Применение стабильных при хранении вязких депо-фосфолипидов для лечения ран - RU2578437C2
Код документа: RU2578437C2
Чертежи







Описание
Уровень техники
Рана - это повреждение организма в результате насилия, несчастного случая или хирургического воздействия, которое обычно включает разрыв или нарушение мембран и обычно приводит к повреждению низлежащих тканей и/или костей. Merriam-Webster′s Collegiate Dictionary, 11e издание, 2004.
Лечение ран зависит от типа, причины, локализации и глубины раны, а также от того, какие другие структуры затронуты, кроме кожи. Тем не менее, обычно лечение включает промывание, обработку антибиотиками, защиту раны путем наложения марлевой прокладки для поглощения жидкостей и предотвращения дальнейшего загрязнения и/или закрытие раны для предотвращения дальнейшего загрязнения. Закрытие раны обычно осуществляют с помощью швов, металлических зажимов, скоб и липких полосок.
Вообще говоря, перед нанесением на рану антибиотиков риск инфекции в ране снижается за счет удаления инородного материала и отмерших тканей в ходе адекватной промывки раны и/или санации раневой полости.
Для удаления раневой жидкости и отмерших тканей, для уменьшения количества бактерий в ране, для улучшения кровоснабжения в раневом ложе и в окружающей ткани и/или для стягивания краев раны и стимуляции роста клеток могут применяться закрывающие устройства на основе вакуумных систем, которые постоянно или периодически прикладывают к поверхности раны субатмосферное давление. Такие закрывающие рану вакуумные системы описаны, например, в патентах США 4949880, 5100396, 5261893, 5527293, 6071267, 7799004, а также в Gestring, Negative Pressure Wound Therapy, UpToDate, May 2010, содержание которых включено в настоящее описание в виде ссылок.
Депо - способ введения активного ингредиента, такого как антибиотик, в организм пациента для системного или локального действия. Обычно его вводят с помощью подкожных или внутримышечных инъекций или вливаний в другие ткани организма, сосуды или полости. Депо-формы могут наноситься на рану до остановки кровотечения, зашивания, бинтования или много закрытия. В отличие от удаляемых депо, биодеградируемые депо-формы через определенный промежуток времени распадаются или деградируют. Обычно это происходит после выхода всего заключенного в него активного фармацевтического ингредиента. В других вариантах биодеградируемые инъектируемые депо-формы высвобождают свой активный фармацевтический ингредиент практически одновременно с постепенной деградацией или в результате такой деградации. Важным преимуществом некоторых биодеградируемых высвобождающих депо-форм является их способность высвобождать лекарственный препарат непосредственно в целевое место действия, обеспечивая повышенные локальные концентрации лекарственного препарата по сравнению с системными значениями.
Депо-формы могут также модулировать высвобождение лекарственного препарата для получения различных профилей высвобождения. Профиль высвобождения может характеризоваться немедленным высвобождением (всплеск) с последующим стационарным состоянием, может иметь, помимо прочего, постоянную (или «нулевого порядка») скорость высвобождения, может обеспечивать медленный рост до установления стационарного состояния или даже обеспечивать задержанное высвобождение. Кроме того, преимуществом депо-форм является возможность с помощью однократного введения обеспечить высвобождение в течение длительного периода времени. В таком случае, например, на концентрации вещества в крови не могут повлиять проблемы, связанные с несоблюдением пациентом предписанного режима лечения.
Депо-формы могут состоять из отдельных систем, таких как депо на основе микросфер и депо на основе наносфер, или из биодеградируемого геля, обычно состоящего из растворимых матриксных компонентов (полимеров, липидов, углеводов) и либо из органического растворителя, либо из смеси смешивающихся и не смешивающихся с водой растворителей.
Фосфолипиды уже применялись для изготовления депо-форм, содержащих липофильный фармацевтически активный агент. Фосфолипиды растворимы в жирах или в органических растворителях, но не растворимы в воде. Для образования депо часто требуется высокая концентрация образующих депо-форму фосфолипидов. Это может влиять на объем и вязкость получающихся депо-форм, и, как следствие, доступные в настоящее время фосфолипидные депо с трудом можно вводить через обычную иглу или шприц. Ниже приведены ссылки, описывающие основанные на фосфолипидах лекарственные формы WO 89/00077, WO 02/32395, EP 0282405 и патенты США №№5,863,549, 4,252,793, 5,660,854, 5,693,337, а также Wang et al., Lyophilization Of Water-In-Oil Emulsions To Prepare Phospholipid-Based Anhydrous Reverse Micelles For Oral Peptide Delivery, 39 European Journal of Pharmaceutical Sciences, 373-79 (2010).
Ванкомицин представляет собой гликопептидный антибиотик, применяемый для профилактики и лечения инфекций, вызванных грамположительными бактериями. Обычно он является лекарственным препаратом выбора при серьезных инфекциях и эндокардитах, вызванных S. Aureus (золотистым стрептококком), не чувствительными к коагулазе стрептококками, пневмококками, β-гемолитическими стрептококками, корнебактериями группы JK, стрептококками viridans или энтерококками, в тех случаях, когда β-лактамы не могут использоваться из-за лекарственной аллергии или резистентности. Ванкомицин можно комбинировать с другими антимикробными препаратами при лечении в том числе, устойчивого к метициллину коагулаза-отрицательного стафилококкового эндокардита, вызванного протезом клапана, и энтерококкового эндокардита. Его использовали также в качестве альтернативного агента при пневмококковом менингите, вызванном штаммами с пониженной чувствительностью к пенициллину. Ванкомицин использовался в сердечнососудистой хирургии для предотвращения постоперационных инфекций. См. Rybak et al., Vancomycin Therapeutic Guidelines: A Summary of Consensus Recommendations From The Infectious Diseases Society of America, The American Society Of Health-System Pharmacists, and The Society Of Infectious Disease Pharmacists, CID 2009:49 (1 Августа), стр. 325.
Гентамицин представляет собой аминогликозидный антибиотик, используемый для лечения многих типов бактериальных инфекций, особенно вызываемых грамотрицательными бактериями. Его использовали в хирургии, поскольку он действует против таких патогенов, как анаэробы и энтерококки. Гентамицин использовался и в других хирургических областях применения (например, при ортопедических операциях) и в настоящее время его применяют в биодеградируемых коллагеновых имплантах.
Как ванкомицин, так и гентамицин в их доступных в настоящее время солевых формах, а именно в форме гидрохлорида ванкомицина и сульфата гентамицина, являются очень гидрофильными антибиотиками. Кроме того, эти антибиотики, особенно в их солевой форме, с трудом переводятся в пригодные для инъекций депо-формы на основе фосфолипидов или другие лекарственные формы с высоким содержанием жировой фазы, поскольку они не склонны к растворению в фосфолипидах и жирах.
Кроме того, в результате проведения нескольких тестов на стабильность было показано, что ванкомицин и гентамицин деградируют по различным механизмам. Ванкомицин теряет стабильность вследствие гидролиза, а гентамицин деградирует в результате окисления или образования продуктов присоединения. Поэтому лекарственные формы, содержащие хотя бы одно из этих соединений, в целом, чувствительны к таким условиям. Кроме того, и ванкомицин, и гентамицин являются термочувствительными, и их нельзя стерилизовать нагреванием, автоклавированием или гамма-облучением.
Соответственно, попытки создать депо-формы, содержащие соль ванкомицина, соль гентамицина или и то, и другое вместе с фосфолипидом и жиром, встречают множество практических трудностей. Одна из них заключается в том, что требуемая лекарственная форма не должна иметь высокую вязкость, поскольку ее придется стерилизовать путем фильтрации через стерилизующую мембрану с порами около 0.22 микрон или меньше. Имеются также некоторые проблемы с взаимоисключающими требованиями. Например, указанные активные агенты также имеют проблемы с совместимостью с фосфолипидами, которые, как и в случае вязкости, приводят к необходимости низкого содержания фосфолипидов. Однако необходимость получения устойчивого и однородного геля и соответствующих характеристик высвобождения выдвигают противоположные требования.
Соответственно, остается неразрешенная потребность в создании стабильного при хранении вязкого фосфолипидного депо, содержащего ванкомицин, гентамицин, их фармацевтическую соль или их смесь, которое можно вводить путем подкожных или внутримышечных инъекций или инъекций или вливаний в рану или в другие ткани организма, сосуды или полости для лечения и/или предотвращения раневых инфекций.
Раскрытие изобретения
Один аспект настоящего изобретения относится к способу получения депо-форм, включающих гидрофильные водорастворимые фармацевтически активные агенты, включающий: (1) растворение гидрофильной формы ванкомицина, гентамицина или их смеси в воде; (2) формирование эмульсии типа масло-в-воде, содержащей фосфолипид, масло и водный раствор, содержащий ванкомицин, гентамицин или их смесь («эмульсия»); (3) гомогенизацию эмульсии с помощью гомогенизатора высокого давления, такого как MICROFLUIDIZER, для получения «монофазного раствора», (4) обеспечение значения pH эмульсии и/или монофазного раствора в диапазоне между приблизительно 3 и приблизительно 6, в диапазоне между приблизительно 3 и приблизительно 5 или в диапазоне между приблизительно 3 и приблизительно 4, (5) лиофилизацию раствора с установленным значением pH, (6) добавление агента, модифицирующего вязкость, в количестве, достаточном для получения требуемого значения вязкости, (7) предварительную фильтрацию раствора с модифицированной вязкостью для получения прозрачного раствора, (8) удаление некоторого количества, модифицирующего вязкость агента из прозрачного раствора для получения депо-формы, имеющей от приблизительно 5,5 до приблизительно 7,5 масс. % агента, модифицирующего вязкость, в расчете на совокупную массу депо-формы, (9) стерилизацию депо-формы без ее нагревания. В другом воплощении предварительная фильтрация и удаление агента, модифицирующего вязкость, являются необязательными стадиями. Можно использовать любые формы ванкомицина и/или гентамицина, которые являются гидрофильными или могут быть сделаны гидрофильными. В некоторых воплощениях фармацевтически активные агенты представляют собой фармацевтически приемлемые соли ванкомицина или гентамицина. В изобретение включены также депо-формы, содержащие гидрофильные формы ванкомицина, гентамицина или их смесь, независимо от того, изготовлены они с помощью данного способа или какого-либо иного способа.
В одном воплощении указанный способ дополнительно содержит стадию асептического заполнения вязкой депо-формой шприца, колбы или другой подходящей емкости, пригодной для хранения и/или доставки депо-формы к месту лечения или к ране.
В соответствии с другим аспектом изобретения в воде вместе с фармацевтически приемлемыми ингредиентами может быть растворен стабилизирующий агент. Примеры стабилизирующих агентов включают динатриевую соль ЭДТА, глицин, L-гистидин, лимонную кислоту, метионин, аскорбиновую кислоту, L-цистеин, альфа-токоферол и их смеси, но не ограничиваются ими. В еще одном аспекте изобретения депо-формы не включают стабилизирующий агент.
В еще одном воплощении осуществляется стадия гомогенизации эмульсии, которая приводит к получению первичной эмульсии, в которой средний диаметр липидной/масляной прерывистой фазы равен приблизительно 200 нм или меньше, чем приблизительно 100 нм, или меньше, чем приблизительно 80 нм.
Считается (без какого-либо ограничения такой теорией), что уменьшение среднего диаметра липидных капель уменьшает вязкость образующегося гомогенного раствора, что позволяет осуществлять стерилизацию через фильтр, вместо того чтобы использовать нагревательную стерилизующую систему, такую как автоклавирование или стерилизацию гамма-облучением, которые могут влиять на стабильность ванкомицина и/или гентамицина.
Перед стадией гомогенизации эмульсия обычно представляет собой белую, непрозрачную, густую йогуртоподобную массу. После гомогенизации образовавшийся однофазный раствор обычно чистый, прозрачный и по вязкости и текучести напоминает воду.
Хотя настоящее изобретение не ограничивается какой-либо одной теорией, считается, что очень гидрофильные формы ванкомицина, гентамицина или их смеси могут вместе с фосфолипидами образовывать монофазный раствор, как показано в настоящем документе, что дает стабильную при хранении вязкую депо-форму с требуемыми характеристиками. Считается, что чрезвычайно мелкие липидные капельки, возникающие при гомогенизации, а также другие факторы, могут определять окончательные свойства образованной депо-формы.
В соответствии с другим воплощением настоящего изобретения значение pH монофазного раствора находится в интервале от приблизительно 3 до приблизительно 6, в интервале от приблизительно 3 до приблизительно 5 или в интервале от приблизительно 3 до приблизительно 4 в других воплощениях изобретения.
В соответствии с еще одним воплощением настоящего изобретения значение pH вязкой депо-формы - конечного продукта - находится в интервале от 3 до приблизительно 6, в интервале от приблизительно 3 до приблизительно 5, или в интервале от приблизительно 3 до приблизительно 4.
Другой аспект настоящего изобретения относится к депо-форме, содержащей по меньшей мере один гидрофильный водорастворимый фармацевтически активный(е) агент(ы), выбранный из группы, состоящей из фармацевтически приемлемой соли ванкомицина, гентамицина и их смеси, воду, фосфолипид и один или большее число масел с определенным установленным значением pH, а также модифицирующий вязкость агент, где количество воды в депо-форме не превышает приблизительно 4 масс. %, не превышает приблизительно 2 масс. %, не превышает по меньшей мере 1 масс. % или не превышает приблизительно 0,5 масс. % в расчете на совокупную масс депо-формы. В другом воплощении депо-форму можно использовать в шприцах.
В одном воплощении настоящего изобретения депо-форма содержит гидрофильную форму одного или обоих антибиотиков: ванкомицина и гентамицина. В другом воплощении депо-форма включает фармацевтические соли одного или обоих антибиотиков: ванкомицина и гентамицина. В другом воплощении депо-форма включает фармацевтически приемлемые соли ванкомицина или гентамицина. В еще одном воплощении депо-форма содержит фармацевтическую соль ванкомицина или гентамицина.
В одном воплощении в соответствии с настоящим изобретением депо-форма является «прозрачной». Это дает преимущества, связанные с возможностью заметить попавший в раствор воздух, частицы примесей, что позволяет предотвратить случайное введение такого препарата в организм. Интересно то, что, как оказалось, если в депо-форме присутствуют фармацевтически приемлемые соли как ванкомицина, так и гентамицина, то депо-форма по настоящему изобретению оказывается более прозрачной, чем в том случае, если она содержит фармацевтически приемлемую соль только одного из ванкомицина или гентамицина. В таком воплощении, в котором фармацевтически приемлемые соли ванкомицина и гентамицина присутствуют в депо-форме одновременно, такая депо-форма называется «ультрапрозрачной», как определено в настоящем документе. В том случае, если депо-форма содержит фармацевтически приемлемую соль только одного из ванкомицина или гентамицина, такая депо-форма называется «полупрозрачной» или «прозрачной», как определено в настоящем документе.
В другом воплощении модифицирующий вязкость агент представляет собой этанол, причем количество этанола в депо-форме составляет от приблизительно 3 до приблизительно 25,0 масс. %, от приблизительно 4 до приблизительно 10 масс. %, или от приблизительно 5 до приблизительно 6.5 масс. % в расчете на совокупную массу композиции.
В еще одном воплощении количество фосфолипида в депо-форме составляет от приблизительно 5 до приблизительно 95 масс. %, от приблизительно 25 до приблизительно 75 масс. % или от приблизительно 35 до приблизительно 60 масс. % в расчете на совокупную массу композиции.
В соответствии с другим воплощением настоящего изобретения количество присутствующего в депо масла составляет от приблизительно 5 до приблизительно 95 масс. %, от приблизительно 25 до приблизительно 75 масс. % или от приблизительно 35 до приблизительно 60 масс. % в расчете на совокупную массу композиции.
В соответствии с одним из воплощений настоящего изобретения за два часа при измерении по методу USP I с использованием в качестве среды 500 мл деионизованной воды высвобождается не более, чем приблизительно 80%, не более, чем приблизительно 50%, или не более, чем приблизительно 20% ванкомицина и/или гентамицина.
В соответствии с другим аспектом изобретения для улучшения стабильности ванкомицина, гентамицина или и того, и другого депо-форма может содержать стабилизирующий агент. Примеры стабилизирующих агентов включают (без ограничения) ЭДТА (этилендиаминтетрауксусную кислоту), динатриевую соль этилендиаминтетрауксусной кислоты, глицин, L-гистидин, лимонную кислоту, метионин, аскорбиновую кислоту, L-цистеин, альфа-токоферол и их смеси. В соответствии с еще одним аспектом настоящего изобретения вязкая депо-форма не включает стабилизирующий агент. В еще одном воплощении количество использованного стабилизирующего агента, если он присутствует, не будет отрицательно влиять на стабильность каждого из активных агентов депо-формы: ванкомицина или гентамицина.
В другом аспекте изобретения депо-форма находится в аппликаторе, шприце, ампуле или любом другом приспособление, способном хранить депо-форму и/или доставлять ее к месту лечения, месту локализации депо-формы или к ране.
Еще один аспект настоящего изобретения относится к способу внутрикожного, внутримышечного, подкожного, капельного или местного введения вязкой депо-формы по изобретению, содержащей фармацевтически приемлемую соль ванкомицина, гентамицина или их смесь, воду, фосфолипид, масло, агент, контролирующий pH и агент, модифицирующий вязкость.
Еще один аспект настоящего изобретения относится к способу профилактики и/или лечения постоперационных инфекций путем введения депо-формы по настоящему изобретению.
Один из аспектов настоящего изобретения относится к способу профилактики и/или лечения раневой инфекции путем введения в рану депо-формы по настоящему изобретению. В одном из воплощений настоящего изобретения рана выбрана из группы, состоящей из хирургических ран, ортопедических ран, травматических ран, военных ранений и любых их комбинаций.
В соответствии с одним из аспектов настоящего изобретения хирургические раны включают (без ограничения) резаные раны, в которых чистый разрез сделан острым инструментом. В другом аспекте настоящего изобретения ортопедические раны включают (без ограничения) повреждения опорно-двигательного аппарата, конечностей, включая таз, позвоночник и связанные с ним структуры, и любых их комбинаций.
В еще одном аспекте настоящего изобретения травматические раны включают (без ограничения) открытые раны головы, лица, груди, живота, конечностей (включая таз) и/или наружные и/или повреждения с рваными или неровными краями с наличием посторонних примесей и/или омертвевших фрагментов ткани, такие как рваные, колотые раны, ссадины и любые их комбинации.
В другом аспекте настоящего изобретения ранения включают (без ограничения) повреждения, нанесенные взрывными устройствами и/или оружием, стреляные раны, любые упомянутые выше травматические раны и любые их комбинации.
Краткое описание фигур
Фиг. 1 представляет собой диаграмму способа получения композиции по изобретению в соответствии с одним из аспектов настоящего изобретения.
Фиг. 2 иллюстрирует тест на выход ванкомицина и гентамицина для формы из примера 1 после автоклавирования.
Фиг.3 представляет собой профиль высвобождения in vitro гентамицина и ванкомицина для формы из примера 6, полученный методом USP I.
Фиг. 4 иллюстрирует концентрации ванкомицина в плазме кроликов для формы из примера 1.
Фиг. 5 иллюстрирует концентрации ванкомицина в ткани кроликов для формы из примера 1.
Фиг. 6 иллюстрирует концентрации гентамицина в плазме кроликов для формы из примера 1.
Фиг. 7 иллюстрирует концентрации гентамицина в ткани кроликов для формы из примера 1.
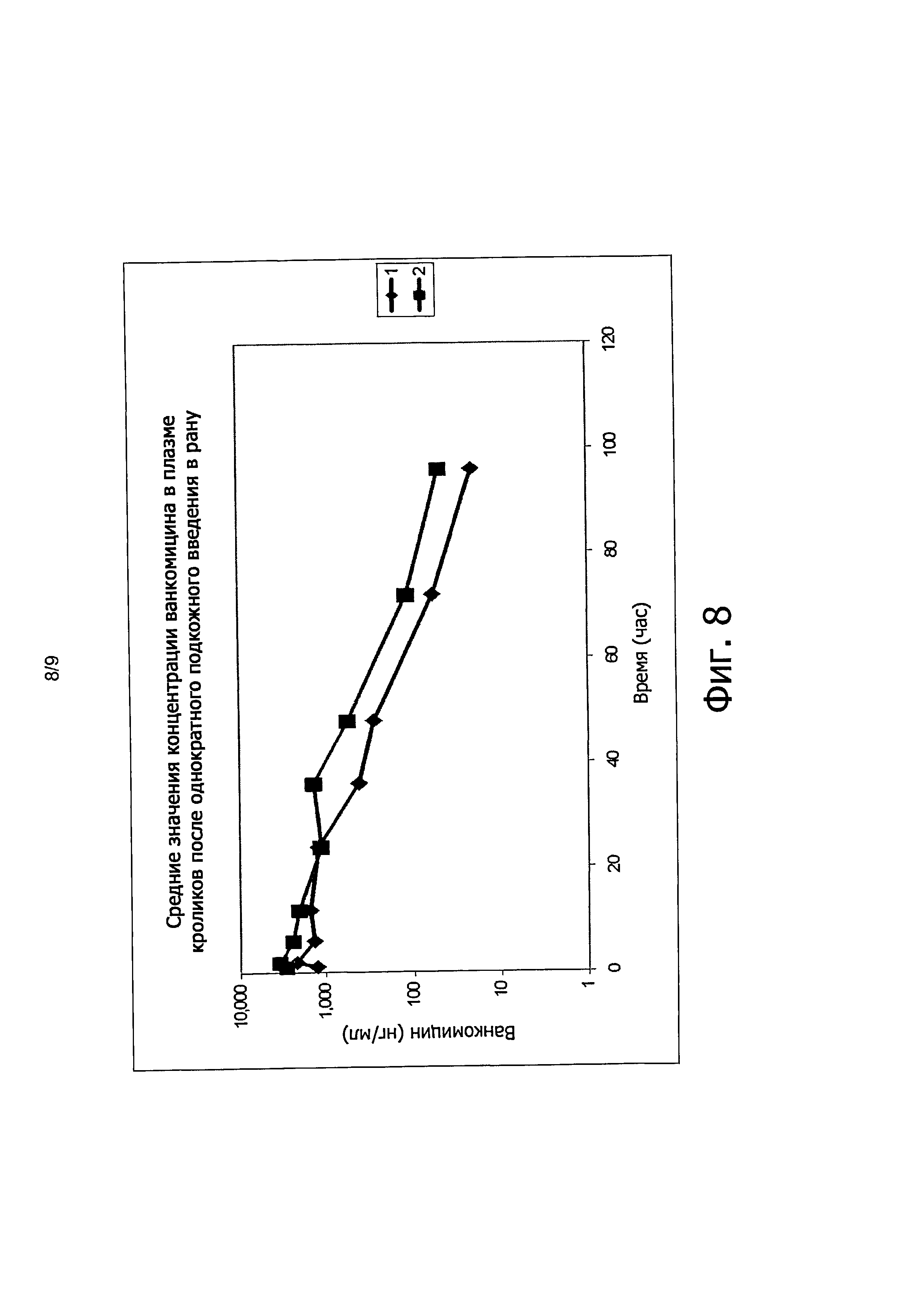
Фиг. 8 иллюстрирует среднюю концентрацию ванкомицина в плазме кроликов после однократного подкожного введения формы из примера 6.
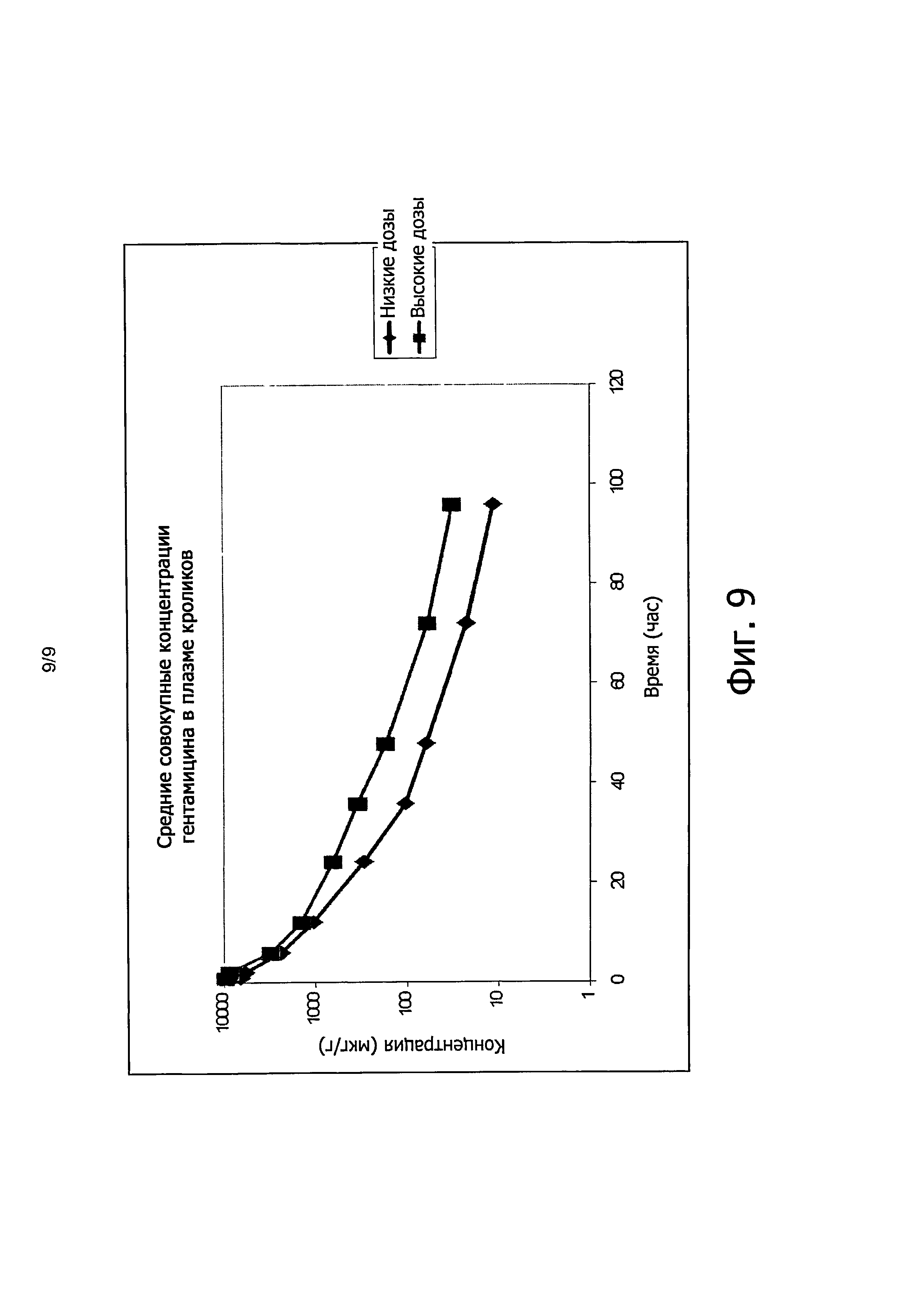
Фиг. 9 иллюстрирует среднюю совокупную концентрацию гентамицина для формы из примера 6 у кроликов.
Подробное описание
Ниже настоящее изобретение будет описано более подробно.
Хотя настоящее описание завершается формулой изобретения, в которой четко описано и заявлено настоящее изобретение, предполагается, что настоящее изобретение будет более понятно из следующего описания. Все приведенные здесь процентные значения и отношения относятся к общей массе композиции, и все измерения, если не указано иное, сделаны при 25°C и нормальном давлении. Все температуры даны в градусах Цельсия, если не указано иное. Настоящее изобретение может содержать (т.е. неисчерпывающий список) или может по существу состоять из компонентов по настоящему изобретению, а также из других описанных здесь ингредиентов или элементов. Используемый в данной заявке термин «содержит» означает присутствие указанного или эквивалентного ему по структуре или функции элемента, а также любого другого элемента или любых других элементов, которые не были перечислены. Термины «имеющий», «содержащий» и «состоящий из» также следует понимать как неисчерпывающие множества, если контекст не предполагает иного. Если в заявке использован термин «состоящий по существу из», то это означает, что изобретение если и включает ингредиенты в дополнение к перечисленным, то только если эти дополнительные ингредиенты по существу не меняют основных и новых свойств заявленного изобретения. Вообще говоря, таких дополнительных ингредиентов может не быть вовсе или они могут присутствовать в следовых количествах. Однако можно включить в состав вплоть до приблизительно 10% по массе материалов, которые могут существенно влиять на основные и новые свойства изобретения, до тех пор, пока соединения сохраняют свою полезность. Все интервалы, перечисленные в настоящем документе, включают крайние точки, включая те случаи, когда интервал задан как лежащий «между» двумя значениями. Такие термины как «приблизительно», «вообще говоря», «главным образом» и тому подобные должны пониматься как то, что предваряемый ими термин или значение не являются абсолютно точными. Значения таких терминов будут зависеть от обстоятельств, и те термины, которые они модифицируют, будут понятны специалистами в данной области техники. Сюда относятся, по крайней мере, величина ожидаемой погрешности эксперимента, технической ошибки и погрешности прибора, характерных для методики, используемой для измерения той или иной величины.
Стоит отметить, что, хотя в описании и формуле изобретения конечный продукт, например, такой как депо-форма или другая дозированная форма по изобретению, может описываться как, например, имеющая такой-то pH на какой-то промежуточной стадии, трудно с уверенностью сказать, что указанное значение справедливо и для конечной дозированной формы. Тем не менее, такое указание может быть оправдано, если указанным свойством обладают материалы, используемые до завершения процесса производства. Аналогичным образом, количество ингредиентов, введенных, например, в эмульсию, в том случае, когда оно описывается с помощью весовых процентов, может измениться на какой-нибудь другой стадии производства, например, для финальной депо-формы, в которой масса может быть больше или меньше. Достаточно того, что такие весовые проценты были правильными на какой-либо из стадий процесса производства и/или для какого-нибудь промежуточного продукта. Действительно, для каждого свойства или характеристики конечного продукта, которые нельзя установить непосредственно для дозированной формы, достаточно, чтобы это свойство определялось компонентами, перечисленными до этапов, относящихся к конечным этапам процесса производства.
Использованный в заявке термин «эмульсия» означает систему из двух несмешиваемых жидких фаз. Одна из двух фаз (внутренняя фаза, прерывистая фаза или дискретная фаза) распределена в виде капель/глобул во второй фазе (внешняя или непрерывная фаза). Использованные в настоящем изобретении эмульсии включают эмульсии типа масло-в-воде (О/W), в которых менее полярная жидкость, обычно обозначаемая как масло, является внутренней фазой; и эмульсии типа вода-в-масле (W/O), в которых водная или другая относительно полярная жидкость является внутренней фазой.
Термин «монофазный раствор» и «наноэмульсия» являются в настоящем изобретении взаимозаменяемыми. Отметим, что термин «раствор» в выражении «монофазный раствор» вовсе не означает, что он является гомогенной смесью двух или большего числа веществ. Он означает лишь результирующий продукт стадии гомогенизации с использованием гомогенизатора высокого давления, такого как MICROFLUIDIZER.
Термин «монофазный» «однофазный» и «подобный однофазному» используют, чтобы обозначить, что результирующий продукт остается в виде одной фазы без разделения фаз или преципитации даже после центрифугирования при 6000 g в течение 10 минут при 25°C в образце, содержащем 1 г вещества, с помощью центрифуги производства Heraeus, Model Biofuge Fresco или любой эквивалентной ей.
Термин «вязкий» в настоящем изобретении означает, что вязкость композиции составляет от приблизительно 1 сантипуаз до приблизительно 5000 сантипуаз, от приблизительно 10 сантипуаз до приблизительно 2000 сантипуаз или от 100 сантипуаз до 1500 сантипуаз.
Использованный в настоящей заявке термин «пригодный для использования в шприцах» означает, что композицию можно вводить с помощью шприца или катетера или набирать из емкости в шприц. Однако это не значит, что композиция по данному изобретению действительно должна находиться в шприце или должна обязательно вводиться с помощью шприца, если только это не предполагается специальным указанием или контекстом.
Термины «пропускающий свет» и «прозрачный» используются взаимозаменяемым образом, чтобы обозначить, что композиция по настоящему изобретению не является мутной или непрозрачной и что в ней не содержится визуально заметных суспендированных частиц. В ней не должно быть также и пузырьков. Кроме того, термин «пропускающий свет» означает, что депо-форма не содержит визуально заметных суспендированных частиц и должно быть также свободным и от пузырьков. Кроме того, термины «пропускающий свет» и «прозрачный» означают также, что депо-формы по настоящему изобретению имеют светопропускание более 90% при 800 нм (Т800) при измерении в кварцевой кювете с оптическим путем 1 см и спиртом в качестве контроля с помощью УФ-видимого спектрофотометра, такого как производимая Pharmacia модель Ultrospec III.
Термины «мутный» или «непрозрачный» означают, что величина Т800 депо-формы меньше, чем приблизительно 90%.
Термин «ультрапрозрачный» означает, что величина Т800 депо-формы больше, чем приблизительно 92 или 95%.
Использованный в данном изобретении термин «стабильный» означает, что (1) композиция остается прозрачной при 25°C в течение по меньшей мере одного года, или (2) композиция остается прозрачной и не разделяется и не преципитирует после центрифугирования, когда композицию выдерживают при 40°C в течение 1 недели.
В настоящем изобретении термины «гель» и «депо-форма» взаимозаменяемы.
Один аспект настоящего изобретения относится к вязкой депо-форме, содержащей гидрофильный водорастворимый фармацевтически активный агент(ы), выбранный(е) из группы, состоящей из ванкомицина, гентамицина, фармацевтически активных солей этих соединений и их смеси, воду, фосфолипид, масло, агент для контроля pH и агент, модифицирующий вязкость, где количество воды в вязкой депо-форме не превышает приблизительно 4 масс. %, не превышает приблизительно 2 масс. % или не превышает приблизительно 0,5 масс. % в расчете на совокупную массу депо-формы.
В соответствии с другим аспектом изобретения вязкая депо-форма может содержать стабилизирующий агент, улучшающий стабильность ванкомицина, гентамицина или и того, и другого. В другом аспекте настоящего изобретения эта вязкая депо-форма находится в шприце, ампуле или любом другом приспособлении, способном доставить депо-форму к месту лечения, к месту локализации депо-формы или к ране.
Фармацевтически активный ингредиент
Фармацевтически активный ингредиент по настоящему изобретению представляет собой гидрофильную форму ванкомицина, гентамицина или их смеси. Это могут быть ванкомицин и гентамицин в форме свободных оснований, солей и их сольватов, которые, как и некоторые из известных солей этих соединений, являются гидрофильными. Они также включают формы, которые не являются гидрофильными сами по себе, например, свободные основания, но которые становятся гидрофильными до, в процессе или после создания депо-формы, в результате, например, образования комплексов. В одном воплощении фармацевтически активный ингредиент по настоящему изобретению представляет собой гидрохлорид ванкомицина, сульфат гентамицина или их смесь. В другом воплощении фармацевтически активные ингредиенты по настоящему изобретению представляют собой гидрохлорид ванкомицина и сульфат гентамицина. В еще одном воплощении фармацевтически активный ингредиент по настоящему изобретению представляет собой либо гидрохлорид ванкомицина, либо сульфат гентамицина.
Примеры фармацевтически приемлемых солей включают, не ограничиваясь только ими, соли с любыми кислотами, которые могут образовывать соли с ванкомицином или гентамицином, такими как уксусная кислота, соляная кислота, лимонная кислота, муравьиная кислота, молочная кислота, янтарная кислота, серная кислота.
Количество фармацевтически активных ингредиентов, которое может присутствовать в вязкой депо-форме, может изменяться в зависимости от числа параметров, включая совокупную величину назначенной дозы, длительность введения, размер депо-формы, а также место и время введения, тип вводимого активного ингредиента, характер введения (например, постоянное, задержанное и т.п.) и т.д. Однако в целом, совокупное количество фармацевтически приемлемого ингредиента может составлять от приблизительно 0,001 до приблизительно 50 масс. %, от приблизительно 0,01 до приблизительно 10 масс. %, или от 0,1 до приблизительно 5 масс. % в расчете на совокупную массу вязкой депо-формы.
Масло
Масло по настоящему изобретению может включать, например, природные масла, такие как растительные масла, животные жиры, витамин Е, сложный эфир витамина Е и подобное, и/или синтетические или полусинтетические масла или их смеси.
Растительные масла означают масла, извлекаемые из семян или орехов растений. Примеры растительных масел включают миндальное масло, бурачниковое масло, масло семян черной смородины, касторовое масло, сафлоровое масло, соевое масло, кунжутное масло, масло из семян хлопка, из косточек винограда, из семян подсолнечника, масло канола, масло из кокосовых орехов, пальмовое масло, апельсиновое масло, кукурузное масло, оливковое масло и т.д., но не ограничиваются ими.
Термин животный жир относится к жирным триглицеридам животного происхождения. Примерами животных жиров могут быть рыбий жир или жиры из других источников, такие как сало, свиное сало и т.д.
Примерами синтетических или полусинтетических жиров являются моно-, ди-, или триглицериды, содержащие C6-C20 насыщенные и/или ненасыщенные жирные кислоты, CAPTEX® (эфиры пропилен гликоля, такие как глицерил трикаприлат/капрат, с различными степенями чистоты); MIGLYOL® (триглицериды каприловой/каприновой кислоты; или триглицериды каприловой/каприновой/линолевой кислоты; или триглицериды каприловой/каприновой/янтарной кислоты; или диэфир пропилен гликоля и каприловой/каприновой кислоты, а также их смеси с другими агентами); CAPMUL® (доступны в разных сортах, например Capmul МСМ. Обычно он является моно- и ди-эфиром глицерина и пропиленгликоля, таким как глицерил моно-олеат и пропилен гликоль монокаприлат. Другой сорт состоит из пропилен гликоля глицерил стеарата). В одном воплощении масло, используемое в соответствии с настоящим изобретением, является кунжутным маслом.
Количество масла, которое может присутствовать в вязкой депо-форме, составляет от приблизительно 5 до приблизительно 95 масс. %, от приблизительно 25 до приблизительно 75 масс. % или от приблизительно 35 до приблизительно 60% от совокупной массы вязкой депо-формы.
В некоторых воплощениях отношение масел к фосфолипидам в вязкой депо-форме может находиться в интервале от приблизительно 20:1 до приблизительно 1:20, от приблизительно 3:1 до приблизительно 1:3 или от приблизительно 1:2 до приблизительно 1:1 по массе.
Фосфолипиды
Фосфолипиды по настоящему изобретению означают липидные молекулы, содержащие одну или несколько фосфатных групп, включая те, которые получены из глицерина (фосфоглицериды, глицерофосфолипиды) или сфингозина (сфинголипиды).
В нескольких воплощениях фосфолипиды являются производными триглицеридов, в которых одна из жирных кислот заменена на фосфатную группу и одну из нескольких азотсодержащих молекул. Цепи жирных кислот гидрофобны, а заряды фосфатных и амино групп делают эту часть молекулы гидрофильной. В результате получается амфифильная молекула.
Согласно Фармакопее США (USP), лецитин - это непатентованное название, относящееся к сложной смеси не растворимых в ацетоне фосфолипидов, состоящих, главным образом, из фосфатидилхолина, фосатидилэтаноламина, фосфатидилсерина и фосфатидилинозитола, скомбинированных с различными количествами других соединений, таких как триглицериды, жирные кислоты и углеводы. Состав лецитина и, как следствие, его физические свойства изменяются в зависимости от источника происхождения лецитина и фосфолипидного состава, например, от содержания фосфатидилхолина и т.д.
В соответствии с одним из воплощений настоящего изобретения используются фармацевтически чистые лецитины, полученные из яиц или сои, которые используются в парентеральных продуктах и которые по существу свободны от раздражающих, аллергенных, воспалительных агентов, или от агентов, вызывающих другие побочные биологические реакции.
В соответствии с практикой настоящего изобретения выбор фосфолипидов для приготовления вязкой депо-формы делали, основываясь на способности фосфолипидов (1) быть химически совместимыми с гидрофильными водорастворимыми фармацевтически активными агентами, выбранными из группы, состоящей из фармацевтически приемлемых солей ванкомицина, гентамицина и их смеси, (2) образовывать монофазный раствор и сохранять малый размер капель в процессе изготовления и в ходе хранения и (3) давать требуемую депо-форму и обеспечивать требуемое высвобождение фармацевтически активного агента.
Примеры фосфолипидов включают сфинголипиды в форме сфингозина и производных (полученные из сои, яиц, мозга и молока), ганглиозиды и фитосфингозиды и производные (полученные из дрожжей), но не ограничиваются ими.
Фосфолипиды могут также быть синтезированы, и примеры общих синтетических фосфолипидов включают (но не ограничиваются ими) диглицерины, такие как 1,2-диауроил-sn-глицерин (DLG), 1,2-димиристоил-sn-глицерин (DMG), 1,2-дипальмитоил-sn-глицерин (DPG), 1,2-дистеароил-sn-глицерин (DSG); фосфатидные кислоты, такие как натриевая соль 1,2-димиристоил-sn-глицеро-3-фосфатидной кислоты (DMPA, Na), натриевая соль 1,2-дипальмитоил-sn-глицеро-3-фосфатидной кислоты (DPPA, Na), натриевая соль 1,2-дистеароил-sn-глицеро-3-фосфатидной кислоты (DSPA, Na); фосфохолины, такие как 1,2-дидеканоил-sn-глицеро-3-фосфохлолин (DDPC), 1,2-дилауроил-sn-глицеро-3-фосфохолин (DLPC), 1,2-димиристоил-sn-глицеро-3-фосфохолин (DMPC), 1,2-дипальмитоил-sn-глицеро-3-фосфохолин (DPPC), 1,2-дистеароил-sn-глицеро-3-фосфохолин (DSPC), 1,2-диолеоил-sn-глицеро-3-фосфохолин (DOPC), 1,2-дилинолеил-sn-глицеро-3-фосфохолин (DLOPC), 1,2-диэрукоил-sn-глицеро-3-фосфохолин (DEPC), 1,2-диэйкозопентаеноил-sn-глицеро-3-фосфохолин (EPA-PC), 1,2-дидокозагексаенил-sn-глицеро-3-фосфохолин (DHA-PC), 1-миристоил-2-пальмитоил-sn-глицеро-3-фосфохолин (МРРС), 1-миристоил-2-стеароил-sn-глицеро-3-фосфохолин (MSPC), 1-пальмитоил-2-миристоил-sn-глицеро-3-фосфохолин (PMPC), 1-пальмитоил-2-стеароил-sn-глицеро-фосфохолин (PSPC), 1-стеароил-2-миристоил-sn-глицеро-3-фосфохолин (SMPC), 1-стеароил-2-пальмитоил-sn-глицеро-3-фосфохолин (SPPC), 1-миристоил-2-олеоил-sn-глицеро-3-фосфохолин (MOPC), 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфохолин (POPC), 1-стеароил-2-олеоил-sn-глицеро-3-фосфохолин (POPC); фосфоэтаноламины, такие как гидрированный соевый фосфоэтаноламин (HSPE), негидрированный фосфоэтаноламин из яиц (EPE), 1,2-дилауроил-sn-глицеро-3-фосфоэтаноламин (DLPE); 1,2-димиристоил-sn-глицеро-3-фосфоэтаноламин (DMPE); 1,2-дипальмитоил-sn-глицеро-3-фосфоэтаноламин (DPPE); 1,2-дистеароил-sn-глицеро-3-фосфоэтаноламин (DSPE); 1,2-диолеоил-sn-глицеро-3-фосфоэтаноламин (DOPE); 1,2-дилинолеил-sn-глицеро-3-фосфоэтаноламин (DLoPE); 1,2-диэруцил-sn-глицеро-3-фосфоэтаноламин (DEPE), 1,2-пальмитоил-sn-глицеро-3-фосфоэтаноламин (POPE); фосфоглицерины, такие как натриевая соль гидрированного соевого фосфатидилглицерина (HSPG, Na), натриевая соль негидрированного яичного фосфатидилглицерина (EPG, Na), натриевая соль 1,2-дилауроил-sn-глицеро-3-фосфоглицерина, (DLPG, Na), натриевая соль 1,2-димиристоил-sn-глицеро-3-фосфоглицерина, (DMPG, Na), аммониевая соль 1,2-димиристоил-sn-глицеро-3-sn-1-фосфоглицерина (DMP-sn-1-G, NH4), натриевая соль 1,2-дипальматоил-sn-глицеро-3-фосфоглицерина (DPPG, Na), натриевая соль 1,2-дистеароил-sn-глицеро-3-фосфоглицерина (DSPG, Na), натриевая соль 1,2-дистеароил-sn-глицеро-3-фосфо-sn-1-глицерина (DSP-sn-1G, Na), натриевая соль 1,2-диолеоил-sn-глицеро-3-фосфоглицерина (DOPG, Na), натриевая соль 1,2-диэруцил-sn-глицеро-3-фосфоглицерина (DEPG, Na), натриевая соль 1,2-пальмитоил-sn-глицеро-3-фосфоглицерина (POPG, Na); фосфатидилсерины, такие как натриевая соль 1,2-димиристоил-sn-глицеро-3-фосфо-L-зина (DMPS, Na), натриевая соль 1,2-дипальмитоил-sn-глицеро-3-фосфо-L-зина (DPPS, Na), натриевая соль 1,2-дистеарил-sn-глицеро-3-фосфо-L-зина (DSPS, Na), натриевая соль 1,2-диолеоил-sn-глицеро-3-фосфо-L-зина (DOPS, Na), натриевая соль 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфо-L-зина (POPS, Na); смешанные фосфолипидные цепи, такие как 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфохолин (POPC), натриевая соль 1-пальмитоил-2-олеолил-sn-глицеро-3-фосфоглицерина (POPG, Na), аммониевая соль 1-пальмитоил-2-олеоил-sn-глицеро-3-фосфоглицерина (POPG, NH4); лизофосфолипиды, такие как 1-миристоил-2-лизо-sn-глицеро-3-фосфохолин (S-lyso-PC), 1-пальмитоил-2-лизо-sn-глицеро-3-фосфохолин (P-lyso-PC), 1-стеароил-2-лизо-sn-глицеро-3-фосфохолин (S-лизо-PC); и пегилированные фосфолипиды, такие как N-(карбонил-метоксиполиэтиленгликоль 2000)-MPEG-2000-DPPE, натриевая соль 1,2-дипальмитоил-sn-глицеро-3-фосфоэтаноламина, натриевая соль N-(карбонилметоксиполиэтиленгликоля-5000)-MPEG-5000-DSPE, натриевая соль 1-2-дистеароил-sn-глицеро-3-фосфоэтаноламина, натриевая соль N-(карбонил-метоксиполиэтиленгликоля 5000)-MPEG-5000-DPPE, натриевая соль 1,2-дипальмитоил-sn-глицеро-3-фосфоэтаноламина, N-(карбонил-метоксиполиэтиленгликоль 750)-MPEG-750-DSPE, натриевая соль 1,2-дистеароил-sn-глицеро-3-фосфоэтаноламина, N-(карбонил-метоксиполиэтиленгликоль 2000)-MPEG-2000-DSPE, натриевая соль 1,2-дистеароил-sn-глицеро-3-фосфоэтаноламина.
Количество фосфолипидов, которое может присутствовать в вязкой депо-форме, может изменяться в зависимости от ряда параметров, включающих вязкость конечной формы, длительность введения, размер депо-формы и способ введения, тип вводимого активного ингредиента, характер введения (например, постоянное, отложенное и т.д.) и т.п. Однако в целом количество фосфолипидов, которое может присутствовать в вязкой депо-форме может составлять от приблизительно 5 до приблизительно 95% в расчете на совокупную массу композиции или от приблизительно 35 до приблизительно 60% в расчете на совокупную массу композиции.
Вода
Вода, которая подходит для использования в настоящем изобретении, включает (но не ограничивается этим) дистиллированную и деионизированную воду или любую другую жидкость, способную растворять гидрофильный водорастворимый ванкомицин и/или гентамицин и способную возгоняться/испаряться в процессе лиофилизации.
Для получения монофазного раствора с помощью гомогенизатора высокого давления, такого как MICROFLUIDIZER, эмульсия типа масло-в-воде должна содержать от приблизительно 10 до приблизительно 80% воды, от приблизительно 30 до приблизительно 80% воды или от приблизительно 60 до приблизительно 80% воды от совокупной массы эмульсии масло-в-воде, чтобы получить при обработке в таком гомогенизаторе, как MICROFLUIDIZER, желаемые свойства.
Однако после получения монофазного раствора большая часть воды может быть удалена с помощью, например, лиофилизации, сублимации и/или испарения.
Ванкомицин разлагается в результате гидролиза, и количество остающейся в конечной вязкой депо-форме воды влияет на долгосрочную стабильность ванкомицина. Когда ванкомицин осаждается, вязкая депо-форма из полупрозрачной превращается в мутную или разделяется на две фазы, как показано в примере 2 настоящего описания.
Соответственно, в настоящем изобретении количество остающейся воды должно быть меньше чем приблизительно 4 масс. %, меньше чем приблизительно 2 масс. % или меньше чем приблизительно 0.5 масс. % воды от совокупной массы вязкой прозрачной депо-формы, чтобы сохранять ванкомицин стабильным при хранении.
Агент для контроля pH
Агент для контроля pH по настоящему изобретению является любой нетоксичной кислотой, щелочью или солью. Примеры агентов для контроля pH включают соляную кислоту, уксусную кислоту, серную кислоту, гидроксид натрия, гидроксид калия, гидроксид аммония, лизин, аргинин и т.д., но не ограничиваются только ими.
Как отмечалось выше, гентамицин разлагается в результате окисления или образования аддуктов. Как показано в примере 4 настоящей заявки, pH влияет на долговременную стабильность гентамицина, и когда гентамицин осаждается, вязкая депо-форма превращается из полупрозрачной в мутную.
Соответственно, pH вязкой депо-формы может составлять от приблизительно 3 до приблизительно 6, в диапазоне от приблизительно 3 до приблизительно 5, или в диапазоне от приблизительно 3 до приблизительно 4.
Стабилизирующий агент
Стабилизирующий агент по настоящему изобретению - это материал, который уменьшает каталитический эффект иона металла в отношении окисления, гидролиза или любых других реакций деградации, и/или увеличивает стабильность гидрофильного водорастворимого фармацевтически активного агента. Примеры таких стабилизирующих агентов включают (но не ограничиваются только ими) ЭДТА (этилен-диаминтетрауксусную кислоту), динатрий эдетат, глицин, L-гистидин, лимонную кислоту, метионин, аскорбиновую кислоту, L-цистеин, альфа-токоферол и их смеси. В некоторых воплощениях количество присутствующего стабилизирующего агента в вязкой депо-форме равно от приблизительно 0,001 до приблизительно 5,0% в расчете на совокупную массу композиции, или от приблизительно 0,01 до приблизительно 1,0% в расчете на совокупную массу композиции. В другом воплощении вязкая депо-форма не включает стабилизирующий агент.
Агент, модифицирующий вязкость
Согласно настоящему изобретению агент, модифицирующий вязкость, представляет собой водную или неводную (кроме примесных количеств воды) жидкость, способную растворять сухую пасту, полученную в результате лиофилизации, сублимации и/или испарения.
Примеры модифицирующих вязкость агентов включают этанол, изопропанол и их смеси, но не ограничиваются только ими. В одном воплощении модифицирующий вязкость агент представляет собой этанол.
Модифицирующий вязкость агент добавляется к сухой пасте, пока она полностью не растворится в этом агенте. Образовавшийся раствор с модифицированной вязкостью может также стать «мутным». В одном воплощении модифицирующий вязкость агент и сухую пасту смешивают при температуре от приблизительно 10 до 80°C или в интервале от приблизительно 50 до приблизительно 70°C, или в интервале от приблизительно 25 до 60°C.
Модифицирующий вязкость агент добавляется к сухой пасте, пока количество модифицирующего вязкость агента не станет равным приблизительно 10, 20, 25 или 30 масс. % в расчете на совокупную массу получившегося раствора. Результирующая вязкость раствора может составлять от приблизительно 10 до приблизительно 200 сантипуаз, от 15 до 100 сантипуаз или от приблизительно 20 сантипуаз до приблизительно 50 сантипуаз.
Вязкость можно определять с помощью цифрового программируемого реометра Brookfield с валом SP-40 или любого другого эквивалентного реометра. Более конкретно, начальная скорость RPM реометра может быть от 0,1 до 1,0, затем RPM снижают до 0,1 со скоростью 0,1 RPM каждые 30 секунд. Измерение вязкости может осуществляться при 0,8 RMP при комнатной температуре приблизительно 30°C.
После этого некоторое количество модифицирующего вязкость агента, используемого для растворения сухой пасты, может быть удалено. Удаление модифицирующего вязкость агента может производиться до тех пор, пока количество оставшегося модифицирующего вязкость агента, присутствующего в вязкой депо-форме, не примет значение от приблизительно 1% до приблизительно 20%, от приблизительно 2% до приблизительно 18% или от приблизительно 5,0% до приблизительно 6,5% от совокупной массы вязкой депо-формы. В случае пересушивания модифицирующий вязкость агент при необходимости может быть добавлен снова.
Вязкость полученной вязкой депо-формы по настоящему изобретению составляет от приблизительно 1 до приблизительно 5000 сантипуаз, от приблизительно 10 до приблизительно 2000 сантипуаз или от приблизительно 100 до приблизительно 1500 сантипуаз.
Описание технологического процесса
Как показано на Фиг. 1, один из аспектов настоящего изобретения относится к процессу получения вязкой депо-формы, содержащей гидрофильный водорастворимый фармацевтически активный агент, выбранный из группы, состоящей из ванкомицина, гентамицина и их смеси, включающий: (1) растворение фармацевтически приемлемой соли ванкомицина или гентамицина или их смеси в воде с образованием водного раствора; (2) формирование эмульсии, содержащей фосфолипид, масло и водный раствор, содержащий фармацевтически приемлемую соль ванкомицина, гентамицина или их смесь; (3) гомогенизацию эмульсии с помощью гомогенизатора высокого давления, такого как MICROFLUIDIZER, для получения монофазного раствора, (4) обеспечение значения pH эмульсии и/или монофазного раствора между приблизительно 3 и приблизительно 6, в диапазоне от приблизительно 3 до приблизительно 5 или в диапазоне от приблизительно 3 до приблизительно 4, (5) лиофилизацию монофазного раствора для получения сухой пасты, (6) добавление агента, модифицирующего вязкость, в количестве, достаточном для достижения требуемой вязкости, (7) предварительную фильтрацию раствора с модифицированной вязкостью для получения прозрачного раствора, (8) удаление некоторого количества модифицирующего вязкость агента из прозрачного раствора для получения вязкой депо-формы, содержащей от приблизительно 1 до приблизительно 20 масс. %, от приблизительно 2 до приблизительно 18 масс. % или от приблизительно 5,5 до приблизительно 7,5 масс. % модифицирующего вязкость агента в расчете на совокупную массу вязкой депо-формы, и (9) стерилизацию вязкой депо-формы без использования нагрева. В другом воплощении предварительная фильтрация и удаление модифицирующего вязкость агента являются необязательными стадиями. В других воплощениях фармацевтически активные агенты являются гидрофильной формой ванкомицина, гентамицина, их фармацевтически приемлемыми солями, их фармацевтически приемлемыми сольватами и/или их смесью.
Растворение солей ванкомицина и/или гентамицина в воде
Первым делом гидрохлорид ванкомицина, сульфат гентамицина или оба этих соединения растворяют в воде для получения водного раствора.
Начальная лекарственная концентрация гидрохлорида ванкомицина в воде составляет от приблизительно 1 до приблизительно 50 мг/мл или от приблизительно 20 до приблизительно 30 мг/мл, а начальная лекарственная концентрация сульфата гентамицина в воде составляет от приблизительно 1 до приблизительно 75 мг/мл или от приблизительно 10 до приблизительно 30 мг/мл.
Формирование эмульсии типа масло-в-воде
Затем водный раствор фармацевтически приемлемых солей ванкомицина и/или гентамицина, фосфолипид, жир, необязательно агент для контроля pH и необязательно стабилизирующий агент смешивают для образования эмульсии типа масло-в-воде.
Гомогенизация для получения монофазного раствора
Затем эмульсия может быть гомогенизирована с помощью миксера с высокой скоростью сдвига и/или гомогенизатора высокого давления, такого как MICROFLUIDIZER, для снижения первичного размера липидов эмульсии до среднего диаметра менее 200 нм, менее 100 нм или менее 80 нм и получения монофазного раствора.
Чтобы создать такой монофазный раствор, эмульсия типа масло-в-воде должна предпочтительно содержать от приблизительно 10 до приблизительно 80% воды, от приблизительно 30 до приблизительно 80% воды или от приблизительно 60 до 80% воды от совокупной массы эмульсии типа масло-в-воде, чтобы получить с помощью гомогенизатора, такого как MICROFLUIDIZER, желаемые реологические характеристики.
Контроль pH
pH может контролировать путем добавления агента для контроля pH до и/или после стадии гомогенизации, так чтобы pH композиции составлял от приблизительно 3 до приблизительно 6, в диапазоне от приблизительно 3 до приблизительно 5, или в диапазоне от приблизительно 3 до приблизительно 4.
В другом воплощении эта стадия осуществляется путем добавления к эмульсии соответствующего количества агента для контроля pH с последующим перемешиванием при высоком усилии сдвига в течение приблизительно 1 минуты. Затем после стадии гомогенизации pH композиции проверяют и при необходимости величина pH может быть снова откорректирована.
Лиофилизация, сублимация или испарение
В результате удаления воды фармацевтически приемлемые соли гентамицина и/или ванкомицина равномерно диспергируются в фосфолипидно/масляных переносчиках. Затем воду из монофазного раствора удаляют путем лиофилизации, сублимации и/или испарения, так что количество остающейся в получившейся сухой пасте или в конечной пригодном для набора в шприц депо-форме воды оказывается меньше, чем приблизительно 4 масс. %, меньше чем приблизительно 2 масс. %, или меньше чем приблизительно 0.5 масс. % в расчете на совокупную массу сухой пасты вязкой прозрачной депо-формы. В другом воплощении эмульсию лиофилизировали с помощью проточного лиофилизатора.
Добавление агента, модифицирующего вязкость.
Агент, модифицирующий вязкость, добавляют к сухой пасте до тех пор, пока сухая паста не растворяется полностью с образованием мутного раствора. Агент, модифицирующий вязкость, можно добавлять к сухой пасте до тех пор, пока количество модифицирующего вязкость агента не станет равным приблизительно 75, приблизительно 50, приблизительно 30 или приблизительно 25 масс. % в расчете на совокупную массу мутного раствора. В одном воплощении модифицирующий вязкость агент и сухая паста могут смешиваться при температуре от приблизительно 10 до 80°C или от приблизительно 25 до 60°C.
Предварительная фильтрация
Мутный раствор затем фильтруют с помощью, например, 0.65 микронного фильтра для получения прозрачного раствора. Непрозрачные компоненты, удаляемые на стадиях предварительной фильтрации, состоят из небольшой фракции ванкомицина (приблизительно 2% согласно целевому анализу) и гентамицина (3-4% согласно целевому анализу). Эту потерю можно компенсировать путем корректировки исходной загрузки или изменения целевых значений. Это необязательная стадия, которая не требуется в некоторых из воплощений изобретения.
Удаление агента, модифицирующего вязкость
Далее удаляют модифицирующий вязкость агент, который был добавлен для растворения сухой пасты. Удаление модифицирующего вязкость агента может осуществляться до тех пор, пока количество модифицирующего вязкость агента, присутствующего в вязкой депо-форме, не составит от приблизительно 1 до приблизительно 50%, от приблизительно 2 до приблизительно 18% или от приблизительно 5 до приблизительно 6,5% от совокупной массы вязкой депо-формы.
В случае пересушивания модифицирующий вязкость агент может быть при необходимости добавлен снова. Удаление модифицирующего вязкость агента может быть выполнено с помощью ротационного испарителя или путем продувки азотом или воздухом. Для измерения количества модифицирующего вязкость агента, удаленного из прозрачного раствора, предназначенного для получения вязкой депо-формы, может применяться термогравиметрический анализ (TGA).
Вязкость получаемой вязкой депо-формы по настоящему изобретению составляет от приблизительно 1 до приблизительно 5000 сантипуаз, от приблизительно 10 до приблизительно 2000 сантипуаз или от приблизительно 100 до приблизительно 1500 сантипуаз. Измерения вязкости могут быть выполнены с помощью любых общепринятых методов, включая использование цифрового программируемого реометра Brookfield, модель № DV-III с валом № SP-40. Это необязательная стадия, которая в некоторых воплощениях изобретения не требуется.
Стерилизующая фильтрация
После этого вязкую депо-форму стерилизуют посредством фильтрации через стерилизующую мембрану с порами размером приблизительно 0.22 микрон или меньше..
Другой аспект настоящего изобретения относится к способу внутрикожного, внутримышечного, подкожного, капельного или местного введения вязкой депо-формы, содержащей фармацевтически приемлемую соль ванкомицина, гентамицина или их смесь, воду, фосфолипид, масло, агент для контроля pH, и агент, модифицирующий вязкость.
Лечение и/или профилактика инфицирования раны
Один из аспектов настоящего изобретения относится к способу профилактики и/или лечения раневой инфекции путем введения в рану депо-формы по настоящему изобретению. В другом воплощении предлагается способ обработки локализованной ткани с целью сделать ее непригодной для обитания патогенных микроорганизмов, посредством введения в рану депо-формы по настоящему изобретению.
Раны включают хронические раны, острые ранения, операционные раны, ортопедические раны, травматические раны, военные ранения и их любые комбинации, но не ограничиваются только ими.
Ортопедические раны включают повреждения опорно-двигательного аппарата, конечностей, включая таз, позвоночник и связанные с ним структуры, а также любые их комбинации, но не ограничиваются только ими.
Травматические раны включают открытые раны головы, лица, грудной клетки, живота, конечностей (включая таз) и/или наружные и/или повреждения с рваными и неровными краями с инородными загрязнителями и/или омертвевшими фрагментами ткани, такие как разрывы, ссадины, колотые раны, проникающие раны и любые их комбинации, но не ограничиваются только ими.
Военные ранения включают повреждения, вызванные взрывными устройствами и/или оружием, стреляные раны, любые из упомянутых выше травматических ран, а также любые их комбинации, но не ограничиваются только ими.
В другом аспекте настоящего изобретения способы дополнительно содержат стадии промывки раны для по существу полного удаления инородного материала и/или омертвевших тканей; и закрытия раны.
В одном из воплощений настоящего изобретения стадия закрытия раны осуществляется с помощью хирургического шва, металлических скреп, скобок или липких полосок.
В другом аспекте настоящего изобретения способы включают введение в рану депо-формы по настоящему изобретению и закрытие раны с использованием системы для закрытия ран с помощью вакуумного насоса.
В еще одном аспекте настоящего изобретения способы включают промывку раны для по существу полного удаления инородного материала и/или омертвевших тканей; использование на ране системы для закрытия ран с помощью вакуумного насоса; введение в рану депо-формы по настоящему изобретению; и закрытие раны.
Кроме того, настоящим изобретением предусматривается способ закрытия раны с использованием депо-формы по настоящему изобретению. Такой способ включает стадию введения депо-формы по настоящему изобретению до, во время или после закрытия раны, с использованием, например, швов, металлических скреп, скоб, бинтов, повязок и/или липких полосок/тесемок.
ПРИМЕРЫ
ПРИМЕР 1: Лекарственная форма, изготовленная в соответствии с настоящим изобретением, и способ получения такой лекарственной формы.
Сто (100) граммов лекарственной формы по настоящему изобретению готовили следующим образом: в начале в лабораторный стакан на 500 мл помещали 0,36 г сульфата гентамицина, 0,24 г гидрохлорида ванкомицина, 53,3 г PL90G, 40 г кунжутного масла и 0,1 г L-гистидина. Затем добавляли воду (мл) для инъекций (WFI), и смесь гомогенизировали с помощью миксера с высоким усилием сдвига при 5000 RPM (об./мин) в течение 15 мин. Полученный монофазный раствор лиофилизировали для удаления воды до уровня менее 0,2% остаточной влаги и для получения сухой пасты. Эту сухую пасту смешивали с водой и/или этанолом для получения лекарственной формы и использовали в нескольких исследованиях, включая примеры 2-5, как показано ниже.
ПРИМЕР 2: Влияние содержания воды на внешний вид лекарственной формы из примера 1
Различные количества воды (от 1.1 до 4.1 масс. %) и этанола (6 масс. %) добавляли в сухую пасту из примера 1 для получения нескольких образцов лекарственной формы из примера 1. Образцы тщательно перемешивали шариковым миксером BeadBeater, центрифугировали для удаления воздушных пузырьков и затем фиксировали начальный внешний вид («Исходный образец»). Далее образцы пропускали через фильтр 0.45 мкм, и фильтраты хранили при 2-8°C для последующего наблюдения («Фильтрованный образец»). Таблица 2 иллюстрирует влияние содержания воды на внешний вид лекарственных форм. Было обнаружено, что содержание воды существенно влияет на внешний вид лекарственных форм:
ПРИМЕР 3: Влияние содержания воды на стабильность гентамицина и ванкомицина
Влияние количеств остаточной воды на стабильность форм гентамицина и ванкомицина из примера 1 оценивали с помощью обработки автоклавированием в течение 60 мин. Как показано в приведенной ниже таблице 3, было обнаружено, что ванкомицин имеет пониженную стабильность в том, что касается восстановления или чистоты при большем содержании остаточной воды. Сколько-нибудь существенного влияния воды на стабильность гентамицина в том же интервале обнаружено не было.
ПРИМЕР 4: Профили стабильности и растворимости гентамицина и ванкомицина в лекарственных формах из примера 1 в зависимости от pH.
Лекарственные формы с установленным значением pH из примера 1 (без добавления воды) помещали в условия 2-8°C для изучения их внешнего вида. (См. приведенную ниже таблицу 4).
Профиль pH-стабильности исследовался путем нагревания образца в течение 60 минут путем автоклавирования (см. таблицу 5 ниже).
Указанные результаты показывают что:
1) pH оказывал влияние на внешний вид лекарственных форм из примера 1. Формы были прозрачными при pH 3,2;
2) pH оказывал влияние на стабильность гентамицина в лекарственной форме. Низкое значение pH (например, pH от 3 до 4) предпочтительно для стабильности гентамицина; и
3) pH не оказывал существенного влияния на стабильность ванкомицина.
ПРИМЕР 5: Профиль стабильности гентамицина в лекарственной форме из примера 1 при pH от 3,0 до 5,5
Готовили образцы лекарственных форм из примера 1 (без добавления воды) при трех разных значениях pH в интервале от 3,0 до 5,5. Кроме того, оценивали также эффект L-гистидина на стабильность лекарственной формы из примера 1 путем сравнения результатов лекарственных форм, содержащих L-гистидин и не содержащих L-гистидина. Стабильность гентамицина и ванкомицина оценивали тем же способом, что и описанный далее в примере 3. Было показано, что:
(1) Стабильность гентамицина в лекарственной форме зависит от pH (для гентамицина предпочтительны низкие значения pH (например, от 3 до 4));
(2) Стабильность ванкомицина в лекарственной форме менее чувствительна к pH в исследованном интервале pH;
(3) L-гистидин увеличивает стабильность гентамицина в исследованном интервале pH; и
(4) L-гистидин уменьшает стабильность ванкомицина в исследованном интервале pH.
Фиг. 2 иллюстрирует исследование восстановления после автоклавирования.
ПРИМЕР 6: Другая лечебная форма, приготовленная в соответствии с настоящим изобретением и способов получения лекарственной формы
Готовили прозрачную желтую стерильную вязкую депо-форму (величина порции: 1500 г), содержащую меньше чем 0.5 масс. %» остаточной воды и имеющую pH 3,3, с помощью многостадийного способа, включающего стадии: (1) эмульсификации, (2) микрофлюидизации/гомогенизации, (3) лиофилизации, (4) разбавления этанолом, (5) предварительной фильтрации, (6) удаления этанола и (7) фильтрации. Простое перемешивание всех перечисленных ингредиентов не может привести к лекарственной форме по настоящему изобретению, так как оно не приводит к прозрачной депо-форме.
Подробные методики для каждой из вышеупомянутых стадий следующие: в начале к сульфату гентамицина и гидрохлориду ванкомицина добавляли воду, чтобы осуществить полное растворение сульфата гентамицина и гидрохлорида ванкомицина. Затем добавляли PHOSPHOLIPON® 90G (от Phospholipid GmbH) и кунжутное масло. Затем осуществляли перемешивание с высоким усилием сдвига при 5000 оборотов в минуту для получения гомогенной эмульсии. Затем pH эмульсии доводили до 3,3±0,2 добавлением 1N HCl. Это делают путем добавления к эмульсии подходящего количества 1N HCl и последующего перемешивания с высоким усилием сдвига в течение 1 минуты. Затем выполняли измерение pH, чтобы убедиться, что эмульсия имеет pH 3,3±0,2.
Затем эмульсию помещали в MICROFLUIDIZER для получения монофазного раствора. Средний размер капель эмульсии определяли с помощью лазерного прибора для измерения светорассеяния.
Затем эмульсию лиофилизовали для удаления воды и получения сухой пасты, содержащей менее 0,5% остаточной воды. Затем сухую пасту смешивали с обезвоженным спиртом. Затем смесь обрабатывали ультразвуком в воде при 60-70°C, до тех пор пока не получали раствор. Затем этот раствор охлаждали до комнатной температуры и предварительно фильтровали через стерильный фильтр 0.65 мкм.
Затем с помощью продувки азотом из раствора удаляли спирт, до тех пор пока остаточное количество спирта не составляло 6,5-7 масс. %, для получения вязкого и прозрачного геля. В случае пересушивания при необходимости вновь добавляли обезвоженный спирт.
В биозащитном вытяжном шкафу в присутствии 40 psi аргона вязкую депо-форму фильтровали через фильтр с порами 0.2 микрон для стерилизации получаемой лекарственной формы. Затем в биозащитном вытяжном шкафу отфильтрованную вязкую депо-форму разливали в стеклянные ампулы.
ПРИМЕР 7: Профиль высвобождения in vitro
Профиль высвобождения лекарственной формы из примера 6, содержащей гентамицин и ванкомицин, in vitro измеряли в соответствии с методом USP I, используя корзиночные центрифуги (100 оборотов в мин при 37°C). 1,36 г лекарственной формы из примера 6 помещали в капсулы размером 000, а заполненные капсулы помещали в корзинки на 40 отделений с перегородками. Фиг. 3 иллюстрирует профиль высвобождения гентамицина и ванкомицина лекарственной формы из примера 6 in vitro, измеренный по методу USP I.
ПРИМЕР 8: Исследование фармакинетики на кроликах
Для выполнения исследования фармакинетики («РК») использовали новозеландских белых кроликов, чтобы оценить доставку лекарственных форм, изготовленных в соответствии с настоящим изобретением. Две лекарственных формы готовили в соответствии с методиками, представленными в примере 1 и примере 6, соответственно, и вводили в хирургическую рану или в подкожный карман. Приведенная ниже таблица 7 показывает результаты исследования фармакинетики на кроликах более подробно:
В первом эксперименте тестировали двух новозеландских белых кроликов. После введения в рану лекарственных форм из примера 1 ванкомицин и гентамицин быстро поглощались с величиной Tmax порядка 1-2 часов. Концентрации Cmax были аналогичны значениям, наблюдаемым у мышей. Концентрации в плазме снижались до почти предельного значения к 36 часам. Концентрации ванкомицина в ткани была максимальной при 72 часах и была выше минимальной ингибирующей концентрации (MIC) в течение 168 часов, как показано на Фиг. 4 и 5.
Концентрации гентамицина в ткани достигала максимума за 72 часа и имела концентрацию на уровне MIC и выше в течение 168 часов, как показано на Фиг. 6 и 7.
Анализ плазмы и ткани выполняли с помощью метода LC-MS/MS (жидкостная хроматография/масс-спектрометрия), и результаты исследования фармакинетики (РК) лекарственной формы из примера 1 суммировали в таблицах 8 и 9, соответственно:
Во втором эксперименте шесть новозеландских кроликов (Группа I) исследовали путем нанесения на рану лекарственной формы из примера 6, содержащей 12,6 мг/кг ванкомицина и 11,46 мг/кг гентамицина; а шесть других новозеландских кроликов (Группа И) исследовали путем введения в рану лекарственной формы из примера 6, содержащей 25,2 мг/кг ванкомицина и 22,9 мг/кг гентамицина.
Гель с меньшей концентрацией (Группа I) привел к среднему значению в месте раны 4 мкг/г как для ванкомицина, так и для гентамицина, в то время как более гель с высокой концентрацией (Группа II) привел к средним значениям 26 и 19, 4 мкг/г для ванкомицина и гентамицина, соответственно, что более чем вчетверо превышает значения MIC (минимальные ингибирующие концентрации). Концентрации ванкомицина и гентамицина в плазме на основе исследований MPI лекарственной формы из примера 6 показывают, что для ванкомицина отношение AUC/MIC (площадь под кривой концентрации/минимальная ингибирующая концентрация) оказалось больше чем 400, а для гентамицина отношение Cmax/MIC (максимальная концентрация/минимальная ингибирующая концентрация) оказалось больше чем 800 при обеих дозах.
Фиг. 8 иллюстрирует средние концентрации ванкомицина в плазме кроликов после однократного подкожного (SC) введения в рану, а Фиг. 9 иллюстрирует среднюю полную концентрацию гентамицина в плазме кроликов. Анализ в плазме и в ткани выполняли с помощью метода LC-MS/MS, а результаты PK лекарственной формы из примера 6 суммированы в приведенной ниже таблице 10.
Принципиальная разница между лекарственными формами из примера 6 (высокая концентрация) и из примера 1 (низкая концентрация) заключается в том, что фармокинетические профили этих двух форм у мелких животных, таких как мыши, одинаковы, а при исследованиях на более крупных животных, таких как кролики, разница оказывается больше, поскольку концентрации в ткани для формы из примера 1 падает ниже значения, равного учетверенной MIC, быстрее, давая меньшую площадь под кривой концентрации (AUC) по отношению к время/площадь.
Сравнительные примеры 1 осуществлялись по тем же методикам, что и примеры 1 или 6, за исключением стадий гомогенизации, удаления этанола и/или предварительной фильтрации.
Сравнительный пример 1 после лиофилизации давал непрозрачную твердую пасту, непригодную для инъекций даже после добавления этанола.
ПРИМЕР 9: Исследование лечения ран у крыс
В этом исследовании использовали взрослых самцов крыс Sprague-Dawley. Это исследование выполняли по протоколу, соответствующему акту об условиях содержания животных, с выполнением правил условия содержания животных и в соответствии с принципами Руководства по уходу за лабораторными животными. Имелось три группы для исследования, каждая из которых включала десять животных. Первая группа не получала лечения, и после промывания рану закрывали. Вторая группа, получавшая четыре 3 мм РММА шарика, содержащих приблизительно 28 мг ванкомицина и 32 мг торбамицина, которые помещались в рану после промывания и перед закрытием. Третья группа получала 1 мл формы, приготовленной в соответствии с примером 1, содержащей приблизительно 17 мг ванкомицина и 19 мг гентамицина, помещенной в рану после промывания и перед закрытием.
Инфицированный открытый дефект бедра был создан с использованием следующей техники: взрослых самцов крыс Sprague-Dawley анестезировали изофлураном и подготавливали для операции; правые бедренные кости экспонировали и стабилизировали подготовленной полиоксиметиленовой пластиной, защищенной шестью нарезными спицами Киршнера. Затем на середине кости сетчатой пилой, охлажденной физиологическим раствором, создавали 6-мм дефект. К дефекту добавляли 30 мг стерильного бычьего коллагена, пропитанного 1×105 КОЕ (CFUs) Staphylococcus aureus в 0,5 мл физиологического раствора. Штамм S. aureus (Xenogen 36), был получен из АТСС 49525, исходно полученного от человека с сепсисом (Caliper LifeSciences, СА USA).
Раны послойно закрывали, и животные восстанавливались. Через шесть часов после исходного «ранения» животных повторно анестезировали, раны открывали, тщательно очищали от всех загрязнителей и промывали 60 мл стерильного физиологического раствора, подаваемого под низким давлением. Раны опять послойно закрывали. Животные восстанавливались. Им обеспечивали полную подвижность, пищу и воду.
Через четырнадцать дней после повреждения животных забивали. Бедро и оборудование обнажали, отделяли друг от друга. Костную ткань замораживали в жидком азоте и измельчали. Образцы кости и импланта по отдельности отправляли для стандартного количественного микробиологического анализа. Вкратце, измельченные образцы кости гомогенизировали с 10 мл физиологического раствора в мешалке. Аналогичным образом, образцы импланта споласкивали 10 мл физиологического раствора в мешалке, затем пробы от отдельных образцов последовательно разбавляли и размещали на триптических соево-агаровых пластинках. После инкубации в течение ночи при 37°C, подсчитывали бактериальные колонии; предел обнаружения составлял 30 КОЕ/г.
РММА шарики с антибиотиками изготавливали в стерильных условиях с помощью цемента для артропластики Palacos R (Zimmer, Dover OH). 40 г ММА сополимерного порошка смешивали с 2,0 г ванкомицина и 2.4 г тобрамицина (Sigma-Aldrich), затем смешивали с 20 мл жидкого ММА-мономера. 3 мм формы затем использовали для получения шариков весом приблизительно 200 мг, содержащих 7 мг ванкомицина и 8 мг тобрамицина каждый. Таким образом, четыре шарика представляли дозу в 28 мг ванкомицина и 32 мг тобрамицина.
Все животные контрольной группы, не получавшие антибиотиков (группа исследований 1) содержали бактерии, в то время как только половина животных, обработанных лекарственной формой по примеру 1, содержала заметное количество бактерий (группа исследований 3). Лекарственная форма по примеру 1 (группа исследований 3) показала значительно более высокие показатели по сравнению с содержащими антибиотики шариками (группа исследований 2) в том, что касалось числа животных, содержащих детектируемое количество бактерий на костях или на имплантах. Подсчет бактерий показал превосходство лекарственной формы, приготовленной в соответствии с примером 1 (группа исследований 3), по сравнению с содержащими антибиотики шариками (группа исследований 2). Количества бактерий у животных, обработанных содержащими антибиотики шариками (группа исследований 2) и у животных, не получавших лечения (группа исследований 1), были весьма схожи.
Хотя настоящее изобретение было описано в настоящем документе со ссылками на конкретные варианты осуществления, следует понимать, что эти варианты только иллюстрируют принципы и области применения настоящего изобретения. Таким образом, следует иметь в виду, что в иллюстративные варианты осуществления настоящего изобретения могут быть внесены многочисленные изменения, и что могут быть разработаны другие решения, не отклоняющиеся от духа и рамок настоящего изобретения, определенных в прилагаемой формуле изобретения.
Реферат
Настоящее изобретение описывает способ профилактики и/или лечения раневой инфекции с помощью введения в рану прозрачной депо-формы, содержащей по меньшей мере два гидрофильных водорастворимых фармацевтически активных агента, выбранных из группы, состоящей из фармацевтически приемлемых солей ванкомицина, гентамицина и их смеси, воду, фосфолипид, масло, агент для контроля pH и агент, модифицирующий вязкость. Масло является выбранным из растительных масел, животных жиров и их смесей. Агент, модифицирующий вязкость, выбран из этанола, изопропанола и их смеси. Содержание воды в вязкой депо-форме не превышает 2 мас.% от совокупной массы депо-формы. Указанная депо-форма вводится посредством подкожной или внутримышечной инъекции или инъекции или вливания в рану. Указанная депо-форма имеет рН от 3 до 5. Вязкая фосфолипидная депо-форма по изобретению является прозрачной и стабильной при хранении. 9 з.п. ф-лы, 9 ил., 11 табл., 9 пр.
Комментарии