Гидроксилированные пиримидилциклопентаны в качестве ингибиторов протеинкиназы (акт) - RU2504542C2
Код документа: RU2504542C2
Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к новым ингибиторам сериновых/треониновых протеинкиназ (например, киназ AKT и родственных им), к фармацевтическим композициям, содержащим указанные ингибиторы, и способам получения указанных ингибиторов. Данные ингибиторы пригодны, например, для лечения гиперпролиферативных заболеваний, таких как рак и воспаление, у млекопитающих.
Уровень техники
Протеинкиназы (РК) представляют собой ферменты, катализирующие фосфорилирование гидроксильных групп белковых остатков тирозина, серина и треонина путем переноса концевого (гамма) фосфата от АТФ. Через пути передачи сигнала, указанные ферменты регулируют рост, дифференцировку и пролиферацию клеток, т.е. по существу все аспекты жизни клетки, так или иначе зависящие от активности протеинкиназ (Hardie, G. and Hanks, S. (1995) The Protein Kinase Facts Boot I and II, Academic Press, San Diego, CA). Кроме того, установили связь между аномальной активностью ПК и множеством нарушений, от заболеваний, относительно не угрожающих жизни, таких как псориаз, до крайне вирулентных заболеваний, таких как глиобластома (рак головного мозга). Протеинкиназы являются важным классом мишеней терапевтической модуляции (Cohen, Р. (2002) Nature Rev. Drug Discovery 1:309).
Существенно, что атипичное фосфорилирование и/или экспрессия белков часто оказываются одной из причин аномального деления клетки, метастазирования и выживаемости клетки при раке. Нарушенную регуляцию и/или экспрессию различных киназ, включая AKT, VEGF, ILK, ROCK, p70S6K, Bcl, PKA, PKC, Raf, Src, PDK1, ErbB2, MEK, IKK, Cdk, EGFR, BAD, CHK1, CHK2 и GSK3, а также множества других, напрямую связывают с раковыми заболеваниями.
Протеинкиназы включают два класса: протеинтирозинкиназы (РТК) и серинтреонинкиназы (STK). Ферменты Протеинкиназы B/Akt представляют собой группу серин/треонинкиназ, экспрессия которых повышена в различных видах злокачественных опухолей у человека. Одной из наиболее охарактеризованных мишеней липидных продуктов PI3K является серин/треонинкиназа Akt массой 57 КД, расположенная после PI3K на пути передачи сигнала (Hemmings, B.A. (1997) Science 275: 628; Hay N. (2005) Cancer Cell 8: 179-183). Akt является гомологом протоонкогена v-akt быстро трансформирующегося ретровируса AKT8 человека. Из-за высокой гомологии последовательностей с протеинкиназами А и С Akt также называют протеинкиназой В (РКВ), а также родственной А и С (RAC). Известны три изоформы Akt, обозначаемые Akt1, Akt2 and Akt3, имеющие общую гомологию 80% (Staal, S.P. (1987) Proc. Natl. Acad. Sci. 84: 5034; Nakatani, K. (1999) Biochem. Biophys. Res. Commun. 257: 906; Li et al (2002) Current Topics in Med. Chem. 2: 939-971; WO 2005/113762). Изоформы Akt имеют общую структуру доменов, которая состоит из домена гомологии Pleckstrin на N-конце, каталитического домена киназы, и короткого регуляторного участка на С-конце. Кроме того, для Akt2 и Akt3 существуют варианты сплайсирования. После внедрения в клеточную мембрану под действием PtdInd(3,4,5)P3, Akt фосфорилируется (активируется) PDK1 на Т308, Т309 и Т305 в случае изоформ Akt1 (РКВα), Akt2 (РКВβ) и Akt3 (РКВγ) соответственно, и на S473, S474 и S472 для изоформ Akt1, Akt2 and Akt3 соответственно. Указанное фосфорилирование осуществляется неизвестной на данный момент киназой (предварительно обозначенной PDK2), хотя предполагают, что в указанном процессе могут играть роль PDK1 (Balendran, А., (1999) Curr. Biol. 9: 393), автофосфорилирование (Toker, A. (2000) J. Biol. Chem. 275: 8271) и интегрин-ассоциированная киназа (ILK) (Delcommenne, M. (1998) Proc. Natl. Acad. Sci. USA, 95: 11211). Активация Akt требует ее фосфорилирования по остатку Ser 473 гидрофобного мотива С-конца (Brodbeck et al (1999) J. Biol. Chem. 274: 9133-9136; Coffer et al (1991) Eur. J. Biochem. 201: 475-481; Alessi et al (1997) Curr. Biol. 7: 261-269). Несмотря на то, что монофосфорилирование Akt активирует киназу, для максимальной активизации киназы требуется бис-фосфорилирование.
Считают, что Akt действует на раковые клетки, подавляя апоптоз и усиливая как ангиогенез, так и деление (Toker et al. (2006) Cancer Res. 66(S): 3963-3966). Экспрессия Akt повышена при множестве раковых заболеваний у человека, включая, без ограничения, рак кишечника (Zinda et al (2001) Clin. Cancer Res. 7: 2475), рак яичника (Cheng et al (1992) Proc. Natl. Acad. Sci. USA 89: 9267), рак мозга (Haas Kogan et al (1998) Curr. Biol. 8: 1195), рак легких (Brognard et al (2001) Cancer Res. 61: 3986), рак поджелудочной железы (Bellacosa et al (1995) Int. J. Cancer 64: 280-285; Cheng et al (1996) Proc. Natl. Acad. Sci. 93: 3636-3641), рак предстательной железы (Graft et al (2000) J. Biol. Chem. 275: 24500), и рак желудка (Staal et al (1987) Proc. Natl. Acad. Sci. USA 84: 5034-5037).
Мишень РI3K/Akt/млекопитающих рапамицинового (mTOR) пути исследовали в качестве средства терапии с использованием низкомолекулярных ингибиторов (Georgakis, G. and Younes, A. (2006) Expert Rev. Anticancer Ther. 6(1): 131-140; Granville et al (2006) Clin. Cancer Res. 12(3): 679-689). Ингибирование сигналов PI3K/Akt вызывает апоптоз и подавляет рост опухолевых клеток, имеющих повышенный уровень Akt (Kim et al (2005) Current Opinion in Investig. Drugs 6(12): 1250-1258; Luo et al (2005) Molecular Cancer Ther. 4(6): 977-986).
Разработка ингибиторов киназ, нацеленных на пути с нарушенной регуляцией, и в итоге приводящих к заболеванию, представляет огромный этический и коммерческий интерес для медицинского и фармацевтического сообщества. Соединение, ингибирующее (1) пополнение (привлечение) Akt в клеточной мембране, (2) активацию под действием PDK1 или PDK2, (3) фосфорилирование субстрата, или (4) одну из расположенных ниже мишеней Akt, может стать важным противораковым агентом, который можно применять либо в качестве отдельного средства лечения, либо в комбинации с другими допустимыми процедурами.
Заявка на патент США 2005/0130954 описывает, среди прочего, ряд соединений, действующих как ингибиторы AKT. Указанные соединения отмечены как пригодные для лечения гиперпролиферативных заболеваний, таких как рак.
Раскрытие изобретения
Согласно настоящему изобретению предложено новое соединение, ингибирующее протеинкиназы AKT. Соединение согласно настоящему изобретению можно применять в качестве терапевтического агента для лечения заболеваний и патологических состояний, которые можно лечить путем ингибирования протеинкиназ AKT.
В частности, настоящее изобретение включает соединения общей формулы I:
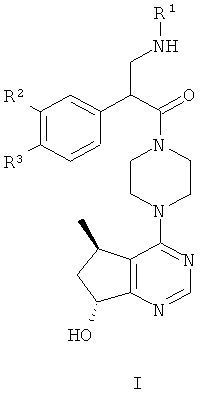
и их фармацевтически приемлемые соли, где R1, R2 и R3 как определено в настоящей заявке.
Настоящее изобретение также обеспечивает фармацевтические композиции, включающие соединения формулы I, или его энантиомер, сольват, метаболит или их фармацевтически приемлемую соль или пролекарство.
Кроме того, настоящее изобретение обеспечивает способ лечения заболеваний или патологических состояний у млекопитающих, опосредованных протеинкиназами AKT, включающий введение указанному млекопитающему одного или более соединений формулы I, или его энантиомера, сольвата, метаболита, или их фармацевтически приемлемой соли или пролекарства, в количестве, эффективном для лечения или предотвращения указанного нарушения. Патологические состояния, опосредуемые протеинкиназами AKT, которые можно лечить способами согласно настоящему изобретению, включают, но не ограничены перечисленными: воспалительные, гиперпролиферативные, сердечнососудистые, нейродегенеративные, гинекологические и дерматологические заболевания и нарушения.
Кроме того, настоящее изобретение обеспечивает способы ингибирования активности протеинкиназ AKT у млекопитающих, включающие введение указанному млекопитающему соединения формулы I или его энантиомера, сольвата, метаболита, или их фармацевтически приемлемой соли или пролекарства в количестве, эффективном для ингибирования выработки протеинкиназы AKT.
Кроме того, настоящее изобретение обеспечивает способы ингибирования активности протеинкиназ AKT, включающие приведение указанной киназы в контакт с соединением формулы I.
Соединение согласно настоящему изобретению может быть полезно в комбинации с другими известными терапевтическими агентами. Следовательно, настоящее изобретение также обеспечивает фармацевтические композиции, содержащие соединение формулы I или его энантиомер, сольват, метаболит, фармацевтически приемлемую соль или пролекарство, в сочетании со вторым терапевтическим агентом.
Настоящее изобретение также обеспечивает соединение формулы I, и его фармацевтически приемлемые соли для применения в качестве средства для лечения патологических состояний, опосредуемых протеинкиназами AKT.
Дополнительным аспектом изобретения является применение соединения формулы I или его энантиомера, сольвата, метаболита, фармацевтически приемлемой соли или пролекарства, для терапии. Согласно одному варианту реализации, терапевтическое лечение включает лечение патологического состояния, опосредуемого протеинкиназами AKT.
Настоящее изобретение дополнительно обеспечивает наборы для лечения заболевания или нарушения, опосредуемого протеинкиназами AKT, включающие соединение формулы I или его энантиомер, сольват, метаболит, фармацевтически приемлемую соль или пролекарство, контейнер, а также, возможно, вкладыш или ярлык с описанием способа лечения. Наборы могут дополнительно включать второе соединение или состав, включающий второй лекарственный агент, пригодный для лечения указанного заболевания или нарушения.
Дополнительно, согласно настоящему изобретению предложено применение соединения формулы I в лечении гиперпролиферативного заболевания. В другом аспекте настоящего изобретения гиперпролиферативное заболевание представляет собой рак.
Настоящее изобретение дополнительно включает способы приготовления, способы разделения, и способы очистки соединений согласно настоящему изобретению.
Дополнительные преимущества и новые признаки настоящего изобретения будут раскрыты в нижеследующем описании, и частично будут очевидны для специалиста при ознакомлении с нижеследующим описанием, либо станут понятны при осуществлении изобретения. Преимущества настоящего изобретения могут быть реализованы и оценены при помощи средств, комбинаций, композиций и способов, конкретно указанных в прилагающейся формуле изобретения.
Осуществление изобретения
Ниже следует подробное описание конкретных вариантов осуществления настоящего изобретения, примеры которых проиллюстрированы в сопутствующих формулах и структурах. Несмотря на то, что ниже изобретение описано подробно и приведены варианты его осуществления, следует понимать, что изобретение не ограничивается указанными вариантами осуществления. Напротив, изобретение включает все альтернативные варианты, модификации и эквиваленты, которые могут быть включены в объем настоящего изобретения, определяемый формулой. Для специалиста будут очевидны многочисленные методы и материалы, аналогичные или эквивалентные описанным здесь, которые могут быть применены при реализации настоящего изобретения на практике. Настоящее изобретение никоим образом не ограничено описанными способами и материалами. В случае если один или несколько включенных в описание источников и сходных материалов отличается или противоречит настоящему описанию, включая, но не ограничиваясь определением терминов, использованием терминов, описанными техническими приемами и т.п., приоритетное значение имеет данное описание.
ОПРЕДЕЛЕНИЯ
В настоящем описании термины в единственном числе обозначают "один или более".
В настоящем описании термины "соединение согласно изобретению" "соединение согласно настоящему изобретению" и "соединение формулы I" включают соединение формулы I и его фармацевтически приемлемые соли.
Фраза "эффективное количество" обозначает количество соединения, которое, при введении млекопитающему, нуждающемуся в соответствующем лечении, достаточно для (i) лечения или предотвращения определенного заболевания, патологического состояния или нарушения, опосредуемого активностью одной или нескольких протеинкиназ AKT, тирозинкиназ, дополнительных серин/треонинкиназ, и/или киназ двойной специфичности, (ii) ослабления, снижения выраженности или устранения одного или нескольких симптомов определенного заболевания, патологического состояния или нарушения, или (iii) предотвращения или замедления проявления одного или нескольких симптомов определенного заболевания, патологического состояния или нарушения, описанного здесь. В случае ракового заболевания, эффективное количество лекарственного средства может снижать число раковых клеток; уменьшать размер опухоли; ингибировать (т.е. в некоторой степени замедлять, и предпочтительно останавливать) проникновение раковых клеток в периферические органы; ингибировать (т.е. в некоторой степени замедлять, и предпочтительно останавливать) метастазирование опухоли; подавлять, в некоторой степени, рост опухоли; и/или в некоторой степени снижать выраженность одного или нескольких симптомов, связанных с раковым заболеванием. Лекарственное средство может в некоторой степени предотвращать рост и/или уничтожать существующие раковые клетки, т.е. оно может обладать цитостатическими и/или цитотоксическими свойствами. При лечении рака эффективность может быть измерена, например, на основе оценки прогрессирования заболевания во времени (ТТР) и/или определения степени ответа (RR).
Предполагается, что "лечение" обозначает, по меньшей мере, облегчение состояния при заболевании у млекопитающего, такого как человек, которое зависит, по меньшей мере частично, от активности одной или нескольких протеинкиназ AKT, тирозинкиназ, дополнительных серинтреонинкиназ, и/или киназ двойной специфичности. Термины "лечить" и "лечение" относятся как к терапевтическому лечению, так и к профилактическим или превентивным мерам, целью которых является предотвращение или замедление (снижение) нежелательного физиологического изменения или нарушения. В рамках настоящего изобретения, полезные или желательные клинические результаты включают, но не ограничены перечисленными: снятие симптомов, снижение степени заболевания, стабилизацию (т.е. отсутствие ухудшений) состояния больного, задержку или замедление прогрессирования заболевания, улучшение или временное снижение выраженности болезненного состояния, и ремиссию (частичную или полную), определяемые или неопределяемые. "Лечение" также может обозначать продление продолжительности жизни по сравнению с ожидаемой продолжительностью жизни при отсутствии лечения. Нуждающиеся в лечении включают как уже имеющих патологическое состояние или расстройство, и тех, кто предрасположен к заболеванию, но у кого оно еще не диагностировано; модулирование и/или подавление патологического состояния. Термины "лечить" и "лечение" включают как превентивное, например, профилактическое, так и паллиативное лечение.
Термин "млекопитающее" в настоящем описании относится к теплокровному животному, имеющему, или находящемуся в группе риска возникновения описанного здесь заболевания, и включает, но не ограничен перечисленными: морских свинок, собаки, кошек, крыс, мышей, хомяков и приматов, включая человека.
"Химиотерапевтический агент" представляет собой химическое соединение, пригодное для лечения рака вне зависимости от механизма действия. Химиотерапевтические агенты включают соединения, используемые в "направленной терапии" и обычной химиотерапии.
Примеры химиотерапевтических агентов включают эрлотиниб (TARCEVA®, Genentech/OSI Pharm.), ботрезомиб (VELCADE®, Millennium Pharm.), фульвестрант (FASLODEX®, AstraZeneca), сутент (SU11248, Pfizer), летрозоль (FEMARA®, Novartis), иматиниба месилат (GLEEVEC®, Novartis), PTK787/ZK 222584 (Novartis), оксалиплатин (ELOXATIN®, Sanofi), 5-FU (5-флюороурацил), лейковорин, рапамицин (Sirolimus, RAPAMUNE®, Wyeth), лапатиниб (TYKERB®, GSK572016, Glaxo Smith Kline), лонафарниб (SCH 66336), сорафениб (BAY43-9006, Bayer Labs), иринотекан (CAMPTOSAR®, Pfizer) и гефитиниб (IRESSA®, AstraZeneca), AG1478, AG1571 (SU 5271; Sugen), алкилирующие агенты, такие как тиотепа и CYTOXAN® циклоспорамид; алкилсульфонаты, такие как бусульфан, импросульфан и пипосульфан; азиридины, такие как бензодопа, карбокон, метуредопа и уредопа; этиленимины и метиламеламины, включая альтретамин, триэтиленемеламин, триэтиленфосфорамид, триэтилентиофосфорамид и триметиломеламин; ацетогенины (особенно буллатацин и буллатицинон); камптотецин (включая синтетический аналог топотекан); бриостатин; каллистатин; СС-1065 (включая его синтетические аналоги адозелезин, карзелезин и бизелезин); криптофицины (в частности крисптофицин 1 и криптофицин 8); доластатин; дуокармицин (включая синтетические аналоги, KW-2189 и СВ1-ТМ1); элеутеробин; панкратистатин; саркодиктин;спонгистатин; азотистые иприты, такие как хлорамбуцил, хлорнафазин, хлорофосфамид, эстрамустин, ифостамид, мехлоретамин, мехлоретамин оксид гидрохлорид, мелфалан, новембицин, фенестерин, преднимустин, трофосфамид, иприт урацила; производные нитрозомочевины, такие как кармустин, хлорзотоцин, фотемустин, ломустин, нимустин и ранимустин; антибиотики, такие как антибиотики энедина (например, калихемицин, особенно калихемицин гамма и калихемицин омега (Angew Chem. Intl. Ed. Engl. (1994) 33: 183-186); динемицин, включая динемицин А; бифосфонаты, такие как хлодронат; эсперамицин; а также хромофор неокарциностатин и родственные хромофоры из хромопротеинов антибиотиков из ряда энедина), аклациномизины, актиномицин, аутрамицин, азасерин, блеомицины, кактиномицин, карабицин, карминомицин, карзинофилин, хромомицины, дактиномицин, даунорубицин, детоубицин, 6-диазо-5-оксо-L-норлейцин, АДРИАМИЦИН® (доксорубицин), морфолинодоксорубицин, цианоморфолинодоксорубицин, 2-пирролино-доксорубицин и дезоксидоксорубицин), эпирубицин, эзорубицин, идарубицин, марцелломицин, митомицины такие как митомицин С, микофенольная кислота, ногаламицин, оливомицины, пепломицин, порфиромицин, пиромицин, квеламицин, родорубицин, стрептонигрин, стрептозоцин, туберцидин, убенимекс, зиностатин, зорубицин; антиметаболиты, такие как метотрексат и 5-фторурацил (5-ФУ); аналоги фолиевой кислоты, такие как деноптерин, метотрексат, птероптерин, триметриксат; аналоги пуринов, такие как флударабин, 6-меркаптопурин, тиамиприн, тиогуанин; аналоги пиримидинов, такие как анцитабин, азацитидин, 6-азауридин, кармофур, цитарабин, дидезоксиуридин, доксифлуридин, эноцитабин, флоксуридин; андрогены, такие как калустерон, дромостанолон пропионат, эпитиостанол, мепитиостан, тестолактон; противонадпочечниковые средства, такие как аминоглютетимид, митотан, трилостан; пополнитель фолиевой кислоты, такой как флориновая кислота; ацеглатон; гликозид альдофосфамид; аминолевулиновая кислота; энилурацил; амсакрин; бестрабуцил; бисантрен, эдатраксат; дефофамин; демекольцин; диазиквон; эльфорнитин; эллиптина ацетат; эпотилон; этоглюцид; нитрат галлия; гидроксимочевина; лентинан; лонидаинин; майтансиноиды, такие как майтанзин и ансамитоцин; митагуазон; митоксантрон; мопиданмол; нитраэрин; пентостатин; фенамет; пирарубицин; лозоксантрон; подофиллиновая кислота; 2-этилгидразид; прокарбазин; PSK® комплекс полисахаридов (JHS Natural Products, Юджин, Орегон); разоксан; ризоксин; сизофиран; спирогерманиум; тенуазоновая кислота; триазиквон; 2,2',2"-трихлортриэтиламин; трихотецины (особенно токсин Т-2, верракурин А; роридин А и ангуидин); уретан; виндезин; дакарбазин; манномустин; митобронитол; митолактол; пипоброман; гацитозин; арабинозид ("Ara-С"); циклофосфамид; тиотепа; таксоиды, например TAXOL® (паклитаксель; Bristol-Myers Squibb Oncology, Princeton, N.J.), ABRAXENE™ (без кремофора); сформированные на альбумине образования наночастиц паклитакселя (American Pharmaceutical Partners, Schaumberg, Illinois), и TAXOTERE® (доксетаксель; Rhone-Poulenc Rorer, Antony, France); хлорамбуцил; GEMZAR® (гемцитабин); 6-тиогуанин; меркаптопурин; метотрексат; аналоги платины, такие как цисплатин и карбоплатин; винбластин; этопозид (VP-16); ифосфамид; митоксантрон; винкристин; NAVELBINE® (винорельбин); новантрон; тенипозид; эдатрексат; дауномицин; аминоптерин; капецитабин (XELODA®); ибандронат; СРТ-11; ингибитор топоизомеразы RFS 2000; дифторметилорнитин (DMFO); ретиноиды, такие как ретиноевая кислота; и фармацевтически приемлемые соли, кислоты и производные любого из вышеперечисленных соединений.
Также в определение "химиотерапевтический агент" включены: (i) антигормональные агенты, регулирующие или ингибирующие действие гормонов на опухоли, такие как антиэстрогены и избирательные модуляторы рецепторов эстрогена (SERMs), включая, например, тамоксифен (в т.ч. NOLVADEX®; цитрат тамоксифена), ралоксифен, дролоксифен, 4-гидрокситамоксифен, триоксифен, кеоксифен, LY 117018, онапристон и FARESTON® (цитрат торемифина); (ii) ингибиторы ароматазы, которые ингибируют фермент ароматазу, регулирующий выработку эстрогена в надпочечниках, такие как, например, 4(5)-имидазолы, аминоглютетимид, MEGASE® (ацетат мегестрола), AROMASIN® (эксеместан; Pfizer), форместаин, фадрозол, RIVISOR® (ворозол), FEMARA® (летрозол; Novartis), и ARIMIDEX® (анастрозол; AstraZeneca); (iii) антиандрогены, такие как флутамид, нилутамид, бикалутамид, лейпролид и госерелин; а также троксацитабин (аналог 1-3-диоксолан нуклеозид цитозина); (iv) ингибиторы протеинкиназы; (v) ингибиторы липидкиназы; (vi) антисмысловые олигонуклеотиды, в частности, ингибирующие экспрессию генов в сигнальных путях, связанных с аномальным делением клетки, такие как, например, РКС-альфа, Ralf and H-Ras; (vii) рибозимы, такие как ингибиторы экспрессии VEGF (например, ANGIOZYME®) и ингибиторы экспрессии HER2; (viii) вакцины, такие как вакцины генной терапии, например, ALLOVECTIN®, LEVVECTIN® и VAXID®; PROLEVKLIN® rIL-2; ингибитор топоизомеразы 1, такой как LURTOTECAN®; ABARELIX® rmRH; (ix) антиангиогенные агенты, такие как бевацизумаб (AVASTIN®, Genentech); и (х) фармацевтически приемлемые соли, кислоты и производные любого из вышеуказанных соединений.
Также в определение "химиотерапевтические агенты" включены терапевтические антитела, такие как алемтузумаб (Campath), бевацизумаб (AVASTIN®, Genentech); цетуксимаб (ERBITUX®, Imclone); панитумумаб (VECTIBIX®, Amgen), ритуксимаб (RITUXAN®, Genentech/Biogen Idee), пертузумаб (OMNITARG®, 2C4, Genentech), трастузумаб (HERCEPTIN®, Genentech), тоситумомаб (Bexxar, Corixia), и конъюгат антитела с лекарственным средством, гемтузумаб озогамицин (MYLOTARG®, Wyeth).
Гуманизированные моноклональные антитела, имеющие терапевтический потенциал в качестве химиотерапевтических агентов в комбинации с ингибиторами PI3K согласно настоящему изобретению, включают: алемтузумаб, аполизумаб, аселизумаб, атлизумаб, бапинеузумаб, бевацизумаб, биватузумаб мертанзин, кантузумаб мертанзин, цеделизумаб, цертолизумаб пеголь, цидфузитузумаб, цидтузумаб, даклизумаб, экулизумаб, эфализумаб, эпратузумаб, эрлизумаб, фелвизумаб, фонтолизумаб, гемтузумаб озогамицин, инотузумаб озогамицин, ипилимумаб, лабетузумаб, линтузумаб, матузумаб, меполизумаб, мотавизумаб, мотовизумаб, натализумаб, нимотузумаб, ноловизумаб, нумавизумаб, окрелизумаб, омализумаб, паливизумаб, пасколизумаб, пекфузитузумаб, пектузумаб, пертузумаб, пекселизумаб, раливизумаб, ранибизумаб, ресливизумаб, реслизумаб, ресивизумаб, ровелизумаб, руплизумаб, сибротузумаб, сиплизумаб, сонтузумаб, такатузумаб тетраксетан, тадокизумаб, тализумаб, тефибазумаб, токилизумаб, торализумаб, трастузумаб, тукотузумаб целмолейкин, тукуситузумаб, умавизумаб, уртоксазумаб и визилизумаб.
ИНГИБИТОРЫ AKT
Предложенные соединения формулы I применимы при ингибировании протеинкиназ AKT. Такие соединения можно применять в качестве терапевтического агента в лечении заболеваний, которые, можно лечить путем ингибирования сигнального пути протеинкиназы AKT, а также путей рецепторных тирозинкиназы и серин/треонинкиназы.
В частности, обнаружено, что соединения формулы I, содержащие 7-гидроксигруппу на циклопента[d]пиримидине, обладают, по меньшей мере, в 50 раз большей селективностью в отношении AKT по сравнению с протеинкиназой А (РКА). Например, по меньшей мере, в 100 раз, и в качестве еще одного примера, по меньшей мере, в 150 раз более селективны в отношении AKT по сравнению с РКА. Селективность превышающая селективность к РКА предпочтительна, поскольку РКА участвует во многих клеточных процессах, важных для нормального функционирования и физиологии многих типов клеток. Кроме того, предполагают, что ингибирование РКА не способствует антипролиферативным и проапоптотическим эффектам ингибирования AKT. Таким образом, ингибирование РКА может приводить к нежелательным явлениям, не ассоциированным с ингибированием AKT, без осуществления полезного модифицирования заболевания, характерного для ингибирования AKT.
Соединения формулы I, можно также применять в качестве ингибиторов тирозинкиназ, а также серин- и треонинкиназ, в дополнение к ингибированию AKT.
В целом, один аспект настоящего изобретения относится к соединениям формулы I:
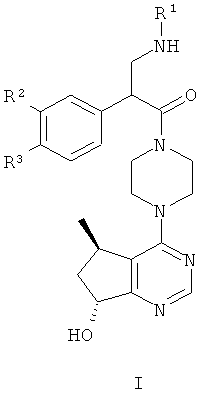
и их фармацевтически приемлемым солям, причем:
R1 представляет собой C1-C6алкил, содержащий один заместитель ОН;
R2 представляет собой водород или F; и
R3 представляет собой Cl или CF3.
Согласно определенным вариантам реализации R1 представляет собой C1-C6алкил,
содержащий один заместитель ОН.
Согласно определенным вариантам реализации R1 представляет собой C4алкил, содержащий один заместитель ОН.
Согласно определенным вариантам реализации R1 представляет собой -СН2С(CH3)2OН. Согласно определенным вариантам реализации R1 представляет собой -С(CH3)2CH2OH.
Согласно определенным вариантам реализации R2 представляет собой водород.
Согласно определенным вариантам реализации R2 представляет собой F.
Согласно определенным вариантам реализации R3 представляет собой Cl.
Согласно определенным вариантам реализации R3 представляет собой CF3.
Согласно определенным вариантам реализации R2 представляет собой водород, а R3 представляет собой Cl.
Согласно определенным вариантам реализации R2 представляет собой F, а R3 представляет собой CF3.
Согласно дополнительным вариантам реализации соединение формулы I имеет структуру формулы II:
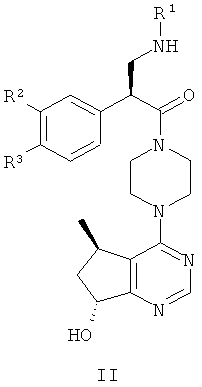
причем R1, R2 и R3 соответствуют определениям, данным в настоящей заявке.
Соединения формулы I, также включают и соли (в т.ч. фармацевтически приемлемые соли) таких соединений.
Фраза "фармацевтически приемлемые" указывает на то, что вещество или соединение химически и/или токсикологически совместимо с другими ингредиентами, входящими в состав, и/или с млекопитающим, которое лечат указанным средством.
Дополнительно, соединение согласно настоящему изобретению может образовывать соль. Примеры солей включают соли, получаемые путем осуществления взаимодействия соединений согласно настоящему изобретению с органической или неорганической кислотой, или неорганическим основанием; подобные соли включают, но не ограничены перечисленными: сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, фосфаты, моногидрогенфосфаты, дигидрогенфосфаты, метафосфаты, пирофосфаты, хлориды, бромиды, иодиды, ацетаты, пропионаты, деканоаты, каприлаты, акрилаты, формиаты, изобутираты, капроаты, гептаноаты, пропиолаты, оксалаты, малонаты, сукцинаты, субераты, себацаты, фумараты, малеаты, бутин-1,4-диоаты, гексин-1,6-диоаты, бензоаты, хлорбензоаты, метилбензоаты, динитробензоаты, гидроксибензоаты, метоксибензоаты, фталаты, сульфонаты, ксиленсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, γ-гидроксибутираты, гликоляты, тартраты, метансульфонаты, пропансульфонаты, нафтален-1-сульфонаты, нафтален-2-сульфонаты и манделаты. Так как одно соединение согласно настоящему изобретению может включать более чем одну кислую или щелочную группу, соединения согласно настоящему изобретению могут включать одинарные, двойные и тройные соли в одном соединении.
В некоторых вариантах выполнения изобретения, соль является "фармацевтически приемлемой солью", что включает, если не указано обратное, соли, сохраняющие биологическую эффективность соответствующей свободной кислоты или основания конкретного соединения, а указанные соли не являются биологически (или иначе) нежелательными.
Соединения формулы I также включают другие соли, которые не обязательно являются фармацевтически приемлемыми, и которые могут быть пригодны в качестве промежуточных веществ для приготовления и/или очистки соединений формулы I, и/или разделении энантиомеров соединений формулы I.
Настоящее изобретение также включает меченые изотопами соединения согласно настоящему изобретению, которые идентичны указанным здесь, за тем исключением, что один или несколько атомов заменены на атомы, имеющие атомную массу или массовое число, отличное от атомной массы или массового числа соответствующих атомов, которые обычно встречаются в природе. Все изотопы любого конкретного атома или элемента, а также их применение, соответственно считаются относящимися к соединениям согласно настоящему изобретению. Примеры изотопов, которые могут быть включены в соединения согласно настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, хлора и йода, такие как2H,3Н,11С,13С,14С,13N,15N,15O,17O,18О,32Р,33Р,35S,18F,36Cl,123I и125I. Конкретные меченые изотопами соединения согласно настоящему изобретению (например, меченные3H и14С) можно использовать при анализе распределения соединения и/или субстрата в тканях. Тритиевые (т.е.3H) изотопы и изотопы углерода-14 (т.е.14С) полезны благодаря легкости их приготовления и детектирования. Кроме того, замена атомов на более тяжелые изотопы, такие как дейтерий (т.е.2Н), может обеспечить определенные терапевтические преимущества, обусловленные большей метаболической стабильностью (т.е. увеличенным периодом полураспада, или более низкой необходимой дозировкой), и по этой причине замена может быть предпочтительной в некоторых ситуациях. Позитронно-активные изотопы, такие как15О,13N,11С и18F можно применять в исследованиях, осуществляемых при помощи позитронно-эмиссионной томографии (PET) для изучения заполнения рецепторов субстрата. Меченые изотопами соединения согласно настоящему изобретению могут быть приготовлены по существу при помощи процедур, аналогичных описанным в схемах и/или примерах, изложенных ниже, путем замены не меченого изотопом реагента на меченый изотопом реагент.
СИНТЕЗ СОЕДИНЕНИЙ ФОРМУЛЫ I
Соединения согласно настоящему изобретению могут быть синтезированы с использованием путей синтеза, которые включают процессы, аналогичные широко известным в области химии, в частности, в свете приведенного ниже описания. Исходные материалы обычно можно приобрести в коммерческих источниках, таких как Aldrich Chemicals (Milwaukee, WI), либо они могут быть легко приготовлены с использованием методов, хорошо известных специалисту (например, приготовлены методами, сущность которых описана в Louis F. Fieser, Mary Fieser, Reagents for Organic Synthesis, v.1-19, Wiley, N.Y. (1967-1999 ed.), или Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, включая дополнительные материалы).
Соединения формулы I, можно готовить отдельно или в качестве библиотеки соединений, содержащей, по меньшей мере, 2, например, 5-1000 соединений, или 10-100 соединений. Библиотеки соединений формулы I можно приготовить при помощи комбинаторного подхода «разделения и смешивания» или при помощи множественного параллельного синтеза с применением либо жидкофазной, либо твердофазной химии, при помощи процедур, известных специалистам в данной области техники. Так, в соответствии с дополнительным аспектом изобретения предложена библиотека соединений, включающая, по меньшей мере, 2 соединения формулы I, или их соли.
При приготовлении соединений формулы I, может быть необходима защита различных функциональных групп (например, первичных или вторичных аминов, и т.д.) промежуточных веществ. Необходимость в подобной защите может варьировать в зависимости от природы функциональной группы и условий методов приготовления. Подходящие группы-аминопротекторы (NH-Pg) включают ацетил, трифторацетил, t-бутоксикарбонил (ВОС), бензилоксикарбонил (CBz) и 9-фторэнилметиленоксикарбонил (Fmoc). Необходимость в подобной защите может быть легко определена специалистом. Общее описание защитных групп и их использования, можно найти в Т.W.Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
МЕТОДЫ РАЗДЕЛЕНИЯ
При использовании любого из методов синтеза для приготовления соединений формулы I может быть желательно отделить продукты реакции друг от друга и/или от исходных материалов. Желаемые продукты каждого этапа или ряда этапов разделяют и/или очищают до желаемой степени гомогенности при помощи широко известных в данной области приемов. Обычно, подобное разделение включает в себя многофазное экстрагирование, кристаллизацию из раствора или смеси растворов, дистилляцию, сублимацию или хроматографию. Хроматография может включать любое количество методов, в т.ч., например: хроматографию с обращенной фазой и нормальной фазой; эксклюзионную хроматографию; ионообменную хроматографию; методы и устройства для жидкостной хроматографии высокого, среднего и низкого давления; малообъемную аналитическую хроматографию; псевдоподвижный слой ("SMB") и препаративную тонкослойную или толстослойную хроматографию, а также методы малообъемной тонкослойной и флэш-хроматографии.
Другая группа способов разделения включает обработку реакционной смеси реагентом, выбранным с целью связывания желаемого продукта, или обеспечения возможности разделения непрореагировавшего исходного материала, побочного продукта реакции, и т.п. иным образом. Подобные реагенты включают адсорбенты или абсорбенты, такие как активированный уголь, молекулярные сита, ионообменные вещества и т.п. Кроме того, реагенты могут представлять собой кислоты в случае, если материал является основанием, основания в случае, если материал является кислотой, связывающие реагенты, такие как антитела, связывающие белки, отдельные хелаторы, такие как краун-эфиры, реагенты выделения ионов жидкость-жидкость ("LIX") и т.п.
Выбор подходящих способов разделения зависит от природы используемых материалов. Например, температура кипения и молекулярный вес имеют значение при дистилляции и сублимации, наличие или отсутствие противоположных функциональных групп имеют значение при хроматографии, стабильность материалов в кислой и щелочной среде имеют значение при многофазном экстрагировании, и т.п. Специалисту будет нетрудно выбрать способы, с помощью которых можно с наибольшей вероятностью достичь желаемого разделения.
Смеси диастереомеров могут быть разделены на отдельные диастереомеры на основании их базовых физико-химических различий методами, хорошо известными специалисту, такими как хроматография и/или фракционная кристаллизация. Энантиомеры могут быть разделены путем превращения смеси энантиомеров в смесь диастереоизомеров путем реакции с подходящим оптически активным соединением (например, хиральным вспомогательным агентом, таким как хиральный спирт или хлорид кислоты Мошера), разделения диастереомеров и превращения (например, гидролиза) отдельных диастереоизомеров в соответствующие чистые энантиомеры. Также, некоторые соединения согласно настоящему изобретению могут быть атропоизомерами (например, замещенные биарилы), и считаются частью настоящего изобретения. Энантиомеры также могут быть разделены при помощи колонки хиральной ВЭЖХ.
Отдельный стереоизомер, например, энантиомер, по существу свободный от своего стереоизомера, может быть получен путем разделения рацемической смеси с использованием такого метода, как образование диастереомеров при использовании оптически активных разделяющих агентов (Eliel, E. and Wilen, S. "Stereochemistry of Organic Compounds," John Wiley & Sons, Inc., New York, 1994; Lochmuller, C.H., J. Chromatogr., (1975) 113(3): 283-302). Рацемические смеси хиральных соединений согласно настоящему изобретению могут быть разделены и выделены любым подходящим методом, включая: (1) образование ионных диастереоизомерных солей с хиральными соединениями, и разделение путем фракционной кристаллизации или иных методов, (2) образование диастереоизомерных соединений с хиральными реагентами, используемьми для получения производных, разделение диастереомеров, и превращение в чистые стереоизомеры, и (3) разделение по существу чистых или обогащенных стереоизомеров напрямую в хиральных условиях. См.: "Drug Stereochemistry, Analytical Methods and Pharmacology," Irving W. Wainer, Ed., Marcel Dekker, Inc., New York (1993).
При использовании метода (1), диастереоизомерные соли могут быть получены путем реакции энантиомерно чистых хиральных оснований, таких как бруцин, хинин, эфедрин, стрихнин, α-метил-β-фенилэтиламин (амфетамин), и т.п., с асимметричными соединениями, работающими как кислоты, такими как карбоновая кислота и сульфоновая кислота. Диастереоизомерные соли могут быть разделены путем фракционной кристаллизации или ионной хроматографии. При разделении оптических изомеров аминосоединений, добавление хиральных карбоновых или сульфоновых кислот, таких как камфорсульфоновая кислота, винная кислота, миндальная кислота, или молочная кислота, может привести к образованию диастереоизомерных солей.
В другом варианте, согласно методу (2), подвергаемый разделению субстрат вводят в реакцию с одним энантиомером хирального соединения с целью образования пары диастереоизомеров (Е. and Wilen, S. "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., 1994, p.322). Диастереоизомерные соединения могут быть получены путем реакции асимметричных соединений с энантиомерно чистыми хиральными реагентами, используемыми для получения производных, такими как производные ментила, с последующим разделением диастереомеров, и гидролизом с получением чистого или обогащенного энантиомера. Метод определения оптической чистоты включает в себя приготовление сложных хиральных эфиров, таких как сложный эфир ментила, например, (-)ментилхлорформиат, в присутствии основания, или сложного эфира Мошера, α-метокси-α-(трифторметил)фенилацетат (Jacob III. J. Org. Chem., (1982) 47: 4165) рацемической смеси, и анализ спектра1H ЯМР на наличие двух атропизомерных энантиомеров или диастереомеров. Стабильные диастереомеры атропизомерных соединений могут быть разделены и выделены при помощи хроматографии с обращенной фазой и нормальной фазой, после применения методов разделения атропизомерных нафтилизохинолинов (WO 96/15111). При использовании метода (3), рацемическая смесь двух энантиомеров может быть разделена при помощи хроматографии с использованием хиральной неподвижной фазы ("Chiral Liquid Chromatography" (1989) W.J.Lough, Ed., Chapman and Hall, New York; Okamoto, J. of Chromatogr., (1990) 513: 375-37S). Обогащенные или очищенные энантиомеры могут быть разделены с помощью методов, используемых для разделения других хиральных молекул с асимметричными атомами углерода, например, оптическим вращением или круговым дихроизмом.
СПОСОБЫ ЛЕЧЕНИЯ СОЕДИНЕНИЯМИ ФОРМУЛЫ I
Соединения, предложенные в настоящем изобретении, можно применять в качестве профилактических или терапевтических агентов для лечения заболевания или нарушений, опосредуемых модуляцией или регуляцией протеинкиназ AKT, тирозинкиназ, дополнительных серин/треониновых киназ и/или киназ с двойной специфичностью. Опосредуемые протеинкиназой AKT патологические состояния, которые можно лечить в соответствии со способами согласно изобретению, включают без ограничения воспалительные, гиперпролиферативные, сердечнососудистые, нейродегенеративные, гинекологические и дерматологические заболевания и нарушения.
В одном варианте осуществления, указанную фармацевтическую композицию применяют для лечения гиперпролиферативных расстройств, включая раковые заболевания следующих категорий: (1) сердца: саркома (ангиосаркома, фибросаркома, рабдомиосаркома, липосаркома), миксома, рабдомиома, фиброма, липома и тератома; (2) легких: бронхогенная карцинома (плоскоклеточная, недифференцированная мелкоклеточная, недифференцированная крупноклеточная, аденокарцинома), альвеолярная (бронхиолярная) карцинома, бронхиальная аденома, саркома, лимфома, хондроматозная гамартома, мезотелиома, немелкоклеточный рак легких, мелкоклеточный рак легких; (3) желудочно-кишечныого тракта: рак пищевода (плоскоклеточная карцинома, аденокарцинома, лейомиосаркома, лимфома), желудка (карцинома, лимфома, лейомиосаркома), поджелудочной железы (протоковая аденокарцинома, инсулинома, глюкагонома, гастринома, карциноидные опухоли, випома), тонкого кишечника (аденокарцинома, лимфома, карциноидные опухоли, саркома Карпози, лейомиома, гемангиома, липома, нейрофиброма, фиброма), толстого кишечника (аденокарцинома, тубулярная аденома, ворсинчатая аденома, гамартома, лейомиома); (4) мочеполового тракта: рак почек (аденокарцинома, опухоль Вильмса [нефробластома], лимфома, лейкемия), мочевого пузыря и уретры (плоскоклеточная карцинома, переходно-клеточная карцинома, аденокарцинома), простаты (аденокарцинома, саркома), яичек (семинома, тератома, эмбриональная карцинома, тератокарцинома, хориокарцинома, саркома, карцинома гиюсных клеток, фиброма, фиброаденома, аденоматоидные опухоли, липома); (5) печени: гепатома (карцинома клеток печени), холангиокарцинома, гепатобластома, ангиосаркома, аденома клеток печени, гемангиома; (6) скелета: остеогенная саркома (остеосаркома), фибросаркома, злокачественная фиброзная гистиоцитома, хондросаркома, саркома Юинга, злокачественная лимфома (ретикулоклеточная саркома), множественная миелома, злокачественная гигантоклеточная хордома, остеохронфрома (костнохрящевые экзостозы), доброкачественная хондрома, хондробластома, хондромиксофиброма, остеоид-остеома и гигантоклеточные опухоли; (7) нервной системы: рак черепа (остеома, гемангиома, гранулома, ксантома, деформирующий остоз), мягких мозговых оболочек (менингиома, менингиосаркома, глиоматоз), мозга (астроцитома, медуллобластома, глиома, эпендимома, герминома [пинеалома], мультиформная глиобластома, олигодендроглиома, шваннома, ретинобластома, врожденные опухоли), спинного мозга (нейрофиброма, менингиома, глиома, саркома); (S) гинекологические: рак матки (эндометриальная карцинома), шейки матки (карцинома шейки матки, предопухолевая дисплазия шейки матки), яичников (карцинома яичников [серозная цистаденокарцинома, муцинозная цистаденокарцинома, неклассифицированная карцинома], опухоли зернистых клеток, опухоли клеток Сертоли-Лейдига, дисгерминома, злокачественная тератома), вульвы (плоскоклеточная карцинома, интраэпителиальная карцинома, аденокарцинома, фибросаркома, меланома), влагалища (светлоклеточная карцинома, плоскоклеточная карцинома, ботриоидная саркома (эмбриональная рабдомиосаркома), фаллопиевых труб (карцинома); (9) гематологические: рак крови (миелоидная лейкемия [острая и хроническая], острая лимфобластическая лейкемия, хронический лимфоцитарный лейкоз, миелопролиферативный синдром, множественная миелома, миелодиспластический синдром), болезнь Ходжкина, не-ходжкинская лимфома [злокачественная лимфома]; (10) кожные: меланома в поздней стадии, злокачественная меланома, карцинома базальных клеток, плоскоклеточная карцинома, саркома Карпози, диспластия родинок, липома, ангиома, дерматофиброма, келоиды, псориаз; (11) надпочечников: нейробластома; (12) молочной железы: метастазы рака молочной железы; аденокарцинома молочной железы; (13) толстой кишки; (14) ротовой полости; (15) лейкоз ворсистых клеток; (16) головы и шеи; (17) и прочие, включая резистентное метастазирование; саркому Карпози; синдром Баннаян-Зонана; и болезнь Каудена или болезнь Лермитта-Дуклоса, а также прочие гиперпролиферативные нарушения.
Соединения и способы согласно настоящему изобретению, также могут быть использованы при лечении заболеваний и патологических состояний, таких как ревматоидный артрит, остеоартрит, болезнь Хрона, ангиофиброма, глазные заболевания (например, васкуляризация сетчатки, диабетическая ретинопатия, возрастная дегенерация желтого пятна, дегенерация желтого пятна и т.д.), множественный склероз, ожирение, болезнь Альцгеймера, рестеноз, аутоиммунные заболевания, аллергия, астма, эндометриоз, атеросклероз, стеноз венозного трансплантата, перианостоматический стеноз протетического трансплантата, гиперплазия простаты, хронические обструктивные легочные заболевания, псориаз, ингибирование нейрологических повреждений из-за восстановления тканей, формирование рубцовой ткани (может также помогать при заживлении ран), множественный склероз, воспалительные заболевания кишечника, инфекции, в частности, бактериальные, вирусные, ретровирусные или паразитарные инфекции (благодаря усилению апоптоза), заболевания легких, новообразования, болезнь Паркинсона, отторжение трансплантата (действует в качестве иммунодепрессанта), септический шок, и т.д.
Соответственно, еще один аспект настоящего изобретения относится к способу лечения заболеваний или патологических состояний, опосредуемых протеинкиназами AKT, у млекопитающих, включающий введение указанному млекопитающему одного или более соединений формулы I, или их фармацевтически приемлемой соли или пролекарства в количестве, эффективном при лечении или профилактике указанного нарушения.
Количество соединения формулы I, которое будет соответствовать подобному количеству, варьируют в зависимости от таких факторов, как конкретное соединение, тяжесть заболевания и его опасность, и характеристики (например, вес) млекопитающего, нуждающегося в лечении, но тем не менее, может быть установлено специалистом в обычном порядке.
Настоящее изобретение также обеспечивает соединения формулы I для применения в лечении патологических состояний, опосредуемых протеинкиназами AKT.
Дополнительным аспектом изобретения является применение соединений формулы I, для приготовления лекарственного препарата для терапии, такой как лечение или профилактика патологических состояний, опосредованных протеинкиназами AKT.
КОМБИНИРОВАННАЯ ТЕРАПИЯ
Соединения согласно настоящему изобретению, может быть использовано в комбинации с одним или несколькими дополнительными лекарственными средствами, как описано ниже. Дозировка второго лекарственного средства может быть соответствующим образом выбрана на основе дозировки, используемой в клинике. Соотношение между соединением согласно настоящему изобретению и вторым лекарственным средством может быть определено в соответствии с субъектом, которому вводят средства, путем введения средства, целевым заболеванием, клиническим состоянием, характером комбинации, и прочими факторами. В случае, если субъектом, которому вводят средство, является человек, второе лекарственное средство может быть использовано, например, в количестве от 0,01 до 100 частей по массе на часть к массе соединения согласно настоящему изобретению.
Второе соединение комбинированного фармацевтического состава или режим дозировки предпочтительно оказывает действие, дополняющее соединение согласно настоящему изобретению, таким образом, что соединения не оказывают друг на друга отрицательного влияния. Подобные лекарственные средства, соответственно, присутствуют в количествах, являющихся эффективными для достижения поставленной цели. Соответственно, согласно еще одному аспекту настоящего изобретения предложена композиция, включающая соединение согласно настоящему изобретению в комбинации со вторым лекарственным средством, описанным в настоящем тексте.
Соединение согласно настоящему изобретению и дополнительный фармацевтически активный агент или агенты можно вводить вместе, в виде единой фармацевтической композиции, или отдельно, и в случае, если агенты вводят раздельно, введение можно осуществлять одновременно или последовательно в любом порядке. Подобное последовательное введение можно осуществлять на коротком или длительном промежутке времени. Количество соединения согласно настоящему изобретению и второй агент или агенты, и относительное время введения выбирают таким образом, чтобы достичь желаемого комбинированного терапевтического эффекта.
Комбинированная терапия может обеспечить эффект "синергии" и оказаться "синеретической", т.е. эффект, достигнутый при совместном использовании активных ингредиентов, выше, чем сумма эффектов, получаемых при использовании данных соединений по отдельности. Синергетический эффект может быть получен, если активные ингредиенты: (1) комбинируют, и вводят или доставляют одновременно в виде комбинированной лекарственной формы, (2) доставляют поочередно или параллельно в виде разных составов; или (3) при ином режиме введения. При поочередной доставке, синергетический эффект может быть получен, если соединения вводят или доставляют в определенной последовательности, т.е. путем разных инъекций в отдельных шприцах. В целом, при чередующейся терапии, эффективные дозы каждого из активных ингредиентов вводят в определенной последовательности, т.е. сериями, а при комбинированной терапии, эффективные дозы двух или более активных ингредиентов вводят вместе.
ПУТИ ВВЕДЕНИЯ
Соединения согласно изобретению, можно вводить любым путем, подходящим для лечения конкретного патологического состояния. Подходящие пути включают оральный, парентеральный (включая подкожный, внутримышечный, внутривенный, внутриартериальный, внутрикожный, интратекальный и эпидуральный), трансдермальный, ректальный, назальный, локальный (включая трансбуккальный и подъязычный), вагинальный, внутрибрюшинный, внутрилегочный и интраназальный. Нужно отметить, что предпочтительный способ может варьировать в зависимости, например, от состояния реципиента. Если соединение вводят перорально, оно может быть изготовлено в виде пилюли, капсулы, таблетки, и т.д. с фармацевтически приемлемым носителем или наполнителем. Если соединение вводят парентерально, оно может быть выполнено в фармацевтически приемлемом парентеральном носителе, и в виде состава для инъекций в лекарственной форме, как подробно описано ниже.
ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ (ЛЕКАРСТВЕННЫЕ ФОРМЫ)
Для применения соединения согласно настоящему изобретению для терапевтического лечения (включая профилактическое лечение) млекопитающих, включая человека, оно обычно выполнено в соответствии со стандартной фармацевтической практикой в форме фармацевтической композиции. Согласно данному аспекту настоящего изобретения, предложена фармацевтическая композиция, которая включает соединение согласно настоящему изобретению. В определенных вариантах осуществления фармацевтическая композиция включает соединение формулы I совместно с фармацевтически приемлемым разбавителем или носителем.
Фармацевтические композиции согласно настоящему изобретению изготавливают, дозируют и вводят согласно соответствующей медицинской практике в отношении количества, концентрации, режима приема, курса лечения, носителя и пути введения. Факторы, которые необходимо учитывать в данном контексте, включают: конкретное нарушение, которое лечат, конкретное млекопитающее, которое лечат, клиническое состояние конкретного пациента, причину нарушения, место введения агента, путь введения, режим введения, и прочие факторы, известные медицинским работникам. Терапевтически эффективное количество вводимого соединения зависит от указанных факторов, и является минимальным количеством, необходимым для предотвращения, снижения выраженности или лечения нарушения. Соединение согласно настоящему изобретению обычно выполнено в виде дозированной лекарственной формы, чтобы обеспечить легко контролируемое дозированное введение лекарственного средства, и приемлемость предписанного режима приема для пациента.
Композиция согласно настоящему изобретению предпочтительно стерильна. В частности, средства, вводимые in vivo, должны быть стерильными. Подобная стерилизация легко достигается, например, путем фильтрации через стерильные фильтрационные мембраны. Соединение обычно можно хранить в твердом виде, в виде лиофилизированного состава или в виде водного раствора.
Фармацевтические композиции, содержащие соединения согласно настоящему изобретению могут быть приготовлены с учетом различных путей и типов введения. Например, соединение согласно настоящему изобретению, имеющее достаточную степень чистоты, может по желанию быть смешано с фармацевтически приемлемыми разбавителями, носителями, наполнителями или стабилизаторами (Remington's Pharmaceutical Sciences (1980) 16е издание, Osol, A. Ed.), приготовлено в форме лиофилизированной композиции, молотого порошка или водного раствора. Состав может быть приготовлен путем смешивания при комнатной температуре и подходящем уровне рН, а также при желаемой степени чистоты, с физиологически приемлемыми носителями, т.е. носителями, в выбранных дозах и концентрациях, не являющихся токсичными для реципиента. Уровень рН композиции зависит главным образом от конкретного способа применения и концентрации соединения, но может варьировать от примерно 3 до примерно 8. Состав в ацетатном буфере при рН 5 является подходящим вариантом осуществления. Композиции могут быть приготовлены с использованием известных процедур растворения и смешивания. Например, партию лекарственного вещества (т.е. соединения согласно настоящему изобретению либо стабилизированной формы соединения (например, комплекс с производным циклодекстрина или иным известным комплексообразующим агентом)) растворяют в подходящем растворителе в присутствии одного или нескольких наполнителей.
Конкретный используемый носитель, разбавитель или наполнитель зависит от средств и цели, для достижения которой используется соединение согласно настоящему изобретению. Растворители обычно выбирают на основе растворителей, которые специалисты считают безопасными (GRAS) для введения млекопитающему. В целом, безопасными растворителями называют нетоксичные водные растворители, такие как вода и прочие нетоксичные растворители, растворимые в воде или смешивающиеся с водой. Подходящие водные растворители включают воду, этанол, пропиленгликоль, полиэтиленгликоли (например, PEG 400, PEG 300), и т.д., а также их смеси. Приемлемые разбавители, носители, наполнители и стабилизаторы являются нетоксичными для реципиентов в используемых дозах и концентрациях, и включают буферы, такие как фосфат, цитрат и прочие органические кислоты; антиоксиданты, включая аскорбиновую кислоту и метионин; консерванты (такие как хлорид октадецилдиметилбензиламмония; хлорид гексаметония; хлорид бензалкония, хлорид бензетония; феноловый, бутиловый или бензиловый спирт; парабены алкилов, такие как парабен метила или пропила; катехол; резорцин; циклогексанол; 3-пентанол; и m-крезол); полипептиды низкой молекулярной массы (менее примерно 10 остатков); белки, такие как серумальбумин, желатин, или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; хелирующие агенты, такие как ЭДТА; сахара, такие как сахароза, маннитол, трегалоза или сорбитол; солеформирующие противоионы, такие как натрий; комплексы металлов (например, цинкопротеиновые комплексы); и/или неионные сурфактанты, такие как TWEENE™ PLURONICS™, или полиэтиленгликоль (PEG). Композиции могут также включать один или несколько стабилизирующих агентов, сурфактантов, смачивающих агентов, смазывающих агентов, эмульгаторов, суспендирующих агентов, консервантов, антиоксидантов, кроющих агентов, глидантов, технологических добавок, красителей, подсластителей, ароматизирующих агентов, вкусовых добавок и прочих известных добавок для удобной формы композиции (т.е. соединения согласно настоящему изобретению или фармацевтической композиции на его основе), или помощи при производстве фармацевтического продукта (т.е. лекарственного средства). Активные фармацевтические ингредиенты также могут быть заключены в микрокапсулы, приготовленные, например, методом коацервации или путем межфазной полимеризации, например, гидроксиметилцеллюлозы или желатиновых микрокапсул, и микрокапсул из поли-(метилметакрилата) соответственно, в коллоидных системах доставки (например, липосомах, альбуминовых микросферах, микроэмульсиях, наночастицах и нанокапсулах) или в макроэмульсиях. Подобные технологии описаны в Remington's Pharmaceutical Sciences 16th edition, Osol, A. Ed. (1980). "Липосома" представляет собой маленький пузырек, состоящий из различных типов липидов, фосфолипидов и/или поверхностно-активного вещества, который можно использовать для доставки лекарственного средства (такого, как соединение формулы I и, возможно, дополнительный терапевтический агент) в организм млекопитающего. Компоненты липосомы обычно имеют двухслойную структуру, сходную с организацией липидов в биологических мембранах.
Из соединений согласно настоящему изобретению могут быть приготовлены формы замедленного высвобождения. Подходящие примеры форм замедленного высвобождения включают полупроницаемые матрицы из твердых гидрофобных полимеров, содержащие соединение формулы I, где указанные матрицы выполнены в виде изделия, имеющего определенную форму, например, пленки или микрокапсулы. Примеры материалов матриц замедленного высвобождения включают полиэфиры, гидрогели (например, поли(2-гидроксиэтилметакрилат) или поливиниловый спирт, полилактиды (US 3,773,919), сополимеры L-глутаминовой кислоты и гаммаэтил-L-глутамат, неразлагающийся этиленвинилацетат, разлагающиеся сополимеры молочной кислоты-гликолевой кислоты, такие как LUPRON DEPOT™ (инъецируемые микросферы, выполненные из сополимера молочной кислоты-гликолевой кислоты и лейпролид ацетата), и поли-D-(-)-3-гидроксимасляная кислота.
Фармацевтические композиции, содержащие соединения согласно настоящему изобретению, могут быть выполнены в форме стерильного инъецируемого состава, такого как стерильная суспензия для инъекций в воде или масле. Указанная суспензия может быть изготовлена согласно известным способам с использованием подходящих диспергирующих или смачивающих агентов, а также суспендирующих агентов, которые были указаны выше. Стерильный состав для инъекций может также представлять собой стерильный раствор для инъекций в нетоксичном разбавителе или растворителе, пригодном для парентерального введения, такой как раствор в 1,3-бутандиоле, или может быть приготовлен в виде лиофилизированного порошка. Некоторые из приемлемых носителей и растворителей, которые могут быть использованы для указанной цели: вода, раствор Рингера и изотоничный раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды могут быть использованы стерильные нелетучие масла. С этой целью может быть использовано любое легкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, жирные кислоты, такие как олеиновая кислота, также могут быть использованы в приготовлении составов для инъекций.
Композиции, подходящие для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостатики, и растворенные вещества, которые обеспечивают изотоничность состава с кровью желаемого реципиента; а также водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители.
Композиции согласно настоящему изобретению могут также быть выполнены в форме, подходящей для перорального введения (например, в виде таблеток, пастилок, твердых или мягких капсул, водных или масляных суспензий, эмульсий, дисперсных порошков или гранул, сиропов или эликсиров), для наружного применения (например, в виде кремов, мазей, гелей, либо водных или масляных растворов или суспензий), для введения путем ингаляции (например, в виде пылеобразного порошка или жидкого аэрозоля), или для введения путем инсуффляции (например, в виде пылеобразного порошка).
Подходящие фармацевтически приемлемые наполнители для композиции в форме таблетки включают, например, инертные разбавители, такие как лактоза, карбонат натрия, фосфат кальция или карбонат кальция, гранулирующие вещества и дезинтегрирующие вещества, такие как кукурузный крахмал или альгиновая кислота; связывающие агенты, такие как крахмал; смазывающие агенты, такие как стеарат магния, стеариновая кислота, или тальк; консерванты, такие как этил или пропил p-гидроксибензоат, и антиоксиданты, такие как аскорбиновая кислота. Составы в форме таблеток могут быть глазированными или неглазированными, либо, для изменения скорости их распада и последующего всасывания активного ингредиента в желудочно-кишечном тракте, или для повышения их стабильности и/или улучшения внешнего вида; в любом случае могут быть использованы глазирующие агенты и процедуры, известные науке.
Композиции для перорального введения могут быть выполнены в форме твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, либо в виде мягких желатиновых капсул, в которых активный ингредиент смешан с водой или маслом, таким как арахисовое масло, жидкий парафин или оливковое масло.
Водные суспензии обычно содержат активный ингредиент в виде тонкодисперсного порошка вместе с одним или несколькими суспендирующими агентами, такими как натрий-карбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и гуммиарабик; диспергирующими или смачивающими агентами, такими как лецитин или продукты конденсации оксида алкилена с жирными кислотами (например, полиоксетилен стеарат), или продукты конденсации оксида этилена с длинноцепочечными алифатическими спиртами, например, гептадекаэтиленоксикетанол, или продукты конденсации оксида этилена с неполными сложными эфирами, производными жирных кислот и гекситола, такие как полиоксиэтиленсорбитолмоноолеат, или продукты конденсации оксида этилена с неполными сложными эфирами, производными жирных кислот и ангидридов гекситола, например, полиэтиленсорбитанмоноолеат. Водные суспензии могут также содержать один или несколько консервантов (таких как этил или пропил р-гидроксибензоат), антиоксидантов (таких как аскорбиновая кислота), красителей, вкусовых добавок, и/или подсластителей (таких как сахароза, сахарин или аспартам).
Масляные суспензии могут быть приготовлены путем суспендирования активного ингредиента в растительном масле (таком как арахисовое масло, оливковое масло, кунжутное масло или кокосовое масло) или в минеральном масле (таком, как жидкий парафин). Масляные суспензии также могут содержать загуститель, такой как пчелиный воск, твердый парафин или цетиловый спирт. Подсластители, такие как указаны выше, и вкусовые добавки могут быть добавлены для обеспечения приятного вкуса композиции для перорального введения. Указанные композиции можно консервировать путем добавления антиоксиданта, такого как аскорбиновая кислота.
Сыпучие порошки и гранулы, подходящие для приготовления жидкой суспензии путем добавления воды, обычно содержат активный ингредиент вместе с диспергирующим или смачивающим агентом, суспендирующим агентом и одним или более консервантами. Примеры подходящих диспергирующих или смачивающих агентов приведены выше. Также могут присутствовать дополнительные наполнители, такие как подсластители, вкусовые добавки и красители.
Фармацевтические композиции согласно настоящему изобретению могут также иметь форму эмульсии типа "масло в воде". Масляная фаза может быть растительным маслом, таким как арахисовое масло или оливковое масло, или минеральным маслом, таким как жидкий парафин, или смесью любых вышеназванных масел. Подходящими эмульгирующими агентами могут быть, например, природные камеди, такие как гуммиарабик или трагакантовая камедь, встречающиеся в природе фосфатиды, такие как соя, лецитин, сложные эфиры или неполные сложные эфиры, производные жирных кислот и ангидридов гекситола (например, сорбитанмоноолеат), и продукты конденсации указанных неполных сложных эфиров с оксидом этилена, такие как полиоксиэтиленсорбитанмоноолеат. Эмульсии также могут содержать подсластители, вкусовые добавки и консерванты.
Сиропы и эликсиры могут быть приготовлены с использованием подсластителей, таких как глицерол, пропиленгликоль, сорбитол, аспартам или сахароза, и также могут содержать средство, смягчающие раздражение, консервант, вкусовую добавку и/или краситель.
Композиции в форме суппозитория могут быть приготовлены путем смешивания активного ингредиента с подходящим нераздражающим наполнителями, находящимся в твердом состоянии при комнатной температуре, и в жидком состоянии при температуре прямой кишки, благодаря чему он будет плавиться в прямой кишке, выделяя лекарственное средство. Подходящие эксципиенты включают, например, какао-масло и полиэтиленгликоли. Композиции, подходящие для вагинального введения, могут иметь вид пессария, тампона, крема, геля, пасты, пены или спрея, содержащего в дополнение к активному ингредиенту носители, которые в данной области известны как пригодные.
Композиции для наружного применения, такие как кремы, мази, гели и жидкие или масляные растворы или суспензии, могут в целом быть получены путем приготовления активного ингредиента в традиционной, пригодной для наружного применения среде или разбавителе, с использованием хорошо известных процедур.
Композиции для трансдермального введения могут быть выполнены в форме трансдермальных кожных пластырей, которые хорошо известны специалисту.
Композиции, подходящие для внутрилегочного или назального введения, имеют размер частиц, например, в пределах от 0,1 до 500 микрон (включая размеры частиц в пределах от 0,1 до 500 микрон с шагом в, например, 0,5, 1, 30 микрон, 35 микрон, и т.д.), который вводится путем частого вдыхания через носовой ход, или путем вдыхания через рот для доставки в альвеолярные мешочки. Подходящие композиции включают жидкие или масляные растворы активного ингредиента. Композиции, подходящие для аэрозольного введения или введения в виде сухого порошка, могут быть приготовлены по известным способам, и могут доставляться вместе с иными терапевтическими агентами, такими как соединения, до этого использовавшиеся в ходе лечения или профилактики расстройств, как описано ниже.
Для использования фармацевтическая композиция может быть упакована различными способами, в зависимости от пути введения композиции. Например, распространяемое изделие может включать контейнер, в котором расположена фармацевтическая композиция в соответствующей форме. Подходящие контейнеры общеизвестны, и включают материалы, такие как бутылки (пластиковые и стеклянные), саше-пакеты, ампулы, пластиковые пакеты, металлические цилиндры, и т.п. Контейнер также может включать защищенные от неумелого использования устройства для предотвращения неосторожного обращения с содержимым упаковки. Кроме того, на контейнере размещен ярлык, на котором описано содержимое контейнера. Ярлык может также содержать соответствующие предупреждения. Композиции могут также быть упакованы в однодозовые или многодозовые контейнеры, например, герметичные ампулы и флаконы, и может храниться в лиофилизированном (сублимированном) состоянии, при котором для осуществления инъекции требуется лишь добавление стерильного жидкого носителя, например воды, непосредственно перед использованием. Изготавливаемые для немедленного приема инъекционные растворы и суспензии приготавливаются из стерильных порошков, гранул и таблеток любого описанного выше типа. Предпочтительные лекарственные формы представляют собой лекарственные формы, содержащие ежедневную дозу или ежедневную долю дозы, как описано выше, или подходящую часть дозы активного ингредиента.
Изобретение также предусматривает ветеринарные композиции, включающие по меньшей мере один активный ингредиент, определенный выше, в комбинации с применяемым в ветеринарии носителем. Ветеринарные носители представляют собой материалы, используемые с целью введения лекарственного средства, и могут быть твердыми, жидкими или газообразными материалами, которые в остальных случаях являются инертными или приемлемыми в ветеринарной науке, а также совместимы с активным ингредиентом. Указанные ветеринарные композиции могут быть введены парентерально, орально, или любым другим желаемым способом.
Количество соединения согласно настоящему изобретению, комбинируемого с одним или несколькими эксципиентами с целью приготовления одной дозы лекарственного средства, будет варьироваться в зависимости от реципиента, опасности нарушения или патологического состояния, частоты введения, расположения соединения и выбора предписывающего данное средство врача. В одном варианте осуществления, подходящее количество соединения согласно настоящему изобретению вводится млекопитающему, нуждающемуся в данном соединении. В одном варианте осуществления, введение соединения осуществляют в количестве от примерно 0,001 мг/кг веса тела до примерно 60 мг/кг веса тела в сутки. В другом варианте осуществления, введение соединения происходит в количестве от 0,5 мг/кг веса тела до примерно 40 мг/кг веса тела в сутки. В некоторых случаях уровень дозировки ниже нижнего порога указанных выше пределов может быть более чем достаточным, а в других случаях без каких-либо неблагоприятных эффектов могут быть введены более крупные дозы, если указанные более крупные дозы предварительно разделены на несколько маленьких доз, вводимых в течение суток. Для получения дополнительной информации о способах введения и дозировании, см. главу 25.3 тома 5 издания Comprehensive Medicinal Chemistry (Corwin Hansch; Chairman of Editorial Board), Pergamon Press 1990, которое особо отмечено здесь посредством ссылки.
ИЗДЕЛИЯ ПРОИЗВОДСТВА
В другом варианте осуществления изобретения представлено готовое изделие, или "набор", содержащий материалы, используемые для лечения нарушений, описанных выше. В одном варианте осуществления набор включает контейнер, содержащий соединение согласно настоящему изобретению. Подходящие контейнеры включают, например, бутылки, флаконы, шприцы, блистерную упаковку, и т.д. Контейнер может быть выполнен из ряда материалов, таких как стекло или пластик. Контейнер может содержать соединение согласно настоящему изобретению, или композицию на его основе, являющийся эффективным при лечении патологического состояния, и может иметь стерильный канал доступа (например, контейнер может представлять собой пакет с внутривенным раствором или флакон, снабженный заглушкой, которую можно проколоть иглой шприца).
Набор может дополнительно включать ярлык или вкладыш на контейнере или связанную с ним. Термин "вкладыш" используется для обозначения инструкций, традиционно включенных в коммерческие упаковки терапевтических продуктов, которые содержат информацию о показаниях к применению, использовании, дозировке, правилах приема, противопоказаниях и/или предупреждениях, касающихся использования подобных терапевтических продуктов. В одном варианте осуществления, ярлык или вкладыш указывает, что композиция, содержащая соединение согласно настоящему изобретению может быть использован для лечения нарушения, опосредуемого, например, киназой AKT. Ярлык или вкладыш может также указывать, что композиция может быть использована для лечения других расстройств.
В определенных вариантах выполнения, наборы подходят для доставки твердых оральных форм соединения согласно настоящему изобретению, таких как таблетки или капсулы. Подобный набор предпочтительно включает несколько доз лекарственного средства. Подобные наборы могут включать карточку, на которой отображены дозы, расположенные в порядке предписанного использования. Примером подобного набора является "блистерная упаковка". Блистерные упаковки хорошо известны в упаковочной промышленности, и широко используются для упаковки доз фармацевтических композиций. При желании, в набор может быть включено напоминание в виде чисел, букв или иных обозначений, либо вкладыша-календаря для обозначения дней режима лечения, по которым могут вводиться дозы.
Согласно другому варианту осуществления, набор может включать (a) первый контейнер с содержащимся в нем соединением согласно настоящему изобретению; и (b) второй контейнер с содержащейся в нем второй фармацевтической композицией, причем вторая фармацевтическая композиция содержит второе соединение, пригодное для лечения нарушения, опосредуемого киназой AKT. В другом варианте или дополнительно, набор может дополнительно включать третий контейнер, содержащий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекций (BWFI), фосфатный буферный раствор, раствор Рингера и раствор декстрозы. Набор может дополнительно включать другие материалы, желательные с коммерческой и пользовательской точки зрения, включая другие буферы, разбавители, фильтры, иглы и шприцы.
Набор может дополнительно включать указания по введению соединения согласно настоящему изобретению и второй фармацевтической композиции, в случае, если она присутствует. Например, если набор включает первую композицию, содержащую соединение согласно настоящему изобретению и вторую фармацевтическую композицию, набор может дополнительно включать указания по одновременному, последовательному или раздельному введению первой и второй фармацевтических композиций пациенту, нуждающемуся в указанных композициях.
В определенных других вариантах выполнения, где набор включает композицию согласно настоящему изобретению и второй терапевтический агент, набор может включать контейнер для содержания компонентов по отдельности, такой как разделенная бутылка, или разделенный пакет из фольги, однако, отдельные компоненты могут также находиться в одном неразделенном контейнере. В определенных вариантах выполнения набор включает указания по введению отдельных компонентов. Форма набора имеет особое преимущество, когда отдельные компоненты предпочтительно вводятся в различных формах (например, орально и парентерально), вводятся на различных временных интервалах, либо если титрирование отдельных компонентов комбинации желательно с точки зрения предписывающего врача.
Соответственно, еще один аспект настоящего изобретения обеспечивает набор для лечения заболевания или нарушения, опосредуемого киназой AKT, где указанный набор включает а) первую фармацевтическую композицию, содержащую соединение согласно настоящему изобретению или его фармацевтически приемлемую соль; и Ь) инструкции по применению.
В некоторых вариантах осуществления, набор дополнительно включает (с) вторую фармацевтическую композицию, причем вторая фармацевтическая композиция содержит второе соединение, пригодное для лечения нарушения или заболевания, опосредуемого киназой AKT. В конкретном варианте осуществления, включающем вторую фармацевтическую композицию, набор дополнительно включает инструкции по одновременному, последовательному или раздельному введению указанных первой и второй фармацевтических композиций пациенту, нуждающемуся в этом. В некоторых вариантах осуществления, указанные первая и вторая фармацевтические композиции находятся в отдельных контейнерах. В других вариантах осуществления, указанные первая и вторая фармацевтические находятся в одном и том же контейнере
Несмотря на то, что соединение формулы I в первую очередь ценно в качестве терапевтического агента для млекопитающих, его также можно применять в любом случае, когда необходим контроль протеинкиназ AKT, тирозинкиназ, дополнительных серинтреонинкиназ, и/или киназ двойной специфичности. Таким образом, соединение можно применять в качестве фармакологического стандарта для использования в сфере разработки новых биологических тестов и поиска новых фармакологических агентов.
Активность соединения согласно настоящему изобретению может быть оценена путем теста с протеинкиназами AKT, тирозинкиназами, дополнительными серин/треонинкиназами, и/или киназами двойной специфичности in vitro, in vivo или в клеточной линии. Исследования in vitro включают исследования, в которых определяют ингибирование активности киназ. Другие исследования in vitro обеспечивают количественный анализ способности ингибитора связывать киназы, которую можно измерять либо путем радиоактивного мечения ингибитора до связывания, выделением комплекса ингибитор/киназа и определением количества связанных меток, либо путем проведения конкурентного эксперимента, где новые ингибиторы инкубируют с известными радиолигандами. Эти и другие подходящие тесты in vitro и тесты в культурах клеток хорошо известны специалисту.
Несмотря на то, что изобретение было описано и проиллюстрировано с определенной степенью уточнения, нужно понимать, что настоящее описание приведено лишь в качестве примера, и что множество изменений в комбинациях и расположении частей может сделано специалистом, не выходя за рамки объема настоящего изобретения, определенного в приведенной ниже формуле изобретения.
БИОЛОГИЧЕСКИЕ ПРИМЕРЫ
Тест киназы AKT-1
Активность соединений, описанных в настоящем изобретении, может быть определена при помощи следующего теста с киназой, который позволяет измерить фосфорилирование флуоресцентно-меченого пептида активной полноразмерной рекомбинантной AKT-1 человека путем флуоресцентной поляризации с использованием коммерчески доступного набора IMAP.
Материалы для исследования получены из полного набора IMAP AKT Assay Bulk Kit, продукт #R8059 от Molecular Devices, Sunnyvale (Калифорния, США). Материалы набора включают реакционный буфер IMAP (5х). Разбавленный реакционный буфер IMAP 1х включал 10 мМ Tris-HCl, pH 7,2, 10 мМ MgCl2, 0,1% БСА, 0,05% NaN3. Дитиотреитол обычно добавляют непосредственно перед использованием до конечной концентрации 1 мМ. Также включен связывающий буфер IMAP (5х), и связывающий реагент IMAP. Связывающий раствор готовят в виде раствора 1:400 связывающего реагента IMAP в связывающем буфере IMAP 1х.
Флюоресцентно-меченый субстрат AKT (Crosstide) имеет последовательность (F1)-GRPRTSSFAEG. Готовили исходный раствор с концентрацией 20 в реакционном буфере IMAP 1х.
Использовали следующие чашки Петри: Costar 3657 (382, из полипропилена, с белым V-образным дном), используется для разбавления соединения, и для приготовления смеси соединения и АТФ. Чашка Петри для анализа - Packard ProxyPlate™-384 F.
Используемая AKT-1 представляла собой полноразмерную рекомбинантную AKT-1 человека, активируемую при PDK-1 и киназой MAP - 2.
Для выполнения анализа готовили основные растворы соединений при 10 мМ в диметилсульфоксиде ("ДМСО"). Основные растворы и контрольное соединение последовательно разбавляли в пропорции 1:2 девять раз в ДМСО (10 мкл соединения+10 мкл ДМСО) с получением серии разбавлений 50х в желаемом диапазоне дозировки. Затем, аликвоты соединений в ДМСО объемом 2,1 мкл переносятся в чашку Петри Costar 3657, содержащую 50 мкл 10,4 мкМ АТФ в 1х реакционном буфере IMAP, содержащем 1 мМ дитиотреитола. После тщательного смешивания, аликвоты объемом 2,5 мкл переносят в чашку Петри ProxyPlate™-384 F.
Анализ инициируют путем добавления аликвот раствора, содержащего 200 нМ флуоресцентно-меченого пептидного субстрата и 4 нМ AKT-1, объемом 2,5 мкл. Чашку Петри подвергают центрифугированию в течение 1 минуты при 1000 g, и выдерживают в течение 60 минут при комнатной температуре. Затем, реакцию гасят путем добавления 15 мкл связывающего раствора, вновь подвергают центрифугированию, и выдерживают в течение еще 30 минут при комнатной температуре, после чего анализируют на приборе Victor 1420 Multilabel HTS Counter, настроенном на измерение флуоресцентной поляризации.
Соединения, описанные в примерах 1-3, тестировали при помощи описанного выше анализа и обнаружили, что они обладают 1Сзо менее 500 нМ.
ПРИМЕРЫ ПРИГОТОВЛЕНИЯ
Нижеследующий пример приведен с целью проиллюстрировать изобретение. Тем не менее, следует понимать, что данный пример не ограничивает изобретение, и является лишь примером практического осуществления изобретения. Специалисту будет понятно, что химические реакции, описанные в данном примере, могут быть легко адаптированы для альтернативных способов приготовления соединения согласно настоящему изобретению, и входят в объем настоящего изобретения. Например, синтез соединения согласно настоящему изобретению может быть успешно проведен с изменениями, очевидными для специалиста, т.е. при соответствующей защите групп, мешающих протеканию реакции, с использованием подходящих реагентов, отличных от указанных в описании, известных в данной области, и/или путем внесения обычных изменений в условия реакций. В другом варианте, иные реакции, известные в данной области, могут применяться для приготовления соединения согласно настоящему изобретению.
В описанном ниже примере все значения температур приведены в градусах Цельсия, если не указано иное. Реагенты были приобретены у коммерческих поставщиков, таких как Aldrich Chemical Company, Lancaster, TCl или Maybridge, и были использованы без дополнительного очищения, если не указано иное. Тетрагидрофуран ("ТГФ"), дихлорометан ("ДХМ"), толуол и диоксан были приобретены у компании Aldrich в герметичных бутылках, и использованы в том виде, в котором были получены.
Реакции, описанные ниже, обычно проводили при положительном давлении азота или аргона, или с использованием осушителя (если не указано иное) в безводных растворителях, а реакционные сосуды снабжали резиновой мембраной для введения субстратов и реагентов при помощи шприца. Стеклотару сушили в печи и/или путем тепловой сушки.
1H ЯМР спектр регистрировали на приборе Varian на частоте 400 МГц.1H-ЯМР спектр получали для растворов COCl3, CD3OD, D2O или d6-ДМСО (приведены в мкг/г) с использованием тетраметилсилана (0.00 ч/млн.) или остаточного растворителя (CDCL3: 7,25 мкг/г; CD3OD: 3,31 мкг/г; D2O: 4,79 ч/млн; d6-ДМСО: 2,50 мкг/г) в качестве контрольного стандарта. Для описания мультиплетов пиков используют следующие аббревиатуры: s (синглет), d (дуплет), t (триплет), m (мультиплет), br (широкий), dd (дуплет дуплетов), dt (дуплет триплетов). Если даны константы взаимодействия приведены в герцах (Гц).
Пример 1
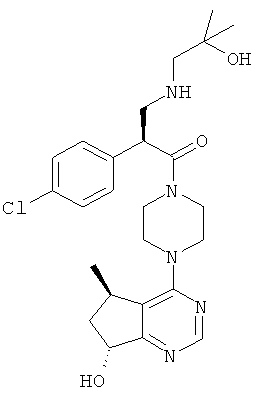
(S)-2-(4-хлорфенил)-3-(2-гидрокси-2-метилпропиламино)-1-(4-((5R,7R-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]-пиримидин-4-ил^)пиперазин-1-ил)пропан-1-он
Стадия 1: Метил 2-(4-хлорфенил)ацетат (36,7 г, 199 ммоль) и параформальдегид (6,27 г, 209 ммоль) растворяли/суспендировали в ДМСО (400 мл) и обрабатывали NaOMe (537 мг, 9,94 ммоль). Смесь оставляли перемешиваться при комнатной температуре в течение 2 часов, за это время реакция завершалась, что определяли при помощи ТСХ сырья. Реакционную смесь заливали в ледяную воду (700 мл; эмульсия) и нейтрализовали путем добавления раствора 1М HCl. Водный слой экстрагировали при помощи этилацетата (3 X), и объединяли органические вещества. Органический слой промывали водой (2 X), насыщенным солевым раствором (1 X), разделяли, сушили над MgSO4, фильтровали, и концентрировали под вакуумом для получения необработанного продукта в виде масла. Остаток загружали на большой фриттованный фильтр с силикагелем и элюировали с гексанами : этилацетатом 9:1 и затем с гексаном : этилацетатом 1:1 для получения метил-2-(4-хлорфенил)-3-гидроксипропаноата в виде масла (39,4 г, 92%).
Стадия 2: Метил-2-(4-хлорфенил)-3-гидроксипропаноат (39,4 г, 184 ммоль) растворяли в DCM (500 мл) и обрабатывали TEA (64,0 мл, 459 ммоль). Раствор охлаждали до 0°С. Раствор затем медленно обрабатывали MsCl (15,6 мл, 202 ммоль) и оставляли перемешиваться в течение 30 минут. Раствор разделяли раствором IN HCl, и водный слой экстрагировали один раз в DCM. Объединенные органические слои промывали еще раз раствором IN HCl, разделяли, промывали разбавленным раствором NaHCO3 и разделяли. Органический слой сушили над MgSO4, фильтровали и концентрировали под вакуумом для получения масла, которое загружали на большой фриттованный фильтр с силикагелем и элюировали с гексаном : этилацетатом 9:1 для получения метил 2-(4-хлорфенил)акрилата в виде масла (30,8 г, 85%).
Стадия 3: 1-амино-2-метил-пропан-2-ол (0,408 г, 4,58 ммоль) добавляли к раствору метил-2-(4-хлорфенил)акрилата (0,300 г, 1,52 ммоль) в тетрагидрофуране (3,84 мл). Реакцию оставляли перемешиваться при комнатной температуре в течение ночи и получали неочищенный метил-2-(4-хлорфенил)-3-(2-гидрокси-2-метилпропиламино)пропаноат (436 мг, 100%). ЖХ-МС: М+1 286,5.
Стадия 4: Калия триметилсиланолат (0,228 г, 1,60 ммоль) добавляли к раствору метил 2-(4-хлорфенил)-3-(2-гидрокси-2-метилпропиламино)пропаноата (0,436 г, 1,52 ммоль) в тетрагидрофуране (3,5 мл), Смесь перемешивали при комнатной температуре в течение ночи. ТГФ удаляли при пониженном давлении. Неочищенный материал очищали при помощи хроматографии с хиральной колонкой для получения двух продуктов: (S)-2-(4-хлорфенил)-3-(2-гидрокси-2-метилпропиламино)пропановой кислоты (43 мг, 10,4%) и (R)-2-(4-хлорфенил)-3-(2-гидрокси-2-метилпропиламино)пропановой кислоты (46 мг, 11,2%). ЖХ-МС: М+1 272,5.
Стадия 5: Этилпулегенат (130 г, 662 ммоль) в этилацетате ("EtOAc"; 900 мл) охлаждали до -78°С при помощи бани сухого льда-изопропанола. Эту смесь подвергали озонолизу до тех пор, пока реакционная смесь не становилась фиолетовой. В этот момент, подачу озона прекращали, и реакцию удаляли из бани на сухом льду. Кислород барботировали через реакционную смесь, пока она не стала желтой. Реакционную смесь концентрировали под вакуумом, и образующийся остаток растворяли в ледяной уксусной кислоте (400 мл). Раствор охлаждали до 0°С, и добавляли Zn пыль (65 г, 993 ммоль) порционно в течение 30 минут. Затем реакционную смесь оставляли перемешиваться в течение 2 часов, после чего реакционную смесь фильтровали через слой целита, чтобы удалить цинковую пыль. Уксусную кислоту нейтрализовали до рН 7 при помощи водного NaOH и NaHCO3 и экстрагировали при помощи простого эфира (3 Х 800 мл). Объединенные органические вещества высушивали насыщенным солевым раствором, MgSO4 и концентрировали для получения (2K)-этил 2-метил-5-оксоциклопентанкарбоксилата в виде жидкости (107 г, 95%).
Стадия 6: КОН (8,3 г, 147,9 ммоль) в воде (60 мл) добавляли к раствору смеси (2R)-этил 2-метил-5-оксоциклопентанкарбоксилата (20 г, 117,5 ммоль) и тиомочевины (9,2 г, 120,9 ммоль) в этаноле (100 мл). Смесь нагревали с обратным холодильником в течение 10 часов. После охлаждения растворитель удаляли. Образующийся остаток нейтрализовали концентрированной HCl (12 мл) при 0°С и затем экстрагировали при помощи DCM (3×150 мл). Растворитель удаляли, и образующийся остаток очищали хроматографией на силикагеле, элюируя гексаном/этилацетатом (2:1) для получения (R)-2-меркапто-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ола (12 г, 56%). МС (APCI+) [М+Н]+183.
Стадия 7: Никелевый катализатор Ренея (15 г) и NH4OH (20 мл) добавляли к суспензии (R)-2-меркапто-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ола (12 г, 65,8 ммоль) в дистиллированной воде (100 мл). Смесь нагревали с обратным холодильником в течение 3 часов и затем фильтровали. Фильтрат концентрировали для получения (R)-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ола (9,89 г, 99%). МС(APCI+)[М+Н]+151.
Стадии 8 и 9 описаны для альтернативного синтеза (R)-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ола, начиная с (R)-этил 2-метил-5-оксоциклопентанкарбоксилата.
Стадия 8: Аммония ацетат (240 г, 3114 ммоль) добавляли к раствору (R)-этил-2-метил-5-оксоциклопент-1-енкарбоксилата (106,0 г, 622,8 ммоль) в МеОН (1.2 L). Реакционную смесь перемешивали при комнатной температуре при действии азота в течение 20 часов, и реакция завершалась, что определяли посредством ТСХ и ВЭЖХ. Реакционную смесь концентрировали, чтобы удалить МеОН. Образующийся остаток растворяли в DCM, промывали H2O (2 X), солевым раствором (1 X), высушивали (Na2SO4), фильтровали, и концентрировали для получения (R)-этил-2-амино-5-метилциклопент-1-енкарбоксилата (102 г, выход 97%) в виде масла. ЖХ/МС(APCI+) m/z 170 [М+Н]+.
Стадия 9: Раствор, содержащий (R)-этил 2-амино-5-метилциклопент-1-ен-карбоксилат (161,6 г, 955 ммоль) и аммония формат (90,3 г, 1433 ммоль) в формамиде (303,5 мл, 7640 ммоль), нагревали до внутренней температуры 150°С и перемешивали в течение 17 часов. Реакционную смесь охлаждали и переносили в отдельную колбу для экстрагирования объемом 2 л. Затем избыток формамида удаляли путем дистилляции под высоким вакуумом. Как только формамид прекращал отгоняться, оставшееся масло в перегонном кубе растворяли в DCM и промывали солевым раствором (3×200 мл). Объединенные водные смывы экстрагировали при помощи DCM. Объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали. Полученное масло растворяли в минимальном DCM, и этот раствор добавляли при помощи делительной воронки к перемешанному раствору простого эфира (примерно 5 объемов эфира на раствор DCM), вызывая образование осадка. Этот осадок отделяли путем фильтрации через среднюю фриттованную воронку, которую промывали простым эфиром и удаляли. Фильтрат концентрировали, и растирание из простого эфира повторяли еще два раза. Затем продукт высушивали при высоком вакууме для получения (R)-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ола (93,23 г, выход 65,0%) в виде пастообразного твердого вещества. ЖХ/МС (APCI-) m/z 149,2.
Стадия 10: Очищенный POCl3 (463,9 мл, 5067 ммоль) добавляли медленно при помощи капельной воронки при 0°С к раствору (R)-5-метил-6,7-дигидро-5Н-циклопенто-[d]пиримидин-4-ола (152,2 г, 1013 ммоль) в DCE (1,2 л). После завершения добавления реакционную смесь нагревали до комнатной температуры и затем нагревали с обратным холодильником при перемешивании в течение 70 минут. Реакция завершалась, что определяли при помощи ВЭЖХ. The реакционная смесь охлаждали до комнатной температуры, и избыток POCl3 гасили в 4 частях следующим образом: Реакционную смесь переносили в разделительную воронку и прокапывали в лабораторный стакан, содержащий лед и насыщенный раствор NaHCO3, охлажденный на ледяной бане. Когда завершали добавление каждой части реакционной смеси, гашенную смесь перемешивали 30 минут, чтобы гарантировать полное разложение POCl3 перед переносом в разделительную воронку. Смесь переносили в разделительную воронку и экстрагировали при помощи DCM (2 X). Объединенные экстракты высушивали (Na2SO4), фильтровали и концентрировали. Неочищенный материал очищали на силикагеле следующим образом: силикагель (1 кг) разжижали в гексан : этилацетате 9:1 на 3 л фриттованной воронке, кремний осаждали под вакуумом, покрывали песком. Неочищенный материал нагружали смесью DCM/гексан, и соединение элюировали при помощи 1 л колб с боковыми горловинами под вакуумом. Биопродукты с высокой Rf элюировали первыми, затем (R)-4-хлор-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин (104,4 г, выход 61,09%) в виде масла. Триэтиламин (93,0 мл, 534 ммоль) и третбутил-пиперазин-1-карбоксилат (34,8 г, 187 ммоль) добавляли к раствору (R)-4-хлор-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидина (30,0 г, 178 ммоль) в n-BuOH (250 мл). Реакционную смесь нагревали с обратным холодильником при действии азота и перемешивали в течение ночи (17 часов), после чего концентрировали на ротационном испарителе. Получаемое масло растворяли в DCM, промывали H2O, высушивали (Na2SO4), фильтровали и концентрировали. Получаемое масло очищали на силикагеле, сначала элюируя с гексанами : этилацетатом 2:1 до извлечения чистого продукта, затем в градиенте ЭСМ : этилацетата 1:1-1:5 для получения (R)-третбутил 4-(5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (42,0 г, выход 74,1%) в виде порошка. ЖХ/МС(APCI+) m/z 319,1 [M+H]+.
Стадия 11: Твердую м-хлорпербензойную кислоту с максимальным содержанием твердого вещества 77% ("m-CPBA"; 23,9 г, 107 ммоль) добавляли частями при 0°С к раствору (R)-третбутил-4-(5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (20,0 г, 62,8 ммоль) в CHCl3 (310 мл). Реакционную смесь перемешивали в течение 5 минут, затем нагревали комнатной температуры и перемешивали в течение еще 90 минут. Результаты ВЭЖХ выглядели аналогично через 7,5 часов. Реакционную смесь охлаждали до 0°С, и затем добавляли NaHCO3 (13,2 г, 157 ммоль) и другие 0,5 эквивалентов m-CPBA. Реакционную смесь перемешивали в течение ночи (14 часов). Реакционную смесь охлаждали до 0°С, и раствор Na2S2O3 (29,8 г, 188 ммоль) в H2O (50 мл) добавляли по каплям через капельную воронку. После этого добавляли раствор Na2CO3 (24,6 г, 232 ммоль) в H2O (10 мл) через воронку для добавления (смесь становилась однородной). Реакционную смесь перемешивали в течение 30 минут, и затем смесь экстрагировали при помощи CHCl3 (3×150 мл). Объединенные экстракты высушивали (Na2SO4), фильтровали и концентрировали для получения (R)-4-(4-(трет-бутоксикарбонил)пиперазин-1-ил)-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-1-оксид (21,0 г, 100%). ЖХ/МС(APCI+) m/z 335,1 [М+Н]+.
Стадия 12: Ac2O (77,0 мл, 816 ммоль) добавляли к (R)-4-(4-(трет-бутоксикарбонил)пиперазин-1-ил)-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-1-оксиду (21,0 г, 62,8 ммоль). Реакционную смесь нагревали при действии азота в песчаной бане при 90°С и перемешивали в течение 100 минут. Реакционную смесь охлаждали до комнатной температуры, и избыток уксусного ангидрида удаляли на ротационном испарителе. Полученное масло растворяли в DCM, которое затем аккуратно выливали в лед, насыщенный Na2CO3. Смесь экстрагировали при помощи DCM, и объединенные экстракты высушивали (Na2SO4), фильтровали и концентрировали для получения (5R)-трет-бутил-4-(7-ацетокси-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)-пиперазин-1-карбоксилата (23,6 г, 100%) в виде пены. ЖХ/МС(APCI+) m/z 377,1. [М+Н]+.
Стадия 13: LiOH-H2O (6,58 г, 157 ммоль) добавляли при 0°С к раствору (5R)-трет-бутил-4-(7-ацетокси-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (23,6 г, 62,69 ммоль) в THF:H2O 2:1 (320 мл). Реакционную смесь перемешивали в течение 10 минут, и затем нагревали до комнатной температуры. ЖХ/МС выглядели аналогично через 3 часа и 4.5 часа. Реакционную смесь охлаждали до 0°С, и затем к смеси добавляли насыщенный NH4Cl. Смесь перемешивали в течение 5 минут, и большая часть ТГФ удаляли на ротационном испарителе; смесь экстрагировали с EtOAc (3×250 мл), и объединенные экстракты высушивали (Na2SO4), фильтровали и концентрировали. Неочищенный материал быстро пропускали через Biotage 65M:4:1 ОСМ : этил ацетат, затем через градиент ВСМ : этилацетата 1:1-1:4. Как только продукт элюировал, этилацетат прогоняли через колонку. Затем DCM:MeOH 30:1 элюировали остаток продукта (8,83 г). Смешанные фракции повторно пропускались через Biotage 40M при тех же условиях для получения другой порции (2,99 г), которая давала объединенный выход (5R)-третбутил 4-(7-гидрокси-5-метил-6,7-дигидро-5Н-циклопента[d]пиридин-4-ил)пиперазин-1-карбоксилата (11,82 г, выход 56,38% yield) в виде пены. ЖХ/МС(APCI+) m/z 335.1 [М+Н]+.
Стадия 14: Раствор DMCO (5,45 мл, 76,8 ммоль) в DCM (50 мл) добавляли по каплям при помощи капельной воронки при -78°С к раствору оксалилхлорида (3,35 мл, 38,4 ммоль) в DCM (150 мл). Реакционную смесь перемешивали в течение 35 минут, и затем добавляли раствор (5R)-третбутил-4-(7-гидрокси-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (9,17 г, 27,4 ммоль) в DCM (80 мл) медленно при помощи капельной воронки. Реакционную смесь перемешивали еще 1 час при -78°С, после чего к смеси добавляли чистый триэтиламин (18,0 мл, 129 ммоль). Затем реакционную смесь оставляли нагреваться до комнатной температуры, и затем ее перемешивали в течение 30 минут. Добавляли Н2О. Смесь экстрагировали при помощи DCM (3×200 мл), и объединенные экстракты высушивали (Na2SO4), фильтровали и концентрировали под вакуумом. Неочищенный материал очищали на силикагеле (Biotage 65M) : колонку промывали 800 мл DCM:EtOAc 4:1, затем градиентом DCM : этилацетата 1:1. пока продукт не элюировали, затем DCM:EtOAc 1:4 элюировали продукт для получения (R)-третбутил-4-(5-метил-7-оксо-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилат (7,5 г, выход 82,3%) в виде пены. Пену концентрировали из DCM/гексанов (3 X), что также давало пену. ВЭЖХ>95% площади. ЖХ/МС(APCI+) m/z 333 [М+Н]+.
Стадия 15: Триэтиламин (4,33 мл, 31,1 ммоль; дегазированный при помощи азота в течение 30 минут перед применением) и муравьиную кислоту (1,36 мл, 36,1 ммоль; дегазированную при помощи азота в течение 30 минут перед применением) добавляли к раствору (R)-третбутил-4-(5-метил-7-оксо-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (9,75 г, 29,3 ммоль) в DCM (210 мл; дегазированный при помощи азота в течение 30 минут перед применением). Смесь перемешивали в течение 5 минут, и затем добавляли катализатор Ru (0,0933 г, 0,147 ммоль). Реакционную смесь перемешивали при положительном давлении азота в течение ночи (18 часов). Реакционную смесь концентрировали до сухого состояния и высушивали под высоким вакуумом. Примесный материал пропускали через Biotage 65M, нагруженный ОСМ : этилацетатом 1:1, 500 мл, затем ВСМ : этилацетатом 1:4 до появления продукта (2-ое пятно), затем градиентом чистого этилацетата, затем DCM:MeOH 25:1 элюировали остаток продукта. Фракции объединяли и концентрировали на ротационном испарителе. Остаток снова концентрировали из DCM/гексанов для получения смеси третбутил-4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилат (основной) и третбутил-4-((5R,7S)-7-гидрокси-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (минорный) (9,35 г, выход 95,3%) в виде пены. ЖХ/МС(APCI+) m/z 335 [М+Н+].1H ЯМР (CDCl3) показал 88% диастереоселективность по интеграции карбинола метина.
Стадия 16: 4-нитробензоил хлорид (4,27 г, 23,0 ммоль) добавляли при 0°С к раствору третбутил-4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (7,0 г, 20,9 ммоль) и триэтиламина (4,38 мл, 31,4 ммоль) в DCM (110 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, после чего добавляли насыщенный NaHCO3. Смесь перемешивали 10 минут и затем экстрагировали DCM. Объединенные экстракты высушивали (Na2SO4), фильтровали и концентрировали. Неочищенный материал пропускали через Biotage 65M (гексаны/гэтилацетат 3:1, нагруженные неочищенным веществом, затем гексаны/гэтилацетат 2:1 элюировали трет-бутил-4-((5R,7R)-5-метил-7-(4-нитробензоилокси)-6,7-дигидро-5Н-циклопента[4]пиримидин-4-ил)пиперазин-1-карбоксилат и несколько смешанных фракций). Затем третбутил-4-((5R,7S)-5-метил-7-(4-нитробензоилокси)-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилат элюировали при помощи гексанов/гэтилацетата 1:2. Фракции с продуктом концентрировали на ротационном испарителе для получения третбутил-4-((5R,7R)-5-метил-7-(4-нитробензоилокси)-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (8,55 г, выход 84.5%) в виде пены. ЖХ/МС(APCI+) m/z 484 [М+Н+.1H ЯМР (CDCl3) показывает один диастереомер). Фракции с другим диастереомером концентрировали на ротационном испарителе для получения третбутил-4-((5R,7S)-5-метил-7-(4-нитробензоилокси)-6,7-дигидро-5Н-циклопенто[а]пиримидин-4-ил)пиперазин-1-карбоксилата (0,356 г, выход 3,52%) в виде пены. ЖХ/МС(APCI+) m/z 484 [М+Н]+.
Стадия 17 описывает альтернативное приготовление третбутил-4-((5R,7S)-5-метил-7-(4-нитробензоилокси)-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата и трет-бутил-4-((5R,7R)-5-метил-7-(4-нитробензоилокси)-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата из (5R)-третбутил-4-(7-гидрокси-5-метил-6,7-dдигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (Стадия 13).
Стадия 17: 4-нитробензоил хлорид (15,78 г, 85,03 ммоль) при 0°С добавляли к раствору (R)-третбутил-4-(7-гидрокси-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (25,85 г, 77,30 ммоль) и NEt3 (11,73 г, 16,16 мл, 115,9 ммоль) в DCM (400 мл). Реакционную смесь перемешивали в течение 5 минут. Смесь затем нагревали до комнатной температуры и перемешивали в течение ночи (17 часов), после чего добавляли насыщенный NaHCO3. Реакционную смесь перемешивали в течение 10 минут и переносили в разделительную воронку. Органические слои собирали, а водные экстракты промывали в DCM (2 X). Объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали. Неочищенный материал пропускали через кремний (silica), нагруженный гексанами/этилацетатом 7:1 (градиент гексаны/этилацетат 5:1 до градиента гексаны/этилацетат 2:1 до гексан/этилацетат 1:1) Выделяли некоторое количество чистого третбутил-4-((5R,7S)-5-метил-7-(4-нитробензоилокси)-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата, некоторое количество чистого трет-бутил-4-((5R,7S)-5-метил-7-(4-нитробензоилокси)-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата и некоторое количество смешанных фракций. Смешанные фракции повторно пропускали через колонку и объединяли с ранее выделенным материалом для получения третбутил-4-((5R,7R)-5-метил-7-(4-нитробензоилокси)-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (14,27 г, 38%) и третбутил-4-((5R,7S)-5-метил-7-(4-нитробензоилокси)-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (12,58 г, 34%). Применение 4-бромбензоилхлорида показало несколько лучшее разделение изомеров.
Стадия 18: LiOH-H2O (0,499 г, 11,9 ммоль) добавляли при 0°С к раствору трет-бутил-4-((5R,7R)-5-метил-7-(4-нитробензоилокси)-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (2,30 г, 4,76 ммоль) в 2:1 ТГФ:Н2О (40 мл). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. ТГФ удаляли на ротационном испарителе. Затем добавляли насыщенный NaHCO3, и смесь экстрагировали при помощи этилацетата. Объединенные экстракты промывали (1 X) насыщенным NaHCO3, высушивали (Na2SO4), фильтровали и концентрировали с образованием третбутил-4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (1,59 г, выход 100.0%) в виде пены. ВЭЖХ после пропускания простого продукта показывала более чем 98% площади чистого вещества. ЖХ/МС(APCI+) m/z 335 [М+Н]+.
Стадия 19: 4М HCl/диоксана (11,2 мл, 44,9 ммоль) добавляли к раствору трет-бутил-4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (0,600 г, 1,79 ммоль) в диоксане (15 мл). Реакционную смесь перемешивали при комнатной температуре при действии азота в течение ночи (20 часов). Смесь концентрировали до сухого состояния и высушивали под высоким вакуумом. Неочищенный материал суспендировали в простом эфире, воздействовали ультразвуком и перемешивали в течение 5 минут.Твердые вещества выделяли путем фильтрации через среднюю фриттованную воронку под давлением азота, промывали простым эфиром, высушивали при действии азота под давлением и высушивали далее под высоким вакуумом для получения (5R,7R.)-5-метил-4-(пиперазин-1-ил)-6,7-дигидро-5Н-циклопента[d]пиримдин-7-ол дигидрохлорида (0,440 г, выход 79,8%) в виде порошка. ЖХ/МС(APCI+) m/z 235.
Стадия 20: O-(Бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат (0,100 г, 0,265 ммоль) добавляли к раствору (5R,7R)-5-метил-4-(пиперазин-1-ил)-6,7-дигидро-5Н-циклопента[d]пиримидин-7-ола (31 мг, 0,13 ммоль), N,N-диизопропилэтиламина (0,231 мл, 1,32 ммоль) и (S)-2-(4-хлорфенил)-3-(2-гидрокси-2-метилпропиламино)пропановой кислоты (43 мг, 0,16 ммоль) в метилен хлориде (1,2 мл, 19 ммоль). Смесь перемешивали в течение 1 часа при комнатной температуре, после чего реакцию гасили насыщенным водным NRiCl. Смесь экстрагировали при помощи DCM (3×10 мл). Объединенные органические экстракты высушивали (Na2SO4), фильтровали и концентрировали. Неочищенный материал очищали при помощи обращено-фазовой ВЭЖХ для получения (S)-2-(4-хлорфенил)-3-(2-гидрокси-2-метилпропиламино)-1-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)пропан-1-она (33 мг, 51%),1H ЯМР (CDCl3, 500 МГц) 6 8,59 (s, 1H), 7,50 (d, J=8,5 Гц, 2H), 7,33 (d, J- 8,5 Гц, 2H), 5,06 (m, 1H), 4,48 (m, 1H), 3,92 (m, 2H), 3,15 (m, 2H), 2,94 (m, 2H), 2,02 (m, 2H), 1,19 (m, 6H), 1,06 (d, 3H), ЖХ-МС М+1 488,3.
Пример 2
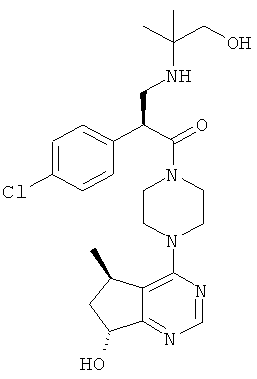
(S)-2-(хлорфенил)-3-(1-гидрокси-2-метилпропан-2-иламино-1-(4-((5R,7R-7-гидрокси-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)пропан-1-он
(S)-2-(хлорфенил)-3-(1-гидрокси-2-метилпропан-2-иламино-1-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)пропан-1-он готовили при помощи процедур, описанных в Примере 1, с применением 2-амино-2-метилпропан-1-ола вместо 1-амино-2-метил-пропан-2-ола.1H ЯМР (CDCl3,500 МГц) 68,42 (s, 1Н), 7,39 (d, J=8,6 Гц, 2Н), 7,33 (d, J=8,6 Гц, 2Н), 4,82 (m, 1Н), 4,37 (m, 1H), 4,31 (m3 2H), 4,07 (m, 1Н), 3,77 (m, 2Н), 3,68-3,43 (m, 6H), 1,97-1,89 (m, 2Н), 1,04 (s, 3H), 1,03 (s, 3H), 0,88 (d, 3H), ЖХ-МС M+1 488,2.
Пример 3
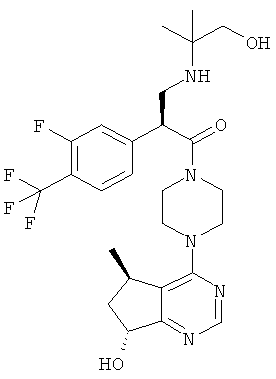
(S)-2-(3-фтор-4-(трифторметил)-фенил)-3-(1-гидрокси-2-метилпропан-2-иламино)-1-(4-((5R,7R-гидрокси-5-метил-6,7-дигидро-5Н-циклопента[d]пиридин-4-ил)пиперазин-1-ил)пропан-1-он
(S)-2-(3-фтор-4-(трифторметил)-фенил)-3-(1-гидрокси-2-метилпропан-2-иламино)-1-(4-((5R,7R)-гидрокси-5-метил-6,7-дигидро-5Н-циклопента[d]пиридин-4-ил)пиперазин-1-ил)пропан-1-он готовили в соответствии с процедурами, описанными в Примере 1, заменяя метил-2-(4-хлорфенил)ацетат на метил 2-(3-фтор-4-трифторметилфенил)ацетат.1Н ЯМР (CDCl3, 500 МГц) 88,58 (s, 1Н), 7,86 (t, J=7,9 Гц, 1Н), 7,50 (d, J=8,5 Гц, 1Н), 7,37 (d, J=7,9 Гц, 1Н), 5,04 (m, 1Н), 4,50 (m, 2Н), 3,93 (m, 2Н), 3,07 (m, 2Н), 2,01 (m, 2Н), 1,23 (s, 3H), 1,22 (s, 3H), 1,06 (d, 3H), ЖХ-МС M+1 540,3.
Предшествующее описание рассматривается как пояснительное в отношении принципов изобретения. Кроме того, поскольку специалисты в данной области техники способны представить множество модификаций и изменений, изобретение не ограничивается конкретными конструкциями и процессами, показанными выше. Соответственно, все подходящие модификации и эквиваленты можно рассматривать входящими в область притязаний изобретения, как определено в формуле изобретения, представленной ниже.
Слова «содержит», «содержащий», «включают», «включающий» и «включает», применяемые в данном описании и в последующей формуле изобретения, обозначают наличие указанных признаков, целого, компонентов, стадий или групп.
Реферат
Изобретение относится к новым гидроксилированным пиримидилциклопентанам общей формулы I и их фармацевтически приемлемым солям. Соединения обладают свойствами селективных ингибиторов АКТ протеинкиназы. Изобретение также относится к фармацевтической композиции, содержащей эффективное количество соединения формулы I, применению соединений для производства лекарственного препарата для терапии патологических состояний, опосредованных АКТ протеинкиназой, таких как гиперпролиферативного заболевания, в частности рак.В формуле I:Rпредставляет собой C-Cалкил, содержащий один заместитель OH; Rпредставляет собой водород или F; и Rпредставляет собой C1 или CF3. Объектом изобретения является набор, содержащий фармацевтическую композицию и инструкцию по ее применению. 4 н. и 11 з.п. ф-лы, 3 пр.
Формула
и его фармацевтически приемлемая соль, где
R1 представляет собой C1-C6алкил, содержащий один заместитель OH;
R2 представляет собой водород, или F; и
R3 представляет собой Cl или CF3.
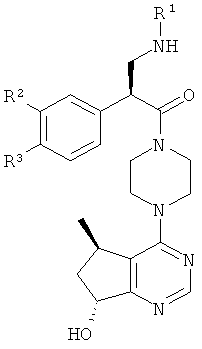
где переменные имеют те же значения, что указаны по п.1.
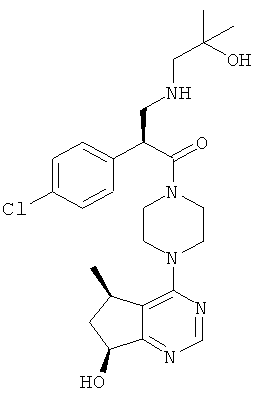
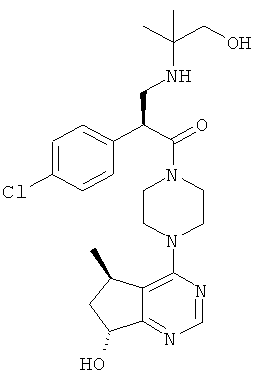
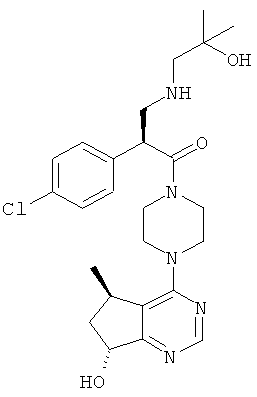
Документы, цитированные в отчёте о поиске
Гидроксилированные и метоксилированные циклопента[d]пиримидины в качестве ингибиторов акт протеинкиназ
Пиримидилциклопентаны в качестве ингибиторов akt-протеинкиназы
Комментарии