Замещенные тетрагидро-, или гексагидроциклопент /в/ индолы, или их фармацевтически приемлемые аддитивные соли и способы их получения - RU2077530C1
Код документа: RU2077530C1
Чертежи


Описание
Изобретение относится к соединениям формулы:
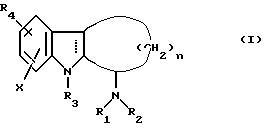
в которой n равно 2, 3, 4 или 5; Х представляет собой водород, низший алкил, низшую алкокси, гидрокси, галоген, трифторметил или нитро группу; R1 представляет собой водород, низший алкил, низший алкенил, низший алкинил, амино-низший алкил, низший алкил-аминонизший алкил, ди-низший алкиламино низший алкил, циклоалкил, циклоалкил низший алкил, циклоалкенил, арил, арил низший алкил, арилциклоалкил,

причем группа "Alk" обозначает двухвалентную низшую алкиленовую группу, а Y обозначает водород, низший алкил, арил или арил низший алкил; R2 представляет собой водород, низший алкил, формил, низший алкилкарбонил, бензилоксикарбонил или низший алкиламинокарбонил; или альтернативно группа

в целом представляет собой


R3 представляет собой водород, низший алкил, арил низший алкил, низший алкилкарбонил или низший алкоксикарбонил;
R4 представляет собой водород, -ОН,

или


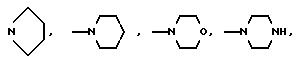

а R7 представляет собой низший алкил, арил или арил низший алкил; такие соединения полезны для облегчения различных дисфункций памяти, характеризующихся холинергическим дефицитом, таких как болезнь Альцгеймера. Соединения формулы I изобретения также ингибируют моноаминоксидазу и/или действуют на центральные α2 -адренергические рецепторы и, следовательно, могут использоваться в качестве антидепрессантов.
В сферу настоящего изобретения включены также соединения формулы II, в которой R3, R4, Х и n имеют указанные выше значения, которые полезны в качестве прямых предшественников целевых соединений настоящего изобретения.
В сферу изобретения включены также соединения формулы III, в которой R8 представляет собой гидрокси, амино низшая алкокси, низший алкил, циклоалкил, циклоалкенил, арил низший алкил, арилциклоалкил, низший алкилкарбонилокси или низший аминокарбонилокси, которые полезны для облегчения различных дисфункций памяти, характеризующихся холинергическим дефицитом, таких как болезнь Альцгеймера. Соединения III настоящего изобретения также ингибируют моноамин оксидазу и/или действуют, как антагонисты пресинаптического α2-адренергического рецептора и, следовательно, являются полезными в качестве антидепрессантов.

Если не оговорено или не указано иное, то следующие ниже определения применимы ко всему тексту описания и прилагаемой формуле изобретения.
Термин низший алкил подразумевает алкильную группу нормального или изостроения, содержащую 1-6 углеродных атомов. Примерами указанного низшего алкила могут служить метил, этил, -н-пропил, изо-пропил, н-бутил, изо-бутил, втор. -бутил, трет.-бутил, а также пентил и гексил нормального или разветвленного строения.
Термин циклоалкил подразумевает циклоалкильную группу из 3-7 углеродных атомов.
Под термином галоген подразумевается фтор, хлор, бром или иод.
Термин арил подразумевает фенильную группу, замещенную 0,1 или 2 заместителями, каждый из которых независимо друг от друга представляет собой низший алкил, низшую алкокси, галоген, трифторметил, гидрокси или нитро группу.
Везде в тексте описания и прилагаемой формулы изобретения химическая формула или название охватывает все стерео и таутомерные изомеры в том случае, когда такие изомеры существуют.
Соединения настоящего изобретения получают путем использования одной или более стадий синтеза, описанных ниже.
В тексте описания стадий синтеза символы n, X, Y и R1 R8 имеют соответствующие значения, приведенные выше, если не оговорено или не указано иное.
Стадия А.
Соединения формулы IV, в которой R9 представляет собой водород или -ОСН3, подвергают циклизации с получением соединения формулы V. Такую реакцию обычно проводят в водной серной кислоте при температуре 25-150oC.

Стадия В.
Производят реакцию между соединением V и сульфатным соединением формулы (R10О)2SO2, в которой R10 представляет собой низший алкил или арил низший алкил, с помощью традиционного способа, известного в этой области, с получением соединения формулы VI. Согласно другой методике, проводят реакцию между соединением V и галогенидным соединением формулы R10 -Наl, где R10 имеет указанные выше значения, с помощью традиционного способа, известного в этой области, с получением соединения формулы VI.

Стадия С.
Проводят реакцию между соединением V и ди низшим
алкилпирокарбонатом формулы R11
-О-СО-О-СО-О-R11, где R11 представляет низшую алкильную группу, в присутствии подходящего катализатора, предпочтительно
4-диметиламинопиридина, с получением соединения формулы
VII

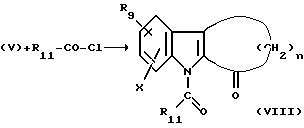
Стадия D.
Проводят реакцию между соединением V и ацилхлоридом формулы R11-СО-Сl, с помощью общепринятого способа, известного в данной области, с получением соединения формулы VIII.
Стадия Е.
Соединение формулы IX, полученное на стадии В, подвергают реакции расщепления с получением соединения формулы Х. Обычно в конце такой реакции соединение IX реагирует с комплексом ВВr3 (тетрагидрофуран, и полученный в результате продукт гидролизуют обычным способом, известным из литературы.

Стадия F.
В специальном случае проводят реакцию между соединением ХI и хлористым хлорацетилом в присутствии хлористого алюминия, с помощью обычного метода, известного в литературе, с получением соединения формулы XII (реакция Фриделя-Крафтса).

Cтадия G.
Обычным методом, известным из литературы, проводят реакцию между соединением XII и надкислотой, предпочтительно м-хлорнадбензойной кислотой, с получением соединения формулы XIII (реакция Байера-Виллигера).

Стадия Н.
Соединение XIII гидролизуют предпочтительно в присутствии такого основания, как гидроксид натрия с получением соединения формулы XIV.

Стадия I.
Соединение формулы XV, в которой R12 представляет собой водород, метокси или гидрокси, которое получено на одной из предыдущих стадий, подвергают реакции с гидрохлоридом гидроксиламина с помощью традиционного способа, известного из литературы, с получением соединения формулы XVI. Обычно такую реакцию проводят путем суспендирования соединения XV в этаноле с последующим добавлением водного раствора ацетата натрия и водного раствора гидрохлорида гидроксиламина, после чего суспензию перемешивают при температуре 25-150oC.

Стадия J.
Стандартным способом проводили реакцию между соединением XVI и бромистым аминонизшим алкилом формулы Вr-R13-NH2, где -R13-NH2 представляет собой аминонизшую алкильную группу, с получением соединения формулы XVII.

Стадия К.
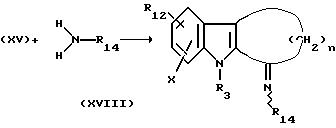
Общепринятым способом, известным из литературы, проводили реакцию между соединением XV и
первичным амином формулы

где R14 представляет собой низший алкил, низший алкенил, низший алкинил, циклоалкил, циклоалкенил, арилнизший алкил, арилциклоалкил или арил, с получением имина формулы XVIII.
Такую реакцию предпочтительно проводят в присутствии изопропилата титана (IV) в среде такого подходящего растворителя, как ацетонитрил. Обычно такую реакцию проводят при температуре 0-80oC. Такой способ более предпочтителен, чем метод с использованием TiCl4 или метод, в котором реакцию проводят в запаянной трубке при повышенной температуре с помощью молекулярных сит, используемых в качестве водоудаляющего средства.
Стадия L.
Соединение XVI восстанавливают с помощью сплава Ренея и раствора гидроксида натрия в соответствии со способом, описанным Б.Стаскуном и Т.ван Эсом (J. Chem. Soc. C, 531, 1966) с получением соединения формулы XIX.

Стадия М.
Проводят реакцию между соединением XV и изопропилатом титана и вторичным амином формулы




с последующим восстановлением цианоборгидридом натрия в условиях, аналогичных описаниям Р.Дж.Маттсоном с сотр. J.Org. Chem. 55- 2552-4 (1990), с получением соединения формулы ХХ.
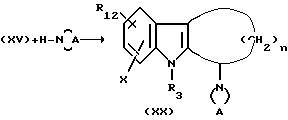
Стадия N.
Традиционным способом, известным из литературы, соединение XVIII восстанавливали боргидридом натрия, цианоборгидридом натрия или комплексом боран/тетрагидрофуран с получением соединения формулы XXI.

Стадия О.
Соединением XIX восстанавливали с помощью комплекса боран/тетрагидрофуран и трифторуксусная кислота с получением соединения формулы XXII.

Cтадия Р.
Проводили реакцию между соединением формулы ХХIII, которое получали на стадии L или О, и галогенидным соединением формулы R15-Наl, в которой R15 представляет собой низший алкил, низший алкенил, низший алкинил, циклоалкинилнизший алкил, арилнизший алкил,


с получением соединения формулы XXIV.

Стадия Q.
Проводили реакцию между соединением формулы XXV, в которой R16 представляет собой водород, низший алкил, низший алкенил, низший алкинил, циклоалкил, циклоалкенил, арилнизший алкил или арилциклоалкил, и муравьиной кислотой в присутствии 1-(3-диметиламинопропил)-3-этилкарбодиимида и 4-диметиламинопиридина, или смешанного ангидрида, полученного из муравьиной кислоты и уксусного ангидрида, с получением соединения формулы XXVI.

Стадия R.
Общепринятым методом, известным из литературы, проводили реакцию между соединением XXV и хлористым ацилом формулы R17 СО-Сl, в которой R17 представляет собой низшую алкильную группу, с получением соединения формулы XXVII.

Стадия S.
Общепринятым способом, известным из литературы, проводили реакцию между соединением формулы XXV, в которой R12 не является гидрокси группой, и бензилхлорформиатом с получением соединения формулы XXVIII.

Стадия Т.
Проводили реакцию между соединением формулы XXV, в
которой R12 не является гидрокси, и изоцианатом формулы R17-N=C=O, в которой R17 представляет собой низший алкил, арил или арилнизшую алкильную группу, с получением
соединения формулы XXIX. Обычно такую реакцию проводят в присутствии такого подходящего катализатора, как 1,8-диазобицикло[5.4.0]ундец-7-ен

Стадия U.
Проводили реакцию между соединением формулы XVI, в которой R12 не является гидрокси, и изоцианатом практически тем же способом, что и на стадии Т, с получением соединения формулы ХХХ.

Стадия Y.
Общепринятым методом, известным из литературы, проводили реакцию между соединением XVI и ацилхлоридом формулы R17-СО-Сl или ангидридом кислоты формулы (R17-СО)2О с получением соединения формулы ХХХI.

Стадия W.
Общепринятым методом, известным из литературы, проводили реакцию между соединением формулы XXXII, в которой R2 не является низшим алкиламинокарбонилом, полученным на одной из предыдущих стадий, с хлорформиатом формулы



Стадия Х.
Проводили реакцию между соединением формулы XXXIIIa, которое получали на стадии W с изоцианатом формулы R17-N=C=O, с использованием того же способа, что применяли на стадии Т, в результате чего получали соединение формулы ХХХIV. После этого соединение XXXIV подвергали гидрогенолизу обычным способом, известным в технике, с помощью подходящего катализатора, такого как палладий на угле, с получением соединения формулы XXXV.
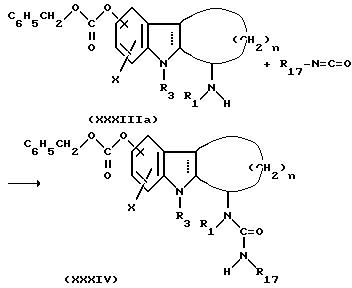

Стадия Y:
В соответствии со способом, описанным для стадии Т, проводили реакцию между соединением формулы XXVI, которое получали на предыдущих стадиях, и изоцианатом формулы R17-N=C=O с получением соединения формулы XXVII.

Соединения формул I и III настоящего изобретения полезны для лечения различных расстройств памяти, характеризующихся пониженной холинергической функцией, таких как болезнь Альцгеймера. Соединения настоящего изобретения также ингибируют моноаминооксидазу и/или воздействуют на центральные α2- адренаргические рецепторы и, следовательно, полезны как антидепрессанты.
Активность в отношении облегчения таких расстройств памяти демонстрируется способностью таких соединений ингибировать фермент ацетилхолинэстеразу и тем самым повышать уровень содержания ацетилхолина в головном мозге.
Испытание на ингибирование холинэстеразы.
Холинэстераза обнаруживается в различных участках тела, как в головном мозге, так и в сыворотке. Однако лишь распределение ацетилхолинэстеразы в головном мозге коррелирует с центральной холинергической иннервацией. Предполагается, что подобная иннервация ослабляется у пациентов с болезнью Альцгеймера. В настоящей работе определялось ингибирование ин витро ацетилхолинэстеразной активности в крысином стриатуме (Striatunu).
Ингибирование ин витро ацетилхолинэстеразной активности в крысином стриатуме.
Ацетилхолинэстераза (АСhE), которую иногда называют истинной или специфической холинэстеразной, обнаружена в нервных клетках, скелетных мышцах, гладких мышцах, различных железах и в красных кровяных клетках. АChE можно отличить от других холинэстераз по субстратной и ингибиторной специфичности и по региональному распределение. Ее распределение в головном мозге строго коррелирует с холинэргической иннервацией, и субфракционирование обнаруживает наивысший уровень содержания в нервных окончаниях.
Общепринятым является тот факт, что физиологическая роль АСhE заключается в быстром гидролизе и инактивации ацетилхолина Ингибиторы АСhE проявляют ярко выраженное холиномиметическое действие в холинергически-иннерватированных эффекторных органах, и их используют терапевтически при лечении глаукомы, миастении гравис и паралитической кишечной непроходимости. Однако результаты последних исследований позволили предположить, что АChE ингибиторы могут также с успехом применяться для лечения болезни Альцгеймера.
Описанный ниже способ использовали в изобретении для оценки холинэстеразной активности. Этот метод является модификацией метода Эллмана с сотр. Biochem Phasm. 7, 88 (1961).
Методика:
А. Реагенты
1. 0,05 М фосфатный буфер, рН
7,
2.
(a) 6,85 г NaH2PO4•H2O/100 мл дистиллированной воды
(b) 13,40 г Na2HPO4•7H2O/100 мл
дистиллированной H2
(c) добавляют (а) к (b) до рН 7,2
(d) осуществляют разбавление 1:10
2. Субстрат в буфере
(a) 198 мг хлористого ацетилтиохолина (10
мМ)
(b) доведение до объема 100 мл с помощью 0,05 М фосфатного буфера,
рН 7,2 (реагент 1)
3. ДТNB в буфере
(a) 19,8 мг 5,5-дитиобиснитробензойной кислоты (ДТNB) (0,
5 мМ)
(b) доведение до объема 100 мл с помощью 0,05 М фосфатного буфера,
рН 7,2 (реагент 1)
4. Приготавливают 22 мМ исходный раствор испытуемого лекарства в подходящем
растворителе и доводят до необходимого объема с помощью 0,5 мМ ДТNB (реагент 3). Лекарства серийно разбавляют (1:10) так, что конечная концентрация (в кювете) составляет 10-4 М, и
подвергают скринингу на активность. В случае наличия активности по ингибиторной активности последующих концентраций определяли значения IC50.
В. Препарат ткани
Самцов крыс разновидности Вистар подвергали декапитации, быстро удаляли мозги, препарировали полоски основания мозга, взвешивали и гомогенизировали их в 19 объемах (примерно 7 мг протеина/мл) 0,05 М
фосфатного буфера, рН 7,2 с использованием гомогенизатора Роtter Elvehjem. Аликвоту гомогената объемом 25 мкл добавляли к 1,0 мл среды для лекарства или испытуемого лекарства различных концентраций и
в течение 10 мин предынкубировали при 37oC.
O. Анализ.
Энзимную активность измеряли с использованием спектрофотометра Бекмана марки ДU-50. Этот метод может использоваться для определения IC50 и измерения кинетических констант.
Набор инструментов
Кинетический модуль Софт-Пак N 598273(10);
Программа N 6
Киндата:
Источник визуальный;
Длина волны 412 нм;
Дозатор отсутствует;
Кюветы 2 мл кюветы, использующие автоматический пробоотборник на 6 образцов;
Слепой
опыт 1 для каждой концентрации субстрата;
Временной интервал 15 с (15 или 30 с для кинетики);
Общее время 5 мин (5 или 10 мин для кинетики);
График имеется;
Интервал автоматическая шкала;
Наклон увеличивающийся;
Результаты положительные (имеется наклон);
В пустые кюветы и кюветы с образцами добавляли следующие реагенты:
Пустые кюветы: 0,8 мл фосфатный буфер/ДТNB; 0,8 мл буфер/субстрат
Контрольные кюветы: 0,8 мл фосфатный буфер/ДТNB/энзим (фермент); 0,8 мл фосфатный буфер/субстрат
Лекарство: 0,8 мл
фосфатный буфер (ДТNB) лекарство/энзим; 0,8 мл фосфатный буфер/субстрат
Для каждого опыта определяли значения в пустых кюветах с целью контроля неэнзимного гидролиза субстрата и эти значения
автоматически вычитались по программе Киндата, которой располагает кинетический модуль Софт-Пак. По этой программе также рассчитывается скорость изменения поглощения в каждой кювете.
Для определения IC50:
При определениях концентрацию субстрата равную 10 мМ разбавляли в соотношении 1:2 с получением конечной концентрации 5 мМ. Концентрация ДТNB составляла 0,5
мМ,
что давало конечную концентрацию 0,25 мМ.

Значения IC50 рассчитывали на с помощью log-пробит анализа
Результаты испытания некоторых соединений настоящего изобретения и физостигмина (ссыльное соединение) представлены в табл. 1.
Данная полезность дополнительно демонстрируется способностью этих соединений восстанавливать недостаток памяти, связанный с холинергическим дефицитом, в испытании на избегание темноты, описанном ниже.
Испытание на избегание темноты
В этом опыте мышей испытывали на их способность запоминать неприятный раздражитель в течение 24 ч. Мышей помещали в камеру с темным отделением; в
результате
воздействия сильного освещения от лампы накаливания мыши перебегали в темное отделение, в котором применяли электрошок через металлические пластины на полу. Животных удаляли из
испытательного
устройства и снова подвергали испытанию через 24 ч на их способность запоминать действие электрошока.
Если скополамин, антихолинергетик, который как известно вызывает ухудшение памяти, применяется до первоначального испытания животного в испытательной камере, то животное повторно входит в темное отделение вскоре после помещения в испытуемую камеру через 24 ч. Данный эффект скополамина блокируется активным испытуемым соединением, что приводит к большему интервалу времени перед повторным вхождением в темное отделение.
Результаты действия активного соединения выражали в процентах животных в группе с блокированным эффектом скополамина, что выражалось увеличением интервала времени между помещением в испытуемую камеру и повторным входом в темное отделение.
Результаты такого испытания для некоторых соединений изобретения и такрина, а также пилокарпина (ссылочного соединения) представлены в табл. 2.
Данная полезность дополнительно демонстрируется способностью соединений изобретения ингибировать фермент моноаминоксидазу, повышать уровни содержания в мозге биогенных аминов и действием в качестве антидепрессантов.
Ингибирование активности моноаминоксидазы типа А и типа В в синаптосомах головного мозга крыс.
Цель установление селективного ингибирования двух указанных форм формоноаминоксидазы (МАО).
Введение.
Для двух указанных выше форм моноаминоксидазы, которые называют тип А и тип В, известно метаболическое дезаминирование аминов. Существование двух таких форм базируется на различных специфичностях в отношении субстрата и ингибитора. Серотонин (5НТ) и норепинефрин (NE) являются субстратами для МАО типа А, β-фенэтиламин (РЕА) и бензиламин являются субстратами для МАО типа В, тогда как допамин (ДА) и тирамин представляют собой субстраты для обоих типов. Клорилин является селективным ингибитором для энзима типа А, депренил и паргилин селективные ингибиторы для энзима типа В, а транилципромин и ипрониазид не являются селективными ингибиторами. Установлено, что ингибиторы МАО обладают антидепрессантными свойствами.
Хотя существуют разнообразные методы измерения МАО активности, описанный способ включает экстракцию радиомеченых дезаминированных метаболитов (3H)-5НТ или (14C)-b-фенэтиламина. Такой метод позволяет измерять активности МАО-А и МАО-В-одновременно или по отдельности.
Методика
А. Реагенты
1.
Фосфатный буфер (0,5 М), рН 7,4: 134 г NaH2PO4•7H2O доводят до объема 1 л с помощью дистиллированной воды (А), 17,3 г Na2HPO4 доводят
до
объема 250 мл с помощью необходимого количества дистиллированной воды (В).
рН А доводят до 7,4 путем медленного добавления В (используя необходимые объемы реагентов).
Проводят разбавление в соотношении 1:10 дистиллированной водой (0,05 М РO4 буфер, рН 7,4).
2. 0,25 М сахароза (в буфере РO4):
21,4 г сахарозы
доводят до
объема 250 мл с помощью 0,05 М буфера РO4
3. Субстрат для МАО-А:
а. Серотонин креатин SO4 (5НТ) получали от Сигма Кемикал Компани. Готовили 5 мМ
исходный
раствор в 0,01 н. НСl. Этот раствор использовали для разбавления специфической активности (3H)-5НТ.
b. [3Н] -5-гидрокситриптамин биноксалат (20-30 Сi, ммоль) получали от Нью-Инглэнд Ньюклеар.
с. 12 мкл (3H)-5НТ добавляли к 2 мл 5 мМ 5 НТ раствора (конечная концентрация амина в анализе составляла 200 мкМ, см. ниже).
4. Субстрат для МАО-В.
а. b-фенэтиламин (РЕА) получали от Сигма Кэмикал Компани. Готовили 5 мМ исходный раствор в 0,01 н. НСl. Этот раствор использовали для разбавления специфической активности (14C)-РЕА.
b. b-[этил-1-14-С] фенэтиламин гидрохлорид (40-50 Сi, ммоль) получали от Нью Инглэнд Ньюклеар.
с. 12 мкл (14 C)-РЕА добавляли к 2 мл 5 мМ раствора РЕА. (Конечная концентрация амина в анализе составляла 200 мкМ; см. ниже).
5. Равные количества субстратов МАО-А (5НТ) и МАО-В(РЕА) объединяли для одновременного испытания обоих типов МАО, т.е. получали смешанный раствор 2,5 мМ 5НТ и 2,5 мМ РЕА, 40 мкл такого смешанного раствора дает конечную концентрацию каждого амина в анализе 200 мкМ. При испытании только одного типа МАО индивидуальные 5 мМ исходные растворы следует разбавлять в соотношении 1:1 дистиллированной водой перед добавлением 40 мкл в инкубационную смесь, т.е. имеет место та же конечная концентрация амина равная 200 мкМ.
В. Тканевый препарат.
Самцов крыс разновидности Вистар весом 15-250 г умертвляли и быстро извлекали мозги. Всю мозговую массу без мозжечка гомогенизировали в 30 об. охлажденной льдом 0,25 М сахарозы с фосфатным буфером с использованием гомогенизатора Роtter Elvejhem. Гомогенат центрифугировали при 1000 g в течение 10 мин и надосадочный слой (S1) декантировали и проводили повторное центрифугирование при 18000 g в течение 20 мин. Полученный в результате осадок (Р2) повторно суспендировали в 0,25 М свежей сахарозе. Полученный продукт служил тканевым источником для митохондриальной МАО.
С. Анализ
10 мкл 0,5 М РО4 буфера, рН 7,4.
50 мкл Н2О или соответствующая концентрация лекарства
400 мкл тканевой суспензии.
Пробирки предварительно инкубировали в течение 15 мин при 37oC и начинали анализ путем добавления 40 мкл объединенного субстрата ([3H]-5НТ и [14C] -РЕА) через 15-секундные интервалы. Пробирки инкубировали в течение 30 мин при 37oC, и реакцию прекращали путем добавления 0,3 мл 2 норм. НСl. Тканевые холостые значения определяли путем добавления кислоты перед радиоактивным субстратом. Окислительные продукты реакции экстрагировали смесью этилацетат/толуол (1: 1). 5 мл такой смеси добавляли в пробирки. Полученную смесь перемешивали в течение 15 мин с помощью турбинной мешалки с целью экстракции дезаминированных метаболитов с переводом их в органическую фазу, которой давали возможность отделяться от водной фазы. Пробирки помещали в ванну со смесью ацетон/сухой лед с целью вымораживания водного слоя. После вымораживания данного слоя верхний органический слой переливали в сцинтилляционную ампулу. Добавляли 10 мл ликвисцинта и проводили подсчет проб с использованием устройства с окошками для14C в одном канале и3H во втором канале. Значения IС50 определяли с помощью log-пробит анализа.
Результаты анализа по ингибированию моноаминооксидазы для соединений настоящего изобретения представлены в табл. 3.
Авторами изобретения был также проведен анализ на связывание клонидина, описанный ниже, с целью установления взаимодействия соединений изобретения с a2-рецепторами.
Связывание 3Н-клонидина: α2-рецептор.
Введение.
Было показано, что ряд антидепрессантов усиливает нейрональное выделение норэпинефрина в результате предполагаемой блокады персинаптического α2-рецептора, и такое свойство может иметь большое значение в отношении механизма действия таких соединений. Взаимодействие соединения с центральными α2-рецепторами оценивали в анализе на связывание 3Н-клонидина.
Методика.
А. Реагенты
1. а. 57,2 г Трис НСl
16,2 г Трис основания доводят до объема 1 л (0,5 М Трис буфера, рН 7,7)
b. Проводили разбавление в
соотношении 1:10 дистиллированной водой (0,05 М Трис
буфера, рН 7,7)
2. Трис буфер, содержащий физиологические ионы
а. исходный буфер
NaCl 7,014 г
КСl 0,372 г
СаСl2 0,222 г доводили до 100 мл в 0,5
М Трис буфере
МgCl2 0,204 г
b. Проводили разбавление дистиллированной Н2О в соотношении 1:10. В
результате этого получали 0,05 М трис НСl, рН 7,7; содержащий
NaCl (120 мМ), КСl (5 мМ), СаСl2 (2 мМ) и МgCl2 (1 мМ)
3. [4-3H]-Клонидин гидрохлорид (20-30
Сi/ммоль) получали от Нью Инглэнд Ньюклвар. Определение значений
IC50: раствор3H-клонидина доводили до концентрации 120 нМ, и в каждую пробирку добавляли по 50 мкл (в
результате в анализиpуемом объеме 2 мл получали конечную концентрацию 3
нМ).
4. Клонидин-НСl получали от Берингер-Ингельхайм. Готовили исходный раствор 0,1 мМ клонидина с целью определения неспецифического связывания. В анализе это давало конечную концентрацию 1 мкМ (20 мкл на 20 мл).
5. Испытуемые соединения. В большинстве анализов в подходящем растворителе готовили 1 мМ исходный раствор и подвергали его серийному разбавлению так, чтобы конечная концентрация в анализе составляла от 10-5 до -10-8 М. В каждом анализе использовали по семь концентраций, причем в зависимости от активности лекарства могут применяться более высокие или более низкие концентрации.
В. Тканевый препарат.
Крыс разновидности Вистар умерщвляли обезглавливанием и быстро рассекали корковую ткань. Эту ткань гомогенизировали в 50 мл 0,05 М Трис буфера, рН 7,7 (буфер 1b) с помощью Политрона Бринкмана и затем центрифугировали в течение 15 мин при 40000 g. Верхний слой отбрасывали, и осадок повторно гомогенизировали в первоначальном объеме 0,05 М Трис буфера рН 7,7 и повторно центрифугировали, как описано выше. Верхний слой отбрасывали и конечный осадок повторно гомогенизировали в 50 об. буфера 2b. Такую тканевую суспензию затем хранили на льду. Конечная концентрация ткани составила 10 мг/мл. Специфическое связывание составляло 1% от общего количества добавленного лиганда и 80% от общего количества связанного лиганда.
С. Анализ.
100 мкл 0,5 М
Трис-физиологических солей, рН 7,7 (буфер 2а)
830 мкл Н2О
20 мкл среды для
лекарства (для общего связывания) или 0,01 мМ клонидина (для неспецифического связывания) или
соответствующая концентрация лекарства
50 мкл3H-клонидинового сырья
1000 мкл
тканевой суспензии
Тканевые гомогенаты инкубировали в течение 20 мин при 25oC с 3 нМ3H-клонидином и различными концентрациями лекарств, после чего их немедленно
фильтровали при пониженном давлении на фильтрах Ватман СF/B. Фильтры трижды промывали 5 мл
охлажденного на льду 0,05 М Трис буфера, рН 7,7, после чего переносили в сцинтилляционные ампулы. К каждому
образцу добавляли по 10 мл раствора Ликвисцинта, после чего осуществляли подсчет с помощью
жидкостной сцинтилляционной спектроскопии. Специфическое связывание клонидина определяли как разность между
общим связыванием и тем, что было установлено с помощью log-пробит анализа. Указанный
процент ингибирования при каждой концентрации лекарства представляет собой среднее значение из трех
определений.
Результаты анализа на связывание 3Н-клонидина соединений изобретения представлены в табл. 4.
С целью оценки степени безопасности соединений настоящего изобретения ниже приводятся данные по токсичности ряда соединений, выраженные в виде 50% -ной острой летальной дозы ALD50, при испытании на мышах.
Испытываемые соединения
вводились внутрибрюшинно, а в некоторых случаях
подкожно.
В частности, соединения примеров 2, 8, 10, 12, 13, 16, 17, 18, 19 при внутрибрюшинном введении и примеров 22, 23, 30, 38, 39 и 41 при подкожном введении показали значения ALD50 >80 (мг/кг). Данный показатель для соединения примера 6 составил >40, но<80.
Эффективные количества соединений изобретения могут применяться на пациентах любым из многочисленных методов, например орально путем приема капсул или таблеток, парентерально в виде стерильных растворов или суспензий, и в некоторых случаях внутривенно в виде стерильных растворов. Конечные продукты в виде свободных оснований, хотя и обладают эффективностью сами по себе, могут быть сформированы и приниматься в виде их фармацевтически приемлемых солей присоединения кислот в виду стабильности, удобства кристаллизации, увеличения растворимости и т.п.
Кислоты, используемые для получения фармацевтически приемлемых солей присоединения кислот настоящего изобретения, включают такие неорганические кислоты, как соляную, бромистоводородную, серную, азотную, фосфорную и надхлорную кислоту, а также такие органические кислоты, как винная, лимонная, уксусная, янтарная, малеиновая, фумаровая, 2-нафталинсульфокислота и щавелевая.
Активные соединения настоящего изобретения могут применяться орально, например, в присутствии инертного разбавителя или съедобного носителя либо они могут быть помещены в желатиновые капсулы или запрессованны в таблетки. В целях орального терапевтического применения активные соединения могут вводиться в присутствии эксципиентов и использоваться в виде таблеток, капсул, эликсиров, суспензий, сиропов, облаток жевательной резинки и т.п. Такие препараты должны содержать по крайней мере 0,5% активного соединения, однако это количество может изменяться в зависимости от конкретной формы и может составлять 4-70% от веса дозировочной формы. Количество активного соединения в таких композициях является таким, чтобы получались подходящие дозировки. Предпочтительные композиции и препараты согласно настоящему изобретению получают таким образом, что оральная дозировочная форма содержит 1,0-300 мг активного соединения.
Таблетки, пилюли и т. п. могут также содержать следующие ингредиенты: связующие, такие как микрокристаллическая целлюлоза, аравийская камедь или желатин; эксципиенты, как крахмал или лактоза, дезинтегрирующий агент, как альгиновая кислота, Примогель, пшеничный крахмал и т.п. смазочные агенты, как стеарат магния или Стеротекс; агенты скольжения, такие как коллоидный диоксид кремния, а также подслащивающие агенты, как сахароза или сахарин, при этом могут также добавляться такие отдушки, как перечная мята, метилсалицилат или апельсиновая отдушка. В том случае, когда единичная дозировочная форма представляет собой капсулу, последняя может содержать, помимо указанных выше материалов, жидкий носитель, такой как жирное масло. Другие единичные дозировочные формы могут содержать различные другие материалы, которые модифицируют физическую форму единичной дозировки, например, путем образования покрытия. Так, например, таблетки или пилюли могут быть покрыты сахаром, шеллаком, или другими желудочными покрывающими агентами. Сироп может содержать, помимо активных соединений, сахарозу в качестве подслащивающего агента, а также некоторые предохраняющие агенты, красители, окрашивающие вещества и отдушки. Используемые материалы, применяемые при приготовлении таких различных композиций, должны быть фармацевтически чистыми и нетоксичными в используемых количествах.
В целях парентерального терапевтического применения активные соединения настоящего изобретения могут вводиться в раствор или суспензию. Такие препараты должны содержать по крайней мере 0,1% активного соединения, однако это количество может изменяться в интервале 0,5-30 мас. Количество активного соединения в таких композициях является таким, чтобы получалось подходящая дозировка. Предпочтительные композиции и препараты согласно настоящему изобретению готовят так, чтобы единичная парентеральная доза содержала 0,5-100 мг активного соединения.
Растворы и суспензии могут также включать следующие компоненты: стерильные разбавители, как вода, солевой раствор, фиксированные масла, полиэтиленгликоли, пропиленгликоль или другие синтетические растворители; антибактериальные агенты, как бензиловый спирт или метилпарабены; антиоксиданты, как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, как этилендиаминтетрауксусная кислота; такие буферы, как ацетаты, цитраты или фосфаты, а также агенты для регулирования тоничности такие, как хлористый натрий или декстроза. Парентеральный препарат может быть помещен в имеющиеся в распоряжении шприцы или ампулы с многократной дозой, выполненные из стекла или пластмассы.
Примеры соединений изобретения включают следующие вещества:
1,2,3,4-тетрагидроциклопент[b]индол-3-амин;
4-метил-1,2,3,4-тетрагидроциклопент[b]индол-3-амин;
1,2,3,
4-тетрагидроциклопент[b]индол-3-циклопропиламин;
4-метил-1,2,3,
4-тетрагидроциклопент[b]индол-3-циклопропиламин;
1,2,3,4-тетрагидроциклопент[b]индол-3-(2-пропинил)амин;
1,2,3,
4-тетрагидроциклопент[b]индол-3-(N-формил)амин;
1,2,3,
4-тетрагидроциклопент[b]индол-3-(N-фенилметилоксикарбонил);
1,2,3,3а,4,8b-гексагидроциклопент[b]индол-3-амин;
1,2,3,3а,
4,8b-гексагидро-4-метилциклопент[b]индол-3-амин;
1,2,
3,3а,4,8b-гексагидро-4-метилциклопент[b]индол-3-(2-пропинил)- 4-метил-3-фенилметиламино-1,2,3,4-тетрагидроциклопент(b)- индол-7-ил
метилкарбамат;
3-(N-циклопропил)-амино-4-метил-1,2,3,
4-тетрагидроциклопент-[b] - индол-7-ол;
3-(N-циклопропил)амино-4-метил-1,2,3,4-тетрагидроциклопент-[b] индол-7-ил метилкарбамат;
3-циклопропиламино-1,2,3,3а,
8b-гексагидро-4-метилциклопент [b]-индол-7-ол;
3-циклопропиламино-1,2,3,3а, 4,8b-гексагидроциклопент[b] индол-7-ил фенилметилкарбонат;
3-(N-циклопропил-N-метиламинокарбонил)амино-1,
2,3,3а, 4,8b- гексагидроциклопент[b] индол-7-ил фенилметилкарбонат; 3-(N-циклопропил-N-метиламинокарбонил)амино-1,2,3,3а,4,
8b-гексагидро-4- метилциклопент[b]индол-7-ол;
3-циклопропиламино-4-метил-1,2,3,4-тетрагидроциклопент [b] -индол-7-ил метилкарбамат;
1,2,3,3а,4,
8b-гексагидро-4-метил-3-фенилметиламино-циклопент-[b]- индол-7-ол;
1,2,3,3а,4,
8b-гексагидро-4-метил-3-аминоциклопент[b] индол-7-ол;
1,2,3,3а, 4,
8b-гексагидро-4-метил-3-фенилметилоксикарбониламиноциклопент [b]индол-7-ол;
1,2,3,3а,4,
8b-гексагидро-4-метил-3-(N-фенилметилоксикарбонил)- аминоциклопент[b]индол-7-ил метилкарбамат;
1,2,3,3а,4,8b-гексагидро-4-метил-3-метиламинокарбонил-аминоциклопент [b] индол-7-ол;
1,
2,3,3а, 4,8b-гексагидро-4-метил-3-(N-фенилметил-N-метиламинокарбонил) аминоциклопент[b]индол-7-ол;
4-трет.-бутилоксикарбонил-1,4-дигидроциклопент[b] индол-3(2Н)-он;
7-хлорацетил-1,
4-дигидро-4-метилциклопент[b]индол-3-(2Н)-он;
7-хлорацетилокси-1,
4-дигидро-4-метилциклопент[b]индол-3(2Н)-он;
1,4-дигидро-7-гидрокси-4-метилциклопент[b] индол-3-(2Н)-он; 1,
4-дигидро-7-метиламинокарбонилокси-4-метилциклопент[b]индол-3(2Н)-он;
3-гидроксиимино-7-метокси-1,2,3,4-тетрагидроциклопент[b]индол;
3-гидрокисилимино-1,2,3,
4-тетрагидроциклопент[b]индол;
3-гидроксилимино-4-метил-1,2,3,4-тетрагидроциклопент[b]индол;
3-(2-аминоэтил)оксимино-4-метил-1,2,3,4-тетрагидроциклопент[b]индол;
3-циклопропиламино-1,2,3,4-тетрагидроциклопент[b]индол;
3-циклопропиламино-4-метил-1,2,3,
4-тетрагидроцикломент[b]индол;
3-гидроксилимино-4-метил-1,2,3,
4-тетрагидроциклопент[b]индол-7- ацетат;
4-метил-3-фенилметилимино-1,2,3,4-тетрагидроциклопент[b]индол;
4-метил-3-фенилметилимино-1,2,3,4-тетрагидроциклопент[b]индол метилкарбамат;
4-метил-3-[(2-фенилциклопропил)имино] -1,2,3,4-тетрагидроцикло[b] индол-7-ол;
4-метил-3-[(2-фенилциклопропил)имино] -1,2,3,4-тетрагидроцикло[b] индол-7-ил метилкарбамат;
3-циклопропилимино-4-метил-1,2,3,4-тетрагидроциклопент[b]индол-7-ол;
3-метиламинокарбонилоксиимино-4-метил-1,2,3,4-тетрагидроциклопент[b] индол-7-ил метилкарбамат;
3-циклопропиламино-1,2,3,3а,4,8b-гексагидро-4-метилциклопент[b]индол7-ил метилкарбамат;
3-амино-1,2,3,3а,4,8b-гексагидро-4-метилциклопент[b]индол-7-ил;
-1,2,3,
4-тетрагидроизохинолилкарбамат;
5-бром-3-циклопентиламино-1,2,3,3а, 4,8а-гексагидро-4-метилциклопент [b]
индол-7-ил фенилметилкарбамат;
3-[2-морфолиноэтиламино]-4-метил-1,2,3,
4-тетрагидроциклопент[b]индол7-ил фенилэтилкарбамат;
4-метил-3-(4-пиперидинил)-амино-1,2,3,
4-тетрагидроциклопент[b] индол7-ил фенилэтилкарбамат.
П р и м е р 1.
3-гидроксиимин-7-метокси-1,2,3,4-тетрагидроциклопент[b]индол.
Перемешиваемый раствор 1,2-циклопентадион моно-4-метоксифенилгидразона (6,0 г) в 100 мл 10%-ного водного раствора Н2SО нагревали на паровой бане в течение 4 ч и после этого давали охлаждаться до комнатной температуры и отфильтровывали с получением 1, 4-дигидро-7-метокси-циклопент[b]индол-3(2Н)-она в виде твердого вещества. К индолу (2,6 г) в 25 мл 95%-ного раствора ЕtОН добавляли гидрохлорид гидроксиламина (1,7 г) в 15 мл воды и затем ацетат натрия (2,1 г) в 15 мл воды. Полученную смесь нагревали с обратным холодильником в течение 2,5 ч и после этого оставляли стоять в течение ночи. ЕtОН удаляли в вакууме и образовавшийся твердый материал собирали и очищали методом флеш-хроматографии с получением 0,8 г смеси изомеров оксима, т.пл. 169-175oC (разл.).
Элементный анализ:
Вычислено для С12H12N2O2:
С 66,65% H 5,95% N 12,95%
Найдено: C 66,39% H 5,51, N 12,91%
П р и м е р
2.
3-гидроксиимино-1,2,3, 4-тетрагидроциклопент[b]индол.
К перемешиваемому раствору 1,4-дигидроциклопент[b]индол-3-(2Н)-она (10 г) в 100 мл 95% ЕtОН добавляли гидрохлорид гидроксиламина (8,3 г) в 20 мл воды, после чего добавляли ацетат натрия (9,7 г) в 20 мл воды. Полученную смесь нагревали с обратным холодильником в течение 2 ч, после этого оставляли стоять в течение ночи при комнатной температуре. ЕtОН удаляли в вакууме, и образовавшийся твердый материал собирали и перекристаллизовывали из 95% ЕtОН с получением 4,5 г преимущественно одного изомера оксима в первой партии и 3,0 г смеси изомеров оксима во второй партии. 1,5 г образца одного изомера перекристаллизовывали с получением 0,9 г аналитически чистого материала, т.пл. 185-189oC.
Элементный анализ:
Вычислено для С11H10N2O2:
С 70,95% H 5,41, N 15,04%
Найдено: C 70,71% H 5,32% N 14,
94%
Элкс с сотр. J. Chem. Soc. 624 (1944).
П р и м е р 3.
3-гидроксиимино-4-метил-1,2,3,4-тетрагидроциклопент[b]индол.
К перемешиваемому раствору 1,4-дигидро-4-метилциклопент[b]индол-3(2Н)-она (3,0 г) в 30 мл 95% ЕtОН добавляли гидрохлорид гидроксиламина (2,25 г) в 9 мл воды с последующим добавлением ацетата натрия (4,4 г) в 9 мл воды. Смесь нагревали с обратным холодильником в течение 4 ч, после чего добавляли еще 1,1 г гидрохлорида гидроксиламина в 5 мл воды и 2,2 г ацетата натрия в 5 мл воды. Через 2 ч дополнительного кипячения с обратным холодильником смесь оставляли стоять в течение ночи при комнатной температуре. Осажденный материал собирали и перекристаллизовывали из 95% ЕtОН с получением 1,9 г аналитически чистого материала, т.пл. 197-199oC.
Элементный анализ:
Вычислено для С12H12N2O2:
С 71,98% Н 6,04% N 13,99%
Найдено: C 72,18% H 6,14% N 14,00%
П р и м е р
4.
3-(2-аминоэтил)оксимино-4-метил-1,2,3,4-тетрагидроциклопент[b]индол.
К перемешиваемой суспензии 3-гидроксимина-4-метил-1,2,3,4-тетрагидроциклопент[b] индола (5,0 г) в хлористом метилене (50 мл) добавляли 50%-ный раствор NaОН (50 мл) с последующим добавлением бромистого тетрабутиламмония (800 мг) и гидробромида бромэтиламина (7,6 г). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Слои разделялись, и водный слой экстрагировали хлористым метиленом (50 мл). Органические слои объединяли, сушили (NaSО4) и концентрировали. Продукт очищали методом флеш-хроматографии на силикагеле, проводя элюирование смесью 10% метанол/хлористый метилен с получением 1,1 г очищенного материала.
П р и м е р 5.
1,2,3,4-тетрагидроциклопент[b]индол-3-амин.
К перемешиваемому раствору 3-гидроксимина -1,2,3, 4-тетрагидроциклопент[b]индола (6 г) в 16 мл 95% ЕtОН при 0oC добавляли сплав никеля (Harshaw Ni-100ОР, 10 г), после чего добавляли 12,9 г гидроксида натрия в 150 мл воды. Баню со льдом удаляли через 0,5 ч, и полученную смесь перемешивали еще 1 ч и фильтровали. ЕtОН удаляли в вакууме, и полученный продукт кристаллизовался, давая 5,0 г твердого вещества. Образец такого вещества перекристаллизовывали из толуола с получением аналитически чистого материала, т.пл. 158-160oC.
Элементный анализ:
Вычислено для С11H12N2:
C 76,71% H 7,02% N 16,26%
Найдено: С 76,
44% Н 6,98% N 15,99%
П р и м е р 6.
4-метил-1,2,3,4-тетрагидроциклопент[b]индол-3-амингидрохлорид.
К перемешиваемому раствору 3-гидроксимина-4-метил-1,2,3, 4-тетрагидроциклопент[b]индола (5 г) в 200 мл 95% ЕtОН при 0oC добавляли сплав никеля (9 г), после чего добавляли 11 г гидроксида натрия в 200 мл воды. Ледяную баню удаляли через 0,25 ч, и полученную смесь перемешивали еще в течение часа. Добавляли дополнительное количество никелевого сплава (2 х 1 г), и полученную смесь перемешивали в течение 2 ч. Катализатор удаляли фильтрацией, ЕtОН удаляли в вакууме, и продукт экстрагировали в СН2Cl2 (2 х 100 мл). СН2Cl2 экстракты высушивали (Na2SO4) и концентрировали с получением масла (4,5 г). Масло (2,0 г) растворяли в диэтиловом эфире (100 мл) и добавляли эфирный НСl до легкого подкисления раствора. Образовавшееся твердое вещество отфильтровывали и сушили в течение ночи с получением 1,6 г 4-метил-1,2,3,4-тетрагидроциклопент[b]индол-3-амина в виде дихлорида, т.пл. 169-173oC (разл.).
Элементный анализ
Вычислено для
С12H14N2, НСl:
C 64,72% H 6,79% N 12,58%
Найдено: C 64,41% H 6,82% N 12,18%
П р и м е
р 7.
4-трет-бутилоксикарбонил-1,
4-дигидроциклопент[b]индол-3-(2Н)-он
К перемешиваемому раствору 1,4-дигидроциклопент[b]индол-3-(2Н)-она (10,0 г) в ацетонитриле (100 мл)
добавляли ди-трет.бутилпирокарбонат (15 г) с
последующим добавлением 4-диметиламинопиридина (700 мг). Полученную смесь перемешивали в течение ночи при комнатной температуре в атмосфере азота.
Растворитель удаляли при пониженном давлении,
остаток очищали методом испарительной колонной хроматографии с получением 4-трет.-бутилокси карбонил-1,4-дигидроциклопент[b]индол-3-(2Н)она (4,5 г) в виде
твердого вещества.
Элементный анализ:
Вычислено для С16H17NO3: С 70,83% Н 6,32% N 5,16%
Найдено: С 71,04% Н 6,35% N 5,16%
П
р и м е р 8.
1,2, 3,4-тетрагидроциклопент[b]индол-3-циклопропиламин гидрохлорид.
1,4-Дигидроциклопент[b] индол-3(2Н)-он (5,0 г) разделяли на две части и помещали в запаянные пробирки, каждая из которых содержала толуол (20 мл), циклопропиламин (2,0 мл) и молекулярные сита ЗА (1 г). Смеси помещали на масляную баню и нагревали с обратным холодильником в течение 7 ч. Каждой пробирке давали охлаждаться при окружающей температуре, молекулярные сита отфильтровывали, и фильтр концентрировали с получением коричневого твердого вещества, которое идентифицировали, как имин методом ЯМР/МС. Объединенный иминный продукт растворяли в изопропаноле (125 мл) и метаноле (25 мл) и после этого добавляли боргидрид натрия (2,66 г), и полученную смесь перемешивали в атмосфере азота при температуре окружающей среды в течение ночи. Смесь охлаждали при 0oC, медленно добавляли воду, и смесь перемешивали в течение 0,5 ч. Смесь экстрагировали с помощью ЕtAc (2 х 200 мл), слой ЕtОАс экстрагировали 10% НСl (2 х 200 мл), и кислотные экстракты нейтрализовали (10% NaОН) и экстрагировали ЕtOAc (3 х 200 мл). ЕtOAc экстракты сушили (NaSO4) и концентрировали в вакууме с получением 3,5 г продукта. Образец в количестве 1,5 г растворяли в Еt2О (100 мл) и добавляли эфирный раствор НСl, осадок собирали и сушили с получением 1,2,3, 4-тетрагидроциклопент[b]индол-3-циклопропиламин гидрохлорида, т.пл. 165-167oC.
Элементный анализ:
Вычислено для С14H16N2•
HCl: C 67,60% H 6,89, N 26%
Найдено: C 67,22% H 6,87% N 10,79%
П р и м е р 9.
4 -метил-1,2,3,4-тетрагидроциклопент[b]индол-3-циклопропиламин-2-нафталин сульфонат.
1,4-Дигидро-4-метил-циклопент[b] индол-3(2Н)-он (2 г) и циклопропиламин (3,0 г) растворяли в 30 мл толуола и охлаждали до -10oC. Четырехлористый титан (0,70 мл) растворяли в 10 мл толуола и добавляли к первому раствору. Реакционной смеси давали нагреваться до комнатной температуры и перемешивали в течение ночи. Имин отделяли фильтрацией смеси через слой оксида кремния, и растворитель удаляли в вакууме. Имин (2,4 г) растворяли в 100 мл смеси изо-РrОН/МеОН в соотношении 5:1, и после этого добавляли боргидрид натрия (1,2 г). Реакционную смесь перемешивали в течение ночи. Растворители удаляли в вакууме, и продукт реакции, очищенный методом хроматографии, выделяли в виде желтого масла (1,6 г).
Часть циклопропиламиноиндола (0,75 г) растворяли в 75 мл Еt2О и перемешивали, медленно добавляя раствор 0,69 г 2-нафталин сульфокислоты в 50 мл Еt2О. Образовывался белый осадок, который отфильтровывали в атмосфере N2, промывали 2 х 50 мл Еt2О и сушили с получением 1,04 г 4-метил-1,2,3,4-тетрагидроциклопент[b] индол-3-циклопропиламин-2-нафталин сульфоната, т.пл. 140-142oC.
Элементный анализ:
Вычислено для С15H18N2•C10H8O3: C 69,10% H 6,03% N 6,45%
Найдено: C 68,98% H 6,04% N 6,
39%
П р и м е р 10.
1,2,3,4-тетрагидроциклопент[b]индол-3-(2-пропинил) амин.
К перемешиваемому раствору 1,2,3, 4-тетрагидроциклопент[b]индол-3-амина (5,0 г) в тетрагидрофуране (30 мл) в атмосфере азота добавляли триэтиламин (2,9 г), после чего прикапывали бромистый пропаргил (4,45 г 80%-ного раствора в толуоле), растворенный в тетрагидрофуране (20 мл). Смесь перемешивали в течение ночи. Добавляли дополнительное количество бромистого пропаргила (0,01 моля), растворенного в тетрагидрофуране (10 мл), и смесь перемешивали в течение 3 ч. Смесь концентрировали в вакууме, добавляли СН2Cl2 (150 мл), и смесь экстрагировали 10% НСl (2 х 50 мл).
Органическую фазу сушили (Na2SO4) и концентрировали с получением 0,85 г продукта. Реакцию повторяли в том же масштабе с использованием идентичных условий. Продукты объединяли и подвергали хроматографической очистке на силикагеле, проводя элюирование 5% МеОН/CH2Cl2 с получением 1,2,3,4-тетрагидроциклопент[b]индол-3-(2-пропинил)амина (1,6 г), т.пл. 110-112oC.
Элементный
анализ:
Вычислено для С14H14N2: С 79,97% H 6,71% N 13,32%
Найдено: C 79,70% H 6,77% N 13,14%
П р и м
е р 11.
1,2,3, 4-тетрагидроциклопент[b]индол-3-(N-формил)амин.
К перемешиваемому раствору 1,2,3,4-тетрагидроциклопент[b]индол-3-амина (2,0 г) в 25 мл хлористого метилена при комнатной температуре добавляли 4-диметиламинопиридин (1,4 г), после чего добавляли 0,46 мл муравьиной кислоты. Добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид (2,24 г), и смесь перемешивали в течение ночи в атмосфере азота. Реакционную смесь разбавляли СН2Cl2 (100 мл), экстрагировали водой (3х50 мл), сушили (Na2SO4) и концентрировали в вакууме с получением твердого вещества, которое перекристаллизовывали из ЕtОН и толуола с получением 1,2,3,4-тетрагидроциклопент[b]индол-3-(N-формил)амина (1,1 г), т.пл. 167-169oC.
Элементный
анализ:
Вычислено для С12H12N2O: С 71,98% Н 6,04, N 13,99%
Найдено: C 71,91% H 5,86, N 13,54%
П р и м
е р 12.
1,2,3, 4-тетрагидроциклопент[b]индол-3-(N-фенилметилоксикарбонил)амин.
К перемешиваемому раствору 1,2,3,4-тетрагидроциклопент[b]индол-3-амина (5 г) в 50 мл СН2Cl2 при комнатной температуре добавляли триэтиламин (3,2 г), после чего добавляли 5,4 г бензилхлорформиата в 25 мл СН2Cl2. Смесь перемешивали в течение 2 ч и после этого последовательно промывали водой (50 мл) 10% НСl (50 мл) и водой (50 мл). Раствор СН2Cl2 сушили (Na2SO4), концентрировали в вакууме и очищали методом флеш-хроматографии, проводя элюирование смесью гексан/ацетон 2:1, с получением 2,0 г 1,2,3,4-тетрагидроциклопент[b]индол-3-(N-фенилметилоксикарбонил)амина, т.пл. 145-146o C.
Элементный
анализ:
Вычислено для С19H18N2O2: С 74,49% Н 5,92% N 9,14%
Найдено: C 74,23% H 5,99% N 8,96%
П р
и м е р 13.
1,2, 3,3а, 4,8b-гексагидроциклопент[b] индол-3-амин-2-нафталинсульфонат полугидрат.
1,2,3,4-тетрагидроциклопент[b]индол-3-амин (2,0 г) помещали в трехгорлую колбу в атмосфере азота и с помощью шприца прикапывали 34 мл 1,0 М комплекса боран-тетрагидрофуран в тетрагидрофуране. Смесь перемешивали в течение 0,5 ч при 0oC и после этого прикапывали трифторуксусную кислоту (34 мл). После перемешивания в течение 2 ч тетрагидрофуран удаляли в вакууме и остаток подщелачивали 10% NaОН, экстрагировали СН2Cl2 (2х75 мл) и концентрировали в масле (2 г). Образец такого масла в количестве 1,0 г растворяли в эфире (200 мл) и при перемешивании прикапывали раствор (1,3 г) 2-нафталин сульфокислоты в эфире. Образовавшийся осадок собирали фильтрацией в токе азота, т.пл. 120oC (разл.).
Элементный анализ:
Вычислено для С11H15N2:C10H8O3•5H2O: C 64,61% H 5,93, N 7,15%
Найдено: C 64,34% H 5,33% N 6,73%
П р и м е р 14.
1,2,3,3а, 4, 8b- гексагидро-4-метилциклопент[b] индол-3-амин-2-нафталинсульфонат.
4-метил-1,2,3,4-тетрагидроциклопент[b] индол-3-амин (10,2 г) помещали в трехгорлую колбу в атмосфере азота и шприцем прикапывали 17 мл 1,0 М комплекса боран-тетрагидрофуран в тетрагидрофуране. Смесь перемешивали в течение 0,5 ч при 0oC и после этого добавляли трифторуксусную кислоту (185 мл) с помощью капельной воронки для работы под давлением. После перемешивания в течение 1 ч тетрагидрофуран удаляли в вакууме, и остаток подщелачивали 10% NaОН (рН 8), экстрагировали СН2Cl2 (2х5000 мл) сушили над Na2SO4 и концентрировали до масла (10,3 г). Сырой материал очищали методом колонной хроматографии.
1,7 г образца 1,2,3,3а,4, 8b-гексагидро-4-метилциклопент[b]индол-3-амина растворяли в 150 мл Еt2О, и при перемешивании прикапывали раствор 1,9 г 2-нафталинсульфокислоты в эфире. Твердое вещество собирали фильтрацией в токе N2, т.пл. 185-190oC.
Элементный анализ:
Вычислено для С12H17N2•C10H8
O3:
C 66,64% H 6,10% N 7,10%
Найдено: C 66,74, H 6,66% N 6,77%
П р и м е р 15.
1,2,3,3а, 4,8b-гексагидро-4-метилциклопент[b] индол-3-(2-пропинил)-амин гидрохлорид.
1,2,3,3а, 4,8b-гексагидро-4-метилциклопент[b] индол-3-амин (5,0 г) растворяли в 50 мл тетрагидрофурана с триэтиламином (2,7 г). Раствор охлаждали до 0oC и медленно добавляли бромистый пропаргил (3,2 г) в 20 мл тетрагидрофурана. После добавления смесь доводили до комнатной температуры и перемешивали в течение ночи. Отпаривали тетрагидрофуран, и остаток переносили в 200 мл СН2Cl2. Органический слой экстрагировали 10% НСl (2 х70 мл). Водные фракции объединяли и подщелачивали 10% NaОН. Водный слой экстрагировали 2х200 мл СН2Cl2 и органические слои объединяли и сушили над сульфатом натрия. Растворитель удаляли в вакууме. В результате очистки методом флеш-хроматографии на силикагеле получали 1,2,3,3а,4, 8b-гексагидро-4-метилциклопент[b]индол-3-(2-пропинил)амин (2,0 г) в виде красновато-коричневого масла.
1,45 г образца индолина растворяли в эфире и интенсивно перемешивали. Эфирный раствор НСl добавляли к полученному раствору до нейтрального значения рН (рН 6). Затем твердое вещество отфильтровывали и сушили над N2 с получением 1,2,3,3а, 4, 8b-гексагидро-4-метил-циклопент[b] индол-3-(2-пропинил)амин гидрохлорида в виде тонкозернистого белого порошка (1,46 г), т.пл. 195-200oC.
Элементный анализ:
Вычислено для С15H18N2•HCl: С 68,56% Н 7,30% N 10,68%
Найдено: C 68,21% H 7,27% N 10,54%
П р и м е р 16.
7-хлорацетил-1, 4-дигидро-4-метилциклопент[b]индол-3-(2Н)-он.
Хлористый алюминий (8,5 г) суспендировали в СН2Cl2 (20 мл) при 0oC, медленно добавляли хлористый хлорацетил (7,2 г), и смесь перемешивали в течение 5 мин. Смесь прикапывали к перемешиваемому раствору 1,4-дигидро-4-метил-циклопент[b] индол-3(2Н)-она (6,0 г) в 100 мл СН2Cl2 при 0oC. Полученную смесь перемешивали при 0oC в течение 45 мин, и после этого прикапывали дополнительный экстракт предварительно полученного раствора хлористого алюминия и хлористого хлорацетила. Через 30 мин реакционную смесь медленно переливали в перемешиваемую смесь лед/вода. Слои разделяли и СН2Cl2 слой промывали NaHCO3, сушили (Na2SO4) и концентрировали до масла. В результате очистки методом флеш-хроматографии на силикагеле, проводя элюирование смесью гексан/ацетон, получали 7-хлорацетил-1, 4-дигидро-4-метил-циклопент-[b]индол-3-(2Н)-он (4,5 г).
Элементный анализ:
Вычислено для С14H12ClNO2: C 64,25% H 4,62% N 5,35%
Найдено: C 64,35% H 4,61% N 5,24%
П р и м е р 17.
7-Хлорацетилокси-1,4-дигидро-4-метилциклопент[b]индол-3(2Н)-он.
К перемешиваемому раствору 7 хлорацетил-1, 4-дигидро-4-метил-циклопент[b] индол-3(2Н)-она (2,0 г) в хлороформе (100 мл) добавляли фосфат натрия (1,02 г), после чего добавляли м-хлорнадбензойную кислоту (2,5 г, 50-60%-ной чистоты). Смесь перемешивали при комнатной температуре в атмосфере азота в течение 14 ч. Добавляли насыщенный водный раствор NaHCO3 (50 мл), слои разделяли и органический слой промывали водой (2х50 мл). Раствор сушили (Na2SO4), фильтровали и концентрировали с получением желтого масла, которое кристаллизовалось при стоянии. Перекристаллизацией из ЕtОН получали 7-хлорацетилокси-1, 4-дигидро-4-метилциклопент[b]индол-3(2Н)-он (1,1 г).
Элементный анализ:
Вычислено для С14H12NСlO3: C 60,55% H 4,36% N 5,
04%
Найдено:
C 60,47% H 4,33% N 4,98%
П р и м е р 18.
1,4-Дигидро-7-метиламинокарбонилокси-4-метилциклопент[b]индол-3(2Н)-он.
7-Хлорацетилокси-1, 4-дигидро-4-метилциклопент[b] индол-3(2Н)-он (5,0 г) суспендировали в ЕtОН (100 мл) и затем добавляли 10%-ный раствор NaOH, и полученную смесь перемешивали в течение 3 ч при комнатной температуре. Смесь концентрировали в вакууме, добавляли СН2Cl2 (100 мл), после чего добавляли 10% НСl до нейтрализации водного слоя. Слои разделяли и водную фазу экстрагировали СН2 Cl2 (2х100 мл). Органическую часть сушили (Na2SO4) и концентрировали, а остаток перекристаллизовывали из 95% ЕtОН с получением 1, 4-дигидро-7-гидрокси-4-метилциклопент[b] индол-3(2Н)-она в виде белого твердого вещества. В СН2Cl2 растворяли фенол (100 мл) и после этого добавляли 1, 8-диазабицикло-/5.4.0/ундец-7-он (0,4 г), после чего добавляли метилизоцианат (1,4 г), и полученную смесь перемешивали в течение ночи. Смесь концентрировали в вакууме с получением маслянистого твердого вещества, которое перекристаллизовывали из ЕtОН с получением 1,4-дигидро-7-метил-аминокарбонилокси-4-метил-циклопент[b]индол-3(2Н)-она (1,1 г).
Элементный анализ:
Вычислено для С14H14N2O3: С 65,11% Н 5,46% N 10,85%
Найдено: C 65,20% H 5,32% N 10,74%
П р и м е р 19.
3-ацетилоксиимино-4-метил-1,2,3,4-тетрагидроциклопент[b]индол-7-ил ацетат
7-хлорацетилокси-1,4, -дигидро-4-метилциклопент[b]индол-3(2Н)-он (8,0 г) суспендировали в ЕtОН (100 мл), и добавляли
раствор NaOAc (15,6 г) в воде (25 мл) и раствор гидроксиламин гидрохлорида (8,0 г) в воде (25 мл), и полученную смесь нагревали в течение 3 ч с обратным холодильником. Полученную смесь
концентрировали
в вакууме, и остаток перекристаллизовывали из 95% ЕtОН с получением 3-гидроксиимино-4-метил-1,2,3,4-тетрагидроциклопент[b]индол-7-ола в виде белого твердого вещества. Оксим растворяли
в
тетрагидрофуране (100 мл), и после этого добавляли уксусный ангидрид (8,1 г) и 4-диметиламинопиридин (400 мг), и полученную смесь перемешивали в токе азота при комнатной температуре в течение ночи.
Смесь концентрировали в вакууме, добавляли СН2Cl2 (100 мл), и раствор промывали водой (50 мл), 5% NaHCO3 (50 мл) и водой (50 мл). После сушки (Na2SO4) растворитель удаляли в вакууме, и продукт реакции перекристаллизовывали на ЕtОН с получением 3-ацетилоксиимино-4-метил-1,2,3,4-тетрагидроциклопент[b] индол-7-ацетата (1,7 г), т.пл.
150-152oC.
Элементный анализ:
Вычислено для С16H16N2O2: С 63,99% Н 5,37% N 9,33%
Найдено: C 63,56% H 5,37% N
9,
29%
П р и м е р 20.
4-метил-3-фенилметилимино-1,2,3,4-тетрагидроциклопент[b]индол-7-ол.
К перемешиваемой суспензии 7-хлорацетилокси-1, 4-дигидро-4-метилциклопент[b] -индол-3(2Н)-она (6,0 г) в толуоле (50 мл) добавляли бензиламин (9,2 г), и смесь нагревали с обратным холодильником при температуре дефлегмации с азеотропным удалением воды при использовании ловушки Дина-Старка. Через 4 ч с помощью анализа методом ТСХ было показано, что произошла полная конверсия в продукт реакции. Смеси давали охлаждаться до комнатной температуры и ее фильтровали, а твердый материал промывали ацетонитрилом. Фильтрат и промывные жидкости объединяли, концентрировали и очищали методом флеш-хроматографии на силикагеле (элюент гексан/ацетон в соотношении 2:1). Кристаллы, образовавшиеся во фракциях, содержащих продукт, собирали фильтрацией с получением 4-метил-3-фенилметилимино-1,2,3,4-тетрагидроциклопент[b]индол-7-ола (1,1 г), и фильтрат концентрировали с получением масла (3,0 г), которое кристаллизовалось при стоянии, т.пл. 172-173oC.
Элементный анализ:
Вычислено для С19H18
N2O: С 78,58% Н 6,25% N 9,65%
Найдено: С 78,26% Н 6,21% N 9,63%
П р и м е р 21.
4-метил-3-фенилметилимино-1,2,3,4-тетрагидроциклопент[b]индол-7-ол
К перемешиваемому раствору, полученному из 4-метил-3-фенилметилимино-1,2,3,4-тетрагидроциклопент[b] индол-7-ола (16,0 г), изопропанола (200 мл) и метанола (50 мл), добавляли борогидрид натрия
(4,8 г),
и смесь перемешивали в токе азота при температуре окружающей среды в течение 3 ч. Смесь охлаждали до 0oC, медленно добавляли воду и перемешивали в течение 0,5 ч. Полученную смесь
экстрагировали с помощью СН2Cl2 (2х200 мл) и CH2Cl2 экстракты сушили (Na2SO4), концентрировали и подвергали хроматографической
очистке
на силикагеле, проводя элюирование смесью гексана/ацетон в соотношении 2:1. Фракции, содержащие продукт, объединяли с получение 4,25 г 4-метил-3(фенилметиламино)-1,2,3,
4-тетрагидроциклопент[b]
индол-7-ола, т. пл. 159-160oC.
Элементный анализ:
Вычислено для С19H20N2O: С 78,05% Н 6,89% N 9,58%
Найдено: С 78,20% Н 6,
97% N 9,54%
П р и м е р 22.
4-метил-3-фенилметилимино-1,2,3,4-тетрагидроциклопент[b] индол-7-ил метилкарбамат.
К перемешиваемому раствору 4-метил-3-фенилметилимино-1,2,3,4-тетрагидроциклопент[b] индол-7-ола (2,0 г) в СН2Cl2 (40 мл) добавляли 1,8-диазабицикло (5.4.0)ундец-7-ен (0,16 г), после чего прикапывали метилизоцианат (0,39 г) в СН2Cl2 (10 мл). За ходом реакции следили методом ТСХ, и через 3 ч раствор концентрировали с получением 4-метил-3-фенилметилимино-1,2,3, 4-тетрагидроциклопент[b]индол-7-ил метилкарбамата (1,85 г), т.пл. 184-185oC.
Элементный анализ:
Вычислено для С21H21N3O2
: С 72,60% Н 6,09% N 12,09%
Найдено: C 72,59% H 6,01% N 12,05%
П р и м е р 23.
4-метил-3-фенилметилимино-1,2,3,4-тетрагидроциклопент[b]индол-7-ил
метилкарбамат
малеат
К перемешиваемому раствору 4-метил-3-фенилметил-1,2,3,4-тетрагидроциклопент[b] индол-7-ил метилкарбамата (1,8 г) в уксусной кислоте (25 мл) добавляли цианоборгидрид
натрия (0,8 г). За
ходом реакции следили методом ТСХ, и через 2 ч добавляли CH2Cl2 (50 мл) и раствор промывали насыщенным раствором NaHCO3 до нейтрального значения
рН. СН2
Cl2 слой сушили (Na2SO4, фильтровали и концентрировали с получением масла, которое очищали методом флеш-хроматографии, проводя элюирование смесью
гексан/ацетон в
соотношении 2:1. Фракции, содержащие продукт, собирали и концентрировали до масла, которое растворяли в эфире, и после этого добавляли эфирный раствор малеиновой кислоты для
подкисления смеси.
Собирали малеат 4-метил-3-фенилметиламино-1,2,3,4-тетрагидроциклопент[b]индол-7-ил метил карбамата (0,8 г), который осаждался в виде бесцветного твердого вещества, т.пл.
104-106oC.
Элементный анализ:
Вычислено для С21H21N3O2•C4H4O4: C 64,51% H 5,85%
N 9,03%
Найдено: С
64,13% Н 5,75% N 8,97%
П р и м е р 24.
4-метил-3-[(2-фенилциклопропил)имино] -1,2,3,4-тетрациклопент[b] индол-7-ил метилкарбамит
К
перемешиваемой суспензии 1,
4-дигидро-7-гидрокси-4-метилциклопент[b]индол-3(2Н)-она (5 г) в ацетонитриле (100 мл) добавляли фенилциклопропиламингидрохлорид (4,2 г) и после этого добавляли триэтиламин
(2,5 г). Раствор
перемешивали при комнатной температуре в атмосфере азота, прикапывая при этом изопропилат титана (IV). Смесь перемешивали в течение 3 ч, после чего быстро охлаждали системой
лед/вода. Смесь
отфильтровывали, твердые вещества промывали СН2Cl2, слои разделяли и органическую часть сушили (Na2O4). После концентрирования сырой
продукт очищали методом
флеш-хроматографии, проводя элюирование смесью гексан/ацетон (2: 1) с получением 4-метил-3-[(2-фенилциклопропил)имино]-1,2,3,4-тетрагидроциклопент[b] индола.
К перемешиваемому раствору этого продукта (1,0 г) в СН2Cl2 (9,0 мл) добавляли 1,8-диазабицикло-/5.4.0/ундец-7-ен (68 мг) с последующим прикапыванием метилизоцианата (0,18 г) в СН2Cl2 (1,0 мл). За ходом реакции следили методом ТСХ, и через 0,5 ч раствор концентрировали и осадок собирали и перекристаллизовывали дважды из ацетонитрила с получением 4-метил-3-[(2-фенилциклопропил)имино] -1,2,3,4-тетрагидроциклопент[b] индол-7-ил метилкарбамата (0,55 г), т.пл. 166oC (разл.).
Элементный анализ:
Вычислено для
С21H21M3O2: C 73,97% H 6,21% N 11,25%
Найдено: C 73,57% H 6,25% N 11,13%
П р и м е р 25.
3-циклопропилимино-4-метил-1,2,
3,
4-тетрагидроциклопент[b]индол-7-ил
7-хлорацетилокси-1,4-дигидро-4-метилциклопент[b] индол-3(2Н)-он (15,0 г) и циклопропиламин (9,6 г) растворяли в 300 мл толуола и охлаждали до -10o
C. Четыреххлористый титан (6,3 г) растворяли в 50 мл толуола, и смесь медленно добавляли к первому раствору. Реакционную смесь доводили до комнатной температуры и перемешивали в течение ночи.
На
следующий день к реакционной смеси добавляли 1,5 эквивалента амина (4,6 г) и смесь перемешивали в течение часа. Реакционную смесь фильтровали через слой силикагеля, проводя элюирование смесью
гексан/этилацетат в соотношении 3:1, получая после удаления растворителей желтое масло. После очистки методом флеш-хроматографии и перекристаллизации из этилацетата в виде светло-желтого твердого
вещества выделяли 3-циклопропилимино-4-метил-1,2,3,4-тетрагидроциклопент[b] индол-7-ол, т.пл. 176-179oC.
Элементный анализ:
Вычислено для С15H16N2O: C 74,97% H 6,71, N 11,66%
Найдено: C 74,57% H 6,54% N 11,37%
П р и м е р 26.
3-(N-циклопропил)амино-4-метил-1,2,3,
4-тетрагидроциклопент[b]индол -7-ол малеат
3-циклопропилимино-4-метил-1,2,3,4-тетрагидроциклопент[b] -7-ол (17,3 г) растворяли в смеси изопропанол/метанол в соотношении 5:1 (250 мл) в токе
N2 и перемешивали при комнатной температуре. Добавляли боргидрид натрия (8,2 г) и реакционную смесь перемешивали в течение ночи. Методом тонкослойной хроматографии было установлено, что
реакция завершилась. Раствор охлаждали до 0oC и медленно добавляли воду (100 мл). Добавляли этилацетат (250 мл), и после разделения слоев органическую часть последовательно промывали
рассолом (2х100 мл), водой (2х100 мл) и сушили над Na2SO4, после чего растворитель удаляли в вакууме. Сырой материал очищали методом препаративной ВЭЖХ с использованием системы
растворителей гексан/этилацетат в соотношении 2:1. Свободное основание выделяли в виде светлого коричневато-желтого масла (7,8 г). Перемешиваемый раствор свободного основания (0,6 г) в эфире (200 мл)
медленно обрабатывали раствором, приготовленным из 0,3 мг малеиновой кислоты, 50 мл Еt2О и 5 мл ЕtОН.
3-(N-циклопропил)амино-4-метил-1,2,3,4-тетрагидроциклопент[b] индол-7-ол малеат выделяли в виде светло-желтого твердого вещества (0,8 г) после фильтрации и сушки в токе N2, т.пл. 135oC (разл.).
Элементный анализ:
Вычислено для С15H18N2O•C4 H4O4: C 63,68% H 6,19% N 7,82.
Найдено: C 63,47% H 6,13% N 7,69%
П р и м
е
р 27.
3-(N-циклопропил)амино-4-метил-1,2,3,4-тетрагидроциклопент[b] индол-7-ол (5,5 г) растворяли в 250 мл СН2Cl2 совместно с триэтиламином (2,8 г), и смесь охлаждали до 0oC при перемешивании. К первому раствору медленно добавляли бензилхлорформиат (3,9 г), растворенный в 50 мл СН2Cl2. После завершения реакции реакционной смеси давали нагреваться до комнатной температуры, промывали Н2О (2 х 150 мл) и сушили над Na2SO4 и растворитель удаляли в вакууме. Сырой материал очищали методом флеш-хроматографии на колонке с использованием в качестве растворителя ЕtOAc. 3-(N-циклопропил)-амино-4-метил-1,2,3,4-тетрагидроциклопент[b] индол-7-ил фенилметилкарбонат выделяли в виде желтовато-коричневой аморфной пены (4,1 г).
Элементный анализ:
Вычислено для С23H24N2O3: C 73,38% H 6,43% N 7,44%
Найдено: C 73,41% H 6,80, N 7,48%
П р и м е р 28.
3-(N-циклопропил)амино-1,2,3,3а, 4,8b-гексагидро-4-метил-циклопент[b] индол-7-ол.
3-циклопропилимино-4-метил-1,2,3,3а,4,8b-гексагидро-4-метил-циклопент[b] индол-7-ол (8,7 г) помещали в 3-горлую колбу и охлаждали до 0oC на бане со смесью лед-вода. Прикапывали 1М раствор борана в ТГФ (540 мл). Смесь перемешивали в течение 1 ч при медленном нагревании до комнатной температуры. Смесь охлаждали до 0oC и прикапывали трифторуксусную кислоту (119 мл). Раствор перемешивали в течение 15 мин и ТГФ удаляли в вакууме. Смесь нейтрализовали 10% -ным раствором NaОН, экстрагировали хлористым метиленом (4 х 500 мл), сушили (Na2SO4) и концентрировали с получением 3-(N-циклопропил)амино-1,2,3,3а, 4,8b-гексагидро-4-метил-циклопент[b] индол-7-ола (8,8 г), т.пл. 155oC (разл.).
П р и м е р 29.
3-(N-циклопропил-N-метиламинокарбонил)амино-1,2,3,3а-4,8b-гексагидро-4- метилциклопент[b]индол-7-ил гидрохлорид.
3-(N-циклопропил)амино-1,2,3,3а, 4, 8b-гексагидро-4-метил-циклопент [b] индол-7-ол (8,8 г) растворяли в СН2CH2 (400 мл) совместно с триэтиламином (4,4 г). Раствор охлаждали до 0oC и перемешивали в токе азота. К первому раствору медленно добавляли бензилхлорформиат (6,1 г), растворенный в СН2Cl2 (50 мл). За ходом реакции следили методом тонкослойной хроматографии при добавлении дополнительного эквивалента (6,1 г) хлорформиата до тех пор, пока не завершится реакция. Раствор подогревали до комнатной температуры перед промыванием водой (2х 100 мл), сушили над Na2 SO4 и концентрировали до масла, которое затем очищали методом препаративной ВЭЖХ с использованием системы растворителей гексан/ацетон в соотношении 3: 1. Выделяли 3-(N-циклопропил)амино-1, 2,3,3а,4, 8b-гексагидро-4-метилциклопент[b] индол-7-ил фенилметилкарбонат (7,0 г), который охарактеризовывали методами ЯМР, МС и ИК. Этот материал растворяли в СН2Cl2 (250 мл), и раствор обрабатывали 1,8-диазабицикло(5.4.0)ундец-7-еном (ДБУ; 0,4 г). Смесь охлаждали до 0oC и перемешивали, добавляя при этом раствор метилизоцианата (1,1 г) в 50 мл СН2Cl2. За ходом реакции следили методом ТСХ (гексан/ацетон в соотношении 1:1), добавляя при этом еще 2,5 эквивалента (2,7 г) метилизоцианата для завершения реакции. Раствор нагревали до комнатной температуры, промывали рассолом (2х100 мл) и водой (1х100 мл), сушили над Na2SO4 и концентрировали. Масло очищали методом колонной флеш-хроматографии с использованием в качестве растворителя этилацетата. Выделяли 3-(N-циклопропилметиламинокарбонил)амино-1,2,3,3а,4,8b-гексагидро4- метилциклопент[b] индол-7-ил фенилметилкарбонат (4,5 г). Этот материал растворяли в абсолютном этаноле (190 мл) и добавляли 10% палладия на угле (10 мас. 0,4 г). Раствор помещали во встряхиватель Парра, пропускали Н2 (45 фунт/дюйм2) и в течение 2 ч проводили встряхивание. Катализатор отфильтровывали и фильтрат концентрировали. Масло обрабатывали ЕtОАс (50 мл) и СН2Cl2 (5 мл) с получением белого твердого вещества (1,05 г).
3-(N-циклопропил-N-метиламинокарбонил)амино-1,2,3,3а-4,8b-гексагидро-4- метилциклопент[b]индол-7-ол охарактеризовывали методами ЯМР, МС и ИК. Твердое вещество вначале растворяли в смеси Еt2 О/EtОН (200 мл) в соотношении 8:1 и медленно добавляли эфирный раствор хлористого водорода до нейтрального значения рН раствора, после чего добавляли дополнительное количество Еt2О (800 мл). 3-(N-циклопропил-N-метиламинокарбонил)амино-1,2,3,3а-4,8b-гексагидро-4- метилциклопент[b] индол-7-ил гидрохлорид выделяли в виде белого твердого вещества после фильтрации и сушки в токе N2 (0,66 г), т.пл. 155oC (разл.).
Элементный анализ:
Вычислено для С17H23N2O3•HCl: C 60,44% H 7,16% N 12,
44%
Найдено: C 60,77% H 7,41% N 12,67%
П р и м е р 30.
3-(N-циклопропил)амино-4-метил-1,2,3,4-тетрагидроциклопент[b] индол7-ил метилкарбамат.
3-(N-циклопропил)амино-4-метил-1,2,3,4-тетрагидроциклопент[b] индол7-ол (2,2 г) растворяли в СН2Cl2 (200 мл) в присутствии 1,8-диазобицикло/5.4.0/ундец-7-ена (ДБУ, 0,21 г) и раствор охлаждали до 0oC. К охлажденному раствору медленно добавляли раствор метилизоцианата (0,52 г в 30 мл СН2Cl2), и за ходом реакции следили методом тонкослойной хроматографии (силикагель, 11: 1 гексан/этилацетат). После нагревания до комнатной температуры смесь последовательно промывали водой (2х100 мл), рассолом (1х100 мл) и снова водой (1х100 мл). Органический слой сушили над Na2SO4 и растворитель удаляли в вакууме. Сырой материал перекристаллизовывали из ацетонитрила. 3-(N-циклопропил)амино-4-метил-1,2,3, 4-тетрагидроциклопент[b] индол7-ил метилкарбамат выделяли в виде светло-желто-белых пластин (1,0 г), т.пл. 159-160oC.
Элементный анализ:
Вычислено для С17H21N3O2: C 68,21% H 7,07% N 14,04%
Найдено: C 68,08% H 6,57% N 13,97%
П р и м е р 31.
1,2,3,3a, 4, 8b-гексагидро-4-метил-3-(N-фенилметил-N-метиламинокарбонил) аминоциклопент[b]индол-7-ил фенилметилкарбонат.
К перемешиваемому раствору 1,2,3,3а,4, 8b-гексагидро-4-метил-3-фенилметиламиноциклопент[b]индол -7-ола (12,0 г) в СН2Cl2 (125 мл) добавляли триэтиламин (4,08 г). Смесь охлаждали до 0oC и медленно прикапывали раствор бензилхлорформиата (6,8 г) в СН2Cl2 (50 мл). Через три часа реакционную смесь промывали водой (2х100 мл), сушили над Na2SO4 и концентрировали, получая 17,0 г масла. Сырой 4-метил-3-фенилметиламино-1,2,3,3а,4,8b-гексагидроциклопент[b] индол-7-ил фенилметилкарбонат (17,0 г) растворяли в СН2Cl2 (125 мл) и добавляли 1,8-диазабицикло(5.4.0) ундец-7-ен (0,9 г), после чего прикапывали раствор метилизоцианата (2,0 г) в СН2Cl2 (25 мл). Реакционную смесь перемешивали в течение 2 ч и добавляли еще 0,5 г метилизоцианата. Реакционную смесь перемешивали еще в течение 15 мин и после этого концентрировали в вакууме с получением масла, которое очищали методом флеш-хроматографии на силикагеле, проводя элюирование смесью гексан/этилацетат в соотношении 2:1. Фракции, содержащие продукт, собирали и концентрировали с получением масла (5,5 г).
Элементный анализ:
Вычислено для С22H25N3O4: C 71,73% H 6,43% N 8,65%
Найдено: C 71,67% H 6,59% N 8,67%
П р и м е р 32.
1,2,3,3а,4,
8b-гексагидро-4-метил-3-фенилметилоксикарбониламино циклопент[b]индол-7-ол
Раствор 4-метил-3-фенилметилимино-1,2,3,4-тетрагидроциклопент[b]индол-7-ола (14,0 г) помещали в 3-горлую колбу и
охлаждали до 0oC на бане со смесью лед-вода. Прикапывали раствор 1 М боран (ТГФ и ТГФ (145 мл). Смесь перемешивали в течение 1 ч по мере ее медленного нагревания до комнатной температуры.
Смесь снова охлаждали до 0oC и прикапывали трифтоуксусную кислоту. Раствор перемешивали в течение 15 мин, нейтрализовали 10% NaОН, экстрагировали СН2Cl2, сушили
(Na2SO4) и концентрировали с получением 1,2,3,3а, 4,8b-гексагидро-4-метил-3-фенилметиламиноциклопент[b] индол-7-ола (14 г), этот материал растворяли в этаноле (100 мл) и смесь
гидрировали при давлении Н2 45 фунт/дюйм2 (3,167 кг/кв•см) в аппарате Парра в течение 5 ч при 50oC. Смесь фильтровали и раствор концентрировали с получением
3-амино-1,2,3,3а,4,8b-гексагидро-4-метилциклопент[b]индол-7-ола (10,7 г).
К раствору 3-амино-1,2,3,3а,4,8b-гексагидро-4-метилциклопент[b]индол-7-ола (10,7 г) в хлористом метилене (125 мл) добавляли триэтиламин (5,6 г) с последующим прикапыванием бензилхлорформиата (10,0 г) в хлористом метилене (25 мл). Смесь перемешивали в течение 2 ч, экстрагировали водой, сушили (Na2 SO4) и концентрировали. Продукт очищали методом хроматографии на силикагеле, проводя элюирование смесью гексан/ацетон в соотношении 2:1 с получением 2,2,3,3а, 4, 8b-гексагидро-4-метил-3-фенил-метил-оксикарбониламиноциклопент[b]индол-7-ола в виде масла.
П р и м е р 33.
1,2,3,3а,4, 8b-гексагидро-4-метил-3-(11-фенилметилоксикарбонил)аминоциклопент[b] индол-7-ил метилкарбамат.
К перемешиваемому раствору 1,2,3,3а,4, 8b-гексагидро-4-метил-3-(N-фенилметилоксикарбонил) аминоциклопент[b] индол-7-ола (1,8 г) в СН2Cl2 (75 мл) добавляли 1,8-диазобицикло/5.4.0/ундец-7-ен (0,12 г) с последующим прикапыванием раствора метилизоцианата (0,36 г) в СН2Cl2 (25 мл). Реакционную смесь перемешивали в течение 2 ч и добавляли дополнительное количество (0,1 г) метилизоцианата. Реакционную смесь перемешивали еще в течение 15 мин и концентрировали в вакууме с получением масла, которое очищали методом флеш-хроматографии на силикагеле, проводя элюирование смесью гексан/этилацетат в соотношении 2:1. Продукт, который кристаллизовался из чистых фракций, собирали фильтрованием с получением 600 мг вещества, и фильтрат концентрировали с получением масла (800 мг), которое кристаллизовалось при стоянии, т.пл. 180-181oC.
Элементный анализ:
Вычислено для С22H25N3O4: C 66,82% H 6,73%
N 10,63%
Найдено: C 66,91% H 6,47% N
10,66%
П р и м е р 34.
3-метиламинокарбонилоксимино-4-метил-1,2,3,4-тетрагидроциклопент[b] индол-7-ил метилкарбамат полугидрат.
К перемешиваемой суспензии 3-гидроксиимино-4-метил-1,2,3,4-тетрагидроциклопент[b] индол-7-ола (3,0 г) в СН2Cl2 (100 мл) добавляли 1, 8-диазобицикло/5.4.0/ундец-7-ен (630 мг), после чего добавляли метилизоцианат (109 г) и смесь перемешивали в течение ночи при комнатной температуре. Полученную смесь концентрировали в вакууме, и полученное в результате твердое вещество перекристаллизовывали из этанола с получением 3-метиламинокарбонилоксимино-4-метил-1,2,3,4-тетрагидроциклопент [b]индол-7-ил метилкарбамата полугидрата (1,7 г), т.пл. 198-200oC.
Элементный
анализ:
Вычислено для С16H18N4O4•0,5H2O: C 56,68% H 5,66% N 16,
53%
Найдено: C 56,57% H 5,64% N 16,68%
П р и м е
р 35.
1,2,3,3а, 4,8b-гексагидро-4-метил-3-метиламинокарбониламиноциклопент [b] индол-7-ол гидрохлорид моногидрат.
Раствор 1,2,3,3а, 4, 8b-гексагидро-4-метил-3-(N-фенилметил-N-метиламинокарбонил) аминоциклопент[b]индол-7-ил фенилметил карбоната (1,7 г) в ледяной уксусной кислоте (100 мл) гидрировали при давлении Н2 45 фунт/дюйм2 (3,164 кг/кв•см) и 50oC в присутствии 20% гидроксида Рd на угле с использованием аппарата Парра. Через 4 ч методом ТСХ было установлено, что реакция завершилась с образованием как основного, так и побочного продукта. Рd-катализатор отфильтровывали в токе азота, и фильтрат концентрировали в вакууме. Материал подвергали хроматографической очистке на силикагеле, проводя элюирование 10% МеОН/СН2Cl2. Фракции, содержащие продукт, собирали и концентрировали. Полученное в результате масло растворяли в ЕtОН (25 мл) и Еt2О (150 мл), раствор отфильтровывали, и к фильтрату добавляли эфирный раствор НСl для подкисления раствора. Образовавшееся бесцветное твердое вещество собирали в токе азота и сушили в вакууме с образованием 1,2,3, 3а,4,8b-гексагидро-4-метил-3-метиламинокарбонил-аминоциклопент [b] индол-7-ол гидрохлорид моногидрата (0,25 г), т.пл. 120oC (разл.).
Элементный анализ:
Вычислено
для С14H19N3O2•НCl•H2O: C 53,25% H 7,02% N 13,31%
Найдено: C 53,26% H 6,54% N 12,78%
П р и м е р 36.
1,2,3,3а, 4,8b-гексагидро-4-метил-3-(N-фенилметил-N-метиламинокарбонил) аминоциклопент[b]индол-7-ол гидрохлорид.
Раствор 1,2,3,3а, 4, 8b-гексагидро-4-метил-3-(N-фенилметил-N-метиламинокарбонил) аминоциклопент[b]индол-7-ил фенилметил карбоната (2,0 г) в абсолютированном этаноле (100 мл) гидрировали при давлении водорода 45 фунт/дюйм2 (3,164 кг/кв•см) в присутствии 5% Рd на угле с использованием аппарата Парра. Через 2 час методом ТСХ было установлено, что реакция завершена с образованием одного продукта. Рd-катализатор отфильтровывали в токе азота, и фильтрат концентрировали в вакууме. Полученное в результате масло растворяли в ЕtОАс (25 мл) и Еt2О (150 мл), раствор отфильтровывали, и к фильтрату добавляли эфирный раствор НСl для подкисления раствора. Образовавшееся бесцветное твердое вещество собирали в токе N2 и сушили в течение ночи при 40oC в вакууме с получением 1,2,3,3а,4,8b-гексагидро-4-метил-3-(N-фенилметил-N-метиламинокарбонил) аминоциклопент[b]индол-7-ол гидрохлорида (1,4 г), т.пл. 130oC (разл.).
Элементный анализ:
Вычислено для С21H25N3O2•HCl: C 65,02% H 6,76% N 10,83%
Найдено: C 64,95% H 6,85% N 10,84%
П р и м е р
37.
4-метил-3-фенилэтиламино-1,2,3,4-тетрагидроциклопент[b]индол-7-ол полугидрат.
К перемешиваемому раствору 7-гидрокси-4-метил-1, 4-дигидроциклопент[b]-3-она (5,0 г) в ацетонитриле (100 мл) добавляли фенилэтиламин (6,0 г) и изопропилат титана (14,1 г), и полученную в результате смесь перемешивали в атмосфере азота при температуре окружающей среды в течение 3 ч. Смесь переливали в смесь воды со льдом (200 мл) и после этого добавляли СН2Cl2 (500 мл). Смесь отфильтровывали и органический слой отделяли от фильтрата, сушили над сульфатом натрия и концентриpовали в вакууме. Перекристаллизацией из СН2Cl2/гексана получали 4-метил-3-фенил-этиламино-1,2,3, 4-тетрагидроциклопент[b]индол-7-ол полугидрат (3,0 г), т.пл. 75-77oC.
Элементный анализ:
Вычислено для С20H20N2O•0,5H2O: C 76,65% H 6,75% N 8,94%
Найдено: C 76,53%
H 6,38% N 8,89%
П р и м е р 38.
4-метил-3-фенилэтилимино-1,2,3,4-тетрагидроциклопент[b] индол-7-ил метилкарбамат.
К перемешиваемому раствору 4-метил-3-фенилэтилимино-1,2,3,4-тетрагидроциклопент[b] индол-7-ол полугидрата (1,43 г) в СН2Cl2 (25 мл) добавляли 1, 8-диазабицикло/5.4.0/ундец-7-ен (0,11 г). К реакционной смеси добавляли метилизоцианат (0,27 г) в СН2Cl2 (20 мл). За ходом реакции следили методом ТСХ, через 3 ч выпаривали СН2Cl2. Коричневый осадок перекристаллизовывали из ацетонитрила, т.пл. 164-166oC.
Элементный анализ:
Вычислено для С22H23N3O2: C 73,11% H 6,41% N
11,63%
Найдено: C 73,03% H 6,35% N 11,65%
П р и м е р 39.
4-метил-3-(2-фенилэтил)амино-1,2,3, 4-тетрагидроциклопент-индол-7-ил метилкарбамат гидрохлорид полугидрат.
К перемешиваемому раствору 4-метил-3-(2-фенилэтил)имино-1,2,3,4-тетрагидроциклопент[b] индол7-метил карбамата (0,80 г) в уксусной кислоте (8 мл), этаноле (8 мл), изопропаноле (8 мл) и тетрагирофуране (8 мл) добавляли цианоборгидрид натрия (0,35 г) в токе азота. За ходом реакции следили методом ТСХ, и через 1 час раствор нейтрализовали с помощью насыщенного раствора NAHCO3, экстрагировали ЕtАОс, сушили над Na2SO4 и концентрировали в вакууме. Полученное в результате желтое масло растворяли в минимальном количестве ЕtОАс, разбавляли эфиром и после этого добавляли эфирный раствор НСl. Полученное в результате белое твердое вещество собирали фильтрованием. В результате перекристаллизации из этанола получали 4-метил-3-(2-фенилэтил)-амино-1,2,3,4-тетрагидроциклопент[b]индол-7-ил метил карбамат гидрохлорид полугидрат (0,68 г). Реакция повторялась и вещества объединяли, т.пл. 155-157oC.
Элементный анализ:
Вычислено для С22H25N3O2•HCl•0,5H2O: C 64,62% H 6,65% N
10,28%
Найдено: C 64,56% H
6,72, N 9,91%
П р и м е р 40.
4-метил-3-(2-пропинил)амино-1,2,3,4-тетрагидроциклопент[b] индол-7-ил метилкарбамат.
К перемешиваемой суспензии 7-гидрокси-4-метил-1,4-дигидроциклопент[b] индол-3-(2Н)-она (5,5 г) в ацетонитриле (100 мл) добавляли пропаргиламин (3,0 г), раствор перемешивали при комнатной температуре в атмосфере азота при прикапывании изопропилата титана (IV) (15:6 г). Смесь перемешивали в течение 16 ч и охлаждали водой. Полученную смесь отфильтровывали, твердые вещества промывали СН2 Cl2, слои разделяли, и органическую часть сушили (Na2SO4). После концентрирования сырой продукт очищали методом флеш-хроматографии, проводя элюирование смесью гексан/ацетон (2:1) с получением 4-метил-3-(2-пропинил)имино-1,2,3,4-тетрагидроциклопент[b]индол-7-ола.
К перемешиваемому раствору 4-метил-3-(2-пропинил)имино-1,2,3, 4-тетрагидроциклопент[b] индол-7-ола (3, 4 г) в СН2Cl2 (15,0 мл) добавляли 1,8-диазобицикло /5.4.0.ундец/7-ен (326 мг), после чего прикапывали метилизоцианат (0,8 г) в СН2Cl2 (5,0 мл). За ходом реакции следили методом ТСХ, и через 1 ч раствор концентрировали, и сырой продукт очищали методом флеш-хроматографии, проводя элюирование смесью гексан/ацетон (2:1). Продукт, который осаждался из чистых фракций, собирали с получением 4-метил-3-(2-пропионил)имино-1,2,3,4-тетрагидроциклопент[b] индол-7-ил метилкарбамата (1,2 г) и фракции концентрировали с получением дополнительного количества вещества (0,9 г).
Элементный анализ:
Вычислено для С17H17N3O2: C 69,14% H
5,80% N 14,23%
Найдено: C 68,94% H 5,81% N 13,
94%
П р и м е р 41.
4-метил-3-(2-пропинил)амино-1,2,3,4-тетрагидроциклопент[b]индол-7-ил метилкарбамат гидрохлорид моногидрат.
К перемешиваемому раствору 4-метил-3-(2-пропинил)имино-1,2,3,4-тетрагидроциклопент[b] индол-7-ил метилкарбамата (1:1 г) в уксусной кислоте (10 мл) добавляли цианоборгидрид натрия (0,57 г). За ходом реакции следили методом ТСХ, и через 2 ч добавляли хлористый метилен (50 мл), и раствор промывали насыщенным раствором NaHCO3 до нейтрального значения рН. Метилен-хлоридный слой сушили (Na2SO4), фильтровали и концентрировали. Полученный в результате материал подвергали хроматографической очистке на силикагеле, проводя элюирование смесью гексан/ацетон 2:1, а чистые фракции собирали и концентрировали. Полученное в результате твердое вещество растворяли в минеральном количестве ЕtАОс, разбавляли эфиром, и после этого добавляли эфирный раствор НСl. Полученное в результате твердое вещество собирали фильтрованием в токе азота с получением 4-метил-3-(2-пропинил)амино-1,2,3,4-тетрагидроциклопент[b] индол-7-ил метилкарбамат гидрохлорид моногидрата (0,4 г). Реакцию повторяли и продукты объединяли, т.пл. 119-120oC (разл.).
Элементный анализ:
Вычислено для С17
H19N3O2•HCl•H2O: C 58,04% H 6,30% N 11,94%
Найдено: C 57,99% H 6,03% N 11,82%
П р и м е р 42.
4-метил-3-(2-фенилэтил)амино-1,2,3,4-тетрагидроциклопент[b]индол7-ил бензилкарбамат гидрохлорид полугидрат.
К перемешиваемому раствору 4-метил-3-(2-фенилэтил)амино-1,2,3, 4-тетрагидроциклопент[b] индол-7-ола (2,70 г) в СН2Cl2 (100 мл) добавляли 1,8-диазабицикло/5.4.0/ундецен (0,21 г). К реакционной смеси шприцем добавляли бензилизоцианат (0,83 г) и смесь перемешивали в атмосфере азота. Через 120 и 180 мин добавляли дополнительное количество бензилизоцианата в количестве 1/4 и 1/2 эквивалента соответственно. За ходом реакции следили методом ТСХ и через 185 мин раствор концентрировали в вакууме. Сырой остаток (2, 72) в соответствии с анализом методами ЯМР и МС представлял собой карбамат. Этот остаток растворяли в ледяной уксусной кислоте (75 мл) при перемешивании в токе азота. При добавлении цианоборгидрида натрия образовывался желтый осадок, который растворялся через 30 мин. Через 3 ч добавляли один эквивалент цианоборгидрида натрия. Через 30 мин, согласно анализу методом ТСХ, было установлено, что реакция завершена. Реакционную смесь нейтрализовали насыщенным раствором бикарбоната натрия, экстрагировали этилацетатом, сушили над Na2SO4 и концентрировали в вакууме. Свободное основание растворяли в эфире и добавляли эфирный раствор НСl. Полученное в результате белое твердое вещество собирали фильтрованием, т.пл. 165-166oC.
Элементный анализ:
Вычислено для С28H30N3O2•HCl•0,5H2O: C 69,
34% H 6,44% N 8,66%
Найдено: C 69,45% H 6,30, N 8,72%
П р и м е р
43.
4-метил-3-[2-(4-морфолинил)этил]имино-1,2,3,4-тетрагидроциклопент [b]индол-7-ол.
К перемешиваемому раствору 7-гидрокси-4-метил-1,4-дигидроциклопент[b]индол-3-она (8,00 г) в ацетонитриле (125 мл) в атмосфере азота добавляли 4-(2-аминоэтил)морфолин (10,35 г) и изопропилат титана (22,60 г). За ходом реакции следили методом ТСХ и через 2 ч добавляли дополнительные количества 4-(2-аминоэтил)морфолина (5,17 г) и изопропилата титана (11,30 г). Через 14 ч реакционную смесь охлаждали водой (200 мл). Добавляли ЕtОАс (200 мл) и смесь перемешивали в течение 15 мин, после чего фильтровали. Слои разделялись, водный слой экстрагировали этилацетатом и объединенные органические экстракты сушили (Na2SO4) и концентрировали в вакууме. Полученное в результате желтое твердое вещество сушили с получением 6,65 г продукта. Образец такого продукта в количестве 2 г дополнительно очищали перекристаллизацией из смеси СН2Cl2 (гексан с получением 1,2 г 4-метил-3-(2-морфолиноэтил)имино-1,2,3, 4-тетрагидроциклопент[b]индол-7-ола, т.пл. 158-159oC.
Элементный анализ:
Вычислено для С18
H23N3O2: C 68,98% H 7,40% N
13,41%
Найдено: C 68,78% H 7,52% N 13,26%
Реферат
Использование: в медицине для лечения различных расстройств памяти, характеризующихся пониженной холинергической функцией. Сущность изобретения: продукты: замещенные тетрагидроили гексагидроциклопент (b) индолы ф-лы I, где R1 водород, низший алкиламинокарбонил, бензилоксикарбонил, R2 - водород, низший алкил, циклоалкил, фенилзамещенный низший алкил, алкинил, формил, бензилоксикарбонил, R3 - водород, низший алкил, R4 - водород, гидрокси, низший алкиламинокарбокси, - - - обозначает отсутствие связи или одинарную связь. Реагент I. Соединение ф-лы 2, которое подвергают восстановлению. Условия реакции: восстановление осуществляют с помощью сплава Ренея в растворе гидроксида натрия. 2 с. и 4 з.п. ф-лы, 4 табл.
Формула

где R1 водород, низший алкиламинокарбонил, бензилоксикарбонил;
R2 водород, низший алкил, циклоалкил, фенилзамещенный низший алкил, алкинил, формил, бензилоксикарбонил, низший алкиаминокарбонил;
R3 водород, низший алкил;
R4 водород, гидрокси, низший алкиламинокарбоксил;
обозначает отсутствие связи или одинарную связь,
или их фармацевтически приемлемые аддитивные соли.
где R1 и R2 водород;
R3 водород, низший алкил;
R4 водород, гидрокси,
отличающийся тем, что производное индола общей формулы

где R3 и R4 имеют указанные значения,
подвергаются восстановлению.
где R1 водород;
R2 незамещенный или замещенный низший алкил, циклоалкил;
R3 водород, низший алкил;
R4 водород, гидрокси,
отличающийся тем, что производное индола общей формулы

где R3 и R4 имеют указанные значения,
подвергают взаимодействию с первичным амином и изопропилатом титана с последующим восстановлением образующегося имина боргидридом натрия.
где R1 водород;
R2 низший алкил, циклоалкил, фенилзамещенный низший алкил, алкинил;
R3 водород, низший алкил;
R4 водород, гидрокси,
отличающийся тем, что производное индола общей формулы

где R2 R4 имеют указанные значения,
подвергают восстановлению.

где R3 низший алкил, алкоксикарбонил;
R4 водород, гидрокси, галогензамещенный низший алканоил, галогензамещенный низший алкилкарбонилокси.
Комментарии