Фосфолипидный депо-препарат - RU2595865C2
Код документа: RU2595865C2
Чертежи









Описание
Перекрестная ссылка на родственные заявки
Настоящая заявка является продолжением Международной заявки №PCT/US2010/061, 015, поданной 17 декабря 2010, которая испрашивает приоритет даты подачи предварительной заявки на патент Соединенных Штатов №61/375, 502, поданной 20 августа 2010 года, которые обе включены в данную заявку посредством ссылки в полном объеме.
Уровень техники
Депо-препарат представляет собой способ введения активного ингредиента в тело пациента для системного или местного действия. Как правило, его вводят подкожной или внутримышечной инъекцией или инстилляцией в другие ткани, сосуды или полости тела. Депо-препарат также может прикладываться к ране до того, как остановят кровотечение из нее, ее зашьют, перевяжут или закроют иным способом. В отличие от съемных депо-препаратов, биоразлагаемые депо-препараты разлагаются или деградируют в течение заранее определенного времени, обычно после доставки включенного в них активного фармацевтического ингредиента. В других конструкциях биоразлагаемый инъекционный депо-препарат высвобождает свой активный фармацевтический ингредиент почти одновременно с его постепенной деградацией или в зависимости от его постепенной деградации. Ключевым преимуществом некоторых биоразлагаемых депо-препаратов доставки является их способность доставлять лекарственный препарат непосредственно к предполагаемому месту действия, обеспечивая повышенные локальные концентрации лекарственного препарата по сравнению с уровнями концентрации в системе.
Депо-препараты также могут модулировать доставку лекарственного препарата для осуществления различных профилей высвобождения. Профиль высвобождения может представлять собой немедленное освобождение (выброс) с последующим стационарным состоянием, он может обеспечивать, в частности, скорость "нулевого порядка" или постоянную скорость доставки, может обеспечивать медленное возрастание высвобождения до достижения стационарного состояния или может даже обеспечивать высвобождение с задержкой. Кроме того, депо-препараты обладают преимуществом, которое заключается в том, что они обеспечивают высвобождение в течение длительного периода времени, достигаемое одним введением. Показатели крови не подвергаются риску, например, из-за несоблюдения пациентом условий лечения.
Депо-препараты могут состоять из систем частиц, например, депо-препараты на основе микросфер и депо-препараты на основе наносфер, или могут также состоять из биоразлагаемого геля, как правило, сделанного из растворимых соединений, способных образовывать матрикс (полимеры, липиды, углеводы), и либо органического растворителя, либо смеси смешивающихся и не смешивающихся с водой растворителей.
Для получения депо-препаратов, содержащих липофильный фармакологический активный агент, использовались фосфолипиды. Фосфолипиды растворяются в маслах и органических растворителях, но не растворяются в воде. Для образования депо-препарата часто требуется высокая концентрация фосфолипидов, образующих депо-препарат. Это может влиять на объем и вязкость полученного депо-препарата и, соответственно, доступные в настоящее время фосфолипидные депо-препараты могут быть введены посредством инъекции через обычную иглу или шприц с большими затруднениями. Ссылки, описывающие композиции на основе фосфолипидов, включают в себя WO 89/00077, WO 02/32395, ЕР 0282405 и патенты США №№5863549, 4252793, 5660854, 5693337 и Wang et al., Lyophilization Of Water-In-Oil Emulsions To Prepare Phospholipid-Based Anhydrous Reverse Micelles For Oral Peptide Delivery, 39 European Journal of Pharmaceutical Sciences, at 373-79 (2010).
Ванкомицин представляет собой гликопептидный антибиотик, используемый для профилактики и лечения инфекций, вызванных грамположительными бактериями. Как правило, данный препарат выбирают в случае серьезных инфекций и эндокардита, вызванных S. aureus, коагулазонегативными стафилококками, пневмонийными стрептококками Streptococcus pneumoniase, β-гемолитическими стрептококками, коринебактериями группы JK, стрептококками виридэнс или энтерококками, когда не могут быть использованы β-лактамы из-за аллергии или устойчивости к лекарственному препарату. Ванкомицин можно сочетать с другими антимикробными препаратами при лечении, в частности, метициллин-резистентного коагулазонегативного стафилококкового эндокардита протеза клапана и энтерококкового эндокардита. Он также был использован в качестве альтернативного средства для лечения пневмококкового менингита, вызванного штаммами с пониженной чувствительностью к пенициллину. Ванкомицин был использован в сердечной и сосудистой хирургии для предотвращения послеоперационной инфекции. См. Rybak et al., Vancomycin Therapeutic Guidelines: A Summary of Consensus Recommendations From The Infectious Diseases Society of America, The American Society Of Health-System Pharmacists, и The Society Of Infectious Disease Pharmacists, CID 2009: 49 (1 August), pg. 325.
Гентамицин представляет собой аминогликозид, используемый для лечения многих видов бактериальных инфекций, особенно тех, которые вызываются восприимчивыми к нему грамотрицательными бактериями. Он использовался при хирургических вмешательствах, поскольку он действует против патогенных микроорганизмов, таких как Pseudomonas aeroginosa и Escherichia coli. Гентамицин был использован в других приложениях, связанных с хирургией (например, будучи смешанным с костным цементом в ортопедических имплантатах). Пропитанный гентамицином имплантат с биоразлагаемым коллагеном (губка) в настоящее время используется на нескольких рынках за пределами США для профилактики хирургических инфекций (surgical site infections (SSI)). Однако два больших ключевых клинических исследования фазы III показали, что у пациентов, получавших гентамициновую губку (колоректальная хирургия), наблюдалась более высокая частота SSI, и не было никакого различия во встречаемости SSI по сравнению со стандартной медицинской помощью (кардиохирургия). В общем случае, см. Е. Bennett-Guerrero, NEJM, 2010, 1-10; и Е. Bennett-Guerrero, JAMA, August 18, 2010, 755-762.
Как ванкомицин, так и гентамицин являются очень гидрофильными антибиотиками. Их также трудно включать в инъекционные депо-препараты на основе фосфолипидов или других препаратов с высоким содержанием масляной фазы, поскольку они не являются свободно растворимыми в фосфолипиде или масле.
Кроме того, путем проведения ряда тестов на стабильность, в настоящее время установлено, что ванкомицин и гентамицин деградируют в результате действия различных механизмов. Ванкомицин теряет устойчивость в результате гидролиза, в то время как гентамицин деградирует из-за окисления или образования аддукта. Таким образом, препараты, содержащие один из данных активных агентов, как правило, чувствительны к таким условиям. Кроме того, как ванкомицин, так и гентамицин чувствительны к перегреву и не могут быть стерилизованы с использованием тепла, например, автоклавированием или использованием гамма-излучения.
Таким образом, попытка разработать депо-препарат, содержащий ванкомицин, гентамицин или оба антибиотика вместе с фосфолипидом и маслом, приводит к необходимости решать многие практические проблемы. Одна из таких проблем заключается в том, что препарат не должен отличаться высокой вязкостью, поскольку данный препарат должен быть стерилизован путем фильтрации через стерилизующую мембрану, такую как мембрану, имеющую поры диаметром около 0,2 микрон или менее. Так же остаются некоторые дихотомические проблемы. Например, у данных двух конкретных активных агентов имеются проблемы совместимости с фосфолипидами, что, как и в случае вязкости, указывает на необходимость поддерживать низкое содержание фосфолипидов. Однако необходимость в образовании когерентного и когезивного геля и надлежащих характеристиках высвобождения предполагает как раз обратное.
Соответственно, остается настоятельная потребность в стабильных при хранении фосфолипидных депо-препаратах, содержащих ванкомицин, гентамицин, их фармацевтическую соль или их смесь, которые можно вводить подкожной или внутримышечной инъекцией, инъекцией в рану или помещением в хирургическую рану или другие ткани, сосуды или полости тела.
Сущность изобретения
Одна задача настоящего изобретения относится к способу получения депо-препарата, содержащего по меньшей мере одно гидрофильное водорастворимое фармацевтически активное вещество, включающему: (1) смешивание по меньшей мере одного гидрофильного водорастворимого фармацевтически активного вещества, выбранного из группы, состоящей из ванкомицина, гентамицина, их фармацевтически приемлемой соли и их смеси, воды, фосфолипида и масла для образования эмульсии масло-в-воде, (2) гомогенизацию данной эмульсии для получения "первичной эмульсии"; (3) применение микрофлюидизатора для получения из первичной эмульсии "монофазного раствора"; (4) обеспечение того, чтобы значение pH первичной эмульсии и/или монофазного раствора находилось в пределах от около 3 до около 6, и в одном варианте осуществления от около 3 до около 5, и в другом варианте осуществления от около 3 до около 4, регулируя pH по мере необходимости, (5) лиофилизацию монофазного раствора с желаемым значением pH для получения сухой пасты, (6) добавление модификатора вязкости в сухую пасту в количестве, достаточном для получения прозрачного раствора, (7) удаление, по меньшей мере, некоторого количества модификатора вязкости из прозрачного раствора для получения депо-препарата, содержащего от около 5,5% масс. до около 7,5% масс. модификатора вязкости по отношению к общей массе депо-препарата, и (8) стерилизацию депо-препарата фильтрацией.
В одном варианте осуществления стадии образования эмульсии и первичной эмульсии могут быть объединены в одну стадию при условии, что конечный продукт представляет собой первичную эмульсию. В другом варианте осуществления стадии образования первичной эмульсии и монофазного раствора могут быть объединены в одну стадию при условии, что конечный продукт представляет собой монофазный раствор. В еще одном варианте осуществления стадии образования эмульсии, первичной эмульсии и монофазного раствора могут быть объединены в одну стадию, приводящую непосредственно к получению монофазного раствора.
В одном варианте осуществления вода, присутствующая в депо-препарате, составляет не более 4% масс. по отношению к общей массе депо-препарата. В другом варианте осуществления содержание воды в депо-препарате составляет не более 2% масс., а в еще одном варианте осуществления составляет не более 1% масс. В еще одном дополнительном варианте осуществления имеется не более 0,5% масс. воды по отношению к общей массе депо-препарата. В других вариантах осуществления фармакологически активные вещества представляют собой гидрохлорид ванкомицина и сульфат гентамицина. В других вариантах осуществления депо-препарат является прозрачным, и еще в других вариантах осуществления депо-препарат является сверхпрозрачным (ultra clear).
Другая задача настоящего изобретения относится к способу получения прозрачного депо-препарата, содержащего по меньшей мере одно гидрофильное водорастворимое фармацевтически активное вещество, содержащему: (1) растворение по меньшей мере одного гидрофильного водорастворимого фармацевтически активного вещества, выбранного из группы, состоящей из ванкомицина, гентамицина, их фармацевтически приемлемой соли и их смеси в воде, с образованием водного раствора; (2) образование эмульсии масло-в-воде, содержащей фосфолипид, масло и водный раствор; (3) гомогенизацию эмульсии для получения первичной эмульсии; (4) применение микрофлюидизатора для получения из первичной эмульсии "монофазного раствора"; (5) доведение значения pH эмульсии, первичной эмульсии и/или монофазного раствора до интервала от около 3 до около 6, в другом варианте осуществления от около 3 до около 5, и в еще одном варианте осуществления от около 3 до около 4, по мере необходимости, (6) лиофилизацию монофазного раствора с желаемым значением pH для получения сухой пасты, (7) добавление модификатора вязкости в сухую пасту в количестве, достаточном для получения желаемой вязкости и/или желаемой прозрачности, (8) предварительную фильтрацию раствора измененной вязкости до получения прозрачного раствора, (9) удаление, по меньшей мере, некоторого количества модификатора вязкости из прозрачного раствора для получения депо-препарата, содержащего от около 5,5% масс. до около 7,5% масс. модификатора вязкости по отношению к общей массе депо-препарата, и (10) стерилизацию депо-препарата без существенного нагрева. Такие стерилизационные процедуры, наряду с другими методами, могут быть проведены фильтрацией. В другом варианте осуществления предварительная фильтрация и удаление модификатора вязкости являются необязательными стадиями. В одном варианте осуществления по меньшей мере одно гидрофильное водорастворимое фармацевтически активное вещество представляет собой ванкомицин, гентамицин, их фармацевтически приемлемую соль и их смесь.
Еще одна задача настоящего изобретения относится к способу изготовления депо-препарата, включающему: (1) образование эмульсии масло-в-воде, включающей в себя фосфолипид, масло, по меньшей мере одно гидрофильное водорастворимое фармацевтически активное вещество, выбранное из группы, состоящей из ванкомицина, гентамицина, их фармацевтически приемлемой соли или их смеси, и воду; (2) превращение эмульсии в монофазный раствор, имеющий значение pH в пределах от около 3 до около 6; (3) лиофилизацию монофазного раствора для получения сухой пасты, (4) добавление модификатора вязкости в сухую пасту в количестве, достаточном для получения желаемой вязкости и/или желаемой прозрачности, (5) удаление, по меньшей мере, некоторого количества модификатора вязкости для получения депо-препарата, и (6) стерилизацию депо-препарата, причем депо-препарат является прозрачным.
В одном варианте осуществления способ дополнительно содержит стадию асептического заполнения депо-препаратом шприца, флакона или любого другого соответствующего устройства, способного хранить и/или доставлять данный депо-препарат к месту лечения или к ране.
В соответствии с другой задачей изобретения стабилизирующий агент является необязательно растворенным в воде наряду с фармацевтически приемлемым(и) ингредиентом(ами). Еще одна задача изобретения относится к стабилизирующему агенту, который необязательно смешивается вместе с фармацевтически приемлемым(и) ингредиентом(ами), водой, фосфолипидом и маслом. Примеры стабилизирующего агента включают в себя, но не ограничиваются ими, динатриевую соль ЭДТА, глицин, L-гистидин, лимонную кислоту, метионин, аскорбиновую кислоту, L-цистеин, альфа-токоферол и их смеси. Еще одна задача изобретения относится к депо-препарату, не включающему в себя стабилизирующий агент.
В одном варианте осуществления, на стадии образования эмульсии масло-в-воде, количество добавляемой воды составляет от около 60% масс. до около 80% масс. по отношению к общей массе полученной эмульсии. В другом варианте осуществления количество воды в эмульсии на стадии образования эмульсии масло-в-воде около двух раз больше массы эмульсии.
В еще одном варианте осуществления, после стадии обработки первичной эмульсии с помощью микрофлюидизатора, в результате чего получают монофазный раствор, также называемый здесь "наноэмульсией", средний диаметр капли наноэмульсии составляет менее чем около 120 нм, менее чем около 100 нм или менее чем около 80 нм.
Считается, что уменьшение среднего диаметра капли наноэмульсии/монофазного раствора, в частности, снижает вязкость полученного монофазного раствора, позволяя проводить стерилизацию через фильтр, а не с использованием системы стерилизации, основанной на нагревании, такой, как автоклавирование или гамма-лучевая стерилизация, которые могут повлиять на стабильность ванкомицина и/или гентамицина.
Перед стадией микрофлюидизации первичная эмульсия представляет собой, как правило, белую, непрозрачную, густую, как йогурт, массу. Полученный после микрофлюидизации монофазный раствор обычно прозрачен на свет и похож по таким свойствам, как вязкость и текучесть на воду.
Хотя настоящее изобретение не ограничивается каким-либо конкретным теоретическим механизмом, предполагается, что очень гидрофильные ванкомицин, гентамицин, их фармацевтически приемлемая соль или их смесь могут быть объединены с фосфолипидами в лекарственную форму с образованием монофазного раствора, как определено в настоящем документе, с образованием стабильных при хранении депо-препаратов с желаемыми свойствами. Считается, что капли наноэмульсии очень маленького размера, получаемые в ходе микрофлюидизации, могут сыграть важную роль в окончательных свойствах производимых депо-препаратов, наряду с прочими факторами, которые могут иметь значение.
В соответствии с другим вариантом осуществления настоящего изобретения значение pH эмульсии, первичной эмульсии и/или монофазного раствора составляет от около 3 до около 6, от около 3 до около 5, или от около 3 до около 4. А если это не так, то значение pH может быть установлено так, чтобы попадать в желаемый интервал.
В соответствии с еще одним вариантом осуществления настоящего изобретения значение pH депо-препарата, конечного продукта, составляет от около 3 до около 6, от около 3 до около 5, а в другом варианте осуществления от около 3 до около 4.
Другая задача настоящего изобретения относится к депо-препарату, содержащему по меньшей мере одно гидрофильное водорастворимое фармацевтически активное вещество, выбранное из группы, состоящей из ванкомицина, гентамицина, их фармацевтически приемлемой соли и их смеси, воду, фосфолипид и одно или более масло, необязательно агент, регулирующий pH, и модификатор вязкости, причем вода, присутствующая в депо-препарате, составляет не более 4% масс., не более 2% масс, не более 1% масс. или не более 0,5% воды по отношению к общей массе депо-препарата. В другом варианте осуществления депо-препарат является подходящим для введения посредством шприца (предназначен для введения шприцом, syringeable).
В одном варианте осуществления изобретения депо-препарат содержит как ванкомицин, так и гентамицин. В другом варианте осуществления депо-препарат содержит фармацевтические соли одного из или обоих ванкомицина и гентамицина. В другом варианте осуществления депо-препарат содержит либо ванкомицин либо гентамицин. В еще одном варианте осуществления депо-препарат содержит фармацевтическую соль либо ванкомицина или гентамицина.
В одном варианте осуществления депо-препараты в соответствии с настоящим изобретением являются "прозрачными". Это дает преимущества в возможности видеть захваченный воздух, инородные тела и тому подобное, чтобы предотвратить непреднамеренное введение их в организм. Интересно, что также было обнаружено, что, когда в депо-препарате присутствуют как ванкомицин, так и гентамицин, то депо-препарат по изобретению является более прозрачным, чем когда депо-препарат содержит только либо ванкомицин или гентамицин. В таком варианте осуществления, когда и ванкомицин и гентамицин присутствуют в депо-препарате, прозрачность такого депо-препарата достигает степени "сверхпрозрачности" ("ultra clear"), как определено в настоящем документе. В одном варианте осуществления, где депо-препарат содержит или ванкомицин или гентамицин, прозрачность такого депо-препарата может быть охарактеризована как «прозрачная» или «полупрозрачная», как определено в настоящем документе.
В одном варианте осуществления модификатор вязкости представляет собой этанол, причем количество этанола, присутствующего в депо-препарате, составляет от около 3% масс. до около 25,0% масс., от около 4% масс. до около 10% масс. В еще одном варианте осуществления количество присутствующего этанола варьирует в интервале от около 5% масс. до около 6,5% масс. по отношению к общей массе депо-препарата. В еще одном варианте осуществления модификатор вязкости представляет собой абсолютный этанол.
В одном варианте осуществления модификатор вязкости может добавляться к сухой пасте до тех пор, пока количество модификатора вязкости не составит около 75% масс. или более от количества раствора измененной вязкости. В других вариантах осуществления количество модификатора вязкости составляет около 50% масс. или более, а в еще одном варианте осуществления около 30% масс. или более. Наконец, количество модификатора вязкости составляет около 25% масс. или более по отношению к общей массе раствора измененной вязкости.
В еще одном варианте осуществления количество фосфолипида, присутствующего в депо-препарате, составляет от около 5% масс. до около 95% масс., а в другом варианте осуществления от около 25% масс. до около 75% масс. по отношению к общей массе депо-препарата. В другом варианте осуществления количество фосфолипида варьирует в интервале от около 35% масс. до около 60% масс. по отношению к общей массе депо- препарата.
В соответствии с другим вариантом осуществления настоящего изобретения количество масла, присутствующего в депо-препарате, составляет от около 5% масс. до около 95% масс., а в другом варианте осуществления от около 25% масс. до около 75% масс. по отношению к общей массе депо-препарата. В еще одном варианте осуществления количество масла варьирует в интервале от около 35% масс. до около 60% масс. по отношению к общей массе депо-препарата.
В соответствии с одним вариантом осуществления настоящего изобретения, не более чем около 80% ванкомицина и/или гентамицина высвобождаются через два часа при измерении в соответствии с методом USP I (метод Фармакопеи США (The United States Pharmacopeial Convention (USP)) с использованием 500 мл деионизированной воды в качестве среды. В другом варианте осуществления не более чем около 50%, а в еще одном варианте осуществления не более чем около 20% ванкомицина и/или гентамицина высвобождаются через два часа при измерении в соответствии с фармакопейным методом USP I с использованием 500 мл деионизированной воды в качестве среды.
В соответствии с другой задачей изобретения, депо-препарат необязательно содержит стабилизирующий агент для повышения стабильности ванкомицина, гентамицина или их обоих. Примеры стабилизирующего агента включают, но не ограничиваются ими, ЭДТА (динатриевая соль), глицин, L-гистидин, лимонную кислоту, метионин, аскорбиновую кислоту, L-цистеин, альфа-токоферол и их смеси. В соответствии с еще одной задачей изобретения депо-препарат не содержит стабилизирующий агент. В еще одном варианте осуществления количество стабилизирующего агента, если таковой содержится, не будет негативно влиять на стабильность каждого активного агента, ванкомицина и гентамицина, в депо-препарате.
Другая задача настоящего изобретения относится к депо-препарату, описанному в настоящем документе, который предоставляется в аппликаторе, шприце, флаконе или любом другом устройстве, способном хранить и/или доставлять депо-препарат к месту лечения, месту хранения или к ране.
Еще одна задача настоящего изобретения относится к способу введения, внутрикожно, внутримышечно, через разрез, подкожно, инстилляцией или наложением, депо-препарата по изобретению, содержащего гидрофильное водорастворимое фармацевтически активное вещество, выбранное из группы, состоящей из ванкомицина, гентамицина, их фармацевтически приемлемой соли и их смеси, воду, фосфолипид и масло, необязательно агент, регулирующий pH, и модификатор вязкости, пациенту, нуждающемуся в этом.
Еще одна задача настоящего изобретения относится к способу профилактики и/или лечения послеоперационных инфекций путем введения депо-препарата по настоящему изобретению.
Еще одна задача настоящего изобретения относится к способу профилактики и/или лечения инфекции, содержащему введение депо-препарата по настоящему изобретению, достигающего достаточно высоких локальных концентраций в ткани, достаточных для лечения и/или профилактики инфекции в локальном месте, не токсичных для почек и/или других органов и не способствующих появлению лекарственно-устойчивых штаммов бактерий.
Другая задача настоящего изобретения относится к способу придания локализованной ткани способности не поддерживать патогенные микроорганизмы путем введения депо-препарата по настоящему изобретению в рану.
Еще одна задача настоящего изобретения относится к способу придания локализованной ткани способности не поддерживать патогенные микроорганизмы путем введения депо-препарата по настоящему изобретению, не вызывая токсичности для почек и других органов и не вызывая появления лекарственно-устойчивых штаммов бактерий.
Краткое описание чертежей
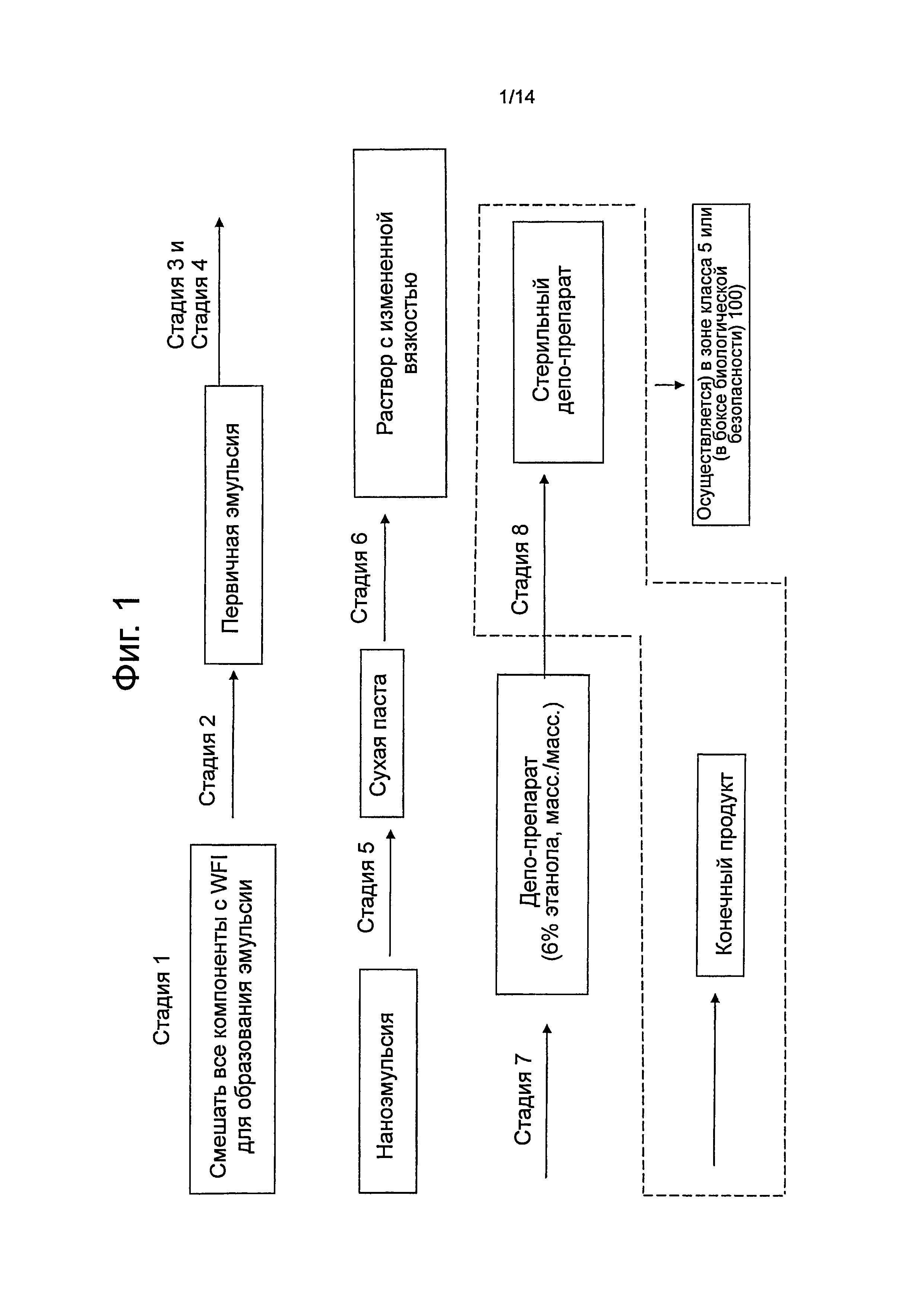
Фиг.1 представляет собой блок-схему процесса для варианта осуществления способа изготовления композиции по изобретению в соответствии с задачей настоящего изобретения.
Фиг.2 представляет собой график, показывающий аналитически определенное остаточное количество ванкомицина и гентамицина из препарата, описанного в примере 1, после автоклавной обработки.
Фиг.3 представляет собой график, показывающий профиль in vitro высвобождения гентамицина и ванкомицина из препарата, описанного в примере 6, полученный с использованием фармакопейного метода USP I.
Фиг.4 собой график, показывающий плазменные концентрации ванкомицина для препарата, описанного в примере, у кроликов.
Фиг.5 представляет собой график, показывающий тканевые концентрации ванкомицина для препарата, описанного в примере 1, у кроликов.
Фиг.6 представляет собой график, показывающий плазменные концентрации гентамицина для препарата, описанного в примере 1, у кроликов.
Фиг.7 представляет собой график, показывающий тканевые концентрации гентамицина для препарата, описанного в примере 1, у кроликов.
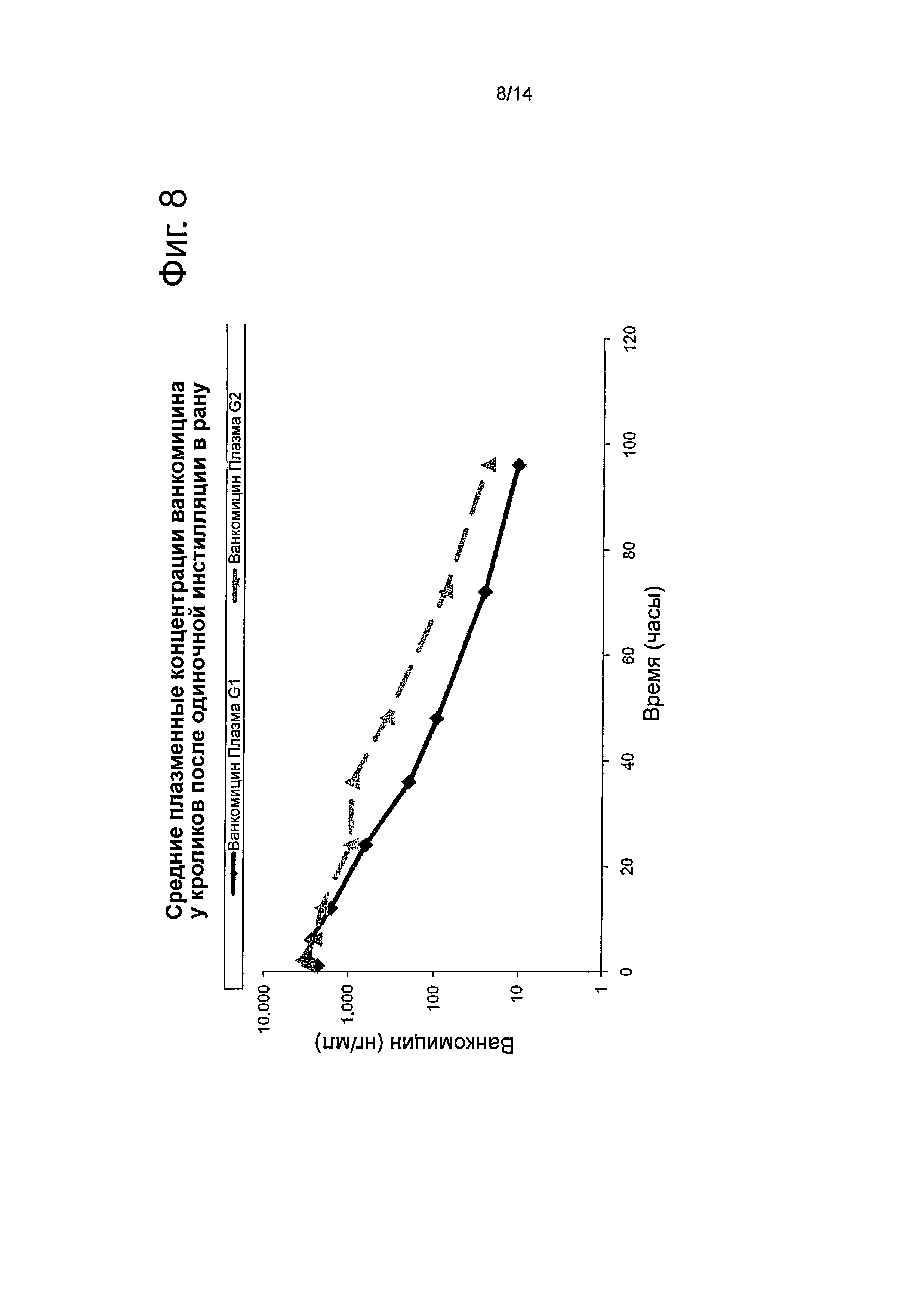
Фиг.8 представляет собой график, показывающий средние плазменные концентрации ванкомицина у кроликов после однократной SC инстилляции в рану препарата, описанного в примере 6.
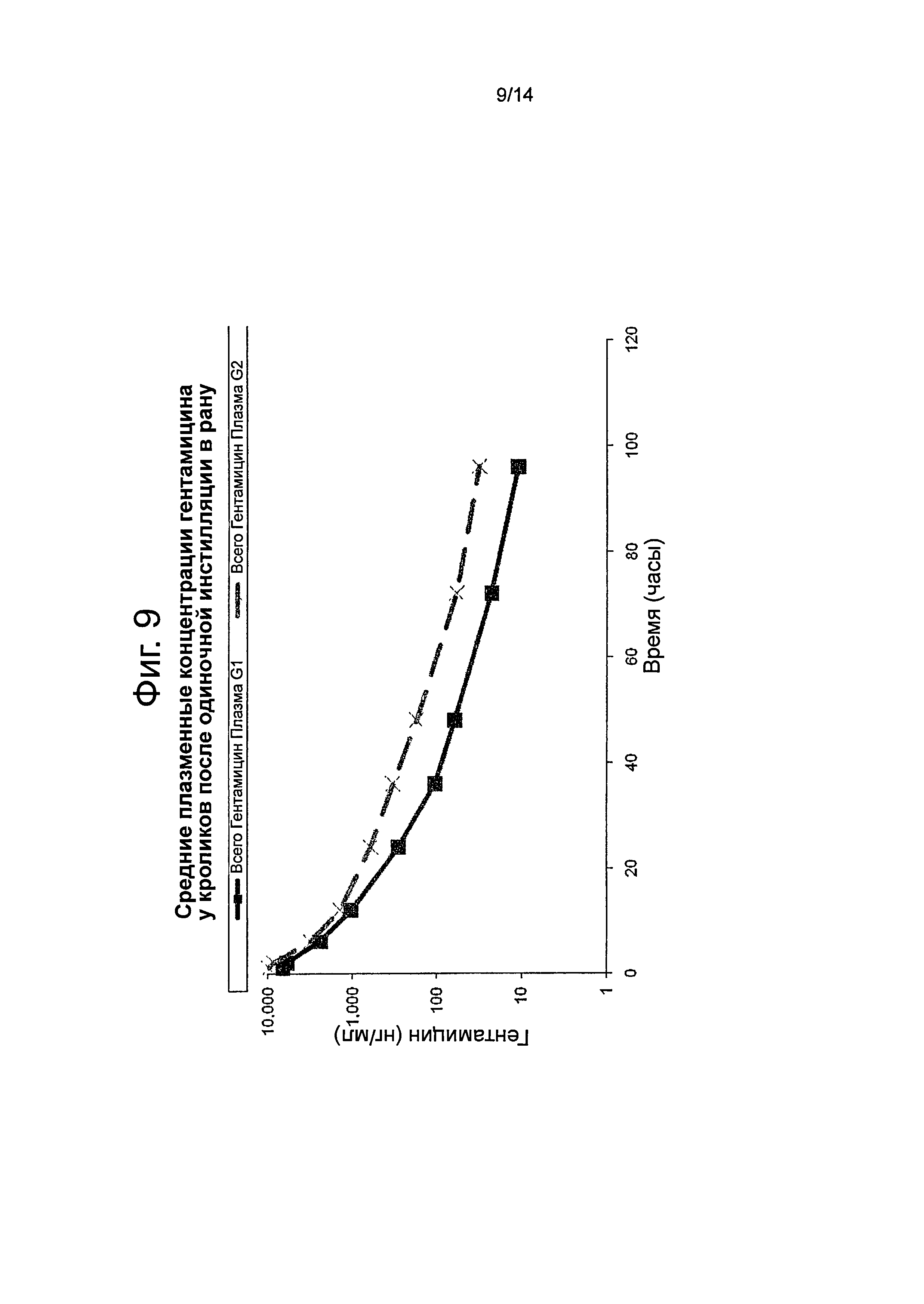
Фиг.9 представляет собой график, показывающий среднюю общую плазменную концентрацию гентамицина для препарата, описанного в примере 6, у кроликов.
Фиг.10 представляет собой гистограмму, показывающую тканевую концентрацию наиболее частых возбудителей хирургических инфекций (surgical site infection (SSI)) у свиньи после введения через разрез депо-препарата по настоящему изобретению по сравнению с MIC90 (минимальная ингибирующая концентрация (Minimum Inhibitory Concentration)), необходимая для подавления роста 90% организмов).
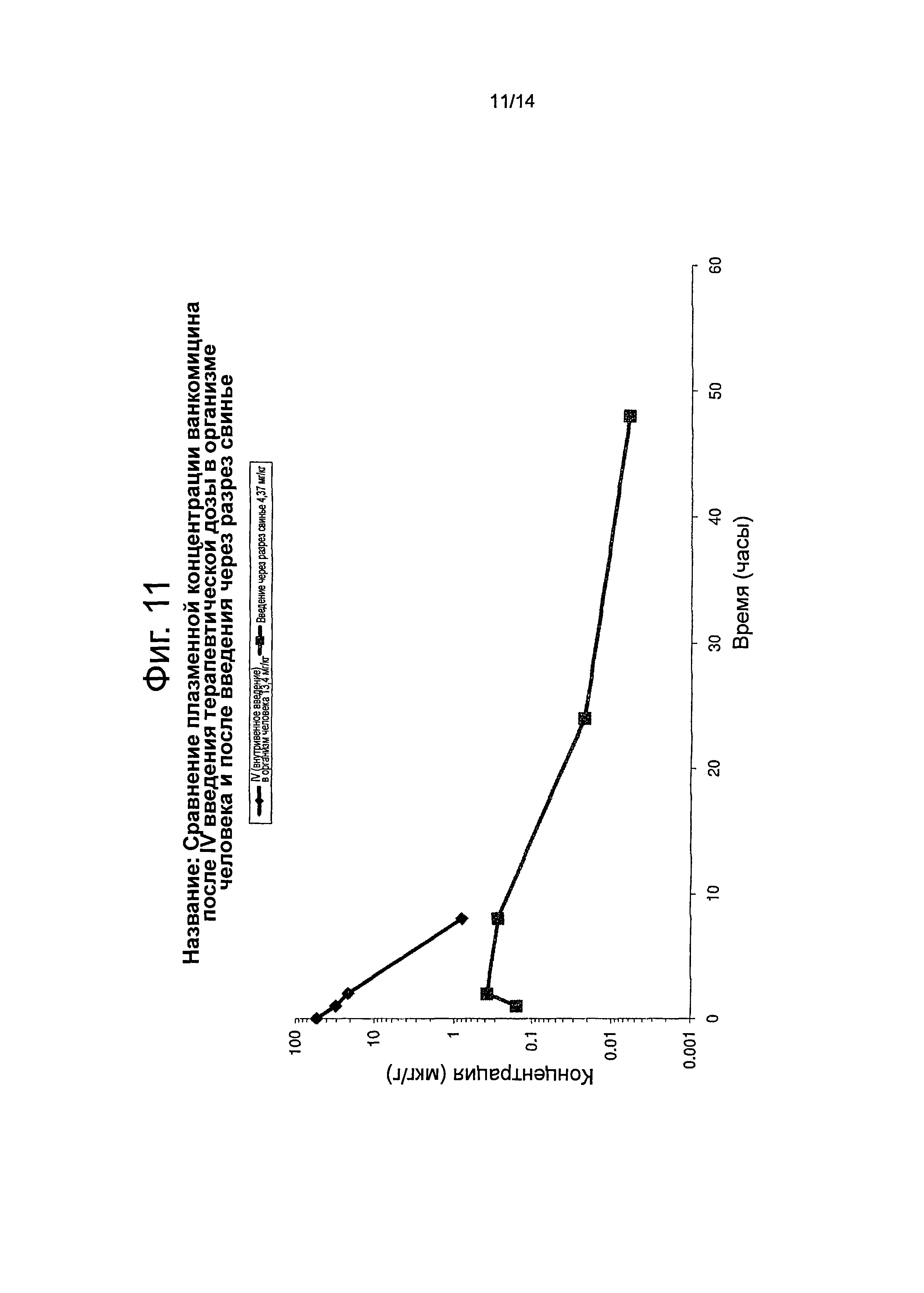
Фиг.11 представляет собой график, показывающий сравнение плазменной концентрации ванкомицина после внутривенного введения терапевтической дозы (intravenous (IV)) в организм человека и после введения через разрез препарата в соответствии с настоящим изобретением свинье.
Фиг.12 представляет собой дифрактограммы малоуглового рассеяния рентгеновских лучей (small angle X-ray diffraction (SAXS)) для примеров от 10А до 10F.
Фиг.13 представляет собой график, показывающий результаты термогравиметрического анализа для примеров 10А и 10D.
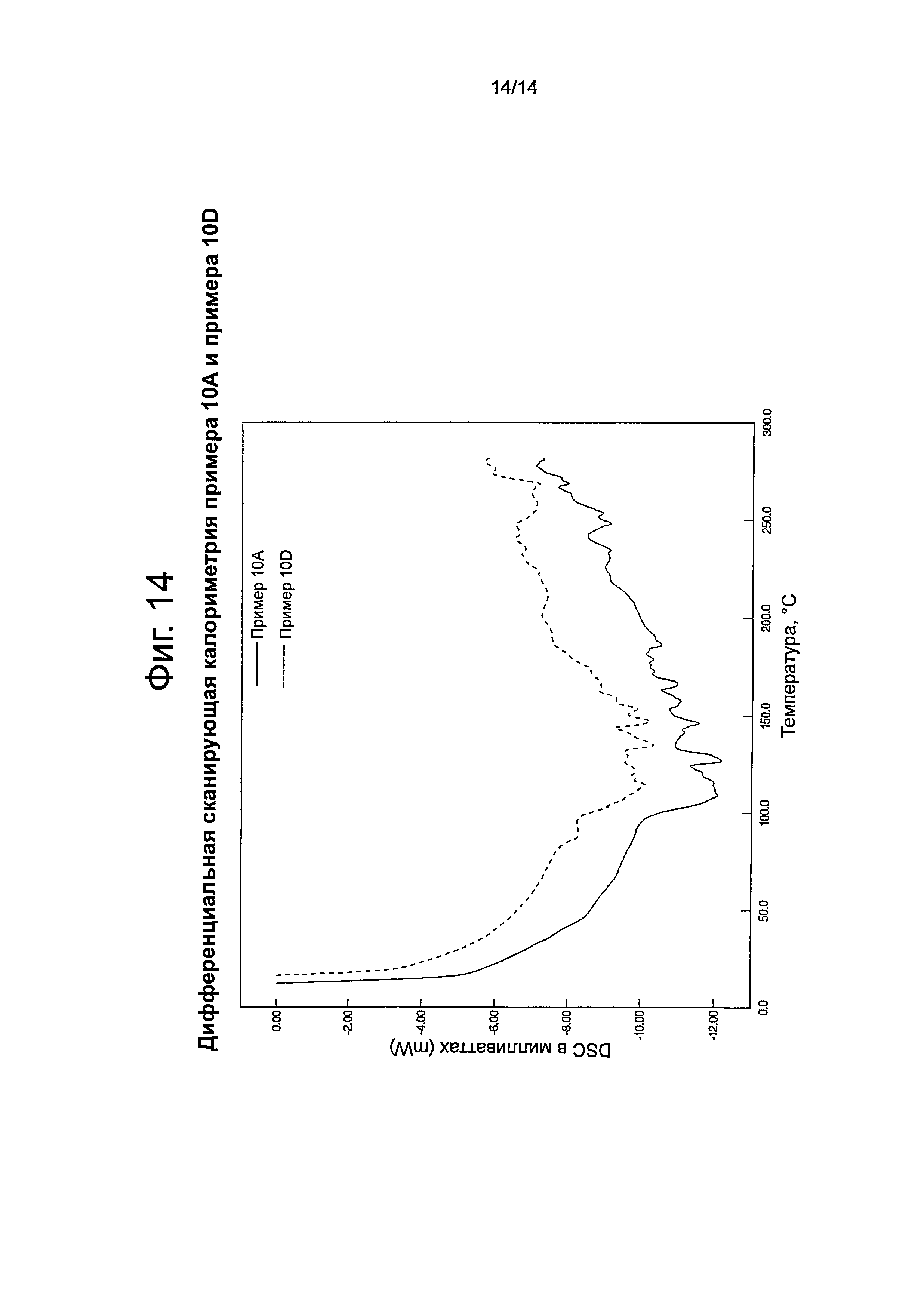
Фиг.14 представляет собой график, показывающий результаты анализа методом дифференциальной сканирующей калориметрии (differential scanning calorimetry (DSC)) для примеров 10А и 10D.
Подробное описание
Настоящее изобретение будет описано более подробно ниже.
Хотя данное описание заканчивается формулой изобретения, которая в деталях и отчетливо очерчивает претензию на изобретение, считается, что настоящее изобретение будет лучше понято из нижеследующего описания. Все проценты и соотношения, используемые в настоящем документе, пересчитаны на массу общей композиции, и все измерения выполнены при 25°С и нормальном давлении, если не обозначено иное. Все температуры указаны в градусах Цельсия, если не указано иное. Настоящее изобретение может "содержать" (без ограничения) или "состоять в основном из" компонентов по настоящему изобретению, а также других ингредиентов или элементов, описанных в настоящем документе. Для целей настоящего изобретения "содержащий" означает перечисленные элементы или их эквивалент по структуре или функции, а также любой другой элемент или элементы, которые не перечислены. Понятия "имеющий", "включающий" и "состоящий из" также следует рассматривать как не ограничивающие, если контекст не говорит об обратном. Для целей настоящего изобретения "состоящий в основном из" означает, что изобретение может включать в себя ингредиенты в дополнение к тем, которые перечислены в формуле изобретения, но только в том случае, если такие дополнительные ингредиенты существенно не изменяют основные и новые характеристики заявленного изобретения. Как правило, такие добавки могут не присутствовать вообще или присутствовать только в следовых количествах. Однако, может оказаться возможным включать до около 10% масс. материалов, которые могут существенно изменить основные и новые характеристики изобретения до тех пор, пока сохраняется применимость соединений (в отличие от степени применимости). Все интервалы, перечисленные в настоящем документе, включают в себя конечные значения, включая в себя те, которые обозначают интервал "между" двумя значениями. Такие термины, как "около", "обычно", "по существу" и тому подобное, должны толковаться как изменение понятия или значения так, что оно не является абсолютным. Такие термины будут определяться обстоятельствами и понятиями, которые они изменяют в соответствии с тем, как данные термины понимаются специалистами в данной области. Это включает в себя, по меньшей мере, степень ожидаемой погрешности эксперимента, ошибку методики и погрешность прибора для данной методики, используемой для измерения величины.
Следует отметить, что, в то время как описание и формула изобретения могут относиться к конечному продукту, такому как, например, депо-препарат или другая лекарственная форма по изобретению, которая, например, характеризуется определенным значением рН на одной из промежуточных стадий, может оказаться затруднительным заключить, исходя из конечной лекарственной формы, что она удовлетворяет этой характеристике из перечня свойств. Однако соответствие с перечнем свойств можно считать удовлетворительным, если исходные материалы, использованные для получения конечного продукта, удовлетворяют данному перечню свойств. Аналогичным образом, количество ингредиентов, введенных, например, в эмульсию, если они описываются в пересчете на массу, может измениться относительно массы продукта на одной из фаз производства, например, при получении конечного депо-препарата, который может весить больше или меньше. Достаточно того, чтобы такие массовые проценты были правильными на одной из стадий производства и/или в одном из промежуточных продуктов. Действительно, что касается любого свойства или характеристики конечного продукта, которое не может быть установлено непосредственно из лекарственной формы, достаточно, если данное свойство присутствует в компонентах, указанных для стадий производства, предшествующих конечным стадиям.
Понятие эмульсия для целей настоящего изобретения представляет собой систему из двух несмешивающихся жидких фаз. Одна из данных двух фаз (внутренняя фаза, дисперсная фаза или дискретная фаза) распределяется в виде капель/шариков во второй фазе (внешняя или непрерывная фаза). Для целей настоящего изобретения эмульсии включают в себя эмульсии масло-в-воде (O/W), в которых менее полярная жидкость, обычно называемая маслом, содержится во внутренней фазе; и эмульсии вода-в-масле (W/O), в которых водная или другая относительно полярная жидкость образует внутреннюю фазу.
Понятие "первичная эмульсия" для целей настоящего изобретения относится к конечному продукту стадии гомогенизации, на которой может использоваться, например, смеситель с большим усилием сдвига.
Понятия "монофазный раствор" и "наноэмульсия" используются в настоящем документе взаимозаменяемо. Следует отметить, что понятие "раствор" в понятии "монофазный раствор" означает не то, что он представляет собой однородную смесь двух или более веществ, но то, что он является продуктом, полученным на стадии микрофлюидизации, на которой может использоваться, например, микрофлюидизатор высокого давления.
Понятия "монофазный", "одна фаза" и "похожий на одну фазу" используются, чтобы обозначить, что конечный продукт будет оставаться в виде одной фазы без разделения фаз или преципитации даже после центрифугирования при 6000 g в течение 10 минут при 25°С образца в количестве 1 г, используя центрифугу, произведенную Heraeus, модель Fresco Biofuge, или любой эквивалент ее.
Понятие "вязкий" для целей настоящего изобретения означает, что вязкость композиции составляет от около 1 сантипуаз до около 5000 сантипуаз, от около 200 сантипуаз до около 2000 сантипуаз, или от около 300 сантипуаз до около 1500 сантипуаз.
Понятие "пригодный для введения шприцем" ("syringeable") для целей настоящего изобретения означает, что композицию можно вводить с помощью шприца или катетера или набирать из флакона в шприц. Это не означает, однако, что композиция по изобретению должна на самом деле находиться в шприце или вводиться с помощью шприца, если конкретное изложение или контекст не предполагает такого смысла.
Понятия "полупрозрачный" и "прозрачный" используются в настоящем документе взаимозаменяемо для обозначения того, что конечный депо-препарат или любая композиция промежуточной стадии, такая как раствор, эмульсия, первичная эмульсия, наноэмульсия и/или гель, не являются мутными или непрозрачными, и что они не содержат определяемых визуально взвешенных частиц. В них также должны отсутствовать пузырьки. Более того, под полупрозрачным понимают, что депо-препарат и/или любая промежуточная композиция, такая как раствор, эмульсия, первичная эмульсия, наноэмульсия и/или гель, не содержат определяемых визуально взвешенных частиц и что в них также должны отсутствовать пузырьки. Более того, "полупрозрачный" или "прозрачный" также означает, что депо-препарат и/или любая промежуточная композиция, такая как раствор, эмульсия, первичная эмульсия, наноэмульсия и/или гель по настоящему изобретению, пропускает более чем около 90% света, измеряемого при 800 нм (Т800) в кварцевой кювете с длиной оптического пути 1 см, используя спирт в качестве бланка (пробы сравнения), при измерении с помощью УФ-видимого спектрофотометра, такого, как спектрофотометр, произведенный Pharmacia, модель Ultrospec III.
Под "мутным" или "непрозрачным" понимают, что значение Т800 депо-препарата составляет менее чем около 90%.
Под "сверхпрозрачным (ultra clear)" понимают, что значение Т800 депо-препарата составляет более чем около 92%, или 95%.
Понятие "стабильный" для целей настоящего изобретения означает, что (1) данный препарат остается прозрачным при 25°С в течение не менее одного года, или (2) данный препарат остается прозрачным и не отделяется или не преципитирует после центрифугирования после того, как данный препарат подвергается воздействию при 40°С в течение одной недели.
Понятия "гель" и "депо-препарат" используются в настоящем документе взаимозаменяемо.
Технологическое описание
Как показано на фиг.1, одна задача настоящего изобретения относится к способу получения депо-препарата, содержащего гидрофильное водорастворимое фармацевтически активное вещество, выбранное из группы, состоящей из ванкомицина, гентамицина, их фармацевтически приемлемой соли и их смеси, содержащему: (1) смешивание по меньшей мере одного гидрофильного водорастворимого фармацевтически активного вещества, выбранного из группы, состоящей из ванкомицина, гентамицина, их фармацевтически приемлемой соли и их смеси, воды, фосфолипида и масла с образованием эмульсии масло-в-воде (см. фиг.1, стадия 1); (2) гомогенизацию эмульсии для получения первичной эмульсии (см. фиг.1, стадия 2); (3) применение микрофлюидизатора для получения из первичной эмульсии "монофазного раствора", также называемого в настоящем документе и на фиг.1 наноэмульсией (см. фиг.1, стадия 3); (4) обеспечение того, чтобы значение pH первичной эмульсии и/или монофазного раствора находилось в интервале от около 3 до около 6, в интервале от около 3 до около 5 или в интервале от около 3 до около 4, с регулированием значения pH по мере необходимости (см. фиг.1, стадия 4); (5) лиофилизацию монофазного раствора с желаемым значением pH для образования сухой пасты (см. фиг.1, стадия 5), (6) добавление модификатора вязкости в сухую пасту в количестве, достаточном для получения прозрачного раствора (см. фиг.1, стадия 6), (7) удаление, по меньшей мере, некоторого количества модификатора вязкости из прозрачного раствора для получения депо-препарата, содержащего от около 5,5% масс. до около 7,5% масс. модификатора вязкости по отношению к общей массе депо-препарата (см. фиг.1, стадия 7), и (8) стерилизацию депо-препарата без нагревания депо-препарата (см. фиг.1, стадия 8).
В одном варианте осуществления настоящего изобретения стадия смешивания по меньшей мере одного гидрофильного водорастворимого фармацевтически активного вещества, выбранного из группы, состоящей из ванкомицина, гентамицина, их фармацевтически приемлемой соли и их смеси, воды, фосфолипида и масла с образованием эмульсии масло-в-воде содержит (1) растворение ванкомицина, гентамицина, их фармацевтически приемлемой соли и их смеси в воде с образованием водного раствора; и (2) образование эмульсии, содержащей фосфолипид, масло и водный раствор, содержащий гидрофильный водорастворимый фармацевтически приемлемый ингредиент(ы), выбранный(е) из группы, состоящей из ванкомицина, гентамицина, их фармацевтически приемлемой соли или их смеси.
В альтернативном варианте осуществления модификатор вязкости добавляют в сухую пасту в количестве, достаточном для получения требуемой вязкости, и затем раствор с измененной вязкостью предварительно фильтруется для получения прозрачного раствора.
В еще одном варианте осуществления вода, присутствующая в депо-препарате, составляет не более 4% масс., не более 2% масс, не более 1% масс. или не более 0,5% воды по отношению к общей массе депо-препарата. В других вариантах осуществления фармакологически активные вещества представляют собой гидрохлорид ванкомицина и сульфат гентамицина. В другом варианте осуществления депо-препарат является прозрачным, и в еще одном варианте осуществления депо-препарат является сверхпрозрачным.
Образование эмульсии масло-в-воде
По меньшей мере одно гидрофильное водорастворимое фармацевтически активное вещество, выбранное из группы, состоящей из ванкомицина, гентамицина, их фармацевтически приемлемой соли и их смеси, воду, фосфолипид и масло смешивают с образованием эмульсии масло-в-воде.
В другом варианте осуществления сначала гидрохлорид ванкомицина, сульфат гентамицина или оба вещества растворяют в воде с образованием водного раствора.
Начальная концентрация лекарственного препарата гидрохлорида ванкомицина в воде составляет от около 1 мг/мл до около 50 мг/мл или от около 20 мг/мл до около 30 мг/мл, а начальная концентрация лекарственного препарата сульфата гентамицина в воде составляет от около 1 мг/мл до около 75 мг/мл или от около 10 мг/мл до около 30 мг/мл.
Затем водный раствор ванкомицина и/или гентамицина, фосфолипид, масло, необязательно агент, регулирующий pH, и, необязательно стабилизирующий агент смешивают с образованием эмульсии масло-в-воде.
Гомогенизация для получения первичной эмульсии
Затем эмульсия может быть гомогенизирована с использованием смесителя с большим усилием сдвига (такого, как, например, смеситель Silverson Model L5M) с образованием первичной эмульсии.
Микрофлюидизация для получения монофазного раствора
Первичную эмульсию затем подвергали микрофлюидизации, используя, например, микрофлюидизатор высокого давления, чтобы получить наноэмульсию/монофазный раствор. Средний диаметр частиц полученной наноэмульсии/монофазного раствора составляет менее 120 нм, менее 100 нм и менее 80 нм, чтобы образовать монофазный раствор/наноэмульсию. Установлено, что размер капель, составляющий более 180 нм, может привести к образованию мутного раствора.
Считается, что уменьшение среднего диаметра капель наноэмульсии, в частности, снижает вязкость полученного монофазного раствора, позволяя проводить стерилизацию через фильтр, а не используя систему стерилизации, основанную на нагреве, такую как автоклавирование или стерилизация гамма-излучением, которая может повлиять на стабильность ванкомицина и/или гентамицина.
Перед стадией микрофлюидизации первичная эмульсия обычно представляет собой белую, непрозрачную, густую, как йогурт, массу. Полученный после микрофлюидизации монофазный раствор обычно прозрачен на свет и похож по таким свойствам, как вязкость и текучесть, на воду.
Для того чтобы получить прозрачный монофазный раствор, эмульсия масло-в-воде предпочтительно содержит от около 10% до около 80% воды, от около 30% до около 80% воды или от около 60% до около 80% воды относительно к общей массе эмульсии масло-в-воде для того с тем, чтобы иметь требуемые характеристики текучести, необходимые для обработки в гомогенизаторе высокого давления, таком как микрофлюидизатор.
Регулирование pH
Значение pH эмульсии, первичной эмульсии или монофазного раствора может быть отрегулировано путем добавления агента, регулирующего pH, так, что значение pH эмульсии, первичной эмульсии или монофазного раствора составляет от около 3 до около 6, в диапазоне от около 3 до около 5 или в диапазоне от около 3 до около 4.
В другом варианте осуществления данная стадия выполняется путем добавления соответствующего количества агента, регулирующего pH, к эмульсии, с последующей стадией гомогенизации путем смешивания с большим усилием сдвига в течение 1 минуты. Затем, после стадии гомогенизации, значение pH композиции проверяется и может быть отрегулировано снова, если это необходимо.
Лиофилизация, сублимация или испарение
При удалении воды гентамицин и/или ванкомицин становятся равномерно распределенными в носителе фосфолипид/масло. Воду из монофазного раствора затем удаляют лиофилизацией, сублимацией и/или испарением, так что количество остаточной воды в полученной сухой пасте или в конечном, пригодном для введения шприцем прозрачном депо-препарате составляет менее чем около 4%, менее чем около 2% масс., или менее чем около 0,5% масс. воды по отношению к общей массе сухой пасты или вязкого прозрачного депо-препарата.
В другом варианте осуществления монофазный раствор подвергают сублимационной сушке с использованием полочного лиофилизатора. В еще одном варианте осуществления лоток лиофилизатора сделан из нержавеющей стали.
В еще одном варианте осуществления высота жидкости, заполняющей лоток лиофилизатора из нержавеющей стали, составляет не более чем около 3 см. В одном варианте осуществления после стадии лиофилизации полученный продукт, который представляет собой сухую пасту, содержит не более 1% масс. воды по отношению к общей массе сухой пасты.
Добавление модификатора вязкости
Модификатор вязкости добавляют в сухую пасту до тех пор, пока сухая паста полностью не растворится. Модификатор вязкости может добавляться к сухой пасте до тех пор, пока количество модификатора вязкости не составит около 75% масс. или более, около 50% масс. или более, около 30% масс. или более или около 25% масс. или более по отношению к общей массе раствора с измененной вязкостью. В одном варианте осуществления модификатор вязкости и сухая паста могут быть смешаны при температуре, равной от около 10°С до около 80°С или от около 25°С до около 60°.
Предварительная фильтрация
Это необязательная стадия, и она не требуется для некоторых вариантов осуществления изобретения. Если раствор с измененной вязкостью, полученный после добавления модификатора вязкости, является мутным, то раствор с измененной вязкостью может быть профильтрован, с использованием, например, фильтра с диаметром пор 0,65 мкм, для получения прозрачного раствора. Мутный компонент, удаленный стадиями предварительной фильтрации, состоит из небольшой фракции ванкомицина (около 2% искомого компонента) и гентамицина (3-4% искомого компонента). Данная потеря может быть компенсирована подгонкой исходных количеств или отказом от необходимости жесткого соблюдения требований по количеству данных компонентов. Это необязательная стадия, и она не требуется при некоторых вариантах осуществления изобретения.
Удаление модификатора вязкости
Впоследствии модификатор вязкости, который был добавлен для растворения сухой пасты, удаляется. Удаление модификатора вязкости может быть осуществлено до достижения остаточного количества модификатора вязкости, который может присутствовать в депо-препарате, от около 1% до около 50%, от около 2% до около 18% или от около 5% до около 6,5% по отношению к общей массе депо-препарата.
В случае пересушивания модификатор вязкости может быть добавлен по мере необходимости. Удаление модификатора вязкости может быть осуществлено с помощью роторного испарителя или продувкой азотом или воздухом. Для измерения количества модификатора вязкости, удаленного из прозрачного раствора для образования депо-препарата, может быть использован термогравиметрический анализ (TGA).
Вязкость полученного депо-препарата в соответствии с настоящим изобретением составляет от около 100 сантипуаз до около 5000 сантипуаз, от около 200 сантипуаз до около 2000 сантипуаз или от около 300 сантипуаз до около 1500 сантипуаз. Измерение вязкости может быть выполнено с помощью любого обычного метода, в том числе, используя цифровой программируемый реометр (Brookfield) (Brookfield Digital Programmable Rheometer) модели No. DV-III со шпинделем No. SP-40. Это необязательная стадия, и она не требуется для некоторых вариантов осуществления изобретения.
Стерильная фильтрация
Депо-препарат затем стерилизуют фильтрацией через стерилизующую мембрану, такую, как мембрану, имеющую поры диаметром около 0,2 микрон или менее.
Депо-препарат
Еще одна задача настоящего изобретения относится к депо-препарату, содержащему по меньшей мере одно гидрофильное водорастворимое фармацевтически активное вещество, выбранное из группы, состоящей из ванкомицина, гентамицина, их фармацевтически приемлемой соли и их смеси, воду, фосфолипид, масло, агент, регулирующий pH, и модификатор вязкости, причем вода присутствует в депо-препарате в количестве, составляющем не более 4% масс., не более 2% масс. или не более 0,5% масс. воды по отношению к общей массе депо-препарата.
В соответствии с другой задачей изобретения, депо-препарат необязательно содержит стабилизирующий агент для повышения стабильности ванкомицина, гентамицина или обоих веществ. Другая задача изобретения относится к данному депо-препарату, предоставляемому в шприце, флаконе или любом другом соответствующем устройстве, способном хранить и/или доставлять данный депо-препарат к месту лечения, хранения или к ране.
Фармацевтический активный ингредиент
Фармацевтический активный ингредиент в соответствии с настоящим изобретением представляет собой ванкомицин, гентамицин, их фармацевтически приемлемую соль или их смесь. В одном варианте осуществления фармацевтический активный ингредиент в соответствии с настоящим изобретением представляет собой гидрохлорид ванкомицина, сульфат гентамицина или их смесь. В другом варианте осуществления фармацевтический активный ингредиент в соответствии с настоящим изобретением представляет собой гидрохлорид ванкомицина и сульфат гентамицина. В еще одном варианте осуществления фармацевтический активный ингредиент в соответствии с настоящим изобретением представляет собой либо гидрохлорид ванкомицина или сульфат гентамицина.
Примеры фармацевтически приемлемой соли включают, но не ограничиваются ими, любые кислоты, которые могут образовывать соли либо с ванкомицином или с гентамицином, такие как уксусная кислота, соляная кислота, бромистоводородная кислота, лимонная кислота, муравьиная кислота, молочная кислота, янтарная кислота, серная кислота и тому подобное.
Количество фармацевтических активных ингредиентов, которые могут присутствовать в депо-препарате, может меняться в зависимости от ряда параметров, включая величину общей предназначенной дозы, продолжительность приема, размер депо-препарата, где и как его будут вводить, тип активного вещества, которое будут вводить, тип введения (например, непрерывное, с задержкой, и т.д.) и тому подобное. Однако обычно, общее количество фармацевтически приемлемого ингредиента может составлять от около 0,001% масс. до около 20% масс., от около 0,01% до около 10% масс. или от около 0,1% масс. до около 5% масс. по отношению к общей массе депо-препарата.
Масло
Масло в соответствии с настоящим изобретением может представлять собой, например, природные масла, такие как растительные масла, животное масло, витамин Е, сложный эфир витамина Е и тому подобное и/или синтетические или полусинтетические масла или их смеси.
Растительное масло относится к маслу, полученному из семян растений или орехов. Примеры растительных масел включают, но не ограничиваются ими, миндальное масло, масло бурачника, масло семян черной смородины, касторовое масло, сафлоровое масло, соевое масло, кунжутное масло, хлопковое масло, масло из виноградных косточек, подсолнечное масло, рапсовое масло, кокосовое масло, пальмовое масло, апельсиновое масло, кукурузное масло, оливковое масло и тому подобное.
Животное масло относится к маслу на основе триглицеридов, полученному из животных источников. Примерами животного масла могут служить рыбий жир или масла из других источников, таких как жир, сало и тому подобное.
Примерами синтетических или полусинтетических масел являются моно-, ди- или триглицериды, чьи кислотные компоненты представляют собой насыщенные и/или ненасыщенные жирные кислоты С6-С20, САРТЕХ® (различные классы сложных эфиров пропиленгликоля, такие как пропиленгликоль-дидеканоат, и сложные эфиры глицерина, такие как трикаприлат/капрат глицерина); MIGLYOL® (триглицериды каприловой/каприновой кислоты, или триглицериды каприловой/каприновой/линолевой кислоты, или триглицериды каприловой/каприновой/янтарной кислоты, или диэфир пропиленгликоля и каприловой/каприновой кислоты и примеси с другими веществами; CAPMUL® (доступны в разных категориях, например, Capmul MCM. Это, в основном, моно- и ди-эфиры глицерина и пропиленгликоля, такие как моноолеат глицерина и монокаприлат пропиленгликоля. Другой класс состоит из моностеарата полиэтиленгликоля и глицерина. В одном варианте осуществления масло, используемое в соответствии с настоящим изобретением, представляет собой кунжутное масло.
Количество масла, которое может присутствовать в депо-препарате, может составлять от около 5% масс. до около 95% масс., от около 25% масс. до около 75% масс. или от около 35% до около 60% по отношению к общей массе депо-препарата.
В некоторых вариантах осуществления соотношение масла к фосфолипиду в депо-препарате может находиться в интервале от около 20:1 до около 1:20, от около 3:1 до около 1:3, или от около 1:2 около 1:1 в пересчете на массу.
Фосфолипид
Фосфолипид в соответствии с настоящим изобретением относится к липидной молекуле, содержащей одну или более фосфатных групп, включая липидные молекулы на основе либо глицерина (фосфоглицериды, глицерофосфолипиды) или сфингозина (сфинголипиды).
В некоторых вариантах осуществления фосфолипиды представляют собой производные триглицеридов, в которых одна жирная кислота была заменена на фосфатную группу и одну из нескольких азотсодержащих молекул. Цепи жирных кислот являются гидрофобными, и заряды на фосфатных группах и аминогруппах делают соответствующую часть молекулы гидрофильной. Результатом является амфифильная молекула.
Согласно Фармакопеи США (United States Pharmacopoeia (USP)), понятие "лецитин" представляет собой непатентованное название, описывающее сложную смесь нерастворимых в ацетоне фосфолипидов, которые состоят, в основном, из фосфатидилхолина, фосфатидилэтаноламина, фосфатидилсерина и фосфатидилинозитола, в сочетании с различными количествами других веществ, таких как триглицериды, жирные кислоты и углеводы. Препарат лецитина и, следовательно, его физические свойства различаются в зависимости от источника лецитина и фосфолипидного препарата, например, содержания фосфатидилхолина, и т.д.
В соответствии с вариантом осуществления настоящего изобретения, лецитины для целей настоящего изобретения представляют собой фармацевтические лецитины, полученные из яиц или сои, которые были использованы в парентеральных продуктах и, по существу, свободны от раздражающих, аллергенных, воспалительных агентов или агентов, которые вызывают другие неблагоприятные биологические реакции.
В соответствии с практикой настоящего изобретения, выбор фосфолипидов для получения депо-препарата определяется на основе способности фосфолипидов: (1) быть химически совместимыми с по меньшей мере одним гидрофильным водорастворимым фармацевтически активным веществом, выбранным из группы, состоящей из ванкомицина, гентамицина и их смеси, (2) образовывать монофазный раствор и поддерживать малый размер капель во время процесса производства и во время хранения, и (3) обеспечивать получение желаемого депо-препарата и желаемое высвобождение фармацевтически активного вещества.
Примеры фосфолипидов включают, но не ограничиваются ими, сфинголипиды в виде сфингозина и производные (полученные из сои, яйца, мозга и молока), ганглиозиды и фитосфингозин и производные (полученные из дрожжей).
Фосфолипиды также могут быть синтезированы, и примеры общеизвестных синтетических фосфолипидов включают, но не ограничиваются ими, диглицериды, такие как 1,2-дилауроил-sn-глицерин (DLG), 1,2-димиристоил-sn-глицерин (DMG), 1,2-дипальмитоил-sn-глицерин (DPG), 1,2-дистеароил-sn-глицерин (DSG); фосфатидные кислоты, такие как 1,2-димиристоил-sn-глицеро-3-фосфатидная кислота, натриевая соль (DMPA, Na), 1,2-дипальмитоил-sn-глицеро-3-фосфатидная кислота, натриевая соль (DPPA, Na), 1,2-дистеароил-sn-глицеро-3-фосфатидная кислота, натриевая соль (DPPA, Na), фосфохолины, такие как 1,2-дидеканоил-sn-глицеро-3-фосфохолин (DDPC), 1,2-дилауроил-sn-глицеро-3-фосфохолин (DLPC), 1,2-димиристоил-sn-глицеро-3-фосфохолин (DMPC), 1,2-дипальмитоил-sn-глицеро-3-фосфохолин (DPPC), 1,2-дистеароил-sn-глицеро-3-фосфохолин (DSPC), 1,2-диолеоил-8П-глицеро-3-фосфохолин (DOPC), 1,2-дилинолеоил-sn-глицеро-3-фосфохолин (DLOPC), 1,2-диэрукоил-sn-глицеро-3-фосфохолин (DEPC), 1,2-диэйкосапентаноил-sn-глицеро-3-фосфохолин (ЕРА-РС), 1,2-дидокосагексанил-sn-глицеро-3-фосфохолин (DHA-PC), 1-миристоил-2-пальмитоил-sn-глицеро-3-фосфохолин (МРРС), 1-миристоил-2-стеароил-sn-глицеро-3-фосфохолин (MSPC), 1-пальмитоил-2-миристоил-sn-глицеро-3-фосфохолин (РМРС), 1-пальмитоил-2-стеароил-sn-глицеро-3-фосфохолин (PSPC), 1-стеароил-2-миристоил-sn-глицеро-3-фосфохолин (SMPC), 1-стеароил-2-пальмитоил-sn-глицеро-3-фосфохолин (SPPC), 1-миристоил-2-олеоил-sn-глицеро-3-фосфохолин (МОРС), 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфохолин (РОРС), 1-стеароил-2-олеоил-sn-глицеро-3-фосфохолин (РОРС), фосфоэтаноламины, такие как гидрогенизированный соевый фосфоэтаноламин (HSPE), негидрированный яичный фосфоэтаноламин (ЕРЕ), 1,2-дилауроил-sn-глицеро-3-фосфоэтаноламин (DLPE); 1,2-димиристоил-sn-глицеро-3-фосфоэтаноламин (DMPE); 1,2-дипальмитоил-sn-глицеро-3-фосфоэтаноламин (DPPE), 1,2-дистеароил-sn-глицеро-3-фосфоэтаноламин (DSPE); 1,2-диолеоил-sn-глицеро-3-фосфоэтаноламин (DOPE), 1,2-дилинолеоил-sn-глицеро-3-фосфоэтаноламин (DLoPE); 1,2-диэруцил-sn-глицеро-3-фосфоэтаноламин (DEPE), 1,2-пальмитоил-sn-глицеро-3-фосфоэтаноламин (POPE); фосфоглицерины, такие как гидрогенизированный соевый фосфатидилглицерин, натриевая соль (HSPG, Na), негидрированный яичный фосфатидилглицерин, натриевая соль (EPG, Na), 1,2-дилауроил-sn-глицеро-3-фосфоглицерин, натриевая соль (DLPG, Na), 1,2-димиристоил-sn-глицеро-3-фосфоглицерин, натриевая соль (DMPG, Na), 1,2-димиристоил-sn-глицеро-3-фосфо-SN-1-глицерин, аммониевая соль (DMP-Sn-1-G, NH4), 1,2-дипальмитоил-sn-глицеро-3-фосфоглицерин, натриевая соль (DPPG, Na), 1,2-дистеароил-sn-глицеро-3-фосфоглицерин, натриевая соль (DSPG, Na), 1,2-дистеароил-sn-глицеро-3-фосфо-sn-1-глицерин, натриевая соль (DSP-SN-1G, Na), 1,2-диолеоил-sn-глицеро-3-фосфоглицерин, натриевая соль (DOPG, Na), 1,2-диэруцил-sn-глицеро-3-фосфоглицерин, натриевая соль (DEPG, Na), 1,2-пальмитоил-sn-глицеро-3-фосфоглицерин, натриевая соль (POPG, Na); фосфотидилсерины, такие как 1,2-димиристоил-sn-глицеро-3-фосфо-L-син, натриевая соль (DMPS, Na), 1,2-дипальмитоил-sn-глицеро-3-фосфо-L-син, натриевая соль (DPPS, Na), l,2-дистеарил-sn-глицеро-3-фосфо-L-син, натриевая соль (DSPS, Na), 1,2-диолеоил-sn-глицеро-3-фосфо-L-син, натриевая соль (DOPS, Na), 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфо-L-син, натриевая соль (POPS, Na); смешанные цепочечные фосфолипиды, такие как 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфохолин (РОРС), 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфоглицерин, натриевая соль (POPG, Na), 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфоглицерин, соль аммония (POPG, NH4); лизофосфолипиды, такие как 1-миристоил-2-лизоз-глицеро-3-фосфохолин (S-лизо-РС), 1-пальмитоил-2-лизо-8П-глицеро-3-фосфохолин (Р-лизо-РС), 1 -стеароил-2-лизо-sn-глицеро-3-фосфохолин (S-лизо-РС), и пегилированные фосфолипиды, такие как N-(карбонил-метоксиполиэтиленгликоль 2000)-MPEG-2000-DPPE, 1,2-дипальмитоил-sn-глицеро-3-фосфоэтаноламин, натриевая соль, N-(карбонил-метоксиполиэтиленгликоль 5000), MPEG-5000-DSPE, 1-2-дистеароил-sn-глицеро-3-фосфоэтаноламин, натриевая соль, N-(карбонил-метоксиполиэтиленгликоль 5000), MPEG-5000-DPPE, 1,2-дипальмитоил-sn-глицеро-3-фосфоэтаноламин, натриевая соль, N-(карбонил-метоксиполиэтиленгликоль 750) MPEG-750-DSPE, 1,2-дистеароил-sn-глицеро-3-фосфоэтаноламин, натриевая соль, N-(карбонил-метоксиполиэтиленгликоль 2000)-MPEG-2000-DSPE, 1,2-дистеароил-sn-глицеро-3-фосфоэтаноламин, натриевая соль.
Количество фосфолипидов, которые могут присутствовать в депо-препарате, может меняться в зависимости от ряда параметров, включая вязкость конечной композиции, продолжительность применения препарата, размер депо-препарата и обстоятельства его применения, тип активного вещества, которое будет вводиться, тип введения (например, непрерывное, с задержкой, и т.д.) и тому подобное. Однако обычно, количество фосфолипида, которое может присутствовать в депо-препарате, может составлять от около 5% до около 95% по отношению к общей массе композиции или от около 35% до около 60% по отношению к общей массе композиции.
Вода
Вода, которая может быть использована в соответствии с настоящим изобретением, включает, но не ограничивается ими, дистиллированную и деионизированную воду или любую другую водную жидкость, которая способна растворять гидрофильные водорастворимые ванкомицин и/или гентамицин и способна к сублимации/испарению во время стадии лиофилизации.
Для того чтобы получить монофазный раствор, например, используя микрофлюидизатор высокого давления, эмульсия масло-в-воде может содержать от около 50% до около 90% воды, от около 60% до около 80% воды или от около 70% до около 80% воды по отношению к общей массе эмульсии масло-в-воде, чтобы обладать требуемым свойством текучести, необходимым для обработки в гомогенизаторе, таком как микрофлюидизатор.
Однако как только получен монофазный раствор, большая часть воды может быть удалена, например, лиофилизацией, сублимацией и/или испарением.
Ванкомицин распадается в результате гидролиза, и количество остаточной воды в конечном депо-препарате влияет на долгосрочную стабильность ванкомицина. Когда ванкомицин преципитирует, депо-препарат становится из полупрозрачного мутным или разделяется на две фазы, как показано в примере 3 настоящего документа.
Таким образом, в соответствии с настоящим изобретением, количество остаточной воды должно поддерживаться на уровне менее чем около 4%, менее чем около 2% масс. или менее чем около 0,5% масс. воды по отношению к общей массе вязкого прозрачного депо-препарата в целях поддержания стабильности ванкомицина во время хранения.
Агент, регулирующий pH
Агент, регулирующий pH, в соответствии с настоящим изобретением представляет собой любую нетоксичную кислоту, основание или соль. Примеры агентов, регулирующих значение pH, включают, но не ограничиваются ими, соляную кислоту, уксусную кислоту, серную кислоту, гидроксид натрия, гидроксид калия, гидроксид аммония, лизин, аргинин и тому подобное.
Как уже упоминалось выше, гентамицин распадается из-за окисления или образования аддукта. Как показано ниже в примере 4 настоящего документа, значение pH влияет на долгосрочную стабильность гентамицина, и когда гентамицин преципитирует, депо-препарат становится из полупрозрачного мутным.
Соответственно, значение pH депо-препарата может быть от около 3 до около 6, в интервале от около 3 до около 5 или от около 3 до около 4.
Стабилизирующий агент
Стабилизирующий агент в соответствии с настоящим изобретением представляет собой материал, который уменьшает каталитическое действие иона металла, оказываемое на окисление, гидролиз или другие реакции распада и/или повышает устойчивость гидрофильного водорастворимого фармацевтически активного вещества. Примеры такого стабилизирующего агента включают, но не ограничиваются ими, ЭДТА (динатриевая соль), глицин, L-гистидин, лимонную кислоту, метионин, аскорбиновую кислоту, L-цистеин, альфа-токоферол и их смеси. В некоторых вариантах осуществления количество стабилизирующего агента, присутствующего в депо-препарате, составляет от около 0,001% до около 5,0% по отношению к общей массе композиции или от около 0,01% до около 1,0% по отношению к общей массе композиции. В другом варианте осуществления депо-препарат не содержит стабилизирующего агента.
Модификатор вязкости
Модификатор вязкости в соответствии с настоящим изобретением представляет собой водную или неводную (за исключением содержащую воду на уровне примеси) жидкость, которая способна растворять сухую пасту, образующуюся после лиофилизации, сублимации и/или испарения.
Примеры модификатора вязкости включают, в частности, этанол, изопропанол и их смеси. В одном варианте осуществления модификатор вязкости является, по существу, неводным. В другом варианте осуществления модификатор вязкости представляет собой этанол.
Модификатор вязкости добавляют в сухую пасту до тех пор, пока сухая паста не растворится полностью в данном веществе. В результате изменения вязкости раствор может также стать "мутным". В одном варианте осуществления модификатор вязкости и сухую пасту смешивают при температуре от около 10°С до около 80°С или в диапазоне от около 50°С до около 70°С или в диапазоне от около 25°С до около 60°С.
Модификатор вязкости добавляют в сухую пасту до тех пор, пока количество модификатора вязкости не составит около 10% масс., 20% масс., 25% масс. или 30% масс. по отношению к общей массе полученного раствора. В результате вязкость раствора может составлять от около 10 до около 200 сантипуаз, от около 15 до около 100 сантипуаз или около 20 сантипуаз до около 50 сантипуаз.
Вязкость можно определить с помощью цифрового программируемого реометра Brookfield со шпинделем SP-40 или любого другого эквивалентного реометра. В частности, начальное число оборотов в минуту (RPM) реометра может составлять от около 0,1 до около 1,0, с последующим уменьшением RPM до 0,1 путем изменения RPM на 0,1 каждые 30 секунд. Измерение вязкости может осуществляться при 0,8 RPM при температуре окружающей среды, равной около 30°С.
Впоследствии некоторое количество модификатора вязкости, используемое для растворения сухой пасты, может быть удалено. Удаление модификатора вязкости может проводиться до тех пор, пока остаточное количество модификатора вязкости, которое может присутствовать в депо-препарате, не составит от около 1% масс. до около 20% масс., от около 2% масс. до около 18% масс. или от около 5% масс. до около 6,5% масс. по отношению к общей массе депо-препарата. При пересушивании модификатор вязкости может быть добавлен обратно по мере необходимости.
Вязкость полученного депо-препарата в соответствии с настоящим изобретением составляет от около 100 сантипуаз до около 5000 сантипуаз, от около 200 сантипуаз до около 2000 сантипуаз или от около 300 сантипуаз до около 1500 сантипуаз.
Способ лечения
Еще одна задача настоящего изобретения относится к способу введения, внутрикожно, внутримышечно, через разрез, подкожно, инстилляцией или через местное введение, депо-препарата по настоящему изобретению, содержащего ванкомицин, гентамицин, их фармацевтически приемлемую соль и их смесь, воду, фосфолипид и масло, необязательно агент, регулирующий pH, и модификатор вязкости. Депо-препарат можно дозировать в желаемом месте с использованием различных доз и в различных интервалах дозирования в зависимости от необходимости. То есть депо-препарат должен быть достаточным для высвобождения фармацевтически активного вещества в течение около по меньшей мере одного дня с объемом дозирования от около 0,1 мл до около 100 мл. Например, могут быть использованы интервалы дозирования один раз в день, один раз каждый второй день, один раз каждый третий день, один раз в неделю или один раз в месяц с объемом дозирования от около 0,1 мл до около 100 мл. Как правило, депо-препарат может быть использован в одном применении и обычно инсталлируется в место раны до зашивания раны.
Еще одна задача настоящего изобретения относится к способу профилактики и/или лечения инфекции, включая, в частности, хирургические инфекции, содержащему введение депо-препарата по настоящему изобретению, тканевые концентрации которого достигают значений, достаточно высоких для лечения и/или профилактики инфекции в локальном месте, не токсичных для почек и/или других органов и не способствующих появлению лекарственно-устойчивых штаммов бактерий.
Еще одна задача настоящего изобретения относится к способу придания локализованной ткани способности не поддерживать патогенные микроорганизмы путем введения депо-препарата по настоящему изобретению в рану.
В другом варианте осуществления это достигается, не вызывая токсичности для почек и/или других органов и не вызывая и/или способствуя появления(ю) лекарственно-устойчивых штаммов бактерий.
В каждом из вышеизложенных способов доза ванкомицина, гентамицина или обоих веществ должна быть такой, чтобы при высвобождении из депо-препарата локализованная ткань не была способна поддерживать патогенные бактерии в течение по меньшей мере 24 часов и в другом варианте осуществления по меньшей мере 48 часов. В еще одном варианте осуществления локализованная ткань не способна поддерживать патогенные бактерии в течение по меньшей мере 3 дней, в течение по меньшей мере одной недели или в течение одного месяца.
Как показано на фиг.10 и в соответствии с опубликованными данными, минимальная ингибирующая концентрация, необходимая для подавления роста 90% организмов (MIC90 (мкг/мл)), для известных патогенных микроорганизмов, вызывающих хирургические инфекции (surgical site infection (SSI), таких как золотистый стафилококк Staphylococcus aureus, коагулазонегативные стафилококки, энтерококки, синегнойная палочка Pseudomonas aeruginosa и кишечная палочка Escherichia coli, находится в диапазоне от 1 до 4 мкг/мл. В общем случае см. М.J. Rybak, et al., Vancomycin Therapeutic Guidelines, CID 2009: 49 (1 August), 325-327; и A.I. Hidron, et al., Infection and Hospital Epidemiology, November 2008, vol. 29, No. 11, 996-1011. Приведенные данные основаны на использовании ванкомицина и гентамицина индивидуально, как показано на фигуре. Когда депо-препарат по настоящему изобретению, содержащий ванкомицин (4,37 мг/кг) и гентамицин (3,89 мг/кг), вводили свинье, локализованная концентрация ванкомицина в ткани свиньи, достигаемая депо-препаратом по настоящему изобретению, составляла более 19 мкг/мл через 48 часов, что в рамках научных представлений, больше, чем MIC90 для вышеуказанных возбудителей SSI. Аналогично, концентрация ванкомицина в ткани свиньи, достигаемая введением депо-препарата по настоящему изобретению, составляла около 12 мкг/мл через 48 часов.
В связи с этим следует отметить, что авторы не заражали свиней указанными выше возбудителями SSI и не вводили затем депо-препарат в локальное место, чтобы определить эффективность данного изобретения. Тем не менее, опубликованные данные по MIC90 для указанных выше возбудителей SSI и достигаемая тканевая концентрация препарата по настоящему изобретению, содержащего ванкомицин и гентамицин, показывают, что применение депо-препарата по настоящему изобретению было бы весьма эффективным в обеспечении локализованных уровней лекарственных препаратов, эффективных для лечения и/или. профилактики инфекции путем превращения локализованной ткани в не способную поддерживать патогенные микроорганизмы.
Из фиг.11 видно, что депо-препарат по настоящему изобретению при введении через разрез свиньям обеспечивает высокие локальные концентрации в тканях обоих активных веществ, ванкомицина и гентамицина, и низкие их системные концентрации (в плазме). Низкие системные концентрации гентамицина, наблюдаемые с использованием депо-препарата по настоящему изобретению, обеспечивают значительный запас безопасности, поскольку токсичность гентамицина (почечная и ототоксичность), как известно, связана с концентрациями в плазме, составляющими более 10 мг/л. См. в целом, D.S. Reeves, Infection 8 (1980) Suppl. 3, S 313-S320.
Почечная токсичность ванкомицина также связана с чрезмерным воздействием лекарственного препарата, но не может быть легко соотнесена с определенной пиковой концентрацией. Однако низкие системные концентрации, наблюдаемые при использовании введения депо-препарата по настоящему изобретению, показали отсутствие системной токсичности на модели свиньи для каждого из данных лекарственных препаратов и находятся явно значительно ниже опубликованных значений для гентамицина.
Вторая проблема, связанная с ванкомицином, представляет собой развитие у бактерий устойчивости к этому препарату. Устойчивость к ванкомицину может развиваться, когда ткани-мишени или вспомогательные ткани, колонизируемые бактериями, подвергаются действию недостаточно эффективных концентраций ванкомицина в течение значительных периодов времени. После системного введения только ванкомицина желаемые минимальные плазменные концентрации в стационарном состоянии составляют по меньшей мере 10 мг/л или могут находиться в диапазоне от около 15 мг/л до около 20 мг/л. Учитывая низкое поглощение ванкомицина тканями из крови, данные концентрации являются достаточными, чтобы добиться установления терапевтически эффективных концентраций в ткани и достигнуть терапевтического эффекта. Концентрации в крови служат в качестве суррогатного маркера для тканей-мишеней, причем целью является достижение соотношения AUC0-t (площадь под фармакокинетической кривой (area under the curve) "концентрация/время")/MIC90 в плазме, составляющего от около 400 до около 300,000, или соотношения AUC0-t/MIC90, составляющего более 400, или отношения AUC0-t/MIC90, составляющего более 1600. Данное соотношение достигается, когда минимальные концентрации поддерживаются на уровнях, указанных выше.
С депо-препаратом по настоящему изобретению, очень низкие концентрации ванкомицина наблюдаются в циркулирующей плазме, в то время как концентрации в месте разреза превышают терапевтические концентрации при введении геля, как показано на фиг.10. Учитывая низкий уровень поглощения ванкомицина из плазмы в ткань, низкие системные концентрации ванкомицина приводят к незначительным уровням ванкомицина в тканях, удаленных от места разреза. Единственным механизмом транспорта является транспорт через плазму и количества, поглощаемые тканями из плазмы, являются небольшими. Таким образом, вероятность развития устойчивости к ванкомицину на участках, отдаленных от места разреза, должна быть очень низкой.
Таким образом, другая задача настоящего изобретения относится к способу лечения пациента, содержащему введение указанному пациенту терапевтически эффективной дозы ванкомицина отдельно или в комбинации с гентамицином или их фармацевтически приемлемыми солями так, что для ванкомицина достигается отношение AUC0-t/MIC90 в плазме, равное более 400, чтобы предотвратить появление устойчивости к золотистому стафилококку S. aureus. Еще одна задача настоящего изобретения относится к пациенту, которому препарат вводится как указано выше и у которого наблюдается 1/10 от минимального значения стационарной концентрации в сыворотке крови, что позволяет полностью избежать нефротоксичности, проявляющейся при введении высокой дозы ванкомицина обычными способами. См. в целом, М.J. Rybak, Vancomycin Therapeutic Guidelines, CID 2009: 49 (1 August), 325-327.
Примеры
Пример 1. Депо-препарат в соответствии с настоящим изобретением
Сначала в стакан емкостью 500 мл добавили 0,36 г сульфата гентамицина, 0,24 г ванкомицина гидрохлорида, 53,3 г PL90G, 40 г кунжутного масла и 0,1 г L-гистидина. К этому добавили воду для инъекций (Water for Injection (WFI)), и смесь гомогенизировали в смесителе с большим усилием сдвига при 5000 RPM в течение 15 мин. Полученный монофазный раствор лиофилизировали, чтобы удалить воду для получения сухой пасты с менее чем 0,2% остаточной влажности.
Пример 2. Влияние содержания воды на внешний вид примера 1
Данную сухую пасту смешивали с водой и/или этанолом для образования раствора с измененной вязкостью и использовали в нескольких исследованиях, включая примеры 2-5, как указано ниже в данном документе.
Различные количества воды (от 1,1% масс. до 4,1% масс.) и этанола (при 6% масс.) были добавлены в сухую пасту примера 1 для получения нескольких образцов. Образцы тщательно смешивали смесителем BeadBeater, центрифугировали для удаления пузырьков воздуха и затем немедленно подвергали визуальной инспекции ("Начальный образец"), Кроме того, образцы пропускали через фильтр с диаметром пор, равным 0,45 мкм, и фильтраты хранили при температуре от 2°С до 8°С для дополнительной визуальной инспекции ("Фильтрованный образец"). В таблице 2 показано влияние содержания воды на внешний вид препаратов. Было установлено, что содержание воды существенно влияет на внешний вид препаратов.
Пример 3. Влияние содержания воды на стабильность гентамицина и ванкомицина
Влияние остаточного содержания воды на стабильность гентамицина и ванкомицина препаратов примера 1 оценивали автоклавной обработкой в течение 60 мин. Как представлено в таблице 3 ниже, было установлено, что ванкомицин обладал пониженной стабильностью, оценивавшейся по остаточному количеству или чистоте, при более высоком уровне остаточной воды. Не наблюдалось существенного влияния воды на стабильность гентамицина при варьировании содержания воды в том же диапазоне.
Пример 4. Профили pH-стабильности и pH-растворимости гентамицина и ванкомицина в препарате примера 1
Препараты с подведенными значениями pH из примера 1 инкубировались при температуре от 2°С до 8°С для визуальной инспекции (см. таблицу 4 ниже).
Профиль pH-стабильности получали нагреванием образцов из примера 1 в ходе 60-минутной автоклавной обработки (см. таблицу 5 ниже).
Данные результаты показали, что:
(1) значение pH влияет на внешний вид препаратов примера 1. Препарат был прозрачен при значении pH, равном 3,2;
(2) значение pH влияет на стабильность гентамицина в препарате. Низкое значение pH (например, значение pH в интервале от 3 до 4) является предпочтительным для стабильности гентамицина; и
(3) значение pH не оказывает значительного влияния на стабильность ванкомицина.
Пример 5. Профиль pH-стабильности гентамицина в препарате из примера 1 при значении pH, равном от 3,0 до 5,5
Были приготовлены образцы препарата примера 1 при трех различных значениях pH, равных от 3,0 до 5,5. Кроме того, было также проверено влияние L-гистидина на стабильность препарата примера 1 путем сравнения препарата, содержащего L-гистидин, с таковыми, которые не содержат L-гистидин. Стабильность гентамицина и ванкомицина оценивалась таким же образом, как указано в примере 3. Было установлено, что
(1) Стабильность гентамицина в препарате зависит от pH (гентамицин предпочитал низкое значение pH (например, значение pH, равное от 3 до 4));
(2) Стабильность ванкомицина в препарате является менее pH-чувствительной в изученном диапазоне значений pH;
(3) L-гистидин увеличивал стабильность гентамицина в изученном диапазоне значений pH; и
(4) L-гистидин снижал стабильность ванкомицина в изученном диапазоне значений pH.
Фиг.2 представляет собой график, показывающий аналитически определенное остаточное количество после автоклавной обработки.
Пример 6. Другой депо-препарат в соответствии с настоящим изобретением и процесс приготовления препарата
Прозрачный желтый стерильный депо-препарат (размер партии: 1500 г), который содержал менее 0,5% масс. остаточной воды, имеющий значение pH, равное 3,3, был приготовлен многостадийным процессом, содержащим следующие стадии: (1) эмульгирование, (2) гомогенизация/микрофлюидизация, (3) лиофилизация, (4) разведение этанолом, (5) предварительная фильтрация, (6) удаление этанола и (7) фильтрация. Простое смешивание всех ингредиентов, перечисленных выше, не приводит к образованию прозрачного депо-препарата.
Подробные процедуры для каждой из вышеизложенных стадий заключаются в следующем: во-первых, к сульфату гентамицина, гидрохлориду ванкомицина добавляли воду, чтобы обеспечить полное растворение сульфата гентамицина и гидрохлорида ванкомицина. Затем добавляли Фосфолипон® 90G (от Phospholipid GmbH) и кунжутное масло с последующим смешиванием с большим усилием сдвига при 5000 оборотах в минуту в течение 60 минут до получения однородной первичной эмульсии. Затем значение pH первичной эмульсии доводили до значения, равного 3,3±0,2 добавлением IN HCl. Это достигалось добавлением соответствующего количества IN HCl к эмульсии с последующим смешиванием с большим усилием сдвига в течение 1 минуты. Затем измеряли значение pH для того, чтобы удостовериться, что первичная эмульсия имеет значение pH, равное 3,3±0,2.
Впоследствии первичную эмульсию помещали в микрофлюидизатор для получения монофазного раствора. Средний диаметр капель монофазного раствора измеряли с помощью устройства, регистрирующего рассеяния света лазера.
Затем монофазный раствор лиофилизировали, чтобы удалить воду для получения сухой пасты с менее чем 0,5% остаточной воды. Затем сухую пасту смешивали с дегидратированным спиртом. Затем смесь обрабатывали ультразвуком на водяной бане при 60-70°С до получения прозрачного раствора (с измененной вязкостью). Затем раствор охлаждали до комнатной температуры и предварительно фильтровали через стерильный фильтр с диаметром пор 0,65 мкм.
Затем спирт из раствора удаляли продувкой азотом, пока остаточное количество дегидратированного спирта не составило от 6,5% масс. до 7% масс., чтобы получить вязкий и прозрачный гель. Дегидратированный алкоголь добавляли обратно по мере необходимости, если гель был пересушенным.
В боксе биологической безопасности применяли аргон при 40 psi (фунтов на квадратный дюйм), чтобы профильтровать депо-препарат через фильтр с диаметром пор 0,2 мкм для стерилизации препарата. Затем в боксе биологической безопасности профильтрованным депо-препаратом наполняли стеклянный флакон.
Пример 7. Профиль высвобождения in vitro
Профиль высвобождения in vitro препарата примера 6, содержащего гентамицин и ванкомицин, измеряли с помощью фармакопейного метода USP I с использованием "корзинки" (basket apparatus) (100 оборотов в минуту при 37°С). Капсулу размера 000 наполняли препаратом примера 6 в количестве 1,36 г и заполненную капсулу помещали в корзинку с ячейками 40 меш с перегородками. Фиг.3 представляет собой график, показывающий профиль высвобождения in vitro гентамицина и ванкомицина препарата примера 6 с использованием фармакопейного метода USP I.
Пример 8. Фармакокинетические исследования на кроликах Новозеландские белые кролики были использованы для проведения фармакокинетических (pharmacokinetic "PK") исследований для оценки доставки препаратов в соответствии с настоящим изобретением. Два препарата были приготовлены в соответствии с процедурами, изложенными в примере 1 и примере 6, соответственно, и вводились в хирургическую рану или в подкожный карман. Дизайн PK исследования на кролике "Rabbit PK study" представлен более подробно в приведенной ниже таблице 7.
В первом эксперименте были протестированы два новозеландских белых кролика. После инстилляции в рану препарата примера 1 ванкомицин (Ванко) и гентамицин (Гента) были быстро абсорбированы со временем достижения плазменной максимальной концентрации (Tmax), равным от 1 до 2 часа. Максимальные плазменные концентрации Cmax были подобны тем, которые наблюдаются у мышей. Плазменные концентрации снизились практически до предела их количественного определения за 36 часов. Тканевые концентрации ванкомицина достигли пикового значения через 72 часов и были выше минимальной ингибирующей концентрации (Minimum Inhibitory Concentration (MIC)) через 168 часов, как представлено на фиг.4 и 5.
Тканевые концентрации гентамицина достигли пикового значения через 72 часа и находились на уровне MIC или ниже MIC через 168 часов, как представлено на фиг.6 и 7.
Анализ плазмы и тканей проводился способом жидкостной хроматографии/масс-спектрометрии (LC-MS/MS), и фармакокинетические (PK) результаты препарата примера 1 приведены в таблицах 8 и 9, соответственно, ниже.
Во втором эксперименте шесть новозеландских кроликов (группа I) были протестированы инстилляцией в рану препарата примера 6, содержащего дозу, составлявшую 12,6 мг/кг ванкомицина и 11,46 мг/кг гентамицина; и шесть дополнительных новозеландских кроликов (II группа) были протестированы инстилляцией в рану препарата примера 6, содержащего дозу, составлявшую 25,2 мг/кг ванкомицина и 22,9 мг/кг гентамицина.
В случае геля более низкой концентрации (группа I) средняя концентрация в месте раны составляла всего 4 мкг/г для обоих ванкомицина и гентамицина, в то время как в случае геля более высокой концентрации (II группа) средняя концентрация составляла 26 мкг/г ванкомицина и 19,4 мкг/г гентамицина, соответственно, что превышало более чем в четыре раза значения минимальных ингибирующих концентраций MIC (minimum inhibitory concentration). Плазменные концентрации ванкомицина и гентамицина из исследования MPI (myocardial perfusion imaging) препарата примера 6 показали соотношения AUC/MIC (площадь под концентрационной кривой/минимальная ингибирующая концентрация) для обеих доз ванкомицина, превышающие 400, и значения соотношений Cmax/MIC (максимальная концентрация/минимальная ингибирующая концентрация) для обеих доз гентамицина, превышающие 800.
Фиг.8 представляет собой график, показывающий средние плазменные концентрации ванкомицина у кроликов после однократной подкожной (subcutaneous (SC)) инстилляции в рану, и фиг.9 представляет собой график, показывающий среднюю плазменную концентрацию гентамицина у кроликов.
Анализ плазмы и тканей был проведен с помощью анализа LC-MS/MS, и РК результаты препарата примера 6 приведены в таблице 10 ниже
Основное различие между препаратом примера 6 (высокой силы) и препаратом примера 1 (низкой силы) заключается в том, что хотя РК профили данных двух препаратов, полученные для небольших животных, таких как мыши, были похожи, имелось большое различие в производительности при испытании на более крупных животных, таких как кролики, поскольку уровень тканевых концентраций препарата примера 1 упал ниже значения, превышающего в 4 раза значение MIC, быстрее, таким образом описывая меньшую площадь под фармакокинетической кривой концентрация/время (AUC), по отношению к соотношению время/площадь превышая данный параметр в 4 раза по сравнению с MIC.
Сравнительный пример 1
Сравнительный пример 1 был получен с использованием тех же способов, что и примеры 1 и 6, за исключением того, что не проводились стадии гомогенизации, удаления этанола и/или предварительной фильтрации.
Сравнительный пример 1 образовывал непрозрачную твердую пасту после лиофилизации и не был прозрачным и не фильтровался после добавления модификатора вязкости (этанола).
Пример 9. Данный пример дополнительно иллюстрирует процесс изготовления депо-препарата по настоящему изобретению.
Сульфат гентамицина, гидрохлорид ванкомицина, PL90G и кунжутное масло и воду добавляли в стакан, перемешивали и гомогенизировали в смесителе с большим усилием сдвига при 500 оборотов в минуту в течение 30 минут для получения первичной эмульсии. Значение pH первичной эмульсии доводили до 3,3 посредством 1 N HCl.
Микрофлюидизатор (М-110ЕН, Microfluidics Corp) был применен для уменьшения размера капель первичной эмульсии. Рабочее давление было создано при 25000 psi (фунтов на квадратный дюйм). После 6 пассажей размер капли (Z-Ave) монофазного раствора составлял менее 80 нм по данным, полученным изменением рассеивания света лазера (Nano-ZS, Malvem). Значение pH монофазного раствора проверяли и доводили до 3,3 по мере необходимости.
Монофазный раствор переносили в контейнер из нержавеющей стали с высотой заполнения менее 3 см и затем лиофилизировали для удаления воды до содержания остаточной воды в количестве менее 1% (титрованием по методу Карла Фишера), чтобы получить сухую пасту. После лиофилизации сухую пасту собирали в стакан емкостью 2 л. Дегидратированный спирт добавляли в пасту до конечной концентрации 25% (масс./масс.). Смесь растворяли перемешиванием при комнатной температуре для образования прозрачного желтого раствора.
Прозрачный раствор выпаривали, чтобы уменьшить содержание спирта, продувкой газом азотом для получения вязкого и прозрачного депо-препарата с 6%-ным спиртом (масс./масс.). Затем депо-препарат стерилизовали пропусканием через два фильтра SARTOPORE® с диаметром пор 0,2 мкм.
Сравнительный пример 2. Препарат, полученный без стадии микрофлюидизации.
Первичную эмульсию готовили таким же образом, как описано в примере 9. Данную первичную эмульсию дополнительно встряхивали в течение ночи или гомогенизировали дополнительным перемешиванием с большим усилием сдвига при 5000 оборотов в минуту в течение 2 часов, а затем лиофилизировали таким же образом, как описано в примере 9. Для данного примера не использовали стадию микрофлюидизации. После лиофилизации в пасту добавляли дегидратированный спирт так, что количество дегидратированного спирта составляло около 25% (масс./масс.). Полученная смесь не была прозрачной даже после перемешивания или нагрева в течение длительного периода времени. Данный процесс не мог быть продолжен, поскольку не был получен прозрачный или фильтруемый раствор.
Сравнительный пример 3. Препарат, полученный с использованием для лиофилизации контейнера не из нержавеющей стали.
Первичную эмульсию и монофазный раствор готовили таким же образом, как описано в примере 9. Наноэмульсию лиофилизировали таким же образом, как описано в примере 9, за исключением того, что вместо контейнера из нержавеющей стали был использован стеклянный контейнер. После лиофилизации в сухую пасту добавляли дегидратированный спирт так, что количество дегидратированного спирта составляет около 25% (масс./масс.) по отношению к общей массе полученного раствора с измененной вязкостью. Полученная смесь не была прозрачной даже после перемешивания или нагрева в течение длительного периода времени. Данный процесс не мог быть продолжен, поскольку не был получен прозрачный или фильтруемый раствор.
Сравнительный пример 4. Препарат, приготовленный без добавления дегидратированного спирта до количества, составляющего около 25% (масс./масс.).
Первичную эмульсию, монофазный раствор и сухую пасту готовили таким же образом, как описано в примере 9. После лиофилизации в пасту добавляли дегидратированный спирт так, что количество дегидратированного спирта составляло около 6% (масс./масс.) по отношению к общей массе полученного раствора с измененной вязкостью, а не 25% масс./масс. Полученная смесь была мутной и непрозрачной даже после перемешивания или нагрева в течение длительного периода времени.
Как уже упоминалось выше, прозрачность раствора измеряется по внешнему виду, например, по тому, что он свободен от определяемых визуально взвешенных частиц, и раствор промежуточного продукта пропускает более чем около 90% света, измеряемого при 800 нм (Т800) в кварцевой кювете с длиной оптического пути 1 см, используя спирт в качестве бланка (пробы сравнения), при измерении с помощью УФ-видимого спектрофотометра, такого, как спектрофотометр, произведенный Pharmacia, модель Ultrospec III.
Примеры 10А-10F
Пример 10А, указанный выше, был приготовлен в соответствии со способом по настоящему изобретению.
Пример 10В, указанный выше, был также приготовлен в соответствии со способом по настоящему изобретению.
Пример 10С, указанный выше, был также приготовлен в соответствии со способом по настоящему изобретению, без содержания любого гидрофильного водорастворимого фармацевтически активного вещества.
Пример 10D был приготовлен смешиванием препарата примера 10С с сульфатом гентамицина и гидрохлоридом ванкомицина. Пример 10D, следовательно, не был приготовлен в соответствии со способом по настоящему изобретению.
Пример 10Е был приготовлен смешиванием препарата примера 10С только с сульфатом гентамицина. Пример 10Е, следовательно, не был приготовлен в соответствии со способом по настоящему изобретению.
Пример 10F был приготовлен без стадии микрофлюидизационной стадии. Соответственно, пример 10F также не был приготовлен в соответствии с настоящим изобретением. Пропуск стадии флюидизации привел к преципитации в депо-препарате. Полученный депо-препарат, следовательно, не был прозрачным.
Пример 11. Структурная характеристика примеров 10A-10F методом малоугловой дифракции рентгеновских лучей (small angle X-ray diffraction (SAXS))
Процедура: Данные анализа малоуглового рассеяния рентгеновских лучей (SAXS) были собраны в гелиевой камере с использованием генератора с вращающимся анодом (Bruker M18XHF22), работающего при 50 кВ и 50 мА, дающего пучок излучения CuKa (λ=1,541838 Å), который был коллимирован с помощью однощелевого коллиматора (типа "пинхоул", pinhole). Излучение КВ отфильтровывали Ni-фильтром. Для сбора данных был использован многопроволочный детектор Highstar. Образцы загружали без изменения в капилляры из боросиликатного стекла, имеющие диаметр, равный 0,9 мм, и заливали эпоксидной смолой. Образцы устанавливали в гелиевой камере на расстоянии от автоматизированного гониометра на образце до детектора, равном 64,55 см. Чтобы предотвратить рассеяние от воздуха, в камеру в течение 1 часа продували гелиевый газ, и затем спектр каждого образца был собран в течение 7200 секунд. Полученные данные были сглажены и интегрированы в угловом диапазоне 360°χ, в области углов рассеяния 29, равной от 0,8 до 4,7°, с угловым шагом, составляющим 0,1 и 0,02 градуса. Дифрактограммы были сопоставлены, и для уточнений положений пиков были использованы интегрирования с угловым шагом, составляющим 0,1 градуса.
Результаты: фиг.12 представляет собой дифрактограммы малоуглового рассеяния рентгеновских лучей (SAXS) для примеров 10A-10F. Дифрактограммы содержат два различных типа дифракционных пиков. На дифрактограммах примеров 10А, 10В и 10F наблюдается при малых значениях углов дифракционный пик при около 2 Theta (градусы), и на дифрактограммах двух физических смесей (примеры 10D и 10Е) и носителя депо-препарата без активного вещества (пример 10С) наблюдается гораздо более широкий дифракционный пик при около 2,5 Theta (градусы).
На дифрактограммах препаратов, полученных с использованием способа по настоящему изобретению (примеры 10А и 10 В), наблюдался уникальный дифракционный пик SAXS, образуемый при около 2 Theta (градусы), который не был найден в случае примера 10С или физических смесей носителя депо-препарата с теми же препаратами (примеры 10D и 10Е). Пример 10С характеризовался меньшим параметром решетки, чем примеры 10А, 10В и 10F.
Когда гентамицин и ванкомицин включены в депо-препарат в соответствии с настоящим изобретением, имеется равное приблизительно от 8 до 9 Å увеличение параметра решетки, приводящее к образованию такой уникальной структуры (далее именуемой "2-Theta структура"),
Параметры решетки для первичного дифракционного пика дифрактограмм двух физических смесей (примеры 10D и 10Е) соответствовали таковому для первичного дифракционного пика дифрактограммы носителя депо-препарата (пример 10С), что указывает на то, что физическое смешивание носителя депо-препарата с гентамицином и ванкомицином не меняет структуру носителя.
Только после того, как ванкомицин и/или гентамицин включаются в носитель депо-препарата с помощью способа по настоящему изобретению, образуется 2-Theta структура.
Это ясно указывает на то, что композиции по настоящему изобретению имеют уникальную 2-Theta структуру и такая структура может быть получена только с помощью способа приготовления по настоящему изобретению.
Наличие пониженной интенсивности дифракции при 2 Theta (градусы), наблюдаемой для примера 10F, дает основания предполагать, что в препарате, полученном без стадии микрофлюидизации, частично существует "2-Theta структура".
Вывод: композиции по настоящему изобретению, примеры 10А и 10В, содержат уникальную особую 2-Theta структуру.
Пример 12. Структурная характеристика примеров 10А и 10D методом термогравиметрического анализа (Thermal Gravimetric Analysis (TGA))
Процедура: TGA-эксперименты проводились на измерительной ячейке TG/DTA 220 (Seiko Instruments). Калибровки по температуре и энтальпии были проведены с использованием индия и олова в качестве стандартов. Сканирование было выполнено с шагом подъема температуры 10°С/мин в диапазоне температур от 25°С до 300°С, со скоростью продувки азотом 80 мл/мин, в открытых тиглях и с массой образца от 5 до 10 мг.
Результаты: Как показано на фиг.13, результаты TGA показали небольшое различие в кривой профиля потери массы между примером 10А, который был приготовлен в соответствии со способом настоящего изобретения, и примером 10D, который не был приготовлен в соответствии со способом настоящего изобретения.
Пример 13. Структурная характеристика примеров 10А и 10D методом дифференциальной сканирующей калориметрии (DSC)
Процедура: DSC-эксперименты проводились с использованием прибора DSC 120 single cell Modulated DSC с RSC (refrigerated cooling) ячейкой (Seiko Instruments). Прибор DSC был откалиброван по температуре и константе ячейки с использованием индия в качестве стандарта. Сканирование проводилось в обычном режиме DSC с шагом подъема температуры 10°С/мин, в закрытых тиглях, со скоростью продувки азотом 40 мл/мин, с массами от 5 до 10 мг, используемыми для каждого образца. Сканирование было выполнено в диапазоне температур от 25°С до 300°С.
Результаты: Как показано на фиг.14, кривые DSC-профилей для обоих образцов (примеры 10А и 10D) характеризуются основным эндотермическим процессом, протекающим в интервале до около 100°С, что, вероятно, связано с десольватацией образцов. Кривая DSC-профиля для примера 10D, однако, содержит дополнительный эндотермический пик при температуре около 80°С, возможно, обусловленный плавлением твердого лекарственного препарата, т.е. сульфата гентамицина и/или гидрохлорида ванкомицина.
Хотя изобретение было описано в настоящем документе со ссылкой на определенные варианты осуществления, следует понимать, что данные варианты осуществления являются лишь иллюстрацией принципов и применений по настоящему изобретению. Поэтому следует понимать, что в иллюстративные варианты осуществления могут быть внесены многочисленные изменения, и что могут быть разработаны другие механизмы без отступления от сущности и объема настоящего изобретения, как определено прилагаемой формулой изобретения.
Реферат
Изобретение относится к медицине и заключается в способе получения депо-препарата, способ включает образование эмульсии масло-в-воде, включающей воду, фосфолипид, масло и ванкомицин и/или гентамицин; гомогенизацию; микрофлюидизацию; обеспечение рН от 3 до 6; лиофилизацию для получения сухой пасты; добавление этанола или изопропанола в количестве 25 мас.% или более от массы полученного раствора; удаление этанола или изопропанола для получения депо-препарата, содержащего от 1 до 20 мас.% этанола или изопропанола по отношению к массе депо-препарата, и стерилизацию. Депо-препарат содержит не более 4 мас.% воды и предназначен для введения внутрикожно, внутримышечно, подкожно, через разрез или местно. Изобретение касается также депо-препаратов, содержащих ванкомицин и/или гентамицин, и способов их введения. Технический результат заключается в стабильности при хранении, в возможности введения подкожно, внутримышечно или помещением в рану, а также в более длительном высвобождении активного агента. 7 н. и 39 з.п. ф-лы, 13 пр., 11 табл., 14 ил.
Формула
(1) образование эмульсии масло-в-воде, включающей в себя фосфолипид, масло, по меньшей мере одно гидрофильное водорастворимое фармацевтически активное вещество, выбранное из группы, состоящей из ванкомицина, гентамицина, их фармацевтически приемлемой соли или их смеси, и воду;
(2) гомогенизацию эмульсии для получения первичной эмульсии;
(3) обработку первичной эмульсии с помощью микрофлюидизатора для получения монофазного раствора;
(4) обеспечение того, чтобы значение рН первичной эмульсии и/или монофазного раствора находилось в интервале от около 3 до около 6, подводя значение рН по мере необходимости,
(5) лиофилизацию монофазного раствора в лотке из нержавеющей стали для получения сухой пасты,
(6) добавление модификатора вязкости в сухую пасту в количестве, составляющем около 25 мас.% или более от общей массы полученного раствора с измененной вязкостью,
(7) удаление по меньшей мере некоторого количества модификатора вязкости для получения депо-препарата, содержащего от около 1 до около 20 мас.% модификатора вязкости по отношению к общей массе депо-препарата, и
(8) стерилизацию депо-препарата,
в котором модификатор вязкости выбран из группы, состоящей из этанола и изопропанола, где масло выбрано из группы, состоящей из растительного масла, животного масла и их смесей; в котором вода, присутствующая в депо-препарате, составляет не больше, чем около 4 мас.% относительно общей массы депо-препарата и указанный депо-препарат предназначен для введения внутрикожно, внутримышечно, подкожно, через разрез или местно.
(1) образование эмульсии масло-в-воде, включающей в себя фосфолипид, масло, по меньшей мере два гидрофильных водорастворимых фармацевтически активных вещества, выбранных из группы, состоящей из ванкомицина, гентамицина, их фармацевтически приемлемой соли или их смеси, и воду;
(2) гомогенизацию эмульсии для получения первичной эмульсии;
(3) обработку первичной эмульсии с помощью микрофлюидизатора для получения монофазного раствора;
(4) обеспечение того, чтобы значение рН первичной эмульсии и/или монофазного раствора находилось в интервале от около 3 до около 6, подводя значение рН по мере необходимости,
(5) лиофилизацию монофазного раствора в лотке из нержавеющей стали для получения сухой пасты,
(6) добавление модификатора вязкости в сухую пасту в количестве, составляющем около 25 мас.% или более от общей массы полученного раствора с измененной вязкостью,
(7) удаление по меньшей мере некоторого количества модификатора вязкости для получения депо-препарата, содержащего от около 1 до около 20 мас.% модификатора вязкости по отношению к общей массе депо-препарата, и
(8) стерилизацию депо-препарата, который является прозрачным,
в котором модификатор вязкости выбран из группы, состоящей из этанола и изопропанола, где масло выбрано из группы, состоящей из растительного масла, животного масла и их смесей, в котором вода присутствует в депо-препарате составляет не больше чем около 4 мас.% относительно общей массы депо-препарата и указанный депо-препарат предназначен для введения внутрикожно, внутримышечно, подкожно, через разрез или местно.
Комментарии