Способ получения производных гидроксиалкилкрахмала - RU2329274C2
Код документа: RU2329274C2
Чертежи























Описание
Настоящее изобретение относится к производным гидроксиалкилкрахмала, в частности к производным гидроксиалкилкрахмала, получаемым в соответствии со способом, согласно которому гидроксиалкилкрахмал подвергают реакции с первичной или вторичной аминогруппой в сшивающем соединении или с двумя сшивающими соединениями, где образуемое производное гидроксиалкилкрахмала содержит по меньшей мере одну функциональную группу X, которая способна взаимодействовать с функциональной группой Y в другом соединении, и где указанная группа Y представляет собой альдегидную группу, кетогруппу, гемиацетальную группу, ацетальную группу или тиогруппу. В соответствии с особенно предпочтительным вариантом настоящее изобретение относится к производным гидроксиалкилкрахмала, получаемым по способу, в рамках которого гидроксиалкилкрахмал подвергают реакции с первичной или вторичной аминогруппой в сшивающем соединении, полученный реакционный продукт далее необязательно подвергают реакции со вторым сшивающим соединением, так что образуемое производное гидроксиалкилкрахмала включает по меньшей мере одну функциональную группу X, которая способна взаимодействовать с функциональной группой Y в другом соединении, и где указанная группа Y представляет собой альдегидную группу, кетогруппу, гемиацетальную группу, ацетальную группу или тиогруппу, и далее указанный реакционный продукт подвергают реакции с полипептидом, предпочтительно с полипептидом, таким как AT III, IFN-бета или эритропоэтин, и особенно предпочтительно с эритропоэтином, который включает по меньшей мере одну из указанных функциональных групп Y. Гидроксиалкилкрахмал, который является особенно предпочтительным, представляет собой гидроксиэтилкрахмал. Согласно настоящему изобретению гидроксиалкилкрахмал и предпочтительно гидроксиэтилкрахмал подвергают реакции с линкерным соединением по его восстановленному концу, который необязательно может быть подвергнут окислению перед проведением данной реакции.
Гидроксиэтилкрахмал (HES) представляет собой производное природного амилопектина, который подвергается деградации в организме под действием α-амилазы. HES представляет собой замещенное производное углеводного полимера амилопектина, который присутствует в кукурузном крахмале в концентрации до 95 мас.%. HES демонстрирует полезные биологические свойства и используется в качестве кровезаменителя и при лечении по методу гемодилюции в клинических условиях (Sommermeyer et al., 1987, Krankenhauspharmazie, 8(8), 271-278, и Weidler et al., 1991, Arzneim. - Forschung/Drug Res., 41, 494-498).
Амилопектин состоит из двух фрагментов глюкозы, причем в главной цепи имеются альфа-1,4-гликозидные связи, а в сайтах ветвления обнаружены альфа-1,6-гликозидные связи. Физико-химические свойства данной молекулы в основном определяются типом гликозидных связей. Из-за точечных разрывов альфа-1,4-гликозидных связей образуются спиральные структуры с шестью глюкозными мономерами на виток структуры. Физико-химические, а также биохимические свойства полимера могут быть модифицированы путем замещения. Введение гидроксиэтильной группы может быть осуществлено путем щелочного гидроксиэтилирования. При соответствующей адаптации реакционных условий можно использовать различную реакционную способность в отношении гидроксиэтилирования соответствующей гидроксильной группы в незамещенном глюкозном мономере. В этой связи любой специалист со средним уровнем знаний в данной области может в некотором ограниченном диапазоне оказывать воздействие на характер замещения.
Некоторые способы получения производного гидроксиэтилкрахмала описаны в данной области техники.
В DE 2616086 описан процесс конъюгирования гемоглобина с гидроксиэтилкрахмалом, где на первой стадии сшивающее соединение, например бромциан, связывают с гидроксиэтилкрахмалом и затем гемоглобин связывают с промежуточным продуктом.
Одной важной областью, в которой HES нашел применение, является стабилизация полипептидов, используемых, например, для введения в систему кровообращения с целью достижения определенного физиологического эффекта. Одним конкретным примером таких полипептидов является эритропоэтин, кислый гликопротеин с массой примерно 34000 кДа, который необходим для регулирования уровня эритроцитов в кровотоке.
Хорошо известная в данной области проблема, связанная с применением полипептидов и ферментов, заключается в том, что указанные белки зачастую проявляют неудовлетворительную стабильность. В особенности известно, что эритропоэтин обладает относительно коротким периодом полувыведения из плазмы (Spivak and Hogans, 1989, Blood 73, 90; McMahon et al., 1990, Blood 76, 1718). Данный факт указывает на то, что терапевтические уровни в плазме быстро теряются и должны проводиться повторные внутривенные введения. Кроме того, в некоторых случаях наблюдается иммунный ответ против вводимых пептидов.
В основном считается, что стабильность полипептидов может быть улучшена и иммунный ответ против указанных полипептидов снижен, если полипептид связать с полимерными молекулами. В WO 94/28024 показано, что физиологически активные полипептиды, модифицированные полиэтиленгликолем (PEG), демонстрируют сниженную иммуногенность и антигенность и циркулируют в кровотоке значительно дольше, чем неконъюгированные белки, то есть имеют более длительный клиренс.Однако конъюгатам PEG-лекарственное средство свойственны некоторые недостатки, определяемые, в частности, тем, что они не имеют природной структуры, которая может распознаваться элементами путей деградации in vivo. В этой связи, кроме PEG-конъюгатов были разработаны другие конъюгаты и белковые полимерные образования. В литературе описано множество методов сшивания различных белков и макромолекул, таких как метод с использованием полимеразы (см., например, Wong, Chemistry of protein conjugation and cross-linking, 1993, CRCS, Inc.).
В целом, следует отметить, что имеется потребность в более усовершенствованных полипептидах с улучшенной стабильностью и/или биологической активностью. Указанное особенно относится к эритропоэтину, в случае которого изоформы с высоким уровнем присоединенных сиаловых кислот и, в этой связи с более высокой активностью, должны быть отделены при очистке от изоформ с низким уровнем содержания сиаловых кислот (см. ЕР 0428267 В1). В этой связи было бы очень полезно, если бы были доступны способы производства, которые обеспечивали бы получение высокоактивных полипептидов без потребности в тщательной очистке. К сожалению, получение полипептидов в бактериях или в клетках насекомых часто затруднено, поскольку полипептиды зачастую не продуцируются в таких случаях в соответствующей складчатой, нативной конформации и не имеют соответствующего гликозилирования.
В WO 02/08079 А2 описаны соединения, включающие конъюгат активного средства и гидроксиалкилкрахмала, где активное средство и гидроксиалкилкрахмал соединяются либо непосредственно, либо через линкерное соединение. В случае непосредственного соединения реакцию активного средства и гидроксиалкилкрахмала проводят в водной среде, которая включает по меньшей мере 10 мас.% воды. В настоящее время отсутствуют примеры, относящиеся к производному гидроксиалкилкрахмала, которое присоединялось бы к карбонильной группе, входящей в состав активного реагента, ни в случае альдегидной группы или кетогруппы, ни в случае ацетальной или гемиацетальной группы.
Следовательно, объектом настоящего изобретения является получение производных гидроксиалкилкрахмала, которые способны сформировать химическую связь с другим соединением, например с полипептидом, который включает в качестве функциональной группы тиогруппу или альдегидную группу, кетогруппу, гемиацетальную группу или ацетальную группу. Предпочтительно альдегидная группа, кетогруппа, гемиацетальная группа или ацетальная группа включена в углеводный фрагмент другого соединения.
В этой связи настоящее изобретение относится к способу получения производного гидроксиалкилкрахмала, где указанный гидроксиалкилкрахмал имеет структуру, соответствующую указанной ниже формуле (I)

включающему взаимодействие
- гидроксиалкилкрахмала формулы (I) по его необязательно окисленному восстановленному концу или
- производного гидроксиалкилкрахмала, получаемого в результате реакции гидроксиалкилкрахмала формулы (I) по его необязательно окисленному восстановленному концу с соединением (D), где указанное соединение (D) включает
- по меньшей мере одну функциональную группу Z1, способную взаимодействовать с необязательно окисленным восстановленным концом гидроксиалкилкрахмала, и
- по меньшей мере одну функциональную группу W с соединением (L), включающим
- по меньшей мере одну функциональную группу Z1, способную взаимодействовать с указанным гидроксиалкилкрахмалом, или по меньшей мере одну функциональную группу Z2, способную взаимодействовать с функциональной группой W, входящей в состав указанного производного гидроксиалкилкрахмала, и
- по меньшей мере одну функциональную группу X, способную взаимодействовать с функциональной группой Y другого соединения (М),
где указанную функциональную группу Y выбирают из группы, состоящей из альдегидной группы, кетогруппы, гемиацетальной группы, ацетальной группы или тиогруппы.
В контексте настоящего описания термин «гидроксиалкилкрахмал» (HAS) относится к производным крахмала, которые содержат замещение по меньшей мере одной гидроксиалкильной группой. В этой связи термин «гидроксиалкилкрахмал» в контексте настоящего описания не ограничивается соединениями, в которых терминальный углеводный фрагмент включает гидроксиалкильные группы R1, R2 и/или R3, как показано, например, для ясности в формуле (I), но также относится к соединениям, в которых по меньшей мере одна гидроксильная группа присутствует в любом месте, или на терминальном углеводном фрагменте и/или в оставшейся части молекулы, HAS', при замещении гидроксиалкильной группой R1, R2 или R3.
В контексте настоящего описания алкильная группа может быть линейной или разветвленной алкильной группой, которая может быть соответствующим образом замещена. Предпочтительно гидроксиалкильная группа содержит от 1 до 10 атомов углерода, более предпочтительно от 1 до 6 атомов углерода, более предпочтительно от 1 до 4 атомов углерода, и еще более предпочтительно от 2 до 4 атомов углерода. Выражение «гидроксиалкилкрахмал» также предпочтительно включает гидроксиэтилкрахмал, гидроксипропилкрахмал и гидроксибутилкрахмал, где гидроксиэтилкрахмал и гидроксипропилкрахмал являются особенно предпочтительными.
Гидроксиалкилкрахмал, включающий две или более разных гидроксиалкильных групп, также входит в область настоящего изобретения.
По меньшей мере одна гидроксиалкильная группа, входящая в HAS, может содержать две или более гидроксигрупп. В соответствии с предпочтительным вариантом осуществления настоящего изобретения, по меньшей мере, одна гидроксиалкильная группа, входящая в HAS, содержит одну гидроксигруппу.
Выражение «Гидроксиалкилкрахмал» также включает производные, в которых алкильная группа является моно- или полизамещенной группой. В данном контексте предпочтительно, чтобы алкильная группа была замещена галогеном, в особенности атомом фтора, или арильной группой, при условии, что HAS остается водорастворимым. Кроме того, терминальная гидроксигруппа гидроксиалкила может быть этерифицирована с образованием простого или сложного эфира.
Кроме того, вместо алкила могут также использоваться линейные или разветвленные, замещенные или незамещенные алкеновые группы.
Гидроксиалкилкрахмал представляет собой эфирное производное крахмала. В контексте настоящего изобретения, кроме указанных производных в виде простого эфира, могут также использоваться другие производные крахмала. Например, могут использоваться производные, которые включают этерифицированные гидроксигруппы. Такие производные могут представлять собой, например, производные незамещенных моно- или дикарбоновых кислот, включающих 2-12 атомов углерода, или их замещенных производных. Особенно подходят для использования производные незамещенных монокарбоновых кислот, включающие 2-6 атомов углерода, в особенности уксусной кислоты. В контексте настоящего изобретения предпочтительны ацетилкрахмал, бутилкрахмал и пропилкрахмал.
Кроме того, предпочтительны производные незамещенных дикарбоновых кислот, включающих 2-6 атомов углерода.
В случае производных дикарбоновых кислот полезно, если вторая карбоксигруппа в дикарбоновой кислоте также этерифицирована. Кроме того, в контексте настоящего изобретения также пригодны производные сложных моноалкиловых эфиров дикарбоновых кислот.
В случае замещенных моно- или дикарбоновых кислот могут быть предпочтительны те же замещающие группы, что и указанные выше для случая замещенных алкильных остатков.
Методики этерификации известны в данной области техники (см., например, Klemm D. et al. Comprehensive Cellulose Chemistry Vol.2, 1998, Whiley-VCH, Weinheim, New York, cm. особенно главу 4.4, Esterification of Celulose (ISBN 3-527-29489-9)).
Гидроксиэтилкрахмал (HES) наиболее предпочтителен для всех вариантов осуществления настоящего изобретения.
В этой связи настоящее изобретение также относится к описанному выше способу, в котором гидроксиалкилкрахмал представляет собой гидроксиэтилкрахмал.
HES преимущественно может быть охарактеризован определенным распределением по молекулярной массе и степенью замещения. Имеются две возможности характеристики степени замещения:
1. Степень замещения может быть описана относительно части замещенных глюкозных мономеров в сравнении с соответствующим показателем для всех глюкозных фрагментов (DS).
2. Степень замещения может быть охарактеризована как «молярное замещение» (MS), когда описывают количество гидроксиэтильных групп, приходящихся на фрагмент глюкозы.
Растворы HES присутствуют в виде полидисперсных композиций, где каждая молекула отличается от другой степенью полимеризации, числом и характером сайтов ветвления, а также характером замещения. В этой связи HES представлен смесью соединений с разной молекулярной массой. Следовательно, конкретный раствор HES может быть определен по средней молекулярной массе с помощью статистических методов. В данном контексте Mn вычисляют как среднее арифметическое значение, зависящее от числа молекул. Альтернативно, Mw как среднее значение молекулярной массы отражает единицу, которая зависит от массы HES.
В контексте настоящего описания гидроксиэтилкрахмал может иметь значение средней молекулярной массы (средний вес) от 1 до 300 кДа, где средняя молекулярная масса, равная 5-100 кДа, является более предпочтительной. Гидроксиэтилкрахмал может также демонстрировать степень молярного замещения от 0,1 до 0,8 и соотношение между С2:C6-замещением в диапазоне 2-20 относительно гидроксиэтильных групп.
Что касается остатков R1, R2 и R3 в формуле (I), то отсутствуют какие-либо конкретные ограничения, при условии, что соединение (I) сохраняет способность взаимодействовать с соединением (D) или соединением (L). В соответствии с предпочтительным вариантом осуществления настоящего изобретения R1, R2 и R3 обозначают, независимо, атом водорода или гидроксиалкильную группу, гидроксиарильную группу, гидроксиаралкильную группу или гидроксиалкарильную группу, включающую от 1 до 10 атомов углерода. Водород и гидроксиалкильные группы, включающие от 1 до 6 атомов углерода, являются предпочтительными. Алкильная, арильная, аралкильная и/или алкарильная группа может быть линейной или разветвленной и может быть соответствующим образом замещена.
В этой связи настоящее изобретение также относится к описанному выше способу, в котором R1 , R2 и R3 обозначают независимо атом водорода или линейную или разветвленную гидроксиалкильную группу, включающую от 1 до 6 атомов углерода.
Таким образом, R1, R2 и R3 могут представлять собой гидроксигексил, гидроксипентил, гидроксибутил, гидроксипропил, такой как 1-гидроксипропил, 2-гидроксипропил, 3-гидроксипропил, 1-гидроксиизопропил, 2-гидроксиизопропил, гидроксиэтил, такой как 1-гидроксиэтил, 2-гидроксиэтил, или гидроксиметил. Водород и гидроксиэтильные группы являются предпочтительными, тогда как водород и 2-гидроксиэтильная группа являются особенно предпочтительными.
В этой связи настоящее изобретение относится также к описанному выше способу, в соответствии с которым R1, R2 и R3 обозначают независимо атом водорода или 2-гидроксиэтильную группу.
В соответствии с настоящим изобретением соединение (D) или соединение (L) подвергают реакции с восстановленным концом гидроксиалкилкрахмала посредством реакции функциональной группы Z1 с восстановленным концом, где Z1 включен в соединение (D) или соединение (L).
В соответствии с первым предпочтительным вариантом осуществления настоящего изобретения соединение (D) или соединение (L) подвергают реакции с восстановленным концом гидроксиалкилкрахмала, где указанный восстановленный конец окисляют перед проведением данной реакции.
Окисление восстановленного конца приводит к получению гидроксиалкилкрахмала, в котором терминальная углеводная группа включает лактоновую группу, или в котором терминальная углеводная группа, в зависимости от условий химической реакции и/или природы окислителей, имеет нециклическую структуру, включающую карбоксигруппу.
В соответствии с одним вариантом осуществления настоящего изобретения гидроксиалкилкрахмал, который был окислен по его восстановленному концу, присутствует в виде смеси соединения, включающего лактоновую группу, и соединения, включающего карбоксигруппу. В данной смеси соответствующие соединения могут быть представлены в любом реально достижимом соотношении.
В этой связи настоящее изобретение относится также к описанному выше способу, в соответствии с которым восстановленный конец гидроксиалкилкрахмала окисляют перед проведением реакции с соединением (D) или соединением (L), где указанный гидроксиалкилкрахмал имеет структуру, соответствующую формуле (IIa)

и/или соответствующую формуле (IIb)
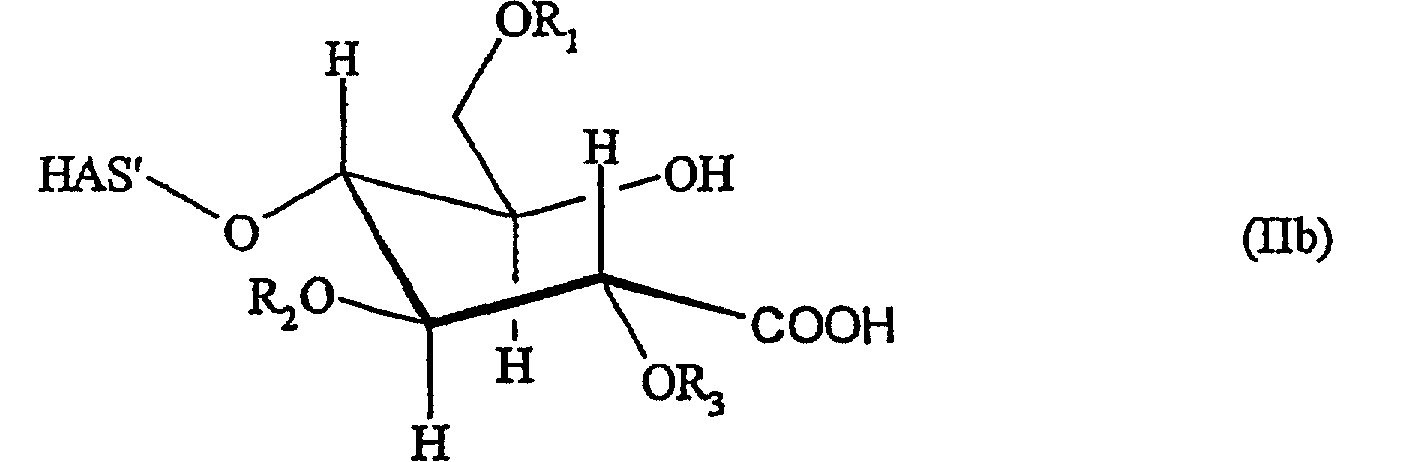
Окисление восстановленного конца гидроксиалкилкрахмала может осуществляться по любому из методов, который(ые) может(гут) приводить к образованию соединений, имеющих указанные выше структуры (IIa) и/или (IIb).
Несмотря на то что окисление гидроксиалкилкрахмала может осуществляться по любому из приемлемых методов, который(ые) может(гут) приводить к окислению восстановленного конца гидроксиалкилкрахмала, предпочтительно осуществлять его с использованием щелочного раствора йода, как описано, например, в 19628705 А1.
В этой связи настоящее изобретение относится к описанному выше способу, в котором восстановленный конец окисляют щелочным раствором йода.
В соответствии со вторым предпочтительным вариантом осуществления настоящего изобретения соединение (D) или соединение (L) подвергают реакции с восстановленным концом гидроксиалкилкрахмала, где указанный восстановленный конец не окисляют перед проведением данной реакции.
В этой связи настоящее изобретение также относится к указанному выше способу, в соответствии с которым восстановленный конец гидроксиалкилкрахмала не окисляют перед проведением реакции с соединением (D) или соединением (L), где указанный гидроксиалкилкрахмал имеет структуру, соответствующую формуле (I)

Образование химической связи между соединением (L) и гидроксиалкилкрахмалом или соединением (D) и гидроксиалкилкрахмалом достигается в ходе реакции функциональной группы Z1 с необязательно окисленным восстановленным концом гидроксиалкилкрахмала.
В качестве функциональной группы Z1 может использоваться любая функциональная группа, способная к образованию химической связи с необязательно окисленным восстановленным концом гидроксиалкилкрахмала.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения функциональная группа Z1 включает химическую структуру -NH-.
В этой связи настоящее изобретение относится к описанному выше способу, в соответствии с которым функциональная группа Z1 включает химическую структуру -NH-.
В одном предпочтительном варианте осуществления настоящего изобретения функциональная группа Z1 представляет собой группу структуры R'-NH-, где R' обозначает водород или алкильный, циклоалкильный, арильный, аралкильный, арилциклоалкильный, алкарильный или циклоалкиларильный остаток, где указанный циклоалкильный, арильный, аралкильный, арилциклоалкильный, алкарильный или циклоалкиларильный остаток может быть присоединен непосредственно к NH-группе или в соответствии с другим вариантом осуществления настоящего изобретения может быть присоединен через кислородный мостик к NH-группе. Алкильный, циклоалкильный, арильный, аралкильный, арилциклоалкильный, алкарильный или циклоалкиларильный остатки могут быть соответствующим образом замещены. В качестве предпочтительных заместителей можно отметить галогены, такие как F, Cl или Br. Особенно предпочтительными остатками R' являются водород, алкильные и алкоксигруппы и еще более предпочтительными являются водород и незамещенные алкильные и алкоксигруппы.
В числе алкильных и алкоксигрупп предпочтительными являются группы, включающие 1, 2, 3, 4, 5 или 6 атомов С. Более предпочтительными являются метильная, этильная, пропильная, изопропильная, метокси, этокси, пропокси и изопропоксигруппы. Особенно предпочтительными являются метил, этил, метокси, этокси, и особое предпочтение отдается метилу или метоксигруппе.
В этой связи настоящее изобретение относится к описанному выше способу, в соответствии с которым R' обозначает водород или метильную или метоксигруппу.
В соответствии с другим предпочтительным вариантом осуществления настоящего изобретения функциональная группа Z1 имеет структуру R'-NH-R'', где R'' предпочтительно включает структурную единицу -NH- и/или структурную единицу -(C=G)-, где G обозначает О или S, и/или структурную единицу -SO2-. В соответствии с более предпочтительными вариантами функциональную группу R'' выбирают из группы, состоящей из

и, в том случае когда G присутствует дважды, обозначает независимо О или S.
В этой связи настоящее изобретение относится к описанному выше способу, в соответствии с которым функциональную группу Z1 выбирают из группы, состоящей из
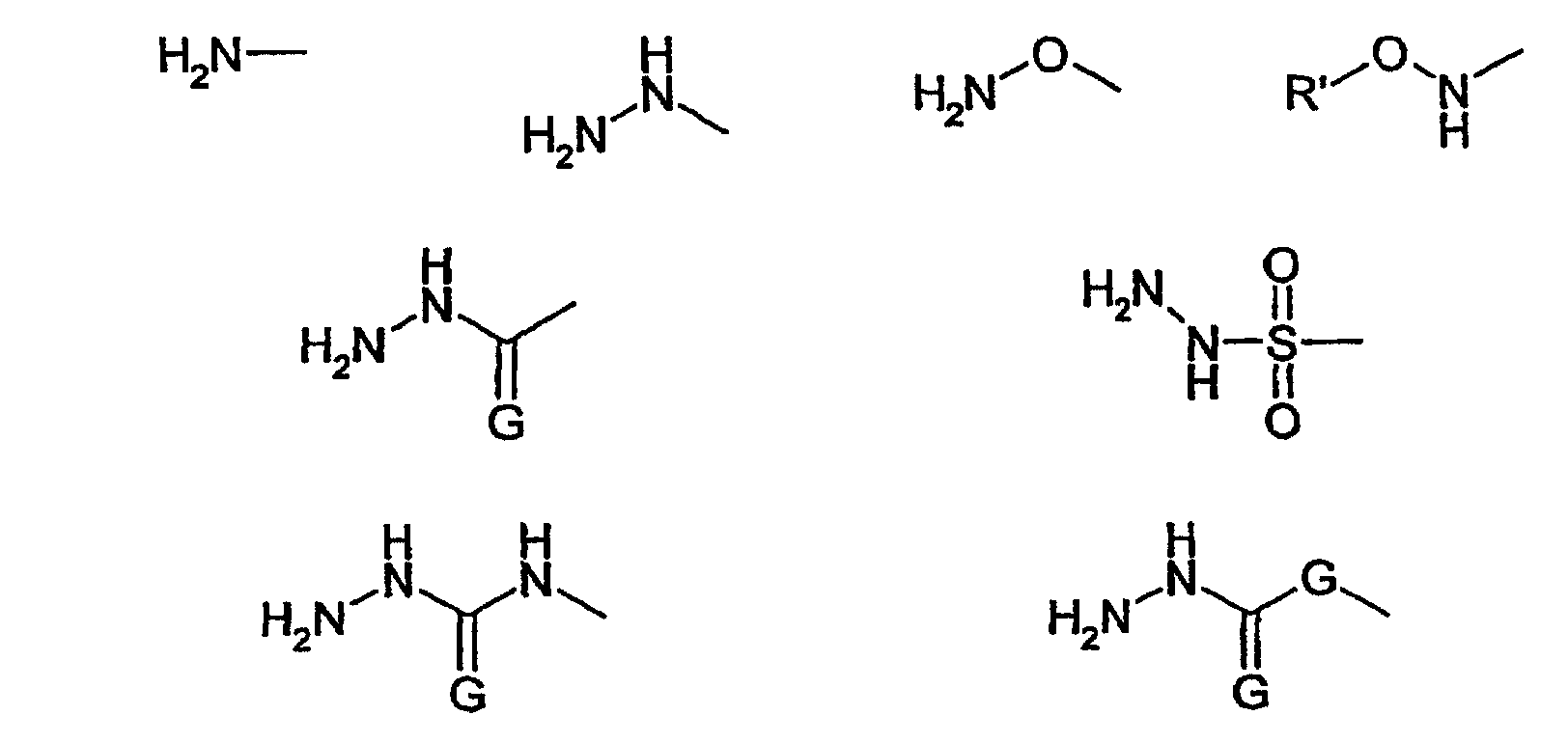
где G обозначает О или S и, если присутствует дважды, обозначает независимо О или S, и R' обозначает метил.
Другим объектом настоящего изобретения является получение производного гидроксиалкилкрахмала, которое включает функциональную группу X, способную взаимодействовать с функциональной группой Y другого соединения (М) с образованием в качестве реакционного продукта производного гидроксиалкилкрахмала, которое включает гидроксиалкилкрахмал, соединение (L), необязательно соединение (D) и другое соединение.
Что касается функциональной группы X, то отсутствуют какие-либо конкретные ограничения, при условии что может быть образована химическая связь с функциональной группой Y, которая включена в другое соединение (М).
Если функциональную группу Y выбирают из группы, состоящей из альдегидной группы, кетогруппы, гемиацетальной группы, ацетальной группы, функциональной группы X, то указанная функциональная группа Х предпочтительно включает химическую структуру -NH-.
В этой связи настоящее изобретение относится к описанному выше способу, в соответствии с которым функциональную группу Y выбирают из группы, состоящей из альдегидной группы, кетогруппы, гемиацетальной группы, ацетальной группы, а функциональная группа Х включает химическую структуру -NH-.
В соответствии с одним предпочтительным вариантом осуществления настоящего изобретения функциональная группа Х представляет собой группу структуры R'-NH-, где R' обозначает водород или алкильный, циклоалкильный, арильный, аралкильный, арилциклоалкильный, алкарильный или циклоалкиларильный остаток, где указанный циклоалкильный, арильный, аралкильный, арилциклоалкильный, алкарильный или циклоалкиларильный остаток может быть присоединен непосредственно к NH-группе или в соответствии с другим вариантом осуществления настоящего изобретения может быть присоединен через кислородный мостик к NH-группе. Алкильный, циклоалкильный, арильный, аралкильный, арилциклоалкильный, алкарильный или циклоалкиларильный остатки могут быть соответствующим образом замещены. В качестве предпочтительных заместителей можно отметить галогены, такие как F, Cl или Br. Особенно предпочтительными остатками R' являются водород, алкильные и алкоксигруппы и еще более предпочтительными являются водород и незамещенные алкильные и алкоксигруппы.
В числе алкильных и алкоксигрупп предпочтительными являются группы, включающие 1, 2, 3, 4, 5 или 6 атомов С. Более предпочтительными являются метил, этил, пропил, изопропил, метокси, этокси, пропокси и изопропоксигруппы. Особенно предпочтительными являются метил, этил, метокси, этокси, и особое предпочтение отдается метилу или метоксигруппе.
В этой связи настоящее изобретение также относится к описанному выше способу, в соответствии с которым R' обозначает водород или метильную или метоксигруппу.
В соответствии с другим предпочтительным вариантом осуществления настоящего изобретения функциональная группа Х имеет структуру R'-NH-R'', где R'' предпочтительно включает структурную единицу -NH- и/или структурную единицу -(C=G)-, где G обозначает О или S, и/или структурную единицу -SO2-. В соответствии с более предпочтительными вариантами функциональную группу R'' выбирают из группы, состоящей из

и, в том случае когда G присутствует дважды, обозначает независимо О или S.
В этой связи настоящее изобретение также относится к описанному выше способу, в соответствии с которым функциональную группу Х выбирают из группы, состоящей из

где G обозначает О или S и, если присутствует дважды, обозначает независимо О или S, и R' обозначает метил.
Если функциональная группа Y обозначает тиогруппу, то функциональную группу Х выбирают из группы, состоящей из

где Hal обозначает Cl, Br или I, предпочтительно Br или I.
В этой связи настоящее изобретение также относится к описанному выше способу, в соответствии с которым функциональная группа Y обозначает -SH, а функциональную группу Х выбирают из группы, состоящей из

где Hal обозначает Cl, Br или I.
В соответствии с одним вариантом осуществления настоящего изобретения гидроксиалкилкрахмал подвергают реакции с соединением (D) и далее полученный реакционный продукт подвергают реакции с соединением (L) с образованием при этом химической связи между соединением (L) и реакционным продуктом посредством реакции функциональной группы Z2, входящей в соединение (L), и функциональной группы W, входящей в соединение (D), представляющее собой часть продукта.
Что касается функциональных групп Z2 и W, то в основном отсутствуют какие-либо ограничения в их значениях, при условии что происходит образование химической связи.
В качестве возможных функциональных групп W или Z2 можно указать, в числе других, следующие приведенные ниже функциональные группы:
- С-С-двойные связи или С-С-тройные связи или ароматические С-С-связи;
- тиогруппа или гидроксигруппы;
- гидразид алкилсульфоновой кислоты, гидразид арилсульфоновой кислоты;
- 1,2-диолы;
- 1,2-аминоспирты;
- аминогруппа -NH2 или производные аминогруппы, включающие структурную единицу -NH, такие как аминоалкильные группы, аминоарильная группа, аминоаралкильные группы или алкариламиногруппы;
- гидроксиламиногруппа -O-NH2 или производные гидроксиламиногруппы, включающие структурную единицу -O-NH, такие как гидроксилалкиламиногруппы, гидроксилариламиногруппы, гидроксиларалкиламиногруппы или гидроксилалкариламиногруппы;
- алкоксиаминогруппы, арилоксиаминогруппы, аралкилоксиаминогруппы или алкарилоксиаминогруппы, каждая из которых включает структурную единицу -NH-O;
- остатки, содержащие карбонильную группу, -Q-C(=G)-M, где G обозначает О или S, и М обозначает, например,
- ОН или -SH;
- алкоксигруппу, арилоксигруппу, аралкилоксигруппу или алкарилоксигруппу;
- алкилтиогруппу, арилтиогруппу, аралкилтиогруппу или алкарилтиогруппу;
- алкилкарбонилоксигруппу, арилкарбонилоксигруппу, аралкилкарбонилоксигруппу, алкарилкарбонилоксигруппу;
- активированные сложные эфиры, такие как сложные эфиры гидроксиламинов, имеющие имидную структуру, такие как N-гидроксисукцинимид, или имеющие структурную единицу O-N, где N является частью гетероарильного соединения, или в случае, когда G=0 и Q отсутствует, например, арилоксисоединения с замещенным арильным остатком, такие как пентафторфенил, паранитрофенил или трихлорфенил;
где Q отсутствует или обозначает NH или гетероатом, такой как S или О;
- -NH-NH2 или -NH-NH-;
- -NO2;
- нитрильную группу/карбонильные группы, такие как альдегидная группа или кетогруппа;
- карбоксигруппу;
- -N=C=O группу или -N=C=S группу;
винилгалогенидные группы, такие как винилиодидная или винилбромидная группа, или трифлат;
- -C≡C-H;
-(C=NH2Cl)-O-алкил;
- группы -(С=O)-СН2-Hal, где Hal обозначает Cl, Br или I;
- -CH=CH-SO2-;
- дисульфидную группу, включающую структуру -S-S-;
- группу
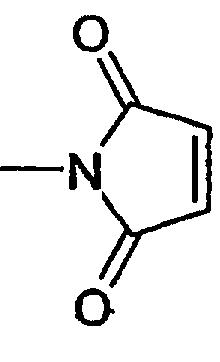
- группу

где Z2 и W обозначают соответственно группу, способную к образованию химической связи с одной из указанных выше групп.
В соответствии с предпочтительными вариантами осуществления настоящего изобретения W и Z2 обозначают группы, выбранные из приведенного выше перечня групп.
В соответствии с особенно предпочтительным вариантом осуществления настоящего изобретения Z2 или W обозначает тиогруппу. В данном конкретном случае функциональную группу W предпочтительно выбирают из группы, состоящей из

где Hal обозначает Cl, Br или I, предпочтительно Br или I. В этой связи настоящее изобретение также относится к описанному выше способу, в соответствии с которым функциональная группа W или функциональная группа Z2 обозначает -SH и функциональную группу Z2 или функциональную группу W выбирают из группы, состоящей из

где Hal обозначает Cl, Br или I.
В соответствии со вторым особенно предпочтительным вариантом осуществления настоящего изобретения Z2 или W выбирают из группы, состоящей из активированного сложного эфира, описанного выше, или карбоксигруппы, которая необязательно может быть преобразована в активированный сложный эфир. В данном случае функциональная группа W или Z2 соответственно включает химическую структуру -NH-.
В этой связи настоящее изобретение также относится к описанному выше способу, в соответствии с которым Z2 или W выбирают из группы, состоящей из активированного сложного эфира, описанного выше, или карбоксигруппы, которая необязательно может быть преобразована в активированный сложный эфир, и функциональная группа W или Z2 соответственно включает химическую структуру -NH-.
В соответствии с одним предпочтительным вариантом осуществления настоящего изобретения функциональная группа W или Z2, включающая структуру -NH-, представляет собой группу структуры R'-NH-, где R' обозначает водород или алкильный, циклоалкильный, арильный, аралкильный, арилциклоалкильный, алкарильный или циклоалкиларильный остаток, где указанный циклоалкильный, арильный, аралкильный, арилциклоалкильный, алкарильный или циклоалкиларильный остаток может быть присоединен непосредственно к NH-группе или в соответствии с другим вариантом осуществления настоящего изобретения может быть присоединен через кислородный мостик к NH-группе. Указанный алкильный, циклоалкильный, арильный, аралкильный, арилциклоалкильный, алкарильный или циклоалкиларильный остаток может быть соответствующим образом замещен. В качестве предпочтительных заместителей можно отметить F, Cl или Br. Особенно предпочтительно остатками R' являются водород, алкильные и алкоксигруппы, и еще более предпочтительными являются водород и незамещенные алкильные и алкоксигруппы.
В числе алкильных и алкоксигрупп предпочтительными являются группы, включающие 1, 2, 3, 4, 5 или 6 атомов С. Более предпочтительными являются метильные, этильные, пропильные, изопропильные, метокси, этокси, пропокси и изопропоксигруппы. Особенно предпочтительными являются метил, этил, метокси, этокси, и особое предпочтение отдается метилу или метоксигруппе.
В этой связи настоящее изобретение также относится к описанному выше способу, в соответствии с которым W или Z2 выбирают из группы, состоящей из активированного сложного эфира, описанного выше, или карбоксигруппы, которая необязательно может быть преобразована в активированный сложный эфир, и функциональная группа W или Z2 соответственно обозначает R'-NH-, где R' обозначает водород или метил или метоксигруппу.
В соответствии с другим предпочтительным вариантом осуществления настоящего изобретения функциональная группа W или Z2 имеет структуру R'-NH-R'', где R'' предпочтительно включает структурную единицу -NH- и/или структурную единицу -(C=G)-, где G обозначает О или S, и/или структурную единицу -SO2-. В соответствии с более предпочтительными вариантами функциональную группу R'' выбирают из группы, состоящей из
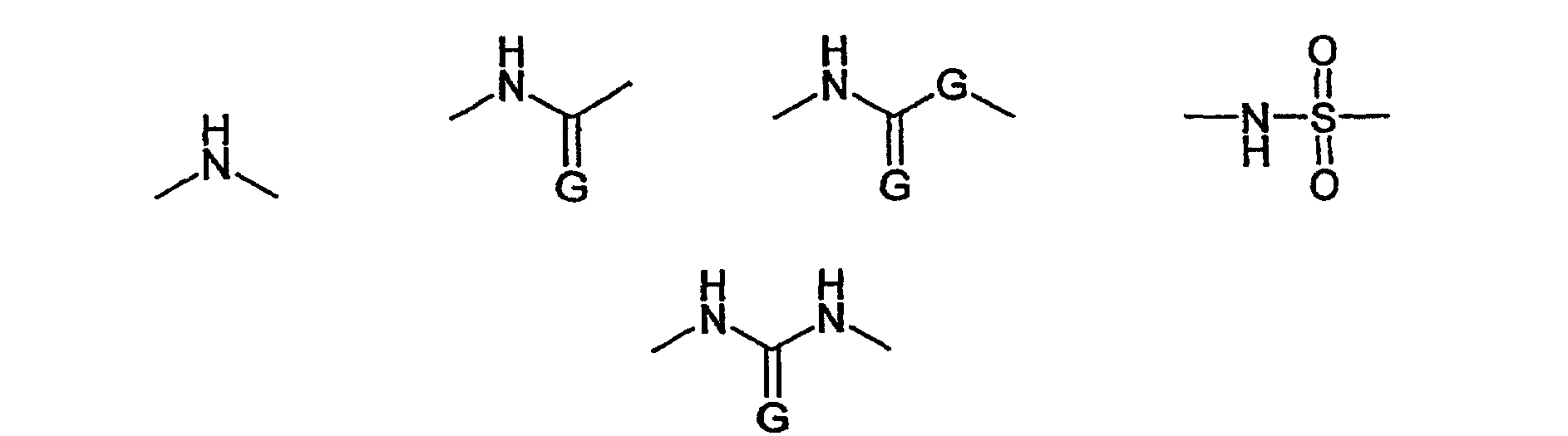
и, в том случае когда G присутствует дважды, обозначает независимо О или S.
В этой связи настоящее изобретение также относится к описанному выше способу, в соответствии с которым W или Z2 выбирают из группы, состоящей из

где G обозначает О или S и, если присутствует дважды, обозначает независимо О или S, и R' обозначает метил.
В соответствии с еще одним аспектом настоящего изобретения, по меньшей мере, одна функциональная группа X, Z2 и/или W может представлять группу, которая не способна взаимодействовать непосредственно с данным другим соединением, но которая может быть химически модифицирована с целью придать ей способность взаимодействовать желательным образом.
В качестве примера такой функциональной группы, которая подлежит модификации перед проведением реакции с другим соединением, является 1,2-аминоспирт или 1,2-диол, который модифицируют, например, посредством окисления с образованием альдегидной или кетогруппы.
Другим примером такой функциональной группы, которая подлежит модификации перед проведением реакции с другим соединением, является группа -NH2, которую модифицируют путем взаимодействия, например, с соединением следующей формулы

с получением структуры следующей формулы

которая является, в частности, реакционноспособной в отношении тиогруппы.
Другим примером функциональной группы, которая подлежит модификации перед проведением реакции с другим соединением, является группа -NH2, которую модифицируют путем взаимодействия, например, с соединением следующей формулы

с получением структуры следующей формулы

которая является, в частности, реакционноспособной в отношении тиогруппы.
Еще одним примером функциональной группы, которая подлежит модификации перед проведением реакции с другим соединением, является аминогруппа, которую подвергают взаимодействию с бромуксусным ангидридом или -N-сукцинимидилйодацетатом.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения соединение (L) имеет структуру Z1-L'-X или Z2-L'-X, где L' представляет собой органический остаток, отделяющий функциональную группу, который может необязательно отсутствовать, и где структура зависит от того, может соединение (D) взаимодействовать с гидроксиалкилкрахмалом или нет.
В соответствии с первым предпочтительным вариантом осуществления настоящего изобретения соединение (D) не участвует в рассматриваемой реакции и Y выбирают из группы, состоящей из альдегидной группы, кетогруппы, гемиацетальной группы и ацетальной группы.
В данном конкретном случае приведенные ниже соединения, имеющие структуру Z1-L'-X, где L' отсутствует, являются, в числе других, предпочтительными в качестве соединения (L):
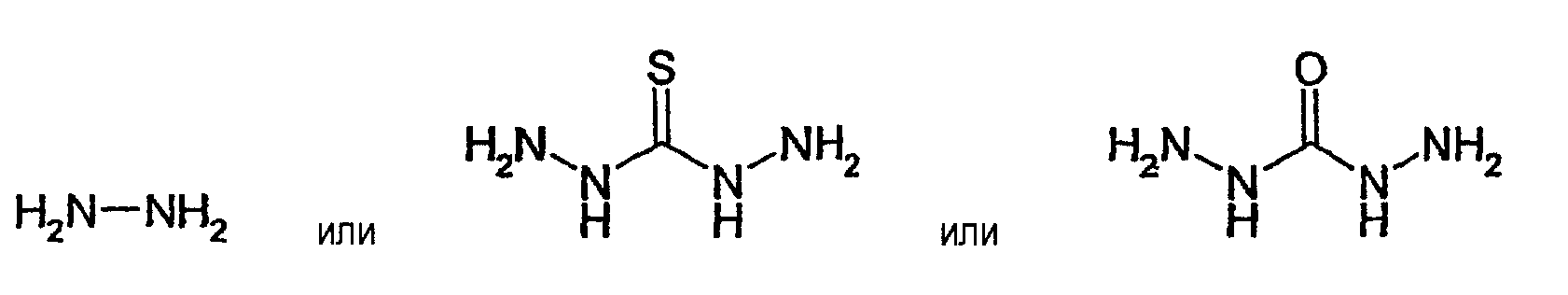
Если в данном конкретном случае L' присутствует, то L' может обозначать линейную или разветвленную алкильную, или циклоалкильную, или арильную, или аралкильную, или арилциклоалкильную, или алкарильную, или циклоалкиларильную группу, где L' может включать по меньшей мере один гетероатом, такой как N, О, S, и где L' группа может быть соответствующим образом замещена. Размер группы L' может быть адаптирован к конкретным потребностям. В целом отделяющая группа L' содержит, как правило, от 1 до 60, предпочтительно от 1 до 40, более предпочтительно от 1 до 20, более предпочтительно от 1 до 10, более предпочтительно от 1 до 6 и особенно предпочтительно от 1 до 4 атомов углерода. Если имеются гетероатомы, то указанная отделяющая группа включает в основном от 1 до 20, предпочтительно от 1 до 8 и особенно предпочтительно от 1 до 4 гетероатомов. В соответствии с особенно предпочтительными вариантами осуществления настоящего изобретения указанная отделяющая группа L' включает от 1 до 4 атомов кислорода. Отделяющая группа L' может включать необязательно разветвленную алкильную цепь или арильную группу или циклоалкильную группу, содержащую, например, от 5 до 7 атомов углерода, или представляет собой аралкильную группу или алкарильную группу, в которых алкильная часть может быть линейной и/или циклической алкильной группой. В соответствии с еще более предпочтительным вариантом осуществления настоящего изобретения указанная отделяющая группа представляет собой алкильную цепь, содержащую от 1 до 20, предпочтительно от 1 до 8, более предпочтительно от 1 до 6, более предпочтительно от 1 до 4 и особенно предпочтительно от 2 до 4 атомов углерода. В том случае если имеются гетероатомы, особенно предпочтительна цепь, включающая от 1 до 4 атомов кислорода.
Что касается того конкретного случая, когда Y выбирают из группы, состоящей из альдегидной группы, кетогруппы, гемиацетальной группы и ацетальной группы, то приведенные ниже соединения будут предпочтительными, в числе других, в качестве соединения (L) со структурой Z1-L'-X, в которой имеется L':

В соответствии со вторым предпочтительным вариантом осуществления настоящего изобретения соединение (D) участвует в реакции.
В соответствии с другим предпочтительным вариантом осуществления настоящего изобретения соединение (D) имеет структуру Z1-D'-W, где D' представляет собой органический остаток, отделяющий функциональную группу, который может необязательно отсутствовать.
В данном конкретном случае приведенные ниже соединения, имеющие структуру Z1-D'-W, где D' отсутствует, являются, в числе других, предпочтительными в качестве соединения (D):

Конкретным примером соединения D, где D' отсутствует, является NH3.
Если в данном конкретном случае D' присутствует, то D' может обозначать линейную или разветвленную алкильную, или циклоалкильную, или арильную, или аралкильную, или арилциклоалкильную, или алкарильную, или циклоалкиларильную группу, где D' может включать по меньшей мере один гетероатом, такой как N, О, S, и где группа D' может быть соответствующим образом замещена. Размер группы D' может быть адаптирован к конкретным потребностям. В основном отделяющая группа D' содержит, как правило, от 1 до 60, предпочтительно от 1 до 40, более предпочтительно от 1 до 20, более предпочтительно от 1 до 10, более предпочтительно от 1 до 6 и особенно предпочтительно от 1 до 4 атомов углерода. Если имеются гетероатомы, то указанная отделяющая группа включает в основном от 1 до 20, предпочтительно от 1 до 8 и особенно предпочтительно от 1 до 4 гетероатомов. В соответствии с особенно предпочтительными вариантами осуществления настоящего изобретения указанная отделяющая группа D' включает от 1 до 4 атомов кислорода.
Отделяющая группа D' может включать необязательно разветвленную алкильную цепь или арильную группу или циклоалкильную группу, содержащую, например, от 5 до 7 атомов углерода, или представляет собой аралкильную группу или алкарильную группу, в которых алкильная часть может быть линейной и/или циклической алкильной группой. В соответствии с еще более предпочтительным вариантом осуществления настоящего изобретения указанная отделяющая группа представляет собой алкильную цепь, содержащую от 1 до 20, предпочтительно от 1 до 8, более предпочтительно от 1 до 6, более предпочтительно от 1 до 4 и особенно предпочтительно от 2 до 4 атомов углерода. В том случае если имеются гетероатомы, особенно предпочтительна цепь, включающая от 1 до 4 атомов кислорода.
Что касается данного конкретного случая, то приведенные ниже соединения будут предпочтительными, в числе других, в качестве соединения (D) со структурой Z1-D'-W, в которой имеется D':

В зависимости от химической природы функциональной группы W, включенной в соединение (D), и функциональной группы Y могут использоваться конкретные соединения (L), соответствующие конкретным потребностям.
Например, если функциональная группа Y обозначает тиогруппу и функциональная группа W включает структуру -NH-, как было подробно описано выше, то предпочтительными, в числе других, являются следующие типы соединений (L):
Если, например, функциональную группу Y выбирают из группы, состоящей из альдегидной группы, кетогруппы, гемиацетальной группы и ацетальной группы, и функциональная группа W обозначает тиогруппу, то предпочтительными, в числе других, являются следующие типы соединений (L):
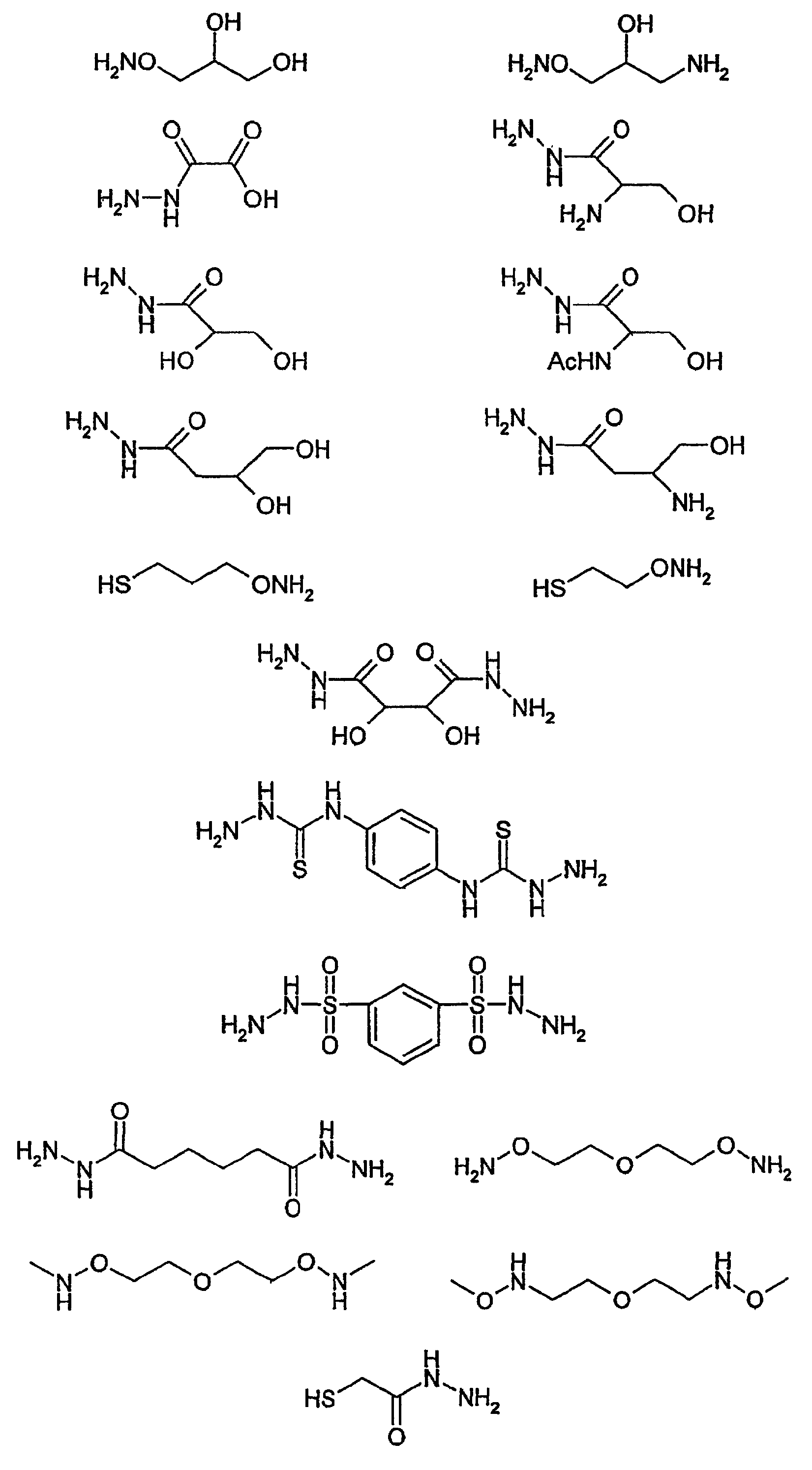
В таблице 1, приведенной в конце описания, перечислены некоторые предпочтительные примеры соединений (L), соответствующих указанным выше типам.
Отделяющие группы L' и/или D' могут быть соответствующим образом замещены. Предпочтительные заместители включают, например, галогены, такие как F, Cl, Br или I.
Отделяющие группы L' и/или D' могут включать один или более сайтов расщепления, таких как

которые позволяют легко расщеплять полученное соединение в заданном сайте.
Особенно предпочтительными примерами соединений (L), которые могут быть присоединены к гидроксиалкилкрахмалу, где получаемое производное гидроксиалкилкрахмала включает функциональную группу X, способную взаимодействовать с функциональной группой Y, включенной в другое соединение (М), и где указанную функциональную группу Y выбирают из группы, состоящей из альдегидной группы, кетогруппы, гемиацетальной группы и ацетальной группы, являются:

соединения (L)

являются особенно предпочтительными. Особенно предпочтительными примерами соединений (D), которые могут быть присоединены к гидроксиалкилкрахмалу, где получаемое производное гидроксиалкилкрахмала включает функциональную группу W, способную взаимодействовать с функциональной группой Z2, включенной в соединение (L), и где получаемое производное гидроксиалкилкрахмала, которое включает гидроксиалкилкрахмал, соединение (D) и соединение (L), способно взаимодействовать с функциональной группой Y в другом соединении (М) и где указанная функциональная группа Y в другом соединении (М) обозначает тиогруппу, являются:

соединения (D)

являются особенно предпочтительными.
Вместе с указанными выше предпочтительными соединениями (D) приведенные ниже соединения (L)

являются предпочтительными, соединения (L)

являются особенно предпочтительными.
В соответствии с первым предпочтительным вариантом осуществления настоящего изобретения соединение (D) или соединение (L) подвергают реакции с восстановленным концом гидроксиалкилкрахмала, который не окисляют.
В зависимости от условий реакции, таких как используемые растворитель или смесь растворителей, температура, давление или рН реакционной смеси, реакционный продукт, получаемый при взаимодействии соединения (D) или соединения (L) с восстановленным концом гидроксиалкилкрахмала, который не окисляют, может иметь разную структуру.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения указанную реакцию проводят в водной системе.
Термин «водная система» в контексте настоящего изобретения относится к растворителю или смеси растворителей, включающих воду в количестве, равном по меньшей мере 10 мас.%, предпочтительно, по меньшей мере 50 мас.%, более предпочтительно, по меньшей мере 80 мас.% и еще более предпочтительно, по меньшей мере 90 мас.% или до 100 мас.%, относительно веса участвующих в реакции растворителей. В качестве дополнительных растворителей можно отметить такие растворители, как ДМСО, ДМФ, этанол или метанол.
Если реакцию проводят в водной системе и функциональная группа Z1 обозначает R'-NH-, как описано выше, то производное гидроксиалкилкрахмала может иметь структуру, соответствующую формуле (IIIa)

Если реакцию проводят в водной системе и функциональная группа Z1 обозначает R'-NH- при R'=H, как описано выше, то производное гидроксиалкилкрахмала может иметь структуру, соответствующую формуле (IIIa) или формуле (IIIb), или может представлять собой смесь соединений формул (IIIa) и (IIIb)
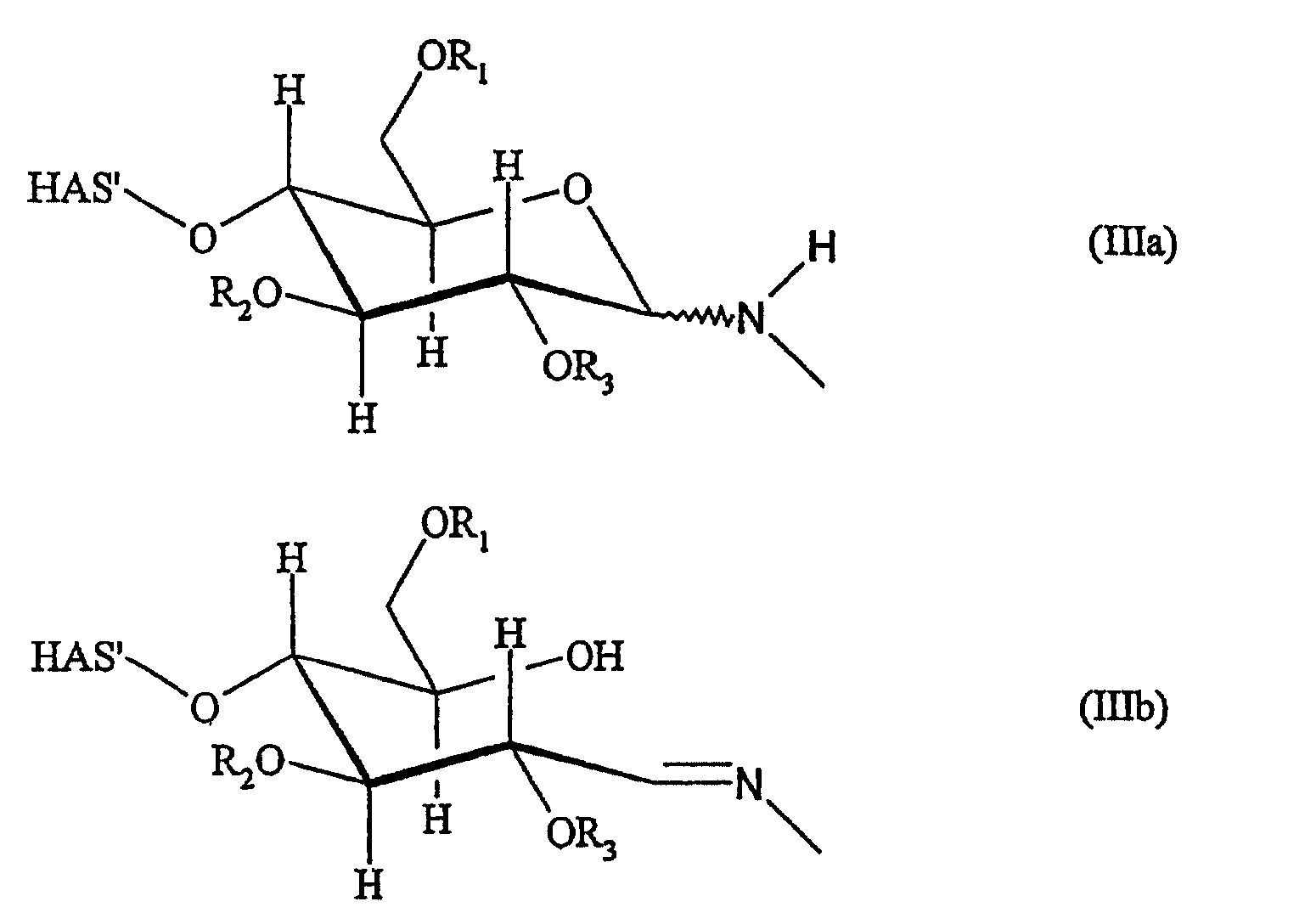
В зависимости от условий реакции и/или химической природы соединений (L) или соединения (D), используемых в реакции, соединения формулы (IIIa) могут иметь атом N в экваториальном или аксиальном положении, а также может присутствовать смесь обеих форм в состоянии их определенного равновесного распределения.
В зависимости от условий реакции и/или химической природы соединений (L) или (D), используемых в реакции, могут присутствовать соединения формулы (IIIb), в которых имеется двойная C-N связь в Е или Z конформации, и может присутствовать смесь обеих форм в состоянии их определенного равновесного распределения.
В этой связи настоящее изобретение также относится к описанному выше производному гидроксиалкилкрахмала, имеющему структуру, соответствующую формуле (IIIb) или (IIIa), или формулам (IIIa) и (IIIb).
В некоторых случаях может быть желательно стабилизировать соединение формулы (IIIa). Это особенно справедливо в том случае, когда соединение формулы (IIIa) получают и/или используют в водном растворе. В качестве способа стабилизации особенно предпочтительно проведение ацилирования соединения формулы (IIIa), особенно в том случае, когда R' обозначает водород. В качестве ацилирующего реагента могут использоваться все приемлемые реагенты, которые приводят к получению желательного производного гидроксиалкилкрахмала формулы (IVa)

В соответствии с особенно предпочтительными вариантами осуществления настоящего изобретения остаток Ra, представляющий собой часть ацилирующего реагента, является метилом. В качестве ацилирующих реагентов предпочтительно используют ангидриды карбоновых кислот, галогенангидриды карбоновых кислот и активированные сложные эфиры карбоновых кислот.
Реакцию ацилирования проводят в температурном диапазоне от 0 до 30°С, предпочтительно в диапазоне от 2 до 20°С и особенно предпочтительно в диапазоне от 4 до 10°С.
В этой связи настоящее изобретение также относится к производному гидроксиалкилкрахмала, получаемому по описанному выше способу, где указанное производное имеет структуру формулы (IVa).
В других случаях может быть желательно осуществлять стабилизацию соединения формулы (IIIb). Такой подход особенно справедлив в том случае, когда соединение формулы (IIIb) получают и/или используют в водном растворе. В качестве способа стабилизации особенно предпочтительно проведение реакции восстановления соединения формулы (IIIb), особенно в том случае, когда R' обозначает водород. В качестве восстановителя могут использоваться все подходящие реагенты, которые приводят к получению желательного производного гидроксиалкилкрахмала формулы (IVb)

В соответствии с особенно предпочтительными вариантами осуществления настоящего изобретения в качестве восстановителей используют боргидриды, такие как NaCNBH3 или NaBH4.
Реакцию восстановления проводят в температурном диапазоне от 4 до 100°С, предпочтительно в диапазоне от 10 до 90°С, и особенно предпочтительно в диапазоне от 25 до 80°С.
В этой связи настоящее изобретение также относится к производному гидроксиалкилкрахмала, получаемому по описанному выше способу, где указанное производное имеет структуру формулы (IVb).
Настоящее изобретение также относится к смесям соединений, имеющих структуры (IIIa) и (IIIb), (IVa) и (IVb), (IIIa) и (IVa), (IIIa) и (IVb), (IIIb) и (IVa), (IIIb) и (IVb), (IIIa) и (IIIb) и (IVa), (IIIa) и (IIIb) и (IVb), (IVa) и (IVb) и (IIIa), и (IVa) и (IVb) и (IIIb), где (IIIa) и/или (IVa) могут независимо присутствовать в конформации, в которой атом N может находиться в экваториальном или аксиальном положении, и/или где может присутствовать (IIIb), в котором имеется двойная C-N связь в Е или Z конформации.
В соответствии со вторым аспектом настоящего изобретения соединение (D) или соединение (L) подвергают реакции с восстановленным концом гидроксиалкилкрахмала, который окисляют.
В данном случае используют предпочтительно полярные апротонные растворители, которые могут также содержать определенное количество воды, например, до 10 мас.%. Предпочтительные апротонные растворители включают, в числе других, ДМСО или ДМФ. Примером предпочтительного температурного диапазона является температура от 20 до 65°С, а время реакции в основном составляет от 1 минуты до нескольких часов и до нескольких дней в зависимости от химической природы функциональной группы, которая вступает в реакцию с подвергнутым окислению восстановленным концом гидроксиалкилкрахмала, а также от других условий реакции.
Если в данном случае функциональная группа Z1 обозначает группу R'-NH-, описанную выше, то производное гидроксиалкилкрахмала может иметь структуру, соответствующую формуле (Va)

В этой связи настоящее изобретение также относится к производному гидроксиалкилкрахмала, получаемому по описанному выше способу, где указанное производное имеет структуру, соответствующую формуле (Va).
Что касается реакций гидроксиалкилкрахмала с соединением (D) или соединением (L), а также с соединением (М), то все возможные последовательности входят в область настоящего изобретения.
Предпочтительный вариант осуществления настоящего изобретения относится к описанному выше способу, в соответствии с которым гидроксиалкилкрахмал подвергают реакции с соединением (L) через взаимодействие функциональной группы Z1 с необязательно окисленным восстановленным концом гидроксиалкилкрахмала, и полученный реакционный продукт подвергают реакции с другим соединением (М) через взаимодействие функциональной группы X, включенной в соединение (L), с функциональной группой Y, включенной в соединение (М).
Другой вариант осуществления настоящего изобретения относится к описанному выше способу, в соответствии с которым гидроксиалкилкрахмал подвергают реакции с соединением (L) через взаимодействие функциональной группы Z1 с необязательно окисленным восстановленным концом гидроксиалкилкрахмала, где соединение (L), перед проведением реакции с гидроксиалкилкрахмалом подвергают реакции с другим соединением (М) через взаимодействие функциональной группы X, включенной в соединение (L), с функциональной группой Y, включенной в соединение (М).
Еще один вариант осуществления настоящего изобретения относится к описанному выше способу, в соответствии с которым гидроксиалкилкрахмал подвергают реакции с соединением (D) через взаимодействие функциональной группы Z1, включенной в соединение (D), с необязательно окисленным восстановленным концом гидроксиалкилкрахмала с получением первого гидроксиалкилкрахмального производного и в рамках которого указанное первое гидроксиалкилкрахмальное производное подвергают реакции с соединением (L) через взаимодействие функциональной группы Z2, включенной в соединение (L), с функциональной группой W, включенной в соединение (D) с получением второго гидроксиалкилкрахмального производного.
Еще один вариант осуществления настоящего изобретения относится к указанному способу, в соответствии с которым второе гидроксиалкилкрахмальное производное подвергают реакции с другим соединением (М) через взаимодействие функциональной группы X, включенной в соединение (L), с функциональной группой Y, включенной в соединение (М).
Еще один вариант осуществления настоящего изобретения относится к описанному выше способу, в соответствии с которым гидроксиалкилкрахмал подвергают реакции с соединением (D) через взаимодействие функциональной группы Z1, включенной в соединение (D), с необязательно окисленным восстановленным концом гидроксиалкилкрахмала с получением первого гидроксиалкилкрахмального производного и в рамках которого указанное первое гидроксиалкилкрахмальное производное подвергают реакции через взаимодействие функциональной группы W, включенной в соединение (D), и функциональной группы Z2, включенной в соединение (L), с соединением (L), где соединение (L) перед проведением реакции с указанным первым производным гидроксиалкилкрахмала подвергают реакции с другим соединением (М) через взаимодействие функциональной группы X, включенной в соединение (L), с функциональной группой Y, включенной в соединение (М).
Что касается условий реакции в рамках всех указанных выше реакционных стадий, то все параметры, такие как температура, давление, рН, или используемые также растворитель или смесь растворителей, могут быть адаптированы к конкретным потребностям и химической природе реагирующих соединений.
В соответствии с особенно предпочтительным вариантом осуществления настоящего изобретения в качестве растворителя используют воду, либо одну, либо в сочетании по меньшей мере с одним растворителем. В числе примеров такого по меньшей мере одного растворителя можно отметить ДМСО, ДМФ, метанол и этанол. В данном варианте осуществления изобретения гидроксиалкилкрахмал предпочтительно подвергают реакции через необязательно окисленный восстановленный конец.
Если гидроксиалкилкрахмал подвергают реакции с соединением (D) или соединением (L) в водной среде и соединение (D) или соединение (L) представляет собой гидроксиламин или гидразид, то температуру реакции поддерживают предпочтительно в диапазоне от 5 до 45°С, более предпочтительно в диапазоне от 10 до 30°С и особенно предпочтительно в диапазоне от 15 до 25°С.
Если гидроксиалкилкрахмал подвергают реакции с соединением (D) или соединением (L) в водной среде и указанная реакция представляет собой восстановительное аминирование, то температуру реакции поддерживают предпочтительно на уровне до 100°С, более предпочтительно в диапазоне от 70 до 90°С и особенно предпочтительно в диапазоне от 75 до 85°С.
В ходе реакции температурой можно варьировать предпочтительно в указанных диапазонах или поддерживать по существу на постоянном уровне.
Длительность реакции гидроксиалкилкрахмала с соединением (D) или соединением (L) может быть адаптирована к конкретным потребностям и составляет в основном от 1 часа до 7 дней.
В том случае когда соединение (D) или соединение (L) представляет собой гидроксиламин или гидразид, то длительность данной реакции предпочтительно составляет от 1 часа до 3 дней и более предпочтительно от 2 часов до 48 часов.
В том случае когда реакция гидроксиалкилкрахмала с соединением (D) или соединением (L) представляет собой восстановительное аминирование, то длительность данной реакции предпочтительно составляет от 2 часов до 7 дней.
Величина рН в реакции гидроксиалкилкрахмала с соединением (D) или соединением (L) может быть адаптирована к конкретным потребностям, таким как химическая природа реагирующих соединений.
В том случае когда соединение (D) или соединение (L) представляет собой гидроксиламин или гидразид, то величина рН составляет предпочтительно от 4,5 до 6,5.
В том случае когда реакция гидроксиалкилкрахмала с соединением (D) или соединением (L) представляет собой восстановительное аминирование, то величина рН составляет предпочтительно от 8 до 12.
Соответствующее значение рН в реакционной смеси может поддерживаться путем его корректировки на каждой стадии реакции добавлением по меньшей мере одного подходящего буфера. В числе предпочтительных буферов можно отметить натрий-ацетатный буфер, фосфатный или боратный буферы.
При необходимости, по меньшей мере, одна функциональная группа Х может быть защищена с помощью по меньшей мере одной защитной группы перед проведением реакции гидроксиалкилкрахмала с соединением (L), или перед проведением реакции соединения (D) с соединением (L), или перед проведением реакции соединения (L) с продуктом реакциигидроксиалкилкрахмала с соединением (D). В данном случае возможны для использования все реально доступные защитные группы, которые препятствуют реакции соединения (L) через по меньшей мере одну функциональную группу X. В этой связи защитная группа может быть выбрана в зависимости от химической природы функциональной группы X, подлежащей защите, включая, например, растворитель, в котором проходит реакция, или рН реакционной смеси. Предпочтительные защитные группы включают, в числе других, бензилоксикарбонильную группу, трет-бутоксикарбонильную группу, метоксифенильную группу, 2,4-диметоксифенильную группу, триарилметильные группы, тритильную группу, монометокситритильную группу, диметокситритильную группу, монометилтритильную группу, диметилтритильную группу, трифторацетильную группу, фталиминовые соединения, 2-(триалкилсилил)этоксикарбонильные соединения, Fmoc-группу, трет-бутильную группу или триалкилсилильные группы.
Если в соединении (L) имеются две или более функциональных групп X, то по меньшей мере одна группа может быть защищена, тогда как другую группу можно оставить незащищенной.
После реакции соединения (L) по меньшей мере одна защитная группа может быть оставлена в реакционном продукте или удалена соответствующими способами, такими как традиционные методики, известные специалистам в данной области. Если две разные функциональные группы Х подвергают защите подходящими защитными группами, возможно удалить по меньшей мере одну защитную группу, оставляя, таким образом, по меньшей мере одну функциональную группу Х доступной для последующей реакции по меньшей мере с одним другим соединением (М) и сохраняя по меньшей мере одну другую функциональную группу в защищенном состоянии до реакции продукта взаимодействия, включающего соединение (L), с другим соединением (М). После этого защитная группа от все еще защищенной функциональной группы может быть удалена, что делает оставшуюся функциональную группу Х доступной для реакции с еще одним соединением (М).
Использование по меньшей мере одной защитной группы может быть важно для предотвращения реакции, приводящей к образованию производного гидроксиалкилкрахмала, включающего соединение (L) или соединение (D), как результат реакции с двумя или более молекулами гидроксиалкилкрахмала, т.е. соединения (L) или (D) с множественным замещением молекулами HAS. Тот же результат, однако, может быть достигнут посредством реакции гидроксиалкилкрахмала с избытком соединения (L) или (D). Если в способе настоящего изобретения используют избыток соединения (L) или (D), то молярное отношение (L) или (D) к гидроксиалкилкрахмалу составляет предпочтительно в диапазоне от 2 до 100.
Образуемый на соответствующей стадии реакции реакционный продукт может быть выделен из реакционной смеси с использованием по меньшей мере одного приемлемого для этого способа. При необходимости, реакционный продукт перед выделением может быть осажден с использованием по меньшей мере одного подходящего способа.
Если реакционный продукт вначале осаждают, то возможно, например, привести в контакт реакционную смесь по меньшей мере с одним растворителем или смесью растворителей, отличных от растворителя или смеси растворителей, присутствующих в реакции, при определенных температурах. В соответствии с особенно предпочтительным вариантом осуществления настоящего изобретения, если в качестве растворителя используется водная система, то реакционную смесь приводят в контакт со смесью этанола и ацетона, взятых предпочтительно в соотношении 1:1, что означает использование равных объемов указанных соединений при температуре, составляющей предпочтительно от -20 до+50°С и особенно предпочтительно от 0 до 25°С.
Выделение реакционного продукта может быть осуществлено подходящим способом, который может включать одну или несколько стадий. В соответствии с предпочтительным вариантом осуществления настоящего изобретения реакционный продукт вначале отделяют от реакционной смеси или смеси реакционной смеси, например, со смесью этанол-ацетон, соответствующим способом, таким как центрифугирование или фильтрование. На второй стадии отделенный реакционный продукт может быть подвергнут дополнительной обработке, такой как пост-обработка типа диализа, фильтрования при центрифугировании или фильтрования под давлением, ионообменной хроматографии, ВЭЖХ, MPLC, гель-фильтрации и/или лиофилизации. В соответствии с еще более предпочтительным вариантом осуществления настоящего изобретения отделенный реакционный продукт вначале диализуют, предпочтительно против воды, и затем лиофилизируют до того момента, когда содержание растворителя в реакционном продукте станет достаточно низким, в соответствии со спецификациями для данного продукта. Лиофилизация может осуществляться при температуре от 20 до 35°С, предпочтительно от 25 до 30°С.
В соответствии с еще более предпочтительным вариантом осуществления настоящего изобретения производное гидроксиалкилкрахмала, включающее гидроксиалкилкрахмал и соединение (L), или включающее гидроксиалкилкрахмал, соединение (D) и соединение (L), далее подвергают реакции с другим соединением (М), которое включает по меньшей мере одну функциональную группу Y.
В основном отсутствуют какие-либо ограничения относительно соединения (М). Предпочтительно в контексте настоящего изобретения в качестве соединения (М) используют полипептид. Однако возможны также другие варианты в качестве соединения (М), представляющие собой полимеры, или олигомеры, или мономолекулярные соединения или смеси двух или более таких форм.
Термин «полипептид» в контексте настоящего описания относится к соединению, которое включает по меньшей мере 2 аминокислоты, которые соединены через пептидную связь, т.е. связь структуры -(С=O)-NH-. Полипептид может быть природным или полипептид может быть неприродным, последний вариант включает природную аминокислоту и/или по меньшей мере одну аминокислоту, которая не существует в природе. Скелет полипептида, полипептидная цепь, может быть далее замещен по меньшей мере одним заместителем, так что он будет иметь по меньшей мере одну боковую цепь. По меньшей мере одна функциональная группа У может быть частью полипептидного скелета или по меньшей мере одного заместителя в указанном скелете, при этом возможны такие варианты осуществления настоящего изобретения, в соответствии с которыми включается по меньшей мере одна функциональная группа, которая представляет собой часть полипептидного скелета, и по меньшей мере, одна функциональная группа, представляющая собой часть по меньшей мере одного заместителя в полипептидном скелете.
Что касается полипептида, то отсутствуют какие-либо ограничения, при условии что полипептид включает по меньшей мере одну функциональную группу Y. Указанная функциональная группа Y может быть присоединена непосредственно к полипептидному скелету или может представлять собой часть боковой цепи полипептидного скелета. Данная боковая цепь, или функциональная группа Y, или обе они могут быть частью природного полипептида или же они могут быть введены перед проведением реакции с функциональной группой X в природный полипептид или в полипептид, который, по меньшей мере частично, не является природным.
Кроме того, рассматриваемый полипептид может быть, по меньшей мере частично, любого происхождения: из организма человека или животного. В предпочтительном варианте полипептид имеет в качестве источника человеческий организм.
Полипептид может представлять собой цитокин, в особенности эритропоэтин, антитромбин (AT), такой как AT III, интерлейкин, в особенности интерлейкин-2, IFN-бета, IFN-альфа, G-CSF, CSF, интерлейкин-6 и терапевтические антитела.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения полипептид представляет собой антитромбин (AT), предпочтительно AT III (Levy JH, Weisinger A, Ziomek CA, Echelard У, Recombinant Antithrombin: Production and Role in Cardiovascular Disorder, Seminars in Thrombosis and Hemostasis 27, 4 (2001) 405-416; Edmunds T, Van Patten SM, Pollock J, Hanson E, Bernasconi R, Higgins E, Manavalan P, Ziomek C, Meade H, McPherson J, Cole ES, Transgenically Produced Human Antithrombin: Structural and Functional Comparison to Human Plasma-Derived Antithrombin, Blood 91, 12 (1998) 4661-4671; Minnema MC, Chang ACK, Jansen PM, Lubbers YTP, Pratt BM, Whittaker BG, Taylor FB, Hack CE, Friedman B, Recombinant human antithrombin III improves survival and attenuates inflammatory responses in baboons lethally challenged with Escherichia coil; Blood, 95, 4 (2000) 1117-1123; Van Patten SM, Hanson EH, Bernasconi R, Zhang K, Manavain P, Cole ES, McPherson JM, Edmunds T, Oxidation of Methionine Residues in Antithrombin, J Biol. Chemistry 274, 15 (1999) 10268-10276).
В соответствии с другим предпочтительным вариантом полипептид представляет собой человеческий IFN-бета, в частности IFN-бета 1а (например, Avonex®, REBIF® и IFN-бета 1b (например, BETASERON®).
Другой предпочтительный полипептид представляет человеческий G-CSF (гранулоцитарный колониестимулирующий фактор) (см., например, Nagata et al.. The chromosomal gene structure and two mRNAs for human granulocyte colony-stimulating factor, EMBO J. 5: 575-581, 1986; Souza et al., Recombinant human granulocyte colony-stimulating factor: effects on normal and leukemic myeloid cells. Science 232 (1986) 61-65; and Herman et al., Characterization, formulation, and stability of Neupogen® (Filgrastim), a recombinant human granulocyte colony-stimulating factor, in: Formulation, characterization, and stability of protein drugs, Rodney Pearlman and Y. John Wang, eds., Plenum Press, New York, 1996, 303-328).
Если используется смесь по меньшей мере двух разных полипептидов, то по меньшей мере два полипептида могут различаться, например, по молекулярной массе, по числу и/или последовательности аминокислот, по числу и/или химической природе заместителей или по числу полипептидных цепей, присоединяемых посредством соответствующих связей, таких как дисульфидные связи.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения реакционный продукт, получаемый при взаимодействии гидроксиалкилкрахмала и соединения (L), или реакционный продукт, получаемый при взаимодействии гидроксиалкилкрахмала и соединения (D), который далее подвергают реакции с соединением (L), выделяют предпочтительно с использованием одного из указанных выше способов и затем подвергают реакции с полипептидом, включающим по меньшей мере одну функциональную группу Y. В соответствии с предпочтительным вариантом осуществления настоящего изобретения функциональная группа Y входит в состав углеводного фрагмента полипептида.
В контексте настоящего описания термин «углеводный фрагмент» относится к гидроксиальдегидам или гидроксикетонам, а также к их химически модифицированным формам (см. Rompp Chemielexikon, Thieme Verlag Stuttgart, Germany, 9th edition 1990, Volume 9, pages 2281-2285 и цитированную в работе литературу). Кроме того, указанный термин также относится к производным природных углеводных фрагментов типа глюкозы, галактозы, маннозы, сиаловой кислоты и т.п. Указанный термин также включает химически окисленные природные углеводные фрагменты. Структура окисленного углеводного фрагмента может быть линейной или циклической.
Углеводный фрагмент может быть присоединен непосредственно к полипептидному скелету. Предпочтительно углеводный фрагмент представляет собой часть углеводной боковой цепи. Более предпочтительно углеводный фрагмент представляет собой терминальный фрагмент боковой цепи углевода.
В еще более предпочтительном варианте осуществления настоящего изобретения углеводный фрагмент представляет собой галактозный остаток в боковой цепи углевода, предпочтительно терминальный галактозный остаток в углеводной боковой цепи. Указанный галактозный остаток можно сделать доступным для проведения реакции с функциональной группой X, включенной в реакционный продукт, получаемый при взаимодействии гидроксиалкилкрахмала и соединения (L), или реакционный продукт, получаемый при взаимодействии гидроксиалкилкрахмала и соединения (D), который далее подвергают реакции с соединением (L), за счет удаления терминальных сиаловых кислот, с последующим окислением, как описано ниже.
В еще одном предпочтительном варианте реакционный продукт, получаемый при взаимодействии гидроксиалкилкрахмала и соединения (L), или реакционный продукт, получаемый при взаимодействии гидроксиалкилкрахмала и соединения (D), который далее подвергают реакции с соединением (L), присоединяют к остатку сиаловой кислоты в углеводных боковых цепях, предпочтительно к терминальному остатку сиаловой кислоты в углеводной боковой цепи.
Окисление терминальных углеводных фрагментов может быть осуществлено химическим или ферментативным способом.
Способы химического окисления углеводных фрагментов полипептидов известны в данной области техники и включают обработку периодатом (Chamow et al., 1992, J. Biol. Chem., 267, 15916-15922).
При проведении химического окисления становится принципиально возможным окислить любой углеводный фрагмент независимо от того, локализован он в концевом положении или нет. Однако при выборе мягких условий обработки (1 мМ периодат, 0°С, тогда как жесткие условия включают: 10 мМ периодат, 1 час при комнатной температуре) достигается возможность окислить предпочтительно терминальный остаток сиаловой кислоты в боковой цепи углевода.
Альтернативно, углеводный фрагмент может быть окислен ферментативным способом. Ферменты, применяемые для окисления отдельных углеводных фрагментов, известны в технике и включают, например, в случае галактозы, фермент галактозооксидазу. В случае проведения окисления терминальных фрагментов галактозы возникает необходимость постепенно удалять терминальные сиаловые кислоты (частично или полностью), если полипептид продуцируется в клетках, способных осуществлять реакцию присоединения сиаловых кислот к углеводным цепям, например в клетках млекопитающих или в клетках, которые были генетически модифицированы, с тем чтобы придать им способность присоединять сиаловые кислоты к углеводным цепям. Химические или ферментативные способы удаления сиаловых кислот известны в данной области техники (Chaplin and Kennedy (eds.), 1986, Carbohydrate Analysis: a practical approach, в особенности Chapter 5 Montreuill, Glycoproteins, pages 175-177; IRL Press Practical approach series (ISBN 0-947946-44-3)).
В соответствии с другим предпочтительным вариантом осуществления настоящего изобретения указанная функциональная группа в полипептиде представляет собой тиогруппу. Соответственно этому продукт реакции гидроксиалкилкрахмала и соединения (L) или продукт реакции гидроксиалкилкрахмала и соединения (D), который далее подвергают реакции с соединением (L), может быть присоединен к полипептиду через тиоэфирную группу, где атом S может быть получен из любой тиогруппы, входящей в состав полипептида.
В контексте данного варианта особенно предпочтительно проводить реакцию полипептида с продуктом взаимодействия гидроксиалкилкрахмала и соединения (D), который далее подвергают реакции с соединением (L).
В этой связи настоящее изобретение также относится к описанному выше способу, в соответствии с которым реакционный продукт, получаемый при взаимодействии гидроксиалкилкрахмала и соединения (D), после его реакции с соединением (L) подвергают реакции с полипептидом через тиогруппу, включенную в полипептид.
В этой связи настоящее изобретение также относится к описанному выше способу, в соответствии с которым реакционный продукт, получаемый при взаимодействии гидроксиалкилкрахмала и соединения (D), после его реакции с соединением (L) подвергают реакции с полипептидом через участие окисленного углеводного фрагмента и тиогруппы, входящей в полипептид.
Тиогруппа может присутствовать в полипептиде как таковая. Кроме того, возможно встроить тиогруппу в полипептид с помощью подходящего метода. В числе других в качестве подходящих для данной цели можно отметить химические методы. Если в полипептиде имеется дисульфидный мостик, возможно восстановить -S-S-структуру с получением тиогруппы. Возможно также осуществить преобразование аминогруппы, имеющейся в полипептиде, в SH-группу посредством реакции полипептида через его аминогруппу с соединением, которое имеет по меньшей мере две разных функциональных группы, одна из которых обладает способностью взаимодействовать с аминогруппой, а вторая представляет собой ЗН-группу или предшественник SH-группы. Указанную модификацию аминогруппы можно рассматривать как пример способа, когда белок сначала подвергают реакции с соединением (L), которое имеет по меньшей мере две разных функциональных группы, одна из которых обладает способностью взаимодействовать с аминогруппой, а вторая представляет собой SH-группу, и затем полученный реакционный продукт подвергают реакции, например, с производным HAS, включающим HAS и соединение (D), где указанное производное включает функциональную группу, способную взаимодействовать с SH-группой. Возможно также встроить SH-группу путем мутации полипептида, такой как введение цистеина или соответствующей SH-функциональной аминокислоты в полипептид, или такой как удаление цистеина из полипептида, так что провоцированный таким образом другой цистеин в полипептиде образует дисульфидную связь.
В качестве особенно предпочтительного полипептида используют эритропоэтин (ЕРО).
В этой связи настоящее изобретение относится к описанному выше способу, в соответствии с которым указанный полипептид представляет собой эритропоэтин.
ЕРО может быть человеческого происхождения (см., например, Inoue, Wada, Takeuchi, 1994, An improved method for the purification of human erythropoietin with high in vivo activity from the urine of anemic patients, Biol Pharm Bull. 17(2), 180-4; Miyake, Kung, Goldwasser, 1977, Purification of human erythropoietin., J. Biol. Chem., 252(15), 5558-64) или может происходить от другого млекопитающего в качестве источника и может быть получен путем выделения и очистки из природных источников, таких как почка человека, эмбриональная печень человека или животного, предпочтительно из почки обезьяны. Кроме того, выражение «эритропоэтин» или "ЕРО" относится также к варианту ЕРО, где одна или более аминокислот (например, от 1 до 25, предпочтительно от 1 до 10, более предпочтительно от 1 до 5, наиболее предпочтительно 1 или 2) заменены на другую аминокислоту и который проявляет эритропоэтическую активность (см., например, ЕР 640619 В1). Определение эритропоэтической активности описано в литературе (см., например, описание измерения активности in vitro: Fibi et al., 1991, Blood, 77, 1203 ff; Kitamura et al., 1989, J. Cell Phys., 140, 323-334; описание метода измерения активности ЕРО in vivo см. в Ph. Eur. 2001, 911-917; Ph. Eur. 2000, 1316 Erythropoietini solutio concentrata, 780-785; European Pharmacopoeia (1996/2000); European Pharmacopoeia, 1996, Erythropoietin concentrated solution, Pharmaeuropa., 8, 371-377; Fibi, Hermentin, Pauly, Lauffer, Zettlmeissi., 1995, N- and O-glycosylation muteins of recombinant human erythropoietin secreted from BHK-21 cells, Blood, 85(5), 1229-36) (ЕРО и модифицированные формы ЕРО инъецируют самкам мышей NMRI (что эквивалентно по белку 50 нг/мышь) в дни 1, 2 и 3, на 4 день отбирают образцы крови и определяют число ретикулоцитов). Другие публикации, в которых описаны тесты для измерения активности ЕРО, включают: Barbone, Aparicio, Anderson, Natarajan, Ritchie, 1994, Reticulocytes measurements as a bioassay for erythropoietin, J. Pharm. Biomed. Anal., 12(4), 515-22; Bowen, Culligan, Beguin, Kendal, Villis, 1994, Estimation of effective and total erythropoiesis in myelodysplasia using serum transferrin receptor and erythropoietin concentrations, with automated reticulocyte parameters, Leukemi, 8(1), 151-5; Delorme, Lorenzini, Giffin, Martin, Jacobsen, Boone, Elliott, 1992, Role of glycosylation on the secretion and biological activity of erythropoietin, Biochemistry, 31(41), 9871-6; Higuchi, Oheda, Kuboniwa, Tomonoh, Shimonaka, Ochi, 1992; Role of sugar chains in the expression of the biological activity of human erythropoietin, J. Biol. Chem., 267(11), 7703-9; Yamaguchi, Akai, Kawanishi, Ueda, Masuda, Sasaki, 1991, Effects of site-directed removal of N-glycosylation sites in human erythropoietin on its production and biological properties, J. Biol. Chem., 266(30), 20434-9; Takeuchi, Inoue, Strickland, Kubota, Wada, Shimizu, Hoshi, Kozutsumi, Takasaki, Kobata, 1989, Relationship between sugar chain structure and biological activity of recombinant human erythropoietin produced in Chinese hamster ovary cells, Proc. Natl. Acad. Sci. USA, 85(20), 7819-22; Kurtz, Eckardt, 1989, Assay methods for erythropoietin, Nephron., 51(1), 11-4 (German); Zucali, Sulkowski, 1989, Purification of human urinary erythropoietin on controlled-pore glass and silicic acid, Exp.Hematol., 13(3), 833-7; Krystal, 1983, Physical and biological characterization of erythroblast enhancing factor (EEF), a late acting erythropoietin stimulator in serum distinct from erythropoietin, Exp.Hematol., 11(1), 18-31.
Предпочтительно ЕРО получают рекомбинантными способами. Указанный вариант включает продукцию в эукариотических или прокариотических клетках, предпочтительно в клетках млекопитающих, насекомых, дрожжей, в бактериальных клетках или в клетках любого другого типа, которые удобны для получения ЕРО рекомбинантными методами. Кроме того, ЕРО может экспрессироваться в трансгенных животных (например, в жидкостях организма, таких как молоко, кровь и т.п.), в яйцах трансгенных птиц, в особенности сельскохозяйственных птиц, предпочтительно кур, или в трансгенных растениях.
Получение полипептидов по рекомбинантной технологии известно специалистам в данной области техники. В целом такая продукция включает трансфекцию в хозяйскую клетку с помощью соответствующего вектора экспрессии, культивирование хозяйских клеток в условиях, позволяющих осуществлять продукцию полипептида и выделение и очистку полипептида из хозяйских клеток. Подробная информация содержится в ряде публикаций (см., например, Krystal, Pankratz, Farber, Smart, 1986, Purification of human erythropoietin to homogeneity by a rapid five-step procedure. Blood, 67(1), 71-9; Quelle, Caslake, Burkert, Wojchowski, 1989, High-level expression and purification of a recombinant human erythropoietin produced using a baculovirus vector. Blood, 74(2), 652-7; EP 640619 B1 and EP 668351 Bl).
В предпочтительном варианте осуществления настоящего изобретения ЕРО имеет аминокислотную последовательность человеческого ЕРО (см. EP 148605 В2).
ЕРО может включать одну или несколько углеводных боковых цепей, предпочтительно от 1 до 12, более предпочтительно от 1 до 9, еще более предпочтительно от 1 до 6, в частности, предпочтительно от 1 до 4, и особенно предпочтительно 4 боковых углеводных цепи, присоединяемых к ЕРО через N- и/или O-связанное гликозилирование, т.е. ЕРО гликозилируется. Обычно в случае продукции ЕРО в эукариотических клетках полипептид подвергается посттрансляционному гликозилированию. Следовательно, углеводные боковые цепи могут присоединяться к ЕРО в процессе биосинтеза в клетках млекопитающего, особенно человека, клетках насекомого или клетках дрожжей. Структура и свойства гликозилированного ЕРО широко исследовались в данной области (см. ЕР 428267 В1; ЕР 640619 В1, Rush, Derby, Smith, Merry, Rogers, Rohde, Katta, 1995, Microheterogeneity of erythropoietin carbohydrate structure. Anal Chem., 67(8), 1442-52; Takeuchi, Kobata, 1991, Structures and functional roles of the sugar chains of human erythropoietins, Glycobiology, 1(4), 337-46 (обзор)).
В этой связи гидроксиалкилкрахмальные производные по настоящему изобретению могут включать по меньшей мере одну, предпочтительно от 1 до 12, более предпочтительно от 1 до 9, еще более предпочтительно от 1 до 6 и особенно предпочтительно от 1 до 4 молекул HAS на одну молекулу ЕРО. Количество HAS-молекул на одну молекулу ЕРО может быть определено путем количественного анализа углеводного состава с использованием ГХ-МС после гидролиза продукта и дериватизации полученных моносахаридов (см. Chaplin and Kennedy (eds.), 1986, Carbohydrate Analysis: a practical approach, IRL Press Practical approach series (ISBN 0-947946-44-3), в особенности Chapter 1, Monosaccharides, page 1-36; Chapter 2, Oligosaccharides, page 37-53, Chapter 3, Neutral Polysaccharides, page 55-96).
В соответствии с особенно предпочтительным вариантом осуществления настоящего изобретения углеводный фрагмент, присоединенный к ЕРО, представляет собой часть углеводной боковой цепи. Более предпочтительно углеводный фрагмент представляет собой терминальный фрагмент углеводной боковой цепи. В еще более предпочтительном варианте углеводный фрагмент представляет собой остаток галактозы в углеводной боковой цепи, предпочтительно терминальный остаток галактозы в углеводной боковой цепи. Указанный галактозный остаток можно сделать доступным для проведения реакции с реакционным продуктом, получаемым при взаимодействии соединения (I) и соединения (II), за счет удаления терминальных сиаловых кислот с последующим окислением, как описано ниже. В еще одном предпочтительном варианте осуществления изобретения реакционный продукт, получаемый при взаимодействии соединения (I) и соединения (II), присоединяют к остатку сиаловой кислоты в боковых углеводных цепях, предпочтительно к терминальному остатку сиаловой кислоты в углеводной боковой цепи. Сиаловую кислоту окисляют, как описано ниже.
В особенно предпочтительном варианте указанный галактозный остаток делают доступным для проведения реакции с реакционным продуктом, получаемым при взаимодействии гидроксиалкилкрахмала и соединения (L), или с реакционным продуктом, получаемым при взаимодействии гидроксиалкилкрахмала и соединения (D), который далее подвергают реакции с соединением (L), через функциональную группу Х за счет удаления терминальной сиаловой кислоты с последующим окислением.
Более предпочтительно указанный галактозный остаток делают доступным для проведения реакции с реакционным продуктом, получаемым при взаимодействии гидроксиалкилкрахмала и соединения (L), или с реакционным продуктом, получаемым при взаимодействии гидроксиалкилкрахмала и соединения (D), который далее подвергают реакции с соединением (L), через функциональную группу Х путем окисления, когда терминальный остаток сиаловой кислоты не удаляют.
Как указывалось выше, реакционный продукт, получаемый при взаимодействии гидроксиалкилкрахмала и соединения (L), или реакционный продукт, получаемый при взаимодействии гидроксиалкилкрахмала и соединения (D), который далее подвергают реакции с соединением (L), может быть далее подвергнут реакции с тиогруппой, входящей в ЕРО.
Возможно также осуществлять реакцию реакционного продукта, получаемого при взаимодействии гидроксиалкилкрахмала и соединения (L), или реакционного продукта, получаемого при взаимодействии гидроксиалкилкрахмала и соединения (D), который далее подвергают реакции с соединением (L), с тиогруппой, а также с углеводным фрагментом, каждый из которых входит в состав по меньшей мере одного другого соединения, предпочтительно полипептида, более предпочтительно эритропоэтина.
В соответствии с предпочтительным вариантом указанная SH-группа может быть присоединена к предпочтительно окисленному углеводному фрагменту, например посредством использования производного гидроксиламина, например гидрохлорида 2-(аминоокси)этилмеркаптана (Bauer L. et al., 1965, J. Org. Chem., 30, 949) или с использованием гидразидного производного, например гидразида тиогликолевой кислоты (Whitesides et al., 1977, J. Org. Chem., 42, 332).
В соответствии с еще одним предпочтительным вариантом осуществления настоящего изобретения тиогруппу предпочтительно вводят в окисленный углеводный фрагмент ЕРО, более предпочтительно в окисленный углеводный фрагмент, который представляет собой часть боковой углеводной цепи ЕРО.
Предпочтительно тиогруппу получают из природного цистеина или из добавленного цистеина. Более предпочтительно ЕРО имеет аминокислотную последовательность человеческого ЕРО, а природные цистеины представляют собой цистеин 29 и/или 33. В более предпочтительном варианте реакционный продукт, получаемый при взаимодействии гидроксиалкилкрахмала и соединения (L), или реакционный продукт, получаемый при взаимодействии гидроксиалкилкрахмала и соединения (D), который далее подвергают реакции с соединением (L), подвергают реакции с цистеином 29, а цистеин 33 замещают другой аминокислотой. Альтернативно реакционный продукт, получаемый при взаимодействии гидроксиалкилкрахмала и соединения (L), или реакционный продукт, получаемый при взаимодействии гидроксиалкилкрахмала и соединения (D), который далее подвергают реакции с соединением (L), подвергают реакции с цистеином 33, а цистеин 29 замещают другой аминокислотой.
В контексте настоящего описания термин «добавленные цистеины» указывает на то, что полипептиды, предпочтительно ЕРО, включают цистеиновый остаток, который не присутствует в полипептиде дикого типа.
В контексте данного аспекта изобретения цистеин может представлять собой дополнительную аминокислоту, добавляемую на N- или С-конце ЕРО.
Кроме того, добавленный цистеин может быть встроен путем замещения природной аминокислоты цистеином или соответствующим образом замещенным цистеином. Предпочтительно в контексте данного аспекта изобретения ЕРО представляет собой человеческий ЕРО, и замещенный аминокислотный остаток представляет собой серин 126.
Что касается условий, относящихся к реакции реакционного продукта, получаемого при взаимодействии гидроксиалкилкрахмала и соединения (L), необязательно с соединением (D), с другим соединением (М), то отсутствуют какие-либо конкретные ограничения, и указанные условия реакции могут быть адаптированы к конкретным потребностям. В соответствии с особенно предпочтительным вариантом осуществления настоящего изобретения в качестве растворителя используют воду, одну или в сочетании по меньшей мере с одним другим растворителем. В качестве такого по меньшей мере одного другого растворителя можно отметить ДМСО, ДМФ, метанол или этанол. Предпочтительные растворители из числа отличных от воды включают метанол и этанол. В соответствии с другими предпочтительными вариантами в качестве растворителя могут использоваться ДМСО, или ДМФ, или метанол, или этанол, или смесь двух или более таких растворителей.
Если, например, гидроксиалкилкрахмал подвергают реакции с соединением (L) в водной системе, как в том случае, когда, например, гидроксиэтилкрахмал подвергают реакции с гидроксиламином, таким как О-[2-(2-аминооксиэтокси)этил]гидроксиламин, посредством взаимодействия неокисленного восстановленного конца крахмала, и далее полученный реакционный продукт подвергают реакции с полипептидом, предпочтительно эритропоэтином, через альдегидную группу, кетогруппу, ацетальную или гемиацетальную группу, температуру реакции предпочтительно поддерживают в диапазоне от 4 до 37°С, более предпочтительно от 10 до 30°С и особенно предпочтительно от 15 до 25°С.
Выделение реакционного продукта, включающего другое соединение (М), предпочтительно полипептид и особенно предпочтительно эритропоэтин, может быть осуществлено с помощью известных процедур, применяемых для очистки природных и рекомбинантных ЕРО (например, вытеснительная хроматография, ионообменная хроматография, ОФ-ВЭЖХ, хроматография на гидроксиапатите, хроматография, основанная на гидрофобном взаимодействии, или их сочетание). Выделение реакционного продукта может быть осуществлено подходящим способом, который может включать одну или более стадий. В соответствии с предпочтительным вариантом осуществления настоящего изобретения реакционный продукт отделяют от реакционной смеси или смеси реакционных смесей, например, смесью этанол-ацетон, соответствующим способом, таким как центрифугирование или фильтрование. На второй стадии отделенный реакционный продукт может быть подвергнут дополнительной обработке, такой как постобработка типа диализа, фильтрования при центрифугировании или фильтрования под давлением, ионообменной хроматографии, такой как, например, хроматография на колонке с сефарозой (Q-Sepharose), ВЭЖХ, MPLC, гель-фильтрации и/или лиофилизации. В соответствии с одним предпочтительным вариантом осуществления настоящего изобретения отделенный реакционный продукт вначале диализуют, предпочтительно против воды, и затем лиофилизируют до того момента, когда содержание растворителя в реакционном продукте станет достаточно низким, в соответствии со спецификациями для данного продукта. Лиофилизация может осуществляться при температуре от 20 до 35°С, предпочтительно от 25 до 30°С. В соответствии с другим предпочтительным вариантом осуществления настоящего изобретения реакционную смесь, включающую реакционный продукт, наносят на колонку с сефарозой (Q-Sepharose), получая элюат, который концентрируют, например, путем фильтрования при центрифугировании.
Другим объектом настоящего изобретения являются производные гидроксиалкилкрахмала, получаемые по одному или более из указанных выше способов.
В этой связи настоящее изобретение относится к производному гидроксиалкилкрахмала, получаемому согласно способу продукции производного гидроксиалкилкрахмала, где указанный гидроксиалкилкрахмал имеет структуру, соответствующую указанной ниже формуле (I)

включающему взаимодействие
- гидроксиалкилкрахмала формулы (I) по его необязательно окисленному восстановленному концу или
- производного гидроксиалкилкрахмала, получаемого в результате реакции гидроксиалкилкрахмала формулы (I) по его необязательно окисленному восстановленному концу с соединением (D), где указанное соединение (D) включает
- по меньшей мере одну функциональную группу Z1, способную взаимодействовать с необязательно окисленным восстановленным концом гидроксиалкилкрахмала, и
- по меньшей мере одну функциональную группу W с соединением (L), включающим
- по меньшей мере одну функциональную группу Z1, способную взаимодействовать с указанным гидроксиалкилкрахмалом, или по меньшей мере одну функциональную группу Z2, способную взаимодействовать с функциональной группой W, входящей в состав указанного производного гидроксиалкилкрахмала, и
- по меньшей мере одну функциональную группу X, способную взаимодействовать с функциональной группой Y другого соединения (М),
где указанную функциональную группу Y выбирают из группы, состоящей из альдегидной группы, кетогруппы, гемиацетальной группы, ацетальной группы или тиогруппы.
В соответствии с предпочтительным вариантом настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, в котором R1, R2 и R3 обозначают независимо водород или линейную или разветвленную гидроксиалкильную группу.
В соответствии с еще более предпочтительным вариантом настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, в котором R1, R2 и R3 обозначают независимо водород или 2-гидроксиэтильную группу.
В соответствии с другим предпочтительным вариантом настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, где указанный гидроксиалкилкрахмал представляет собой гидроксиэтилкрахмал.
В соответствии с другим предпочтительным вариантом настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, в котором функциональная группа Z1 включает структуру -NH-.
В соответствии с особенно предпочтительным вариантом настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, для которого функциональную группу Z1 выбирают из группы, состоящей из

где G обозначает О или S и, если присутствует дважды, обозначает независимо О или S, и R' обозначает метил.
В соответствии с другим предпочтительным вариантом настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, для которого функциональную группу Y выбирают из группы, состоящей из альдегидной группы, кетогруппы, гемиацетальной группы и ацетальной группы, а функциональная группа X включает структуру -NH-.
В соответствии с особенно предпочтительным вариантом настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, для которого функциональную группу Х выбирают из группы, состоящей из

где G обозначает О или S и, если присутствует дважды, обозначает независимо О или S, и R обозначает метил.
В соответствии с другим предпочтительным вариантом настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, в котором функциональная группа Y обозначает -SH, а функциональную группу Х выбирают из группы, состоящей из
где Hal обозначает Cl, Br или I.
В соответствии с другим предпочтительным вариантом настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, в котором функциональная группа W или функциональная группа Z2 обозначает -SH, и функциональную группу Z2 или функциональную группу W выбирают из группы, состоящей из

где Hal обозначает Cl, Br или I.
В соответствии с другим предпочтительным вариантом настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, в котором функциональную группу W или функциональную группу Z2 выбирают из группы, состоящей из активированного сложного эфира, описанного выше, или карбоксигруппы, которая необязательно может быть преобразована в активированный сложный эфир, и функциональную группу Z2 или функциональную группу W выбирают из группы, состоящей из

где G обозначает О или S и, если присутствует дважды, обозначает независимо О или S, и R обозначает метил.
В соответствии с особенно предпочтительным вариантом настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, где восстановленный конец гидроксиалкилкрахмала не окисляют перед проведением реакции с соединением (D) или с соединением (L), при этом указанный гидроксиалкилкрахмал имеет структуру, соответствующую формуле (I)

В соответствии с другим особенно предпочтительным вариантом настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, где восстановленный конец гидроксиалкилкрахмала подвергают окислению перед проведением реакции с соединением (D) или с соединением (L), при этом указанный гидроксиалкилкрахмал имеет структуру, соответствующую формуле (На)

и/или соответствующую формуле (IIb)

В соответствии с другим предпочтительным вариантом настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, где восстановленный конец гидроксиалкилкрахмала подвергают окислению щелочным раствором йода.
В соответствии с другим предпочтительным вариантом настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, где гидроксиалкилкрахмал подвергают реакции с соединением (L) через взаимодействие функциональной группы Z1 с необязательно окисленным восстановленным концом гидроксиалкилкрахмала, и полученный реакционный продукт подвергают реакции с другим соединением (М) через взаимодействие функциональной группы X, входящей в соединение (L), с функциональной группой Y, входящей в соединение (М).
В соответствии с еще одним предпочтительным вариантом настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, где гидроксиалкилкрахмал подвергают реакции с соединением (L) через взаимодействие функциональной группы Z1 с необязательно окисленным восстановленным концом гидроксиалкилкрахмала, при этом соединение (L) перед проведением реакции с гидроксиалкилкрахмалом подвергают реакции с другим соединением (М) через взаимодействие функциональной группы X, входящей в соединение (L), с функциональной группой Y, входящей в соединение (М).
В соответствии с еще одним предпочтительным вариантом настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, где гидроксиалкилкрахмал подвергают реакции с соединением (D) через взаимодействие функциональной группы Z1, входящей в соединение (D), с необязательно окисленным восстановленным концом гидроксиалкилкрахмала с получением первого производного гидроксиалкилкрахмала, и где указанное первое производное гидроксиалкилкрахмала подвергают реакции с соединением (L) через взаимодействие функциональной группы Z2, входящей в соединение (L), с функциональной группой W, входящей в соединение (D) с получением второго производного гидроксиалкилкрахмала.
В соответствии с особенно предпочтительным вариантом настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, где указанное второе производное гидроксиалкилкрахмала подвергают реакции с другим соединением (М) через взаимодействие функциональной группы X, входящей в соединение (L), с функциональной группой Y, входящей в соединение (М).
В соответствии с еще одним предпочтительным вариантом настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, где гидроксиалкилкрахмал подвергают реакции с соединением (D) через взаимодействие функциональной группы Z1, входящей в соединение (D), с необязательно окисленным восстановленным концом гидроксиалкилкрахмала с получением первого производного гидроксиалкилкрахмала, и где указанное первое производное гидроксиалкилкрахмала подвергают реакции через взаимодействие функциональной группы W, входящей в соединение (D), и функциональной группы Z2, входящей в соединение (L), с соединением (L), где соединение (L) перед проведением реакции с первым производным гидроксиалкилкрахмала подвергают реакции с другим соединением (М) через взаимодействие функциональной группы X, входящей в соединение (L), с функциональной группой Y, входящей в соединение (М).
В соответствии с особенно предпочтительным вариантом настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, где по меньшей мере одно другое соединение (М) представляет собой полипептид.
В конкретном предпочтительном варианте настоящее изобретение относится к описанному выше производному гидроксиалкилкрахмала, где указанный полипептид представляет собой эритропоэтин.
Производное гидроксиалкилкрахмала, которое далее в описании будет обозначаться как конъюгат HAS-EPO и который образуется в результате реакции гидроксиалкилкрахмала с соединением (L) и необязательно соединением (D) и эритропоэтином, обладает преимуществом, связанным с тем, что он демонстрирует повышенную биологическую стабильность в сравнении с эритропоэтином до конъюгирования. Кроме того, он демонстрирует более высокую биологическую активность, чем стандартный препарат ЕРО BRP. Указанное свойство связано, главным образом, с тем, что данное производное гидроксиалкилкрахмала в меньшей степени или вовсе не распознается системами удаления в печени и почках, и в этой связи указанный конъюгат присутствует в системе кровообращения в течение более длительного периода времени. Кроме того, поскольку HAS присоединяется сайт-специфичным образом, минимизируется риск нарушения биологической активности ЕРО in vivo путем конъюгирования HAS с ЕРО.
Конъюгат HAS-EPO по настоящему изобретению может демонстрировать по существу ту же биологическую активность in vitro, что и рекомбинантный нативный ЕРО, поскольку его биологическая активность in vitro основана на измерении лишь аффинности по связыванию с рецептором ЕРО. Способы определения биологической активности in vitro известны в данной области техники.
Кроме того, HAS-EPO демонстрирует более высокую активность in vivo, чем ЕРО, используемый в качестве исходного материала для конъюгирования (неконъюгированный ЕРО). Способы определения биологической активности in vivo известны в технике.
Конъюгат HAS-EPO может демонстрировать активность in vivo, составляющую от 110 до 500%, предпочтительно от 300 до 400% или предпочтительно от 110 до 300%, более предпочтительно от 110% до 200%, более предпочтительно от 110% до 180% или от 110% до 150%, наиболее предпочтительно от 110% до 140%, если активность in vivo неконъюгированного ЕРО принять за 100%.
В сравнении с высоко сиалилированной формой ЕРО в составе препарата Amgen (см. ЕР 428267 В1), HAS-EPO демонстрирует активность, равную предпочтительно по меньшей мере 50%, более предпочтительно по меньшей мере 70%, еще более предпочтительно по меньшей мере 85% или по меньшей мере 95%, по меньшей мере 150%, по меньшей мере 200% или по меньшей мере 300% от активности высоко сиалилированного ЕРО, если активность in vivo высоко сиалилированного ЕРО принять за 100%. Наиболее предпочтительно указанная форма демонстрирует активность по меньшей мере на уровне 95% от активности in vivo высоко сиалилированного ЕРО.
Высокая биологическая активность in vivo конъюгата HAS-EPO по настоящему изобретению в основном связана с тем фактом, что конъюгат HAS-EPO остается дольше в кровотоке, чем неконъюгированный ЕРО, поскольку он хуже распознается системами удаления печени и поскольку снижен почечный клиренс из-за более высокой молекулярной массы. Способы определения in vivo периода полувыведения ЕРО из кровотока известны в данной области техники (Sytkowski, Lunn, Davis, Feldman, Siekman, 1998, Human erythropoietin dimers with markedly enhanced in vivo activity, Proc. Natl. Acad. Sci. USA, 95(3), 1184-8).
В этой связи большим преимуществом настоящего изобретения является тот факт, что конъюгат HAS-EPO представляет собой форму, которая может вводиться реже, чем препараты ЕРО, коммерчески доступные в настоящее время. В то время как стандартные препараты ЕРО должны вводиться по меньшей мере каждые 3 дня, конъюгат HAS-EPO по настоящему изобретению предпочтительно вводится два раза в неделю, более предпочтительно один раз в неделю.
Кроме того, способ по настоящему изобретению имеет то достоинство, что может быть получено эффективное производное ЕРО со сниженной стоимостью, поскольку данный способ не включает дорогостоящие и длительные по времени стадии очистки, приводящие к низкому конечному выходу, то есть в данном способе нет необходимости очищать продукт от недостаточно сиалилированных форм ЕРО, которые, как известно, характеризуются низкой биологической активностью in vivo или вовсе ее отсутствием. В частности, приведенный пример 8.11(d) иллюстрирует тот факт, что HAS-EPO в случае введения небольших модификаций на стадиях его получения характеризуется активностью, превышающей в 3 раза активность стандартного препарата ЕРО BRP.
Еще одним объектом настоящего изобретения является фармацевтическая композиция, которая включает в терапевтически эффективном количестве конъюгат HAS-EPO по настоящему изобретению.
Кроме того, настоящее изобретение относится к фармацевтической композиции, включающей в терапевтически эффективном количестве конъюгат HAS-полипептид, предпочтительно конъюгат HAS-EPO, более предпочтительно конъюгат HES-EPO по настоящему изобретению. В предпочтительном варианте фармацевтическая композиция включает также по меньшей мере один фармацевтически приемлемый разбавитель, адъювант и/или носитель, используемый в терапии эритропоэтином.
В этой связи настоящее изобретение относится к фармацевтической композиции, включающей в терапевтически эффективном количестве описанное выше производное гидроксиалкилкрахмала, получаемую по способу, включающему продукцию производного гидроксиалкилкрахмала, где указанный гидроксиалкилкрахмал имеет структуру, соответствующую указанной ниже формуле (I)
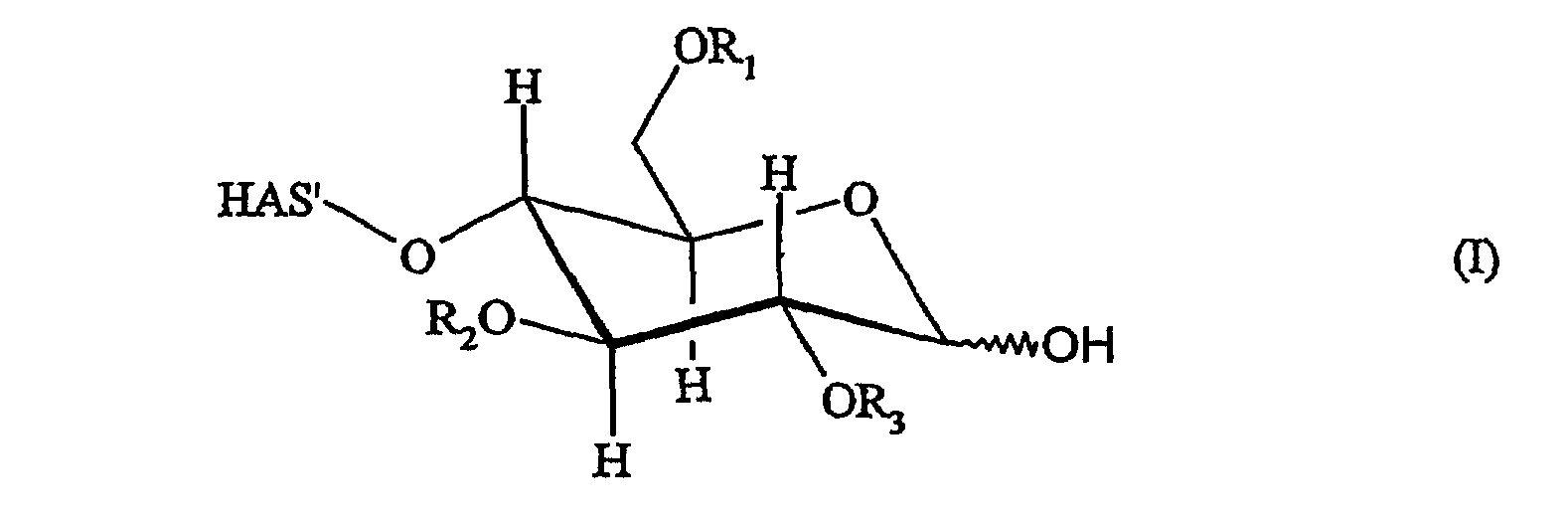
и где указанный способ включает взаимодействие
- гидроксиалкилкрахмала формулы (I) по его необязательно окисленному восстановленному концу или
- производного гидроксиалкилкрахмала, получаемого в результате реакции гидроксиалкилкрахмала формулы (I) по его необязательно окисленному восстановленному концу с соединением (D), где указанное соединение (D) включает
- по меньшей мере одну функциональную группу Z1, способную взаимодействовать с необязательно окисленным восстановленным концом гидроксиалкилкрахмала, и
- по меньшей мере одну функциональную группу W, с соединением (L), включающим
- по меньшей мере одну функциональную группу Z1, способную взаимодействовать с указанным гидроксиалкилкрахмалом, или по меньшей мере одну функциональную группу Z2, способную взаимодействовать с функциональной группой W, входящей в состав указанного производного гидроксиалкилкрахмала, и
- по меньшей мере одну функциональную группу X, способную взаимодействовать с функциональной группой Y другого соединения (М),
где указанную функциональную группу Y выбирают из группы, состоящей из альдегидной группы, кетогруппы, гемиацетальной группы, ацетальной группы или тиогруппы,
где указанный способ получения производного гидроксиалкилкрахмала включает далее взаимодействие реакционного продукта, включающего гидроксиалкилкрахмал, соединение (L) и необязательно соединение (D), с другим соединением (М), и где по меньшей мере одно другое соединение представляет собой полипептид.
Кроме того, настоящее изобретение относится также к использованию описанного выше производного гидроксиалкилкрахмала в производстве лекарственного средства, предназначенного для лечения анемических расстройств или дисфункций гематопоэза или связанных с ними заболеваний.
В соответствии с предпочтительным вариантом осуществления настоящее изобретение относится к фармацевтической композиции, где указанный полипептид представляет собой антитромбин (AT), предпочтительно AT III (Levy JH, Weisinger A, Ziomek CA, Echelard Y, Recombinant Antithrombin: Production and Role in Cardiovascular Disorder, Seminars in Thrombosis and Hemostasis 27, 4 (2001) 405-416; Edmunds T, Van Patten SM, Pollock J, Hanson E, Bernasconi R, Higgins E, Manavalan P, Ziomek C, Meade H, McPherson J, Cole ES, Transgenically Produced Human Antithrombin: Structural and Functional Comparison to Human Plasma-Derived Antithrombin, Blood 91, 12 (1998) 4661-4671; Minnema MC, Chang ACK, Jansen PM, Lubbers YTP, Pratt BM, Whittaker BG, Taylor FB, Hack CE, Friedman B, Recombinant human antithrombin III improves survival and attenuates inflammatory responses in baboons lethally challenged with Escherichia coli; Blood, 95, 4 (2000) 1117-1123; Van Patten SM, Hanson EH, Bernasconi R, Zhang K, Manavain P, Cole ES, McPherson JM, Edmunds T, Oxidation of Methionine Residues in Antithrombin, J. Biol. Chemistry 274, 15 (1999) 10268-10276).
В соответствии с другими предпочтительными вариантами осуществления настоящее изобретение относится к фармацевтическим композициям, где указанный полипептид представляет собой G-CSF или IFN-бета.
В соответствии с особенно предпочтительным вариантом настоящее изобретение относится к описанной выше фармацевтической композиции, в которой полипептид представляет собой эритропоэтин.
В соответствии с другим вариантом настоящее изобретение относится к описанной выше фармацевтической композиции, в которой функциональная группа Y обозначает -SH и соединение (L) представляет собой соединение общей формулы Z1-L'-X, где функциональную группу Z1 выбирают из группы, состоящей из

где G обозначает О или S и, если присутствует дважды, обозначает независимо О или S, и R' обозначает метил, и где функциональную группу Х выбирают из группы, состоящей из

где Hal обозначает Cl, Br или I, и где L' обозначает органическую цепь, соединяющую мостиковой связью Z1 и X, или где L' отсутствует.
В соответствии с предпочтительным вариантом настоящее изобретение относится к описанной выше фармацевтической композиции, в которой функциональную группу Y выбирают из группы, состоящей из альдегидной группы, кетогруппы, гемиацетальной группы и ацетальной группы, и соединение (L) представляет собой соединение общей формулы Z1-L'-X, где функциональную группу Z1 выбирают из группы, состоящей из

где G обозначает О или S и, если присутствует дважды, обозначает независимо О или S, и R' обозначает метил, и где функциональную группу Х выбирают из группы, состоящей из

где G обозначает О или S и, если присутствует дважды, обозначает независимо О или S, и R' обозначает метил, и где L' обозначает органическую цепь, соединяющую мостиковой связью Z1 и X, или где L' отсутствует.
В соответствии с другим предпочтительным вариантом настоящее изобретение относится к описанной выше фармацевтической композиции, в которой функциональная группа Y обозначает -SH, соединение (D) представляет собой соединение общей формулы Z1-D'-W, и соединение (L) представляет собой соединение общей формулы Z2-L'-X, где функциональную группу Z1 выбирают из группы, состоящей из

где G обозначает О или S и, если присутствует дважды, обозначает независимо О или S, и R' обозначает метил, и где функциональную группу Х выбирают из группы, состоящей из

где Hal обозначает Cl, Br или I, где функциональная группа W или функциональная группа Z2 обозначает -SH, и где указанную функциональную группу Z2 или функциональную группу W выбирают из группы, состоящей из

где Hal обозначает Cl, Br или I, или где функциональную группу W или функциональную группу Z2 выбирают из группы, состоящей из указанного выше активированного сложного эфира или карбоксигруппы, которую необязательно преобразуют в активированный сложный эфир, и где функциональную группу Z2 или функциональную группу W выбирают из группы, состоящей из

где G обозначает О или S и, если присутствует дважды, обозначает независимо О или S, и R' обозначает метил, и где D' обозначает органическую цепь, соединяющую мостиковой связью Z1 и W или где D' отсутствует и где L' обозначает органическую цепь, соединяющую мостиковой связью Z2 и X, или где L' отсутствует.
В соответствии с еще одним вариантом настоящее изобретение относится к описанной выше фармацевтической композиции, в которой функциональную группу Y выбирают из группы, состоящей из альдегидной группы, кетогруппы, гемиацетальной группы и ацетальной группы, соединение (D) представляет собой соединение общей формулы Z1-D'-W и соединение (L) представляет собой соединение общей формулы Z2-L'-X, где функциональную группу Z1 выбирают из группы, состоящей из

где G обозначает О или S и, если присутствует дважды, обозначает независимо О или S, и R' обозначает метил, и где функциональную группу Х выбирают из группы, состоящей из

где G обозначает О или S и, если присутствует дважды, обозначает независимо О или S, и R' обозначает метил, и где функциональная группа W или функциональная группа Z2 обозначает -SH, и где указанную функциональную группу Z2 или функциональную группу W выбирают из группы, состоящей из

где Hal обозначает Cl, Br или I, или где функциональную группу W или функциональную группу Z2 выбирают из группы, состоящей из указанного выше активированного сложного эфира или карбоксигруппы, которую необязательно преобразуют в активированный сложный эфир, и где функциональную группу Z2 или функциональную группу W выбирают из группы, состоящей из

где G обозначает О или S и, если присутствует дважды, обозначает независимо О или S, и R' обозначает метил, и где D' обозначает органическую цепь, соединяющую мостиковой связью Z1 и W или где D' отсутствует и где L' обозначает органическую цепь, соединяющую мостиковой связью Z2 и X, или где L' отсутствует.
В соответствии с особенно предпочтительным вариантом настоящее изобретение относится к описанной выше фармацевтической композиции, отличающейся тем, что гидроксиэтилкрахмал подвергают реакции в водной среде с соединением формулы

и реакционный продукт подвергают реакции с эритропоэтином.
В соответствии с еще более предпочтительным вариантом настоящее изобретение относится к описанной выше фармацевтической композиции, отличающейся тем, что эритропоэтин перед проведением данной реакции подвергают окислению периодатом натрия.
В соответствии с другим предпочтительным вариантом настоящее изобретение относится к описанной выше фармацевтической композиции, отличающейся тем, что эритропоэтин перед проведением данной реакции подвергают частичному десиалилированию с последующим окислением периодатом натрия.
В соответствии с другим предпочтительным вариантом из настоящего изобретения исключаются фармацевтические композиции, включающие гидроксиалкилкрахмальное производное, которое получают согласно методике примера 6 на основе полностью восстановленного тио-ЕРО.
Указанная выше фармацевтическая композиция особенно полезна для лечения анемических расстройств или дисфункций гематопоэза или связанных с ними заболеваний.
Термин «терапевтически эффективное количество» в контексте настоящего описания относится к количеству, которое обеспечивает терапевтический эффект в данных условиях и в данном режиме введения. Введение изоформ эритропоэтина предпочтительно проводится парентеральными способами. Конкретный выбираемый способ зависит от состояния пациента, которого предстоит лечить.
Введение изоформ эритропоэтина предпочтительно проводят в виде композиции, содержащей соответствующий носитель, такой как человеческий сывороточный альбумин, соответствующий разбавитель, такой как забуференный солевой раствор, и/или соответствующий адъювант. Требуемая дозировка будет представлять собой количества, достаточные для повышения гематокрита у пациентов, и будет варьировать в зависимости от тяжести состояния, подлежащего лечению, выбранного способа введения и т.п.
Объектом лечения фармацевтической композицией по настоящему изобретению предпочтительно является повышение уровня гемоглобина более чем 6,8 ммоль/л в крови. Для этой цели фармацевтическая композиция может вводиться таким способом, чтобы содержание гемоглобина повышалось от 0,6 ммоль/л до 1,6 ммоль/л в неделю. Если содержание гемоглобина превышает уровень 8,7 ммоль/л, то предпочтительно лечение следует прервать до снижения уровня гемоглобина ниже 8,1 ммоль/л.
Композиция по настоящему изобретению предпочтительно используется в составе препарата, пригодного для подкожной или внутривенной или парентеральной инъекции. Пригодные для этой цели наполнители и носители включают, например, дигидрофосфат натрия, гидрофосфат динатрия, хлорат натрия, полисорбат 80, HSA и воду для инъекций. Указанная композиция может вводиться три раза в неделю, предпочтительно два раза в неделю, более предпочтительно один раз в неделю и наиболее предпочтительно через неделю.
Предпочтительно фармацевтическую композицию вводят в количестве 0,01-10 мкг/кг веса тела пациента, более предпочтительно от 0,1 до 5 мкг/кг, от 0,1 до 1 мкг/кг или 0,2-0,9 мкг/кг, наиболее предпочтительно 0,3-0,7 мкг/кг и еще наиболее предпочтительно 0, 4-0,6 мкг/кг веса тела.
В целом, в составе дозы вводят предпочтительно от 10 мкг до 200 мкг, предпочтительно от 15 мкг до 100 мкг.
Настоящее изобретение также относится к применению HAS-полипептида по настоящему изобретению при получении лекарственного средства, применяемого для лечения человека или животного.
Настоящее изобретение также относится к применению HAS-EPO по настоящему изобретению при получении лекарственного средства, применяемого для лечении анемических расстройств или гемопоэтических дисфункций или связанных с ними заболеваний.
В случае использования соединения (L) по настоящему изобретению один или более хиральных центров в соединении (II) могут находиться в R или S конфигурации или в виде рацемического соединения относительно каждого хирального центра.
В случае использования соединения (D) по настоящему изобретению один или более хиральных центров в соединении (D) могут находиться в R или S конфигурации или в виде рацемического соединения относительно каждого хирального центра.
Ниже настоящее изобретение иллюстрируется с помощью поясняющих его примеров, таблиц и чертежей, которые ни в коей мере не следует рассматривать как ограничивающие область настоящего изобретения.
Краткое описание чертежей
Фиг.1
На фиг.1 показаны результаты анализа в SDS-PAGE конъюгата HES-EPO, полученного по методике примера 5.1.
Линия А: Белковый маркер Roti®-Mark PRESTAINED (Carl Roth GmbH+Co, Karlsruhe, D); значения молекулярной массы (кД) белкового маркера сверху вниз: 245, 123, 77, 42, 30, 25,4 и 17.
Линия В: Неочищенный продукт, полученный при конъюгировании по методике примера 5.1.
Линия С: Исходный материал ЕРО.
Фиг.2
На фиг.2 показаны результаты анализа в SDS-PAGE конъюгата HES-EPO, полученного по методике примера 5.3.
Линия А: Неочищенный продукт, полученный при конъюгировании по методике примера 5.3.
Линия В: Исходный материал ЕРО.
Линия С: Белковый маркер Roti® -Mark PRESTAINED (Carl Roth GmbH+Co, Karlsruhe, D); значения молекулярной массы (кД) белкового маркера сверху вниз: 245, 123, 77, 42, 30, 25,4 и 17.
Фиг.3
На фиг.3 показаны результаты анализа в SDS-PAGE конъюгатов HES-EPO, полученных по методике примеров 5.4 и 5.5.
Линия А: Белковый маркер Roti®-Mark PRESTAINED (Carl Roth GmbH+Co, Karlsruhe, D); значения молекулярной массы (кД) белкового маркера сверху вниз: 245, 123, 77, 42, 30, 25,4 и 17.
Линия В: Неочищенный продукт, полученный при конъюгировании по методике примера 5.4.
Линия С: Неочищенный продукт, полученный при конъюгировании по методике примера 5.5.
Линия D: Исходный материал ЕРО.
Фиг.4
На фиг.4 показаны результаты анализа в SDS-PAGE конъюгатов HES-EPO, полученных по методике примеров 7.1 и 7.4.
Линия А: Белковый маркер Roti®-Mark PRESTAINED (Carl Roth GmbH+Co, Karlsruhe, D); значения молекулярной массы (кД) белкового маркера сверху вниз: 245, 123, 77, 42, 30, 25,4 и 17.
Линия В: Неочищенный продукт, полученный при конъюгировании по методике примера 7.4.
Линия С: Неочищенный продукт, полученный при конъюгировании по методике примера 7.1.
Линия D: Исходный материал ЕРО.
Фиг.5
На фиг.5 показаны результаты анализа в SDS-PAGE конъюгатов HES-EPO, полученных по методике примеров 7.2, 7.3, 7.5 и 7.6.
Линия А: Белковый маркер Roti®-Mark PRESTAINED (Carl Roth GmbH+Co, Karlsruhe, D); значения молекулярной массы (кД) белкового маркера сверху вниз: 245, 123, 77, 42, 30, 25,4 и 17.
Линия В: Неочищенный продукт, полученный при конъюгировании по методике примера 7.6, на основе примера 1.3b).
Линия С: Неочищенный продукт, полученный при конъюгировании по методике примера 7.5, на основе примера 1.1b).
Линия D: Неочищенный продукт, полученный при конъюгировании по методике примера 7.6, на основе примера 1.3а).
Линия Е: Неочищенный продукт, полученный при конъюгировании по методике примера 7.5, на основе примера 1.1a).
Линия F: Неочищенный продукт, полученный при конъюгировании по методике примера 7.2.
Линия G: Неочищенный продукт, полученный при конъюгировании по методике примера 7.3.
Линия К: Исходный материал ЕРО.
Фиг.6
На фиг.6 показаны результаты анализа в SDS-PAGE конъюгатов HES-EPO, полученных по методике примеров 7.7, 7.8, 7.9, 7.10, 7.11 и 7.12.
Линия А: Белковый маркер Roti®-Mark PRESTAINED (Carl Roth GmbH+Co, Karlsruhe, D); значения молекулярной массы (кД) белкового маркера сверху вниз: 245, 123, 77, 42, 30, 25,4 и 17.
Линия В: Неочищенный продукт, полученный при конъюгировании по методике примера 7.11.
Линия С: Неочищенный продукт, полученный при конъюгировании по методике примера 7.10.
Линия D: Неочищенный продукт, полученный при конъюгировании по методике примера 7.7.
Линия Е: Неочищенный продукт, полученный при конъюгировании по методике примера 7.8.
Линия F: Неочищенный продукт, полученный при конъюгировании по методике примера 7.12.
Линия G: Исходный материал ЕРО.
Линия К: Неочищенный продукт, полученный при конъюгировании по методике примера 7.9.
Фиг.7
Результаты анализа в SDS-PAGE EPO-GT-1, подвергнутого мягкой кислотной обработке: в течение 5 минут=линия 2; 10 минут=линия 3; 60 минут=линия 4 и необработанный ЕРО=линия 1; показано смещение мобильности ЕРО после удаления N-гликанов (+PNGASE).
Фиг.8
Результаты анализа в HPAEC-PAD олигосахаридов, выделенных из необработанного ЕРО и из ЕРО, инкубированных в течение 5 минут, 10 минут и 60 минут в условиях мягкого кислотного гидролиза. Римские цифры I-V указывают положение при элюировании: I=десиалилированной диантенной структуры, II=трисиалилированных трехантенных структур (два изомера); III=тетрасиалилированной тетраантенной структуры+2N-ацетиллактозаминовых повтора; IV=тетрасиалилированной тетраантенной структуры+1N-ацетиллактозаминовый повтор; V=тетрасиалилированной тетраантенной структуры без N-ацетиллактозаминового повтора. В скобках показана область элюирования олигосахаридных структур без сиаловой кислоты и при наличии 1-4 сиаловых кислот.
Фиг.9
Результаты анализа в HPAEC-PAD N-связанных олигосахаридов после десиалилирования; показано положение в ходе элюирования N-ацетилнейраминовой кислоты; номера 1-9 указывают положение при элюировании стандартных олигосахаридов: 1=диантенный; 2=триантенный (2-4 изомера), 3=триантенный (2-6 изомеров), 4=тетрантенный; 5=триантенный плюс 1 повтор; 6=тетраантенный плюс 1 повтор; 7=триантенный плюс 2 повтора; 8=тетраантенный плюс 2 повтора и 9=тетраантенный плюс 3 повтора.
Фиг.10
Результаты анализа в SDS-PAGE ЕРО после обработки в мягких условиях и необработанного ЕРО, которые подвергают окислению периодатом по остаткам сиаловой кислоты. 1=окисление периодатом, но без кислотной обработки; 2=окисление периодатом плюс 5 минут кислотной обработки; 3=окисление периодатом плюс 10 минут кислотной обработки; 4=окисление периодатом, но без кислотной обработки; 5=стандарт ЕРО ВНР без окисления периодатом и без кислотной обработки.
Фиг.11
Результаты анализа в HPAEC-PAD нативных олигосахаридов, выделенных из необработанного ЕРО и из ЕРО, инкубированных в течение 5 минут и 10 минут в условиях мягкого кислотного гидролиза, с последующей обработкой периодатом. В скобках 1-5 показана область элюирования олигосахаридных структур без сиаловой кислоты и при наличии 1-4 сиаловых кислот.
Фиг.12
Результаты анализа в SDS-PAGE HES-модификации с течением времени EPO-GT-1-A: аликвоты по 20 мкл EPO-GT-1-A подвергают реакции с производным X, полученным с использованием модифицированного гидроксиламином HES, в течение 30 минут, 2, 4 и 17 часов. Линия 1=время реакции 30 минут; линия 2=время реакции 2 часа; линия 3=время реакции 4 часа; линия 4=время реакции 17 часов; линия 5=EPO-GT-1-A без модификации HES. На левом чертеже показано смещение подвижности EPO-GT-1-A при увеличении времени инкубации в присутствии модифицированного гидроксиламином HES-производного (скорость течения: 1 мл/мин); X: линия 1=время реакции 30 минут; линия 2=время реакции 2 часа; линия 3=время реакции 4 часа; линия 4=время реакции 17 часов; линия 5=EPO-GT-1-A при наличии HES-модификации. Чертеж в правой стороне показывает результат анализа тех же образцов после их обработки N-гликозидазой.
Фиг.13
Результаты анализа в SDS-PAGE фракций конъюгатов HES-EPO, полученных при элюировании с сефарозой (Q-Sepharose). Концентрируют в концентраторе Speed Vac 1% фракции, полученной при прохождении потока через колонку, и 1% фракции, элюируемой при высокой концентрации соли, и затем наносят на гели в буфере для образца. ЕРО белок окрашивают красителем кумасси блю. А=образец I; В=образец II; С=образец III; К=контроль EPO-GT-1; А1, В1, Cl и К1 относятся к фракциям, полученным при свободном прохождении потока; А2, В2, С2 и К2 указывают на фракции, элюируемые при высокой концентрации соли.
Фиг.14а
Результаты анализа в SDS-PAGE HES-модифицированного образца ЕРО А2 (см. фиг.13), контрольного образца ЕРО К2 и EPO-GT-1-A препарата ЕРО, расщепленных в присутствии N-гликозидазы, для удаления N-связанных олигосахаридов. Все образцы ЕРО демонстрируют смещение мобильности в сторону форм с низкой молекулярной массой, которые не содержат или содержат O-гликан. Наблюдается сниженный коэффициент соотношения полос для O-гликозированного и негликозилированного белка в случае HES-модифицированного образца ЕРО А2 после де-N-гликозилирования, а также отмечается размытая, белковая полоса в области 30 кДа, что преимущественно отражает наличие HES-модификации по сиаловой кислоте O-гликанового остатка (см. стрелку, помеченную звездочкой).
Фиг.14b
Результаты анализа в SDS-PAGE HES-модифицированного образца ЕРО А2 после гидролиза в мягких условиях (см. фиг.13), контрольного образца ЕРО К2 и EPO-GT-1A, необработанных или расщепленных в присутствии N-гликозидазы для удаления N-связанных олигосахаридов (см. фиг.14а). Как высокомолекулярная форма А2 до, так и форма А после N-гликозидазной обработки (см. участки со скобками, помеченными и не помеченными стрелками), исчезают при кислотной обработке образцов. Стандарт ЕРО BRP, который анализируют для сравнения, не подвергают кислотной обработке в мягких условиях.
Фиг.15
Результаты анализа в HPAEC-PAD N-связанного олигосахаридного материала, высвобождаемого из HES-модифицированного образца А, из EPO-GT-1-A и из контрольного образца ЕРО, инкубированного с немодифицированным HES (К). Римские цифры I-V указывают положение при элюировании: I=дисиалилированной диантенной структуры, II=трисиалилированных триантенных структур (два изомера); III=тетрасиалилированной тетраантенной структуры+2N-ацетиллактозаминовых повтора; IV=тетрасиалилированной тетраантенной структуры+1N-ацетиллактозаминовый повтор; V=тетрасиалилированной тетраантенной структуры без N-ацетиллактозаминового повтора; в скобках показана область элюирования ди-, три- и тетрасиалилированных N-гликанов, как описано в пояснении к фиг.8 и 11.
Фиг.16
Результаты анализа в HPAEC-PAD N-связанного олигосахаридного материала, высвобождаемого из HES-модифицированного образца А, из EPO-GT-1A и из контрольного образца ЕРО (К), инкубированного с немодифицированным HES. Показано время удерживания смеси стандартных олигосахаридов: номера 1-9 указывают положение при элюировании стандартных олигосахаридов: 1=диантенного; 2=триантенного (2-4 изомера); 3=триантенного (2-6 изомеров); 4=тетраантенного; 5=триантенного плюс 1 повтор; 6=тетраантенного плюс 1 повтор; 7=триантенного плюс 2 повтора; 8=тетраантенного плюс 2 повтора и 9=тетраантенного плюс 3 повтора.
Фиг.17-23
На фиг.17-23 показаны масс-спектры MALDI/TOF энзиматически высвобожденных и химически десиалилированных N-гликанов, выделенных из препаратов HES-модифицированного ЕРО и контрольного ЕРО. Основные сигналы при значениях т/т., равных 1809,7, 2174,8, 2539,9, 2905,0 и 3270,1 ([M+Na:T), соответствуют ди - тетраантенным N-гликановым структурам комплексного типа в отсутствие или при наличии одного или двух N-ацетиллактозаминовых повторов, сопровождаемых слабыми сигналами из-за потери фукозы или галактозы, вызванной условиями кислотной обработки, используемыми для десиалилирования образцов, подвергаемых МС анализу.
Фиг.17
MALDI/TOF спектр: десиалилированные олигосахариды HES-модифицированного ЕРО А2.
Фиг.18
MALDI/TOF спектр: десиалилированные олигосахариды ЕРО GT-1-А.
Фиг.19
MALDI/TOF спектр: десиалилированные олигосахариды ЕРО К2.
Фиг.20
MALDI/TOF спектр: десиалилированные олигосахариды EPO-GT-1.
Фиг.21
MALDI/TOF спектр: десиалилированные олигосахариды EPO-GT-1, подвергнутые кислотному гидролизу в течение 5 минут.
Фиг.22
MALDI/TOF спектр: десиалилированные олигосахариды EPO-GT-1, подвергнутые кислотному гидролизу в течение 10 минут.
Фиг.23
MALDI/TOF спектр: десиалилированные олигосахариды EPO-GT-1, подвергнутые кислотному гидролизу в течение 60 минут.
Фиг.24
На фиг.24 показаны результаты анализа в SDS-PAGE двух конъюгатов HES-EPO.
ММ: маркер
Линия 1: HES-EPO, полученный по методике примера 8: ЕРО подвергают конъюгированию с гидразидо-НЕЗ 12KD L.
Линия 2: HES-EPO, полученный по методике примера 9: ЕРО подвергают конъюгированию с гидроксиламино-HES 12KD К.
С: Контроль (неконъюгированный ЕРО); верхняя полоса соответствует димеру ЕРО.
Фиг.25
Фиг.25 демонстрирует, что HES подвергают конъюгированию с углеводным фрагментом углевода боковой цепи углевода, при этом показано расщепление HAS-модифицированных форм ЕРО N-гликозидазой полипептидов.
Линия 1: HES-EPO, полученный по методике примера 8, после расщепления N-гликозидазой.
Линия 2: HES-EPO, полученный по методике примера 9, после расщепления N-гликозидазой.
Линия 3: Стандарт BRP ЕРО.
Линия 4: Стандарт BRP ЕРО после расщепления N-гликозидазой. ММ.: маркер (стандарты для SDS-PAGE Bio-Rad Low range Catalog No 161-0305, Bio-Rad Laboratories, Hercules, CA, USA)
Примеры
Пример 1: Получение производных гидроксиэтилкрахмала путем восстановительного аминирования по неокисленному восстановленному концу
Пример 1.1. Реакция гидроксиэтилкрахмала с 1,3-диамино-2-гидроксипропаном

а) К раствору 200 мг гидроксиэтилкрахмала (HES18/0.4 (ММ=18000 Д, DS=0,4) в 5 мл воды добавляют 0,83 ммоль 1,3-диамино-2-гидроксипропана (Sigma Aldrich, Taufkirchen, D) и 50 мг цианборгидрида натрия NaCNBH3. Полученную смесь инкубируют при 80°С в течение 17 часов. Реакционную смесь добавляют к 160 мл охлажденной смеси ацетона и этанола, 1:1 (объем/объем). Осадок собирают центрифугированием, диализуют в течение 4 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, D) и лиофилизируют.
b) Инкубирование смеси, полученной при добавлении 0,83 ммоль 1,3-диамино-2-гидроксипропана и 50 мг цианборгидрида натрия NaCNBH3 к раствору 200 мг гидроксиэтилкрахмала, также возможно и проводится при 25°С в течение 3 дней.
Пример 1.2. Реакция гидроксиэтилкрахмала с 1,2-дигидрокси-3-аминопропаном

a) К раствору 200 мг гидроксиэтилкрахмала (HES18/0,4 (ММ=18000 Д, 03=0,4) в 5 мл воды добавляют 0,83 ммоль 1, 2-дигидрокси-3-аминопропана (Sigma Aldrich, Taufkirchen, D) и 50 мг цианборгидрида натрия NaCNBH3. Полученную смесь инкубируют при 80°С в течение 17 часов. Реакционную смесь добавляют к 160 мл охлажденной смеси ацетона и этанола, 1:1 (объем/объем). Осадок собирают центрифугированием, диализуют в течение 4 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, D) и лиофилизируют.
b) Инкубирование смеси, полученной при добавлении 0,83 ммоль 1, 2-дигидрокси-3-аминопропана и 50 мг цианборгидрида натрия NaCNBH3 к раствору 200 мг гидроксиэтилкрахмала, также возможно и проводится при 25°С в течение 3 дней.
Осуществление реакции 1,2-дигидрокси-3-аминопропана с HES подтверждается опосредованно при определении количества формальдегида, образованного при окислительном расщеплении 1,2-диола в реакционном продукте периодатом, как описано в работе G Avigad, Anal. Biochem. 134 (1983) 449-504.
Пример 1.3.Реакция гидроксиэтилкрахмала с 1,4-диаминобутаном

a) К раствору 200 мг гидроксиэтилкрахмала (НЕ318/0,4 (ММ=18000 Д, DS=0,4) в 5 мл воды добавляют 0, 83 ммоль 1,4-диаминобутана (Sigma Aldrich, Taufkirchen, D) и 50 мг цианборгидрида натрия NaCNBH3. Полученную смесь инкубируют при 80°С в течение 17 часов. Полученную смесь добавляют к 160 мл охлажденной смеси ацетона и этанола, 1:1 (объем/объем). Осадок собирают центрифугированием, диализуют в течение 4 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, D) и лиофилизируют.
b) Инкубирование смеси, полученной при добавлении 0,83 ммоль 1,4-диаминобутана и 50 мг цианборгидрида натрия NaCNBH3 к раствору 200 мг гидроксиэтилкрахмала, также возможно и проводится при 25°С в течение 3 дней.
Пример 1.4. Реакция гидроксиэтилкрахмала с 1-меркапто-2-аминоэтаном

а) К раствору 200 мг гидроксиэтилкрахмала (HES18/0, 4 (ММ=18000 Д, DS=0,4)) в 5 мл воды добавляют 0,83 ммоль 1-меркапто-2-аминоэтана (Sigma Aldrich, Taufkirchen, D) и 50 мг цианборгидрида натрия NaCNBH3. Полученную смесь инкубируют при 80°С в течение 17 часов. Реакционную смесь добавляют к 160 мл охлажденной смеси ацетона и этанола, 1:1 (объем/объем). Осадок собирают центрифугированием, диализуют в течение 4 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, D) и лиофилизируют.
b) Инкубирование смеси, полученной при добавлении 0,83 ммоль 1-меркапто-2-аминоэтана и 50 мг цианборгидрида натрия NaCNBH3 к раствору 200 мг гидроксиэтилкрахмала, также возможно и проводится при 25°С в течение 3 дней.
Пример 2: Получение производных гидроксиэтилкрахмала путем конъюгирования с неокисленным восстановленным концом
Пример 2.1: Реакция гидроксиэтилкрахмала с карбогидразидом

0,96 г HESIS/0,4 (MM=18000 Д, DS=0,4) растворяют в 8 мл водного 0,1 М натрий-ацетатного буфера, рН 5,2 и добавляют 8 ммоль карбогидразида (Sigma, Aldrich, Taufkirchen, D). После перемешивания в течение 18 часов при 25°С реакционную смесь добавляют к 160 мл охлажденной смеси ацетона и этанола, 1:1 (объем/объем). Осажденный продукт собирают центрифугированием, перерастворяют в 40 мл воды, диализуют в течение 3 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, D) и лиофилизируют.
Пример 2.2: Реакция гидроксиэтилкрахмала с адипиновым дигидразидом

0,96 г HES18/0,4 (MM=18000 Д, DS=0,4) растворяют в 8 мл водного 0, 1 М натрий-ацетатного буфера, рН 5,2 и добавляют 8 ммоль адипинового дигидразида (Lancaster Synthesis, Frankfurt/Main, D). После перемешивания в течение 18 часов при 25°С реакционную смесь добавляют к 160 мл охлажденной смеси ацетона и этанола, 1:1, (объем/объем). Осажденный продукт собирают центрифугированием, перерастворяют в 40 мл воды, диализуют в течение 3 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, D) и лиофилизируют.
Пример 2.3: Реакция гидроксиэтилкрахмала с 1, 4-фенилен-бис-3-тиосемикарбазидом

0,96 г HES18/0,4 (ММ=18000 Д, DS=0,4) растворяют в 8 мл водного 0,1 М натрий-ацетатного буфера, рН 5,2 и добавляют 8 ммоль 1,4-фенилен-бис-3-тиосемикарбазида (Lancaster Synthesis, Frankfurt/Main, D). После перемешивания в течение 18 часов при 25°С к реакционной смеси добавляют 8 мл воды и суспензию центрифугируют в течение 15 минут при 4500 об/мин. Прозрачный супернатант декантируют и затем добавляют к 160 мл охлажденной смеси ацетона и этанола, 1:1, (объем/объем). Осажденный продукт собирают центрифугированием, перерастворяют в 40 мл воды и центрифугируют в течение 15 минут при 4500 об/мин. Прозрачный супернатант диализуют в течение 3 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, D) и лиофилизируют.
Пример 2.4: Реакция гидроксиэтилкрахмала с О-[2-(2-аминооксиэтокси)этил]гидроксиламином
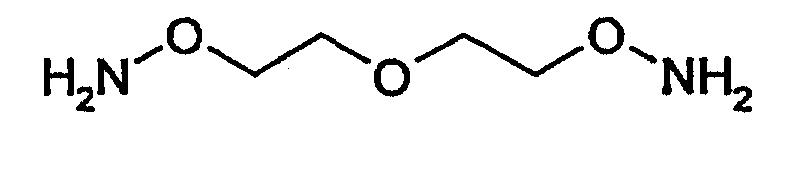
О-[2-(2-Аминооксиэтокси)этил]гидроксиламин синтезируют по методу Boturyn et al. Tetrahedron 53 (1997) p.5485-5492 в 2 стадии из коммерчески доступного материала.
0,96 г HES18/0,4 (MM=18000 Д, DS=0,4) растворяют в 8 мл водного 0,1 М натрий-ацетатного буфера, рН 5,2 и добавляют 8 ммоль О-[2-(2-аминооксиэтокси)этил]гидроксиламина. После перемешивания в течение 18 часов при 25°С реакционную смесь добавляют к 160 мл охлажденной смеси ацетона и этанола, 1:1 (объем/объем). Осажденный продукт собирают центрифугированием, перерастворяют в 40 мл воды, диализуют в течение 3 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, D) и лиофилизируют.
Пример 3. Получение гидроксиэтилкрахмальных производных при реакции с восстановленным концом при его окислении
Пример 3.1. Реакция гидроксиэтилкрахмала с карбогидразидом

0,12 ммоль Оксо-HES 10/0,4 (MM=10000 Д, DS=0,4, получен в соответствии с DE 19628705 1А) растворяют в 3 мл абсолютного диметилсульфоксида (ДМСО) и добавляют по каплям в атмосфере азота к смеси 15 ммоль карбогидразида (Sigma, Aldrich, Taufkirchen, D) в 15 мл ДМСО. После перемешивания в течение 88 часов при 65°С реакционную смесь добавляют к 160 мл охлажденной смеси ацетона и этанола, 1:1, (объем/объем). Осажденный продукт собирают центрифугированием, диализуют в течение 4 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, D) и лиофилизируют.
Пример 3.2. Реакция гидроксиэтилкрахмала с 1,4-фенилен-бис-3-тиосемикарбазидом

0,12 ммоль Оксо-HES 10/0,4 (ММ=10000 Д, DS=0,4, получают в соответствии с DE 19628705 1А) растворяют в 3 мл абсолютного диметилсульфоксида (ДМСО) и добавляют по каплям в атмосфере азота к смеси 15 ммоль 1, 4-фенилен-бис-3-тиосемикарбазида
(Lancaster Synthesis, Frankfurt/Main, D) в 15 мл ДМСО. После перемешивания в течение 88 часов при 65°С реакционную смесь добавляют к 160 мл охлажденной смеси ацетона и этанола, 1:1, (объем/объем). Осажденный продукт собирают центрифугированием, диализуют в течение 4 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, D) и лиофилизируют.
Пример 3.3. Реакция гидроксиэтилкрахмала с гидразином
H2 N-NH2
1,44 г (0,12 ммоль) Оксо-HES 10/0,4 (ММ=10000 Д, DS=0,4, получают в соответствии с DE 19628705 1А) растворяют в 3 мл абсолютного диметилсульфоксида (ДМСО) и добавляют по каплям в атмосфере азота к смеси 0,47 мл (15 ммоль) гидразина в 15 мл ДМСО. После перемешивания в течение 19 часов при 40°С реакционную смесь добавляют к 160 мл смеси ацетона и этанола, 1:1, (объем/объем). Осажденный продукт собирают центрифугированием, перерастворяют в 40 мл воды, диализуют в течение 2 дней против 0,5% (объем/объем) триэтиламина в водном растворе и в течение 2 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, D) и лиофилизируют.
Пример 3.4. Реакция гидроксиэтилкрахмала с гидроксиламином

О-[2-(2-Аминооксиэтокси)этил]гидроксиламин синтезируют по описанной выше методике Boturyn в 2 стадии из коммерчески доступного материала (Boturyn, Boudali, Constant, Defrancq, Lhomme, 1997, Tetrahedron 53, 5485).
1,44 г (0,12 ммоль) Оксо-HES 10/0,4 (MM=10000 Д, DS=0,4, получают в соответствии с DE 19628705 1А) растворяют в 3 мл абсолютного диметилсульфоксида (ДМСО) и добавляют по каплям в атмосфере азота к смеси 2,04 мл (15 ммоль) О-[2-(2-аминооксиэтокси)этил]гидроксиламина в 15 мл ДМСО. После перемешивания в течение 48 часов при 65°С реакционную смесь добавляют к 160 мл смеси ацетона и этанола, 1:1, (объем/объем). Осажденный продукт собирают центрифугированием, перерастворяют в 40 мл воды, диализуют в течение 4 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, D) и лиофилизируют.
Пример 3.5. Реакция гидроксиэтилкрахмала с адипиновым дигидразидом

1,74 г (15 ммоль) адипинового дигидразида (Lancaster Synthesis, Frankfurt/Main, D) растворяют в 20 мл абсолютного диметилсульфоксида (ДМСО) при 65°С и добавляют по каплям в атмосфере азота 1,44 г (0,12 ммоль) Оксо-HES 10/0,4 (ММ=10000 Д, DS=0,4, получают в соответствии с DE 19628705 1А), растворенного в 3 мл ДМСО. После перемешивания в течение 68 часов при 60°С реакционную смесь добавляют к 200 мл воды. Раствор, содержащий реакционный продукт, диализуют в течение 2 дней против 0,5% (объем/объем) триэтиламина в водном растворе и в течение 2 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, D) и лиофилизируют.
Пример 3.6. Реакция гидроксиэтилкрахмала с 1,4-диаминобутаном

1,44 г (0,12 ммоль) Оксо-HES 10/0,4 (ММ=10000 Д, DS=0,4, получают в соответствии с DE 19628705 1А) растворяют в 3 мл сухого диметилсульфоксида (ДМСО) и добавляют по каплям в атмосфере азота к смеси 1,51 мл (15 ммоль) 1,4-диаминобутана (Sigma Aldrich, Taufkirchen, D) в 15 мл ДМСО. После перемешивания в течение 19 часов при 40°С реакционную смесь добавляют к 160 мл смеси этанола и ацетона, 1:1, (объем/объем). Осадок амино-HES10KD/O,4 собирают центрифугированием, перерастворяют в 40 мл воды, диализуют в течение 4 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, D) и лиофилизируют.
Пример 4. Окисление эритропоэтина
Окисленный эритропоэтин получают по методике, описанной в примере 8. В качестве окисленного эритропоэтина используют ЕРО-GT-1-A, описанный в примере 8.11(с) (EPO-GT-1, полученный без кислотного гидролиза путем обработки в условиях мягкого окисления периодатом).
Пример 5: Конъюрирование гидроксиэтилкрахмальных производных с окисленным эритропоэтином из примера 4
Пример 5.1. Реакция окисленного эритропоэтина с реакционным продуктом из примера 2.1
Окисленный ЕРО (1,055 мкг/мкл) в 20 мМ буфера PBS доводят до значения рН 5,3 с использованием 5 М натрий-ацетатного буфера, рН 5,2. К 19 мкл раствора ЕРО добавляют 18 мкл раствора производного HES, полученного по процедуре примера 2.1 (MM 18 кД; 18,7 мкг/мкл в 0,1 М натрий-ацетатном буфере, рН 5,2), и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт очищают в SDS-PAGE с использованием буфера NuPAGE 10% Bis-Tris гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Invitrogen. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента проиллюстрирован на фиг.1. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокой молекулярной массы. Расширение полосы связано с распределением по молекулярной массе используемых производных HES и количеством производных HES, присоединенных к белку.
Пример 5.2. Реакция окисленного эритропоэтина с реакционным продуктом из примера 2.3
Окисленный ЕРО (1,055 мкг/мкл) в 20 мМ буфера PBS доводят до значения рН 5,3 с использованием 5 М натрий-ацетатного буфера, рН 5,2. К 19 мкл раствора ЕРО добавляют 18 мкл раствора производного HES, полученного по процедуре примера 2.3 (MM 18 кД; 18,7 мкг/мкл в 0,1 М натрий-ацетатном буфере, рН 5,2) и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом SDS-PAGE с использованием буфера NuPAGE 10% Bis-Tris гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Invitrogen.
Пример 5.3. Реакция окисленного эритропоэтина с реакционным продуктом из примера 2.4
Окисленный ЕРО (1,055 мкг/мкл) в 20 мМ буфера PBS доводят до значения рН 5,3 с использованием 5 М натрий-ацетатного буфера, рН 5,2. К 19 мкл раствора ЕРО добавляют 18 мкл раствора производного HES, полученного по процедуре примера 2.4 (MM 18 кД; 18,7 мкг/мкл в 0,1 М натрий-ацетатном буфере, рН 5,2) и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом SDS-PAGE с использованием буфера NuPAGE 10% Bis-Tris гели/MOPS (Invitrogen, Carlsbad, CA, USA) как описано в инструкциях компании Invitrogen. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.2. Удачный результат конъюгирования показан в виде перемещения белковой полосы в сторону более высокой молекулярной массы. Расширение полосы связано с распределением по молекулярной массе используемых производных HES и с числом производных HES, присоединенных к белку.
Пример 5.4. Реакция окисленного эритропоэтина с реакционным продуктом из примера 3.1
Окисленный ЕРО (1,055 мкг/мкл) в 20 мМ буфера PBS доводят до значения рН 5,3 с использованием 5 М натрий-ацетатного буфера, рН 5,2. К 19 мкл раствора ЕРО добавляют 18 мкл раствора производного HES, полученного по процедуре примера 3.1 (MM 10 кД; 18,7 мкг/мкл в 0,1 М натрий-ацетатном буфере, рН 5,2), и смесь инкубируют в течение 16 часов при 25° С. После лиофилизации неочищенный продукт анализируют методом SDS-PAGE с использованием буфера NuPAGE 10% Bis-Tris гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Invitrogen. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.3. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокой молекулярной массы. Расширение полосы связано с распределением по молекулярной массе используемых производных HES и с числом производных HES, присоединенных к белку.
Пример 5.5. Реакция окисленного эритропоэтина с реакционным продуктом из примера 3.2
Окисленный ЕРО (1,055 мкг/мкл) в 20 мМ буфера PBS доводят до значения рН 5,3 с помощью 5 М натрий-ацетатного буфера, рН 5,2. К 19 мкл раствора ЕРО добавляют 18 мкл раствора производного HES, полученного по процедуре примера 3.1 (MM 10 кД; 18,7 мкг/мкл в 0,1 М натрий-ацетатном буфере, рН 5,2) и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом SDS-PAGE с использованием буфера NuPAGE 10% Bis-Tris гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Invitrogen. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.3. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокой молекулярной массы. Расширение полосы связано с распределением молекулярной массы используемых производных HES и с числом производных HES, присоединенных к белку.
Пример 6. Образование тио-ЕРО путем восстановления эритропоэтина
241,5 мкг эритропоэтина (EPO-GT-1, см. пример 8) в 500 мкл 0,1 М натрий-боратного буфера, 5 мМ ЭДТА, 10 мМ DTT (Lancaster, Morcambe, UK), pH 8,3, инкубируют в течение 1 часа при 37°С. DTT удаляют фильтрованием при центрифугировании с использованием концентратора на 0,5 мл VIVASPIN, 10 KD MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин с последующей промывкой 3 раза боратным буфером и дважды фосфатным буфером (0,1 М, 9,15 М NaCl, 50 мМ ЭДТА, pH 7,2). Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Пример 7. Конъюгирование производных гидроксиэтилкрахмала с тио-эритропоэтином с использованием сшивающего соединения
В каждом из приведенных ниже примеров в качестве сшивающего соединения используют сложный N-(альфа-малемидоацетокси)сукцинимидный эфир (AMAS).
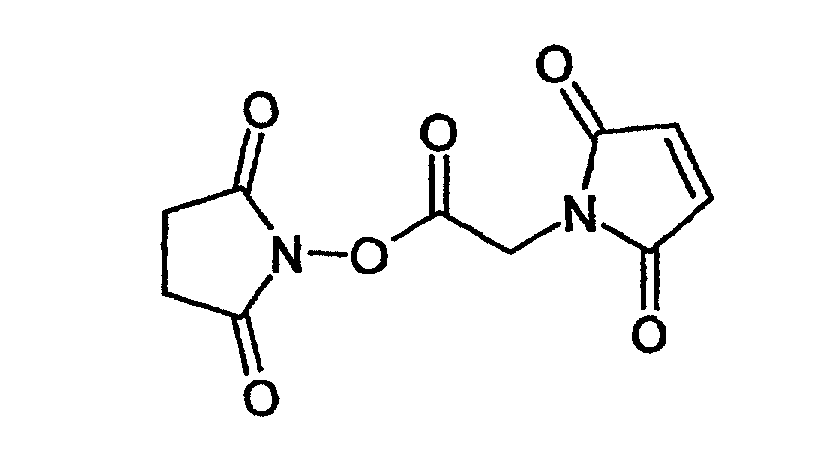
Пример 7.1. Реакция тио-эритропоэтина с реакционным продуктом из примера 2.1 и сшивающим соединением
К 50 нмоль HES-производного, полученного по методике примера 2.1 и растворенного в 200 мкл 0,1 М натрий-фосфатного буфера (0,1 М, 9,15 М NaCl, 50 мМ ЭДТА, pH 7,2), добавляют 10 мкл 2,5 мкмоль раствора AMAS (Sigma Aldrich, Taufkirchen, D) в ДМСО. Прозрачный раствор инкубируют в течение 80 минут при температуре 25°С и 20 минут при 40°С. Оставшийся AMAS удаляют фильтрованием при центрифугировании с использованием концентратора на 0,5 мл VIVASPIN, 5 кД MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин с промывкой 4 раза в течение 30 минут фосфатным буфером.
К оставшемуся раствору добавляют 15 мкг тиоЕРО, полученного по методике примера 5 (1 мкг/мкл в фосфатном буфере) и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом SDS-PAGE с использованием буфера NuPAGE 10% Bis-Tris гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Invitrogen. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.4. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокой молекулярной массы. Расширение полосы связано с распределением по молекулярной массе используемых производных HES и с числом производных HES, присоединенных к белку.
Пример 7.2. Реакция тио-эритропоэтина с реакционным продуктом из примера 2.2 и сшивающим соединением
К 50 нмоль HES-производного, полученного по методике примера 2.2 и растворенного в 200 мкл 0,1 М натрий-фосфатного буфера (0,1 М, 9,15 М NaCl, 50 мМ ЭДТА, рН 7,2), добавляют 10 мкл раствора 2,5 мкмоль AMAS (Sigma Aldrich, Taufkirchen, D) в ДМСО. Прозрачный раствор инкубируют в течение 80 минут при температуре 25°С и 20 минут при 40° С. Оставшийся AMAS удаляют фильтрованием при центрифугировании с использованием концентратора на 0,5 мл VIVASPIN, 5 кД MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин с промывкой 4 раза в течение 30 минут в фосфатном буфере.
К оставшемуся раствору добавляют 15 мкг тиоЕРО, полученного по методике примера 5 (1 мкг/мкл в фосфатном буфере), и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют в SDS-PAGE с использованием буфера NuPAGE 10% Bis-Tris гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Invitrogen. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.5. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокой молекулярной массы. Расширение полосы связано с распределением по молекулярной массе используемых производных HES и с числом производных HES, присоединенных к белку.
Пример 7.3. Реакция тио-эритропоэтина с реакционным продуктом из примера 2.3 и сшивающим соединением
К 50 нмоль HES-производного, полученного по методике примера 2.3 и растворенного в 200 мкл 0,1 М натрий-фосфатного буфера (0,1 М, 9,15 М NaCl, 50 мМ ЭДТА, рН 7,2), добавляют 10 мкл раствора 2,5 мкмоль AMAS (Sigma Aldrich, Taufkirchen, D) в ДМСО. Прозрачный раствор инкубируют в течение 80 минут при температуре 25°С и 20 минут при 40°С. Оставшийся AMAS удаляют фильтрованием при центрифугировании с использованием концентратора на 0,5 мл VIVASPIN, 5 кД MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин с промывкой 4 раза в течение 30 минут в фосфатном буфере.
К оставшемуся раствору добавляют 15 мкг тиоЕРО, полученного по методике примера 5 (1 мкг/мкл в фосфатном буфере), и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом SDS-PAGE с использованием буфера NuPAGE 10% Bis-Tris гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Invitrogen. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.5. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокой молекулярной массы. Расширение полосы связано с распределением по молекулярной массе используемых производных HES и с числом производных HES, присоединенных к белку.
Пример 7.4. Реакция тио-эритропоэтина с реакционным продуктом из примера 2.4 и сшивающим соединением
К 50 нмоль HES-производного, полученного по методике примера 2.4 и растворенного в 200 мкл 0,1 М натрий-фосфатного буфера (0,1 М, 9,15 М NaCl, 50 мМ ЭДТА, рН 7,2), добавляют 10 мкл раствора 2,5 мкмоль AMAS (Sigma Aldrich, Taufkirchen, D) в ДМСО. Прозрачный раствор инкубируют в течение 80 минут при температуре 25°С и 20 минут при 40°С. Оставшийся AMAS удаляют фильтрованием при центрифугировании с использованием концентратора на 0,5 мл VIVASPIN, 5 кД MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин с промывкой 4 раза в течение 30 минут в фосфатном буфере.
К оставшемуся раствору добавляют 15 мкг тиоЕРО, полученного по методике примера 5 (1 мкг/мкл в фосфатном буфере), и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом SDS-PAGE с использованием буфера NuPAGE 10% Bis-Tris гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Invitrogen. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.4. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокой молекулярной массы. Расширение полосы связано с распределением по молекулярной массе используемых производных HES и с числом производных HES, присоединенных к белку.
Пример 7.5. Реакция тио-эритропоэтина с реакционным продуктом из примера 1.1 и сшивающим соединением
К 50 нмоль HES-производного, полученного по методике примера 1.1, при инкубации в условиях, включающих 80°С в течение 17 часов, а также 25°С в течение 3 дней, и растворенного в 200 мкл 0,1 М натрий-фосфатного буфера (0,1 М, 9,15 М NaCl, 50 мМ ЭДТА, рН 7,2) добавляют 10 мкл раствора 2,5 мкмоль AMAS (Sigma Aldrich, Taufkirchen, D) в ДМСО. Прозрачный раствор инкубируют в течение 80 минут при температуре 25°С и 20 минут при 40°С. Оставшийся AMAS удаляют фильтрованием при центрифугировании с использованием концентратора на 0, 5 мл VIVASPIN, 5 кД MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин с промывкой 4 раза в течение 30 минут в фосфатном буфере.
К оставшемуся раствору добавляют 15 мкг тио-ЕРО, полученного по методике примера 5 (1 мкг/мкл в фосфатном буфере), и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом SDS-PAGE с использованием буфера NuPAGE 10% Bis-Tris гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Invitrogen. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.5. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокой молекулярной массы. Расширение полосы связано с распределением по молекулярной массе используемых производных HES и с числом производных HES, присоединенных к белку.
Пример 7.6. Реакция тио-эритропоэтина с реакционным продуктом из примера 1.3 и сшивающим соединением
К 50 нмоль HES-производного, полученного по методике примера 1.3, при инкубации в условиях, включающих 80°С в течение 17 часов, а также 25°С в течение 3 дней, и растворенного в 200 мкл 0,1 М натрий-фосфатного буфера (0,1 М, 9,15 М NaCl, 50 мМ ЭДТА, рН 7,2), добавляют 10 мкл раствора 2,5 мкмоль AMAS (Sigma Aldrich, Taufkirchen, D) в ДМСО. Прозрачный раствор инкубируют в течение 80 минут при температуре 25°С и 20 минут при 40°С. Оставшийся AMAS удаляют фильтрованием при центрифугировании с использованием концентратора на 0,5 мл VIVASPIN, 5 кД MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин с промывкой 4 раза в течение 30 минут в фосфатном буфере.
К оставшемуся раствору добавляют 15 мкг тио-ЕРО, полученного по методике примера 5 (1 мкг/мкл в фосфатном буфере), и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом SDS-PAGE с использованием буфера NuPAGE 10% Bis-Tris гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Invitrogen. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.5. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокой молекулярной массы. Расширение полосы связано с распределением по молекулярной массе используемых производных HES и с числом производных HES, присоединенных к белку.
Пример 7.7. Реакция тио-эритропоэтина с реакционным продуктом из примера 3.1 и сшивающим соединением
К 50 нмоль HES-производного, полученного по методике примера 3.1 и растворенного в 200 мкл фосфатного буфера (0,1 М, 9,15 М NaCl, 50 мМ ЭДТА, рН 7,2), добавляют 10 мкл раствора 2,5 мкмоль AMAS (Sigma Aldrich, Taufkirchen, D) в ДМСО и прозрачный раствор инкубируют в течение 80 минут при температуре 25°С и 20 минут при 40°С. AMAS удаляют фильтрованием при центрифугировании с использованием концентратора на 0,5 мл VIVASPIN, 5 кД MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин с промывкой 4 раза в течение 30 минут в фосфатном буфере.
К оставшемуся раствору добавляют 15 мкг тио-ЕРО (1 мкг/мкл в фосфатном буфере) и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом SDS-PAGE с использованием буфера NuPAGE 10% Bis-Tris гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Invitrogen. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.6. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокой молекулярной массы. Расширение полосы связано с распределением по молекулярной массе используемых производных HES и с числом производных HES, присоединенных к белку.
Пример 7.8. Реакция тио-эритропоэтина с реакционным продуктом из примера 3.2 и сшивающим соединением
К 50 нмоль HES-производного, полученного по методике примера 3.2, растворенного в 200 мкл фосфатного буфера (0,1 М, 9,15 М NaCl, 50 мМ ЭДТА, рН 7,2), добавляют 10 мкл раствора 2,5 мкмоль AMAS (Sigma, Aldrich, Taufkirchen, D) в ДМСО и прозрачный раствор инкубируют в течение 80 минут при температуре 25°С и 20 минут при 40°С. AMAS удаляют фильтрованием при центрифугировании с использованием концентратора на 0,5 мл VIVASPIN, 5 кД MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин с промывкой 4 раза в течение 30 минут в фосфатном буфере.
К оставшемуся раствору добавляют 15 мкг тио-ЕРО (1 мкг/мкл в фосфатном буфере) и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом SDS-PAGE с использованием буфера NuPAGE 10% Bis-Tris гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Invitrogen. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.6. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокой молекулярной массы. Расширение полосы связано с распределением по молекулярной массе используемых производных HES и с числом производных HES, присоединенных к белку.
Пример 7.9. Реакция тио-эритропоэтина с реакционным продуктом из примера 3.3 и сшивающим соединением
К 50 нмоль HES-производного, полученного по методике примера 3.3 и растворенного в 200 мкл фосфатного буфера (0,1 М, 9,15 М NaCl, 50 мМ ЭДТА, рН 7,2), добавляют 10 мкл раствора 2,5 мкмоль AMAS (Sigma Aldrich, Taufkirchen, D) в ДМСО и прозрачный раствор инкубируют в течение 80 минут при температуре 25°С и 20 минут при 40°С. AMAS удаляют фильтрованием при центрифугировании с использованием концентратора на 0,5 мл VIVASPIN, 5 кД MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин с промывкой 4 раза в течение 30 минут в фосфатном буфере.
К оставшемуся раствору добавляют 15 мкг тио-ЕРО (1 мкг/мкл в фосфатном буфере) и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом SDS-PAGE с использованием буфера NuPAGE 10% Bis-Tris гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Invitrogen. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.6. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокой молекулярной массы. Расширение полосы связано с распределением по молекулярной массе используемых производных HES и с числом производных HES, присоединенных к белку.
Пример 7.10. Реакция тио-эритропоэтина с реакционным продуктом из примера 3.4 и сшивающим соединением
К 50 нмоль HES-производного, полученного по методике примера 3.4 и растворенного в 200 мкл фосфатного буфера (0,1 М, 9,15 М NaCl, 50 мМ ЭДТА, рН 7,2), добавляют 10 мкл раствора 2,5 мкмоль AMAS (Sigma Aldrich, Taufkirchen, D) в ДМСО и прозрачный раствор инкубируют в течение 80 минут при температуре 25°С и 20 минут при 40°С. AMAS удаляют фильтрованием при центрифугировании с использованием концентратора на 0,5 мл VIVASPIN, 5 кД MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин с промывкой 4 раза в течение 30 минут в фосфатном буфере.
К оставшемуся раствору добавляют 15 мкг тио-ЕРО (1 мкг/мкл в фосфатном буфере) и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом SDS-PAGE с использованием буфера NuPAGE 10% Bis-Tris гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Invitrogen. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.6. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокой молекулярной массы. Расширение связано с распределением по молекулярной массе используемых производных HES и с числом производных HES, присоединенных к белку.
Пример 7.11. Реакция тио-эритропоэтина с реакционным продуктом из примера 3.5 и сшивающим соединением
К 50 нмоль HES-производного, полученного по методике примера 3.5 и растворенного в 200 мкл фосфатного буфера (0,1 М, 9,15 М NaCl, 50 мМ ЭДТА, рН 7,2), добавляют 10 мкл раствора 2,5 мкмоль AMAS (Sigma, Aldrich, Taufkirchen, D) в ДМСО и прозрачный раствор инкубируют в течение 80 минут при температуре 25°С и 20 минут при 40°С. AMAS удаляют фильтрованием при центрифугировании с использованием концентратора на 0,5 мл VIVASPIN, 5 кД MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин с промывкой 4 раза в течение 30 минут в фосфатном буфере.
К оставшемуся раствору добавляют 15 мкг тио-ЕРО (1 мкг/мкл в фосфатном буфере) и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом SDS-PAGE с использованием буфера NuPAGE 10% Bis-Tris гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Invitrogen. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.6. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокой молекулярной массы. Расширение полосы связано с распределением по молекулярной массе используемых производных HES и с числом производных HES, присоединенных к белку.
Пример 7.12. Реакция тио-эритропоэтина с реакционным продуктом из примера 3.6 и сшивающим соединением
К 50 нмоль HES-производного, полученного по методике примера 3.6 и растворенного в 200 мкл фосфатного буфера (0,1 М, 9,15 М NaCl, 50 мМ ЭДТА, рН 7, 2), добавляют 10 мкл раствора 2,5 мкмоль AMAS (Sigma, Aldrich, Taufkirchen, D) в ДМСО и прозрачный раствор инкубируют в течение 80 минут при температуре 25°С и 20 минут при 40°С. AMAS удаляют фильтрованием при центрифугировании с использованием концентратора на 0,5 мл VIVASPIN, 5 кД MWCO (VIVASCIENCE, Hannover, D) при 13000 об/мин с промывкой 4 раза в течение 30 минут в фосфатном буфере.
К оставшемуся раствору добавляют 15 мкг тио-ЕРО (1 мкг/мкл в фосфатном буфере) и смесь инкубируют в течение 16 часов при 25°С. После лиофилизации неочищенный продукт анализируют методом SDS-PAGE с использованием буфера NuPAGE 10% Bis-Tris гели/MOPS (Invitrogen, Carlsbad, CA, USA), как описано в инструкциях компании Invitrogen. Гель окрашивают красящим реактивом кумасси (Roti-Blue) (Roth, Karlsruhe, D) в течение ночи.
Результат эксперимента приведен на фиг.6. Удачный результат конъюгирования показан в виде миграции белковой полосы в сторону более высокой молекулярной массы. Расширение полосы связано с распределением по молекулярной массе используемых производных HES и с числом производных HES, присоединенных к белку.
Пример 8. Препаративное получение конъюгатов HES-EPO
Краткое описание
Конъюгаты HES-EPO синтезируют путем связывания HES-производных (среднее значение ММ составляет 18000 Дальтон/степень замещения гидроксиэтилом составляет 0,4) с частично (в условиях мягкой обработки периодатом) окисленными остатками сиаловой кислоты на олигосахаридных цепях рекомбинантного человеческого ЕРО. На основании структурного анализа углеводов было показано, что введенные модификации не воздействуют на структурную целостность ядерных олигосахаридных цепей, поскольку анализ методом MALDI/TOF-MS HES-модифицированных гликанов в условиях мягкой обработки кислотой выявил наличие интактных нейтральных цепей N-ацетиллактозаминового типа, которые не отличались от соответствующих цепей, наблюдаемых в немодифицированном ЕРО-продукте. Полученные результаты указывают на то, что по меньшей мере 3 модифицированных HES-остатка присоединяются к молекуле ЕРО в препарате ЕРО, который подвергается модификации без предшествующего частичного удаления сиаловой кислоты. Вариант ЕРО, потерявший примерно 50% остатков сиаловой кислоты относительно предшествующего белка, демонстрирует аналогичную мобильность в SDS-PAGE, явно указывающую на высокую молекулярную массу (60-110 КДа против 40 кДа, характерного для стандарта BRP ЕРО). HES-модифицированный ЕРО стабилен в стандартных условиях ионообменной хроматографии при комнатной температуре и рН 3-10.
Биотест на ЕРО в системе нормоцитных мышей указывает на то, что HES-модифицированный ЕРО обладает в 2,5-3,0 раза более высокой удельной активностью (МЕ/мг) в данном тесте в сравнении с международным эталонным стандартом BRP ЕРО, что следует из результатов определения белка по величине поглощения в УФ области в соответствии с методом Европейской Фармакопеи и результатов определения белка ЕРО методом ОФ-ВЭЖХ, калиброванным относительно стандартного препарата BRP ЕРО.
Пример 8.1. Материалы и методы
(а) Высвобождение N-связанных олигосахаридов путем расщепления N-гликозидазой
Образцы инкубируют с 25 единицами (в соответствии со спецификацией от производителя, Roche Diagnostics, Germany) рекомбинантной PNG-азы F в течение ночи при 37°С. Полноту расщепления отслеживают по специфическому смещению мобильности белка в SDS-PAGE. Высвобожденные N-гликаны отделяют от полипептида путем добавления 3 объемов холодного 100% этанола с последующей инкубацией при -20°С в течение по меньшей мере 2 часов (Schroeter S et al., 1999). Осажденный белок удаляют центрифугированием в течение 10 минут при 4°С со скоростью 13000 об/мин. Полученный осадок затем промывают два раза с использованием 500 мкл охлажденного льдом 75% этанола. Олигосахариды в объединенных супернатантах сушат в вакуумной центрифуге (концентратор Speed Vac, Savant Instruments Inc., USA). Образцы гликана перед использованием обессоливают с помощью картриджей Hypercarb (25 мг или 100 мг HyperCarb) следующим образом: колонки промывают 3х500 мкл 80% ацетонитрила (объем/объем) в 0,1% ТФУ с последующими промывками 3×500 мкл воды. Образцы разбавляют водой до конечного объема 300 мкл-600 мкл перед внесением в картридж, который затем тщательно промывают водой. Олигосахариды элюируют с использованием 1,2 мл (картриджи на 25 мг; берут 1,8 мл в случае картриджей на 100 мг) 25% ацетонитрила в воде, содержащей 0,1% трифторуксусной кислоты (объем/объем). Элюированные олигосахариды нейтрализуют с использованием 2 М NH4 OH и сушат в концентраторе Speed Vac. В некоторых случаях обессоливание олигосахаридов, высвобожденных N-гликозидазой, проводят путем адсорбции расщепленной смеси из образцов, содержащих<100 мкг общего (глико)протеина, на картриджах Hypercarb на 100 мг.
(b) Анализ олигосахаридов с помощью лазерной десорбции на основе матричной/ионизационной масс-спектрометрии по времени пролета (MALDI/TOF/TOF-MS)
Используют прибор Bruker ULTRAFLEX для анализа по времени пролета (TOF-TOF): нативные десиалилированные олигосахариды анализируют с использованием 2, 5-дигидроксибензойной кислоты в качестве УФ-поглощающего материала как в варианте положительного, так и отрицательного иона с использованием в обоих случаях рефлектрона. В случае анализов по методу MS-MS выбранные парные ионы подвергают индуцированной лазером диссоциации (LID) и полученные фрагменты ионов разделяют с помощью второго блока TOF прибора (LIFT). Образец растворов по 1 мкл в соответствующей концентрации 1-10 пмоль/мкл смешивают с равным количеством соответствующей матрицы. Указанную смесь наносят капельно на мишень из нержавеющей стали и сушат при комнатной температуре перед проведением анализа.
Пример 8.2. Получение и характеристика рекомбинантного человеческого ЕРО (EPO-GT-1)
ЕРО получают из рекомбинантных СНО клеток при его экспрессии в них, как описано в литературе (Mueller PP et al., 1999, Dorner AJ et al., 1984) и исследуют характеристики полученных препаратов в соответствии с методами Европейской Фармакопеи (Ph. Еиг. 4, Monography 01/2002:1316: Erythropoietin concentrated solution). Конечный продукт характеризуется содержанием сиаловой кислоты на уровне 12 нмоль (± 1,5 нмоль) на нмоль белка. Определяют структуры N-связанных олигосахаридов методами НРАЕС-PAD и MALDI/TOF-MS, как описано в литературе (Nimtz et al., 1999, Grabenhorst, 1999). Полученные ЕРО препараты содержат ди-, три- и тетрасиалилированные олигосахариды (2-12%, 15-28% и 60-80% соответственно, сульфатированные и пентасиалилированные цепи присутствуют в небольших количествах). Было показано, что в целом характеристики гликозилирования препаратов ЕРО аналогичны таковым для международного стандартного препарата BRP ЕРО.
Результаты изоэлектрофокусирования рекомбинантного ЕРО были сравнимы с результатами соответствующего анализа международного стандартного препарата BRP ЕРО, которые демонстрировали соответствующие изоформы. У 25% белка ЕРО отсутствовало O-гликозилирование в положении Serize в полипептидной цепи.
Пример 8.3. Получение частично десиалилированных форм ЕРО Белок EPO-GT-1 (2,84 мг/мл) нагревают до 80°С в 20 мМ Na-фосфатном буфере, рН 7,0 и затем добавляют 100 мкл 1 N H2SO4 в расчете на 1 мл раствора ЕРО, инкубацию продолжают в течение 5 мин, 10 мин. и 60 мин соответственно, что приводит к получению препаратов ЕРО с разной степенью сиалилирования. Количественное определение олигосахаридов с наличием 0-4 сиаловых кислот проводят после высвобождения олигосахаридов N-гликозидазой полипептида и выделение N-связанных цепей проводят при обессоливании с использованием картриджей Hypercarb (25 мг HyperSep Hypercarb; ThermoHypersil-Keystone, UK). Препараты ЕРО нейтрализуют путем добавления 1 N NaOH, затем замораживают в жидком N2 и хранят при температуре -20°С до дальнейшего использования.
Пример 8.4. Окисление периодатом сиалилированных форм ЕРО
К 10 мг необработанного или обработанного кислотой в мягких условиях ЕРО, который растворен в 3,5 мл 20 мМ Na-фосфатного буфера, рН 7,0, добавляют 1,5 мл 0,1 М Na-ацетатного буфера, рН 5,5 и смесь охлаждают до 0°С на ледяной бане; добавляют 500 мкл 10 мМ периодата Na и реакционную смесь выдерживают в темноте в течение 60 минут при 0°С. Затем добавляют 10 мкл глицерина и инкубацию продолжают еще в течение 10 минут в темноте. Частично окисленные формы ЕРО отделяют от реагентов путем обессоливания с использованием концентраторов VIVASPIN (10000 MWCO, PES Vivascience AG, Hannover, Germany) в соответствии с рекомендациями производителя со скоростью 3000 об/мин в лабораторной центрифуге, снабженной фиксированным угловым ротором. После замораживания в жидком азоте препараты ЕРО хранят в конечном объеме 4 мл при температуре -20°С.
Аликвоты частично окисленного препарата ЕРО по 100 мкг подвергают обработке N-гликозидазой и выделяют олигосахариды с использованием картриджей Hypercarb, как описано. Олигосахариды подвергают десиалилированию путем мягкой обработки кислотой и анализируют методом HPAEC-PAD, сравнивая полученное значение времени удерживания с соответствующими показателями аутентичных препаратов стандартных олигосахаридов, как описано в литературе (Nimtz et al., 1990 и 1993).
Пример 8.5. Восстановление дисульфидов ЕРО с использованием дитиоэритрейтола
5 мг EPO-GT-1 инкубируют с 5 мл 0,1 М Трис/HCl буфера, рН 8,1, в присутствии 30 мМ дитиоэритрейтола (DTT) при 37°С в течение 60 минут, удаление DTT достигается путем использовании концентратора Vivaspin при 4°С с проведением 4 циклов замены буфера. Конечный восстановленный препарат ЕРО замораживают в жидком азоте и хранят при -20°С в 50 мМ Na-ацетатном буфере, рН 5,5.
Пример 8.6. Анализ препарата ЕРО
Количественное определение белка в препарате ЕРО проводят путем измерения УФ поглощения при длине волны 280 нм в соответствии с методикой Европейской Фармакопеи (European Pharmacopeia 4, Monography 01/2002; 1316: erythropoietin concentrated solution) в кювете с длиной пробега 1 см. Кроме того, количественное ЕРО проводят методом ОФ-ВЭЖХ с использованием колонки RP-C4 (Vydac Protein C4, Cat#214TP5410, Grace Vydac, Ca, US); методика ВЭЖХ была откалибрована с использованием эталонного препарата эритропоэтина BRP 1 (European Pharmacopeia, Conseil, de Г Europe В.Р. 907-F67029, Strasbourg Cedex 1).
Пример 8.7.Окисление десиалилированного ЕРО галактозооксидазой
4,485 мг полностью десиалилированного ЕРО инкубируют в 20 мМ Na-фосфатном буфере, рН 6,8, в присутствии 16 мкл каталазы (6214 единиц/200 мл) и 80 мкл галактозооксидазы (2250 единиц/мл из Dactylium dendroides (Sigma-Aldrich, Steinheim, Germany));
инкубацию проводят при 37°С в течение ночи; добавляют 2 раза по 20 мкл галактозооксидазы через 4 часа и через 8 часов после начала инкубации.
Пример 8.8. Подготовка образцов ЕРО для биотестирования
Выделение к очистка образцов ЕРО из инкубационных сред, содержащих белковые препараты ЕРО с активированным JSSS, обработанные периодатом или окисленные галахтозоохсидазой
Очистку препаратов ЕРО (удаление непрореагировавших производных HES) проводят при комнатной температуре. Смеси для инкубации ЕРО (примерно 5 мг по белку ЕРО) разбавляют 1:10 буфером А (20 мМ N-морфолинпропансульфоновой кислоты [MOPS/NaOH] в бидистиллированной НзО, рН 8,0) и наносят на колонку, содержащую 3 мл сефарозы (Q-Sepharose HP (Pharmacia, код No. 17-1014-03, Лот No. 220211)), уравновешенную буфером А в количестве, равном 10 объемам колонки (CV), со скоростью течения 0,5 мл/мин. Колонку промывают 6-8 CV буфера А (скорость течения=0,8 мл/мин) и проводят элюирование с использованием буфера В (20 мМ морфолинэтансульфоновой кислоты [MES/NaOH], 0,5 М NaCl в бидистиллированной Н2O, рН 6,5) со скоростью течения 0,5 мл/мин. ЕРО определяют по УФ поглощению при длине волны 280 нм и элюируют примерно в объеме 6 мл. Колонку регенерируют с использованием 3 CV буфера С (20 мМ MES, 1,5 М NaCl в Н2O, доведенной до рН 6,5) и подвергают повторному уравновешиванию с использованием 10 CV буфера А (скорость течения=0,7 мл/мин).
Буферную замену в элюатах ЕРО, полученных на стадии элюирования с Q-Sepharose, проводят с помощью концентратора Vivaspin и фосфатно-буферного раствора (PBS) в течение 3 циклов центрифугирования на образец; объем образцов доводят до 2 мл с помощью PBS, после чего образцы хранят при -20°С.
Получают лишь<25% частично десиалилированных и впоследствии окисленных в условиях мягкой обработки периодатом форм ЕРО, которые подверглись НЕЗ-модификации из элюата с колонок с сефарозой (Q-Sepharose), поскольку в использованных условиях основные формы ЕРО не связываются с сефарозой (Q-Sepharose) и обнаруживаются в протекающем потоке вместе с непрореагировавшими НЕЗ-производными.
Пример 8.9. Анионообменная хроматография при высоком рН с пульсовым амперометрическим обнаружением (HPAEC-PAD)
Очищенные нативные и десиалилированные олигосахариды анализируют с помощью анионообменной хроматографии при высоком рН (НРАЕ) в системе Dionex BioLC (Dionex, USA), снабженной колонкой CarboPac PA1 (0,4×25 см) в сочетании с системой пульсового амперометрического выявления (PAD) (Schroter et al., 1999; Nimtz et al., 1999). Значения потенциалов детектора (Е) и длительность пульсовой обработки (Т) составляют: Е1:+50 мВ, 11: 480 мсек; Е2:+500 мВ, Т2: 120 мсек; Е3: -500 мВ, Т3: 60 мсек, и диапазон выхода составляет 500-1500 нА. Затем олигосахариды инъецируют в колонку CarboPac PA1, уравновешенную 100% растворителем А. В случае элюирования десиалилированных олигосахаридов (скорость течения: 1 мл/мин) указанное элюирование проводят при использовании линейного градиента (0-20%) растворителя В в течение 40 минут с последующим линейным повышением уровня растворителя В от 20 до 100% в течение 5 минут. Растворитель А представляет собой 0,2 М NaOH в бидистиллированной воде, растворитель В состоит из 0,6 М NaOAc в растворителе А. В случае нативных олигосахаридов колонку уравновешивают 100% растворителем С (0,1 М NaOH в бидистиллированной HzO) и элюирование (скорость течения: 1 мл/мин) проводят с использованием линейного градиента (0-35%) растворителя D в течение 48 минут с последующим линейным повышением уровня растворителя D от 35 до 100% в течение 10 минут. Растворитель D представляет собой 0,6 М NaAc в растворителе С.
Пример 8.10. Анализ моносахаридного состава N-гликанов, HES-модифицированных N-гликанов и белка ЕРО методом ГХ-МС
Моносахариды анализируют в виде соответствующих метилгликозидов после проведения метанолиза, N-реацетилирования и триметилсилилирования по методу ГХ-МС [Chaplin, M.F. (1982) А rapid and sensitive method for the analysis of carbohydrate. Anal. Biochem. 123, 336-341]. Анализы проводят в масс-спектрометре, действующем по механизму захвата ионов, Finnigan GCQ (Finnigan MAT Corp., San Jose, CA), работающем в режиме положительных ионов EI, который снабжен капиллярной колонкой DB5 длиной 30 м. Прибор работает по следующей температурной программе: 2 минуты изотермический режим при 80°С, затем температуру повышают со скоростью 10 градусов/мин до 300°С.
Моносахариды идентифицируют по значению времени удерживания и показателям характерных фрагментов. Для количественного определения используют некорректированные результаты интеграции электронных пиков. Моносахарид, дающий более чем один пик, в связи с аномерностью и/или присутствием фураноидных и пираноидных форм количественно определяют путем сложения всех основных пиков. Используют 0,5 мкг миоинозита в качестве внутреннего стандартного соединения.
Пример 8.11 Результаты
Пример 8.11 (а) Характеристика N-гликанов в EPO-GT-1 после его обработки кислотой в мягких условиях (частично десиалилированного)
Препараты EPO-GT-1 подвергают обработке кислотой в мягких условиях в течение 5, 10 или 60 минут и затем анализируют в SDS-PAGE до и после высвобождения N-связанных олигосахаридов при инкубации с N-гликозидазой, как показано на фиг.7. N-связанные олигосахариды подвергают картированию олигосахаридов по методу HPAEC-PAD (фиг.8). Необработанный EPO-GT-1 содержит более 90% N-связанных олигосахаридов с 3 или 4 остатками сиаловой кислоты, тогда как после 5-минутной инкубации в присутствии слабой кислоты менее 40% углеводных цепей содержат 3 или 4 остатка сиаловой кислоты. Анализ по методу HPAEC-PAD десиалилированных N-гликанов показал, что соотношение нейтральных олигосахаридов, которые были обнаружены в необработанном EPO-GT-1, остается стабильным в препаратах, подвергнутых кислотной обработке в течение 5, 10 или 60 минут. Анализ методом MALDI/TOF-MS десиалилированных гликанов показал, что менее 90% проксимальной фукозы присутствует после обработки слабой кислотой данного белка.
Пример 8.11 (b) Характеристика EPO-GT-1, обработанного периодатом
На фиг.10 приведены данные, показывающие мобильность в SDS-PAGE обработанных в мягких условиях периодатом форм ЕРО, которые были предварительно подвергнуты обработке кислотой в течение 5 и 10 минут или которые не обрабатывались. Условия, использованные для периодатного окисления сиаловых кислот, не влияют на поведение препаратов ЕРО в SDS-PAGE (в сравнении с фиг.7). Окисление сиаловых кислот приводит к смещению олигосахаридов в рамках анализа HPAEC-PAD, к более ранним показателям по времени элюирования (для сравнения см. фиг.8 и 11).
Пример 8.11 (с) Характеристика HES-модифицированных производных ЕРО
(аа) HES-модификация с течением времени EPO-GT-1-A с использованием модифицированного гидроксиламином HES-производного X, полученного по методике примера 2.4
400 мкг модифицированного гидроксиламином HES-производного Х добавляют к 20 мкг EPO-GT-1-A (ЕРО, окисленный в мягких условиях периодатом, но не подвергнутый кислотному гидролизу перед указанным окислением периодатом в мягких условиях) в 20 мкл 0,5 М NaOAc буфера, рН 5,5, и реакцию останавливают через 30 минут, 2, 4, и 17 часов соответственно путем замораживания образцов в жидком азоте. Впоследствии образцы хранят при -20°С до проведения дальнейшего анализа.
Добавляют буфер для проведения анализа образцов в SDS-PAGE и нагревают указанные образцы до 90°С, после чего наносят на SDS-содержащие гели. Как показано на фиг.12, увеличение времени инкубации приводит к более сильному смещению образцов в сторону более высокой молекулярной массы белка. После 17 часов инкубации в присутствии модифицированного гидроксиламином HES-производного Х обнаруживается диффузная окрашиваемая кумасси белковая полоса, мигрирующая в зоне между 60 и 11 кДа относительно положения стандартных препаратов для определения молекулярной массы (см. правую часть фиг.12). При обработке N-гликозидазой большая часть белков мигрирует в сторону положения де-N-гликозилированного ЕРО (см. фиг.14, правый гель; стрелка А указывает на положение перемещения N-гликозидазы, стрелка В указывает на миграцию де-N-гликозилированного ЕРО; диффузная белковая полоса, видимая на участке между положениями стандартных препаратов для определения молекулярной массы в диапазоне между 28 кДа и 36 кДа, преимущественно отражает те ЕРО формы, которые включают модификации, определяемые HES и сайтом O-гликозилирования молекулы). В связи со специфичностью действия N-гликозидазы авторы на основании данного эксперимента пришли к выводу, что фактически HES-модификация происходит на окисленных периодатом остатках сиаловой кислоты гликанов в белке ЕРО.
(bb) Характеристика конъюгатов HES-EPO
Конъюгаты HES-EPO I (получаемые из EPO-GT-1 после окисления периодатом в мягких условиях, например на основе EPO-GT-1-A), II (получаемые из EPO-GT-1, подвергнутого 5-минутному кислотному гидролизу и окислению периодатом в мягких условиях) и III (получаемые из EPO-GT-1, подвергнутого 10-минутному кислотному гидролизу и окислению периодатом в мягких условиях) синтезируют, как было описано ранее. Методика включает вариант контрольной инкубации (К), содержащий немодифицированный EPO-GT-1 в том же буфере, к которому добавляют эквивалентное количество немодифицированного HES. Инкубационные смеси подвергают дальнейшей очистке для проведения последующего биохимического анализа производных HES-EPO.
Полученные при инкубации конъюгаты HES-EPO I, II и III, a также продукт контрольной инкубации К подвергают очистке на сефарозе (Q-Sepharose), как было описано в разделе «Материалы и методы» (Пример 8.8) с целью удаления избытка непрореагировавшего HES-реагента, который, как ожидается, выходит с протекающим потоком из ионообменной колонки. В связи с большими количествами основных ЕРО форм, содержащихся в предварительно обработанных кислотой образцах II и III, авторы ожидали появления в потоке значительных количеств модифицированного ЕРО продукта как результат указанных вариантов инкубации. Как видно на фиг.13, практически весь ЕРО материал из образцов I удерживается на колонке с сефарозой (Q-Sepharose), тогда как лишь примерно 20-30% образцов II и III выходят во фракции, элюируемой при высокой концентрации соли. Весь белковый материал из инкубационных сред, включающих HES-производное X, как в потоке, так и во фракциях, элюируемых при высокой концентрации соли, характеризуется явно более высокой молекулярной массой при анализе в SDS-PAGE в сравнении с контрольным препаратом ЕРО.
С целью более детальной характеристики HES-модифицированного ЕРО в образцах А и К (см. фиг.11), проводят сравнение полученных для них результатов с окисленной периодатом формой EPO-GT-1-A. Образцы подвергают обработке N-гликозидазой и, как видно из фиг.14а и 14b, высвобождение N-гликанов приводит к получению двух полос с низкой молекулярной массой в положении О-гликозилированной и негликозилированной форм ЕРО стандартного препарата ЕРО. В случае образца А обнаруживается другая полоса, мигрирующая в положение, соответствующее ММ 28 кДа, что предположительно указывает на наличие HES-модификации по O-гликану в данном варианте ЕРО (см. пример 8.11 (с) (аа)). Указанная полоса (а также высокомолекулярная форма N-гликозилированного ЕРО с высокой степенью HES-модификации, см. фиг.14а и 14b) исчезает после проведения мягкого гидролиза образцов, что согласуется с точкой зрения, согласно которой HES-модификация осуществляется по окисленным периодатом остаткам сиаловой кислоты в эритропоэтине.
Аликвоты инкубационных смесей для N-гликозидазной обработки гидролизуют с использованием условий, позволяющих полностью удалить остатки сиаловой кислоты (а также производное сиаловой кислоты с присоединенным HES) из олигосахаридов; после нейтрализации смеси сорбируют на небольшие колонки Hypercarb для обессоливания. Колонки тщательно промывают водой и затем проводят элюирование связанных нейтральных олигосахаридов 40% ацетонитрилом в H2O, содержащей 0,1% трифторуксусную кислоту. Полученные олигосахариды подвергают анализу по методу MALDI/TOF-MS. Спектры десиалилированных фракций олигосахаридов из образца A, EPO-GT-1-A и образца К демонстрируют идентичную массу относительно олигосахаридов комплексного типа со значениями m/z=1810 Да (диантенный), 2175=триантенный, 2540=тетраантенный, 2906=тетраантенный плюс 1 N-ацетиллактозаминовый повтор и 3271=тетраантенный плюс 2 N-ацетиллактозаминовых повтора; обнаруживаются небольшие сигналы, соответствующие отсутствию фукозы (-146) и галактозы (минус 162), которые определяются условиями кислотного гидролиза, применяемого для удаления сиаловой кислоты (см. MALDI-фиг.17, 18 и 19).
В параллельном эксперименте смесь после расщепления N-гликозидазой абсорбируют на картридже RP-C18 на 1 мл (без предварительного кислотного гидролиза олигосахаридов) и проводят элюирование 5% ацетонитрилом в воде, содержащей 0,1% ТФУ; в указанных условиях белок ЕРО полностью удерживается на RP-материале и олигосахариды вымываются из колонки 5% ацетонитрилом в Н2O, содержащей 0,1% ТФУ. Белок в де-N-гликозилированном ЕРО элюируют 70% ацетонитрилом в Н2О, содержащей 0,1% ТФУ. Олигосахаридные фракции на стадии RP-C18 обработанного N-гликозидазой образца A, EPO-GT-1-A и образца К нейтрализуют и подвергают обессоливанию с использованием картриджей Hypercarb, как было описано ранее. Выделенные олигосахариды подвергают картированию по методу анализа HPAEC-PAD до (см. фиг.15) и после обработки кислотой в мягких условиях, позволяющей осуществить количественное удаление сиаловых кислот из гликанов (см. фиг.16).
Профиль, выявляемый при анализе по методу HPAEC-PAD нативного материала, полученного из HES-модифицированного образца А, демонстрирует лишь ничтожные сигналы, характерные для олигосахаридов, тогда как олигосахариды из EPO-GT-1-A демонстрирует тот же профиль гликанов, что и вариант, показанный на фиг.11 (образец, обозначенный как EPO-GT-1 после обработки периодатом в мягких условиях). Профиль элюирования олигосахаридов, полученных из контрольного образца ЕРО (К), соответствует ожидаемым характеристикам (в сравнении с профилем, показанным на фиг.8). Для сравнения приведен профиль нативного олигосахарида в международном стандартном препарате BRP-EPO, выполняющем функцию эталонного стандарта.
После проведения кислотного гидролиза в мягких условиях все олигосахаридные препараты демонстрируют идентичный профиль элюирования нейтральных олигосахаридных структур (см. фиг.16) с ожидаемым количественным и качественным составом ди-, три- и тетраантенных углеводных цепей комплексного типа, как описано в методическом разделе относительно препарата ЕРО, который использовался в качестве исходного материала для настоящего исследования. Данный результат показывает, что HES-модификация образца ЕРО приводит к ковалентному связыванию HES-производного, которое отделяется от ЕРО белка с помощью N-гликозидазы и которое является кислотолабильным, поскольку удаляется из N-гликанов в условиях мягкого кислотного гидролиза, приводящего, как известно, к десиалилированию углеводов (см. фиг.14а+b).
(ее) Анализ моносахаридного состава HES-EPO и N-гликанов HES-EPO по методу ГХ-МС
Для того чтобы дополнительно подтвердить наличие HES-модификации ЕРО по N-гликанам в молекуле, образцы ЕРО расщепляют N-гликозидазой и белок ЕРО адсорбируют на картриджах RP-C18, тогда как олигосахаридный материал вымывается, как было описано выше. Как показано в таблице 2, глюкоза и гидроксиэтилированные производные глюкозы обнаруживаются только в белке ЕРО, который был подвергнут НЕЗ-модификации по цистеиновым остаткам, и в олигосахаридных фракциях ЕРО в образце А2.
Пример 8.11 (d) Тестирование in vivo биологической активности HES-модифицированного ЕРО
Тестирование ЕРО в системе нормоцитных мышей проводят в соответствии с процедурами, описанными в Европейской Фармакопее;
в лаборатории, проводящей ЕРО-тестирование, используется международный стандартный препарат BRP ЕРО. В случае препарата HES-модифицированного ЕРО А2 было определено среднее значение удельной активности, равное 294600 единиц на мг белка ЕРО, указывающее на наличие примерно в 3 раза более высокой удельной активности, чем в международном стандартном препарате BRP ЕРО, который был включен для оценки активности вместе с исследуемыми образцами.
Результаты исследования приведены в таблице 3.
Ссылки к примерам 1-8:
Nimtz M, Noll G, Paques EP, Conradt HS.
Carbohydrate structures of a human tissue plasminogen activator expressed in recombinant Chinese hamster ovary cells.
FEES Lett. 1990 Oct. 1; 271 (1-2):14-8.
Dorner AJ, Wasley LC, Kaufman RJ.
Increased synthesis of secreted proteins induced expression of glucose-regulated proteins in butyrate-treated Chinese hamster ovary cells.
J. Eiol. Chem. 1989 Dec 5; 264 (34):20602-7.
Mueller PP, Schlenke P, Nimtz M, Conradt HS, Hauser H
Recombinant glycoprotein quality in proliferation-controlled BHK-21 cells.
Biotechnol Bioeng. 1999 Dec 5; 65(5):529-36
Nimtz M, Martin W, Wray V, Kloppel KD, Augustin J, Conradt HS.
Structures of sialylated oligosaccharides of human erythropoietin expressed in recombinant BHK-21 cells.
Eur J Biochem. 1993 Apr. 1; 213(1):39-56
Hermentin P, Witzel R, Vliegenthart JF, Kamerling JP, Nimtz M, Conradt HS.
A strategy for the mapping of N-glycans by high-ph anion-exchange chromatography with pulsed amperometric detection.
Anal Biochem. 1992 Jun; 203(2):281-9
Schroter S, Derr P, Conradt HS, Nimtz M, Hale G, Kirchhoff C.
Male specific modification of human CD52.
J Biol Chem. 1999 Oct. 15; 274(42):29862-73
Пример 9
Получение рекомбинантного ЕРО
А) Продукция в клетках млекопитающего
Рекомбинантный ЕРО получают в СНО клетках следующим образом:
Плазмиду, несущую кДНК для человеческого ЕРО, клонируют в эукариотическом векторе экспрессии (pCR3, обозначенном впоследствии как pCREPO). Свит-направленный мутагенез проводят по описанным стандартным методикам (Grabenhorst, Nimtz, Costa et al., 1998. In vitro specificity of human alpha 1,3/4-fucosyltransferases III-VII in the biosynthesis of Lewis(x) and sialyl Lewis(x) motifs on complex-type N-glycans - Coexpression studies from BHK-21 cells together with human beta-trace protein, J. Biol. Chem., 273(47), 30985-30994).
СНО клетки, стабильно экспрессирующие человеческий ЕРО или его аминокислотные варианты (например, Cys-29→Ser/Ala или Cys-33→Ser/Ala, Ser-126→Ala и т.п.), получают по методике осаждения фосфатом кальция и отбирают с помощью С418-сульфата, как описано в литературе (Grabenhorst et al.). Через три дня после трансфекции клетки подвергают субкультивированию в пропорции 1:5 и отбирают на среде DMEM, содержащей 10% PBS и 1,5 г/л G418-сульфата.
При использовании для селекции указанной методики обычно имеют уровень выживания 100-500 клонов, которые далее размножают в селективной среде еще в течение 2-3 недель. Супернатанты клеточных культур из выросших до слияния монослоев анализируют далее на уровень экспрессии ЕРО по методике вестерн-блоттинга и в рамках IEF/вестерн-блоттинг анализа.
ЕРО получают из стабильных субклонов в роллерных колбах или в перфузионных реакторах на 2 л. Различные гликоформы ЕРО с варьирующими количествами NeuAc (например, 2-8, 4-10, 8-12 остатками NeuAc) выделяют по известным методикам с использованием сочетаний различных хроматографических процедур, как описано ниже.
Литература:
Grabenhorst, Conradt, 1999, The cytoplasmic, transmembrane, and stem regions of glycosyltransferases specify their in vivo functional sublocalization and stability in the Golgi., J Biol Chem., 274(51), 36107-16; Grabenhorst, Schlenke, Pohl, Nimtz, Conradt, 1999, Genetic engineering of recombinant glycoproteins and the glycosylation pathway in mammalian host cells, Glycoconj J., 16(2), 81-97; Mueller, Schlenke, Nimtz, Conradt, Hauser, 1999, Recombinant glycoprotein product quality in proliferation-controlled BHK-21 cells. Biotechnology and bioengineering, 65(5), 529-536; Schlenke, Grabenhorst, Nimtz, Conradt, 1999, Construction and characterization of stably transfected BHK-21 cells with human-type sialylation characteristic, Cytotechnology, 30(1-3), 17-25.
В) Получение в клетках насекомого
Рекомбинантный ЕРО получают в клетках клеточной линии насекомых SF9 и SF21 после инфицирования клеток рекомбинантным бакуловирусным вектором, содержащим кДНК для человеческого ЕРО, под контролем полигедринового промотора, как описано в литературе.
Клетки, выращенные в бессывороточной культуральной среде, инфицируют с плотностью клеток 2×106 или ×107 клеток на мл и определяют титр ЕРО каждый день в супернатантах клеточных культур. ЕРО очищают хроматографией на сефарозе (Blue Sepharose), ионообменной хроматографией на сефарозе (Q-Sepharose) и в конце методом ОФ-ВЭЖХ в С4-фазе.
Чистоту продукта проверяют в SDS-PAGE и проводят анализ путем N-терминального секвенирования. Детальный структурный анализ углеводов (N- и O-гликозилирования) проводят в соответствии с опубликованными процедурами.
Литература:
Grabenhorst, Hofer, Nimtz, Jager, Conradt, 1993, Biosynthesis and secretion of human interleukin 2 glycoprotein variants from baculovirus-infected Sf21 cells. Characterization of polypeptides and posttranslational modifications, Eur J Biochem., 215(1), 189-97; Quelle, Caslake, Burkert, Wojchowski, 1989, High-level expression and purification of a recombinant human erythroproietin produced using a baculovirus vector, Blood, 74(2), 652-7.
Пример 10 Получение реактивных HES производных
1. SH-реактивный HES
1.1 Реакция ЕМСН с оксо-НЕ312КР с образованием SH-реактивного HES12KD В

0,144 г (0,012 ммоль) оксо-HES12KD (Fresenius German патент DE 19628705 Al) растворяют в 0,3 мл абсолютного диметилсульфоксида (ДМСО) и добавляют по каплям в атмосфере азота к смеси 34 мг (0,15 ммоль) ЕМСН (Perbio Science Deutschland GmbH, Bonn, Germany) в 1,5 мл ДМСО. После перемешивания в течение 19 часов при 60°С реакционную смесь добавляют к 16 мл смеси этанола и ацетона, 1:1. Осадок собирают центрифугированием, перерастворяют в 3 мл ДМСО и снова осаждают, как описано. SH-реактивный HES12KD В получают после центрифугирования и сушки в вакууме. Реакция конъюгации с тио-ЕРО описана в примере 11, 2.2.
Альтернативы:
В данной реакции могут использоваться все сшивающие линкеры, которые демонстрируют наличие гидразидной или малеимидной функций, разделенных спейсером. Другие примеры молекул с данной группой, доступные от компании Perbio Science, Deutschland GmbH, Bonn, Germany, показаны в таблице 1 со знаком «А». Кроме того, может использоваться другая группа сшивающих линкеров, демонстрирующих наличие активированной дисульфидной функции вместо малеимидной функции.
1.2 Галогенацетамидные производные HES-гликозиламинов
а) Образование гликозиламина (Manger, Wong, Rademacher, Dwek, 1992, Biochemistry, 31, 10733-10740; Manger, Rademacher, Dwek, 1992, Biochemistry, 31, 10724-10732)
1 мг образца HES12KD растворяют в 3 мл насыщенного бикарбоната аммония. Добавляют дополнительное количество бикарбоната аммония для поддержания насыщенности раствора в ходе инкубации в течение 120 часов при 30°С. Амино-НЕ312КО С обессоливают путем непосредственной лиофилизации реакционной смеси.
b) Ацилирование гликозиламина С ангидридом хлоруксусной кислоты
1 мг амино-НЕ312КО С растворяют в 1 мл 1 М бикарбоната натрия и охлаждают на льду. К указанной смеси добавляют кристалл твердого ангидрида хлоруксусной кислоты (˜5 мг) и реакционной смеси дают нагреться до комнатной температуры. Отслеживают рН реакционной смеси и если значение рН опускается ниже 7,0, добавляют дополнительное количество основания. После двухчасового выдерживания при комнатной температуре добавляют вторую аликвоту основания и ангидрида. Через шесть часов продукт хлорацетамид-HES D1 (Х=Cl) обессоливают пропусканием через смешанный слой ионообменных смол Amberlite МВ-3(Н)(ОН)).
c) Ацилирование гликозиламина бромуксусным ангидридом [Black, Kiss, Tull, Withers, 1993, Carbohydr., Res., 250, 195)
Получают бромуксусный ангидрид по методике, описанной Thomas (Thomas, 1977, Methodes Enzymol., 46, 362). 1 мг образца амино-HE312KD С растворяют в 0,1 мл сухого ДМФ, охлаждают на льду и добавляют 5 мг бромуксусного ангидрида. Реакционную смесь медленно доводят до комнатной температуры и раствор перемешивают в течение 3 часов. Далее указанную реакционную смесь добавляют к 1 мл смеси этанола и ацетона, 1:1, при -20°С. Осадок собирают центрифугированием, перерастворяют в 0,1 мл ДМФ и снова осаждают, как описано. Бромацетамид-HES D2 (Х=Br) получают после центрифугирования и сушки в вакууме. Реакция конъюгации с тио-ЕРО описана в примере 11, 1.2.
d) Соответствующее йодистое производное D3 (Х=1) синтезируют по методике получения D2. Вместо бромуксусного ангидрида используют N-сукцинимидилиодацетат и все стадии синтеза проводят в темноте.

Альтернативы:
С целью ацилирования аминогрупп могут использоваться другие галогенсодержащие производные кислот, например бромангидриды или хлорангидриды; сложные эфиры, например сложный N-гидроксисукцинимидный эфир, сложные эфиры на основе замещенных фенолов (п-нитрофенол, пентафторфенол, трихлорфенол и т.п.).
Кроме того, могут использоваться все сшивающие линкеры, которые имеют реактивную аминогруппу и галогенацетильную функцию, разделенные спейсером. Примером таких линкеров является SBAP. Указанная молекулярная форма, как и ряд других, доступна от компании Perbio Science Deutschland GmbH, Bonn, Germany. Данные молекулы помечены в таблице 1 знаком "D". Относительно использования сшивающих линкеров для лигирования амино-HES с тио-ЕРО без выделения производных галогенацетамид-HES см примечания, приведенные в примере 11, 1.2.
1.3 Галогенацетамидные производные амино-HES Е (Manger, Wohg, Rademacher, Dwek, 1592, Biochemistry, 32, 10733-I0740/ Manger, Rademacher, Dwek, 1992, Biochemistry, 31, 10724-10732)
а) Реакция 1,4-диаминобутана с оксо-HES12KD с образованием амино-НЕ312КО Е (S.Frie, Diplomarbeit, Fachhochschule Hamburg, 1998)
1,44 г (0,12 ммоль) оксо-HES12KD растворяют в 3 мл сухого диметилсульфоксида (ДМСО) и добавляют по каплям в атмосфере азота к смеси 1,51 мл (15 ммоль) 1,4-диаминобутана в 15 мл ДМСО. После перемешивания в течение 19 часов при 40°С реакционную смесь добавляют к 160 мл смеси этанола и ацетона, 1:1. Осадок амино-HES12KD Е собирают центрифугированием, перерастворяют в 40 мл воды, диализуют в течение 4 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, Germany) и лиофилизируют.

b) Хлорацетамид-HES12KD Fl получают по методике, описанной выше в пункте 1.3, для хлорацетамид-HES12KD Dl.
c) Бромацетамид-HES12KD F2 (Х=Br) получают по методике, описанной выше в пункте 1.3, для бромацетамид-HES12KD D2. Реакция конъюгирования с тио-ЕРО описана в примере 11, 1.2.
d) Соответствующее йодистое производное F3 (Х=1) не выделяют перед его реакцией с тио-ЕРО. Указанный эксперимент описан в примере 11, 1.1.
Альтернативы:
См. пункт 1.2, приведенный выше.
2. СНО-реактивный HES
2.1 Гидразид-HES

а) Реакция гидразина с оксо-HES12KD
1,44 г (0,12 ммоль) оксо-HES12KD растворяют в 3 мл абсолютного диметилсульфоксида (ДМСО) и добавляют по каплям в атмосфере азота к смеси 0,47 мл (15 ммоль) гидразина в 15 мл ДМСО. После перемешивания в течение 19 часов при 40°С реакционную смесь добавляют к 160 мл смеси этанола и ацетона, 1:1. Осажденный продукт J собирают центрифугированием, перерастворяют в 40 мл воды, диализуют в течение 2 дней против 0,5% (объем/объем) водного раствора триэтиламина и в течение 2 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, Germany) и лиофилизируют. Реакция конъюгирования с окисленным глико-ЕРО описана в примере 12, 2.2.

b) Реакция дигидразида адипиновой кислоты с оксо-HES12KD
1,74 г (15 ммоль) дигидразида адипиновой кислоты растворяют в 20 мл абсолютного диметилсульфоксида (ДМСО) при 65°С и добавляют по каплям в атмосфере азота 1,44 мл (0,12 ммоль) оксо-HES12KD, растворенного в 3 мл абсолютного ДМСО. После перемешивания в течение 68 часов при 60°С реакционную смесь добавляют к 200 мл воды. Раствор, содержащий L, диализуют в течение 2 дней против 0,5% (объем/объем) водного раствора триэтиламина и в течение 2 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, Germany) и лиофилизируют. Реакция конъюгирования с оксиленным глико-ЕРО описана в примере 12, 2.2.
Альтернативы:
Кроме того, могут использоваться производные, в которых 2 гидразидные группы разделены любым спейсером.
3. Другие амино-НЕ812КВ производные I и Н (Manger, Wong, Rademacher, Dwek, 1992, Biochemistry, 31, 10733-10740; Manger, Rademacher, Dwek, 1992, Biochemistry, 31, 10724-10732)
Аммонолиз D или F проводят отдельно путем растворения образца по 1 мг каждого из галогенацетамидов в 0,1 мл насыщенного карбоната аммония. Затем добавляют дополнительное количество твердого карбоната аммония для поддержания состояния насыщенности раствора в ходе инкубации в течение 120 часов при 30°С. Реакционную смесь добавляют к 1 мл смеси этанола и ацетона, 1:1, при -20°С. Осадок собирают центрифугированием, перерастворяют в 0,05 мл воды и снова осаждают, как описано. Продукт амино-НЕЗ Н или I получают путем центрифугирования и последующей сушки в вакууме. Реакция конъюгирования с окисленным глико-ЕРО описана в примере 12, 4.1.

4. HES12KD К, модифицированный гидроксиламином
О-[2-(2-Аминооксиэтокси)этил]гидроксиламин синтезируют по методике Boturyn et al. в 2 стадии из коммерчески доступного материала (Boturyn, Boudali, Constant, Defrancq, Lhomme, 1997, Tetrahedron, 53, 5485). 1,44 г (0,12 ммоль) оксо-HES12KD растворяют в 3 мл абсолютного диметилсульфоксида (ДМСО) и добавляют по каплям в атмосфере азота к смеси 2,04 г (15 ммоль) О-[2-(2-аминооксиэтокси)этил]гидроксиламина в 15 мл ДМСО. После перемешивания в течение 48 часов при 65°С реакционную смесь добавляют к 160 мл смеси этанола и ацетона, 1:1. Осажденный продукт К собирают центрифугированием, перерастворяют в 40 мл воды, диализуют в течение 4 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, Germany) и лиофилизируют. Реакция конъюгирования с окисленным глико-ЕРО описана в примере 12, 3.1.
Альтернативы:
Кроме того, могут использоваться производные, в которых две гидроксиламиновые группы разделены любым спейсером.
5. THO-HES12KD
5.1 Реакция присоединения к оксо-НЕ312КР

1,44 г (0,12 ммоль) оксо-HES12KD растворяют в 3 мл абсолютного диметилсульфоксида (ДМСО) и добавляют по каплям в атмосфере азота к смеси 1,16 г (15 ммоль) цистеамина в 15 мл ДМСО. После перемешивания в течение 24 часов при 40°С реакционную смесь добавляют к 160 мл смеси этанола и ацетона, 1:1. Осажденный продукт М собирают центрифугированием, перерастворяют в 40 мл воды, диализуют в течение 2 дней против 0,5% (объем/объем) водного раствора триэтиламина и в течение 2 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, Germany) и лиофилизируют. Реакция конъюгирования с окисленным глико-ЕРО описана в примере 12, 2.1.
Альтернативы:
Могут использоваться производные, в которых аминогруппа и тиофункция разделены любым спейсером. Кроме того, аминогруппа в таких производных может быть замещена гидразином, гидразидом или гидроксиламином. Тиофункция может быть защищена, например, за счет создания дисульфидного производного или формы тритильного производного. Однако в данном случае далее должна быть проведена стадия удаления защитной группы перед конъюгированием, которая приведет к высвобождению компонента, аналогичного М.
5.2 Модификация амино-НЕ312КР Е, Н или I
а) Модификация с использованием SATA/SATP
1,44 г (0,12 ммоль) амино-НЕ312КО Е, Н или I растворяют в 3 мл абсолютного диметилсульфоксида (ДМСО) и добавляют по каплям в атмосфере азота к смеси 139 мг (0,6 ммоль) SATA в 5 мл ДМСО. После перемешивания в течение 24 часов при комнатной температуре реакционную смесь добавляют к 160 мл смеси этанола и ацетона, 1:1. Осажденный продукт N собирают центрифугированием, перерастворяют в 40 мл воды, диализуют в течение 2 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, Germany) и лиофилизируют.
Реакцию удаления защитной группы проводят в 50 мМ натрий-фосфатном буфере, содержащем 25 мМ ЭДТА и 0,5 М гидроксиламин, рН 7,5 в течение 2 часов при комнатной температуре и продукт О очищают путем диализа против 0,1 М натрий-ацетатного буфера, рН 5,5, содержащего 1 мМ ЭДТА. Реакцию удаления защитной группы проводят непосредственно перед реакцией конъюгирования, которая описана в примере 12, 2.1.

b) Модификация с использованием SPDP
1,44 г (0,12 ммоль) амино-НЕ312КО Е, Н или I растворяют в 3 мл абсолютного диметилсульфоксида (ДМСО) и добавляют по каплям в атмосфере азота к смеси 187 мг (0,6 ммоль) SPDP в 5 мл ДМСО. После перемешивания в течение 24 часов при комнатной температуре реакционную смесь добавляют к 160 мл смеси этанола и ацетона, 1:1. Осажденный продукт Р собирают центрифугированием, перерастворяют в 40 мл воды, диализуют в течение 2 дней против воды (трубка для диализа SnakeSkin с границей отсечения молекулярной массы на уровне 3,5 кДа, Perbio Science Deutschland GmbH, Bonn, Germany) и лиофилизируют.
Реакцию удаления защитной группы проводят в растворе 12 мг дитиотрейтола (DTT) в 0,5 мл 100 мМ натрий-ацетатного буфера, содержащего 100 мМ хлорида натрия, при рН 4,5 в течение 30 минут при комнатной температуре и продукт О очищают путем диализа против 0,1 М натрий-ацетатного буфера, рН 5,5, содержащего 1 мМ ЭДТА. Реакцию удаления защитной группы проводят непосредственно перед реакцией конъюгирования, которая описана в примере 12, 2.1.
Альтернативы:
Для целей превращения амино- и тиольных групп как в свободной форме, так и в защищенном виде может использоваться ряд доступных реагентов. После модификации продукты могут быть выделены. Альтернативно, как принято в случае использования сшивающих линкеров, они могут непосредственно использоваться для реакции конъюгирования, предпочтительно после стадии очистки. Для выделения и хранения тио-HES производных может осуществляться синтез тио-HES производных в защищенной форме. С этой целью могут использоваться все аналогичные SATA производные, которые содержат активную сложноэфирную функцию и тиоэфирную функцию, разделенные спейсером. SATP как другой представитель данной группы приведен в таблице 1 с пометкой "Н". Производные, которые аналогичны SPDP, могут содержать активную сложноэфирную функцию и дисульфидную функцию, разделенные спейсером. Другие представители данной группы приведены в таблице 1 с пометкой "F". Дополнительные аналогичные производные могут иметь активную сложноэфирную функцию и тиольную функцию, защищенную в виде тритильного производного, которые разделены спейсером.
Пример 11 Реакции конъюгирования с тио-ЕРО
1. Реакция тио-ЕРО с модифицированным галогенацетамидом SH-реактивным HES
1.1 Пример проведения процедуры 1
Конъюгирование ТиоЕРО с амино-НЕ312КО (Е, Н или I) с использованием сшивающего линкера, содержащего NHS-активный сложный эфир, например, SIA (Cumber, Forrester, Foxwell, Ross, Thorpe, 1985, Methods Enzymol., 112, 207)
Материалы
А. Боратный буфер. Композиция включает 50 мМ борат натрия, рН 8,3 и 5 мМ ЭДТА.
В. PBS, фосфатно-буферный раствор: 10 мМ фосфат натрия, 150 мМ NaCI, pH 7,4.
С. Амино-НЕ312КО Е, Н или I. Готовят в концентрации 1 мг/мл в боратном буфере.
D. Раствор сшивающего линкера для хранения: 14 мг SIA растворяют в 1 мл ДМСО.
Е. Колонки для обессоливания с декстраном D-Salt™, рабочий объем 2 х 5 мл (Perbio Science Deutschland GmbH, Bonn, Germany)
F. Реактив для количественного определения белка кумасси (Coomassie®) (Perbio Science Deutschland GmbH, Bonn, Germany)
G. Раствор тиоЕРО: 5 мг/мл тиоЕРО 1 в боратном буфере.
Н. Микроконцентратор Microcon УМ-3 (amicon, Milipore GmbH, Eschborn, Germany).
Метод
100 мкл раствора SIA добавляют к 400 мкл раствора аминоHES12KD Е и оставляют для реакции при перемешивании в течение 0,5 часа при комнатной температуре. Избыток сшивающего линкера удаляют центрифугированием образца при 14000×g в течение 60 минут с использованием микроконцентратора. После центрифугирования объем образца доводят до исходного значения боратным буфером и указанный процесс повторяют еще два раза. Оставшийся раствор добавляют к 1 мл раствора тиоЕРО и реакционную смесь инкубируют в течение 16 часов при комнатной температуре. В конце инкубационного периода реакционную способность избытка йодацетамида гасят добавлением цистеина до конечной концентрации 10 мМ. Реакционную смесь наносят на колонку для обессоливания, уравновешенную буфером PBS, и во фракциях отслеживают содержание белка по методу анализа, включающему использование кумасси. Объединяют все фракции, содержащие белковый конъюгат, и получают конъюгат путем лиофилизации после диализа против воды в течение ночи.
Альтернативы:
В данной реакции могут использоваться все сшивающие линкеры, которые содержат сукцинимидную или сульфосукцинимидную функцию и йодацетамидную функцию, разделенные спейсером. Другие примеры приведены в таблице 1. Они помечены знаком "С" и доступны от компании Perbio Science Deutschland GmbH, Bonn, Germany.
1.2 Пример проведения процедуры 2
Конъюгирование тиоЕРО 1 с бромацетамидом D2, F2 или йодацетамидом D3 3Н-реактивного HES12KD (de Valasco, Merkus, Anderton, Verheui, Lizzio, Van der Zee, Van Eden, Hoffmann, Verhoef, Snippe, 1995, Infect. Immun., 63, 961)
Материалы
А. Фосфатный буфер. Композиция включает 100 мМ фосфат натрия, рН 6,1 и 5 мМ ЭДТА.
В. PBS, фосфатно-буферный раствор: 10 мМ фосфат натрия, 150 мМ NaCl, pH 7,4.
С. Бромацетамид SH-реактивного HES12KD D2. Готовят в концентрации 10 мг/мл в фосфатном буфере.
D. Колонки для обессоливания с декстраном D-Salt™, рабочий объем 2 х 5 мл (Perbio Science Deutschland GmbH, Bonn, Germany)
E. Реактив для количественного определения белка кумасси (Coomassie®) (Perbio Science Deutschland GmbH, Bonn, Germany)
F. Раствор тиоЕРО: 5 мг/мл тиоЕРО 1 в фосфатном буфере.
Метод
Объединяют 1 мл раствора бромацетамида SH-реактивного HES12KD D2 и 1 мл раствора тиоЕРО и реакционную смесь инкубируют в течение 48 часов при комнатной температуре. В конце инкубационного периода реакционную способность избытка бромацетамида гасят добавлением цистеина до конечной концентрации 10 мМ. Реакционную смесь наносят на колонку для обессоливания, уравновешенную буфером PBS. Отслеживают во фракциях содержание белка по методу анализа, включающему использование кумасси, и все фракции, содержащие белковый конъюгат, объединяют и получают конъюгат путем лиофилизации после диализа против воды в течение ночи.
Альтернативы:
Вместо выделения бромацетамида 3Н-реактивного HES12KD D2 может быть проведено связывание HES12KD (E, Н, I) с линкером через сукцинимидную и бромацетамидную функцию (см. пункт 1.1, описанный выше). SBAP как представитель данной группы сшивающих линкеров приведен в таблице 1 с пометкой знаком "D".
2. Реакция тио-ЕРО с модифицированным малеимидом SH-реактивным HES
2.1 Пример проведения процедуры 4
Конъюгирование тиоЕРО с малеимидо-НЕ312КО В. Материалы
А. Малеимидо-НЕ312КО В: в концентрации 10 мг/мл в 0,1 М натрий-ацетатном буфере, рН 5,5
В. Раствор тиоЕРО: 5 мг/мл тиоЕРО в фосфатном буфере, содержащем NaCl
С. Фосфат/NaCl: 0,1 М раствор фосфата натрия, 50 мМ NaCl, рН 7,0
D. Колонка для гель-фильтрации: например, с сефадексом (Sephadex® G-200) (1,5×45 см)
Е. Реактив кумасси (Coomassie®) для количественного определения белка (Perbio Science Deutschland GmbH, Bonn, Germany)
F. PBS, фосфатно-буферный раствор: 10 мМ фосфат натрия, 150 мМ NaCl, рН 7,4.
Метод
Объединяют 1 мл раствора SH-реактивного HES12KD В и 1 мл раствора тиоЕРО 1 и реакционную смесь инкубируют в течение 2 часов при комнатной температуре. В конце инкубационного периода реакционную способность избытка малеимида гасят добавлением цистеина до конечной концентрации 10 мМ. Реакционную смесь наносят на колонку с сефадексом (Sephadex® G-200) (1,5×45 см), уравновешенную буфером PBS, и собирают фракции по 1 мл. Во фракциях отслеживают содержание белка по методу анализа, включающему использование реактива для количественного определения белка кумасси. Объединяют все фракции, содержащие белковый конъюгат, и получают конъюгат путем лиофилизации после диализа против воды в течение ночи.
2.2 Пример проведения процедуры 12
Конъюгирование тиоЕРО с аминоНЕ312КО (Е, Н, I) с использованием сшивающего линкера, содержащего NHS-активный сложный эфир и малеимидную группу, например SMCC.
Материалы
А. Микроконцентратор Microcon YM-3 (amicon, Milipore GmbH, Eschborn, Germany)
B. PBS, фосфатно-буферный раствор: 10 мМ фосфат натрия, 150 мМ NaCl, pH 7,4
С. АминоНЕ312КО Е, Н или I. Готовят в концентрации 10 мг/мл в буфере PBS.
D. Раствор SMCC: 1 мг SMCC растворяют в 50 мкл ДМСО.
Е. Колонки для обессоливания с декстраном D-Salt™, рабочий объем 2х5 мл (Perbio Science Deutschland GmbH, Bonn, Germany)
F. Реактив для количественного определения белка кумасси (Coomassie® (Perbio Science Deutschland GmbH, Bonn, Germany)
G. Раствор тиоЕРО 1:5 мг/мл тиоЭПО 1 в буфере PBS.
Метод
К 50 мкл раствора SMCC добавляют 400 мкл раствора аминоHES12KD Е и реакционную смесь оставляют для реакции в течение 80 минут при комнатной температуре и 10 минут при 46°С. Избыток сшивающего линкера удаляют центрифугированием образца при 14000×g в течение 60 минут с использованием микроконцентратора. Объем доводят до отметки 450 мкл буфером PBS и указанный процесс повторяют еще два раза. После последнего центрифугирования объем оставшегося раствора доводят до 450 мкл с помощью PBS и добавляют к 1 мл раствора тиоЕРО, после чего реакционную смесь инкубируют в течение 16 часов при комнатной температуре. В конце инкубационного периода реакционную способность избытка малеимида гасят добавлением цистеина до конечной концентрации 10 мМ. Реакционную смесь наносят на колонку для обессоливания, уравновешенную буфером PBS. Во фракциях отслеживают содержание белка по методу анализа, включающему использование реактива для количественного определения белка кумасси, все фракции, содержащие белковый конъюгат, объединяют и получают конъюгат путем лиофилизации после диализа против воды в течение ночи.
Альтернативы:
В данной реакции могут использоваться все сшивающие линкеры, которые содержат сукцинимидную или сульфосукцинимидную функцию и малеимидную функцию, разделенные спейсером. Другие примеры молекул, которые относятся к данной группе, доступные от компании Perbio Science Deutschland GmbH, Bonn, Germany, приведены в таблице 1 с пометкой "Е". Имеется еще одна группа сшивающих линкеров, которые содержат вместо малеимидной функции активированную дисульфидную функцию. Указанные сшивающие линкеры также могут использоваться для конъюгирования. Однако дисульфидная связь в конъюгате расщепляется в восстановительных условиях. Представители данной группы отмечены в таблице 1 знаком "F". Третья группа сшивающих линкеров в качестве SH-реактивной группы использует вместо малеимидной функции винилсульфоновую функцию. Представитель такой группы "SVSB" помечен в таблице 1 знаком "G".
Пример 12 Реакции конъюгирования с окисленным ЕРО
1. Окисление глико-ЕРО
1.1 Окисление глико-ЕРО мета-периодатом натрия: пример проведения процедуры 5
Материалы
А. Раствор глико-ЕРО: 10 мг/мл глико-ЕРО в ацетатном буфере
В. Раствор мета-периодата натрия: 10 мМ или 100 мМ периодата натрия в ацетатном буфере, используют в свежеприготовленном виде. Держат в темноте. При использовании данных растворов конечная концентрация периодата натрия в смеси для окисления составляет 1 мМ или 10 мМ соответственно.
С. Ацетатный буфер: 0,1 М натрий-ацетатный буфер, рН 5,5
D. Глицерин
Е. Микроконцентратор Microcon YM-3 (amicon, Milipore GmbH, Eschborn, Germany)
Метод
Все стадии выполняют в темноте.
К 1 мл охлажденного раствора глико-ЕРО добавляют 0,1 мл охлажденного раствора мета-периодата натрия и проводят реакцию окисления в течение 1 часа в темноте. Если подвергаемый окислению глико-ЕРО содержит остатки сиаловой кислоты, то условия окисления включают следующее: 1 мМ периодат натрия, 0°С. В ином случае используют 10 мМ периодат натрия в условиях комнатной температуры. Для остановки реакции окисления добавляют глицерин до конечной концентрации 15 мМ и инкубируют в течение 5 минут при температуре 0°С. Избыток реагентов и побочных продуктов реакции удаляют центрифугированием продукта при 14000×g в течение 60 минут с использованием микроконцентратора. После центрифугирования объем образца доводят до исходного значения буфером, используемым на следующей стадии, т.е. ацетатным буфером. Указанный процесс повторяют еще два раза.
1.2 Ферментативное окисление глико-ЕРО: пример проведения процедуры 6
Ферментативное окисление ЕРО описано в литературе (Chamow et al., 1992, J. Biol. Chem., 267, 15916-15922).
2. Конъюгирование с производными гидразина/гидразида
2.1 Пример проведения процедуры 7
Конъюгирование окисленного глико-ЕРО с тио-НЕ312КО М, О или Q с использованием сшивающего линкера, содержащего гидразидную и малеимидную функциональную группу, например M3C3H (Perbio Science, Deutschland, GmbH, Bonn, Germany).
Материалы
А. Раствор М2С2Н для хранения: 10 мг/мл М2С2Н в ДМСО, используют в свежеприготовленном виде
В. Раствор окисленного глико-ЕРО из 6.1.1: 5 мг/мл глико-ЕРО в ацетатном буфере
С. Тио-НЕ312КО М, О или Q: 10 мг/мл в фосфатном буфере/NaCl
D. Ацетатный буфер: 0,1 М натрий-ацетатный буфер, рН 5,5
Е. Фосфат/NaCl: 0,1 М раствор фосфата натрия, 50 мМ NaCl, рН 7,0
F. Микроконцентратор: Microcon УМ-3 (amicon, Milipore GmbH, Eschborn, Gerniany)
G. Колонка для гель-фильтрации: например, с сефадексом (Sephadex® G-200) (1,5×45 см)
Н. Реактив для количественного определения белка кумасси (Coomassie®) (Perbio Science Deutschland GmbH, Bonn, Germany)
I. PBS, фосфатно-буферный раствор: 10 мМ фосфат натрия, 150 мМ NaCl, рН 7,4
Метод
Раствор для хранения M2C2H добавляют к 1 мл окисленного глико-ЕРО до конечной концентрации 1 мМ и оставляют для реакции в течение 2 часов с перемешиванием при комнатной температуре. Избыток сшивающего линкера удаляют центрифугированием образца при 14000×g в течение 60 минут с использованием микроконцентратора. После центрифугирования объем образца доводят до исходного значения фосфатным буфером/NaCl и указанный процесс повторяют еще два раза. К М2С2Н-модифицированному глико-ЕРО добавляют 1 мл раствора тио-НЕ312КО М, О или Q и реакционную смесь инкубируют в течение 16 часов при комнатной температуре. В конце инкубационного периода реакционную способность избытка малеимида гасят добавлением цистеина. Реакционную смесь наносят на колонку с сефадексом (Sephadex® G-200) (1,5×45 см), уравновешенную буфером PBS, и собирают фракции по 1 мл. Во фракциях отслеживают содержание белка по методу анализа, включающему использование реактива для количественного определения белка кумасси, все фракции, содержащие белковый конъюгат, объединяют и получают конъюгат путем лиофилизации после диализа против воды в течение ночи.
Примечания по процедуре осуществления методики
Гидразоновый аддукт отличается несколько сниженной стабильностью при экстремальных значениях рН. В случае тех направлений применения, которые включают обработку при низком рН, авторы проводят восстановление гидразона с использованием 30 мМ цианборгидрида натрия в буфере PBS до гидразина. Однако для большинства направлений применения такая дополнительная стадия является необязательной.
2.2 Пример проведения процедуры 8
Реакция прямого конъюгирования окисленного глико-ЕРО с гидразидо-НЕ312КО L.
Материалы
А. Раствор окисленного глико-ЕРО из 6.1.1: 5 мг/мл глико-ЕРО в ацетатном буфере
В. Гидразидо-HESlSKD L или J: 10 мг/мл в ацетатном буфере
С. Ацетатный буфер: 0,1 М натрий-ацетатный буфер, рН 5,5
D. Колонка для гель-фильтрации: например, с сефадексом (Sephadex® G-200) (1, 5×45 см)
Е. Реактив для количественного определения белка кумасси (Coomassie®) (Perbio Science Deutschland GmbH, Bonn, Germany)
F. PBS, фосфатно-буферный раствор: 10 мМ фосфат натрия, 150 мМ NaCl, pH 7,4
Метод
Объединяют 1 мл раствора гидразидо-НЕ312КО L и добавляют 1 мл раствора окисленного глико-ЕРО и реакционную смесь оставляют для реакции при перемешивании в течение 16 часов при комнатной температуре. Реакционную смесь наносят на колонку с сефадексом (Sephadex® G-200) (1,5×45 см), уравновешенную буфером PBS, и собирают фракции по 1 мл. Во фракциях отслеживают содержание белка по методу анализа, включающему использование реактива для количественного определения белка кумасси, все фракции, содержащие белковый конъюгат, объединяют и получают конъюгат путем лиофилизации после диализа против воды в течение ночи. Результат конъюгирования показан на фиг.24. Наблюдаемое смещение молекул указывает на то, что конъюгирование прошло удачно. Некоторое размазывание полос связано с гетерогенностью HES. Из фиг.25 видно, что HES конъюгирует с углеводным фрагментом боковой цепи углевода.
Примечания по процедуре осуществления методики
Гидразоновый аддукт отличается несколько сниженной стабильностью при экстремальных значениях pH. В случае тех направлений применения, которые включают обработку при низком pH, авторы проводят восстановление гидразона с использованием 30 мМ цианборгидрида натрия в буфере PBS до гидразина. Однако для большинства направлений применения такая дополнительная стадия является необязательной.
3. Конъюгирование с производными гидроксиламина (Rose, 1994, Am. Chem. Soc., 116, 30)
3.1 Пример проведения процедуры 9
Реакция конъюгирования окисленного глико-ЕРО с гидроксиламино-НЕ312КО К
Материалы
А. Раствор окисленного глико-ЕРО из 6.1.1: 5 мг/мл глико-ЕРО в ацетатном буфере
В. Гидроксиламино-НЕ312КО К: 10 мг/мл в ацетатном буфере
С. Ацетатный буфер: 0,1 М натрий-ацетатный буфер, рН 5,5
D. Колонка для гель-фильтрации, наример, с сефадексом (Sephadex® G-200) (1,5×45 см)
Е. Реактив для количественного определения белка кумасси (Coomassie®) (Perbio Science Deutschland GmbH, Bonn, Germany)
F. PBS, фосфатно-буферный раствор: 10 мМ фосфат натрия, 150 мМ NaCl, рН 7,4.
Метод
Объединяют 1 мл раствора гидроксиламино-НЕ312КО К и 1 мл раствора окисленного глико-ЕРО и реакционную смесь оставляют для реакции при перемешивании в течение 16 часов при комнатной температуре. Реакционную смесь наносят на колонку с сефадексом (Sephadex® G-200) (1,5×45 см), уравновешенную буфером PBS, и собирают фракции по 1 мл. Во фракциях отслеживают содержание белка по методу анализа, включающему использование реактива для количественного определения белка кумасси, все фракции, содержащие белковый конъюгат, объединяют и получают конъюгат путем лиофилизации после диализа против воды в течение ночи. Результат конъюгирования показан на фиг.24. Наблюдаемое молекулярное смещение в полосе 2 указывает на то, что конъюгирование прошло удачно. Некоторое размазывание полос связано с гетерогенностью HES. Из фиг.25 видно, что HES конъюгирует с углеводным фрагментом боковой цепи углевода.
Пример 13 Характеристика N-гликанов ЕРО, обработанных галактозооксидазой
Рекомбинантный ЕРО или частично десиалилированные формы ЕРО (полученные путем ограниченного кислотного гидролиза) инкубируют с галактозооксидазой в присутствии каталазы при 37°С от 30 минут до 4 часов в 0,05 М Na-фосфатном буфере, рН 7,0. Ход реакции отслеживают путем отбора аликвот ЕРО по 50 мкл с последующей обработкой белка N-гликаназой полипептидов.
Высвобожденные N-связанные олигосахариды (тестирование проводят путем выявления в SDS-PAGE де-N-гликозилированного полипептида) подвергают картированию по методу HPAEC-PAD, как описано в литературе (Grabenhorst et al., 1999, Nimtz et al., 1993/1994; Schlenke et al., 1999) до и после удаления сиаловых кислот. Количественное определение окисленных остатков галактозы в индивидуальных олигосахаридах ЕРО проводят на основании определения типичного смещения, отмечаемого в процессе HPAEC-PAD, и подтверждают результаты при исследовании олигосахаридных смесей по методу MALDI/TOF MS.
Пример 14 Характеристика HAS-модифицированного ЕРО
Отделение HAS-модифицированных форм ЕРО от непрореагировавшего ЕРО и молекул-предшественников HAS достигается путем гель-фильтрации с использованием, например, Ultrogel AcA 44/54 или аналогичных сред, применяемых для фильтрования. Альтернативно, непрореагировавший HAS удаляют путем выделения ЕРО иммуноаффинным методом на колонке объемом 4 мл, содержащей моноклональные антитела, связанные с гелем Affigel (BioRad), с последующим отделением немодифицированного ЕРО гель-фильтрацией (например, с использованием матрицы, позволяющей осуществлять отделение глобулярных белков с молекулярной массой от 20 кДа до 200 кДа).
HAS-модифицированный ЕРО идентифицируют при анализе в SDS-PAGE (с использованием 12,5 или 10% гелей акриламида) путем определения их повышенных молекулярных масс в сравнении с молекулярной массой немодифицированного ЕРО при окрашивании гелей красителем кумасси бриллиантовый синий. Более высокая молекулярная масса HAS-модифицированных полипептидов ЕРО может быть также идентифицирована посредством вестерн-блот-анализа образцов с использованием поликлональных антител, образованных против рекомбинантного человеческого ЕРО.
N-гликановая модификация форм ЕРО была продемонстрирована при успешном удалении ЕРО белка N-гликаназой полипептидов (рекомбинантная N-гликозидаза от компании Roche, Germany, используемая в количестве 25 единиц/мг ЕРО белка при 37°С в течение 16 часов); анализ в SDS-PAGE выявляет наличие типичного смещения белка ЕРО в положении миграции обработанного N-гликозидазой немодифицированного ЕРО с массой примерно 20 кДа.
Модификация одного десиалилированного и обработанного галактозооксидазой O-гликана ЕРО в положении Ser 126 была продемонстрирована в SDS-PAGE по миграции де-N-гликозилированного продукта путем выявления положения его миграции в сравнении с непрореагировавшим де-N-гликозилированным ЕРО. Если требуется, модифицированный ЕРО перед анализом в SDS-PAGE подвергают фракционированию методом ОФ-ВЭЖХ в СВ-фазе. O-гликановую модификацию HAS в ЕРО также анализируют путем β-элиминации O-гликана и выявления де-0-гликозилированной формы ЕРО методом вестерн-блоттинга с использованием поликлональных антител, образованных против рекомбинантного человеческого ЕРО.
Пример 15
Количественное определение ЕРО и модифицированных форм ЕРО
Количественное определение ЕРО проводят на основе измерения УФ поглощения в соответствии с процедурой Европейской Фармакопеи (Phr. Eur, 2000, Erythropoietini solutio concentrata, 1316, 780-785) и сравнивают с международным стандартом BRP ЕРО. Альтернативно концентрации ЕРО определяют методом ОФ-ВЭЖХ с использованием колонки RP-C4 и при измерении уровня абсорбции при длине волны 254 нм с использованием для калибровки 20, 40, 80 и 120 мкг стандартного препарата BRP ЕРО.
Пример 16
Биологическая активность in-vitro HES-модифицированного рекомбинантного человеческого ЕРО
Активность очищенного HES-модифицированного ЕРО определяют в рамках биотеста на эритропоэтин, как описано Krystal [Krystal, 1984, Exp.Heamatol., 11, 649-660].
Анемию индуцируют у мышей NMRI обработкой гидрохлоридом фенилгидразина, собирают клетки селезенки и используют, как описано в литературе [Fibi et al., 1991, Blood, 77, 1203, ff].
Варианты с разбавлениями ЕРО инкубируют с 3×105 клеток/ячейку в 96-ячеечных микротитрационных планшетах. После инкубирования в течение 24 часов при 37°С в увлажненной атмосфере (5% CO2) клетки в течение 4 часов подвергают мечению 1 мкCi3 H-тимидина в расчете на ячейку. Уровень включенной радиоактивности определяют путем счета в жидком сцинтилляторе. Для сравнения используют Международный стандартный препарат ЕРО (BRP-стандарт).
Альтернативно биологическую активность ЕРО определяют в тесте in vitro с использованием ЕРО-чувствительной клеточной линии TF-1 (Kitamura et al., [J. cell Phys., 140, 323-334]). Экспоненциально растущие клетки отмывают от ростовых факторов и инкубируют в присутствии серийных разбавлений ЕРО еще в течение 48 часов. Пролиферацию клеток оценивают с использованием теста на восстановление, как описано Mosmann [Mosmann, 1983, J.Immunol. Methods, 65, 55-63].
Пример 17
Определение in-vivo активности ЕРО и HAS-модифицированных форм ЕРО
Определение in vivo активности проводят с использованием нормоцитных мышей путем измерения уровня повышения количества ретикулоцитов через 4 дня после введения животным предварительно определенной дозы ЕРО или модифицированных форм ЕРО. Тесты проводят с использованием стандарта BPR ЕРО, который калибруют против стандартного препарата WHO на ЕРО в тесте с использованием полицитемичных мышей. Образцы ЕРО разбавляют фосфатно-буферным раствором, содержащим 1 мг/мл бычьего сывороточного альбумина (Sigma).
0,5 мл исследуемого раствора ЕРО в буферном растворе Дульбекко (который по эквиваленту белка ЕРО соответствует 100, 80, 40 или 20 МЕ/мл стандартного препарата BPR ЕРО) инъецируют подкожно мышам. Через 4 дня после инъекции отбирают образцы крови и окрашивают ретикулоциты акридином оранжевым, количественное определение ретикулоцитов проводят методом проточной цитометрии, подсчитывая в целом 30000 клеток крови в течение 5 часов после отбора образца (см. Ph. Eur, 2000, Erythropoietini solutio concentrata, 1316, pages 780-785 и European Pharmacopoeia (1996/2000, приложение 2002)).
Пример 18 Определение периода полувыведения in vivo
Кроликам инъецируют внутривенно заданные количества HAS-модифицированной формы ЕРО. Через определенные интервалы времени отбирают образцы крови и получают сыворотку. Определяют уровень эритропоэтина в сыворотке с помощью биотеста in vivo или с использованием ЕРО-специфичного коммерческого набора для ELISA.
Пример 19 Фармакокинетика in vivo
При определении на мышах: каждому животному вводят подкожно 300 ME ЕРО/кг. Через семь дней после обработки у каждого животного определяют гематокрит. У 9 из всех животных, которым был введен модифицированный ЕРО, отмечалось существенное повышение гематокрита, что представляет собой ожидаемый результат, определяемый относительно коротким периодом полувыведения необработанного ЕРО. Средний показатель изменения гематокрита в группе, принимавшей модифицированный ЕРО, существенно отличался от соответствующего показателя в группе приема необработанного ЕРО и в контрольной группе.
При исследовании на кроликах: Кроликам вводят однократную дозу немодифицированного или HAS-модифицированного ЕРО в дозе, соответствующей 200 или до 800 нг/кг массы тела. Через 2, 6, 16, 24 и 48 часов образцы крови анализируют с использованием коммерческого ЕРО-специфичного набора ELISA для определения концентраций в плазме. Определяют среднее значение концентраций ЕРО в плазме и вычисляют средние значения исходных показателей периода полувыведения (α-фаза) и конечных показателей периода полувыведения (β-фаза) на основании результатов анализа методом ELISA, как описано: (Zettlmissi et al., 1989, J. Biol. Chem., 264, 21153-21159).
Литература:
Sytkowski, Lunn, Risinger, and Davis, 1999, An Erythropoietin Fusion Protein Comprised of Identical Repeating Domains Exhibitis Enhanced Biological Properties, J. Biol. Chem., 274, 24773-24778.
Пример 20
Оценка биологической активности in vitro HES-модифицированиого рекомбинантного человеческого IL-2
Модифицированный IL-2 выделяют гель-фильтрацией в Ultrogel АсА 54. Аликвоты соответствующей фракции стерильно фильтруют и определяют биологическую активность IL-2 при использовании IL-2-зависимой клеточной линии клеток мышей CTLL-2 [Gillis, Ferm, On, and Smith, 1978, J. Immunol., 120, 2027-2032]. Полученное значение активности сравнивают со стандартным препаратом IL-2, используемым в качестве международного эталона.





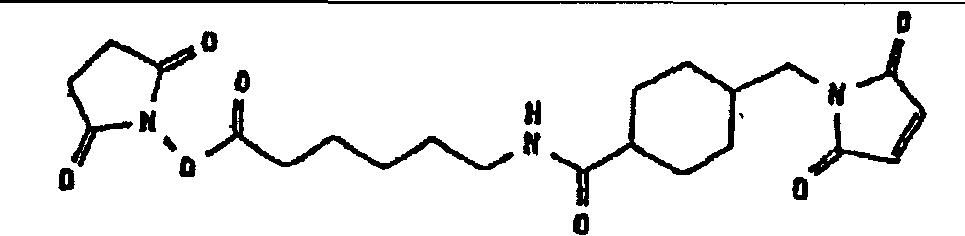










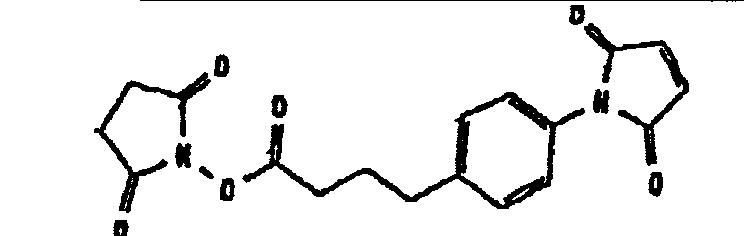













* эквивалентное количество Cys-HES-модифицированного белка ЕРО подвергают анализу для определения состава; ЕРО белок выделяют из инкубационной смеси с HES хроматографией на колонке с сефарозой (Q-Sepharose), как было описано выше, и подвергают обессоливанию в процессе центрифугирования с использованием устройства для разделения Vivaspin 5.
** Определение количества моносахаридов проводят при однократном пропускании в системе ГХ пертриметилсилилированных метилгликозидов; значения пиков при их электронной интеграции приводятся без коррекции на потери в ходе процедур дериватизации и восстановления каждого соединения.
Реферат
Способ получения производного гидроксиалкилкрахмала, где указанное производное гидроксиалкилкрахмала имеет структуру, соответствующую формуле (I), который включает взаимодействие гидроксиалкилкрахмала формулы (I) по его окисленному восстановленному концу, или производного гидроксиалкилкрахмала, получаемого при взаимодействии гидроксиалкилкрахмала формулы (I) по его окисленному восстановленному концу с соединением (D), где указанное соединение (D) включает по меньшей мере одну функциональную группу Z1, способную взаимодействовать с окисленным восстановленным концом гидроксиалкилкрахмала, и по меньшей мере одну функциональную группу W, с соединением (L), включающим по меньшей мере одну функциональную группу Z1, способную взаимодействовать с указанным гидроксиалкилкрахмалом, или по меньшей мере одну функциональную группу Z2, способную взаимодействовать с функциональной группой W, включенной в указанное производное гидроксиалкилкрахмала, и по меньшей мере одну функциональную группу X, способную взаимодействовать с функциональной группой Y в другом соединении (М), где указанную функциональную группу Y выбирают из группы, состоящей из альдегидной группы, кетогруппы, гемиацетальной группы, ацетальной группы и тиогруппы, где образование химической связи между соединением (L) и гидроксиалкилкрахмалом или соединением (D) и гидроксиалкилкрахмалом достигается взаимодействием функциональной группы Z1 с окисленным восстановленным концом гидроксиалкилкрахмала, и где функциональная группа Z1 включает структуру -NH. Изобретение также относится к производным гидроксиалкилкрахмала, а также к фармацевтической композиции, включающей производные гидроксиалкилкрахмала. 4 н. и 50 з.п. ф-лы, 25 ил., 3 табл.
Формула










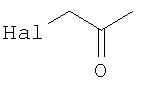


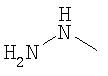













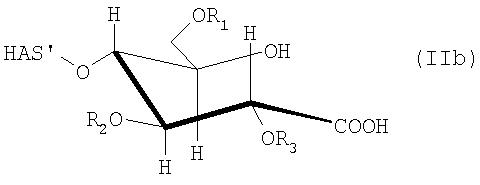






Документы, цитированные в отчёте о поиске
Композиции длительного высвобождения, способ их получения и применение
Комментарии