Ингибиторы fgfr4 - RU2715708C2
Код документа: RU2715708C2
Чертежи


Описание
УРОВЕНЬ ТЕХНИКИ
Факторы роста фибробластов (FGF) являются семейством более чем 20 структурно связанных белков с различной биологической активностью. Их основные рецепторы, рецепторы факторов роста фибробластов (FGFR1, FGFR2, FGFR3 и FGFR4), являются семейством рецепторных тирозинкиназ, которые связывают FGF и принимают участие в процессах пролиферации и дифференциации клеток. Нарушение регуляции сети передачи сигнала FGFR включает в себя ряд патофизиологических состояний, включая многие типы раковых заболеваний человека.
Известно, что “рецептор 4 фактора роста фибробластов” или “FGFR4” регулирует пролиферацию и антиапоптоз и экспрессируется или сильно экспрессируется при многих раковых заболеваниях. См., например, Dieci et al. 2013, Cancer Discovery, OF1-OF16. Исследования показали, что экспрессия FGFR4 является предсказанием более агрессивного фенотипа рака и дефицит или снижение экспрессии FGFR4 вызывает уменьшение пролиферации и промотирование апоптоза. См., например, Wesche et al. 2011, BioChem J 437:199-213.
Например, экспрессия или сверхэкспрессия FGFR4 связана с раковой агрессивностью при раке желудка (Ye et al. 2011, Cancer, 5304-5313), раке простаты (Xu et al. 2011, BMC Cancer, 11;84), саркоме, такой как рабдомиосаркома (Taylor VI et al. 2009, J Clin Invest, 119(11):3395-3407), раке кожи, таком как меланома (Streit et al. 2006, British J Cancer, 94:1879-1886), раке печени, таком как холангиокарцинома (Sia et al. 2013, Gastroenterology 144:829-840) и гепато-клеточной карциноме (French et al. 2012, PLoS ONE 7(5): e367313; Miura et al. 2012, BMC Cancer 12:56; Chiang et al. 2008, Cancer Res 68(16):6779-6788; Sawey et al. 2011, Cancer Cell 19:347-358), раке поджелудочной железы, таком как панкреатическая интраэпителиальная неоплазия и панкреатическая дуктальная аденокарцинома (Motoda et al. 2011, Int'l J Oncol 38:133-143), раке легкого, таком как немелкоклеточный рак легкого (Fawdаr et al. 2013, PNAS 110(30):12426-12431), колоректальном раке (Pelaez-Gарcia et al. 2013, PLoS ONE 8(5): e63695; Bаrderas et al. 2012, J Proteomics 75:4647-4655) и овариальном раке (Zaid et al. 2013, Clin Cancer Res 19:809-820).
Клиническая разработка различных ингибиторов FGFR подтвердила их эффективность в качестве противоопухолевых агентов. Dieci et al. 2013, Cancer Discovery, 0F1-0F16. Однако, требуются новые агенты, которые являются применимыми для “нацеливания” на FGFR, и в частности, на FGFR4.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является предоставление соединения формулы I

в которой
R3 выбирают из группы, состоящей из C1-6алкила, C1-6алкоксиC1-6алкила, NR10R11C1-6aлкила, R10гетероциклилC1-6алкила, R10арилC1-6алкила и R10гетероарилC1-6алкила, где R10 и R11, каждый независимо, выбирают из группы, состоящей из водорода и C1-6алкила;
E выбирают из группы, состоящей из
–NR13C(O)CR14=CHR15 и
–NR13C(O)C≡CR14,
где R13 выбирают из группы, состоящей из водорода и метила и R14 и R15, каждый независимо, выбирают из группы, состоящей из водорода, метила, фтора и хлора;
R12 выбирают из группы, состоящей из водорода, галогена, C1-6алкила, C1-6алкокси, гидроксиC1-6алкила, гидроксиC1-6алкокси, C1-6алкокси,C1-6алкокси, C1-6алкоксиC1-6алкила, R5R6гетероциклила, –C(O)гетероциклилR5R6, R5R6гетероциклилC1-6алкила, NR5R6, NR5R6C1-6алкила, –C(O)NR5R6 и NR5R6C1-6алкокси, где R5 и R6, каждый независимо, выбирают из группы, состоящей из водорода, C1-6алкила, гидроксиC1-6алкила, аминоC1-6алкила, -C(O)C1-6алкила и C1-6алкилсульфонила, и
R1 представляет собой фенил, где указанный фенил замещен 2, 3 или 4 заместителями, независимо выбранными из галогена или C1-6алкокси,
и его фармацевтически приемлемой соли.
В некоторых вариантах изобретения R3 представляет собой C1-6алкил.
В некоторых вариантах изобретения R3 выбирают из группы, состоящей из метила, метоксиэтила, 4-пиридилметила, 3-пиридилметила, 2-пиридилметила, бензила, N,N-диметиламинопропила, 3-метилизоксазол-5-илметила и 4-метилпиперазин-1-илпропила.
В некоторых вариантах изобретения E представляет собой –NR13C(O)CH=CHR15 или –NR13C(O)CF=CH2, где R13 и R15 имеют значения, указанных выше. В некоторых вариантах изобретения E представляет собой –NHC(O)CH=CH2.
В некоторых вариантах изобретения R12 выбирают из группы, состоящей из водорода, фтора, хлора, метила, метокси, N,N-диметиламиноэтила, пиперазин-1-ила, 4-этилпиперазин-1-ила, 4-этилпиперазин-1-илметила, 1-метилпиперидин-4-ила, 1-этилпиперидин-4-ила, N,N-диметиламинометила, N,N-диметиламинопропила, пиперидин-4-ила, морфолино, 3,5-диметилпиперазин-1-ила, 4-(метилсульфонил)пиперазин-1-ила, N,N-диметиламиноэтокси, 4-(2-гидроксиэтил)пиперазин-1-ила, гидроксиэтокси, метоксиэтокси, гидроксиметила, метоксиметила, 2-метоксипропила, 2-гидроксипропила, 2-аминопропила, 4-метилпиперазин-1-илкарбонила, 4-этилпиперазин-1-илкарбонила, 4-[2-пропил]пиперазин-1-ила, 4-ацетилпиперазин-1-ила, N-метил-N-гидроксиэтиламино, N,N-диметиламидо и 4-(2-аминоэтил)пиперазин-1-ила.
В некоторых вариантах осуществления R12 выбирают из группы, состоящей из водорода, C1-6алкила, гидроксиC1-6алкила, R5R6гетероциклила, R5R6гетероциклилC1-6алкила, –C(O)NR5R6, NR5R6C1-6алкила, NR5R6C1-6алкокси, C1-6алкокси и C1-6алкоксиC1-6алкила, где R5 и R6, каждый независимо, выбирают из группы, состоящей из водорода, C1-6алкила, гидроксиC1-6алкила, –C(O)C1-6алкила и C1-6алкилсульфонила.
В некоторых вариантах осуществления R12 представляет собой R5R6гетероциклил, где R5 и R6 имеют значения, указанные выше.
В некоторых вариантах осуществления R5R6гетероциклил представляет собой R5R6пиперазинил, где R5 и R6 имеют значения, указанные выше.
В некоторых вариантах осуществления R12 представляет собой -4-этилпиперазин-1-ил.
В некоторых вариантах осуществления R12 не является водородом.
В некоторых вариантах осуществления R1 представляет собой 2,6-дихлор-3,5-диметоксифенил.
В некоторых вариантах осуществления соединение представляет собой соединение формулы I(a)

в которой R3, E, R12 и R1 имеют значения, указанные выше,
или его фармацевтически приемлемую соль.
Следующей целью является фармацевтическая композиция, содержащая соединение или соль, описываемую в контексте, и фармацевтически приемлемый носитель. В некоторых вариантах осуществления композицию изготовляют для перорального или парентерального введения.
Следующей целью является способ лечения гепато-клеточной карциномы у субъекта, нуждающегося в этом, содержащий введение указанному субъекту эффективного для лечения количества соединения или соли, или композиции, описываемой в контексте. В некоторых вариантах осуществления гепато-клеточная карцинома имеет измененный статус FGFR4 и/или FGF19 (например, повышенную экспрессию FGFR4 и/или FGF19).
Следующей целью является способ лечения гепато-клеточной карциномы у субъекта, нуждающегося в этом, содержащий детектирование измененного статуса FGFR4 и/или FGF19 (например, повышенной экспрессии FGFR4 и/или FGF19) в биологическом образце, содержащем клетки указанной гепато-клеточной карциномы, и, если указанная гепато-клеточная карцинома имеет измененный статус FGFR4 и/или FGF19, введение соединения или композиции, описанной в контексте, указанному субъекту в эффективном для лечения количестве.
Следующей целью является применение соединения или соли или композиции, описываемой в контексте, в способе лечения гепато-клеточной карциномы.
Следующей целью является применение соединения или соли, описываемой в контексте, при получении лекарственного средства для лечения гепато-клеточной карциномы.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
ФИГ. 1 представляет собой результаты тестирования эффективности in vivo в модели гепато-клеточной карциномы с применением клеток HUH7. Соединение 108 (25 мг/кг или 37,5 мг/кг) или наполнитель как контроль вводили посредством внутрибрюшинной инъекции и объем опухоли измеряли два раза еженедельно на протяжении курса 15 дней.
ФИГ. 2 представляет собой результаты тестирования эффективности in vivo в модели гепато-клеточной карциномы с применением клеток НЕР3В. Соединение 108 (12,5 мг/кг, 25 мг/кг или 37,5 мг/кг) или наполнитель как контроль вводили посредством внутрибрюшинной инъекции и объем опухоли измеряли два раза еженедельно на протяжении курса 15 дней.
ФИГ. 3 представляет собой результаты тестирования эффективности in vivo в модели гепато-клеточной карциномы с применением клеток JHH7. Соединение 108 (12,5 мг/кг, 25 мг/кг или 37,5 мг/кг) или наполнитель как контроль вводили посредством внутрибрюшинной инъекции и объем опухоли измеряли два раза еженедельно на протяжении курса 15 дней.
ФИГ. 4 представляет собой результаты сравнения тестирования эффективности in vivo в модели гепато-клеточной карциномы с применением клеток HEP3B. Соединение 108 (25 мг/кг, 37,5 мг/кг или 50 мг/кг) вводили дважды ежедневно посредством внутрибрюшинной инъекции или BGJ398 (30 мг/кг или 60 мг/кг) вводили перорально дважды ежедневно.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
В контексте представлены соединения, пригодные в качестве ингибиторов FGFR4. В некоторых вариантах осуществления соединения являются селективными ингибиторами FGFR4, поскольку они обладают более высоким сродством связывания FGFR4 и/или ингибирующим FGFR4 действием по сравнению со сродством связывания и/или ингибирующим действием FGFR1 и/или FGFR2 и/или FGFR3 (например, 10-кратным, 100-кратным или 1000-кратным или более).
A. Определения
Соединения, применимые в качестве активных агентов согласно настоящему описанию, включают в себя соединения, описанные в общем выше и ниже, и дополнительно иллюстрированные вариантами осуществления, субвариантами осуществления и разновидностями, описанными в контексте. Следует применять используемые в контексте нижеследующие определения, если не оговорено особо.
Описываемые в контексте соединения изобретения могут быть необязательно замещены одним или несколькими заместителями, такими как заместители, указанные в общем в контексте или указываемые в качестве примеров конкретными классами, подклассами и разновидностями изобретения. В общем, термин "замещенный" относится к замене водорода в данной структуре указанным заместителем. Если не указано иначе, замещенная группа может иметь заместитель в каждом замещаемом положении группы, и когда более чем одно положение в любой данной структуре может быть замещено более чем одним заместителем, выбранным из указанной группы, заместители могут быть либо одинаковыми, либо разными в каждом положении. Комбинации заместителей, рассмотренных в данном изобретении, являются предпочтительно комбинациями, которые приводят к образованию стабильных соединений. Слово "стабильное", применяемое в контексте, относится к химически возможному соединению, которое по существу не изменяется при выдерживании при температуре 40°C или менее в отсутствие влаги или в других химически реакционных условиях в течение по меньшей мере недели.
Как должно быть понятно специалисту в данной области, применяемое в контексте обозначение "H" является водородом, "C" является углеродом, "N" является азотом, "S" является серой и "O" является кислородом.
"Aлкил" или "алкильная группа", применяемая в контексте, означает цепь нормального строения (т.е. неразветвленную) или разветвленную углеводородную цепь, которая является полностью насыщенной. В некоторых вариантах осуществления алкил имеет 1, 2, 3, 4, 5 или 6 атомов углерода. В некоторых вариантах осуществления алкильные группы содержат 1-6 атома углерода (С1-6алкил). В некоторых вариантах осуществления алкильные группы содержат 1-4 атома углерода (С1-4алкил). В некоторых вариантах осуществления алкильные группы содержат 1-3 атома углерода (С1-3алкил). В других вариантах осуществления алкильные группы содержат 2-3 атома углерода (C2-3алкил) и еще в других вариантах осуществления алкильные группы содержат 1-2 атома углерода (C1-2алкил).
"Алкенил" или "алкенильная группа", применяемая в контексте, относится к цепи нормального строения (т.е. неразветвленной) или разветвленной углеводородной цепи, которая имеет одну или несколько двойных связей. В некоторых вариантах осуществления алкенил имеет 2, 3, 4, 5 или 6 атомов углерода. В некоторых вариантах осуществления алкенильные группы содержат 2-8 атомов углерода (C2-8алкенил). В некоторых вариантах осуществления алкенильные группы содержат 2-6 атомов углерода (C2-6алкенил). В других вариантах осуществления алкенильные группы содержат 3-4 атома углерода (C3-4алкенил) и еще в других вариантах осуществления алкенильные группы содержат 2-3 атома углерода (C2-3алкенил). Согласно другому аспекту, термин алкенил относится к углеводородной группе с неразветвленной цепью, имеющей две двойные связи, называемой также "диеном". Неограничивающие примеры примерных алкенильных групп включают в себя
–CH=CH2, –CH2CH=CH2, –CH=CHCH3, –CH2CH2CH=CH2,
–CH2CH=CHCH3, –CH=CHCH2CH3 и –CH=CHCH=CH2.
Термин "алкинил" или "алкинильная группа", применяемый в контексте, относится к цепи нормального строения (т.е. неразветвленной) или разветвленной углеводородной цепи, которая имеет одну или более тройных связей. В некоторых вариантах осуществления алкинил имеет 2, 3, 4, 5 или 6 атомов углерода. В некоторых вариантах осуществления алкинильные группы содержат 2-8 атомов углерода (C2-8алкинил). В некоторых вариантах осуществления алкинильные группы содержат 2-6 атомов углерода (C2-6алкинил). В других вариантах осуществления алкинильные группы содержат 3-4 атома углерода (C3-4алкинил) и еще в других вариантах осуществления алкинильные группы содержат 2-3 атома углерода (C2-3алкинил).
"Ар" или "арил" относится к ароматическому карбоциклическому остатку, имеющему одно или несколько замкнутых колец. Примеры их включают в себя, без ограничения, фенил, нафтил, антраценил, фенантраценил, бифенил и пиренил.
"Галоген" относится к хлору (Cl), фтору (F), брому (Br) или иоду (I).
"Галогеналкил" относится к одному или нескольким атомам галогенов, присоединенным к исходному молекулярному остатку посредством алкильной группы. Примеры его включают в себя, но не ограничиваются указанным, хлорметил, фторметил, трифторметил, и т.д.
"Гетероарил" относится к циклическому остатку, имеющему одно или несколько замкнутых колец с одним или несколькими гетероатомами (кислорода, азота или серы) по меньшей мере в одном из колец, где по меньшей мере одно из колец является ароматическим и где кольцо или кольца могут быть независимо конденсированными и/или мостиковыми. Примеры их включают в себя, без ограничения, хинолинил, изохинолинил, индолил, фурил, тиенил, пиразолил, хиноксалинил, пирролил, индазолил, тиено[2,3-c]пиразолил, бензофурил, пиразоло[1,5-a]пиридил, тиофенилпиразолил, бензотиенил, бензотиазолил, тиазолил, 2-фенилтиазолил и изоксазолил.
"–OR" или "окси" относится к группе R, присоединенной к основному молекулярному остатку через атом кислорода, где R представляет собой Н, алкил, алкенил, алкинил и тому подобное.
"Алкокси" относится к алкильной группе, определяемой в контексте, которая присоединена к основной углеродной цепи через атом кислорода ("алкокси"). Репрезентативные примеры "алкокси" включают в себя, но не ограничиваются указанным, метокси, этокси, пропокси, фенокси, 2-пропокси, бутокси, трет-бутокси, пентилокси, гексилокси и тому подобное.
"Гидрокси" относится к группе –OH.
"Карбонил" представляет собой группу, имеющую атом углерода, связанный двойной связью с атомом углерода (C=O), часто изображаемую в химической формуле как C(O).
"Ацетил" представляет собой группу –C(O)CH3.
"Амин" или "амино" относится к группе –NH2, у которой один или два атома водорода могут быть замещены подходящим заместителем, таким как aлкил, алкенил, алкинил и т.д.
"Амид" или "амидо" относится к группе, имеющей карбонил, связанный с атомом азота, такой как –C(O)NH2, у которой один или два атома водорода могут быть заменены подходящим заместителем, описываемым в контексте, таким как алкил, алкенил, алкинил и т.д.
"–SR" относится к группе R, присоединенной к основному молекулярному остатку через атом серы, где R представляет собой алкил, алкенил, алкинил, арил, циклоалкил, гетероцикло или гетероарил. Репрезентативные примеры "–SR" включают в себя, но не ограничиваются указанными, этантиол, 3-метил-1-бутантиол, фенилтиол и тому подобное.
Термин “циклоалкил'” применяемый в контексте, относится к насыщенной циклической углеводородной группе, содержащей от 3 до 8 атомов углерода или больше. Репрезентативные примеры циклоалкила включают в себя, но не ограничиваются указанными, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил.
Термин “циклоалкенил”, применяемый в контексте, относится к ненасыщенной циклической углеводородной группе, содержащей от 3 до 8 атомов углерода или более и имеющей одну или несколько двойных связей.
Термин “циклоалкинил”, применяемый в контексте, относится к ненасыщенной циклической углеводородной группе, содержащей от 3 до 8 атомов углерода или больше и имеющей одну или несколько тройных связей.
Термин “электрофил, применяемый в контексте, относится к группе, имеющей пониженную электронную плотность, обычно содержащей атом углерода, который непосредственно связан с более электроотрицательным атомом, таким как атом кислорода, азота или галогена. Примерные электрофилы включают в себя, но не ограничиваются указанными, диазометан, триметилсилилдиазометан, алкилгалогениды, такие как, например, метилиодид, бензилбромид и тому подобное, алкилтрифлаты, такие как, например, метилтрифлат и тому подобно, алкилсульфонаты, такие как, например, этилтолуолсульфонат, бутилметансульфонат и тому подобное, ацилгалогениды, такие как, например, ацетилхлорид, бензoилбромид и тому подобное, и ангидриды кислот, такие как, например, уксусный ангидрид, янтарный ангидрид, малеиновый ангидрид и тому подобное, изоцианаты, такие как, например, метилизоцианат, фенилизоцианат и тому подобное, изотиоцианаты, такие как, например, метилизотиоцианат, фенилизотиоцианат и тому подобное, хлорформиаты, такие как, например, метилхлорформиат, этилхлорформиат, бензилхлорформиат и тому подобное, сульфонилгалогениды, такие как, например, метансульфонилхлорид, метансульфонилфторид, п-толуолсульфонилхлорид и тому подобное, силилгалогениды, такие как, например, триметилсилилхлорид, трет-бутилдиметилсилилхлорид и тому подобное, фосфорилгалогениды, такие как, например, диметилхлорфосфат и тому подобное, эпоксиды, такие как, например, 2-метилоксиран, азиридины, такие как, например, 2-метилазиридин, альфа-галогенкетон, такой как, например, 1-хлор-2-пропанон, альфа-бета-ненасыщенные карбонильные соединения, такие как, например, акролеин, метилвинилкетон, коричный альдегид, N,N-диметилакриламид и тому подобное, и гамма-галоген-альфа-бета-ненасыщенные карбонильные соединения, такие как, например, (E)-6-хлоргекс-4-ен-3-он. В некоторых вариантах осуществления электрофилами являются альфа-галогенкетоны, изотиоцианаты, эпоксиды, азиридины, сульфонилгалогениды или альфа-бета-ненасыщенные карбонилы.
В некоторых вариантах осуществления электрофилом является акцептор Михаэля. Как известно в данной области, “акцептором Михаэля” является алкен или алкин формулы


где электроноакцепторные группы Z, Z' и Z" описаны выше. В некоторых вариантах осуществления акцепторами Михаэля являются альфа-бета-ненасыщенные карбонильные соединения, включающие в себя, но не ограничивающиеся перечисленным, альфа-бета-ненасыщенные амиды, альфа-бета-ненасыщенные кетоны, альфа-бета-ненасыщенные сложные эфиры, сопряженные алкинилкарбонилы и альфа-бета-ненасыщенные нитрилы.
"Альфа-бета-ненасыщенный амид" или "ненасыщенный амид", применяемый в контексте, относится к амиду, содержащему алкен или алкин, связанный непосредственно с карбонильной группой амида, и представлен структурой

"Гетероатом" относится к O, S или N.
"Гетероцикл" или гетероциклил", применяемый в контексте, означает моноциклический гетероцикл, бициклический гетероцикл или трициклический гетероцикл, содержащий по меньшей мере один гетероатом в кольце.
Моноциклический гетероцикл представляет собой 3-, 4-, 5-, 6-, 7 или 8-членное кольцо, содержащее по меньшей мере один гетероатом, независимо выбранный из групп, состоящей их O, N, и S. В некоторых вариантах осуществления гетероциклом является 3- или 4-членное кольцо, содержащее один гетероатом, выбранный из группы, состоящей из O, N и S. В некоторых вариантах осуществления гетероциклом является 5-членное кольцо, содержащее ноль или одну двойную связь и один, два или три гетероатома, выбранные из группы, состоящей из O, N и S. В некоторых вариантах осуществления гетероциклом является 6-, 7- или 8-членное кольцо, содержащее ноль, одно или две двойные связи и один, два или три гетероатома, выбранные из группы, состоящей из O, N и S. Репрезентативные примеры моноциклического гетероцикла включают в себя, но не ограничиваются перечисленным, азетидинил, азепанил, азиридинил, диазепанил, 1,3-диоксaнил, 1,3-диоксoланил, дигидропиранил (включая 3,4-дигидро-2H-пиран-6-ил), 1,3-дитиоланил, 1,3-дитианил, имидазолинил, имидазолидинил, изотиазолинил, изотиазолидинил, изоксaзолинил, изоксазолидинил, морфолинил, оксaдиазолинил, оксaдиазолидинил, оксaзолинил, оксaзолидинил, пиперазинил, пиперидинил, пиранил, пиразолинил, пиразолидинил, пирролинил, пирролидинил, тетрагидрофуранил, тетрагидропиранил (включая тетрагидро-2H-пиран-4-ил), тетрагидротиенил, тиадиазолинил, тиадиазолидинил, тиазолинил, тиазолидинил, тиоморфолинил, 1,1-диоксидотиоморфолинил (тиоморфолинсульфон), тиопиранил и тритианил.
Примерами бициклических гетероциклов настоящего изобретения может быть моноциклический гетероцикл, конденсированный с арильной группой, или моноциклический гетероцикл, конденсированный с моноциклическим циклоалкилом, или моноциклический гетероцикл, конденсированный с моноциклическим циклоалкенилом, или моноциклический гетероцикл, конденсированный с моноциклическим гетероциклом. Репрезентативные примеры бициклических гетероциклов включают в себя, но не ограничиваются перечисленным, 3,4-дигидро-2H-пиранил, 1,3-бензoдиоксолил, 1,3-бензoдитиолил, 2,3-дигидро-1,4-бензoдиоксинил, 2,3-дигидро-1-бензoфуранил, 2,3-дигидро-1-бензoтиенил, 2,3-дигидро-1Н-индолил, 3,4-дигидрохинолин-2(1H)-он и 1,2,3,4- тетрагидрохинолинил.
Трициклический гетероцикл представляет собой бициклический гетероцикл, конденсированный с арильной группой, или бициклический гетероцикл, конденсированный с моноциклическим циклоалкилом, или бициклический гетероцикл, конденсированный с моноциклическим циклоалкенилом, или бициклический гетероцикл, конденсированный с моноциклическим гетероциклом. Репрезентативные примеры трициклических гетероциклов включают в себя, но не ограничиваются перечисленным, 2,3,4,4a,9,9a-гексaгидро-1H-карбазолил, 5a,6,7,8,9,9a-гексaгидродибензo[b,d]фуранил и 5a,6,7,8,9,9a-гексaгидродибензo[b,d]тиенил.
В указанных выше гетероариле и гетероциклах атомы азота или серы могут быть необязательно окислены в различные степени окисления. В конкретном примере группа S(O)0-2 относится к –S– (сульфиду), –S(O)– (сульфоксиду) и –SO2– (сульфону) соответственно. Имеется в виду, что для удобства атомы азота, в частности, но не исключительно, которые определяются как аннелярные ароматические атомы азота, включают в себя атомы азота, соответствующие формам N-оксидов.
Термин "фармацевтически приемлемая соль", применяемый в контексте, относится к кислотно-аддитивным солям или основно-аддитивным солям соединений настоящего описания. Фармацевтически приемлемой солью является любая соль, которая сохраняет активность исходного соединения и не оказывает никакого чрезмерного повреждающего или нежелательного действия на субъект, которому ее вводят, и в контексте, в котором ее вводят. Фармацевтически приемлемые соли включают в себя, но не ограничиваются перечисленным, комплексы металлов и соли как неорганических, так и карбоновых кислот. Фармацевтически приемлемые соли включают в себя также соли металлов, таких как алюминий, кальций, железо, магний, марганец, и комплексные соли. Кроме того, фармацевтически приемлемые соли включают в себя, но не ограничиваются перечисленным, соли кислот, таких как уксусная, аспарагиновая, алкилсульфоновая, арилсульфоновая, аксетиловая, бензолсульфоновая, бензойная, дикарбоновая, дисерная, дивинная, масляная, этилендиаминотетрауксусная (кальциевая соль), камсиловая, угольная, хлорбензoйная, лимонная, этилендиаминтетрауксусная, эдисиловая, эстоловая, эсиловая, муравьиная, фумаровая, глюцептовая, глюконовая, глутаминовая, гликолевая, гликолиларсаниловая, гексaмовая, гексилрезорциновая, гидрабаминовая, бромистоводородная, хлористоводородная, иодистоводородная, гидроксинафтойная, изетионовая, молочная, лактобионовая, малеиновая, яблочная, малоновая, миндальная, метансульфоновая, метилазотная, метилсерная, слизиевая, муконовая, напсиловая, азотная, щавелевая, п-нитрометансульфоновая, памоевая, пантотеновая, фосфорная, моногидрофосфорная, дигидрофосфорная, фталевая, полигалактоуроновая, пропионовая, салициловая, стеариновая, янтарная, сульфаминовая, сульфаниловая, сульфоновая, серная, дубильная, винная, теоклевая, толуолсульфоновая кислоты и т.д. Фармацевтически приемлемые соли можно получать из аминокислот, включающих в себя, но не ограничивающихся указанным, цистеин. Способы получения соединений в виде солей известны специалисту в данной области техники (см., например, Stahl et al., Handbook of Pharmаceutical Salts: Properties, Selection, and Use, Wiley-VCH; Verlag Helvetica Chimica Аcta, Zurich, 2002; Berge et al., J. Phаrm. Sci. 66: 1, 1977).
Если не оговорено иначе, номенклатура, применяемая для описания химических групп или остатков, применяемых в контексте, соответствует конвенции, в которой название читается слева направо, точкой присоединения к остатку молекулы является правая сторона названия. Например, группа "арилC1-6алкил" присоединяется к остатку молекулы у алкильного конца.
Если не оговорено иначе, когда химическая группа описывается ее химической формулой, включающей концевой остаток, указанный как "–," должно быть понятно, что присоединение читается слева направо. Например, –C(O)C1-6алкил присоединяется к остатку молекулы у карбонильного конца.
Если не оговорено иначе, имеется в виду, что структуры, указанные в контексте, включают в себя все энантиомерные, диастереомерные и геометрические (или конформационные) формы структуры, например, R- и S-конфигурации для каждого асимметричного центра, изомеры с двойной связью (Z) и (E) и конформационные изомеры (Z) и (E). Следовательно, индивидуальные стереохимические изомеры, а также энантиомерные, диастереомерные и геометрические (или конформационные) смеси настоящих соединений находятся в объеме настоящего изобретения. Если не оговорено иначе, все таутомерные формы соединений изобретения находятся в объеме изобретения. Кроме того, если не оговорено иначе, все ротамерные формы соединений изобретения находятся в пределах объема изобретения. Если не оговорено иначе, имеется также в виду, что структуры, указанные в контексте, включают в себя соединения, которые отличаются только присутствием одного или нескольких обогащенных изотопами атомов. Например, соединения, имеющие настоящие структуры, за исключением замещения водорода дейтерием или тритием или замещения атома углерода13C- или14C-обогащенным атомом углерода, находятся в пределах объема данного изобретения. Такие соединения являются применимыми, например, в качестве аналитических средств или проб в биологических анализах.
Термин "изомеры" относится к соединениям, имеющим одинаковое число и одинаковый тип атомов и поэтому одинаковую молекулярную массу, но имеющим различия в отношении расположения или конфигурации атомов. Однако, должно быть понятно, что некоторые изомеры или рацематы или другие смеси изомеров могут проявлять большую активность, чем другие изомеры. "Стереоизомеры" относятся к изомерам, которые различаются только расположением атомов в пространстве. "Диaстереоизомеры" относятся к стереoизомерам, которые не являются зеркальными изображениями друг друга. “Энантиомеры” относятся к стереоизомерам, которые являются зеркальными изображениями, которые не накладываются друг на друга.
В некоторых вариантах осуществления энантиомерные соединения, изучаемые в контексте, могут быть "энантиомерно чистыми" изомерами, которые содержат по существу один изомер, например, в количестве, которое больше или равно 90%, 92%, 95%, 98% или 99%, или равно 100% индивидуального энантиомера.
В некоторых вариантах осуществления энантиомерные соединения, изучаемые в контексте, могут быть стереомерно чистыми. Термин "стереомерно чистый", применяемый в контексте, означает соединение или его композицию, которая содержит один стереоизомер соединения и по существу не содержит другие стереоизомеры этого соединения. Например, стереомерно чистая композиция соединения, имеющего один хиральный центр, может быть по существу свободной от противоположного энантиомера соединения. Стереоизомерно чистая композиция соединения, имеющего два хиральных центра, может быть по существу свободной от диастереомеров и по существу свободной от противоположного энантиомера соединения. Типичное стереомерно чистое соединение содержит больше приблизительно 80 масс.% одного стереомера соединения и меньше приблизительно 20 масс.% других стереоизомеров соединения, более предпочтительно больше приблизительно 90 масс.% одного стереоизомера соединения и меньше приблизительно 10 масс.% других стереоизомеров соединения, еще более предпочтительно больше приблизительно 95 масс.% одного стереоизомера соединения и меньше приблизительно 5 масс.% других стереоизомеров соединения и наиболее предпочтительно больше приблизительно 97 масс.% одного стереоизомера соединения и меньше приблизительно 3 масс.% других стереоизомеров соединения. См., например, патент США No. 7189715.
"R" и "S" в качестве терминов, описывающих изомеры, являются обозначениями стереохимической конфигурации у асимметрично замещенного атома углерода. Обозначение асимметрично замещенного атома углерода как “R” или “S” осуществляют посредством применения правил приоритета Кана-Ингольда-Прелога, которые являются хорошо известными специалистам в данной области и описаны в the International Union of Pure and Applied Chemistry (IUPAC) Rules for the Nomenclature of Organic Chemistry. Section E, Stereochemistry.
"Энантиомерным избытком" (ee) энантиомера является [(молярная часть основного энантиомера) минус (молярная часть меньшего энантиомера)] × 100.
B. Соединения
В качестве активных агентов согласно некоторым вариантам осуществления предложено соединение формулы I

в которой
R3 выбирают из группы, состоящей из C1-6алкила, C1-6алкoксиC1-6алкила, NR10R11C1-6алкила, R10гетероциклилC1-6алкила, R10арилC1-6алкила и R10гетероарилC1-6алкила, где R10 и R11, каждый независимо, выбирают из группы, состоящей из водорода и C1-6алкила;
E выбирают из группы, состоящей из
-NR13C(O)CR14=CHR15 и
–NR13C(O)C≡CR14,
где R13 выбирают из группы, состоящей из водорода и метила, и R14 и R15, каждый независимо, выбирают из группы, состоящей из водорода, метила, фтора и хлора;
R12выбирают из группы, состоящей из водорода, галогена, C1-6алкила, C1-6алкoкси, гидроксиC1-6алкила, гидроксиC1-6алкoкси, C1-6алкoксиC1-6алкoкси, C1-6алкoксиC1-6алкила, R5R6гетероциклила, –C(O)гетероциклилR5R6, R5R6гетероциклилC1-6алкила, NR5R6, NR5R6C1-6алкила, –C(O)NR5R6 и NR5R6C1-6алкoкси, где R5 и R6, каждый независимо, выбирают из группы, состоящей из водорода, C1-6алкила, гидроксиC1-6алкила, аминоC1-6алкила, –C(O)C1-6алкила и C1-6алкилсульфонила; и
R1 представляет собой фенил, где указанный фенил замещен 2, 3 или 4 заместителями, независимо выбранными из галогена или C1-6алкoкси,
или его фармацевтически приемлемая соль.
В некоторых вариантах осуществления R3 представляет собой С1-6алкил.
В некоторых вариантах осуществления R3 выбирают из группы, состоящей из метила, метоксиэтила, 4-пиридилметила, 3-пиридилметила, 2-пиридилметила, бензила, N,N-диметиламинопропила, 3-метилизоксазол-5-илметила и 4-метилпиперазин-1-илпропила.
В некоторых вариантах осуществления E представляет собой –NR13C(O)CH=CHR15 или –NR13C(O)CF=CH2, где R13 и R15 имеют значения, указанные выше. В некоторых вариантах осуществления E представляет собой –NHC(O)CH=CH2.
В некоторых вариантах осуществления R12 выбирают из группы, состоящей из водорода, фтора, хлора, метила, метокси, N,N-диметиламиноэтила, пиперазин-1-ила, 4-этилпиперазин-1-ила, 4-этилпиперазин-1-илметила, 1-метилпиперидин-4-ила, 1-этилпиперидин-4-ила, N,N-диметиламинометила, N,N-диметиламинопропила, пиперидин-4-ила, морфолинo, 3,5-диметилпиперазин-1-ила, 4-(метилсульфонил)пиперазин-1-ила, N,N-диметиламиноэтокси, 4-(2-гидроксиэтил)пиперазин-1-ила, гидроксиэтокси, метоксиэтокси, гидроксиметила, метоксиметила, 2-метоксипропила, 2-гидроксипропила, 2-аминопропила, 4-метилпиперазин-1-илкарбонила, 4-этилпиперазин-1-илкарбонила, 4-[2-пропил]пиперазин-1-ила, 4-ацетилпиперазин-1-ила, N-метил-N-гидроксиэтиламино, N,N-диметиламидо и 4-(2-аминоэтил)пиперазин-1-ила
В некоторых вариантах осуществления R12 выбирают из группы, состоящей из водорода, C1-6алкила, гидроксиC1-6алкила, R5R6гетероциклила, R5R6гетероциклилC1-6алкила, –C(O)NR5R6, NR5R6C1-6алкила, NR5R6C1-6алкилoкси, C1-6алкoкси и C1-6алкoксиC1-6алкила, где R5 и R6, каждый независимо, выбирают из группы, состоящей из водорода, C1-6алкила, гидроксиC1-6алкила, –C(O)C1-6алкила and C1-6алкилсульфонила.
В некоторых вариантах осуществления R12 представляет собой R5R6гетероциклил, где R5 и R6 имеют значения, указанные выше.
В некоторых вариантах осуществления R5R6гетероциклил представляет собой R5R6пиперазинил, у которого R5 и R6 имеют значения, указанные выше.
В некоторых вариантах осуществления R12 представляет собой 4-этилпиперазин-1-ил.
В некоторых вариантах осуществления R12 не является водородом.
В некоторых вариантах осуществления R1 представляет собой 2,6-дихлор-3,5-диметоксифенил.
В некоторых вариантах осуществления соединение является соединением формулы I(a)

в которой R3, E, R12 и R1 имеют значения, указанные выше,
или его фармацевтически приемлемой солью.
C. Фармацевтические препараты
Активные агенты настоящего изобретения можно комбинировать с фармацевтически приемлемым носителем для получения их фармацевтических препаратов. Конкретный выбор носителя и препарата будет зависеть от конкретного пути введения, который предназначен для композиции.
Применяемый в контексте термин ”фармацевтически приемлемый носитель” относится к нетоксичному носителю, адъюванту или наполнителю, который не нарушает фармакологическую активность соединения, с которым его изготовляют. Фармацевтически приемлемые носители, адъюванты или наполнители, которые можно применять в композициях данного изобретения, включают в себя, но не ограничиваются указанными компонентами, сорбиновую кислоту, сорбат калия, смеси неполных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, вторичный кислый фосфат натрия, вторичный кислый фосфат калия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натриевую соль карбоксиметилцеллюлозы, полиакрилаты, воски, полиэтиленгликоль и ланолин.
Композиции настоящего изобретения могут быть подходящими для парентерального, перорального, ингаляционного, местного, ректального, назального, трансбуккального, вагинального введения или введения в виде аэрозоля или из имплантированного резервуара и т.д. В некоторых вариантах осуществления препарат содержит ингредиенты из природных или неприродных источников. В некоторых вариантах осуществления препарат или носитель может быть представлен в стерильной форме. Неограничивающие примеры стерильного носителя включают в себя воду без эндотоксинов или воду без пирогенов.
Термин “парентеральный”, применяемый в контексте, включает в себя подкожные, внутривенные, внутримышечные, внутрисуставные, внутрисиновиальные, внутригрудинные, внутрикапсульные, внутрипеченочные, внутрираневые и внутрикраниальные способы инъекции или инфузии. В конкретных вариантах осуществления соединения вводят внутривенным, пероральным, подкожным или внутримышечным введением. Стерильные инъецируемые формы композиций данного изобретения могут быть водной или масляной суспензией. Эти суспензии можно изготовлять согласно способам, известным в данной области с применением подходящих диспергирующих или смачивающих агентов или суспендирующих агентов. Стерильный инъецируемые препарат может также быть стерильным инъецируемым раствором или суспензией в нетоксичном, парентерально приемлемом разбавителе или растворителе. Среди приемлемых наполнителей и растворителей, которые можно применять, имеется вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно применяют стерильные нелетучие масла.
Для этой цели можно применять любое легкое нелетучее масло, включая синтетические моно- или диглицериды. Жирные кислоты и их глицеридные производные применимы при получении инъецируемых препаратов, когда они являются природными фармацевтически приемлемыми маслами, такими как оливковое масло или касторовое масло, особенно в их полиоксиэтилированных формах. Эти масляные растворы или суспензии могут содержать также спирт с длинной цепью в качестве разбавителя или диспергатор, такой как карбоксиметилцеллюлоза, или аналогичные диспергирующие агенты, которые обычно применяют при изготовлении фармацевтически приемлемых лекарственных форм, включающих в себя эмульсии и суспензии. Другие обычно применяемые поверхностно-активные вещества, такие как твины, спаны, и другие эмульгирующие агенты, которые обычно применяют при изготовлении фармацевтически приемлемых твердых, жидких или других лекарственных форм, можно также применять для целей изготовления препаратов.
Для перорального введения соединение или соль можно применять в приемлемой пероральной лекарственной форме, включающей в себя, но не ограничивающейся указанным, капсулы, таблетки, водные суспензии или растворы. В случае таблеток для перорального применения, обычно применяемые носители включают в себя лактозу и кукурузный крахмал. Можно также добавлять смазывающие агенты, такие как стеарат магния. Для перорального введения в форме капсулы применимые разбавители включают в себя лактозу и высушенный кукурузный крахмал. Когда водные суспензии требуются для перорального применения, активный ингредиент можно сочетать с эмульгирующими или суспендирующими агентами. При желании можно добавлять некоторые подслащивающие, ароматизирующие или окрашивающие агенты. Кроме того, можно также добавлять консерванты. Подходящие примеры фармацевтически приемлемых консервантов включают в себя, но не ограничиваются указанными, различные антибактериальные и антигрибковые агенты, такие как растворители, например, этанол, пропиленгликоль, бензиловый спирт, хлорбутанол, четвертичные аммониевые соли и парабены (такие как метилпарабен, пропилпарабен и т.д.)
D. Субъекты и способы применения
Активные агенты настоящего изобретения можно применять для лечения гепато-клеточной карциномы.
Термин “лечение” относится к реверсии, ослаблению, задержке начала, ингибированию развития или же уменьшению интенсивности симптомов заболевания или нарушения, описываемого в контексте. В некоторых вариантах осуществления активные агенты можно вводить после развития одного или нескольких симптомов. В других вариантах осуществления активные агенты можно вводить при отсутствии симптомов. Например, активные агенты можно вводить восприимчивому индивидууму до появления симптомов (например, в свете изучения симптомов и/или в свете генетических или других факторов восприимчивости). Лечение можно также продолжать после устранения симптомов, например, для предотвращения или задержки их рецидива.
Применяемый в контексте термин “пациент” или “субъект”, означает субъект-животное, предпочтительно субъект-млекопитающее и особенно субъекты-люди (включая как мужских, так и женских субъектов и включая новорожденных, младенцев, юношеских, взрослых и гериатрических субъектов). Термин субъекты может включать в себя также другие млекопитающие (например, собаку, кошку, лошадь, корову, овцу, козу, обезьяну, птицу и т.д.) для лабораторных или ветеринарных целей.
В некоторых вариантах осуществления лечение предназначается для субъекта, имеющего гепато-клеточную карциному с измененным статусом FGFR4 и/или FGF19 (фактора роста фибробластов 19).
В некоторых вариантах осуществления лечение может включать в себя анализ FGFR4 и/или FGF19 в биологическом образце, содержащем клетки указанной гепато-клеточной карциномы, или лечение можно проводить в сочетании с таким анализом, и, если указанная гепато-клеточная карцинома имеет изменение FGFR4 и/или FGF19, лечение субъекта проводят эффективным количеством активного агента, описываемого в контексте.
"Измененный статус", применяемый в контексте со ссылкой на FGFR4 и/или FGF19, включает в себя повышенную их экспрессию (например, повышенные уровни мРНК или повышенные уровни белка), повышенное число копий в геноме и/или повышенную активность закодированного белка в результате мутации и т.д. по сравнению с соответствующей неконцерогенной тканью. В некоторых вариантах осуществления измененный статус FGFR4 и/или FGF19 включает в себя мутации гена и/или закодированного белка, которые приводят к повышению активности или в противном случае связаны с более агрессивной формой гепато-клеточной карциномы.
“Экспрессия” FGFR4 и/или FGF19 означает, что ген, кодирующий их, транскрибируется и предпочтительно транслируется. Обычно экспрессия кодирующей области приводит к продуцированию закодированного полипептида.
Белки FGFR4 и FGF19 являются известными и их измененный статус и/или экспрессию можно измерить с помощью способов, стандартных в данной области, например, геномным анализом мутаций и/или аберрациями числа копий, такими как амплификация нуклеиновых кислот, секвенирующим анализом и/или способами на основе гибридизации, анализом экспрессии РНК, таким как назерн-блоттинг или qR-PCR-, вестерн-блоттинг или другой иммуноблоттинг или иммуноанализ, клеточный сортер с возбуждением флуоресценции (FACS) и т.д.
Для более полного понимания описанного в контексте изобретения предлагаются следующие примеры. Должно быть понятно, что эти примеры предназначены только для иллюстративных целей и не должны истолковываться как его ограничение.
ПРИМЕРЫ
Микроволновое нагревание проводили с помощью Biotage Emrys Liberator или микроволнового инициатора. Колоночную хроматографию проводили с применением Isco Rf00d. Удаление растворителей проводили с применением либо роторного испарителя Buchi, либо центробежного испарителя Gienevaс. Препаративную ЖХ/МС проводили с применением автоочистителя Waters и колонки 19×100 мм Xterra 5 микрон MС С18 в условиях кислотной подвижной фазы. Спектры ЯМР получали с применением спектрометра Varian 400 МГц.
Когда применяют термин “инертный” для описания реактора (например, реакционного сосуда, колбы, стеклянного реактора и тому подобного), это означает, что воздух в реакторе заменяли по существу не содержащим влагу или сухим инертным газом (таким как азот, аргон и т.д.).
Общие методы и эксперименты для получения соединений настоящего изобретения приводятся выше. В некоторых случаях получение соединения описывается посредством примера. Однако, должно быть понятно, что в каждом случае ряд соединений настоящего соединения получали согласно схемам и экспериментам, описанным выше.
Условия препаративной ВЭЖХ для очистки целевых соединений
Условия хроматографирования
Инструмент: препаративная система Waters 2767-SQD Mass trigger
Колонка: Waters Xbridge C18 150 мм*19 мм*5 мкм
Детектор: VWD SQD
Скорость потока: 15 мл/мин
Градиент состава по времени:
Репрезентативная подвижная фаза
1)
Подвижная фаза А: 0,1% ТFА в воде
Подвижная фаза В: ACN
2)
Подвижная фаза А: 0,1% NH4HCO3 в воде
Подвижная фаза В: ACN
3)
Подвижная фаза А: 0,1% NH4ОАс в воде
Подвижная фаза В: ACN
4)
Подвижная фаза А: 0,1% NH4ОН в воде
Подвижная фаза В: ACN
Определения: следующие аббревиатуры имеют указанные значения
ACN:ацетонитрил
Вос2О: ди-трет-бутилдикарбонат
Brettphos: 2-(дициклогексилфосфино)-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил
tBuONa: трет-бутоксид натрия
CH3I: иодметан
Cs2CO3: карбонат цезия
DCC: N,N’-дициклогексилкарбодиимид
DCM: дихлорметан
ДИEA: N,N-диизопропилэтиламин
DIPEA: N,N-диизопропилэтиламин
DMAP: 4-(диметиламино)пиридин
DME: диметиловый простой эфир
DMF: диметилформамид
DМСО: диметилсульфоксид
EGTA: этиленгликольтетрауксусная кислота
ESI-MС: ионизация электрораспылением-масс-спектрометрия
EtOH: этaнол
HATU: гексафторфосфат 3-оксида 1-[бис(диметиламино)метилeн]-1H-1,2,3-триазоло[4,5-b]пиридиния
H2SO4: серная кислота
iPrOH: изопропанол
K2CO3: карбонaт калия
KHMDS: бис(триметилсилил)амид калия
KOH: гидроксид калия
ЖХ-MС: жидкостная хроматография–масс-спектрометрия
MeOH: метанол
MsCl: метaнсульфонилхлорид
NaBH3CN: цианоборогидрид натрия
NaBH(OАс)3: триацетoксиборогидрид натрия
NH4Cl: хлорид аммония
NH4HCO3: бикарбонат аммония
NaI: иодид натрия
NaNO3: нитрат натрия
NaOАс: ацетaт натрия
nBuOH: н-бутaнол
преп-ВЭЖХ: препаративная высокоэффективная жидкостная хроматография
преп-ТСХ: препаративная тонкослойная хроматография
TBAF: фторид тетрабутиламмония
TBDMS-CL: трет-бутилдиметилсилилхлорид
TBSCl: трет-бутилдиметилсилилхлорид
TBSOTf: трет-бутилдиметилсилилтрифторметансульфонат
TEA: триэтиламин
TESCl: хлортриэтилсилaн
TFA: трифторуксусная кислота
THF: тетрагидрофуран
Ti(OiPr)4: изопропоксид титана
TСХ: тонкослойная хроматография
PPTS: п-толуолсульфонат пиридиния
PE: петролейный эфир
PЭГ: поли(этиленгликоль)
PtO2: диоксид платины
EtOАс: этилацетaт
Pd/C: палладий(0) на угле
Pd2(dba)3: трис(дибензилиденацетон)дипалладий(0)
Pd(dppf)2Cl2: [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II)
Ruphos: 2-дициклогексилфосфино-2',6'-диизопропoксибифенил
Xantphos: 4,5-бис(дифенилфосфино)-9,9-диметилксантен
Вещества: следующие соединения являются коммерчески доступными и/или их можно получить рядом способов, известных специалисту в области органического синтеза. Более конкретно, описанные соединения можно получить при помощи реакций и способов, описанных в контексте. Должно быть понятно, что в описании синтетических способов, указанных ниже, все предложенные условия реакции, включая выбор растворителя, атмосферы реакции, температуры реакции, продолжительности эксперимента и способы обработки, можно выбрать так, чтобы они были условиями, стандартными для такой реакции, если не указано иначе. Специалисту в области органического синтеза должно быть понятно, что функциональная группа, присутствующая в различных положениях молекулы, должна быть совместима с предложенными реагентами и реакциями. Заместители, не совместимые с условиями реакции, должны быть известны специалисту в данной области, и поэтому указываются альтернативные методы. Исходные вещества для примеров являются либо коммерчески доступными, либо легко получаются стандартными способами из известных веществ.
СИНТЕЗ И ИСПЫТАНИЕ СОЕДИНЕНИЙ ПРИМЕРОВ
Соединения таблицы I получали способами 2А-2L.
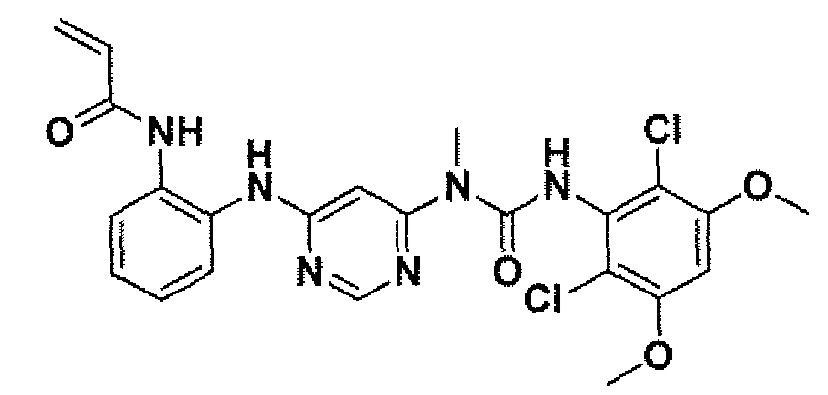












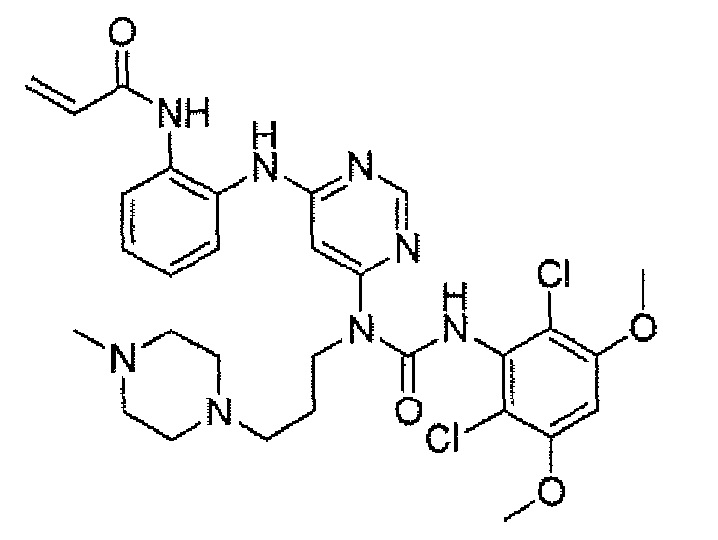









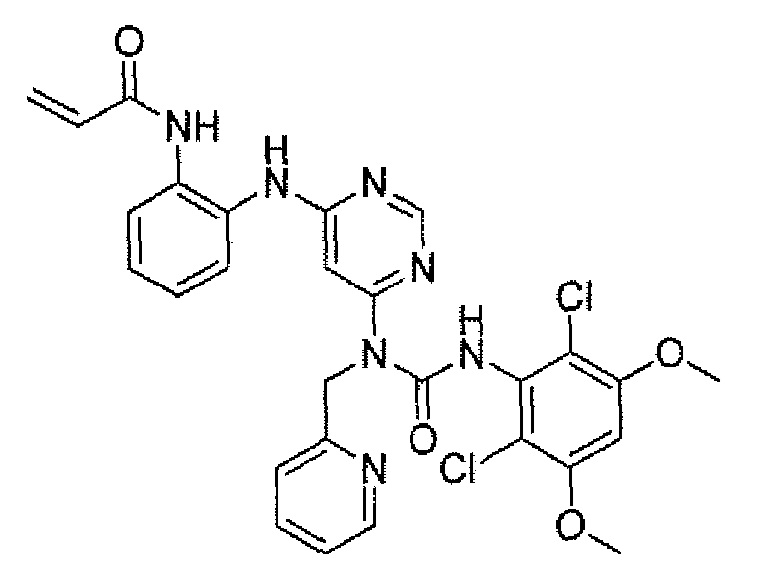














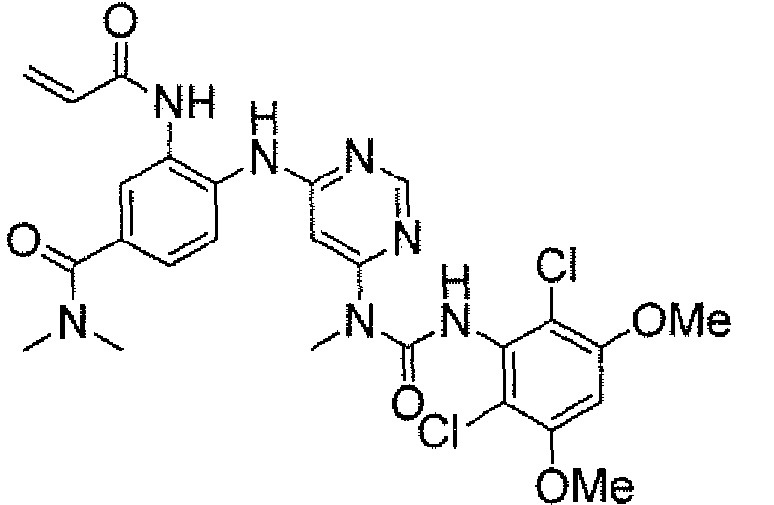



















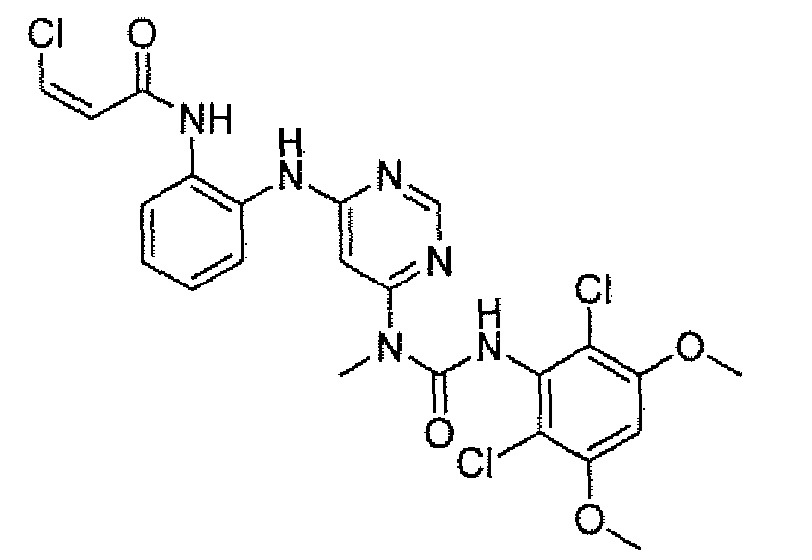







Способ 2A: - пример 100
Схема 1

N-(2-{6-[3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо]пиримидин-4-иламино}фенил)акриламид
Схема 2

a. N-(3,5-Диметоксифенил)ацетамид
К раствору 3,5-диметоксифениламина (20 г, 0,131 моль) в толуоле добавляли уксусный ангидрид (14 г, 0,137 ммоль) при комнатной температуре. Образовавшуюся смесь перемешивали в течение 18 часов при комнатной температуре. Добавляли РЕ (55 мл), осадок фильтровали и промывали PE (100 мл), получая при этом указанное в заголовке соединение (24,2 г, выход: 95%).1H-ЯМР(400 МГц, CDCl3) δ 2,16 (с, 3H), 3,77 (с, 6H), 6,23 (с, 1H), 6,75 (с, 2H), 7,20 (с, 1H).

b.N-(2,6-Дихлор-3,5-диметоксифенил)ацетамид
К раствору N-(3,5-диметоксифенил)ацетамида (5 г, 25,6 ммоль)в АСN (75 мл) добавляли сульфурилхлорид (6,9 г, 51,2 ммоль) при 0°C в атмосфере азота. Образовавшуюся смесь перемешивали в течение 30 минут при такой же температуре и гасили насыщенным водным раствором NaHCO3 (40 мл). Осадок фильтровали, промывали водой и сушили, получая при этом указанное в заголовке соединение (2,3 г, выход 34%).1H-ЯМР(400 МГц, CDCl3) δ 2,25 (с, 3H), 3,86 (с, 6H), 6,54 (с, 1H), 6,90 (с, 1H).
Схема 3

c.2,6-Дихлор-3,5-диметоксифениламин
Раствор N-(2,6-дихлор-3,5-диметоксифенил)ацетамида (3,6 г, 13,7 ммоль) в EtOH (130 мл) и KOH (2M, 75 мл) нагревали для кипячения с обратным холодильником в течение 24 час. Реакционную смесь охлаждали до 0°C и перемешивали в течение 1 час при такой температуре. Осадок фильтровали и сушили, получая при этом указанное в заголовке соединение (2,3 г, выход 76%).1H-ЯМР(400 МГц, CDCl3) δ 3,90 (с, 6H), 4,57 (ушир. с, 2H), 6,05 (с, 1H).
Схема 4

d.2,4-Дихлор-3-изоцианато-1,5-диметоксибензол
Смесь 2,6-дихлор-3,5-диметоксифениламина (500 мг, 2,25 ммоль), трифосгена (335 мг, 1,12 ммоль) и TEA (342 мг, 3,38 ммоль) в диоксaне (15 мл) нагревали до 130°C в течение 2 час при микроволновом облучении. Реакционную смесь концентрировали и остаток очищали флэш-хроматографией на диоксиде кремния с элюированием DCM, получая при этом указанное в заголовке соединение (450 мг, выход 80%).1H-ЯМР(400 МГц, CDCl3) δ 3,92 (с, 6H), 6,42 (с, 1H).
Схема 5

e. (6-Хлорпиримидин-4-ил)метиламин
К раствору 4,6-дихлорпиримидина (7,45 г, 50 ммоль) в iPrOH (50 мл) добавляли раствор метиламина в ТГФ (2M, 30 мл, 60 ммоль) при комнатной температуре. Образовавшуюся смесь перемешивали в течение 18 час. Смесь концентрировали и остаток очищали флэш-хроматографией на диоксиде кремния с элюированием смесью DCM:EtOАс = 6:1~1:1, получая при этом указанное в заголовке соединение (4,4 г, выход: 62%) в виде белого твердого вещества.1H ЯМР(400 МГц, CDCl3) δ 2,96 (д, 3H), 5,22-5,36 (ушир. с, 1H), 6,35 (с, 1H), 8,35 (с, 1H); МС (ESI) 144[M+H]+.

f.N-Метил-N'-(2-нитрофенил)пиримидин-4,6-диамин
Смесь (6-хлр-пиримидин-4-ил)метиламина (1 г, 7 ммоль), 2-нитрофениламина (965 мг, 7 ммоль), Brettphos (279 мг, 0,35 ммоль) и tBuONa (2 г, 21 ммоль) в DME (50 мл) нагревали до 90°C в течение 1 час в атмосфере азота. Раствор концентрировали остаток очищали флэш-хроматографией на диоксиде кремния с элюированием смесью DCM:EtOАс = 10:1~1:1, получая при этом указанное в заголовке соединение (600 мг, выход 35%) в виде желтого твердого вещества.1H ЯМР(400 МГц, CDCl3) δ 2,94 (д, 3H), 4,99 (ушир. с, 1H), 5,82 (с, 1H), 7,04 (т, 1H), 7,60 (т, 1H), 8,21 (д, 1H), 8,33 (с, 1H), 8,75 (д, 1H), 9,91 (с, 1H); МС (ESI): 246[M+H]+.
Схема 6

g. 3-(2,6-Дихлор-3,5-диметоксифенил)-1-метил-1-[6-(2-нитрофениламино)пиримидин-4-ил]мочевина
К раствору N-метил-N'-(2-нитрофенил)пиримидин-4,6-диамина (150 мг, 0,61 ммоль) в ТГФ (15 мл) добавляли NaH (60%, 60 мг, 1,5 ммоль) при 0°C и смесь перемешивали в течение 30 минут при комнатной температуре. При комнатной температуре по каплям добавляли раствор 2,4-дихлор-3-изоцианато-1,5-диметоксибензола (180 мг, 0,73 ммоль). Образовавшуюся смесь перемешивали в течение 2 часов. Для гашения реакции добавляли воду (2 мл). Смесь концентрировали и остаток очищали флэш-хроматографией на диоксиде кремния с элюированием смесью DCM:EtOАс = 6:1~1:1, получая при этом указанное в заголовке соединение (54 мг, выход 18%) в виде желтого твердого вещества.1H ЯМР(400 МГц, ДМСО-d6) δ 3,38 (с, 3H), 3,93 (с, 6H), 6,75 (с, 1H), 6,91 (с, 1H), 7,34 (т, 1H), 7,72 (т, 1H), 7,79 (д, 1H), 8,01 (д, 1H), 8,38 (с, 1H), 9,99 (с, 1H), 11,78 (с, 1H); MС (ESI) 493[M+H]+.
Схема 7

h. 1-[6-(2-Аминофениламино)пиримидин-4-ил]-3-(2,6-дихлор-3,5-диметоксифенил)-1-метилмочевина
К раствору 3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-1-[6-(2-нитрофениламино) пиримидин-4-ил]мочевины (50 мг, 0,1 ммоль) в ТГФ (10 мл) и MeOH (10 мл) добавляли Ni Ренея (суспензия в воде) при комнатной температуре, образовавшуюся смесь перемешивали в течение 2 часов в атмосфере водорода. Реакционную смесь фильтровали и концентрировали, получая при этом указанное в заголовке соединение (38 мг, выход: 82%), которое применяли непосредственно в следующей стадии.1H ЯМР(400 МГц, CDCl3) δ 3,28 (с, 3H), 3,85 (с, 2H), 3,94 (с, 6H), 5,86 (с, 1H), 6,52 (с, 1H), 6,78-6,87 (м, 3H), 7,16-7,20 (м, 2H), 8,39 (с, 1H), 12,62 (с, 1H); МС (ESI) 463[M+H]+.
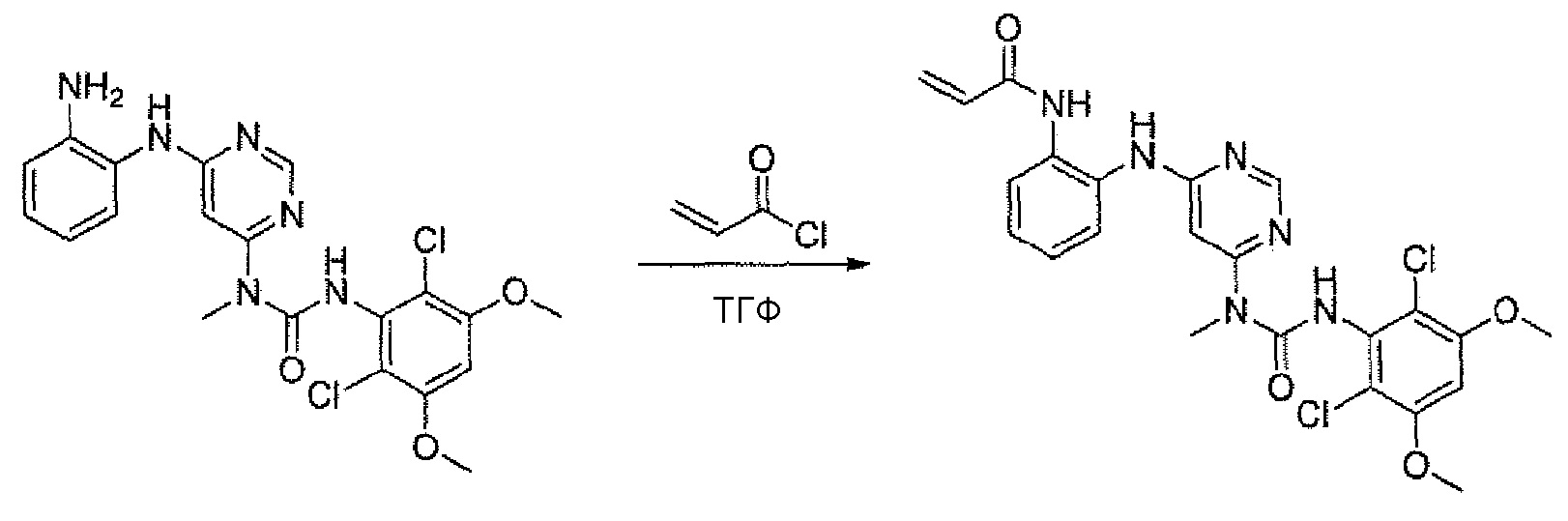
i. N-(2-{6-[3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо]пиримидин-4-иламино}фенил)акриламид
К раствору 1-[6-(2-аминофениламино)пиримидин-4-ил]-3-(2,6-дихлор-3,5-диметоксифенил)-1-метилмочевины(25 мг, 0,05 ммоль) в ТГФ (10 мл) добавляли раствор акрилоилхлорида в ТГФ (20 мг/мл, 0,5 мл, 0,1 ммоль) при -10°C и образовавшуюся смесь перемешивали в течение 1 час при такой температуре. Для гашения реакции добавляли MeOH (1 мл). Смесь концентрировали и остаток очищали преп-ТСХ, получая при этом указанное в заголовке соединение (12 мг, выход 43%).1H ЯМР(400 МГц, ДМСО-d6) δ 3,26 (с, 3H), 3,94 (с, 6H), 5,74 (д, 1H), 6,24 (д, 1H), 6,37 (с, 1H), 6,47-6,54 (м, 1H), 6,90 (с, 2H), 7,20 (д, 2H), 7,56-7,58 (м, 1H), 7,66-7,68 (м, 1H), 8,38 (с, 1H), 9,99 (с, 1H), 9,70 (с, 1H), 11,99 (с, 1H); МС (ESI): 517[M+H]+.
Соединения 102, 103 и 105 синтезировали таким же способом, как соединение 100.
Методика 2B: пример – 107
Схема 8

N-(2-{6-[3-(2,6-Дихлор-3,5-диметоксифенил)-1-пиридин-3-илметилуреидо]-пиримидин-4-иламино}фенил)акриламид
Схема 9

a. (6-Хлорпиримидин-4-ил)пиридин-3-илметиламин
К раствору 4,6-дихлорпиримидина (1 г, 6,71 ммоль) в диоксaне (20 мл) добавляли раствор пиридин-3-илметиламина (745 мг, 6,9 ммоль) при комнатной температуре. Образовавшуюся смесь перемешивали при комнатной температуре на протяжении ночи. Смесь концентрировали и остаток очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (680 мг, выход 46%). МС (ESI): 221[M+H]+.
Схема 10

b. N-(2-Нитрофенил)-N'-пиридин-3-илметилпиримидин-4,6-диамин
Дегазированную смесь (6-хлорпиримидин-4-ил)пиридин-3-илметиламина (300 мг, 1,36 ммоль), 2-нитрофениламина (188 мг, 1,36 ммоль), Pd2(dba)3 (128 мг, 0,14 ммоль), Xantphos (161 мг, 0,28 ммоль) и Cs2CO3 (913 мг, 2,8 ммоль) в толуоле (10 мл) нагревали при 100°C в течение 4 часов. Реакционную смесь концентрировали и остаток очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (150 мг, выход 34%). МС (ESI): 323[M+H]+.
Схема 11
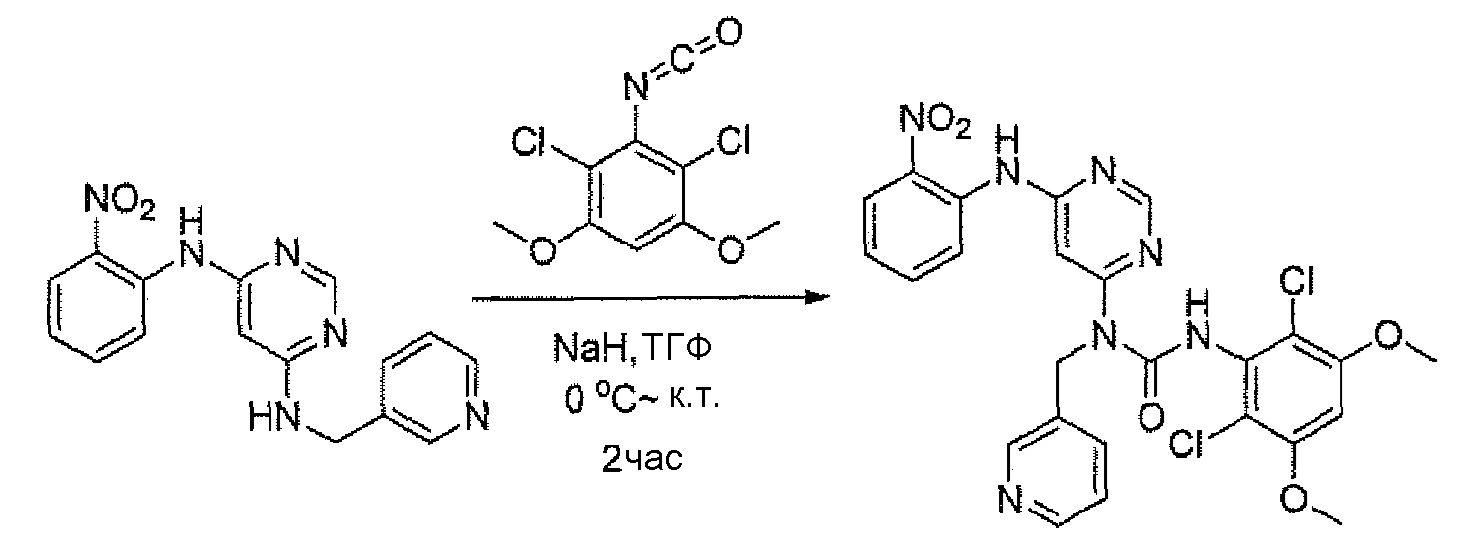
c. 3-(2,6-Дихлор-3,5-диметоксифенил)-1-[6-(2-нитрофениламино)пиримидин-4-ил]-1-пиридин-3-илметилмочевина
К раствору N-(2-нитрофенил)-N'-пиридин-3-илметилпиримидин-4,6-диамина (150 мг, 0,467 ммоль) в ТГФ (15 мл) добавляли NaH (60%, 48 мг, 1,2 ммоль) при 0°C и смесь перемешивали в течение 30 минут при комнатной температуре. При комнатной температуре по каплям добавляли 2,4-дихлор-3-изоцианато-1,5-диметоксибензол (методика 2A, стадии a-d; 180 мг, 0,73 ммоль). Образовавшуюся смесь перемешивали в течение 2 часов. Для гашения реакции по каплям добавляли воду (2 мл). Смесь концентрировали и остаток очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (85 мг, выход 32%). МС (ESI): 570[M+H]+.

d. 1-[6-(2-Аминофениламино)пиримидин-4-ил]-3-(2,6-дихлор-3,5-диметоксифенил)-1-пиридин-3-илметилмочевина
Смесь 3-(2,6-дихлор-3,5-диметоксифенил)-1-[6-(2-нитрофениламино)пиримидин-4-ил]-1-пиридин-3-илметилмочевины (85 мг, 0,149 ммоль) и Fe (84 мг, 1,5 ммоль) в АсOH (5 мл) нагревали при 50°C в течение 2 часов. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме, получая при этом сырой продукт, который очищали колоночной хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (53 мг, выход: 66%). МС (ESI) 540[M+H]+.

e. N-(2-{6-[3-(2,6-Дихлор-3,5-диметоксифенил)-1-пиридин-3-илметилуреидо]пиримидин-4-иламино}фенил)акриламид
К раствору 1-[6-(2-аминофениламино)пиримидин-4-ил]-3-(2,6-дихлор-3,5-диметоксифенил)-1-пиридин-3-илметилмочевины (53 мг, 0,1 ммоль) в ТГФ (10 мл) добавляли раствор акрилоилхлорида в ТГФ (20 мг/мл, 0,5 мл, 0,1 ммоль) при -10°C и смесь перемешивали в течение 1 часа при такой температуре. Для гашения реакции добавляли MeOH (1 мл). Смесь концентрировали и остаток очищали преп-ТСХ, получая при этом указанное в заголовке соединение (9 мг, выход 15%).1H ЯМР(400 МГц, CDCl3) δ 3,84 (с, 6H), 5,01 (с, 2H), 5,69 (д, 1H), 5,75 (с, 1H), 6,10 (дд, 1H), 6,34 (д, 1H), 6,47 (с, 1H), 7,00 (д, 1H), 7,09-7,24 (м, 2H), 7,28 (т, 1H), 7,32 (с, 1H), 7,47 (д, 1H), 7,69-7,71 (м, 2H), 8,31-8,34 (м, 2H), 8,40-8,42 (м, 1H), 12,60 (с, 1H); МС (ESI): 594 [M+H]+.
Методика 2C: пример – 108
Схема 12

N-[2-{6-[3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо]пиримидин-4-иламино}-5-(4-этилпиперазин-1-ил)фенил]акриламидметан
схема 13

a.трет-Бутил-4-бром-2-нитрофенилкарбамат
Смесь 4-бром-2-нитроанилина(4 г, 18,4 ммоль), (Boc)2O (4,4 г, 20,24 ммоль) в ТГФ (50 мл) нагревали при кипячении с обратным холодильником на протяжении ночи. Смесь концентрировали и остаток очищали флэш-хроматографией на диоксиде кремния с элюированием смесью PE:EtOAc = 20:1, получая при этом указанное в заголовке соединение (5,4 г, выход 93%). МС (ESI): 317, 319 [M+H]+.
Схема 14

b.трет-Бутил 4-(4-этилпиперазин-1-ил)-2-нитрофенилкарбамат
Дегазированную смесьтрет-бутил-4-бром-2-нитрофенилкарбамата(5,4 г, 17 ммоль), 1-этилпиперазина (2,91 г, 25,5 ммоль), Pd2(dba)3 (2,1 г, 3,4 ммоль), xantphos (3,92 г, 6,8 ммоль) и Cs2CO3 (11,1 г, 34 ммоль) в толуоле (85 мл) нагревали при 100°C в течение 4 часов. Реакционную смесь концентрировали и остаток очищали флэш-хроматографией на диоксиде кремния с элюированием смесью MeOH:DCM = 1:50~1:20, получая при этом указанное в заголовке соединение (3,3 г, выход: 55%). МС (ESI) 351 [M+H]+.
Схема 15

c. 4-(4-Этилпиперазин-1-ил)-2-нитроанилин
К раствору трет-бутил-4-(4-этилпиперазин-1-ил)-2-нитрофенилкарбамата (3,3 г, 9,43 ммоль) в DCM (50 мл) добавляли TFA (20 мл) при 0°C, образовавшуюся смесь перемешивали в течение 3 часов при комнатной температуре. После удаления всех летучих компонентов в вакууме остаток снова растворяли в DCM, нейтрализовали насыщенным водным раствором K2CO3 и экстрагировали DCM. Объединенные экстракты концентрировали, получая при этом указанное в заголовке соединение (2,1 г, выход 90%), которое непосредственно применяли в следующей стадии.1H ЯМР(400 МГц, ДМСО-d6) δ 1,02 (т, 3H), 2,36 (кв. 2H), 2,47-2,49 (м, 4H), 2,97-3,00 (м, 4H), 6,97 (д, 1H), 7,20 (с, 2H), 7,25 (с, 1H), 7,34 (дд, 1H); МС (ESI) 251[M+H]+.
Схема 16

d.N4-(4-(4-Этилпиперазин-1-ил)-2-нитрофенил)-N6-метилпиримидин-4,6-диамин
Дегазированную смесь 4-(4-этилпиперазин-1-ил)-2-нитроанилина (2,1 г, 8,4 ммоль), 6-хлор-N-метилпиримидин-4-амина (методика 2A, стадия e; 1,2 г, 8,4 ммоль), Pd2(dba)3 (1,54 г, 1,68 ммоль), xantphos (1,94 г, 3,36 ммоль) и Cs2CO3 (5,48 г, 16,8 ммоль) в толуоле (45 мл) нагревали при 100°C в течение 1 часа. Реакционную смесь концентрировали и остаток очищали флэш-хроматографией на диоксиде кремния с элюированием смесью MeOH:DCM = 1:40~1:20, получая при этом указанное в заголовке соединение (870 мг, выход 29%). МС (ESI) 358[M+H]+.

e. 3-(2,6-Дихлор-3,5-диметоксифенил)-1-(6-(4-(4-этилпиперазин-1-ил)-2-нитрофениламино)пиримидин-4-ил)-1-метилмочевина
К раствору N4-(4-(4-этилпиперазин-1-ил)-2-нитрофенил)-N6-метилпиримидин-4,6-диамина (870 мг, 2,44 ммоль) в ТГФ (15 мл) добавляли NaH (60%, 200 мг, 5 ммоль) при 0°C и смесь перемешивали в течение 30 минут при комнатной температуре. По каплям при 0°C добавляли раствор 2,4-дихлор-3-изоцианато-1,5-диметоксибензол (методика 2A, стадии a-d; 908 мг, 3,66 ммоль) в ТГФ. Образовавшуюся смесь перемешивали при комнатной температуре в течение 2 часов. Для гашения реакции добавляли насыщенный водный раствор NH4Cl (2 мл). Смесь концентрировали и экстрагировали DCM. Объединенные экстракты промывали насыщенным раствором соли, сушили над безводным Na2SO4 и концентрировали, получая при этом сырой продукт, который очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (330 мг, выход 21%) в виде красного масла.1H ЯМР(400 МГц, CDCl3) δ 1,44 (т, 3H), 3,01 (т, 2H), 3,21 (кв, 2H), 3,41-3,49 (м, 5H), 3,73-3,80 (м, 4H), 3,92 (с, 6H), 6,27 (с, 1H), 6,55 (с, 1H), 7,25 (д, 1H), 7,69 (с, 1H), 8,32 (д, 1H), 8,52 (с, 1H), 10,28 (ушир. с, 1H), 12,05 (ушир. с, 1H); МС (ESI): 605[M+H]+.
Схема 17

f.1-(6-(2-Амино-4-(4-этилпиперазин-1-ил)фениламино)пиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метилмочевина
К раствору 3-(2,6-дихлор-3,5-диметоксифенил)-1-(6-(4-(4-этилпиперазин-1-ил)-2-нитрофениламино)пиримидин-4-ил)-1-метилмочевины (330 мг, 0,546 ммоль) в ТГФ (20 мл) и MeOH (20 мл) добавляли Ni Ренея (суспензия в воде) при комнатной температуре, образовавшуюся смесь перемешивали в течение 3 часов в атмосфере водорода (1 атм). Реакционную смесь фильтровали и концентрировали. Остаток промывали дважды MeOH, получая при этом указанное в заголовке соединение (280 мг, чистота 90%), которое применяли непосредственно в следующей стадии. МС (ESI) 575[M+H]+.

g.N-(2-(6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-иламино)-5-(4-этилпиперазин-1-ил)фенил)акриламид
К раствору 1-(6-(2-амино-4-(4-этилпиперазин-1-ил)фениламино)пиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метилмочевины (280 мг, чистота 90%, 0,44 ммоль) в ТГФ (30 мл) добавляли раствор акрилoилхлорида в ТГФ (20 мг/мл, 2 мл, 0,44 ммоль) при -10°C и образовавшуюся смесь перемешивали в течение 1 часа при такой температуре. Для гашения реакции добавляли MeOH (1 мл). Смесь концентрировали и остаток очищали преп-ВЭЖХ и преп-ТСХ, получая при этом указанное в заголовке соединение (20 мг, выход 7%).1H ЯМР(400 МГц, CDCl3) δ 1,31 (т, 3H), 2,65 (кв, 2H), 2,62-2,68 (м, 4H), 3,27 (с, 3H), 3,36-3,38 (м, 4H), 3,91 (с, 6H), 5,76 (д, 1H), 5,90 (с, 1H), 6,24 (дд, 1H), 6,41 (д, 1H), 6,52 (с, 1H), 6,74 (дд, 1H), 7,07 (ушир. с, 1H), 7,23 (д, 1H), 7,72 (ушир. с, 1H), 7,98 (ушир. с, 1H), 8,37 (с, 1H), 12,52 (с, 1H); МС (ESI) 629 [M+H]+.
Пример – 110

N-(2-(6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-иламино)фенил)-2-фторакриламид
Соединение синтезировали по способу, описанному в методике 2A (пример 100), модифицируя стадию (i) следующим образом: к раствору 1-[6-(2-аминофениламино)пиримидин-4-ил]-3-(2,6-дихлор-3,5-диметоксифенил)-1-метилмочевины (130 мг, смешанной с тетрахлоранилином) и DCC (118 мг, 0,56 ммоль) в хлороформе (100 мл), добавляли раствор 2-фторакриловой кислоты (50 мг, 0,56 ммоль) в хлороформе (50 мл) при 0°C и образовавшуюся смесь перемешивали при комнатной температуре на протяжении ночи. Для гашения реакции добавляли воду (1 мл). Смесь концентрировали и остаток очищали на колонке с обращенной фазой и преп-ТСХ, получая при этом указанное в заголовке соединение (4 мг, выход 5%).1H ЯМР(400 МГц, CDCl3) δ 12,17 (с, 1H), 8,45 (с, 1H), 8,35 (с, 1H), 7,84 (д, 1H), 7,37 (д, 1H), 7,29 (т, 1H), 7,26 (т, 1H), 6,47 (с, 1H), 5,94 (с, 1H), 5,78 (дд, 1H), 5,21 (дд, 1H), 3,85 (с, 6H), 3,25 (с, 3H); МС (ESI): 535 [M+H]+.
Методика 2E: пример – 111
Схема 18

N-(2-(6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-иламино)-5-(1-этилпиперидин-4-ил)фенил)акриламид
Схема 19

а. трет-Бутил-4-(4-амино-3-нитрофенил)-5,6-дигидропиридин-1(2H)-карбоксилат
К дегазированной смеси 4-бром-2-нитроанилина (1 г, 4,6 ммоль), трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксaборoлан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (1,42 г, 4,6 ммоль), тригидрата фосфата калия (3,9 г, 14,64 ммоль) в диоксане и воде (30 мл, 8:1) добавляли Pd(dppf)2Cl2 (337 мг, 0,46 ммоль). Смесь кипятили с обратным холодильником при 110°C в течение 3 часов. Фильтрование и концентрирование давали сырой продукт, который очищали колоночной хроматографией на силикагеле, получая при этом указанное в заголовке соединение (1,1 г, выход 75%). МС (ESI) 320[M+H]+.
Схема 20

b.1-(6-Хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)мочевина
К раствору 6-хлор-N-метилпиримидин-4-амина (методика 2A, стадия e; 460 мг, 3,21 ммоль) в ДМФА (15 мл) добавляли NaH (60%, 193 мг, 4,81 ммоль) при 0°C и смесь перемешивали в течение 30 минут при комнатной температуре. При комнатной температуре по каплям добавляли раствор 2,4-дихлор-3-изоцианато-1,5-диметоксибензола (методика H, стадии a-d; 1,03 г, 4,17 ммоль) в ДМФА (5 мл). Образовавшуюся смесь перемешивали в течение 0,5 часа. Добавляли SEMCl (804 мг, 4,81 ммоль) в ДМФА (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Для гашения реакции добавляли насыщенный водный раствор NH4Cl. Смесь разбавляли водой и экстрагировали EtOАс. Объединенные экстракты промывали водой и насыщенным раствором соли, сушили над безводным Na2SO4 и фильтровали. Фильтрат упаривали в вакууме, получая при этом сырой продукт, который очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (470 мг, выход: 28%). МС (ESI) 521[M+H]+.
Схема 21

c. трет-Бутил-(4-(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)уреидо)пиримидин-4-иламино)-3-нитрофенил)-5,6-дигидропиридин-1(2H)-карбоксилат
Дегазированную смесь 1-(6-хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)мочевины (470 мг, 0,9 ммоль), трет-бутил-4-(4-амино-3-нитрофенил)-5,6-дигидропиридин-1(2H)-карбоксилата (320 мг, 1 ммоль), Pd2(dba)3 (92 мг, 0,1 ммоль), xantphos (115 мг, 0,2 ммоль) и Cs2CO3 (652 мг, 2 ммоль) в толуоле (10 мл) нагревали при 100°C в течение 5 часов. Реакционную смесь концентрировали и остаток очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (400 мг, выход 57%). МС (ESI) 804[M+H]+.

d. трет-Бутил-4-(3-амино-4-(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)уреидо)пиримидин-4-иламино)фенил)пиперидин-1-карбоксилат
К раствору трет-бутил-4-(4-(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)уреидо)пиримидин-4-иламино)-3-нитрофенил)-5,6-дигидропиридин-1(2H)-карбоксилата (380 мг, 0,473 ммоль) в MeOH (10 мл) добавляли PtO2 (38 мг, 10 масс.%) и одну каплю хлорбензола при комнатной температуре, образовавшуюся смесь перемешивали в атмосфере водорода (1 атм) на протяжении ночи. Реакционную смесь фильтровали и концентрировали. Остаток очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (130 мг, выход 37%). МС (ESI) 776 [M+H]+.

e. Соль N-(2-(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-иламино)-5-(пиперидин-4-ил)фенил)акриламида с TFA
К раствору трет-бутил-4-(3-амино-4-(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)уреидо)пиримидин-4-иламино)фенил)пиперидин-1-карбоксилата (130 мг, 0,168 ммоль) в ТГФ (15 мл) по каплям добавляли раствор акрилoилхлорида (10 мг/мл, 1,7 мл, 0,19 ммоль) при -10°C и образовавшуюся смесь перемешивали при 0°C в течение 1 часа. Анализ ЖХ-МС показал, что реакция была завершена. Для гашения реакции добавляли MeOH (5 мл) и реакционную смесь концентрировали. Остаток в DCM (2 мл) добавляли по каплям к смеси DCM/TFA (2/1, об./об., 3 мл). Смесь перемешивали при комнатной температуре в течение 1 часа и затем концентрировали в вакууме. Остаток (50 мг, колич.) применяли непосредственно для следующей стадий без дополнительной очистки. МС (ESI) 600 [M+H]+.

f.N-(2-(6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-иламино)-5-(1-этилпиперидин-4-ил)фенил)акриламид
К раствору соли N-(2-(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-иламино)-5-(пиперидин-4-ил)фенил)акриламид с TFA (35 мг, 0,049 ммоль) в EtOH (1 мл) добавляли NaOАс (4 мг, 0,05 ммоль) и водный ацетальдегид (1 мл, 0,9 ммоль, 40%). После перемешивания смеси при комнатной температуре в течение 1 часа добавляли NaBH3CN (12 мг, 0,18 ммоль)и раствор перемешивали при комнатной температуре в течение еще 3 часов. После удаления всех летучих компонентов в вакууме остаток распределяли между DCM и водой. Водный слой экстрагировали хлороформом дважды. Комбинированные экстракты промывали насыщенным раствором соли, сушили над безводным Na2SO4 и фильтровали. Фильтрат упаривали в вакууме, получая при этом сырой продукт, который очищали преп-ВЭЖХ, получая при этом указанное в заголовке соединение (3 мг, выход 10%).1H ЯМР(400 МГц, MeOH-d4) δ 8,38 (с, 1H), 7,68 (с, 1H), 7,54 (д, 1H), 7,24 (дд, 1H), 6,83 (с, 1H), 6,46-6,35 (м, 3H), 5,81 (д, 1H), 3,97 (с, 6H), 3,74-3,70 (м, 2H), 3,37 (с, 3H), 3,26 (кв, 2H), 3,17-3,11 (м, 2H), 2,99-2,96 (м, 1H), 2,26-2,22 (м, 2H), 2,06-1,99 (м, 2H), 1,43 (т, 3H); МС (ESI) 628 [M+H]+.
Пример – 112

N-(2-(6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-иламино)-5-(1-метилпиперидин-4-ил)фенил)акриламид
Соединение синтезировали по способу, описанному в методике 2Е (пример 111), с применением формальдегида в стадии (f), получая при этом указанное в заголовке соединение (1,5 мг, выход 11,6%).1H ЯМР(400 МГц, MeOH-d4) δ 8,26 (с, 1H), 7,55 (с, 1H), 7,41 (д, 1H), 7,11 (д, 1H), 6,71 (с, 1H), 6,34-6,22 (м, 3H), 5,68 (д, 1H), 3,84 (с, 6H), 3,55-3,52 (м, 2H), 3,25 (с, 3H), 3,21-3,08 (м, 2H), 2,90-2,83 (м, 4H), 2,11-2,08 (м, 2H), 1,89-1,85 (м, 2H); МС (ESI) 614 [M+H]+.
Методика 2F: пример – 113
Схема 22

N-(2-{6-[3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо]пиримидин-4-иламино}-5-диметиламинометилфенил)акриламид
схема 23

a. 4-Фтор-3-нитробензальдегид
К перемешиваемому раствору (4-фтор-3-нитрофенил)метaнола (750 мг, 4,4 ммоль) в DCM (40 мл) при 0°C добавляли реагент Десс-Мартина (3,0 г, 7 ммоль). Раствор перемешивали при комнатной температуре в течение 4 часов. ТСХ показала исчезновение исходного вещества. Реакцию гасили 10% NaHCO3 и 10% водным раствором Na2S2O3 и слой DCM отделяли и промывали водой (100 мл) и насыщенным раствором соли (50 мл). Реакционную смесь концентрировали и остаток очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (570 мг, выход: 75%).1H-ЯМР(400 МГц, ДМСО-d6) δ 10,09 (д, 1H), 8,36 (т, 1H), 8,06 (дд, 1H), 7,97 (м, 1H).

b. 4-Амино-3-нитробензальдегид
К раствору 4-фтор-3-нитробензальдегида (570 мг, 3,3 ммоль) в ТГФ (20 мл) добавляли NH4OH (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Образовавшееся желтое твердое вещество собирали и промывали водой, сушили в вакууме, получая при этом указанное в заголовке соединение (300 мг, выход: 53%).1H-ЯМР(300 МГц, ДМСО-d6) δ 9,76 (с, 1H), 8,57 (д, 1H), 8,18 (ушир. с, 2H), 7,80 (дд, 1H), 7,10 (д, 1H).

c. 4-Диметиламинометил-2-нитрофениламин
К перемешиваемому раствору диметиламина (4,0 мл, 2 M, 8,0 ммоль) в MeOH (4 мл) добавляли Ti(OiPr)4 (1,15 g, 4 ммоль) и раствор перемешивали при комнатной температуре в течение 15 минут. Затем добавляли 4-амино-3-нитробензальдегид (160 мг, 1,0 ммоль) в MeOH (2 мл) и раствор перемешивали при комнатной температуре на протяжении ночи. Затем добавляли NaBH4 (78 мг, 2 ммоль) и раствор перемешивали при комнатной температуре в течение 1 часа. Анализ ЖХ-МС показал пик основного продукта. Раствор разбавляли EtOАс (60 мл) и промывали водой (2×100 мл) и насыщенным раствором соли (50 мл), сушили над безводным Na2SO4. Раствор упаривали досуха и собирали 130 мг сырого продукта, который применяли для следующей стадии без дополнительной очистки.1H ЯМР(400 МГц, ДМСО-d6) δ 7,82 (с, 1H), 7,35 (ушир. с, 2H), 7,31 (дд, 1H), 6,97 (д, 1H), 3,26 (с, 2H), 2,12 (с, 6H).
Схема 24

d. 1-(2,6-Дихлор-3,5-диметоксифенил)-3-[6-(4-диметиламинометил-2-нитрофениламино)пиримидин-4-ил]-3-метил-1-(2-триметилсилaнилэтоксиметил)мочевина
К перемешиваемому раствору-(6-хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-(2-триметилсилaнилэтоксиметил)мочевины (методика L, стадия b; 260 мг, 0,5 ммоль) в толуоле (5 мл) добавляли 4-диметиламинометил-2-нитрофениламин (100 мг, 0,5 ммоль), Cs2CO3 (400 мг, 1,25 ммоль), Pd2(dba)3 (46 мг, 0,05 ммоль), xantphos (90 мг, 0,15 ммоль). Раствор перемешивали при 100°C на протяжении ночи. Анализ ЖХ-МС показал пик основного продукта. Раствор упаривали с силикагелем и очищали флэш-хроматографией на диоксиде кремния с элюированием EtOАс (0,5 масс.% TEA):MeOH (0,5 масс.% TEA) = 10~10:0.5, получая при этом требуемый продукт (100 мг, выход: 30%). МС (ESI) 680[M+H]+.

e. [6-(2-Амино-4-диметиламинометилфениламино)пиримидин-4-ил]-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-(2-триметилсилaнилэтоксиметил)мочевина
К перемешиваемому раствору 1-(2,6-дихлор-3,5-диметоксифенил)-3-[6-(4-диметиламинометил-2-нитрофениламино)пиримидин-4-ил]-3-метил-1-(2-триметилсилaнилэтоксиметил)мочевины (100 мг, 0,15 ммоль) в MeOH (10 мл) добавляли 4 капли хлорбензола и затем PtO2 (30 мг, 30 масс. %). Раствор перемешивали в атмосфере водорода при комнатной температуре на протяжении ночи. Раствор фильтровали и концентрировали. Остаток применяли в следующей стадии без дополнительной очистки. МС (ESI) 650[M+H]+.

f. 1-[6-(2-Амино-4-диметиламинометилфениламино)пиримидин-4-ил]-3-(2,6-дихлор-3,5-диметоксифенил)-1-метилмочевина
К перемешиваемому раствору 1-[6-(2-амино-4-диметиламинометилфениламино)пиримидин-4-ил]-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-(2-триметилсилaнилэтоксиметил)мочевины в безводном DCM (10 мл) добавляли TFA (10 мл) при комнатной температуре. Раствор перемешивали при комнатной температуре в течение 3 часов. Анализ ЖХ-МС показал пик основного продукта. Раствор выпаривали досуха, разбавляли DCM (40 мл) и промывали 10% насыщенным раствором Na2CO3 (10 мл). Слой DCM сушили над безводным Na2SO4. Концентрирование в вакууме давало сырой продукт, который очищали колоночной хроматографией на диоксиде кремния (10% MeOH/DCM с 0,5% Et3N), получая при этом указанное в заголовке соединение (45 мг, выход: 58% в двух стадиях). МС (ESI) 520[M+H]+.
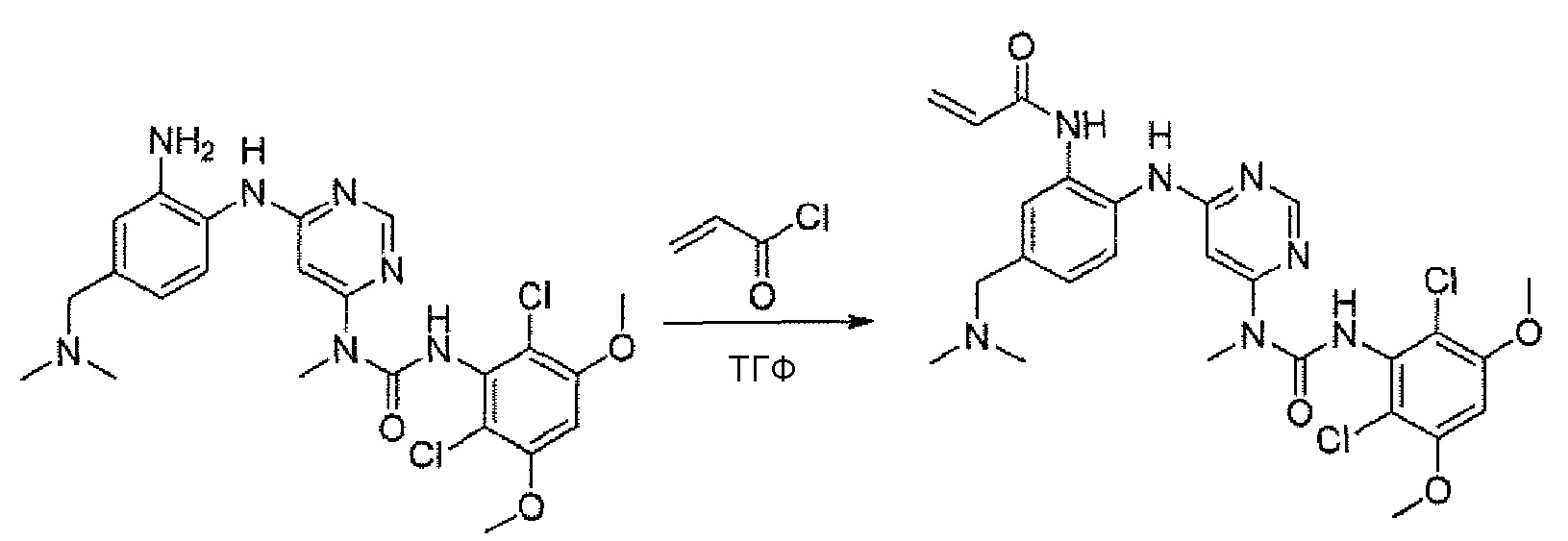
g. N-(2-{6-[3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо]пиримидин-4-иламино}-5-диметиламинометилфенил)акриламид
К перемешиваемому раствору 1-[6-(2-амино-4-диметиламинометилфениламино)пиримидин-4-ил]-3-(2,6-дихлор-3,5-диметоксифенил)-1-метилмочевины (45 мг, 0,11 ммоль) в ТГФ (40 мл) при -10°C добавляли акрилoилхлорид (30 мг, 0,33 ммоль) в ТГФ (3 мл). Раствор перемешивали при -10°C в течение 5 часов. Анализ ЖХ-MС показал пик основного продукта. Реакцию гасили MeOH (3 мл) и смесь упаривали. Остаток очищали преп-ВЭЖХ (вода/АСN в NH4HCO3), получая при этом указанное в заголовке соединение (6 мг, выход 15%).1H-ЯМР(400 МГц, MeOH-d4) δ 8,25 (с, 1H), 7,47 (д, 1H), 7,44 (с, 1H), 7,16 (дд, 1H), 6,70 (с, 1H), 6,33-6,20 (м, 3H), 5,67 (дд, 1H), 3,84 (с, 6H), 3,43 (с, 2H) 3,22 (с, 3H), 2,20 (м, 6H); МС (ESI): 574[M+H]+.
Пример – 114

N-(2-(6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-иламино)фенил)пропиоламид
Соединение синтезировали по способу, описанному в методике 2C (пример 108) с изменением стадии (g) следующим образом: к раствору 1-[6-(2-аминофениламино)пиримидин-4-ил]-3-(2,6-дихлор-3,5-диметоксифенил)-1-метилмочевины (50 мг, 0,108 ммоль) и DCC (46 мг, 0,22 ммоль) в хлороформе (50 мл) добавляли раствор пропиоловой кислоты (16 мг, 0,22 ммоль) в хлороформе (50 мл) при 0°C и образовавшуюся смесь перемешивали при комнатной температуре на протяжении ночи. Для гашения реакции добавляли воду (1 мл). Смесь концентрировали и остаток очищали на колонке с обращенной фазой и преп-ТСХ, получая при этом указанное в заголовке соединение (5 мг, выход 9,1%).1H ЯМР(400 МГц, ДМСО-d6) δ 12,04 (с, 1H), 10,36 (с, 1H), 9,03 (с, 1H), 8,45 (с, 1H), 7,67 (д, 1H), 7,59 (д, 1H), 7,30-7,25 (м, 2H), 6,96 (с, 1H), 6,50 (с, 1H), 4,42 (с, 1H), 4,00 (с, 6H), 3,35 (с, 3H); МС (ESI) 515 [M+H]+.
Пример – 116

N-(2-{6-[3-(2,6-Дихлор-3,5-диметоксифенил)-1-(2-метоксиэтил)уреидо]пиримидин-4-иламино}фенил)акриламид
Соединение синтезировали по способу, описанному в методике 2G (пример 123), с применением 1-(6-хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-(2-метоксиэтил)-3-(2-триметилсилaнилэтоксиметил)мочевины (получение, указанное ниже)в стадии (d), получая при этом указанное в заголовке соединение (40 мг, выход 16% после пяти стадий).1H ЯМР(400 МГц, ДМСО-d6) δ 11,28 (с, 1H), 9,71 (с, 1H), 8,85 (с, 1H), 8,38 (с, 1H), 7,70-7,68 (м, 1H), 7,55-7,53 (м, 1H), 7,20-7,18 (м, 2H), 6,89 (с, 1H), 6,69 (с, 1H), 6,50 (дд, 1H), 6,26 (д, 1H), 5,75 (д, 1H), 4,03 (т, 2H), 3,94 (с, 6H), 3,56 (т, 2H), 3,24 (с, 3H); МС (ESI) 437 [M+H]+.
Получение 1-(6-хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-(2-метоксиэтил)-3-(2-триметилсилaнилэтоксиметил)мочевины

a. (6-Хлорпиримидин-4-ил)-(2-метоксиэтил)амин
К раствору 4,6-дихлорпиримидина (2 г, 14 ммоль) в iPrOH (70 мл) и DIPEA (1,94 г, 15 ммоль) добавляли раствор 2-метоксиэтиламина (1,13 г, 15 ммоль) при комнатной температуре. Образовавшуюся смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли воду и смесь экстрагировали DCM. Объединенные экстракты промывали насыщенным раствором соли, сушили над безводным Na2SO4 и концентрировали, получая при этом сырой продукт, который очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (1,95 г, выход: 82%). MС (ESI): 188[M+H]+.
Схема 25

b.1-(6-Хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-(2-метоксиэтил)-3-(2-триметилсилaнилэтоксиметил)мочевина
К раствору (6-хлорпиримидин-4-ил)-(2-метоксэтил)амина (300 мг, 1,6 ммоль) в ДМФА (10 мл) добавляли NaH (60%, 96 мг, 2,4 ммоль) при 0°C и смесь перемешивали в течение 10 минут при комнатной температуре. По каплям при 0°C добавляли раствор 1-изоцианато-3,5-диметоксибензола (590 мг, 2,4 ммоль) в ДМФА (5 мл). Образовавшуюся смесь перемешивали в течение 30 минут. Добавляли SEMCl (400 мг, 2,4 ммоль) в ДМФА (2 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Для гашения реакции добавляли насыщенный водный раствор NH4Cl. Смесь разбавляли водой и экстрагировали EtOAc. Объединенные экстракты промывали водой и насыщенным раствором соли, сушили над безводным Na2SO4 и концентрировали, получая при этом сырой продукт, который очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (720 мг, выход 78%). МС (ESI) 565 [M+H]+.
Пример – 120

Соль N-(2-{6-[3-(2,6-дихлор-3,5-диметоксифенил)-1-(3-диметиламинопропил)уреидо]пиримидин-4-иламино}фенил)акриламида с трифторуксусной кислотой
Соединение синтезировали по способу, описанному в методике 2I (пример 142), с применением 2-нитроанилина и N-(3-диметиламинопропил)-N'-(2-нитрофенил)пиримидин-4,6-диамина (получен способом, описанным ниже) в стадии (c), получая при этом указанное в заголовке соединение (11 мг, выход 7,5% после шести стадий).1H ЯМР(300 МГц, ДМСО-d6) δ 11,30 (с, 1H), 9,80 (с, 1H), 9,28 (м, 1H), 8,92 (с, 1H), 8,42 (с, 1H), 7,64 (д, 1H), 7,55 (д, 1H), 7,24-7,16 (м, 2H), 6,90 (с, 1H), 6,53 (с, 1H), 6,49 (дд, 1H), 6,25 (д, 1H), 5,76 (д, 1H), 3,95 (с, 6H), 3,91-3,88 (м, 2H), 3,11-3,05 (м, 2H), 2,74 (д, 6H), 1,97-1,92 (м, 2H); МС (ESI) 588 [M+H]+.
Получение N-(3-диметиламинопропил)-N'-(2-нитрофенил)пиримидин-4,6-диамина

a. N'-(6-Хлорпиримидин-4-ил)-N,N-диметилпропан-1,3-диамин
К раствору 4,6-дихлорпиримидина (1 г, 6,7 ммоль) и DIPEA (1,03 г, 8 ммоль) в iPrOH (20 мл) добавляли N,N-диметилпропан-1,3-диамин (714 мг, 7 ммоль) при комнатной температуре. Образовавшуюся смесь перемешивали в течение 2 часов. Добавляли воду и смесь экстрагировали DCM. Объединенные экстракты промывали насыщенным раствором соли, сушили над Na2SO4 и концентрировали, получая при этом сырой продукт, который очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (1,15 г, выход 80%). МС (ESI) 215[M+H]+.
Схема 27

b.N-(3-Диметиламинопропил)-N'-(2-нитрофенил)пиримидин-4,6-диамин
Дегазированную смесь N'-(6-хлорпиримидин-4-ил)-N,N-диметилпропан-1,3-диамина (800 мг, 3,74 ммоль), нитроанилина (525 мг, 3,8 ммоль), Pd2(dba)3 (348 мг, 0,38 ммоль), Xantphos (438 мг, 0,76 ммоль) и Cs2CO3 (3,05 г, 9,35 ммоль) в толуоле (15 мл) нагревали при 100°C в течение 4 часов. Реакционную смесь концентрировали и остаток очищали хроматографией с обращенной фазой, получая при этом указанное в заголовке соединение (530 мг, выход 45%). МС 317 [M+H]+.
Пример – 121

N-[2-(6-{3-(2,6-Дихлор-3,5-диметоксифенил)-1-[3-(4-метилпиперазин-1-ил)пропил]уреидо}пиримидин-4-иламино)фенил]акриламид
Соединение синтезировали по способу, описанному в методике 2I (примерe 142), с применением 2-нитроанилина и N-[3-(4-метилпиперазин-1-ил)пропил]-N'-(2-нитрофенил)пиримидин-4,6-диамина (получение показано ниже) в стадии (e), получая при этом указанное в заголовке соединение (8 мг, выход 1,2% после шести стадий).1H ЯМР(300 МГц, метaнол-d4) δ 8,17 (с, 1H), 7,46 (д, 1H), 7,32 (д, 1H), 7,12-7,08 (м, 2H), 6,62 (с, 1H), 6,29-6,14 (м, 3H), 5,58 (д, 1H), 3,82 (т, 2H), 3,75 (с, 6H), 2,27-2,19 (м, 8H), 2,06 (с, 3H), 1,67 (т, 2H); МС (ESI) 643 [M+H]+.
Получение N-[3-(4-метилпиперазин-1-ил)пропил]-N'-(2-нитрофенил)пиримидин-4,6-диамина

a. (6-Хлорпиримидин-4-ил)-[3-(4-метилпиперазин-1-ил)пропил]амин
К раствору 4,6-дихлорпиримидина (1,5 г, 10 ммоль) и DIPEA (1,55 г, 12 ммоль) в iPrOH (50 мл) добавляли раствор 3-(4-метилпиперазин-1-ил)пропиламина (1,73 г, 11 ммоль) при комнатной температуре. Образовавшуюся смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли воду и смесь экстрагировали DCM. Объединенные экстракты промывали насыщенным раствором соли, сушили над безводным Na2SO4 и концентрировали, получая при этом указанное в заголовке соединение (1,4 г, 51%), которое применяли непосредственно в следующей стадии без дополнительной очистки. МС (ESI) 270[M+H]+.
Схема 28

b. N-[3-(4-Метилпиперазин-1-ил)пропил]-N'-(2-нитрофенил)пиримидин-4,6-диамин
Дегазированную смесь (6-хлорпиримидин-4-ил)-[3-(4-метилпиперазин-1-ил)пропил]амина (600 мг, 2,22 ммоль), нитроанилина (317 мг, 2,3 ммоль), Pd2(dba)3 (210 мг, 0,23 ммоль), Xantphos (265 мг, 0,46 ммоль) и Cs2CO3 (1,81 г, 5,55 ммоль) в толуоле (15 мл) нагревали при 100°C в течение 4 часов. Реакционную смесь концентрировали и остаток очищали хроматографией с обращенной фазой, получая при этом указанное в заголовке соединение (400 мг, выход 48%). МС (ESI) 372 [M+H]+.
Пример – 122

Соль N-(2-{6-[3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо]пиримидин-4-иламино}-5-пиперидин-4-илфенил)акриламида с трифторуксусной кислотой
Соединение синтезировали по способу, указанному в методике 2E (пример 111), заменяя стадии (e) и (f) следующей процедурой:крастворутрет-бутил-4-(3-амино-4-(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)уреидо)пиримидин-4-иламино)фенил)пиперидин-1-карбоксилата (методика 2E, пример 111, 65 мг, 0,084 ммоль) в ТГФ (15 мл) добавляли раствор акрилoилхлорида (10 мг/мл, 0,9 мл, 0,1 ммоль) по каплям при -10°C и реакционную смесь перемешивали при 0°C в течение 1 часа. Для гашения реакции добавляли MeOH (5 мл) и реакционную смесь концентрировали. Остаток растворяли в DCM (2 мл) и добавляли по каплям к смеси DCM/TFA (2:1, 3 мл). Смесь перемешивали при комнатной температуре в течение 1 часа и затем концентрировали. Остаток очищали преп-ВЭЖХ, получая при этом указанное в заголовке соединение (23 мг, выход 47%).1H ЯМР(300 МГц, ДМСО-d6) δ 11,92 (с, 1H), 9,64 (с, 1H), 8,98 (с, 1H), 8,60 (м, 1H), 8,36 (с, 1H), 8,25 (м, 1H), 7,66 (с, 1H), 7,51 (д, 1H), 7,05 (д, 1H), 6,90 (с, 1H), 6,52 (дд, 1H), 6,41 (с, 1H), 6,24 (д, 1H), 5,74 (д, 1H), 3,96 (с, 6H), 3,49 (д, 2H), 3,28 (с, 3H), 3,02 (кв., 2H), 2,85 (т, 1H), 1,96 (д, 2H), 1,78 (кв, 2H); МС (ESI) 600 [M+H]+.
Методика 2G: пример – 123
Схема 29

N-(2-{6-[3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо]пиримидин-4-иламино}-5-морфолин-4-илфенил)акриламид
Схема 30

a. Ди-трет-бутил-4-бром-2-нитрофенилкарбамат
Смесь 4-бром-2-нитроанилина(10 г, 46 ммоль), (Boc)2O (20,7 г, 95 ммоль) в ТГФ (250 мл) нагревали при кипячении с обратным холодильником на протяжении ночи. Смесь концентрировали, получая при этом указанное в заголовке соединение (19,2 г, выход колич.) которое применяли непосредственно в следующей стали без дополнительной очистки.

b.Ди-трет-бутиловый эфир(4-морфолин-4-ил-2-нитрофенил)карбаминовой кислоты
Дегазированную смесьтрет-бутил-4-бром-2-нитрофенилкарбамата(1 г, 2.4 ммоль), морфолина (314 мг, 3,6 ммоль), Pd2(dba)3 (220 мг, 0,24 ммоль), Xantphos (278 мг, 0,48 ммоль) и Cs2CO3 (1,56 г, 4,8 ммоль) в толуоле (30 мл) нагревали при 100°C в течение 1 часа. Реакционную смесь концентрировали и остаток очищали флэш-хроматографией на диоксиде кремния, получая при этом сырую смесь указанного заголовке соединения и моно-Boc-продукта (744 мг). Смесь применяли непосредственно в следующей стадии без дополнительной очистки. МС (ESI): 324 [M-Boc+H]+.

c. 4-Морфолин-4-ил-2-нитрофениламин
К раствору ди-трет-бутилового эфира (4-морфолин-4-ил-2-нитрофенил)карбаминовой кислоты и mono-Boc-продукта (744 мг) в DCM (20 мл) добавляли TFA (10 мл) при 0°C, образовавшуюся смесь перемешивали в течение 2 час при комнатной температуре. После удаления всех летучих компонентов в вакууме, остаток снова растворяли в DCM, нейтрализовали насыщенным водным раствором NaHCO3 и экстрагировали DCM. Объединенные экстракты концентрировали и остаток очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (290 мг, выход 54% после двух стадий). МС (ESI) 251[M+H]+.

d. 1-(2,6-Дихлор-3,5-диметоксифенил)-3-метил-3-[6-(4-морфолин-4-ил-2-нитрофениламино)пиримидин-4-ил]-1-(2-триметилсилaнилэтоксиметил)мочевина
Дегазированную смесь 4-морфолин-4-ил-2-нитрофениламина (290 мг, 1,3 ммоль), 1-(6-хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-(2-триметилсилaнилэтоксиметил)мочевины (методика 2E , стадия b; 624 мг, 1,2 ммоль), Pd2(dba)3 (110 мг, 0,12 ммоль), Xantphos (139 мг, 0,24 ммоль) и Cs2CO3 (782 мг, 2,4 ммоль) в толуоле (15 мл) нагревали при 100°C в течение 2,5 часа. Реакционную смесь концентрировали и остаток очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (440 мг, выход 49%) в виде красного твердого вещества. МС (ESI) 708[M+H]+.
Схема 31

e. трет-Бутиловый эфир {6-[3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-(2-триметилсилaнилэтоксиметил)уреидо]пиримидин-4-ил}-(4-морфолин-4-ил-2-нитрофенил)карбаминовой кислоты
Смесь 1-(2,6-дихлор-3,5-диметоксифенил)-3-метил-3-[6-(4-морфолин-4-ил-2-нитрофениламино)пиримидин-4-ил]-1-(2-триметилсилaнилэтоксиметил)мочевины (200 мг, 0,28 ммоль), (Boc)2O (93 мг, 0,42 ммоль) и каталитического количества DMAP в ТГФ (10 мл) нагревали при кипячении с обратным холодильником в течение 1 часа. Смесь концентрировали и остаток применяли для следующей стадии без дополнительной очистки. МС (ESI): 808[M+H]+.
Схема 32

f.трет-Бутиловый эфир(2-амино-4-морфолин-4-илфенил)-{6-[3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-(2-триметилсилaнилэтоксиметил)уреидо]пиримидин-4-ил}карбаминовой кислоты
К раствору трет-бутилового эфира {6-[3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-(2-триметилсилaнилэтоксиметил)уреидо]пиримидин-4-ил}-(4-морфолин-4-ил-2-нитрофенил)карбаминовой кислоты (неочищенного, полученного выше) в MeOH (20 мл) добавляли Ni Ренея (суспензия в воде) при комнатной температуре, полученную смесь перемешивали в течение 2 часов в атмосфере водорода (1 атм). Реакционную смесь фильтровали и концентрировали. Остаток промывали дважды MeOH, получая при этом указанный в заголовке продукт (160 мг, выход 70%). МС (ESI):778[M+H]+.

g.трет-Бутиловый эфир(2-акрилoиламино-4-морфолин-4-илфенил)-{6-[3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-(2-триметилсилaнилэтоксиметил)уреидо]пиримидин-4-ил}карбаминовой кислоты
К раствору трет-бутилового эфира (2-амино-4-морфолин-4-илфенил)-{6-[3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-(2-триметилсилaнилэтоксиметил)уреидо]пиримидин-4-ил}карбаминовой кислоты (80 мг, 0,103 ммоль) в DCM (5 мл) добавляли по каплям раствор TEA (10 мг/мл, 1,2 мл, 0,12 ммоль) и раствор акрилoилхлорида (10 мг/мл, 1 мл, 0,11 ммоль) при 0°C и образовавшуюся смесь перемешивали при комнатной температуре в течение 1 часа. Анализ ЖХ-МС показал, что реакция была завершена. Для гашения реакции добавляли воду (5 мл) и реакционную смесь экстрагировали DCM. Объединенные экстракты промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат выпаривали в вакууме, получая при этом сырой продукт, который применяли для следующей стадии без дополнительной очистки.

h. N-(2-{6-[3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо]пиримидин-4-иламино}-5-морфолин-4-илфенил)акриламид
К раствору трет-бутилового эфира (2-акрилoиламино-4-морфолин-4-илфенил)-{6-[3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-(2-триметилсилaнилэтоксиметил)уреидо]пиримидин-4-ил}карбаминовой кислоты (сырой продукт, получен выше) в CH2Cl2 (10 мл) добавляли TFA (3 мл) при 0°C, образовавшуюся смесь перемешивали в течение 1 часа при комнатной температуре. Реакционную смесь концентрировали и нейтрализовали NH3.H2O, получая при этом сырое соединение, которое очищали преп-ВЭЖХ, получая при этом указанное в заголовке соединение (10 мг, выход 16% в двух стадиях).1H ЯМР(300 МГц, ДМСО-d6) δ 12,05 (с, 1H), 9,60 (с, 1H), 8,74 (с, 1H), 8,32 (с, 1H), 7,33-7,30 (м, 2H), 6,89 (с, 1H), 6,83 (д, 1H), 6,48 (дд, 1H), 6,25-6,21 (м, 2H), 5,72 (д, 1H), 3,96 (с, 6H), 3,73 (ушир., 4H), 3,44 (с, 3H), 3,10 (ушир., 4H); МС (ESI) 602 [M+H]+.
Пример – 124

N-[2-{6-[3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо]пиримидин-4-иламино}-5-(3,5-диметилпиперазин-1-ил)фенил]акриламид
Соединение синтезировали по способу, описанному в методике 2G (пример 123), с применением 4-(3,5-диметилпиперазин-1-ил)-2-нитрофениламина (получение показано ниже) в стадии d), получая при этом указанное в заголовке соединение (26 мг, выход 30% после трех стадий).1H ЯМР(300 МГц, метaнол-d4) δ 8,32 (с, 1H), 7,36-7,33 (м, 2H), 6,95 (д, 1H), 6,80 (с, 1H), 6,43-6,37 (м, 2H), 6,15 (с, 1H), 5,77 (д, 1H), 3,95 (с, 6H), 3,70 (д, 2H), 3,29 (с, 3H), 3,18-3,13 (м, 2H), 2,45 (т, 2H), 1,24 (д, 6H); МС (ESI) 629 [M+H]+.
Получение 4-(3,5-Диметилпиперазин-1-ил)-2-нитрофениламина
Схема 33

a. Ди-трет-бутил-4-бром-2-нитрофенилкарбамат
Смесь 4-бром-2-нитроанилина(10 г, 46 ммоль), (Boc)2O (20,7 г, 95 ммоль) в ТГФ (250 мл) нагревали при кипячении с обратным холодильником на протяжении ночи. Смесь концентрировали, получая при этом указанное в заголовке соединение (19,2 г, колич.), которое применяли для следующей стадии без дополнительной очистки.

b.Ди-трет-бутиловый эфир[4-(3,5-диметилпиперазин-1-ил)-2-нитрофенил]карбаминовой кислоты
Дегазированную смесь ди-трет-бутил-4-бром-2-нитрофенилкарбамата(1 г, 2,4 ммоль), 2,6-диметилпиперазина (410 мг, 3,6 ммоль), Pd2(dba)3 (220 мг, 0,24 ммоль), Xantphos (278 мг, 0,48 ммоль) и Cs2CO3 (1,56 г, 4,8 ммоль) в толуоле (30 мл) нагревали при 100°C в течение 1 часа. Реакционную смесь концентрировали и остаток очищали флэш-хроматографией на диоксиде кремния, получая при этом смесь указанного в заголовке соединения и моно-Вос-продукта (600 мг). Смесь применяли непосредственно в следующей стадии без дополнительной очистки. МС (ESI) 350 [M-Boc+H]+.

c. 4-(3,5-Диметилпиперазин-1-ил)-2-нитрофениламин
К раствору ди-трет-бутилового эфира [4-(3,5-диметилпиперазин-1-ил)-2-нитрофенил]карбаминовой кислоты и моно-Boc-продукта(600 мг) в DCM (20 мл) добавляли TFA (10 мл) при 0°C, образовавшуюся смесь перемешивали в течение 3 часов при комнатной температуре. После удаления всех летучих компонентов в вакууме остаток снова растворяли в DCM, нейтрализовали насыщенным водным раствором NaHCO3 и экстрагировали DCM. Объединенные экстракты концентрировали и остаток очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (233 мг, выход 39% после двух стадий). МС (ESI) 251[M+H]+.
Пример - 125

N-[2-{6-[3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо]пиримидин-4-иламино}-5-(4-метансульфонилпиперазин-1-ил)фенил]акриламид
Соединение синтезировали по способу, описанному в методике 2G (пример 123), с применением 4-(4-метансульфонилпиперазин-1-ил)-2-нитрофениламина (получение показано ниже) в стадии (d), получая при этом указанное в заголовке соединение (15 мг, выход 9,6% в пяти стадиях).1H ЯМР(300 МГц, ДМСО-d6) δ 12,03 (с, 1H), 9,62 (с, 1H), 8,76 (с, 1H), 8,33 (с, 1H), 7,37-7,32 (м, 2H), 6,89-6,83 (м, 2H), 6,46 (дд, 1H), 6,26-6,21 (м, 2H), 5,72 (д, 1H), 3,93 (с, 6H), 3,34-3,16 (м, 11H), 2,93 (с, 3H); МС (ESI) 679 [M+H]+.
Получение 4-(4-метансульфонилпиперазин-1-ил)-2-нитрофениламина

a. Ди-трет-бутил-4-бром-2-нитрофенилкарбамат
Смесь 4-бром-2-нитроанилина(10 г, 46 ммоль), (Boc)2O (20,7 г, 95 ммоль) в ТГФ (250 мл) нагревали при кипячении с обратным холодильником на протяжении ночи. Смесь концентрировали, получая при этом указанное в заголовке соединение (19,2 г, колич.), которое применяли в следующей стадии без дополнительной очистки.

b.Ди-трет-бутиловый эфир[4-(4-метансульфонилпиперазин-1-ил)-2-нитрофенил]карбаминовой кислоты
Дегазированную смесьди-трет-бутил-4-бром-2-нитрофенилкарбамата(1 г, 2,4 ммоль), 1-метансульфонилпиперазина (590 мг, 3,6 ммоль), Pd2(dba)3 (220 мг, 0,24 ммоль), Xantphos (278 мг, 0,48 ммоль) и Cs2CO3 (1,56 г, 4,8 ммоль) в толуоле (30 мл) нагревали при 100°C в течение 1 часа. Реакционную смесь концентрировали и остаток очищали флэш-хроматографией на диоксиде кремния, получая при этом смесь указанного в заголовке соединения и моно-Boc-продукта (755 мг). Смесь применяли непосредственно в следующей стадии без дополнительной очистки. МС (ESI) 400 [M-Boc+H]+.

c. 4-(4-Метансульфонилпиперазин-1-ил)-2-нитрофениламин
К раствору ди-трет-бутилового эфира [4-(4-метансульфонилпиперазин-1-ил)-2-нитрофенил]карбаминовой кислоты и моно-Boc-продукта (755 мг) в DCM (20 мл) добавляли TFA (10 мл) при 0°C, полученную смесь перемешивали в течение 3 часов при комнатной температуре. После удаления всех летучих компонентов в вакууме остаток снова растворяли в DCM, нейтрализовали насыщенным водным раствором NaHCO3 и экстрагировали DCM. Объединенные экстракты концентрировали и остаток очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (290 мг, выход 40% в двух стадиях). МС (ESI) 301 [M+H]+.
Пример - 126

N-[2-{6-[3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо]пиримидин-4-иламино}-5-(3-диметиламинопропил)фенил]акриламид
Соединение синтезировали по способу, описанному в методике 2G (пример 123), с применением 4-(3-диметиламинопроп-1-инил)-2-нитрофениламина (получение показано ниже) в стадии (d) и оксида платины в стадии (f), получая при этом указанное в заголовке соединение (4 мг, выход 1,4% в пяти стадиях).1H-ЯМР(300 МГц, ДМСО-d6) δ 11,98 (с, 1H), 9,66 (д, 1H), 8,88 (с, 1H), 8,36 (с, 1H), 7,52 (д, 1H), 7,44 (д, 1H), 7,04 (дд, 1H), 6,90 (с, 1H), 6,48 (дд, 1H), 6,34 (с, 1H), 6,24 (дд, 1H), 5,72 (дд, 1H), 3,94 (с, 6H), 3,25 (с, 3H), 2,59 (т, 2H), 2,24 (т, 2H), 2,11 (с, 6H), 1,71 (м, 2H); МС (ESI) 602[M+H]+.
Получение 4-(3-диметиламинопроп-1-инил)-2-нитрофениламина
Схема 34

a. 4-(3-Диметиламинопроп-1-инил)-2-нитрофениламин
К перемешиваемому раствору 4-бром-2-нитрофениламина (1,08 г, 5 ммоль) и диметилпроп-2-иниламина (1,0 г, 12 ммоль) в TEA (20 мл) добавляли Pd(PPh3)2Cl2 (0,7 г, 1 ммоль) и CuI (360 мг, 2 ммоль). Раствор перемешивали при 60°C в атмосфере азота в течение 3 часов. Раствор упаривали с силикагелем и очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (1,1 г, чистота 70%). Указанное в заголовке соединение применяли непосредственно в следующей стадии без дополнительной очистки. МС (ESI) 221[M+H]+.
Пример - 127

N-[2-{6-[3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо]пиримидин-4-иламино}-5-(2-диметиламиноэтокси)фенил]акриламид
Соединение синтезировали по способу, описанному в методике 2G для соединения 123, с применением 4-(2-диметиламиноэтокси)-2-нитрофениламина (получение показано ниже) в стадии (d) и оксида платины в стадии (f), получая при этом указанное в заголовке соединение (4,3 мг, выход 8% после пяти стадий).1H-ЯМР(300 МГц, ДМСО-d6) δ 12,04 (с, 1H), 9,58 (с, 1H), 8,81 (с, 1H), 8,33 (с, 1H), 7,43 (д, 1H), 7,33 (д, 1H), 6,89 (с, 1H), 6,79 (дд, 1H), 6,51 (дд, 1H), 6,26~6,20 (м, 2H), 5,73 (дд, 1H), 4,04 (т, 2H), 3,96 (с, 6H), 3,27 (с, 3H), 2,67 (т, 2H), 2,24 (с, 6H); МС (ESI) 604[M+H]+.
Получение 4-(2-диметиламиноэтокси)-2-нитрофениламина
Схема 35

a. 4-(2-Диметиламиноэтокси)-2-нитрофениламин
К перемешиваемому раствору 4-амино-3-нитрофенoла (1,54 г, 10 ммоль) и хлоргидрата (2-хлорэтил)диметиламина (1,43 г, 10 ммоль) в бутaноне (40 мл) добавляли Cs2CO3(10 г, 30 ммоль) и NaI (150 мг, 1 ммоль). Раствор медленно нагревали до 80°C на протяжении одного часа. Затем раствор перемешивали при 80°C на протяжении 2 часов. Раствор фильтровали через целит® и промывали ацетоном. Раствор упаривали с силикагелем и очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (1,0 г, выход 45%) в виде коричневого твердого вещества.1H-ЯМР(300 МГц, ДМСО-d6) δ 7,38 (д, 1H), 7,24 (с, 2H), 7,16 (дд, 2H), 6,99 (д, 1H), 3,98 (т, 2H), 2,58 (т, 2H), 2,20 (с, 6H); МС (ESI) 226[M+H]+.
Пример - 128

(2-{6-[3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо]пиримидин-4-иламино}фенил)амид бут-2-иновой кислоты
Соединение синтезировали по способу, описанному в методике 2G (пример 123), с применением 2-нитрофениламина в стадии (d), опусканием стадии (e) и заменой стадии (g) следующей процедурой, указанной ниже, получая при этом указанное в заголовке соединение (7 мг, выход 4,8% после пяти стадий).1H ЯМР(300 МГц, ДМСО-d6) δ 11,97 (с, 1H), 10,07 (с, 1H), 8,89 (с, 1H), 8,39 (с, 1H), 7,60 (д, 1H), 7,53 (д, 1H), 7,24-7,15 (м, 2H), 6,90 (с, 1H), 6,44 (с, 1H), 3,93 (с, 6H), 3,29 (с, 3H), 2,22 (с, 3H); МС (ESI) 529 [M+H]+.

К раствору трет-бутилового эфира (2-аминофенил)-{6-[3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-(2-триметилсилaнилэтоксиметил)уреидо]пиримидин-4-ил}карбаминовой кислоты (50 мг, 0,075 ммоль) и DCC (42 мг, 0,2 ммоль)в DCM (50 мл) добавляли раствор бут-2-иновой кислоты (13 мг, 0,15 ммоль) в DCM (10 мл) при 80°С и полученную смесь перемешивали при комнатной температуре на протяжении ночи. Для гашения реакции добавляли воду (1 мл). Смесь концентрировали и остаток очищали хроматографией с обращенной фазой, получая при этом указанное в заголовке соединение (20 мг, выход 34%). MS (ESI) 659[M+H]+.
Соединение 129 синтезировали способом, аналогичным способу синтеза соединения 100.
Пример - 130

N-(2-{6-[1-Бензил-3-(2,6-дихлор-3,5-диметоксифенил)уреидо]пиримидин-4-иламино}фенил)акриламид
Соединение синтезировали по способу, описанному в методике 2G (пример 123), с применением 2-нитрофениламина и 1-бензил-1-(6-хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-3-(2-триметилсилaнилэтоксиметил)мочевины (получение указано ниже) в стадии (d), получая при этом указанное в заголовке соединение (26 мг, выход 20% после пяти стадий).1H ЯМР(300 МГц, ДМСО-d6) δ 12,27 (с, 1H), 9,68 (с, 1H), 8,90 (с, 1H), 8,39 (с, 1H), 7,69 (д, 1H), 7,33-7,19 (м, 6H), 6,92 (с, 1H), 6,45 (дд, 1H), 6,24 (д, 1H), 6,18 (с, 1H), 5,74 (д, 1H), 5,07 (с, 2H), 3,95 (с, 6H); MS (ESI) 593 [M+H]+.
Получение 1-бензил-1-(6-хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-3-(2-триметилсилaнилэтоксиметил)мочевины

a. Бензил-(6-хлорпиримидин-4-ил)амин
К раствору 4,6-дихлорпиримидина (1,5 г, 10 ммоль) в iPrOH (40 мл) и DIPEA (1,55 г, 12 ммоль) добавляли раствор бензиламина (1,28 г, 12 ммоль) при комнатной температуре. Полученную смесь перемешивали при комнатной температуре в течение 2,5 часа. Добавляли воду и смесь экстрагировали DCM. Объединенные экстракты промывали насыщенным раствором соли, сушили над безводным Na2SO4 и концентрировали, получая при этом сырой продукт, который очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (1,5 г, выход 68%) в виде белого твердого вещества.1H ЯМР(300 МГц, CDCl3) δ 8,35 (ушир. с, 1H), 7,20-7,38 (м, 5 H), 6,35 (с, 1H), 4,52 (с, 2H); МС (ESI) 220[M+H]+.
Схема 36

b.1-Бензил-1-(6-хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-3-(2-триметилсилaнилэтоксиметил)мочевина
К раствору бензил-(6-хдорпиримидин-4-ил)амина (800 мг, 3,64 ммоль) в ДМФА (15 мл) добавляли NaH (60%, 218 мг, 5,45 ммоль) при 0°С и смесь перемешивали в течение 10 минут при комнатной температуре. При 0°С по каплям добавляли раствор 1-изоцианато-3,5-диметоксибензол (методика 2A , стадии a-d; 1,35 г, 5,45 ммоль) в ДМФА (2 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Для гашения реакции добавляли насыщенный водным раствор NH4Cl (2 мл). Смесь концентрировали и экстрагировали DCM. Объединенные экстракты промывали насыщенным водным раствором соли, сушили над безводным Na2SO4 и концентрировали, получая при этом сырой продукт, который концентрировали флэш-хроматографией на силикагеле, получая при этом сырой продукт, который очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (1,7 g, выход 77%). MS (ESI) 599[M+H]+.
Пример - 131

N-(2-{6-[3-(2,6-Дихлор-3,5-диметоксифенил)-1-пиридин-2-илметилуреидо]пиримидин-4-иламино}фенил)акриламид
Соединение синтезировали по способу, описанному в методике 2G (пример 123) с применением 2-нитрофениламина и 1-(6-хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-пиридин-2-илметил-3-(2-триметилсилaнилэтоксиметил)мочевины (получение показано ниже) в стадии (d), получая при этом указанное в заголовке соединение (35 мг, выход 13% после пяти стадий).1H ЯМР(300 МГц, ДМСО-d6) δ 12,15 (с, 1H), 9,65 (с, 1H), 8,83 (с, 1H), 8,50 (с, 1H), 8,39 (с, 1H), 7,80 (т, 1H), 7,66 (д, 1H), 7,31-7,25 (м, 3H), 7,17 (т, 1H), 7,09 (т, 1H), 6,91 (с, 1H), 6,45 (дд, 1H), 6,39 (с, 1H), 6,23 (д, 1H), 5,74 (д, 1H), 5,17 (с, 2H), 3,94 (с, 6H); МС (ESI) 594 [M+H]+.
Получение 1-(6-хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-пиридин-2-илметил-3-(2-триметилсилaнилэтоксиметил)мочевины

a. (6-Хлорпиримидин-4-ил)пиридин-2-илметиламин
К раствору 4,6-дихлорпиримидина (1 г, 7 ммоль) в iPrOH (40 мл) и DIPEA (1,16 г, 9 ммоль) добавляли раствор 2-пиридинилметaнамина (970 мг, 9 ммоль) при комнатной температуре. Полученную смесь перемешивали при комнатной температуре в течение 2,5 часа. Добавляли воду и смесь экстрагировали DCM. Объединенные экстракты промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали, получая при этом сырой продукт, который очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (1,2 г, выход 78%). МС (ESI) 221[M+H]+.

b.1-(6-Хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-пиридин-2-илметил-3-(2-триметилсилaнилэтоксиметил)мочевина
К раствору (6-хлорпиримидин-4-ил)пиридин-2-илметиламина (200 мг, 0,91 ммоль) в ДМФА (5 мл) добавляли NaH (60%, 55 мг, 1,37 ммоль) при 0°С и смесь перемешивали в течение 10 минут при комнатной температуре. При 0°С по каплям добавляли раствор 1-изоцианато-3,5-диметоксибензола (методика 2A, стадии a-d; 337 мг, 1,37 ммоль) в ДМФА (2 мл). Образовавшуюся смесь перемешивали в течение 30 минут. Добавляли SEMCl (230 мг, 1,37 ммоль) в ДМФА (2 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Для гашения реакции добавляли насыщенный водный раствор NH4Cl. Смесь разбавляли водой и экстрагировали EtOАс. Объединенные экстракты промывали водой и насыщенным раствором соли, сушили над безводным Na2SO4 и фильтровали. Фильтрат концентрировали, получая при этом сырой продукт, который очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (420 мг, выход 78%). МС (ESI) 598 [M+H]+.
Пример - 132

N-(2-{6-[3-(2,6-Дихлор-3,5-диметоксифенил)-1-пиридин-4-илметилуреидо]пиримидин-4-иламино}фенил)акриламид
Соединение синтезировали способом, указанным в методике 2G (пример 123), с применением 3-(2,6-дихлор-3,5-диметокси-фенил)-1-[6-(2-нитрофениламино)пиримидин-4-ил]-1-пиридин-4-илметилмочевины в стадии (e), получая при этом указанное в заголовке соединение (14 мг, выход 8,1%).1H ЯМР(300 МГц, ДМСО-d6) δ 12,03 (с, 1H), 9,68 (с, 1H), 8,92 (с, 1H), 8,49 (д, 2H), 8,39 (с, 1H), 7,67 (д, 1H), 7,27-7,17 (м, 4H), 7,10 (т, 1H), 6,92 (с, 1H), 6,48 (дд, 1H), 6,24 (д, 1H), 6,15 (с, 1H), 5,74 (д, 1H), 5,09 (с, 2H), 3,94 (с, 6H); МС (ESI) 594 [M+H]+.
Получение 3-(2,6-дихлор-3,5-диметоксифенил)-1-[6-(2-нитрофениламино)пиримидин-4-ил]-1-пиридин-4-илметилмочевины

a. (6-Хлорпиримидин-4-ил)пиридин-4-илметиламин
К раствору 4,6-дихлорпиримидина (1,5 г, 10,5 ммоль) и DIPEA (1,62 г, 12,6 ммоль) в iPrOH (40 мл) добавляли 4-пиридинилметaнамин (1,2 г, 11 ммоль) при комнатной температуре. Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли воду и смесь экстрагировали DCM. Объединенные экстракты промывали насыщенным раствором соли, сушили над безводным Na2SO4 и концентрировали, получая при этом сырой продукт, который очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (1,8 г, выход 80%). МС (ESI) 221[M+H]+.
Схема 37

b.N-(2-Нитрофенил)-N'-пиридин-4-илметилпиримидинe-4,6-диамин
Дегазированную смесь (6-хлорпиримидин-4-ил)пиридин-4-илметиламина(500 мг, 2,27 ммоль), 2-нитроанилина (317 мг, 2,3 ммоль), Pd2(dba)3 (200 мг, 0,22 ммоль), Xantphos (253 мг, 0,44 ммоль) и Cs2CO3 (1,48 г, 9,35 ммоль) в толуоле (10 мл) нагревали при 100°С в течение 3 часов. Реакционную смесь концентрировали и остаток очищали хроматографией с обращенной фазой с последующей флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (330 мг, выход 45%). МС (ESI) 323 [M+H]+.

c. 3-(2,6-Дихлор-3,5-диметоксифенил)-1-[6-(2-нитрофениламино)пиримидин-4-ил]-1-пиридин-4-илметилмочевина
К раствору N-(2-нитрофенил)-N'-пиридин-4-илметилпиримидин-4,6-диамина (330 мг, 1,02 ммоль) в ДМФА (10 мл) добавляли NaH (60%, 56 мг, 1,4 ммоль) при 0°С и смесь перемешивали в течение 30 минут при комнатной температуре. При 0°С по каплям добавляли раствор 1-изоцианато-3,5-диметоксибензола (методика 2A, стадии a-d; 345 мг, 1,4 ммоль) в ДМФА (2 мл). Образовавшуюся смесь перемешивали при комнатной температуре в течение 2 часов. Для гашения реакции добавляли насыщенный водный раствор NH4Cl (2 мл). Смесь концентрировали и экстрагировали DCM. Объединенные экстракты промывали насыщенным раствором соли, сушили над безводным Na2SO4 и концентрировали, получая при этом сырой продукт, который очищали преп-ТСХ, получая при этом указанное в заголовке соединение (190 мг, выход 33%). МС (ESI) 570[M+H]+.
Пример - 133

Получение 5-(4-этилпиперазин-1-ил)-2-нитрофениламина
Схема 38

1. 5-(4-Этилпиперазин-1-ил)-2-нитрофениламин
Смесь 1-этилпиперазина (1,2 мл, 9,6 ммоль), 5-фтор-2-нитрофениламина (1 г, 6,4 ммоль), DIPEA (1,24 г, 9,6 ммоль) в ДМФА (15 мл) нагревали при 80°С на протяжении ночи. Реакционную смесь выливали в ледяную воду и экстрагировали EtOАс. Объединенные экстракты промывали насыщенным раствором соли, сушили над безводным Na2SO4 и концентрировали, получая при этом сырой продукт, который очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (1 г, выход 63%). ESI-МС 251 [M+H]+.
Соль N-[2-{6-[3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо]пиримидин-4-иламино}-4-(4-этилпиперазин-1-ил)фенил]акриламида с трифторуксусной кислотой
Соединение синтезировали по способу, описанному в методике 2G (пример 123), с применением 5-(4-этилпиперазин-1-ил)-2-нитрофениламина в стадии(a), получая при этом указанное в заголовке соединение (20 мг, выход 39%).1H ЯМР(300 МГц, метaнол-d4) δ 8,37 с, 1H), 7,49 (d, 1H), 7,25 (с, 1H), 6,98 (д, 1H), 6,82 (с, 1H), 6,43-6,38 (м, 3H), 5,78 (д, 1H), 3,96-3,88 (м, 8H), 3,68-3,64 (м, 2H), 3,37 (с, 3H), 3,33-3,08 (м, 6H), 1,40 (т, 3H); МС (ESI) 629 [M+H]+.
Пример – 135

N-[2-{6-[3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо]пиримидин-4-иламино}-5-(4-этилпиперазин-1-илметил)фенил]акриламид
Соединение синтезировали по способу, описанному в методике 2G (пример 123), с применением 4-(4-этилпиперазин-1-илметил)-2-нитрофениламина (получение показано ниже) в стадии (d), получая при этом указанное в заголовке соединение (30 мг, выход 31% после пяти стадий).1H ЯМР(300 МГц, ДМСО-d6) δ 11,97 (s, 1H), 9,71 (с, 1H), 8,95 (с, 1H), 8,37 (с, 1H), 7,60 (с, 1H), 7,50 (д, 1H), 7,12 (д, 1H), 6,90 (с, 1H), 6,50 (дд, 1H), 6,39 (с, 1H), 6,25 (д, 1H), 5,73 (д, 1H), 3,93 (с, 6H), 3,48 (с, 2H), 3,32 (с, 3H), 2,50-2,25 мm, 10H), 0,98 (т, 3H); Mс (ESI) 643 [M+H]+.
Получение 4-(4-этилпиперазин-1-илметил)-2-нитрофениламина

a. 4-(4-Этилпиперазин-1-илметил)-2-нитрофениламин
К перемешиваемому раствору 1-этилпиперазина (1,37 г, 12 ммоль) в MeOH (30 мл) добавляли Ti(OiPr)4 (1,73 г, 6 ммоль). Затем раствор перемешивали при комнатной температуре при комнатной в течение 15 мин. Затем добавляли 4-амино-3-нитро-бензальдегид (методика 2F, стадии a-b, 1,5 г, 9 ммоль) в MeOH (10 мл) и раствор перемешивали при комнатной температуре на протяжении ночи. Затем добавляли NaBH4 (380 мг, 10 ммоль) и раствор перемешивали при комнатной температуре в течение 1 часа. Раствор разбавляли EtOАс и фильтровали. Остаток промывали водой и насыщенным раствором соли, сушили над безводным Na2SO4. Концентрирование давало продукт, который очищали флэш-хроматографией на силикагеле, получая при этом указанное в заголовке соединение (800 мг, выход 34%). МС (ESI) 265 [M+H]+.
Пример - 136

N-(2-{6-[3-(2,6-Дихлор-3,5-диметоксифенил)-1-(3-метилизоксазол-5-илметил)уреидо]пиримидин-4-иламино}фенил)акриламид
Соединение синтезировали по способу, описанному в методике 2G (примерe 123), с применением 1-(6-хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-(3-метилизоксазол-5-илметил)-3-(2-триметилсилaнилэтоксиметил)мочевины (получение показано ниже) в стадии (d) и смеси железо/уксусная кислота при 60°С в стадии (f), получая при этом указанное в заголовке соединение (26 мг, выход 12% в пяти стадиях).1H ЯМР(300 МГц, ДМСО-d6) δ 11,78 (c, 1H), 9,72 (с, 1H), 8,97 (с, 1H), 8,40 (с, 1H), 7,66 (д, 1H), 7,45 (д, 1H), 7,21-7,15 (м, 2H), 6,90 (с, 1H), 6,52-6,43 (м, 2H), 6,26 (д, 1H), 6,09 (с, 1H), 5,74 (д, 1H), 5,16 (с, 2H), 3,93 (с, 6H), 2,18 (с, 3H); МС (ESI): 598 [M+H]+.
Получение 1-(6-хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-(3-метилизоксазол-5-илметил)-3-(2-триметилсилaнилэтоксиметил)мочевины

a. (6-Хлорпиримидин-4-ил)-(3-метилизоксазол-5-илметил)амин
К раствору 4,6-дихлорпиримидина (660 мг, 4,46 ммоль) в iPrOH (40 мл) и DIEA (690 мг, 5,35 ммоль) добавляли раствор C-пиридин-2-илметиламина (560 мг, 5 ммоль) при комнатной температуре. Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли воду и смесь экстрагировали DCM. Объединенные экстракты промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали в вакууме, получая при этом сырой продукт. Сырой продукт очищали флэш-хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (650 мг, выход 65%). МС (ESI) 225[M+H]+.

b.1-(6-Хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-(3-метилизоксазол-5-илметил)-3-(2-триметилсилaнилэтоксиметил)мочевина
К раствору (6-хлорпиримидин-4-ил)-(3-метилизоксaзол-5-илметил)амина (300 мг, 1,34 ммоль) в ДМФА (5 мл) добавляли NaH (60%, 80 мг, 2 ммоль) при 0°С и смесь перемешивали в течение 10 минут при комнатной температуре. При 0°С по каплям добавляли раствор 1-изоцианато-3,5-диметоксибензола (методика 2A, стадии a-d, 337 мг, 1,37 ммоль) в ДМФА (2 мл). Реакционную смесь перемешивали в течение 0,5 часа. Добавляли SEMCl (230 мг, 1,37 ммоль) в ДМФА (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Для гашения реакции добавляли насыщенный водный раствор NH4Cl. Смесь разбавляли водой и экстрагировали EtOАс. Объединенные экстракты промывали водой и насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали в вакууме, получая при этом сырой продукт, который очищали флэш-хроматографией на диоксиде кремния, получая при этом указанный в заголовке продукт (440 мг, выход 55%). МС (ESI) 604 [M+H]+.
Пример - 137

Соль N-[2-{6-[3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо]пиримидин-4-иламино}-4-(2-диметиламиноэтил)фенил]акриламида с TFA
Соединение синтезировали по способу, указанному в методике 2G, с заменой 5-(2-диметиламиноэтил)-2-нитрофениламина (получен способом, указанным ниже) в стадии (d), получая при этом указанное в заголовке соединение (39 мг, выход 29%) в виде соли с TFA.1H ЯМР(300 МГц, ДМСО-d6) δ 11,86 (с, 1H), 9,76-9,70 (м, 2H), 9,02 (с, 1H), 8,40 (с, 1H), 7,86 (д, 1H), 7,51 (с, 1H), 7,12 (д, 1H), 6,90 (с, 1H), 6,57-6,48 (м, 2H), 6,24 (д, 1H), 5,74 (д, 1H), 3,94 (с, 6H), 3,35-3,28 (м, 5H), 2,98-2,92 (м, 2H), 2,79 (с, 6H); МС (ESI) 588 [M+H]+.
Получение 5-(2-диметиламиноэтил)-2-нитрофениламина

a. [2-(3-Фтор-4-нитрофенил)винил]диметиламин
Смесь 2-фтор-4-метил-1-нитробензола (3 г, 19,3 ммоль), диметилацеталя N,N-диметилформамида (10 мл) и 3 мл ДМФА (30 мл) нагревали при 125°С в течение 1 часа. Смесь охлаждали и концентрировали при пониженном давлении, получая при этом темно-красное твердое вещество. Растирание с гексанами давало указанный в заголовке продукт (2,5 г, выход 63%). МС (ESI) 211[M+H]+.

b. [2-(3-Фтор-4-нитрофенил)этил]диметиламин
К раствору [2-(3-фтор-4-нитрофенил)винил]диметиламина (1,7 г, 8 ммоль) в MeOH добавляли NaBH3CN (770 мг, 12 ммоль) и одну каплю АсOH. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и гасили водой. После удаления всех летучих компонентов в вакууме остаток экстрагировали 10% метанолом в DCM дважды. Объединенные экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия. Концентрирование в вакууме давало сырой продукт, который очищали колонкой с обращенной фазой, получая при этом указанное в заголовке соединение (1,08 г, выход 63%). МС (ESI) 213[M+H]+.

c. 5-(2-Диметиламиноэтил)-2-нитрофениламин
К раствору [2-(3-фтор-4-нитрофенил)этил]диметиламина (800 мг, 3,76 ммоль) в MeOH (20 мл) добавляли гидроксид аммония (5 мл). Реакционную смесь нагревали при 100°С микроволновым излучением в течение 1,5 часаs. Полученное желтое твердое вещество собирали и промывали водой, сушили в вакууме и комбинировали с предыдущей порцией, получая при этом чистое твердо соединение (560 мг, выход 55%).МС (ESI) 210[M+H]+.
Методика 2H: пример – 139
Схема 39

N-(2-((6-(3-(3,5-Диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид
Схема 40

a. 1-(6-Хлорпиримидин-4-ил)-3-(3,5-диметоксифенил)-1-метилмочевина
К перемешиваемому раствору 6-хлор-N-метилпиримидин-4-амина (1 г, 6,965 ммоль) в диоксaне (10 мл) добавляли DIPEA (3,6 мл, 20,895 ммоль) и трифосген (0,81 г, 2,786 ммоль) в атмосфере аргона при 0°С. Полученную смесь перемешивали в течение 1 час при 70°С и затем давали ей возможность охладиться до комнатной температуры. Полученную смесь добавляли через канюлю к раствору 3,5-диметоксианилина (1,2 г, 8,358 ммоль) и DIPEA (1,2 мл, 6,965 ммоль) в диоксaне (4 мл) в атмосфере аргона при 0°С. Реакционной смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 12 час. После завершения реакции по данным ТСХ (гексaны:EtOАс, 7:3), реакционную смесь разбавляли этилацетатом и насыщенным водным раствором NaHCO3. Водный слой отделяли и экстрагировали этилацетатом (2×50 мл). Объединенную органическую фазу промывают насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (гексaны:EtOАс, 70:30), получая при этом 1-(6-хлорпиримидин-4-ил)-3-(3,5-диметоксифенил)-1-метилмочевину (1,1 г, выход 50%) в виде белого твердого вещества.1H ЯМР (CDCl3, 300 МГц): δ 12,38 (с, 1H), 8,71 (с, 1H), 6,99 (с, 1H), 6,79 (д, 2H), 6,26 (т, 1H), 3,81 (с, 6H), 3,45 (с, 3H); МС (ESI) 323,10 [M+H]+.

b. 1-(6-Хлорпиримидин-4-ил)-3-(3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтмочевина
NaH (0,12 г, 3,098 ммоль) добавляли к перемешиваемому раствору 1-(6-хлорпиримидин-4-ил)-3-(3,5-диметоксифенил)-1-метилмочевины (0,5 г, 1,549 ммоль) в безводном ДМФА (4 мл) в атмосфере аргона при 0°С. Образовавшуюся смесь перемешивали в течение 15 мин и добавляли ди-трет-бутилдикарбонaт (0,50 мл, 2,323 ммоль) при 0°С. Полученной реакционной смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 16 час. Реакционную смесь разбавляли этилацетатом и избытком холодной воды. Водный слой отделяли и экстрагировали этилацетатом (3x25 мл). Объединенную органическую фазу промывали насыщенным раствором соли, сушили над N2SO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (гексаны:EtOАc, 80:20), получая при этом указанное в заголовке соединение (0,45 г, выход 69%) в виде белого твердого вещества.1H ЯМР (CDCl3, 300 МГц): δ 8,72 (д, 1H), 8,78 (д, 1H), 6,43-6,37 (м, 3H), 3,77 (с, 6H), 3,49 (с, 3H), 1,40 (с, 9H); МС (ESI) 424,10 [M+2]+.
1H ЯМР (CDCl3, 300 МГц): δ 8,72 (д, 1H), 8,78 (д, 1H), 6,43-6,37 (м, 3H), 3,77 (с, 6H), 3,49 (с, 3H), 1,40 (с, 9H); МС (ESI) 424,10 [M+2]+.

c. 1-(3,5-Диметоксифенил)-3-(6-((4-(4-этилпиперазин-1-ил)-2-нитрофенил)амино)пиримидин-4-ил)-3-метил-1-трет-бутилкарбонaтмочевина
Pd2(dba)3(0,095 г, 0,104 ммоль) и Xantphos (0,1202 г, 0,208 ммоль) помещали в 10 мл сухого толуола в герметичной трубке в атмосфере аргона при комнатной температуре. Аргон продолжали пропускать в течение дополнительных 5-10 мин. Затем добавляли 1-(6-хлорпиримидин-4-ил)-3-(3,5-диметоксифенил)-1-диметил-3-трет-бутилкарбонaтмочевину(0,43 г, 1,042 ммоль) и 4-(4-этилпиперазин-1-ил)-2-нитроанилин (методика 2C, стадии а – c; 0,317 г, 1,25 ммоль) и через полученную реакционную смесь пропускали газообразный аргон в течение 5 мин и затем добавляли Cs2CO3 (0,676 г, 2,08 ммоль). Пропускание газообразного аргона продолжали в течение дополнительных 5 мин перед герметизацией реакционного сосуда. Затем реакционную смесь нагревали при 100°С в течение 12 час. После завершения реакции по анализу ТСХ (DCM:MeOH, 98:2), реакционную массу распределяли между EtOАс и водой. Водный слой экстрагировали EtOАс (2×25 мл) и объединенный органический слой промывали водой, насыщенным раствором соли, сушили над Na2SO4 и упаривали в вакууме. Сырой остаток очищали колоночной хроматографией на силикагеле (DCM:MeOH/97:3), получая при этом указанное в заголовке соединение (0,250 г, выход 37%).1H ЯМР (CDCl3, 300 МГц): δ 9,30 (с, 1H), 8,58 (с, 1H), 8,17 (д, 1H), 7,59 (д, 1H), 7,14 (с, 1H), 7,08 (дд, 1H), 6,42 (д, 2H), 6,36 (т, 1H), 3,75 (с, 6H), 3,49 (с, 3H), 3,23 (т, 4H), 2,62 (т, 4H), 2,49 (кв, 2H), 1,39 (с, 9H), 1,14 (т, 3H); МС (ESI) 637,4 [M+H]+.

d. трет-Бутил-(6-(3-(3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтуреидо)пиримидин-4-ил)(4-(4-этилпиперазин-1-ил)-2-нитрофенил)карбамат
NaH (0,0314 г, 0,786 ммоль) добавляли к перемешиваемому раствору 1-(3,5-диметоксифенил)-3-(6-((4-(4-этилпиперазин-1-ил)-2-нитрофенил)амино)пиримидин-4-ил)-3-метил-1-трет-бутилкарбонaтмочевины(0,25 г, 0,393 ммоль) в безводном ДМФА (4 мл) в атмосфере аргона при 0°С. Полученную смесь перемешивали в течение 15 мин. Затем при 0°С добавляли ди-трет-бутилдикарбонaт (0,12 мл, 0,589 ммоль). Полученной реакционной смеси давали возможность нагреться добавляли комнатной температуры и перемешивали в течение 18 час. Реакционную смесь разбавляли этилацетатом и холодной водой. Водный слой отделяли и экстрагировали (3×20 мл). Объединенную органическую фазу промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (DCM:MeOH, 97:3), получая при этом указанное в заголовке соединение (0,280 г, выход 96%) в виде коричневого твердого вещества.1H ЯМР (CDCl3, 300 МГц): δ 8,44 (с, 1H), 8,30 (с, 1H), 7,60 (д, 1H), 7,14-7,03 (м, 2H), 6,48 (д, 1H), 6,44-6,21 (м, 2H), 3,75 (с, 6H), 3,49 (с, 3H), 3,49 (т, 4H), 2,62 (т, 4H), 2,49 (кв, 2H), 1,38 (д, 18H), 1,14 (т, 3H); МС (ESI) 737,5 [M+H]+.
Схема 42

e. трет-Бутил-(2-амино-4-(4-этилпиперазин-1-ил)фенил)(6-(3-(3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтуреидо)пиримидин-4-ил)карбамат
Смесь трет-бутил-(6-(3-(3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтуреидо)пиримидин-4-ил)(4-(4-этилпиперазин-1-ил)-2-нитрофенил)карбамата (0,280 г, 0,380 ммоль) и никеля Ренея (0,05 г) в смеси МeOH и ТГФ (1:1) (10 мл) перемешивали в течение 36 час при комнатной температуре в атмосфере водорода (баллон). Реакционную смесь фильтровали через подушку целита. Фильтрат концентрировали, получая при этом указанное в заголовке соединение (0,14 г, выход 52%). МС (ESI) 707,7 [M+H]+.
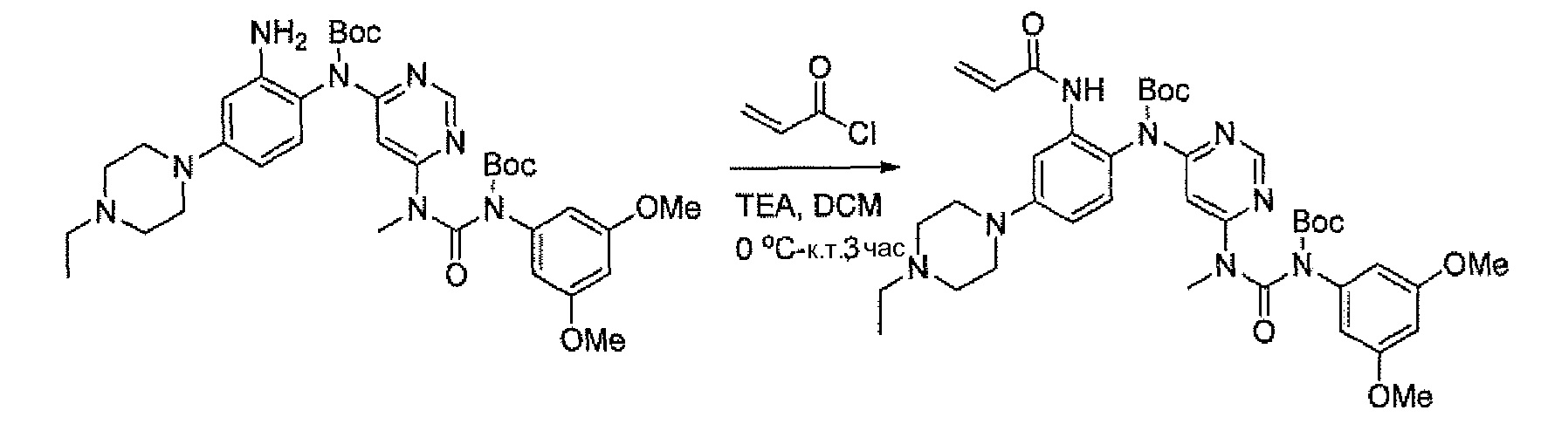
f. трет-Бутил-(2-акриламидо-4-(4-этилпиперазин-1-ил)фенил)(6-(3-(3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтуреидо)пиримидин-4-ил)карбамат
К перемешиваемому раствору трет-бутил-(2-амино-4-(4-этилпиперазин-1-ил)фенил)(6-(3-(3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтуреидо)пиримидин-4-илкарбамата(0,14 г, 0,183 ммоль) в безводном DCM (5 мл) добавляли TEA (0,08 мл, 0,594 ммоль) в атмосфере аргона при 0°С. Полученную смесь перемешивали в течение 15 мин и медленно добавляли акрилoилхлорид (0,03 мл, 0,396 ммоль) при 0°С. Полученной реакционной смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 3 час. Реакционную смесь разбавляли DCM и водой. Водный слой отделяли и экстрагировали DCM (3×20 мл). Органическую фазу промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (DCM:MeOH, 97:3), получая при этом указанное в заголовке соединение (0,070 г, выход 46%) в виде коричневого твердого вещества. МС (ESI) 761,4 [M+H]+.

g.N-(2-((6-(3-(3,5-Диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид
TFA (0,35 мл, 5 об.) медленно добавляли к перемешиваемому раствору трет-бутил-(2-акриламидо-4-(4-этилпиперазин-1-ил)фенил)(6-(3-(3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтуреидо)пиримидин-4-ил)карбамата(0,070 г, 0,124 ммоль) в сухом DCM (2 мл) в атмосфере аргона при 0°С. Полученной реакционной смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 24 час. Мониторинг реакции проводили ЖХ-МС, после завершения реакции избыточные растворителей удаляли при пониженном давлении. Полученный остаток разбавляли DCM и насыщенным водным раствором NaHCO3. Водный слой отделяли и экстрагировали DCM (3×10 мл). Органическую фазу промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (DCM/MeOH, 97:3), получая при этом 45 мг требуемого продукта с чистотой по данным ВЭЖХ 80%, который очищали препаративной ВЭЖХ (условия: колонка XBRIDGE-C18 (19,0×150 мм, 5 микрон); (подвижная фаза: A; 0,1% TFA в воде, B; АCN), получая при этом указанное в заголовке соединение (19 мг, выход 37%) в виде не совсем белого твердого вещества.1H ЯМР (CDCl3, 300 МГц): δ 12,85 (с, 1H), 8,39 (с, 1H), 7,72 (с, 1H), 7,55 (с, 1H), 7,19 (д, 1H), 6,79 (с, 2H), 6,75 (д, 1H), 6,64 (с, 1H), 6,39 (д, 1H), 6,22-6,13 (м, 2H), 5,80-5,73 (м, 2H), 3,78 (с, 6H), 3,28 (т, 4H), 3,21 (с, 3H), 2,60 (т, 4H), 2,47 (кв, 2H), 1,12 (т, 3H); МС (ESI) 561,60 [M+H]+; ВЭЖХ 96,04%, rt 6,40 мин.
Пример - 140

N-(2-((6-(3-(2,6-Дихлорфенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид
Соединение синтезировали по способу, указанному в методике 2H (пример 139), с применением 2,6-дихлоранилина в стадии (a) и (2-(хлорметокси)этил)триметилсилaна в стадии b), получая при этом указанное в заголовке соединение (18 мг, выход 3,2%) в виде не совсем белого соединения.1H ЯМР (CDCl3, 300 МГц): δ 12,58 (с, 1H), 8,39 (с, 1H), 7,75 (с, 1H), 7,61 (с, 1H), 7,37 (д, 2H), 7,22 (д, 1H), 7,14 (т, 1H), 6,77 (дд, 1H), 6,67 (с, 1H), 6,42 (д, 1H), 6,25-6,16 (м, 1H), 5,85 (с, 1H), 5,78 (д, 1H), 3,35-3,24 (м, 7H), 2,62 (т, 4H), 2,49 (кв, 2H), 1,14 (т, 3H); МС (ESI) 569,10 [M]+; ВЭЖХ 96,98%, rt 3,49 мин.
Пример – 141

N-(2-((6-(3-(2-Хлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид
Соединение синтезировали по способу, указанному в методике 2H (пример 139), с применением 2-хлор-3,5-диметоксианилина (методика показана ниже) в стадии (a), получая при этом указанное в заголовке соединение (20 мг, выход 6,8%) в виде не совсем белого твердого вещества. 1H-ЯМР (CDCl3, 400 МГц): δ 13,46 (с, 1H), 8,43 (с, 1H), 7,79-7,71 (м, 2H), 7,56 (с, 1H), 7,18 (д, 1H), 6,74 (дд, 1H), 6,61 (с, 1H), 6,38 (д, 1H), 6,26 (д, 1H), 6,21-6,15 (м, 1H), 5,79-5,74 (м, 2H), 3,86 (с, 3H), 3,81 (с, 3H), 3,28 (т, 4H), 3,23 (с, 3H), 2,59 (т, 4H), 2,47 (кв, 2H), 1,12 (т, 3H); МС (ESI) 595,15 [M]+; ВЭЖХ 98,14%, rt 3,49 мин.
Получение 2-хлор-3,5-диметоксианилина

a. N-(3,5-Диметоксифенил)ацетамид
Уксусный ангидрид (6,5 мл) медленно добавляли к насыщенному раствору 3,5-диметоксианилина(10 г, 65,359 ммоль) в толуоле (50 мл) в атмосфере аргона при комнатной температуре и образовавшуюся реакционную смесь перемешивали в течение 15 час. После завершения реакции реакционную смесь разбавляли гексаном и образовавшийся осадок собирали фильтрованием и сушили в вакууме, получая при этом указанное в заголовке соединение (12,5 г, выход 98%) в виде не совсем белого твердого вещества.1H ЯМР (CDCl3, 300 МГц):δ 7,38 (с, 1H), 6,75 (д, 2H), 6,23 (с, 1H), 3,76 (с, 6H), 2,15 (с, 3H); МС (ESI) 196,1 [M+H]+.

b. N-(2-Хлор-3,5-диметоксифенил)ацетамид
К перемешиваемому раствору N-(3,5-диметоксифенил)ацетамида(5 г, 25,64 ммоль) в уксусной кислоте (17 мл) добавляли 32% водный раствор хлористоводородной кислоты (14 мл) с последующим добавлением раствора хлората натрия (1,16 г, 11 ммоль) в воде (1,5 мл) при 0°С. Полученную реакционную смесь перемешивали в течение 30 мин при 0°С. После этого реакционную смесь выливали в ледяную воду и делали ее основной добавлением порошка K2CO3. Осадок фильтровали и промывали водой. Остаток очищали колоночной хроматографией на силикагеле (гексан/EtOАс, 88:12), получая при этом указанное в заголовке соединение (1,8 г, выход 31%) в виде белого твердого вещества.1H ЯМР (ДМСО-d6, 300 МГц) δ 9,36 (с, 1H), 7,03 (д, 1H), 6,53 (д, 1H), 3,84 (с, 3H), 3,74 (с, 3H), 2,08 (с, 3H); МС (ESI) 230,2 [M+H]+.
Схема 43

c. 2-Хлор-3,5-диметоксианилин
Гидроксид калия (2,19 г, 39,18 ммоль) добавляли к раствору N-(2-хлор-3,5-диметоксифенил)ацетамида (1,8 г, 7,837 ммоль) в EtOH (100 мл) и воде (10 мл) и реакционную смесь нагревали для кипячения с обратным холодильником в течение 12 час. Избыточный EtOH удаляли при пониженном давлении, получая при этом остаток. Остаток затем распределяли между водой и диэтиловым эфиром. Органический слой отделяли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая при этом указанное в заголовке соединение (1,2 г, выход 82%) в виде белого твердого вещества.1H ЯМР (CDCl3, 300 МГц): δ 5,97 (с, 2H), 4,08 (ушир., 2H), 3,84 (с, 3H), 3,75 (с, 3H); МС (ESI) 188,1 [M+H]+.
Методика 2I: пример – 142
Схема 44

N-(2-((6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(2-метоксиэтокси)фенил)акриламид

a. 4-(2-Метоксиэтокси)-2-нитроанилин
Порошкообразный и высушенный карбонат калия (3,58 г, 0,025 моль) добавляли к раствору 4-амино-3-нитрофенoла(2 г, 0,012 моль) в ДМФА (20 мл) при 0°С в атмосфере азота К раствору по каплям добавляли 1-бром-2-метоксиэтaн (1,34 мл, 0,014 моль) и полученную реакционную смесь кипятили с обратным холодильником в течение ночи. Затем реакционную смесь фильтровали через целит. Фильтрат концентрировали и разбавляли этилацетатом и водой. Водный слой отделяли и экстрагировали этилацетатом (3×30 мл). Органическую фазу промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, получая при этом указанное в заголовке соединение (0,6 г, выход 24%) в виде твердого вещества. МС (ESI) 213,15 [M+H]+.

b. N4-(4-(2-Метоксиэтокси)-2-нитрофенил)-N6-метилпиримидин-4,6-диамин
4-(2-Метоксиэтокси)-2-нитроанилин (0,6 г, 2,830 ммоль) и 6-хлор-N-метилпиримидин-4-амин(0,404 г, 2,830 ммоль) помещали в 10 мл сухого толуола в герметичной трубке в атмосфере аргона при комнатной температуре. Пропускание аргона продолжали в течение дополнительных 5-10 мин. Затем добавляли Cs2CO3 (2,3 г, 7,075 ммоль, 2,5 экв) и Xantphos (0,490 г, 0,849 ммоль) и через образованную реакционную смесь пропускали газообразный аргон в течение 5 мин с последующим добавлением Pd2(dba)3(0,518 г, 0,566 ммоль). Пропускание аргона продолжали в течение дополнительных 5 мин перед герметизацией реакционного сосуда. Затем реакционную смесь нагревали при 100°С в течение 7 час. После завершения реакции по данным ТСХ (DCM:MeOH, 98:2), реакционную массу фильтровали через целит и фильтрат упаривали в вакууме, получая при этом сырой остаток. Сырой остаток очищали колоночной хроматографией на силикагеле, получая при этом указанное в заголовке соединение (0,93 г, выход 70%); МС (ESI) 320,3 [M+H]+.
Схема 45

c. 3-(2,6-Дихлор-3,5-диметоксифенил)-1-(6-((4-(2-метоксиэтокси)-2-нитрофенил)амино)пиримидин-4-ил)-1-метилмочевина
К перемешиваемому раствору 2,6-дихлор-3,5-диметоксианилина(500 мг, 2,252 ммоль) в диоксане (10 мл) добавляли 20% фосгена в толуоле (4,4 мл, 9,0 ммоль) в атмосфере аргона при 0°С. Полученную смесь перемешивали в течение 6 час при 90°С и затем давали ей возможность охладиться до комнатной температуры. Растворители удаляли и остаток растворяли в толуоле (10 мл). К раствору добавляли N4-(4-(2-метоксиэтокси)-2-нитрофенил)-N6-метилпиримидин-4,6-диамин(0,718 г, 8,358 ммоль, 1,0 экв). Полученную реакционную смесь затем кипятили с обратным холодильником в течение 6 час. После завершения реакции по данным ТСХ (гексaны:EtOАс, 7:3), реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме, получая при этом сырую реакционную смесь. Твердый осадок, осажденный при добавлении этилацетата к сырой реакционной смеси, отделяли фильтрованием, промывали эфиром и пентаном, получая при этом указанное в заголовке соединение (0,285, выход 32%) в виде белого твердого вещества. МС (ESI) 567,0 [M+H]+.

d. трет-Бутил-(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтуреидо)пиримидин-4-ил)(4-(2-метоксиэтокси)-2-нитрофенилкарбамат
DMAP (0,025 г, 0,2 ммоль) и ди-трет-бутилдикарбонaт (0,438 г, 2,009 ммоль) добавляли к перемешиваемому раствору 3-(2,6-дихлор-3,5-диметоксифенил)-1-(6-((4-(2-метоксиэтокси)-2-нитрофенил)амино)пиримидин-4-ил)-1-метилмочевины(0,285 г, 0,502 ммоль) в безводном ТГФ (10 мл)в атмосфере аргона при 0°С. Полученную смесь кипятили с обратным холодильником в течение 3-4 час. После завершения реакции по данным ТСХ (гексaны:EtOАс, 1:1), реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме, получая при этом сырой остаток. Остаток очищали колоночной хроматографией на силикагеле, получая при этом указанное в заголовке соединение (0,35 г, выход 88%) в виде не совсем белого твердого вещества. МС (ESI) 767,1 [M+H]+.

e. трет-Бутил-(2-амино-4-(2-метоксиэтокси)фенил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтуреидо)пиримидин-4-ил)карбамат
Никель Ренея (0,05 г) добавляли к раствору трет-бутил-(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтуреидо)пиримидин-4-ил)(4-(2-метоксиэтокси)-2-нитрофенил)карбамата (0,350 г, 0,456 ммоль) в смеси ТГФ и MeOH (10 мл) и полученную реакционную смесь перемешивали в течение 14 час при комнатной температуре в атмосфере водорода (баллон). Реакционную смесь фильтровали через подушку целлита. Фильтрат концентрировали, получая при этом сырой остаток. Остаток очищали колоночной хроматографией на силикагеле (MeOH:DCM, 5:95), получая при указанное в заголовке соединение (0,26 г, выход 77%) в виде твердого вещества. МС (ESI): 737,2 [M+H]+.

f. трет-Бутил-(2-акриламидо-4-(2-метоксиэтокси)фенил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтуреидо)пиримидин-4-ил)карбамат
К перемешиваемому раствору трет-бутил-(2-амино-4-(2-метоксиэтокси)фенил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтуреидо)пиримидин-4-ил)карбамата(0,26 г, 0,352 ммоль) в безводном DCM (6 мл) добавляли TEA (0,09 мл, 0,704 ммоль) в атмосфере аргона при 0°С. Полученную смесь перемешивали в течение 15 мин и медленно добавляли акрилоилхлорид (0,04 мл, 0,528 ммоль) при 0°С. Полученной реакционной смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 2 час. Реакционную смесь разбавляли DCM и водой. Водный слой отделяли и экстрагировали DCM (3×30 мл). Органическую фазу промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией на силикагеле, получая при этом указанное в заголовке соединение (0,200 г, выход 72%) в виде твердого вещества. МС (ESI) 791,2 [M+H]+.

g. N-(2-((6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(2-метоксиэтокси)фенил)акриламид
TFA (0,38 мл, 5,05 ммоль) медленно добавляли к перемешиваемому раствору трет-бутил-(2-акриламидо-4-(2-метоксиэтокси)фенил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтуреидо)пиримидин-4-ил)карбамата(0,2 г, 0,252 ммоль) в сухом DCM (2 мл) в атмосфере аргона при 0°С. Полученной реакционной смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 12 час. Мониторинг развития реакции проводили ЖХ-МС. После завершения реакции избыточные растворители удаляли при пониженном давлении. Сырое твердое вещество промывали эфиром, получая при этом указанное в заголовке соединение (86 мг, выход 58%) в виде белого твердого вещества.1H ЯМР ДМСО-d6, 400 МГц) δ 12,01 (с, 1H), 9,60 (с, 1H), 8,85 (с, 1H), 8,33 (с, 1H); 7,42 (с, 1H), 7,34 (д, 1H), 6,89 (с, 1H), 6,79 (д, 1H), 6,51 (дд, 1H), 6,23 (д, 2H), 5,72 (д, 1H), 4,08 (м,2H), 3,93 (с, 6H), 3,66 (м, 2H), 3,31 (с, 3H), 3,23 (с, 3H). МС (ESI) 591,3 [M+H]+; ВЭЖХ 96,04%, rt 3,72 мин.
Пример 144
N-(2-((6-(3-(2-Хлор-6-фтор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид
Указанное в заголовке соединение синтезировали по способу, указанному в методике 2I (пример 142), с применением N4-(4-(4-этилпиперазин-1-ил)-2-нитрофенил)-N6-метилпиримидин-4,6-диамина (методика 2C, пример 108, стадии a–d) и 2-хлор-6-фтор-3,5-диметоксианилина (методика показана ниже) в стадии (c), получая при этом предыдущее указанное в заголовке соединение (0,46 мг, выход 2,6 % после пяти стадий) в виде не совсем белого твердого вещества. МС (ESI) 813,1 [M+H]+. Свободный амин (46 мг, 0,075 ммоль) растворяли в смеси этилацетaт:DCM:MeOH и обрабатывали фосфорной кислотой (7 мг, 0,07 ммоль). После перемешивания в течение 1 час осажденное твердое вещество фильтровали и промывали эфиром и пентаном, получая при этом указанное в заголовке соединение (34,7 мг, выход 65%) в виде не совсем белого твердого вещества.1H ЯМР (ДМСО-d6, 300 МГц) δ 12,15 (с, 1H), 9,60 (с, 1H), 8,76 (с, 1H), 8,32 (с, 1H), 7,29 (д, 2H), 6,9 (д, 2H), 6,81 (дд, 1H), 6,51-6,45 (м, 1H), 6,25-6,20 (м, 2H), 5,72 (д, 1H), 3,91 (с, 3H), 3,89 (с, 3H), 3,23 (т, 4H), 3,14 (т, 4H), 2,54-2,50 (м, 5H), 1,05 (т, 3H); МС (ESI) 613,2 (M-H3PO4]+; ВЭЖХ 98,6%, rt 6,13 мин.
Получение 2-хлор-6-фтор-3,5-диметоксианилина
Схема 46

a. Метил-2-фтор-3,5-диметоксибензoат
Суспензию фтора (48,9 г, 0,15 моль) в ацетoнитриле (1,1 л) добавляли к раствору метил-3,5-диметоксибензoла (20 г, 0,10 моль) в ацетoнитриле при 0°С в атмосфере азота. Полученную реакционную смесь нагревали до комнатной температуры и перемешивали на протяжении ночи. Реакционную смесь концентрировали в вакууме, разбавляли насыщенным раствором карбонaта натрия и этилацетaтом. Водный слой отделяли и экстрагировали этилацетатом (3×200 мл). Органическую фазу промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (градиент гексaн/эфир, от 30:1 до 4:1), получая при этом указанное в заголовке соединение (4 г, выход 16,9%).

b. Метил-2-хлор-6-фтор-3,5-диметоксибензоат
SO2Cl2 (2,20 г, 0,016 моль) добавляли по каплям к раствору метил-2-фтор-3,5-диметоксибензoата (3,5 г, 0,016 моль) в ацетoнитриле (40 мл) при 0°С в атмосфере азота. Образовавшуюся реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 1 часа. Реакционную смесь гасили насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом (3×30 мл). Органическую фазу промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией на силикагеле с применением элюента (градиент от смеси гексaн/эфир (20:1) до гексaн/эфир (5:1)), получая при этом указанное в заголовке соединение (2,7 г, выход 67%) в виде твердого вещества.
Схема 47

c. 2-Хлор-6-фтор-3,5-диметоксибензойная кислота
Суспензию метил-2-хлор-6-фтор-3,5-диметоксибензoaта (2,7 г, 0,010 моль) и гидроксида натрия (1,088 г, 0,0272) в безводном этaноле (30 мл) кипятили с обратным холодильником в течение 24 час. Полученную реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме, получая при этом сырой остаток. Сырой остаток растворяли в воде и экстрагировали эфиром (3×30 мл). Водный слой подкисляли конц. HCl и осажденное твердое вещество фильтровали, промывали холодной водой и сушили в вакууме, получая при этом указанное в заголовке соединение (1,8 г, выход 71%) в виде твердого вещества.
Схема 48

d. 2-Хлор-6-фтор-3,5-диметоксианилин
Суспензию 2-хлор-6-фтор-3,5-диметоксибензойной кислоты (1,8 г, 0,0077 моль) и триэтиламина (0,934 г, 0,0092 моль)в трет-BuOH (50 мл) перемешивали в течение 5 мин. К полученной реакционной смеси добавляли дифенилфосфорилазид (2,3 г, 0,0092 моль) и смесь нагревали до 82°С и выдерживали при такой температуре в течение ночи. Реакционную смесь затем концентрировали в вакууме, получая при этом сырой остаток. Сырой остаток растворяли в дихлорметане (20 мл) и охлаждали до 0°С. К реакционной смеси добавляли TFA (4 мл) и полученную реакционную смесь затем перемешивали при комнатной температуре в течение 2 час. Растворители удаляли в вакууме и сырой остаток разбавляли этилацетатом и насыщенным раствором карбоната натрия. Водный слой отделяли и экстрагировали этилацетатом (3×30 мл). Органическую фазу промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией на силикагеле с применением элюента (градиент от гексана до смеси гексан:эфир (65:35)), получая при этом указанное в заголовке соединение (0,95 г, выход 60%) в виде твердого вещества. МС (ESI) 205,7 [M+H]+.
Методика 2J – пример 145
Схема 49

N-(2-((6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(гидроксиметил)фенил)акриламид
Схема 50

a. 1-(6-Хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонатмочевина
DMAP (0,080 г, 0,655 ммоль) и ди-трет-бутилдикарбонaт (2,9 мл, 12,6 ммоль) добавляли к перемешиваемому раствору 1-(6-хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метилмочевины (методика 2E, стадия b; 2,6 г, 6,632 ммоль) в безводном ТГФ (20 мл) в атмосфере аргона при температуре 0°С. Полученную смесь затем кипятили с обратным холодильником в течение 2 час. После завершения реакции по данным ТСХ (EtOАс:гексaн, 3:7), реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме, получая при этом сырой остаток. Остаток очищали колоночной хроматографией на силикагеле, получая при этом указанное в заголовке соединение (2,5 г, выход 69,4%) в виде твердого вещества.1H-ЯМР (CDCl3, 300 МГц) δ 8,73 (д, 1H), 7,95 (д, 1H), 6,61 (с, 1H), 3,95 (с, 6H), 3,63 (с, 3H), 1,35 (с, 9H).

b. Метил 4-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтуреидо)пиримидин-4-ил)амино)-3-нитробензoат
Метил-4-амино-3-нитробензoат (0,956 г, 0,004 моль) и 1-(6-хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонатмочевину (2 г, 0,004 моль) помещали в 10 мл сухого толуола в герметичной трубке при комнатной температуре и пропускали газообразный аргон в течение 5-10 мин. Затем добавляли Cs2CO3 (3,25 г, 0,01 моль) и Xantphos (0,46 г, 0,0008 моль)и через полученную реакционную смесь пропускали газообразный аргон в течение 5 мин с последующим добавлением Pd2(dba)3(0,36 г, 0,0004 моль). Пропускание газообразного аргона продолжали в течение дополнительных 5 мин перед герметизацией реакционного сосуда. Затем реакционную смесь нагревали при 100°С в течение 12 час. После завершения реакции по данным ТСХ, реакционную массу фильтровали через целит и фильтрат упаривали в вакууме, получая при этом сырой остаток. Сырой остаток очищали колоночной хроматографией на силикагеле, получая при этом указанное в заголовке соединение (2,10 г, выход 79%);1H-ЯМР (CDCl3, 300 МГц) δ 10,3 (с, 1H), 9,00-8,95 (м, 2H), 8,75 (с, 1H), 8,22 (дд, 1H), 7,63 (с, 1H), 6,61 (с, 1H), 3,95 (с, 6H), 3,94 (с, 3H), 3,67 (с, 3H), 1,39 (с, 9H).

c. Метил-4-((трет-бутoксикарбонил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутилуреидо)пиримидин-4-ил)амино)-3-нитробензoат
DMAP (0,039 г, 0,322 ммоль) и ди-трет-бутилдикарбонaт (1,48 мл, 6,45 ммоль) добавляли к перемешиваемому раствору метил-4-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтуреидо)пиримидин-4-ил)амино)-3-нитробензoата (2,1 г, 3,22 ммоль) в безводном ТГФ (5 мл) в атмосфере аргона при 0°С. Полученную смесь кипятили с обратным холодильником в течение 12 час. После завершения реакции по данным ТСХ (EtOАс:гексан 40:60), реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме, получая при этом сырой остаток. Остаток очищали колоночной хроматографией на силикагеле, получая при этом указанное в заголовке соединение (2,1 г, выход 87%) в виде твердого вещества. МС (ESI) 751,0 [M]+.

d. трет-Бутил-(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтуреидо)пиримидин-4-ил)(4-(гидроксиметил)-2-нитрофенил)карбамат
Борогидрид лития(0,049 г, 2,26 ммоль) добавляли к раствору метил-4-((трет-бутoксикарбонил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутилуреидо)пиримидин-4-ил)амино)-3-нитробензoата (1,7 г, 2,26 ммоль) в безводном ТГФ (18 мл) в атмосфере аргона при температуре 0°С. Полученной реакционной смеси затем давали возможность нагреться до комнатной температуре в течение 2,5 час. Реакционную смесь затем гасили смесью лед-вода и разбавляли этилацетaтом. Водный слой отделяли и экстрагировали этилацетaтом (3×20 мл). Органическую фазу промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, получая при этом указанное в заголовке соединение (0,480 г, выход 29%) в виде твердого вещества. МС (ESI) 723.2 [M]+.
Схема 51

e. трет-Бутил-(4-(((трет-бутилдиметилсилил)oкси)метил)-2-нитрофенил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтуреидо)пиримидин-4-ил)карбамат
Имидазол (0,071 г, 1,051 ммоль) и TBDMS-Cl (0,118 г, 0,787 ммоль) добавляли к перемешиваемому раствору трет-бутил-(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонатуреидо)пиримидин-4-ил)(4-(гидроксиметил)-2-нитрофенил)карбамата (0,380 г, 0,525 ммоль) в безводном дихлорметане (5 мл) в атмосфере аргона при 0°С. Полученную реакционную смесь перемешивали в течение 4 час при комнатной температуре. Реакционную смесь затем разбавляли DCM и водой. Водный слой отделяли и экстрагировали DCM (3×10 мл). Органическую фазу промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, получая при этом указанное в заголовке соединение (0,330 г, выход 75%) в виде твердого вещества. МС (ESI) 837,4 [M]+.
Схема 52

f. трет-Бутил-(2-амино-4-(((трет-бутилдиметилсилил)oкси)метил)фенил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутил карбонaтуреидо)пиримидин-4-ил)карбамат
Никель Ренея (0,06 г) добавляли к раствору трет-бутил-(4-(((трет-бутилдиметилсилил)oкси)метил)-2-нитрофенил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтуреидо)пиримидин-4-ил)карбамата (0,3 г, 0,358 ммоль) в MeOH (5 мл) и полученную реакционную смесь перемешивали в течение 12 час при комнатной температуре в атмосфере водорода (баллон). Реакционную смесь фильтровали через подушку целлита. Фильтрат концентрировали, получая при этом сырой остаток, который затем очищали колоночной хроматографией, получая при этом указанное в заголовке соединение (0,180 г, выход 64%) в виде твердого вещества. МС (ESI) 807,2 [M]+.

g. трет-Бутил-(2-акриламидо-4-(((трет-бутилдиметилсилил)oкси)метил)фенил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтуреидо)пиримидин-4-ил)карбамат
К перемешиваемому раствору трет-бутил-(2-амино-4-(((трет-бутилдиметилсилил)oкси)метил)фенил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутилкарбонaтуреидо)пиримидин-4-ил)карбамата (0,180 г, 0,223 ммоль) в безводном DCM (5 мл) добавляли TEA (0,08 мл, 0,557 ммоль) в атмосфере аргона при 0°С. Полученную смесь перемешивали в течение 15 мин и медленно добавляли акрилoилхлорид (0,03 мл, 0,334 ммоль) при 0°С. Полученной реакционной смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 4 час. Реакционную смесь затем концентрировали и остаток очищали колоночной хроматографией на силикагеле, получая при этом указанное в заголовке соединение (0,130 г, выход 68%) в виде твердого вещества. МС (ESI) 861,3 [M]+.

h. N-(2-((6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(гидроксиметил)фенил)акриламид
TFA (1,0 мл) медленно добавляли к перемешиваемому раствору трет-бутил-(2-акриламидо-4-(((трет-бутилдиметилсилил)oкси)метил)фенил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-трет-бутил карбонaтуреидо)пиримидин-4-ил)карбамата (0,130 г, 0,150 ммоль) в сухом DCM (3 мл) в атмосфере аргона при 0°С. Полученной реакционной смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 13 час. Мониторинг развития реакции проводили ЖХ-МС, после завершения реакции избыточные растворители удаляли при пониженном давлении. Полученный остаток разбавляли DCM и насыщенным водным раствором NaHCO3. Водный слой отделяли и экстрагировали DCM (3×10 мл). Органическую фазу промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, получая при этом 95 мг требуемого продукта с чистотой по данным ВЭЖХ, который затем очищали препаративной ВЭЖХ (условия: колонка X bridge C18 (19 мм × 150 мм, 5 мкм); (подвижная фаза A; 0,01% TFA в воде, B; АCN), получая при этом указанное в заголовке соединение (16 мг, выход 16%) в виде белого твердого вещества.1H-ЯМР (ДМСО-d6, 400 МГц) δ 12,08 (с, 1H), 9.70 (с, 1H), 8,95 (с, 1H), 8,37 (с, 1H), 7,64 (с, 1H), 7,49 (д, 1H), 7,15 (с, 1H), 6,90 (с, 1H), 6,53-7,46 (кв, 1H), 6,32 (с, 1H), 6,25 (д, 1H), 5,74 (д, 1H), 5,42 (с, 1H), 5,35 (с, 1H), 4,49 (с, 2H), 3,94 (с, 6H), 3,25 (с, 3H); МС (ESI) 547,0 [M+H]+; ВЭЖХ 97,4%, rt 3,83 мин.
Пример 147

N-(2-((6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(метоксиметил)фенил)акриламид
Указанное в заголовке соединение синтезировали по способу, указанному в методике 2I (пример 142), с применением N4-(4-(метоксиметил)-2-нитрофенил)-N6-метилпиримидин-4,6-диамина (методика описана ниже) в стадии (c), получая при этом указанное в заголовке соединение (5,0 мг, выход 1,8%) в виде не совсем белого твердого вещества. 1H-ЯМР (CDCl3, 400 МГц): δ 12,30 (с, 1H), 8,41 (с, 1H), 7,86 (с, 1H), 7,81 (с, 1H), 7,52 (д, 1H), 7,49 (с, 1H), 7,30 (д, 1H), 6,54 (с, 1H), 6,46 (д, 1H), 6,26 (дд, 1H), 6,07 (с, 1H), 5,83 (д, 1H), 5,18 (с, 2H), 3,93 (с, 6H), 3,82 (с, 3H), 3,34 (с, 3H); МС (ESI) 561,1 [M+H]+; ВЭЖХ 95,76%, rt 4,36 мин.
Получение N4-(4-(метоксиметил)-2-нитрофенил)-N6-метилпиримидин-4,6-диамина

a. Метил-4-амино-3-нитробензoат
Тионилхлорид (19,4 г, 164,85 ммоль) добавляли к раствору 4-амино-3-нитробензoйной кислоты (20 г, 109,89 ммоль) в метаноле (200 мл) при 0°С. Полученную смесь затем кипятили с обратным холодильником в течение 12 час. Реакционной смеси давали возможность охладиться до комнатной температуры. Осажденное желтое твердое вещество отделяли фильтрованием и сушили, получая при этом указанное в заголовке соединение (22 г, выход 100%) в виде твердого вещества. 1H-ЯМР (CDCl3, 300 МГц): δ 8,85 (д, 1H), 7,99 (дд, 1H), 6,83 (д, 1H), 6,40 (с, 2H), 3,90 (с, 3H).

b. Метил-4-((6-(метиламино)пиримидин-4-ил)амино)-3-нитробензoат
6-Хлор-N-метилпиримидин-4-амин (3 г, 20,97 ммоль) и метил-4-амино-3-нитробензoат (3,9 г, 20,97 ммоль) помещали в толуол (5 мл) в герметичной трубке в атмосфере аргона при комнатной температуре. Газообразный аргон пропускали в течение 5-10 мин. Затем добавляли Cs2CO3 (17,0 г, 52,4 ммоль) и Xantphos (3,6 г, 6,29 ммоль) и полученную реакционную смесь продували газообразным аргоном в течение 5 мин с последующим добавлением Pd2(dba)3(3,8 г, 4,19 ммоль). Пропускание газообразного аргона продолжали в течение дополнительных 5 мин перед герметизацией реакционного сосуда. Затем реакционную смесь нагревали при 110°С в течение 16 час. После завершения реакции по данным ТСХ, реакционную смесь охлаждали до комнатной температуры, фильтровали через слой целита и фильтрат упаривали в вакууме, получая при этом сырой остаток. Сырой остаток очищали колоночной хроматографией на силикагеле, получая при этом указанное в заголовке соединение (3,2 г, выход 51%) в виде твердого вещества; МС (ESI) 304,2 [M+H]+.

c. Метил-4-((трет-бутoксикарбонил)(6-((трет-бутoксикарбонил)(метил)амино)пиримидин-4-ил)амино)-3-нитробензoат
DMAP (0,497 г, 4,078 ммоль) и ди-трет-бутилдикарбонaт (8,89 г, 40,78 ммоль) добавляли к перемешиваемому раствору промежуточного соединения 4 (3,1 г, 10,197 моль) в безводном ТГФ (35 мл) в атмосфере аргона при комнатной температуре. Полученную смесь кипятили с обратным холодильником в течение 2-3 час. После завершения реакции по данным ТСХ, реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме, получая при этом сырой остаток. Остаток очищали колоночной хроматографией на силикагеле, получая при этом указанное в заголовке соединение (1,9 г, выход 37%) в виде твердого вещества. MS (ESI): 504,0 [M+H]+.

d. трет-Бутил-(6-((трет-бутoксикарбонил)(4-(гидроксиметил)-2-нитрофенил)амино)пиримидин-4-ил)(метил)карбамат
Борогидрид лития (0,157 г, 7,14 ммоль) добавляли к раствору метил-4-((трет-бутoксикарбонил)(6-((трет-бутoксикарбонил)(метил)амино)пиримидин-4-ил)амино)-3-нитробензoат (1,8 г, 3,57 ммоль) в ТГФ (20 мл) в атмосфере аргона при 0°С. Полученной смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 12 час. Реакционную смесь затем гасили смесью льда с водой и разбавляли этилацетатом. Водный слой отделяли и экстрагировали этилацетатом (3×40 мл). Органическую фазу промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали, получая при этом сырой остаток. Остаток очищали колоночной хроматографией на силикагеле, получая при этом промежуточное соединение 6 (1,2 г, 75%) в виде твердого вещества. МС (ESI) 476,1 [M+H]+.
Схема 53

e. трет-Бутил-(6-((трет-бутoксикарбонил)(4-(метоксиметил)-2-нитрофенил)амино)пиримидин-4-ил)(метил)карбамат
К раствору трет-бутил-(6-((трет-бутoксикарбонил)(4-(гидроксиметил)-2-нитрофенил)амино)пиримидин-4-ил)(метил)карбамата (1,5 г, 3.488 ммоль) в ацетоне (20 л) в атмосфере аргона при 0°С добавляли карбонат калия (0,48 г, 6,97 ммоль) с последующим добавлением диметилсульфата (0,87 г, 6,97 ммоль). Полученную реакционную смесь затем кипятили с обратным холодильником в течение 24 час и затем давали ей возможность охладиться до комнатной температуры. Реакционную смесь концентрировали в вакууме, получая при этом сырой остаток. Остаток очищали колоночной хроматографией на силикагеле, получая при этом указанное в заголовке соединение (0,3 г, выход 19%) в виде твердого вещества. МС (ESI) 490,55 [M+H]+.

f. N4-(4-(Метоксиметил)-2-нитрофенил)-N6-метилпиримидин-4,6-диамин
TFA (5 мл) медленно добавляли к перемешиваемому раствору трет-бутил-(6-((трет-бутoксикарбонил)(4-(метоксиметил)-2-нитрофенил)амино)пиримидин-4-ил)(метил)карбамата (0,85 г) в сухом DCM (10 мл) в атмосфере аргона при 0°С. Полученной реакционной смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 1 час. После завершения реакции Избыточные растворители удаляли при пониженном давлении и остаток промывали эфиром, получая при этом указанное в заголовке соединение (0,7 г, сырой продукт) в виде твердого вещества. МС (ESI) 290,2 [M+H]+.
Методика 2K - пример 148
Схема 54

N-(2-((6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-метилпиперазин-1-карбонил)фенил)акриламид

a. 4-((трет-Бутoксикарбонил)(6-(3-(трет-бутoксикарбонил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-3-нитробензoйная кислота
Гидроксид лития (0,08 г, 2,99 ммоль) добавляли к раствору метил-4-((трет-бутoксикарбонил)(6-(3-(трет-бутoксикарбонил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-3-нитробензoата (1,5 г, 1,997 ммоль) (методика 2J, стадия c) в смеси ТГФ (10 мл) и воды (2 мл) при 0°С. Полученной реакционной смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 5 час. После завершения реакции по данным ТСХ (EtOАс:гексaн 3:7), реакционную смесь концентрировали в вакууме, получая при этом сырой остаток. Сырой остаток растворяли в воде и экстрагировали эфиром (3×30 мл). Водный слой подкисляли 10% раствором лимонной кислоты и осажденное твердое вещество отделяли фильтрованием, промывали холодной водой и сушили в вакууме, получая при этом указанное в заголовке соединение (1,3 г, выход 93%) в виде светло-коричневого твердого вещества. МС (ESI) 737,2 [M+H]+.

b.трет-Бутил-(2-нитро-4-(4-метилпиперазин-1-карбонил)фенил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-3-трет-бутoксикарбонил-1-метилуреидо)пиримидин-4-ил)карбамат
DIPEA (0,3 мл, 1,62 ммоль), HATU (0,515 г, 1,355 ммоль) и N-метилпиперазин (0,09 мл, 0,813 ммоль) добавляли к раствору 4-((трет-бутoксикарбонил)(6-(3-(трет-бутoксикарбонил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-3-нитробензойной кислоты (0,4 г, 0,542 ммоль) в ДМФА (5 мл) при 0°С в атмосфере аргона. Полученной реакционной смеси затем давали возможность нагреться до комнатной температуры и перемешивали в течение 18 час. После завершения реакции по данным ТСХ (MeOH:DCM 1:19), к реакционной смеси добавляли воду. Осажденное сырое твердое вещество фильтровали, сушили и очищали колоночной хроматографией на силикагеле, получая при этом указанное в заголовке соединение (0,27 г, выход 61%) в виде твердого вещества. МС (ESI) 819,1 [M+H]+.

c. трет-Бутил-(2-амино-4-(4-метилпиперазин-1-карбонил)фенил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-3-трет-бутoксикарбонил-1-метилуреидо)пиримидин-4-ил)карбамат
Никель Ренея (0,06 г) добавляли к раствору трет-бутил-(2-нитро-4-(4-метилпиперазин-1-карбонил)фенил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-3-трет-бутoксикарбонил-1-метилуреидо)пиримидин-4-илкарбамата (0,27 г, 0,329 ммоль) в смеси MeOH (4 мл) и ТГФ (4 мл) и полученную реакционную смесь перемешивали в течение 20 час при комнатной температуре в атмосфере водорода. Полученную смесь фильтровали через слой целита. Фильтрат концентрировали, получая при этом указанное в заголовке соединение (0,22 г, выход 84%) в виде твердого вещества. МС (ESI) 789,4 [M+H]+.

d. трет-Бутил-(2-акриламидо-4-(4-метилпиперазин-1-карбонил)фенил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-3-трет-бутoксикарбонил-1-метилуреидо)пиримидин-4-ил)карбамат
К перемешиваемому раствору трет-бутил-(2-амино-4-(4-метилпиперазин-1-карбонил)фенил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-3-трет-бутoксикарбонил-1-метилуреидо)пиримидин-4-ил)карбамата (0,22 г, 0,278 ммоль) в безводном DCM (5 мл) добавляли TEA (0,08 мл, 0,557 ммоль) в атмосфере аргона при 0°С. Полученную смесь перемешивали в течение 15 мин и медленно добавляли акрилoилхлорид (0,037 г, 0,417 ммоль) при 0°С. Полученной реакционной смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 2 час. Реакционную смесь гасили насыщенным раствором бикарбоната натрия и разбавляли DCM. Водный слой отделяли и экстрагировали DCM (3×20 мл). Органическую фазу промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали, получая при этом сырой остаток. Остаток очищали колоночной хроматографией на силикагеле, получая при этом указанное в заголовке соединение (0,060 г, выход 25%) в виде твердого вещества. МС (ESI) 843,3 [M+H]+.

e. N-(2-((6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-метилпиперазин-1-карбонил)фенил)акриламид
TFA (0,2 мл) медленно добавляли к перемешиваемому раствору трет-бутил-(2-акриламидо-4-(4-метилпиперазин-1-карбонил)фенил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-3-трет-бутилкарбонил-1-метилуреидо)пиримидин-4-ил)карбамата (0,060 г, 0,0711 ммоль) в сухом DCM (2 мл) в атмосфере аргона при 0°С. Полученной реакционной смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 18 час. Мониторинг развития реакции проводили ЖХ-МС, после завершения реакции избыточные растворители удаляли при пониженном давлении. Полученный остаток разбавляли DCM и гасили насыщенным водным раствором NaHCO3. Водный слой отделяли и экстрагировали DCM (3×10 мл). Органическую фазу промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая при этом сырой остаток. Остаток очищали колоночной хроматографией на силикагеле, получая при этом 70 мг требуемого продукта с чистoтой по данным ВЭЖХ 35%, который затем очищали препаративной ВЭЖХ (условия: колонка Gemini NX C18 (21,2 мм × 150 мм, размер частицы 5 мкм); (подвижная фаза: A; 0,1% раствор бикарбоната аммония в воде, B; ACN), получая при этом требуемое соединение. Соединение затем разбавляли дихлорметаном и водой. Водный слой отделяли и экстрагировали DCM (3×10 мл). Органическую фазу промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая при этом указанное в заголовке соединение (0,005 г, выход 11%) в виде белого твердого вещества. 1H-ЯМР (CD3OD, 400 МГц) δ 8,41 (с, 1H), 7,79-7,76 (м, 2H), 7,36 (дд, 1H), 6,82 (с, 1H), 6,47-6,42 (м, 3H), 5,82 (д, 1H), 3,96 (с, 6H), 3,81-3,55 (м, 8H), 2,54 (с, 3H), 2,37 (с, 3H); МС (ESI) 643,1 [M+H]+; ВЭЖХ 97,6%, rt 6,19 мин.
Пример 149

3-Акриламидо-4-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-N,N-диметилбензамид
Указанное в заголовке соединение синтезировали по способу, указанному в методике 2K (пример 148), с применением диметиламина в стадии (b), получая при этом указанное в заголовке соединение (12,0 мг, выход 6,1%) в виде не совсем белого твердого вещества. 1H-ЯМР (CD3OD, 400 МГц) δ 8,38 (с, 1H), 7,78-7,72 (м, 2H), 7,34 (дд, 1H), 6,80 (с, 1H), 6,46-6,38 (м, 3H), 5,80 (дд, 1H), 3,94 (с, 6H), 3,36 (с, 3H), 3,10 (с, 6H); МС (ESI) 587,9 [M+H]+; ВЭЖХ 99,04%, rt 3,98 мин.
Пример 150

N-(2-((6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-карбонил)фенил)акриламид
Указанное в заголовке соединение синтезировали по способу, указанному в методике 2K (пример 148), с применением 1-этилпиперазина в стадии (b), получая при этом указанное в заголовке соединение (10,0 мг, выход 9,7%) в виде не совсем белого твердого вещества 1H-ЯМР (CD3OD, 400 МГц) δ 8,41 (с, 1H), 7,76-7,79 (м, 2H), 7,36 (д, 1H), 6,82 (с, 1H), 6,38-6,47 (м, 3H), 5,82 (д, 1H), 3,96 (с, 6H), 3,72-3,82 (м, 2H), 3,53-3,65 (м, 3H), 3,39 (с, 4H), 2,49-2,54 (м, 6H), 1,15 (т, 3H); МС (ESI) 657,0 [M+H]+; ВЭЖХ 95,98%, rt 6,25 мин.
Пример 151

Соль N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-изопропилпиперазин-1-ил)фенил)акриламида с фосфорной кислотой
Указанное в заголовке соединение синтезировали способом, указанным в методике 2J (пример 145), с применением 4-(4-изопропилпиперазин-1-ил)-2-нитроанилина (методика показана ниже) в стадии (b) и с исключением стадий (d) и (e), получая при этом свободное основание указанного в заголовке соединения (0,1 г, общий выход 9,7%) в виде не совсем белого твердого вещества. МС (ESI) 643,1 [M+H]+. К раствору N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-изопропилпиперазин-1-ил)фенил)акриламида (0,1 г) в 95% растворе ТГФ-MeOH (5 мл) медленно добавляли 85% H3PO4. Полученную реакционную смесь затем перемешивали при комнатной температуре в течение 30 мин, выпаривали растворитель и остаток растирали с диэтиловым эфиром, сушили в вакууме, получая при этом указанное в заголовке соединение (0,16 г) в виде не совсем белого твердого вещества.1H ЯМР(ДМСО-d6, 400 МГц): δ 12,08 (с, 1H), 9,62 (с, 1H), 8,76 (с, 1H), 8,32 (с, 1H), 7,30 (д, 2H), 6,89 (д, 1H), 6,81 (д, 1H), 6,46-6,49 (м, 1H), 6,21-6,25 (м, 2H), 5,72 (д, 1H), 3,93 (с, 6H), 3,35-3,41 (м, 1H), 3,23 (с, 3H), 3,12-3,17 (м, 4H), 2,67-2,70 (м, 4H), 1,05-1,11 (м, 6H); MS (ESI) 643,3 (M + 1); ВЭЖХ 95,41%, rt 6,48 мин.
Получение-(4-изопропилпиперазин-1-ил)-2-нитроанилина

a. трет-Бутил-4-изопропилпиперазин-1-карбоксилат
Boc-пиперазин (10 г, 53,76 ммоль) и ацетон (4 л) помещали в смесь сухого DCM (100 мл) и уксусной кислоты (3,2 мл). Смесь перемешивали при комнатной температуре в течение 20 мин. Добавляли Na(OАc)3BH (17 г, 80,2 ммоль) и перемешивание продолжали при комнатной температуре в течение 12 час. Реакционную смесь разбавляли водой, экстрагировали этилацетатом (3×25 мл). Органический слой промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая при этом сырое указанное в заголовке соединение (13 г, сырой продукт). МС (ESI) 229,2 [M+H]+.

b. 1-Изопропилпиперазин
TFA (15 мл) медленно добавляли к перемешиваемому раствору трет-бутил-4-изопропилпиперазин-1-карбоксилата (13 г, сырой продукт) в сухом DCM (20 мл) в атмосфере аргона при 0°С. Полученной реакционной смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 18 час. Реакционную массу концентрировали в вакууме и остаток растирали с н-гексаном и диэтиловым эфиром. Высушивание в вакууме давало указанное в заголовке соединение (7 г, выход 97%). МС (ESI) 129,1 [M+H]+.
Схема 55
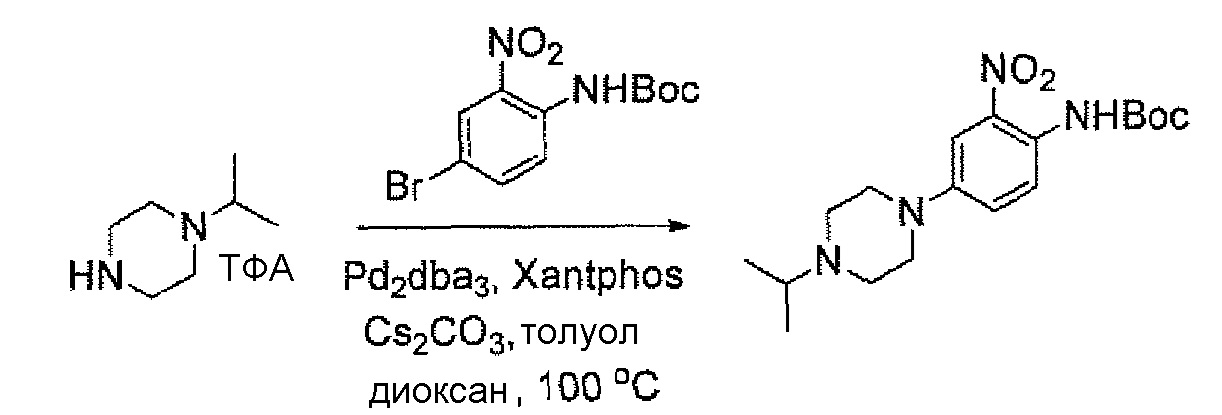
с.трет-Бутил-(4-(4-изопропилпиперазин-1-ил)-2-нитрофенил)карбамат
1-Изопропилпиперазин (1,29 г, 5,66 ммоль) и трет-бутил-(4-бром-2-нитрофенил)карбамат (1,5 г, 4,71 ммоль) помещали в смесь сухого толуола (15 мл) и диоксaна (2 л) в герметичной трубке в атмосфере аргона при комнатной температуре. Аргон пропускали в течение 5-10 мин. Затем добавляли Сs2CO3 (3,06 г, 9,43 ммоль) и Xantphos (0,54 г, 0,94 моль) и через полученную смесь пропускали газообразный аргон в течение 5 мин с последующим добавлением Pd2(dba)3(0,3 г, 0,47 ммоль). Пропускание аргона продолжали в течение дополнительных 5 мин перед герметизаций реакционного сосуда. Затем реакционную смесь нагревали до 100°С в течение 12 час. После завершения реакции по данным ТСХ, реакционную смесь охлаждали до комнатной температуры, фильтровали через слой целита и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (DCM:MeOH/97:3), получая при этом указанное в заголовке соединение (1,2 г, 70,5%). МС (ESI) 365,5 [M+H]+.

d. 4-(4-Изопропилпиперазин-1-ил)-2-нитроанилин
TFA (3 мл) медленно добавляли к перемешиваемому раствору трет-бутил-(4-(4-изопропилпиперазин-1-ил)-2-нитрофенил)карбамата (1,2 г, 3,29 моль) в сухом DCM (5 мл) в атмосфере аргона при 0°С. Полученной реакционной смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 12 час. Мониторинг развития реакции проводили ЖХ-МС, после завершения реакции избыточные растворители удаляли при пониженном давлении, получая при этом сырой остаток. Сырой остаток несколько раз промывали эфиром, получая при этом указанное в заголовке соединение (1 г, выход 80,6%) в виде красного твердого вещества. МС (ESI) 265,1 [M+H]+.
Пример - 154
схема 56

Получение N-(5-(4-ацетилпиперазин-1-ил)-2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)фенил)акриламида
N-(2-((6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(пиперазин-1-ил)фенил)акриламид (методика 2L, пример 157) (8,6 мг, 0,014 ммоль) и DIEA (7,5 мк, 0,043 ммоль) перемешивали в DCM (1,0 мл) при комнатной температуре в атмосфере азота. Добавляли раствор ацетилхлорида (1,0 мкл, 0,016 ммоль) в DCM (11 мкл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель выпаривали и полученное вещество растворяли в 400 мкл ДМСО. Раствор в ДМСО разбавляли 1,0 мл MeOH и очищали преп-ВЭЖХ (вода/ACN в муравьиной кислоте), получая при этом указанное в заголовке соединение (1,4 мг, выход 15%).1H-ЯМР(400 МГц, MeOH-d4) δ 2,16 (с, 3 H), 3,65–3,80 (м, 4 H), 3,94 (с, 6 H), 5,76 (дд, 1 H), 6,15 (с, 1 H), 6,28–6,49 (м, 2 H), 6,78–6,82 (м, 1 H), 6,95 (дд, 2,89 Гц, 1 H), 7,28–7,38 (м, 2 H), 8,31 (д, 1 H), 8,58 (ушир. с, 1 H); ESI-MС 643 [M+H]+.
Пример – 155 и 156

Получение N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(1-метоксиэтил)фенил)акриламида и N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(1-гидроксиэтил)фенил)
Соединение синтезировали по способу, указанному в методике 2G (пример 123) с заменой 4-(1-метоксиэтил)-2-нитроанилина (методика показана ниже) в стадии (d), получая при этом указанное в заголовке соединение после очистки с применением флэш-хроматографии на диоксиде кремния и элюирования смесью от 90% до 100% EtOАс/гексaн. Неполярной фракцией был N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(1-метоксиэтил)фенил)акриламид(16 мг, выход 6% в пяти стадиях)1H-ЯМР(400 МГц, MeOH-d4) δ 1,46 (д, 3 H), 3,29 (с, 3 H), 3,96 (с, 6 H), 4,12 (д, 1 H), 4,40 (кв, 1 H), 5,78–5,82 (м, 1 H), 6,33–6,50 (м, 3 H), 6,82 (с, 1 H), 7,27 (дд, 1 H), 7,55–7,64 (м, 2 H), 8,37 (д, 1 H); ESI-MМ 575 [M+H]+. Полярной фракцией был N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(1-гидроксиэтил)фенил)акриламид(22 мг, выход 8% в пяти стадиях).1H-ЯМР(400 МГц, MeOH-d4) δ 1,53 (д, 3 H) 4,00 (с, 6 H) 5,83 (дд, 1 H) 6,29–6,56 (м, 3 H) 6,86 (с, 1 H) 7,38 (дд, 1 H) 7,57 (д, 1 H) 7,69 (с, 1 H) 8,40 (д, 1 H); ESI-MС 661 [M+H]+.
Получение 4-(1-метоксиэтил)-2-нитроанилина

a. 1-(4-Фтор-3-нитрофенил)этaнол
1-(4-фтор-3-Нитрофенил)этaнон (1,0 г, 5,46 ммоль) растворяли в MeOH (15,0 мл) и раствор перемешивали при охлаждении на ледяной бане. NaBH4 (0,62 г, 16,0 ммоль) добавляли порциями. После завершения добавления реакционную смесь перемешивали при комнатной температуре в течение 10 минут. Реакционную смесь выливали в EtOАс и насыщенный раствор соли. Органический слой отделяли, сушили над MgSO4 и упаривали. Полученное вещество очищали флэш-хроматографией на диоксиде кремния с элюированием 20%-70% EtOАс/гексaн, получая при этом указанное в заголовке соединение (881 мг, выход 87%). %).1H-ЯМР(400 МГц, CDCl3) δ 1,53 (д, 4 H), 4,99 (дд, 1 H), 7,22–7,33 (м, 1 H), 7,66 (ддд, 1 H), 8,09 (дд, 1 H).
Схема 58

b. 1-Фтор-4-(1-метоксиэтил)-2-нитробензол
1-(4-Фтор-3-нитрофенил)этaнол (870 мг, 4,7 ммоль) растворяли в MeOH (10 мл) и осторожно добавляли концентрированную серную кислоту (2,5 мл, 47 ммоль). Реакционную смесь нагревали при 150°С при помощи микроволнового излучения (Biotage Initiator) в течение 5 минут. После охлаждения до комнатной температуры реакционную смесь выливали в смесь EtOАс/вода. Органический слой промывали насыщенным раствором соли, сушили над МgSO4 и упаривали. Полученное вещество очищали флэш-хроматографией на диоксиде кремния с элюированием смесью от 0% до 50% EtOАс/гексaн, получая при этом указанное в заголовке соединение (664 мг, выход 71%).1H-ЯМР(400 МГц, CDCl3) δ 1,45 (д, 3 H), 3,27 (с, 3 H), 4,36 (кв, 1 H), 7,28–7,32 (м, 1 H), 7,56–7,63 (м, 1 H), 7,98–8,04 (м, 1 H).

c. 4-(1-Метоксиэтил)-2-нитроанилин
1-Фтор-4-(1-метоксиэтил)-2-нитробензол (664 мг, 3,33 ммоль) перемешивали в ТГФ (10 мл). Добавляли NH4OH (0,39 мл, 10,0 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли дополнительно 780 мкл NH4OH и реакционную смесь нагревали при 120°С в течение 10 минут при помощи микроволнового излучения (Biotage Initiator) и затем при 140°С в течение 50 минут. После охлаждения до комнатной температуры реакционную смесь выливали в смесь EtOАс/вода. Органический слой промывали насыщенным раствором соли, сушили над МgSO4 и упаривали. Полученное вещество очищали флэш-хроматографией на диоксиде кремния с элюированием смесью от 0% до 50% EtOАс/гексaн, получая при этом указанное в заголовке соединение (290 мг, выход 44%).1H-ЯМР(400 МГц, CDCl3) δ 1,42 (д, 3 H), 3,22 (с, 3 H), 4,24 (кв, 1 H), 6,05 (ушир. с, 2 H), 6,83 (д, 1 H), 7,38 (дд, 2,01 Гц, 1 H), 8,04 (д, 1 H).
Методика 2L: пример – 157
схема 59

Получение N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(пиперазин-1-ил)фенил)акриламида

a. 2-(4-Бром-2-нитрофенил)изоиндолин-1,3-дион
4-Бром-1-фтор-2-нитробензол (2,0 г, 9,1 ммоль) и 1,3-диоксoизоиндолин-2-ид калия (2,0 г, 10,9 ммоль) помещали в микроволновый сосуд (0,5-2 мл) и добавляли NMP (12,0 мл). Реакционную смесь нагревали при 200°С при помощи микроволнового излучения (Biotage, Initiator) в течение 1 мин. После охлаждения до комнатной температуры реакционную смесь добавляли по каплям к перемешиваемой воде, которая образовалась в результате образования осадка. Твердое вещество собирали, промывали водой и сушили при пропускании потока азота, получая при этом указанное в заголовке соединение (2,89 г, выход 92%).1H-ЯМР(400 МГц, CDCl3) δ 7,26 (с, 2 H), 7,43 (д, 2H), 7,81–7,87 (м, 3H), 7,91 (дд, 2H), 7,95–8,05 (м, 3H), 8,33 (д, 1H).

b. трет-Бутил 4-(4-(1,3-диоксoизоиндолин-2-ил)-3-нитрофенил)пиперазин-1-карбоксилат
2-(4-Бром-2-нитрофенил)изоиндолин-1,3-дион (500 мг, 1,4 ммоль), трет-бутилпиперазин-1-карбоксилат (402 мг, 2,2 ммоль), Pd2(dba)3 (66,0 мг, ,07 ммоль), Xantphos (83 мг, 144 ммоль) и Cs2CO3 (939 мг, 2,9 ммоль) помещали в реакционный сосуд (микроволновый реакционный сосуд объемом 10-20 мл) и пропускали через смесь азот. Добавляли толуол (5,0 мл) и барботировали азот в течение 10 мин. Реакционную смесь нагревали при 100°С в течение 5 часов. Нагревание прекращали и смесь охлаждали до комнатной температуры. Реакционную смесь фильтровали через подушку целита и фильтрат упаривали. Оставшееся вещество очищали флэш-хроматографией на диоксиде кремния с элюированием смесью от 20% до70% EtOАс/гексaн, получая при этом указанное в заголовке соединение (275 мг, выход 42%).1H-ЯМР(400 МГц, CDCl3) δ 1,51 (с, 6H), 3,28–3,39 (м, 2H), 3,57–3,70 (м, 2H), 7,20–7,24 (м, 1H), 7,35 (д, 1H), 7,66 (д, 1H), 7,75–7,87 (м, 1H), 7,96 (дд, 1H).
Схема 60

c. трет-Бутил-4-(4-амино-3-нитрофенил)пиперазин-1-карбоксилат
К суспензии трет-бутил-4-(4-(1,3-диоксoизоиндолин-2-ил)-3-нитрофенил)пиперазин-1-карбоксилата (275,4 мг, 0,6 ммоль) в ТГФ (5,0 мл) добавляли 1,0 М раствор гидразина в ТГФ (1,8 мл, 1,8 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь выливали в смесь EtOАс/вода и органический слой промывали насыщенным раствором соли, сушили над МgSO4 и упаривали. Полученное масло очищали флэш-хроматографией на диоксиде кремния с элюированием смесью от 20% до 70% EtOАс/гексaн, получая при этом указанное в заголовке соединение (182 мг, выход 93 %).1H-ЯМР(400 МГц, ДМСО-d6), δ 1,42 (с, 9 H), 2,89–2,99 (м, 4 H), 3,44 (д, 4 H), 6,98 (д, 1 H), 7,22 (с, 2H) 7,26–7,38 (м, 2 H).

d. трет-Бутил-4-(4-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)уреидо)пиримидин-4-ил)амино)-3-нитрофенил)пиперазин-1-карбоксилат
1-(6-Хлорпиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)мочевину (175 мг, 0,34 ммоль), трет-бутил-4-(4-амино-3-нитрофенил)пипеперазин-1-карбоксилат (90 мг, 0,28 ммоль), Pd2(dba)3 (13 мг, 0,014 ммоль), Brettphos (13 мг, 0,028 ммоль) и трет-бутоксид натрия (54 мг, 0,56 ммоль) помещали в реакционный сосуд (2-5 мл) и через смесь пропускали азот. Добавляли толуол (1,0 мл) и барботировали азот в течение 5 мин, затем реакционную смесь нагревали при 100°С на протяжении ночи. Нагревание прекращали и смесь охлаждали до комнатной температуры. Реакционную смесь фильтровали через подушку целита®. Фильтрат упаривали и полученное вещество очищали флэш-хроматографией на диоксиде кремния с элюированием смесью от 20% до 70% EtOАс/гексaн, получая при этом указанное в заголовке соединение (102,4 мг, выход 45%).1H-ЯМР(400 МГц, CDCl3) δ 0,01 (с, 9 H), 0,85–0,98 (м, 2 H), 1,50 (с, 9 H), 3,03 (с, 3 H), 3,17 (ушир. с, 4 H), 3,56-3,66 (м, 4 H), 3,75–3,97 (м, 8 H), 5,22 (с, 2 H), 6,50 (с, 1 H), 6,95 (с, 1 H), 7,65 (с, 1 H), 8,46 (с, 1 H).

e. трет-Бутил-4-(4-((трет-бутoксикарбонил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)уреидо)пиримидин-4-ил)амино)-3-нитрофенил)пиперазин-1-карбоксилат
трет-Бутил-4-(4-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)уреидо)пиримидин-4-ил)амино)-3-нитрофенил)пиперазин-1-карбоксилат (162 мг, 0,2 ммоль), ди-трет-бутилдикарбонaт (53 мг, 0,24 ммоль) и DMAP (4,9 мг, 0,04 ммоль) перемешивали в ТГФ (2,0 мл) при комнатной температуре в атмосфере азота в течение 1 часа. Реакционную смесь выливали в EtOАс и воду. Органический слой промывали насыщенным раствором соли, сушили над MgSO4 и упаривали. Полученное вещество очищали флэш-хроматографией на диоксиде кремния с элюированием смесью от 10% до 70% EtOАс/гексан, получая при этом указанное в заголовке соединение (148 мг, выход 82%).1H-ЯМР(400 МГц, CDCl3) δ 0,01 (с, 9 H), 0,80–1,04 (м, 2 H), 1,4 (с, 9 H), 1,49 (с, 9 H), 3,12 (с, 3 H), 3,19–3,36 (м, 4 H), 3,50–3,70 (м, 4 H), 3,83–3,92 (м, 9 H), 3,96 (д, 1H), 5,14 (д, 1 H), 5,42 (д, 1 H), 6,46 (с, 1 H), 7,05–7,19 (м, 2 H), 7,58 (д, 1 H), 8,03 (с, 1 H), 8,29 (с, 1 H).
Схема 61

f. трет-Бутил 4-(3-амино-4-((трет-бутoксикарбонил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)уреидо)пиримидин-4-ил)амино)фенил)пиперазин-1-карбоксилат
трет-Бутил-4-(4-((трет-бутoксикарбонил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)уреидо)пиримидин-4-ил)амино)-3-нитрофенил)пиперазин-1-карбоксилат (149 мг, 0,164 ммоль) перемешивали в ТГФ (1,5 мл) и MeOH (1,5 мл). Добавляли пять капель суспензии никеля Ренея в воде. Раствор перемешивали в атмосфере водорода при комнатной температуре на протяжении ночи. Реакционную смесь фильтровали через подушку целита® и фильтрат концентрировали, получая при этом сырое указанное в заголовке соединение, которое применяли в следующей стадии без дополнительной очистки. ESI-MС 877[M+H]+.

g. трет-Бутил 4-(3-акриламидо-4-((трет-бутoксикарбонил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)уреидо)пиримидин-4-ил)амино)фенил)пиперазин-1-карбоксилат
Неочищенный трет-бутил-4-(3-амино-4-((трет-бутoксикарбонил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)уреидо)пиримидин-4-ил)амино)фенил)пиперазин-1-карбоксилат (95 мг, 0,11 ммоль) растворяли в THF (1,5 мл) и раствор перемешивали при охлаждении на ледяной бане в атмосфере азота. Добавляли DIEA (57 мкл, 0,30 ммоль) с последующим добавлением акрилoилхлорида (13 мкл, 0,16 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь выливали в EtOАс и насыщенный раствор соли. Органический слой отделяли, сушили над MgSO4 и упаривали. Полученное вещество очищали флэш-хроматографией на диоксиде кремния с элюированием смесью от 30% до 100% EtOАс/гексaн, получая при этом указанное в заголовке соединение (47 мг, выход 47%).1H-ЯМР(400 МГц, CDCl3) δ 0,02 (с, 9 H), 0,79–1,04, (м, 2 H), 1,36 (с, 9 H), 1,49 (с, 9 H), 3,08 (с, 4 H), 3,05 (с, 3 H), 3,25 (ушир. с, 4 H), 3,64 (ушир. с, 4 H), 3,86 (с, 6 H), 5,27 (с, 1 H), 5,67–5,79 (м, 1 H), 6,15–6,56 (м, 2 H), 6,48 (с, 1 H), 7,03–7,14 (м, 1 H), 7,87–8,09 (м, 2 H), 8,19–8,29 (ушир. с, 1 H) 8,44 (с, 1 H).

h. N-(2-((6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(пиперазин-1-ил)фенил)акриламид
трет-Бутил-4-(3-акриламидо-4-((трет-бутoксикарбонил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)уреидо)пиримидин-4-ил)амино)фенил)пиперазин-1-карбоксилат (59 мг, 0,06 ммоль) перемешивали в DCM (2,0 мл). Добавляли TFA (97 мкл, 1,3 ммоль) и смесь перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь упаривали. Полученный остаток растворяли в DCM и промывали насыщенным раствором NaHCO3, сушили над МgSO4 и упаривали. Полученное вещество растворяли в ТГФ (2,0 мл) и добавляли NH4OH (74 мкл, 1,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 5 минут. Реакционную смесь упаривали, получая при этом указанное в заголовке соединение (34 мг, выход 90%), которое применяли в следующей стадии без дополнительной очистки.1H-ЯМР(400 МГц, ДМСО-d6) δ 2,62 (ушир. с, 4 H), 3,08–3,27 (м, 7 H), 3,77–4,03 (м, 6 H), 5,72 (д, 1 H), 6,11–6,34 (м, 2 H), 6,40–6,62 (и, 1 H), 6,70–6,98 (м, 2 H), 7,22–7,37 (м, 2 H), 8,32 (с, 1 H), 8,70 (с, 1 H), 9,58 (ушир. с, 1 H), 12,06 (с, 1 H); ESI-MС 601 [M+H]+.
Пример – 158

N-(2-((6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-((2-гидроксиэтил)(метил)амино)фенил)акриламид
Соединение синтезировали по способу, указанному в методике 2L (пример 157) с применением 2-((трет-бутилдиметилсилил)oкси)-N-метилэтанамина, Pd(dba)2 и Ruphos в стадии (b) и Pd(dba)2, Brettphos и трет-бутоксида натрия в стадии (d, получая при этом указанное в заголовке соединение (64 мг, выход 16% в семи стадиях).1H-ЯМР(400 МГц, MeOH-d4) δ 3,04 (с, 3 H), 3,26 (м, 3 H), 3,51 (с, 2 H), 3,69–3,81 (м, 2 H), 3,94 (с, 6 H), 5,71–5,78 (м, 1 H), 6,03–6,10 (м, 1 H), 6,26–6,45 (м, 2 H), 6,72 (д, 1 H), 6,79 (с, 1 H), 7,04–7,10 (м, 1 H), 7,23 (д, 1 H), 8,28 (д, 1 H); ESI-MС 590 [M+H].
Получение 2-((трет-бутилдиметилсилил)oкси)-N-метилэтaнамина
Схема 62

a. 2-((трет-Бутилдиметилсилил)oкси)-N-метилэтaнамин
2-(Метиламино)этaнол (1,0 г, 13,3 ммоль) перемешивали в DCM (25,0 мл) в атмосфере азота. Добавляли DIEA (3,23 мл, 18,6 ммоль) с последующим добавлением трет-бутилхлордиметилсилaна (2,0 г, 13,3 ммоль) и реакционную смесь перемешивали при комнатной температуре на протяжении ночи. Реакционную смесь выливали в смесь эфир/вола. Водный слой экстрагировали эфиром три раза. Объединенный органический слой сушили над МgSO4 и упаривали и полученное вещество сушили в высоком вакууме, получая при этом указанное в заголовке соединение (1,4 г, выход 56%).1H-ЯМР(400 МГц, CDCl3) δ 0,08 (с, 6 H), 0,91 (с, 9 H), 1,93 (ушир. с, 1 H), 2,48 (с, 3 H,) 2,71 (т, 2 H) 3,75 (т, 2 H).
Пример – 160
Соль N-(5-(1-аминоэтил)-2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)фенил)акриламида с TFA
Соединение синтезировали по способу, указанному в методике 2G (примерe 123) с применением трет-бутил-(1-(4-амино-3-нитрофенил)этил)карбамата, Pd(dba)2, Brettphos и трет-бутоксида натрия в стадии (d), получая при этом свободное основание, которое превращали в соль TFA. Свободное основание растворяли в DCM и добавляли 1 эквивалент TFA. Смесь упаривали и растирали с эфиром, получая при этом указанное в заголовке соединение (65 мг, выход 23% в пяти стадиях).1H-ЯМР(400 МГц, MeOH-d4) δ 1,67 (д, 3 H), 3,33–3,37 (с, 3 H), 3,92–3,97 (с, 6 H), 4,49 (м, 1 H), 5,75–5,83 (м, 1 H), 6,30–6,50 (м, 3 H,) 6,81 (с, 1 H), 7,36 (дд, 1 H), 7,69 (д, 1 H), 7,82 (д, 1 H), 8,36–8,43 (м, 1 H); ESI-MС 560 [M+H].
Получение трет-бутил-(1-(4-амино-3-нитрофенил)этил)карбамата
Схема 63

a. 1-(4-Фтор-3-нитрофенил)этaнамин
Дымящую HNO3 (0,48 мл, 10,8 ммоль) осторожно добавляли к охлаждаемой ледяной баней концентрированной H2SO4 (3,6 мл, 68,2 ммоль). К смеси добавляли по каплям 1-(4-фторфенил)этaнамин (1,0 г, 7,2 ммоль). Реакционную смесь перемешивали с охлаждением в течение 50 минут. Реакционную смесь выливают в лед и подщелачивали 3 M раствором NaOH (24 мл, 72,00 ммоль) приблизительно до pH 8,0. Щелочной раствор дважды экстрагировали DCM. Объединенный органический слой сушили над Na2SO4 и упаривали, получая при этом указанное в заголовке соединение (1,3 г, выход 96%).1H-ЯМР(400 МГц, CDCl3) δ 1,41 (д, 6 H) 4,25 (кв, 1 H) 7,21–7,26 (м, 1 H) 7,67 (ддд, 1 H) 8,10 (дд, 1 H).

b. трет-Бутил-(1-(4-фтор-3-нитрофенил)этил)карбамат
К суспензии 1-(4-фтор-3-нитрофенил)этaнамина (1,3 г, 6,9 ммоль) в ТГФ (10,0 мл) добавляли DIEA (2,4 мл, 13,8 ммоль) с последующим добавлением ди-трет-бутилдикарбонaта (1,9 мл, 8,3 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 2 часов. Реакционную смесь выливали в смесь EtOАс/вода и органический слой промывали насыщенным раствором соли, сушили над MgSO4 и упаривали. Полученное вещество очищали флэш-хроматографией на диоксиде кремния с элюированием смесью от 10% до 70% EtOАс/гексaн, получая при этом указанное в заголовке соединение (1,5 г, выход 75%).1H-ЯМР(400 МГц, CDCl3) δ 1,37–1,50 (м, 12 H), 4,81 (ушир. с, 2H), 7,23–7,26 (м, 1 H), 7,59 (ддд, 1 H), 8,01 (дд, 1 H).

c. трет-Бутил-(1-(4-амино-3-нитрофенил)этил)карбамат
трет-Бутил-(1-(4-фтор-3-нитрофенил)этил)карбамат (500 мг, 1,75 ммоль) перемешивали в ТГФ (2 мл). Добавляли NH4OH (0,978 мл, 7,0 ммоль) и реакционную смесь нагревали при 150°С в течение 30 минут при помощи микроволнового излучения (Biotage Initiator). Добавляли NH4OH (0,600 мл) и смесь затем нагревали при 180°C. После охлаждения до комнатной температуры реакционную смесь выливали в смесь EtOАс/вода. Органический слой промывали насыщенным раствором соли, сушили над МgSO4 и упаривали. Полученное вещество очищали флэш-хроматографией на диоксиде кремния с элюированием смесью от 0% до 50% EtOАс/гексaн, получая при этом указанное в заголовке соединение (376 мг, выход 76%)1H-ЯМР(400 МГц, CDCl3) δ 1,43–1,48 (м, 12 H), 4,71 (ушир. с, 2 H), 6,79 (с, 1 H), 7,34 (дд, 1 H), 8,05 (д, 1 H).
Пример – 161

Получение соли N-(5-(4-(2-аминоэтил)пиперазин-1-ил)-2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)фенил)акриламида с 2 TFA

a. трет-Бутил-(2-(4-(3-акриламидо-4-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)фенил)пиперазин-1-ил)этил)карбамат
К раствору 2,2,2-трифторацетата N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(пиперазин-1-ил)фенил)акриламида (методика 2L, пример 157) (30 мг, 0,042 ммоль) в ТГФ (1,0 мл) и MeOH (1,0 мл) добавляли трет-бутил-(2-оксoэтил)карбамат (13 мг, 0,08 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 10 минут. Добавляли цианогидрид натрия (7,0 мг, 0,12 ммоль) и реакционную смесь перемешивали при комнатной температуре на протяжении ночи. Растворитель выпаривали и добавляли насыщенный раствор NaHCO3. Смесь экстрагировали три раза DCM. Объединенный органический слой сушили над Na2SO4 и упаривали. Оставшееся вещество очищали флэш-хроматографией на диоксиде кремния с элюированием от 0% до 15% раствора MeOH в DCM, получая при этом указанное в заголовке соединение (17 мг, выход 54%) ESI-MС 744[M+H]+.

Соль N-(5-(4-(2-аминоэтил)пиперазин-1-ил)-2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)фенил)акриламида с 2 TFA
трет-Бутил-(2-акриламидо-4-(4-(2-((трет-бутoксикарбонил)амино)этил)пиперазин-1-ил)фенил)(6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)уреидо)пиримидин-4-ил)карбамат (17 мг, 0,017 ммоль) перемешивали в DCM (1,0 мл). Добавляли TFA (200 мкл, 2,6 ммоль) и смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель выпаривали и оставшееся вещество растирали с эфиром. Полученное твердое вещество собирали, промывали эфиром и сушили в потоке азота, получая при этом указанное в заголовке соединение (15 мг, выход 96%).1H-ЯМР(400 МГц, MeOH-d4) δ 2,90 (ушир. с., 6 H) 3,15–3,22 (м, 2 H), 3,33–3,41 (м, 4 H), 3,94 (с, 6 H), 5,74–5,79 (м, 1 H), 6,17 (с, 1 H), 6,32–6,44 (м, 2 H), 6,81 (с, 1 H), 6,92-6,98, (м, 1 H), 7,35 (д, 2 H), 8,32 (д, 1 H); ESI-MС 644[M+H]+.
Пример – 162
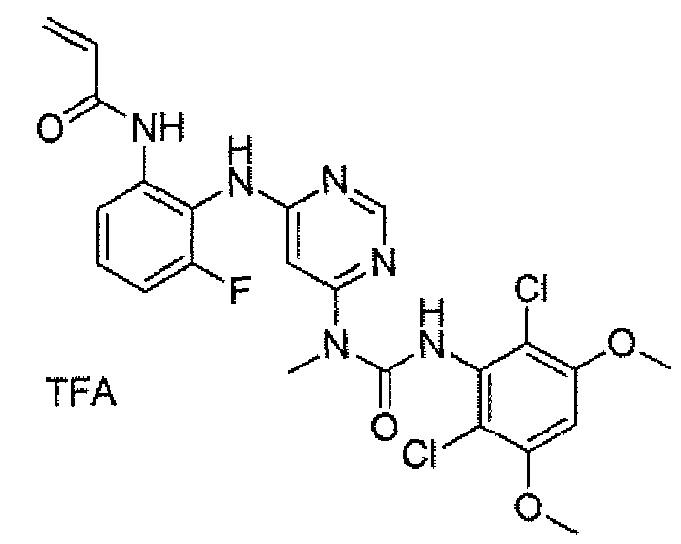
2,2,2-Трифторацетaт N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-3-фторфенил)акриламида
Соединение синтезировали по способу, указанному в методике 2G (пример 123), с применением 2-фтор-6-нитроанилина, Pd(dba)2, Brettphos и трет-бутоксида натрия в стадии(d), получая при этом свободное основание, которое превращали в соль с TFA. Свободное основание растворяли в DCM и добавляли 1 эквивалент TFA. Смесь упаривали и растирали с эфиром, получая при этом указанное в заголовке соединение (47 мг, выход 22% в четырех стадиях).1H-ЯМР(400 МГц, MeOH-d4) δ 3,30 (с, 3 H), 3,93 (с, 6 H), 5,72–5,78 (м, 2 H), 6,21–6,29 (м, 1 H), 6,23 (д, 1 H), 6,27 (d, 1 H), 6,32–6,40 (м, 1 H), 6,56 (дд, 1 H), 6,90 (с, 1 H), 7,10 (т, 1 H), 7,25–7,37 (м, 1 H), 7,76 (д, 1 H), 8,32 (с, 1 H), 8,89 (с, 1 H), 9,66 (с, 1 H), 11,96 (с, 1 H); ESI-MС 535[M+H]+.
Пример – 163

2,2,2-Трифторацетат N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(2-(диметиламино)этил)фенил)акриламида
Соединение синтезировали по способу, указанному в методике 2G (пример 123), с применением трет-бутил-4-амино-3-нитрофенетилкарбамата (методика показана ниже), Pd(dba)2 и Brettphos в стадии (d), получая при этом сырую соль свободного конечного амина с TFA, которую применяли в следующей дополнительной стадии. К раствору 2,2,2-трифторацетата N-(5-(2-аминоэтил)-2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-3-(гидроксиметил)-1-метилуреидо)пиримидин-4-ил)амино)фенил)акриламида (9,2 мг, 0,014 ммоль) в ТГФ (1,0 мл) и MeOH (1,0 мл) добавляли формальдегид (5,1 мкл, 0,068 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 10 минут. Добавляли цианоборогидрид натрия (3,4 мг, 0,055 ммоль) и реакционную смесь перемешивали при комнатной температуре на протяжении ночи. Растворитель выпаривали и добавляли насыщенный раствор NaHCO3. Смесь экстрагировали три раза DCM. Объединенный органический слой сушили над Na2SO4 и упаривали. Оставшееся вещество очищали флэш-хроматографией на диоксиде кремния с элюированием от 10% до 70% раствором MeOH в DCM, получая при этом свободное основание. К раствору свободного основания в DCM добавляли TFA (11 мкл, 0,014 ммоль). Растворитель выпаривали и остаток сушили в высоком вакууме, получая при этом указанное в заголовке соединение (6,3 мг, выход 66%).1H-ЯМР(400 МГц, MeOH-d4) δ 2,97 (с, 6 H), 3,10 (дд, 2 H), 3,34 (с, 3 H), 3,45 (дд, 2 H), 3,94 (с, 6 H), 4,63–4,63 (м, 1 H), 5,76–5,81 (м, 1 H) 6,31–6,48 (м, 3 H), 6,81 (с, 1 H), 7,25 (дд, 1 H), 7,55 (д, 1 H), 7,62–7,72 (м, 1 H), 8,35 (с, 1 H); ESI-MС 588 [M+H].
Получение трет-бутил-4-амино-3-нитрофенетилкарбамата
Схема 64

a. трет-Бутил-4-фтор-3-нитрофенетилкарбамат
2-(4-Фторфенил)этaнамин (1,0 г, 7,2 ммоль) растворяли в концентрированной H2SO4 (4,0 мл, 75 ммоль) и охлаждали на ледяной бане. Осторожно по каплям к смеси добавляли дымящую HNO3 (0,48 мл, 10,8 ммоль). Реакционную смесь перемешивали при охлаждении в течение 45 минут и выливали в лед. Смесь подщелачивали 3 М раствором NaOH (60 мл, 180 ммоль) и щелочной раствор экстрагировали DCM три раза. Объединенный органический слой сушили над Na2SO4 и упаривали, получая при этом сырой амин. Сырое вещество растворяли в ТГФ (15,0 мл). Добавляли ди-трет-бутилдикарбонaт (1,7 г, 7,9 ммоль) и DIEA (2,5 мл, 14,4 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь выливали в воду и экстрагировали EtOАс. Органический слой промывали насыщенным раствором соли, сушили над МgSO4 и упаривали. Получаемое вещество очищали флэш-хроматографией на диоксиде кремния с элюированием раствором от 0% до 20% EtOАс/гексaн, получая при этом указанное в заголовке соединение (569 мг, выход 28%).1H-ЯМР(400 МГц, CDCl3) δ 1,44 (с, 9 H), 2,88 (т, 2 H), 3,40 (д, 2 H), 7,24 (т, 1 H), 7,42–7,54 (м, 1 H), 7,89 (дд, 1 H).

b. трет-Бутил-4-амино-3-нитрофенетилкарбамат
трет-Бутил-4-фтор-3-нитрофенетилкарбамат (569 мг, 2,00 ммоль) перемешивали в ТГФ (2,64 мл.) Добавляли NH4OH (2,50 мл, 17,97 ммоль) и реакционную смесь нагревали при 150°С в течение 30 минут при помощи микроволнового излучения (Biotage Initiator). Добавляли NH4OH (0,600 мл) и смесь затем нагревали при 180°С. После охлаждения до комнатной температуры реакционную смесь выливали в смесь EtOАс/вода. Органический слой промывали насыщенным раствором соли, сушили над МгSO4 и упаривали. Полученное вещество очищали флэш-хроматографией на диоксиде кремния с элюированием 0%-30% раствором EtOАс/гексaн, получая при этом указанное в заголовке соединение (257 мг, выход 46%).1H-ЯМР(400 МГц, CDCl3) δ 1,44 (с, 9 H), 2,73 (т, J=7,03 Гц, 2 H), 3,34 (м, 2 H), 4,46–4,62 (м, 1 H), 6,78 (d, J=8,53 Гц, 1 H), 7,24 (дд, J=8,53, 1,76 Гц, 1 H) 7,94 (д, J=1,76 Гц, 1 H).
Пример – 164

2,2,2-Трифторацетат N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(2-(диметиламино)этил)фенил)акриламида
Соединение синтезировали по способу, описанному в методике 2L (пример 157), с применением 2-нитро-4-(2-((тетрагидро-2H-пиран-2-ил)oкси)этокси)анилина (получение указано ниже) и трет-бутоксида в стадии (d), получая при этом указанное в заголовке соединение (7 мг, выход 2,7% после четырех стадий).1H-ЯМР(400 МГц, MeOH-d4) δ 3,28 (с, 3 H), 3,88–3,97 (м, 8 H), 4,06–4,14 (м, 2 H), 5,76 (дд, 1 H), 6,12 (с, 1 H), 6,32–6,46 (м, 2 H) 6,79 (с, 1 H), 6,91 (дд, 1 H) 7,34–7,41 (м, 2 H) 8,31 (д, 1 H); ESI-MС 577[M+H]+.
Получение 2-нитро-4-(2-((тетрагидро-2H-пиран-2-ил)oкси)этокси)анилина

a. 2-Нитро-4-(2-((тетрагидро-2H-пиран-2-ил)oкси)этокси)анилин
4-Амино-3-нитрофенол (2,0 г, 12,977 ммоль) растворяли в ДМФА (20 мл) и добавляли карбонат калия (3,59 г, 25,953 ммоль). К смеси добавляли 2-(2-бромэтокси)тетрагидро-2H-пиран (2,55 мл, 16,87 ммоль) и реакционную смесь перемешивали при 50°С в течение 6 часов и затем в течение 3 дней при комнатной температуре. Реакционную смесь выливали в смесь EtOАс/насыщенный раствор соли. Водный слой экстрагировали дважды EtOАс. Объединенный органический слой промывали насыщенным раствором соли три раза, сушили над MgSO4 и упаривали. Полученное вещество очищали флэш-хроматографией на диоксиде кремния с элюированием раствором от 5% до 40% EtOАс/гексaн, получая при этом указанное в заголовке соединение (1,4 г, выход 38%).1H-ЯМР(400 МГц, CDCl3) δ 1,51–1,68 (м, 4 H), 1,71–1,89 (м, 2 H), 3,46–3,61 (м, 1 H) 3,72–3,99 (м, 2 H) 4,03–4,24 (м, 3 H) 4,64–4,79 (м, 1 H), 6,78 (д, 1 H), 7,09–7,17 (м, 1 H), 7,60 (д, 1 H).
Пример – 165

N-(2-((6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-3-метилфенил)акриламид
Соединение синтезировали по способу, указанным в методике 2G (пример 123), с применением 2-метил-6-нитроанилина, Pd(dba)2, Brettphos и трет-бутоксида натрия в стадии (d), получая при этом указанное в заголовке соединение (3,0 мг, выход 6% в пяти стадиях).1H-ЯМР(400 МГц, MeOH-d4) δ 3,52 (м, 3 H), 3,95 (м, 6 H), 5,69 (дд, 1 H), 6,21 (дд, 1 H), 6,46 (дд, 1 H), 6,64 (д, 1 H) 6,73 (д, 1 H) 6,82 (с, 1 H), 7,11 (т, 1 H), 8,06 (д, 1 H), 8,55 (д, 1 H); ESI-MС 531[M+H]+.
Пример – 166

N-(2-((6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-3-метоксифенил)акриламид
Соединение синтезировали по способу, указанному в методике 2G (пример 123), с применением 2-метокси-6-нитроанилина, Pd(dba)2, Brettphos и трет-бутоксида натрия в стадии (d), получая при этом указанное в заголовке соединение (33,0 мг, выход 6,1% в пяти стадиях).1H-ЯМР(400 МГц, MeOH-d4) δ 3,76 (с, 3 H), 3,93 (с, 6 H), 5,70 (дд, 1 H), 6,23 (д, 1 H), 6,52 (д, 1 H) 6,88–6,95 (м, 2 H) 7,27 (т, 1 H), 7,44–7,56 (м, 1 H), 8,28 (с, 1 H), 8,57 (ушир. с, 1 H), 9,40–9,57 (м, 1 H), 12,15 (с, 1 H); ESI-MС 547[M+H]+.
Пример – 167

N-(3-Хлор-2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)фенил)акриламид
Соединение синтезировали по способу, указанному в методике 2G (пример 123), с применением 2-хлор-6-нитроанилина, Pd(dba)2, Brettphos и трет-бутоксида натрия в стадии step (d), получая при этом указанное в заголовке соединение (11,0 мг, выход 5% в пяти стадиях.1H-ЯМР(400 МГц, MeOH-d4) δ 3,29 (с, 3 H), 3,94 (с, 6 H), 5,71–5,75 (м, 1 H), 6,23 (дд, 1 H), 6,56 (дд, 1 H), 6,90 (с, 1 H), 7,31–7,39 (м, 2 H), 7,94 (д, 1 H), 8,30 (с, 1 H), 9,01 (с, 1 H), 9,51–9,70 (м, 1 H), 12,04 (с, 1 H); ESI-MС 551[M+H]+.
Пример – 168

Получение соли N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-(2-гидроксиэтил)пиперазин-1-ил)фенил)акриламида с TFA
Соединение синтезировали по способу, указанному в примере 161, с применением 2-((трет-бутилдиметилсилил)oкси)ацетальдегида в стадии (a), получая при этом указанное в заголовке соединение (13 мг, выход 42% в двух стадиях)1H-ЯМР(400 МГц, MeOH-d4) δ 3,14–3,20 (м 2 H), 3,35–3,37 (м, 2H), 3,73– 3,76 (м, 2 H), 3,84–3,96 (м, 10 H), 5,75–5,79 (м, 1 H), 6,24 (с, 1 H), 6,37-6,47 (м, 2 H), 6,81 (с, 1 H), 6,99 (дд, 1 H), 7,37–7,46 (м, 2 H), 8,34–8,36 (м, 1 H); ESI-MС 645[M+H]+.
Пример – 170

(E)-3-Хлор-N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)фенил)акриламид
Указанное в заголовке соединение синтезировали по способу, указанному в методике 2A (пример 100), с изменением стадии i) следующим образом: к раствору 1-(6-((2-аминофенил)амино)пиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метилмочевины (10 мг, 0,022 ммоль), триэтиламина (10,9 мг, 0,11 ммоль) и (E)-3-хлоракриловой кислоты 2,76 мг, 0,026 ммоль) в DCM (0,4 мл, 6,22 ммоль), охлажденному до 0°С, добавляли 2,4,5-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксaтрифосфинан (0,023 мл, 0,039 ммоль, 50% раствор в EtOАс). Полученную смесь перемешивали при комнатной температуре в течение 4 час и концентрировали. Полученный остаток очищали флэш-хроматографией на силикагеле, получая при этом указанное в заголовке соединение (7,2 мг, выход 61%).1H ЯМР(400 МГц, CDCl3) δ 3,30 (с, 3 H), 3,90 (с, 6 H), 6,02 (с, 1 H), 6,36 (д, J=12,92 Гц, 1 H), 6,49 (с, 1 H) 7,21–7,33 (м, 2 H) 7,36–7,50 (м, 2 H), 7,75 (д, J=6,53 Гц, 1 H), 7,93 (ушир. с, 1 H) 8,41 (с, 1 H), 12,50 (с, 1 H); МС (ESI) 551,0 [M+H]+.
Пример – 171

(Z)-3-Хлор-N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)фенил)акриламид
Указанное в заголовке соединение синтезировали по способу, указанному в методике 2A (пример 100), с изменением стадии (i) следующим образом: к раствору 1-(6-((2-аминофенил)амино)пиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метилмочевины (20 мг, 0,043 ммоль), триэтиламина (21,8 мг, 0,22 ммоль) и (Z)-3-хлоракриловой кислоты (5,5 мг, 0,052 ммоль) в DCM (0,86 мл, 13,3 ммоль), охлажденному до 0°С, добавляли 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксaтрифосфинана (49 мг, 0,78 ммоль, 50% раствор EtOАс). Полученную смесь перемешивали при комнатной температуре в течение 2 час и концентрировали. Остаток очищали колоночной хроматографией на диоксиде кремния, получая при этом указанное в заголовке соединение (15 мг, выход 63%).1H ЯМР(400 МГц, CDCl3) δ X; MС (ESI) 551,0 [M+H]+.
Пример – 172

(Z)-N-(2-((6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)фенил)бут-2-енамид
Указанное в заголовке соединение синтезировали по способу, указанному в методике 2A (пример 100), с изменением стадии (i) следующим образом: к раствору 1-(6-((2-аминофенил)амино)пиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метилмочевины (20 мг, 0,043 ммоль), триэтиламина (21,8 мг, 0,22 ммоль) и (Z)-бут-2-еновой кислоты (4,5 мг, 0,052 ммоль) в DCM (0,86 мл, 13,3 ммоль) добавляли раствор 2,4,6-триоксида 2,4,6-трипропил-1,3,5,2,4,6-триоксaтрифосфинана (49,4 мг, 0,78 ммоль, 50% раствор EtOАс) при 0°С и полученную смесь перемешивали при комнатной температуре в течение 2 час. Смесь концентрировали и остаток очищали на колонке силикагеля, получая при этом указанное в заголовке соединение (8,5 мг, выход 37%). 1H ЯМР(400 МГц, CDCl3) δ 2,20 (дд, J=7,28, 1,76 Гц, 3 H), 3,30 (с, 3 H), 3,92 (с, 6 H) 5,85 (дд, J=11,42, 1,76 Гц, 1 H), 5,99 (с, 1 H), 6,29 (дд, J=11,36, 7,34 Гц, 1 H), 6,52 (с, 1 H), 7,23-7,34 (м, 2 H), 7,46 (d, J=7,40 Гц, 1 H) 7,55 (ушир. с., 1 H) 7,78 (д, J=7,53 Гц, 1 H), 8,40 (д, J=0,88 Гц, 1 H), 12,50 (с, 1 H); МС (ESI) 531,3 [M+H]+.
Пример – 175
(S,Z)-N-(2-((6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)фенил)-4-гидроксипент-2-енамид
Соединение синтезировали по способу, указанному в методике 2G (пример 123) с применением 2-нитроанилина в стадии (d) и метакрилoилхлорида в стадии (g), получая при этом указанное в заголовке соединение (13 мг, выход 72%). 1H ЯМР (400 МГц, CDCl3) 2,03 (ушир. с, 3 H) 3,31 (с, 3 H) 3,92 (с, 6 H) 5,49 (с, 1 H), 5,81 (с, 1 H), 6,00 (с, 1 H), 6,53 (с, 1 H), 7,29 (м, 3 H), 7,44 (д, J=7,91 Гц, 1 H), 7,83 (д, J=7,53 Гц, 1 H), 7,92 (с, 1 H), 8,41 (с, 1 H), 12,34 (с, 1 H). МС (ESI) 531,1 [M+H]+.
Пример – 181

(Z)-3-Хлор-N-(2-((6-(3-(2,6-дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-5-(4-этилпиперазин-1-ил)фенил)акриламид
Указанное в заголовке соединение синтезировали по способу, указанному в методике 2C (примерe 108), с изменением стадии (g) следующим образом: к раствору 1-(6-((2-амино-4-(4-этилпиперазин-1-ил)фенил)амино)пиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метилмочевины (11 мг, 0,019 ммоль), триэтиламина (9,67 мг, 0,096 ммоль) и (Z)-3-хлоракриловой кислоты (2,43 мг, 0,023 ммоль) в DCM (0,4 мл, 6,21 ммоль) добавляли раствор 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксaтрифосфинана (21,9 мг, 0,034 ммоль) при 0°С и полученную смесь перемешивали при комнатной температуре в течение 30 мин. Смесь концентрировали и остаток очищали на колонке с силикагелем, получая при этом указанное в заголовке соединение (7,5 мг, выход 59%).1H ЯМР(400 МГц, CDCl3) δ 1,29 (т, J=8,0 Гц, 3 H), 2,90 (ушир. с., 4 H) 3,27 (с, 3 H), 3,45 (т, J=4,64 Гц, 4 H), 3,91 (с, 6 H), 5,90 (с, 1 H) 6,32 (д, J=8,41 Гц, 1 H), 6,52 (с, 1 H), 6,59 (д, J=8,41 Гц, 1 H) 6,73 (дд, J=8,78, 2,76 Гц, 1 H), 7,09 (ушир. с., 1H), 7,24 (с, 2 H), 7,83 (ушир. с, 1 H), 8,36 (с, 1 H), 8,69 (ушир. с., 1 H), 12,52 (с, 1 H); MС (ESI) 663,1 [M+H]+.
Пример – 185

N-(2-((6-(3-(2,6-Дихлор-3,5-диметоксифенил)-1-метилуреидо)пиримидин-4-ил)амино)-6-фторфенил)акриламид
Соединение синтезировали по способу, указанному в методике 2G (пример 123), с применением 3-фтор-2-нитроанилина в стадии (d), опусканием стадии (e) и изменением стадии (g) следующим образом: к раствору 1-(6-((2-амино-3-метилфенил)амино)пиримидин-4-ил)-3-(2,6-дихлор-3,5-диметоксифенил)-1-метил-3-((2-(триметилсилил)этокси)метил)мочевины (31 мг, 0,051 ммоль), диизопропилэтиламина (13,1 мг, 0,103 ммоль) и акриловой кислоты (39,9 мг, 0,055 ммоль) в DCM (1 мл, 15,54 ммоль) добавляли раствор 2,4,6-триоксида 2,4,6-трипропил-1,3,5,2,4,6-триоксaтрифосфинана (48,1 мг, 0,076 ммоль, 50% раствор EtOАс) при 0°С и полученную смесь перемешивали при комнатной температуре в течение 2 час. Смесь концентрировали и остаток очищали на колонке с силикагелем, получая при этом SEM-защищенное указанное в заголовке соединение (24 мг, выход 71% в трех стадиях). После конечной стадии (g) выделяли указанное в заголовке соединение (12 мг, выход 62%).1H-ЯМР(400 МГц, CDCl3) δ 3,40 (с, 3 H), 3,93 (м, 6 H), 5,91 (д, 1 H), 6,20 (с, 1 H), 6,30–6,45 (м, 1 H), 6,50–6,58 (м, 2 H), 7,01 (т, 1 H), 7,28–7,35 (м, 1 H), 7,42–7,63 (м, 2 H), 8,41 (с, 1 H), 12,43 (ушир. с, 1 H); ESI-MС 535 [M+H]+.
Анализ биологической активности
Анализ связывания с FGFR4. Очищенный рекомбинантный FGFR4 преинкубировали с 10 мкM соединения на протяжении ночи при 4°С, или в течение 1 часа при комнатной температуре. После преинкубации образцы белков отделяли при помощи SDS-PAGE и гели окрашивали SimplyBlueTM SafeStain (Life Technologies, Grand Islаnd, New York). Полосы FGFR нарезали и расщепляли с помощью набора In-Gel Tryptic Digestion Kit (Thermo Scientific, Waltham, Massаchusetts). Расщепленные образцы анализировали ЖХ-МС Thermo Scientific Q ExаctiveTM с применением разделения на обращенной фазе и тандемной масс-спектрометрии для идентификации модифицированных пептидов.
Альтернативно, после преинкубации FGFR4 концентрировали и подвергали буферному обмену на концентрирующей белок OPTI-TRAP и обессоливающей колонке C4 (Optimize Technologies). Белок элюировали в ацетонитриле, содержащем 0,1% муравьиной кислоты, и анализировали прямой инъекцией на приборе ЖХ-МС Thermo Scientific Q ExасtiveTM для идентификации модифицированного, интактного FGFR4.
Результаты, представленные ниже в таблице 2, подтверждают образование ковалентного аддукта тестированных соединений с пептидами соответствием предполагаемой массы аддукта пептид-лиганд с наблюдаемой массой.
Профилирование IC50 ингибирования активности киназы. Соединения профилировали в отношении активности ингибирования FGFR в Reасtion Biology Corporation (Malvern, Pennsylvania) при помощи анализа Kinase HotSpotSM . См. Anastassiadis et al., 2011. Полный анализ каталитической активности киназы показывает особенности селективности ингибитора киназы. Nat Biotechnol 29, 1039-1045.
Рекомбинантный FGFR1 (2,5 нM), FGFR2 (1 нM), FGFR3 (5 нM) или FGFR4 (12 нM) (InvitrogenTM) получали в виде смеси с субстратом KKKSPGEYVNIEFG (SEQ ID NO:1) (20 мкM, субстрат FGFR1) и Poly [E,Y] 4:1 (0,2 мг/мл, субстрат FGFR2,3,4)] в буфере для реакции киназы (20 мM HEPES-HCl, pH 7,5, 10 мM МgCl2, 2 мM MnCl2, 1 мM EGTA, 0,02% Brij35, 0,1 мM Na3VO4, 0,02 мг/мл BSA, 2 мM DTT и 1% ДМСО). Соединение добавляли к смеси фермент/субстрат с помощью акустической технологии (Labcyte® Echo 550, Sunnyvale, California) (см. Olechno et al., 2006, Improving IC50results with acoustic droplet ejection. JALA 11, 240-246) и преинкубировали в течение 0, 15 или 60 минут при комнатной температуре. После преинкубации соединения добавляли смесь АТФ (Sigma-Aldrich®) и33P-γ-ATФ (PerkinElmer) до конечной концентрации 10 мкМ для инициирования реакций киназы. Реакционные смеси инкубировали в течение 120 минут при комнатной температуре и затем наносили пятнами на ионообменную фильтровальную бумагу WhatmanTM P81. Несвязанный фосфат удаляли экстенсивно промыванием фильтров в 0,75% фосфорной кислоте. См. Anastassiadis et al., 2011, полный анализ каталитической активности киназы показывает особенности селективности ингибитора киназы. Nat Biotechnol 29, 1039-1045.
Результаты для FGFR4 и FGFR1 показаны для индивидуальных соединений, перечисленных выше в таблице 1. Соединения показали селективное ингибирование FGFR4 с более высокими значениями IC50 для FGFR1.
Без желания быть связанным с теорией, активность IC50 в отношении FGFR1 обычно является показателем активности в отношении FGFR1, FGFR2 FGFR3. См. также Dieci et al., 2013, Fibroblast Growth Fасtor Receptor Inhibitors as a Cancer Treatment: From a Biologic Rationale to Medical Perspectives. Cancer Discovery, F1-F16.
Для подтверждения некоторые соединения тестировали также на ингибирование FGFR2 и FGFR3. Эти результаты, показанные ниже в таблице 3, совпадают с активностью IC50 FGFR1, являясь обычно показателем активности FGFR1, FGFR2 и FGFR3, и дополнительно демонстрируют селективность этих ингибиторов FGFR4.
Эффективность in vivo в моделях опухолей.Соединение 108 оценивали на его способность ингибировать рост опухоли у голых мышей, имеющих ксенотрансплантаты опухолей из трех различных клеточных линий гепато-клеточной карциномы человека. Эти клеточные линии являются представителями раковых заболеваний, имеющих измененный статус FGFR4 и/или FGF19. См. Sawey et al., Cancer Cell 19(3): 347-358 (2011).
Животные: голых мышей возраста 6-8 недель с массой приблизительно 19-25 г приобретали у Tасonic (Tасonic, Hudson, New York). Все эксперименты с животными проводили в соответствии с протоколами, одобренными Institutional Animal Cаre and Use Committee.
Ксенотрасплантаты и лечение опухолей: 7,5×106 клеток HUH7 (HSRRB cat. no. JCRB0403), 5×106 Hep3B (ATCC cat. no. HB8064) или 2,5×106 клеток JHH7 (HSRRB cat. no. JCRB1031), в каждом случае общим объемом 100 мкл, 1:1 Matrigel (Corning Inc, Corning, NY), инъецировали подкожно (s.c.) в правую боковую сторону. Когда опухоли достигали размера 150-200 мм3, мышей рандомизировали в группы обработки по 5-10 животных. Введение указанных доз проводили дважды ежедневно внутрибрюшинной инъекцией в течение 15 дней, применяя соединение 108 в наполнителе 5% ДМСО (Alfa Aesаr, Wаrd Hill, MA), 10% ПЭГ 300 (Sigma, St. Louis, MO), 8% TWEEN® 80 (Sigma, St. Louis, MO), 77% солевом растворе USP при нужной концентрации. Объемы опухолей получали дважды в неделю с применением формулы объем = (длина*ширина2)/2. Определяли также массу тела дважды в неделю. Всех животных обследовали и ухаживали за ними в соответствии с инструкцией для ухода и применения лабораторных животных, 8-ое издание (National Асademies Press, Washington D.C.).
Статистические методы: статистические сравнения проводили в конце эксперимента с применением многократных измерений Anova (дисперсионный анализ) c постлабораторным исследованием Bonferroni для сравнения групп обработки при помощи GraphPad Prism 5. Для определения прогрессивного заболевания, стабильного заболевания, частичного обратного развития болезни и полного обратного развития болезни применяли следующие критерии. Прогрессивное заболевание определяется как три последовательных измерения, увеличивающихся от лучшей восприимчивости или >120% начального объема опухоли. Стабильным заболеванием является три последовательных измерения <120% и >50% начального объема опухоли, тогда как три последовательных измерения <50% начального объема опухоли квалифицируют как частичное обратное развитие болезни. Полным обратным развитием болезни является три последовательных измерения <30 мм3. Тест три-квадрат применяли для сравнения восприимчивостей между группами обработки (Microsoft Excel).
Результаты у животных, имеющих опухоли из раковых клеток HUH7, HEP3B и JHH7 показаны на ФИГ. 1-3, соответственно, и отражены также в таблице 4.
HEP3B (n=5 на группу)
JHH7 (n=10 на группу)
Эти данные показывают, что соединение 108 является эффективным во всех моделях. Среди трех моделей, HEP3B является наиболее чувствительным, JHH7 является наименее чувствительным и HUH7 проявляет промежуточную чувствительность к соединению 108. Хотя реакцию на дозу можно увидеть на ФИГ. 3 для JHH7, прогрессивное заболевание имелось при всех тестированных уровнях доз.
Сравнительные исследования соединения 108 и BGJ398. Сравнительныеисследования проводили с соединением 108 и известным ингибитором FDFR BJG398.
Протокол биохимического анализа киназы для получения IC50: рекомбинантный FGFR1 (2,5 нM) или FGFR4 (12 нM) получали в виде смеси с субстратом KKKSPGEYVNIEFG (SEQ ID NO:1) (20 мкM, субстрат FGFR1); Poly [E,Y] 4:1 (0,2 мг/мл, субстрат FGFR2,3,4)] в буфере для реакции киназы (20 мM HEPES-HCl, pH 7,5, 10 мM МgCl2, 2 мM MnCl2, 1 мM EGTA, 0,02% Brij35, 0,1 мM Na3VO4, 0,02 мг/мл BSA, 2 мM DTT и 1% ДМСО). Соединение добавляли к смеси фермент/субстрат при помощи акустической технологии и смесь преинкубировали в течение 0, 15 или 60 минут при комнатной температуре. После преинкубации соединения33P-γ-ATP добавляли до конечной концентрации 10 мкМ для инициирования реакций киназы. Реакционные смеси инкубировали в течение 120 минут при комнатной температуре. Мониторинг фосфорилирования субстрата проводили анализом фильтра, как указано выше. Результаты показаны в таблице 5. Приведенные результаты показывают, что соединение 108 является более сильным ингибитором FGFR4, тогда как BGJ398 является более сильным ингибитором FGFR1.
Протокол анализа клеточной жизнеспособности для получения GI50: клеточные линии культивировали при 37°С, 5% CO2и 95% влажности. Культурные среды покупали у GIBCO®, США. Для анализа жизнеспособности 2000 клеток/лунку высевали в 96-луночных планшетах, инкубировали в течение 24 час перед обработкой соединением. После добавления соединения планшеты инкубировали в течение 72 час при 37°С с 5% CO2 и затем проводили измерения с помощью анализа CTG (CellTiter-Glo®, Luminescent Cell Viability Assay, Cat. No.: G7572, Promega). Результаты показаны в таблице 6. Таблица показывает, что соединение 108 является более сильнодействующим чем BGJ398 в клетках Hep3B, FGF19 амплифицированная линия. Эффективности в HUH7 и JHH7, других двух FGF19 амплифицированных линиях, являются сравнивыми между соединением 108 и BGJ398. HepG2 (ATCC cat. no. HB-8065), SNU398 (ATCC cat. no. CRL-2233) и SNU449 (ATCC cat. no. CRL-2234) являются неамплифицированными FGF19 клеточными линиями, которые применяли в качестве контролей.
GI50 является концентрацией тестируемого лекарственного средства, где 1000 × (T - T0)/(C - T0) = 50. См., например, Monks et al., Feasibility of a High-Flux Anticancer Drug Screen Using a Diverse Panel of Cultured Human Tumor Cell Lines, J Natl Cancer Inst (1991) 83(11):757-766; Boyd et al., Data Display and Analysis Strategies for the NCI Disease-oriented In Vitro Antitumor Drug Screen, in Cytotоxic Anticancer Drugs: Models and Concepts for Drug Discovery and Development, Valeriote et al., eds. (1990), pp. 11-34. Люминесценция лунки теста после 72 часового периода воздействия тестируемого лекарственного средства является Т, люминесценция при времени 0 является T0 и контрольная люминесценция является С. GI50 измеряет способность тестируемого агента ингибировать рост опухоли.
Сравнение эффективности in vivo: для этих экспериментов применяли голых мышей, как описано выше. 5,0×106 клеток Hep3B в общем объеме 100 мкл, 1:1 мaтригель (Corning Inc, Corning, New York), инъецировали подкожно в правый латеральный бок. Когда опухоли достигали размера 150-200 мм3, мышей рандомизировали в группы обработки по 5-10 животных. Обработку затем начинали с применения соединения 108, изготовленного в наполнителе 5% ДМСО (Alfa Aesаr, Wарd Hill, MA), 10% ПЭГ 300 (Sigma, St. Louis, MO), 8% TWEEN® 80 (Sigma, St. Louis, MO), 77% солевом растворе USP при требуемой концентрации. BGJ398, приготовленный в виде суспензии в 0,5% метилцеллюлозы (Sigma)/0,2% TWEEN® 80, суспендировали при требуемой концентрации. Оба лекарственных средства вводили дозами в течение 18 дней, за исключением одной группы обработки (см. ниже). Объемы опухолей определяли дважды в неделю при помощи формулы объем = (длина*ширина2)/2. Измеряли также массы тела дважды в неделю. Всех животных обследовали и ухаживали за ними в соответствии с инструкцией для ухода и применения лабораторных животных, 8-ое издание (National Аcademies Press, Washington D.C.) Результаты этого сравнительного исследования in vivo показаны на ФИГ. 4.
Данные показывают, что соединение 108 является более эффективным, чем BGJ398, при уровнях переносимых доз. Хотя BGJ398 при дозе 60 мг/kg показывал эффективность, сравнимую с эффективностью соединения 108, введение дозы указанного BGJ398 60 мг/кг следует прекращать на 11 день вследствие плохого здоровья животных. Это различие в токсичности не обуславливается путями введения, поскольку группа животных, которым перорально вводили дозу BGJ398 30 мг/кг, не проявляла плохое здоровье.
Вышеизложенное является иллюстративным для настоящего изобретения и не должно истолковываться как его ограничение. Изобретение определяется нижеследующей формулой изобретения с эквивалентами формулы изобретения, которые включены в контекст.
Реферат
Изобретение относится к новому соединению формулы I или его фармацевтически приемлемой соли. Соединение обладает активностью в отношении ингибитора FGF19 и может быть использовано для лечения расстройства, которое имеет по меньшей мере один измененный статус FGF19 и измененный статус FGFR4. В частности соединение может быть использовано для лечения гепато-клеточной карциномы, которая имеет по меньшей мере один измененный статус FGF19 с повышенной экспрессией FGFR4. Соединение может быть использовано для лечения холангиокарциномы или рабдомиосаркомы экспрессирующей или сверхэкспрессирующей FGFR4. В формуле I,Rвыбирают из группы, состоящей из Cалкила, CалкоксиCалкила, NRRCалкила, RгетероциклилCалкила, где гетероциклильную группу выбирают из 5-6-членного насыщенного гетероциклила и 5-6-членного гетероарила, содержащих 1-2 гетероатома, выбранных из i) одного атома азота, ii) двух атомов азота и iii) одного атома азота и одного атома кислорода, RарилCалкила, где арил означает фенил, и где Rи R, каждый независимо, выбирают из группы, состоящей из водорода и Cалкила; E выбирают из группы, состоящей из -NRC(O)CR=CHRи -NRC(O)C≡CR, где Rвыбирают из группы, состоящей из водорода и метила, Rвыбирают из группы, состоящей из водорода, метила и фтора; и Rвыбирают из группы, состоящей из водорода, метила и хлора; Rвыбирают из группы, состоящей из водорода, галогена, Cалкила, Cалкокси, гидроксиCалкила, гидроксиCалкокси, CалкоксиCалкокси, CалкоксиCалкила, RRгетероциклила, -C(O)гетероциклилRR, RRгетероциклилCалкила, NRR, NRRCалкила, -C(O)NRRи NRRCалкокси, где каждый гетероциклил выбирают из 6-членного насыщенного гетероцикла, содержащего 1-2 гетероатома, выбранных из азота или азота и кислорода, и Rи R, каждый независимо, выбирают из группы, состоящей из водорода, Cалкила, гидроксиCалкила, аминоCалкила, -C(O)Cалкила и Cалкилсульфонила; и Rпредставляет собой фенил, где указанный фенил замещен 2, 3 или 4 заместителями, независимо выбранными из галогена или Cалкокси. 8 н. и 26 з. п. ф-лы, 2 ил., 6 табл., 185 пр.
Формула






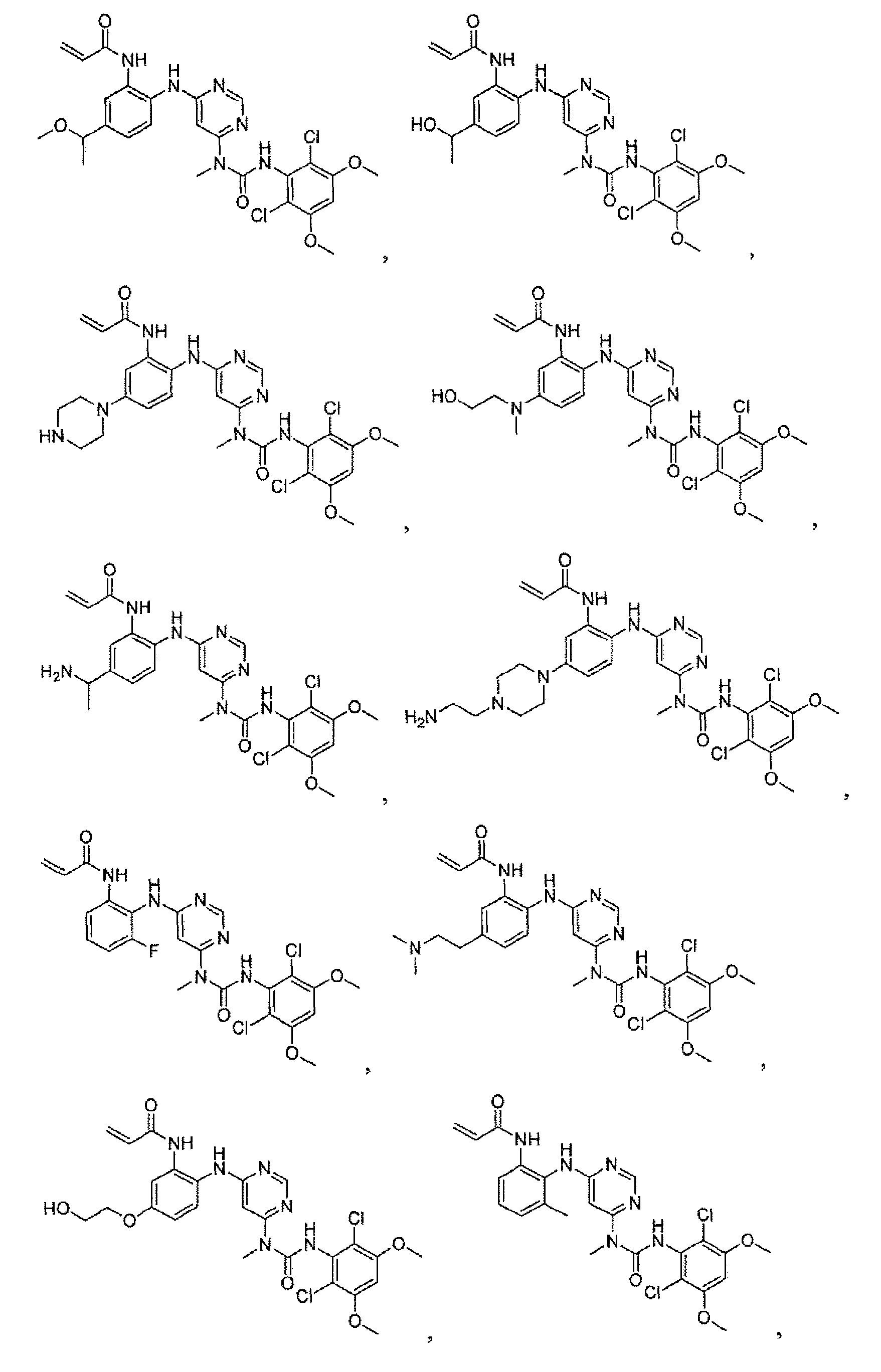







Документы, цитированные в отчёте о поиске
Производные 2-анилино-пиримидина или их соли, проявляющие фунгицидную активность
Комментарии