Органические соединения - RU2733975C2
Код документа: RU2733975C2
Описание
Перекрестная ссылка на родственные заявки
Настоящая заявка испрашивает приоритет и преимущество предварительной патентной заявки США с серийным № 62/313629, поданной 25 марта 2016 года, содержание которой включено в настоящую заявку посредством ссылки во всей полноте.
Область, к которой отностся изобретение
Изобретение относится к конкретным дейтерированным гетероциклическим конденсированным гамма-карболинам в свободной форме, в форме фармацевтически приемлемой соли и/или по существу чистой форме, как описано в настоящей заявке, к их фармацевтическим композициям и способам применения в лечении заболеваний, связанных с 5-HT2Aрецептором, транспортером серотонина (SERT) и/или путями с вовлечением сигнальных систем допаминовых D1/D2 рецепторов, например, заболеваний или расстройств, таких как беспокойство, психоз, шизофрения, расстройства сна, сексуальные расстройства, мигрень, состояния, связанные с головной болью, социальные фобии, желудочно-кишечные расстройства, такие как нарушение моторики желудочно-кишечного тракта и ожирение; депрессия и расстройства настроения, связанные с психозом или болезнью Паркинсона; психоз, такой как шизофрения, связанная с депрессией; биполярное расстройство; и другие психиатрические и неврологические состояния, а также к комбинациям с другими средствами.
Предпосылки создания изобретения
Психоз, особенно шизофрения и шизоаффективное расстройство, поражают приблизительно 1-2% населения во всем мире. Шизофрения состоит из трех фаз: продромальная фаза, активная фаза и остаточная фаза. Продромальная фаза является ранней фазой, в которой наблюдаются субклинические признаки и симптомы. Эти симптомы могут включать потерю интереса к обычным занятиям, отчуждение от друзей и членов семьи, путаницу сознания, проблемы с концентрацией, чувство вялости и апатии. Активная фаза характеризуется обострениями положительных симптомов, таких как бред, галлюцинации и подозрительность. Остаточная фаза характеризуется негативными симптомами, такими как эмоциональное отчуждение, пассивное социальное отчуждение и стереотипное мышление; и общими психопатологическими симптомами, включая активную социальную изоляцию, беспокойство, напряжение и соматические проблемы. Симптомы остаточной фазы также часто сопровождаются депрессией, когнитивной дисфункцией и бессонницей. В совокупности эти симптомы остаточной фазы не очень хорошо лечатся многими антипсихотическими препаратами, которые в настоящее время доступны на рынке, и поэтому обычно наблюдаются после того, как симптомы активной фазы утихают после лечения. Эта фаза болезни заключается в том, что пациенты хотели бы вернуться к более продуктивной и полноценной жизни, но так как остаточные отрицательные симптомы и когнитивные нарушения не вылечены должным образом, это препятствует возврату к такой функции. Существует настоятельная необходимость в антипсихотическом средстве, которое может лечить не только симптомы активной или острой фазы, но также симптомы остаточной фазы психоза, например, шизофрении. Кроме того, существует потребность в лекарственных средствах для лечения этих симптомов, которые не имеют нежелательных побочных эффектов, вызываемых нецелевым взаимодействием с гистаминовыми H1 и мускариновыми ацетилхолиновыми рецепторными системами.
Замещенные гетероциклические конденсированные гамма-карболины известны как агонисты или антагонисты 5-HT2 рецепторов, в частности, 5-HT2A рецепторов, для лечения расстройств центральной нервной системы. Эти соединения были раскрыты в патентах США №№ 6548493; 7238690; 6552017; 6713471; 7183282; U.S. RE39680 и U.S. RE39679, как новые соединения полезные для лечения расстройств, связанных с модуляцией 5-HT2A рецептора, таких как ожирение, беспокойство, депрессия, психоз, шизофрения, расстройства сна, сексуальные расстройства, мигрень, состояния, связанные с головной болью, социальные фобии, желудочно-кишечные расстройства, такие как нарушение моторики желудочно-кишечного тракта, и ожирение.
PCT/US08/03340 (WO 2008/112280) и заявка США с серийным № 10/786935 раскрывают способы получения замещенных гетероциклических конденсированных гамма-карболинов и применения этих гамма-карболинов в качестве агонистов и антагонистов серотонина, полезных для контроля и профилактики расстройств центральной нервной системы, таких как аддиктивное поведение и расстройства сна.
WO/2009/145900 раскрывает применение конкретных замещенных гетероциклических конденсированных гамма-карболинов для лечения комбинации психоза и депрессивных расстройств, а также расстройств сна, депрессивных расстройств и/или расстройств настроения у пациентов с психозом или болезнью Паркинсона. Помимо расстройств, связанных с психозом и/или депрессией, эта патентная заявка раскрывает и заявляет применение этих соединений при низкой дозе в качестве селективных антагонистов 5-HT2A рецепторов, не затрагивая при этом или минимально затрагивая допаминовые D2 рецепторы, таким образом, полезных для лечения расстройства сна без побочных эффектов, затрагивающих допаминовые D2 пути, или побочных эффектов, затрагивающих другие пути (например, GABAAрецепторы), связанных с традиционными седативно-гипнотическими средствами (например, бензодиазепинами), включая, но не ограничиваясь этим, седативно-гипнотические средства (например, бензодиазепины), включающих, но не ограничивающихся этим, развитие лекарственной зависимости, мышечную гипотонию, слабость, головную боль, помутнение зрения, головокружение, тошноту, рвоту, эпигастральный дистресс, диарею, боли в суставах и боли в груди.
Кроме того, было обнаружено, что эти конкретные замещенные гетероциклические конденсированные гамма-карболиновые соединения (соединения, описанные ниже) эффективны при лечении не только острых симптомов, но и остаточных симптомов психоза. Таким образом, настоящее изобретение обеспечивает способы применения конкретных замещенных гетероциклических конденсированных гамма-карболиновых соединений (соединений, описанных ниже), либо отдельно, либо в качестве дополнительной терапии для лечения остаточных симптомов психоза, особенно шизофрении.
WO 2009/114181 раскрывает способы получения кристаллов аддитивной соли толуолсульфоновой кислоты конкретных замещенных гетероциклических конденсированных гамма-карболинов, например, соли присоединения толуолсульфоновой кислоты 4-((6bR,10aS)-3-метил-2,3,6b,9,10,10a-гексагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8(7H)-ил)-1-(4-фторфенил)-1-бутанона.
WO 2011/133224 раскрывает пролекарства/метаболиты замещенного гетероциклического конденсированного гамма-карболина для улучшенной композиции, например, композиции с пролонгированным/контролируемым высвобождением. Эта заявка раскрывает, что гетероциклический конденсированный гамма-карболин, N-замещенный 4-фторфенил(4-гидрокси)бутильной группой, имеет высокую селективность в отношении серотонинового транспортера (SERT) по сравнению с гетероциклическим конденсированным гамма-карболином, содержащим 4-фторфенилбутанон. Однако гидроксигруппа на этих соединениях взаимопревращается в кетон и из кетона в плазме и в головном мозге, что позволяет ему служить в качестве резервуара для 4-фторфенилбутанонового лекарственного средства. Хотя замещенные гетероциклические конденсированные гамма-карболины и их применение известны, авторы настоящего изобретения неожиданно обнаружили, что, хотя конкретные замещенные гетероциклические конденсированные гамма-карболины менее активны в испытаниях in vitro, происходит взаимопреобразование между этими менее активными соединениями и высокоактивным кетоновым лекарственным средством в плазме и головном мозге. Авторы настоящего изобретения также предоставили пролекарства конкретных замещенных гетероциклических конденсированных гамма-карболинов, которые имеют измененный фармакокинетический профиль, например, измененные механизмы и/или скорость абсорбции и дистрибуцию, и поэтому могут быть полезны для улучшенной композиции и/или для контроля продолжительности эффекта лекарственного средства в организме (например, для длительного или контролируемого высвобождения).
WO 2013/155505 раскрывает соединения, которые блокируют in-in vivo взаимопреобразование между гидроксилом и кетоном путем включения алкильного заместителя по углероду, несущему гидроксильную группу, таким образом получая соединения, которые антагонизируют 5-HT2A рецепторы, а также ингибируют повторное поглощение серотонина.
Основными путями метаболизма ранее описанных соединений являются N-деметилирование, катализируемое CYP 3A4, и восстановление кетона, катализируемое кеторедуктазой. Известно, что N-деалкилирование ферментами цитохромоксидазами происходит путем первоначального окисления одного или нескольких атомов углерода в альфа-положении относительно атома азота. Семейство ферментов, катализирующих восстановление кетона, велико и разнообразно, и механизм полностью не выяснен. Интересно то, что, механически, восстановление кетона может происходить либо посредством енольного таутомера кетона, либо кето-таутомера.
WO 2015/154025 раскрывает типичные дейтерированные гетероциклические конденсированные гамма-карболины с целью снижения метаболического разложения путем частичного ограничения метаболизма кетона и/или N-метильного заместителя.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Авторы настоящего изобретения неожиданно обнаружили, что основные пути метаболизма конденсированного гетероциклического гамма-карболина формулы Q включают N-деалкилирование и альфа-окисление на пиперазиновом кольце и восстановление карбонила с получением соединений формулы Q-1, Q-2 и Q-3, как показано ниже:
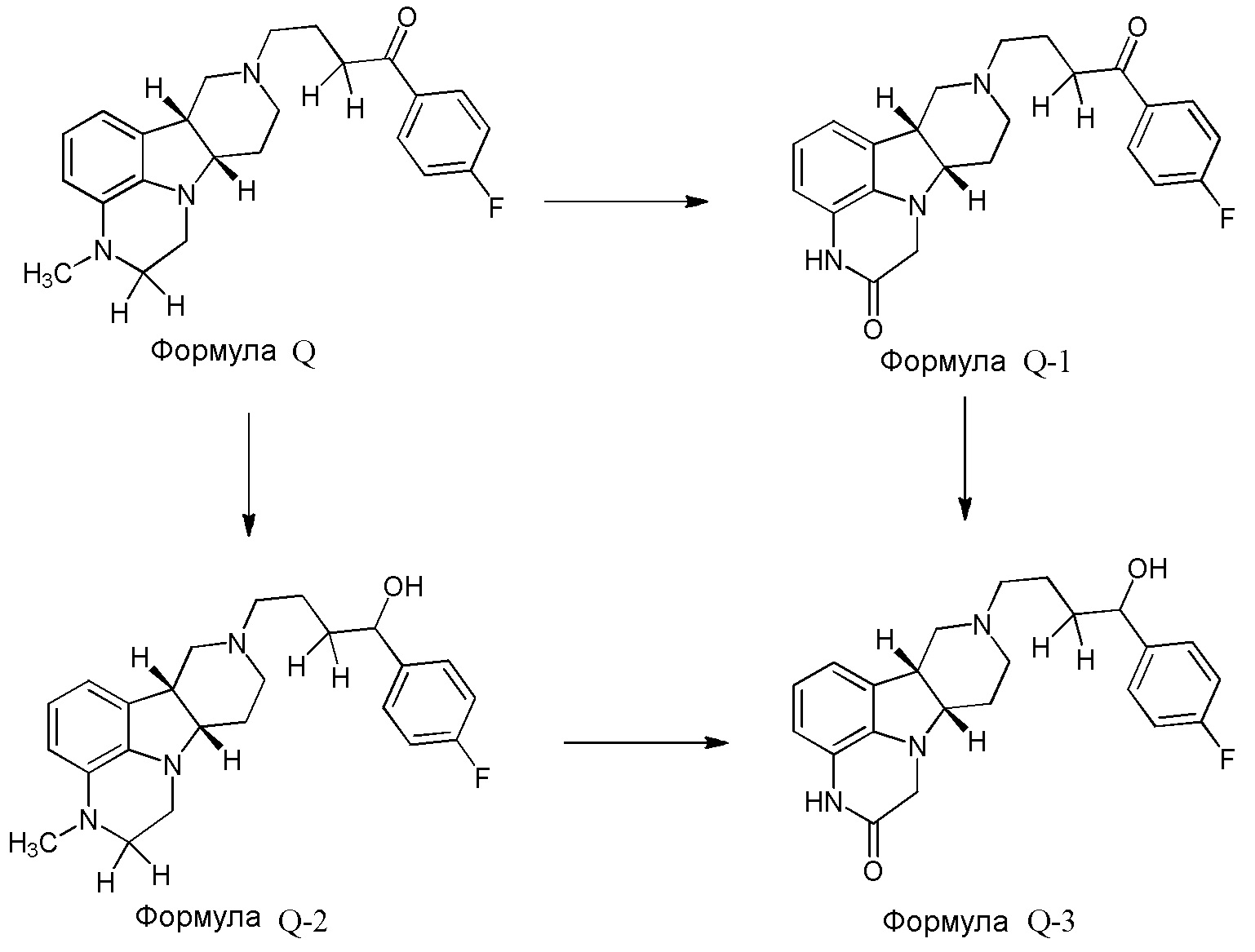
Авторы настоящего изобретения также обнаружили, что спиртовой метаболит формулы Q-2 сохраняет существенную фармакологическую активность.
Не связывая это с теорией, настоящее изобретение относится к соединениям, которые специфически ограничивают и/или предотвращают метаболизм, происходящий этими путями. Считается, что из-за очень сходных свойств атомов дейтерия (2Н) по сравнению с нормальными атомами водорода (1Н) лекарственные соединения, в которых дейтерий замещает водород, как правило, имеют сходную биологическую активность с недейтерированным аналогом, но потенциально имеют улучшенные фармакокинетические свойства. Степень, в которой такая замена приведет к улучшению фармакокинетических свойств без чрезмерной потери фармакологической активности, варьируется. Таким образом, в некоторых случаях полученное дейтерированное соединение имеет только умеренное повышение фармакокинетической стабильности, в то время как в других обстоятельствах полученное дейтерированное соединение может иметь значительно улучшенную стабильность. Более того, возможно будет трудно предсказать с уверенностью эффекты одновременных замещений дейтерием. Они могут приводить или не приводить к аддитивному (синергетическому) улучшению метаболической стабильности.
Настоящее изобретение обеспечивает соединения, содержащие тридетерированный N-метил и/или ди-дейтерированный метилен, смежный с N-метилом. Эти новые соединения антагонизируют рецепторы 5-НТ2А, ингибируют траспортер повторного поглощения серотонина и модулируют допаминергическое фосфорилирование белка таким же образом, как их природные водородные аналоги. Однако эти соединения демонстрируют неожиданно улучшенную метаболическую стабильность.
В первом варианте осуществления изобретение обеспечивает соединение формулы I:
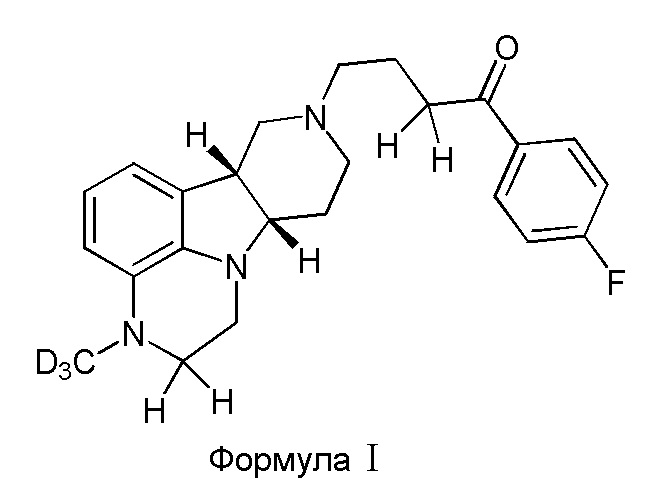
в свободной или солевой форме, например, в форме фармацевтически приемлемой соли (например, тозилата).
Во втором варианте осуществления изобретение обеспечивает соединение формулы II:
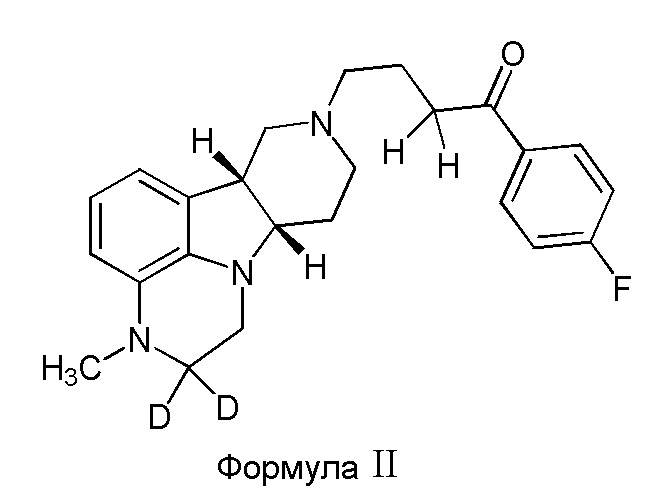
в свободной или солевой форме, например, в форме фармацевтически приемлемой соли (например, тозилата).
В третьем варианте осуществления изобретение обеспечивает соединение формулы III:
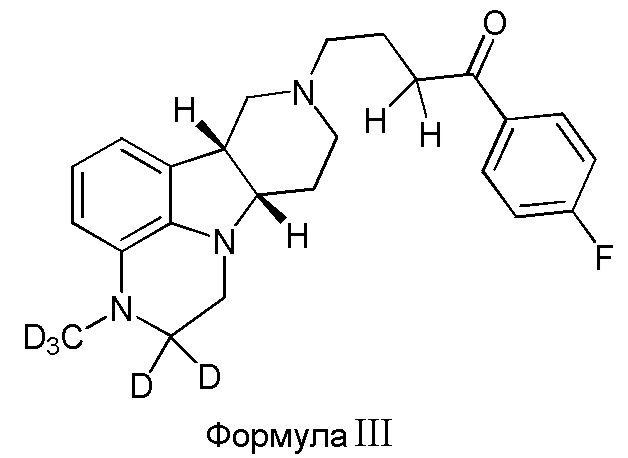
в свободной или солевой форме, например, в форме фармацевтически приемлемой соли (например, тозилата).
В четвертом варианте осуществления изобретение обеспечивает соединение формулы IV,
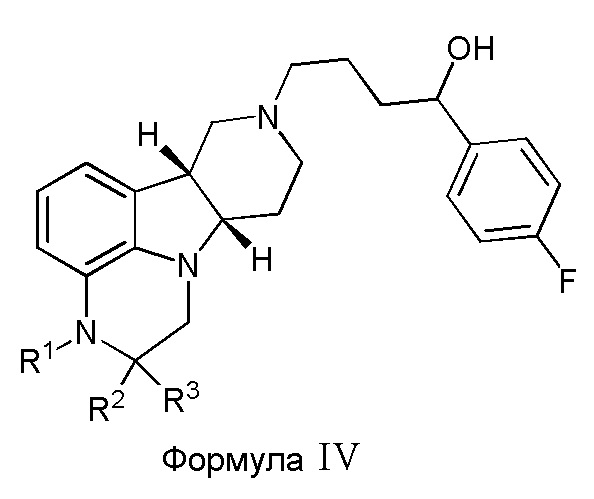
где:
R1 представляет собой CH3илиCD3;
R2 и R3 либо оба представляют собой H, либо оба представляют собой D;
при условии, что, когда R1 представляет собой CH3, R2 и R3 оба представляют собой D;
в свободной или солевой форме, например, в форме фармацевтически приемлемой соли (например, тозилата).
В дополнительных вариантах осуществления изобретение обеспечивает следующие соединения:
1.1. Соединение любой из формул I - IV, где соединение находится в свободной форме или в форме фармацевтически приемлемой соли;
1.2. Соединение формулы 1.1, где солевая форма представляет собой кислотно-аддитивную соль фармацевтически приемлемой кислоты;
1.3. Соединение формулы 1.2 где кислота представляет собой толуолсульфоновую кислоту;
1.4. Соединение любой из формул I - IV или 1.1-1.3, где Соединение находится в по существу чистой диастереомерной форме (т.е. по существу свободной от других диастереомеров);
1.5. Соединение любой из формул I - IV или 1.1-1.4, где соединение имеет диастереомерный избыток больше чем 70%, предпочтительно больше чем 80%, более предпочтительно больше чем 90%, и наиболее предпочтительно больше чем 95%;
1.6. Соединение любой из формул I - IV или 1.1-1.5, где соединение имеет больше чем природное включение дейтерия в указанных положениях дейтерия структуры (т.е. больше чем 0,0156%);
1.7. Соединение любой из формул I - IV или 1.1-1.6, где соединение имеет по существу больше чем природное включение дейтерия в указанных положениях дейтерия структуры (например, больше чем 0,1%, или больше чем 0,5%, или больше чем 1%, или больше чем 5%);
1.8. Соединение любой из формул I - IV или 1.1-1.7, где соединение имеет больше чем 50% включение дейтерия в указанных дейтерированных положениях структуры (т.е. больше чем 50 атом% D), например, больше чем 60%, или больше чем 70%, или больше чем 80%, или больше чем 90%, или больше чем 95%, или больше чем 96%, или больше чем 97%, или больше чем 98%, или больше чем 99%.
Во втором аспекте изобретение обеспечивает фармацевтическую композицию, включающую соединение любой из формул I - IV или 1.1-1.8 (соединения по изобретению) в свободной форме или в форме фармацевтически приемлемой соли, в смеси с фармацевтически приемлемым разбавителем или носителем, например, для обеспечения немедленного высвобождения или для обеспечения пролонгированного или замедленного высвобождения.
Еще в одном варианте осуществления второго аспекта фармацевтическая композиция по изобретению предназначена для пролонгированного или замедленного высвобождения, например, депо препарата. В одном варианте осуществления депо препарат включает соединения по изобретению в полимерной матрице. В другом варианте осуществления соединения по изобретению диспергированы или растворены в полимерной матрице. В другом варианте осуществления полимерная матрица включает стандартные полимеры, используемые в депо препаратах, такие как полимеры, выбранные из полиэфира гидрокси-жирной кислоты и его производных или полимера алкил альфа-цианоакрилата, полиалкиленоксалата, поли(ортоэфира), поликарбоната, полиортокарбоната, поли(аминокислоты), эфира гиалуроновой кислоты и их смесей. В другом варианте осуществления полимер выбран из группы, состоящей из полилактида, поли d,l-лактида, полигликолида, PLGA 50:50, PLGA 75:25, PLGA 85:15 и PLGA 90:10 полимера. В другом варианте осуществления полимер выбран из поли(гликолевой кислоты), поли-D,L-молочной кислоты, поли-L-молочной кислоты, coполимеров вышеуказанных, поли(алифатических карбоновых кислот), сополиоксалатов, поликапролактона, полидиоксонона, поли(ортокарбонатов), поли(ацеталей), поли(молочной кислоты-капролактоны), полиортоэфиров, поли(гликолевой кислоты-капролактоны), полиангидридов и природных полимеров, включая альбумин, казеин и воски, например, глицерин моно- и дистеарата и т.п. В конкретном варианте осуществления полимерная матрица включает поли (d,l-лактид-ко-гликолид). Любая из композиций, описанных выше, может представлять собой фармацевтическую композицию, где указанная композиция находится в смеси с фармацевтически приемлемым разбавителем или носителем.
(Фармацевтические) депо препараты, как описано выше, особенно полезны для пролонгированного или замедленного высвобождения, где соединения по изобретению высвобождаются при разрушении полимерной матрицы. Эти композиции можно сформулировать для контролируемого и/или пролонгированного высвобождения соединений по изобретению (например, в виде депо композиции) в течение периода вплоть до 180 дней, например, от около 14 до около 30 до около 180 дней. Например, полимерная матрица может разрушаться и высвобождать соединения по изобретению в течение периода of около 30, около 60 или около 90 дней. В другом примере полимерная матрица может разрушаться и высвобождать соединения по изобретению в течение периода около 120 или около 180 дней.
Еще в одном дополнительном варианте осуществления фармацевтические композиции по изобретению, в частности, депо композиции по изобретению, формулируют для введения путем инъекции.
В третьем аспекте изобретение обеспечивает соединения по изобретению, описанные выше, в пероральной лекарственной форме пролонгированного или замедленного высвобождения. Например, изобретение обеспечивает пероральную систему доставки с осмотически контролируемым высвобождением (OROS) для доставки соединений по изобретению, например, аналогичную системам, описанным в WO 2000/35419 и EP 1 539 115 (U.S. Pub. No. 2009/0202631), содержание каждой из этих заявок включено посредством ссылки во всей полноте. Поэтому в одном варианте осуществления этого аспекта изобретение обеспечивает фармацевтическую композицию или устройство, включающее (a) желатиновую капсулу, содержащую соединение по изобретению в свободной форме или в форме фармацевтически приемлемой соли или фармацевтическую композицию по изобретению, описанную выше; (b) многослойную оболочку, напластованную на желатиновую капсулу, включающую, в направлении наружу от капсулы: (i) барьерный слой, (ii) расширяемый слой и (iii) полупроницаемый слой; и (c) и отверстие, образованное или формируемое через оболочку. (Композиция P.1)
В другом варианте осуществления этого аспекта изобретение обеспечивает композицию, включающую желатиновую капсулу, содержащую жидкость, соединения по изобретению в свободной форме или в форме фармацевтически приемлемой соли или фармацевтическую композицию по изобретению, описанную выше, при этом желатиновая капсула окружена слоистой оболочкой, включающей барьерный слой, контактирующий с внешней поверхностью желатиновой капсулы, расширяемый слой, контактирующий с барьерным слоем, полупроницаемый слой, охватывающий расширяемый слой, и выходное отверстие, образованное или формируемое в оболочке. (Композиция P.2)
Еще в одном варианте осуществления третьего аспекта изобретение обеспечивает композицию, включающую желатиновую капсулу, содержащую жидкость, соединение по изобретению в свободной форме или в форме фармацевтически приемлемой соли или фармацевтическую композицию по изобретению, описанную выше, при этом желатиновая капсула окружена слоистой оболочкой, включающей барьерный слой, контактирующий с внешней поверхностью желатиновой капсулы, расширяемый слой, контактирующий с барьерным слоем, полупроницаемый слой, охватывающий расширяемый слой, и выходное отверстие, образованное или формируемое в оболочке, где барьерный слой образует уплотнение между расширяемым слоем и окружающей средой на выходном отверстии. (Композиция P.3)
Еще в одном варианте осуществления третьего аспекта изобретение обеспечивает композицию, включающую желатиновую капсулу, содержащую жидкость, соединение по изобретению в свободной форме или в форме фармацевтически приемлемой соли или фармацевтическую композицию по изобретению, описанную выше, при этом желатиновая капсула окружена барьерным слоем, контактирующим с внешней поверхностью желатиновой капсулы, расширяемым слоем, контактирующим с частью барьерного слоя, полупроницаемым слоем, охватывающим по меньшей мере расширяемый слой и выходное отверстие, образованное или формируемое в лекарственной форме, проходящее от внешней поверхности желатиновой капсулы до среды, в которой ее используют. (Композиция P.4). Расширяемый слой может быть образован в одной или нескольких отдельных секциях, например, двух секциях, расположенных на противоположных сторонах или концах желатиновой капсулы.
В специальном варианте осуществления третьего аспекта соединения по изобретению в пероральной системе доставки с осмотически-контролируемым высвобождением (т.е. в Композиции P.1-P.4) находятся в жидкой лекарственной форме, при этом лекарственная форма может представлять собой чистое соединение без растворителя, жидкое активное вещество, жидкое активное вещество в растворе, супензию, эмульсию или самоэмульгирующуюся композицию или т.п.
Более подробную информацию о пероральной системе доставки с осмотически-контролируемым высвобождением, включая характеристики желатиновой капсулы, барьерного слоя, расширяемого слоя, полупроницаемого слоя и отверстия, можно найти в WO 2000/35419, содержание которой включено посредством ссылки во всей полноте. Другие пероральные системы доставки с осмотически-контролируемым высвобождением для соединения или фармацевтической композиции по изобретению можно найти в EP 1 539 115 (U.S. Pub. No. 2009/0202631), содержание которой включено посредством ссылки во всей полноте.
Поэтому в другом варианте осуществления третьего аспекта изобретение обеспечивает композицию или устройство, включающее (a) два или более слоев, указанные два или более слоев включают первый слой и второй слой, указанный первый слой включает соединение по изобретению в свободной форме или в форме фармацевтически приемлемой соли или фармацевтическую композицию, описанную выше, указанный второй слой включает полимер; (b) внешнюю оболочку, окружающую указанные два или более слоев; и (c) отверстие в указанной внешней оболочке. (Композиция P.5)
В Композиции P.5 предпочтительно используют полупроницаемую мембрану, окружающую трехслойное ядро: в этих вариантах осуществления первый слой указан как первый лекарственный слой и содержит небольшие количества лекарственного средства (например, соединения по изобретению) и осмотический агент, такой как соль, средний слой указан как второй лекарственный слой и содержит бóльшие количества лекарственного средства, эксципиенты и никакой соли; и третий слой указан как слой осмотического вещества и содержит осмотическое вещество и никакого лекарственноого средства. По меньшей мере одно отверстие просверлено через мембрану на конце первого лекарственного слоя таблетки в форме капсулы. (Композиция P.6)
Композиция Р.5 или Р.6 может включать мембрану, ограничивающую отсек, при этом мембрана окружает внутренний защитный подслой, по меньшей мере одно выходное отверстие, сформированное или формируемое в нем, и по меньшей мере часть мембраны является полупроницаемой; расширяемый слой, расположенный в отсеке вдали от выходного отверстия и находящийся в жидкостном сообщении с полупроницаемой частью мембраны; первый лекарственный слой, расположенный рядом с выходным отверстием; и второй лекарственный слой, расположенный внутри отсека между первым лекарственным слоем и расширяемым слоем, при этом лекарственные слои содержат соединение по изобретению в свободной форме или в форме его фармацевтически приемлемой соли. В зависимости от относительной вязкости первого лекарственного слоя и второго лекарственного слоя получают различные профили высвобождения. Необходимо определить оптимальную вязкость для каждого слоя. В настоящем изобретении вязкость модулируется добавлением соли, хлорида натрия. Профиль доставки из ядра зависит от массы, композиции и толщины каждого из слоев лекарственного средства.
В конкретном варианте осуществления изобретение обеспечивает композицию Р.7, где первый лекарственный слой содержит соль, а второй лекарственный слой не содержит соли. Композиция P.5-P.7 необязательно может включать промотирующий текучесть слой между мембраной и лекарственными слоями. Композиции P.1-P.7 обычно указываются как композиция системы доставки с осмотически-контролируемым высвобождением.
В четвертом аспекте изобретение обеспечивает способ (способ I) для лечения или профилактики расстройства центральной нервной системы, включающий введение пациенту, нуждающемуся в этом, соединения формул I-IV или 1.1-1.8 в свободной форме или в форме фармацевтически приемлемой соли или фармацевтической композиции, описанной выше, и в котором соединение формул I-IV или 1.1-1.8 необязательно вводят в эффективной дозе, которая ниже, чем эффективная доза для лечения этого же расстройства с использованием соединения формулы Q.
Еще в одном варианте осуществления четвертого аспекта изобретение обеспечивает Способ I, который более подробно описан в следующих формулах:
7.1 Способ I, где расстройство центральной нервной системы представляет собой одно или несколько расстройств, ассоциированных с деменцией, например, расстройства, ассоциированные с легким когнитивным расстройством и деменцией, включая старческую деменцию, болезнь Альцгеймера, болезнь Пика, фронто-темпоральную деменцию, парасупрануклеарный паралич, деменцию с тельцами Леви, сосудистую деменцию, болезнь Гентингтона, болезнь Паркинсона, рассеянный склероз, амиотрофический боковой склероз, синдром Дауна, старческую депрессию, синдром Вернике-Корсакова, кортико-базальные дегенерации и прионовую болезнь, аутизм и синдром дефицита внимания и гиперактивности;
7.2 Способ I или 7.1, где расстройства, ассоциированные с деменцией, выбраны из группы, состоящей из следующих: (1) поведенческие расстройства или расстройства настроения, такие как возбуждение/раздражение, агрессивное/буйное поведение, гнев, физические или эмоциональные всплески; (2) психоз; (3) депрессия; и (4) расстройства сна;
7.3. Способ I или 7.1, где расстройство центральной нервной системы представляет собой возбуждение/раздражение, агрессивное/буйное поведение, гнев, физические или эмоциональные всплески;
7.4. Способ I, где расстройство центральной нервной системы представляет собой расстройство, выбранное из группы, включающей ожирение, беспокойство, депрессию (например, рефракторную депрессию и большое депрессивное расстройство (MDD)), психоз, шизофрению, расстройства сна (в частности, расстройства сна, связанные с шизофренией и другими психиатрическими и неврологическими заболеваниями), сексуальные расстройства, мигрень, состояния, связанные с головной болью, социальные фобии, возбуждение при деменции (например, возбуждение при болезни Альцгеймера), возбуждение при аутизме и подобных аутических расстройствах, и желудочно-кишечные расстройства, такие как нарушение моторики желудочно-кишечного тракта;
7.5 Способ I или любой из 7.2-7.4, где расстройство центральной нервной системы представляет собой расстройство, связанное с серотонином 5-HT2A, системой допаминовых D1/D2 рецепторов и/или путями транспортера обратного захвата серотонина (SERT), как аналогичным образом описано в WO/2009/145900, содержание которой включено в настоящую заявку посредством ссылки во всей полноте;
7.6 Способ I или в соответствии с любой из формул 7.2-7.5, где расстройство центральной нервной системы представляет собой расстройство, связанное с путями транспортера обратного захвата серотонина (SERT);
7.7 Способ I или в соответствии с любой из формул 7.2-7.6, где расстройство центральной нервной системы представляет собой расстройство, выбранное из следующих: (i) психоз, например, шизофрения, у пациента, страдающего депрессией; (2) депрессия у пациента, страдающего психозом, например, шизофренией; (3) расстройства настроения, связанные с психозом, например, шизофренией, или с болезнью Паркинсона; и (4) расстройства сна, связанные с психозом, например, шизофренией или болезнью Паркинсона; (5) депрессия; (6) беспокойство; (7) пост-травматическое стрессовое расстройство; или (8) расстройство импульсного контроля, например, интермиттирующее эксплозивное расстройство;
7.8 Способ I или в соответствии с любой из формул 7.2-7.7, где расстройство центральной нервной системы представляет собой психоз, например, шизофрению, и указанный пациент представляет собой пациента, страдающего депрессией; 7.2-7.7
7.9 Способ I или в соответствии с любой из формул 7.2-7.8, где указанный пациент не может переносить побочные эффекты традиционных антипсихотических лекарственных средств, например, таких как хлорпромазин, галоперидол, дроперидол, флуфеназин, локсапин, мезоридазин, молиндон, перфеназин, пимозид, прохлорперазин, промазин, тиоридазин, тиотиксен, трифлуоперазин, клозапин, арипипразол, оланзапин, кветиапин, рисперидон и зипразидон;
7.10 Способ I или в соответствии с любой из формул 7.2-7.9, где указанный пациент не может переносить побочные эффекты традиционных антипсихотических лекарственных средств, например, таких как галоперидол, арипипразол, клозапин, оланзапин, кветиапин, рисперидон и зипразидон;
7.11 Способ I или в соответствии с любой из формул 7.2-7.10, где указанное расстройство представляет собой депрессию и указанный пациент представляет собой пациента, страдающего психозом, например, шизофренией, или болезнью Паркинсона;
7.12 Способ I или в соответствии с любой из формул 7.2-7.6, где указанное расстройство представляет собой расстройство сна, и указанный пациент страдает депрессией;
7.13 Способ I или любой из 7.2-7.6, где указанное одно или несколько расстройств представляет собой расстройство сна, и указанный пациент страдает психозом, например, шизофренией;
7.14 Способ I или любой из 7.2-7.6, где указанное одно или несколько расстройств представляет собой расстройство сна, и указанный пациент страдает болезнью Паркинсона;
7.15 Способ I или любой из 7.2-7.6, где указанное одно или несколько расстройств представляет собой расстройство сна, и указанный пациент страдает депрессией и психозом, например, шизофренией, или болезнью Паркинсона;
7.16 Способ I или любой из 7.1-7.6, где расстройство центральной нервной системы представляет собой остаточные симптомы психоза, например, шизофрении (например, остаточный подтип), бредового расстройства (например, соматического типа), большой депрессии с психозом, биполярного расстройства с психотическими симптомами, кратковременного психотического расстройства, шизофрениформного расстройства, шизоаффективного расстройства или психоза, вызванного медицинским состоянием или употреблением психоактивных веществ. Предпочтительно, пациент страдает остаточными симптомами шизофрении;
7.17 Способ I или любой из 7.1-7.6, где симптомы остаточной фазы включают: негативные симптомы, такие как притупленный аффект, эмоциональное отчуждение, трудности в общении, пассивное или апатическое социальное отчуждение, нарушение абстрактного мышления, недостаток спонтанного и плавного течения беседы и стереотипное мышление; общие психопатологические симптомы, такие как соматическая озабоченность, беспокойство, чувство вины, напряженность, манерность и застывание в определенной позе, депрессия, двигательная заторможенность, неконтактность, необычное содержание мышления, дезориентация, снижение внимания, нарушение суждений и критики, волевые расстройства, ослабление контроля импульсивности, озабоченность и активная социальная изоляция; когнитивное расстройство и расстройства сна (например, бессонница);
7.18 Любой из вышеуказанных способов, где эффективное количество составляет 1 мг-1000 мг, предпочтительно 2,5 мг-50 мг, еще предпочтительнее 1-40 мг, например, 1-10 мг, например, 10 мг, 20 мг или больше чем 20 мг, например, 30 мг, 40 мг;
7.19 Любой из вышеуказанных способов, где эффективное количество составляет 1 мг-100 мг в день, предпочтительно 2,5 мг-50 мг в день, еще предпочтительнее 1-40 мг/день, например, 1-10 мг/день, например, 10 мг/день, 20 мг/день или больше чем 20 мг/день, например, 30 мг/день, 40 мг/день;
7.20 Любой из вышеуказанных способов где состояние, подлежащее лечению, представляет собой дискинезию, например, у пациента, принимающего допаминергические средства, например, лекарственные средства, выбранные из леводопа и вспомогательных средств к леводопа (карбидопа, ингибиторы COMT, ингибиторы MAO-B), допаминовых агонистов, например, леводопа, и антиходинергических средств;
7.21 Любой из вышеуказанных способов где пациент страдает болезнью Паркинсона;
7.22 Любой из вышеуказанных способов, где пациент не отвечает на селективный ингибитор обратного захвата серотонина, например, выбранный из одного или нескольких из следующих: циталопрам (Целекса, Ципрамил, Ципрам, Далсан, Рецитал, Эмокал, Сепрам, Серопрам, Цитокс, Цитал); дапоксетин (Прилиджи); эсциталопрам (Лексапро, Ципралекс, Сероплекс, Esertia); флуоксетин (Депекс, Прозак, Фонтекс, Серомекс, Серонил, Сарафем, Ладоз, Мотивест, Флутоп, Флуктин (EUR), Флуокс (NZ), Депресс (UZB), Лован (AUS), Продеп (IND)); флувоксамин (Лувокс, Феварин, Фаверин, Думирокс, Фавоксил, Мовокс); индалпин (Upstene); пароксетин (Паксил, Сероксат, Sereupin, Аропакс, Дероксат, Дивариус, Рексетин, Ксетанор, Пароксат, Локсамин, Депарок); сертралин (Золофт, Лустрал, Серлаин, Асентра); вилазодон (Виибрид); или зимелидин (Зелмид, Нормуд);
7.23 Любой из вышеуказанных способов, где пациент также принимает селективный ингибитор обратного захвата серотонина, например, выбранный из одного или нескольких из следующих: циталопрам (Целекса, Ципрамил, Ципрам, Далсан, Рецитал, Эмокал, Сепрам, Серопрам, Цитокс, Цитал); дапоксетин (Прилиджи); эсциталопрам (Лексапро, Ципралекс, Сероплекс, Esertia); флуоксетин (Депекс, Прозак, Фонтекс, Серомекс, Серонил, Сарафем, Ладоз, Мотивест, Флутоп, Флуктин (EUR), Флуокс (NZ), Депресс (UZB), Лован (AUS), Продеп (IND)); флувоксамин (Лувокс, Феварин, Фаверин, Думирокс, Фавоксил, Мовокс); индалпин (Upstene); пароксетин (Паксил, Сероксат, Sereupin, Аропакс, Дероксат, Дивариус, Рексетин, Ксетанор, Пароксат, Локсамин, Депарок); сертралин (Золофт, Лустрал, Серлаин, Асентра); вилазодон (Виибрид); или зимелидин (Зелмид, Нормуд);
7.24 Любой из вышеуказанных способов, где пациент страдает расстройством аутического спектра, например, аутизмом или синдромом Аспергера;
7.25 Любой из вышеуказанных способов, где пациенты страдают деменцией, например, расстройствами, связанными с легким когнитивным расстройством и деменцией, включая старческую деменцию, болезнь Альцгеймера, болезнь Пика, фронто-темпоральную деменцию, парасупрануклеарный паралич, деменцию с тельцами Леви, сосудистую деменцию, болезнь Гентингтона, болезнь Паркинсона, рассеянный склероз, амиотрофический боковой склероз, синдром Дауна, старческую депрессию, синдром Вернике-Корсакова, кортико-базальные дегенерации и прионовую болезнь, аутизм и синдром дефицита внимания и гиперактивности;
7.26 Любой из вышеуказанных способов, где пациент также принимает ингибитор холинэстеразы (например, ингибитор ацетилхолинэстеразы) или антагонист рецептора N-Метил-D-Аспартата (NMDA), в свободной форме или в форме фармацевтически приемлемой соли;
7.27 Способ 7.26, где ингибитор холинэстеразы (например, ингибитор ацетилхолинэстеразы) выбран из группы, состоящей из Такрина, ривастигмина (Экселон), допенезила (Арисепт) и галантамина (Разадин, ранее называвшийся Реминил)) в свободной форме или в форме фармацевтически приемлемой соли;
7.28 Способ 7.26, где ингибитор холинэстеразы (например, ингибитор ацетилхолинэстеразы) представляет собой допенезил в свободной форме или в форме фармацевтически приемлемой соли;
7.29 Способ 7.26, где антагонист NMDA-рецептора представляет собой мемантин в свободной форме или в форме фармацевтически приемлемой соли;
7.30 Любой из вышеуказанных способов, дополнительно включающий введение одного или нескольких других терапевтических средств, таких как дополнительные антипсихотические средства и/или антидепрессивные средства и/или гипнотические средства;
7.31 Способ 7.30, где одно или несколько других терапевтических средств выбраны из антидепрессивных средств, таких как соединения, которые модулируют активность GABA (например, усиливают активность и способствуют GABA трансмиссии), агонист GABA-B, модулятор 5-HT (например, агонист 5-HT1A, антагонист 5-HT2A, обратный агонист 5-HT2A и т.д.), агонист мелатонина, модулятор ионных каналов (например, блокатор), антагонист/ингибитор обратного захвата серотонина-2 (SARIs), антагонист орексинового рецептора, агонист H3, норадренергический антагонист, агонист галанина, антагонист CRH, человеческий гормон роста, агонист гормона роста, эстроген, агонист эстрогена, нейрокинин-1 лекарственное средство; и антипсихотических средств, например, атипичных антипсихотических средств, в свободной форме или в форме фармацевтически приемлемой соли;
7.32 Способ 7.30 или 7.31, где одно или несколько других терапевтических средств представляют собой антипсихотические средства, например, хлорпромазин, галоперидол, дроперидол, флуфеназин, локсапин, мезоридазин, молиндон, перфеназин, пимозид, прохлорперазин промазин, тиоридазин, тиотиксен, трифлуоперазин, клозапин, арипипразол, оланзапин, кветиапин, рисперидон, зипразидон, палиперидон, азенапин, луразидон, илоперидон, карипразин, амисулприд, зотепин, сертиндол, где одно или несколько других терапевтических средств вводят в качестве вспомогательного средства для соединения формул I - IV или 1.1-1.8, или соединение формул I - IV или 1.1-1.8 представляет собой вспомогательное средство для одного или нескольких других терапевтических средств.
В специальном варианте осуществления четвертого аспекта изобретение обеспечивает способ (Способ IP) для лечения или профилактики расстройства центральной нервной системы, описанного выше, включающий введение пациенту, нуждающемуся в этом:
7.4P соединения формул I - IV или 1.1-1.8, в свободной форме или в форме фармацевтически приемлемой соли;
7.8P фармацевтической или депо композиции, описанной выше; или
7.11P композиции пероральной системы доставки с осмотически-контролируемым высвобождением, описанной выше.
Еще в одном варианте осуществления четвертого аспекта изобретение обеспечивает Способ IP, где способ более подробно описан в любой из формул 7.1-7.32.
В специальном варианте осуществления четвертого аспекта изобретение обеспечивает Способ I, IP или любой из 7.1-7.32, где расстройство представляет собой шизофрению или расстройство сна.
В специальном варианте осуществления четвертого аспекта изобретение обеспечивает Способ I, IP или любой из 7.1-7.32, где расстройство представляет собой депрессию или беспокойство.
В специальном варианте осуществления четвертого аспекта изобретение обеспечивает Способ I, IP или любой из 7.1-7.32, где расстройство представляет собой пост-травматическое стрессовое расстройство или расстройство импульсного контроля, например, интермиттирующее эксплозивное расстройство.
В специальном варианте осуществления четвертого аспекта изобретение обеспечивает Способ I, IP или любой из 7.1-7.32, где расстройство представляет собой пост-травматическое стрессовое расстройство или расстройство импульсного контроля, например, интермиттирующее эксплозивное расстройство у пациента, страдающего деменцией, например, старческой деменцией, болезнью Альцгеймера, болезнью Пика, фронто-темпоральной деменцией, парасупрануклеарным параличем, деменцией с тельцами Леви, сосудистой деменцией, болезнью Гентингтона, болезнью Паркинсона, рассеянным склерозом, амиотрофическим боковым склерозом, синдромом Дауна, старческой депрессией, синдромом Вернике-Корсакова, кортико-базальной дегенерацией, прионовой болезнью, аутизмом и/или синдромом дефицита внимания и гиперактивности.
Еще в одном варианте осуществления четвертого аспекта изобретение обеспечивает Способ I, IP или любой из 7.1-7.32, где депо композицию по изобретению вводят для контролируемого и/или пролонгированного высвобождения соединений по изобретению в течение периода от около 14 дней, около 30 до около 180 дней, предпочтительно в течение периода около 30, около 60 или около 90 дней. Контролируемое и/или пролонгированное высвобождение особенно полезно, чтобы избежать преждевременного прерывания лечения, в частности, для терапии антипсихотическими лекарственными средствами, где часто бывают случаи некомплаентности или несоблюдения режима лечения.
В пятом аспекте изобретение обеспечивает способ (Способ II) для профилактики или лечения одного или нескольких из расстройств сна, возбуждения, агрессивных поведений, пост-травматического стрессового расстройства и/или расстройства импульсного контроля, например, интермиттирующего эксплозивного расстройства, включающий введение пациенту, нуждающемуся в этом, соединения, описанного в следующих формулах:
8.1 соединение формул i - iv или 1.1-1.8 в свободной форме или в форме фармацевтически приемлемой соли;
8.2 фармацевтическая или депо композиция, описанная выше;
8.3 композиция пероральной системы доставки с осмотически-контролируемым высвобождением, описанная выше.
В одном варианте осуществления пятого аспекта изобретение обеспечивает Способ II или любой из 8.1-8.3, где расстройство представляет собой расстройства сна. В другом варианте осуществления пятого аспекта изобретение обеспечивает Способ II, где расстройство представляет собой возбуждение, агрессивные поведения, пост-травматическое стрессовое расстройство и/или расстройство импульсного контроля, например, интермиттирующее эксплозивное расстройство.
Еще в одном варианте осуществления пятого аспекта изобретение обеспечивает Способ II, 8.1-8.3, где расстройство сна включает бессонницу поддержания сна, частые пробуждения и чувство, что проснулся неотдохнувшим;
8.11 Любой из вышеуказанных способов, где расстройство сна представляет собой бессонницу поддержания сна;
8.12 Любой из вышеуказанных способов, где эффективное количество составляет 1 мг-10 мг в день, например, l-5 мг, предпочтительно 2,5-5 мг, в день, еще предпочтительнее 10 мг в день;
8.13 Любой из вышеуказанных способов, где эффективное количество составляет 2,5 мг или 5 мг, в день или 10 мг в день;
8.14 Любой из вышеуказанных способов где расстройство сна присутствует у пациента, страдающего от дискинезии или имеющего повышенный риск дискинезии, например, у пациента, принимающего допаминергические средства, например, выбранные из леводопа и вспомогательных средств к леводопа (карбидопа, ингибиторы COMT, ингибиторы MAO-B), допаминовых агонистов, например, принимающего леводопа и антиходинергические средства;
8.15 Любой из вышеуказанных способов, где пациент страдает болезнью Паркинсона.
Соединения по изобретению (например, соединение формул I - IV или 1.1-1.8) обеспечивают эффективное лечение связанных с 5-HT2A, SERT и/или D2 рецепторами расстройств с минимальными экстрапирамидальными побочными эффектами или без них, как аналогичным образом раскрыто и заявлено в WO 2009/145900, содержание которой включено посредством ссылки во всей полноте. Поэтому соединения по изобретению, фармацевтические композиции по изобретению или депо композиции по изобретению можно использовать в комбинации со вторым терапевтическим средством, в частности, при более низких дозах, чем когда отдельные средства используются в качестве монотерапии, чтобы повысить терапевтическую активность комбинированных средств, не вызывая при этом нежелательных побочных эффектов, обычно возникающих при обычной монотерапии. Следовательно, соединения по изобретению можно одновременно, последовательно или совместно вводить с другими антидепрессантами, антипсихотическими средствами, другими снотворными средствами и/или средствами для лечения болезни Паркинсона или расстройств настроения или деменции. В другом примере побочные эффекты могут быть уменьшены или сведены к минимуму путем введения соединения по изобретению в сочетании с одним или несколькими терапевтическими средствами в свободной или солевой форме, где дозировки (i) второго терапевтического средства(средств) или (ii) и соединения по изобретению и второго терапевтического средства ниже, чем если средство/соединение вводят в виде монотерапии. В конкретном варианте осуществления соединения по изобретению являются полезными для лечения дискинезии у пациента, получающего допаминергические препараты, например, выбранные из леводопа и вспомогательных средства к леводопа (карбидопа, ингибиторы COMT, ингибиторы МАО-В), агонистов допамина, например, таких как используемые при лечении болезни Паркинсона, и антихолинергических средств, используемых для лечения побочных эффектов лечения болезни Паркинсона.
Поэтому в шестом аспекте настоящее изобретение обеспечивает Способ I или IP, например, или любой из формул 7.1-7.32, или Способ II или любой из 8.1-8.15, дополнительно включающий одно или несколько терапевтических средств, выбранных из соединений, которые модулируют активность GABA (например, усиливают активность и способствуют GABA трансмиссии), агониста GABA-B, модулятора 5-HT (например, агониста 5-HT1A, антагониста 5-HT2A, обратного агониста 5-HT2A и т.д.), агониста мелатонина, модулятора ионных каналов (например, блокатора), антагониста рецептора/ингибитора обратного захвата серотонина-2 (SARIs), антагониста орексинового рецептора, агониста или антагониста H3, норадренергического агониста или антагониста, агониста галанина, антагониста CRH, человеческого гормона роста, агониста гормона роста, эстрогена, агониста эстрогена, воздействующего на нейрокинин-1 лекарственного средства, антидепрессанта и антипсихотического средства, например, атипичного антипсихотического средства, в свободной форме или в форме фармацевтически приемлемой соли (Способ I-A и II-A, соответственно).
В другом варианте осуществления шестого аспекта Способ I-A и II-A, Способ I, Способ IP, например, или любой из формул 7.1-7.32, или Способ II или любой из 8.1-8.15 дополнительно включает одно или несколько терапевтических средств, выбранных из ингибитора холинэстеразы (например, ингибитора ацетилхолинэстеразы) или антагониста рецептора N-Метил-D-Аспартата (NMDA), в свободной форме или в форме фармацевтически приемлемой соли. В конкретном варианте осуществления ингибитор холинэстеразы (например, ингибитор ацетилхолинэстеразы) выбран из группы, состоящей из Такрина, ривастигмина (Экселон), допенезила (Арисепт) и галантамина (Разадин, ранее называвшийся Реминил)) в свободной форме или в форме фармацевтически приемлемой соли. В другом варианте осуществления ингибитор холинэстеразы (например, ингибитор ацетилхолинэстеразы) представляет собой допенезил в свободной форме или в форме фармацевтически приемлемой соли. В другом варианте осуществления антагонист NMDA-рецептора представляет собой мемантин в свободной форме или в форме фармацевтически приемлемой соли.
Еще в одном варианте осуществления шестого аспекта изобретение обеспечивает Способ I-A или II-A, как представлено ниже, дополнительно включающий одно или несколько терапевтических средств.
9.1 Способ I-A или II-A, где терапевтическое средство(средства) представляет собой соединение, которое модулирует активность GABA (например, усиливает активность и способствует GABA трансмиссии);
9.2 Способ I-A или II-A или 9.1, где GABA соединение выбрано из группы, состоящей из одного или нескольких из следующих: доксепина, алпразолама, бромазепама, клобазама, клоназепама, клоразепата, диазепама, флунитразепама, флуразепама, лоразепама, мидазолама, нитразепама, оксазепама, темазапама, триазолама, индиплона, зопиклона, эсзопиклона, залеплона, Золпидема, габаксадола, вигабатрина, тиагабина, EVT 201 (Evotec Pharmaceuticals) и эстазолама;
9.3 Способ I-A или II-A, где терапевтическое средство является дополнительным 5HT2A антагонистом;
9.4 Способ I-A или II-A или 9.3, где указанный дополнительный 5HT2A антагонист выбран из одного или нескольких из следующих: кетансерина, рисперидона, эпливансерина, волинансерина (Sanofi-Aventis, France), прувансерина, MDL 100907 (Sanofi-Aventis, France), HY 10275 (Eli Lilly), APD 125 (Arena Pharmaceuticals, San Diego, CA) и AVE8488 (Sanofi-Aventis, France); Способ I-A или II-A, 9.3 или 9.4, дополнительно выбранные из примавансерина (ACP-103) и пизотифена;
9.5 Способ I-A или II-A, где терапевтическое средство представляет собой агонист мелатонина;
9.6 Способ I-A или II-A или 9.5, где агонист мелатонина выбран из группы, состоящей из одного или нескольких из следующих: мелатонин, рамелтеон (ROZEREM®, Takeda Pharmaceuticals, Japan), VEC-162 (Vanda Pharmaceuticals, Rockville, MD), PD-6735 (Phase II Discovery) и агомелатин;
9.7 Способ I-A или II-A, где терапевтическое средство представляет собой блокатор ионных каналов;
9.8 Способ I-A или II-A или 9.7, где указанный блокатор ионных каналов представляет собой одно или несколько из следующих средств: ламотригин, габапентин и прегабалин.
9.9 Способ I-A или II-A, где терапевтическое средство представляет собой антагонист орексинового рецептора;
9.10 Способ I-A или II-A или 9.9, где антагонист орексинового рецептора выбран из группы, состоящей из орексина, 1,3-биарилмочевины, SB-334867-a (GlaxoSmithKline, UK), GW649868 (GlaxoSmithKline) и бензамидного производного;
9.11 Способ I-A или II-A, где терапевтическое средство представляет собой антагонист рецептора/ингибитор обратного захвата серотонина-2 (SARI);
9.12 Способ I-A или II-A или 9.11, где антагонист рецептора/ингибитор обратного захвата серотонина-2 (SARI) выбран из группы, состоящей из одного или нескольких из следующих: Org 50081 (Organon-Netherlands), ритансерин, нефазодон, серзон и тразодон;
9.13 Способ I-A или II-A, где терапевтическое средство представляет собой агонист 5HT1a;
9.14 Способ I-A или II-A или 9.13, где агонист 5HT1a выбран из группы, состоящей из одного или нескольких из следующих: репинотан, саризотан, эптапирон, буспирон и MN-305 (MediciNova, San Diego, CA);
9.15 Способ I-A или II-A, где терапевтическое средство представляет собой воздействующее на нейрокинин-1 лекарственное средство;
9.16 Способ I-A или II-A или 9.15, где воздействующее на нейрокинин-1 лекарственное средство представляет собой Касопитант (GlaxoSmithKline);
9.17 Способ I-A или II-A, где терапевтическое средство представляет собой антипсихотическое средство;
9.18 Способ I-A или II-A или 9.17, где антипсихотическое средство выбрано из группы, состоящей из хлорпромазина, галоперидола, дроперидола, флуфеназина, локсапина, мезоридазина, молиндона, перфеназина, пимозида, прохлорперазина промазина, тиоридазина, тиотиксена, трифлуоперазина, клозапина, арипипразола, оланзапина, кветиапина, рисперидона, зипразидона и палиперидона;
9.19 Способ I-A или II-A, где терапевтическое средство представляет собой антидепрессант;
9.20 Способ I-A или II-A или 9.19, где антидепрессант выбран из амитриптилина, амоксапина, бупропиона, циталопрама, кломипрамина, дезипрамина, доксепина, дулоксетина, эсциталопрама, флуоксетина, флувоксамина, имипрамина, изокарбоксазида, мапротилина, миртазапина, нефазодона, нортриптилина, пароксетина, фенелазина сульфата, протриптилина, сертралина, транилципромина, тразодона, тримипрамина и венлафаксина;
9.21 Способ I-A или II-A, 9.17 или 9.18, где антипсихотическое средство представляет собой атипичное антипсихотическое средство;
9.22 Способ I-A или II-A, или любой из 9.17-9.21, где атипичное антипсихотическое средство выбрано из группы, состоящей из клозапина, арипипразола, оланзапина, кветиапина, рисперидона, зипразидона и палиперидона;
9.23 Способ I-A или II-A, где терапевтическое средство выбрано из любого из способов 9.1-9.22, например, выбрано из группы, состоящей из модафинила, армодафинила, доксепина, алпразолама, бромазепама, клобазама, клоназепама, клоразепата, диазепама, флунитразепама, флуразепама, лоразепама, мидазолама, нитразепама, оксазепама, темазапама, триазолама, индиплона, зопиклона, эсзопиклона, залеплона, Золпидема, габаксадола, вигабатрина, тиагабина, EVT 201 (Evotec Pharmaceuticals)а, эстазолама, кетансерина, рисперидона, эпливансерина, волинансерина (Sanofi-Aventisа, France), прувансерина, MDL 100907 (Sanofi- Aventisа, France), HY 10275 (Eli Lilly), APD 125 (Arena Pharmaceuticals, San Diego, CA), AVE8488 (Sanofi-Aventis, France), репинотана, саризотана, эптапирона, буспирона, MN-305 (MediciNova, San Diego, CA), мелатонина, рамелтеона (ROZEREM®, Takeda Pharmaceuticals, Japan), VEC- 162 (Vanda Pharmaceuticals, Rockville, MD), PD-6735 (Phase II Discovery)а, агомелатина, ламотригина, габапентина, прегабалина, орексина, 1,3-биарилмочевины, SB-334867-a (GlaxoSmithKline, UK), GW649868 (GlaxoSmithKline), бензамидного производного, Org 50081 (Organon -Netherlands), ритансерина, нефазодона, серзона, тразодона, Касопитанта (GlaxoSmithKline), амитриптилина, амоксапина, бупропиона, циталопрама, кломипрамина, дезипрамина, доксепина, дулоксетина, эсциталопрама, флуоксетина, флувоксамина, имипрамина, изокарбоксазида, мапротилина, миртазапина, нефазодона, нортриптилина, пароксетина, фенелазин сульфата, протриптилина, сертралина, транилципромина, тразодона, тримипрамина, венлафаксина, хлорпромазина, галоперидола, дроперидола, флуфеназина, локсапина, мезоридазина молиндона, перфеназина, пимозида, прохлорперазина промазина, тиоридазина, тиотиксена, трифлуоперазина, клозапина, арипипразола, оланзапина, кветиапина, рисперидона, зипразидона и палиперидона; в дополнение к терапевтическим средствам, перечисленным выше, Способа I-A или II-Aа, кроме того, выбрано из примавансерина (ACP-103) и пизотифена;
9.24 Способ I-A или II-A, где терапевтическое средство представляет собой агонист H3;
9.25 Способ I-A или II-A, где терапевтическое средство представляет собой антагонист H3;
9.26 Способ I-A или II-A, где терапевтическое средство представляет собой норадренергический агонист или антагонист;
9.27 Способ I-A или II-A, где терапевтическое средство представляет собой агонист галанина;
9.28 Способ I-A или II-A, где терапевтическое средство представляет собой антагонист CRH;
9.29 Способ I-A или II-A, где терапевтическое средство представляет собой человеческий гормон роста;
9.30 Способ I-A или II-A, где терапевтическое средство представляет собой агонист гормона роста;
9.31 Способ I-A или II-A, где терапевтическое средство представляет собой эстроген или агонист эстрогена;
9.32 Способ I-A или II-A, где терапевтическое средство представляет собой антагонист 5-HT6 рецептора;
9.33 Способ I-A или II-A, где терапевтическое средство представляет собой воздействующее на нейрокинин-1 лекарственное средство;
9.34 Способ I-A или II-A, где терапевтическое средство комбинируют с соединениями формулы (I) и терапевтическое средство представляет собой средство от болезни Паркинсона, такое как L-допа, ко-карелдопа, дуодопа, сталова, Симметрел, бензотропин, бипериден, бромкриптин, энтакапон, перголид, прамипексол, проциклидин, ропинирол, селегилин и толкапон;
9.35 Способ I-A или II-A, где соединения формулы (I) можно использовать для лечения расстройств сна, депрессии, психоза или любой их комбинации у пациентов, страдающих перечисленными заболеваниями и/или болезнью Паркинсона;
9.36 Способ I-A или II-A, где расстройство выбрано из по меньшей мере одного или нескольких из следующих: психоз, например, шизофрения, депрессия, расстройства настроения, расстройства сна (например, поддержания сна и/или наступления сна), или любой комбинации таких расстройств;
9.37 Любой из вышеуказанных способов, где расстройство представляет собой расстройство сна;
9.38 Любой из вышеуказанных способов, где расстройство представляет собой расстройство сна, связанное с психозом, например, шизофренией, или болезнью Паркинсона; в свободной форме или в форме фармацевтически приемлемой соли.
В другом варианте осуществления шестого аспекта настоящее изобретение обеспечивает Способ IP или Способ II, описанный выше, дополнительно включающий одно или несколько терапевтических средств, выбранных из соединений, которые модулируют GABA активность (например, усиливают активность и способствуют GABA трансмиссии), агониста GABA-B, модулятора 5-HT (например, агониста 5-HT1A, антагониста 5-HT2A, обратного агониста 5-HT2A и т.д.), агониста мелатонина, модулятора ионных каналов (например, блокатора), антагониста/ингибитора обратного захвата серотонина-2 (SARIs), антагониста орексинового рецептора, агониста или антагониста H3, норадренергического агониста или антагониста, агониста галанина, антагониста CRH, человеческого гормона роста, агониста гормона роста, эстрогена, агониста эстрогена, воздействующего на нейрокинин-1 лекарственного средства, антидепрессанта и антипсихотического средства, например, атипичного антипсихотического средства, в свободной форме или в форме фармацевтически приемлемой соли (Способ IP-A и II-A, соответственно). В другом варианте осуществления этого аспекта изобретение обеспечивает Способ IP-A или II-A, как аналогичным образом описано в любой из формул 9.1-9.38.
Еще в одном варианте осуществления шестого аспекта Способ IP или Способ II, описанный выше, дополнительно включает одно или несколько терапевтических средств, выбранных из ингибитора холинэстеразы (например, ингибитора ацетилхолинэстеразы) или антагониста рецептора N-Метил-D-Аспартата (NMDA), в свободной форме или в форме фармацевтически приемлемой соли. В конкретном варианте осуществления ингибитор холинэстеразы (например, ингибитор ацетилхолинэстеразы) выбран из группы, состоящей из Такрина, ривастигмина (Экселон), допенезила (Арисепт) и галантамина (Разадин, ранее называвшийся Реминил)) в свободной форме или в форме фармацевтически приемлемой соли. В другом варианте осуществления ингибитор холинэстеразы (например, ингибитор ацетилхолинэстеразы) представляет собой допенезил в свободной форме или в форме фармацевтически приемлемой соли. В другом варианте осуществления антагонист NMDA-рецептора представляет собой мемантин в свободной форме или в форме фармацевтически приемлемой соли.
В седьмом аспекте изобретения комбинацию соединения по изобретению и одного или нескольких вторых терапевтических средств, описанных в Способах I-A, II-A или любом из 9.1-9.38, можно вводить в виде фармацевтической композиции или депо композиции, описанной выше. Подобным образом, комбинацию соединения по изобретению и одного или нескольких вторых терапевтических средств, описанных в Способах Ip-A, II-A или любом из 9.1-9.38, можно вводить в виде фармацевтической композиции или депо композиции, описанной выше. Композиции в комбинации могут включать смеси комбинированных лекарственных средств, а также две или более отдельные композиции лекарственных средств, при этом отдельные композиции можно, например, совместно вводить пациенту.
В специальном варианте осуществления Способы I-A, II-A, Ip-A, II-A или любой из 9.1-9.38 включают введение пациенту, нуждающемуся в этом, соединения по изобретению в комбинации с атипичным антипсихотическим средством, например, соединением, выбранным из клозапина, арипипразола, оланзапина, кветиапина, рисперидона, зипразидона или палиперидона, в свободной форме или в форме фармацевтически приемлемой соли, например, где доза атипичного антипсихотического средства снижена и/или побочные эффекты уменьшаются.
В другом варианте осуществления Способы I-A, II-A, Способы Ip-A, II-A или любой из 9.1-9.38 включают введение пациенту, нуждающемуся в этом, соединения по изобретению в комбинации с антидепрессантом, например, амитриптилином, амоксапином, бупропионом, циталопрамом, кломипрамином, дезипрамином, доксепином, дулоксетином, эсциталопрамом, флуоксетином, флувоксамином, имипрамином, изокарбоксазидом, мапротилином, миртазапином, нефазодоном, нортриптилином, пароксетином, фенелазином сульфатом, протриптилином, сертралином, транилципромином, тразодоном, тримипрамином или венлафаксином, в свободной форме или в форме фармацевтически приемлемой соли. Альтернативно, антидепрессант можно использовать в качестве вспомогательного лечения в дополнение к соединению по изобретению.
Еще в одном варианте осуществления Способы I-A, II-A, Ip-A, II-A или любой из 9.1-9.38 включают введение пациенту, нуждающемуся в этом, соединения по изобретению в комбинации с соединением, которое модулирует GABA активность, например, соединением, выбранным из доксепина, алпразолама, бромазепама, клобазама, клоназепама, клоразепата, диазепама, флунитразепама, флуразепама, лоразепама, мидазолама, нитразепама, оксазепама, темазапама, триазолама, индиплона, зопиклона, эсзопиклона, залеплона, Золпидема, габаксадола, вигабатрина, тиагабина, EVT 201 (Evotec Pharmaceuticals), эстазолама или любой их комбинации, в свободной форме или в форме фармацевтически приемлемой соли.
В другом конкретном варианте осуществления Способы I-A, II-A, Ip-A, II-A или любой из 9.1-9.38 включают введение пациенту, нуждающемуся в этом, соединения по изобретению в комбинации с доксепином в свободной форме или в форме фармацевтически приемлемой соли. Дозы доксепина могут варьироваться в любых пределах, известных среднему специалисту в данной области. В одном примере 10 мг дозу доксепина можно комбинировать с любой дозой соединения по изобретению.
В другом варианте осуществления Способы I-A, II-A, Ip-A, II-A или любой из 9.1-9.38 включают введение пациенту, нуждающемуся в этом, соединения по изобретению в комбинации (в том числе в виде части ежедневного режима дозирования) с атипичным стимулятором, например, модафинилом, адрафинилом или армодафинилом. Режим, включающий соединение по изобретению с такими лекарственными средствами, способствует более регулярному сну и избежанию побочных эффектов, таких как психоз или мания, связанные с более высокими уровнями таких лекарственных средств, например, при лечении биполярной депрессии, когнитивного расстройства, связанного с шизофренией, и чрезмерной сонливости и слабости при состояниях, такие как болезнь Паркинсона и рак.
В восьмом аспекте изобретение обеспечивает применение соединения, как описано в следующих формулах:
11.1 Соединение формулы I или любой из формул 1-1.9 в свободной форме или в форме фармацевтически приемлемой соли;
11.2 фармацевтическая композиция, описанная выше;
11.3 депо композиция, описанная выше; или
11.4 композиция пероральной системы доставки с осмотически-контролируемым высвобождением, описанная выше,
(для получения лекарственного средства) для лечения или профилактики одного или нескольких расстройств, раскрытых выше, например, в любом из Способа I, любого из 7.1-7.32, Способа II, любого из 8.1-8.15, Способов I-A, II-A, любого из 9.1-9.38, Способа IP, Способов IP-A или любых способов, описанных в шестом или седьмом аспекте изобретения.
В девятом аспекте изобретение обеспечивает фармацевтическую композицию, описанную выше, например, в следующих формулах:
12.1 фармацевтическая композиция, описанная выше;
12.2 депо композиция, описанная выше; или
12.3 композиция пероральной системы доставки с осмотически-контролируемым высвобождением, описанная выше,
для применения в лечении или профилактики одного или нескольких расстройств, раскрытых выше, например, в любом из Способа I, любого из 7.1-7.32, Способа II, любого из 8.1-8.15, Способов I-A, II-A, любого из 9.1-9.38, Способа IP, Способов IP-A или любых способов, описанных в шестом или седьмом аспекте изобретения.
В конкретных вариантах осуществления любого из способов, описанных выше, включая любые предшествующие варианты осуществления четвертого аспекта (включая Способ I и любой из Способов 7.1-7.32), пятого аспекта (включая Способ II и любой из Способов 8.1-8.15), Способа IP, Способа IP-A, шестого аспекта (включая Способ I-A, II-A и любой из Способов 9.1-9.38) и седьмого аспекта, указанные расстройства и состояния имеют такое значение, которое определено в American Psychiatric Association's Diagnostic and Statistical Manual of Mental Disorsers, Fifth Edition (DSM-V) (2013).
В других конкретных вариантах осуществления любого из способов, описанных выше, включая любые предшествующие варианты осуществления четвертого аспекта (включая Способ I и любой из Способов 7.1-7.32), пятого аспекта (включая Способ II и любой из Способов 8.1-8.15), Способа IP, Способов IP-A, шестого аспекта (включая Способ I-A, II-A и любой из Способов 9.1-9.38) и седьмого аспекта, указанные расстройства и состояния имеют такое значение, которое определено в World Health Organization's International Classification of Diseases, Tenth Revision (ICD-10), Chapter V (Mental and Behavioral Disorders) (1992).
Подробное описание изобретения
Если не указано иное или не ясно из контекста, следующие термины, как они используются в настоящей заявке, имеют следующие значения:
a. ʺОстаточные симптомыʺ в контексте настоящей заявки включают негативные симптомы и общие психопатологические симптомы, описанные в Positive and Negative Symptom Scale (PANSS) for Shizophrenia, Kay et al., Schizophr. Bull. (1987) 13(2):261-276, содержание которого включено посредством ссылки во всей полноте. Негативные симптомы включают: притупленный аффект, эмоциональное отчуждение, трудности в общении, пассивное/апатическое социальное отчуждение, нарушение абстрактного мышления, недостаток спонтанного и плавного течения беседы и стереотипное мышление. Общие психопатологические симптомы включают следующие: соматическая озабоченность, беспокойство, чувство вины, напряженность, манерность и застывание в определенной позе, депрессия, двигательная заторможенность, неконтактность, необычное содержание мышления, дезориентация, снижение внимания, нарушение суждений и критики, волевые расстройства, ослабление контроля импульсивности, озабоченность и активная социальная изоляция. Остаточные симптомы также могут включать депрессию, когнитивное расстройство и расстройства сна (например, бессонницу). Из этих остаточных симптомов, соединения по изобретению особенно полезны для лечения пассивного социального отчуждения, стереотипности мышления, соматической озабоченности, беспокойства, напряженности, активной социальной изоляции и депрессии. Поэтому соединения по настоящему изобретению особенно полезны для улучшения социальной интеграции и социальной функции у пациентов, страдающих шизофренией. Лечение этих остаточных симптомов также особенно эффективно у пациентов с шизофренией, также страдающих депрессией.
b. Как используется в формуле, например, в структуре любой из формул I-IV или 1.1-1.8, и в термине, таком как ʺCD3ʺ, ʺDʺ относится к атому водорода, который содержит больше чем природное включение изотопного дейтерия (2H). Все природные химические соединения включают атомы водорода, содержащие приблизительно 0,0156 атом% дейтерия для каждого атома водорода. Использование ʺDʺ и ʺдейтерийʺ в настоящем раскрытии относится к любому обогащению дейтерием выше этого природного относительного содержания, например, выше 0,1%, или выше 1%, вплоть до любого значения почти до 100% (например, 99%, или 99,9%, или 99,99%, или 99,999%). Использование ʺHʺ в качестве атома водорода относится к атому водорода в химической структуре, имеющей не больше чем природное относительное содержание дейтерия, например, не больше чем 0,0156 атом% дейтерия.
Если не указано иное, соединения по изобретению, например, соединение формул I - IV или 1.1-1.8, могут существовать в свободной или солевой форме, например, в виде кислотно-аддитивных солей. Кислотно-аддитивная соль соединения по изобретению, которое является достаточно щелочным, представляет собой, например, кислотно-аддитивную соль с, например, неорганической или органической кислотой. В конкретном варианте осуществления соль соединений по изобретению представляет собой аддитивную соль толуолсульфоновой кислоты.
Соединения по изобретению предназначены для использования в качестве фармацевтических препаратов, поэтому предпочтительны фармацевтически приемлемые соли. Соли, которые непригодны для фармацевтических целей, могут быть полезны, например, для выделения или очистки свободных соединений по изобретению, и поэтому также включены.
Соединения по изобретению могут содержать один или несколько хиральных атомов углерода. Таким образом, соединения существуют в индивидуальной изомерной, например, энантиомерной или диастереомерной форме или в виде смесей отдельных форм, например, рацемических/диастереомерных смесей. Может присутствовать любой изомер, в котором асимметричный центр находится в конфигурации (R) -, (S) - или (R, S). Изобретение следует понимать как охватывающее отдельные оптически активные изомеры, а также смеси (например, рацемические/диастереомерные смеси). Соответственно, соединения по изобретению могут представлять собой рацемическую смесь или могут быть преимущественно, например, в чистой или по существу чистой изомерной форме, например, более 70% энантиомерного/диастереомерного избытка («эи»), предпочтительно более 80% эи, более предпочтительно более 90% эи, наиболее предпочтительно более 95% эи. Очистку указанных изомеров и разделение указанных изомерных смесей можно осуществить стандартными методами, известными в данной области (методом например, колоночной хроматографии, препаративной ТСХ, препаративной ВЭЖХ, псевдодвижущегося слоя и т.п.).
Геометрические изомеры в зависимости от природы заместителей относительно двойной связи или кольца могут присутствовать в цис (Z) или транс (Е) форме, и обе изомерные формы охватываются объемом настоящего изобретения.
Альтернативно и/или дополнительно, соединения по изобретению могут быть включены в виде депо-композиции, например, путем диспергирования, растворения или инкапсулирования соединений по изобретению в полимерной матрице, как описано во втором и третьем аспектах, чтобы соединение непрерывно высвобождалось по мере разрушения полимера. Высвобождение соединений по изобретению из полимерной матрицы обеспечивает контролируемое и/или замедленное и/или пролонгированное высвобождение соединений, например, из фармацевтической депо композиции, в организм субъекта, например, теплокровного животного, такого как человек, которому вводят фармацевтический депо препарат. Таким образом, фармацевтический депо препарат доставляет соединения по изобретению субъекту в концентрациях, эффективных для лечения конкретного заболевания или медицинского состояния в течение длительного периода времени, например, 14-180 дней, предпочтительно около 30, около 60 или около 90 дней.
Полимеры, полезные для полимерной матрицы в композиции по изобретению (например, депо композиции по изобретению), могут включать полиэфир гидрокси-жирной кислоты и ее производных или другие вещества, такие как полимолочная кислота, полигликолевая кислота, полилимонная кислота, полияблочная кислота, поли-бета.-гидроксимасляная кислота, эпсилон.-капро-лактоновое кольцо-раскрывающий полимер, сополимер молочной кислоты-гликолевой кислоты, сополимер 2-гидроксимасляной кислоты-гликолевой кислоты, сополимер полимолочной кислоты-полиэтиленгликоля или сополимер полигликолевой кислоты-полиэтиленгликоля), полимер алкил альфа-цианоакрилата (например, поли(бутил 2-цианоакрилат)), полиалкиленоксалат (например, политриметиленоксалат или политетраметиленоксалат), полиортоэфир, поликарбонат (например, полиэтиленкарбонат или полиэтиленпропиленкарбонат), полиорто-карбонат, полиаминокислота (например, поли-гамма.-L-аланин, поли-.гамма.-бензил-L-глутаминовая кислота или поли-y-метил-L-глутаминовая кислота), эфир гиалуроновой кислоты и т.п., и можно использовать один или несколько из этих полимеров.
Если полимеры являются сополимерами, они могут представлять собой любой из статистических, блок- и/или графт-сополимеров. Когда вышеуказанные альфа-гидроксикарбоновые кислоты, гидроксидикарбоновые кислоты и гидрокситрикарбоновые кислоты обладают оптической активностью в их молекулах, можно использовать любой из D-изомеров, L-изомеров и/или DL-изомеров. Среди прочих, можно использовать полимер альфа-гидроксикарбоновой кислоты (предпочтительно полимер молочной кислоты-гликолевой кислоты), ее сложный эфир, эфиры поли-альфа-цианоакриловой кислоты и т.д., и сополимер молочной кислоты-гликолевой кислоты (также указан как поли(лактид-альфа-гликолид) или поли(молочная-ко-гликолевая кислота) и далее указанный как PLGA) является предпочтительным. Таким образом, в одном аспекте полимер, полезный для полимерной матрицы, представляет собой PLGA. В контексте настоящей заявки термин PLGA включает полимеры молочной кислоты (также указаны как полилактид, поли(молочная кислота), или PLA). Наиболее предпочтительно, полимер представляет собой биоразлагаемый поли(d,l-лактид-ко-гликолид) полимер.
В предпочтительном варианте осуществления полимерная матрица по изобретению представляет собой биосовместимый и биоразлагаемый полимерный материал. Термин «биосовместимый» определяется как полимерный материал, который не токсичен, не является канцерогенным и по существу не индуцирует воспаление в тканях организма. Материал матрицы должен быть биоразлагаемым, при этом полимерный материал должен разлагаться в результате происходящих в организме процессов на продукты, которые легко удаляются организмом и не накапливаются в организме. Продукты биоразложения также должны быть биосовместимы с организмом, поскольку полимерная матрица биосовместима с организмом. Конкретные полезные примеры материалов полимерной матрицы включают поли(гликолевую кислоту), поли-D,L-молочную кислоту, поли-L-молочную кислоту, сополимеры вышеуказанных, поли(алифатические карбоновые кислоты), сополиоксалаты, поликапролактон, полидиоксонон, поли(ортокарбонаты), поли(ацетали), поли(молочную кислоту-капролактон), полиортоэфиры, поли(гликолевую кислоту-капролактон), полиангидриды и природные полимеры, включая альбумин, казеин и воски, такие как, глицерин моно- и дистеарат и т.п. Предпочтительным полимером для применения в осуществлении настоящего изобретения является dl-(полилактид-ко-гликолид). Предпочтительно, чтобы молярное отношение лактида к гликолиду в таком сополимере находилось в пределах от около 75:25 до 50:50.
Полезные PLGA полимеры могут иметь средневесовую молекулярную массу от около 5000 до 500000 дальтон, предпочтительно около 150000 дальтон. В зависимости от скорости разложения, которая должна достигаться, можно использовать полимеры с разной молекулярной массой. Для диффузионного механизма высвобождения лекарственного средства полимер должен оставаться неповрежденным до тех пор, пока все лекарственное средство не высвободится из полимерной матрицы, а затем разлагаться. Лекарственное средство также может высвобождаться из полимерной матрицы по мере биоразложения полимерного эксципиента.
PLGA можно получить любым обычным способом, или они могут быть коммерчески доступны. Например, PLGA можно получить путем полимеризации с раскрытием кольца с подходящим катализатором из циклического лактида, гликолида и т.д. (См. EP-0058481B2; Эффекты переменных полимеризации на свойства PLGA: молекулярная масса, композиция и структура цепи).
Считается, что PLGA является биоразлагаемым путем разложения всей твердой полимерной композиции из-за разрушения гидролизуемых и ферментативно расщепляемых сложноэфирных связей в биологических условиях (например, в присутствии воды и биологических ферментов в тканях теплокровных животных, таких как человек) с образованием молочной кислоты и гликолевой кислоты. Как молочная кислота, так и гликолевая кислота являются водорастворимыми, нетоксичными продуктами нормального метаболизма, которые могут далее разлагаться, образуя углекислый газ и воду. Другими словами, PLGA, как полагают, разлагается посредством гидролиза его сложноэфирных групп в присутствии воды, например, в теле теплокровного животного, такого как человек, с образованием молочной кислоты и гликолевой кислоты и создания кислотного микроклимата. Молочная и гликолевая кислоты являются побочными продуктами различных метаболических путей в организме теплокровного животного, такого как человек, в нормальных физиологических условиях, и поэтому хорошо переносятся и вызывают минимальную системную токсичность.
В другом варианте осуществления полимерная матрица, полезная для настоящего изобретения, может содержать звездчатый полимер, в котором структура сложного полиэфира имеет звездную форму. Эти сложные полиэфиры имеют один полиольный остаток в виде центральной части, окруженной цепями кислотных остатков. Молекула полиола может представлять собой, например, глюкозу или, например, маннит. Эти сложные эфиры известны и описаны в патенте США № 2145422 и в патенте США № 5538739, содержание которых включено посредством ссылки.
Звездчатые полимеры можно получить с использованием полигидрокси-соединений, например, полиола, например, глюкозы или маннита в качестве инициатора. Полиол содержит по меньшей мере 3 гидроксигруппы и имеет молекулярную массу до около 20000 дальтон, с по меньшей мере 1, предпочтительно по меньшей мере 2, например, в среднем 3 гидроксильными группами полиола, находящимися в форме сложноэфирных групп, которые содержат полилактидные или ко-полилактидные цепи. Разветвленные полиэфиры, например, поли(d,l-лактид-ко-гликолид), имеют центральную глюкозную часть, имеющую лучи из линейных полилактидных цепей.
Депо композиция по изобретению, как описано выше, может содержать полимер в форме микрочастиц или наночастиц или в жидкой форме, с соединениями изобретения, диспергированными или инкапсулированными в нем. «Микрочастицы» означают твердые частицы, которые содержат соединения по изобретению либо в растворе, либо в твердой форме, где такое соединение диспергировано или растворено в полимере, который служит матрицей, состоящей из частиц. При соответствующем подборе полимерных материалов можно получить композицию микрочастиц, в которой полученные микрочастицы обладают как свойствами диффузионного высвобождения, так и высвобождения путем биоразложения.
В конкретном варианте осуществления соединение по изобретению сформулировано в виде микрочастиц соответствующего размера для обеспечения кинетики медленного высвобождения после внутримышечной инъекции.
Когда полимер находится в форме микрочастиц, микрочастицы можно получить с использованием любого подходящего способа, такого как выпаривание растворителя или экстракция растворителем. Например, в способе выпаривания растворителя соединения по изобретению и полимер можно растворить в летучем органическом растворителе (например, в кетоне, таком как ацетон, галогенированном углеводороде, таком как хлороформ или метиленхлорид, галогенированном ароматическом углеводороде, циклическом эфире, таком как диоксан, сложном эфире, такой как этилацетат, нитриле, таком как ацетонитрил, или спирте, таком как этанол) и диспергировать в водной фазе, содержащей подходящий стабилизатор эмульсии (например, поливиниловый спирт, PVA). Затем органический растворитель выпаривают с получением микрочастиц с соединениями по изобретению, инкапсулированными в них. В способе экстракции растворителем соединения по изобретению и полимер можно растворить в полярном растворителе (таком как ацетонитрил, дихлорметан, метанол, этилацетат или метилформиат) и затем диспергировать в водной фазе (такой как раствор вода/PVA). Получают эмульсию для обеспечения микрочастиц с соединениями по изобретению, инкапсулированными в них. Распылительная сушка является альтернативной технологией для получения микрочастиц.
Другой способ получения микрочастиц по изобретению также описан в Патенте США № 4389330 и Патенте США № 4530840, содержание которых включено посредством ссылки.
Микрочастицы по настоящему изобретению можно получить любым способом, способным обеспечивать микрочастицы в диапазоне размеров, приемлемом для использования в инъекционной композиции. Одним из предпочтительных способов получения является способ, описанный в патенте США № 4389330. В этом способе активное вещество растворяют или диспергируют в подходящем растворителе. К среде, содержащей это вещество, добавляют полимерный матричный материал в количестве относительно активного ингредиента, которое обеспечивает продукт, имеющий желаемую нагрузку активного вещества. Необязательно, все ингредиенты продукта микрочастиц могут быть смешаны вместе в среде растворителя.
Растворители для соединений по изобретению и полимерного матричного материала, которые можно использовать для осуществления настоящего изобретения, включают органические растворители, такие как ацетон; галогенированные углеводороды, такие как хлороформ, метиленхлорид и т.п.; ароматические углеводородные соединения; галогенированные ароматические углеводородные соединения; циклические простые эфиры; спирты, такие как бензиловый спирт; этилацетат; и т.п. В одном варианте осуществления растворитель для использования при осуществлении настоящего изобретения может представлять собой смесь бензилового спирта и этилацетата. Дополнительную информацию для получения микрочастиц, полезных для изобретения, можно найти в патентной публикации США № 2008/0069885, содержание которой включено в настоящую заявку посредством ссылки во всей полноте.
Количество соединений по изобретению, включенное в микрочастицы, обычно варьируется от около 1% масс. до около 90% масс., предпочтительно 30-50% масс., более предпочтительно 35-40% масс. Под % масс. подразумевается доля соединений по изобретению в расчете на общую массу микрочастиц.
Фармацевтические депо композиции могут включать фармацевтически приемлемый разбавитель или носитель, такой как смешиваемый с водой разбавитель или носитель.
Подробную информацию о композициях пероральной системы доставки с осмотически-контролируемым высвобождением можно найти в EP 1 539 115 (U.S. Pub. No. 2009/0202631) и WO 2000/35419, содержание каждой из которых включено посредством ссылки во всей полноте.
"Терапевтически эффективное количество" представляет собой любое количество соединений по изобретению (например, содержащихся в фармацевтической депо композиции), которое при введении субъекту, страдающему заболеванием или расстройством, является эффективным, чтобы вызвать облегчение, ремиссию или регрессию заболевания или расстройства в течение периода времени, предполагаемого для лечения.
Дозировки, применяемые при практическом осуществлении настоящего изобретения, будут, разумеется, варьироваться в зависимости, например, от конкретного заболевания или состояния, подлежащего лечению, конкретных соединений по изобретению, способа введения и желаемой терапии.
Соединения по изобретению можно вводить любым приемлемым путем, в том числе перорально, парентерально (внутривенно, внутримышечно или подкожно) или чрескожно, но предпочтительно их вводят перорально. В некоторых вариантах осуществления соединения по изобретению, например, в депо композиции, предпочтительно вводят парентерально, например, путем инъекции.
Как правило, удовлетворительные результаты для Способа I или любой из формул 7.1-7.32 или Способа IP или применения соединений по изобретению, описанного выше, например, для лечения комбинации заболеваний, такой как комбинация по меньшей мере депрессии, психоза, например, (1) психоза, например, шизофрении, у пациента, страдающего депрессией; (2) депрессии у пациента, страдающего психозом, например, шизофренией; (3) расстройств настроения, связанных с психозом, например, шизофренией, или болезнью Паркинсона; и (4) расстройств сна, связанных с психозом, например, шизофренией, или болезнью Паркинсона, как описано выше, получают при пероральном введении при дозах порядка от около 1 мг до l00 мг один раз в день, предпочтительно около 2,5 мг-50 мг, например, 2,5 мг, 5 мг, l0 мг, 20 мг, 30 мг, 40 мг или 50 мг, один раз в день, предпочтительно при пероральном введении.
Удовлетворительные результаты для Способа II или любого из 8.1-8.15, Способа II или применения соединений по изобретению, описанного выше, например, для лечения расстройства сна только или возбуждения, агрессивного поведения, пост-травматического стрессового расстройства или расстройства импульсного контроля только, например, интермиттирующего эксплозивного расстройства только, как показано, получают при пероральном введении при дозах около 1 мг-10 мг один раз в день, например, около 2,5 мг-5 мг, например, 2,5 мг, 3 мг, 4 мг, 5 мг или 10 мг соединения по изобретению, в свободной форме или в форме фармацевтически приемлемой соли, один раз в день, предпочтительно при пероральном введении.
Удовлетворительные результаты для Способа I-A или любого из 9.1-9.38 или Способа IP-A, как показано, получают при дозах меньше чем l00 мг, предпочтительно меньше чем 50 мг, например, меньше чем 40 мг, меньше чем 30 мг, меньше чем 20 мг, меньше чем 10 мг, меньше чем 5 мг, меньше чем 2,5 мг, один раз в день. Удовлетворительные результаты для Способа II-A или любого из 9.1-9.38, как показано, получают при меньше чем 10 мг, например, меньше чем 5 мг, или предпочтительно меньше чем 2,5 мг.
Для лечения расстройств, описанных в настоящей заявке, где депо композицию используют для достижения большей продолжительности действия, дозы будут выше по сравнению с композицией с более короткой продолжительностью действия, например, больше чем 1-100 мг, например, 25 мг, 50 мг, 100 мг, 500 мг, 1000 мг или больше чем 1000 мг. В конкретном варианте осуществления режим дозирования для депо композиции включает первоначальную пероральную немедленную высвобождаемую дозу вместе с депо высвобождением, чтобы обеспечить стабильный уровень содержания в крови лекарственного средства. Продолжительность действия соединений по изобретению можно контролировать путем манипуляций с полимерной композицией, то есть варьируя соотношение полимер:лекарственное средство и размер микрочастиц. В случае, когда композиция по изобретению представляет собой депо композицию, предпочтительным является введение путем инъекции.
Фармацевтически приемлемые соли соединений по изобретению можно синтезировать из исходного соединения, которое содержит основную или кислотную часть, обычными химическими методами. Как правило, такие соли могут быть получены путем взаимодействия форм свободного основания этих соединений со стехиометрическим количеством подходящей кислоты в воде или в органическом растворителе или в смеси этих двух растворителей; обычно предпочтительны неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил. Дополнительные подробности получения этих солей, например, солей толуолсульфоновой кислоты в аморфной или кристаллической форме, можно найти в PCT/US 08/03340 и/или в предварительной патентной заявке США № 61/036069.
Фармацевтические композиции, содержащие соединения по изобретению, можно получить с использованием обычных разбавителей или эксципиентов (пример включает, но не ограничивается этим, кунжутное масло), и методов, известных в области получения галеновых препаратов. Таким образом, пероральные лекарственные формы могут включать таблетки, капсулы, растворы, суспензии и т.п.
Все ссылки на дозу, дозировку или терапевтически эффективное количество соединения или композиции по изобретению относятся к эквивалентному свободному основанию в дозе, исключающей любые соли.
Способы получения соединений по изобретению
Промежуточные соединения для соединений по изобретению обычно получают, как описано в WO PCT/US08/03340 (WO 2008/112280); заявке США с серийным № 10/786935; Патентах США №№ 6548493; 7238690; 6552017; 6713471; 7183282; U.S. RE39680 и U.S. RE39679 и WO 2015/154025, содержание каждого из этих документов включено посредством ссылки во всей полноте. Соли соединений по изобретению также можно получить, как аналогичным образом описано в Патентах США №№ 6548493; 7238690; 6552017; 6713471; 7183282; U.S. RE39680; U.S. RE39679; и WO 2009/114181, содержание каждого из этих документов включено посредством ссылки во всей полноте.
Выделение или очистку диастереомеров соединений по изобретению можно осуществить традиционными способами, известными в данной области техники, например, колоночной очисткой, препаративной тонкослойной хроматографией, препаративной ВЭЖХ, кристаллизацией, растиранием в порошок, методом псевдодвижущегося слоя и т.п.
ПРИМЕР 1
1-(4-Фторфенил)-4-((6bR,10aS)-3-метил-d3-2,3,6b,7,10,10a-гексагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8(9H)-ил)бутан-1-он п-толуолсульфонат
К супензии этилового эфира (6bR, 10aS)-2-оксо-2,3,6b,9,10,10a-гексагидро-1H,7H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8-карбоновой кислоты (4,27 г, 14,2 ммоль) в DMF (70 мл) добавляют NaH (560 мг, 95%, 21,3 ммоль) по порциям при комнатной температуре. Супензию затем перемешивают при комнатной температуре в течение 30 минут до получения прозрачного ярко-красного раствора. После охлаждения раствора до 0-5°C добавляют CD3I (982 мкл, 17,0 ммоль) в DMF (1 мл). Реакционную смесь перемешивают при 0-5°C, пока не израсходуется все исходное вещество. После гашения льдом смесь подкисляют до pH 3-5 при помощи HCl (12 N, 0,2 мл) и затем концентрируют при пониженном давлении. Полученный остаток суспендируют в смеси дихлорметана (100 мл) и H2O (50 мл) и затем доводят до pH ≥ 14 при помощи 50% NaOH. Органическую фазу отделяют и затем концентрируют досуха с получением 5 г неочищенного (6bR,10aS)-этил 3-метил-d3-2-оксо-2,3,6b,7,10,10a-гексагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8(9H)-карбоксилата в виде коричневого твердого вещества, которое используют непосредственно на следующей стадии без дополнительной очистки. MS (ESI) m/z 319,2 [M+H]+.
К перемешиваемому раствору этилового эфира (6bR,10aS)-3-метил-2-оксо-2,3,6b,9,10,10a-гексагидро-1H,7H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8-карбоновой кислоты (3,09 г, 9,71 ммоль) в THF (20 мл) добавляют BH3 в THF (50 мл, 1,0 M, 50,0 ммоль) при комнатной температуре. Смесь перемешивают при комнатной температуре в течение 36 часов и затем охлаждают до 0-5°C, с последующим гашением при помощи MeOH (5 мл). Растворители удаляют при пониженном давлении с получением светло-желтого остатка. К остатку добавляют HCl (12 N, 35 мл) при комнатной температуре. Полученную смесь перемешивают при 95°C в течение 30 часов, охлаждают до 0-5°C и затем доводят до pH ≥14 при помощи NaOH (10 N). Смесь экстрагируют дихлорметаном (100 мл). Объединенную органическую фазу сушат над K2CO3 и затем концентрируют досуха с получением (6bR,10aS)-3-метил-d3-2,3,6b,7,8,9,10,10a-октагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалина в виде коричневого масла, которое используют непосредственно на следующей стадии без дополнительной очистки. MS (ESI) m/z 233,2 [M+H]+.
Супензию (6bR,10aS)-3-метил-d3-2,3,6b,7,8,9,10,10a-октагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалина (1,6 г, 6,89 ммоль), K2CO3 (2,0 г), KI (1,7 г) и 4-хлор-4'-фторбутирофенона (2,3 мл) в 3-пентаноне (80 мл) дегазируют путем барботирования аргоном в течение 10 минут. После добавления N,N-диизопропилэтиламина (1,2 мл, 6,89 ммоль) реакционную смесь перемешивают при 75°C в течение 36 часов. После охлаждения смеси до комнатной температуры растворитель удаляют. Остаток суспендируют в дихлорметане (500 мл) и затем промывают при помощи H2O два раза (160 мл). Органическую фазу сушат над K2CO3 и затем упаривают досуха. Остаток очищают флэш-хроматографией на силикагеле с использованием градиента 0-100% этилацетата в смеси этилацетата и метанола (10:1) с 1% TEA в качестве элюента, с получением 1-(4-фторфенил)-4-((6bR,10aS)-3-метил-d3-2,3,6b,7,10,10a-гексагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8(9H)-ил)бутан-1-она в виде коричневого масла (1,66 г, выход 61%). MS (ESI) m/z 397,2 [M+H]+.
К раствору 1-(4-фторфенил)-4-((6bR,10aS)-3-метил-d3-2,3,6b,7,10,10a-гексагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8(9H)-ил)бутан-1-она (1,52 г, 3,83 ммоль) в изопропиловом спирте (5,4 мл) медленно добавляют раствор моногидрата п-толуолсульфоновой кислоты (656 мг, 3,45 ммоль) в 2,1 мл изопропилового спирта при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре до получения гелеобразной супензии. Добавляют изопропиловый спирт (5,0 мл) и смесь перемешивают при комнатной температуре еще в течение 2 часов. После фильтрования фильтровальную лепешку промывают изопропиловым спиртом (2,5 мл). Лепешку сушат в вакууме с получением указанного в заголовке соединения в виде белого порошка (1,75 г, выход 80%).1H ЯМР (500 МГц, DMSO-d6) δ 9,1 (с, 1H), 8,1 (ддд, J=2,73, 5,44, 8,68 Гц, 2H), 7,6-7,4 (м, 2H), 7,4-7,3 (м, 2H), 7,1 (д, J=7,81 Гц, 2H), 6,6 (т, J=7,62 Гц, 1H), 6,4 (д, J=7,89 Гц, 1H), 3,6 (дд, J=6,34, 12,15 Гц, 1H), 3,5-3,4 (м, 3H), 3,4-3,3 (м, 2H), 3,3-3,2 (м, 1H), 3,2-3,0 (м, 5H), 2,7 (td, J=3,04, 10,27 Гц, 1H), 2,7-2,5 (м, 1H), 2,3 (с, 3H), 2,3-2,2 (м, 1H), 2,0 (м, 3H).
ПРИМЕР 2
2,2-D2-1-(4-фторфенил)-4-((6bR,10aS)-3-метил-2,3,6b,7,10,10a-гексагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8(9H)-ил)бутан-1-он
К супензии этилового эфира (6bR,10aS)-3-метил-2-оксо-2,3,6b,9,10,10a-гексагидро-1H,7H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8-карбоновой кислоты (945 мг, 3 ммоль) в THF (5 мл) медленно добавляют BD3-THF (1,0 M в THF, 10 мл, 10 ммоль) при комнатной температуре. После завершения добавления реакционную смесь перемешивают при комнатной температуре в течение ночи и затем осторожно гасят при помощи D2O (2,0 мл). Растворитель удаляют в вакууме и остаток суспендируют в HCl (12 N, 9 мл). После перемешивания при 95°C в течение 20 часов реакционную смесь охлаждают до комнатной температуры и затем доводят до pH 12 при помощи 50% NaOH. Смесь концентрируют досуха с получением 2,2-d2-(6bR,10aS)-3-метил-2,3,6b,7,8,9,10,10a-октагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалина в виде коричневого твердого вещества, которое используют непосредственно на следующей стадии без дополнительной очистки. MS (ESI) m/z 232,2 [M+H]+.
К раствору 2,2-d2-(6bR, 10aS)-3-метил-2,3,6b,7,8,9,10,10a-октагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалина (200 мг, 0,87 ммоль) в 3-пентаноне (6 мл) добавляют KI (290 мг, 1,75 ммоль) и 4-хлор-4'-фторбутирофенон (0,29 мл, 1,75 ммоль), с последующим добавлением N,N-диизопропилэтиламина (0,16 мл, 1,75 ммоль). Полученную смесь перемешивают при 75°C в течение 20 часов. Растворитель удаляют при пониженном давлении, затем полученный остаток очищают колоночной хроматографией на силикагеле с использованием градиента 0-100% этилацетата в смеси этилацетата и метанола (10:1) с 2% TEA в качестве элюента, с получением указанного в заголовке соединения в виде коричневого масла. (47 мг, 14% выход).1H ЯМР (500 МГц, CDCl3) δ 8,00 (дд, J=8,9, 5,4 Гц, 2H), 7,13 (т, J=8,6 Гц, 2H), 6,65 (т, J=7,6 Гц, 1H), 6,51 (д, J=6,6 Гц, 1H), 6,41 (д, J=7,8 Гц, 1H), 3,29 (д, J=10,1 Гц, 1H), 3,25-3,14 (м, 2H), 3,02 (т, J=7,1 Гц, 2H), 2,98-2,90 (м, 1H), 2,86 (с, 3H), 2,84-2,69 (м, 2H), 2,61-2,23 (м, 3H), 2,17-1,86 (м, 5H). ). MS (ESI) m/z 396,2 [M+H]+.
ПРИМЕР 3
2,2-D2-1-(4-фторфенил)-4-((6bR,10aS)-3-метил-d3-2,3,6b,7,10,10a-гексагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8(9H)-ил)бутан-1-он п-толуолсульфонат
К раствору (6bR,10aS)-этил 3-метил-d3-2-оксо-2,3,6b,7,10,10a-гексагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8(9H)-карбоксилата (1,2 г, 3,77 ммоль) в THF(7,0 мл) медленно добавляют BD3·THF (10 мл, 1M в THF). Полученную смесь перемешивают при комнатной температуре в течение ночи. Добавляют CD3OD (1,0 мл) по каплям для гашения реакции, с последующим добавлением D2O (2,0 мл). Растворители удаляют при пониженном давлении и остаток суспендируют в HCl (12 N, 12 мл). Коричневую супензию перемешивают при 95°C в течение 24 часов и затем охлаждают до 0-5°C. Полученную смесь доводят до pH ≥14 при помощи NaOH (10 N) и затем экстрагируют дихлорметаном три раза (90 мл). Объединенную органическую фазу сушат над K2CO3, упаривают при пониженном давлении и затем сушат в вакууме с получением 2,2-d2-(6bR,10aS)-3-метил-d3-2,3,6b,7,8,9,10,10a-октагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалина в виде коричневого масла (770 мг, 87% выход ). MS (ESI) m/z 235,2 [M+H]+.
Смесь 2,2-d2-(6bR, 10aS)-3-метил-d3-2,3,6b,7,8,9,10,10a-октагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалина (500 мг 2,13 ммоль), KI (720 мг, 4,34 ммоль), 4-хлор-4'-фторбутирофенона (0,7 мл, 4,26 ммоль) в DMF (14 мл) барботируют аргоном в течение 10 минут. Добавляют N,N-диизопропилэтиламин (0,7 мл, 4,02 ммоль) и смесь перемешивают при 95°C, пока не израсходуется все исходное вещество. Реакционную смесь охлаждают до комнатной температуры и затем концентрируют при пониженном давлении. Остаток суспендируют в дихлорметане (50 мл) и затем промывают при помощи H2O (30 мл). Полученный дихлорметановый раствор сушат над K2CO3 и концентрируют досуха. Остаток очищают колоночной хроматографией на силикагеле с использованием градиента 0-100% этилацетата в смеси этилацетата и метанола (10:1) с 1,5% TEA в качестве элюента, с получением 2,2-d2-1-(4-фторфенил)-4-((6bR,10aS)-3-метил-d3-2,3,6b,7,10,10a-гексагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8(9H)-ил)бутан-1-она в виде коричневого масла (446 мг, 53% выход). MS (ESI) m/z 399,2 [M+H]+..
2,2-D2-1-(4-фторфенил)-4-((6bR,10aS)-3-метил-d3-2,3,6b,7,10,10a-гексагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8(9H)-ил)бутан-1-он (201 мг, 0,51 ммоль) растворяют в изопропаноле (2 мл). К раствору добавляют п-толуолсульфоновую кислоту (86,2 мг, 0,45 ммоль) в изопропаноле (1 мл). Полученный прозрачный раствор перемешивают при комнатной температуре до получения молочно-белой смеси. Смесь охлаждают до 0-5°C и затем фильтруют. Фильтровальную лепешку промывают холодным изопропанолом (2 мл) и затем сушат в высоком вакууме с получением указанного в заголовке продукта в виде белого твердого вещества (180 мг, выход 63%).1H ЯМР (500 МГц, DMSO-d6) δ 9,1 (с, 1H), 8,0 (ддд, J=2,70, 5,52, 8,77 Гц, 2H), 7,5-7,4 (м, 2H), 7,4-7,3 (м, 2H), 7,1 (д, J=7,83 Гц, 2H), 6,4 (д, J=7,32 Гц, 1H), 3,6 (с, 1H), 3,5-3,2 (м, 5H), 3,1 (дт, J=7,74, 14,91 Гц, 4H), 2,8-2,5 (м, 1H), 2,3 (с, 4H), 2,2-1,9 (м, 4H).
СРАВНИТЕЛЬНЫЙ ПРИМЕР 4
1-(4-Фтор(2,3,5,6-d4)фенил)-4-((6bR,10aS)-3-метил-2,3,6b,7,10,10a-гексагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8(9H)-ил)бутан-1-он п-толуолсульфонат
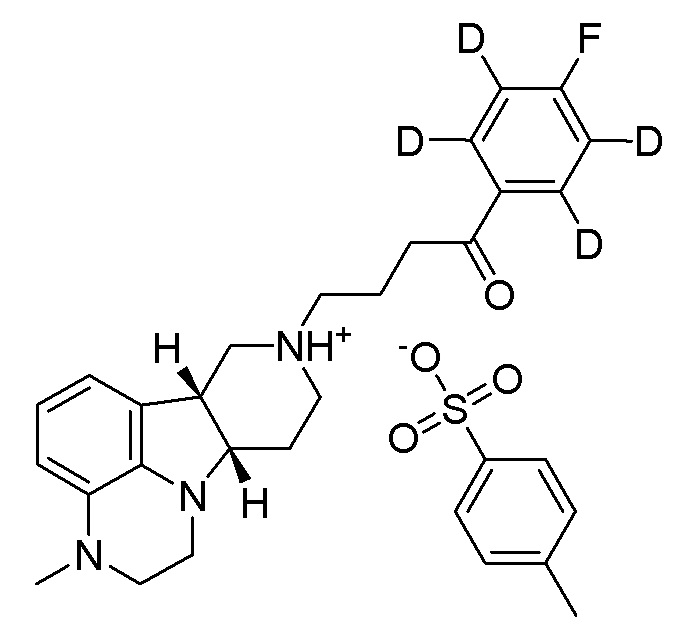
3-Пентанон (4 мл) добавляют в смесь (6bR, 10aS)-3-метил-2,3,6b,7,8,9,10,10a-октагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалина (460 мг, 2,0 ммоль), 2',3',5',6'-d4-4-хлор-4'-фторбутирофенона (428 мг, 2,0 ммоль), KI (335 мг, 2,0 ммоль) и K2CO3 (300 мг, 2,2 ммоль). Полученную смесь барботируют аргоном в течение 10 минут и затем перемешивают при 75°C в течение 20 часов. Реакционную смесь охлаждают до комнатной температуры, затем добавляют дихлорметан (30 мл) и H2O (15 мл). Органическую фазу отделяют и затем экстрагируют 1N раствором HCl (30 мл). Полученную водную фазу промывают дихлорметаном (5 мл) и затем медленно добавляют к смеси дихлорметанаа (20 мл) и NaOH (50%, 10 мл) при 0-5°C. После завершения добавления органическую фазу отделяют и концентрируют досуха. Остаток затем очищают колоночной хроматографией на щелочном Al2O3 с использованием градиента 0-40% этилацетата в гексане в качестве элюента с получением 1-(4-фтор(2,3,5,6-d4)фенил)-4-((6bR,10aS)-3-метил-2,3,6b,7,10,10a-гексагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8(9H)-ил)бутан-1-она в форме свободного основания (200 мг, 25% выход ). MS (ESI) m/z 398,2 [M+H]+.
К очищенному свободному основанию (125 мг, 0,31 ммоль) в изопропаноле (2 мл) добавляют п-толуолсульфоновую кислоту (52 мг, 0,28 ммоль) в изопропаноле (1 мл) при комнатной температуре. Полученный прозрачный раствор перемешивают при комнатной температуре до образования молочно-белой супензии. Раствор охлаждают до 0-5°C и затем фильтруют. Фильтровальную лепешку промывают холодным изопропанолом (2 мл) и затем сушат в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества (120 мг, выход 68%).1H ЯМР (500 МГц, DMSO-d6) δ 9,1 (с, 1H), 7,6-7,4 (м, 2H), 7,1 (д, J=7,82 Гц, 2H), 6,6 (т, J=7,64 Гц, 1H), 6,5 (д, J=7,30 Гц, 1H), 6,4 (д, J=7,87 Гц, 1H), 3,6 (дд, J=6,36, 12,51 Гц, 1H), 3,5-3,4 (м, 3H), 3,4-3,3 (м, 2H), 3,3-3,2 (м, 1H), 3,2-3,0 (м, 5H), 2,8 (с, 3H), 2,7 (тд, J=2,95, 10,27 Гц, 1H), 2,7-2,5 (м, 1H), 2,3 (с, 3H), 2,3 (д, J=15,10 Гц, 1H), 2,1-1,9 (м, 3H).
ПРИМЕР 5: Измерение уровней исходного соединения и метаболита у мышей
Мышам вводят соединения примеров 1-3 и соединение формулы Q и исследуют уровни исходных соединений и основных амидных метаболитов. Процедуры для синтеза соединения формулы Q можно найти в WO 2008/112280. Соединение сравнительного примера 4 используют в качестве внутреннего стандарта в каждом испытании для контроля разницы в скорости естесственного метаболизма в каждой испытываемой группе животных. После одной пероральной дозы испытываемого соединения и внутреннего стандарта уровни в плазме исходных соединений и метаболитов измеряют через 0,25, 0,5, 1, 2, 4 и 6 часов. Определяют максимальную концентрацию, время до достижения максимальной концентрации и площадь под кривой (AUC) как для исходного соединения, так и для основного амидного метаболита. AUC значение для каждого испытываемого соединения нормализуют к AUC внутреннего стандарта (Пр. 4). Относительное образование амида, таким образом, рассчитывают следующим образом для каждого Примера X (т.е. Пр. 1, Пр. 2, Пр. 3 и Пр. Q):
Относительное образование амида (Пр. X) =
AUCX-Исходн.соед./AUCX-Амид
AUCСтд.-Исходн.соед./AUCСтд.-Амид
Результаты представлены в Таблице 1 ниже.
Обнаружено, что степень конверсии исходного соединения в амидный метаболит существенно ниже для соединений Примера 1, 2 и 3 по сравнению с недейтерированным соединением формулы Q. После нормализации к степени метаболизма внутреннего стандарта было обнаружено, что степень образования амида для соединений примеров 1, 2 и 3 существенно ниже, чем для недейтерированного соединения Q.
Исследования связывания с рецептором указывают на то, что соединения Примера 1, Примера 2 и Примера 3 показывают по существу такой же профиль связывания с рецептором, как и недейтерированное соединение формулы Q (включая, например, связывание с серотониновым рецептором (например, 5-HT2A), допаминовым рецептором (например, D2) и транспортером серотонина). Например, соединение Примера 2 показывает 98% ингибирование человеческого серотонинового 5-HT2A рецептора при концентрации 0,1 мкМ.
ПРИМЕР 6: Сравнение фармакокинетики между дейтерированными и недейтерированными соединениями у крыс
In vivo метаболизм (деметилирование/окисление) дейтерированного соединения примера 2 (соединение формулы I, тозилатная соль) сравнивают с его недейтерированным аналогом соединением формулы Q (тозилатная соль). Фармакокинетику каждого соединения определяют после перорального (PO) и внутривенного (IV) введения в перекрестном испытании на крысах.
PO Введение: Шесть самцов крыс Sprague-Dawley делят на две группы по 3 крысы в каждой для PO введения соединения в день 1 испытания. Крысам в группе 1 вводят 10 мг/кг (в пересчете на свободное основание) соединения формулы Q, а крысам в группе 2 вводят 10 мг/кг (в пересчете на свободное основание) соединения примера 2. Образцы крови собирают через 0,25, 0,5, 1, 2, 4, 6, 8, 12 и 24 часов после введения дозы и анализируют для определения концентрации в плазме введенного соединения и его метаболитов. После трехдневного периода вымывания крысам группы 1 и группы 2 меняют введение и наоборот вводят, соответственно, 10 мг/кг (в пересчете на свободное основание) соединения примера 2 и 10 мг/кг (в пересчете на свободное основание) соединения формулы Q. Образцы крови собирают и анализируют, как описано выше, за исключением взятия дополнительного образца до введения дозы.
IV Введение: Шесть самцов крыс Sprague-Dawley делят на две группы по 3 крысы в каждой для IV введения соединения в день 1 испытания. Крысам в группе 1 вводят 1 мг/кг (в пересчете на свободное основание) соединения формулы Q, а крысам в группе 2 вводят 1 мг/кг (в пересчете на свободное основание) соединения примера 2. Образцы крови собирают через 2 минуты, 5 минут, 0,25, 0,5, 2, 4, 6, 8 и 12 часов после введения дозы и анализируют для определения концентрации в плазме введенного соединения и его метаболитов. После 72-часового периода вымывания крысам группы 1 и группы 2 меняют введение и наоборот вводят, соответственно, 1 мг/кг (в пересчете на свободное основание) соединения примера 2 и 1 мг/кг (в пересчете на свободное основание) соединения формулы Q. Образцы крови собирают и анализируют, как описано выше, за исключением взятия дополнительного образца до введения дозы.
Все образцы крови обрабатывают для получения плазмы и анализируют на концентрации исходного соединения и метаболита с использованием жидкостной хроматографии в тандеме с масс-спектрометрией (LC-MS/MS). Анализируемые метаболиты включают N-деметилированное амидное соединение Q-1 (обсуждаемое выше). Площадь под кривой (AUC) исходного соединения и метаболитов на основании данных концентрации в плазме против времени рассчитывают с использованием программы Prism 5.04 (GraphPad Software, Inc.).
Результаты представлены в Таблице 2 ниже (значение AUC показано для 0-24 часов, измеренное в нг-час/мл):
Обнаружено, что после PO введения соединения формулы Q исходное соединение интенсивно метаболизируется с существенным образованием N-деметилированного/альфа-окисленного амида (Формула Q-1). AUC метаболита Q-1 в 2,2 больше, чем AUC исходного соединения. В отличие от этого, IV введение приводит к значительно менее интенсивному метаболизму. После IV введения AUC Q-1 метаболита составляет только около 2% от AUC исходного соединения. Это показывает на высокую степень пресистемного (печеночного) метаболизма, который происходит преимущественно путем N-деметилирования и альфа-N окисления.
В отличие от этого, PO введение соединения примера 2 приводит к существенно меньшему метаболизму в метаболит Q-1 по сравнению с его недейтерированным аналогом. AUC метаболита Q-1 только в 1,2 больше, чем AUC исходного соединения, по сравнению с в 2,2 раза большим AUC в случае введения соединения формулы Q. Таким образом, имеет место 55% снижение относительного метаболизма в деметилированное амидное производное. Подобные результаты получают для IV введения, где AUC Q-1 метаболита составляет около 1% от AUC исходного соединения. Также показано, что при сравнении AUC плазмы от эквивалентного PO введения соединения формулы Q с соединением примера 2, последнее приводит приблизительно к половине AUC для метаболита Q-1 в плазме (67,8 нг-час/мл против 128,2 нг-час/мл).
ПРИМЕР 7: Сравнение фармакокинетики между дейтерированным и недейтерированным соединением у собак
In vivo метаболизм (деметилирование и альфа-окисление) дейтерированного соединения примера 2 (соединение формулы I, тозилатная соль) сравнивают метаболизмом его недейтерированного аналога, т.е. соединения формулы Q (тозилатная соль). Фармакокинетику каждого соединения определяют после сублингвального (SL) и подкожного (SC) введения в не-перекрестных последовательных испытаниях на собаках.
SC Введение: Шесть самцов собак породы бигль возраста от 2 до 5 лет рандомизируют в две группы по три собаки в каждой. Собакам в группе 1 вводят соединение формулы Q при дозе 1 мг/кг (в пересчете на свободное основание) в носителе 0,5% метилцеллюлозы/дистиллированная вода. Собакам в группе 2 вводят соединение примера 2 при дозе 1 мг/кг (в пересчете на свободное основание) в носителе 0,5% метилцеллюлозы/дистиллированная вода. Введение осуществляют подкожно в межлопаточную область через иглу размера 22 или 23. Образцы цельной крови собирают через головную вену собак до введения дозы и после введения дозы в точках времени 5, 15 и 30 минут, 1, 2, 4, 6, 8 и 24 часов. После минимум 7-дневного периода вымывания собак переводят в сублингвальную часть испытания.
SL Введение: Собакам группы 1 вводят соединение формулы Q при дозе 1 мг/кг (в пересчете на свободное основание) в носителе 0,5% метилцеллюлозы/дистиллированная вода. Собакам в группе 2 вводят соединение примера 2 при дозе 1 мг/кг (в пересчете на свободное основание) в носителе 0,5% метилцеллюлозы/дистиллированная вода. Животных анестезируют перед введением дозы с использованием пропофола (6 мг/кг) и анестезию поддерживают в течение 30 минут с использованием 3-4,5% изофлурана. Введение осуществляют сублингвально и дозу наносят в течение 30 минут, затем вытирают с использованием нетканой марли. Образцы цельной крови собирают через головную вену собак до введения дозы и после введения дозы в точках времени 5, 15 и 30 минут, 1, 2, 4, 6, 8, 24, 36 и 48 часов.
Все образцы крови обрабатывают для получения плазмы и анализируют на концентрации исходного соединения и метаболитов с использованием жидкостной хроматографии в тандеме с масс-спектрометрией (LC-MS/MS). Анализируемые метаболиты включают N-деметилированное соединение Q-1A (показано ниже) и N-деметилированное/альфа-окисленное амидное соединение Q-1 (обсуждаемое выше). Площадь под кривой (AUC) исходного соединения и метаболитов на основании данных концентрации в плазме против времени рассчитывают с использованием Prism программы 5.04 (GraphPad Software, Inc.).
Результаты представлены в Таблице 2 ниже (значение AUC показано для 0-24 часов, измеренное в нг-час/мл):
N.Q.=не поддающийся количественному определению
N.D.=не определяли
Обнаружено, что SL введение соединения примера 2 приводит к AUC исходного соединения примерно на 72% больше по сравнению с введением соединения формулы Q. AUC деметилированного метаболита Q-1A составляет около 3% от AUC исходного соединения для соединения формулы Q и около 8% от AUC исходного соединения для соединения примера 2. Концентрация амидного метаболита Q-1 поддается определению при меньше чем 1 нг/мл в кажой точке времени для SL введения соединения формулы Q (AUC количественно не определяется), но не поддается определению для SL введения соединения примера 2 (< 0,1 нг/мл).
В отличие от этого, SC введение приводит к более сопоставимым результатам между этими двумя соединениями. Для соединения формулы Q, AUC Q-1A метаболита составляет около 3% от исходного соединения, тогда как для соединения примера 2, AUC Q-1A метаболита составляет около 6% от исходного соединения. Для SC введения, метаболит Q-1 не поддается определению (< 0,1 нг/мл) для обоих соединений. Было найдено, что значение AUC исходного соединения сопоставимо для дейтерированных и недейтерированных соединений.
Сравнение SC с SL результатами для соединения формулы Q показывает, что SL введение приводит к AUC исходного соединения на 10% меньше по сравнению с SC введением. В отличие от этого, введение дейтерированного соединения примера 2 приводит к AUC исходного соединения на 61% больше для SL по сравнению с SC. Не привязывая это к какой-либо теории, считается, что эта разница связана с разницей в скорости абсорбции из подкожного пространства между дейтерированными и недейтерированными соединениями.
Взятые вместе, эти результаты показывают, что дейтерирование метиленовой группы смежной с азотом пиперазина снижало метаболизм соединения по изобретению по сравнению с его недейтерированным аналогом, приводя в более высоким и более пролонгированным концентрациям в плазме исходного лекарственного средства. Поскольку обнаружено, что концентрация деметилированного Q-1A метаболита выше для дейтерированного соединения по сравнению с недейтерированным соединением, результаты дают основание предположить, как это можно наблюдать у крыс, что дейтерирование ингибирует последующее окисление деметилированного амина до его амидного производного (Q-1).
Считают, что образование метаболита Q-1 происходит через посредство промежуточного метаболита Q-1A, показаного ниже:
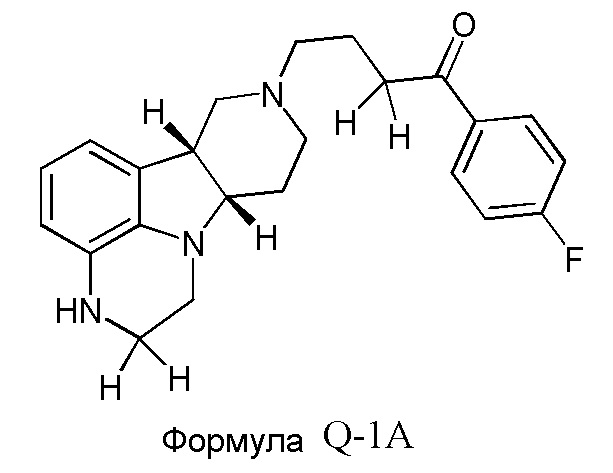
Таким образом, исходное соединение Q подергается деметилированию с образованием амина, с последующим окислением метилена смежного с амином с образованием амидного метаболита Q-1A. Результаты, представленные в Примерах 6 и 7, демонстрируют, что как для собак, так и для крыс, дейтерирование соединения формулы Q в указанном положении, для получения соединения примера 2, существенно снижает окисление N-метилпиперазиновой группы, таким образом, указывая на блокаду этого метаболического пути.
Уменьшение метаболизма исходного соединения Q до Q-1 метаболита может иметь важные клинические последствия, поскольку известно, что исходное соединение Q, Q-1 метаболит и Q-1A метаболиты все являются фармакологически активными соединениями, но с разными профилями селективности к рецептору. Например, Таблица 2 показывает некоторые различия в активности в отношении рецепторов между этими соединениями (указанные значения представляют собой Ki (нМ)):
Таблица 2 показывает, что хотя все три соединения активны на рецепторах серотонина, допамина D1, допамина D2 и транспортера серотонина, их относительная активность на этих рецепторах различается. В то время как метаболит Q-1A имеет наиболее схожий фармакологический профиль с исходным соединением Q, Таблица 2 показывает, что метаболит Q-1 имеет существенные отличия, заключающиеся в том, что он имеет существенно более низкую относительную активность на допаминовом D2 рецепторе и транспортере серотонина. Кроме того, обнаружено, что соединение формулы Q-1, в отличие от Q и Q-1A, является сильным антагонистом опиоидного рецептора мю (Ki около 22 нМ). Из-за их разных профилей активности в отношении рецепторов, каждый из релевантных метаболитов имеет отличающиеся функциональные фармакологические эффекты по сравнению с исходным лекарственным средством Q. Таким образом, блокируя путь метаболизма, который превращает соединения Q и Q-1A в метаболит Q-1, можно получить существенный эффект на фармакологическую функцию. Настоящее изобретение поэтому является полезным для ингибирования этого метаболического пути для модуляции общего фармакологического профиля, обеспечиваемого исходными лекарственными средствами.
Реферат
Изобретение относится к дейтерированным гетероциклическим конденсированным гамма-карболинам формул I, II, III, в свободной или солевой форме, а также их фармацевтическим композициям на основе данных соединений, к способам лечения заболеваний, связанных с 5-HTрецептором, транспортером серотонина(SERT) и/или путями с вовлечением сигнальных систем допаминовых D/Dрецепторов, и/или лечения остаточных симптомов. Получены и описаны соединения, которые демонстрируют улучшенную метаболическую стабильность и могут найти свое применение в лечении расстройств центральной нервной системы. 5 н. и 15 з.п. ф-лы, 2 табл., 7 пр.
Формула
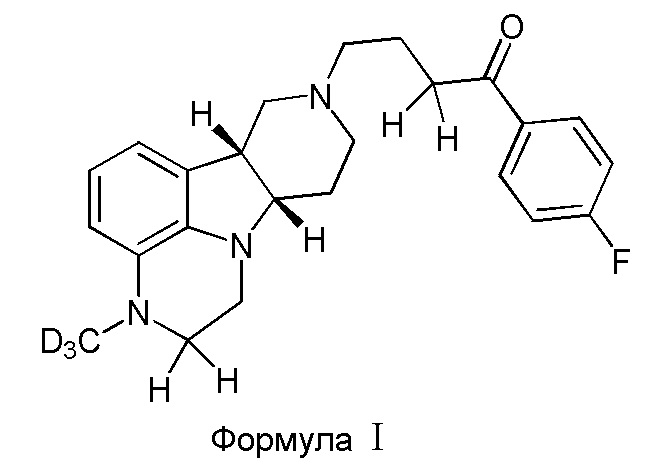
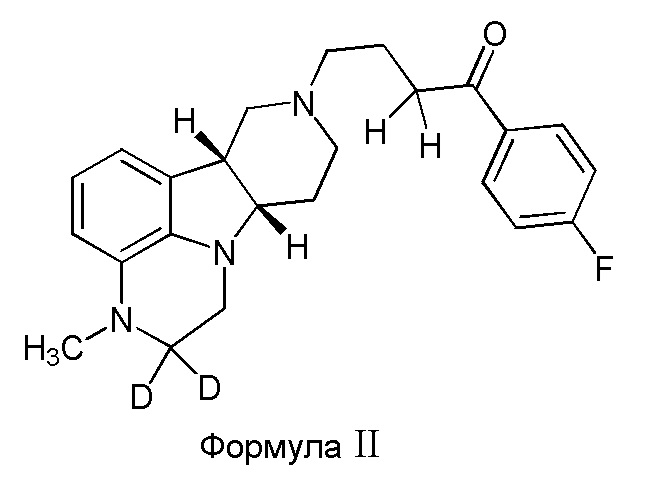
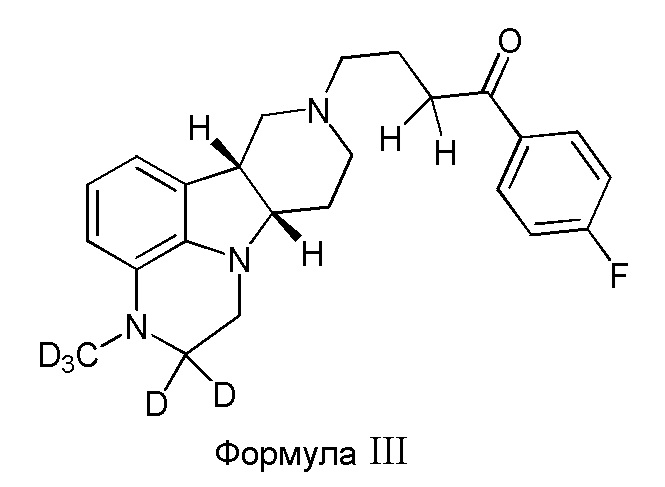
Документы, цитированные в отчёте о поиске
Органические соединения
Комментарии