Липидный преконцентрат с замедленным высвобождением фармакологически активного вещества и фармацевтическая композиция, содержащая его - RU2632433C2
Код документа: RU2632433C2
Чертежи


Описание
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к липидному преконцентрату с замедленным высвобождением фармакологически активного вещества и к фармацевтической композиции, содержащей его.
УРОВЕНЬ ТЕХНИКИ
Препаративные формы с замедленным высвобождением предназначены для высвобождения одноразовой дозы фармакологически активного вещества с заданной скоростью для поддержания эффективной концентрации вещества в плазме циркулирующей крови в течение определенного периода времени при минимизации побочных эффектов, вызванных множественными дозами.
PLGA [ПМГК, сополимер молочной и гликолевой кислоты] является представителем используемых в настоящее время биоразлагаемых материалов, которые разрешены для использования при замедленном высвобождении Администрацией пищевых продуктов и лекарственных средств США (FDA). В патенте США № 5480656 описано замедленное высвобождение фармакологически активного вещества путем разложения ПМГК на молочную кислоту и гликолевую кислоту в течение определенного периода времени in vivo. Однако продукты кислотного разложения ПМГК вызывают воспаление и уменьшение клеточного роста (K. Athanasiou, G.G. Niederauer and C.M. Agrawal, Biomaterials, 17, 93 (1996)).
Для замедленного высвобождения необходимо производить инъекцию твердых частиц диаметром от 10 до 100 мкм, включая содержащееся в них лекарственное средство. Инъекция твердых частиц ПМГК сопровождается болью или воспалением, потому что твердая частица диаметром от 10 до 100 мкм должна вводиться посредством подкожной или внутримышечной инъекции и разлагается в течение периода до нескольких месяцев в участке инъекции. Поэтому существует потребность в новой препаративной форме замедленного высвобождения, которая доставляет эффективную концентрацию в плазме фармакологически активного вещества в течение продолжительного периода времени при улучшенном соблюдении пациентом назначенной схемы введения.
В результате интенсивных и тщательных исследований заявителей настоящего изобретения по поиску препаративной формы замедленного высвобождения было обнаружено, что липидный преконцентрат, содержащий a) сложный эфир ненасыщенной жирной кислоты сорбитана, имеющий полярную головку по меньшей мере с двумя или более -OH (гидроксильными) группами; b) фосфолипид; и c) жидкокристаллический отвердитель, не содержащий ионизируемую группу, имеющую гидрофобную часть из 15-40 атомов углерода, с триацильной группой или углеродной кольцевой структурой, существует в жидком состоянии в отсутствие водной жидкости и переходит в форму гелеподобного жидкого кристалла после воздействия водной жидкости, проявляя превосходный профиль замедленного высвобождения, и что преконцентрат безопасен для организма и обладает высокой способности биоразложения.
Ниже описаны документы уровня техники, релевантные к настоящему изобретению.
В Международном патентной публикации WO 2005/117830 описан предварительный состав, содержащий имеющую низкую вязкость, не жидкокристаллическую смесь: по меньшей мере одной нейтральной диацильной жидкости и/или по меньшей мере одного токоферола, по меньшей мере одного фосфолипида и по меньшей мере одного биосовместимого, содержащего кислород, имеющего низкую вязкость органического растворителя. В Международной публикации WO 2006/075124 описаны предварительные составы имеющей низкую вязкость смеси, содержащей по меньшей мере один диацилглицерин, по меньшей мере один фосфатидилхолин, по меньшей мере один содержащий кислород органический растворитель и по меньшей мере один аналог соматостатина. Все эти прекомпозиции высвобождают фармакологически активные материалы in vivo в течение двух недель или дольше, но диацильный липид, компонент, существенный для прекомпозиций, в качестве фармацевтического компонента не используется, и необходимо доказать, что он достаточно безопасен. Другое отличие от настоящего изобретением состоит в том, что, как было обнаружено, органические растворители, использовавшиеся в публикациях уровня техники, снижали активность некоторых лекарственных средств (H. Ljusberg-Wahre, F.S. Nielse, 298, 328-332 (2005); H. Sah, Y. bahl, Journal of Controlled Release 106, 51-61(2005)).
В патенте США № 7731947 описана композиция, содержащая: интерферон, сахарозу, метионин и цитратный буфер, и суспендирующую основу, содержащую растворитель, такой как бензилбензоат, причем композиция в форме частиц диспергирована в суспендирующей основе. В одном примере описано, что фосфатидилхолин растворяют вместе с витамином E (токоферолом) в органическом растворителе и используют для диспергирования в нем композиции в форме частиц. Однако эта композиция отличается от прозрачного и фильтруемого состава в форме раствора по настоящему изобретению тем, что композиция используется для диспергирования твердых частиц и не допускает образования жидких кристаллов.
В патенте США № 7871642 описан способ получения дисперсии для доставки фармакологически активного средства, включающий диспергирование однородной смеси фосфолипида, полиоксиэтиленового соэмульгатора, триглицерида и этанола в воде, причем полиоксиэтиленовый соэмульгатор выбран между сложными эфирами жирных кислот сорбитана (полисорбатом) и полиэтоксилированными производными витамина E. Полиэтоксилированные сложные эфиры жирных кислот сорбитана и полиэтоксилированные производные витамина E, полученные конъюгированием гидрофильного полимера полиоксиэтилена со сложным эфиром жирной кислоты сорбитана и витамином E, соответственно, совершенно отличны по структуре от сложного эфира жирной кислоты сорбитана и витамина E. Они обычно применяются в качестве гидрофильных поверхностно-активных веществ, использующих свойство полиоксиэтилена, который отличается от компонента по настоящему изобретению.
В патенте США № 5888533 описана текучая композиция для образования твердого биоразлагаемого имплантата in situ внутри организма, содержащая: не полимерный, нерастворимый в воде, биоразлагаемый материал; и биосовместимый, органический растворитель, который по меньшей мере частично, солюбилизирует не полимерный, нерастворимый в воде материал и является смешиваемым или диспергируемым в биологических жидкостях и способен диффундировать или вытекать из композиции в биологическую жидкость после помещения внутрь организма, после чего не полимерный материал коагулируется или осаждается для образования твердого имплантата. В данной композиции стерины, сложные эфиры холестерила, жирные кислоты, глицериды жирных кислот, сложные эфиры жирных кислот и сахарозы, сложные эфиры жирных кислот сорбитана, жирные спирты, сложные эфиры жирных спиртов с жирными кислотами, ангидриды жирных кислот, фосфолипиды, ланолин, ланолиновые спирты и их смеси описаны в качестве не полимерного материала, и этанол используется в качестве растворителя. Однако отличия от настоящего изобретения состоят в том, что данная композиция не может образовывать жидкие кристаллы и предназначено для образования твердых имплантатов простой коагуляцией или осаждением нерастворимых в воде материалов, и что необходимо использование большого количества органического растворителя.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Техническая проблема
Целью настоящего изобретения является получение жидкого преконцентрата на основе ненасыщенного сложного эфира сорбитана, имеющего полярную головку по меньшей мере с двумя -OH (гидроксильными) группами, который характеризуется высокой безопасностью и биоразлагаемостью и существует в жидком состоянии, предпочтительном для инъекционного применения лекарственной формы, в то же время образуя жидкий кристалл после воздействия водной жидкости, таким образом, увеличивая замедленное высвобождение лекарственного средства in vivo.
Другой целью настоящего изобретения является получение липидного преконцентрата, который может инъецироваться, не вызывая боль или воспаление, проблем, связанных с обычными составами.
Еще одной целью настоящего изобретения является получение фармацевтической композиции, дополнительно содержащей фармакологически активный ингредиент плюс преконцентрат по настоящему изобретению.
Разрешение проблемы
В соответствии с одним его аспектом, настоящее изобретение относится к липидному преконцентрату для замедленного высвобождения, содержащей a) сложный эфир ненасыщенной жирной кислоты сорбитана, имеющий полярную головку по меньшей мере с двумя или более -OH (гидроксильными) группами; b) фосфолипид; и c) жидкокристаллический отвердитель, не содержащий ионизируемую группу, имеющую гидрофобную часть из 15-40 атомов углерода с триацильной группой или углеродной кольцевой структурой, причем указанный липидный преконцентрат существует в виде жидкой фазы в отсутствие водной жидкости и формируется в жидкий кристалл в присутствии водной жидкости.
Сложный эфир ненасыщенной жирной кислоты сорбитана, имеющий полярную головку по меньшей мере с двумя или более -OH (гидроксильными) группами, полезный для настоящего изобретения, представлен следующей химической формулой 1:
Химическая формула 1
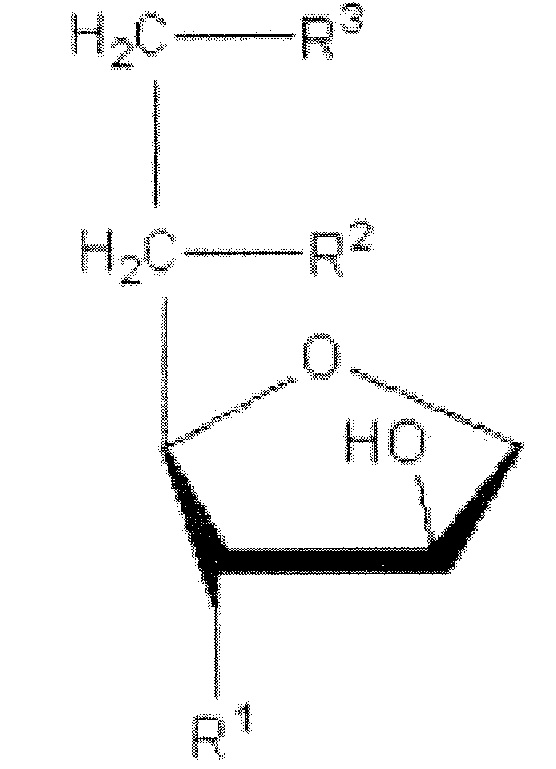
где R1 обозначает OH, R2 обозначает OH или R и R3 обозначает R, причем R обозначает сложный алкиловый эфир из 4-30 атомов углерода с одной или несколькими ненасыщенными связями.
Сложный эфир жирной кислоты сорбитана, который ответствен за образование жидкого кристалла в настоящем изобретении, отличается от обычных совместно используемых ингредиентов, таких как олеилглицерат (OG), фитанилглицерат (PG), и глицерин моноолеат (GMO), глицерин диолеат (GDO, вид диацилглицерола) следующей химической формулы 2. То есть, обычные молекулы, ответственные за жидкокристаллические фазы, разделяют общую структуру, состоящую из полярной головки и неполярного хвоста, полученного из липидного спирта или жирной кислоты.
Химическая формула 2

Однако обычные молекулы, ответственные за жидкокристаллические фазы, несколько затруднительно применять для разработки лекарственных препаратов ввиду следующих недостатков. Хотя олеилглицерат (OG) и фитанилглицерат (PG) способны легко формироваться в жидкие кристаллы, они редко используются в качестве фармацевтических эксципиентов в клинической медицине ввиду их относительно высокой токсичности. С другой стороны, глицерин моноолеат используется в качестве фармацевтически приемлемого эксципиента, но имеет слабую кристалличность для образования жидких кристаллов, необходимых для модификаций замедленного высвобождения.
Глицеролдиолеат, который используется в Международной патентной публикации WO 2005/117830, как описано выше, представляет собой диацильный липид с глицерином, функционирующим в качестве полярной головки. Эта молекула в целом не используется в качестве фармацевтического эксципиента, потому что ее безопасность еще не была доказана. Кроме того, она обладает очень слабой биоразлагаемостью.
В результате интенсивных и тщательных исследований, авторы обнаружили, что сложные эфиры ненасыщенных жирных кислот сорбитана имеют преимущества перед обычно используемыми жидкокристаллическими молекулами, глицерином или производными глицериновой кислоты в том, что они очень эффективно образуют жидкие кристаллы для замедленного высвобождения активных ингредиентов, при превосходстве в отношении безопасности и биоразлагаемости, и их можно применять для разработки медицинских продуктов, преодолевая проблемы, встречавшиеся в уровне техники. Для использования в композициях для лекарственных препаратов должно быть гарантировано, что материалы являются безопасными и биоразлагаемыми. Кроме того, является очень важным фактором для материала, который ответствен за замедленное высвобождение в организме. Если инъекционный препарат замедленного высвобождения с использованием ПМГК предназначен для высвобождения активного ингредиента в течение одной недели, то в идеале, ПМГК должна разлагаться in vivo через одну неделю после инъекции. Однако в действительности, ПМГК остается интактной в течение периода от одного до нескольких месяцев даже после окончании осуществления функции замедленного высвобождения. Поэтому, сложный эфир ненасыщенной жирной кислоты сорбитана по настоящему изобретению, который обладает превосходным свойством замедленного высвобождения, безопасности и биоразлагаемости, может применяться в фармацевтической промышленности для нового материала, вызывающего образование в большом количестве жидких кристаллов.
Жирная кислота сложного эфира ненасыщенной жирной кислоты сорбитана по настоящему изобретению может быть получена из растительного масла (например, пальмового масла, касторового масла, оливкового масла, арахисового масла, прованского масла, кукурузного масла, кунжутного масла, масла хлопковых семян, соевого масла, подсолнечного масла, масла сафлора, масла льняных семян), животного жира и масла (например, молочного жира, свиного сала, говяжьего сала и т.д.), китового жира и рыбьего жира. Сложный эфир ненасыщенной жирной кислоты сорбитана по настоящему изобретению может быть выбран из сложных моноэфиров сорбитана, сложных полуторных эфиров сорбитана, сложных диэфиров сорбитана и их смесей. Сложный моноэфир сорбитана представляет собой молекулу сорбитана с одной группой жирной кислоты, присоединенной к нему через сложноэфирную связь и может быть выбран из сорбитана моноолеата, сорбитана монолинолеата, сорбитана монопальмитолеата, сорбитана мономиристолеата и их смесей. Сложный полуторный эфир сорбитана представляет собой молекулу сорбитана, к которой присоединены 1,5 группы жирной кислоты в среднем через сложноэфирную связь. Репрезентативными среди сложных полуторных эфиров сорбитана, полезных в настоящем изобретении, являются сорбитана полуторный олеат, сорбитана полуторный линолеат, сорбитана полуторный пальмитолеат, сорбитана полуторный миристолеат и их смесь. Сложный диэфир сорбитана представляет собой молекулу сорбитана с двумя группами жирной кислоты, присоединенными к нему через сложноэфирную связь, и могут быть выбраны из сорбитана диолеата, сорбитана дилинолеата, сорбитана дипальмитолеата, сорбитана димиристолеата и их смеси.
Фосфолипиды существенны для конструкции слоистых структур, таких как липосомы, но сами по себе не могут образовывать структуру не слоистой фазы, такую как жидкий кристалл. Однако фосфолипиды могут осаждаться при запускаемом сложным эфиром ненасыщенной жирной кислоты сорбитана образовании структур не слоистой фазы, служащей для стабилизации полученных в результате жидких кристаллов. Фосфолипид, полезный в настоящем изобретении, содержит группу сложного алкилового эфира насыщенной или ненасыщенной жирной кислоты из 4-30 атомов углерода с полярной головкой. Фосфолипид может быть выбран из группы, состоящей из фосфатидилхолина, фосфатидилэтаноламина, фосфатидилсерина, фосфатидилглицерина, фосфатидилинозитола, фосфатидиновой кислоты, сфингомиелина и их смеси. Фосфолипиды обнаруживаются в растениях и животных, таких как соя и яйца. В фосфолипидах длинные жирнокислотные углеводородные цепи, которые ответственны за гидрофобные хвосты, включают цепи насыщенных жирных кислот, такие как моно- и дипальмитоил, моно- и димиристоил, моно- и дилаурил и моно- и дистеарил, цепи ненасыщенных жирных кислот, такие как моно- или дилинолеил, моно- и диолеил, моно- и дипальмитолеил, моно- или димиристолеил и их смесь.
Жидкокристаллический отвердитель сам по себе не может образовывать ни не слоистую структуру (жидкий кристалл), в отличие от сложных эфиров ненасыщенных жирных кислот сорбитана, ни слоистую структуру (липосому), в отличие от фосфолипидов. Однако жидкокристаллический отвердитель участвует в запускаемом сложными эфирами ненасыщенных жирных кислот сорбитана образовании структур не слоистой фазы увеличением кривизны не слоистых структур для увеличения упорядоченного сосуществования масла и воды в наномасштабе. В интересах данной функции, требуется, чтобы жидкокристаллический отвердитель имел очень ограниченную полярную часть и объемную неполярную часть внутри его молекулярной структуры.
На практике биосовместимые молекулы, которые могут инъецироваться в организм, могут быть выбраны в качестве жидкокристаллического отвердителя по настоящему изобретению только путем экспериментального «опыта и ошибки». В результате, жидкокристаллические отвердители, подходящие для композиции по настоящему изобретению, имеют молекулярные структуры, которые отличаются друг от друга, и, таким образом, не могут рассматриваться как одна молекулярная структура. Общий структурный признак, выведенный из всех выбранных жидкокристаллических отвердителей состоит в том, что они не содержат ионизируемые группы, такие как карбоксильная и аминная группы, и имеют гидрофобные части из 15-40 атомов углерода, содержащие объемную углеродную кольцевую структуру или триацильную группу. Предпочтительные примеры жидкокристаллического отвердителя по настоящему изобретению могут не содержать ионизируемые группы, такие как карбоксильная и аминная группы, и имеющие не более одной сложноэфирной и -OH (гидроксильной) группы в качестве полярной головки, и имеющие гидрофобные части из 20-40 атомов углерода, содержащие объемную углеродную кольцевую структуру или триацильную группу. Предпочтительные примеры жидкокристаллического отвердителя по настоящему изобретению могут включать без ограничения триглицерид, ретинилпальмитат, токоферилацетат, холестерин, бензилбензоат и смесь.
В композиции по настоящему изобретению массовое соотношение между компонентами a) и b) находится в диапазоне от 10:1 до 1:10 и предпочтительно в диапазоне от 5:1 до 1:5. Массовое отношение a) + b) к c) укладывается в пределы диапазона от 100:1 до 1:1 и предпочтительно в пределы диапазона от 50:1 до 2:1. Образуя желаемые жидкие кристаллы, компоненты в таких массовых отношениях гарантируют эффективное замедленное высвобождение.
Используемый в настоящем описании термин «водная жидкость» предназначен для включения воды и биологической жидкости, такой как раствор на слизистых оболочках, слезная жидкость, пот, слюна, жидкость желудочно-кишечного тракта, внесосудистая жидкость, внеклеточная жидкость, интерстициальная жидкость и плазма. При вступлении в контакт с поверхностями, областями или полостями организма (например, внутри тела), чьи внешние окружения составлены водными жидкостями, композиция по настоящему изобретению подвергается переходу из подобной золю водной фазы в подобную гелю жидкокристаллическую фазу. То есть композиция по настоящему изобретению представляет собой преконцентрат, который существует в жидком состоянии перед нанесением на человеческое тело, и сдвигается в жидкокристаллическую фазу, давая надежду на замедленное высвобождение внутри организма.
Жидкие кристаллы, образованные композицией по настоящему изобретению, имеют не ламеллярную фазовую структуру, в которой в которой масло и находятся в упорядоченной смеси и расположении без различения между внутренней и внешней фазами. Упорядоченное расположение масла и воды придает не ламеллярную фазовую структуру мезофазы, которая представляет собой состояние вещества, промежуточное между жидким и твердым. Преконцентрат по настоящему изобретению отличается от обычных композиций, которые образуют ламеллярные структуры, такие как мицеллы, эмульсии, микроэмульсии, липосомы и липидные бислои, которые широко использовались в составлении фармацевтических препаративных форм. Такие ламеллярные структуры относятся к типу масла в воде (м/в) или воды в масле (в/м), в которых имеется четкая дифференциация между внутренней и внешней фазами.
Используемый в настоящем описании термин «кристаллизация жидкости» относится к образованию жидких кристаллов, имеющих не ламеллярную фазовую структуру, из преконцентрата после воздействия водной жидкости.
Липидный преконцентрат по настоящему изобретению может быть получен при комнатной температуре из композиции, содержащей по меньшей мере один сложный эфир ненасыщенной жирной кислоты сорбитана, имеющий полярную головку по меньшей мере с двумя или более -OH (гидроксильных) групп, по меньшей мере один фосфолипид и по меньшей мере один жидкокристаллический отвердитель, если необходимо, нагреванием или использованием гомогенизатора.
Гомогенизатор может представлять собой гомогенизатор высокого давления, ультразвуковой гомогенизатор, бисерный мельничный гомогенизатор и т.д.
Как описано выше, ввиду того, что липидный преконцентрат по настоящему изобретению может представлять собой фармацевтическую композицию, которая существует в виде жидкой фазы в отсутствие водной жидкости, и формируется в жидкие кристаллы в присутствии водной жидкости в организме, ее можно вводить, используя способ, выбранный из инъекции, покрытия, капания, плюсования, перорального введения и распыления. Преконцентрат по настоящему изобретению может включаться в состав различных лекарственных форм, включая инъекционные препараты, мази, гели, лосьоны, капсулы, таблетки, жидкие формы, суспензии, аэрозоли, ингаляционные препараты, глазные капли, пластыри и трансдермальные системы.
В частности, когда предпринимается инъекционный путь, преконцентрат по настоящему изобретению может вводиться подкожной или внутримышечной инъекцией или другими инъекционными путями, в зависимости от свойств используемого фармакологически активного ингредиента.
Фармакологически активный ингредиент, применимый в преконцентрате по настоящему изобретению, может быть выбран из группы, состоящей из белка, пептида, вакцина, гена, не пептидного гормона, синтетического химического соединения и их комбинации.
Примеры белка или пептида в качестве фармакологически активного ингредиента в композиции по настоящему изобретению включают эритропоэтин, гормоны роста (человеческие, свиные, коровьи и т.д.), факторы, высвобождающие гормоны роста, факторы роста нервов, G-CSF (фактор, стимулирующий образование колоний гранулоцитов), GM-CSF (фактор, стимулирующий образование колоний гранулоцитов-макрофагов), M-CSF (фактор, стимулирующий образование колоний макрофагов), факторы свертывания крови, инсулин, окситоцин, вазопрессин, адренокортикотропный гормон, эпидермальный фактор роста, тромбоцитарный фактор роста, пролактин, соматостатин, глюкагон, интерлейкин-2 (IL-2), интерлейкин-11 (IL-11), гастрин, тетрагастрин, пентагастрин, урогастрон, секретин, кальцитонин, энкефалин, эндорфин, ангиотензин, тиреолиберин, фактор некроза опухолей, лиганд, вызывающий апоптоз, связанный с фактором некроза опухолей, гепариназа, костный морфогенный белок, hANP (человеческий предсердный натрийуретический пептид), подобный глюкагону пептид, реннин, брадикинин, бацитрацин, полимиксин, колистин, тироцидин, грамицидин, циклоспорин, полиэтиленгликоль-конъюгированные белки и их синтетические аналоги, моноклональные антитела, ферменты, цитокины и их комбинацию, но не ограничиваются ими.
Не пептидные гормоны представляют собой класс гормонов, которые не являются белками или пептидами и могут быть без ограничения выбраны из тестостерона, эстрадиола, прогестерона, простагландина, финатерида, дутастерида, их синтетических аналогов и их комбинаций.
Примеры гена, заключенного внутрь преконцентрата по настоящему изобретению, включают плазмидную ДНК, siРНК (малую интерфирирующую РНК), полинукелеотиды, олигодеоксинуклеотиды, антисмысловые олигонуклеотиды и их смеси, но не ограничиваются ими.
Синтетическое химическое соединение может быть выбрано из группы, состоящей из такролимуса, анатрозола, оланзапина, арипипразола, рисперидона, медроксипрогестерона, налтрексона, метотрексата, пинитола, олопатадина, латанопроста, анекортава, трипторелина памоата, миноксидила, тиболона, солифенацина, тадалафила, варениклина, ропинирола, фентанила, кетотифена, монтелукаста и их комбинации, но не ограничиваются ими.
Соответственно, в его другом аспекте, настоящее изобретение относится к фармацевтической композиции, содержащей d) фармакологически активный ингредиент, выбранный из белков, пептидов, вакцин, генов, не пептидных гормонов, синтетических химических соединений и их комбинации, в дополнение к липидному преконцентрату по настоящему изобретению.
Описание ингредиентов с a) по c) и жидкого кристалла, используемых в фармацевтической композиции, может относиться к тем, которые приведены в отношении липидного преконцентрата.
Кроме того, описание фармакологически активного ингредиента d) фармацевтической композиции может быть таким же, как описание, представленное в отношении липидного преконцентрата.
Фармацевтическая композиция может быть предпочтительно составлена в виде инъекционного препарата, мази, геля, лосьона, капсулы, таблетки, жидкого препарата, суспензии, аэрозоля, ингаляционного препарата, глазных капель, лейкопластыря и трансдермальной системы, но не ограничивается ими. Предпочтительнее, она может предпочтительно составлена в виде инъекционного препарата.
Содержание фармакологически активного ингредиента в фармацевтической композиции по настоящему изобретению варьируется в зависимости от его вида и подлежащей применению препаративной формы, и в целом находится в диапазоне от 0,0001 до 90 масс.%, на основании общей массы фармацевтической композиции.
Фармацевтическая композиция по настоящему изобретению может быть получена добавлением фармакологически активного ингредиента к преконцентрату по настоящему изобретению. При необходимости, нагревание или гомогенизатор могут использоваться при получении фармацевтической композиции по настоящему изобретению, но это не представляет собой ограничивающий фактор для настоящего изобретения.
Доза фармацевтической композиции по настоящему изобретению соответствует хорошо известной дозе используемого фармакологически активного ингредиента и может варьироваться в зависимости от различных факторов, включая состояние, возраст и пол пациента. Она может вводиться перорально или парентерально.
В соответствии с дополнительным его аспектом, настоящее изобретение предусматривает способ поддержания фармацевтической эффективности посредством замедленного высвобождения фармакологически активного ингредиента введением фармацевтической композиции по настоящему изобретению млекопитающему, включая человека, и применение фармацевтической композиции для замедленного высвобождения фармакологически активного ингредиента.
ПРЕВОСХОДЯЩИЕ УРОВЕНЬ ТЕХНИКИ ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
Как описано в настоящей заявке, липидный преконцентрат по настоящему изобретению, основанный на сложном эфире ненасыщенной жирной кислоты сорбитана, является высоко безопасным и биоразлагаемым и представлен в виде жидкой фазы в отсутствие водной жидкости, но быстро изменяется в жидкие кристаллы после воздействия водной жидкости внутри тела. Поэтому, при включении в состав с фармакологически активным ингредиентом, преконцентрат в жидкой фазе улучшает соблюдение пациентом предписанной схемы введения и проявляет превосходное замедленное высвобождение без побочных эффектов, таких как боль и воспаление, по сравнению с обычными препаративными формами замедленного высвобождения в фазах твердых частиц.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Указанные выше и другие цели, признаки и другие преимущества настоящего изобретения станут лучше понятны из следующего подробного описания, взятого в сочетании с сопровождающими чертежами, в которых:
На фиг. 1 показана биоразлагаемость in vivo композиций примеров 4 и 5 и сравнительных примеров 1-3.
На фиг. 2 показаны характеристики высвобождения in vitro лекарственного средства композиции примера 14;
На фиг. 3 показан фармакокинетический профиль, показывающий характеристики высвобождения in vitro лекарственного средства композиций примера 16 и сравнительного примера 5;
На фиг. 4 показаны фазовые изменения композиций примера 4 и сравнительного примера 4 после воздействия водной жидкости; и
На фиг. 5 показаны жидкокристаллические структуры композиции примера 4 на микрофотографиях, полученных аппаратом Cryo TEM.
ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Следующие не ограничивающие примеры служат для иллюстрации выбранных вариантов осуществления изобретения. Следует понимать, что для специалистов в данной области техники будут очевидны изменения пропорций и альтернативы элементов, которые охватываются объемом вариантов осуществления настоящего изобретения.
Добавки и эксципиенты, используемые в настоящем изобретении, удовлетворяли требованиям Корейской Фармакопеи и приобретались у компаний Aldrich, Lipoid и Croda.
ПРИМЕРЫ С 1 ПО 11: Получение липидных преконцентратов
Сложные эфиры ненасыщенных жирных кислот сорбитана, имеющие полярную головку по меньшей мере с двумя -OH группами, фосфолипиды и жидкокристаллический отвердитель смешивали в массовых соотношениях, показанных ниже в таблице 1, необязательно, в растворителе. В примерах 1-4 ингредиенты смешивали в водяной бане, поддерживаемой при 25~45°C, с использованием гомогенизатора (PowerGen модель 125. Fisher) в течение примерно 10 мин при 3000 об/мин. Ингредиенты примеров 5 и 6 перемешивали в течение 3 часов в водяной бане, поддерживаемой при 25~50°C. В примерах 7-11 ингредиенты смешивали в водяной бане, поддерживаемой при 45~75°C, с использованием гомогенизатора (PowerGen модель 125. Fisher) в течение примерно 20 мин при 3000 об/мин. Затем, полученные липидные растворы оставляли при комнатной температуре для получения термического равновесия при 25°C перед загрузкой в одноразовые шприцы емкостью 1 см3. Липидные преконцентраты, полученные описанным выше способом, инъецировали в воду (2 г дистиллированной воды) и формировали в жидкокристаллическую фазу.
ПРИМЕРЫ С 12 ПО 21: Получение фармацевтических композиций, содержащих фармакологически активные ингредиенты
Сложные эфиры ненасыщенных жирных кислот сорбитана, имеющие полярную головку по меньшей мере с двумя -OH группами, фосфолипиды и жидкокристаллический отвердитель смешивали в массовых соотношениях, показанных ниже в таблице 2.
В примерах с 12 по 15, ингредиенты смешивали в водяной бане, поддерживаемой при 30~60°C с использованием гомогенизатора (PowerGen модель 125. Fisher) в течение примерно 10 мин при 3000 об/мин. В примерах с 16 по 21, ингредиенты смешивали в водяной бане, поддерживаемой при 25~50°C с использованием гомогенизатора (PowerGen модель 125. Fisher) в течение примерно 5 мин при 3000 об/мин. Полученные липидные растворы оставляли при комнатной температуре для получения термического равновесия при 25°C с последующим добавлением к ним фармакологически активных ингредиентов. В качестве фармакологически активных ингредиентов, использовали генные лекарственные средства siРНК (Bioneer) и конъюгированную с флуоресцирующим маркером siРНК (Invitrogen, Block-iT Fluorescent oligo), пептидное лекарственное средство эксенатид (Teva) и синтетическое лекарственное средство тамсулосин. В последующем, ингредиенты гомогенизировали с использованием гомогенизатора при 3000 об/мин в течение 5 мин получения фармацевтической композиции в фазе раствора. В случае генных лекарственных средств (siРНК, конъюгированной с флуоресцирующим маркером siРНК), их смешивали в количествах, показанных в таблице 2, вместе с раствором хитозана в дистиллированной воде для образования комплексов перед внесением в липидные растворы.
СРАВНИТЕЛЬНЫЕ ПРИМЕР С 1 ПО 4
В сравнительных примерах 1-3, диолеилглицерид, класс диацилглицеридов, использовали в количествах, указанных в таблице 3, вместе с фосфатидилхолином, токоферолом и/или этанолом с последующей гомогенизацией в течение примерно 10 мин при 3000 об/мин в гомогенизаторе (PowerGen модель 125. Fisher).
В сравнительном примере 4, полиоксиэтилен сорбитан моноолеат, фосфатидилхолин и токоферилацетат использовали в количествах, показанных в таблице 3, с последующей гомогенизацией в течение примерно 30 мин при 3000 об/мин в гомогенизаторе. В данном случае, полиоксиэтилен сорбитан моноолеат имеет полиоксиэтиленовую группу, заместившую -OH группу на полярной головке сорбитана и отличается от сорбитана моноолеата, используемого в настоящем изобретении. Полиоксиэтилен сорбитан моноолеат в целом используется в качестве гидрофобного поверхностно-активного вещества вследствие объемной полиоксиэтиленовой части.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 5
К 1 мл физиологического солевого раствора добавляли 20 мкг эксенатида с последующей гомогенизацией при комнатной температуре.
ЭКСПЕРИМЕНТАЛЬНЫЙ ПРИМЕР 1: Сравнение безопасности in vitro
Безопасность композиций по настоящему изобретению исследовали in vitro выполнением теста цитотоксичности экстракционным анализом колоний следующим образом. В 18 мл эссенциальной минимальной среды Игла (EMEM) с добавлением 10% фетальной телячьей сыворотки, экстрагировали 2 г каждой из композиций примеров 1, 4 и сравнительных примеров 1 и 2. Клетки L929 (мышиные фибробласты, Американская Коллекция Типовых Культур) высевали при плотности 1×102 клеток/лунку в 6-луночные планшеты и инкубировали в течение 24 часов при 37°C в 5% CO2 в увлажненном инкубаторе. Экстракты разбавляли в EMEM (0, 5, 25, 50%) и затем помещали в количестве 2 мл/лунку в контакт со стабилизированными клетками L929. После инкубации в течение 7 дней при 37°C в 5% CO2 в увлажненном инкубаторе, клетки фиксировали 10% раствором формалина и окрашивали раствором Гимзы для подсчета колоний. Результаты суммированы ниже в таблице 4.
Как видно из таблицы 4, в группах, где вводили композиции примеров 1 и 4, проявились значимо высокие частоты клеточного роста на всех разбавленных средах (5%, 25% и 50%), по сравнению с группами, где вводили композиции сравнительных примеров 1 и 2, указывая на то, что композиции (липидные преконцентраты) по настоящему изобретению гораздо безопаснее, чем обычные композиции (описанные в заявке на Международный патент № WO 2005/117830).
ЭКСПЕРИМЕНТАЛЬНЫЙ ПРИМЕР 2: Сравнение биоразлагаемости in vivo
Композиции по настоящему изобретению оценивали в отношении биоразлагаемости in vivo в следующих экспериментах. Каждую из композиций примеров 4 и 5 подкожно инъецировали в дозе 400 мг в спину крыс SD и проводили мониторинг в течение заданного периода времени. Для сравнения, таким же образом тестировали композиции сравнительных примеров 1-3. Участки инъекции фотографировали через две недели после инъекции, и они показаны на фиг. 1.
Как видно на фиг. 1, наблюдали, что композиции примеров 4 и 5 в большинстве случаев подвергались биодеградации, почти не вызывая ощущения раздражения, тогда как композиции сравнительных примеров 1-3 сохраняли от одной до двух третей их первоначального объема.
Поэтому, композиции примеров 4 и 5 проявляли значимо высокую биоразлагаемость, по сравнению с композициями сравнительных примеров 1-3 (заявка на Международный патент № WO 2005/117830).
Для сравнения, известно, что обычный материал ПМГК [поли(молочная-ко-гликолевая кислота)], который широко использовался для замедленного высвобождения, оставался не разложившимся in vivo даже после двух или трех месяцев.
Соответственно, липидные преконцентраты по настоящему изобретению разрешают проблему того, что даже после полного высвобождения лекарственных средств, обычная система носителя остается внутри организма вследствие ее низкой биоразлагаемости.
ЭКСПЕРИМЕНТАЛЬНЫЙ ПРИМЕР 3: Тест замедленного высвобождения in vitro
Характеристики высвобождения лекарственных средств из композиций по настоящему изобретению исследовали in vitro в следующем тесте. Клетки рака предстательной железы (Рак предстательной железы-3, Корейский Банк Линий Клеток) высевали при плотности 5×104 клеток/лунку в планшеты с вкладышами из полиэфирной мембраны и инкубировали в течение 2 дней при 37°C в 5% CO2 в увлажненном инкубаторе. Композицию примера 14 добавляли в количестве 100 мг на вкладыш, содержащий 3 мл среды RPMI (Мемориального Института Росуэлл Парк) 1640 с добавлением 10% фетальной телячьей сыворотки. Флуоресценцию, испускаемую из композиции примера 14, измеряли, используя флуоресцентный микроскоп (Eclipse Ti-S, Nikon), тогда как вкладыш вставляли через каждые 24 часа в течение семи дней в планшеты с вкладышами из полиэфирной мембраны. Результаты показаны на фиг. 2.
Левые фотографии фиг. 2 получали, используя дифференциальную интерферентно-контрастную микроскопию (DIC), тогда как на правых фотографиях показан внутриклеточный захват связанной с флуоресцентным маркером siРНК. Как понятно из данных, представленных на фиг. 2, композиция по настоящему изобретению постоянно высвобождала фармакологически активный ингредиент в течение по меньшей мере 7 дней.
ЭКСПЕРИМЕНТАЛЬНЫЙ ПРИМЕР 4: Тест замедленного высвобождения in vivo
Характеристики высвобождения из композиций по настоящему изобретению исследовали in vivo в следующем тесте. Композицию примера 16 подкожно инъецировали 6 крысам (самцам) SD в возрасте 9 недель со средней массой тела 300 г в такой дозе, которая соответствует 140 мкг/кг эксенатида.
Концентрации эксенатида в образцах плазмы, взятых у крыс SD, контролировали в течение 14 дней, используя имеющийся в продаже набор (набор иммунного анализа, Bachem) для построения графика PK профиля (фармакокинетического профиля), показанного на фиг. 3. Для сравнения, композицию сравнительного примера 5 вводили в дозе, соответствующей 10 мкг/кг эксенатида (в данном случае, причиной того, почему доза эксенатида примера 16 была в 14 раз больше, чем доза в сравнительном примере 5, состоит в том, что недельная доза (7-дневная) препаративной формы замедленного высвобождения соответствует дозе, в 14 раз бόльшей в целом инъецируемой дозы ввиду применения два раза в день).
Как показано на фиг. 3, композиция примера 16 увеличивала период полувыведения in vivo биологически активного ингредиента примерно в 25 раз, по сравнению с композицией сравнительного примера 1, которая представляет собой общую инъекцию, обеспечивающую свой превосходный эффект замедленного высвобождения (на фиг. 3, на график нанесены средние величины измерений, полученных у 6 крыс).
ЭКСПЕРИМЕНТАЛЬНЫЙ ПРИМЕР 5: Тест фармакологического эффекта in vivo
Фармакологический эффект композиции по настоящему изобретению оценивали в следующем тесте. Композицию примера 16, содержащую эксенатид (антидиабетическое средство), которое может вызвать потерю массы тела, подкожно инъецировали 6 крысам SD (самцам) в возрасте 9 недель со средней массой тела 300 г, в той дозе, которая соответствует 140 мкг/кг эксенатида. Величины средней массы тела рассчитывали в 0-ой и 14-й дни, и результаты представлены ниже в таблице 5.
Как показано в таблице 5, у группы, которой вводили композицию примера 16, потеря массы тела составила примерно 25% в течение двух недель, по сравнению с массой тела у группы, которой вводили физиологический раствор. Поэтому, композиция замедленного высвобождения по настоящему изобретению обеспечивает длительно продолжающуюся фармакологическую эффективность in vivo, а также значительное увеличение периода полувыведения биологически активного ингредиента в тесте замедленного высвобождения in vivo (ЭКСПЕРИМЕНТАЛЬНЫЙ ПРИМЕР 4).
ЭКСПЕРИМЕНТАЛЬНЫЙ ПРИМЕР 6: Образование жидкого кристалла в водной жидкости
Композицию по настоящему изобретению оценивали в отношении способности образовывать жидкий кристалл в водной фазе в следующем тесте. После загрузки в шприцы, композиции примера 4 и сравнительного примера 4 закапывали в 2 г PBS (забуференного фосфатом солевого раствора) (pH 7,4) и результаты представлены на фиг. 4.
Композиция примера 4 на основе сложного эфира ненасыщенной жирной кислоты сорбитана, имеющего полярную головку по меньшей мере с двумя -OH (гидроксильными) группами (сорбитан моноолеат), существовала в виде жидкой фазы в отсутствие водной жидкости, но образовывала жидкие кристаллы после воздействия водной жидкости. С другой стороны, композиция сравнительного примера 4 на основе сложного эфира ненасыщенной жирной кислоты сорбитана (полиоксиэтилен сорбитан моноолеат) существовала в виде жидкой фазы и диспергировалась PBS, но не формировалась в жидкий кристалл даже после воздействия водной жидкости. Следовательно, только композиция по настоящему изобретению быстро формируется в жидкие кристаллы, способствующие эффекту замедленного высвобождения в присутствии водной жидкости, например, в среде внутри тела.
Внутри жидких кристаллов имеется большое число бинепрерывных водных каналов нано размера (менее 20 нм), которые напоминают ленту Мебиуса. Водные каналы окружены бинепрерывными липидными слоями. Таким образом, как только липидная композиция формируется в жидкий кристалл в полутвердой фазе, фармакологически активное вещество может высвобождаться из жидкокристаллической структуры только после того как оно прошло через многочисленные водные каналы, что усиливает эффект замедленного высвобождения фармакологически активного вещества. Поэтому, композиция по настоящему изобретению может применяться для замедленного высвобождения лекарственных препаративных форм.
ЭКСПЕРИМЕНТАЛЬНЫЙ ПРИМЕР 7: Определение внутренней структуры жидкого кристалла с использованием Cryo TEM (электронного блока с криоблоком)
Внутреннюю структуру жидких кристаллов композиция по настоящему изобретению исследовали в следующем эксперименте. Композицию примера 4 в жидкой фазе накапывали на 2 г воды для получения жидкокристаллической структуры. Используя гомогенизатор, жидкие кристаллы в водной фазе в достаточной степени диспергировали и поддерживали в равновесном состоянии при комнатной температуре до анализа. Разбавленные жидкие кристаллы адсорбировали на решетку и замораживали с последующим исследованием структуры в крио-трансмиссионном электронном микроскопе (Cryo TecaiF20G2, FEI). Результаты показаны на фиг. 5.
Как показано на фотографиях фиг. 5, наблюдали, что жидкие кристаллы имеют кристаллические структуры, такие как кубические фазы или шестиугольные фазы. Как правило, ламеллярные структуры, такие как мицеллы, эмульсии, микроэмульсии, липосомы и т.д., обычно существуют в полностью сферических состояниях, тогда как не ламеллярные структуры в соответствии с композицией по настоящему изобретению принимают упорядоченные формы с определенными углами, которые совершенно отличны от сферических форм.
Хотя изобретение было проиллюстрировано и описано в отношении одного или нескольких вариантов исполнений, для других специалистов в данной области будет очевидны возможные эквивалентные изменения и модификации по прочтении и осмысливании данного описания и прилагаемых чертежей. Кроме того, хотя мог быть описан конкретный признак изобретения в отношении одного или нескольких вариантов исполнения, такой признак может комбинироваться с одним или несколькими другими признаками других вариантов исполнения для достижения желательного эффекта и преимуществ для любого данного или конкретного применения.
Реферат
Изобретение относится к медицине, в частности к биоразлагаемому липидному концентрату для замедленного высвобождения, который содержит сложный эфир ненасыщенной жирной кислоты сорбитана, имеющий две или более -OH (гидроксильных) групп на полярной головке, фосфолипид и отвердитель жидких кристаллов, выбранный из группы, состоящей из триглицерида, ретинилпальмитата, токоферилацетата, холестерина, бензилбензоата и их смеси. Изобретение также относится к фармацевтической композиции для замедленного высвобождения активного ингредиента, содержащая фармакологически активный ингредиент и липидный преконцентрат. Липидный преконцентрат характеризуется высокой безопасностью и биоразлагаемостью, а также замедленным высвобождением активного вещества. 2 н. и 10 з.п. ф-лы, 33 пр., 5 табл., 5 ил.
Формула

Комментарии