кислотные концевые группы) и использовали микросита с размером пор 75 мкм в получении микросферы. Получали 0,25 г микросфер PLGA соединения А с распределение размера 75-110 мкм. Содержание лекарственного средства в полученных микросферах составляло 37%, как определяли с помощью ВЭЖХ. Начальное содержание лекарственного средства составляло 40% и эффективность включения лекарственного средства составила 93%.
Пример 7: Получение микросфер PLGA (PLA/PLG=75/25, 0,68 дл/г) соединения A в форме свободного основания (партия F)
Эту партию PLGA микросфер соединения А получали с использованием методики, аналогичной той, которая описана в примере 4, за исключением того, что использовали новый тип полимера PLGA (PLA/PLG=75/25; 0,68 дл/г; молекулярная масса: 113000 Д; кислотные концевые группы) и микросита с размерами пор 53 мкм и 106 мкм, соответственно, использовали в получении микросферы. Получали 1,22 г микросфер PLGA с распределение размера 52-101 мкм. Содержание лекарственного средства в полученных микросферах составляло 37%, как определяли с помощью ВЭЖХ. Начальное содержание лекарственного средства составляло 40% и эффективность включения лекарственного средства составила 93%.
Пример 7-A: Получение микросфер PLGA (PLA/PLG=75/25, 0,32-0,44 дл/г) соединения A в форме свободного основания (партия F)
2 г Поли(винилового спирта) (ПВС, гидролизованный на 87-89%, типичная молекулярная масса 13000-23000) в 400 мл деионизированной воды обрабатывали ультразвуком в ультразвуковой ванне в течение 5-10 минут, а затем фильтровали с получением 0,5% водного раствора ПВС. Фильтрат переносили в 500-мл колбу с плоским дном. 0,40 г Соединения A и 2,1 г полимера PLGA (PLA/PLG=75/25; 0,32-0,44 дл/г; кислотные концевые группы) растворяли в 21 мл дихлорметана с обработкой ультразвуком. Этот раствор добавляли в 0,5% водный раствор ПВС по каплям при интенсивном перемешивании. Использовали механическую верхнеприводную мешалку и скорость перемешивания составляла приблизительно 500 ppm в течение добавления. После завершения добавления смесь перемешивали при приблизительно 550 ppm в течение 5 часов. Поток аргона применяли в течение всего процесса для способствования испарению дихлорметана из водного раствора. Микросита с размером 75 мкм укладывали поверх микросита 30 мкм. Суспензию медленно вливали на расположенные друг над другом микросита, и затем промывали водой по меньшей мере пять раз. Микросферы, собранные на микросите с размером 30 мкм, переносили в стеклянную пробирку 7,5 мл и затем сушили в вакууме с получением 1,43 г PLGA микросферы соединения А в форме свободного основания с распределением по размерам 25-70 мкм. Содержание лекарственного средства в полученных микросферах составляло 8,5%, как определяли с помощью ВЭЖХ. Начальное содержание для этого препарата составляло 16% (рассчитывали на основании 0,4 г соединения А в форме свободного основания в 2,1 г полимера PLGA). Эффективность включения лекарственного средства составила 53%.
Пример 8: Получение микросфер PLGA (PLA/PLG=75/25, 0,68 дл/г) соединения В в форме свободного основания (партия G)
Для целей этого примера, соединение В относится к соединению формулы I, где X представляет собой -N(CH3)- и Y представляет собой -CH(OH)-. 2 г Поли(винилового спирта) (ПВС, гидролизованный на 87-89%, типичная молекулярная масса 13000-23000) в 400 мл деионизированной воды обрабатывали ультразвуком в ультразвуковой ванне в течение 5-10 минут, а затем фильтровали с получением 0,5% водного раствора ПВС. Фильтрат переносили в 500 мл колбу с плоским дном. 0,80 г Соединения B (свободное основание) и 1,2 г полимера PLGA (PLA/PLG=75/25, 0,32-0,44 дл/г, кислотные концевые группы) растворяли в 15 мл дихлорметана. Этот раствор добавляли в 0,5% водный раствор ПВС по каплям при интенсивном перемешивании. Использовали механическую верхнеприводную мешалку и скорость перемешивания составляла приблизительно 700 ppm в течение добавления. После завершения добавления смесь перемешивали при этой скорости в течение часа, а затем перемешивали при приблизительно 550 ppm в течение 4 часов. Поток аргона применяли в течение всего процесса для способствования испарению дихлорметана из водного раствора. Микросита с размером 75 мкм укладывали поверх микросита 20 мкм. Суспензию медленно вливали на расположенные друг над другом микросита, и затем промывали водой по меньшей мере пять раз. Микросферы, собранные на микросите 20 мкм, переносили в 50-мл пробирку фирмы Falcon с деионизированной водой и затем лиофилизировали с получением 1,36 г PLGA микросфер соединения В с распределением по размеру 13-60 мкм. Содержание лекарственного средства в полученных микросферах составляло 29%, как определяли с помощью ВЭЖХ. Начальное содержание для этого препарата составляло 40% (рассчитывали на основании 0,8 г соединения В в форме свободного основания в 1,2 г полимера PLGA). Эффективность включения лекарственного средства составила 72,5%.
Пример 9: Определение содержания инъекционных микросфер длительного действия
Приблизительно 5 мг микросфер растворяли в 10 мл смеси дихлорметана/ацетонитрила в соотношении 1:2 об/об. Переносили 1 мл раствора в 1,5-мл микропробирку и растворитель удаляли, используя оборудование ʺSavant Speed Vacʺ; затем остаток восстанавливали в 95% ацетонитрила/5% воды. Этот раствор фильтровали (фильтрующая насадка на шприц 0,2 мкм Waters Nylon syringe filter) и измерения проводили в трех повторах с помощью системы Waters ACQUITY UPLC с детектором оптической плотности УФ с PDA, установленным на 314 нм. Подвижная фаза представляла собой градиент ацетонитрил-вода с об/об 0,1% муравьиной кислоты. Использовали колонку Waters Acquity UPLC HSS T3 (2,1×50 мм) со скоростью потока 0,3 мл/мин. Стандартную кривую получали, включая приблизительно 0,1-0,7 мг/мл ожидаемой концентрации соединения А.
Содержание лекарственного средства определяли как: процент содержания лекарственного средства=100×(10×концентрация отфильтрованного раствора)/масса используемых микросфер. Результаты представлены в виде среднего значения ± стандартная ошибка.
Пример 10: Определение высвобождения лекарственного средства инъекционных микросфер длительного действия
Эксперименты по высвобождению лекарственного средства микросферы проводили в 0,1 М фосфатном буфере (рН 7,4), содержащем 10 мМ аскорбиновой кислоты. Суспензии микросферы, содержащие известное количество лекарственного средства (приблизительно 30 мг), помещали в регенерированную целлюлозную мембрану диализного устройства (Float-a-Lyzer). Float-a-Lyzer помещали в 45-мл пробирку фирмы Falcon, содержащую 40 мл буфера, среду высвобождения поддерживали при 37°C и встряхивали в горизонтальной плоскости со скоростью 100 оборотов в минуту в печи. Буфер заменяли свежим раствором через заданные интервалы времени. Содержание лекарственного средства среды высвобождения определяли с помощью ВЭЖХ с использованием УФ-VIS оптической плотности при 314 нм с помощью стандартной кривой, построенной с известной концентрацией ITI07 (3~30 мкг/мл).
Пример 11: Фармакокинетика инъекционных микросфер длительного действия
Для определения характеристик in vivo, определяли высвобождения лекарственного средства in vivo (клиническое исследование). Фармакокинетику препарата изучали у взрослых самцов крыс Sprague-Dawley. Крысам (N=12/эксперимент) инъецировали подкожно во внутрилопаточную область с суспензией (2 мл/кг) соединения А в форме свободного основания, сформулированного в биоразлагаемые PLGA микросферы (около 60 мг/крысу), суспендированные в растворе 0,5% карбоксиметилцеллюлозы низкой вязкости в стерильном солевом растворе, содержащем 0,1% Tween-20. Через определенные промежутки времени после инъекции (т.е. от 24 часов до 28 дней) крыс (N=2 или 3/временная точка) проверяли на судорожное движение головой, индуцированное агонистом 5-HT2A, в качестве функционального показателя 5-HT2A-антагонистической активности, вызванное уровнями циркулирующего в крови соединения A. Для этих измерений крысам вводили внутрибрюшинно 2,5-диметокси-4-иодамфетамин (DOI) (2,5 мг/кг) в объеме 2 мл/кг. Через пять минут у крыс наблюдали стереотипное судорожное движения головой, которые вручную подсчитывали и записывали. Крыс умерщвляли декапитацией в определенные моменты времени (т.е., через 24 часа - 28 дней после инъекции препарата) и кровь из туловища и ткани головного мозга, собирали для анализа уровней соединения А и его известных метаболитов (соединение B), используя способ ВЭЖХ-МС/МС.
Используя методику, как описано или аналогично описанным выше, несколько составов инъекционных микросфер длительного действия анализировали на их профиль высвобождения in vivo соединения формулы I, где X представляет собой -N(CH3)- и Y представляет собой -C(O)- (Соединение A в форме свободного основания). Результаты приведены в таблицах ниже. В таблице 2 приведены физические характеристики составов LAI микросфер 2-7 и 7A. Распределение частиц по размеру определяли с использованием фотомикроскопии.
Фактическое содержание лекарственного средства описывается % масс./масс. соединения А в микросферах. Микросферы дозируют животным при любой дозе из 60 мг/кг или 30 мг/кг. Доза соединения А в таблице 2 представляет собой количество лекарственного средства, введенное в виде дозы животному (фактическое содержание лекарственного средства×доза микросферы). Уровни в плазме и головном мозге и соединения А, и основного метаболита соединения А (соединение B), измеряли в День 1, День 3, День 7, День 10, День 14 и День 21 после подкожной инъекции у крыс. Результаты показаны в таблице 3.
Было отмечено, неожиданно, что улучшенный профиль высвобождения (пролонгированное высвобождение) получают с использованием меньшей загрузки микросферы. Состав микросферы, имеющей загрузку 8,5% масс. (пример 7-А) дает профиль высвобождения с пиковым уровнем лекарственного средства в головном мозге на 7 день, с измеряемыми уровнями лекарственного средства через 21 день исследования (11 нг/мл уровень в головном мозге на 21 день). В противоположность этому, микросферы с более высокой загрузкой лекарственного средства приводят к более раннему пику высвобождения лекарственного средства без каких-либо измеримых уровней или низких уровней лекарственного средства, сохраняемых после 10-го дня исследования.
Реферат
Настоящая группа изобретений относится к медицине, а именно к психиатрии, и касается лечения остаточных симптомов психоза. Для этого вводят эффективное количество соединения формулы I, в том числе в виде инъекционной композиции длительного действия. Это обеспечивает эффективное лечение остаточных негативных симптомов психоза. 5 н. и 26 з.п. ф-лы, 11 пр., 3 табл., 4 ил.
Формула
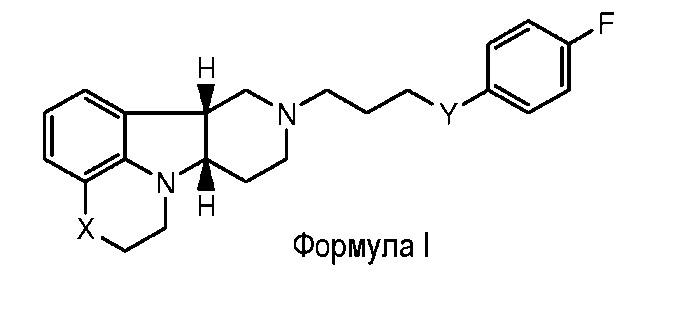
Документы, цитированные в отчёте о поиске
Селективные обратные агонисты серотонин 2а/2с рецептора, применяемые в качестве лекарственных средств при нейродегенеративных заболеваниях
Комментарии