Лекарственная форма c постоянной скоростью высвобождения лекарственного вещества, ядро лекарственной формы и способ обеспечения облегченного высвобождения лекарственного вещества из лекарственной формы - RU2246295C2
Код документа: RU2246295C2
Чертежи












Описание
Область техники, к которой относится изобретение
Изобретение относится к контролируемой доставке фармацевтических агентов и их лекарственных форм. В частности, изобретение направлено на усовершенствованные способы, лекарственные формы и устройства для осуществления практически полного выхода активных агентов, имеющие расширяющийся выталкивающий слой и слой, содержащий лекарственное вещество, которое предназначено для распространения в среде применения.
Уровень техники
Некоторые лекарственные вещества могут предназначаться для доставки в больших дозах, иногда несколько раз в сутки, для достижения желательного терапевтического эффекта. Хотя большие суточные дозы лекарственного вещества могут применяться в виде многократных введений в течение суток, схемы лечения с многократным введением часто не являются предпочтительными вследствие проблем с получением согласия пациента, потенциальных побочных эффектов и опасности передозировки. В соответствии с этим отмечен сдвиг в сторону схем лечения с введением один или два раза в сутки, если это возможно, даже в тех случаях, когда имеется необходимость доставки больших доз лекарственного вещества в течение длительного времени, например, в течение 12-24 часов, как может быть в ряде случаев.
Для высоких значений суточных доз может потребоваться, чтобы нагрузка (доля) лекарственного вещества в композициях лекарственных веществ в лекарственных формах составляла не меньше, чем 20-90% или больше от общей массы композиции. Данные требования к нагрузке могут представлять собой проблемы в плане приготовления композиций и производства лекарственных форм, которые являются пригодными для перорального введения и могут проглатываться без излишних затруднений. Высокий уровень нагрузки лекарственным веществом может являться еще большей проблемой при приготовлении лекарственных форм, которые предназначены для введения ограниченное число раз в день, такого как введение один раз в сутки, поскольку требуется большая унифицированная лекарственная форма.
Были описаны различные устройства и способы, предназначенные для применения в плане использования при высоком уровне нагрузки лекарственным веществом. Например, в Патентах США No 4892778 и 4940465 описаны дозаторы для доставки целебного вещества в среду применения, которые включают полупроницаемую стенку, ограничивающую полость, содержащую слой расширяющегося материала, который выталкивает слой лекарственного вещества из полости, образованной стенкой. Выходное отверстие в устройстве имеет диаметр, практически такой же, как внутренний диаметр полости, образованной стенкой.
В Патенте США No 4915949 описан дозатор для доставки целебного вещества в среду применения, который включает полупроницаемую стенку, имеющую слой расширяющегося материала, который выталкивает слой лекарственного вещества из полости, образованной стенкой. Слой лекарственного вещества содержит отдельные маленькие пилюли, диспергированные в носителе. Выходное отверстие в устройстве имеет диаметр, практически такой же, как внутренний диаметр полости, образованной стенкой.
В Патенте США No 5126142 описано устройство для доставки ионофора скоту, которое включает полупроницаемую оболочку, в которой находятся композиция, содержащая ионофор и носитель, и расширяющийся гидрофильный слой, а также дополнительный элемент, который придает устройству достаточную плотность, чтобы оно сохранялось в мешке сетеобразного рубца жвачного животного. Во время хранения ионофор и носитель находятся в сухом состоянии, и композиция переходит в дозируемое подобное жидкости состояние при контактировании с текучей средой в месте применения. Описан ряд различных выходных приспособлений, включая множество отверстий на конце устройства и одно выходное отверстие переменного диаметра для контроля количества лекарственного вещества, выходящего в единицу времени вследствие диффузии и действия осмотического насоса.
Другие устройства, в которых композиции лекарственного вещества доставляют в виде пасты, суспензии или раствора через маленькое выходное отверстие под действием расширяющегося слоя, описаны в патентах США NoNo 5660861, 5633011, 5190765, 5252338, 5620705, 4931285, 5006346, 5024842 и 5160743. В качестве ближайшего аналога настоящего изобретения принят патент США 5190765. Устройство, раскрытое в этом патенте, а также другие устройства данного типа, включают расширяющийся выталкивающий слой и слой лекарственного вещества, окруженный полупроницаемой мембраной. В ряде случаев представлен слой лекарственного вещества с субпокрытием для защиты композиции лекарственного вещества в тех областях желудочно-кишечного тракта, которые имеют кислый рН, с целью задержки выхода композиции лекарственного вещества в среду применения или с целью образования покрытия, связанного с полупроницаемой мембраной. Однако данные устройства в основном не очень хорошо подходят в качестве лекарственных форм в случае высокой степени нагрузки лекарственным веществом, вследствие требований к размеру, необходимому для размещения больших количеств лекарственного вещества в пасте, суспензии или растворе, и необходимости получения лекарственной формы для перорального применения, имеющей размер, удобный для того, чтобы ее можно было проглотить.
Другая лекарственная форма раскрыта в Патенте США 5536507, где описан трехкомпонентный фармацевтический препарат, в котором используется среди других компонентов рН-чувствительный полимер, необязательно включающий осмотический агент, который будет набухать в областях с повышенным рН нижнего отдела тонкой кишки и в толстой кишке для освобождения лекарственного вещества в данных средах. Дополнительные компоненты лекарственной формы включают задерживающую выход выходом оболочку и энтеросолюбильную оболочку, обеспечивающие лекарственную форму, которая выделяет очень мало, если выделяет сколько-нибудь, лекарственного вещества в желудке, относительно низкое количество в тонкой кишке и, как сообщают, приблизительно 85% или более в толстой кишке. Такая лекарственная форма обеспечивает выход лекарственного вещества в изменяющемся в широком диапазоне периоде времени после введения, который может не начинаться в течение 1-3 часов, пока лекарственная форма не выйдет из желудка, и в течение дополнительных 3 часов или более, в течение которых лекарственная форма проходит в толстую кишку.
В Патенте 5169638 описан плавающий фармацевтический порошковый препарат с контролируемым выходом, предназначенный для наполнения капсул, в которых используют рН-зависимый полимер, образованный из альгиновой кислоты и гидроксипропилметилцеллюлозы, который обеспечивает выход фармацевтических агентов с контролируемой скоростью. Судя по описанию, препарат в капсулах предназначен для имитации характеристик таблетированного препарата.
В случае высокой степени нагрузки лекарственным веществом часто является предпочтительным, когда в дозирующем устройстве имеется большое отверстие, составляющее 50-100% от внутреннего диаметра полости для лекарственного вещества, чтобы слой лекарственного вещества мог дозироваться в нежидком состоянии. Под воздействием среды применения лекарственное вещество освобождается из слоя лекарственного вещества путем эрозии или диффузии. Обычная проблема, связанная с выходом лекарственного вещества из лекарственных форм, соответствующих предшествующему уровню техники, в которых слой лекарственного вещества дозируется из доставляющего устройства в сухом виде, состоит в том, что остаточное количество лекарственного вещества часто остается в устройстве и не выходит в организм субъекта. До 20-30% нагрузки композиции лекарственным веществом может остаться в устройстве, не высвобождаясь. С целью компенсации данного недостатка в способах, соответствующих предшествующему уровню техники, обычно предлагалась сверхнагрузка лекарственного вещества с тем, чтобы доставлялось требуемое количество несмотря на то, что значительное количество остается невысвободившимся в доставляющем устройстве. Нагрузка избыточного количества лекарственного вещества еще более усугубляет проблемы, связанные с лекарственными формами, которые являются большими и которые трудно глотать. Кроме того, дополнительная стоимость может быть значительной для активных веществ, имеющих высокую стоимость сырья и производства. Следовательно, существует потребность в усовершенствованных доставляющих устройствах, имеющих расширяющийся выталкивающий слой и слой лекарственного вещества, пригодный для применения при высокой нагрузке лекарственным веществом, которые высвобождают практически все лекарственное вещество из устройства в среду применения.
Сущность изобретения
Один из объектов изобретения представляет устройство для доставки лекарственного средства, а именно - лекарственную форму, обеспечивающую постоянное высвобождение содержащегося в ней лекарственного вещества. Представленная лекарственная форма включает стенку, ограничивающую полость, при этом стенка имеет выходное отверстие, образованное или образующееся в ней, и по меньшей мере часть стенки является полупроницаемой; расширяющийся слой, расположенный в полости на удалении от выходного отверстия и связанный посредством текучей среды с полупроницаемой частью стенки; слой, содержащий лекарственное вещество, расположенный в полости рядом с выходным отверстием и непосредственно или опосредованно контактирующий с расширяющимся слоем; а также слой, способствующий продвижению, помещенный между внутренней поверхностью стенки и по меньшей мере наружной поверхностью слоя, содержащего лекарственное вещество, расположенного в полости. Предпочтительным является содержание лекарственного вещества по меньшей мере 20% от общей массы слоя, содержащего лекарственное вещество. Более предпочтительно содержание лекарственного вещества по меньшей мере 40% от общей массы слоя, содержащего лекарственное вещество.
В предпочтительном варианте осуществления изобретения слой, содержащий лекарственное вещество, представляет собой прессованную композицию лекарственного вещества, а расширяющийся слой содержит осмотический агент.
Материалы для получения слоя, способствующего продвижению, выбирают, в частности, из следующей группы: гидрогели, желатин, полиэтиленоксиды молекулярной массы меньше, чем 100000, гидроксиалкилцеллюлозы, имеющие среднечисловые молекулярные массы между 9500 и 1250000, и гидроксиалкилалкилцеллюлозы, имеющие среднечисловые молекулярные массы между 80000 и 850000, а также их смеси.
Предпочтительным является выполнение слоя, способствующего продвижению, с возможностью обеспечения высвобождения в среду применения по меньшей мере 80% лекарственного вещества из слоя, содержащего лекарственное вещество.
Другой объект изобретения представляет промышленное изделие, представляющее собой ядро лекарственной формы, включающее слой, содержащий лекарственное вещество, который непосредственно или опосредованно контактирует с расширяющимся слоем, образуя вместе с ним двухслойное ядро, которое дополнительно покрыто способствующим продвижению слоем.
В предпочтительном варианте осуществления изобретения слой ядра, содержащий лекарственное вещество, представляет собой прессованную композицию лекарственного вещества, а способствующий продвижению слой содержит материал, выбранный из гидрогелей, желатина, полиэтиленоксидов молекулярной массы менее, чем 100000, гидроксиалкилцеллюлоз, имеющих среднечисловые молекулярные массы между 9500 и 1250000, и гидроксиалкилалкилцеллюлоз, имеющих среднечисловые молекулярные массы между 80000 и 850000, а также их смесей.
Еще один объект изобретения представляет способ обеспечения облегченного высвобождения лекарственного вещества из устройства, представляющего собой лекарственную форму, содержащую лекарственное вещество, а также полупроницаемую стенку и расширяющийся слой, причем способ предусматривает помещение способствующего продвижению слоя между полупроницаемой стенкой и слоем, содержащим лекарственное вещество.
В предпочтительном варианте осуществления способа слой, способствующий продвижению, формируют на прессованной композиции лекарственного вещества в виде покрытия, которое получают из гидроксиалкилцеллюлозы и низшего алканола.
Перечень фигур чертежей и иных материалов
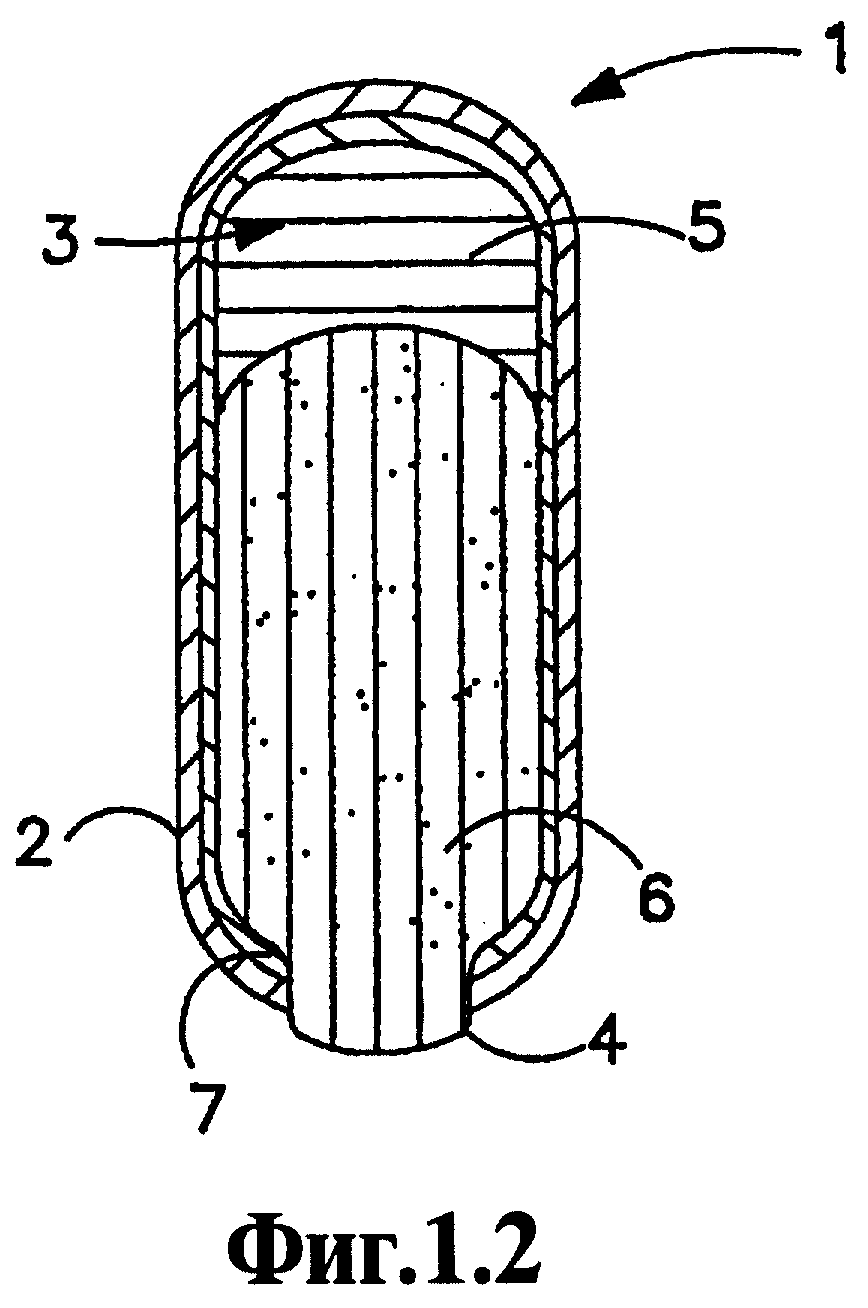
На Фигурах 1.1 и 1.2 проиллюстрирован один вариант осуществления лекарственной формы согласно данному изобретению, где на Фигуре 1.1 представлена лекарственная форма перед введением субъекту, а на Фигуре 1.2 представлена лекарственная форма спустя период времени после введения субъекту.
На Фигуре 2 представлен профиль высвобождения (скорость высвобождения как функция времени) активного агента нефазодона гидрохлорида из репрезентативной лекарственной формы, имеющей основные характеристики, приведенные на Фигуре 1, которая изготовлена с отверстием 4826 мкм (190 мил) и содержит 400 мг нефазодона гидрохлорида.
На Фигуре 3 представлен профиль высвобождения (скорость высвобождения как функция времени) активного агента нефазодона гидрохлорида из репрезентативной лекарственной формы, имеющей основные характеристики, приведенные на Фигуре 1, которая получена с отверстием 4826 мкм (190 мил) и содержит 400 мг нефазодона гидрохлорида.
На Фигуре 4 представлен кумулятивный (совокупный) выход нефазодона гидрохлорида в течение времени для ряда репрезентативных лекарственных форм, содержащих гранулы нефазодона гидрохлорида на основе полиэтиленоксида при нагрузке нефазодона гидрохлорида 100 мг и отверстии 2972 мкм (117 мил).
На Фигуре 5 представлен профиль высвобождения (скорость высвобождения как функция времени) активного агента нефазодона гидрохлорида для репрезентативных лекарственных форм, приготовленных с соответствии с процедурой, описанной в Примере 3.
На Фигуре 6 представлен кумулятивный выход нефазодона гидрохлорида в течение времени для репрезентативных лекарственных форм, приготовленных с соответствии с процедурой, описанной в Примере 3.
На Фигуре 7 представлен профиль высвобождения (скорость высвобождения как функция времени) активного агента нефазодона гидрохлорида для репрезентативных лекарственных форм, приготовленных с соответствии с процедурой, описанной в Примере 4.
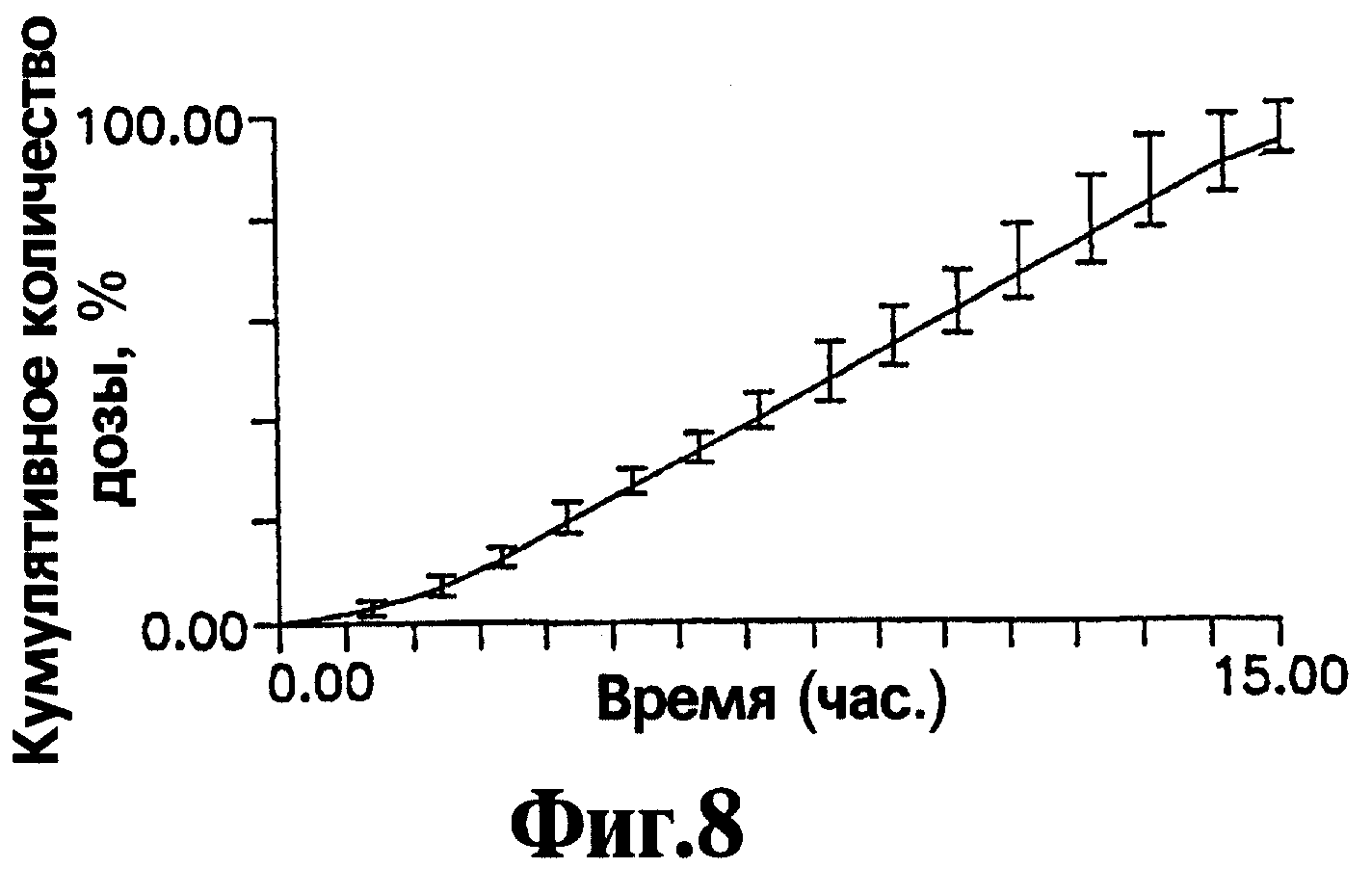
На Фигуре 8 представлен кумулятивный выход нефазодона гидрохлорида в течение времени для репрезентативных лекарственных форм, приготовленных с соответствии с процедурой, описанной в Примере 4.
На Фигуре 9 представлен профиль высвобождения (скорость высвобождения как функция времени) активного агента нефазодона гидрохлорида для репрезентативных лекарственных форм, приготовленных с соответствии с процедурой, описанной в Примере 5.
На Фигуре 10 представлен кумулятивный выход нефазодона гидрохлорида в течение времени для репрезентативных лекарственных форм, приготовленных с соответствии с процедурой, описанной в Примере 5.
На Фигуре 11 представлен профиль высвобождения (скорость высвобождения как функция времени) активного агента нефазодона гидрохлорида для репрезентативных лекарственных форм, приготовленных с соответствии с процедурой, описанной в Примере 6.
На Фигуре 12 представлен кумулятивный выход нефазодона гидрохлорида в течение времени для репрезентативных лекарственных форм, приготовленных с соответствии с процедурой, описанной в Примере 6.
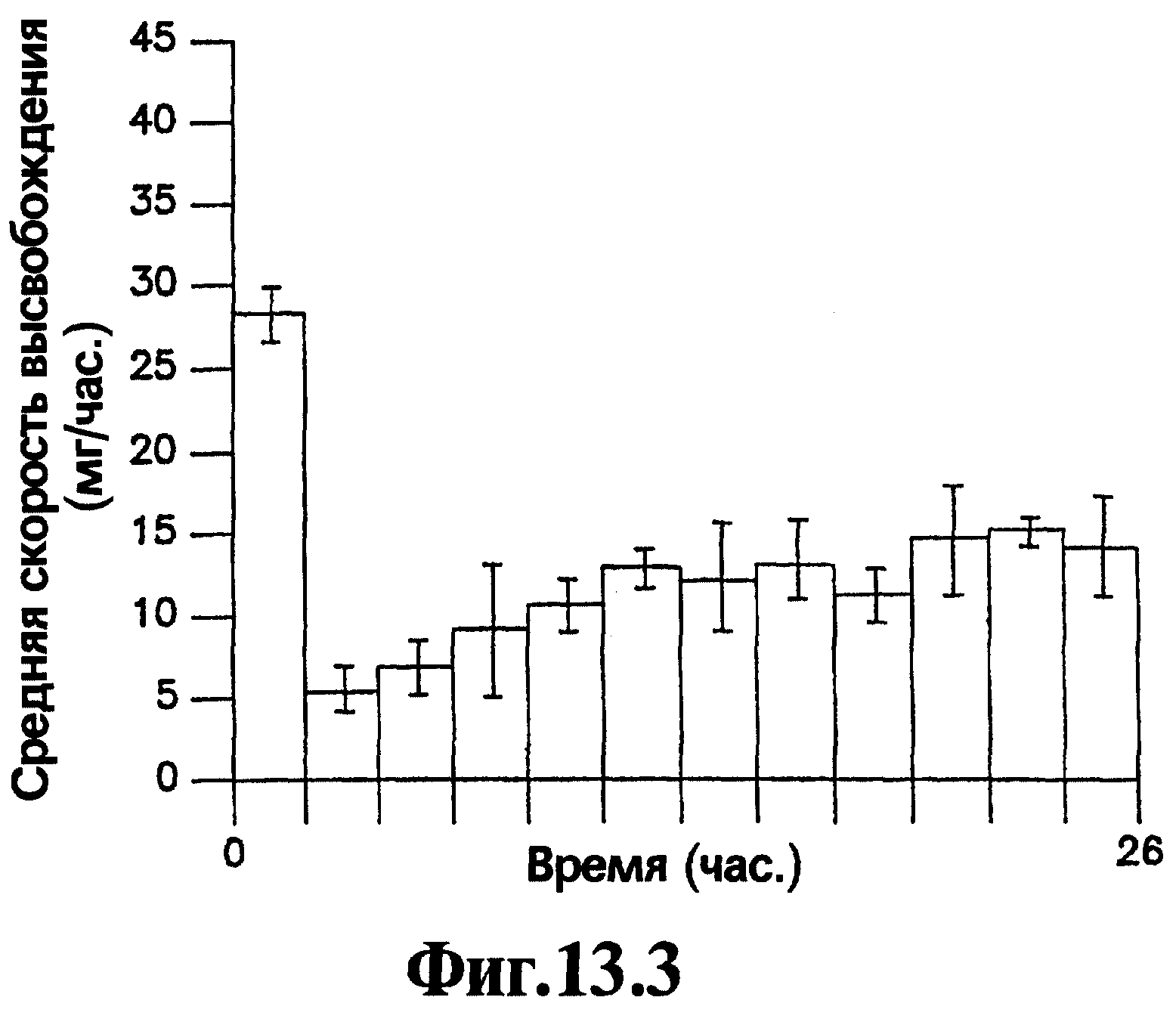
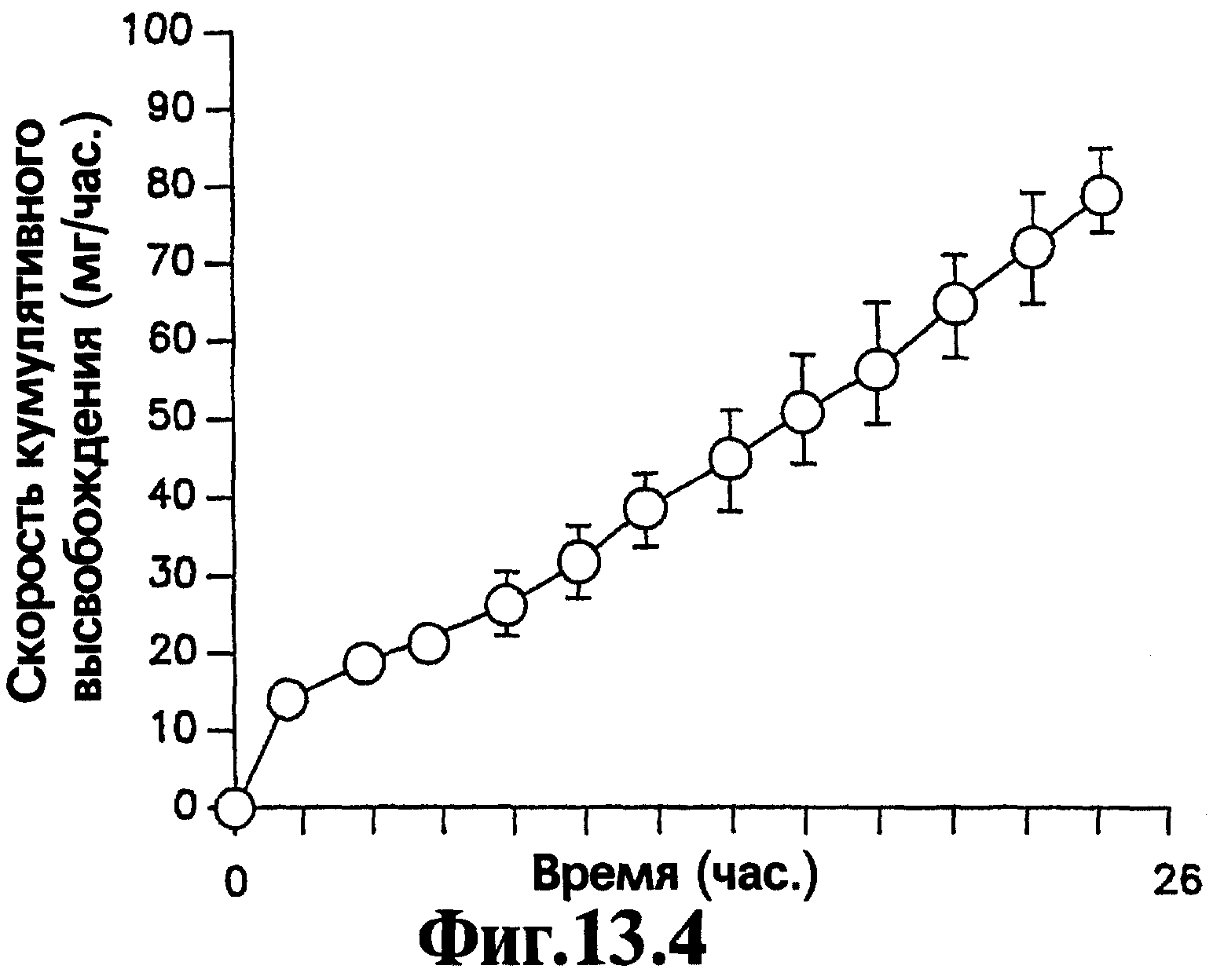
На Фигурах 13.1-13.4 представлено сравнение профиля скорости высвобождения и кумулятивного выхода как функции времени для имеющих оболочку и не имеющих оболочку лекарственных форм, содержащих 400 мг нефазодона гидрохлорида.
Сведения, подтверждающие возможность осуществления изобретения
Данное изобретение является наиболее понятным со ссылкой на следующие определения, чертежи и описание примеров.
Определения
Под терминами "активный агент", "лекарственное вещество" или "соединение", которые в данном контексте употребляют взаимозаменяемо, имеют в виду агент, лекарственное вещество, соединение, композицию материала или их смесь, которые обеспечивают какой-либо физиологический, психологический, биологический или фармакологический и часто благоприятный эффект при введении субъекту.
Терминами "постоянная скорость высвобождения" или "скорость равномерного высвобождения" обозначают скорость высвобождения активного агента из лекарственной формы, которая не отклоняется в сторону увеличения или уменьшения более чем на 30% от среднего значения скорости высвобождения активного агента в течение длительного периода времени, как определено с помощью устройства для определения интервала высвобождения USP типа 7 (USP type 7 Interval Release Apparatus). Предпочтительные постоянные скорости будут изменяться не более чем на 25% (в сторону увеличения или уменьшения) относительно среднего значения скорости высвобождения, определенного в течение длительного периода времени.
Под термином "длительный период времени" или "длительный период" имеют в виду непрерывный период времени 4 часа или больше, в более типичном случае 6 часов или больше.
Термином "лекарственная форма" обозначают фармацевтическую композицию или устройство, содержащие активный фармацевтический агент, при этом композицию или устройство, необязательно содержащие неактивные ингредиенты, такие как фармацевтически приемлемые носители, наполнители, агенты, образующие суспензию, поверхностно-активные вещества, дезинтегрирующие агенты, связующие агенты, разбавители, смазывающие вещества, стабилизаторы, антиоксиданты, осмотические агенты, окрашивающие агенты, пластификаторы и т.п., которые используют для приготовления и доставки активных фармацевтических агентов.
Терминами "фармацевтически приемлемая соль, полученная при добавлении кислоты" или "фармацевтически приемлемая соль", которые в данном контексте употребляют взаимозаменяемо, обозначают такие соли, в которых анион не играет существенную роль в проявлении токсичности или фармакологической активности соли, и в таком случае они являются фармакологическими эквивалентами оснований соединений, к которым относятся. Примеры фармацевтически приемлемых кислот, которые являются подходящими для целей образования солей, включают без ограничения перечисленным гидрохлорную, бромистоводородную, иодистоводородную, лимонную, уксусную, бензойную, миндальную, изэтионовую, пальмитиновую и другие кислоты.
Под термином "задержанное высвобождение (выход)" имеют в виду непрерывное высвобождение активного агента в среду в течение длительного времени.
Под термином "стабильное состояние" имеют в виду состояние, в котором количество лекарственного вещества, присутствующее в плазме крови субъекта, не изменяется существенным образом в течение длительного периода времени.
Сокращение "С" означает концентрацию лекарственного вещества в плазме крови субъекта, обычно выражаемую, как масса/единицу объема, как правило, в нанограммах/миллилитр.
Сокращение "Сmах" означает максимальную концентрацию лекарственного вещества в плазме крови субъекта, обычно выражаемую, как масса/единицу объема, как правило, в нанограммах/миллилитр, в определенном интервале времени после введения лекарственного вещества субъекту.
Сокращение "Сmin" означает минимальную концентрацию лекарственного вещества в плазме крови субъекта, обычно выражаемую, как масса/единицу объема, как правило, в нанограммах/миллилитр, в определенном интервале времени после введения лекарственного вещества субъекту.
Под выражением "анализ скорости высвобождения" имеют в виду стандартизированный анализ, направленный на определение соединения с использованием устройства для определения интервала высвобождения USP типа 7 (USP type 7 Interval Release Apparatus) в основном согласно описанию, приведенному в Примере 2. Понятно, что реагенты эквивалентного качества могут быть заменены в анализе в соответствии с общепринятыми методиками.
Термины "сухое состояние" или "практически сухое состояние" означают, что композиция, образующая слой лекарственного вещества лекарственной формы, выталкивается из лекарственной формы в виде пробки, при этом композиция является практически сухой или имеет настолько высокую вязкость, что не вытекает с легкостью как жидкий поток из лекарственной формы под давлением, оказываемым выталкивающим слоем.
Одним из наиболее подходящих устройств для контролируемого высвобождения лекарственных веществ, для которых требуется высокий уровень нагрузки в лекарственной форме с целью доставки количества лекарственного вещества, имеющего желательный терапевтический эффект, является устройство, имеющее полупроницаемую стенку, ограничивающую полость, расширяющийся выталкивающий слой и слой лекарственного вещества в полости, а также выходное отверстие, образованное в лекарственной форме для обеспечения выделения слоя лекарственного вещества в дозированном виде в практически сухом состоянии в среду применения. При приготовлении данных лекарственных форм обычной практикой является получение прессованной таблетки, содержащей слой лекарственного вещества и выталкивающий слой. Как правило, композицию выталкивающего слоя, находящуюся для удобства в форме гранул или порошка, прессуют в полости штампа вертикального пресса для таблетирования. Затем композицию слоя лекарственного вещества, также находящуюся для удобства в форме гранул или порошка, помещают в полость штампа над выталкивающим слоем и таким же образом прессуют с целью получения двухслойной таблетки. Хотя поверхность полости формы является очень гладкой, образовавшаяся двухслойная таблетка может все-таки иметь дефекты поверхности. Больше проблем существует со слоем лекарственного вещества, чем с выталкивающим слоем, особенно при высоком уровне нагрузки лекарственным веществом, когда используемые количества смазывающего вещества, носителя и связующего агента могут быть ограничены в связи с ограничениями размера таблетки.
Для множества применений дефекты слоя лекарственного вещества, как описано выше, могут не иметь большой важности. Однако, когда слой лекарственного вещества должен выходить в сухом виде из полости, образованной полупроницаемой стенкой, наружная поверхность слоя лекарственного вещества проходит по внутренней поверхности полупроницаемой стенки. Слой лекарственного вещества будет оказывать сопротивление движению, обусловленное силой трения, существующей между двумя поверхностями. Степень сопротивления будет возрастать с ростом числа и степени дефектов на наружной поверхности слоя лекарственного вещества и внутренней поверхности полупроницаемой стенки. Кроме того, поскольку на практике полупроницаемую стенку получают путем покрытия двухслойного ядра лекарственного вещества, то внутренняя поверхность полупроницаемой стенки будет изначально соответствовать дефектам, присутствующим на наружной поверхности слоя лекарственного вещества. Далее, когда слой лекарственного вещества продвигают мимо полупроницаемой стенки, дефекты наружной поверхности слоя лекарственного вещества должны противостоять дефектам внутренней поверхности полупроницаемой стенки. Это создает трение и сопротивление движению соответствующих слоев. Поскольку каждая из поверхностей является практически твердой, удобно рассматривать относительное движение (i) слоя лекарственного вещества или композита слой лекарственного вещества/выталкивающий слой и (ii) полупроницаемой наружной стенки, как "вытекание" слоя лекарственного вещества из устройства по мере расширения выталкивающего слоя. Таким образом, внутренний слой или субоболочка характеризуются как "способствующий прохождению потока слой" или "способствующий продвижению слой". По своему действию слой, способствующий продвижению (в частности, слоя лекарственного вещества), представляет собой слой материала, расположенный между наружной поверхностью слоя лекарственного вещества и внутренней поверхностью полупроницаемой стенки, который снижает трение между данными двумя поверхностями и облегчает их движение друг относительно друга, когда текучая среда проходит через полупроницаемую стенку и впитывается расширяющимся слоем.
В системах без слоя, способствующего продвижению, сопротивление, возникающее между слоем лекарственного вещества и наружной полупроницаемой стенкой, может создать ряд проблем. Одна из них состоит в том, что величина силы, препятствующей продвижению слоя лекарственного вещества, может быть функцией относительных положений слоя лекарственного вещества и наружной стенки в течение любого периода времени. Изменения величины силы сопротивления могут вызывать изменения скорости, с которой слой лекарственного вещества выделяется в среду применения. Это приводило бы затем к изменениям в высвобождении лекарственного вещества из лекарственной формы и потенциальным изменениям уровней лекарственного вещества в плазме субъекта с течением времени. Как можно видеть из профилей высвобождения лекарственных форм, описанных в данном описани, при реализации изобретения активный агент высвобождается с постоянной скоростью из лекарственных форм в течение длительного периода времени. Данный выход с постоянной скоростью обеспечивает значительные фармакологические преимущества при доставке активных агентов.
Во-вторых, при отсутствии слоя, способствующего продвижению, часть слоя лекарственного вещества имеет тенденцию "прилипать" к внутренней поверхности наружной стенки и оставаться в лекарственной форме, когда остальная часть слоя лекарственного вещества выделяется в среду применения под действием расширяющегося слоя. Данное остаточное количество неосвободившегося лекарственного вещества может быть значительным; остаточные количества, превышающие 20-30% от исходной нагрузки слоя лекарственного вещества, были отмечены в условиях высокой степени нагрузки лекарственным веществом.
В изобретении предложена лекарственная форма, промышленное изделие и способ практически полного высвобождения лекарственного вещества из лекарственной формы, в особенности из лекарственных форм, для которых может требоваться высокий уровень нагрузки лекарственным веществом с целью получения желательного фармакологического эффекта. Лекарственные формы, приготовленные в соответствии с данным изобретением, могут в результате образовывать истощенную лекарственную форму, сохраняющую 20% массы или менее, предпочтительно 10% массы или менее и наиболее предпочтительно 5% массы или менее от исходного количества лекарственного вещества, загруженного в лекарственную форму, при тестировании с использованием стандартного анализа скорости высвобождения.
При дозировках лекарственного вещества в диапазоне высоких значений, например, 100-2000 мг лекарственного вещества/однократную дозу, может потребоваться нагрузка лекарственного вещества в предназначенных для введения композициях, составляющая 20-90% или более от общей массы композиции. Такие требования к нагрузке могут представлять проблемы при составлении композиций и производстве устройств, которые являются пригодными для перорального введения и которые можно проглотить без излишних затруднений. Требования к нагрузке представляют собой еще большие проблемы при приготовлении лекарственных форм, которые предназначены для введения ограниченное число раз в день, такого как введение один раз в сутки. Проблемы размера усугубляются, когда не вся композиция лекарственного вещества высвобождается из доставляющего устройства, поскольку сверхнагрузка лекарственного вещества, т.е. обеспечение в доставляющем устройстве количества, превышающего количество, которое будет выходить в организме субъекта, создавая желательный фармакологический эффект, является необходимой для гарантии того, что надлежащее количество лекарственного вещества является доступным субъекту.
Лекарственные формы, соответствующие данному изобретению, высвобождают эффективные количества активного агента в организме пациента в течение длительного периода времени и нередко дают возможность менее частого введения, включая введение один раз в сутки, чем ранее требовалось для композиций с немедленным высвобождением. Лекарственные формы, соответствующие данному изобретению, содержат композицию, содержащую активный агент, причем композиция снаружи покрыта слоем, способствующим продвижению.
Активные агенты включают среди прочих пищевые продукты, пищевые добавки, питательные вещества, лекарственные вещества, антациды, витамины, агенты, аттенюирующие микроорганизмы, и другие агенты, которые являются благоприятными для среды применения. Активные агенты включают любую физиологически или фармакологически активную субстанцию, которая обладает местным или системным эффектом или эффектами в отношении животных, включая теплокровных млекопитающих, человека и приматов, домашних или сельскохозяйственных животных, таких как кошки, собаки, овцы, козы, крупный рогатый скот, лошади и свиньи, лабораторных животных, таких как мыши, крысы и морские свинки, содержащихся в зоопарках и диких животных и т.п. Активные агенты, которые могут быть доставлены, включают неорганические и органические соединения, в том числе, без ограничения перечисленным, активные агенты, которые действуют на периферические нервы, адренергические рецепторы, холинергические рецепторы, скелетные мышцы, сердечно-сосудистую систему, гладкую мускулатуру, систему кровообращения, синоптические участки, функциональные участки нейроэффекторов, эндокринную и гормональную системы, иммунную систему, репродуктивную систему, скелетную систему, системы физиологически активных веществ, пищеварительную и выделительную системы, гистаминную систему и центральную нервную систему.
Подходящие активные агенты могут быть выбраны, например, из белков, ферментов, ингибиторов ферментов, гормонов, полинуклеотидов, нуклеопротеинов, полисахаридов, гликопротеинов, липопротеинов, полипептидов, стероидов, гипнотических и седативных агентов, психостимуляторов, транквилизаторов, противосудорожных агентов, антидепрессантов, мышечных релаксантов, агентов против болезни Паркинсона, анальгетиков, противовоспалительных агентов, антигистаминных агентов, местных анестезирующих агентов, агентов, сокращающих мышцы, антимикробных агентов, противомалярийных агентов, антивирусных агентов, антибиотиков, агентов против ожирения, гормональных агентов, включая контрацептивы, симпатомиметиков, полипептидов и белков, способных вызывать физиологические эффекты, диуретиков, регуляторов липидного обмена, антиандрогенных агентов, антипаразитарных агентов, неопластических (вызывающих неоплазию) агентов, антинеопластических агентов (агентов против новообразований), антигипергликемических агентов, гипогликемических агентов, пищевых агентов и добавок, ростовых добавок, жиров, глазных препаратов, противоэнтеритных агентов, электролитов и диагностических агентов.
Примеры особенно активных агентов, используемых в данном изобретении, включают прохлорперазина эдизилат, сульфат железа, альбутерол, аминокапроновую кислоту, мекамиламина гидрохлорид, прокаинамида гидрохлорид, амфетамина сульфат, метамфетамина гидрохлорид, бензфетамина гидрохлорид, изопротеренола сульфат, фенметразина гидрохлорид, бетанехола хлорид, метахолина хлорид, пилокарпина гидрохлорид, атропина сульфат, скополамина бромид, изопропамида иодид, тридигексетила хлорид, фенформина гидрохлорид, метилфенидата гидрохлорид, теофиллина холинат, цефалексина гидрохлорид, дифенидол, меклозина гидрохлорид, прохлорперазина малеат, феноксибензамин, триэтилперазина малеат, анизиндион, дифенадионэритритила тетранитрат, дигоксин, изофлурофат, ацетазоламид, нифедипин, метазоламид, бендрофлуметиазид, хлорпропамид, глипизид, глибурид, гликлазид, тобутамид, хлорпроамид, толазамид, ацетогексамид, метформин, троглитазон, орлистат, бупропион, нефазодон, толазамид, хлормадинона ацетат, фенагликодол, аллопуринол, алюминат аспирина, метотрексат, ацетилсульфизоксазол, гидрокортизон, гидрокортикостерона ацетат, кортизона ацетат, дексаметазон и его производные, такие как бетаметазон, триамцинолон, метилтестостерон, 17-эстрадиол, этинилэстрадиол, этинилэстрадиола 3-метиловый эфир, преднизолон, 17-гидроксипрогестерона ацетат, 19-нор-прогестерон, норгестрел, норэтиндрон, норэтистерон, норэтиедерон, прогестерон, норгестерон, норэтинодрел, терфандин, фексофенадин, аспирин, ацетаминофен, индометацин, напроксен, фенопрофен, сулиндак, индопрофен, нитроглицерин, изосорбида динитрат, пропранолол, тимолол, атенолол, алпренолол, циметидин, клонидин, имипрамин, леводопа, селегилин, хлорпромазин, метилдопа, дигидроксифенилаланин, глюконат кальция, кетопрофен, ибупрофен, цефалексин, эритромицин, галоперидол, зомепирак, лактат железа, винкамин, феноксибензамин, дилтиазем, милринон, каптоприл, мандол, кванбенз, гидрохлортиазид, ранитидин, флурбипрофен, фенбуфен, флупрофен, толметин, аклофенак, мефенамик, флуфенамик, дифунинал, нимодипин, нитрендипин, нисолдипин, никардипин, фелодипин, лидофлазин, тиапамил, галлопамил, амлодипин, миофлацин, лизиноприл, эналаприл, каптоприл, рамиприл, эналаприлат, фамотидин, низатидин, сукралфат, этинтидин, тетратолол, миноксидил, хлордиазепоксид, диазепам, амитриптилин и имипрамин, а также фармацевтические соли данных активных агентов. Следующие примеры представляют собой белки и пептиды, которые включают без ограничения перечисленным инсулин, колхицин, глюкагон, тиреостимулирующий гормон, гормоны паращитовидной железы и гипофиза, кальцитонин, ренин, пролактин, кортикотропин, тиреотропный гормон, фолликул-стимулирующий гормон, хорионический гонадотропин, гонадотропин-высвобождающий гормон, бычий соматотропин, свиной соматотропин, окситоцин, вазопрессин, десмопрессин, соматостатин, липрессин, панкреозимин, лютеинизирующий гормон, LHRH (лютеинизирующий гормон-высвобождающий гормон), интерфероны, интерлейкины, гормоны роста, такие как человеческий гормон роста, бычий гормон роста и свиной гормон роста, ингибиторы фертильности, такие как простагландины, стимуляторы фертильности, факторы роста и человеческий гормон поджелудочной железы - высвобождающий фактор.
Активные агенты из ряда антидепрессантов могут быть выбраны из группы, состоящей из трициклических третичных аминов, таких как, например, амитриптилин, кломипрамин, доксепин, имипрамин, (+)-тримипрамин, трициклических вторичных аминов, таких как, например, амозапин, десипрамин, мапротилин, нортириптилин, протриптилин, ингибиторов вторичного всасывания серотонина, таких как, например, флуоксетин, флувоксамин, пароксетин, сертралин, венлафазин, и атипических антидепрессантов, таких как брупропион, нефазодон, тразодон, фенелзин, транилципромин, селегилин и их фармацевтически приемлемых солей. Лекарственная форма, как правило, может включать носитель, например, гидрофильный полимер, в сочетании с активным агентом.
На Фигуре 1.1 представлен предпочтительный вариант осуществления лекарственной формы 1 согласно данному изобретению, имеющей конфигурацию "выталкивающийся стержень", до введения субъекту. Лекарственная форма 1 имеет стенку 2, ограничивающую полость 3. Стенка 2 снабжена выходным отверстием 4. Внутри полости 3 на удалении (с промежутком) от выходного отверстия 4 находится выталкивающий слой 5. Слой лекарственного вещества 6 расположен в полости 3 смежно выходному отверстию 4. В соответствии с изобретением, слой, способствующий продвижению 7, функция которого будет описана и который может быть выполнен в виде второй стенки, находится между слоем лекарственного вещества 6 и внутренней поверхностью стенки 2.
Стенка 2 сделана так, чтобы она была проницаемой для прохождения снаружи такой текучей среды, как вода и биологические жидкости, и практически непроницаемой для прохождения активного агента, осмотического агента, осмополимера и т.п. В таком виде ее можно определить, как полупроницаемую. Композиции с избирательной проницаемостью, используемые для образования стенки, являются в основном неразрушающимися, и они не растворяются в биологических жидкостях в период существования лекарственной формы. Не требуется, чтобы стенка 2 была полностью полупроницаемой. Однако, по меньшей мере, часть стенки 2 должна быть полупроницаемой, чтобы позволить текучей среде контактировать или сообщаться с выталкивающим слоем 5 так, чтобы выталкивающий слой 5 впитывал жидкость в процессе использования. Специальные материалы для создания полупроницаемой стенки 2 хорошо известны в области техники, и репрезентативные примеры данных материалов описаны в данном описании далее.
Вторичная стенка 7, которая функционирует как способствующий продвижению слой, находится в контакте с внутренней поверхностью полупроницаемой стенки 2 и по меньшей мере с наружной поверхностью слоя лекарственного вещества, который расположен напротив стенки 2, хотя вторичная стенка 7 может (и предпочтительно будет) проходить, окружать и контактировать с наружной поверхностью выталкивающего слоя. Стенка 7, как правило, будет окружать по меньшей мере ту часть наружной поверхности слоя лекарственного вещества, которая расположена напротив внутренней поверхности стенки 2. Вторичная стенка 7 может быть выполнена в виде покрытия, нанесенного на прессованное ядро, содержащее слой лекарственного вещества и выталкивающий слой. Наружная полупроницаемая стенка 2 окружает и служит оболочкой для внутренней вторичной стенки 7. Вторичная стенка 7 предпочтительно выполнена в виде субоболочки (подслоя) по меньшей мере поверхности лекарственного слоя 6, а при желании полной наружной поверхности спрессованного слоя лекарственного вещества 6 и выталкивающего слоя 5. При выполнении полупроницаемой стенки 2 в виде покрытия композита, образованного из слоя лекарственного вещества 6, выталкивающего слоя 5 и вторичной стенки 7, обеспечивается контактирование полупроницаемой стенки 2 с внутренней оболочкой.
Вторичная стенка 7 облегчает высвобождение лекарственного вещества из лекарственных форм, соответствующих изобретению. Для лекарственных форм, в которых имеется высокий уровень нагрузки лекарственного вещества, т.е. 20% или выше, но более часто 40% или выше активного агента в слое лекарственного вещества от общей массы слоя лекарственного вещества, и отсутствует вторичная стенка, было показано, что значительные остаточные количества лекарственного вещества могут сохраняться в устройстве после окончания периода доставки. В ряде случаев остаточные количества лекарственного вещества, превышающие 20% и даже превышающие 30% от массы исходной нагрузки лекарственного вещества в лекарственной форме, могут оставаться в лекарственной форме в конце двадцатичетырехчасового периода при тестировании с использованием анализа скорости высвобождения. Сравнение высвобождения нефазодона гидрохлорида из соответствующей данному изобретению репрезентативной лекарственной формы, имеющей способствующий продвижению слой, и лекарственной формы, не имеющей способствующего продвижению слоя, (детали которого приведены в Примере 8) представлено на Фигурах 13.1-13.4 для лекарственной формы, имеющей нагрузку лекарственным веществом 83% (400 мг нефазодона гидрохлорида). На Фигурах 13.1 и 13.2 представлена лекарственная форма, соответствующая изобретению, которая имеет способствующий продвижению слой, а на Фигурах 13.3 и 13.4 представлена аналогичная лекарственная форма без способствующего продвижению слоя. Значительная разница средних скоростей выхода в одной точке и скоростей кумулятивного выхода является очевидной для двух лекарственных форм. Кроме того, очевидно, что через 24 часа остается значительно больше лекарственного вещества в лекарственной форме без способствующего продвижению слоя, чем лекарственного вещества в лекарственной форме, имеющей способствующий продвижению слой.
Как замечено выше, количество оставшегося лекарственного вещества может быть эффективно уменьшено введением вторичной стенки 7, которую делают в виде внутренней оболочки из способствующего продвижению слоя, т.е. агента, который снижает силу трения между внешней стенкой в виде полупроницаемой мембраны 2 и наружной поверхностью слоя лекарственного вещества 6. Вторичная стенка или внутренняя оболочка 7 снижает силы трения, возникающие между полупроницаемой стенкой 2 и наружной поверхностью слоя лекарственного вещества, обеспечивая тем самым более полную доставку лекарственного вещества из устройства. Данное усовершенствование представляет значительные экономические преимущества, особенно в случае активных соединений, имеющих высокую стоимость, поскольку отсутствует необходимость нагружать слой лекарственного вещества избытком лекарственного вещества, чтобы гарантировать доставку минимального требуемого количества лекарственного вещества.
Внутренняя субоболочка 7, как правило, может быть толщиной от 0,01 до 5 мм, более часто - толщиной от 0,5 до 5 мм, и она содержит компонент, выбранный из гидрогелей, желатина, низкомолекулярных полиэтиленоксидов, например, молекулярной массы меньше 100000, гидроксиалкилцеллюлоз, например, гидроксиэтилцеллюлозы, гидроксипропилцеллюлозы, гидроксиизопропилцеллюлозы, гидроксибутилцеллюлозы и гидроксифенилцеллюлозы, а также гидроксиалкилалкилцеллюлоз, например, гидроксипропилметилцеллюлозы, и их смесей. Гидроксиалкилцеллюлозы содержат полимеры, имеющие среднечисловую молекулярную массу от 9500 до 1250000. Например, используют гидроксипропилцеллюлозы, имеющие среднечисловые молекулярные массы между 80000 и 850000. Способствующий продвижению слой может быть приготовлен из традиционно используемых растворов или суспензий вышеупомянутых материалов в водных растворителях или инертных органических растворителях. Предпочтительные материалы для субоболочки или способствующего продвижению слоя включают гидроксипропилцеллюлозу, гидроксиэтилцеллюлозу, гидроксипропилметилцеллюлозу, повидон [поли(винилпирролидон)], полиэтиленгликоль и их смеси. Более предпочтительными являются смеси гидроксипропилцеллюлозы и повидона, приготовленные в органических растворителях, в частности, в органических полярных растворителях, таких как низшие алканолы, имеющие 1-8 атомов углерода, предпочтительно этанол, смеси гидроксиэтилцеллюлозы и гидроксипропилметилцеллюлозы, приготовленные в водном растворе, и смеси гидроксиэтилцеллюлозы и полиэтиленгликоля, приготовленные в водном растворе. Наиболее предпочтительно, когда субоболочка состоит из смеси гидроксипропилцеллюлозы и повидона, приготовленной в этаноле. Удобно, когда масса субоболочки, нанесенная на двухслойное ядро, может коррелировать с толщиной субоболочки и остаточным количеством лекарственного вещества, которое остается в лекарственной форме при анализе скорости высвобождения, как описано в данном тексте. В процессе производства толщину субоболочки можно регулировать путем контроля массы субоболочки, нагруженной на стадии покрытия оболочкой.
Когда вторичная стенка 7 сформирована в виде субоболочки, т.е. путем нанесения на таблетированный двухслойный композит слоя лекарственного вещества и выталкивающего слоя, субоболочка может заполнять дефекты поверхности, образованные на двухслойном ядре в процессе таблетирования. Полученная гладкая наружная поверхность облегчает скольжение между покрытым оболочкой двухслойным композитом и полупроницаемой стенкой в процессе дозирования лекарственного вещества, что приводит к снижению количества остаточной композиции лекарственного вещества, которое остается в устройстве в конце периода дозирования. Когда стенка 7 выполнена из гелеобразующего материала, контакт с водой в среде применения облегчает образование гелевой или гелеобразной внутренней оболочки, имеющей вязкость, которая может способствовать и усиливать скольжение между наружной стенкой 2 и слоем лекарственного вещества 6.
Репрезентативные полимеры для образования стенки 2 содержат полупроницаемые гомополимеры, полупроницаемые сополимеры и т.п. Данные материалы включают сложные эфиры целлюлозы, простые эфиры целлюлозы и сложные-простые эфиры целлюлозы. Целлюлозные полимеры имеют степень замещения (СЗ) ангидроглюкозных единиц от больше 0 до 3 включительно. Степень замещения (СЗ) означает среднее число гидроксильных групп, исходно присутствующих на ангидроглюкозной единице, которые замещены группой заместителя или превращены в другую группу. Ангидроглюкозная единица может быть частично или полностью замещена такими группами, какацил, алканоил, алкеноил, ароил, алкил, алкокси группа, галоген, карбоалкил, алкилкарбамат, алкилкарбонат, алкилсульфонат, алкилсульфамат, группами, образующими полупроницаемый полимер, и т.п., где органические структуры содержат от одного до двенадцати атомов углерода и предпочтительно от одного до восьми атомов углерода.
Полупроницаемые композиции, как правило, включают ингредиент, выбранный из группы, состоящей из ацилата целлюлозы, диацилата целлюлозы, триацилата целлюлозы, ацетата целлюлозы, диацетата целлюлозы, триацетата целлюлозы, моно-, ди- и триалканилатов целлюлозы, моно-, ди- и триалкенилатов, моно-, ди- и триароилатов и т.п. Примеры полимеров включают ацетат целлюлозы, имеющий СЗ 1,8-2,3 и содержание ацетила 32-39,9%; диацетат целлюлозы, имеющий СЗ 1-2 и содержание ацетила 21-35%; триацетат целлюлозы, имеющий СЗ 2-3 и содержание ацетила 34-44,8% и т.п. Более специфические целлюлозные полимеры включают пропионат целлюлозы, имеющий СЗ 1,8 и содержание пропионила 38,5%; ацетат пропионат целлюлозы с содержанием ацетила 1,5-7% и содержанием пропионила 39-42%; ацетат пропионат целлюлозы с содержанием ацетила 2,5-3%, средним содержанием пропионила 39,2-45% и содержанием гидроксила 2,8-5,4%; ацетат бутират целлюлозы, имеющий СЗ 1,8, содержание ацетила 13-15% и содержание бутирила 34-39%; ацетат бутират целлюлозы с содержанием ацетила 2-29%, содержанием бутирила 17-53% и содержанием гидроксила 0,5-4,7%, триацилаты целлюлозы, имеющие СЗ 2,6-3, такие как тривалерат целлюлозы, триамилат целлюлозы, трипальмитат целлюлозы, триоктаноат целлюлозы, и трипропионат целлюлозы; сложные диэфиры целлюлозы, имеющие СЗ 2,2-2,6, такие как дисукцинат целлюлозы, дипальмитат целлюлозы, диоктаноат целлюлозы, дикаприлат целлюлозы, и т.п., а также смешанные сложные эфиры целлюлозы, такие как ацетат валерат целлюлозы, ацетат сукцинат целлюлозы, пропионат сукцинат целлюлозы, ацетат октаноат целлюлозы, валерат пальмитат целлюлозы, ацетат гептаноат целлюлозы, и т.п. Полупроницаемые полимеры известны из Патентов США No 4077407 и их можно синтезировать с использованием процедур, описанных в Энциклопедии науки и технологии полимеров (Encyclopedia of Polymer Science and Technology), т.3, с.325-354, (1964), Interscience Publishers Inc., New York, NY.
Дополнительные полупроницаемые полимеры для образования наружной стенки 2 включают ацетальдегиддиметилацетат целлюлозы, ацетатэтилкарбамат целлюлозы, ацетатметилкарбамат целлюлозы, диметиламиноацетат целлюлозы, полупроницаемый полиамид, полупроницаемые полиуретаны, полупроницаемые сульфонированные полистиролы, перекрестно-сшитые избирательно полупроницаемые полимеры, образованные соосаждением аниона и катиона, как описано в Патентах США NoNo 3173876, 3276586, 3541005, 3541006 и 3546142, полупроницаемые полимеры, как описано Loeb и соавт. в Патенте США No 3133132, полупроницаемые производные полистирола, полупроницаемый поли(стиролсульфонат натрия), полупроницаемый поли(хлоридвинилбензилтриметиламмония) и полупроницаемые полимеры с проницаемостью жидкости 25,4· (10-5-10-2) (cм3·мкм/cм· чac· aтм.), которую выражают в атмосферах как разность гидростатического или осмотического давления относительно полупроницаемой стенки. Полимеры являются известными в области техники из Патентов США NoNo 3845770, 3916899 и 4160020 и книги Scott и Rotf, Справочник по известным полимерам (Handbook of Common Polymers) (1971), CRC Press, Cleveland, OH.
Стенка 2 может также содержать агент, регулирующий поток (расход). Агент, регулирующий поток, представляет собой соединение, которое добавляют для того, чтобы способствовать регуляции проницаемости текучей среды или потока через стенку 2. Агент, регулирующий поток, может быть усиливающим или уменьшающим потокагентом. Агент можно предварительно выбрать для увеличения или снижения потока жидкости. Агенты, которые приводят к заметному усилению проницаемости текучей среды, такой как вода, часто являются в основном гидрофильными, тогда как агенты, приводящие к заметному уменьшению текучести сред, таких как вода, являются в основном гидрофобными. Количество регулятора в стенке (в случае введения в нее) в основном составляет от приблизительно 0,01 до 20 мас.% или более. В одном варианте осуществления агенты, регулирующие поток, которые его усиливают, включают многоатомные спирты, полиалкиленгликоли, полиалкилендиолы, сложные полиэфиры алкиленгликолей и т.п. Типичные усилители потока включают полиэтиленгликоль 300, 400, 600, 1500, 4000, 6000 и т.п., низкомолекулярные гликоли, такие как полипропиленгликоль, полибутиленгликоль и полиамиленгликоль; полиалкилендиолы, такие как поли(1,3-пропандиол), поли(1,4-бутандиол), поли(1,6-гександиол) и т.п.; алифатические диолы, такие как 1,3-бутиленгликоль, 1,4-пентаметиленгликоль, 1,4-гексасметиленгликоль и т.п.; алкилентриолы, такие как глицерин, 1,2,3-бутантриол, 1,2,4-гексантриол, 1,3,6-гексантриол и т.п., сложные эфиры, такие как этиленгликольдипропионат, этиленгликольбутират, бутиленгликольдипропионат, сложные эфиры глицеролацетата и т.п. Репрезентативные агенты, уменьшающие поток, включают фталаты, замещенные алкильной или алкоксигруппой или обеими - алкильной и алкоксигруппой, такие как диэтилфталат, диметоксиэтилфталат, диметилфталат и ди(2-этилгексил)фталат, арилфталаты, такие как трифенилфталат и бутилбензилфталат, нерастворимые соли, такие как сульфат кальция, сульфат бария, фосфат кальция и т.п.; нерастворимые оксиды, такие как оксид титана; полимеры в виде порошка, гранул и подобных формах, такие как полистирол, полиметилметакрилат, поликарбонат и полисульфон, сложные эфиры, такие как сложные эфиры лимонной кислоты, этерифицированной алкильными группами с длинной цепью; инертные и практически водонепроницаемые наполнители; материалы для образования стенки на основе совместимых с целлюлозой смол и т.п.
Другие материалы, которые могут быть использованы для образования стенки 2, придающие стенке свойства эластичности и растяжения, делающие стенку 2 менее хрупкой и устойчивой к разрыву, включают фталатные пластификаторы, такие как дибензилфталат, дигексилфталат, бутилоктилфталат, фталаты с неразветвленной цепью из шести-одиннадцати атомов углерода, диизононилфталат, диизодецилфталат и т.п.
Пластификаторы включают нефталаты, такие как триацетин, диоктилазелат, эпоксидированный таллат, триизооктилтримеллитат, триизононилтримеллитат, сахарозоацетатизобутират, эпоксидированное соевое масло и т.п. Количество пластификатора в стенке при введении в нее составляет приблизительно 0,01-20 мас.% или выше.
Слой лекарственного вещества 6 содержит композицию, образованную из активного агента и носителя, такого как гидрофильный полимер. Гидрофильный полимер представляет собой частицу гидрофильного полимера в композиции лекарственного вещества, которая вносит вклад в обеспечение постоянства скорости высвобождения активного агента и систему контролируемой доставки. Репрезентативными примерами данных полимеров являются полиалкиленоксид со среднечисловой молекулярной массой 100000-750000, включая полиэтиленоксид, полиметиленоксид, полибутиленоксид и полигексиленоксид, а также поликарбоксиметилцеллюлоза со среднечисловой молекулярной массой 40000-400000, представленная солью щелочного металла поликарбоксиметилцеллюлозы, натриевой солью поликарбоксиметилцеллюлозы, калиевой солью поликарбоксиметилцеллюлозы и литиевой солью поликарбоксиметилцеллюлозы. Композиция лекарственного вещества может содержать гидроксипропилалкилцеллюлозу со среднечисловой молекулярной массой 9200-125000 для улучшения доставляющих свойств лекарственной формы, представленную гидроксипропилэтилцеллюлозой, гидроксипропилметилцеллюлозой, гидроксипропилбутилцеллюлозой и гидроксипропилпентилцеллюлозой, и поливинилпирролидон со среднечисловой молекулярной массой 7000-75000 для улучшения свойств потока в лекарственной форме. Предпочтительными среди данных полимеров являются полиэтиленоксид со среднечисловой молекулярной массой 100000-300000. Носители, которые разрушаются в среде желудка, т.е. биоразрушающиеся носители, являются особенно предпочтительными.
Поверхностно-активные вещества и дезинтегрирующие агенты также могут быть использованы в составе носителя. Примерами поверхностно-активных веществ являются поверхностно-активные вещества, имеющие значение HLB между приблизительно 10 и 25, такие как полиэтиленгликоль 400-моностеарат, полиоксиэтилен-4-сорбитан-монолаурат, полиоксиэтилен-20-сорбитан-моноолеат, полиоксиэтилен-20-сорбитан-монопальмитат, полиоксиэтилен-20-сорбитан-монолаурат, полиоксиэтилен-40-стеарат, олеат натрия и т.п. Дезинтегрирующие агенты могут быть выбраны из крахмалов, глин, целлюлоз, альгинов и камедей, а также перекрестно-сшитых крахмалов, целлюлоз и полимеров. Репрезентативные дезинтегранты включают кукурузный крахмал, картофельный крахмал, кроскармелозу, кросповидон, натриевую соль гликолата крахмала, Veegum HV, метилцеллюлозу, агар, бентонит, карбоксиметилцеллюлозу, альгиновую кислоту, гуаровую камедь и т.п.
Слой лекарственного вещества 6 получают в виде смеси, содержащей активный агент и носитель. Слой лекарственного вещества может быть образован из частиц путем измельчения, при котором получают такой размер частиц лекарственного вещества и размер сопутствующего полимера, который используют для приготовления слоя лекарственного вещества, как правило, в виде ядра, содержащего соединение, в соответствии с вариантом осуществления изобретения и согласно способу по изобретению. Методы получения частиц включают гранулирование, распылительную сушку, просеивание, лиофилизацию, дробление, растирание, перемалывание на струйной мельнице или мельнице для сверхтонкого размола и крошения с целью получения намеченного микронного размера частиц. Способ можно реализовать с использованием оборудования для уменьшения размера, такого как мельница с распылением для сверхтонкого помола, мельница с использованием энергии жидкости, дробилка, валковая мельница, молотковая мельница, фрикционная мельница, бегуны, шаровая мельница, виброшаровая мельница, ударная мельница тонкого помола, центрифугальный распылитель, дробилка грубого помола и дробилка тонкого помола. Размер частиц можно установить просеиванием, в том числе с помощью колосникового грохота, плоского сита, виброгрохота, барабанного вращающегося грохота, качающегося грохота, вибрационного сита и сита, совершающего возвратно-поступательное движение. Процессы и оборудование для приготовления частиц лекарственного вещества и носителя описаны в работах Remington "Справочник по фармацевтическим наукам" (Pharmaceutical Sciences), 17-е изд., с. 1585-1594 (1985); Perry "Справочник для инженеров-химиков" (Chemical Engineers Handbook), 6-ое изд., с.21-13 - 21-19 (1984); Parrot, Journal of Pharmaceutical Sciences, т.61, No 6, с.813-829, (1974); Hixon "Справочник для инженера-химика" (Chemical Engineer), с.94-103 (1990).
Активное соединение может быть введено в слой лекарственного вещества в количествах от 1 мкг до 5000 мг/лекарственную форму в зависимости от требующегося уровня дозировки, который должен поддерживаться в течение периода доставки, т.е. в течение времени между последовательными введениями лекарственных форм. Как правило, нагрузка соединения в лекарственных формах будет обеспечивать дозы соединения в организме субъекта, лежащие в интервале от 1 мкг до 2500 мг/сутки, более часто от 1 мг до 2500 мг/сутки. Во многих случаях может быть предпочтительным ограничение количества лекарственного вещества в каждой лекарственной форме до менее чем 1000 мг и обеспечение потребности суточной дозировки, превышающей данное количество, путем введения субъекту более, чем одной лекарственной формы для удовлетворения суточной потребности. Слой лекарственного вещества, как правило, будет представлять собой сухую композицию, образованную прессованием носителя и лекарственного вещества в качестве одного слоя и расширяющегося и выталкивающего слоя в качестве второго слоя. Расширяющийся слой будет выталкивать слой лекарственного вещества из выходного отверстия, поскольку выталкивающий слой впитывает жидкость из среды применения, и подвергшийся воздействию слой лекарственного вещества будет разрушаться с высвобождением лекарственного вещества в среду применения. Это можно видеть, обратившись к Фигуре 1.2.
Выталкивающий слой 5 представляет собой расширяющийся слой, имеющий выталкивающую-замещающую композицию, причем слои размещены так, что слой лекарственного вещества 6 расположен непосредственно или опосредованно контактирующим с расширяющимся слоем 5. При размещении опосредованно контактирующих слоев инертный, между слоем лекарственного вещества и выталкивающим слоем может быть помещен элемент (не показан), такой как разделительный слой или диск.
Выталкивающий слой 5 содержит полимер, который впитывает водную или биологическую жидкость и набухает, чтобы вытолкнуть композицию лекарственного вещества из выходного средства устройства. Примеры набухающих в текучей среде замещающих полимеров включают их представители, выбранные из полиалкиленоксида со среднечисловой молекулярной массой 1 миллион - 15 миллионов, который представлен полиэтиленоксидом и солью щелочного металла поликабоксиметилцеллюлозы со среднечисловой молекулярной массой 500000-3500000, где щелочной металл является натрием, калием или литием. Примерами дополнительных полимеров, используемых для получения выталкивающей-замещающей композиции, являются осмополимеры, включающие полимеры, которые образуют гидрогели, такие как кислый карбоксиполимер Carbopol®, акриловый полимер, перекрестно-сшитый с полиаллилсахарозой, известный также как карбоксиполиметилен, и карбоксивиниловый полимер, имеющий молекулярную массу 250000-4000000; полиакриламиды Cyanamer®; перекрестно-сшитые набухающие в воде полимеры инденмалеинового ангидрида; полиакриловая кислота Good-rite® молекулярной массы 80000-200000; Aqua-Keeps® - акрилатные полимерные полисахариды, содержащие конденсированные глюкозные единицы, например, сложный диэфир перекрестно-сшитого полиглюкана и т.п. Репрезентативные полимеры, которые образуют гидрогели, известны в предшествующем уровне техники из Патента США No 3865108, выданного Hartop; Патента США No 4002173, выданного Manning; Патента США No 4207893, выданного Michaels, и книги Scott и Roff Справочник по известным полимерам (Handbook of Common Polymers) (1971), CRC Press, Cleveland, OH.
Осмагент, известный также как осмотическое растворенное вещество и осмотически эффективный агент, который демонстрирует градиент осмотического давления относительно наружной стенки и субоболочки, включает представитель, выбранный из группы, состоящей из хлорида натрия, хлорида калия, хлорида лития, сульфата магния, хлорида магния, сульфата калия, сульфата натрия, сульфата лития, кислого фосфата калия, маннита, мочевины, инозита, сукцината магния, винной кислоты, рафинозы, сахарозы, глюкозы, лактозы, сорбита, неорганических солей, органических солей и углеводов.
Примеры растворителей, пригодных для производства соответствующих стенок, слоев, оболочек и субоболочек, применяемых в лекарственных формах, соответствующих изобретению, включают водные и инертные органические растворители, которые не повреждают материалы, используемые для приготовления лекарственных форм. В растворители входит широкий круг их представителей, выбранных из группы, состоящей из водных растворителей, спиртов, кетонов, сложных эфиров, простых эфиров, алифатических углеводородов, галогенированных растворителей, циклоалифатических соединений, ароматических соединений, гетероциклических растворителей и их смесей. Типичные растворители включают ацетон, диацетоновый спирт, метанол, этанол, изопропиловый спирт, метилацетат, этилацетат, изопропилацетат, н-бутилацетат, метилизобутилкетон, метилпропилкетон, н-гексан, н-гептан, простой моноэтиловый эфир этиленгликоля, моноэтилацетат этиленгликоля, метилендихлорид, этилендихлорид, пропилендихлорид, четыреххлористый углерод, нитроэтан, нитропропан, тетрахлорэтан, этиловый эфир, изопропиловый эфир, циклогексан, циклооктан, бензол, толуол, нафта, 1,4-диоксан, тетрагидрофуран, диглим, воду, водные растворители, содержащие неорганические соли, такие как хлорид натрия, хлорид кальция и т.п., а также их смеси, такие как ацетон и вода, ацетон и метанол, ацетон и этиловый спирт, метилендихлорид и метанол и этилендихлорид и метанол.
Для получения заполненной лекарственной формы может быть удобным использовать нанесение покрытия в форме, за исключением выходного отверстия. В системе нанесения покрытия в форме субболочку из образующих стенку композиций наносят путем последовательного напыления соответствующей композиции на двухслойное ядро, содержащее слой лекарственного вещества и выталкивающий слой, которое сопровождается переворачиванием во вращающейся форме. Устройство для нанесения покрытия в форме благодаря его доступности используют в промышленном масштабе. Для покрытия ядра лекарственного вещества могут быть использованы другие технологии. Покрытая лекарственная форма может быть высушена в печи с поддувом или в печи с контролем температуры и влажности до получения лекарственной формы, не содержащей растворитель. Условия сушки будут, как это принято, выбраны с учетом имеющегося оборудования, условий среды, растворителей, покрытий, толщины покрытия и т.п.
Возможно также использование других технологий нанесения покрытия. Например, полупроницаемая стенка и субоболочка лекарственной формы могут быть выполнены в рамках одной технологии с использованием воздушно-суспензионной процедуры. Данная процедура состоит в суспендировании и переворачивании в токе воздуха двухслойного ядра, композиции внутренней субоболочки и композиции, образующей наружную полупроницаемую стенку, пока (в любой операции) субоболочка и покрытие наружной стенки не будут нанесены на двухслойное ядро. Воздушно-суспензионная процедура хорошо подходит для независимого образования стенки лекарственной формы. Воздушно-суспензионная процедура описана в Патенте США No 2799241, в J. Am. Pharm. Assoc., т.48, с 451-459, (1959) и там же т.49, с.82-84, (1960). Лекарственная форма может быть также покрыта водно-суспензионным материалом для оболочки Wurster® с использованием, например, метилендихлоридметанола в качестве сорастворителя. Возможно использование воздушно-суспензионного материала для оболочки Aeromatic® с применением данного сорастворителя.
Лекарственная форма, соответствующая изобретению, может быть приготовлена стандартными способами. Например, лекарственная форма может быть приготовлена способом мокрой грануляции. В способе мокрой грануляции лекарственное вещество и ингредиенты, включающие первый слой или композицию лекарственного вещества, смешивают, используя органический растворитель, такой как денатурированный безводный этанол в качестве жидкости для грануляции. Ингредиенты, образующие первый слой или композицию лекарственного вещества, пропускают через предварительно выбранное сито, а затем тщательно перемешивают в смесителе. После этого другие ингредиенты, содержащие первый слой, могут быть растворены в части жидкости для грануляции, такой как растворитель, описанный выше. Затем последнюю приготовленную мокрую смесь медленно добавляют к смеси лекарственного вещества при непрерывном перемешивании в смесителе. Жидкость для грануляции добавляют до тех пор, пока не получат мокрую смесь, затем массу данной мокрой смеси пропускают через предварительно выбранное сито, помещая на подносы печи. Смесь сушат в течение 18-24 часов при 24-35° С в печи с поддувом. Высушенные гранулы затем сортируют по размеру. Далее к грануляту лекарственного вещества добавляют стеарат магния, затем помещают в вибрационные мельницы и смешивают в вибрационной мельнице в течение 10 минут. Композицию прессуют в виде слоя, например, с помощью пресса Manesty®. Устанавливают скорость пресса 20 об/мин, при максимальной нагрузке 2 тонны. Первый слой спрессовывают с композицией, образующей второй слой, и двухслойные таблетки вводят в пресс для получения покрытия сухим способом Kilian® и окружают не содержащей лекарственное вещество оболочкой с последующим покрытием растворителем наружной стенки.
В другом способе производства полезное лекарственное вещество и другие ингредиенты, содержащие первый слой, находящийся против выхода, смешивают и прессуют в виде твердого слоя. Слой имеет размеры, которые соответствуют внутренним размерам площади, которую слой должен занимать в лекарственной форме, а также имеет размеры, соответствующие второму слою, с целью образования с ним контактирующей структуры. Лекарственное вещество и другие ингредиенты также могут быть смешаны с растворителем и перемешаны с образованием твердой или полутвердой формы с помощью традиционных способов, таких как измельчение на шаровой мельнице, каландрование, перемешивание или вальцевание с последующим прессованием в заданной форме. Далее расширяющийся слой, например, слой композиции осмополимера, помещают так, чтобы он подобным образом контактировал со слоем лекарственного вещества. Получение слоев препарата лекарственного вещества и осмополимера можно осуществить с помощью известных технологий двухслойного прессования. Два контактирующих слоя сначала покрывают способствующей продвижению субоболочкой, а затем наружной полупроницаемой стенкой. Воздушно-суспензионная процедура или процедура в воздушном вращающемся барабане состоят в суспендировании и переворачивании прессованных контактирующих первого и второго слоев в токе воздуха, содержащем композицию с замедленным образованием до тех пор, пока первый и второй слои не будут окружены композицией стенки.
Другой процесс производства, который может быть использован для получения композиции, образующей полость, предусматривает смешивание порошков ингредиентов в грануляторе с псевдоожиженным слоем. После перемешивания в грануляторе порошков ингредиентов в сухом виде на порошки распыляют жидкость для грануляции, например, поливинилпирролидон в воде. Покрытые порошки затем высушивают в грануляторе. С помощью данного процесса гранулируют все присутствующие в смеси ингредиенты, добавляя жидкость для грануляции. После высушивания гранул смазывающий агент, такой как стеариновая кислота или стеарат магния, смешивают с гранулятом, используя поддон или V-смеситель. Затем гранулы прессуют вышеописанным способом.
Лекарственную форму, соответствующую изобретению, снабжают по крайней мере одним выходным отверстием. Выходное отверстие взаимодействует с ядром лекарственного вещества в плане равномерного выхода лекарственного вещества из лекарственной формы. Выходное отверстие может быть сделано во время приготовления лекарственной формы или во время доставки лекарственного вещества лекарственной формой в текучей среде применения. Выражение "выходное отверстие", как используют в целях данного изобретения, может включать средство, такое как проход, щель, отверстие и канал. Этот термин включает также отверстие, которое образуется из субстанции или полимера, которые разрушаются, растворяются или вытекают из наружной оболочки или стенки или внутренней оболочки с образованием выходного отверстия. Субстанция или полимер могут включать разрушающуюся полигликолевую кислоту или полимолочную кислоту в наружном или внутреннем покрытиях; желатиновое волокно, удаляемый водой поливиниловый спирт, вытекающее соединение, такое как удаляемый жидкостью порообразователь, выбранный из группы, состоящей из неорганической или органической соли, оксида или углевода. Выход или множество выходов может образовываться при вытекании компонента, выбранного из группы, состоящей из сорбита, лактозы, фруктозы, глюкозы, маннозы, галактозы, талозы, хлорида натрия, хлорида калия, цитрата натрия и маннита, что приводит к образованию выходного отверстия в виде пор с таким размером, который обеспечивает равномерный выход. Выходное отверстие может иметь любую форму, такую как круглая, треугольная, квадратная, эллиптическая и т.п. для равномерного дозированного высвобождения лекарственного вещества из лекарственной формы. Может быть сконструирована лекарственная форма с одним или более выходов, разделенных пространственно или одной или более поверхностями лекарственной формы. Выходное отверстие можно сделать прошивкой, включая механическую или лазерную прошивку, наружной оболочки, внутренней оболочки или обеих оболочек. Выходы и оборудование для формирования выходов описаны в Патентах США NoNo 3845770 и 3916899, выданных Theeuwes и Higuchi, Патенте США No 4063064, выданном Saunders и соавт. и Патенте США No 4088864, выданном Theeuwes и соавт. Выходное отверстие может составлять от 10 до 100% от внутреннего диаметра полости, образованной стенкой 2, предпочтительно от 30 до 100% и наиболее предпочтительно от 50 до 100%.
Хотя для некоторых лекарственных форм, соответствующих изобретению, может потребоваться высокая степень нагрузки лекарственным веществом для получения реакции пациента, лекарственные формы, соответствующие данному изобретению, которые обеспечивают равномерную скорость выхода активного соединения, предоставляют возможность использовать меньшее количество соединения/лекарственную форму/сутки, чем это было бы рассчитано на основании простого умножения дозы активного агента в продукте с немедленным выходом на число раз/сутки, рекомендованное для введения продукта с немедленным выходом.
Даже при высоком уровне дозировки, при котором активное соединение присутствует в количестве от 40 до 90 мас.% от массы композиции слоя лекарственного вещества, данные лекарственные формы и устройства способны эффективно высвобождать требующееся количество активного соединения в течение длительного периода времени с равномерной скоростью высвобождения. Предпочтительно, когда массовый процент активного соединения в лекарственных формах, соответствующих изобретению, будет составлять 75% или меньше и наиболее предпочтительно - меньше, чем 70%, но больше или равно 40%, наиболее предпочтительно больше, чем 60% от массы композиции слоя лекарственного вещества, чтобы обеспечить получение лекарственных форм, которые можно легко проглотить.
В условиях, когда желательно введение лекарственного вещества в количестве, которое превышало бы 75% от композиции слоя лекарственного вещества, обычно предпочтительным является одновременный прием двух или более таблеток лекарственной формы с общей нагрузкой лекарственного вещества, равной или превышающей количество, которое было бы использовано в одной таблетке.
Изобретение может быть проиллюстрировано лекарственными формами для приема один раз в сутки, приготовленными со 100 мг, 200 мг, 300 мг, 400 мг и 500 мг нефазодона гидрохлорида/лекарственную форму. В каждом случае в лекарственной форме остается менее чем 10% от исходного количества лекарственного вещества через 24 часа при тестировании с помощью анализа скорости высвобождения. После начального стартового периода (обычно приблизительно 2-3 часа или меньше) лекарственные формы обеспечивают равномерную скорость высвобождения соединения в течение длительного периода времени, как правило, от 4 часов до 20 часов или более часто от 4 часов или 16 часов и более обыкновенно в течение от 4 часов до 10 часов. В конце пролонгированного периода равномерного высвобождения скорость высвобождения лекарственного вещества из лекарственной формы может снижаться приблизительно в течение такого периода времени, как несколько часов. Лекарственные формы обеспечивают терапевтически эффективные количества лекарственного вещества для широкого круга применений и нужд отдельных субъектов.
При исходном введении лекарственные формы могут обеспечить концентрацию лекарственного вещества в плазме субъекта, которая повышается в течение начального периода времени, как правило, в течение нескольких часов или меньше, а затем обеспечивает относительно постоянную концентрацию лекарственного вещества в плазме в течение длительного периода времени, обычно от 4 часов до 24 часов или более. Профили высвобождения лекарственных форм, соответствующих данному изобретению, представляют высвобождение лекарственного вещества в течение целого 24-часового периода, соответствующего введению один раз в сутки, так что стабильная концентрация лекарственного вещества в плазме крови субъекта может поддерживаться на терапевтически эффективных уровнях в течение 24-часового периода после введения лекарственной формы с задержанным выходом. Стабильные уровни лекарственного вещества в плазме могут, как правило, достигаться через двадцать четыре часа или в ряде случаев нескольких дней, например 2-5 дней, у большинства субъектов.
Например, в системах, содержащих 100 мг, 200 мг, 300 мг, 400 мг и 500 мг нефазодона гидрохлорида, приготовленных практически в соответствии с процедурами, описанными в Примере 1, и имеющих Т90 12 часов, нефазодон гидрохлорид высвобождается со средними скоростями высвобождения 8,6, 17,2, 25,8, 34,4 и 43,0 мг/час, соответственно, в течение непрерывного периода времени 4 часа или более, в основном в течение непрерывного периода приблизительно от 4 до 10 часов, как показано при анализе скорости высвобождения, начиная приблизительно через 2-3 часа после начального помещения в раствор. В каждом из данных препаратов процент нагрузки лекарственного вещества от общей массы слоя лекарственного вещества составляет приблизительно 69% для лекарственных форм по 100 мг, 200 мг, 300 мг, 400 мг и 500 мг. В каждом случае нефазодона гидрохлорид высвобождался из лекарственной формы с равномерной скоростью с течение длительного периода времени.
Скорость высвобождения, как функция времени для репрезентативной лекарственной формы, содержащей 400 мг нефазодона гидрохлорида, представлена на Фигуре 2. Лекарственная форма имела Т90, равное 17,7 часов, и среднюю скорость высвобождения приблизительно 22 мг/час. Была изготовлена лекарственная форма с выходным отверстием 4826 мкм (190 мил), субоболочкой массой 40 мг, образованной из 70/30 мас.% Klucel/PVPK29-32 и оболочкой из полупроницаемой мембраны массой 70,4 мг из 90/10 мас.% ацетата целлюлозы 398 и полиэтиленгликоля 3350. На Фигуре 3 представлены скорости высвобождения для аналогичным образом изготовленной лекарственной формы, имеющей Т90 18,5 часов и среднюю скорость высвобождения приблизительно 5,2 мг/час. Была изготовлена лекарственная форма с выходным отверстием 2972 мкм (117 мил), субоболочкой массой 10,6 мг, образованной из 70/30 мас.% Klucel/PVPK29-32 и оболочкой из полупроницаемой мембраны массой 46,9 мг из 97/3 мас.% ацетата целлюлозы 398 и полиэтиленгликоля 3350. В каждом случае слой лекарственного вещества содержал 65% нефазодона гидрохлорида. Как можно видеть из данных фигур, длительный период с равномерной скоростью высвобождения продолжается от приблизительно 4 часов до приблизительно 18 часов для лекарственной формы, соответствующей Фигуре 3.
Относительно лекарственных форм, содержащих 100-400 мг, приготовленных, как описано в данном контексте, было обнаружено, что 100 мг лекарственная форма, имеющая ядро диаметром приблизительно 0,48 см (3/16 дюйма), выходное отверстие 2794-3302 мкм (110-130 мил), предпочтительно 2921-3175 мкм (115-125 мил) и наиболее предпочтительно 3048 мкм (120 мил), обеспечивает эффективный профиль высвобождения. Лекарственная форма на 200 мг, имеющая ядро диаметром приблизительно 0,6 см (15/64 дюйма), выходное отверстие 3683-4191 мкм (145-165 мил), предпочтительно 3810-4064 мкм (150-160 мил) и наиболее предпочтительно 3937 мкм (155 мил), обеспечивает эффективный профиль высвобождения. Лекарственная форма на 300 мг, имеющая ядро диаметром приблизительно 0,67 см (17/64 дюйма), выходное отверстие 4191-4699 мкм (165-185 мил), предпочтительно 4318-4572 мкм (170-180 мил) и наиболее предпочтительно 4445 мкм (175 мил), обеспечивает эффективный профиль высвобождения. Лекарственная форма на 400 мг, имеющая ядро диаметром приблизительно 0,71 см (9/32 дюйма), выходное отверстие 4572-5080 мкм (180-200 мил), предпочтительно 4699-4953 мкм (185-195 мил) и наиболее предпочтительно 4826 мкм (190 мил), обеспечивает эффективный профиль высвобождения. Лекарственные формы высвобождают лекарственное вещество со скоростью, которая изменяется меньше, чем на 30% относительно средней скорости высвобождения, измеренной в течение длительного периода времени. Предпочтительно, когда устройства высвобождают лекарственное вещество со скоростью, которая изменяется меньше, чем на 25% относительно средней скорости высвобождения, измеренной в течение длительного периода времени.
Лекарственные формы, соответствующие изобретению, высвобождают лекарственное вещество с равномерной скоростью выхода в течение длительного периода времени, как определено с помощью стандартного анализа скорости высвобождения, такого как описан в данном контексте. При введении субъекту лекарственные формы, соответствующие изобретению, обеспечивают уровни лекарственного вещества в плазме крови субъекта, которые меньше изменяются в течение длительного периода времени, чем уровни лекарственного вещества, полученные при использовании лекарственных форм с немедленным высвобождением. При регулярном введении лекарственных форм, соответствующих данному изобретению, один раз в сутки лекарственные формы, соответствующие изобретению, обеспечивают стабильные уровни лекарственного вещества в плазме крови, так что разница между Сmax и Сmin в течение 24-часового периода значительно уменьшается относительно разницы, полученной при введении продукта с немедленным выходом, который предназначен для высвобождения такого же количества лекарственного вещества за 24-часовой период, как высвобождающееся из лекарственных форм, соответствующих изобретению.
Лекарственные формы, соответствующие изобретению, адаптированы для высвобождения активного вещества с равномерной скоростью выхода в течение длительного периода времени, предпочтительно 6 часов или более. Измерения скорости высвобождения, как правило, проводят in vitro в подкисленной воде с целью имитации условий желудочных соков и проводят в течение ограниченных увеличивающихся периодов времени для получения приблизительного значения скорости высвобождения в данной точке времени. Информация о таких скоростях высвобождения in vitro в отношении определенной лекарственной дозы может быть использована в качестве вспомогательной при выборе лекарственной формы, которая будет обеспечивать желательные результаты in vivo. Данные результаты могут быть получены имеющимися способами, такими как анализы плазмы крови и клиническое наблюдение, которые используются практикующими врачами при назначении промышленно выпускаемых лекарственных форм с немедленным высвобождением.
Было показано, что соответствующие данному изобретению лекарственные формы, имеющие профили скорости высвобождения, как описано в данном тексте, могут обеспечивать в организме пациента практически постоянную концентрацию в плазме крови и пролонгированный терапевтический эффект активного агента в течение длительного периода времени после введения лекарственной формы. Соответствующие данному изобретению лекарственные формы с задержанным высвобождением демонстрируют меньшую вариабельность концентрации лекарственного вещества в плазме в течение 24-часового периода по сравнению с препаратами с немедленным выходом, которые характеризуются образованием значительных пиков концентрации лекарственного вещества сразу или вскоре после введения субъекту.
Реализация вышеупомянутого способа путем перорального введения субъекту лекарственной формы, соответствующей изобретению, один раз в сутки с целью лечения болезненных состояний или симптомов, чувствительных к активному агенту, содержащемуся в лекарственной форме, является предпочтительной.
Предпочтительный способ изготовления лекарственных форм в соответствии с данным изобретением в основном описан ниже. Проценты представляют собой маc. проценты, если не оговорено иначе.
Пример 1
Приготовление гранулята слоя лекарственного вещества
Связующий раствор готовят путем добавления гидроксипропилцеллюлозы (Klucel MF, Aqualon Company), "HPC" в воду с образованием раствора, содержащего 5 мг НРС/0,995 г воды. Раствор перемешивают до растворения гидроксипропилцеллюлозы. Для загрузки определенного объема в гранулятор с псевдоожиженным слоем ("ГПС") вносят необходимые количества нефазодона HCL (69,0%), полиэтиленоксида (молекулярной массы 200000) (Polyox® N-80, Union Carbide Corporation) (20,3%), гидроксипропилцеллюлозы (Klucel MF) (5%), стеарата полиоксила 40 (3%) и кросповидона (2%). После перемешивания сухих материалов в емкости добавляют связующий раствор, приготовленный, как описано выше. Затем гранулят высушивают в ГПС до консистенции, пригодной для измельчения (<1 мас.% воды) и измельчают, пропуская через сито 7 или 10.
Гранулят переносят в смеситель с поддоном или V-смеситель. Необходимые количества антиоксиданта, бутилированного гидрокситолуола ("БИТ"), (0,01%) и смазывающего агента, стеариновой кислоты, (1%) просеивают на сите 40 и оба компонента смешивают с гранулятом, используя смеситель с поддоном или V-смеситель до равномерного распределения (стеариновую кислоту смешивают приблизительно в течение 1 минуты и БГТ смешивают приблизительно в течение 10 минут).
Приготовление гранулята осмотического выталкивающего слоя
Связующий раствор готовят путем добавления гидроксипропилметилцеллюлозы 2910 ("НРМС") в воду в соотношении 5 мг ГПМЦ на 1 г воды. Раствор перемешивают до растворения ГПМЦ. Порошок хлорида натрия (30%) и красный оксид железа (1,0%) измельчают и просеивают. В гранулятор с псевдоожиженным слоем ("ГПС") загружают необходимые количества полиэтиленоксида (молекулярной массы 7000000) (Polyox®303) (63,7%), ГПМЦ (5,0%), хлорид натрия и красный оксид железа. После перемешивания сухих материалов в емкости добавляют связующий раствор, приготовленный, как описано выше. Гранулят высушивают в ГПС до достижения заданного содержания жидкости (<1 мас.% воды). Гранулят измельчают, пропуская через сито 7, и переносят в смеситель с поддоном или V-смеситель. Необходимые количества антиоксиданта, бутилированного гидрокситолуола (0,08%), просеивают через сито 60. Требующееся количество смазывающего агента, стреариновой кислоты, (0,25%) просеивают через сито 40 и оба материала смешивают с гранулятом, используя смеситель с поддоном или V-смеситель, до равномерного распределения (стеариновую кислоту смешивают приблизительно в течение 1 минуты и БГТ смешивают приблизительно в течение 10 минут).
Прессование двухслойного ядра
Пресс для таблеток удлиненной формы снабжен круглыми глубокими вогнутыми штампами и формами. На прессе помещены два сырьевых бункера. Слой лекарственного вещества, приготовленного, как описано выше, помещают в один из бункеров, тогда как осмотический выталкивающий слой, приготовленный, как описано выше, помещают в другой бункер.
Начальное регулирование параметров таблетирования (слоя лекарственного вещества) проводят с целью получения ядер с одинаковой заданной массой слоя лекарственного вещества, как правило, 100 мг лекарственного вещества в каждой таблетке. Проводят регулирование параметров таблетирования второго слоя (осмотического выталкивающего слоя), который связывает слой лекарственного вещества с осмотическим слоем, с целью получения ядер с одинаковыми конечными массой, толщиной, твердостью и хрупкостью. Вышеуказанные параметры можно регулировать путем изменения наполняемого пространства и/или силовым регулированием. Типичная таблетка, содержащая лекарственное вещество в заданном количестве 100 мг, имеет длину приблизительно 1,18 см (0,465 дюймов) и диаметр приблизительно 0,48 см (0,188 дюйма).
Приготовление раствора для субоболочки и системы с субоболочкой
Раствор для субоболочки готовят в покрытой нержавеющей сталью емкости. Подходящие количества повидона (К29-32) (2,4%) и гидроксипропилцеллюлозы (молекулярная масса 80000) (Klucel EF, Aqualon Company) (5,6%) перемешивают в безводном этиловом спирте (92%) до тех пор, пока полученный раствор не станет прозрачным. Приготовленные, как описано выше, двухслойные ядра помещают во вращающееся перфорированное устройство для нанесения покрытий с перфорированным поддоном. Включают устройство для нанесения покрытий и после достижения температуры покрытия 28-36° С приготовленный, как описано выше, раствор для субоболочки равномерно наносят на слой переворачивающихся таблеток. После нанесения раствора в количестве, достаточном для получения желательного увеличения массы субоболочки, процесс нанесения субоболочки заканчивают. Желательную массу субоболочки выбирают так, чтобы получить приемлемые остаточные количества лекарственного вещества, которое остается в лекарственной форме, что определяют с помощью анализа скорости высвобождения в течение 24-часового периода. В общем желательно, чтобы от исходной нагрузки лекарственного вещества оставалось меньше 10%, более предпочтительно - меньше 5% и наиболее предпочтительно - меньше 3% остаточного лекарственного вещества через 24 часа тестирования с помощью стандартного анализа скорости высвобождения, как описано в данном контексте. Это может быть определено на основании корреляции между массой субоболочки и остаточного лекарственного вещества для ряда лекарственных форм, имеющих одинаковые двухслойные ядра, но различные массы субоболочек с помощью стандартного анализа скорости высвобождения.
Приготовление мембраны для контроля скорости и покрытой мембраной системы
Двухслойные ядра, покрытые субоболочкой, готовят, как описано выше, и помещают во вращающееся устройство для нанесения покрытий с перфорированным поддоном. Включают устройство для нанесения покрытий и после достижения температуры покрытия 28-38° С подходящий раствор для покрытия, приготовленный, как описано ниже в А, В или С, равномерно наносят на слой переворачивающихся таблеток, пока не получают желательное увеличение массы мембраны. Через регулярные интервалы времени в течение процесса покрытия определяют увеличение массы, а образцы изделий, покрытых мембраной, могут быть протестированы с помощью анализа скорости высвобождения с целью определения Т90 для изделий с покрытием. Увеличение массы в анализе скорости высвобождения может коррелировать с Т90для мембран изменяющейся толщины. После нанесения достаточного количества раствора, что удобно определять по соответствию желаемого увеличения массы мембраны желательному значению Т90, процесс покрытия мембраной заканчивают.
А. Раствор для покрытия готовят в покрытой нержавеющей сталью емкости. Соответствующие количества ацетона (565 мг) и воды (29,7 мг) перемешивают с полоксамером 188 (1,6 мг) и ацетатом целлюлозы (29,7 мг) до полного растворения твердых веществ. Во время использования раствор для покрытия содержит приблизительно 5% твердых веществ. Использование мембраны приводит к получению лекарственной формы со значением Т90приблизительно 13 часов согласно анализу скорости высвобождения.
В. Ацетон (505,4 мг) перемешивают с ацетатом целлюлозы (27,72 мг) до полного растворения ацетата целлюлозы. Полиэтиленгликоль 3350 (0,28 мг) и воду (26,6 мг) смешивают в отдельном контейнере. Два раствора перемешивают до образования прозрачного раствора. Во время использования раствор для покрытия содержит приблизительно 5% твердых веществ. Использование мембраны приводит к получению лекарственной формы со значением Т90 приблизительно 13 часов (т.е. приблизительно 90% лекарственного вещества высвобождается из лекарственной формы в течение 13 часов) согласно анализу скорости высвобождения.
С. Ацетон (776,2 мг) перемешивают с ацетатом целлюлозы (42,57 мг) до полного растворения ацетата целлюлозы. Полиэтиленгликоль 3350 (0,43 мг) и воду (40,9 мг) смешивают в отдельном контейнере. Два раствора перемешивают до образования прозрачного раствора. Во время использования раствор для покрытия содержит приблизительно 5% твердых веществ. Использование мембраны приводит к получению лекарственной формы со значением Т90 приблизительно 18 часов (т.е. приблизительно 90% лекарственного вещества высвобождается из лекарственной формы в течение 18 часов) согласно анализу скорости высвобождения.
Просверливание систем, покрытых мембраной
Одно выходное отверстие просверливают в том конце системы, покрытой мембраной, где расположен слой лекарственного вещества. Во время процесса просверленные образцы проверяют через регулярные интервалы времени на размер отверстия, расположение и число выходов.
Высушивание просверленных систем с покрытием
Просверленные системы с покрытием, приготовленные, как описано выше, помещают на перфорированные подносы для печи, ставят на решетку печи в условиях относительной влажности 43-45% и сушат для удаления остаточных растворителей.
Окрашенные и бесцветные оболочки
По выбору окрашенные или бесцветные растворы для оболочек готовят в покрытой нержавеющей сталью емкости. Для окрашенной оболочки 88 частей очищенной воды перемешивают с 12 частями Opadry II (цвет не является важным) до получения гомогенного раствора. Для бесцветной оболочки 90 частей очищенной воды перемешивают с 10 частями Opadry Clear до получения гомогенного раствора. Высушенные ядра, приготовленные, как описано выше, помещают во вращающееся устройство для нанесения покрытий с перфорированным поддоном (ванной). Включают устройство для покрытий и после достижения температуры покрытия 35-45° С раствор для получения окрашенной оболочки равномерно наносят на переворачивающийся слой таблеток. После нанесения достаточного количества раствора, что удобно определять по достижению желательного увеличения массы окрашенной оболочки, процесс нанесения окрашенной оболочки заканчивают. Затем раствор для получения бесцветной оболочки равномерно наносят на переворачивающийся слой таблеток. После нанесения достаточного количества раствора или достижения желательного увеличения массы бесцветной оболочки, процесс нанесения бесцветной оболочки заканчивают. Агент, обеспечивающий сыпучесть (например, воск Саr-nu-bo), наносят на слой таблеток после нанесения бесцветной оболочки.
Пример 2
Скорость высвобождения лекарственного вещества из устройств, содержащих лекарственные формы, соответствующие изобретению, определяют с помощью следующего стандартизованного анализа. Способ включает высвобождение систем в подкисленной воде (рН 3). Аликвоты растворов для определения скорости высвобождения образца вводят в хроматографическую систему для определения количества лекарственного вещества, высвободившегося в течение определенных тест-интервалов. Лекарственное вещество разделяют на колонке C18 и определяют по УФ-поглощению (254 нм для нефазодона гидрохлорида). Определение количества осуществляют с помощью линейного регрессионного анализа площадей пиков с помощью стандартной кривой, имеющей по меньшей мере пять точек.
Образцы готовят с использованием устройства для определения интервала высвобождения USP типа 7 (USP type 7 Interval Release Apparatus). Каждую анализируемую систему (устройство, соответствующее изобретению) взвешивают. Затем каждую систему приклеивают к пластиковому стержню, который имеет заостренный конец, и каждый стержень присоединяют к погружающему держателю для определения скорости высвобождения.
Каждый погружающий держатель для определения скорости высвобождения присоединен к шейкеру возвратно-поступательного движения с движением вверх/вниз (устройство для определения интервала высвобождения USР типа 7 (USP type 7 Interval Release Apparatus)), который работает с амплитудой приблизительно 3 см и 2-4 секунд/цикл. Концы стержня с присоединенными системами непрерывно погружают в калиброванные тест-пробирки на 50 мл, содержащие 50 мл подкисленной H2O (подкисление до рН 3,00±0,05 фосфорной кислотой), уравновешенные в водяной бане до постоянной температуры, отрегулированной на 37° С±0,5° С. В конце каждого интервала времени, определенного, как правило, одним часом или двумя часами, системы переносят в следующий ряд тест-пробирок, содержащих свежую подкисленную воду. Процесс повторяют в течение желаемого числа интервалов до окончания выхода. Затем пробирки с раствором, содержащие освободившееся лекарственное вещество, удаляют и охлаждают при комнатной температуре. После охлаждения каждую пробирку наполняют до отметки 50 мл подкисленной водой, каждый из растворов тщательно перемешивают и затем переносят в сосуды для образцов для анализа с помощью высокоэффективной жидкостной хроматографии ("ВЭЖХ"). Стандартные растворы лекарственного вещества готовят с возрастанием концентраций, охватывающим интервал от 5 мкг до приблизительно 400 мкг и анализируют с помощью ВЭЖХ. Стандартную кривую концентраций получают, используя линейный регрессионный анализ. Образцы лекарственного вещества, полученные в тесте на высвобождение, анализируют с помощью ВЭЖХ и определяют концентрацию с помощью линейного регрессионного анализа. Вычисляют количество лекарственного вещества, освободившегося в течение каждого интервала времени высвобождения. Результаты для различных лекарственных форм, соответствующих изобретению, представлены на Фигурах 2-13.
Пример 3
Используя основную процедуру, соответствующую Примеру 1, и пропорциональные количества материалов (все проценты выражают, как массовые проценты), готовят следующую лекарственную форму, содержащую 100 мг нефазодона гидрохлорида.
Готовят слой лекарственного вещества, имеющий массу 145,0 мг, состоящий из 69% нефазодона гидрохлорида, 20,24% полиэтиленоксида (Polyox N-80), 5% гидроксипропилцеллюлозы (Klucel MF), 3% полиоксил 40 стеарата (MYRJ 528), 2% кросповидона (PVP XL), 0,75% стеариновой кислоты и 0,01% бутилированного гидрокситолуола (ВТК). Получают выталкивающий слой массой 92 мг, состоящий из 63,67% полиэтиленоксида (Polyox 303), 30,0% хлорида натрия, 5% гидроксипропилметилцеллюлозы (ГПМЦ Е-5), 1% красного оксида железа, 0,25% стеариновой кислоты и 0,08% ВТН. Двухслойное ядро, содержащее слой лекарственного вещества и выталкивающий слой, таблетируют, как описано.
Затем готовят субоболочку из 70% Klucel EF и 30% повидона К29-32 с использованием этанола в качестве растворителя. Субоболочка содержит 8% твердых веществ при нанесении. После нанесения количество субоболочки на двухслойном ядре составляет 13,5 мг. Полупроницаемую мембрану готовят из 99% ацетата целлюлозы 398-10 и 1% полиэтиленгликоля 3350 с использованием системы растворителей, состоящей из 95% ацетона и 5% воды. Мембранное покрытие содержит 5% твердых веществ при нанесении, и масса мембраны на двухслойном ядре с субоболочкой после нанесения составляет 43,8 мг.
Отверстие, имеющее диаметр 2895,6 мкм (114 мил), просверливают в лекарственных формах, которые затем высушивают при 45° С и относительной влажности 45% в течение приблизительно 120 часов и сушат в течение дополнительных 5 часов при 45° С или (в противном случае) в условиях окружающей среды.
Проводят оценку лекарственных форм в отношении выхода нефазодона гидрохлорида с помощью анализа, описанного в Примере 2. Скорости высвобождения для двенадцати отдельных лекарственных форм и кумулятивный процент освободившейся дозы представляют на Фигурах 5 и 6, соответственно. Лекарственные формы демонстрируют значение Т90 18,3 часа и среднее значение скорости высвобождения 5,2 мг/час в течение длительного периода времени, продолжающегося практически от интервала 4 до интервала 18. Показано, что лекарственные формы высвобождают нефазодона гидрохлорид с постоянной скоростью выхода в течение длительного периода времени.
При снижении массы ацетата целлюлозы в полупроницаемой мембране до 28,5 мг и замене пластификатора полиэтиленгликоля 1,5 мг полоксамера 188 и нанесении полупроницаемой мембраны при достижении массы около 26 мг/дозу получают лекарственную форму, имеющую Т90 приблизительно 12 часов.
При снижении массы ацетата целлюлозы в полупроницаемой мембране до 27,2 мг, уменьшении количества пластификатора полиэтиленгликоля до 0,28 мг и нанесении полупроницаемой мембраны при достижении массы около 28 мг/дозу получают лекарственную форму, имеющую Т90приблизительно 13 часов.
Пример 4
Используя основную процедуру, соответствующую Примеру 1, и пропорциональные количества материалов (все проценты выражают, как мас. проценты), готовят следующую лекарственную форму, содержащую 200 мг нефазодона гидрохлорида.
Готовят слой лекарственного вещества, имеющий массу 290 мг, состоящий из 69% нефазодона гидрохлорида, 20,24% полиэтиленоксида (Polyox N-80), 5% гидроксипропилцеллюлозы (Klucel MF), 3% полиоксил 40 стеарата (MYRJ 52S), 2% кросповидона (PVPXL), 0,75% стеариновой кислоты и 0,01% бутилированного гидрокситолуола (ВТН). Получают выталкивающий слой массой 145 мг, состоящий из 64,10% полиэтиленоксида (Polyox 303), 30,0% хлорида натрия, 5% гидроксипропилметилцеллюлозы (ГПМЦ Е-5), 0,5% красного оксида железа, 0,25% стеариновой кислоты и 0,08% ВТН. Двухслойное ядро, содержащее слой лекарственного вещества и выталкивающий слой, таблетируют, как описано.
Затем готовят субоболочку из 70% Klucel EF и 30% повидона К29-32 с использованием этанола в качестве растворителя. После нанесения количество субоболочки на двухслойном ядре составляет 23,6 мг. Полупроницаемую мембрану готовят из 90% ацетата целлюлозы 398-10 и 10% полиоксамера (Pluronics F68, BASF Corporation) с использованием системы растворителей, состоящей из 95% ацетона и 5% воды. Масса мембранного покрытия на двухслойном ядре с субоболочкой после нанесения составляет 37,5 мг.
Отверстие, имеющее диаметр 3937 мкм (155 мил), просверливают в лекарственных формах, которые затем высушивают при 45° С и относительной влажности 45% в течение приблизительно 120 часов и сушат в течение дополнительных 5 часов при 45° С или (в противном случае) в условиях окружающей среды.
Проводят оценку лекарственных форм в отношении выхода нефазодона гидрохлорида с помощью анализа, описанного в Примере 2. Скорости высвобождения для пяти отдельных лекарственных форм и кумулятивный процент освободившейся дозы представляют на Фигурах 7 и 8, соответственно. Лекарственные формы демонстрируют значение Т90 15,1 часа и среднее значение скорости высвобождения 13,4 мг/час в течение длительного периода времени, продолжающегося практически от интервала 4 до интервала 10. Лекарственные формы высвобождают нефазодона гидрохлорид с постоянной скоростью выхода в течение длительного периода времени.
Пример 5
Используя основную процедуру, соответствующую Примеру 1, и пропорциональные количества материалов (все проценты выражают, как маc. проценты), готовят следующую лекарственную форму, содержащую 300 мг нефазодона гидрохлорида.
Готовят слой лекарственного вещества, имеющий массу 435 мг, состоящий из 69% нефазодона гидрохлорида, 20,24% полиэтиленоксида (Polyox N-80), 5% гидроксипропилцеллюлозы (Klucel MF), 3% полиоксил 40 стеарата (MYRJ 52S), 2% кросповидона (PVPXL), 0,75% стеариновой кислоты и 0,01% бутилированного гидрокситолуола (ВТН). Получают выталкивающий слой массой 174 мг, состоящий из 64,1% полиэтиленоксида (Polyox 303), 30,0% хлорида натрия, 5% гидроксипропилметилцеллюлозы (ГПМЦ Е-5), 0,5% красного оксида железа, 0,25% стеариновой кислоты и 0,08% ВТН. Двухслойное ядро, содержащее слой лекарственного вещества и выталкивающий слой, таблетируют, как описано.
Затем готовят субоболочку из 70% Klucel EF и 30% повидона К29-32 с использованием этанола в качестве растворителя. После нанесения количество субоболочки на двухслойном ядре составляет 31,4 мг. Полупроницаемую мембрану готовят из 85% ацетата целлюлозы 398-10 и 15% полиоксамера (Pluroncs F68) с использованием системы растворителей, состоящей из 95% ацетона и 5% воды. Масса мембранного покрытия на двухслойном ядре с субоболочкой после нанесения составляет 40,3 мг.
Отверстие, имеющее диаметр 4445 мкм (175 мил), просверливают в лекарственных формах, которые затем высушивают при 45° С и относительной влажности 45% в течение приблизительно 120 часов и сушат в течение дополнительных 5 часов при 45° С или (в противном случае) в условиях окружающей среды.
Проводят оценку лекарственных форм в отношении выхода нефазодона гидрохлорида с помощью анализа, описанного в Примере 2. Скорости высвобождения для пяти отдельных лекарственных форм и кумулятивный процент освободившейся дозы представляют на Фигурах 9 и 10, соответственно. Лекарственные формы демонстрируют значение Т9011,9 часа и среднее значение скорости высвобождения 26,7 мг/час в течение длительного периода времени, продолжающегося практически от интервала 4 до интервала 10. Лекарственные формы высвобождают нефазодона гидрохлорид с постоянной скоростью выхода в течение длительного периода времени.
Пример 6
Используя основную процедуру, соответствующую Примеру 1, и пропорциональные количества материалов (все проценты выражают, как маc. проценты), готовят следующую лекарственную форму, содержащую 400 мг нефазодона гидрохлорида.
Готовят слой лекарственного вещества, имеющий массу 580,0 мг, состоящий из 69% нефазодона гидрохлорида, 20,24% полиэтиленоксида (Polyox N-80), 5% гидроксипропилцеллюлозы (Klucel MF), 3% полиоксил 40 стеарата (MYRJ 52S), 2% кросповидона (PVPXL), 0,75% стеариновой кислоты и 0,01% бутилированного гидрокситолуола (ВТН). Получают выталкивающий слой массой 232,0 мг, состоящий из 64,1% полиэтиленоксида (Polyox 303), 30,0% хлорида натрия, 5% гидроксипропилметилцеллюлозы (ГПМЦ Е-5), 0,5% красного оксида железа, 0,25% стеариновой кислоты и 0,08% ВТН. Двухслойное ядро, содержащее слой лекарственного вещества и выталкивающий слой, таблетируют, как описано.
Затем готовят субоболочку из 70% Klucel EF и 30% повидона К29-32 с использованием этанола в качестве растворителя. После нанесения количество субоболочки на двухслойном ядре составляет 36,3 мг. Полупроницаемую мембрану готовят из 80% ацетата целлюлозы 398-10 и 20% полиоксамера F68 с использованием системы растворителей, состоящей из 95% ацетона и 5% воды. Масса мембранного покрытия на двухслойном ядре с субоболочкой после нанесения составляет 88,7 мг.
Отверстие, имеющее диаметр 4826 мкм (190 мил), просверливают в лекарственных формах, которые затем высушивают при 45° С и относительной влажности 45% в течение приблизительно 120 часов и сушат в течение дополнительных 5 часов при 45° С или (в противном случае) в условиях окружающей среды.
Проводят оценку лекарственных форм в отношении выхода нефазодона гидрохлорида с помощью анализа, описанного в Примере 2. Скорости высвобождения для пяти отдельных лекарственных форм и кумулятивный процент освободившейся дозы представляют на Фигурах 11 и 12, соответственно. Лекарственные формы демонстрируют значение Т90 14 часов и среднее значение скорости высвобождения 29,7 мг/час в течение длительного периода времени, продолжающегося практически от интервала 5 до интервала 13. Лекарственные формы высвобождают нефазодона гидрохлорид с постоянной скоростью выхода в течение длительного периода времени.
Пример 7
Репрезентативные образцы лекарственных форм, соответствующих изобретению, которые содержат 100-600 мг нефазодона гидрохлорида и имеют диаметры отверстия 2794-5080 мкм (110-200 мил), вводят субъектам перорально один раз в сутки. В образцах крови, полученных от субъектов через регулярные интервалы времени (обычно 1-4 часа), и полученных таким образом образцах плазмы крови проводят анализ количества присутствующего нефазодона гидрохлорида. Лекарственные формы, соответствующие изобретению, обеспечивают пролонгированные уровни в плазме крови в интервале между 5 нг/мл и 2500 нг/мл. Стабильные уровни в плазме крови поддерживаются на постоянных терапевтических уровнях, так что частное, полученное при делении [Cmax-Cmin]/Cmin, для нефазодона гидрохлорида в плазме в течение 24-часового периода после введения составляет 3 или меньше.
Неожиданно показано, что способствующая продвижению стенка 7 обеспечивает практически полное высвобождение, т.е. 80 мас.% или больше лекарственного вещества из лекарственных форм, изготовленных в соответствии с данным изобретением. Для лекарственных форм, в которых имеется высокая нагрузка лекарственного вещества, т.е. 40% или более активного агента в слое лекарственного вещества от общей массы слоя лекарственного вещества, и при отсутствии способствующего продвижению слоя 7 показано, что значительные остаточные количества лекарственного вещества сохраняются в устройстве, как описано в данном контексте, после окончания периода доставки. В ряде случаев при отсутствии способствующего продвижению слоя количества, превышающие 20%, остаются в устройстве в конце 24-часового периода. Остаточные количества лекарственного вещества уменьшают добавлением внутреннего покрытия из гидроксиалкилцеллюлозы, которое наносят на слой лекарственного вещества. Когда внутренний слой содержит гидроксипропилцеллюлозу (Klucel EF), имеющую среднечисловую молекулярную массу 80000, массы субоболочки, необходимые для получения остаточного содержания лекарственного вещества 7%, 4% и 3%, составляют в процентах от двухслойного ядра 9%, 12% и 15%, соответственно. Способствующий продвижению слой или внутренняя стенка 7 снижают силу трения между полупроницаемой стенкой 2 и наружной поверхностью слоя лекарственного вещества, обеспечивая таким образом более полную доставку лекарственного вещества из устройства. В частности, в случае активных соединений, имеющих высокую стоимость, данное усовершенствование представляет существенные экономические преимущества, поскольку снимает необходимость избыточной нагрузки слоя лекарственного вещества, чтобы гарантировать доставку минимального количества, которое требуется.
Пример 8
Используя основную процедуру, соответствующую Примеру 1, готовят лекарственные формы с 400 мг нефазодона гидрохлорида, которые содержат 83% слоя лекарственного вещества массы 482 мг и имеют субоболочку массой 14,1 мг, образующую способствующий продвижению слой, и полупроницаемую мембрану массы 81,3 мг, с выходным отверстием 3937 мкм (155 мил) на конце лекарственной формы. Аналогичным образом готовят лекарственные формы, содержащие такое же количество нефазодона гидрохлорида и полупроницаемую мембрану массы 83,7 мг и имеющие выходное отверстие 3937 мкм (155 мил) на конце лекарственной формы, но без субоболочки. Репрезентативные лекарственные формы тестируют, используя анализ скорости высвобождения, и представляют результаты в графической форме на Фигурах 13.1-13.4. Результаты для лекарственных форм с субоболочкой приводят на Фигуре 13.1, представляющей среднюю скорость высвобождения приблизительно 10,3 мг/ч, что является практически нулевым порядком, и Фигуре 13.2, представляющей скорость кумулятивного выхода, которая имеет Т90 приблизительно 26,6 часа. Напротив, результаты для лекарственной формы без покрытия, приведенные на Фигурах 13.3 и 13.4, иллюстрируют изменяющуюся скорость высвобождения при том, что через 26 часов высвобождается лишь приблизительно 55% лекарственного вещества. Лекарственные формы, изготовленные со способствующим продвижению слоем, нанесенным в виде субоболочки, обеспечивают контролируемый выход лекарственного вещества в течение длительного периода времени при минимальном количестве остаточного лекарственного вещества, сохраняющегося в лекарственной форме через 24 часа после введения.
Данное изобретение содержит характеристики и признаки, либо по отдельности, либо в комбинации друг с другом или с несколькими признаками, которые представлены в нижеследующей формуле изобретения.
Реферат
Изобретение относится к лекарственной форме с постоянной скоростью высвобождения лекарственного вещества, ядру лекарственной формы и к способу обеспечения облегченного высвобождения лекарственного вещества из лекарственной формы, включающей слой, содержащий лекарственное вещество, а также полупроницаемую стенку и расширяющийся слой, так что между полупроницаемой стенкой и слоем, содержащим лекарственное вещество, размещают способствующий продвижению слой, а содержание лекарственного вещества составляет по меньшей мере 20% от общей массы слоя, содержащего лекарственное вещество. Технический результат: изобретение обеспечивает высвобождение практически всего лекарственного вещества из лекарственной формы в среду применения. 3 с. и 9 з.п. ф-лы, 13 ил.

Комментарии