Депо-составы гидрофобного активного ингредиента и способы их получения - RU2678433C2
Код документа: RU2678433C2
Чертежи





Описание
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится в целом к депо-составу, который может быть инъецирован или инфильтрирован в требуемую область и который может обеспечивать замедленное высвобождение гидрофобного терапевтического агента. Конкретнее, настоящее изобретение относится к неводному пролипосомному депо-составу, по существу не содержащему синтетических липидов, преимущество которого заключается в том, что он образует липосомы или другие липидные везикулярные структуры in situ при контакте с физиологическими жидкостями.
УРОВЕНЬ ТЕХНИКИ
Применение состава с пролонгированным высвобождением, содержащего терапевтические агенты, может способствовать соблюдению пациентом режима лечения, сокращать время пребывания в стационаре и больничные расходы и, таким образом, обеспечивать экономию средств для пациента и системы здравоохранения. Согласно расчетам, продажи таких составов с пролонгированным высвобождением ежегодно только в США превышают несколько миллиардов долларов.
Подходящий способ, который позволяет достигнуть длительного терапевтического эффекта, представляет собой однократное применение депо-состава. Депо-состав может быть оптимизирован для инъекции, инфильтрации в разрез, имплантации или местного применения. Содержащийся в депо-составе терапевтический агент приготовлен вместе с носителями, что обеспечивает постепенное высвобождение терапевтического агента в течение периода времени, составляющего от нескольких часов до нескольких дней, или дольше. Депо-составы обычно получают на основе биодеградируемой матрицы, которая постепенно подвергается разложению или растворяется с высвобождением терапевтического агента.
Следовательно, преимущество депо-составов заключается в том, что активные терапевтические агенты высвобождаются постепенно в течение длительных периодов времени без необходимости повторного введения дозы. Эти составы, таким образом, особенно подходят для случаев, при которых необходим длительный терапевтический эффект, когда применяют круглосуточное введение с помощью инъекционного насосного дозирующего устройства, обеспечивающего доставку лекарственного средства в вены (внутривенное, наиболее распространенный способ), под кожу (подкожное) или в пространство между твердой мозговой оболочкой и позвоночником (эпидуральное).
Многие депо-составы получают на основе частиц, заключенных в липосомы или микросфер для инкапсуляции терапевтического агента. Липосомные депо-составы, однако, сложны в изготовлении, чрезвычайно чувствительны к действию поверхностно-активных агентов, имеют ограниченный срок годности или требуют хранения в особых условиях и при пониженной температуре. В связи с размером и уязвимостью этих частиц, препятствующими применению стандартных способов стерилизации, таких как фильтрация, облучение или автоклавирование, липосомные мультивезикулярные депо-составы обычно получают в асептических условиях, что делает производственный процесс трудоемким и дорогостоящим. Кроме того, липосомные депо-составы обычно обеспечивают пролонгированное высвобождение терапевтического агента в течение не более чем 12 часов.
Описаны различные продукты, содержащие лекарственные средства, заключенные в микросферы в носителях на масляной основе.
Например, в патенте США №7547452, Atkins и др., предложены композиции с замедленным высвобождением на основе микрочастиц. Может быть получена композиция на основе микрочастиц, которая обеспечивает пролонгированное высвобождение в течение периода времени, составляющего от примерно 7 дней до примерно 200 дней. Микрочастицы могут быть приготовлены с применением биодеградируемого и биосовместимого полимера и активного агента, такого как рисперидон, 9-гидроксирисперидон и их фармацевтически приемлемые кислые соли.
В патенте США №5480656, Okada, предложена микрокапсула, предназначенная для подчиняющегося кинетике нулевого порядка высвобождения физиологически активного полипептида в течение периода времени, составляющего по меньшей мере два месяца, которую получают путем приготовления эмульсии типа "вода-в-масле", содержащей внутренний водный слой, содержащий примерно 20-70% (масс/масс.) указанного полипептида, и масляный слой, содержащий сополимер или гомополимер, имеющий усредненную молекулярную массу от 7000 до 30000, причем, соотношение молочная кислота/гликолевая кислота в составе сополимера или гомополимера составляет от 80/10 до 100/0, и дальнейшего микроинкапсулирования указанной эмульсии типа "вода-в-масле".
В патенте США №5654010, Johnson, предложена композиция и способы получения и применения указанной композиции для замедленного высвобождения биологически активного, стабилизированного гормона роста человека (ГРЧ). Композиция с замедленным высвобождением в соответствии с этим изобретением содержит полимерную матрицу на основе биосовместимого полимера и частицы, содержащие биологически активный, стабилизированный ГРЧ, при этом указанные частицы диспергированы внутри биосовместимого полимера. Предложенный согласно этому изобретению способ получения композиции с замедленным высвобождением биологически активного ГРЧ включает растворение биосовместимого полимера в растворителе для полимера с образованием раствора полимера, диспергирование частиц на основе биологически активного, стабилизированного ГРЧ в растворе полимера и далее отверждение полимера с образованием полимерной матрицы, содержащей дисперсию указанных частиц на основе ГРЧ. Способ применения композиции согласно этому изобретению представляет собой способ обеспечения терапевтически эффективного уровня биологически активного, неагрегированного ГРЧ в крови субъекта в течение длительного периода времени. В соответствии с этим способом, субъекту вводят эффективную дозу композиции с замедленным высвобождением. Способ применения композиции с замедленным высвобождением включает обеспечение терапевтически эффективного уровня биологически активного, неагрегированного гормона роста человека в крови субъекта в течение длительного периода времени путем введения субъекту максимально допустимой дозы, поскольку он может влиять на ЦНС и противопоказан для внутривенного введения. Поскольку инъекционный Наропин часто сочетают с перорально принимаемыми опиатами при введении для лечения послеоперационной боли, оно сохраняет некоторое количество указанной композиции с замедленным высвобождением.
В патенте США №5538739, Bodmer и др., предложены микрочастицы, содержащие полипептид, предпочтительно, соматостатин или его аналог или производное, более предпочтительно, октреотид, внутри полимерной матрицы, предпочтительно, на основе поли(лактид-ко-гликолид)глюкозы. Согласно этому изобретению также предложены составы с замедленным высвобождением, содержащие указанные микрочастицы, и применение указанных составов для лечении акромегалии и рака молочной железы.
В патенте США №6132766, Sankaram и др., предложена мультивезикулярная липосомная композиция, содержащая по меньшей мере одну кислоту, отличную от галогенводородной кислоты, и по меньшей мере одно биологически активное вещество, при этом везикулы характеризуются определенным распределением по размерам, регулируемым средним размером, величиной внутреннего объема и номером и обеспечивают контролируемую скорость высвобождения биологически активного вещества из композиции. Согласно этому изобретению также предложен способ получения композиции, особенность которого заключается в добавлении кислоты, отличной от галогенводородной кислоты, обеспечивающей поддержание и регулирование скорости высвобождения инкапсулированного биологически активного вещества из везикул на терапевтических уровнях in vivo.
В патенте США №5863549, Tarantino, предложен способ получения in vivo геля на основе лецитина, который обеспечивает замедленное высвобождение биологически активного соединения, содержащегося в геле. Это изобретение также относится к способу длительного лечения человека или других млекопитающих с использованием терапевтического количества биологически активного соединения с применением геля для обеспечения замедленного высвобождения биологически активного соединения. Предложенные и приведенные в качестве примеров биологически активные соединения представляют собой пептиды и полипептиды.
В патенте США №2005/0287180, Chen, предложены композиции, содержащие фосфолипидный компонент (который содержит один или более фосфолипидов) и фармацевтически приемлемый жидкий носитель, при этом содержание фосфолипидного компонента находится в диапазоне от примерно 10% до примерно 90% из расчета на общую массу. Композиции могут дополнительно содержать нефосфолипидные наполнители, причем количество нефосфолипидных наполнителей находится в диапазоне от примерно 5% до примерно 50% из расчета на общую массу. В некоторых вариантах реализации композиции могут быть инъекционными, нелипосомными и/или иметь форму геля или пасты. Композиции согласно изобретению могут подходить для заживления и наращивания мягких и/или твердых тканей или для замедленной местной доставки лекарственных средств. Одним из примеров составов на лекарственного средства является бупивакаин в фосфолипидной пасте с добавлением пропиленгликоля.
В патенте США №2012/0046220, Chen и др., предложен прозрачный депо-состав, содержащий по меньшей мере один гидрофильный водорастворимый фармацевтически активный антибактериальный агент, выбранный из группы, состоящей из ванкомицина, гентамицина, их фармацевтически приемлемых солей и их смеси, воду, фосфолипид, масло, необязательно регулятор рН и модификатор вязкости, выбранный из группы, состоящей из этанола, изопропанола и их смеси, при этом содержание воды в конечном депо-составе не превышает примерно 4 масс. % из расчета на общую массу депо-состава и рН депо-состава находится в диапазоне от примерно 3 до примерно 6.
В патенте США №2012/0316108, Chen и др., предложены композиции и способы получения фосфолипидных депо-составов, которые способны проходить через тонкую иглу.
Дополнительные публикации, содержащие описание составов на основе фосфолипидов, включают WO 89/00077, WO 02/32395, ЕР 0282405 и патенты США №№4252793; 5660854; 5693337 и публикацию Wang et al., Lyophilization Of Water-In-Oil Emulsions To Prepare Phospholipid-based Anhydrous Reverse Micelles For Oral Peptide Delivery, 39 European Journal of Pharmaceutical Sciences, 373-79 (2010).
Известным депо-составам на основе фосфолипидов присущи недостатки, заключающиеся в высокой вязкости, затрудняющей их введение, и отсутствии долгосрочной стабильности при обычных температурах. Существует неудовлетворенная потребность в стабильных депо-составах из гидрофобных активных ингредиентов с улучшенными вязкостью и размером частиц, которые обеспечили бы пригодность для доставки в требуемое место действия.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Согласно настоящему изобретению предложен неводный пролипосомный депо-состав гидрофобного активного фармацевтического ингредиента (АФИ), имеющего низкую растворимость в воде, преимущество которого заключается в том, что он образует липосомы или другие липидные везикулярные структуры in situ при контакте с физиологическими жидкостями. Согласно настоящему изобретению также предложен способ получения депо-составов согласно изобретению, в соответствии с которым композицию не подвергают воздействию водной фазы ни на одной из стадий производственного процесса. Композицию не подвергают воздействию водной фазы ни на одной из стадий производственного процесса. Композиция не содержит воды за исключением остаточной влаги, которая может присутствовать во вспомогательных веществах, применяемых для получения композиции.
Кроме того, согласно настоящему изобретению предложен неводный пролипосомный депо-состав, полученный с применением вспомогательных веществ исключительно категории GRAS (признанных в целом безопасными) и по существу не содержащий синтетических фосфолипидов. Таким образом, композиции согласно изобретению обеспечивают улучшенную стабильность, увеличенную продолжительность терапевтического действия АФИ и меньшее количество возможных нежелательных эффектов. Преимуществом является то, что депо-составы согласно изобретению обеспечивают поддержание терапевтически эффективных уровней содержащихся в них АФИ в течение нескольких дней или даже недель. Терапевтические эффекты в предпочтительном варианте могут быть ограничены локальными эффектами или эффектами вблизи места введения депо-состава.
Настоящее изобретение, в частности, основано на неожиданно обнаруженном факте, что масляный раствор, являющийся носителем АФИ, дольше задерживается в ткани и обеспечивает улучшенные характеристики замедленного высвобождения по сравнению с гелем или гелеобразным составом, переносящим то же самое количество АФИ. В соответствии с некоторыми вариантами реализации раствор согласно изобретению образует липосомы или мицеллы или другие типы липидных ассоциатов in vivo после естественного попадания физиологических жидкостей организма в послеоперационную рану, благодаря чему высвобождение АФИ продолжается в течение длительного периода времени. Соответственно, пролипосомный состав является стабильным и может храниться при комнатной температуре, что обуславливает преимущества в отношении транспортировки и хранения составов по сравнению с липосомными составами, требующими хранения при 2-8°C. Состав будет удерживать свой активный ингредиент и не будет демонстрировать взрывного высвобождения при контакте с поверхностно-активными веществами, соответственно, не будет высвобождать все количество АФИ в организм, как может происходить в случае разрыва липосом.
Согласно одному из аспектов изобретения предложен пролипосомный неводный фармацевтический состав, содержащий: гидрофобный АФИ; природный несинтетический фосфолипид или его фармацевтически приемлемую соль; неводный фармацевтически приемлемый носитель и сорастворитель в качестве регулятора вязкости, при этом указанная композиция по существу не содержит воды.
В другом варианте реализации неводный фармацевтический состав состоит по существу из: гидрофобного АФИ, природного несинтетического фосфолипида или его фармацевтически приемлемой соли, неводного фармацевтически приемлемого носителя и сорастворителя в качестве регулятора вязкости, при этом указанная композиция по существу не содержит воды.
В некоторых вариантах реализации неводный фармацевтический состав состоит из: гидрофобного АФИ; природного несинтетического фосфолипида или его фармацевтически приемлемой соли, неводного фармацевтически приемлемого носителя, антиоксиданта и сорастворителя в качестве регулятора вязкости, при этом указанная композиция по существу не содержит воды.
В соответствии с некоторыми вариантами реализации композиция стабильна в течение по меньшей мере 24 месяцев при комнатной температуре. В соответствии с некоторыми вариантами реализации композиция стабильна в течение по меньшей мере 12 месяцев при комнатной температуре. В соответствии с некоторыми вариантами реализации композиция стабильна в течение по меньшей мере 6 месяцев при комнатной температуре. В соответствии с некоторыми вариантами реализации композиция стабильна в течение по меньшей мере 1 месяца при комнатной температуре.
В соответствии с некоторыми вариантами реализации сорастворитель представляет собой неароматический сорастворитель. В соответствии с некоторыми вариантами реализации неароматический сорастворитель представляет собой спирт. В некоторых вариантах реализации спирт представляет собой этанол. В некоторых вариантах реализации этанол присутствует в количестве от примерно 1% до примерно 15% по массе. В некоторых вариантах реализации этанол присутствует в количестве от примерно 2% до примерно 10% по массе. В некоторых вариантах реализации этанол присутствует в количестве от примерно 4% до примерно 6% по массе.
В некоторых вариантах реализации сорастворитель выступает в качестве регулятора вязкости, который обеспечивает пригодность композиции для введения посредством инъекции. В некоторых вариантах реализации вязкость состава составляет менее 2500 сПуаз. В некоторых вариантах реализации вязкость состава составляет менее 2000 сПуаз. В некоторых вариантах реализации вязкость состава находится в диапазоне 1000-2500 сПуаз. В некоторых вариантах реализации вязкость состава находится в диапазоне 1000-2000 сПуаз.
В некоторых вариантах реализации композиция не содержит частиц размером более 100 нм. В некоторых вариантах реализации композиция не содержит частиц размером более 50 нм. В некоторых вариантах реализации композиция не содержит частиц размером более 20 нм. В некоторых вариантах реализации композиция не содержит частиц размером более 10 нм. В некоторых вариантах реализации композиция представляет собой масляный раствор, по существу не содержащий частиц. В некоторых вариантах реализации композиция представляет собой прозрачный раствор.
В некоторых вариантах реализации АФИ присутствует в количестве, эквивалентном от примерно 0,2% до примерно 18% по массе. В некоторых вариантах реализации АФИ присутствует в количестве, эквивалентном от примерно 1% до примерно 12% по массе. В других вариантах реализации АФИ присутствует в количестве, эквивалентном от примерно 2% до примерно 4% по массе. В некоторых вариантах реализации АФИ присутствует в количестве, эквивалентном от примерно 3% до примерно 6% по массе. В некоторых вариантах реализации АФИ представляет собой единственный активный фармацевтический ингредиент, который является гидрофобным.
В некоторых вариантах реализации гидрофобный АФИ выбран из группы, состоящей из: ропивакаина, диклофенака, дексаметазона, кетопрофена и их комбинации. Каждый из возможных вариантов представляет собой отдельный вариант реализации изобретения.
В некоторых вариантах реализации гидрофобный АФИ характеризуется экспериментальным значением параметра гидрофобности LogP, составляющим по меньшей мере 1,5. В некоторых вариантах реализации АФИ характеризуется экспериментальным значением параметра гидрофобности LogP, составляющим по меньшей мере 1,6. В некоторых вариантах реализации АФИ характеризуется экспериментальным значением параметра гидрофобности LogP, составляющим по меньшей мере 2.
В некоторых вариантах реализации содержащийся в композиции растворимость АФИ в чистой воде составляет менее 10 мг/мл при температуре примерно 25°C. В некоторых вариантах реализации содержащийся в композиции растворимость АФИ в чистой воде составляет менее 1 мг/мл при температуре примерно 25°C.
В некоторых вариантах реализации фосфолипид представляет собой природный фосфолипид. В некоторых вариантах реализации фосфолипид присутствует в количестве от примерно 10% до примерно 80% по массе. В некоторых вариантах реализации фосфолипид присутствует в количестве от примерно 40% до примерно 60% по массе. В некоторых вариантах реализации фосфолипид присутствует в количестве от примерно 45% до примерно 55% по массе. В некоторых вариантах реализации фосфолипид не содержит каких-либо синтетических фосфолипидов. В некоторых вариантах реализации фосфолипид не содержит 1,2-димиристоил-sn-глицеро-3-фосфоглицерина (ДМФГ) или его фармацевтически приемлемой соли. В некоторых вариантах реализации природный несинтетический фосфолипид представляет собой фосфатидилхолин (ФХ) или его фармацевтически приемлемую соль.
В некоторых вариантах реализации состав по существу не содержит наполнителей, в частности, водонерастворимых или дисперсных наполнителей. В некоторых вариантах реализации составы не содержат инертных дисперсных или взвешенных материалов, таких как микросферы.
В некоторых вариантах реализации состав по существу не содержит воды. В другом варианте реализации термин "по существу не содержит воды" в настоящем тексте относится к содержанию, составляющему менее 0,5% об./об. или масс/масс, из расчета на общий объем или общую массу состава. В других вариантах реализации термин "по существу не содержит воды" в настоящем тексте относится к содержанию, составляющему 0,2% об./об. или масс/масс, из расчета на общий объем или общую массу состава. В некоторых вариантах реализации композиция не содержит воды за исключением остаточной влаги, которая может присутствовать во вспомогательных веществах, применяемых для получения композиции. В некоторых вариантах реализации содержание остаточной влаги составляет менее 0,3%. В некоторых вариантах реализации содержание остаточной влаги составляет менее 0,15%.
В некоторых вариантах реализации неводный фармацевтически приемлемый носитель содержит кунжутное масло, хлопковое масло, сафлоровое масло или один или более триглицеридов. Каждый из возможных вариантов представляет собой отдельный вариант реализации изобретения. В некоторых вариантах реализации неводный фармацевтически приемлемый носитель представляет собой касторовое масло.
В некоторых вариантах реализации неводный фармацевтически приемлемый носитель присутствует в количестве от примерно 20% до примерно 50% по массе.
В некоторых вариантах реализации соотношение природного несинтетического фосфолипида и неводного фармацевтически приемлемого носителя находится в диапазоне от 2,2:1 до 1,2:1. В другом варианте реализации соотношение природного несинтетического фосфолипида и неводного фармацевтически приемлемого носителя находится в диапазоне от 2:1 до 1:1.
В некоторых вариантах реализации фармацевтический состав дополнительно содержит антиоксидант. В некоторых вариантах реализации антиоксидант представляет собой цистеин или его фармацевтически приемлемую соль.
Согласно другому аспекту изобретения предложен пролипосомный неводный маслянистый фармацевтический состав, содержащий: АФИ в количестве, эквивалентном от примерно 0,2% до примерно 10% по массе, от примерно 40% до примерно 60% по массе фосфатидилхолина (ФХ), от примерно 35% до примерно 55% по массе касторового масла и от примерно 2% до примерно 10% по массе этанола, при этом композиция по существу не содержит воды.
Согласно другому аспекту изобретения предложен пролипосомный неводный маслянистый фармацевтический состав, содержащий: ропивакаин в количестве, эквивалентном от примерно 0,5% до примерно 5% по массе; от примерно 40% до примерно 60% по массе фосфатидилхолина (ФХ); от примерно 35% до примерно 55% по массе касторового масла и от примерно 2% до примерно 10% по массе этанола.
Согласно другому аспекту изобретения предложен пролипосомный неводный маслянистый фармацевтический состав, содержащий: кетопрофен в количестве, эквивалентном от примерно 0,5% до примерно 5% по массе, от примерно 40% до примерно 60% по массе фосфатидилхолина (ФХ), от примерно 35% до примерно 55% по массе касторового масла и от примерно 2% до примерно 10% по массе этанола.
Согласно другому аспекту изобретения предложен пролипосомный неводный маслянистый фармацевтический состав, содержащий: диклофенак в количестве, эквивалентном от примерно 0,5% до примерно 5% по массе; от примерно 40% до примерно 60% по массе фосфатидилхолина (ФХ); от примерно 35% до примерно 55% по массе касторового масла и от примерно 2% до примерно 10% по массе этанола.
Согласно другому аспекту изобретения предложен пролипосомный неводный маслянистый фармацевтический состав, содержащий: дексаметазон в количестве, эквивалентном от примерно 0,5% до примерно 5% по массе, от примерно 40% до примерно 60% по массе фосфатидилхолина (ФХ), от примерно 35% до примерно 55% по массе касторового масла и от примерно 2% до примерно 10% по массе этанола.
В некоторых вариантах реализации фармацевтический состав вводят в виде депо-состава. В некоторых вариантах реализации фармацевтический состав вводят в виде однократной дозы. В некоторых вариантах реализации фармацевтический состав вводят посредством инфильтрации в разрез. В некоторых вариантах реализации фармацевтический состав вводят посредством инъекции в разрез. В некоторых вариантах реализации фармацевтический состав вводят посредством инъекции в разрез с последующим зашиванием указанного разреза.
В некоторых вариантах реализации фармацевтическая композиция подходит для введения с помощью шприца с иглой калибра 18-22 G. В некоторых вариантах реализации фармацевтическая композиция подходит для введения с помощью шприца с иглой калибра 21 G.
Согласно другому аспекту изобретения предложен способ получения пролипосомного неводного маслянистого фармацевтического состава, включающий: (а) смешивание неводного фармацевтически приемлемого носителя с: (i) гидрофобным АФИ; (ii) природным несинтетическим фосфолипидом или его фармацевтически приемлемой солью и (iii) сорастворителем в качестве регулятора вязкости с получением неводного раствора; (b) удаление сорастворителя полностью или частично из неводного раствора; (с) добавление того же самого или другого сорастворителя к неводному раствору до общего количества, составляющего от примерно 2% до примерно 12% по массе.
В соответствии с некоторыми вариантами реализации фармацевтический состав, полученный указанными способом, представляет собой прозрачный раствор, не содержащий частиц размером более 100 нм, стабильный при обычной температуре и по существу не содержащий воды.
В соответствии с некоторыми вариантами реализации фармацевтический состав, полученный указанными способом, представляет собой прозрачный раствор, не содержащий частиц размером более 50 нм, стабильный при обычной температуре и по существу не содержащий воды.
В некоторых вариантах реализации удаление сорастворителя полностью или частично из неводного раствора приводит к получению маслянистого раствора.
В соответствии с настоящим изобретением, способ не включает обработку состава водной фазой и не включает стадий эмульгирования.
В некоторых вариантах реализации неводный фармацевтически приемлемый носитель содержит касторовое масло. В некоторых вариантах реализации способ дополнительно включает смешивание неводного фармацевтически приемлемого носителя с антиоксидантом.
В некоторых вариантах реализации сорастворитель удаляют из неводного раствора путем выпаривания и/или вакуумной сушки. В альтернативных вариантах реализации способ не включает применения избытка сорастворителя в качестве регулятора вязкости и, следовательно, отличается отсутствием необходимости выпаривания или вакуумной сушки.
В некоторых вариантах реализации способ дополнительно включает автоклавирование полученного состава.
Согласно другому аспекту изобретения предложен набор, содержащий: емкость, содержащую фармацевтический состав в соответствии с любым из вышеприведенных вариантов реализации и инструкции по применению.
Согласно другому аспекту изобретения предложен предварительно наполненный шприц, содержащий фармацевтический состав в соответствии с любым из вышеприведенных вариантов реализации.
В соответствии с другим аспектом настоящего изобретения предложен пролипосомный неводный базовый состав, содержащий все вспомогательные вещества депо-состава за исключением АФИ, и способ получения этого состава. В некоторых вариантах реализации пролипосомный неводный базовый состав содержит: природный несинтетический фосфолипид; неводный фармацевтически приемлемый носитель и сорастворитель в качестве регулятора вязкости. В соответствии с некоторыми вариантами реализации базовый состав представляет собой готовый для применения состав, к которому может быть добавлен гидрофобный АФИ. В соответствии с другим вариантом реализации, базовый состав имеет высокую стабильность и может храниться в течение длительных периодов времени до добавления гидрофобного АФИ.
В некоторых вариантах реализации базовый состав по существу не содержит воды. В некоторых вариантах реализации базовый состав не содержит воды за исключением остаточной влаги, которая может присутствовать во вспомогательных веществах, применяемых для получения депо-состава. В некоторых вариантах реализации содержание остаточной влаги составляет менее 0,3%. В некоторых вариантах реализации содержание остаточной влаги составляет менее 0,15%.
В некоторых вариантах реализации вязкость базового состава находится в диапазоне 1000-2500 сПуаз. В некоторых вариантах реализации вязкость базового состава находится в диапазоне 1000-2000 сПуаз.
В некоторых вариантах реализации базовый состав не содержит частиц размером более 100 нм. В некоторых вариантах реализации базовый состав не содержит частиц размером более 50 нм. В некоторых вариантах реализации композиция не содержит частиц размером более 20 нм. В некоторых вариантах реализации базовый состав не содержит частиц размером более 10 нм. В некоторых вариантах реализации базовый состав представляет собой масляный раствор, по существу не содержащий частиц. В некоторых вариантах реализации базовый состав представляет собой прозрачный раствор.
Согласно другому аспекту изобретения предложен способ получения пролипосомного неводного базового состава, содержащего все вспомогательные вещества депо-состава за исключением АФИ. Указанный способ включает: (а) уравновешивание неводного фармацевтически приемлемого носителя и (b) растворение природного несинтетического фосфолипида в указанном неводном фармацевтически приемлемом носителе путем нагревания и перемешивания. В соответствии с некоторыми вариантами реализации способ дополнительно включает добавление сорастворителя на стадии (а). В альтернативном варианте способ включает добавление сорастворителя на стадии (b). Следует понимать, что указанный способ может представлять собой непрерывный процесс, во время которого добавление и обработку всех ингредиентов осуществляют одновременно.
В соответствии с некоторыми вариантами реализации базовый состав, полученный указанными способом, представляет собой прозрачный раствор, не содержащий частиц размером более 100 нм, стабильный при обычной температуре и по существу не содержащий воды.
В соответствии с некоторыми вариантами реализации базовый состав, полученный указанными способом, представляет собой прозрачный раствор, не содержащий частиц размером более 50 нм, стабильный при обычной температуре и по существу не содержащий воды.
Неожиданно было обнаружено, что сочетание нагревания, перемешивания с приложением вращающего момента и большого сдвигового усилия приводит к полному растворению фосфолипида в фармацевтически приемлемом неводном носителе. Соответственно, преимущество является отсутствие необходимости добавления избытка этанола и последующего выпаривания этанола.
В некоторых вариантах реализации антиоксидант добавляют к сорастворителю до смешивания сорастворителя с базовым составом. В альтернативном варианте антиоксидант добавляют отдельно на любой из стадий (а) или (b) указанного способа или одновременно с добавлением всех других ингредиентов.
В некоторых вариантах реализации уравновешивание неводного фармацевтически приемлемого носителя (и необязательно сорастворителя и антиоксиданта) включает нагревание до по меньшей мере примерно 50°C. В другом варианте реализации уравновешивание неводного фармацевтически приемлемого носителя (и необязательно сорастворителя и антиоксиданта) включает нагревание до по меньшей мере примерно 65°C. В другом варианте реализации уравновешивание неводного фармацевтически приемлемого носителя включает нагревание до по меньшей мере примерно 85°C.
В некоторых вариантах реализации ни одна из стадий указанного способа не включает обработки составов водной фазой или эмульгирования.
В некоторых вариантах реализации ни одна из стадий способа получения не включает применения избытка этанола. Следовательно, применение этого способа обеспечивает преимущество, заключающееся в отсутствии необходимость выпаривания этанола в присутствии АФИ, что помогает избежать лишних потерь АФИ.
Настоящее изобретение основано, частично, на неожиданно обнаруженном факте, что базовый состав представляет собой готовый для применения базовый состав, с которым гидрофобный АФИ может легко быть смешан без дополнительной обработки состава. Соответственно, любой гидрофобный АФИ, описанный в настоящем тексте, может быть добавлен к базовому составу с образованием пролипосомной неводной фармацевтической композиции. В альтернативном варианте АФИ растворяют перед добавлением к базовому составу.
В других некоторых вариантах реализации способ дополнительно включает автоклавирование полученного базового состава.
В соответствии с другим аспектом настоящего изобретения предложен способ получения пролипосомной неводной фармацевтической композиции, включающий: (а) уравновешивание неводного фармацевтически приемлемого носителя и (b) растворение природного несинтетического фосфолипида в указанном неводном фармацевтически приемлемом носителе путем нагревания и перемешивания. Указанный способ дополнительно включает добавление сорастворителя на стадии (а) или (b). В соответствии с некоторыми вариантами реализации гидрофобный АФИ может быть добавлен на стадии (а) указанного способа. В соответствии с некоторыми вариантами реализации гидрофобный АФИ может быть добавлен на стадии (b) указанного способа. В соответствии с некоторыми вариантами реализации гидрофобный АФИ может быть добавлен на дополнительной стадии (с) указанного способа.
В соответствии с некоторыми вариантами реализации фармацевтическая композиция, полученная указанным способом, представляет собой прозрачный раствор, не содержащий частиц размером более 100 нм, стабильный при обычной температуре и по существу не содержащий воды. В соответствии с некоторыми вариантами реализации фармацевтическая композиция, полученная указанным способом, представляет собой прозрачный раствор, не содержащий частиц размером более 50 нм, стабильный при обычной температуре и по существу не содержащий воды.
В соответствии с некоторыми вариантами реализации указанный способ обеспечивает возможность добавления гидрофобного АФИ к предварительно полученному базовому составу без дополнительной обработки. В альтернативном варианте указанный способ является непрерывным процессом, что означает одновременное добавление и обработку всех или по меньшей мере части ингредиентов.
В соответствии с другим аспектом изобретения предложен набор, содержащий емкость, содержащую базовый состав согласно любому из вышеприведенных вариантов реализации. В некоторых вариантах реализации набор дополнительно содержит гидрофобный АФИ.
В соответствии с другим аспектом изобретения предложен пролипосомный неводный базовый состав, полученный способом согласно любому из вышеприведенных вариантов реализации.
Согласно другому аспекту изобретения предложен способ введения гидрофобного АФИ субъекту, включающий введение фармацевтического состава в соответствии с любым из вышеприведенных вариантов реализации, содержащего гидрофобный активный фармацевтический ингредиент.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Настоящее изобретение описано со ссылкой на следующие фигуры, которые представлены исключительно в иллюстративных целях и которые не ограничивают изобретение.
На фигуре 1 показаны результаты анализа распределения частиц по размерам, полученные с применением анализатора размера частиц Coulter LS230. На фигуре 1А показаны результаты, полученные для смеси депо-состав А/солевой раствор (1:1), на фигуре 1В показаны результаты, полученные для смеси депо-состав А/плазма свиньи (1:1).
Фигура 2 представляет собой изображения, полученные методом просвечивающей криоэлектронной микроскопии (крио-ПЭМ), для состава А, разбавленного плазмой свиньи в соотношениях 1:1 и 1:2.
Фигура 3 представляет собой графическое изображение результатов исследования терапевтического эффекта при лечении послеоперационной боли у поросят массой 10 кг, получавших различные составы ропивакаина.
Фигура 4 представляет собой графическое изображение результатов фармакокинетического исследования на здоровых добровольцах, получавших различные составы ропивакаина.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Согласно настоящему изобретению предложен неводный пролипосомный депо-состав гидрофобного АФИ и способ получения этого состава, который не включает стадий эмульгирования и в соответствии с которым композицию не обрабатывают водной фазой ни на одной из стадий до введения в организм пациента. Кроме того, согласно настоящему изобретению предложен депо-состав, по существу не содержащий синтетических фосфолипидов и полученный с применением вспомогательных веществ исключительно категории GRAS.
Согласно настоящему изобретению также предложен пролипосомный неводный базовый состав, содержащий все вспомогательные вещества депо-состава за исключением АФИ, и способ получения этого состава. Указанный базовый состав готов для добавления гидрофобного АФИ и по существу не содержит воды.
Определения и сокращения
В настоящем тексте АФИ с низкой растворимостью в воде представляет собой АФИ растворимость которого в чистой воде составляет менее 10 мг/мл при комнатной температуре (т.е. при температуре примерно 25°C), более предпочтительно растворимость которого в чистой воде составляет менее 1 мг/мл. В одном из вариантов реализации АФИ является гидрофобным, что в рамках настоящей заявки определяется как характеризующийся значением LogP (где Р представляет собой коэффициент рапределения октанол/вода), составляющим по меньшей мере 1,5.
В настоящем тексте термин "растворимость" определен в соответствии со стандартами Фармакопеи США (USP): растворимость в чистой воде, составляющая>1000 мг/мл при температуре примерно 25°C, относится к молекулам, которые являются "очень легко растворимыми", растворимость, составляющая 100-1000 мг/мл, относится к молекулам, которые являются "легкорастворимыми", растворимость, составляющая 33-100 мг/мл, относится к молекулам, которые являются "растворимыми", растворимость, составляющая 10-33 мг/мл, относится к молекулам, которые являются "умеренно растворимыми", растворимость, составляющая 1-10 мг/мл, относится к молекулам, которые являются "малорастворимыми", растворимость, составляющая 0,1-1, относится к молекулам, которые являются "очень мало растворимыми", и растворимость, составляющая<0,1, относится к молекулам, которые являются "практически нерастворимыми".
В настоящем тексте термин "гидрофобный активный фармацевтический ингредиент" относится к соединениям, имеющим большую растворимость в органических растворителях с низкой полярностью, таких как длинноцепочечные спирты, чем в водном растворе. Описанные согласно настоящему изобретению составы способствуют солюбилизации гидрофобных соединений, которые легко растворяются в спиртах.
В настоящем тексте термин "гидрофобный" относится к АФИ, характеризующимся экспериментальным значением параметра гидрофобности LogP, составляющим по меньшей мере 1,5, более предпочтительно по меньшей мере 1,6, наиболее предпочтительно по меньшей мере 2, где Р представляет собой коэффициент распределения октанол/вода.
В настоящем тексте термин "сольват" относится к молекулярному комплексу, содержащему соединение или соль соединения и одну или более молекул фармацевтически приемлемого растворителя, например, одну или болеее молекул этанола.
В настоящем тексте термин "гидрат" относится к сольвату, в котором одна или более молекул растворителя представляют собой молекулы воды.
В настоящем тексте термин "неводный" означает "по существу не содержащий воды" или "не содержащий воды" и относится к составу, в котором растворитель не содержит воду. Соответственно, "неводный состав" представляет собой состав, содержащий воду в количестве менее 0,5%, 0,4%, 0,3% или 0,2% масс/масс, или об./об. "Неводный состав", однако, может содержать следовые количества воды (не более 0,5% масс/масс), в частности, воду, присутствующую в одном из растворенных веществ. Также в настоящем тексте термин "неводный состав" относится к составу, не содержащему эмульсии ни на одной из стадий его получения. Растворение АФИ может быть достигнуто путем обработки ультразвуком в водяной ультразвуковой ванне, нагретой до примерно 50°C. В настоящем тексте термин "пролипосомный состав" относится к составу, не содержащему определяемых количеств липосом до применения или в условиях хранения. В другом варианте реализации липосомы образуются при контакте с жидкостями живых тканей. В другом варианте реализации липосомы образуются in vivo. В другом варианте реализации нелипосомный состав представляет собой пролипосомный состав (липосомы образуются in vivo при контакте с физиологическими жидкостями).
В настоящем тексте термин "вязкость" относится к сопротивлению состава постепенной деформации под действием напряжения сдвига или растягивающего напряжения. В соответствии с некоторыми вариантами реализации композиция имеет вязкость в диапазоне 1000-3000 сПуаз, 1250-2500 сПуаз, 1400-2000 сПуаз, 1500-1850 сПуаз. Каждый из возможных вариантов представляет собой отдельный вариант реализации изобретения.
В настоящем тексте термин "инъекционный" относится к составу, который может быть инъецирован или инфильтрирован в рану с применением иглы калибра 18-30 G, 20-25 G, 21-23 G. Каждый из возможных вариантов представляет собой отдельный вариант реализации изобретения.
В настоящем тексте термин "примерно" означает диапазон ±10% от величины, к которой он относится. Например, "примерно 100" означает от 90 до 110, включая 90 и 110; "примерно 5%" означает от 4,5% до 5,5%, включая 4,5% и 5,5%.
В настоящем тексте термин "фосфолипид" относится к молекуле, содержащей по меньшей мере одну фосфатную головную группу и по меньшей мере одну неполярную хвостовую группу. В настоящем тексте термин "фосфолипид" ограничивается природным несинтетическим фосфолипидом. В настоящем тексте термин "фосфолипид" ограничивается природным фосфолипидом.
В настоящем тексте термин "маслянистый раствор" относится к раствору с вязкостью, близкой к вязкости масла. Маслянистый раствор имеет меньшую вязкость по сравнению с гелем, пастой, пастообразным или гелеобразным составом. В настоящем тексте термин "маслянистая жидкость" относится к жидкости с вязкостью, близкой к вязкости масла. Маслянистая жидкость имеет меньшую вязкость, чем гель, паста, пастообразная или гелеобразная жидкость.
В настоящем тексте термин "сорастворитель" относится к веществу, которое увеличивает растворимость терапевтического агента в составе и/или уменьшает вязкость состава, что делает состав пригодным для введения посредством инъекции. В соответствии с некоторыми вариантами реализации сорастворитель представляет собой неароматический сорастворитель.
В настоящем тексте термин "стабильная композиция" относится к композициям, которые не образуют осадков при хранении при обычной температуре.
В настоящем тексте термины " обычная температура" и "комнатная температура" взаимозаменяемо относятся к температуре в диапазоне 20-25°C.
В настоящем тексте термин "прозрачный раствор" относится к по существу прозрачным растворам, не содержащим частиц размером более 100 нм. В альтернативном варианте термин "прозрачный раствор" относится к по существу прозрачным растворам, не содержащим частиц размером более 50 нм. В альтернативном варианте термин "прозрачный раствор" относится к по существу прозрачным растворам, не содержащим частиц размером более 20 нм.
В настоящем тексте термин "не содержащий частиц размером более 100 нм" относится к растворам, содержащим менее 5% частиц размером более 100 нм. В настоящем тексте термин "не содержащий частиц размером более 50 нм" относится к растворам, содержащим менее 5% частиц размером более 50 нм. В настоящем тексте термин "не содержащий частиц размером более 20 нм" относится к растворам, содержащим менее 5% частиц размером более 20 нм.
В настоящем тексте термины "наполнитель" и "нефосфолипидный компонент - наполнитель" взаимозаменяемо относятся к биодеградируемому или бионедеградируемому веществу, такому как, без ограничения, сополимер лактида и гликолида (ПЛГ), гидроксиапатит, микросферы на основе полиметилметакрилата (ПММА), которые могут быть приспособлены для применения в качестве тканевых наполнителей.
В настоящем тексте, если конкретно не указано иное, термин "по массе" относится к отношению масс/масс. В настоящем тексте перечисление диапазона числовых значений для переменной величины подразумевает, что изобретение может быть осуществлено на практике с применением любого значения переменной величины в пределах этого диапазона. Таким образом, в случае переменной величины, которая по сути дискретна, последняя может быть равна любому целому значению в пределах числового диапазона, включая конечные точки диапазона. Аналогично, в случае переменной величины, которая по сути непрерывна, последняя может быть равна любому действительному значению в пределах числового диапазона, включая конечные точки диапазона. В качестве примера и без ограничения, переменная величина, которая описана как имеющая значения от 0 до 2, может принимать значения 0, 1 или 2, если она по сути дискретна, и может принимать значения 0,0, 0,1, 0,01, 0,001 или любые другие действительные значения ≥0 и ≤2, если она по сути непрерывна.
В настоящем тексте, если конкретно не указано иное, слово "или" применяют в смысле включения "и/или", но не в смысле исключения "либо/либо". В одном из вариантов реализации термин "содержащий" включает "состоящий".
В настоящем тексте неопределенная и определенная формы единственного числа также включают определяемые объекты во множественном числе, если контекст явно не предписывает иное.
В настоящем тексте термин "субъект" относится к любому животному, включая, но не ограничиваясь перечисленными, людей, приматов, отличных от человека, и других млекопитающих, рептилий и птиц.
В настоящем тексте использованы следующие сокращения, имеющие указанные определения: ПК означает "подкожный", ВМ означает "внутримышечный", ВВ означает "внутривенный", ФХ означает "фосфатидилхолин", ДМФГ означает "1,2-димиристоил-sn-глицеро-3-фосфоглицерин" или его соль или комбинацию его солей, NAC означает "N-ацетил-L-цистеин", КТМХ означает "2-карбокси-2,5,7,8-тетраметил-6-хроманол", МС означает "морская свинка", ДС означает "домашняя свинья", НБС означает "нейтральный буферный состав", КТ означает "комнатная температура".
Депо-составы с пролонгированным высвобождением
Введение депо-состава подходит для постепенного высвобождения активного терапевтического агента в течение продолжительного периода лечения. В некоторых вариантах реализации введение депо-состава, образующего липосомоподобные структуры in vivo, подходит для постепенного высвобождения активного терапевтического агента. В некоторых вариантах реализации активный терапевтический агент приготовлен вместе с носителями, обеспечивающими постепенное высвобождение активного терапевтического агента в течение периода времени продолжительностью от нескольких часов до нескольких дней. Депо-составы часто получают на основе деградируемой матрицы, которая постепенно растворяется в организме с высвобождением активного терапевтического агента. Депо-составы могут быть разработаны таким образом, что они либо допускают, либо предотвращают первоначальное взрывное высвобождение активного агента. Все компоненты депо-составов являются биосовместимыми и биодеградируемыми. В некоторых вариантах реализации термины "композиция" и "состав" используются взаимозаменяемо. В некоторых вариантах реализации термины: "депо-состав", "депо-композиция", "состав согласно изобретению", "пролипосомный состав", "маслянистый состав" и "неводный состав" используются взаимозаменяемо.
В этом контексте следует понимать, что составы согласно изобретению представляют собой пролипосомные неводные стабильные при КТ однородные растворы от момента завершения процесса получения на протяжении всего срока хранения вплоть до и включая момент применения. После проникновения в организм субъекта, при естественном вступлении в контакт с физиологическими жидкостями, они спонтанно образуют липосомы или другие везикулы или мицеллы in situ.
Также описанные в настоящем тексте депо-составы с пролонгированным высвобождением минимизируют сложность соблюдения пациентом режима лечения, уменьшают частоту посещений лечебного учреждения и размер необходимой клинической поддержки и сокращают общее время лечения и время лечения в стационаре и расходы на лечение. Кроме того, в зависимости от содержащегося в депо-составе активного терапевтического агента, описанный в настоящем тексте депо-состав может снизить риски злоупотребления лекарственными средствами за счет устранения или уменьшения необходимости применения медикаментозной терапии на дому.
В некоторых вариантах реализации описанные в настоящем тексте депо-составы являются нелипосомными. В другом варианте реализации описанные в настоящем тексте депо-составы являются нелипосомными и неводными. В других вариантах реализации описанные в настоящем тексте депо-составы являются маслянистыми, но нелипосомными и неводными. В некоторых вариантах реализации депо-состав образует липосомы при попадании в ткань-мишень in vivo. В другом варианте реализации депо-состав образует липосомы при контакте с живой тканью или физиологическими жидкостями.
Депо-составы с пролонгированным высвобождением Гидрофобные АФИ с низкой растворимостью в воде
В соответствии с некоторыми вариантами реализации АФИ является гидрофобным. В одном из вариантов реализации АФИ характеризуется экспериментальным значением параметра гидрофобности LogP, составляющим по меньшей мере 1,5. В другом варианте реализации АФИ характеризуется экспериментальным значением параметра гидрофобности LogP, составляющим по меньшей мере 1,6. В еще одном варианте реализации АФИ характеризуется экспериментальным значением параметра гидрофобности LogP, составляющим по меньшей мере 2.
В соответствии с некоторыми вариантами реализации АФИ мало растворим в воде, но легко растворим в неводных полярных растворителях, в частности, спиртах.
В соответствии с некоторыми вариантами реализации растворимость АФИ в чистой воде составляет менее 10 мг/мл при комнатной температуре (т.е. при температуре примерно 25°C). В соответствии с некоторыми вариантами реализации растворимость АФИ в чистой воде составляет менее 1 мг/мл.
В соответствии с некоторыми вариантами реализации также может применяться комбинация из двух или более гидрофобных АФИ или их фармацевтически приемлемых солей.
Подходящие гидрофобные активные ингредиенты не ограничены фармакотерапевтической группой и могут представлять собой, например, обезболивающие средства, противовоспалительные агенты, противогельминтные агенты, антиаритмические агенты, антибактериальные агенты, противовирусные агенты, антикоагулянты, антидепрессанты, противодиабетические средства, противоэпилептические средства, противогрибковые агенты, агенты против подагры, гипотензивные агенты, противомалярийные агенты, противомигренозные агенты, антимускариновые агенты, противоопухолевые агенты, средства для лечения эректильной дисфункции, иммуносупрессоры, противопротозойные агенты, антитиреоидные агенты, анксиолитики, успокоительные средства, снотворные, нейролептики, бета-адреноблокаторы, сердечные инотропные агенты, кортикостероиды, мочегонные средства, противопаркинсонические агенты, желудочно-кишечные агенты, антагонисты гистаминовых рецепторов, кератолитики, агенты, регулирующие обмен липидов, местные анестетики, антиангинальные агенты, ингибиторы циклооксигеназы-2 (ЦОГ-2), ингибиторы лейкотриенов, макролиды, миорелаксанты, питательные вещества, ингибиторы протеазы, половые гормоны, стимуляторы, миорелаксанты, средства для лечения остеопороза, агенты против ожирения, средства для улучшения когнитивных функций, средства для лечения недержания мочи, пищевые масла, агенты против доброкачественной гипертрофии предстательной железы, заменимые жирные кислоты и их смеси. Каждый из возможных вариантов представляет собой отдельный вариант реализации изобретения.
Конкретные неограничивающие примеры подходящих гидрофобных активных ингредиентов представляют собой: ацетретин (acetretin), альбендазол, альбутерол, аминоглютетимид, амиодарон, амлодипин, амфетамин, амфотерицин В, аторвастатин, атоваквон, азитромицин, баклофен, беклометазон, бенезеприл (benezepril), бензонатат, бетаметазон, бикалутанид (bicalutanide), будесонид, бупропион, бусульфан, бутенафин, кальцифедиол, кальципотриен, кальцитриол, камптотецин, кандесартан, капсаицин, карбамезепин (carbamezepine), каротины, целекоксиб, церивастатин, цетиризин, хлорфенирамин, холекальциферол, цилостазол, циметидин, циннаризин, ципрофлоксацин, цисаприд, кларитромицин, клемастин, кломифен, кломипрамин, клопидогрел, кодеин, коэнзим Q10, циклобензаприн, циклоспорин, даназол, дантролен, дексаметазон, дексхлорфенирамин, диклофенак, дикумарин, дигоксин, дегидроэпиандростерон, дексаметазон, дигидроэрготамин, дигидротахистерол, диритромицин, донезепил (donezepil), эфавиренз, эпосартан, эргокальциферол, эрготамин, источники незаменимых жирных кислот, этодолак, этопозид, фамотидин, фенофибрат, фентанил, фексофенадин, финастерид, флуконазол, флурбипрофен, флувастатин, фосфенитоин, фроватриптан, фуразолидон, габапентин, гемфиброзил, глибенкламид, глипизид, глибурид, глимепирид, гризеофульвин, галофантрин, ибупрофен, ирбесартан, иринотекан, изосорбида динитрат, изотретиноин, итраконазол, ивермектин, кетоконазол, кетопрофен, кеторолак, ламотриджин, лансопразол, лефлуномид, лизиноприл, лоперамид, лоратадин, ловастатин, L-тироксин, лютеин, ликопен, медроксипрогестерон, мифепристон, мефлокин, мегестрола ацетат, метадон, метоксален, метронидазол, миконазол, мидазолам, миглитол, миноксидил, митоксантрон, монтелукаст, набуметон, налбуфин, наратриптан, нелфинавир, нифедипин, нилсолидипин (nilsolidipine), нилутанид (nilutanide), нитрофурантоин, низатидин, омепразол, опревелкин (oprevelkin), эстрадиол, оксапрозин, оксикодон, паклитаксел, паракальцитол, пароксетин, пентазоцин, пиоглитазон, пизофетин (pizofetin), правастатин, преднизолон, пробукол, прогестерон, псевдоэфедрин, пиридостигмин, рабепразол, ралоксифен, рофекоксиб, ропивакаин, репаглинид, рифабутин, рифапентин, римексолон, ритановир, ризатриптан, росиглитазон, саквинавир, сертралин, сибутрамин, силденафила цитрат, симвастатин, сиролимус, спиронолактон, суматриптан, такрин, такролимус, тамоксифен, тамсулозин, таргретин, тазаротен, телмисартан, тенипозид, тербинафин, теразозин, тетрагидроканнабинол, тиагабин, тиклопидин, тирофибран (tirofibran), тизанидин, топирамат, топотекан, торемифен, трамадол, третиноин, троглитазон, тровафлоксацин, убидекаренон, вальсартан, венлафаксин, вертепорфин, вигабатрин, витамин А, витамин D, витамин Е, витамин К, зафирлукаст, зилеутон, золмитриптан, золпидем и зопиклон. Каждый из возможных вариантов представляет собой отдельный вариант реализации изобретения. Несомненно, также могут применяться соли, изомеры и производные вышеперечисленных гидрофобных активных ингредиентов, а также их смеси.
Среди вышеперечисленных гидрофобных активных ингредиентов, предпочтительные активные ингредиенты включают: ацетретин (acetretin), альбендазол, альбутерол, аминоглютетимид, амиодарон, амлодипин, амфетамин, амфотерицин В, аторвастатин, атоваквон, азитромицин, баклофен, бензонатат, бикалутанид (bicalutanide), бусульфан, бутенафин, кальцифедиол, кальципотриен, кальцитриол, камптотецин, капсаицин, карбамезепин (carbamezepine), каротины, целекоксиб, церивастатин, хлорфенирамин, холекальциферол, циметидин, циннаризин, ципрофлоксацин, цисаприд, цитризин, кларитромицин, клемастин, кломифен, кодеин, коэнзим Q10, циклоспорин, даназол, дантролен, дексхлорфенирамин, диклофенак, дигоксин, дегидроэпиандростерон, дексаметазон, дигидроэрготамин, дигидротахистерол, диритромицин, донепезил, эфавиренз, эргокальциферол, эрготамин, источники незаменимых жирных кислот, этодолак, этопозид, фамотидин, фенофибрат, фентанил, фексофенадин, финастерид, флуконазол, флурбипрофен, флувастатин, фосфенитоин, фроватриптан, фуразолидон, габапентин, гемфиброзил, глибенкламид, глипизид, глибурид, глимепирид, гризеофульвин, галофантрин, ибупрофен, иринотекан, изотретиноин, итраконазол, ивермектин, кетоконазол, кетопрофен, кеторолак, ламотриджин, лансопразол, лефлуномид, лоперамид, лоратадин, ловастатин, L-тироксин, лютеин, ликопен, мифепристон, мефлокин, мегестрола ацетат, метадон, метоксален, метронидазол, миконазол, мидазолам, миглитол, митоксантрон, медроксипрогестерон, монтелукаст, набуметон, налбуфин, наратриптан, нелфинавир, нилутанид (nilutanide), нитрофурантоин, низатидин, омепразол, эстрадиол, оксапрозин, оксикодон, паклитаксел, паракальцитол, пентазоцин, пиоглитазон, пизофетин (pizofetin), правастатин, пробукол, прогестерон, псевдоэфедрин, пиридостигмин, рабепразол, ралоксифен, рофекоксиб, ропивакаин, репаглинид, рифабутин, рифапентин, римексолон, ритановир, ризатриптан, росиглитазон, саквинавир, сибутрамин, силденафила цитрат, симвастатин, сиролимус, спиронолактон, суматриптан, такрин, такролимус, тамоксифен, тамсулозин, таргретин, тазаротен, тенипозид, тербинафин, тетрагидроканнабинол, тиагабин, тизанидин, топирамат, топотекан, торемифен, трамадол, третиноин, троглитазон, тровафлоксацин, вертепорфин, вигабатрин, витамин А, витамин D, витамин Е, витамин К, зафирлукаст, зилеутон, золмитриптан, золпидем, зопиклон, их фармацевтически премлемые соли, изомеры и производные и их смеси. Каждый из возможных вариантов представляет собой отдельный вариант реализации изобретения.
Более предпочтительные гидрофобные активные ингредиенты включают: ацетретин (acetretin), альбутерол, аминоглютетимид, амиодарон, амлодипин, ампренавир, аторвастатин, атоваквон, баклофен, бензонатат, бикалутанид (bicalutanide), бусульфан, кальцифедиол, кальципотриен, кальцитриол, камптотецин, капсаицин, карбамезепин (carbamezepine), каротины, целекоксиб, хлорфенирамин, холекальциферол, циметидин, циннаризин, цисаприд, цетиризин, клемастин, коэнзим Q10, циклоспорин, даназол, дантролен, дексхлорфенирамин, дексаметазон, диклофенак, дегидроэпиандростерон, дигидроэрготамин, дигидротахистерол, эфавиренз, эргокальциферол, эрготамин, источники незаменимых жирных кислот, этодолак, этопозид, фамотидин, фенофибрат, фентанил, фексофенадин, финастерид, флуконазол, флурбипрофен, фосфенитоин, фроватриптан, фуразолидон, глибенкламид, глипизид, глибурид, глимепирид, ибупрофен, иринотекан, изотретиноин, итраконазол, ивермектин, кетоконазол, кетопрофен, кеторолак, ламотриджин, лансопразол, лефлуномид, лоперамид, лоратадин, ловастатин, L-тироксин, лютеин, ликопен, медроксипрогестерон, мифепристон, мегестрола ацетат, метоксален, метронидазол, миконазол, миглитол, митоксантрон, монтелукаст, набуметон, наратриптан, нелфинавир, нилутанид (nilutanide), нитрофурантоин, низатидин, омепразол, эстрадиол, оксапрозин, оксикодон, паклитаксел, паракальцитол, пиоглитазон, пизофетин (pizofetin), пранлукаст, пробукол, прогестерон, псевдоэфедрин, рабепразол, ралоксифен, рофекоксиб, ропивакаин, репаглинид, рифабутин, рифапентин, римексолон, ритановир, ризатриптан, росиглитазон, саквинавир, силденафила цитрат, симвастатин, сиролимус, такролимус, тамоксифен, тамсулозин, таргретин, тазаротен, тенипозид, тербинафин, тетрагидроканнабинол, тиагабин, тизанидин, топирамат, топотекан, торемифен, трамадол, третиноин, троглитазон, тровафлоксацин, вигабатрин, витамин А, витамин D, витамин Е, витамин К, зафирлукаст, зилеутон, золмитриптан, их фармацевтически премлемые соли, изомеры и производные и их смеси. Каждый из возможных вариантов представляет собой отдельный вариант реализации изобретения.
В некоторых вариантах гидрофобный АФИ выбран из группы, состоящей из: ропивакаина, диклофенака, дексаметазона, кетопрофена и любой их комбинации. Каждый из возможных вариантов представляет собой отдельный вариант реализации изобретения.
В соответствии с некоторыми вариантами реализации АФИ присутствует в количестве, эквивалентном от примерно 0,2% до примерно 18% по массе. В соответствии с некоторыми вариантами реализации АФИ присутствует в количестве, эквивалентном от примерно 0,5% до примерно 12% по массе. В соответствии с некоторыми вариантами реализации АФИ присутствует в количестве, эквивалентном от примерно 1% до примерно 10% по массе. В соответствии с некоторыми вариантами реализации АФИ присутствует в количестве, эквивалентном от примерно 3% до примерно 6% по массе.
Фосфолипиды
Фосфолипиды являются подходящими компонентами для описанных в настоящем тексте депо-составов с пролонгированным высвобождением. Фосфолипид содержит по меньшей мере одну полярную головную группу и по меньшей мере одну неполярную хвостовую группу, при этом по меньшей мере одна из полярных головных групп представляет собой фосфатную группу. Неполярные фрагменты молекул могут являться остатками жирных кислот. Фосфолипид обычно содержит две неполярные группы, хотя достаточно единственной неполярной группы. В случае присутствия более одной неполярной группы, эти группы могут быть одинаковыми или разными. Подходящие фосфолипидные полярные головные группы включают, но не ограничиваются перечисленными, головные группы фосфатидилхолина (ФХ), фосфатидилэтаноламина, фосфатидилсерина и фосфатидилинозитола.
Неожиданно было обнаружено, что улучшенные свойства, присущие составу согласно настоящему изобретению, связаны с природным фосфолипидом, но не синтетическим фосфолипидом, таким как, без ограничения, 1,2-димиристоил-sn-глицеро-3-фосфоглицерин (ДМФГ) или его фармацевтически приемлемая соль. Таким образом, в некоторых вариантах реализации фосфолипид представляет собой природный фосфолипид. Подходящие источники фосфолипидов включают яйцо, сердце (например, быка), мозг, печень (например, быка) и растительные источники, включая соевые бобы. Природные фосфолипиды в меньшей степени склонны вызывать воспаление и реакции в организме субъекта. Это не только более удобно для субъекта, но и может обеспечивать увеличение времени удержания полученного депо-состава, в частности, в случае парентеральных депо-составов, поскольку в месте введения наблюдается меньшая активность иммунной системы.
Фосфолипиды включают липидные молекулы, которые представляют собой либо производные глицерина (фосфоглицериды, глицерофосфолипиды), либо производные сфингозина (сфинголипиды). Они включают полярные липиды и некоторые фосфолипиды, имеющие важное значение для структуры и функции клеточных мембран, и являются наиболее распространенными мембранными липидами.
Некоторые фосфолипиды представляют собой производные триглицеридов, в которых один остаток жирной кислоты заменен фосфорилированной группой и остатком одной из нескольких азотсодержащих молекул. Цепи жирных кислот являются гидрофобными. Однако заряды на фосфорилированной группе и аминогруппе придают гидрофильность этому фрагменту молекулы. В результате молекула оказывается амфифильной.
Амфифильные фосфолипиды являются основными составляющими клеточных мембран. Эти молекулы образуют фосфолипидный бислой, в котором их гидрофильные (полярные) головки обращены в сторону водного окружения (например, цитозоля), а их гидрофобные хвосты обращены друг к другу. Наиболее распространенным природным фосфолипидом является фосфатидилхолин (ФХ).
Фосфолипиды получают из природных источников, или они могут быть получены путем органического синтеза. Лецитин представляет собой природную смесь диглицеридов стеариновой, пальмитиновой и олеиновой кислот, связанных со сложным эфиром холина и фосфорной кислоты, обычно называемую фосфатидилхолином. Гидрогенизированный лецитин является продуктом контролируемой гидрогенизации лецитина.
Природные фосфолипиды на основе лецитина
В соответствии с положением Фармакопеи США (USP), лецитин является непатентованным названием, которое описывает сложную смесь нерастворимых в ацетоне фосфолипидов, в основном состоящую из фосфатидилхолина (ФХ), фосфатидилэтаноламина (ФЭ), фосфатидилсерина (Ptd-L-Ser или ФС) и фосфатидилинозитола (Ptdlns или ФИ) в сочетании с различными количествами других веществ, таких как триглицериды, жирные кислоты и углеводы. Состав лецитина и, следовательно, его физические свойства варьируют в зависимости от источника лецитина и точного фосфолипидного состава, например, содержания фосфатидилхолина и т.д. Коммерчески доступные продукты на основе лецитина (лецитины) получают из двух основных источников: яичного желтка и соевых бобов. Лецитины включают: лецитин (общий), лецитин из соевых бобов или соевый лецитин и лецитин из яичного желтка или яичный лецитин.
Лецитин является компонентом клеточных мембран и, соответственно, потребляется как часть нормального рациона человека. Он характеризуется высокой биосовместимостью и фактически отсутствием токсичности в исследованиях на животных с однократным пероральным введением, краткосрочным пероральным введением и субхроническим трансдермальным введением. Лецитин и гидрогенизированный лецитин в составе косметических средств для ухода за кожей животных и человека обычно не обладают раздражающим и сенсибилизирующим действием (см. Fiume Z, 2001 "Final report on the safety assessment of Lecithin and Hydrogenated Lecithin", Int J Toxicol; 20 Suppl 1:21-45).
В фармацевтической промышленности лецитины в основном применяют в качестве диспергирующих, эмульгирующих и стабилизирующих агентов и включают в состав препаратов для внутримышечных (ВМ) и внутривенных (ВВ) инъекций, парентеральных питательных составов и продуктов для местного применения. Лецитин также включен в перечень Руководства по неактивным ингредиентам Федерального управления по контролю качества пищевых продуктов и лекарственных средств США (FDA) для применения в ингаляциях, ВМ и ВВ инъекциях, капсулах для перорального введения, суспензиях и таблетках, ректальных, местных и вагинальных препаратах. В косметической промышленности лецитин и гидрогенизированный лецитин безопасны для применения в составе быстро смываемых косметических средств; их можно безопасно применять в составе несмываемых средств в концентрации не более 15%, являющейся самой высокой концентрацией, проверенной в клинических исследованиях раздражающего и сенсибилизирующего эффекта косметических средств.
Один из источников фосфолипидов на основе лецитина, подходящий для получения описанных в настоящем тексте депо-составов, представляет собой соевый лецитин высокой чистоты, т.е. не содержащий аллергенов, провоспалительных агентов или агентов, вызывающих другие вредные биологические реакции, и соответствующий требованиям для применения в инъекционных продуктах. Такие инъекционные формы соевого лецитина коммерчески доступны под торговыми наименованиями Фосфолипон (Phospholipon®) от Phospholipid GmbH (Cologne, Germany), Липоид С (Lipoid® S) от Lipoid GmbH (Ludwigshafen, Germany), Эпикурон (Epikuron®) от Evonik Industries (Parsippany, NJ - ранее Degussa). Эти очищенные продукты на основе соевого лецитина могут содержать различные концентрации фосфатидилхолина (ФХ) в диапазоне от 30% до 100%. Путем объединения продуктов на основе лецитина с различным содержанием ФХ можно изменять консистенцию имплантата и его стабильность в ткани. Конкретным примером состава на основе соевых лецитинов является Фосфолипон 90G (Phospholipon® 90G), представляющий собой чистый фосфатидилхолин, стабилизированный 0,1% аскорбилпальмитата.
Другие природные фосфолипиды
Другие примеры природных фосфолипидов, которые могут применяться для получения описанных в настоящем тексте депо-составов, включают, но не ограничиваются перечисленными: сфинголипиды в форме сфингозина и его производных (из соевых бобов, яйца, мозга или молока), фитосфингозина и его производных (из дрожжей), фосфатидилэтаноламин, фосфатидилсерин и фосфатидилинозитол.
Общее содержание фосфолипидов в депо-составах
Необязательно, описанные в настоящем тексте депо-составы содержат более одного фосфолипида. Суммарное количество (масс/масс.) всех фосфолипидов в депо-составе называется общим содержанием фосфолипидов.
Общее содержание фосфолипидов в описанных в настоящем тексте депо-составах находится обычно в диапазоне от примерно 10% до примерно 80% из расчета на общую массу депо-состава. В некоторых вариантах реализации минимальное общее содержание (масс/масс.) фосфолипидов в депо-составе составляет примерно 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 60%, 70%, 75% или 80% (включая любое значение между 10% и 80%). В некоторых вариантах реализации максимальное общее содержание (масс/масс.) фосфолипидов в депо-составе составляет примерно 40%, 45%, 50%, 55%, 60% или 70% (включая любое значение между 40% и 70%). Каждый из возможных вариантов представляет собой отдельный вариант реализации изобретения. В некоторых вариантах реализации общее содержание фосфолипидов находится в диапазоне значений от минимального содержания фосфолипидов до максимального содержания фосфолипидов.
Неводные фармацевтически приемлемые носители
В одном из вариантов реализации неводный фармацевтически приемлемый носитель содержит кунжутное масло, хлопковое масло, сафлоровое масло или один или более триглицеридов. В другом варианте реализации неводный фармацевтически приемлемый носитель представляет собой касторовое масло. В некоторых вариантах реализации неводный фармацевтически приемлемый носитель представляет собой поверхностно-активный агент. В некоторых вариантах реализации неводный фармацевтически приемлемый носитель присутствует в количестве от примерно 20% до примерно 60% по массе. В еще одном варианте реализации неводный фармацевтически приемлемый носитель присутствует в количестве от примерно 30% до примерно 50% по массе.
В еще одном варианте реализации соотношение между фосфолипидами и неводным носителем находится в диапазоне 3:1-1:2, 2,5:1-1:5, 2,2:1-1:1,2, 2:1-1:1. Каждый из возможных вариантов представляет собой отдельный вариант реализации изобретения.
Сорастворитель
В одном из вариантов реализации состав дополнительно содержит сорастворитель. В соответствии с некоторыми вариантами реализации сорастворитель может представлять собой, без ограничения: этанол, пропиленгликоль, глицерин, диметилацетамид, диметилизосорбид, диметилсульфоксид, N-метил-2-пирролидинон и т.п. В некоторых вариантах реализации сорастворитель представляет собой глицерин. В некоторых вариантах реализации сорастворитель представляет собой этанол. В некоторых вариантах реализации сорастворитель представляет собой спирт, соответствующий стандартам Фармакопеи США (USP), который содержит от 94,9% до 96,0% об./об. этилового спирта. В другом варианте реализации сорастворитель присутствует в составе в количестве 1-15% по массе. В другом варианте реализации сорастворитель присутствует в составе в количестве 0,5-10% по массе. В другом варианте реализации сорастворитель присутствует в составе в количестве 4-8% по массе. В другом варианте реализации сорастворитель присутствует в составе в количестве 5-7% по массе. В другом варианте реализации сорастворитель присутствует в составе в количестве 5,5-6,5% по массе. Неожиданно было обнаружено, что в случае применения ароматических сорастворителей, таких как, без ограничения, бензиловый спирт, введение композиции в резаную рану морских свинок вызывало неблагоприятные реакции, такие как раздражение кожи в месте применения.
Модификаторы вязкости
В некоторых вариантах реализации сорастворитель выступает в качестве регулятора вязкости, обеспечивающего пригодность композиции для введения путем инъекции через инъекционные иглы калибра 18-25 G. В другом варианте реализации сорастворитель выступает в качестве регулятора вязкости, обеспечивающего пригодность композиции для введения путем инъекции через иглу калибра 21 G.
В соответствии с некоторыми вариантами реализации добавление этанола к депо-составу не оказывает отрицательного влияния ни на активность, ни на концентрацию в крови и ране, ни на стабильность АФИ с низкой растворимостью в воде, но увеличивает текучесть за счет уменьшения вязкости, что обеспечивает повышенную проходимость через иглу при введении состава и позволяет применять инъекционные иглы значительно меньшего диаметра.
Кроме того, как было показано в исследованиях образования липосом, добавление этанола к депо-составу в качестве модификатора вязкости не оказывает отрицательного влияния на способность к образованию липосом при воздействии водного окружения и впоследствии обеспечивает медленное высвобождение гидрофобного АФИ.
В некоторых вариантах реализации вязкость состава составляет менее 2500 сПуаз. В некоторых вариантах реализации вязкость состава составляет менее 2000 сПуаз. В другом варианте реализации вязкость состава предпочтительно находится в диапазоне 1000-3000 сПуаз, 1000-2500 сПуаз, 1000-2000 сПуаз, 1250-2000 сПуаз, 1500-2000 сПуаз, 1500-1850 сПуаз. Каждый из возможных вариантов представляет собой отдельный вариант реализации изобретения.
Неожиданно было обнаружено, что для увеличения времени удержания и продолжительности действия композиция должна иметь вязкость в диапазоне 1000-2000 сПуаз, несмотря на пониженную проходимость через иглу при таких значениях вязкости. Например, как показано в примере 5 ниже, состав в соответствии с настоящим изобретением демонстрировал поддержание более высокой концентрации ропивакаина вблизи от раны даже через четыре дня после инъекции.
Антиоксиданты
В одном из вариантов реализации депо-составы с пролонгированным высвобождением содержат один или более антиоксидантов. Антиоксиданты могут применяться для предотвращения или уменьшения интенсивности процессов окисления фосфолипидов, содержащихся в описанных в настоящем тексте депо-составах. Для этой цели может быть применен любой нетоксичный биосовместимый антиоксидант. Примеры антиоксидантов включают, но не ограничиваются перечисленными, аскорбиновую кислоту (витамин С), цистеин (L-цистеин), N-ацетил-L-цистеин (NAC), L-карнитин, ацетил-L-карнититн, альфа-липоевую кислоту, глутатион, альфа-токоферол (витамин Е), 2-карбокси-2,5,7,8-тетраметил-6-хроманол (КТМХ), аскорбилпальмитат и мочевую кислоту. Фармацевтически приемлемые соли этих или других антиоксидантов также рассматривают как "антиоксиданты", и они могут применяться для получения описанных в настоящем тексте депо-составов. Эти упомянутые выше примеры антиоксидантов являются коммерчески доступными из различных источников.
Вспомогательные вещества
В депо-составы на основе гидрофобных АФИ с низкой растворимостью в воде могут быть включены различные вспомогательные вещества.
Примеры подходящих фармацевтических вспомогательных веществ описаны в публикациях Remington's Pharmaceutical Sciences 1447-1676 (Alfonso R. Gennaro eds., 19th ed. 1995) и Strickley R., "Solubilizing Excipients in Oral and Injectable Formulations", Pharmaceutical Research, Vol. 21, No. 2, Feb 2004, pp. 201-230, полное содержание каждой из которых включено в настоящую заявку посредством ссылки.
Как правило, ингредиенты обеспечивают представлены либо по отдельности, либо смешанными в смеси друг с другом в составе лекарственной формы для однократного введения, например, в виде сухого лиофилизированного порошка или безводного концентрата в герметично закрытой емкости, такой как ампула или саше, с указанием количества активного агента.
Пролипосомные неводные депо-составы
В одном из вариантов реализации депо-составы с пролонгированным высвобождением являются неводными составами, маслянистыми составами или представляют собой любую их комбинацию. Составы могут содержать: (а) гидрофобный АФИ с низкой растворимостью в воде; (b) первый фосфолипид или его фармацевтически приемлемую соль; (с) необязательно, второй фосфолипид или его фармацевтически приемлемую соль; (d) неводный фармацевтически приемлемый носитель, такой как, без ограничения, масло, и (е) сорастворитель, такой как, без ограничения, спирт. В некоторых вариантах реализации депо-состав является пролипосомным и образует липосомы in situ.
В некоторых вариантах реализации композиция не содержит частиц размером более 100 нм. В некоторых вариантах реализации композиция не содержит частиц размером более 50 нм. В некоторых вариантах реализации композиция не содержит частиц размером более 20 нм. В некоторых вариантах реализации композиция не содержит частиц размером более 10 нм. В некоторых вариантах реализации композиция представляет собой масляный раствор, по существу не содержащий частиц. В некоторых вариантах реализации композиция представляет собой прозрачный раствор.
Известные из уровня техники составы (см. патент США №2012/0316108), полученные в соответствии со способом, включающим образование нанодисперсии, которую впоследствии лиофилизируют с получением безводного геля, не образуют прозрачные растворы и содержат частицы. Другими словами, эти составы представляют собой нанодисперсии, а не прозрачные растворы.
В некоторых вариантах реализации АФИ присутствует в количестве, эквивалентном от примерно 0,2% до примерно 18% по массе. В другом варианте реализации АФИ присутствует в количестве, эквивалентном от примерно 1% до примерно 12% по массе. В еще одном варианте реализации АФИ присутствует в количестве, эквивалентном от примерно 2% до примерно 10% по массе. В еще одном варианте реализации АФИ присутствует в количестве, эквивалентном от примерно 3% до примерно 6% по массе.
Несмотря на то, что АФИ может использоваться в высоких дозах, количество АФИ в депо-составе будет зависеть от: вида применяемого АФИ, текущего максимально допустимого разового количества, медицинских показаний, пациента и т.д.
В одном варианте реализации природный несинтетический фосфолипид может представлять собой любой из вышеописанных фосфолипидов. В некоторых вариантах реализации природный несинтетический фосфолипид присутствует в составе в количестве от примерно 10% до примерно 80% по массе. В некоторых вариантах реализации фосфолипид представляет собой несинтетический липид, такой как 1,2-димиристоил-sn-глицеро-3-фосфоглицерин (ДМФГ) или его фармацевтически приемлемая соль. В другом варианте реализации состав согласно изобретению не содержит 1,2-димиристоил-sn-глицеро-3-фосфоглицерина (ДМФГ) или его фармацевтически приемлемой соли. В других вариантах реализации композиции согласно изобретению по существу не содержат ДМФГ. В этом контексте термин "по существу не содержит ДМФГ" относится к концентрации менее 0,5%, предпочтительно менее 0,1%.
В некоторых вариантах реализации несинтетический фосфолипид присутствует в составе в количестве от примерно 40% до примерно 60% по массе. В некоторых вариантах реализации фосфолипид представляет собой фосфатидилхолин (ФХ) или его фармацевтически приемлемую соль.
В некоторых вариантах реализации неводный фармацевтически приемлемый носитель присутствует в составе в количестве от примерно 20% до примерно 50% по массе. В некоторых вариантах реализации неводный фармацевтически приемлемый носитель представляет собой касторовое масло, кунжутное масло, хлопковое масло, сафлоровое масло или один или более триглицеридов.
В некоторых вариантах реализации сорастворитель представляет собой неароматический сорастворитель. В некоторых вариантах реализации сорастворитель представляет собой спирт. В некоторых вариантах реализации спирт представляет собой этанол. Этанол может присутствовать в составах в количестве от примерно 1% до примерно 15% по массе. В некоторых вариантах реализации сорастворитель выступает в качестве регулятора вязкости, обеспечивающего пригодность композиции для введения путем инъекции. В некоторых вариантах реализации вязкость состава составляет менее 2500 сПуаз. В некоторых вариантах реализации вязкость состава составляет менее 2000 сПуаз. В некоторых вариантах реализации вязкость состава находится в диапазоне 1000-2500 сПуаз. В некоторых вариантах реализации вязкость состава находится в диапазоне 1000-2000 сПуаз. Неводные пролипосомные депо-составы необязательно содержат антиоксидант. Подходящие антиоксиданты были описаны выше.
Неводные нелипосомные депо-составы могут необязательно содержать дополнительные ингредиенты, такие как различные вспомогательные вещества, регуляторы рН, хелаторы металлов, такие как ЭДТА или эдетовая кислота, соли, красители и т.п.
В соответствии с некоторыми вариантами реализации состав не содержит Сахаров, таких как, без ограничения, сахароза, декстроза, лактоза, глюкоза, трегалоза, мальтоза, маннит, сорбит, глицерин, амилоза, крахмал, амилопектин или их смесь.
Несмотря на то, что описанный в настоящем тексте пролипосомный неводный депо-состав определен как неводный (или по существу не содержащий воды), может сохраняться присутствие остаточного или следового количества молекул воды, например, из ингредиентов, применяемых для получения состава. Такой депо-состав тем не менее считают неводным. В некоторых вариантах реализации вода вызывает образование липосом и, следовательно, депо-состав не должен содержать воду или по меньшей мере должен содержать незначительное количество воды, как описано в настоящем тексте. В некоторых вариантах реализации содержание остаточной влаги, определенное с помощью метода Карла Фишера, составляет менее 0,3%. В некоторых вариантах реализации содержание остаточной влаги, определенное с помощью метода Карла Фишера, составляет менее 0,15%. Таким образом, в некоторых вариантах реализации липосомы образуются внутри депо-состава при контакте с водными физиологическими жидкостями (например, в контакт с живой тканью, содержащей воду). В некоторых вариантах реализации состав согласно настоящему изобретению получают и хранят в сухих условиях, что обеспечивает по существу "безводную" форму композиций и образование липосом in situ только после введения в жидкости организма.
Способы получения нелипосомных неводных составов
В некоторых вариантах реализации пролипосомные неводные депо-составы согласно изобретению получают следующим образом:
1. АФИ (например, диклофенак), природный несинтетический фосфолипид (например, Фосфолипон 90G), фармацевтически приемлемый неводный носитель (например, касторовое масло) и необязательно антиоксидант (например, гидрохлорид цистеина) растворяют в сорастворителе (таком как спирт, например, этанол) путем нагревания и/или обработки ультразвуком и/или любого другого способа диспергирования и/или смешивания ингредиентов.
2. Избыток спирта удаляют или частично удаляют, например, путем выпаривания и/или высушивания с применением вакуумного насоса. Другие способы также могут применяться для удаления спирта, или состав получают с точным количеством этанола в конечном составе.
3. Необязательно, остаточное количество спирта в готовом продукте, полученном на стадии 2, является заранее определенным.
4. При необходимости дополнительные количества того же самого или другого сорастворителя могут быть добавлены и смешаны с полученным на стадии 2 продуктом до конечной концентрации спирта, составляющей примерно 4-8% (масс/масс). Было определено, что хорошо применимой является конечная концентрация спирта, составляющая 6% (масс/масс).
5. Необязательно, полученный на стадии 4 продукт переносят во флаконы и/или стерилизуют (например, посредством автоклавирования).
Также предусмотрены различные модификации этого способа. Однако в процессе получения пролипосомных неводных депо-составов воду в качестве технологической добавки или в качестве вспомогательного вещества не добавляют. Поскольку в ходе процесса получения воду не добавляют, стадии лиофилизации не требуется. В сущности, получение композиции не требует добавления cахаров, зачастую необходимого для предотвращения агрегации фосфолипидных частиц или капель в процессе удаления воды.
Поскольку согласно предложенному способу не происходит образования ни эмульсии, ни дисперсии, полученный состав по существу не содержит частиц и представляет собой истинный раствор. В случае присутствия частиц такие частицы будут иметь средний размер менее 50 нм, в альтернативном варианте менее 20 нм, в альтернативном варианте менее 10 нм, и раствор может быть определен как масляный раствор, по существу не содержащий частиц. Этот способ является предпочтительным в отличие от других способов, известных в данной области техники, включающих стадию эмульгирования и/или дисперсии. Известные способы, включающие получение эмульсии и/или дисперсии, приводят к получению композиций, содержащих частицы, как показано в нижеприведенных примерах.
Неожиданно было обнаружено, что состав согласно настоящему изобретению может быть стерилизован, например, путем автоклавирования, без потери активности или нарушения консистенции состава. В этом заключается отличие от составов, известных из существующего уровня техники, получаемых способом, включающим получение эмульсии или нанодисперсии, которую впоследствии лиофилизируют с получением безводного геля. Автоклавирование составов, известных из существующего уровня техники, приведет к разрушению присутствующих в геле наночастиц и, фактически, потере активности и нарушению консистенции состава.
Пролипосомные неводные базовые составы
Настоящее изобретение основано, частично, на неожиданно обнаруженном факте, что может быть получен готовый для применения базовый состав, с которым легко смешивается гидрофобный АФИ. Базовый состав содержит природный несинтетический фосфолипид; неводный фармацевтически приемлемый носитель и сорастворитель в качестве регулятора вязкости. В соответствии с некоторыми вариантами реализации базовый состав не содержит воды за исключением остаточной влаги, которая может присутствовать во вспомогательных веществах, применяемых для получения композиции. В некоторых вариантах реализации содержание остаточной влаги, определенное с помощью автоматического метода определения содержания воды по Карлу Фишеру, составляет менее 0,3%. В некоторых вариантах реализации содержание остаточной влаги, определенное с помощью автоматического метода определения содержания воды по Карлу Фишеру, составляет менее 0,15%.
После получения базового состава гидрофобный АФИ может быть сразу добавлен без дополнительной обработки состава за исключением перемешивания. В альтернативном варианте АФИ предварительно растворяют перед добавлением к базовому составу. В соответствии с некоторыми вариантами реализации АФИ растворяют в том же самом или другом фармацевтически приемлемом неводном носителе перед добавлением к базовому составу. В альтернативном варианте АФИ может быть добавлен с последующим нагреванием и перемешиванием конечной композиции.
В некоторых вариантах реализации базовый состав не содержит частиц размером более 100 нм. В некоторых вариантах реализации базовый состав не содержит частиц размером более 50 нм. В некоторых вариантах реализации базовый состав не содержит частиц размером более 20 нм. В некоторых вариантах реализации базовый состав не содержит частиц размером более 10 нм. В некоторых вариантах реализации базовый состав представляет собой масляный раствор, по существу не содержащий частиц. В некоторых вариантах реализации базовый состав представляет собой прозрачный раствор.
Составы, известные из существующего уровня техники и предложенные в виде безводных однофазных гелей (патент США №2012/0316108), имеют вид прозрачных для видимого света суспензий наночастиц, но в действительности не являются истинными растворами. Известные из существующего уровня техники составы, полученные путем а) смешивания компонентов с образованием первичной дисперсии, содержащей один или более фосфолипид(ов) и избыток воды; b) гомогенизации первичной дисперсии с образованием нанодисперсии со средним размером частиц, составляющим менее чем примерно 200 нм в диаметре; с) пропускания нанодисперсии через фильтр с диаметром пор 0,2 или 0,45 мкм и d) удаления воды до содержания менее 5%, предпочтительно менее 3% и более предпочтительно менее 1% по массе, не образуют прозрачные растворы и содержат частицы. Другими словами, эти составы представляет собой нанодисперсии, а не прозрачные растворы.
В соответствии с некоторыми вариантами реализации состав не содержит Сахаров, таких как, без ограничения, сахароза, декстроза, лактоза, глюкоза, трегалоза, мальтоза, маннит, сорбит, глицерин, амилоза, крахмал, амилопектин или их смесь.
В одном из вариантов реализации природный несинтетический фосфолипид может представлять собой любой из вышеописанных фосфолипидов. В другом варианте реализации природный несинтетический фосфолипид присутствует в составе в количестве от примерно 10% до примерно 80% по массе. В еще одном варианте реализации фосфолипид представляет собой несинтетический липид, такой как 1,2-димиристоил-sn-глицеро-3-фосфоглицерин (ДМФГ) или его фармацевтически приемлемая соль. В еще одном варианте реализации состав согласно изобретению не содержит 1,2-димиристоил-sn-глицеро-3-фосфоглицерина (ДМФГ) или его фармацевтически приемлемой соли. В еще одном варианте реализации композиции согласно изобретению по существу не содержат ДМФГ. В этом контексте термин "не содержит ДМФГ" относится к концентрации менее 0,5%, предпочтительно менее 0,1 %.
В одном из вариантов реализации первый фосфолипид присутствует в составе в количестве от примерно 40% до примерно 60% по массе. В другом варианте реализации фосфолипид представляет собой фосфатидилхолин (ФХ) или его фармацевтически приемлемую соль.
В одном из вариантов реализации неводный фармацевтически приемлемый носитель присутствует в составе в количестве от примерно 20% до примерно 50% по массе. В другом варианте реализации неводный фармацевтически приемлемый носитель представляет собой касторовое масло, кунжутное масло, хлопковое масло, сафлоровое масло или один или более триглицеридов. В еще одном варианте реализации соотношение между фосфолипидами и неводным носителем находится в диапазоне 3:1-1:2, 2,5:1-1:5, 2,2:1-1:1,2, 2:1-1:1. Каждый из возможных вариантов представляет собой отдельный вариант реализации изобретения.
В одном из вариантов реализации сорастворитель представляет собой спирт. В другом варианте реализации спирт представляет собой этанол. Этанол может присутствовать в составах в количестве от примерно 1% до примерно 15% по массе. Как объяснено выше, сорастворитель может выступать в качестве регулятора вязкости. В некоторых вариантах реализации вязкость базового состава составляет менее 2500 сПуаз. В некоторых вариантах реализации вязкость базового состава составляет менее 2000 сПуаз. В некоторых вариантах реализации вязкость базового состава находится в диапазоне 1000-2500 сПуаз. В некоторых вариантах реализации вязкость базового состава находится в диапазоне 1000-2000 сПуаз.
Неводные пролипосомные депо-составы необязательно содержат антиоксидант. Подходящие антиоксиданты были описаны выше.
Неводные нелипосомные депо-составы могут необязательно содержать дополнительные ингредиенты, такие как различные вспомогательные вещества, регуляторы рН, хелаторы металлов, такие как ЭДТА или эдетовая кислота, соли, красители и т.п.
Несмотря на то, что описанный в настоящем тексте пролипосомный неводный депо-состав определен как неводный (или по существу не содержащий воды), может сохраняться присутствие остаточного или следового количества молекул воды, например, из ингредиентов, применяемых для получения состава. Такой депо-состав тем не менее считают неводным. В некоторых вариантах реализации содержание остаточной влаги, определенное с помощью автоматического метода определения содержания воды по Карлу Фишеру, составляет менее 0,3%. В некоторых вариантах реализации содержание остаточной влаги, определенное с помощью автоматического метода определения содержания воды по Карлу Фишеру, составляет менее 0,15%.
Способы получения нелипосомных неводных базовых составов
В соответствии с некоторыми вариантами реализации пролипосомные неводные фармацевтические композиции согласно изобретению получают путем добавления гидрофобного АФИ к предварительно приготовленному базовому составу. Применение этого способа предпочтительно исключает необходимость добавления избытка этанола и последующего выпаривания этанола в присутствии АФИ.
Неожиданно было обнаружено, что сочетание нагревания, перемешивания с приложением вращающего момента и большого сдвигового усилия приводит к полному растворению фосфолипида в фармацевтически приемлемом неводном носителе.
Базовый состав получали следующим образом:
1. Фармацевтически приемлемый неводный носитель (например, касторовое масло) и необязательно сорастворитель (такой как спирт, например, этанол), необязательно содержащий антиоксидант (например, гидрохлорид цистеина), приводят в равновесие при 65°C.
2. Добавляют природный несинтетический фосфолипид (лецитин, например, Фосфолипон 90G) и перемешивают с большим вращающим моментом и большого сдвигового усилия при 65°C.
3. После полного растворения несинтетического фосфолипида смесь охлаждают до комнатной температуры.
4. Необязательно, полученный на стадии 3 продукт переносят во флаконы и/или стерилизуют (например, посредством автоклавирования).
После этого гидрофобный АФИ (например, кетопрофен) может быть добавлен к предварительно приготовленному базовому составу. В альтернативном варианте гидрофобный АФИ может быть добавлен на стадии 1 данного способа, на стадии 2 данного способа, на стадии 3 данного способа или на стадии 4 данного способа. Каждый из возможных вариантов представляет собой отдельный вариант реализации изобретения.
Специалисту в данной области техники будет понятно, что усовершенствованный способ получения устраняет необходимость добавления избытка этанола в процессе растворения. Это позволяет избежать последующего удаления избытка этанола из состава путем выпаривания в присутствии АФИ.
Неожиданно было обнаружено, что состав согласно настоящему изобретению может быть подвергнут стерилизации, например, без ограничения, автоклавированию, без потери активности или нарушения консистенции состава. В этом заключается отличие от сходных составов, получаемых способом, включающим образование нанодисперсии, которую впоследствии лиофилизируют с получением безводного геля. Автоклавирование последнего приведет к разрушению присутствующих в геле наночастиц и, фактически, потере активности и нарушению консистенции состава.
Введение пролипосомных неводных депо-составов
Описанные в настоящем тексте композиции подходят для доставки гидрофобного АФИ и медленного его высвобождения (пролонгированного высвобождения).
Описанные в настоящем тексте депо-составы могут применяться в различных терапевтических целях, требующих введения состава с медленным высвобождением.
В некоторых вариантах реализации фармацевтический состав вводят в виде депо-состава. В некоторых вариантах реализации депо-составы могут быть введены парентерально. В некоторых вариантах реализации депо-составы вводят посредством инъекции в разрез. В некоторых вариантах реализации составы вводят посредством подкожной (внутрикожной) инъекции. В некоторых вариантах реализации фармацевтический состав вводят посредством инфильтрации в разрез, или депо-состав вводят посредством многократных внутрикожных инъекций малых доз вдоль пораженных нервных путей или окончаний. Кроме того, депо-состав может быть введен посредством имплантации, местно или с применением трансдермальных кожных пластырей, хорошо известных специалистам в данной области техники. В случае введения с помощью трансдермальной системы доставки, введение дозы может быть скорее непрерывным, чем периодическим, для всей схемы дозирования. Другие типовые препараты для местного введения включают кремы, мази, лосьоны, распыляемые аэрозоли и гели, концентрация АФИ в которых находится в диапазоне от примерно 0,1% до примерно 10% (масс/масс.) или (масс/об.).
В некоторых вариантах реализации депо-состав вводят с применением устройства для доставки лекарственных средств, подходящего для парентеральной доставки, такого как шприц.
В некоторых вариантах реализации фармацевтический состав является терапевтически активным в течение по меньшей мере примерно 24 часов с момента введения. В некоторых вариантах реализации состав является терапевтически активным в течение 24-48 часов. В некоторых вариантах реализации фармацевтический состав является терапевтически активным в течение по меньшей мере примерно 48 часов. В некоторых вариантах реализации фармацевтический состав является терапевтически активным в течение 48-72 часов. В некоторых вариантах реализации фармацевтический состав является терапевтически активным в течение по меньшей мере примерно 72 часов.
Этот продолжительный период доставки лекарственных средств согласно изобретению обеспечивают посредством разовой инъекции с относительно высокой нагрузкой АФИ, содержащегося в описанных в настоящем тексте депо-составах, или посредством введения повторных последовательных малых доз, без риска взрывного высвобождения, которое может встречаться в случаях инъекций липосом, внезапного увеличения дозы до токсичных уровней или уровней, способных оказывать неблагоприятное воздействие на внутренние системы организма. В одном из вариантов реализации доставку АФИ в организм субъекта осуществляют без необходимости повторного применения шприца и/или без необходимости повторного наполнения шприца или повторного введения дозы через некоторый период времени.
В одном из вариантов реализации по существу непрерывная доставка АФИ (например, посредством инфузии, диффузии и т.д.) может быть выполнена с помощью, например, устройства для доставки лекарственных средств в виде внешнего или имплантируемого насоса. Пути доставки, предполагаемые настоящим изобретением, включают, но не ограничиваются перечисленными: имплантаты, парентеральные пути введения (например, подкожную инъекцию, инфильтрацию или капельное введение, внутривенное, внутримышечное, интраспинальное введение, инфильтрацию и т.п), а также местное введение. Парентеральный путь доставки в открытую рану или область около раны (например, инфильтрация в хирургический разрез) представляет особый интерес.
Если необходим более длительный период доставки лекарственного средства, введение депо-состава можно повторять. Как правило, введение депо-состава можно повторять два, три или более раз в течение периода времени в диапазоне от примерно 1 недели до примерно 12 месяцев или более.
Фактическая доставляемая доза лекарственного средства может легко быть рассчитана специалистом в данной области техники и будет варьировать в зависимости от множества факторов, таких как активность и другие свойства выбранного применяемого лекарственного средства (например, гидрофобность).
Количество АФИ, эффективное для лечения или предотвращения заболевания, может быть определено стандартными клиническими методами. Кроме того, для облегчения установления оптимальных диапазонов доз, необязательно могут быть проведены анализы in vitro или in vivo. Точная применяемая доза также может зависеть от пути введения и серьезности состояния, подлежащего лечению, и может быть определена в соответствии с решением лечащего врача и с учетом обстоятельств для каждого субъекта, например, опубликованных данных клинических исследований. Подходящие эффективные дозы, однако, находятся в диапазоне от примерно 0,1% до примерно 10% АФИ из расчета на массу депо-состава. В одном из вариантов реализации эффективная доза составляет примерно 0,2%, 0,5%, примерно 1%, примерно 1,5%, примерно 2%, примерно 2,5%, примерно 3%, примерно 3,5% мг, примерно 4%, примерно 4,5%, примерно 5%, примерно 6% мг, примерно 7%, примерно 8%, примерно 9% или примерно 10%.
Эквивалентные дозы можно вводить через различные периоды времени, включая, но не ограничиваясь перечисленными: примерно каждые 12 часов, примерно каждые 24 часа, примерно каждые 36 часов, примерно каждые 48 часов, примерно каждые 72 часа, примерно каждую неделю, примерно каждые две недели, примерно каждые три недели, примерно каждый месяц и примерно каждые два месяца. Эффективные дозы, описанные в настоящем тексте, относятся к общим вводимым количествам; то есть, если вводят более одного АФИ, эффективные дозы соответствуют общему введенному количеству.
Режим дозирования для описанных в настоящем тексте депо-составов на основе АФИ может быть выбран в соответствии с различными факторами, включая тип, возраст, массу тела, пол и медицинское состояние субъекта; тяжесть состояния, подлежащего лечению; путь введения; показатели почечной или печеночной функции субъекта и вид конкретного применяемого АФИ.
Депо-составы гидрофобного АФИ могут быть введены в виде однократной суточной дозы, или общая суточная доза может быть введена в раздельных дозах с двумя, тремя или четырьмя интервалами за сутки.
Депо-составы могут быть исследованы in vitro или in vivo на наличие необходимой терапевтической или профилактической активности до применения на людях. Для доказательства безопасности и эффективности описанных в настоящем тексте депо-составов могут применяться модельные животные системы.
Согласно настоящему изобретению дополнительно предложено введение профилактического или терапевтического агента субъекту, получающему АФИ. В одном из вариантов реализации другой профилактический или терапевтический агент вводят в эффективном количестве.
В одном из вариантов реализации другой профилактический или терапевтический агент представляет собой агент, подходящий для уменьшения любого возможного побочного эффекта АФИ. Такие возможные побочные эффекты включают, но не ограничиваются перечисленными, тошноту, рвоту, головную боль, низкий уровень лейкоцитов в крови, низкий уровень эритроцитов в крови, низкий уровень тромбоцитов, головную боль, лихорадку, сонливость, боль в мышцах, общую боль, диарею, нейропатию, зуд, стоматит, потерю волос, тревогу или депрессию.
В одном из вариантов реализации депо-состав на основе АФИ может быть введен до, во время или после введения другого профилактического или терапевтического агента, или в тот же день, или с разницей между введениями, составляющей 1 час, 2 часа, 12 часов, 24 часа, 48 часов или 72 часа.
Эффективные количества других профилактических или терапевтических агентов хорошо известны специалистам в данной области техники. Однако определение оптимального диапазона эффективных количеств другого профилактического или терапевтического агента находится в пределах компетенции специалиста в данной области техники. В одном из вариантов реализации изобретения, отличающемся введением субъекту другого профилактического или терапевтического агента, эффективное количество АФИ в депо-составе меньше его эффективного количества для вариантов реализации без введения другого профилактического или терапевтического агента. В этом случае, без привязки к какой-либо теории, полагают, что АФИ и другой профилактический или терапевтический агент действуют синергически.
Предложенные согласно настоящему изобретению фармацевтические композиции для парентерального введения содержат продукт в соответствии с изобретением в сочетании с одним или более фармацевтически приемлемыми стерильными неводными растворами, дисперсиями, суспензиями или эмульсиями или стерильными порошками, которые непосредственно перед применением могут быть восстановлены в стерильные растворы или дисперсии для инъекций, которые могут содержать антиоксиданты, буферы, бактериостатические агенты, растворенные вещества, придающие составу изотоничность крови предполагаемого реципиента, или суспендирующие агенты или загустители.
Продукты, содержащие описанные в настоящем тексте депо-составы
Наборы
Согласно настоящему изобретению предложены наборы, которые могут упростить введение субъекту депо-состава гидрофобного АФИ.
Пример набора содержит лекарственную форму для однократного введения депо-состава гидрофобного АФИ.
В одном из вариантов реализации лекарственная форма для однократного введения представляет собой емкость, которая может быть стерильной, содержащую эффективное количество депо-состава на основе АФИ. Набор может дополнительно содержать листок-вкладыш или печатные инструкции с информацией по применению депо-состава на основе АФИ.
Набор также может содержать емкость, содержащую базовый состав в соответствии с любым из вышеприведенных вариантов реализации. Набор может дополнительно содержать гидрофобный АФИ.
Набор также может дополнительно содержать лекарственную форму для однократного введения другого профилактического или терапевтического агента, например, емкость, содержащую эффективное количество другого профилактического или терапевтического агента. В некоторых вариантах реализации набор содержит емкость, содержащую депо-состав на основе более одного АФИ. Примеры потенциальных АФИ перечислены выше.
Предварительно наполненные шприцы
В соответствии с некоторыми вариантами реализации согласно настоящему изобретению предложен предварительно заполненный шприц, наполненный описанным в настоящем тексте депо-составом. Депо-состав содержит гидрофобный АФИ. Предварительно наполненный шприц также может иметь иглу, подходящую для инъекции депо-составов, или другие средства для введения состава. В одном из вариантов реализации игла представляет собой иглу калибра 18-25 G. В другом варианте реализации игла представляет собой иглу калибра 21 G.
Настоящее изобретение дополнительно проиллюстрировано следующими примерами, которые не являются ограничивающими. Специалисты в данной области техники должны признать или быть способными знают или способны установить, используя лишь рутинные эксперименты, удостовериться в существовании многочисленныхе эквивалентовы конкретных веществ и способов, описанных в настоящей настоящем текстезаявке. Та. Такие эквиваленты находятся в рамках входят в объем формулы изобретения, следующей ниже после примеров. В описании был приведен ряд ссылок, полное содержание которых включено в настоящую заявку посредством ссылки.
ПРИМЕРЫ
Пример 1
Получение пролипосомного неводного маслянистого депо-состава
Пролипосомный неводный маслянистый депо-состав ропивакаина получали следующим образом. Необходимое количество моногидрата ропивакаина гидрохлорида помещали в предварительно взвешенную (масса тары) круглодонную колбу и добавляли необходимое количество гидрохлорида цистеина. В колбу добавляли необходимое количество лецитина (PL90G, фосфатидилхолина) с последующим добавлением необходимого количества касторового масла (порядок добавления ингредиентов не имеет значения). В колбу добавляли абсолютный этанол в количестве, равном необходимому итоговому количеству или превышающем его. Колбу герметично закрывали и взвешивали. Колбу с ингредиентами помещали в водяную ультразвуковую ванну и нагревали до примерно 50°C. Когда все ингредиенты были растворены и количество абсолютного этанола превысило необходимое итоговое количество, колбу присоединяли к подходящему испарительному устройству (например, ротационному испарителю Rotavapor) и поддерживали температуру водяной бани на уровне примерно 50°C. Величину вакуума доводили до 200 мбар, и колбу вращали со скоростью примерно 60 об/мин. Давление постепенно снижали с шагом 10 мбар, пока оно не достигло 40 мбар. Выпаривание продолжали до тех пор, пока масса колбы не свидетельствовала о содержании необходимого итогового количества абсолютного этанола, составлявшего 6% или менее (масс/масс), как рассчитывали путем взвешивания колбы с ее содержимым или определения количества выпаренного и охлажденного спирта, собранного в ловушке вне испарителя. Колбу и ее содержимое оставляли для охлаждения до комнатной температуры. При необходимости добавляли абсолютный этанол для достижения содержания 6% (масс/масс). Если определяли, что раствор содержал менее 6% (масс/масс.) абсолютного этанола, этанол добавляли для получения необходимого процентного содержания (масс/масс). Колбу можно было хранить в холодильнике или при комнатной температуре до момента времени, на который было запланировано наполнение флаконов или шприцев. Колбу вновь нагревали и вращали в нагретой до 50°C водяной ультразвуковой ванне в течение примерно 1 часа перед разливом в емкости для готовой лекарственной формы. Колбу и ее содержимое оставляли для охлаждения до комнатной температуры. Полученным раствором заполняли стеклянные флаконы или другие емкости с применением подходящего разливочного устройства.
Как можно видеть из приведенного выше описания способа, воду не добавляли в течение всего этого процесса.
Пример 2
Пролипосомные неводные маслянистые составы ропивакаина
Неводный пролипосомный маслянистый состав ропивакаина получали в соответствии со способом, описанным в примере 1. В таблице 1А представлены компоненты этого состава (состав А).

В таблице 1В представлены компоненты менее качественных составов, содержащих синтетические фосфолипиды, применяемых для сравнения с составом А.

Исследование стабильности образцов в прозрачных флаконах проводили для образцов, хранившихся в климатических камерах при комнатной температуре (КТ) и в условиях ускоренной оценки стабильности при 40°C в течение периода времени от нескольких недель до нескольких месяцев, посредством визуального осмотра образцов на присутствие какого-либо осадка, изменение цвета и изменение прозрачности с последующим подтверждением величины дозировки химическими методами. Было обнаружено, что составы 3 и 4 образовывали осадки при КТ, которые превращали данные составы в практически непрозрачные гели. Эти осадки не поддавались повторному диспергированию даже при интенсивном встряхивании. Напротив, предпочтительный депо-состав А сохранял химическую и физическую стабильность более 24 месяцев при этих условиях. Было также отмечено, что состав А был прозрачным и бесцветным, без видимых частиц. Таким образом, был сделан вывод о том, что составы, содержащие синтетические фосфолипиды, не подходят для дальнейших коммерческих разработок.
Пример 3
Получение пролипосомного неводного базового депо-состава Был успешно найден способ получения пролипосомного неводного готового для применения депо-состава, не требующий выпаривания этанола в присутствии АФИ. Сочетание нагревания, перемешивания с приложением вращающего момента и большого сдвигового усилия обеспечило получение готового для применения базового 5 состава, который являлся стабильным, не содержал избытка этанола и облегчал добавление требуемого АФИ.
Базовый состав получали следующим образом:
Необходимые количества касторового масла, этанола и цистеина (предварительно растворенного в этаноле) добавляли в емкость и приводили в равновесие при 65°C. Затем добавляли лецитин с последующим перемешиванием с большим вращающим моментом и большого сдвигового усилия при 125 об/мин и 3577 об/мин, соответственно, в течение приблизительно получаса. После полного растворения лецитина смесь выливали в колбы и выдерживали в течение ночи с получением прозрачных масел. Как должно быть понятно из описания способа, на протяжении всего этого процесса воду не добавляли.
Специалисту в данной области техники будет понятно, что базовый состав согласно изобретению облегчает добавление различных гидрофобных АФИ.
Кроме того, следует понимать, что усовершенствованный способ получения устраняет необходимость добавления избытка этанола в процессе растворения. В результате можно избежать последующего удаления избытка этанола из состава путем выпаривания в присутствии АФИ. В таблице 2 приведены компоненты базового состава.

Пример 4
Пролипосомные неводные маслянистые составы ропивакаина
Неводный пролипосомный маслянистый базовый состав получали в соответствии со способом, описанным в примере 3. Ропивакаин добавляли к готовому для применения базовому составу. В таблице 3 представлены составляющие композиций, содержащих различные количества ропивакаина.

Конечный состав был прозрачным и бесцветным, без видимых частиц. Конечный состав был стабилен в течение по меньшей мере одного месяца в условиях ускоренной оценки стабильности. Предпочтительно, состав был стабилен более 2 месяцев, более 4 месяцев, более 6 месяцев или дольше.
Пример 5
Пролипосомные неводные маслянистые композиции на основе диклофенака
Неводный пролипосомный маслянистый базовый состав получали в соответствии со способом, описанным в примере 3. Далее добавляли диклофенак к готовому для применения базовому составу. В таблице 4 представлены составы, содержащие различное количество диклофенака.

Конечный состав был прозрачным и бесцветным, без видимых частиц. Конечный состав был стабилен в течение по меньшей мере одного месяца в условиях ускоренной оценки стабильности. Предпочтительно, состав был стабилен более 2 месяцев, более 4 месяцев, более 6 месяцев или дольше.
Пример 6
Пролипосомные неводные композиции на основе дексаметазона
Неводный нелипосомный маслянистый базовый состав получали в соответствии со способом, описанным в примере 3. Дексаметазон добавляли к готовому для применения базовому составу. В таблице 5 представлены композиции, содержащие различные количества дексаметазона.

Конечный состав был прозрачным и бесцветным, без видимых частиц. Конечный состав был стабилен в течение по меньшей мере одного месяца в условиях ускоренной оценки стабильности. Предпочтительно, состав был стабилен более 2 месяцев, более 4 месяцев, более 6 месяцев или дольше.
Пример 7
Пролипосомные неводные композиции на основе кетопрофена
Неводный нелипосомный маслянистый базовый состав получали в 20 соответствии со способом, описанным в примере 3. Далее добавляли кетопрофен к готовому для применения базовому составу. В таблице 6 представлены композиции, содержащие различные количества кетопрофена.


Конечный состав был прозрачным и бесцветным, без видимых частиц. Конечный состав был стабилен в течение по меньшей мере одного месяца в условиях ускоренной оценки стабильности. Предпочтительно, состав был стабилен более 2 месяцев, более 4 месяцев, более 6 месяцев или дольше.
Пример 8
Пролипосомные неводные маслянистые составы на основе оксикодона
Неводный пролипсомный маслянистый состав на основе оксикодона получали в соответствии со способом, описанным в примере 1. В таблице 7 приведены компоненты состава.

Пример 9
Пролипосомные неводные маслянистые составы на основе фентанила
Неводный пролипсомный маслянистый состав на основе фентанила получали в соответствии со способом, описанным в примере 1. В таблице 8 приведены компоненты состава.


Пример 10
Измерения вязкости депо-композиций ропивакаина
Вязкость депо-состава А ропивакаина, а также составов 3 и 4 ропивакаина измеряли посредством метода вращения шпинделя с применением ротационного вискозиметра Брукфильда (Brookfield, модель DV-II), оснащенного шпинделем №5; при температуре водяной бани 30°C, скоростью вращения 30, 60 и 100 об/мин.
Состав А ропивакаина имел более низкую вязкость (1720 сПуаз) по сравнению с составами 3 и 4, которые содержали ДМФГ (3031 сПуаз). Это различие было явно выражено в повышенных характеристиках текучести депо-состава А, а также более простой работе с ним в целом (с точки зрения проходимости через иглу и возможности введения посредством инъекции), что делает состав А ропивакаина более подходящим для парентерального введения. Составы, не содержащие этанола, имели более высокую вязкость и оказались непригодными для парентерального введения.
Пример 11
Измерения вязкости композиций ропивакаина с различными концентрациями сорастворителя
Вязкость депо-составов ропивакаина, содержащих различное количество этанола, измеряли посредством метода вращения шпинделя с применением ротационного вискозиметра Брукфильда (Brookfield, модель DV-II), оснащенного шпинделем №5; при температуре водяной бани 30°C, скоростью вращения 30, 60 и 100 об/мин. Как следует из данных таблицы 9, измерения вязкости демонстрировали уменьшение вязкости с повышением концентрации этанола.

Пример 12
Проверка проходимости через иглу депо-составов ропивакаина с различными концентрациями сорастворителя
Проходимость через иглу депо-композиций ропивакаина, содержащих различное количество этанола, измеряли путем продавливания через шприц с иглой калибра 21G заданного количества композиции на предварительно взвешенную чашку.
Как следует из данных таблицы 10, скорость прохождения через иглу возрастала с повышением концентрации этанола.
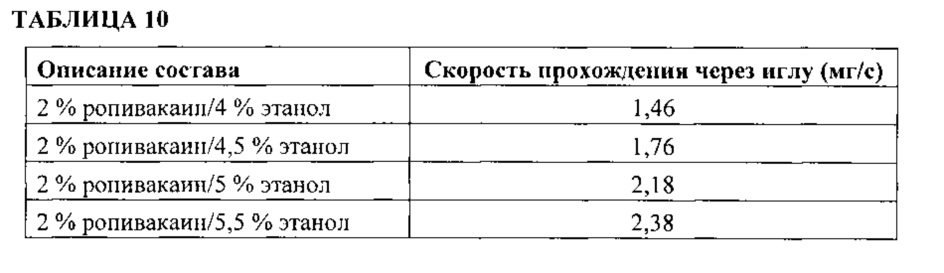
Пример 13
Образование липосомоподобных структур под воздействием солевого раствора или плазмы свиньи
Депо-состав А ропивакаина хранили в сцинтилляционных флаконах, и в состав медленно вводили 0,9% раствор NaCl или плазму свиньи до количества не более 50% (масс/масс.) из расчета на общую массу состава с получением соотношения состав/солевой раствор 1:1. Полученные смеси затем перемешивали при 200 об/мин с применением шейкера с водяной баней при температуре 37°C.
Физические характеристики депо-состава А, содержащего солевой раствор или плазму свиньи в соотношении 1:1, оценивали с применением следующих методов:
1. Распределение частиц по размерам: анализ распределения частиц по размерам выполняли с применением анализатора размера частиц Coulter LS230.
2. Морфологическая оценка с применением просвечивающей криоэлектронной микроскопии (крио-ПЭМ): морфологическую оценку проводили посредством ПЭМ (на микроскопе FEI Technai 12 G2 с ускоряющим напряжением 120 кВ, оснащенном криодержателем Gatan, охлаждаемым до -180°C, и изображения записывали на охлаждаемую камеру с медленной разверткой, представляющую собой устройство с зарядовой связью (CCD), от производителя Gatan).
Результаты анализа распределения частиц по размерам (фигуры 1А и В) свидетельствуют об образовании многослойных липосомных везикул (МЛВ) в обеих смесях. Определенный для смеси состав А/разбавляющий солевой раствор средний/срединный размер частиц, составлявший примерно 1,4 мкм, позволял предположить присутствие большего количества многослойных частиц, чем мицелл или капель эмульсии типа "масло-в-воде", размеры которых находятся в наномасштабном диапазоне. Определенный для смеси депо-состав А/плазма свиньи (1:1) средний/срединный размер частиц составлял примерно 20 мкм, что значительно превышало значение для смеси депо-состав А/солевой раствор (1:1) и указывало на образование МЛВ после контакта с физиологическими жидкостями in vivo.
Для проведения морфологической оценки посредством крио-ПЭМ состав А разбавляли плазмой свиньи с получением разведений 1:1 и 1:2. Затем образцы подвергали исследованию методом крио-ПЭМ. В качестве контроля применяли не смешанную с составом плазму свиньи.
Как можно видеть на фигуре 2, многослойные везикулы и похожие липидные ассоциаты были легко обнаружены после разбавления состава А плазмой свиньи в соотношениях 1:1 и 1:2. Этот результат резко контрастировал с отсутствием таких структур в контрольной пробе плазмы свиньи.
Был сделан вывод о том, что депо-состав А образовывал МЛВ под воздействием плазмы свиньи и что добавление модификатора вязкости этанола неожиданно не влияло на процесс образования этих липосомных везикул при воздействии водных растворов. Более того, эти результаты показывают, что медленное высвобождение и продолжительное облегчение боли, достигаемое при введении состава А согласно настоящему изобретению, наиболее вероятно, обусловлены образованием липосом или других липидных везикулярных структур и не требуют гелевой или гелеобразной консистенции, которую обычно считают необходимой.
Пример 14
Эффективность пролипосомных неводных составов ропивакаина in vivo
В этом исследовании оценивали способность составов ропивакаина облегчать послеоперационную боль на модели молодых свиней с применением методологии фон Фрея. Нити для тестов фон Фрея получают из нейлонового волокна различного диаметра. Нити должны быть вплотную прижаты к коже с достаточным усилием таким образом, чтобы нити сгибались и принимали U-образную форму. Количество единиц грамм-силы, необходимое для сгибания каждого волоска, постоянно, т.е. эти нити могут применяться для приложения очень точного и воспроизводимого усилия при исследовании конкретных, заранее определенных участков кожи, что делает метод фон Фрея подходящим инструментом для диагностики, исследований и скрининга. Этот метод можно легко применять для изучения участков кожи с нормальной чувствительностью, а также гипер- или гипочувствительных участков.
Различные вводимые свиньям составы ропивакаина представлены в таблице 11. Термин "4% ропивакаин" относится к 4% ропивакаину в форме свободного основания.

В таблице 12 приведены экспериментальные группы данного исследования. Термин "4% ропивакаин" относится к 4% ропивакаину в форме свободного основания.

Во время проведения операции поросята находились под наркозом, вызванным с помощью смеси изофлуран/кислород, вводимой через дыхательную маску. На левом боку делали разрез кожи и фасции длиной 6-7 см, не затрагивая мышц. Разрез на коже закрывали с применением стерильного шовного материала. Исследуемые инъецируемые (TI) препараты, плацебо, исследуемый состав в форме геля (состав 3), исследуемый состав в форме масляного раствора (состав А) или положительный контроль осторожно вводили в полученную хирургическим путем полость посредством двух инъекций объемом 2,5 мл (5 мл/животное). Каждую инъекцию выполняли с применением нового шприца объемом 5 мл с фиксатором иглы через иглу калибра 18 G. Каждую обработку проводили в одном и том же направлении (от головы к хвосту). Каждый разрез выполняли с помощью подвергнутого автоклавированию стерильного набора хирургических инструментов. После разрезания кожи свиньи не обязательно получали 2 вида антибиотиков. Животных держали под наркозом в течение всего времени проведения операции и введения препаратов (примерно 20 минут).
Изучаемые препараты вводили только один раз в день исследования 0 в виде подкожной инъекции под шов. Исходную реакцию животных в тестах фон Фрея определяли в день исследования -1. Данные, принятые в качестве исходного уровня, представляли собой данные, зарегистрированные в день исследования -1. Животных включали в исследование, если прикладываемое к коже на боках усилие, при котором наблюдали реакцию отдергивания, на исходном уровне составляло ≤26 г. Предпочтительно, прикладываемое к коже на боках усилие, соответствующее реакции отдергивания, на исходном уровне составляло 60 г на обоих боках. За боль принимали реакцию отдергивания, для получения которой прикладываемое к коже на боках усилие составляло ≤10 г.
Нити фон Фрея (Ugo Basile) прикладывали к поверхности кожи на боках на расстоянии приблизительно 0,5 см от линии разреза. По мере увеличения числа граммов волосков происходило увеличение силы, воздействующей на кожу на боках. Максимальное усилие составляло 60 г. Нити прикладывали до тех пор, пока животное не демонстрировало реакцию отдергивания от раздражителя. Каждый волосок прикладывали 3-5 раз.
Как показано на фигуре 3, состав А, представляющий собой маслянистый раствор, неожиданно обеспечивал более длительное облегчение боли чем все другие составы, исследованные в эксперименте, включая составы, содержащие идентичные концентрации этанола и ропивакаина. Это следует из величины силы, показанной на фигуре 3 через 18 ч после введения дозы. Этот результат был неожиданным, поскольку маслянистый раствор имел меньшую вязкость, чем гелеобразный состав, содержащий ДМФГ. Депо-составы обычно представляют собой твердые имплантаты или гелеобразные вещества, которые не растворяются или не распределяются при введении в ткань. Соответственно, в целом считают, что более вязкие составы дольше сохраняются в ткани, чем менее вязкие составы, так как более вязкое вещество растворяется медленнее, если вообще растворяется.
Было обнаружено, что в случае, когда вязкость состава находилась в диапазоне 1000-2000 сПуаз, время удержания и продолжительность действия были оптимальными, несмотря на пониженную способность состава проходить через иглу.
Обнаруженный факт, что депо-состав в форме маслянистого раствора (состав А) обеспечивал благоприятный профиль замедленного высвобождения, был непредвиденным и неожиданным и позволил предположить, что добавление этанола в качестве уменьшающего вязкость агента действительно не оказывало негативного воздействия ни на эффективность ропивакаина, ни на продолжительность облегчения боли.
Для оценки распределения ропивакаина между тканью раны и кровью образцы ткани раны и крови забирали через 4 дня после введения либо Наропина, либо состава А, и концентрации ропивакаина определяли посредством ВЭЖХ/МС/МС.
Как можно видеть из представленных в таблице 13 результатов, значительно более высокая концентрация ропивакаина в ране была достигнута в случае применения депо-состава А по сравнению с инъекцией препарата Наропин.

Эти результаты свидетельствуют о том, что депо-состав А обеспечивал поддержание более высоких концентраций ропивакаина в непосредственной близости от раны даже четыре дня после инъекции.
На основании всех вышеприведенных результатов был сделан вывод о том, что депо-состав А превосходил коммерческий состав Наропин, а также гелевые составы, и, соответственно, этот состав был выбран для дальнейшего усовершенствования.
Пример 15
Фаза I - Клиническое исследование эффективности на здоровых людях-добровольцах
Цель этого исследования заключалась в оценке начала и продолжительности обезболивания под действием 2,5 мл каждого из следующих препаратов: депо-состава А, раствора ропивакаина (Наропина) и состава-плацебо в форме геля, вводимых путем подкожной (ПК) инъекции в экспериментальной модели боли на человеке.
В часть 1 исследования были включены пятнадцать (15) субъектов-мужчин, состояние которых оценивали в течение 72 часов после инъекции. Каждый субъект сам выступал в качестве своего контроля и получал одновременно все препараты в форме подкожных инъекций объемом 2,5 мл. В таблице 14 представлены препараты, полученные каждым пациентом.

На спине каждого добровольца отмечали четыре ограниченные области, и каждый из этих трех препаратов вводили посредством инъекции в одну произвольно назначенную область, в то время как четвертую произвольно выбранную область использовали в качестве не подвергаемой инъекциям контрольной области. Эффект местного обезболивания, вызываемый исследуемыми и контрольными препаратами, оценивали посредством определения порога тактильной чувствительности (нити фон Фрея), теста уколом булавкой (Pinprick test, PPT), тестов на ощущение холода и переносимость боли при тепловом воздействии. Исследования показали, что продолжительность облегчения боли была больше при введении состава А по сравнению с раствором Наропина. В соответствии с этим, процентная доля субъектов, достигших эффекта обезболивания в течение ≥24 часов, была больше после введения состава А, как показано в таблице 15.

Часть 2 исследования представляла собой фармакокинетический анализ, в который были включены девять субъектов. Каждый субъект получал подкожно в участок на спине посредством однократной инъекции объемом 2,5 мл либо маслянистый депо-состав А (6 субъектов), либо коммерческий раствор ропивакаина для инъекций (0,5% Наропин) (3 субъекта). Образцы венозной крови (объемом 9 мл), для измерения концентрации ропивакаина в плазме, забирали непосредственно перед введением препаратов (момент времени 0) и через 0,5, 1, 1,5, 2, 3, 6, 9, 12, 18, 24, 30, 36, 48 и 72 часа после введения препаратов.
Как показано на фигуре 4, концентрация ропивакаина в крови была значительно выше в случае введения состава А по сравнению с Наропином уже через 3 часа после инъекции. Продолжительность действия состава А превышала 48 часов и была значительно больше таковой для раствора ропивакаина (Наропина), действие которого длилось всего лишь не более 12 часов. В таблице 16 кратко представлены основные фармакокинетические параметры. Как можно видеть из приведенных в таблице данных, значение Cmax для состава А совпадало с аналогичным значением для инъекции Наропина и было различимо ниже значения Cmax, полученного для липосомных составов ропивакаина и составляющего приблизительно 0,87 мг/мл, а также различимо ниже порога токсического действия, составляющего 0,6 мг/мл, что указывало на отсутствие первоначального взрывного высвобождения ропивакаина. Это облегчает введение высокой концентрации ропивакаина в виде однократной инъекции.


По результатам этого исследования был сделан вывод о том, что состав А характеризовался благоприятным профилем пролонгированного высвобождения по сравнению с коммерчески доступным 0,5% раствором ропивакаина и что безопасность состава А не подлежала сомнению.
Пример 16
Распределение частиц депо-составов по размерам
Распределение по размерам частиц депо-состава, не содержащего гидрофобных АФИ (полученного в соответствии с примером 3), измеряли методом динамического рассеяния света с применением анализаторов размера частиц Malvern Zetasizer, Coulter N4plus или Nicomp 300, позволяющих измерять размер частиц в субмикронном диапазоне (анализируемый диапазон ≥0,5 нм ≤1 мкм).
Аналогично, размер частиц известного из существующего уровня техники состава, обозначенного в настоящем тексте "состав 5" (см. патент США №2012/0316108), также измеряли с целью сравнения. Эталонный состав 5 получали в соответствии со способом, включающим стадии а) смешивания компонентов с образованием первичной дисперсии, содержащей один или более фосфолипид(ов) и избыток воды; b) гомогенизации первичной дисперсии с образованием нанодисперсии со средним размером частиц, составляющим менее чем примерно 200 нм в диаметре; с) пропускания нанодисперсии через фильтр с диаметром пор 0,2 или 0,45 мкм и d) удаления воды до содержания менее 5%, предпочтительно менее 3% и более предпочтительно менее 1% по массе. В таблице 17 представлены компоненты известного из существующего уровня техники состава 5.


Известный из существующего уровня техники состав 5 получали следующим образом:
1. Делали навески кунжутного масла, соевого лецитина и бензилового спирта в стеклянную колбу.
2. Добавляли этанол концентрации USP 200 proof и вращали колбу для растворения всех компонентов.
3. Высушивали в вакууме для удаления этанола до содержания менее 1% по массе.
4. Добавляли КН2РО4, ЭДТА и деионизированную воду.
5. Гомогенизировали с образованием нанодисперсии.
6. Доводили рН до 7,0+/- 0,2 с помощью NaOH/HCl.
7. Подвергали нанодисперсию стерилизации посредством фильтрации через фильтр с диаметром пор 0,2 мкм.
8. Подвергали нанодисперсию лиофилизации для удаления воды до содержания менее 2%.
9. Добавляли этанол.
10. Перемешивали с получением безводного геля.
Пример 17
Эффективность in vivo пролипосомных неводных маслянистых составов ропивакаина по сравнению с известными из существующего уровня техники составами
Сравнивали распределение ропивакаина между тканью раны и кровью для различных составов. Содержащие ропивакаин Наропин, состав А или состав 5, полученный в соответствии с описанным в примере 16 способом, капельно вводили в хирургическую рану только один раз в день 0. Образцы ткани раны или экссудата и крови забирали ежедневно в течение 4 дней после введения. Концентрации определяли посредством ВЭЖХ/МС/МС, как описано в примере 13.
Реферат
Изобретение относится к медицине, в частности к пролипосомной фармацевтической депо-композиции, способу ее получения, пролипосомному неводному базовому составу, способу его получения, а также наборам для введения гидрофобного активного фармацевтического ингредиента. Согласно настоящему изобретению предложены пролипосомные неводные фармацевтические депо-композиции, содержащие природный несинтетический фосфолипид на основе лецитина, масло в качестве неводного фармацевтически приемлемого носителя, этанол в качестве регулятора вязкости. Осуществление изобретения позволяет получить пролипосомную неводную депо-композицию, обеспечивающую поддержание терапевтически эффективных уровней АФИ в течение нескольких дней, при этом пролипосомная депо-композиция представляет собой стабильный прозрачный маслянистый раствор, отличающийся низкой вязкостью. 12 н. и 41 з.п. ф-лы, 4 ил., 17 табл., 17 пр.
Комментарии