Фармацевтические композиции для доставки пептидов с замедленным высвобождением - RU2456018C2
Код документа: RU2456018C2
Чертежи



Описание
РОДСТВЕННЫЕ ЗАЯВКИ
В настоящей заявке заявлен приоритет предварительной заявки США №60/830011, поданной 11 июля 2006 года, содержание которой, таким образом, включено в эту заявку путем ссылки.
В этой заявке присутствуют ссылки на различные публикации. Описание этих публикаций, таким образом, включено в эту заявку путем ссылки с целью более полного описания уровня техники, к которому относится настоящее изобретение.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
1. Область изобретения
Это изобретение относится к области доставки терапевтических пептидов с контролируемым высвобождением и к композициям и способам, полезным для доставки терапевтических пептидов с контролируемым высвобождением, которые ковалентно модифицированы одной или более липофильными или амфифильными молекулами.
2. Описание уровня техники, относящегося к настоящему изобретению.
Пептиды, иначе называемые олигопептиды, полипептиды и протеины, имели широкое применение в качестве терапевтических агентов. Пептиды могут быть легко получены по технологии рекомбинантной ДНК или могут быть синтезированы по хорошо изученной технологии пептидного синтеза. Однако многие пептиды чувствительны к ферментативному разрушению и имеют очень короткий период полупревращения in vivo в кровообращении. Поэтому большинство пептидных медикаментов вводили путем инъекции, обычно многократной в течение дня. Было бы чрезвычайно выгодно, если бы такие пептиды с целью улучшения безопасности, эффективности и соблюдения больным режима и схемы лечения можно было бы доставлять контролируемым способом за продолжительные периоды времени.
Для замедленной доставки терапевтических пептидов применяли биодеградируемые полимеры. Пептид обычно включают в полимерную композицию и вне организма придают ей желаемые формы, такие как палочки, пластинки и микрочастицы. Эти твердые композиции затем можно вводить в организм путем надреза или инъекции. Альтернативно и предпочтительно, с целью формирования имплантата in situ некоторые полимерные композиции можно впрыскивать в организм в виде жидкой полимерной композиции. Инъецируемые жидкие биодеградируемые полимерные композиции для формирования имплантатов in situ с целью контролируемой доставки лекарственных средств описаны в патентной литературе. Полагают, что следующие ниже ссылки являются репрезентативными в этой области и включены в данное описание изобретения путем ссылки: патенты США под номерами 6565874; 6528080; RE37, 950; 6461631; 6395293; 6355657; 6261583; 6143314; 5990194; 5945115; 5792469; 5780044; 5759563; 5744153; 5739176; 5736152; 5733950; 5702716; 5681873; 5599552; 5487897; 5340849; 5324519; 5278202; 5278201 и 4938763. В них описано, что с целью обеспечения жидкой композиции биоактивный агент растворяют или диспергируют в растворе биодеградируемого полимера в биосовместимом органическом растворителе. Когда жидкую композицию впрыскивают в организм, растворитель рассеивается в окружающей водной среде, и осаждается полимер с формированием твердого или желеобразного депо, из которого биоактивный агент высвобождается в течение продолжительного периода времени, пока разрушается полимер. Применение такой системы доставки было показано на примере доставки ацетата лейпролида для лечения развившегося рака предстательной железы (Eligard™). Несмотря на некоторый успех, эти методы были не вполне удовлетворительными для большого количества пептидов, чтобы их можно было эффективно доставить таким способом.
У многих терапевтических пептидов во время процесса высвобождения было обнаружено ацилирование и/или разрушение пептидов, инкапсулированных в микросферы поли(DL-лактид-со-гликолида) [например, Na DН, Youn YS, Lee SD, Son МО, Kim WA, DeLuca PP, Lee KC. J Control Release. 2003; 92 (3): 291-9]. Нуклеофильные функциональные группы пептидов могут не только взаимодействовать с биодеградируемым полимером, но также могут катализировать разрушение биодеградируемого полимера. Также было установлено, что ацилирование и/или разрушение могли бы происходить намного быстрее в растворе полимера, чем в твердом состоянии. Например, когда ацетат октреотида смешивали с раствором 50/50 поли(DL-лактид-со-гликолида), имеющего концевую карбоксигруппу, в NMP (N-метил-2-пирролидон), более чем 80% октреотида ацилировалось и/или разрушалось в течение 24 часов. Взаимодействие/реакция между пептидом и полимером или продуктами его разрушения может происходить во время приготовления композиции, хранения и введения. Поэтому для сохранения стабильности композиций пептид обычно поставляют в отдельном шприце, в то время как остальные компоненты упаковывают в другой шприц. Содержимое шприцев смешивают непосредственно перед применением. Однако из-за вязкой природы полимерных композиций, конечным пользователям часто трудно смешать содержимое двух отдельных шприцев. Однородность композиций, приготовленных конечным пользователем, может значительно варьироваться, также может произойти загрязнение, и, таким образом, качество лечения может подвергаться значительной опасности. Более того, формирование твердого имплантата in situ из инъецируемой жидкой полимерной композиции является медленным процессом. Обычно процесс рассеивания/диффузии может занять от нескольких часов до нескольких суток или даже дольше в зависимости от используемого растворителя. В течение этого периода присутствие органического растворителя может способствовать взаимодействию/реакции между пептидом и полимером или продуктами его разрушения.
Кроме того, во время формирования имплантата скорость диффузии пептида из коагулирующей полимерной композиции может быть намного более высокой, чем скорость высвобождения, которое имеет место из сформированного впоследствии твердого имплантата. Первоначальное "взрывное" высвобождение пептида во время формирования имплантата может привести к потере или высвобождению большого количества терапевтических пептидов. Если пептид является, в частности, токсичным или имеет узкое терапевтическое окно, то это первоначальное высвобождение или "взрыв", вероятно, приведет к токсичным побочным эффектам и может повредить соседние ткани. Поэтому медленный процесс формирования твердого имплантата и нестабильность биоактивных агентов и/или эксципиентов является очень существенной проблемой для применения композиций этого типа для доставки терапевтических пептидов с замедленным высвобождением.
Было описано, что ковалентная модификация пептидов липофильными молекулами, такими как жирные кислоты, улучшает терапевтическую эффективность путем увеличения периода полупревращения в кровообращении in vivo посредством связывания с альбумином [ЕР 0708179-А2, ЕР 0699686-А2, US 6268343, Knudsen LB, Nielsen PF, Huusfeldt PO, Johansen NL, Madsen K, Pedersen FZ, Thogersen H, Wilken M, Agerso H. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem. 2000, 43 (9): 1664-9; Kurtzhals P, Havelund S, Jonassen I, Kiehr B, Larsen UD, Ribel U, Markussen J. Albumin binding of insulins acylated with fatty acids: characterization of the ligand-protein interaction and correlation between binding affinity and timing of the insulin effect in vivo. Biochem J. 1995; 312 (3): 725-31, и ссылки, приведенные там]. Хотя липофильно модифицированные пептиды показали пролонгированное действие in vivo по сравнению с чистыми пептидами, время пребывания в плазме модифицированных пептидов ограничено их аффинностью связывания с альбумином. Удачным примером является ацилированный инсулин (Detemir), который имеет период полупревращения в кровообращении 10,2±1,2 ч [Havelund S, Plum A, Ribel U, Jonassen I, Vølund A, Markussen J and Kurtzhals P, Pharmaceutical Research, 2004; 21 (8), 1498-1504]. Этот продукт был одобрен для инъекции пациентам, которые больны диабетом типа I. Однако его все еще необходимо вводить пациентам ежедневно. Поэтому сохраняется значительная потребность в стабильной композиции, с помощью которой можно контролировать более легко скорость доставки некоторых пептидов, особенно пептида, который требует замедленного высвобождения в течение длительного периода времени.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Неожиданно было обнаружено, что может быть приготовлена композиция с пептидами, ковалентно модифицированными одной или более липофильными и/или амфифильными молекулами, и биодеградируемыми полимерами, приводящая в результате к значительно улучшенной стабильности и профилям замедленного высвобождения относительно неконъюгированных пептидов. Липофильно и/или амфифильно модифицированные пептиды могли не только предотвратить неконтролируемое случайное ацилирование и разрушение пептидов во время приготовления композиции, хранения и последующих процессов высвобождения in vivo, но также могли уменьшить нежелательное первоначальное "взрывное" высвобождение пептидов. Такие системы доставки предусматривают более высокие концентрации терапевтического пептида, который должен быть надежно включен в систему доставки с биодеградируемым полимером. Также улучшается эффективность таких продуктов, так как намного более высокий процент неповрежденного активного пептида остается в системе доставки для замедленного высвобождения и не теряется посредством разрушения во время приготовления композиции, хранения и последующего высвобождения in vivo.
Соответственно, в настоящем изобретении предложены новые фармацевтические композиции для контролируемого замедленного высвобождения терапевтических пептидов. Композиции по настоящему изобретению содержат: (а) пептид, который ковалентно конъюгирован с одной или более липофильной(ыми) молекулой(ами); (б) биодеградируемый полимер и (в) фармацевтически приемлемый органический растворитель. Пептиды ковалентно конъюгируют с одной или более липофильной(ыми) молекулой(ами) таким образом, что конъюгированный пептид сохраняет большую часть биологических активностей или все биологические активности неконъюгированного пептида, несмотря на то что обладает повышенной стойкостью к химическому взаимодействию с биодеградируемым полимером как in vitro, так и in vivo относительно неконъюгированного пептида. Затем приготавливают композицию с липофильно модифицированным пептидом путем растворения или диспергирования в растворе биодеградируемого полимера с использованием фармацевтически приемлемого органического растворителя. Композиции по настоящему изобретению не только повышают стабильность пептида во время приготовления композиции, хранения, введения и последующего высвобождения, но также улучшают профили его высвобождения по причине более низких первоначального "взрыва" уровней и длительного периода.
В другом аспекте настоящего изобретения предложена композиция, содержащая: (а) пептид, который ковалентно конъюгирован с одной или более амфифильными группировками; (б) биодеградируемый полимер и (в) фармацевтически приемлемый органический растворитель. Пептиды ковалентно конъюгируют с одной или более амфифильными группировками таким образом, что конъюгированный пептид сохраняет большую часть биологических активностей или все биологические активности неконъюгированного пептида, несмотря на то что обладает повышенной стойкостью к химическому взаимодействию с биодеградируемым полимером как in vitro, так и in vivo относительно неконъюгированного пептида. Затем приготавливают композицию с амфифильно модифицированным пептидом путем растворения или диспергирования в растворе биодеградируемого полимера с использованием фармацевтически приемлемого органического растворителя. Композиции по настоящему изобретению не только повышают стабильность пептида во время приготовления композиции, хранения, введения и последующего высвобождения, но также улучшают профили его высвобождения по причине более низких первоначального "взрыва" уровней и длительного периода. Конъюгированный пептид также уменьшает разрушение полимера, катализируемое нуклеофильными группами пептида.
Каждая из композиций по настоящему изобретению может представлять собой вязкую или невязкую жидкость, гель или полутвердое вещество, которое перемещается как жидкость, так что его можно впрыскивать с использованием шприца. Каждая композиция может быть получена в виде имплантируемой полимерной матрицы in vitro или, альтернативно, она может быть образована in situ в форме геля или твердого имплантата. Композиции можно вводить инъекцией и/или имплантацией подкожным, внутримышечным, внутрибрюшинным или внутрикожным путем. Контролируемое высвобождение пептида при введении субъекту может быть продлено на желательный период времени в зависимости от состава имплантата. Путем подбора биодеградируемого полимера и других эксципиентов можно регулировать продолжительность замедленного высвобождения пептида в течение периода времени от нескольких недель до одного года.
Различные признаки новизны, характеризующие это изобретение, подробно указаны в прилагаемой и составляющей часть описания формуле изобретения. Для лучшего понимания этого изобретения, его функциональных преимуществ и конкретных целей, достигаемых в результате его применения, следует обратиться к графическому и текстовому материалу, где проиллюстрированы и описаны предпочтительные воплощения этого изобретения.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фиг.1. Высвобождение in vitro лизоцима и лизоцима, ацилированного пальмитиновой кислотой, из композиций в растворе RG503H в mPEG350 (метокси-полиэтиленоксигликоль с молекулярной массой 350).
Фиг.2. Высвобождение in vitro грелина и дезацетилированного грелина из композиций в растворе RG503H в mPEG350.
Фиг.3. Высвобождение in vitro октреотида из композиции в растворе DLPLG85/15 [поли(DL-лактид-со-гликолид) при соотношении лактида и гликолида 85/15] (IV 0,28) в NMP.
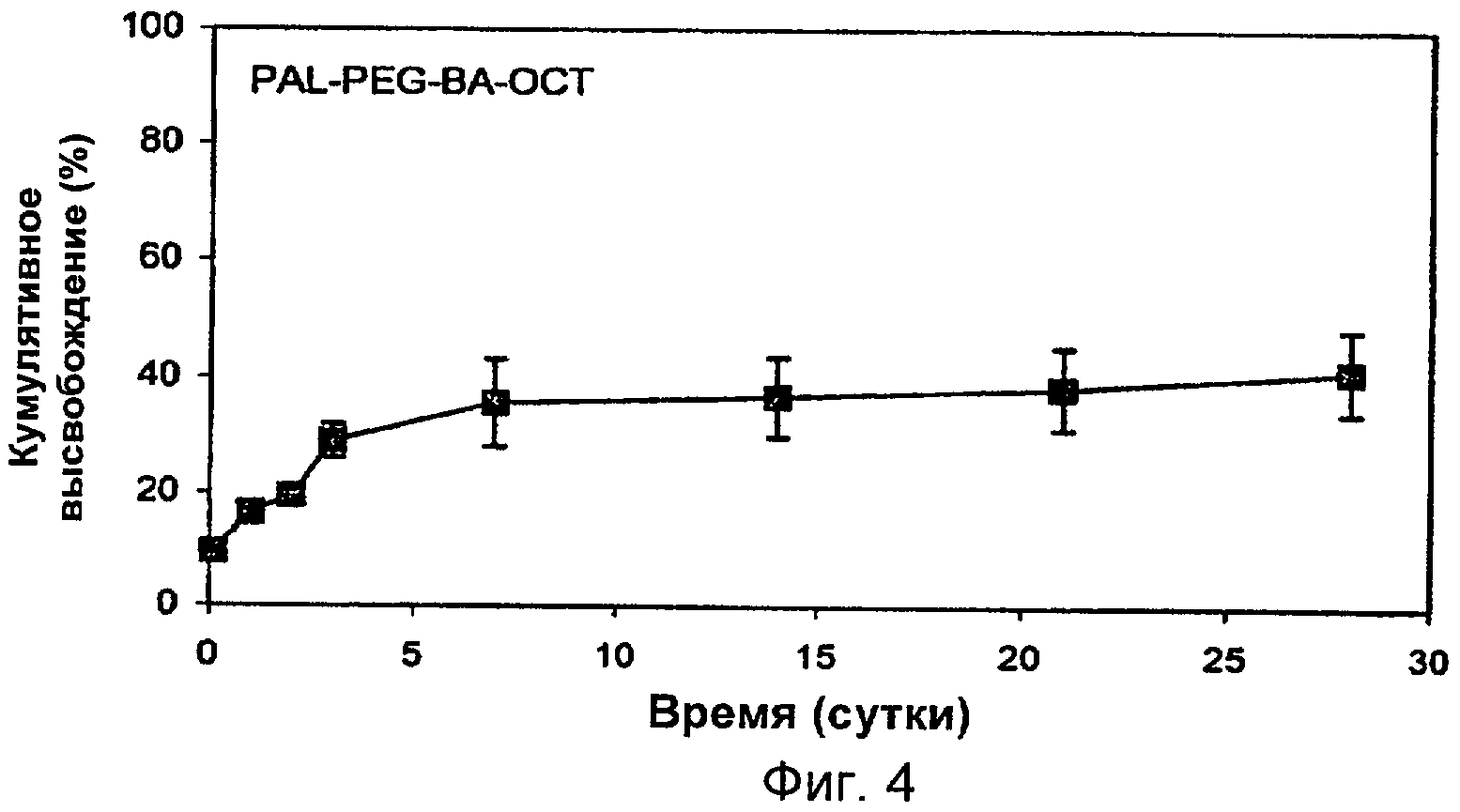
Фиг.4. Высвобождение in vitro модифицированного октреотида (Pal-PEG-ВА-ОСТ) из композиции в растворе DLPLG85/15 (IV 0,28) в NMP.
ПОДРОБНОЕ ОПИСАНИЕ ВОПЛОЩЕНИЙ, ПРЕДПОЧТИТЕЛЬНЫХ В НАСТОЯЩЕЕ ВРЕМЯ
В настоящем изобретении предложены инъецируемые жидкие биодеградируемые полимерные композиции для формирования системы доставки пептидов с контролируемым высвобождением. В настоящем изобретении также предложен способ их изготовления и способ их применения.
Композиции по настоящему изобретению содержат: (а) пептид, который конъюгирован, предпочтительно ковалентно, с одной или более липофильными молекулами; (б) фармацевтически приемлемый, не растворимый в воде, биодеградируемый полимер и (в) фармацевтически приемлемый органический растворитель. Пептиды ковалентно конъюгируют с одной или более липофильными молекулами таким образом, что конъюгированный пептид сохраняет большую часть биологических активностей или все биологические активности неконъюгированного пептида, несмотря на то что обладает повышенной стойкостью к химическому взаимодействию с биодеградируемым полимером как in vitro, так и in vivo относительно неконъюгированного пептида. Затем приготавливают композицию с липофильно модифицированным пептидом путем растворения или диспергирования в растворе биодеградируемого полимера с использованием фармацевтически приемлемого органического растворителя. Композиции по настоящему изобретению не только повышают стабильность пептида во время приготовления композиции, хранения, введения и последующего высвобождения, но также улучшают профили его высвобождения по причине более низких первоначального "взрыва" уровней и длительного периода.
В другом аспекте настоящего изобретения предложена композиция, содержащая: (а) пептид, который конъюгирован, предпочтительно ковалентно, с одной или более амфифильными молекулами; (б) фармацевтически приемлемый, нерастворимый в воде, биодеградируемый полимер и (в) фармацевтически приемлемый органический растворитель. Пептиды ковалентно конъюгируют с одной или более амфифильными молекулами таким образом, что конъюгированный пептид сохраняет большую часть биологических активностей или все биологические активности неконъюгированного пептида, несмотря на то что обладает повышенной стойкостью к химическому взаимодействию с биодеградируемым полимером как in vitro, так и in vivo относительно неконъюгированного пептида. Затем приготавливают композицию с амфифильно модифицированным пептидом путем растворения или диспергирования в растворе биодеградируемого полимера с использованием фармацевтически приемлемого органического растворителя. Композиции по настоящему изобретению не только повышают стабильность пептида во время приготовления композиции, хранения, введения и последующего высвобождения, но также улучшают профили его высвобождения по причине более низких первоначального "взрыва" уровней и длительного периода. Конъюгированный пептид также уменьшает разрушение полимера, катализируемое нуклеофильными группами пептида.
В настоящей заявке термины, указывающие на неопределенное количество, и термин "один" следует понимать как "один или более" и "по меньшей мере один".
Термин "пептид" является синонимом по отношению к терминам "полипептид" и "белок". Неограничивающие примеры терапевтических свойств, которыми может обладать пептид, включают антиметаболические, противогрибковые, противовоспалительные, противоопухолевые, противоинфекционные, антибиотические, питательные, агонистические и антагонистические свойства.
Более конкретно, пептиды по настоящему изобретению могут быть ковалентно модифицированы липофильной(ыми) или амфифильной(ыми) молекулой(ами). Пептиды предпочтительно содержат одну или более чем одну модифицируемую функциональную группу. Пептиды, полезные в приготовлении композиций по изобретению, включают окситоцин, вазопрессин, адренокортикотропный гормон (АСТН), эпидермальный фактор роста (EGF), тромбоцитарный фактор роста (PDGF), пролактин, гормоны, такие как лютеинизирующий гормон, рилизинг-фактор лютеинизирующего гормона (LHRH), агонисты LHRH, антагонисты LHRH, гормоны роста, рилизинг-фактор гормона роста, инсулин, эритропоэтин, соматостатин, глюкагон, интерлейкин, интерферон-альфа, интерферон-бета, интерферон-гамма, гастрин, тетрагастрин, пентагастрин, урогастрон, секретин, кальцитонин, энкефалины, эндорфины, ангиотензины, рилизинг-фактор тиреотропина (TRH), фактор некроза опухолей (TNF), паратиреоидный гормон (РТН), фактор роста нервной ткани (NGF), гранулоцитарный колониестимулирующий фактор (G-CSF), гранулоцитарно-макрофагальный колониестимулирующий фактор (GM-CSF), макрофагальный колониестимулирующий фактор (M-CSF), гепариназу, фактор роста эндотелия сосудов (VEG-F), костный морфогенетический белок (BMP), hANP (предсердный натрийуретический пептид человека), глюкагоноподобный пептид (GLP-1), экзенатид, пептид YY (PYY), грелин, ренин, брадикинин, бацитрацины, полимиксины, колистины, тироцидин, грамицидины, циклоспорины, ферменты, цитокины, антитела, вакцины, антибиотики, гликопротеины, фолликулостимулирующий гормон, киоторфин, тафтсин, тимопоэтин, тимозин, тимостимулин, гуморальный фактор тимуса, сывороточный фактор тимуса, колониестимулирующие факторы, мотилин, бомбезин, динорфин, нейротензин, церулеин, урокиназу, калликреин, аналоги и антагонисты вещества Р, ангиотензин II, факторы свертывания крови VII и IX, лизоцим, грамицидин, меланоцитстимулирующий гормон, рилизинг-фактор тиреоидного гормона, тиреостимулирующий гормон, панкреозимин, холецистокинин, плацентарный лактоген человека, хорионический гонадотропин человека, стимулирующий синтез белка пептид, желудочный ингибиторный пептид, вазоактивный пептид кишечника, тромбоцитарный фактор роста, пигментные гормоны, соматомедин, хорионический гонадотропин, гипоталамический рилизинг-фактор, антидиуретические гормоны, тиреостимулирующий гормон, бифалин и пролактин, но не ограничиваются ими.
Термин "липофильная группировка", используемый в данном описании изобретения, относится к любой группировке, обладающей липофильными свойствами и имеющей растворимость в воде при 20°С менее чем 5 мг/мл, предпочтительно менее чем 0,5 мг/мл. Такую липофильную группировку обычно выбирают из С3-39-алкильных, С3-39-алкенильных, С3-39-алкадиенильных, токоферольных и стероидных остатков. Термины "С3-39-алкил", "С3-39-алкенил" и "С3-39-алкадиенил" предназначены для схватывания прямоцепочечного и разветвленного, предпочтительно прямоцепочечного, насыщенного, мононенасыщенного и диненасыщенного углеводорода из 3-39 атомов углерода.
Ковалентная конъюгация липофильной группировки с пептидом приводит к липофильно модифицированному пептиду, который может обладать улучшенным терапевтическим эффектом по сравнению с чистым пептидом. Обычно ее можно достигнуть путем взаимодействия функциональной группы, такой как аминная группа, в пептиде с кислотной или другими реакционноспособными группами в липофильной молекуле. Альтернативно, конъюгацию между пептидом и липофильной молекулой осуществляют через дополнительную группировку, такую как мостиковая, спейсерная или связывающая группировка, которая может быть деградируемой или недеградируемой. Некоторые примеры раскрыты в предшествующем уровне техники [например, ацилированные жирными кислотами инсулины описаны в заявке на японский патент 1254699; см. также: Hashimoto, M., et al., Pharmaceutical Research, 6: 171-176 (1989) и Lindsay, D. G., et al., Biochemical J., 121: 737-745 (1971)]. Кроме того, описания ацилированных жирными кислотами инсулинов и жирно-ацилированных инсулиновых аналогов и способов их синтеза находятся в патенте США №5693609, WO 95/07931, патенте США №5750497 и WO 96/29342. Дополнительные примеры ацилированных пептидов находятся в WO 98/08871, WO 98/08872 и WO 99/43708. В описаниях этих документов раскрыты липофильно модифицированные пептиды, и эти документы делают возможным получение таких пептидов.
Термин "амфифильная группировка", используемый в данном описании изобретения, относится к любой группировке, обладающей как липофильными, так и гидрофильными свойствами и растворимой как в воде, так и в липофильных растворителях. Амфифильные молекулы, применяемые в настоящем изобретении, состоят из липофильных и гидрофильных группировок. Липофильные группировки предпочтительно представляют собой природные жирные кислоты или алкильные цепи и такие, которые описаны выше. Гидрофильные группировки выбирают из полиэтиленгликоля, поливинилпирролидона, сахара и тому подобного. Гидрофильные группировки предпочтительно представляют собой полиэтиленгликоль (PEG), имеющий менее 1000 этиленгликолевых единиц. Размер и состав липофильных группировок и гидрофильных группировок можно корректировать с целью получения желаемой амфифильности.
Термины "конъюгированный", "соединенный", "связанный" и тому подобные, используемые в данном описании изобретения со ссылкой на пептид и другие компоненты модифицированного пептида по настоящему изобретению, означают, что определенные группировки связаны друг с другом, предпочтительно ковалентно, через линкер, мостик, спейсер или тому подобное.
Термины "линкер", "мостик", "спейсер" или тому подобные, используемые в данном описании изобретения, относятся к атому или группе атомов, которые связывают, предпочтительно ковалентно, например, липофильную группировку с терапевтическим пептидом.
Для осуществления ковалентной конъюгации терапевтический пептид может иметь одну или более подходящих функциональных групп или может быть модифицирован с целью включения одной или более подходящих функциональных групп для ковалентного сочетания с липофильной или амфифильной группировкой. Подходящие функциональные группы включают, например, следующие группы: гидроксильную группу, аминогруппу (первичную аминогруппу или вторичную аминогруппу), тиольную группу и карбоксильную группу. Липофильные или амфифильные группировки по настоящему изобретению могут иметь одну или более подходящих функциональных групп или могут быть модифицированы с целью включения одной или более подходящих функциональных групп для ковалентного сочетания с пептидом. Подходящие функциональные группы включают, например, следующие группы: гидроксильную группу, аминогруппу (первичную аминогруппу или вторичную аминогруппу), тиольную группу, карбоксильную группу, альдегидную группу, изоцианатную группу, группу сульфоновой кислоты, группу серной кислоты, группу фосфорной кислоты, группу фосфоновой кислоты, аллилгалогенидную группу, бензилгалогенидную группу, замещенную бензилгалогенидную группу и оксиранильную группу.
Терапевтический пептид можно непосредственно или косвенно сочетать с одной или более липофильными группировками через сложноэфирную группу, амидную группу, вторичную или третичную аминную группу, карбаматную группу, сульфонатную группу, сульфатную группу, фосфатную группу, фосфонатную группу или простую эфирную группу.
В одном воплощении настоящего изобретения пальмитиновую кислоту активировали N-гидроксисукцинимидом, а затем подвергали взаимодействию с аминными группами на октреотиде, октапептиде, с образованием конъюгата через амидный линкер между пальмитильной липофильной группировкой и пептидом. Октреотид содержит две первичные аминные группы. Обе аминные группы можно было конъюгировать одновременно, или путем подбора реакционных условий можно было селективно конъюгировать только одну аминную группу с последующим разделением.
В другом воплощении деканаль, липофильное соединение с концевой альдегидной группой, подвергали взаимодействию с аминными группами на октреотиде с образованием конъюгата через вторичную аминную связь. Обе аминные группы можно было конъюгировать одновременно, или путем подбора реакционных условий можно было конъюгировать только одну аминную группу с последующим разделением.
В дополнительном воплощении пальмитиновую кислоту конъюгировали с лизоцимом через его шесть аминных групп в нескольких соотношениях. Если отношение пальмитиновой кислоты к лизоциму составляет менее 6, то места конъюгации на лизоциме могут быть случайными в зависимости от реакционной способности каждой аминной группы.
В еще одном воплощении грелин представляет собой пептид, ацилированный через его гидроксильную группу с н-октаноильной группировкой. Грелин представляет собой желудочный пептид, который стимулирует секрецию гормона роста и увеличивает ожирение. Он является первым идентифицированным природным лигандом для ранее клонированного рецептора стимулятора секреции гормона роста, присутствующего в гипофизе и гипоталамической области головного мозга.
Липофильную группировку сначала можно ковалентно сочетать с гидрофильной группировкой с образованием амфифильной молекулы. Амфифильные молекулы по настоящему изобретению могут иметь одну или более подходящих функциональных групп или могут быть модифицированы с целью приобретения одной или более подходящих функциональных групп для ковалентного сочетания с пептидом. Подходящие функциональные группы выбирают из гидроксильной группы, аминогруппы (первичной аминогруппы или вторичной аминогруппы), тиольной группы, карбоксильной группы, альдегидной группы, изоцианатной группы, группы сульфоновой кислоты, группы серной кислоты, группы фосфорной кислоты, группы фосфоновой кислоты, аллилгалогенидной группы, бензилгалогенидной группы, замещенной бензилгалогенидной группы и оксиранильной группы.
Терапевтический пептид можно непосредственно или косвенно сочетать с одной или более амфифильными группировками через сложноэфирную группу, амидную группу, вторичную или третичную аминную группу, карбаматную группу, сульфонатную группу, сульфатную группу, фосфатную группу, фосфонатную группу или простую эфирную группу.
Предпочтительно, терапевтический пептид ковалентно конъюгируют с одной или более амфифильными молекулами, содержащими: (а) гидрофильную группировку и (б) липофильную группировку, где сбалансированные гидрофильные и липофильные свойства амфифильной молекулы придают конъюгату соответствующую растворимость в биологической жидкости или водном растворе.
Более предпочтительно, терапевтический пептид ковалентно конъюгируют с одной или более амфифильными молекулами, содержащими: (а) линейную полиэтиленгликолевую группировку и (б) липофильную группировку, где терапевтический пептид, полиэтиленгликоль и липофильную группировку конформационно располагают таким образом, чтобы иметь доступную с внешней стороны липофильную группировку для взаимодействия с липофильным окружением или клеточными мембранами. Такой амфифильно модифицированный пептид обладает повышенной стойкостью к химическому взаимодействию с биодеградируемым полимером как in vitro, так и in vivo относительно неконъюгированного пептида.
Предпочтительно, амфифильная молекула имеет следующую общую структуру: L-S-(OC2H4)mOH (формула 1), где L представляет собой липофильную группировку, предпочтительно выбранную из С3-39-алкильных, С3-39-алкенильных, С3-39-алкадиенильных, токоферольных и стероидных остатков, и где S представляет собой линкер, выбранный из группы сложноэфирной группы, амидной группы, вторичной или третичной аминной группы, карбаматной группы, сульфонатной группы, сульфатной группы, фосфатной группы, фосфонатной группы или простой эфирной группы.
В одном воплощении алкильную группу из 16 углеродов ковалентно сочетали с полиэтиленгликолевой молекулой через простую эфирную связь. Полученная амфифильная молекула имеет одну гидроксильную группу, которая может быть активирована или дериватизирована для взаимодействия с подходящими функциональными группами на пептидах. В одном воплощении настоящего изобретения амфифильную молекулу дериватизировали с целью приобретения концевой альдегидной группы. Затем амфифильную молекулу ковалентно конъюгировали с октреотидом путем взаимодействия с аминными группами на октреотиде с последующей реакцией восстановления с NaCNBH3. Обе аминные группы на октреотиде можно было конъюгировать одновременно, или путем подбора реакционных условий можно было селективно конъюгировать только одну аминную группу с последующим разделением. Конъюгат образовывали через вторичный амин, который не изменяет зарядные характеристики неконъюгированного октреотида. Это свойство может быть полезно для сохранения активности пептида.
В другом воплощении амфифильную молекулу монопальмитил-поли(этиленгликоля) (среднечисленная молекулярная масса приблизительно 1124) активировали 4-нитрофенилхлорформиатом. Затем амфифильную молекулу ковалентно конъюгировали с октреотидом путем взаимодействия с аминными группами на октреотиде. Обе аминные группы на октреотиде можно было конъюгировать одновременно, или путем подбора реакционных условий можно было селективно конъюгировать только одну аминную группу с последующим разделением.
Пептиды, ковалентно модифицированные одной или более липофильными или амфифильными группировками, включают, например, фармацевтически приемлемые соли и комплексы модифицированного пептида. Модификация может иметь место в одном или более чем одном сайте пептида. Такие пептиды также включают, например, сайт-специфично модифицированные пептиды и смеси пептидов, модифицированных по одному и множеству сайтов.
"Фармацевтически приемлемая соль" означает соль, образованную между любой одной или более заряженными группами пептида и любым одним или более фармацевтически приемлемыми нетоксичными катионами или анионами. Органические и неорганические соли включают, например, соли, полученные из кислот, таких как хлористоводородная, серная, сульфоновая, винная, фумаровая, бромистоводородная, гликолевая, лимонная, малеиновая, фосфорная, янтарная, уксусная, азотная, бензойная, аскорбиновая, п-толуолсульфоновая, бензолсульфоновая, нафталинсульфоновая, пропионовая, угольная и им подобные, или, например, аммония, натрия, калия, кальция или магния.
Термин "биодеградируемый, нерастворимый в воде полимер" включает любой биосовместимый (то есть фармацевтически приемлемый) и биодеградируемый синтетический или природный полимер, который может быть использован in vivo. Этот термин также включает полимеры, которые являются нерастворимыми или становятся нерастворимыми в воде или биологической жидкости при 37°С. Полимеры могут быть очищены, возможно, для удаления мономеров и олигомеров с использованием методик, известных в области техники (например, патент США №4728721; заявка на патент США №2004/0228833). Некоторыми неограничивающими примерами таких полимеров являются полилактиды, полигликолиды, поликапролактоны, полидиоксаноны, поликарбонаты, полигидроксибутираты, полиалкиленоксалаты, полиангидриды, полиамиды, полиэфирамиды, полиуретаны, полиацетали, полиортокарбонаты, полифосфазены, полигидроксивалераты, полиалкенсукцинаты и полиортоэфиры и сополимеры, блоксополимеры, разветвленные сополимеры, тройные сополимеры и их комбинации и смеси.
Подходящие молекулярные массы для полимеров могут быть определены средним специалистом в области техники. Факторы, которые могут быть учтены при определении молекулярных масс, включают желаемую скорость разрушения полимера, механическую прочность и скорость растворения полимера в растворителе. Подходящий диапазон молекулярных масс полимеров обычно составляет от около 2000 дальтон до около 150000 дальтон с полидисперсностью от 1,1 до 2,8, в зависимости, среди других факторов, от того, какой полимер выбран для применения.
Согласно настоящему изобретению, фармацевтические композиции терапевтических пептидов приготавливают в форме инъецируемых растворов или суспензий полимера в фармацевтически приемлемом растворителе, содержащем диспергированные или солюбилизированные, липофильно или амфифильно модифицированные пептиды. Некоторые реакционноспособные группы пептида в результате ковалентного сочетания пептида с липофильной или амфифильной молекулой являются защищенными и недоступными для взаимодействия с полимером в растворе. Таким образом, путем ковалентной модификации пептида улучшают стабильность пептида и полимера в композициях по настоящему изобретению.
Следовательно, в настоящем изобретении предложен способ формирования твердого биодеградируемого имплантата в субъекте in situ, включающий: (а) растворение или диспергирование биосовместимого, нерастворимого в воде, биодеградируемого полимера и терапевтического пептида, ковалентно модифицированного одной или более липофильными или амфифильными группировками в биосовместимом, растворимом в воде органическом растворителе с образованием композиции; причем органический растворитель способен рассеиваться или диффундировать в жидкости организма после помещения в ткань организма; и (б) введение композиции в место размещения имплантата в организме таким образом, чтобы позволить органическому растворителю рассеиваться или диффундировать в жидкостях организма, а полимеру коагулировать или затвердеть с образованием биодеградируемого твердого имплантата.
Дополнительно в настоящем изобретении предложена жидкая фармацевтическая композиция для формирования биодеградируемого имплантата в организме in situ, содержащая эффективное количество биосовместимого, нерастворимого в воде, биодеградируемого полимера и эффективное количество терапевтического пептида, ковалентно модифицированного одной или более чем одной липофильной или амфифильной группировкой, которые растворены или диспергированы в эффективном количестве биосовместимого, растворимого в воде органического растворителя; причем растворитель способен рассеиваться или диффундировать в жидкости организма, а полимер способен коагулировать или затвердевать после контакта с жидкостью организма.
Подходящими пептидами и липофильными или амфифильными молекулами являются такие, которые определены выше. Молярное отношение пептида к липофильной или амфифильной молекуле в конъюгате будут варьировать в зависимости от природы пептида, например от 1:1 до 1:10.
Может быть использован любой биодеградируемый полимер при условии, что полимер является нерастворимым или становится нерастворимым в водной среде или жидкости организма при 37°С. Подходящими биодеградируемыми полимерами являются такие, которые определены выше.
Тип, молекулярная масса и количество биодеградируемого полимера, присутствующего в композициях, могут влиять на продолжительность времени, в течение которого пептид высвобождается из имплантата с контролируемым высвобождением. Выбор типа, молекулярной массы и количества биодеградируемого полимера, присутствующего в композициях, с целью достижения желаемых характеристик контролируемого высвобождения может быть осуществлен средним специалистом в области техники путем проведения обычных экспериментов.
Фармацевтически приемлемые органические растворители включают N-метил-2-пирролидон, N,N-диметилформамид, диметилсульфоксид, пропиленкарбонат, капролактам, триацетин, бензилбензоат, бензиловый спирт, этиллактат, глицерилтриацетат, эфиры лимонной кислоты, полиэтиленгликоли, алкоксиполиэтиленгликоли и полиэтиленгликольацетаты или любую их комбинацию, но не ограничиваются ими.
Критериями для органических растворителей биодеградируемых полимеров являются их фармацевтическая приемлемость и смешиваемость для диспергируемости в водной среде или в жидкости организма. Подходящий органический растворитель должен быть способен диффундировать в жидкость организма таким образом, чтобы жидкая композиция коагулировала или затвердевала с формированием имплантата in situ. Можно применять индивидуальные и/или смешанные растворители, а пригодность таких растворителей может быть легко определена путем проведения обычных экспериментов.
Фармацевтические композиции по изобретению обычно содержат пептиды в диапазоне от 0,1 до 40% (мас./об.). Вообще, оптимальная загрузка лекарственного средства зависит от желаемого периода высвобождения и эффективности пептида. Очевидно, что для пептида с низкой эффективностью и более продолжительным периодом высвобождения требуются более высокие уровни включения.
Вязкость инъецируемых жидких композиций по изобретению определяется молекулярной массой используемого полимера и органического растворителя. Например, если применяют поли(лактид-со-гликолид), то раствор полиэфира в NMP обладает более низкой вязкостью, чем в mPEG350. Обычно, если применяют один и тот же растворитель, то чем выше молекулярная масса и концентрация полимера, тем выше вязкость. Предпочтительно, концентрация полимера в растворе составляет менее 70% по массе. Более предпочтительно, концентрация полимера в растворе составляет между 20 и 60% по массе.
Высвобождение липофильно или амфифильно модифицированных пептидов из таких имплантатов, формирующихся in situ, будет следовать подобным общим правилам для высвобождения лекарственного средства из монолитного полимерного агрегата. На высвобождение липофильно или амфифильно модифицированных пептидов может оказывать влияние размер и форма имплантата, загрузка липофильно или амфифильно модифицированных пептидов в имплантат, факторы проницаемости, касающиеся липофильно или амфифильно модифицированных пептидов и индивидуального полимера, и разрушение полимера. Специалист в области доставки лекарственных средств может корректировать указанные выше параметры в зависимости от количества модифицированных пептидов, выбранных для доставки, с целью получения желаемой скорости и продолжительности высвобождения.
Количество вводимой инъецируемой композиции обычно будет зависеть от желаемых свойств имплантата с контролируемым высвобождением. Например, количество инъецируемой композиции в виде раствора может влиять на продолжительность времени, за которое пептид высвобождается из имплантата с контролируемым высвобождением.
Композиции по настоящему изобретению, содержащие липофильно или амфифильно модифицированные пептиды, можно вводить субъекту, если желательна доставка пептида с замедленным контролируемым высвобождением. Термин "субъект", используемый в данном описании изобретения, предназначен для включения теплокровных животных, предпочтительно млекопитающих, наиболее предпочтительно людей.
Термин "вводимая", используемый в данном описании изобретения, предназначен для того, чтобы сослаться на дозирование, доставку композиции (например, фармацевтической композиции) субъекту или нанесение ее на субъект любым подходящим путем доставки композиции к желаемому месту субъекта, включая доставку инъекцией и/или имплантацией подкожным, внутримышечным, внутрибрюшинным или внутрикожным путем и путем введения к слизистым оболочкам с целью обеспечения желаемой дозировки пептида, основанной на известных параметрах лечения различных медицинских состояний терапевтическими пептидами.
Термин "доставка с контролируемым замедленным высвобождением", используемый в данном описании изобретения, включает, например, непрерывную доставку терапевтического пептида in vivo за период времени после введения, предпочтительно, по меньшей мере в течение от нескольких суток до недель или месяцев. Доставка с контролируемым замедленным высвобождением пептида может быть доказана, например, непрерывным терапевтическим эффектом агента во времени (например, для октреотида замедленная доставка пептида может быть доказана непрерывными уменьшениями GH (гормон роста) во времени). Альтернативно, замедленная доставка агента может быть доказана посредством обнаружения присутствия агента in vivo во времени.
В этой заявке также предусмотрены различные воплощения, изложенные в формуле изобретения, для настоящих жидких фармацевтических композиций, mutatis mutandis, для настоящих способов получения таких композиций и настоящих способов формирования твердых имплантатов.
ПРИМЕРЫ
Следующие ниже примеры иллюстрируют композиции и способы по настоящему изобретению. Следующие ниже примеры не следует рассматривать в качестве ограничений; они будут учить только лишь тому, как создать системы доставки полезных лекарственных средств.
Пример 1. Получение пальмитоил-октреотида (PAL-OCT)
50 мг ацетата октреотида растворяли в 1 мл безводного DMSO (диметилсульфоксид), содержащего 100 мкл триэтиламина (TEA). 40,2 мг N-гидроксисукцинимидового эфира пальмитиновой кислоты (М.м. (молекулярная масса) 353,50) растворяли в 3 мл безводного DMSO и добавляли к раствору пептида. Реакции позволяли протекать в течение 3 часов при комнатной температуре. Смесь вливали в диэтиловый эфир для осаждения пальмитоилированного октреотида. Осадок дважды промывали диэтиловым эфиром и затем сушили под вакуумом. Полученный ацилированный пептид был в виде белого порошка.
Пример 2. Получение пальмитоил-октреотида (PAL-OCT)
50 мг ацетата октреотида растворяли в 1000 мкл безводного DMSO, содержащего 100 мкл TEA. 17,1 мг N-гидроксисукцинимидового эфира пальмитиновой кислоты (М.м. 353,50) растворяли в 3 мл безводного DMSO и добавляли к раствору пептида прямым впрыскиванием. Реакции позволяли протекать в течение ночи при комнатной температуре. Смесь вливали в диэтиловый эфир для осаждения пальмитоилированного октреотида. Осадок дважды промывали диэтиловым эфиром и затем сушили под вакуумом. Полученный ацилированный пептид был в виде белого порошка.
Пример 3. Получение деканаль-октреотида (DCL-OCT)
50 мг октреотида растворяли в 2 мл 20 мМ раствора цианоборгидрида натрия (М.м. 62,84, NaCNBH3) (2,51 мг) в 0,1 М ацетатном буфере при pH 5. 13,7 мг деканаля (М.м. 156,27) (OCT:DCL=1:2) добавляли к раствору пептида прямым впрыскиванием. Реакции позволяли протекать в течение ночи при 4°С. Смесь разделяли центрифугированием. Выпавший в осадок PAL-OCT подвергали лиофилизации.
Пример 4. Получение пальмитоил-лизоцима (PAL-Lyz, 3:1)
302 мг лизоцима (М.м. 14500) растворяли в 1000 мкл безводного DMSO, содержащего 200 мкл TEA. 18,25 мг N-гидроксисукцинимидового эфира пальмитиновой кислоты (М.м. 353,50) растворяли в 3 мл безводного DMSO и добавляли к раствору белка прямым впрыскиванием. Реакции позволяли протекать в течение ночи при КГ (комнатная температура). PAL-Lyz осаждали в диэтиловом эфире и после удаления органического растворителя конечный продукт подвергали лиофилизации.
Пример 5. Высвобождение пальмитоил-лизоцима из инъецируемых полимерных композиций
40% PLGA (поли(DL-лактид-co-гликолид)) RG503H приготавливали соответствующим растворением полимера в mPEG350. Затем пальмитоил-лизоцим и лизоцим смешивали с раствором полимера при приблизительно 7%, соответственно. Композиции тщательно смешивали до получения однородных композиций.
Высвобождение лизоцима и пальмитоилированного лизоцима in vitro из инъецируемого раствора полимера: суспензии композиций (около 100 мг) впрыскивали в 3 мл фосфатно-буферного раствора при pH 7,4 с 0,1% азида натрия и при 37°С. Получившуюся жидкость в определенные моменты времени замещали свежим буферным раствором, а удаленный буферный раствор подходящим образом разбавляли фосфатным буфером при pH 7,4 и анализировали в отношении концентрации лекарственного средства с помощью УФ спектрофотометра при 280 нм относительно стандартных кривых.
На Фиг.1 показаны профили кумулятивного высвобождения как ацилированного, так и чистого лизоцима. Чистый лизоцим показал значительное первоначальное высвобождение по сравнению с ацилированным лизоцимом.
Пример 6. Получение пальмитоил-лизоцима (PAL-Lyz, 5:1)
50 мг лизоцима (М.м. 14500) растворяли в воде и устанавливали pH 9,58. Раствор подвергали лиофилизации. Затем высушенный порошок растворяли в 3 мл DMSO. Затем 322 мкл 20 мг/мл раствора N-гидроксисукцинимидового эфира пальмитиновой кислоты (М.м. 353,50) в безводном DMSO добавляли к раствору белка прямым впрыскиванием. Реакции позволяли протекать в течение ночи при 4°С. PAL-Lyz осаждали в диэтиловом эфире и после удаления органического растворителя конечный продукт подвергали лиофилизации.
Пример 7. Получение пальмитоил-лизоцима (PAL-Lyz, 13:1)
50 мглизоцима (М.м. 14500) растворяли в воде и устанавливали pH 9,58. Раствор подвергали лиофилизации. Затем высушенный порошок растворяли в 3 мл DMSO. Затем 799 мкл 20 мг/мл раствора N-гидроксисукцинимидового эфира пальмитиновой кислоты (М.м. 353,50) в безводном DMSO добавляли к раствору белка прямым впрыскиванием. Реакции позволяли протекать в течение ночи при 4°С. PAL-Lyz осаждали в диэтиловом эфире и после удаления органического растворителя конечный продукт подвергали лиофилизации.
Пример 8. Получение пальмитоил-лизоцима (PAL-Lyz)
Лизоцим добавляли к PAL-NHS (N-гидроксисукцинимидовому эфиру пальмитиновой кислоты) в PBS (фосфатно-буферном растворе) (pH 8,0), содержащем 2% дезоксихолата (DOC). Смесь инкубировали при 37°С в течение 6 часов. Смесь центрифугировали для удаления непрореагировавшего PAL-NHS. Продукт диализировали в отношении PBS, содержащего 0,15% DOC, в течение 48 ч (PAL-NHS:Lyso=15:1).
Пример 9. Высвобождение грелина из инъецируемых полимерных композиций
40% PLGA RG503H приготавливали соответствующим растворением полимера в mPEG350. Затем грелин (человеческий, крысиный 1-5) и дезацилированный грелин (Дез-н-октаноил-[Ser]3-грелин (человеческий, крысиный 1-5)) смешивали с раствором полимера при приблизительно 6%, соответственно. Композиции тщательно смешивали до получения однородных композиций.
Суспензии композиций (около 100 мг) впрыскивали в 3 мл фосфатно-буферного раствора при pH 7,4 с 0,1% азида натрия и при 37°С. Получившуюся жидкость в определенные моменты времени замещали свежим буферным раствором, а удаленный буферный раствор подходящим образом разбавляли фосфатным буфером при pH 7,4 и анализировали в отношении концентрации лекарственного средства с помощью ВЭЖХ (высокоэффективная жидкостная хроматография) с использованием стандартных кривых.
На Фиг.2 показано кумулятивное высвобождение грелина и дезацилированного грелина в PBS. Дезацилированный грелин показал намного более быстрое высвобождение в течение двухнедельного периода испытаний. Грелин с липофильной группировкой показал намного более медленную скорость высвобождения.
Пример 10. Получение монопальмитил-поли(этиленгликоль)-бутиральдегида диэтилацеталя
Смесь монопальмитил-поли(этиленгликоля) (среднее значение М.м. приблизительно 1124) (5,0 г, 4,45 ммоль) и толуола (75 мл) сушили азеотропно путем отгонки толуола при пониженном давлении. Высушенный монопальмитил-поли(этиленгликоль) растворяли в безводном толуоле (50 мл), к которому добавляли 20%-ный (мас./мас.) раствор трет-бутоксида калия в THF (тетрагидрофуран) (4,0 мл, 6,6 ммоль) и диэтилацеталь 4-хлорбутиральдегида (0,96 г, 5,3 ммоль, М.м. 180,67). Смесь перемешивали при 100-105°С в течение ночи в атмосфере аргона. После охлаждения до комнатной температуры смесь фильтровали и добавляли к 150 мл диэтилового эфира при 0-5°С. Выпавший в осадок продукт отфильтровывали и сушили при пониженном давлении.
Пример 11. Конъюгация октреотида по N-концевой аминной группе с монопальмитил-поли(этиленгликолем) (PAL-PEG-BA-OCT)
При обычном получении 201,6 мг монопальмитил-поли(этиленгликоль)-бутиральдегида диэтилацеталя (PAL-PEG-BADA) растворяли в 10 мл 0,1 М фосфорной кислоты (pH 2,1), и полученный раствор нагревали при 50°С в течение 1 ч, затем охлаждали до комнатной температуры. С помощью 1 н. NaOH устанавливали pH раствора 5,5, и полученный раствор добавляли к раствору 195,3 мг октреотида в 3,5 мл 0,1 М буфера фосфата натрия (pH 5,5). Через 1 ч, с целью получения концентрации 20 мМ добавляли 18,9 мг NaCNBH3. Реакцию продолжали в течение ночи при комнатной температуре. Затем реакционную смесь или диализировали с использованием мембраны, имеющей отсечение М.м. 2000 дальтон, или наносили на препаративную ВЭЖХ колонку С-18. Очищенный конъюгированный октреотид главным образом представлял собой единственное соединение с одним первичным амином (лизином) и одним вторичным амином (N-концевым).
Пример 12. Высвобождение и стабильность пептидов и биодеградируемого полимера in vitro в жидких полимерных композициях
Поли(DL-лактид-со-гликолид) (PLGA) при соотношении лактида и гликолида 85/15, имеющий полидисперсность 1,5 (DLPLG85/15, IV: 0,28), растворяли в N-метил-2-пирролидоне (NMP) с получением 50%-ного раствора по массе. Пептиды смешивали с раствором PLGA в NMP с образованием однородной инъецируемой композиции в отношениях, указанных в таблице 1.
Аликвоту из каждой композиции отбирали для высвобождения in vitro в фосфатном буфере при pH 7,4, содержащем 0,1% азида натрия, и 37°С, а оставшуюся композицию использовали для наблюдения за стабильностью пептидов и полимера при комнатной температуре от времени. Моментами времени были 0,125, 1, 2, 5, 7, 14, 21 и 28 суток. Чистоту пептидов в образце определяли с помощью ВЭЖХ. Молекулярную массу полимера определяли с помощью гельпроникающей хроматографии (ГПХ) с использованием полистирольных стандартов с известными молекулярными массами.
Согласно предшествующему уровню техники, наличие нуклеофильной группы в пептиде может привести к взаимодействию между пептидом и биодеградируемым полимером композиции. Нуклеофильные группы пептида могут реагировать с биодеградируемым полимером с образованием ацилированных продуктов и могут катализировать разрушение биодеградируемого полимера. Хорошо известно, что если объединить октреотид и поли(DL-лактид-со-гликолид), в особенности в органическом растворителе, таком как NMP, то октреотид будет ацилироваться, и полимер будет быстро разрушаться. N-концевая конъюгация октреотида по настоящему изобретению содержит одну первичную аминную, одну вторичную аминную и одну С-концевую группу карбоновой кислоты, и ожидается, что взаимодействует и/или реагирует с полимером аналогично самому октреотиду. Однако неожиданно было обнаружено, что ковалентно конъюгированный октреотид по настоящему изобретению предотвратил реакцию ацилирования и значительно снизил скорость разрушения полимера относительно немодифицированного октреотида. Как описано в предшествующем уровне техники и показано в таблице 2, если октреотид был смешан с раствором поли(DL-лактид-со-гликолида) в NMP, то октреотид ацилировался более чем на 80% в пределах 24 часов при комнатной температуре и почти полностью реагировал через 7 суток. Однако ковалентно конъюгированный октреотид был стабилен при тех же условиях даже после 56 суток. Как показано в таблице 3, молекулярная масса полимера в композиции, содержащей октреотид, быстро уменьшалась при комнатной температуре. Через 21 сутки молекулярная масса полимера уменьшалась на 50%. Однако для полимера в композиции, содержащей ковалентно конъюгированный октреотид, сохранялось более 90% первоначальной молекулярной массы.
Как раскрыто в предшествующем уровне техники, для поддержания стабильности биоактивных агентов и эксципиентов в композиции, биоактивный агент обычно упаковывают отдельно от других компонентов композиции, таких как в коммерческой лейпролидной композиции Eligard. Затем все ингредиенты смешивают непосредственно перед применением. Несмотря на то что такой способ получения может предотвратить взаимодействие между пептидами и биодеградируемыми полимерами при хранении, он не предотвращает какое-либо взаимодействие после их смешивания. Взаимодействие между пептидами и полимером может происходить во время введения и последующего высвобождения in vitro или in vivo.
Когда из каждой композиции после приготовления отобрали аликвоту для осуществления высвобождения in vitro в фосфатном буфере при pH 7,4, содержащем 0,1% азида натрия, и 37°С, то неожиданно было обнаружено, что взаимодействие между октреотидом и полимером происходило во время смешивания и последующего высвобождения in vitro. Как показано на Фиг.3, около 30% октреотида, обнаруженного в среде высвобождения, разрушалось или взаимодействовало с полимером в пределах 3 часов. И более чем 50% октреотида, обнаруженного в среде высвобождения, разрушалось или ацилировалось через 28 суток. Через 28 суток полимерную матрицу растворяли в ацетонитриле, и с использованием воды осаждали полимер. Октреотид анализировали с помощью ВЭЖХ. Было обнаружено, что более 50% октреотида, оставшегося в полимерной матрице, также было ацилировано. Такое разрушение и/или ацилирование октреотида могло бы значительно снизить полезность чистого октреотида и может привести к нежелательным токсичным побочным продуктам. Предотвращение такого взаимодействия между пептидом и полимером было бы очень благоприятно.
На Фиг.4 показано высвобождение it vitro ковалентно конъюгированного октреотида Pal-PEG-BA-OCT. Хотя модифицированный октреотид содержит такие же нуклеофилы, что и немодифицированный октреотид, неожиданно было установлено, что за 28 суток не было обнаружено никакого разрушения модифицированного октреотида в среде высвобождения и в полимерной матрице. Результаты показывают, что ковалентная конъюгация пептида с амфифильной группировкой, такой как монопальмитил-поли(этиленгликоль), может предотвратить или значительно уменьшить взаимодействие и/или реакцию между пептидами и биодеградируемыми полимерами.
Пример 13. Получение монопальмитил-поли(этиленгликоля), активированного 4-нитрофенилхлорформиатом (NPC)
Смесь монопальмитил-поли(этиленгликоля) (среднее значение М.м. приблизительно 1124) (10,0 г, 8,9 ммоль) и бензола (100 мл) сушили азеотропно путем отгонки 50 мл бензола при пониженном давлении. Реакционную смесь охлаждали до 30°С, после чего добавляли безводный пиридин (0,809 мл, 10 ммоль) под аргоном и 4-нитрофенилхлорформиат (2,015 г, 10,0 ммоль). После завершения добавления реакционную смесь перемешивали при 45°С в течение 2 ч с последующим перемешиванием при комнатной температуре в течение ночи.
Затем реакционную смесь фильтровали с последующим удалением растворителя из фильтрата путем перегонки в вакууме. Остаток перекристаллизовывали из 2-пропанола с образованием 8,2 г продукта (PAL-PEG-NPC).
Пример 14. Конъюгация октреотида с монопальмитил-поли(этиленгликолем (PAL-PEG-OCT)
236,5 мг PAL-PEG-NPC добавляли к раствору 239 мг октреотида в 10 мл 50 мМ натрийборатного буфера (pH 9). Раствор непрерывно перемешивали с помощью магнитной мешалки в течение ночи. Конечный раствор диализировали с использованием мембраны с отсечением М.м. 2000. Диализированный раствор подвергали лиофилизации и анализировали с помощью ВЭЖХ. Результаты показывают, что модифицированный пептид представляет собой смесь октреотидов, конъюгированных по одному и множеству сайтов.
Это изобретение не ограничено воплощениями, описанными выше, которые приведены только в качестве примеров и могут быть модифицированы различными способами в пределах объема охраны, определяемого прилагаемой формулой изобретения.
Реферат
Изобретение относится к области медицины. Предложена жидкая полимерная фармацевтическая композиция для контролируемого высвобождения терапевтического полипептида, содержащая фармацевтически приемлемый, нерастворимый в воде, биодеградируемый полимер, фармацевтически приемлемый органический растворитель и терапевтический полипептид, конъюгированный с одной или более липофильными группировками и/или с одной или более амфифильными группировками. Предложен способ получения указанной композиции, включающий стадию растворения биодеградируемого полимера в фармацевтически приемлемом органическом растворителе и стадию смешивания полученного раствора полимера с эффективным количеством терапевтического полипептида. Изобретение обеспечивает создание фармацевтической композиции, имеющей повышенную стабильность in vitro и уменьшенное первоначальное "взрывное" высвобождение терапевтического полипептида по сравнению с композицией, в которой терапевтический полипептид не конъюгирован с липофильными и/или амфифильными группировками. 2 н. и 15 з.п. ф-лы, 4 ил., 3 табл., 14 пр.
Формула
(а) фармацевтически приемлемый, нерастворимый в воде, биодеградируемый полимер, выбранный из группы, состоящей из полилактида, полигликолида, поликапролактона, полидиоксанона, поликарбоната, полигидроксибутирата, полиалкиленоксалата, полиангидрида, полиамида, полиэфирамида, полиуретана, полиацеталя, полиортокарбоната, полифосфазена, полигидроксивалерата, полиалкиленсукцината и полиортоэфира и сополимеров, блоксополимеров, разветвленных сополимеров, тройных сополимеров и их комбинаций и смесей;
(б) фармацевтически приемлемый органический растворитель, который растворяет биодеградируемый полимер, и
(в) терапевтический полипептид, конъюгированный с одной или более липофильными группировками и/или конъюгированный с одной или более амфифильными группировками.
(а) растворения фармацевтически приемлемого, нерастворимого в воде, биодеградируемого полимера в фармацевтически приемлемом органическом растворителе с образованием раствора полимера и
(б) смешивания раствора полимера с эффективным количеством терапевтического полипептида, конъюгированного с одной или более липофильными группировками, и/или с эффективным количеством терапевтического полипептида, конъюгированного с одной или более амфифильными группировками, с образованием фармацевтической композиции.
Комментарии