Лечение амиотрофического бокового склероза с использованием полученных из пуповины клеток - RU2612391C2
Код документа: RU2612391C2
Чертежи





















Описание
ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА СМЕЖНЫЕ ЗАЯВКИ
Настоящая заявка испрашивает преимущество заявки на патент США № 13/026995, поданной 14 февраля 2011 года, которая является частичным продолжением заявки на патент США № 12/429849, поданной 24 апреля 2009 года, которая является продолжением заявки на патент США № 10/877269, поданной 25 июня 2004 года, в настоящее время патент США № 7524489, выданный 28 апреля 2009 года, который испрашивает преимущество предварительной заявки на патент США № 60/483264, поданной 27 июня 2003 года, полное содержание которых включено в настоящий документ путем ссылки. Другие смежные заявки включают следующие одновременно находящиеся на рассмотрении заявки тех же заявителей, полное содержание каждой из которых включено в настоящий документ путем ссылки: заявка на патент США № 10/877012, поданная 25 июня 2004 года, в настоящее время патент США № 7510873, выданный 31 марта 2009 года; заявка на патент США № 10/877446, поданная 25 июня 2004 года; заявка на патент США № 10/877445, поданная 25 июня 2004 года; заявка на патент США № 10/877541, поданная 25 июня 2004 года, в настоящее время патент США № 7413734, выданный 19 августа 2008 года; заявка на патент США № 10/877009, поданная 25 июня 2004 года, в настоящее время патент США № 7560276, выданный 14 июля 2009 года; заявка на патент США № 10/876998, поданная 25 июня 2004 года; заявка на патент США № 11/315943, поданная 22 декабря 2005 года, в настоящее время патент США № 7875273, выданный 25 января 2001 года; и предварительная заявка на патент США № 60/555908, поданная 24 марта 2004 года.
ОБЛАСТЬ ПРИМЕНЕНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится по существу к композициям, способам и наборам для клеточной или регенеративной терапии неврологических заболеваний и расстройств, таких как амиотрофический боковой склероз. Более конкретно, настоящее изобретение представляет фармацевтические композиции, устройства и способы для терапии амиотрофического бокового склероза с использованием клеток, полученных из ткани пуповины.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
В ходе настоящего описания упоминаются различные патенты и другие публикации. Каждая из данных публикаций полностью включена в настоящий документ путем ссылки.
Неврологические заболевания и другие расстройства центральной и периферической нервной системы являются одними из самых тяжело протекающих заболеваний для пациента не только из-за их физических проявлений, но также и их необратимости. Одно из таких заболеваний представляет собой амиотрофический боковой склероз (АБС) (также известный как болезнь Лу Герига).
АБС представляет собой прогрессирующее нейродегенеративное заболевание, в основном поражающее двигательные нейроны и приводящее к мышечной слабости и атрофии. Заболевание характеризуется избирательной и преждевременной дегенерацией и гибелью двигательных нейронов. Пораженные нейроны страдают от потери дендритов, изменений цитоскелета и накопления белков и телец включения. АБС приводит к прогрессирующему параличу, как правило, приводящему к летальному исходу в течение нескольких лет из-за остановки дыхания, вызванного параличом дыхательных мышц. Средняя продолжительность заболевания от его появления до смерти больного составляет от трех до пяти лет. Только приблизительно 10% пациентов, страдающих АБС, продолжают жить в течение десяти или более лет. В США АБС страдают от 20000 до 30000 человек, и каждый год такой диагноз ставится еще 5000 человек.
Механизмы патогенеза АБС до конца не выяснены, однако считается, что АБС обусловлен рядом процессов, включая дисфункцию митохондрий, окислительный стресс, эксайтотоксичность, а также изменения в цитоскелете, аксональном транспорте, процессинге белков и кальциевом гомеостазе (Ilieva et al. (2009 г.), J. Cell Biol. 187: 761-72). Несмотря на значительные усилия, в настоящее время имеются лишь очень ограниченные терапевтические возможности замедления течения заболевания, хотя некоторые успехи достигнуты в паллиативной терапии. Аллогенная клеточная терапия может обеспечить эффективную многофакторную терапию при лечении АБС путем стимулирования трофических факторов, что приводит к снижению дегенерации двигательных нейронов, сохранению функций двигательных нейронов и продлению жизни.
В связи с очень тяжелым протеканием АБС и нехваткой подходов к его лечению существует значительная потребность в создании способов лечения АБС у пациента и тем самым повышения качества жизни пациента.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Описанные проблемы решаются с использованием композиций, способов и наборов, представленных в примерах осуществления, описанных в настоящем документе. Настоящее изобретение представляет композиции и способы, применимые к клеточной или регенеративной терапии неврологических заболеваний и расстройств, таких как амиотрофический боковой склероз. В частности, в настоящем изобретении предложены фармацевтические композиции, устройства и способы для регенерации или восстановления нервной ткани с использованием клеток, полученных из ткани пуповины.
В одном аспекте настоящего изобретения предложена выделенная клетка, полученная из ткани пуповины и по существу свободная от крови, причем клетка способна к самообновлению и размножению в культуре и обладает потенциалом к дифференцированию в клетку нейронного фенотипа; причем для роста клетке необходим L-валин, и клетка способна расти в среде, содержащей по меньшей мере приблизительно 5% кислорода. Данная клетка дополнительно обладает одной или более из следующих характеристик: (a) потенциал к по меньшей мере приблизительно 40 удвоениям в культуре; (b) закрепление и размножение на поверхности сосуда для культивирования с покрытием или без покрытия, причем сосуд для культивирования с покрытием содержит покрытие из желатина, ламинина, коллагена, полиорнитина, витронектина или фибронектина; (c) выработка по меньшей мере одного из тканевого фактора, виментина и альфа-актина гладких мышц; (d) выработка по меньшей мере одного из CD10, CD13, CD44, CD73, CD90, PDGFr-альфа, PD-L2 и HLA-A, B, C; (e) отсутствие выработки по меньшей мере одного из CD31, CD34, CD45, CD80, CD86, CD117, CD141, CD178, B7-H2, HLA-G и HLA-DR, DP, DQ по результатам анализа с использованием проточной цитометрии; (f) экспрессия гена, которая, по сравнению с клеткой человека, представляющей собой фибробласт, мезенхимальную стволовую клетку или клетку костного мозга гребня подвздошной кости, повышена для по меньшей мере одного из генов, кодирующих: интерлейкин-8; ретикулон 1; CXCL1 (хемокиновый (мотив C-X-C) лиганд 1/стимулятор активности роста меланомы, альфа); CXCL6 (хемокиновый (мотив C-X-C) лиганд 6/белок хемотаксиса гранулоцитов 2)); CXCL3 (хемокиновый (мотив C-X-C) лиганд 3); TNFAIP3 (индуцируемый фактором некроза опухоли альфа белок 3); (g) экспрессия гена, которая, по сравнению с клеткой человека, представляющей собой фибробласт, мезенхимальную стволовую клетку или клетку костного мозга гребня подвздошной кости, понижена для по меньшей мере одного из генов, кодирующих: SHOX2 (содержащий гомеобокс ген низкорослости 2); HSPB2 (27 кДа белок теплового шока 2); CXCL12 (хемокиновый (мотив C-X-C) лиганд 12/стромальный фактор 1); эластин (надклапанный аортальный стеноз, синдром Вильямса-Бойрена); мРНК Homo sapiens; кДНК DKFZp586M2022 (из клона DKFZp586M2022); Meox2 (содержащий мезенхимальный гомеобокс ген 2, содержащий гомеобокс блокировки роста ген); SIX1 (гомолог содержащего гомеобокс гена sine oculis 1) (Drosophila); кристаллин, альфа B; DAAM2 (ассоциированный с белком Disheveled активатор морфогенеза 2); белок DKFZP586B2420; аналог нейтралина 1; тетранектин (связывающийся с плазминогеном белок); STAC (ген, содержащий домен src-гомологии 3 (SH3) и богатый цистеином домен); холестерин-25-гидроксилаза; RUNX3 (связанный с карликовостью фактор транскрипции 3); рецептор интерлейкина-11, альфа; PCOLCE (усилитель проколлаген-C-эндопептидазы); FZD7 (гомолог frizzled 7) (Drosophila); гипотетический ген BC008967; коллаген, тип VIII, альфа 1; TNC (тенасцин C, гексабрахион); IRX5 (содержащий гомеобокс IRX белок 5); гефестин; интегрин, бета 8; SV2A (гликопротеин синаптического пузырька 2); NBL1 (нейробластома, ген подавления онкогенности 1); IGFBP2 (связывающийся с инсулиноподобным фактором роста белок 2, 36 кДа); кДНК Homo sapiens FLJ12280 fis, клон MAMMA1001744; CRLF1 (подобный цитокиновому рецептору фактор 1); KCNN4 (активируемый кальцием калиевый канал средней/низкой проводимости, подсемейство N, член 4); интегрин, бета 7; транскрипционный коактиватор с мотивом связывания с PDZ (TAZ); SIX2 (гомолог содержащего гомеобокс гена sine oculis 2) (Drosophila); белок KIAA1034; VAMP5 (везикуло-ассоциированный мембранный белок 5/миобревин); EFEMP1 (содержащий EGF фибулинподобный белок внеклеточного матрикса 1); EGR3 (белок раннего ростового ответа 3); DLX5 (содержащий гомеобокс distal-less ген 5); гипотетический белок FLJ20373; AKR1C3 (альдокеторедуктаза семейства 1, член C3/3-альфа-гидроксистероиддегидрогеназа, тип II); бигликан; транскрипционный коактиватор с мотивом связывания с PDZ (TAZ); фибронектин 1; проэнкефалин; интегрин, бета-подобный 1 (с доменами EGF-подобных повторов); мРНК Homo sapiens, полноразмерная вставка кДНК, клон EUROIMAGE 1968422; EphA3; белок KIAA0367; NPR3 (рецептор натрийуретического пептида C/гуанилатциклаза C/рецептор атрионатрийуретического пептида C); гипотетический белок FLJ14054; мРНК Homo sapiens; кДНК DKFZp564B222 (из клона DKFZp564B222); BNIP3L (ген белка, подобного белку 3, взаимодействующему с BCL2/белком аденовируса E1B 19 кДа); AE-связывающий белок 1; COX7A1 (полипептид 1 субъединицы VIIa цитохром c оксидазы) (мышечный); аналог нейтралина 1; BTG1 (ген транслокации B-клеток 1); гипотетический белок FLJ23191; и DKFZp586L151, (h) секреция по меньшей мере одного из MCP-1, IL-6, IL-8, GCP-2, HGF, KGF, FGF, HB-EGF, BDNF, TPO, MIP1a, RANTES и TIMP1; и (i) отсутствие секреции по меньшей мере одного из TGF-бета2, ANG2, PDGFbb, MIP1b, I309, MDC и VEGF по результатам ИФА.
В конкретных вариантах осуществления клетка, полученная из пуповины, имеет все идентификационные признаки любого из: клеточного типа UMB 022803 (P7) (№ доступа ATCC PTA-6067) или клеточного типа UMB 022803 (P17) (№ доступа ATCC PTA-6068).
В определенных вариантах осуществления клетки, полученные из ткани пуповины, выделяют в присутствии одной или более ферментативных активностей, содержащих металлопротеазную активность, муколитическую активность и нейтральнопротеазную активность. Предпочтительно клетки имеют нормальный кариотип, который сохраняется в процессе пассирования клеток при культивировании. В предпочтительных вариантах осуществления клетки, полученные из послеродового материала, содержат каждый из CD10, CD13, CD44, CD73, CD90, PDGFr-альфа и HLA-A, B, C и не содержат любой из CD31, CD34, CD45, CD117, CD141 или HLA-DR, DP, DQ по результатам анализа с использованием проточной цитометрии.
В другом аспекте настоящего изобретения предложена популяция клеток, содержащая клетки, полученные из ткани пуповины, как описано выше. В одном варианте осуществления популяция представляет собой по существу однородную популяцию клеток, полученных из ткани пуповины. В конкретном варианте осуществления популяция содержит клональную клеточную линию клеток, полученных из ткани пуповины. В другом варианте осуществления популяция представляет собой неоднородную популяцию, содержащую клетки, полученные из ткани пуповины, и клетки по меньшей мере одного другого типа. В определенных вариантах осуществления клетки другого типа представляют собой астроциты, олигодендроциты, нейроны, предшественники нейронов, нейронные стволовые клетки или другие мультипотентные или плюрипотентные стволовые клетки. В других вариантах осуществления популяцию клеток культивируют в контакте с одним или более факторами, которые стимулируют дифференцирование стволовых клеток в направлении нейронной линии дифференцирования.
В соответствии с настоящим изобретением также предложен клеточный лизат, приготовленный из клеток, полученных из ткани пуповины. Клеточный лизат можно разделить на фракцию, обогащенную мембранными фрагментами, и растворимую клеточную фракцию. Настоящее изобретение также представляет внеклеточный матрикс, продуцируемый клетками, полученными из ткани пуповины, а также кондиционированную среду, в которой выращивали клетки.
В другом аспекте настоящего изобретения предложен способ лечения пациента с нейродегенеративным состоянием, причем способ содержит введение пациенту клеток, полученных из ткани пуповины, как описано выше, в количестве, эффективном для лечения нейродегенеративного состояния. В определенных вариантах осуществления нейродегенеративное состояние представляет собой острое нейродегенеративное состояние, такое как травма головного мозга, травма спинного мозга или травма периферического нерва. В других вариантах осуществления данное состояние представляет собой хроническое или прогрессирующее нейродегенеративное состояние, такое как болезнь Паркинсона, болезнь Альцгеймера, болезнь Хантингтона, амиотрофический боковой склероз, опухоль, рассеянный склероз или хроническая травма периферического нерва.
В одном варианте осуществления клетки, полученные из ткани пуповины, перед введением индуцируют in vitro для дифференцирования в клетки нейронной линии дифференцирования. В другом варианте осуществления клетки модифицируют методами генетической инженерии для выработки продукта гена, который способствует лечению нейродегенеративного состояния.
В определенных вариантах осуществления клетки вводят вместе с клетками по меньшей мере одного другого типа, такими как астроциты, олигодендроциты, нейроны, предшественники нейронов, нейронные стволовые клетки или иные мультипотентные или плюрипотентные стволовые клетки. В данных вариантах осуществления клетки другого типа можно вводить одновременно, до или после клеток, полученных из ткани пуповины. Аналогичным образом в данных или других вариантах осуществления клетки вводят вместе с по меньшей мере одним другим агентом, таким как лекарственное средство для нейронной терапии или другой полезный вспомогательный агент, такой как противовоспалительный агент, противоапоптозные агенты, антиоксидант или фактор роста. В данных вариантах осуществления другой агент можно вводить одновременно, до или после клеток, полученных из ткани пуповины.
В определенных вариантах осуществления клетки вводят в заранее определенное место в центральной или периферической нервной системе пациента. Их можно вводить путем инъекции или инфузии или инкапсулировать в имплантируемое устройство, либо путем имплантации содержащей клетки матрицы или каркаса.
Один вариант осуществления настоящего изобретения представляет собой способ лечения амиотрофического бокового склероза, содержащий введение пациенту клеток, полученных из ткани пуповины, в количестве, эффективном для лечения амиотрофического бокового склероза. В данном варианте осуществления клетки, полученные из ткани пуповины, выделены из ткани пуповины человека и по существу свободны от крови, способны к самообновлению и размножению в культуре, обладают потенциалом к дифференцированию в клетки других фенотипов, могут произвести по меньшей мере 40 удвоений и обладают следующими характеристиками: (a) экспрессия каждого из CD10, CD13, CD44, CD73, CD90, PDGFr-альфа, PD-L2 и HLA-A, B, C; (b) отсутствие экспрессии любого из CD31, CD34, CD45, CD80, CD86, CD 117, CD141, CD178, B7-H2, HLA-G или HLA-DR, DP, DQ; и (c) повышенная экспрессия интерлейкина-8, ретикулона 1 и CXCL3 (хемокинового (мотив C-X-C) лиганда 3) по сравнению с клеткой человека, представляющей собой фибробласт, мезенхимальную стволовую клетку или клетку костного мозга гребня подвздошной кости. В другом варианте осуществления способа клетки, полученные из ткани пуповины, не экспрессируют hTERT или теломеразу. В еще одном варианте осуществления клетки, полученные из ткани пуповины, вводят путем инъекции (такой как, например, внутривенная или подоболочечная инъекция) или инфузии. В одном варианте осуществления клетки, полученные из ткани пуповины, оказывают трофический эффект на нервную систему пациента.
Другой вариант осуществления настоящего изобретения представляет собой способ лечения амиотрофического бокового склероза, содержащий введение пациенту эффективного количества по существу однородной популяции клеток, полученных из ткани пуповины. В данном варианте осуществления популяция клеток, полученных из ткани пуповины, выделена из ткани пуповины человека и по существу свободна от крови, способна к самообновлению и размножению в культуре, обладает потенциалом к дифференцированию в клетки других фенотипов, может произвести по меньшей мере 40 удвоений и обладает следующими характеристиками: (a) экспрессия каждого из CD10, CD13, CD44, CD73, CD90, PDGFr-альфа, PD-L2 и HLA-A, B, C; (b) отсутствие экспрессии любого из CD31, CD34, CD45, CD80, CD86, CD 117, CD141, CD178, B7-H2, HLA-G или HLA-DR, DP, DQ; и (c) повышенная экспрессия интерлейкина-8, ретикулона 1 и CXCL3 (хемокинового (мотив C-X-C) лиганда 3) по сравнению с клеткой человека, представляющей собой фибробласт, мезенхимальную стволовую клетку или клетку костного мозга гребня подвздошной кости. В одном варианте осуществления клетки, полученные из ткани пуповины, не экспрессируют hTERT или теломеразу. В другом варианте осуществления по существу однородную популяцию клеток, полученных из ткани пуповины, вводят путем инъекции или инфузии. В альтернативном варианте осуществления по существу однородную популяцию клеток, полученных из ткани пуповины, вводят путем внутривенной или подоболочечной инъекции. В другом варианте осуществления по существу однородная популяция клеток, полученных из ткани пуповины, оказывает трофический эффект на нервную систему пациента.
Другой вариант осуществления настоящего изобретения представляет собой способ лечения амиотрофического бокового склероза, содержащий введение пациенту фармацевтической композиции, содержащей клетки, полученные из ткани пуповины, в количестве, эффективном для лечения амиотрофического бокового склероза. В данном варианте осуществления клетки, полученные из ткани пуповины, выделены из ткани пуповины человека и по существу свободны от крови, способны к самообновлению и размножению в культуре, обладают потенциалом к дифференцированию в клетки других фенотипов, могут произвести по меньшей мере 40 удвоений и обладают следующими характеристиками: (a) экспрессия каждого из CD10, CD13, CD44, CD73, CD90, PDGFr-альфа, PD-L2 и HLA-A, B, C; (b) отсутствие экспрессии любого из CD31, CD34, CD45, CD80, CD86, CD 117, CD141, CD178, B7-H2, HLA-G или HLA-DR, DP, DQ; и (c) повышенная экспрессия интерлейкина-8, ретикулона 1 и CXCL3 (хемокинового (мотив C-X-C) лиганда 3) по сравнению с клеткой человека, представляющей собой фибробласт, мезенхимальную стволовую клетку или клетку костного мозга гребня подвздошной кости. В одном варианте осуществления клетки, полученные из ткани пуповины, не экспрессируют hTERT или теломеразу. В еще одном варианте осуществления клетки, полученные из ткани пуповины, вводят путем инъекции (такой как, например, внутривенная или подоболочечная инъекция) или инфузии. В одном варианте осуществления клетки, полученные из ткани пуповины, оказывают трофический эффект на нервную систему пациента.
В другом аспекте настоящего изобретения предложена фармацевтическая композиция для лечения пациента с нейродегенеративным состоянием, таким как амиотрофический боковой склероз, содержащая фармацевтически приемлемый носитель и описанные выше клетки, полученные из ткани пуповины. Нейродегенеративное состояние, нуждающееся в лечении, может представлять собой острое нейродегенеративное состояние или может представлять собой хроническое или прогрессирующее состояние.
В определенных вариантах осуществления фармацевтическая композиция содержит клетки, перед приготовлением композиции индуцированные in vitro для дифференцирования в клетки нейронной линии дифференцирования, или клетки, модифицированные методами генетической инженерии для выработки продукта гена, который способствует лечению нейродегенеративного состояния.
В определенных вариантах осуществления фармацевтическая композиция содержит клетки по меньшей мере одного другого типа, такие как астроциты, олигодендроциты, нейроны, предшественники нейронов, нейронные стволовые клетки или другие мультипотентные или плюрипотентные стволовые клетки. В данных или других вариантах осуществления фармацевтическая композиция содержит по меньшей мере один другой агент, такой как лекарственное средство для нейронной терапии, или другой полезный вспомогательный агент, такой как противовоспалительный агент, противоапоптозные агенты, антиоксидант или фактор роста.
В определенных вариантах осуществления фармацевтическую композицию готовят для введения путем инъекции или инфузии. В альтернативном варианте осуществления композиция может содержать имплантируемое устройство с инкапсулированными клетками или матрицу или каркас, содержащие клетки.
В соответствии с другим аспектом настоящего изобретения предложен набор для лечения пациента с нейродегенеративным состоянием. Набор содержит фармацевтически приемлемый носитель, популяцию описанных выше клеток, полученных из ткани пуповины, и инструкции по использованию набора в соответствии со способом лечения пациента. Набор может дополнительно содержать по меньшей мере один реагент и инструкции по культивированию клеток, полученных из ткани пуповины. Он также может содержать популяцию клеток по меньшей мере одного другого типа или по меньшей мере один другой агент для лечения нейродегенеративного состояния.
В соответствии с другим аспектом настоящего изобретения предложен способ лечения пациента с нейродегенеративным состоянием (таким как, например, амиотрофический боковой склероз), который содержит введение пациенту препарата, изготовленного из описанных выше клеток, полученных из ткани пуповины. Такой препарат может содержать клеточный лизат (или его фракцию) из клеток, полученных из ткани пуповины, внеклеточный матрикс клеток, полученных из ткани пуповины, или кондиционированную среду, в которой выращивали клетки, полученные из ткани пуповины. В другом аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и препарат, изготовленный из клеток, полученных из ткани пуповины, который может представлять собой клеточный лизат (или его фракцию) из клеток, полученных из ткани пуповины, внеклеточный матрикс клеток, полученных из ткани пуповины, или кондиционированную среду, в которой выращивали клетки, полученные из ткани пуповины. Также предложены наборы для реализации на практике данного аспекта настоящего изобретения. Данные наборы могут включать один или более из фармацевтически приемлемого носителя или другого агента или реагента, один или более из клеточного лизата или его фракции, внеклеточного матрикса или кондиционированной среды для клеток, полученных из ткани пуповины, а также инструкции по использованию компонентов набора.
Другие характеристики и преимущества настоящего изобретения станут понятны после изучения следующего подробного описания и примеров.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На фиг.1 представлены результаты контроля веса животных на протяжении исследования.
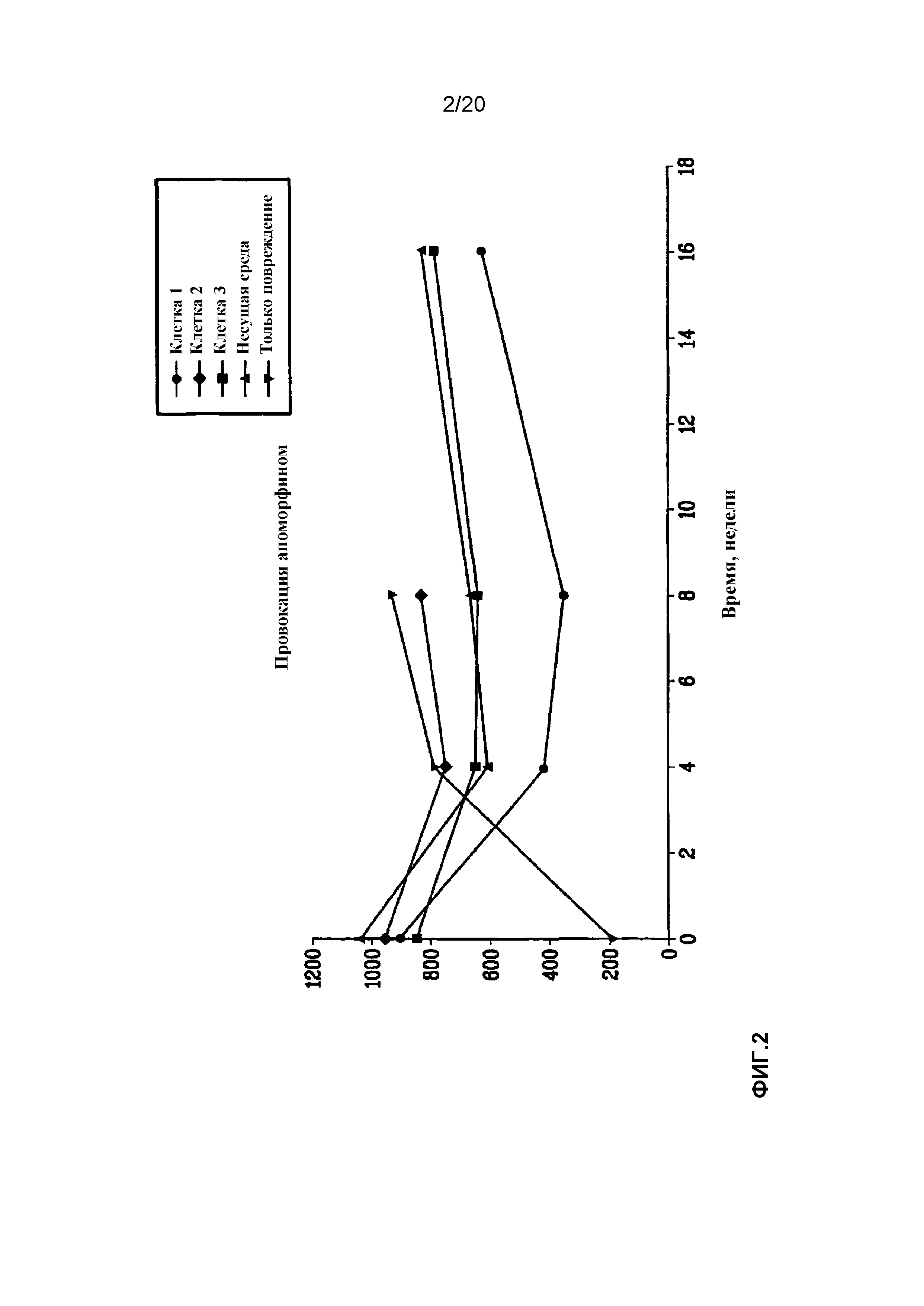
На фиг.2 представлен отклик различных групп клеток на провокационную пробу с апоморфином.
На фиг.3 представлены результаты мониторинга различий в количестве поворотов головы налево и направо на протяжении исследования.
На фиг.4 представлены результаты мониторинга потребления корма животными на протяжении исследования с использованием лестничного испытания.
На фиг.5 представлены гистограммы с результатами качественного анализа окрашивания на (а) Iba-1; (b) ED-1; и (c) DAPI клеточных трансплантатов в соответствии со следующими критериями: 0 = нет (отсутствие клеток); 1 = видимое окрашивание; 2 = достаточное окрашивание; 3 = очень густое окрашивание; 4 = плотное окрашивание.
На фиг.6 представлены гистограммы с результатами качественного анализа окрашивания на (а) GFAP и (b) виментин клеточных трансплантатов в соответствии со следующими критериями: 0 = нет (отсутствие клеток); 1 = видимое окрашивание; 2 = достаточное окрашивание; 3 = очень густое окрашивание; 4 = плотное окрашивание.
На фиг.7 представлен эффект клеток, полученных из ткани пуповины человека (hUTC), на выживаемость в крысиной модели АБС SOD1 (G93A) для (A) групп 1 и 2 и (B) групп 3 и 4.
На фиг.8 представлены продолжительности жизни животных в группах, получавших несущую среду в качестве контроля и получавших hUTC (смотрите пример 21). Гистограмма показывает, что в группах, получавших клетки, особенно в группе исследования 2, продолжительность жизни была на 15,75 суток (2,25 нед., P<0,035) больше, чем в группах, получавших несущую среду, хотя возраст, при котором начиналась болезнь, был аналогичным.
На фиг.9 представлены результаты измерения двигательной активности животных, получавших или не получавших клетки hUTC. На фиг.9А представлены результаты теста по BBB-шкале (Basso-Beattie-Bresnahan) для групп 1 и 2. На фиг.9B представлены результаты теста «наклонная плоскость» для групп 1 и 2. На фиг.9C представлены результаты теста по BBB-шкале для групп 3 и 4. На фиг.9D представлены результаты теста «наклонная плоскость» для групп 3 и 4. Представленные данные показывают разделение по двум главным требованиям мышечной слабости для групп, получавших несущую среду и клетки.
На фиг.10 представлена кривая выживаемости для 10-недельной группы исследования (по всем субъектам).
На фиг.11 представлена кривая выживаемости для 12-недельной группы исследования (по всем субъектам).
На фиг.12 представлена кривая выживаемости для 12-недельной группы исследования (последние две пары).
На фиг.13 представлены результаты теста «наклонная плоскость» для 10-недельной группы исследования.
На фиг.14 представлены результаты теста по BBB-шкале для 10-недельной группы исследования.
На фиг.15 представлены результаты теста «наклонная плоскость» для первых шести пар из 12-недельной группы исследования.
На фиг.16 представлены результаты теста «наклонная плоскость» для последних двух пар из 12-недельной группы исследования.
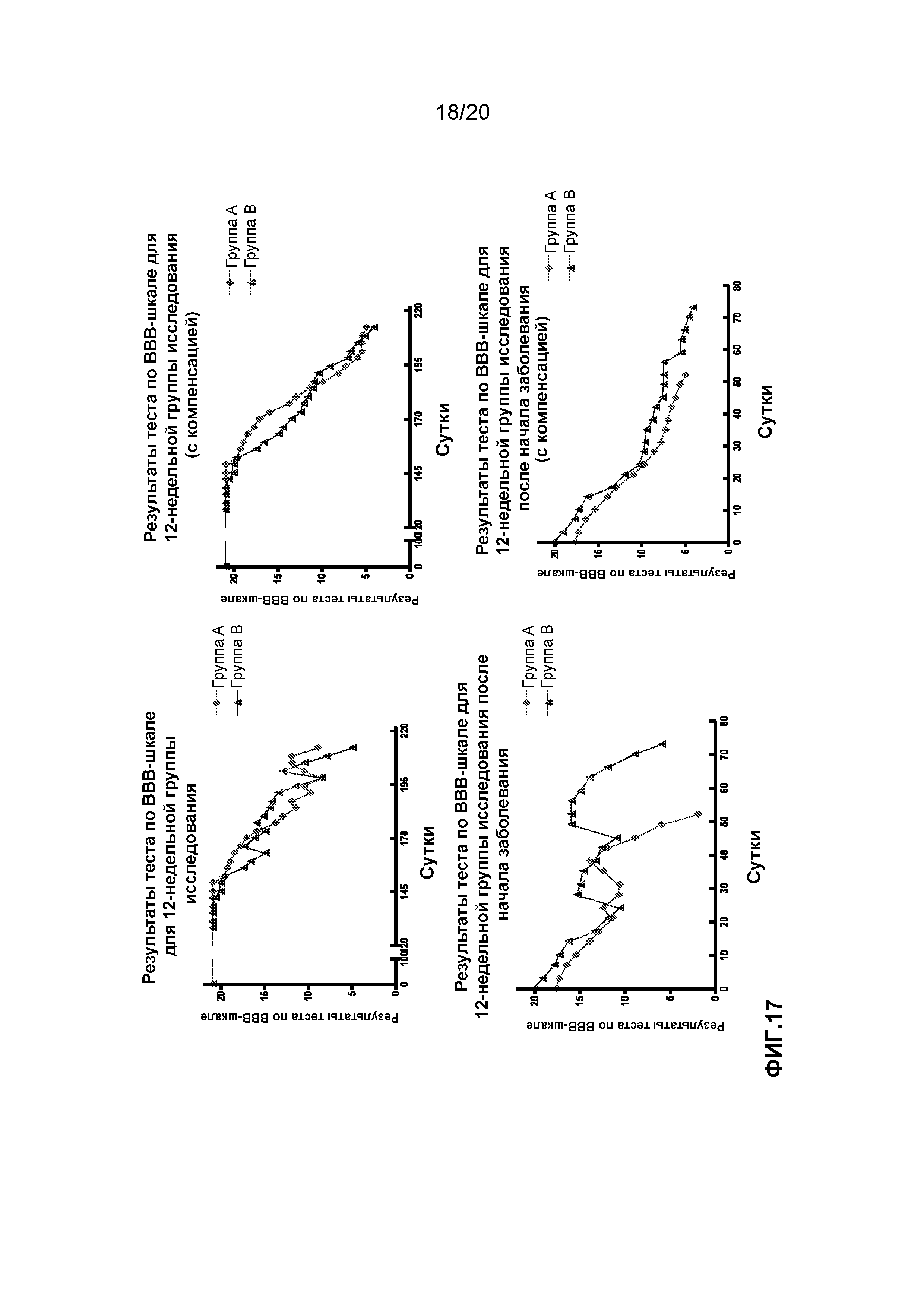
На фиг.17 представлены результаты по BBB-шкале для первых шести пар из 12-недельной группы исследования.
На фиг.18 представлены результаты по BBB-шкале для последних двух пар из 12-недельной группы исследования.
На фиг.19 представлен вид окрашенных крезиловым фиолетовым сечений поясничного отдела спинного мозга крыс SOD1 G93A. На фиг.19А представлен вид окрашенных крезиловым фиолетовым сечений поясничного отдела спинного мозга для животных с нечетными номерами. На фиг.19B представлен вид окрашенных крезиловым фиолетовым сечений поясничного отдела спинного мозга для животных с четными номерами. На фиг.19C представлен вид окрашенных крезиловым фиолетовым сечений поясничного отдела спинного мозга для нормальных животных.
ПОДРОБНОЕ ОПИСАНИЕ ПРИМЕРОВ ОСУЩЕСТВЛЕНИЯ
Определения
Для различных терминов, используемых в описании и формуле изобретения, приняты следующие определения.
Стволовые клетки представляют собой недифференцированные клетки, определяющиеся по способности на уровне единичной клетки как самообновляться, так и дифференцироваться для получения клеток-потомков, включая самообновляющихся предшественников, необновляющихся предшественников и окончательно дифференцированные клетки. Стволовые клетки также характеризуются своей способностью дифференцирования in vitro в функциональные клетки различных клеточных линий дифференцирования из множества зародышевых листков (энтодермы, мезодермы и эктодермы), а также после трансплантации давать начало тканям, происходящим от множества зародышевых листков, и способствовать образованию по существу большинства, если не всех, тканей после инъекции в бластоцисты.
По своему потенциалу развития стволовые клетки разделяются на: (1) тотипотентные; (2) плюрипотентные; (3) мультипотентные; (4) олигопотентные; и (5) унипотентные. Тотипотентные клетки способны преобразовываться во все типы эмбриональных и внеэмбриональных клеток. Плюрипотентные клетки способны преобразовываться во все типы эмбриональных клеток. Мультипотентные клетки включают клетки, способные преобразовываться в подмножество клеточных линий дифференцирования, но в рамках конкретной ткани, органа или физиологической системы (например, гематопоэтические стволовые клетки (ГСК) могут вырабатывать потомков, которые включают ГСК (самообновление), олигопотентные ограниченные клетки-предшественники крови и все типы клеток и элементов (например, тромбоциты), которые представляют собой нормальные компоненты крови). Клетки, которые являются олигопотентными, способны преобразовываться в более ограниченное подмножество клеточных линий дифференцирования, чем мультипотентные стволовые клетки; а клетки, которые являются унипотентными, способны преобразовываться в единственную клеточную линию дифференцирования (например, сперматогенные стволовые клетки).
Стволовые клетки также разделяются на категории по источнику их потенциального получения. Взрослая стволовая клетка по существу представляет собой мультипотентную недифференцированную клетку, присутствующую в ткани, содержащей множество дифференцированных типов клеток. Взрослая стволовая клетка способна к самообновлению. В нормальных условиях они также могут дифференцироваться для получения специализированных типов клеток той ткани, в которой они находятся, а также, возможно, тканей других типов. Эмбриональная стволовая клетка представляет собой плюрипотентную клетку из внутренней клеточной массы эмбриона на стадии бластоцисты. Фетальная стволовая клетка представляет собой клетку, происходящую из тканей или мембран плода. Неонатальная стволовая клетка представляет собой мультипотентную или плюрипотентную клетку, происходящую по существу из доступной после родов внеэмбриональной ткани, а именно плаценты и пуповины. Известно, что данные клетки обладают признаками, характерными для плюрипотентных стволовых клеток, включая быструю пролиферацию и потенциал дифференцирования в клетки многих клеточных линий дифференцирования. Неонатальные стволовые клетки можно получить из крови (например, клетки, полученные из пуповинной крови) или получить не из крови (например, из отличных от крови тканей пуповины и плаценты).
Эмбриональную ткань, как правило, определяют как ткань, происходящую из эмбриона (что для человека обозначает срок развития от оплодотворения до приблизительно шести недель). Ткань плода обозначает ткань, происходящую из плода, что для человека обозначает срок развития от приблизительно шести недель до рождения. Внеэмбриональная ткань представляет собой ткань, связанную с эмбрионом или плодом, но не происходящую из них. Внеэмбриональные ткани включают внеэмбриональные мембраны (хорион, амнион, желточный мешок и аллантоис), пуповину и плаценту (которая самостоятельно образована из хориона и базальной отпадающей материнской плаценты).
Дифференцирование представляет собой процесс, при помощи которого неспециализированная («некоммитированная») или менее специализированная клетка приобретает характеристики специализированной клетки, например, такой как нервная клетка или мышечная клетка. Дифференцированная клетка представляет собой клетку, занявшую более специализированное («коммитированное») положение в линии дифференцирования клетки. Термин «коммитированный» применительно к способу дифференцирования относится к клетке, дошедшей в процессе дифференцирования до стадии, от которой в нормальных условиях она продолжит дифференцирование до конкретного типа клеток или подмножества типов клеток и не сможет в нормальных условиях дифференцироваться в другой тип клеток или вернуться к менее дифференцированному типу. Дедифференцирование обозначает процесс, в ходе которого клетка возвращается к менее специализированному (или коммитированному) положению в линии дифференцирования клетки. В настоящем документе линия дифференцирования клетки определяет наследственность клетки, т.е. из каких клеток она произошла и каким клеткам она может дать начало. В линии дифференцирования клетка помещается в наследственную схему развития и дифференцирования.
В широком смысле клетка-предшественник представляет собой клетку, обладающую способностью образовывать клетки-потомки, которые будут более дифференцированы по сравнению с ней, и при этом сохраняющую способность к восполнению пула клеток-предшественников. По данному определению стволовые клетки также сами являются клетками-предшественниками, так же как и более непосредственные предшественники окончательно дифференцированных клеток. При описании клеток, составляющих предмет настоящего изобретения, как более подробно описано ниже, можно использовать данное широкое определение клетки-предшественника. В более узком смысле клетку-предшественник часто определяют как клетку, которая является промежуточным предшественником в процессе дифференцирования, т.е. она происходит из стволовой клетки и является промежуточным звеном в получении зрелого типа клеток или подмножества типов клеток. Клетки-предшественники данного типа по существу не способны к самообновлению. Соответственно при отсылке к клетке данного типа в настоящем документе она будет называться «необновляющейся клеткой-предшественником» или «промежуточной клеткой-предшественником».
В настоящем документе фраза «дифференцируется в нейронную линию дифференцирования или фенотип» относится к клетке, которая становится частично или полностью коммитированной к конкретному нейронному фенотипу ЦНС или ПНС, т.е. нейрону или глиальной клетке, причем последняя категория включает, без ограничений, астроциты, олигодендроциты, шванновские клетки и микроглии.
Клетки, составляющие предмет настоящего изобретения, по существу называются «клетками, полученными из ткани пуповины» (либо UTC, либо hUTC). Они также могут иногда называться «клетками, полученными из пуповины» (UDC). Кроме того, клетки могут быть описаны как являющиеся стволовыми клетками или клетками-предшественниками, причем последний термин используется в широком смысле. Термин «полученный из» используется для указания того, что клетки были получены из их биологического источника и были выращены или иным образом обработаны in vitro (например, культивированы в ростовой среде для размножения популяции и/или получения клеточной линии). Манипуляции in vitro над пуповинными стволовыми клетками и уникальные характеристики клеток, полученных из пуповины, которые составляют предмет настоящего изобретения, более подробно описаны ниже.
Для описания клеток в процессе культивирования используются различные термины. Культура клеток по существу обозначает клетки, взятые из живого организмам и выращенные в контролируемых условиях («в культуре» или «культивированные»). Первичная культура клеток обозначает культуру клеток, тканей или органов, взятых непосредственно из организма(ов) до первого пересева. Клетки размножают в культуре, когда их помещают в ростовую среду в условиях, облегчающих рост и/или деление клеток, что приводит к большей популяции клеток. При размножении клеток в культуре скорость пролиферации клеток иногда измеряют по количеству времени, которое необходимо клеткам для удвоения численности. Данное время называют временем удвоения.
Клеточная линия представляет собой популяцию клеток, образованную одним или более пересевами из первичной культуры клеток. Каждый цикл пересева называют пассажем. Клетки, которые пересеивают, называются клетками, которые пассированы. Конкретная популяция клеток, или клеточная линия, иногда относится или характеризуется количеством выполненных с ней пассажей. Например, пассированная десять раз культивируемая популяция клеток может называться культурой P10. Первичная культура, т.е. первая культура после выделения клеток из ткани, обозначается P0. После первого пересева клетки описывают как вторичную культуру (P1, или культура первого пассажа). После второго пересева клетки превращаются в третичную культуру (P2, или культура второго пассажа) и т.п. Специалистам в данной области будет понятно, что за период пассирования популяция клеток может многократно удваиваться; следовательно, количество удвоений популяции в культуре больше номера пассажа. Степень размножения клеток (т.е. количество удвоений популяции) за период времени между пассированиями зависит от многих факторов, включая, без ограничений, плотность посева, тип субстрата, тип среды, условия роста и время между пассированиями.
Кондиционированная среда представляет собой среду, в которой культивировали и из которой затем удалили конкретную клетку или популяцию клеток. При культивировании в среде клетки могут секретировать клеточные факторы, которые обеспечивают трофическую поддержку для других клеток. Такие трофические факторы включают, без ограничений, гормоны, цитокины, внеклеточный матрикс (ECM), белки, везикулы, антитела и гранулы. Среду, содержащую клеточные факторы, и называют кондиционированной средой.
По существу трофический фактор определяют как вещество, стимулирующее выживаемость, рост, дифференцирование, пролиферацию и/или созревание клетки или стимулирующее повышенную активность клетки. Взаимодействие между клетками через трофические факторы может происходить между клетками разных типов. Межклеточное взаимодействие через трофические факторы обнаруживается по существу во всех типах клеток и является особенно значимым средством коммуникации между типами нервных клеток. Трофические факторы также могут действовать аутокринным образом, т.е. клетка может вырабатывать трофические факторы, воздействующие на ее собственную выживаемость, рост, дифференцирование, пролиферацию и/или созревание.
При описании культивированных клеток позвоночных термин «старение» (также «репликативное старение» или «клеточное старение») обозначает свойство конечных клеточных культур, а именно, их неспособность расти далее конечного количества удвоений популяции (иногда называется «пределом Хейфлика»). Хотя клеточное старение было впервые описано с использованием фибробластоподобных клеток, клеточное старение характерно для большинства нормальных типов клеток человека, которые можно успешно выращивать в культуре. Продолжительность жизни in vitro различных типов клеток варьируется, однако максимальная продолжительность жизни, как правило, не превышает 100 удвоений популяции (это количество удвоений, за которое все клетки в культуре состарятся, в результате чего культура полностью утратит способность к делению). Старение не зависит от хронологического времени, а измеряется количеством клеточных делений, или удвоений популяции, совершенных культурой. Таким образом, клетки, переведенные в фазу покоя путем удаления существенных факторов роста, могут возобновить свой рост и деление после обратного введения факторов роста в среду и тем самым могут выполнить то же количество удвоений, что и эквивалентные клетки, которые росли непрерывно. Аналогичным образом при заморозке в жидком азоте после варьируемого количества удвоений популяции и последующей разморозке и культивировании клетки совершают по существу то же количество удвоений, что и клетки, которые поддерживали в культуре в незамороженном состоянии. Старые клетки не являются мертвыми или умирающими клетками; на самом деле они устойчивы к запрограммированной смерти клетки (апоптозу) и успешно поддерживались в своем состоянии неделения до трех лет. Данные клетки совершенно живы и метаболически активны, но они не делятся. В настоящее время пока не найдено биологических, химических или вирусных агентов, способных обратить состояние неделения старых клеток.
В настоящем документе термин «нейродегенеративное состояние (или расстройство)» представляет собой общий термин, охватывающий острые и хронические состояния, расстройства или заболевания центральной или периферической нервной системы. Нейродегенеративное состояние может быть обусловлено возрастом или может быть следствием травмы или повреждения, либо может быть связано с конкретным заболеванием или расстройством. Острые нейродегенеративные состояния включают, без ограничений, состояния, связанные с гибелью или потерей функции нервных клеток, включая церебрально-васкулярную недостаточность, фокальную или диффузную травму головного мозга, диффузные повреждения головного мозга, повреждение спинного мозга или травму периферической нервной системы, например в результате химического или физического ожога, глубокого пореза или отсечения конечности. Примеры острых нейродегенеративных расстройств представляют собой ишемию или инсульт головного мозга, включая эмболическую окклюзию и тромботическую окклюзию, реперфузию после острой ишемии, перинатальное гипоксически-ишемическое поражение нервной системы, остановку сердца, а также внутричерепную гематому любого типа (такую как эпидуральная, субдуральная, субарахноидальная и внутрицеребральная), и внутричерепные и межпозвоночные повреждения (такие как ушиб, перфорация, смещение, защемление и разрыв), а также синдром детского сотрясения. Хронические нейродегенеративные состояния включают, без ограничений, болезнь Альцгеймера, болезнь Пика, болезнь диффузных телец Леви, прогрессирующий надъядерный паралич (болезнь Стила-Ричардсона-Ольшевского), мультисистемную дегенерацию (синдром Шая-Дрейджера), хронические эпилептические состояния, связанные с нейродегенерацией, болезни двигательных нейронов, включая амиотрофический боковой склероз, дегенеративные атаксии, корко-базальную дегенерацию, АБС-паркинсонизм-деменцию (комплекс острова Гуам), подострый склерозирующий панэнцефалит, болезнь Хантингтона, болезнь Паркинсона, синуклеопатии (включая множественную системную атрофию), первичную прогрессирующую афазию, стриатонигральную дегенерацию, болезнь Мачадо-Джозефа (спиномозжечковая атаксия типа 3) и оливомостомозжечковые дегенерации, болезнь Жиля де ла Туретта, бульбарный и псевдобульбарный паралич, спинальную и спинобульбарную мышечную атрофию (болезнь Кеннеди), первичный боковой склероз, семейную спастическую параплегию, болезнь Верднига-Гоффмана, болезнь Кугельберга-Веландер, болезнь Тея-Сакса, болезнь Сандхоффа, семейную спастическую болезнь, болезнь Вольфарта-Кугельберга-Веландер, спастический парапарез, прогрессирующую многоочаговую лейкоэнцефалопатию, семейную дизавтономию (синдром Райли-Дея), а также прионные заболевания (включая, без ограничений, болезнь Крейтцфельдта-Якоба, болезнь Герстманна-Штраусслера-Шейнкера, куру, а также фатальную семейную инсомнию), заболевания и расстройства, сопровождаемые демиелинизацией, включая рассеянный склероз и наследственные заболевания, такие как лейкодистрофии.
Другие нейродегенеративные состояния включают опухоли и другие неопластические состояния, затрагивающие ЦНС и ПНС. Хотя основное заболевание считается пролиферативным (а не нейродегенеративным), оно может привести к нарушению функций окружающих тканей. Кроме того, клеточную терапию можно использовать для доставки индуцирующих апоптоз или иных антинеопластических молекул к месту опухоли, например путем доставки генетически модифицированных клеток, вырабатывающих такие агенты.
Другие нейродегенеративные состояния включают различные нейропатии, такие как многоочаговые нейропатии, сенсорные нейропатии, двигательные нейропатии, сенсорно-двигательные нейропатии, связанные с инфекцией нейропатии, вегетативные нейропатии, сенсорно-вегетативные нейропатии, демиелинизирующие нейропатии (включая, без ограничений, синдром Гийена-Барре и хроническую воспалительную демиелинизирующую полирадикулонейропатию), другие воспалительные и иммунные нейропатии, нейропатии, индуцированные лекарственными средствами, нейропатии, индуцированные фармакологической терапией, нейропатии, индуцированные токсинами, травматические нейропатии (включая, без ограничений, компрессионные, раздавливающие, разрывные и сегментирующие нейропатии), метаболические нейропатии, эндокринные и паранеопластические нейропатии и т.п.
Другие нейродегенеративные состояния включают слабоумие независимо от этиологии, включая возрастное слабоумие и другие виды слабоумия, а также состояния, сопровождающиеся потерей памяти, включая слабоумие, связанное с болезнью Альцгеймера, сосудистое слабоумие, диффузное поражение белого вещества (болезнь Бинсвангера), слабоумие эндокринного или метаболического происхождения, слабоумие в результате травмы головы и диффузного повреждения мозга, слабоумие боксеров, а также слабоумие лобной доли.
В настоящем документе термин «лечение нейродегенеративного состояния» обозначает облегчение последствий или задержку, остановку или обращение развития, либо задержку или профилактику начала нейродегенеративного состояния, как описано в настоящем документе.
В настоящем документе термин «эффективное количество» относится к концентрации или количеству реагента или фармацевтической композиции, таких как фактор роста, индуцирующий дифференцирование агент, трофический фактор, популяция клеток или другой агент, которые эффективно обеспечивают получение необходимого результата, включая рост и/или дифференцирование клеток in vitro или in vivo либо лечение нейродегенеративного состояния, как описано в настоящем документе. В отношении факторов роста эффективное количество может означать диапазон от приблизительно 1 нанограмма/миллилитр до приблизительно 1 микрограмма/миллилитр. В отношении клеток, вводимых пациенту in vivo, эффективное количество может означать диапазон от нескольких сотен или менее до нескольких миллионов или более. В конкретных вариантах осуществления эффективное количество может находиться в диапазоне от 103 до 1011, более конкретно составлять по меньшей мере приблизительно 104 клеток. Следует понимать, что количество вводимых клеток будет изменяться в зависимости от конкретного заболевания, терапия которого проводится, включая, без ограничений, размер или общий объем/площадь обрабатываемой поверхности, а также степень близости места введения к обрабатываемой участку, среди других факторов, знакомых медицинскому биологу.
В настоящем документе термины «эффективный период времени (или время)» и «эффективные условия» обозначают период времени или другие контролируемые условия (например, температуру, влажность для способов in vitro), необходимые или предпочтительные для получения предполагаемых результатов от применения агента или фармацевтической композиции.
В настоящем документе термин «пациент «или «субъект» относится к животным, включая млекопитающих, предпочтительно людей, которые подвергаются лечению с помощью фармацевтических композиций или в соответствии со способами, описанными в настоящем документе.
В настоящем документе термин «фармацевтически приемлемый носитель (или среда)», который может использоваться взаимозаменяемо с термином «биологически совместимый носитель (или среда)», относится к реагентам, клеткам, соединениям, материалам, композициям и/или дозированным формам, которые не только совместимы с вводимыми в терапевтических целях клетками и другими агентами, но которые также, в рамках медицинского здравого смысла, подходят для использования в контакте с тканями людей или животных, не вызывая чрезмерную токсичность, раздражение, аллергическую реакцию или другое осложнение, соразмерное с разумным соотношением пользы/риска от применения. Как более подробно описано в настоящем документе, подходящие для целей настоящего изобретения фармацевтически приемлемые носители включают жидкости, полутвердые (например, гели) и твердые материалы (например, клеточные каркасы и матрицы, трубки, листы и другие такие материалы, известные в данной области и более подробно описанные в настоящем документе). Данные полутвердые и твердые материалы могут быть выполнены с возможностью сопротивления деструкции внутри организма (небиоразлагаемые) или могут быть выполнены с возможностью деструкции внутри организма (биоразлагаемые, биоразрушаемые). Биоразлагаемый материал может дополнительно быть саморассасывающимся или биорассасывающимся, т.е. он может растворяться и всасываться жидкостями организма (одним из примеров являются водорастворимые имплантаты) или разлагаться и, в конце концов, выводиться из организма, либо путем превращения в другие материалы, либо путем расщепления и устранения естественными процессами.
Несколько терминов в настоящем документе относятся к области заместительной клеточной терапии. В настоящем документе термины «аутогенный перенос», «аутогенная трансплантация», «аутогенный трансплантат» и т.п. относятся к методам лечения, в которых донор клеток также является и получателем заместительной клеточной терапии. В настоящем документе термины «аллогенный перенос», «аллогенная трансплантация», «аллогенный трансплантат» и т.п. относятся к методам лечения, в которых донор клеток является представителем того же биологического вида, что и получатель заместительной клеточной терапии, но не является тем же индивидом. Перенос клеток, при котором клетки донора были подобраны с точки зрения гистосовместимости с организмом получателя, иногда называется «сингенным переносом». В настоящем документе термины «ксеногенный перенос», «ксеногенная трансплантация», «ксеногенный трансплантат» и т.п. относятся к методам лечения, в которых донор клеток является представителем другого биологического вида по сравнению с получателем заместительной клеточной терапии.
ОПИСАНИЕ
Общий признак нейродегенеративных состояний, которые охватывают острые, хронические и прогрессирующие расстройства и заболевания с самыми различными причинами, представляет собой дисфункцию или утрату конкретной или уязвимой группы нервных клеток. Эта общность позволяет разрабатывать аналогичные терапевтические подходы к восстановлению и регенерации уязвимой или поврежденной нервной ткани, одним из которых является клеточная терапия. В различных вариантах осуществления, описанных в настоящем документе, настоящее изобретение предлагает способы и фармацевтические композиции для восстановления и регенерации нервной ткани, в которых применяются клетки-предшественники и популяции клеток, полученные из ткани пуповины. Настоящее изобретение может использоваться для лечения любого нейродегенеративного состояния, но особенно подходит для ряда неврологических расстройств, лечение которых или излечение от которых до настоящего момента было сложным или совсем отсутствовало. Данные расстройства включают, без ограничений, болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, инсульт, амиотрофический боковой склероз, рассеянный склероз, повреждение спинного мозга и повреждение периферического нерва (например, связанное с диабетической нейропатией). В одном варианте осуществления настоящего изобретения нейродегенеративное состояние представляет собой амиотрофический боковой склероз (АБС).
Как кратко описано выше, в одном из аспектов настоящее изобретение по существу относится к выделенным клеткам, полученным из ткани пуповины, (UTC), которые по существу свободны от крови. UTC способны к самообновлению и размножению в культуре и обладают потенциалом к дифференцированию в клетки нейронных фенотипов. Определенные варианты осуществления предлагают популяции, содержащие такие клетки, фармацевтические композиции, содержащие клетки, или их компоненты или продукты, а также способы использования фармацевтических композиций для лечения пациентов с острыми или хроническими нейродегенеративными состояниями. Для клеток, полученных из ткани пуповины, охарактеризованы ростовые свойства в культуре, маркеры клеточной поверхности, экспрессия генов, способность вырабатывать определенные биохимические трофические факторы и иммунологические качества.
Приготовление клеток, полученных из ткани пуповины и плаценты
В соответствии со способами, описанными в настоящем документе, плаценту и пуповину млекопитающего получают по завершении доношенной или недоношенной беременности, например после отделения последа, или вскоре после этого. Ткань можно транспортировать с места приема родов в лабораторию в стерильном контейнере, таком как колба, лабораторный стакан, чашка для культивирования или пакет. Контейнер может содержать раствор или среду, включая, без ограничений, солевой раствор, такой как, например, модифицированная по способу Дульбекко среда Игла (DMEM) или фосфатно-солевой буфер (ФСБ), либо любой раствор, применяемый для транспортировки органов, используемых для трансплантации, такой как раствор, разработанный в Университете штата Висконсин, или перфторированный раствор. В среду или буферный раствор можно добавить один или более антибиотиков и/или антимикотиков, включая, без ограничений, пенициллин, стрептомицин, амфотерицин B, гентамицин и нистатин. Ткань пуповины или плаценты можно промыть в растворе антикоагулянта, таком как гепаринсодержащий раствор. До экстрагирования клеток ткань предпочтительно поддерживать при температуре приблизительно 4-10°C. Еще более предпочтительно не замораживать ткань до экстрагирования клеток, полученных из плаценты или пуповины.
Выделение клеток предпочтительно проводить в асептических условиях. Пуповину можно отделить от плаценты с использованием средств, известных специалистам в данной области. В альтернативном варианте осуществления пуповину и плаценту используют без отделения. Перед выделением клеток предпочтительно удаляют кровь и продукты распада клеток. Например, ткань можно промыть в буферном растворе, таком как, без ограничений, фосфатно-солевой буфер. Промывочный буфер может также содержать один или более антимикотиков и/или антибиотиков, таких как, без ограничений, пенициллин, стрептомицин, амфотерицин B, гентамицин и нистатин.
Ткань, представляющую собой цельную пуповину, цельную плаценту или их фрагмент или часть, механически измельчают (используя разрезающее или сдвиговое усилие). В предпочтительном в настоящее время варианте осуществления в процедуре выделения клеток также используют способ ферментативного расщепления. Специалистам в данной области известно множество ферментов, которые можно использовать для выделения отдельных клеток из сложных тканевых матриц для облегчения их выращивания в культуре. В продаже доступны следующие ферменты: от слабо расщепляющих (например, дезоксирибонуклеазы и диспаза (нейтральная протеаза)) до сильно расщепляющих (например, папаин и трипсин). Неограничивающий перечень ферментов, совместимых с целями настоящего изобретения, включает ферменты с муколитической активностью, металлопротеазы, нейтральные протеазы, серинпротеазы (такие как трипсин, химотрипсин или эластаза) и дезоксирибонуклеазы. В настоящее время предпочтительными являются ферменты с активностью, выбранные из металлопротеаз, нейтральных протеаз и ферментов с муколитической активностью. Например, известно использование коллагеназ для выделения различных клеток из тканей. Дезоксирибонуклеазы могут расщеплять одноцепочечные ДНК и позволяют свести к минимуму агглютинацию клеток в процессе выделения. Предпочтительные способы включают ферментативную обработку, например, коллагеназой и диспазой или коллагеназой, диспазой и гиалуронидазой, и такие способы предложены там, где в определенных предпочтительных вариантах осуществления на стадии диссоциации используется смесь коллагеназы и диспазы (нейтральной протеазы). Более предпочтительны способы с применением расщепления в присутствии по меньшей мере одной коллагеназы из Clostridium histolyticum и одного из ферментов с протеазной активностью - диспазы или термолизина. Еще более предпочтительны способы с применением расщепления ферментами как с коллагеназной, так и диспазной активностью. Также предпочтительны способы, которые включают расщепление ферментом с гиалуронидазной активностью помимо ферментов с коллагеназной и диспазной активностью. Специалист в данной области определит, что известно множество способов таких ферментативных обработок для выделения клеток из различных тканей-источников. Например, для способов настоящего изобретения подходят комбинации ферментов серии LIBERASE™ Blendzyme (Roche). Известны и другие источники ферментов, и специалист в данной области также может выделить такие ферменты непосредственно из их природных источников. Специалист в данной области также может оценивать возможности новых или дополнительных ферментов или комбинаций ферментов для выделения клеток, составляющих предмет настоящего изобретения. Предпочтительная продолжительность ферментативной обработки составляет 0,5, 1, 1,5 или 2 часа или более. В других предпочтительных вариантах осуществления ткань инкубируют при температуре 37°C в процессе ферментативной обработки на стадии диссоциации.
В некоторых вариантах осуществления настоящего изобретения ткань разделяют на части, содержащие различные аспекты ткани, такие как, например, неонатальный, неонатальный/материнский и материнский аспекты плаценты. Разделенные части затем диссоциируют с использованием механической и/или ферментативной диссоциации в соответствии со способами, описанными в настоящем документе. Клетки неонатальных или материнских линий можно идентифицировать любыми средствами, известными специалистам в данной области, например с использованием анализа кариотипа или гибридизации in situ на Y-хромосому.
Выделенные клетки или ткань, из которых вырастают клетки hUTC, можно использовать для инициирования, или посева, культур клеток. Выделенные клетки переносят в стерильные сосуды для культивирования ткани без покрытия или с покрытием внеклеточным матриксом или лигандами, такими как ламинин, коллаген (нативный, денатурированный или сшитый), желатин, фибронектин и другие белки внеклеточного матрикса. Клетки hUTC культивируют в любой культуральной среде, способной поддерживать рост клеток, такой как, без ограничений, DMEM (с высоким или низким содержанием глюкозы), улучшенная DMEM, DMEM/MCDB 201, основная среда Игла, среда F10 Хэма (F10), среда F-12 Хэма (F12), модифицированная по способу Искова среда Дульбекко, ростовая среда для мезенхимальных стволовых клеток (MSCGM), DMEM/F12, среда RPMI 1640 и среда Cellgro FREE™. В культуральную среду можно дополнительно ввести один или более компонентов, включая, например, эмбриональную бычью сыворотку (FBS), предпочтительно в количестве приблизительно 2-15% (об/об); сыворотку лошади (ES); сыворотку человека (HS); бета-меркаптоэтанол (BME или 2-ME), предпочтительно в количестве приблизительно 0,001% (об/об); один или более факторов роста, например тромбоцитарный фактор роста (PDGF), эпидермальный фактор роста (EGF), фактор роста фибробластов (FGF), фактор роста эндотелия сосудов (VEGF), инсулиноподобный фактор роста-1 (IGF-1), фактор ингибирования лейкоцитов (LIF) и эритропоэтин; аминокислоты, включая L-валин; и один или более антибиотиков и/или антимикотиков для борьбы с микробактериальным заражением, такие как, например, пенициллин G, сульфат стрептомицина, амфотерицин B, гентамицин и нистатин, индивидуально или в комбинации. Культуральная среда предпочтительно представляет собой ростовую среду (DMEM с низким содержанием глюкозы, сыворотку, BME и антибиотик).
Клетки высевают в сосуды для культивирования с плотностью, позволяющей клеткам расти. В предпочтительном варианте осуществления клетки культивируют в атмосфере с содержанием CO2 от приблизительно 0 до приблизительно 5% об. В некоторых предпочтительных вариантах осуществления клетки культивируют в атмосфере с содержанием O2 от приблизительно 2 до приблизительно 25%, предпочтительно в атмосфере с содержанием O2 от приблизительно 5 до приблизительно 20%. Клетки предпочтительно культивируют при температуре от приблизительно 25 до приблизительно 40°C и более предпочтительно культивируют при температуре 37°C. Клетки предпочтительно культивируют в инкубаторе. Среда в сосуде для культивирования может быть неподвижной или возбужденной, например с использованием биореактора. Клетки предпочтительно выращивают в условиях низкого окислительного стресса (например, с добавлением глутатиона, витамина C, каталазы, витамина E, N-ацетилцистеина). В настоящем документе «условия низкого окислительного стресса» обозначают условия отсутствия или минимального свободнорадикального повреждения культивируемых клеток.
Способы выбора наиболее предпочтительной культуральной среды, подготовки среды и методик культивирования клеток хорошо известны в данной области и описаны в ряде источников, включая руководства Doyle et al., (под ред.), 1995 г., Cell & Tissue Culture: Laboratory Procedures, John Wiley & Sons, г. Чичестер; и Ho и Wang (под ред.), 1991 г., Animal Cell Bioreactors, Butterworth-Heinemann, г. Бостон, которые включены в настоящий документ путем ссылки.
После культивирования выделенных клеток или фрагментов ткани в течение достаточного периода времени клетки UTC прорастают в результате миграции из ткани или деления клеток либо по обеим причинам. В некоторых вариантах осуществления настоящего изобретения клетки UTC пассируют или переносят в отдельный сосуд для культивирования, содержащий свежую среду того же типа или другого типа по сравнению с первоначально использованной, в котором популяцию клеток можно митотически размножить. Клетки, составляющие предмет настоящего изобретения, можно применять на любой стадии от пассажа 0 до старения. Клетки предпочтительно пассируют от приблизительно 3 до приблизительно 25 раз, более предпочтительно пассируют от приблизительно 4 до приблизительно 12 раз и предпочтительно пассируют 10 или 11 раз. Для подтверждения выделения клональной популяции клеток можно провести клонирование и/или субклонирование.
В некоторых аспектах настоящего изобретения различные типы клеток, присутствующие в ткани пуповины или плаценты, фракционируют на субпопуляции, из которых можно выделить клетки. Этого можно достигнуть с использованием стандартных методик разделения клеток, включая, без ограничений, ферментативную обработку для диссоциации ткани на составляющие ее клетки с последующим клонированием и отбором клеток конкретных типов, включая, без ограничений, отбор на основе морфологических и/или биохимических маркеров; выборочный рост необходимых клеток (положительный отбор), выборочное разрушение нежелательных клеток (отрицательный отбор); разделение на основе различной способности клеток в смешанной популяции к агглютинации, например с соевым агглютинином; процедуры заморозки-разморозки; различие адгезионных характеристик клеток в смешанной популяции; фильтрование; стандартное и зональное центрифугирование; элютриационное центрифугирование (противоточное центрифугирование); разделение при нормальном поле тяжести; противоточное распределение; электрофорез; и сортировку флуоресцентноактивированных клеток (FACS). Обзор методик клонального отбора и разделения клеток можно найти в руководстве Freshney, 1994 г., Culture of Animal Cells: A Manual of Basic Techniques, 3-е изд., Wiley-Liss, Inc., г. Нью-Йорк, которое включено в настоящий документ путем ссылки.
Культуральную среду меняют по необходимости, например осторожно отсасывая среду из чашки, например с помощью пипетки, и добавляя требуемое количество свежей среды. Инкубацию продолжают до накопления в чашке достаточного количества или плотности клеток. Исходные эксплантированные части ткани можно удалить и трипсинизировать оставшиеся клетки с помощью стандартных методик или с использованием скребка. После трипсинизации клетки собирают, переносят в свежую среду и инкубируют, как описано выше. В некоторых вариантах осуществления среду заменяют по меньшей мере один раз приблизительно через 24 часа после трипсинизации для удаления плавающих клеток. Оставшиеся в культуре клетки считают клетками, полученными из ткани пуповины (UTC).
Клетки UTC можно криоконсервировать. Соответственно, в предпочтительном варианте осуществления, более подробно описанном ниже, клетки UTC для аутогенного переноса (либо для матери, либо для ребенка) можно получить из соответствующих послеродовых тканей после рождения ребенка и затем криоконсервировать их таким образом, чтобы после этого они были доступны в случае возникновения необходимости в трансплантации.
Характеристики клеток, полученных из пуповины и плаценты
Клетки, полученные из пуповины и плаценты, могут быть охарактеризованы, например, по ростовым характеристикам (например, способности к удвоению популяции, времени удвоения, количеству пассажей до старения), анализу кариотипа (например, нормальный кариотип; материнская или неонатальная линия), проточной цитометрией (например, анализ FACS), иммуногистохимически и/или иммуноцитохимически (например, на обнаружение эпитопов), по профилю экспрессии генов (например, с использованием генных чипов; полимеразной цепной реакции (например, ПЦР с обратной транскриптазой, ПЦР в реальном времени и стандартной ПЦР)), по белковым чипам, по секреции белков (например, по анализу коагулирующей активности плазмы или анализу среды кондиционированной плазмацитоидными дендритными клетками (ПДК), например с использованием твердофазного иммуноферментного анализа (ИФА)), реакции смешанной культуры лимфоцитов (например, как меры стимуляции мононуклеарных клеток периферической крови (МКПК)) и/или другими способами, известными в данной области.
Примеры клеток, полученных из ткани плаценты, депонированы в коллекции American Type Culture Collection (ATCC, г. Манассас, штат Вирджиния) под следующими номерами доступа ATCC: (1) штамм PLA 071003 (P8) депонирован 15 июня 2004 года под № доступа PTA-6074; (2) штамм PLA 071003 (P11) депонирован 15 июня 2004 года под № доступа PTA-6075; и (3) штамм PLA 071003 (P16) депонирован 16 июня 2004 года под № доступа PTA-6079. Примеры клеток, полученных из ткани пуповины, депонированы в коллекции American Type Culture Collection 10 июня 2004 года под следующими номерами доступа ATCC: (1) штамму UMB 022803 (P7) присвоен № доступа PTA-6067; и (2) штамму UMB 022803 (P17) присвоен № доступа PTA-6068.
В различных вариантах осуществления клетки обладают одной или более из следующих характеристик роста: (1) для роста в культуре клеткам необходим L-валин; (2) клетки могут расти в атмосфере с содержанием кислорода от приблизительно 5% до по меньшей мере приблизительно 20%; (3) клетки обладают потенциалом к по меньшей мере приблизительно 40 удвоениям в культуре до достижения старения; и (4) клетки закрепляются и размножаются на поверхности сосуда для культивирования с покрытием или без покрытия, причем сосуд для культивирования с покрытием содержит покрытие из желатина, ламинина, коллагена, полиорнитина, витронектина или фибронектина.
В определенных вариантах осуществления клетки обладают нормальным кариотипом, который поддерживается при пассировании клеток. Кариотипирование, в частности, полезно для идентификации и отличия неонатальных клеток от материнских клеток, полученных из плаценты. Способы кариотипирования доступны и известны специалистам в данной области.
В других вариантах осуществления клетки могут быть охарактеризованы по выработке определенных белков, включая (1) выработку по меньшей мере одного из тканевого фактора, виментина и альфа-актина гладких мышц; и (2) выработку по меньшей мере одного из CD10, CD13, CD44, CD73, CD90, PDGFr-альфа, PD-L2 и маркеров клеточной поверхности HLA-A, B, C по результатам анализа с использованием проточной цитометрии. В других вариантах осуществления клетки UTC могут быть охарактеризованы по отсутствию выработки по меньшей мере одного из CD31, CD34, CD45, CD80, CD86, CD117, CD141, CD178, B7-H2, HLA-G и маркеров клеточной поверхности HLA-DR, DP, DQ по результатам анализа с использованием проточной цитометрии. Особенно предпочтительны клетки, которые вырабатывают по меньшей мере два из тканевого фактора, виментина и альфа-актина гладких мышц. Более предпочтительны клетки, которые вырабатывают все три из тканевого фактора, виментина и альфа-актина гладких мышц.
В других вариантах осуществления клетки могут быть охарактеризованы по экспрессии гена, которая, по сравнению с клеткой человека, представляющей собой фибробласт, мезенхимальную стволовую клетку или клетку костного мозга гребня подвздошной кости, повышена для гена, кодирующего по меньшей мере один из интерлейкина-8; ретикулона 1; CXCL1 (хемокиновый (мотив C-X-C) лиганд 1/стимулятор активности роста меланомы, альфа); CXCL6 (хемокиновый (мотив C-X-C) лиганд 6/белок хемотаксиса гранулоцитов 2)); CXCL3 (хемокиновый (мотив C-X-C) лиганд 3); TNFAIP3 (индуцируемый фактором некроза альфа белок 3).
В других вариантах осуществления клетки могут быть охарактеризованы по экспрессии гена, которая, по сравнению с клеткой человека, представляющей собой фибробласт, мезенхимальную стволовую клетку или клетку костного мозга гребня подвздошной кости, снижена для гена, кодирующего по меньшей мере один из: SHOX2 (содержащий гомеобокс ген низкорослости 2); HSPB2 (27 кДа белок теплового шока 2); CXCL12 (хемокиновый (мотив C-X-C) лиганд 12/стромальный фактор 1); эластина (надклапанный аортальный стеноз, синдром Вильямса-Бойрена); мРНК Homo sapiens; кДНК DKFZp586M2022 (из клона DKFZp586M2022); Meox2 (содержащий мезенхимальный гомеобокс ген 2, содержащий гомеобокс блокировки роста ген); SIX1 (гомолог содержащего гомеобокс гена sine oculis 1) (Drosophila); кристаллина, альфа B; DAAM2 (ассоциированный с белком Disheveled активатор морфогенеза 2); белка DKFZP586B2420; аналога нейтралина 1; тетранектина (связывающийся с плазминогеном белок); STAC (ген, содержащий домен src-гомологии 3 (SH3) и богатый цистеином домен); холестерин-25-гидроксилазы; RUNX3 (связанный с карликовостью фактор транскрипции 3); рецептора интерлейкина-11, альфа; PCOLCE (усилитель проколлаген-C-эндопептидазы); FZD7 (гомолог frizzled 7) (Drosophila); гипотетического гена BC008967; коллагена, тип VIII, альфа 1; TNC (тенасцин C, гексабрахион); IRX5 (содержащий гомеобокс IRX белок 5); гефестина; интегрина, бета 8; SV2A (гликопротеин синаптического пузырька 2); NBL1 (нейробластома, ген подавления онкогенности 1); IGFBP2 (связывающийся с инсулиноподобным фактором роста белок 2, 36 кДа); кДНК Homo sapiens FLJ12280 fis, клон MAMMA1001744; CRLF1 (подобный цитокиновому рецептору фактор 1); KCNN4 (активируемый кальцием калиевый канал средней/низкой проводимости, подсемейство N, член 4); интегрина, бета 7; транскрипционного коактиватора с мотивом связывания с PDZ (TAZ); SIX2 (гомолог содержащего гомеобокс гена sine oculis 2) (Drosophila); белка KIAA1034; VAMP5 (везикуло-ассоциированный мембранный белок 5/миобревин); EFEMP1 (содержащий EGF фибулинподобный белок внеклеточного матрикса 1); EGR3 (белок раннего ростового ответа 3);DLX5 (содержащий гомеобокс distal-less ген 5); гипотетического белка FLJ20373; AKR1C3 (альдокеторедуктаза семейства 1, член C3/3-альфа-гидроксистероиддегидрогеназа, тип II); бигликана; транскрипционного коактиватора с мотивом связывания с PDZ (TAZ); фибронектина 1; проэнкефалина; интегрина, бета-подобного 1 (с доменами EGF-подобных повторов); мРНК Homo sapiens, полноразмерная вставка кДНК, клон EUROIMAGE 1968422; EphA3; белка KIAA0367; NPR3 (рецептор натрийуретического пептида C/гуанилатциклаза C/рецептор атрионатрийуретического пептида C); гипотетического белка FLJ14054; мРНК Homo sapiens; кДНК DKFZp564B222 (из клона DKFZp564B222); BNIP3L (ген белка, подобного белку 3, взаимодействующему с BCL2/белком аденовируса E1B 19 кДа); AE-связывающего белка 1; и COX7A1 (полипептид 1 субъединицы VIIa цитохром c оксидазы) (мышечный).
В других вариантах осуществления клетки могут быть охарактеризованы по секреции по меньшей мере одного из MCP-1, IL-6, IL-8, GCP-2, HGF, KGF, FGF, HB-EGF, BDNF, TPO, MIP1a, RANTES и TIMP1. В альтернативных вариантах осуществления клетки могут быть охарактеризованы по отсутствию секреции по меньшей мере одного из TGF-бета2, ANG2, PDGFbb, MIP1b, I309, MDC и VEGF по результатам ИФА.
В предпочтительных вариантах осуществления клетка обладает двумя или более из перечисленных выше характеристик роста, выработки белков/маркеров клеточной поверхности, экспрессии генов или секреции. Более предпочтительны клетки, обладающие тремя, четырьмя или пятью или более характеристиками. Еще более предпочтительны клетки, обладающие шестью, семью или восемью или более характеристиками. Еще более предпочтительны клетки, обладающие всеми из указанных выше характеристик.
В одном варианте осуществления настоящего изобретения клетки, полученные из ткани пуповины, которые выделены из ткани пуповины человека и по существу свободны от крови, способны к самообновлению и размножению в культуре, обладают потенциалом к дифференцированию в клетки других фенотипов, способны произвести по меньшей мере 40 удвоений и обладают следующими характеристиками: (a) экспрессия каждого из CD10, CD13, CD44, CD73, CD90, PDGFr-альфа, PD-L2 и HLA-A, B, C; (b) отсутствие экспрессии любого из CD31, CD34, CD45, CD80, CD86, CD 117, CD141, CD178, B7-H2, HLA-G или HLA-DR, DP, DQ; и (c) повышенная экспрессия интерлейкина-8, ретикулона 1 и CXCL3 (хемокинового (мотив C-X-C) лиганда 3) по сравнению с клеткой человека, представляющей собой фибробласт, мезенхимальную стволовую клетку или клетку костного мозга гребня подвздошной кости. В одном варианте осуществления данные клетки, полученные из ткани пуповины, также обладают одной или более из следующих характеристик: (a) секреция каждого из факторов MCP-1, MIP1-бета, IL-6, IL-8, GCP-2, HGF, KGF, FGF, HB-EGF, BDNF, TPO, RANTES и TIMP1; и (b) отсутствие секреции любого из факторов SDF-1альфа, TGF-бета2, ANG2, PDGFbb, MIP1a и VEGF. В другом варианте осуществления данные клетки, полученные из ткани пуповины, не экспрессируют hTERT или теломеразу.
Среди клеток, которые в настоящее время предпочтительны для использования в целях настоящего изобретения в некоторых его аспектах, встречаются клетки, полученные из ткани пуповины, обладающие описанными выше характеристиками, и, в частности, те из них, которые имеют нормальные кариотипы и поддерживают нормальные кариотипы при пассировании, и дополнительно те из них, которые экспрессируют каждый из маркеров CD10, CD13, CD44, CD73, CD90, PDGFr-альфа и HLA-A, B, C, причем клетки вырабатывают иммунологически детектируемые белки, соответствующие перечисленным маркерам. Еще более предпочтительны клетки, которые, в дополнение к описанному выше, не вырабатывают белки, соответствующие любому из маркеров CD31, CD34, CD45, CD117, CD141 или HLA-DR, DP, DQ по результатам анализа с использованием проточной цитометрии.
Определенные клетки, обладающие потенциалом к дифференцированию по линиям, ведущим к различным фенотипам, неустойчивы и, таким образом, могут спонтанно дифференцироваться. В настоящее время предпочтительными для использования в целях настоящего изобретения являются клетки, которые не дифференцируются спонтанно, например по линиям нейронного дифференцирования. Предпочтительные клетки, при выращивании в ростовой среде, по существу устойчивы по отношению к клеточным маркерам, вырабатывающимся на их поверхности, и по отношению к профилю экспрессии различных генов, например определяемых с использованием генного чипа Affymetrix GENECHIP®. Такие клетки остаются по существу постоянными, например в отношении характеристик маркеров поверхности при пассировании, на протяжении множества удвоений популяции.
Однако одной из отличительных характеристик таких клеток является возможность их намеренного индуцирования для дифференцирования в фенотипы нейронной линии дифференцирования путем создания для них при культивировании условий, индуцирующих такое дифференцирование. Этого можно достичь при помощи одного или более способов, известных специалистам в данной области. Например, как описано в настоящем документе, клетки, полученные из ткани пуповины, можно высеивать на покрытые ламинином колбы в среде Neurobasal-A (Invitrogen, г. Карлсбад, штат Калифорния), содержащей B27 (добавка B27, Invitrogen), L-глутамин и пенициллин/стрептомицин, комбинация которых в настоящем документе будет называться средой размножения клеток-предшественников нейронов (NPE). В среды NPE можно дополнительно добавить bFGF и/или EGF. В альтернативном варианте осуществления клетки UTC можно индуцировать для дифференцирования in vitro путем (1) совместного культивирования клеток UTC с клетками-предшественниками нейронов или (2) выращивания клеток UTC в среде, кондиционированной клетками-предшественниками нейронов.
Дифференцирование клеток UTC можно показать по морфологии биполярной клетки с протяженными отростками. Популяции индуцированных клеток могут давать положительный результат при окрашивании на наличие нестина. Дифференцированные клетки UTC можно оценивать по определению нестина, TuJ1 (бета-III-тубулин), GFAP, тирозингидроксилазы, GABA, O4 и/или MBP. В некоторых вариантах осуществления клетки UTC показывали способность образовывать объемные объекты, характерные для формирования нейросфер из нервных стволовых клеток.
Популяции, модификации, компоненты и продукты клеток UTC
В другом аспекте настоящего изобретения предложены популяции описанных выше клеток UTC. В некоторых вариантах осуществления популяция клеток является неоднородной. Неоднородная популяция клеток, составляющая предмет настоящего изобретения, может содержать по меньшей мере приблизительно 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% или 95% клеток UTC, составляющих предмет настоящего изобретения. Неоднородные популяции клеток, составляющие предмет настоящего изобретения, могут дополнительно содержать стволовые клетки или другие клетки-предшественники, такие как клетки-предшественники нейронов, или могут дополнительно содержать полностью дифференцированные нервные клетки. В некоторых вариантах осуществления популяция является по существу однородной, т.е. содержит по существу только клетки UTC (предпочтительно по меньшей мере приблизительно 96%, 97%, 98%, 99% или более клеток UTC). Однородная популяция клеток, составляющая предмет настоящего изобретения, содержит клетки, полученные из пуповины. Однородные популяции клеток, полученных из пуповины, предпочтительно свободны от клеток материнской линии. Однородность популяции клеток можно получить при помощи любого способа, известного в данной области, например путем сортировки клеток (например, с использованием проточной цитометрии) или путем клонального размножения в соответствии с известными способами. Таким образом, предпочтительные однородные популяции клеток UTC могут содержать клонированную линию клеток, полученных из ткани пуповины. Такие популяции особенно полезны в тех случаях, когда выделен клеточный клон с крайне необходимыми характеристиками.
В настоящем документе также предложены популяции клеток, инкубированных в присутствии одного или более факторов, или в условиях, которые стимулируют дифференцирование стволовых клеток по нейрогенной линии. Такие факторы известны в данной области, и специалист определит, что подбор подходящих условий для дифференцирования можно провести стандартными экспериментами. Оптимизацию таких условий можно получить путем статистического построения эксперимента и его анализа, например, методология поверхности отклика позволяет одновременно провести оптимизацию по множеству переменных, например в биологической культуре. Предпочтительные в настоящее время факторы включают, без ограничений, факторы, такие как факторы роста или трофические факторы, деметилирующие агенты, совместное культивирование с клетками нейронной линии дифференцирования или культивирование в среде, кондиционированной клетками нейронной линии дифференцирования, а также другие условия, известные в данной области, для стимулирования дифференцирования стволовых клеток по нейрогенной линии или линии дифференцирования (смотрите, например, Lang, K.J.D. et al., 2004 г., J. Neurosci. Res. 76: 184-192; Johe, K.K. et al., 1996 г., Genes Devel. 10: 3129-3140; Gottleib, D., 2002 г., Ann. Rev. Neurosci. 25: 381-407).
Клетки UTC можно также генетически модифицировать, например, для выработки продуктов нейротерапевтически полезных генов или для выработки антинеопластических агентов для лечения опухолей. Генетическую модификацию можно провести с использованием любого из доступных векторов, включая, без ограничений, интегрирующие вирусные векторы, например ретровирусный вектор или векторы, связанные с аденовирусами; неинтегрирующие реплицирующиеся векторы, например векторы вируса папилломы, векторы SV40, аденовирусные векторы; или вирусные векторы с нарушенной репликацией. Другие способы введения ДНК в клетки включают применение липосом, электропорации, генной пушки или прямую инъекцию ДНК.
Клетки-хозяева предпочтительно трансформируют или трансфицируют ДНК под контролем или в функциональной связи с одним или более соответствующими элементами управления экспрессией, такими как промоторные или усилительные последовательности, терминаторы транскрипции, сайты полиаденилирования и т.п., и селектируемым маркером. Для индуцирования экспрессии вставленного гена можно использовать любой промотор. Например, вирусные промоторы включают, без ограничений, промотор/усилитель CMV, промоторы SV 40, вирус папилломы, вирус Эпштейна-Барра или промотор гена эластина. В некоторых вариантах осуществления управляющие элементы, используемые для контроля над экспрессией рассматриваемого гена, может позволять осуществление регулируемой экспрессии гена таким образом, чтобы в условиях in vivo продукт синтезировался только в случае необходимости. При необходимости временной экспрессии предпочтительно используют конститутивные промоторы в неинтегрирующем векторе и/или векторе с нарушенной репликацией. В альтернативном варианте осуществления при необходимости для активации экспрессии вставленного гена можно использовать индуцируемые промоторы. Индуцируемые промоторы включают, без ограничений, промоторы, связанные с металлотионеином и белками теплового шока.
После введения экзогенной ДНК клетки, полученные методами генетической инженерии, можно оставить для роста в обогащенных средах и затем перевести в селективные среды. Селектируемый маркер в экзогенной ДНК придает устойчивость к селекции и позволяет клеткам стабильно интегрировать экзогенную ДНК, например вводимую на плазмиде, в свои хромосомы и вырасти для образования очагов, которые затем можно клонировать и размножить в клеточные линии. Данный способ можно эффективно использовать для конструирования клеточных линий, экспрессирующих продукт гена.
Клетки, составляющие предмет настоящего изобретения, можно модифицировать методами генетической инженерии для «выключения» или «выбивания» экспрессии факторов, стимулирующих воспаление или отторжение имплантата в месте введения. Ниже описаны методики отрицательной модуляции для снижения уровней экспрессии целевого гена или активности продукта целевого гена. В настоящем документе «отрицательная модуляция» обозначает снижение уровня и/или активности продукта целевого гена по отношению к уровню и/или активности продукта целевого гена в отсутствии модулирующей обработки. Экспрессию нативного гена нейрона или глиальной клетки можно уменьшить или выключить с помощью ряда методик, включая, например, ингибирование экспрессии путем инактивации гена с помощью методики гомологичной рекомбинации. Как правило, экзон, кодирующий важный участок белка (или экзон в положении 5’ к данному участку), прерывают положительно селектируемым маркером, например neo, тем самым блокируя получение нормальной мРНК с целевого гена, что приводит к инактивации гена. Ген также можно инактивировать путем создания делеции в части гена или путем удаления всего гена. Использование конструкции с двумя участками гомологии к целевому гену, находящимися в геноме далеко от друга, позволяет вырезать соединяющие два участка последовательности (Mombaerts et al., 1991 г., Proc. Nat. Acad. Sci. U.S.A. 88: 3084-3087). Для снижения уровня активности целевого гена также можно использовать антисенсы, ДНК-ферменты, рибозимы, малые интерферирующие РНК (миРНК) и другие такие молекулы, которые ингибируют экспрессию целевого гена. Например, показано, что молекулы антисмысловой РНК, ингибирующие экспрессию главных комплексов генов гистосовместимости (HLA), являются наиболее универсальными в отношении иммунных ответов. Кроме того, для снижения уровня активности целевого гена можно использовать трехцепочечные молекулы. Данные методики подробно описаны в работе L.G. Davis et al. (под ред.), 1994 г., Basic Methods in Molecular Biology, 2-е изд., Appleton & Lange, г. Норуолк, штат Коннектикут.
В других аспектах настоящее изобретение представляет клеточные лизаты и растворимые клеточные фракции, приготовленные из клеток UTC, или неоднородные или однородные популяции клеток, содержащие клетки UTC, а также клетки UTC или их популяции, генетически модифицированные или стимулированные для дифференцирования по нейрогенной линии. Такие лизаты и их фракции имеют множество применений. Использование растворимой фракции лизата клеток UTC (например, по существу свободной от мембран) in vivo, например, позволяет использовать полезное внутриклеточное вещество аллогенно для пациента без введения значительного количества белков клеточной поверхности, которые, скорее всего, могут спровоцировать отторжение или другие нежелательные иммунные ответы. Способы лизиса клеток хорошо известны в данной области и включают различные способы механического разрушения, ферментативного разрушения или химического разрушения или их комбинации. Такие клеточные лизаты можно приготовить из клеток непосредственно в их ростовой среде таким образом, чтобы они содержали секретированные факторы роста и т.п., либо их можно приготовить из отмытых от среды клеток, например, с использованием ФСБ или другого раствора. При необходимости отмытые клетки можно повторно суспендировать в концентрациях, превышающих плотность исходной популяции.
В одном варианте осуществления получают цельноклеточные лизаты, например, путем разрушения клеток без последующего разделения клеточных фракций. В другом варианте осуществления фракцию клеточных мембран отделяют от растворимой клеточной фракции стандартными способами, известными в данной области, например центрифугированием, фильтрованием или аналогичными способами.
Клеточные лизаты или растворимые клеточные фракции, приготовленные из популяций клеток, полученных из ткани пуповины, можно использовать непосредственно, в дополнительно сконцентрированном виде, например с использованием ультрафильтрации или лиофилизации, или даже в высушенном виде, частично высушенном виде, в сочетании с известными специалистам фармацевтически приемлемыми носителями или разбавителями или в сочетании с другими соединениями, такими как биологические соединения, например фармацевтически эффективными белковыми композициями. Клеточные лизаты или их фракции можно использовать in vitro или in vivo, индивидуально или, например, вместе с аутогенными или сингенными живыми клетками. При использовании in vivo лизаты можно вводить локально в месте обработки или удаленно, например чтобы обеспечить пациента необходимыми клеточными факторами роста.
В дополнительном варианте осуществления клетки UTC можно культивировать in vitro для получения биологических продуктов с высоким выходом. Например, такие клетки, которые либо естественным образом вырабатывают конкретный рассматриваемый биологический продукт (например, трофический фактор), либо были модифицированы методами генетической инженерии для выработки биологического продукта, можно клонально размножать с использованием методик культивирования, описанных в настоящем документе. В альтернативном варианте осуществления клетки можно размножать в среде, которая индуцирует дифференцирование клеток по нейронной линии дифференцирования. В любом случае из кондиционированной среды можно легко выделить вырабатываемые клеткой и секретируемые в среду биологические продукты, используя стандартные методики разделения, например, такие как дифференциальное осаждение белков, ионообменная хроматография, гельпроникающая хроматография, электрофорез, ВЭЖХ и т.п. Для реализации проточного способа кормления, например, объемной культуры in vitro можно эффективно использовать «биореактор». По существу по мере прохождения свежей среды через объемную культуру биологический продукт вымывается из культуры и затем может быть выделен из выходящего потока, как описано выше.
В альтернативном варианте осуществления рассматриваемый биологический продукт может оставаться внутри клеток, и, таким образом, для его сбора может потребоваться лизис клеток, как описано выше. Затем биологический продукт можно очистить с использованием любой одной или более из перечисленных выше методик.
В других вариантах осуществления настоящее изобретение представляет кондиционированную среду от культивированных клеток UTC для применения in vitro и in vivo, как описано ниже. Использование кондиционированной клетками UTC среды позволяет использовать секретированные клетками UTC полезные трофические факторы аллогенно для пациента без введения интактных клеток, что может спровоцировать отторжение или другие нежелательные иммунологические ответы. Кондиционированную среду готовят путем культивирования клеток в культуральной среде и последующего удаления клеток из среды.
Кондиционированную среду, приготовленную из популяций клеток, полученных из ткани пуповины, можно использовать непосредственно, в дополнительно сконцентрированном виде, например с использованием ультрафильтрации или лиофилизации, или даже в высушенном виде, частично высушенном виде, в сочетании с известными специалистам фармацевтически приемлемыми носителями или разбавителями или в сочетании с другими соединениями, такими как биологические соединения, например фармацевтически эффективными белковыми композициями. Кондиционированную среду можно использовать in vitro или in vivo, индивидуально или, например, вместе с аутогенными или сингенными живыми клетками. При использовании in vivo кондиционированную среду можно вводить локально в месте обработки или удаленно, например чтобы обеспечить пациента необходимыми клеточными факторами роста или трофическими факторами.
В другом варианте осуществления в качестве альтернативы имплантации живых клеток субъекту, нуждающемуся в восстановлении или замещении ткани, готовят, собирают и используют внеклеточный матрикс (ECM), получаемый культивированием клеток UTC на жидких, твердых или полутвердых субстратах. Клетки UTC культивируют in vitro на объемном каркасе, как описано далее в настоящем документе, в условиях, обеспечивающих секретирование необходимого количества ECM на каркас. Новую ткань удаляют и ECM обрабатывают для последующего использования, например в виде препарата для инъекции. С данной целью клетки на каркасе убивают и с каркаса удаляют продукты распада клеток. Данный процесс можно проводить несколькими различными способами. Например, живую ткань можно резко заморозить в жидком азоте без использования криопротектора, или ткань можно погрузить в стерильную дистиллированную воду таким образом, чтобы клетки лопнули под воздействием осмотического давления.
После умерщвления клеток можно разрушить клеточные мембраны и удалить продукты распада клеток обработкой мягким детергентом, таким как ЭДТА, CHAPS или цвиттер-ионный детергент. В альтернативном варианте осуществления ткань можно ферментативно переварить и/или экстрагировать реагентами, которые разрушают клеточные мембраны и позволяют провести извлечение содержимого клеток. Примеры таких ферментов включают, без ограничений, гиалуронидазу, диспазу, протеазы и нуклеазы. Примеры детергентов включают неионные детергенты, такие как, например, алкиларилполиэфирный спирт (TRITON X-100), октилфеноксиполиэтоксиэтанол (Rohm and Haas, г. Филадельфия, штат Пенсильвания), BRIJ-35, полиэтоксиэтаноллауриловый эфир (Atlas Chemical Co., г. Сан-Диего, штат Калифорния), полисорбат 20 (TWEEN 20), полиэтоксиэтанолсорбитанмонолаурат (Rohm and Haas), полиэтиленлауриловый эфир (Rohm and Haas); и ионные детергенты, такие как, например, додецилсульфат натрия, сульфатированные высшие алифатические спирты, сульфонированные алканы и сульфонированные алкиларены, содержащие от 7 до 22 атомов углерода в разветвленной или неразветвленной цепи.
Сбор ECM можно проводить различными способами в зависимости, например, от того, образована ли новая ткань на объемном каркасе, который является или не является биоразлагаемым. Например, если каркас не является биоразлагаемым, ECM можно удалить обработкой каркаса ультразвуком, водяными струями высокого давления, механическим соскабливанием или мягкой обработкой детергентами или ферментами или любой комбинацией перечисленных способов.
Если используемый каркас является биоразлагаемым, ECM можно собрать, например, позволив каркасу разложиться или раствориться в растворе. В альтернативном варианте осуществления, если биоразлагаемый каркас состоит из материала, который можно вводить пациенту вместе с ECM, каркас и ECM можно обрабатывать in toto для последующей инъекции. В альтернативном варианте осуществления ECM можно удалить с биоразлагаемого каркаса любым из описанных выше способов для сбора ECM с каркаса, который не является биоразлагаемым. Все процедуры сбора внеклеточного матрикса предпочтительно составляют таким образом, чтобы не допустить денатурации ECM.
После сбора можно продолжить обработку ECM. Например, собранный ECM можно гомогенизировать до мелких частиц с использованием методик, известных в данной области, таких как обработка ультразвуком, чтобы обеспечить возможность его прохождения по хирургической игле. При необходимости в компонентах ECM можно провести поперечную сшивку полимеров гамма-облучением. Предпочтительно для стерилизации и поперечной сшивки ECM доза облучения ECM составляет от 0,25 до 2 мегарад. Также возможна, но по существу не является предпочтительной химическая поперечная сшивка с использованием токсичных агентов, таких как глутаральдегид.
Количества и/или соотношения белков, таких как различные типы коллагена, присутствующего в ECM, можно корректировать путем смешивания ECM, выработанного клетками, составляющими предмет настоящего изобретения, с ECM одного или более других типов клеток. Кроме того, в ECM можно ввести биологически активные вещества, такие как белки, факторы роста и/или лекарственные средства. Примеры биологически активных веществ включают тканевые факторы роста, такие как TGF-бета, и т.п., которые стимулируют заживление и восстановление ткани в месте введения. Такие дополнительные агенты можно использовать в любом из описанных выше в настоящем документе вариантов осуществления, например с цельноклеточными лизатами, растворимыми клеточными фракциями или дополнительно очищенными компонентами и продуктами, выработанными клетками UTC.
Фармацевтические композиции, содержащие клетки UTC, компоненты или продукты клеток UTC
В другом аспекте настоящее изобретение представляет фармацевтические композиции, которые используют клетки UTC, популяции клеток UTC, компоненты и продукты клеток UTC в различных способах лечения нейродегенеративных состояний (таких как, например, амиотрофический боковой склероз). Некоторые варианты осуществления охватывают фармацевтические композиции, содержащие живые клетки (только клетки UTC или их смесь с клетками других типов). Другие варианты осуществления охватывают фармацевтические композиции, содержащие клеточные компоненты клеток UTC (например, клеточные лизаты, растворимые клеточные фракции, кондиционированную среду, ECM или компоненты любого из перечисленных выше вариантов) или продукты (например, трофические и другие биологические факторы, вырабатываемые клетками UTC естественно или после генетической модификации, кондиционированную среду после культивирования клеток UTC). В любом случае фармацевтическая композиция может дополнительно содержать другие активные агенты, такие как противовоспалительные агенты, противоапоптозные агенты, антиоксиданты, факторы роста, нейротрофические факторы или нейрорегенеративные или нейропротекторные лекарственные средства, известные в данной области.
Примеры других компонентов, которые можно добавить в фармацевтические композиции на основе клеток UTC, включают, без ограничений: (1) другие нейропротекторные или нейростимулирующие лекарственные средства; (2) выбранные компоненты внеклеточного матрикса, такие как один или более типов коллагенов, известных в данной области, и/или факторы роста, плазму с высоким содержанием тромбоцитов и лекарственные средства (альтернативно клетки UTC можно модифицировать методами генетической инженерии для экспрессии и выработки факторов роста); (3) противоапоптозные агенты (например, эритропоэтин (EPO), миметические антитела EPO, тромбопоэтин, инсулиноподобные факторы роста IGF-I, IGF-II, фактор роста гепатоцитов, ингибиторы каспазы); (4) противовоспалительные соединения (например, ингибиторы p38 MAP-киназы, ингибиторы TGF-бета, статины, ингибиторы IL-6 и IL-1, PEMIROLAST, TRANILAST, REMICADE®, SIROLIMUS и нестероидные противовоспалительные лекарственные средства (НПВС) (такие как TEPOXALIN, TOLMETIN и SUPROFEN); (5) иммуносупрессорные или иммуномодулирующие агенты, такие как ингибиторы кальциневрина, ингибиторы mTOR, антипролиферативные агенты, кортикостероиды и различные антитела; (6) антиоксиданты, такие как пробукол, витамины C и E, кофермент Q-10, глутатион, L-цистеин и N-ацетилцистеин; и (7) местные анестетики и т.п.
Фармацевтические композиции, составляющие предмет настоящего изобретения, содержат клетки UTC или их компоненты или продукты, смешанные с фармацевтически приемлемым носителем или средой. Подходящие для целей настоящего изобретения фармацевтически приемлемые носители включают воду, солевой раствор (такой как раствор Рингера), спирты, масла, желатины и углеводы, такие как лактоза, амилоза или крахмал, эфиры жирных кислот, гидроксиметилцеллюлозу и поливинилпирролидин. Такие препараты можно стерилизовать и при необходимости смешать с вспомогательными агентами, такими как смазывающие вещества, консерванты, стабилизаторы, смачивающие вещества, эмульгаторы, соли для коррекции осмотического давления, буферные растворы и красители. Фармацевтические носители, подходящие для использования в целях настоящего изобретения, известны в данной области и описаны, например, в книге Pharmaceutical Sciences (17-е изд., Mack Pub. Co., Easton, PA) и международной заявке № WO 96/05309.
Как правило, хотя и не всегда, фармацевтические композиции, содержащие компоненты или продукты клеток UTC, но не живые клетки, готовят в виде жидкостей (или в виде твердых таблеток, капсул и т.п. в случае приемлемости перорального введения). Данные композиции можно готовить для введения любым приемлемым и известным специалистам способом для достижения доставки лекарственных средств и биологических молекул к целевой нервной ткани, включая, без ограничений, пероральное, интраназальное, офтальмологическое и парентеральное, включая внутривенное, введение. Конкретные способы парентерального введения включают, без ограничений, внутримышечный, подкожный, интраперитонеальный, интрацеребральный, интравентрикулярный, интрацеребровентрикулярный, подоболочечный, интрацистернальный, интраспинальный и/или периспинальный способы введения с использованием интракраниальных или интравертебральных игл и/или катетеров с помощью насосных устройств или без них.
Фармацевтические композиции, содержащие живые клетки UTC, как правило, готовят в виде жидкостей, полутвердых композиций (например, гелей) или твердых композиций (например, матриц, каркасов и т.п., как требуется для восстановления нервной ткани). Жидкие композиции готовят для введения любым способом, известным специалистам в данной области, для достижения доставки живых клеток к целевым нервным тканям. Как правило, данные способы включают инъекцию или инфузию в ЦНС или ПНС либо диффузным образом, либо целенаправленно в месте неврологического заболевания или нарушения, без ограничений используя в качестве способа введения интраокулярный, интрацеребральный, интравентрикулярный, интрацеребровентрикулярный, подоболочечный, интрацистернальный, интраспинальный и/или периспинальный способы введения с использованием интракраниальных или интравертебральных игл и/или катетеров с помощью насосных устройств или без них.
Фармацевтические композиции, содержащие живые клетки в полутвердом или твердом носителе, как правило, готовят для хирургической имплантации в месте неврологической травмы или нарушения. Следует понимать, что жидкие композиции также можно вводить с использованием хирургических процедур. В конкретных вариантах осуществления полутвердые или твердые фармацевтические композиции могут представлять собой полупроницаемые гели, решетки, клеточные каркасы и т.п., которые могут быть биоразлагаемыми или биологически неразлагаемыми. Например, в определенных вариантах осуществления может быть необходимо или уместно отделить экзогенные клетки от их окружения, при этом позволив клеткам секретировать и поставлять биологические молекулы (например, нейротрофические факторы) окружающим нервным клеткам. В таких вариантах осуществления клетки можно приготовить в виде автономных имплантатов, содержащих живые клетки UTC или содержащие клетки UTC клеточные популяции, окруженные неразлагаемым, селективно проницаемым барьером, который физически отделяет трансплантированные клетки от ткани-хозяина. Такие имплантаты иногда называются «иммунопротекторными», поскольку они обладают способность не допускать, чтобы клетки и макромолекулы иммунной системы убивали трансплантированные клетки в отсутствие фармакологически индуцированной иммуносупрессии (обзор таких устройств и способов приведен, например, в P.A. Tresco et al., 2000 г., Adv. Drug Delivery Rev. 42: 3-27).
В одном варианте осуществления настоящего изобретения фармацевтические композиции, содержащие клетки, полученные из ткани пуповины, по существу однородные популяции, содержащие клетки, полученные из ткани пуповины, или клетки, полученные из ткани пуповины, вводят путем внутривенной и/или подоболочечной инъекции. В другом варианте осуществления фармацевтические композиции, содержащие клетки, полученные из ткани пуповины, по существу однородные популяции, содержащие клетки, полученные из ткани пуповины, или клетки, полученные из ткани пуповины, многократно вводят путем внутривенной и/или подоболочечной инъекции.
В других вариантах осуществления для получения фармацевтических композиций, составляющих предмет настоящего изобретения, используют различные виды разлагаемых гелей и сеток. Например, разлагаемые материалы, в частности, подходящие для получения композиций с замедленным высвобождением, включают биосовместимые полимеры, такие как полимолочная кислота, сополимер молочной и гликолевой кислот, метилцеллюлоза, гиалуроновая кислота, коллаген и т.п. Структура, выбор и применение разлагаемых полимеров в средах доставки лекарственных средств представлены в нескольких публикациях, включая работу A. Domb et al., 1992 г., Polymers for Advanced Technologies, 3: 279.
В других вариантах осуществления, например, предназначенных для восстановления крупных нервных повреждений, таких как повреждение или разрыв спинного мозга или нервного пучка рассеченной конечности, может быть необходимо или уместно вводить клетки на биоразлагаемом, предпочтительно саморассасывающемся или биорассасывающемся каркасе или матриксе или в нем. Такие биоматериалы с, как правило, объемной структурой содержат живые клетки, прикрепленные к каркасу, распределенные внутри каркаса или встроенные во внеклеточный матрикс, удерживаемый в каркасе. После имплантирования в целевой участок организма данные имплантаты интегрируются с тканью-хозяином, в которой постепенно приживаются трансплантированные клетки (смотрите, например, P.A. Tresco et al., 2000 г., supra; смотрите также D.W. Hutmacher, 2001 г., J. Biomater. Sci. Polymer Edn. 12: 107-174).
Примеры материалов каркасов или матриксов (иногда собирательно называемых просто «каркасы»), которые можно использовать в целях настоящего изобретения, включают нетканые маты, пористые пеноматериалы или самособирающиеся пептиды. Нетканые маты могут быть образованы, например, с использованием волокон, образованных из синтетического рассасывающегося сополимера гликолевой и молочной кислот (PGA/PLA), доступных в продаже под торговой маркой VICRYL (Ethicon, Inc., г. Сомервилл, штат Нью-Джерси). Также можно использовать пеноматериалы, состоящие, например, из сополимера поли(эпсилон-капролактона)/полигликолевой кислоты (PCL/PGA), образованные с помощью таких процессов, как сублимационная сушка, или лиофилизация, как описано в патенте США № 6355699. Также можно использовать гидрогели, такие как самособирающиеся пептиды (например, RAD16). Для целей настоящего изобретения также подходят формируемые in situ разлагаемые сетки (смотрите, например, Anseth, K.S. et al., 2002 г., J. Controlled Release 78: 199-209; Wang, D. et al., 2003 г., Biomaterials 24: 3969-3980; публикацию патента США № 2002/0022676, выданного He et al.). Такие материалы, приготовленные в виде подходящих для инъекции жидкостей, затем можно активировать различными способами (например, изменением температуры, pH, освещением) для образования разлагаемых гидрогелевых сеток in situ или in vivo.
В другом варианте осуществления каркас представляет собой войлок, который может состоять из мультифиламентной пряжи, изготовленной из биорассасывающегося материала, например сополимеров или смесей PGA, PLA, PCL или гиалуроновой кислоты. Пряжу сваливают в войлок с использованием стандартных методик обработки текстильных материалов, состоящих из обжатия, нарезки, чесания и простегивания. В другом варианте осуществления клетки высевают на каркасы из пеноматериалов, которые могут представлять собой композитные структуры.
Во многих из указанных выше вариантов осуществления каркасу можно придать подходящую форму, такую как, например, форма спинного мозга с отдельными каналами для восстановления нервного тракта (Friedman JA et al., 2002 г., Neurosurgery 51: 742-51). Кроме того, следует понимать, что клетки UTC можно культивировать на заранее образованных, неразлагающихся хирургических или имплантируемых устройствах, например, способом, соответствующим способу, используемому для получения содержащих фибробласты разделяемых эндоваскулярных спиралей Гульельми (Marx, W.F. et al., 2001 г., Am. J. Neuroradiol. 22: 323-333).
Перед посевом клеток матрикс, каркас или устройство можно обработать для улучшения клеточной адгезии. Например, перед посевом клеток нейлоновые матрицы можно обработать 0,1 молярным раствором уксусной кислоты и инкубировать в полилизине, ФСБ и/или коллагене для покрытия нейлона. Полистирол можно аналогичным образом обработать серной кислотой. Можно также модифицировать внешние поверхности каркаса для улучшения клеточной адгезии или роста и дифференцирования ткани, используя, например, плазменное покрытие каркаса или добавление одного или более белков (например, коллагенов, эластичных волокон, ретикулярных волокон), гликопротеинов, гликозаминогликанов (например, гепаринсульфата, хондроитин-4-сульфата, хондроитин-6-сульфата, дерматансульфата, кератинсульфата), клеточного матрикса и/или других материалов, включая, без ограничений, желатин, альгинаты, агар, агарозу и растительные камеди и т.п.
Каркасы, содержащие клетки UTC, готовят в соответствии со способами, известными в данной области. Например, клетки можно растить свободно в сосуде для культивирования до получения субконфлюентной или конфлюентной культуры, затем изъять из культуры и засеять на каркас. При необходимости в культуральную среду можно добавить факторы роста до, во время или после высевания клеток для инициирования дифференцирования клеток и формирования ткани. В альтернативном варианте осуществления сами каркасы можно модифицировать таким образом, чтобы улучшить рост клеток на них или снизить риск отторжения имплантата. Таким образом, в каркас можно добавить одно или более биологически активных соединений, включая, без ограничений, противовоспалительные агенты, иммуносупрессоры или факторы роста, для их местного высвобождения.
Способы использования клеток UTC, компонентов или продуктов клеток UTC
Клетки UTC, или клеточные популяции, содержащие клетки UTC, или компоненты или продукты, вырабатываемые клетками UTC, можно использовать различными способами для поддержки и облегчения восстановления и регенерации нервных клеток и тканей. Такие возможности охватывают способы in vitro, ex vivo и in vivo. Например, клетки UTC, или клеточные популяции, содержащие клетки UTC, или компоненты или продукты, вырабатываемые клетками UTC, можно использовать для лечения амиотрофического бокового склероза.
Способы in vitro и ex vivo
В одном варианте осуществления клетки UTC можно использовать in vitro для скрининга широкого спектра соединений на эффективность и цитотоксичность фармацевтических агентов, факторов роста, регуляторных факторов и т.п. Например, такой скрининг можно проводить на по существу однородных популяциях клеток UTC для оценки эффективности или токсичности потенциальных соединений для совместного использования в композициях с клетками UTC или совместного введения с ними для лечения нейродегенеративного состояния. В альтернативном варианте осуществления такой скрининг можно проводить на клетках UTC, стимулированных для дифференцирования в нервные клетки или клетки-предшественники нейронов, с целью оценки эффективности новых потенциальных фармацевтических лекарственных средств. В данном варианте осуществления клетки UTC поддерживают in vitro и обрабатывают их тестируемым соединением. Активность потенциально цитотоксичного соединения можно измерить по его способности повреждать или убивать клетки в культуре. Это легко можно оценить с помощью методик прижизненного окрашивания клеток. Эффект факторов роста или регуляторных факторов можно оценить путем анализа количества или устойчивости культивированных клеток по сравнению с клетками, которые не обрабатывались тестируемыми факторами. Это можно сделать с использованием стандартных цитологических и/или гистологических методик, включая использование иммуноцитохимических методик с применением антител, которые определяют типоспецифические клеточные антигены.
В дополнительном варианте осуществления, как описано выше, клетки UTC можно культивировать in vitro для выработки биологических продуктов, которые либо вырабатываются клетками в естественных условиях, либо вырабатываются клетками после индуцирования дифференцирования по нейронным линиям дифференцирования, либо вырабатываются клетками в результате генетической модификации. Например, обнаружена секреция TIMP1, TPO, KGF, HGF, FGF, HBEGF, BDNF, MIP1-бета, MCP1, RANTES, I309, TARC, MDC и IL-8 клетками, полученными из пуповины и выращенными в ростовой среде. Секреция TIMP1, TPO, KGF, HGF, HBEGF, BDNF, MIP1a, MCP-1, RANTES, TARC, эотаксина и IL-8 обнаружена для клеток, полученных из плаценты и культивированных в ростовой среде (смотрите примеры). Некоторые из данных трофических факторов, такие как BDNF и IL-6, играют важную роль в регенерации нервной ткани. Клетки UTC, вероятно, вырабатывают и секретируют в среду и другие, пока невыявленные или неизученные трофические факторы, которые могут быть пригодны для восстановления и регенерации нервной ткани.
В связи с этим в другом варианте осуществления настоящего изобретения предложено использовать клетки UTC для выработки кондиционированной среды, либо от недифференцированных клеток UTC, либо от клеток UTC, которые инкубировали в условиях, стимулирующих дифференцирование по нейронной линии дифференцирования. Предусматривается использование таких кондиционированных сред in vitro или ex vivo при культивировании клеток-предшественников нервных клеток, или in vivo для поддержки трансплантированных клеток, например, однородных популяций клеток UTC или неоднородных популяций, содержащих клетки UTC и клетки-предшественники нейронов.
Еще один вариант осуществления содержит применение клеточных лизатов из клеток UTC, их растворимых клеточных фракций или компонентов либо ECM или его компонентов, в различных целях. Как указано выше, некоторые из данных компонентов можно применять в фармацевтических композициях. В других вариантах осуществления клеточный лизат или ECM используют для нанесения покрытия или другой обработки веществ или устройств, предназначенных для использования при выполнении хирургической операции или для имплантации, или для целей ex vivo, для стимуляции заживления или повышения выживаемости клеток или тканей, с которыми производится контакт при выполнении таких процедур.
Как описано в примерах 13 и 15, клетки, полученные из пуповины или плаценты, продемонстрировали способность поддерживать выживаемость, рост и дифференцирование взрослых клеток-предшественников нейронов при выращивании в совместной культуре с данными клетками. Соответственно, в другом варианте осуществления клетки UTC преимущественно используют в совместных культурах in vitro для обеспечения трофической поддержки другим клеткам, в частности нервным клеткам и клеткам-предшественникам нейронов. При совместном культивировании для клеток UTC и других необходимых клеток может быть необходимо совместное культивирование в условиях, обеспечивающих контакт между клетками двух типов. Этого можно достичь, например, высевая клетки в культуральной среде или на подходящий субстрат в виде неоднородной популяции клеток. В альтернативном варианте осуществления можно сначала нарастить клетки UTC до конфлюентности культуры, которая затем будет служить субстратом для культивирования клеток второго необходимого типа. В данном последнем варианте осуществления клетки можно дополнительно физически разделить, например, с помощью мембраны или аналогичного устройства таким образом, чтобы после периода совместного культивирования клетки другого типа можно было снять и использовать отдельно. Применение клеток UTC при совместном культивировании для стимуляции размножения и дифференцирования типов нервных клеток может быть актуальным в научно-исследовательской работе и в клинической/терапевтической областях. Например, совместное культивирование с клетками UTC можно использовать для облегчения роста и дифференцирования нервных клеток в культуре, например, для научно-исследовательских целей или для применения в скрининговых тестах с использованием лекарственных средств. Совместное культивирование с клетками UTC также можно использовать для размножения ex vivo клеток-предшественников нейронов для последующего введения пациенту в терапевтических целях. Например, у пациента можно взять клетки-предшественники нейронов, размножить их ex vivo совместным культивированием с клетками UTC и затем возвратить их обратно данному пациенту (аутогенный перенос) или ввести другому пациенту (сингенный или аллогенный перенос). В данных вариантах осуществления следует понимать, что после размножения ex vivo смешанную популяцию клеток, содержащую клетки UTC и клетки-предшественники нейронов, можно ввести пациенту, нуждающемуся в лечении. В альтернативном варианте осуществления в ситуациях, когда уместен или необходим аутогенный перенос, совместно культивируемые популяции клеток можно физически разделить при культивировании, что позволяет отделить аутогенные клетки-предшественники нейронов для введения пациенту.
Способы in vivo
Как описано в примерах 16 и 17, для клеток UTC показана возможность эффективной трансплантации в организм с восполнением утраченной нейронной функции в животной модели, принятой за возможность прогнозирования эффективности на людях. Данные результаты поддерживают предпочтительный вариант осуществления настоящего изобретения, в котором клетки UTC используют в клеточной терапии для лечения нейродегенеративного состояния, такого как амиотрофический боковой склероз. После трансплантации в целевое местоположение нервной ткани в организме клетки UTC могут сами дифференцироваться в клетки одного или более нейронных фенотипов, или они могут обеспечивать трофическую поддержку клеток-предшественников нейронов и самих нервных клеток in situ, либо они могут оказывать благоприятный эффект каждым из данных способов и т.п.
Клетки UTC можно вводить отдельно (например, в виде по существу однородных популяций) или в смеси с другими клетками. Как описано выше, клетки UTC можно вводить в составе фармацевтического препарата на основе матрикса или каркаса либо в стандартных фармацевтически приемлемых носителях. При введении клеток UTC вместе с другими клетками их можно вводить одновременно или последовательно с другими клетками (перед другими клетками или после них). Клетки, которые можно вводить в сочетании с клетками UTC, включают, без ограничений, нейроны, астроциты, олигодендроциты, клетки-предшественники нейронов, нейронные стволовые клетки и/или другие мультипотентные или плюрипотентные стволовые клетки. Клетки других типов можно смешивать с клетками UTC непосредственно во время или незадолго перед введением, либо клетки можно совместно культивировать в течение некоторого времени перед введением.
Клетки UTC можно вводить вместе с другими лекарственными средствами, благоприятными для нейронной терапии, или биологическими молекулами, или другими активными агентами, такими как противовоспалительные агенты, противоапоптозные агенты, антиоксиданты, факторы роста, нейротрофические факторы или нейрорегенеративные или нейропротекторные лекарственные средства, известные в данной области. При введении клеток UTC совместно с другими агентами их можно вводить вместе в виде одной фармацевтической композиции или в отдельных фармацевтических композициях, одновременно или последовательно с другими агентами (перед введением других агентов или после него).
Примеры других компонентов, которые можно вводить вместе с клетками UTC, включают, без ограничений: (1) другие нейропротекторные или нейростимулирующие лекарственные средства; (2) выбранные компоненты внеклеточного матрикса, такие как один или более типов коллагенов, известных в данной области, и/или факторы роста, плазму с высоким содержанием тромбоцитов и лекарственные средства (альтернативно клетки UTC можно модифицировать методами генетической инженерии для экспрессии и выработки факторов роста); (3) противоапоптозные агенты (например, эритропоэтин (EPO), миметические антитела EPO, тромбопоэтин, инсулиноподобные факторы роста IGF-I, IGF-II, фактор роста гепатоцитов, ингибиторы каспазы); (4) противовоспалительные соединения (например, ингибиторы p38 MAP-киназы, ингибиторы TGF-бета, статины, ингибиторы IL-6 и IL-1, PEMIROLAST, TRANILAST, REMICADE®, SIROLIMUS и нестероидные противовоспалительные лекарственные средства (НПВС) (такие как TEPOXALIN, TOLMETIN и SUPROFEN); (5) иммуносупрессорные или иммуномодулирующие агенты, такие как ингибиторы кальциневрина, ингибиторы mTOR, антипролиферативные агенты, кортикостероиды и различные антитела; (6) антиоксиданты, такие как пробукол, витамины C и E, кофермент Q-10, глутатион, L-цистеин и N-ацетилцистеин; и (7) местные анестетики и т.п.
В одном варианте осуществления клетки UTC вводят в виде недифференцированных клеток, т.е. в том виде, который получен при культивировании в ростовой среде. В альтернативном варианте осуществления клетки UTC можно вводить после создания в процессе культивирования условий, стимулирующих дифференцирование по линии необходимого нейронного фенотипа, например астроцитов, олигодендроцитов или нейронов, и в частности серотонинергических, дофаминергических, холинергических, GABA-эргических или глутаматергических нейронов (смотрите, например, Isacson, O., 2003 г., The Lancet (Neurology) 2: 417-424).
Клетки, составляющие предмет настоящего изобретения, можно хирургически имплантировать, вводить шприцом, доставлять (например, с помощью катетера или шприца) или иным образом вводить напрямую или опосредованно в место неврологического повреждения или нарушения (например, для лечения амиотрофического бокового склероза). Способы введения клеток, составляющих предмет настоящего изобретения, или содержащих их композиций включают, без ограничений, внутривенный, внутримышечный, подкожный, интраназальный, интрацеребральный, интравентрикулярный, интрацеребровентрикулярный, подоболочечный, интрацистернальный, интраспинальный и/или периспинальный способы введения с использованием интракраниальных или интравертебральных игл и/или катетеров с помощью насосных устройств или без них.
При введении клеток в составе полутвердых или твердых устройств подходящим средством введения, как правило, является хирургическая имплантация в требуемое местоположение в организме. Однако жидкие или текучие фармацевтические композиции можно вводить в более общем местоположении в ЦНС или ПНС (например, по всей диффузно пораженной области, как например, в случае болезни Паркинсона или диффузного ишемического повреждения), поскольку продемонстрирована способность клеток-предшественников нейронов к активной миграции от точки ввода в нервную систему до конкретного местоположения, например, следуя за радиальными глиальными клетками или в ответ на химические сигналы.
Действительно, данная способность нейронных стволовых клеток к миграции открыла новые возможности в лечении злокачественных опухолей головного мозга, т.е. применение клеток-предшественников для доставки терапевтических генов/продуктов генов для лечения данных мигрирующих опухолей. Например, известно, что нейронные стволовые клетки, при их имплантировании in vivo в интракраниальные глиомы у взрослых грызунов, быстро и широко распространяются по ложу опухоли и мигрируют вместе с размножающимися и распространяющимися клетками опухоли, продолжая стабильно экспрессировать чуждый ген (Aboody, K. et al., 2000 г., Proc. Natl. Acad. Sci. USA 97: 12846-12851). Ожидается, что клетки UTC также подходят для использования в таких целях, как, например, генетическое модифицирование клеток UTC для выработки апоптозного или другого противоопухолевого агента, например IL-12 (Ehtesham, M. et al., 2002 г., Cancer Research 62: 5657-5663), или возможность введения связанного с фактором некроза опухоли индуцирующего апоптоз лиганда (Ehtesham, M. et al., 2002 г., Cancer Research 62: 7170-7174) путем инъекции или иным способом в общее место злокачественной опухоли (например, глиобластомы), откуда клетки UTC затем могут мигрировать к клеткам опухоли для местной доставки терапевтического агента.
Другие варианты осуществления охватывают способы лечения нейродегенеративных состояний (таких как, например, амиотрофический боковой склероз) путем введения фармацевтических композиций, содержащих клеточные компоненты UTC (например, клеточные лизаты или их компоненты) или продукты (например, трофические и другие биологические факторы, вырабатываемые клетками UTC в естественных условиях или в результате генетической модификации, в кондиционированной среде от культивирования клеток UTC). Данные способы также могут дополнительно содержать введение других активных агентов, таких как факторы роста, нейротрофические факторы или нейрорегенеративные или нейропротекторные лекарственные средства, известные в данной области.
Формы дозирования и режимы введения клеток UTC или любых других фармацевтических композиций, описанных в настоящем документе, разрабатывают в соответствии с надлежащей медицинской практикой, принимая во внимание состояние конкретного пациента, например, природу и степень нейродегенеративного состояния, возраст, пол, вес тела и общее медицинское состояние, а также другие факторы, известные медработникам. Таким образом, эффективное количество фармацевтической композиции для введения пациенту определяется с учетом данных соображений, как известно в данной области.
Поскольку ЦНС является в некоторой степени иммунопривилегированной тканью, подавление иммунитета у пациента перед началом клеточной терапии с использованием клеток UTC может оказаться ненужным или нежелательным. Кроме того, как показано в примере 11, клетки UTC не стимулируют аллогенные МКПК в реакции смешанной культуры лимфоцитов. Соответственно, в некоторых случаях может допускаться трансплантация аллогенных или даже ксеногенных клеток UTC.
Однако в других случаях может быть необходимо или уместно фармакологическое подавление иммунитета у пациента перед началом клеточной терапии. Этого можно достичь применением местных или системных иммуносупрессорных агентов, или же этого можно достичь доставкой клеток в инкапсулированном устройстве, как описано выше. В данной области известны данные и другие способы снижения или устранения иммунного ответа на трансплантированные клетки. В качестве альтернативы можно генетически модифицировать клетки UTC для снижения их иммуногенности, как указано выше.
Выживаемость трансплантированных клеток UTC у живого пациента можно определить путем использования различных методик сканирования, например с помощью исследования компьютерной аксиальной томографией (КАТ или КТ), магниторезонансной томографией (МРТ) или позитронно-эмиссионной томографией (ПЭТ). Определение выживаемости трансплантированных клеток можно также провести post mortem путем извлечения нервной ткани и изучения ее визуально или с помощью микроскопа. В альтернативном варианте осуществления клетки можно обрабатывать красителями, специфичными на нервные клетки или их продукты, например нейромедиаторы. Трансплантированные клетки также можно идентифицировать по предварительно введенным в них меткам-красителям, таким как меченные родамином или флуоресцеином микросферы, быстрый голубой, микрочастицы железа, бисбензамид или продукты генетически встроенного репортерного гена, такие как бета-галактозидаза или бета-глюкуронидаза.
Степень функционального интегрирования трансплантированных клеток UTC в нервную ткань субъекта можно определить по оценке степени восстановления поврежденной или пораженной нервной функции. Такие функции включают, без ограничений, моторную, когнитивную, сенсорную и эндокринную функции, оценку которых можно проводить в соответствии с процедурами, хорошо известными нейробиологам и терапевтам.
Наборы и банки, содержащие клетки UTC, компоненты или продукты клеток UTC
В другом аспекте в настоящем изобретении предложены наборы, которые используют клетки UTC, популяции клеток UTC, компоненты и продукты клеток UTC в различных способах регенерации и восстановления нервной ткани, как описано выше. В одном варианте осуществления настоящего изобретения наборы можно использовать для лечения амиотрофического бокового склероза. Для использования в лечении нейродегенеративных состояний или при проведении других запланированных курсов лечения наборы могут включать в себя одну или более популяций клеток, включая по меньшей мере клетки UTC и фармацевтически приемлемый носитель (жидкий, полутвердый или твердый). Наборы также могут необязательно включать в себя средство для введения клеток, например, путем инъекции. Наборы могут дополнительно включать в себя инструкции по применению клеток. Наборы, приготовленные для использования в условиях полевого госпиталя, например, в условиях боевых действий, могут включать в себя полный комплект для проведения всей процедуры, включая каркасы тканей, хирургические нити и т.п., где клетки будут использоваться в сочетании с восстановлением острых поражений. Наборы для проведения тестов и исследований in vitro, описанные в настоящем документе, могут содержать одно или более из (1) клеток UTC или компонентов или продуктов клеток UTC, (2) реагентов для осуществления способа in vitro, (3) других клеток или популяций клеток, если это необходимо, и (4) инструкций по осуществлению способа in vitro.
В еще одном аспекте в настоящем изобретении также предложены банки тканей, клеток, клеточных компонентов и популяций клеток, составляющих предмет настоящего изобретения. Как описано выше, клетки можно легко криоконсервировать. Следовательно, в настоящем изобретении предложены способы криоконсервирования клеток в банке, в котором клетки хранятся в замороженном виде с полной характеризацией клеток на основе иммунологических, биохимических и генетических свойств клеток. Замороженные клетки можно разморозить и размножить или использовать непосредственно для аутогенной, сингенной или аллогенной терапии в зависимости от требований процедуры и потребностей пациента. Информацию о каждом криоконсервированном образце предпочтительно хранят в компьютере, выполненном с возможностью поиска на основе требований хирурга, процедуры и пациента, с выдачей подходящих результатов поиска на основе характеризации клеток или популяций. Клетки, составляющие предмет настоящего изобретения, предпочтительно выращивают и размножают до необходимого количества клеток и готовят терапевтические клеточные композиции либо отдельно, либо в виде совместно культивируемых клеток, в присутствии или в отсутствие матрикса или носителя. Хотя для некоторых процедур может быть предпочтительно использовать свежеприготовленные клетки, остальные клетки можно криоконсервировать и поместить в банк путем замораживания клеток и введения информации в компьютер для создания записи, связанной с депонированными в банк образцами. Даже если не требуется подбор соответствия источника или донора таких клеток с получателем, для иммунологических целей система банка облегчает подбор, например, необходимых биохимических или генетических свойств депонированных клеток для терапевтических целей. После подбора хранящегося в банке образца с необходимыми свойствами его извлекают и готовят для терапевтического использования. Клеточные лизаты, ECM или клеточные компоненты, приготовленные, как описано в настоящем документе, также можно криоконсервировать или сохранять иным образом (например, путем лиофилизации) и депонировать в банк в соответствии с настоящим изобретением.
Следующие примеры предназначены для более подробного описания настоящего изобретения. Они призваны проиллюстрировать, но ни в коей мере не ограничить настоящее изобретение.
В следующих примерах и в других местах настоящего описания термин «ростовая среда» по существу относится к среде, достаточной для культивирования клеток UTC. В частности, одной предпочтительной в настоящее время средой для культивирования клеток, составляющих предмет настоящего изобретения, являются модифицированные по способу Дульбекко среды Игла (для обозначения которых в настоящем документе также используется сокращение DMEM). Особенно предпочтительной является среда DMEM с низким содержанием глюкозы (для обозначения которой в настоящем документе также используется сокращение DMEM-LG) (Invitrogen, г. Карлсбад, штат Калифорния). В среду DMEM с низким содержанием глюкозы предпочтительно добавляют 15% (об/об) эмбриональной бычьей сыворотки (например, эмбриональную бычью сыворотку определенного состава, Hyclone, г. Логан, штат Юта), антибиотики/антимикотики (предпочтительно 50-100 единиц/миллилитр пенициллина, 50-100 микрограмм/миллилитр стрептомицина и 0-0,25 микрограмма/миллилитр амфотерицина B; Invitrogen, г. Карлсбад, штат Калифорния) и 0,001% (об/об) 2-меркаптоэтанола (Sigma, г. Сент-Луис, штат Миссури). В приведенных ниже примерах термин «ростовая среда» относится к среде DMEM с низким содержанием глюкозы с добавлением 15% эмбриональной бычьей сыворотки и антибиотиков/антимикотиков (при добавлении пенициллина/стрептомицина их предпочтительные количества составляют 50 ед./миллилитр и 50 микрограмм/миллилитр соответственно; при использовании пенициллина/стрептомицина/амфотерицина B их предпочтительные количества составляют 100 ед./миллилитр, 100 микрограмм/миллилитр и 0,25 микрограмма/миллилитр соответственно). В некоторых случаях используют другие ростовые среды или добавляют в них другие компоненты, обычно они указаны в тексте в качестве добавок в ростовую среду.
Также в приведенных ниже примерах и в других местах настоящего описания термин «стандартные условия роста» обозначает культивирование клеток при 37°C в стандартной атмосфере, содержащей 5% CO2. Хотя описанные выше условия можно использовать при культивировании клеток, следует понимать, что специалист в данной области, принимая во внимание возможности культивирования клеток, известные в данной области, вполне может изменить такие условия.
В примерах и в других местах настоящего описания и формуле изобретения могут встретиться следующие сокращения: ANG2 (или Ang2) - ангиопоэтин 2; АПК - антигенпредставляющая клетка; BDNF - нейротрофический фактор головного мозга; bFGF - основной фактор роста фибробластов; bid (BID) - дважды в сутки; CK18 - цитокератин 18; ЦНС - центральная нервная система; CXC3 - лиганд 3 хемокинового рецептора; DMEM - модифицированная по способу Дульбекко среда Игла; DMEM:lg (или DMEM:Lg, DMEM:LG) - среда DMEM с низким содержанием глюкозы; ЭДТА - этилендиаминтетрауксусная кислота; EGF (или E) - эпидермальный фактор роста; FACS - сортировка флуоресцентноактивированных клеток; FBS - эмбриональная бычья сыворотка; FGF (или F) - фактор роста фибробластов; GCP-2 - гранулоцитарный хемотаксический белок-2; GFAP - глиофибриллярный кислый белок; HB-EGF - связывающийся с гепарином эпидермальный фактор роста; HCAEC - клетки эндотелия коронарной артерии человека; HGF - фактор роста гепатоцитов; hMSC - мезенхимальные стволовые клетки человека; HNF-1альфа - гепатоцит-специфичный фактор транскрипции 1 альфа; HUVEC - клетки эндотелия пуповинной вены человека; I309 - хемокин и лиганд рецептора CCR8; IGF-1 - инсулиноподобный фактор роста 1; IL-6 - интерлейкин-6; IL-8 - интерлейкин-8; K19 - кератин 19; K8 - кератин 8; KGF - фактор роста кератиноцитов; LIF - фактор, ингибирующий лейкемию; MBP - основной миелиновый белок; MCP-1 - моноцитарный хемотаксический белок 1; MDC - хемокин макрофагов; MIP1-альфа - макрофагальный белок воспаления 1 альфа; MIP1-бета - макрофагальный белок воспаления 1 бета; MMP - матриксная металлопротеаза; MSC - мезенхимальные стволовые клетки; NHDF - нормальные фибробласты кожи человека; NPE - среды для размножения клеток-предшественников нейронов; O4 - маркер дифференцирования по линии олигодендроцитов или глиальных клеток O4; МКПК - мононуклеарная клетка периферической крови; ФСБ - фосфатно-солевой буфер; PDGFbb - тромбоцитарный фактор роста; PO - в месяц; ПНС - периферическая нервная система; Rantes (или RANTES) - хемокин, экспрессируемый и выделяемый нормальными T-клетками при активации; rhGDF-5 - рекомбинантный фактор роста и дифференцирования человека 5; SC - подкожно; SDF-1альфа - стромальный фактор 1 альфа; SHH - белок sonic hedgehog; SOP - стандартная операционная процедура; TARC - регулируемый при активации хемокин тимуса; TCP - пластик для культивирования тканей; TCPS - полистирол для культивирования тканей; TGF-бета2 - трансформирующий фактор роста бета-2; TGF-бета3 - трансформирующий фактор роста бета-3; TIMP1 - тканевый ингибитор матриксной металлопротеиназы 1; TPO - тромбопоэтин; TuJ1 - бета-III-тубулин; VEGF - фактор роста эндотелия сосудов; vWF - фактор фон Виллебранда; и альфаFP - альфа-фетопротеин.
ПРИМЕР 1
Получение клеток из ткани пуповины и плаценты
В данном примере описано получение клеток из тканей пуповины и плаценты. Пуповины и плаценты получали при родах после завершения доношенной или недоношенной беременности. Клетки собирали у 5 независимых доноров ткани пуповины и плаценты. Тестировали различные способы выделения клеток для проверки их способности давать на выходе клетки, обладающие: 1) потенциалом дифференцирования в клетки различных фенотипов (данная характеристика является общей для стволовых клеток) или 2) потенциалом выработки трофических факторов, применимых к другим клеткам и тканям.
Способы и материалы
Выделение клеток пуповины. Пуповины получили в Национальном центре по обмену материалами для исследования заболеваний (National Disease Research Interchange/NDRI, г. Филадельфия, штат Пенсильвания). Ткани были получены после нормально прошедших родов. Протокол выделения клеток проводили в асептических условиях в ламинарном боксе. Для удаления крови и продуктов распада клеток пуповину промывали в фосфатно-солевом буфере (ФСБ; Invitrogen, г. Карлсбад, штат Калифорния) в присутствии антимикотика и антибиотика (100 единиц/миллилитр пенициллина, 100 микрограмм/миллилитр стрептомицина, 0,25 микрограмма/миллилитр амфотерицина B). Затем ткани механически диссоциировали в планшетах для культивирования тканей площадью 150 см2 в присутствии 50 миллилитров среды (среды DMEM с низким содержанием глюкозы или среды DMEM с высоким содержанием глюкозы; Invitrogen) до измельчения ткани в мелкую пульпу. Размельченные ткани перенесли в конические пробирки объемом 50 миллилитров (приблизительно по 5 грамм ткани в пробирку). Затем ткани расщепляли либо в среде DMEM с низким содержанием глюкозы, либо в среде DMEM с высоким содержанием глюкозы, в каждом случае с добавлением антимикотика и антибиотика, как описано выше. В некоторых экспериментах использовали ферментативную смесь коллагеназы и диспазы (C:D; коллагеназа (Sigma, г. Сент-Луис, штат Миссури), 500 единиц/миллилитр; и диспаза (Invitrogen), 50 единиц/миллилитр в среде DMEM с низким содержанием глюкозы). В других экспериментах использовали смесь коллагеназы, диспазы и гиалуронидазы (C:D:H) (коллагеназа, 500 единиц/миллилитр; диспаза, 50 единиц/миллилитр; и гиалуронидаза (Sigma), 5 единиц/миллилитр, в среде DMEM с низким содержанием глюкозы). Конические пробирки, содержащие ткань, среду и расщепляющие ферменты, инкубировали при температуре 37°C на орбитальном шейкере (Environ, Бруклин, г. Нью-Йорк) при скорости 225 об/мин в течение 2 ч.
После расщепления ткани центрифугировали при 150×g в течение 5 минут и отсасывали супернатант. Осадок повторно суспендировали в 20 миллилитрах ростовой среды (среда DMEM с низким содержанием глюкозы (Invitrogen), 15 процентов (об/об) эмбриональной бычьей сыворотки (FBS; бычья сыворотка с определенным составом; партия № AND18475; Hyclone, г. Логан, штат Юта), 0,001% (об/об) 2-меркаптоэтанола (Sigma), 1 миллилитр на 100 миллилитров антибиотика/антимикотика, как описано выше. Клеточную суспензию фильтровали через нейлоновый клеточный фильтр с размером пор 70 микрометров (BD Biosciences). Фильтр дополнительно промыли 5 миллилитрами ростовой среды. Затем клеточную суспензию пропустили через нейлоновый клеточный фильтр с размером пор 40 микрометров (BD Biosciences) и фильтр дополнительно промыли 5 миллилитрами ростовой среды.
Фильтрат повторно суспендировали в ростовой среде (до общего объема 50 миллилитров) и центрифугировали при 150×g в течение 5 минут. Супернатант отсасывали и клетки повторно суспендировали в 50 миллилитрах свежей ростовой среды. Описанную процедуру повторили еще дважды.
После последнего центрифугирования супернатант отсосали и клеточный осадок повторно суспендировали в 5 миллилитрах свежей ростовой среды. Количество жизнеспособных клеток определили окрашиванием трипановым синим. Затем клетки культивировали в стандартных условиях.
Клетки, выделенные из пуповин, высевали с плотностью 5000 клеток/см2 на покрытые желатином колбы T-75 см2 (Corning Inc., г. Корнинг, штат Нью-Йорк) в ростовой среде с добавлением антибиотиков/антимикотиков, как описано выше. Через 2 суток (в разных экспериментах клетки инкубировали в течение 2-4 суток) истощенную среду отсасывали из колб. Клетки трижды промывали ФСБ для удаления продуктов распада клеток и клеток крови. Затем клетки снова заливали ростовой средой и давали культуре расти до конфлюентности (приблизительно 10 суток от пассажа 0) до пассажа 1. При последующих пассированиях (от пассажа 1 до пассажа 2 и т.п.) клетки достигали субконфлюентности (75-85 процентов конфлюентности) за 4-5 суток. Для данных последующих пассирований клетки высевали с плотностью 5000 клеток/см2. Клетки растили в увлажняемом инкубаторе в атмосфере с 5 процентами диоксида углерода и атмосферного кислорода при 37°C.
Выделение клеток плаценты. Плацентарную ткань получили из NDRI (г. Филадельфия, штат Пенсильвания). Ткани принадлежали беременной женщине и были получены во время нормальных хирургических родов. Плацентарные клетки выделяли, как описано выше для выделения клеток пуповины.
Следующий пример относится к выделению отдельных популяций материнских и неонатальных клеток из плацентарной ткани.
Протокол выделения клеток проводили в асептических условиях в ламинарном боксе. Для удаления крови и продуктов распада клеток плацентарную ткань промывали в фосфатно-солевом буфере (ФСБ; Invitrogen, г. Карлсбад, штат Калифорния) в присутствии антимикотика и антибиотика, как описано выше. Затем плацентарную ткань рассекли на три части: верхнюю часть (неонатальная сторона или аспект), среднюю часть (выделение смешанных неонатальных и материнских клеток) и нижнюю часть (материнская сторона или аспект).
Разделенные части по отдельности промыли несколько раз в ФСБ с антибиотиком/антимикотиком для дополнительного удаления крови и продуктов распада клеток. Затем каждую часть механически диссоциировали в планшетах для культивирования тканей площадью 150 см2 в присутствии 50 миллилитров среды DMEM с низким содержанием глюкозы до измельчения ткани в мелкую пульпу. Пульпу перенесли в конические пробирки объемом 50 миллилитров. Каждая пробирка содержала приблизительно 5 грамм ткани. Затем ткани расщепляли либо в среде DMEM с низким содержанием глюкозы, либо в среде DMEM с высоким содержанием глюкозы, в каждом случае с добавлением антимикотика и антибиотика (100 единиц/миллилитр пенициллина, 100 микрограмм/миллилитр стрептомицина, 0,25 микрограмма/миллилитр амфотерицина B) и расщепляющих ферментов. В некоторых экспериментах использовали ферментативную смесь коллагеназы и диспазы (C:D), содержащую коллагеназу (Sigma, г. Сент-Луис, штат Миссури) в концентрации 500 единиц/миллилитр и диспазу (Invitrogen) в концентрации 50 единиц/миллилитр в среде DMEM с низким содержанием глюкозы. В других экспериментах использовали смесь коллагеназы, диспазы и гиалуронидазы (C:D:H) (коллагеназа, 500 единиц/миллилитр; диспаза, 50 единиц/миллилитр; и гиалуронидаза (Sigma), 5 единиц/миллилитр, в среде DMEM с низким содержанием глюкозы). Конические пробирки, содержащие ткань, среду и расщепляющие ферменты, инкубировали в течение 2 ч при температуре 37°C на орбитальном шейкере (Environ, Бруклин, г. Нью-Йорк) при скорости 225 об/мин.
После расщепления ткани центрифугировали при 150×g в течение 5 минут и отсасывали полученный супернатант. Осадок повторно суспендировали в 20 миллилитрах ростовой среды с добавлением пенициллина/стрептомицина/амфотерицина B. Клеточную суспензию фильтровали через нейлоновый клеточный фильтр с размером пор 70 микрометров (BD Biosciences) и фильтр дополнительно промыли 5 миллилитрами ростовой среды. Затем общую клеточную суспензию пропустили через нейлоновый клеточный фильтр с размером пор 40 микрометров (BD Biosciences) и фильтр дополнительно промыли 5 миллилитрами ростовой среды.
Фильтрат повторно суспендировали в ростовой среде (до общего объема 50 миллилитров) и центрифугировали при 150×g в течение 5 минут. Супернатант отсасывали и клеточный осадок повторно суспендировали в 50 миллилитрах свежей ростовой среды. Описанную процедуру повторили еще дважды. После последнего центрифугирования супернатант отсосали и клеточный осадок повторно суспендировали в 5 миллилитрах свежей ростовой среды. Количество клеток определили тестом на вытеснение трипанового синего. Затем клетки культивировали в стандартных условиях.
Выделение клеток с использованием ферментативной смеси LIBERASE™. Клетки выделяли из тканей пуповины в среде DMEM с низким содержанием глюкозы с использованием ферментативной смеси LIBERASE™ (Boehringer Mannheim Corp., г. Индианаполис, штат Индиана) (2,5 миллиграмма на миллилитр, Blendzyme 3; Roche Applied Sciences, г. Индианаполис, штат Индиана) и гиалуронидазы (5 единиц/миллилитр, Sigma). Расщепление ткани и выделение клеток проводили как описано выше для расщепления с использованием других протеаз, используя смесь LIBERASE™/гиалуронидаза вместо ферментативной смеси C:D или C:D:H. Расщепление ткани с использованием LIBERASE™ позволило выделить из тканей пуповины и плаценты популяции клеток, которые легко размножались.
Выделение клеток с использованием других комбинаций ферментов. Сравнили между собой процедуры выделения клеток из пуповины с использованием разных комбинаций ферментов. Ферменты, которые сравнивали на расщепление, включали в себя: i) коллагеназу; ii) диспазу; iii) гиалуронидазу; iv) смесь коллагеназа:диспаза (C:D); v) смесь коллагеназа:гиалуронидаза (C:H); vi) смесь диспаза:гиалуронидаза (D:H); и vii) смесь коллагеназа:диспаза:гиалуронидаза (C:D:H). При выделении клеток с использованием данных разных условий ферментативного расщепления наблюдали различия, представленные в таблице 1-1.
Выделение клеток из остаточной крови в пуповинах. Предприняли другие попытки выделения пулов клеток из пуповины различными способами. В одном случае пуповину нарезали и промывали ростовой средой для отделения сгустков крови и студенистого материала. Смесь крови, студенистого материала и ростовой среды собрали и центрифугировали при 150×g. Клеточный осадок повторно суспендировали и высевали в ростовой среде на покрытые желатином колбы. Данные эксперименты позволили выделить популяцию клеток, которые легко размножались.
Выделение клеток из пуповинной крови. Клетки также выделили из образцов пуповинной крови, полученных из NDRI. Использовали протокол выделения, описанный в международной заявке на патент PCT/US2002/029971, авторы Ho et al. (Ho, T. W. et al., WO2003025149 A2). Образцы (объемом 50 миллилитров и 10,5 миллилитра соответственно) пуповинной крови (NDRI, г. Филадельфия, штат Пенсильвания) смешивали с лизирующим буфером (стерилизованный фильтрацией раствор 155 мМ хлорида аммония, 10 мМ бикарбоната калия, 0,1 мМ ЭДТА, буферизованный до pH 7,2 (все компоненты производства Sigma, г. Сент-Луис, штат Миссури)). Клетки лизировали при соотношении пуповинной крови к лизирующему буферу 1:20. Полученную суспензию клеток в течение 5 секунд перемешивали на вортексе и затем инкубировали в течение 2 минут при температуре окружающей среды. Лизат центрифугировали (10 минут при 200×g). Клеточный осадок повторно суспендировали в полной минимальной поддерживающей среде (Gibco, г. Карлсбад, штат Калифорния), содержащей 10 процентов эмбриональной бычьей сыворотки (Hyclone, г. Логан, штат Юта), 4 мМ глутамина (Mediatech, г. Херндон, штат Вирджиния), 100 единиц пенициллина на 100 миллилитров и 100 микрограмм стрептомицина на 100 миллилитров (Gibco, г. Карлсбад, штат Калифорния). Повторно суспендированные клетки центрифугировали (10 минут при 200×g), супернатант отсасывали и клеточный осадок промывали в полной среде. Клетки высевали непосредственно либо в колбы T75 (г. Корнинг, штат Нью-Йорк) либо в покрытые ламинином колбы T75, либо в покрытые фибронектином колбы T175 (все колбы производства Becton Dickinson, г. Бедфорд, штат Массачусетс).
Выделение клеток с использованием различных комбинаций ферментов и условий роста. Чтобы определить возможность выделения популяций клеток в разных условиях и их размножения в различных условиях сразу после выделения клетки расщепляли в ростовой среде с добавлением или без добавления 0,001 процента (об/об) 2-меркаптоэтанола (Sigma, г. Сент-Луис, штат Миссури) с использованием комбинации ферментов C:D:H в соответствии с описанными выше процедурами. Выделенные таким образом клетки плаценты высевали в различных условиях. Все клетки растили в присутствии пенициллина/стрептомицина (таблица 1-2).
Выделение клеток с использованием различных комбинаций ферментов и условий роста. При всех условиях клетки закреплялись и хорошо размножались между пассажами 0 и 1 (таблица 1-2). Для клеток при условиях 5-8 и 13-16 наблюдали хорошую пролиферацию вплоть до 4 пассажей после посева, после чего их криоконсервировали и депонировали в банк.
Результаты
Выделение клеток с использованием различных комбинаций ферментов. Комбинация ферментов C:D:H обеспечила наилучший выход клеток после выделения и дала клетки, которые размножались в культуре в течение большего количества поколений, чем при использовании других условий (таблица 1). При использовании только коллагеназы или только гиалуронидазы не удалось получить способную к размножению популяцию клеток. Попыток проверить, специфичен ли этот результат к конкретному тестируемому коллагену, не предпринималось.
Выделение клеток с использованием различных комбинаций ферментов и условий роста. Клетки закреплялись и хорошо размножались между пассажами 0 и 1 при всех тестируемых условиях по ферментативному расщеплению и условиям роста (таблица 2). Для клеток при экспериментальных условиях 5-8 и 13-16 наблюдали хорошую пролиферацию вплоть до 4 пассажей после посева, после чего их криоконсервировали. Все клетки депонировали в банк для дальнейшего исследования.
Выделение клеток из остаточной крови в пуповинах. Ядросодержащие клетки быстро закреплялись и росли. Данные клетки анализировали методом проточной цитометрии, и они оказались аналогичны клеткам, полученным методом ферментативного расщепления.
Выделение клеток из пуповинной крови. Препараты содержали эритроциты и тромбоциты. Ядросодержащие клетки не закреплялись и не делились в течение первых 3 недель. Среду заменили через 3 недели после посева, не наблюдали никакого закрепления и роста клеток.
Обзор. Популяции клеток из ткани пуповины и плаценты можно эффективно получать с использованием комбинации ферментов: коллагеназы (матриксной металлопротеазы), диспазы (нейтральной протеазы) и гиалуронидазы (муколитический фермент, расщепляющий гиалуроновую кислоту). Также можно использовать смесь LIBERASE™, которая представляет собой смесь ферментов типа Blendzyme. Более конкретно, для выделения клеток также использовали смесь Blendzyme 3, которая представляет собой смесь коллагеназы (4 единицы Вунша/г) и термолизина (1714 единиц казеина/г), вместе с гиалуронидазой. Данные клетки легко размножались на протяжении многих пассирований при культивировании в ростовой среде на покрытом желатином пластике.
Клетки также выделили из остаточной крови в пуповинах, но не из пуповинной крови. Присутствие клеток в сгустках крови, смытых с ткани, которые закрепляются и растут в используемых условиях, может быть обусловлено клетками, высвобожденными в процессе рассечения.
ПРИМЕР 2
Ростовые характеристики клеток, полученных из пуповины и плаценты
Потенциал размножения клеток, полученных из ткани пуповины и плаценты, сравнили с потенциалом размножения других популяций выделенных стволовых клеток. Процесс размножения клеток до старения называется пределом Хейфлика (Hayflick L. (1974 г.) J Am Geriatr Soc. 22: 1-12; Hayflick L. (1974 г.) Geontologist. 14: 37-45). Клетки, полученные из ткани пуповины, очень хорошо подходят для терапевтических целей, поскольку их можно легко размножать для получения достаточного количества клеток.
Материалы и способы
Покрытие колб желатином. Пластиковые колбы для культивирования тканей покрывали путем добавления 20 миллилитров 2% (в/об) раствора свиного желатина (тип B: 225 Bloom; Sigma, г. Сент-Луис, штат Миссури) в колбу T75 (Corning, г. Корнинг, штат Нью-Йорк) на 20 минут при комнатной температуре. После сливания раствора желатина добавляли и затем отсасывали 10 миллилитров фосфатно-солевого буфера (ФСБ) (Invitrogen, г. Карлсбад, штат Калифорния).
Сравнение потенциала размножения клеток UTC и популяций других клеток. Для сравнения потенциала роста размножения использовали следующие популяции клеток: i) мезенхимальные стволовые клетки (MSC; Cambrex, г. Уолкерсвилл, штат Мэриленд); ii) адипозные клетки (патент США № 6555374 B1; публикация заявки на патент США № 2004/0058412); iii) нормальные фибробласты дермы кожи (cc-2509, партия № 9F0844; Cambrex, г. Уолкерсвилл, штат Мэриленд); iv) клетки, полученные из пуповины; и v) клетки, полученные из плаценты (публикация заявки на патент США № 2004/0048372). Клетки исходно высевали с плотностью 5000 клеток/см2 на покрытые желатином колбы T75 в ростовой среде с добавлением пенициллина/стрептомицина/амфотерицина B. Для последующих пассирований клеточные культуры обрабатывали следующим образом. После трипсинизации подсчитывали жизнеспособные клетки после окрашивания трипановым синим. Суспензию клеток (50 микролитров) смешивали с трипановым синим (50 микролитров, Sigma, г. Сент-Луис, штат Миссури). Количество жизнеспособных клеток оценивали с помощью гемоцитометра.
После подсчета клетки высевали с плотностью 5000 клеток/см2 на покрытые желатином колбы T75 в 25 миллилитрах свежей ростовой среды. Клетки растили в стандартных условиях при 37°C. Ростовую среду меняли дважды в неделю. Клетки пассировали при достижении степени конфлюентности приблизительно 85 процентов; данный процесс повторяли до достижения клетками старения.
При каждом пассировании клетки трипсинизировали и подсчитывали. Рассчитывали выход жизнеспособных клеток, количество удвоений популяции [ln (конечное количество клеток/исходное количество клеток)/ln 2] и время удвоения (время в культуре (ч)/количество удвоений популяции). Для целей определения оптимального размножения клеток определяли общий выход клеток за пассаж путем умножения общего выхода клеток для предыдущего пассажа на фактор размножения для каждого пассажа (т.е. фактор размножения = конечное количество клеток/исходное количество клеток).
Потенциал размножения клеток в банках клеток при низкой плотности. Также тестировали потенциал размножения клеток, депонированных в банк на пассаже 10, используя другой набор условий. Тестировали нормальные фибробласты дермы кожи (cc-2509, партия № 9F0844; Cambrex, г. Уолкерсвилл, штат Мэриленд), клетки, полученные из пуповины, и клетки, полученные из плаценты. Данные популяции клеток ранее депонировали в банк на пассаже 10 после культивирования при плотности 5000 клеток/см2 и выращивании до конфлюентности на каждом предыдущем пассаже. Определили эффект плотности клеток на популяции клеток после размораживания клеток на пассаже 10. Клетки размораживали в стандартных условиях и подсчитывали с использованием окрашивания трипановым синим. Затем размороженные клетки высевали с плотностью 1000 клеток/см2 в ростовой среде DMEM с низким содержанием глюкозы с добавлением антибиотика/антимикотика, как описано выше. Клетки растили в стандартных атмосферных условиях при 37°C. Ростовую среду меняли дважды в неделю и клетки пассировали при достижении степени конфлюентности приблизительно 85%. Клетки последовательно пассировали до старения, т.е. до момента, когда их дальнейшее размножение становилось невозможным. При каждом пассировании клетки трипсинизировали и подсчитывали. Для каждого пассирования рассчитывали выход клеток, количество удвоений популяции [ln (конечное количество клеток/исходное количество клеток)/ln 2] и время удвоения (время в культуре (ч)/количество удвоений популяции). Определяли общий выход клеток за пассаж путем умножения общего выхода клеток для предыдущего пассажа на фактор размножения для каждого пассажа (т.е. фактор размножения = конечное количество клеток/исходное количество клеток).
Размножение клеток UTC при низкой плотности из исходного посева клеток. Тестировали потенциал размножения свежевыделенных клеток UTC в условиях низкой плотности посева. Клетки UTC готовили как описано в настоящем документе. Клетки высевали с плотностью 1000 клеток/см2 и пассировали до старения, как описано выше. Клетки растили в стандартных атмосферных условиях при 37°C. Ростовую среду меняли дважды в неделю. Клетки пассировали при достижении степени конфлюентности приблизительно 85%. При каждом пассировании клетки трипсинизировали и подсчитывали окрашиванием трипановым синим. Для каждого пассажа рассчитывали выход клеток, количество удвоений популяции [ln (конечное количество клеток/исходное количество клеток)/ln 2] и время удвоения (время в культуре (ч)/количество удвоений популяции). Определяли общий выход клеток за пассаж путем умножения общего выхода клеток для предыдущего пассажа на фактор размножения для каждого пассажа (т.е. фактор размножения = конечное количество клеток/исходное количество клеток). Клетки растили на покрытых желатином и не покрытых желатином колбах.
Размножение клональных неонатальных клеток, полученных из плаценты. Для размножения популяции неонатальных клеток из ткани плаценты использовали клонирование. После выделения из плаценты трех различных популяций клеток (как описано в настоящем документе) данные популяции клеток размножали в стандартных условиях роста и затем кариотипировали для идентификации популяций выделенных клеток. Поскольку данные клетки выделили из ткани матери, родившей мальчика, различить мужские и женские хромосомы путем проведения метафазного распластывания оказалось очень просто. Данные эксперименты показали, что клетки эмбрионального аспекта были кариотип-положительны на неонатальный фенотип, клетки среднего слоя были кариотип-положительны как на неонатальный, так и на материнский фенотипы, а клетки материнского аспекта были кариотип-положительны на материнские клетки.
Размножение клеток в условиях культивирования с низким содержанием кислорода. Результаты показали, что условия культивирования клеток с низким содержанием кислорода могут в определенных обстоятельствах улучшать размножение клеток (публикация заявки на патент США № 2004/0005704). Для проверки возможности улучшения размножения клеток UTC путем изменения условий культивирования выращивали культуры клеток, полученных из пуповины, в условиях с низким содержанием кислорода. Клетки высевали с плотностью 5000 клеток/см2 в ростовой среде на покрытых желатином колбах. Клетки первоначально культивировали в условиях стандартной атмосферы вплоть до пассажа 5, на котором их перенесли в условия культивирования с низким содержанием кислорода (5% O2).
Другие условия роста. В других протоколах клетки размножали на планшетах без покрытия, покрытых коллагеном, покрытых фибронектином, покрытых ламинином и покрытых белком внеклеточного матрикса. Результаты показали, что культуры хорошо размножаются на данных разных матрицах.
Результаты
Сравнение потенциала размножения клеток UTC и популяций других стволовых клеток и нестволовых клеток. Как клетки, полученные из пуповины, так и клетки, полученные из плаценты, размножались на протяжении более 40 пассажей, при этом на выходе оказалось >1E17 клеток за 60 суток. В отличие от этого клетки MSC и фибробласты достигали старения через <25 суток и <60 суток соответственно. Хотя адипозные клетки размножались в течение почти 60 суток, они дали общие выходы клеток 4,5E12. Таким образом, при посеве с плотностью 5000 клеток/см2 в используемых экспериментальных условиях клетки, полученные из неонатального материала, размножались намного лучше, чем клетки других типов, выращиваемые в тех же условиях (таблица 2-1).
Потенциал размножения клеток в банках клеток при низкой плотности. Клетки, полученные из пуповины, клетки, полученные из плаценты, и фибробласты размножались в течение более 10 пассажей, причем на выходе через 60 суток оказывалось >1E11 клеток (таблица 2-2). После 60 суток при данных условиях фибробласты достигали старения, тогда как популяции клеток, полученных из пуповины, и клеток, полученных из плаценты, достигали старения после 80 суток, совершив >50 и >40 удвоений популяции соответственно.
Размножение клеток UTC при низкой плотности из исходного посева клеток. Клетки UTC размножались при низкой плотности (1000 клеток/см2) на покрытых желатином и не покрытых планшетах или колбах. Потенциал роста данных клеток в данных условиях оказался хорошим. Клетки легко размножались в логарифмической фазе роста. Скорость размножения клеток была аналогична скорости, полученной при высевании полученных из плаценты клеток с плотностью 5000 клеток/см2 на покрытых желатином колбах в ростовой среде. Никаких различий между потенциалом размножения клеток при культивировании на непокрытых колбах или покрытых желатином колбах не обнаружено. Однако на покрытых желатином колбах клетки были фенотипически значительно меньшего размера, а на непокрытых колбах получили клетки гораздо более крупного фенотипа.
Размножение клональных неонатальных или материнских клеток, полученных из плаценты. Популяцию клональных неонатальных или материнских клеток можно размножать из полученных из плаценты клеток, выделенных из неонатального аспекта или материнского аспекта плаценты соответственно. Клетки последовательно разбавляют и затем высевают на покрытые желатином планшеты в ростовой среде для размножения, используя плотность 1 клетка/лунку в 96-луночных покрытых желатином планшетах. Среди данных исходных клонов идентифицируют способных к размножению клонов, трипсинизируют их и пересевают в 12-луночные покрытые желатином планшеты в ростовую среду с последующим пассированием их в покрытые желатином колбы T25 с плотностью 5000 клеток/см2 в ростовой среде. Для подтверждения идентификации клональной популяции клеток проводят субклонирование. Для экспериментов по субклонированию клетки трипсинизируют и высевают с плотностью 0,5 клетки/лунку. Хорошо растущие субклоны размножают на покрытых желатином колбах T25 с плотностью 5000 клеток/см2/колбу. Клетки пассируют с плотностью 5000 клеток/см2/колбу T75. Для демонстрации размножения клеток можно построить график ростовых характеристик клона. С помощью кариотипического анализа можно подтвердить, что клон является либо неонатальным, либо материнским.
Размножение клеток в условиях культивирования с низким содержанием кислорода. Клетки хорошо размножались в условиях с низким содержанием кислорода, однако культивирование в условиях с низким содержанием кислорода не оказало значительного эффекта на размножение клеток UTC в используемых условиях.
Обзор. Условия размножения клеток, содержащие выращивание выделенных клеток, полученных из ткани пуповины, с плотностями приблизительно 5000 клеток/см2 в ростовой среде на покрытых желатином или непокрытых колбах, в условиях стандартного атмосферного кислорода, достаточны для получения большого количества клеток на пассаже 11. Кроме того, данные указывают на то, что клетки можно легко размножать в условиях культивирования с меньшей плотностью (например, 1000 клеток/см2). Размножение клеток, полученных из пуповины, в условиях с низким содержанием кислорода также облегчает размножение клеток, хотя никакое дополнительное улучшение потенциала размножения клеток при использовании данных условий роста пока не обнаружено. В настоящее время для получения больших пулов клеток предпочтительным является культивирование клеток, полученных из ткани пуповины, в условиях стандартной атмосферы. Однако изменение условий культивирования также может изменить размножение клеток, полученных из ткани пуповины. Данную стратегию можно использовать для повышения пролиферативной и дифференцирующей способности данных клеточных популяций.
Хотя при используемых условиях потенциал размножения клеток MSC и адипозных клеток оказывается ограничен, клетки, полученные из ткани пуповины, легко размножаются до большого количества.
ПРИМЕР 3
Оценка ростовых сред для клеток, полученных из плаценты
Оценили способность нескольких сред для культивирования клеток поддерживать рост клеток, полученных из плаценты. Рост клеток, полученных из плаценты, при нормальном (20%) и пониженном (5%) содержании кислорода оценивали через 3 суток с использованием колориметрического теста клеток MTS.
Способы и материалы
Клетки, полученные из плаценты, на пассаже 8 (P8) высевали с плотностью 1×103 клеток/лунку в 96-луночных планшетах в ростовой среде с добавлением пенициллина/стрептомицина. Через 8 часов среду заменяли, как описано ниже, и клетки инкубировали при нормальном (атмосферном) или низком (5%, об/об) содержании кислорода при температуре 37°C, 5% CO2, в течение 48 часов. Затем в культуральную среду добавили MTS (раствор для анализа пролиферации клеток Cell Titer 96® AQueous One Solution Cell Proliferation Assay, Promega, г. Мадисон, штат Висконсин) и через 3 часа измерили коэффициент поглощения на 490 нанометров (Molecular Devices, г. Саннивейл, штат Калифорния).
Результаты
Используя стандартные кривые для анализа MTS, установили линейную корреляцию между увеличением коэффициента поглощения и увеличением количества клеток. Полученные значения коэффициента поглощения перевели в оценочные количества клеток и рассчитали изменение (%) относительно исходного посева.
Эффект сыворотки. Добавление в среды сыворотки при нормальных кислородных условиях привело к воспроизводимому дозозависимому увеличению коэффициента поглощения и, таким образом, количества жизнеспособных клеток. Добавление сыворотки в полную среду MSCGM привело к дозозависимому уменьшению коэффициента поглощения. Из сред без добавления сыворотки клетки заметно росли только в средах Cellgro FREE™, Хэма F10 и DMEM.
Эффект кислорода. Снижение концентрации кислорода привело к увеличению скорости роста клеток в ростовой среде, средах Хэма F10 и MSCGM. Если располагать среды по убыванию роста, то наилучшими средами для роста клеток оказались ростовая среда > среда MSCGM > среда Искова + 10% FBS = среда DMEM-H+10% FBS = среда Хэма F12+10% FBS = среда RPMI 1640+10% FBS.
Обзор. Клетки, полученные из плаценты, можно выращивать в различных культуральных средах при нормальном или низком содержании кислорода. Изучили кратковременный рост клеток, полученных из плаценты, в двенадцати основных средах с добавлением 0, 2 и 10% (об/об) сыворотки при атмосферной или 5% концентрации кислорода. В целом, клетки, полученные из плаценты, не так хорошо росли в бессывороточных условиях, за исключением среды Хэма F10 и среды Cellgro FREE™, которая также не содержит белки. Рост в данных бессывороточных средах составил приблизительно 25-33% от максимального роста, наблюдаемого для сред, содержащих 15% сыворотки.
ПРИМЕР 4
Рост клеток, полученных из ткани пуповины и плаценты, в среде, содержащей D-валин
Как известно, использование среды, содержащей D-валин вместо нормальной изоформы L-валина, позволяет избирательно ингибировать рост фибробластоподобных клеток в культуре (Hongpaisan J. 2000 г. Cell Biol Int. 24: 1-7; Sordillo et al. 1988 г. Cell Biol Int Rep. 12: 355-64). Ранее не было известно, могут ли клетки, полученные из ткани пуповины и плаценты, расти в среде, содержащей D-валин.
Способы и материалы
Клетки, полученные из плаценты, (P3), фибробласты (P9) и клетки, полученные из пуповины, (P5) высевали с плотностью 5×103 клеток/см2 в покрытых желатином колбах T75 (Corning, г. Корнинг, штат Нью-Йорк). Через 24 часа среду удаляли и клетки промывали фосфатно-солевым буфером (ФСБ) (Gibco, г. Карлсбад, штат Калифорния) для удаления остатков среды. Среду заменяли на модифицированную ростовую среду (среда DMEM с D-валином (специальный заказ, Gibco), 15% (об/об) диализованной эмбриональной бычьей сыворотки (Hyclone, г. Логан, штат Юта), 0,001% (об/об) бетамеркаптоэтанола (Sigma), пенициллин/стрептомицин (Gibco)).
Результаты
Клетки, полученные из плаценты, клетки, полученные из пуповины, и клетки фибробластов после посева в содержащую D-валин среду не пролиферировали в отличие от клеток, высеянных в ростовую среду, содержащую диализованную сыворотку. Клетки фибробластов изменились морфологически, увеличившись в размере и изменив форму. Все клетки погибали и, в конце концов, отделялись от поверхности колбы через 4 недели. Данные результаты показывают, что среда, содержащая D-валин, не подходит для избирательного выращивания клеток, полученных из неонатального материала.
ПРИМЕР 5
Среды для криоконсервирования полученных из плаценты клеток
Оценивали разные среды для криоконсервирования в плане возможности их использования для криоконсервирования полученных из плаценты клеток.
Способы и материалы
Клетки, полученные из плаценты и выращенные в ростовой среде в покрытой желатином колбе T75, промыли ФСБ и трипсинизировали с использованием 1 миллилитра смеси трипсин/ЭДТА (Gibco). Трипсинизацию остановили добавлением 10 миллилитров ростовой среды. Клетки центрифугировали при 150×g, удалили супернатант и клеточный осадок повторно суспендировали в 1 миллилитре ростовой среды. Отобрали аликвоту клеточной суспензии, 60 микролитров, и добавили к 60 микролитрам трипанового синего (Sigma). Количество жизнеспособных клеток оценивали с помощью гемоцитометра. Клеточную суспензию разделили на четыре равные аликвоты, каждая из которых содержала по 88×104 клеток. Клеточную суспензию центрифугировали и повторно суспендировали в 1 миллилитре каждой среды, представленной ниже, и затем переносили в криопробирки (Nalgene).
1.) Ростовая среда + 10% (об/об) DMSO (Hybrimax, Sigma, г. Сент-Луис, штат Миссури)
2.) Среда для замораживания клеток с DMSO, с метилцеллюлозой, бессывороточная (C6295, Sigma, г. Сент-Луис, штат Миссури)
3.) Среда для замораживания клеток, бессывороточная (C2639, Sigma, г. Сент-Луис, штат Миссури)
4.) Среда для замораживания клеток с глицерином (C6039, Sigma, г. Сент-Луис, штат Миссури)
Клетки охлаждали в течение ночи со скоростью приблизительно -1°C/мин в холодильнике на -80°C с использованием контейнера для замораживания Mr Frosty в соответствии с инструкциями производителя (Nalgene, г. Рочестер, штат Нью-Йорк). Флаконы с клетками поместили в жидкий азот на 2 суток и затем быстро разморозили на водяной бане при температуре 37°C. Клетки добавили в 10 миллилитров ростовой среды, центрифугировали и затем оценили количество и жизнеспособность клеток. Клетки высевали на покрытые желатином колбы с плотностью 5000 клеток/см2 для определения способности клеток к закреплению и пролиферации.
Результаты
По результатам окрашивания трипановым синим исходная жизнеспособность клеток для криоконсервирования составила 100%. По результатам окрашивания трипановым синим исходная жизнеспособность клеток для криоконсервирования составила 100%.
Для среды C6295 наблюдали соразмерное снижение количества клеток и жизнеспособности из-за лизиса клеток. Во всех четырех растворах жизнеспособные клетки криоконсервировались, делились и за 3 суток давали конфлюентный монослой. Заметных различий в оцениваемой скорости роста не обнаружили.
Обзор. Криоконсервирование клеток является одной из возможных процедур для получения банка клеток или клеточного продукта. Сравнили четыре смеси для криоконсервирования относительно их способности защитить клетки человека, полученные из плаценты, от повреждения при замораживании. Из участвовавших в сравнении смесей предпочтительной для криоконсервирования клеток, полученных из плаценты, является модифицированная по способу Дульбекко среда Игла (DMEM) с добавлением 10% (об/об) диметилсульфоксида (ДМСО).
ПРИМЕР 6
Кариотипический анализ клеток, полученных из ткани пуповины, и клеток, полученных из плаценты
Используемые в клеточной терапии клеточные линии предпочтительно должны быть однородными и свободными от любых примесей клеток других типов. Используемые в клеточной терапии клетки должны иметь нормальное количество (46) и структуру хромосом. Для идентификации клеточных линий клеток, полученных из плаценты и пуповины, однородных и свободных от клеток природы, отличной от клеток тканей неонатального материала, провели анализ кариотипов образцов клеток.
Материалы и способы
Клетки UTC, полученные из тканей неонатального материала новорожденного мальчика, культивировали в ростовой среде, содержащей пенициллин/стрептомицин. Ткань неонатального материала новорожденного мальчика (X,Y) выбрали для возможности различения неонатальных клеток и материнских клеток (X,X). Клетки высевали с плотностью 5000 клеток на кв. сантиметр в ростовой среде в колбе T25 (Corning, г. Корнинг, штат Нью-Йорк) и размножали до степени конфлюентности 80%. Затем содержащую клетки колбу T25 заполнили по горлышко ростовой средой. Образцы доставили курьером в лабораторию клинической цитогенетики (оцениваемое время транспортировки из лаборатории в лабораторию составляет один час). Клетки анализировали во время метафазы, когда хромосомы видны лучше всего. Из двадцати подсчитанных в метафазе клеток пять проанализировали на количество кариотипов для нормальной однородной популяции клеток (два). Образец клеток охарактеризовали как однородный при наблюдении двух кариотипов. Образец клеток охарактеризовали как неоднородный при наблюдении более двух кариотипов. При определении количества кариотипов (четыре), характерного для неоднородной популяции клеток, подсчитывали и анализировали дополнительные клетки в метафазе.
Результаты
Все направленные на хромосомный анализ образцы клеток имели нормальный внешний вид. Три из шестнадцати проанализированных клеточных линий продемонстрировали неоднородный фенотип (XX и XY), указывающий на присутствие клеток как неонатального, так и материнского происхождения (таблица 6-1). Клетки, полученные из ткани плацента-N, были выделены из неонатального аспекта плаценты. На пассаже 0 данная клеточная линия была однородной XY. Однако на пассаже 9 клеточная линия была неоднородной (XX/XY), что указывает на ранее не замеченное присутствие клеток материнского происхождения.
Обзор. В результате хромосомного анализа идентифицированы клетки, полученные из плаценты и пуповины, кариотипы которых были интерпретированы в лаборатории клинической цитогенетики как нормальные. В результате кариотипического анализа также идентифицированы клеточные линии, свободные от материнских клеток, как было определено по однородному кариотипу.
ПРИМЕР 7
Оценка маркеров клеточной поверхности клеток, полученных из ткани пуповины и плаценты человека, методом проточной цитометрии
Характеризацию белков, или «маркеров», клеточной поверхности методом проточной цитометрии можно применять для определения отличительных признаков клеточной линии. Стабильность экспрессии можно определить по множеству доноров и в клетках, культивировавшихся и обрабатывавшихся в разных условиях. Линии клеток, выделенных из плаценты и пуповины, охарактеризовали методом проточной цитометрии, получив профиль экспрессии для установления отличительных признаков данных клеточных линий.
Материалы и способы
Среды и сосуды для культивирования. Клетки культивировали в ростовой среде (Gibco г. Карлсбад, штат Калифорния) с добавлением пенициллина/стрептомицина. Клетки культивировали в обработанных плазмой колбах для культивирования тканей T75, T150 и T225 (Corning, г. Корнинг, штат Нью-Йорк) до конфлюентности. Ростовые поверхности колб покрывали желатином инкубацией с 2% (в/об) раствором желатина (Sigma, г. Сент-Луис, штат Миссури) в течение 20 минут при комнатной температуре.
Окрашивание антителами и анализ методом проточной цитометрии. Закрепившиеся клетки в колбах промывали ФСБ и отделяли с помощью трипсина/ЭДТА. Клетки собирали, центрифугировали и повторно суспендировали в 3% (об/об) FBS в ФСБ при концентрации 1x107 клеток на миллилитр. В соответствии с инструкциями производителя антитело на рассматриваемый маркер клеточной поверхности (смотрите ниже) добавляли к ста микролитрам клеточной суспензии и инкубировали смесь в темноте в течение 30 минут при 4°C. После инкубации клетки промывали ФСБ и центрифугировали для удаления несвязавшихся антител. Клетки повторно суспендировали в 500 микролитрах ФСБ и анализировали методом проточной цитометрии. Анализ методом проточной цитометрии проводили на анализаторе FACScalibur (Becton Dickinson, г. Сан-Хосе, штат Калифорния).
Применяли следующие антитела на маркеры клеточной поверхности.
Сравнение клеток плаценты и пуповины. Клетки, полученные из плаценты, сравнили с клетками, полученными из пуповины, на пассаже 8.
Сравнение клеток разных пассажей. Клетки, полученные из плаценты и пуповины, анализировали на пассажах 8, 15 и 20.
Сравнение клеток разных доноров. Для сравнения различий между донорами сравнивали между собой клетки, полученные из плаценты, от разных доноров и сравнивали между собой клетки, полученные из пуповины, от разных доноров.
Сравнение клеток в зависимости от покрытия поверхности колб. Клетки, полученные из плаценты, культивировавшиеся на покрытых желатином колбах, сравнивали с клетками, полученными из плаценты, культивировавшимися на непокрытых колбах. Клетки, полученные из пуповины, культивировавшиеся на покрытых желатином колбах, сравнили с клетками, полученными из пуповины, культивировавшимися на непокрытых колбах.
Сравнение клеток, полученных с разными расщепляющими ферментами. Сравнили результаты четырех процедур для выделения и подготовки клеток. Сравнили клетки, выделенные из плаценты путем обработки 1) коллагеназой; 2) коллагеназой/диспазой; 3) коллагеназой/гиалуронидазой; и 4) коллагеназой/гиалуронидазой/диспазой.
Сравнение клеток разных плацентарных слоев. Клетки, полученные из материнского аспекта плацентарной ткани, сравнили с клетками, полученными из ворсинчатого участка плацентарной ткани, и клетками, полученными из неонатального эмбрионального аспекта плаценты.
Результаты
Сравнение клеток плаценты и пуповины. Проанализированные методом проточной цитометрии клетки, полученные из плаценты и пуповины, показали положительную экспрессию CD10, CD13, CD44, CD73, CD 90, PDGFr-альфа и HLA-A, B, C, на что указывают повышенные значения флуоресценции по сравнению с используемым в качестве контроля IgG. Данные клетки были отрицательными в отношении определяемой экспрессии CD31, CD34, CD45, CD117, CD141 и HLA-DR, DP, DQ, на что указывают значения флуоресценции в сравнении с используемым в качестве контроля IgG. Во внимание были приняты вариации значений флуоресценции положительных кривых. Средние значения (т.е. CD13) и диапазон значений (т.е. CD90) положительных кривых имели некоторые вариации, но сами кривые имели нормальный вид, что подтверждает однородность популяции. Обе кривые по отдельности показали значения, превышающие значения для используемого в качестве контроля IgG.
Сравнение клеток, полученных из плаценты, разных пассажей. Все проанализированные методом проточной цитометрии клетки, полученные из плаценты, на пассажах 8, 15 и 20 были положительными в отношении экспрессии CD10, CD13, CD44, CD73, CD 90, PDGFr-альфа и HLA-A, B, C, что отражается в увеличенном значении флуоресценции по сравнению с используемым в качестве контроля IgG. Данные клетки были отрицательными в отношении экспрессии CD31, CD34, CD45, CD117, CD141 и HLA-DR, DP, DQ, причем значения флуоресценции согласовывались с используемым в качестве контроля IgG.
Сравнение клеток, полученных из пуповины, разных пассажей. Все проанализированные методом проточной цитометрии клетки, полученные из пуповины, на пассажах 8, 15 и 20 экспрессировали CD10, CD13, CD44, CD73, CD 90, PDGFr-альфа и HLA-A, B, C, на что указывают значения флуоресценции по сравнению с используемым в качестве контроля IgG. Данные клетки были отрицательными в отношении CD31, CD34, CD45, CD117, CD141 и HLA-DR, DP, DQ, на что указывают значения флуоресценции, согласующиеся с используемым в качестве контроля IgG.
Сравнение клеток, полученных из плаценты, разных доноров. Все проанализированные методом проточной цитометрии клетки, полученные из плаценты и выделенные у разных доноров, экспрессировали CD10, CD13, CD44, CD73, CD 90, PDGFr-альфа и HLA-A, B, C с увеличением значений флуоресценции по сравнению с используемым в качестве контроля IgG. Данные клетки были отрицательными в отношении экспрессии CD31, CD34, CD45, CD117, CD141 и HLA-DR, DP, DQ, на что указывает значение флуоресценции, согласующееся с используемым в качестве контроля IgG.
Сравнение клеток, полученных из пуповины, разных доноров. Все проанализированные методом проточной цитометрии клетки, полученные из пуповины и выделенные у разных доноров, показали положительную экспрессию CD10, CD13, CD44, CD73, CD 90, PDGFr-альфа и HLA-A, B, C, на что указывают повышенные значения флуоресценции по сравнению с используемым в качестве контроля IgG. Данные клетки были отрицательными в отношении экспрессии CD31, CD34, CD45, CD117, CD141 и HLA-DR, DP, DQ, причем значения флуоресценции согласовывались с используемым в качестве контроля IgG.
Влияние покрытия поверхности колбы желатином на клетки, полученные из плаценты. Все проанализированные методом проточной цитометрии клетки, полученные из плаценты и размножаемые либо на покрытых желатином, либо на непокрытых колбах, экспрессировали CD10, CD13, CD44, CD73, CD 90, PDGFr-альфа и HLA-A, B, C, что отражается в увеличенных значениях флуоресценции по сравнению с используемым в качестве контроля IgG. Данные клетки были отрицательными в отношении экспрессии CD31, CD34, CD45, CD117, CD141 и HLA-DR, DP, DQ, на что указывают значения флуоресценции, согласующиеся с используемым в качестве контроля IgG.
Влияние покрытия поверхности колбы желатином на клетки, полученные из пуповины. Все проанализированные методом проточной цитометрии клетки, полученные из пуповины и размножаемые на покрытых желатином и на непокрытых колбах, были положительными в отношении экспрессии CD10, CD13, CD44, CD73, CD 90, PDGFr-альфа и HLA-A, B, C, при этом значения флуоресценции повышены по сравнению с используемым в качестве контроля IgG. Данные клетки были отрицательными в отношении экспрессии CD31, CD34, CD45, CD117, CD141 и HLA-DR, DP, DQ, причем значения флуоресценции согласовывались с используемым в качестве контроля IgG.
Влияние процедуры ферментативного расщепления, используемой для приготовления клеток, на профиль экспрессии маркеров клеточной поверхности. Все проанализированные методом проточной цитометрии клетки, полученные из плаценты и выделенные с использованием различных расщепляющих ферментов, экспрессировали CD10, CD13, CD44, CD73, CD 90, PDGFr-альфа и HLA-A, B, C, на что указывают увеличенные значения флуоресценции по сравнению с используемым в качестве контроля IgG. Данные клетки были отрицательными в отношении экспрессии CD31, CD34, CD45, CD117, CD141 и HLA-DR, DP, DQ, на что указывают значения флуоресценции, согласующиеся с используемым в качестве контроля IgG.
Сравнение клеток разных плацентарных слоев. Проанализированные методом проточной цитометрии клетки, выделенные из материнского, ворсинчатого и неонатального слоев плаценты соответственно, показали положительную экспрессию CD10, CD13, CD44, CD73, CD 90, PDGFr-альфа и HLA-A, B, C, на что указывают увеличенные значения флуоресценции по сравнению с используемым в качестве контроля IgG. Данные клетки были отрицательными в отношении экспрессии CD31, CD34, CD45, CD117, CD141 и HLA-DR, DP, DQ, на что указывают значения флуоресценции, согласующиеся с используемым в качестве контроля IgG.
Обзор. Анализ клеток, полученных из плаценты и пуповины, методом проточной цитометрии позволил установить отличительные признаки данных клеточных линий. Клетки, полученные из плаценты и пуповины, положительны в отношении экспрессии CD10, CD13, CD44, CD73, CD90, PDGFr-альфа, HLA-A, B, C и отрицательны в отношении экспрессии CD31, CD34, CD45, CD117, CD141 и HLA-DR, DP, DQ. Данные отличительные признаки стабильны при изменении различных переменных, включая донора, номер пассажа, покрытие поверхности сосуда для культивирования, расщепляющие ферменты и плацентарный слой. Наблюдаются некоторые вариации в средних значениях и диапазонах изменения кривых гистограмм значений флуоресценции, но все положительные кривые при всех тестируемых условиях имели нормальный вид и значения флуоресценции, превышающие значения для используемого в качестве контроля IgG, тем самым подтверждая, что клетки представляют собой однородную популяцию с положительной экспрессией маркеров.
ПРИМЕР 8
Иммуногистохимическая характеризация фенотипов тканей пуповины и плаценты
Проводили иммуногистохимический анализ фенотипов клеток, присутствующих в пуповине и плаценте человека.
Материалы и способы
Подготовка тканей. Ткани пуповины и плаценты человека собирали и фиксировали погружением в 4% (в/об) раствор параформальдегида в течение ночи при температуре 4°C. Иммуногистохимический анализ проводили с использованием антител, направленных на следующие эпитопы: виментин (1:500; Sigma, г. Сент-Луис, штат Миссури), дезмин (1:150, выработанные на кролика; Sigma; или 1:300, выработанные на мышь; Chemicon, г. Темекула, штат Калифорния), альфа-актин гладких мышц (SMA; 1:400; Sigma), цитокератин 18 (CK18; 1:400; Sigma), фактор фон Виллебранда (vWF; 1:200; Sigma) и CD34 (CD34 человека, класс III; 1:100; DAKOCytomation, г. Карпинтерия, штат Калифорния). Кроме того, тестировали следующие маркеры: анти-человеческий GROальфа - PE (1:100; Becton Dickinson, г. Франклин Лейкс, штат Нью-Джерси), анти-человеческий GCP-2 (1:100; Santa Cruz Biotech, г. Санта-Крус, штат Калифорния), рецептор 1 окисленных ЛПНП против человека (ox-LDL R1; 1:100; Santa Cruz Biotech) и анти-человеческий NOGO-A (1:100; Santa Cruz Biotech). Зафиксированные образцы обрезали скальпелем и поместили в герметизирующее вещество OCT (Tissue-Tek OCT; Sakura, г. Торранс, штат Калифорния) на баню с сухим льдом, содержащую этанол. Затем замороженные блоки нарезали на части толщиной 10 мкм с использованием стандартного криостата (Leica Microsystems) и установили на предметные стекла для окрашивания.
Иммуногистохимический анализ. Иммуногистохимический анализ провели аналогично предыдущим исследованиям (например, Messina, et al., 2003 г., Exper. Neurol. 184: 816-829). Части тканей промывали фосфатно-солевым буфером (ФСБ) и обрабатывали белковым блокирующим раствором, содержащим ФСБ, 4% (об/об) козьей сыворотки (Chemicon, г. Темекула, штат Калифорния) и 0,3% (об/об) Тритона (Triton X-100, Sigma), в течение 1 часа для доступа к внутриклеточным антигенам. Когда рассматриваемый эпитоп находился на клеточной поверхности (CD34, ox-LDL R1), Тритон исключали на всех стадиях процедуры, чтобы не допустить потери эпитопа. Кроме того, когда первичное антитело было выработано на козу (GCP-2, ox-LDL R1, NOGO-A), в течение всей процедуры вместо козьей сыворотки использовали 3% (об/об) ослиную сыворотку. Затем части обрабатывали в течение 4 часов при комнатной температуре первичными антителами, разбавленными в блокирующем растворе. Растворы первичных антител удаляли и промывали культуры ФСБ перед добавлением растворов вторичных антител (1 час при комнатной температуре), содержащих блокирующий раствор вместе с конъюгатом козьего антимышиного IgG - Texas Red (1:250; Molecular Probes, г. Юджин, штат Орегон) и/или конъюгатом козьего анти-кроличьего IgG - Alexa 488 (1:250; Molecular Probes), либо конъюгатом ослиного анти-козьего IgG - FITC (1:150; Santa Cruz Biotech). Культуры промывали и обрабатывали 10-микромолярным раствором DAPI (Molecular Probes) в течение 10 минут для визуализации клеточных ядер.
После иммунного окрашивания получали флуоресцентные изображения с использованием соответствующего фильтра для флуоресценции на инвертированном эпи-флуоресцентном микроскопе (Olympus, г. Мелвилл, штат Нью-Йорк). Положительное окрашивание представляло собой флуоресцентный сигнал выше сигнала окрашивания контроля. Представительные изображения получали с использованием цветной цифровой видеокамеры и программного обеспечения ImagePro (Media Cybernetics, г. Карлсбад, штат Калифорния). Для образцов с тройным окрашиванием каждое изображение получали с использованием только одного эмиссионного фильтра. Затем получили послойные монтажи с использованием программного обеспечения Adobe Photoshop (Adobe, г. Сан-Хосе, штат Калифорния).
Результаты
Характеризация пуповины. Маркеры виментин, дезмин, SMA, CK18, vWF и CD34 экспрессировали подмножеством клеток, присутствующих в пуповине. В частности, экспрессия vWF и CD34 ограничивалась кровеносными сосудами, находящимися внутри пуповины. Клетки CD34+ находились на самом внутреннем слое (со стороны просвета). Экспрессию виментина обнаружили по всему матриксу и кровеносным сосудам пуповины. Экспрессия SMA ограничивалась матриксом и внешними стенками артерии и вены, но не проходила внутри самих сосудов. CK18 и дезмин наблюдали только внутри сосудов, причем экспрессия дезмина ограничивалась только средним и внешними слоями.
Характеризация плаценты. Каждый из виментина, дезмина, SMA, CK18, vWF и CD34 наблюдали внутри плаценты и были региоспецифичными.
Экспрессия GROальфа, GCP-2, ox-LDL R1 и NOGO-A в тканях. Ни один из данных маркеров не обнаружили в ткани пуповины или плаценты.
Обзор. Виментин, дезмин, альфа-актин гладких мышц, цитокератин 18, фактор фон Виллебранда и CD 34 экспрессируются в клетках пуповины и плаценты человека.
ПРИМЕР 9
Анализ клеток с использованием олигонуклеотидных чипов
Для сравнения профилей экспрессии генов клеток, полученных из пуповины и плаценты, с профилями экспрессии для фибробластов, мезенхимальных стволовых клеток человека и другой клеточной линии, полученной из костного мозга человека, использовали чипы Affymetrix GENECHIP®. Данный анализ позволил охарактеризовать клетки и идентифицировать уникальные молекулярные маркеры данных клеток.
Материалы и способы
Выделение и культивирование клеток. Пуповины и плаценту человека получили в Национальном центре по обмену материалами для исследования заболеваний (NDRI, г. Филадельфия, штат Пенсильвания) после нормально прошедших родов при доношенной беременности с согласия пациента. Ткани получили и выделили из них клетки, как описано в примере 1. Клетки культивировали в ростовой среде (используя среду DMEM-LG) на покрытых желатином пластиковых колбах для культивирования тканей. Культуры инкубировали при 37°C в атмосфере с 5% CO2.
Фибробласты дермы человека приобрели в компании Cambrex Incorporated (г. Уолкерсвилл, штат Мэриленд; партия № 9F0844) и ATCC CRL-1501 (CCD39SK). Обе линии культивировали в среде DMEM/F12 (Invitrogen, г. Карлсбад, штат Калифорния) с добавлением 10% (об/об) эмбриональной бычьей сыворотки (Hyclone) и пенициллина/стрептомицина (Invitrogen). Клетки растили на стандартном обработанном тканью пластике.
Мезенхимальные стволовые клетки человека (hMSC) приобрели в компании Cambrex Incorporated (г. Уолкерсвилл, штат Мэриленд; партии № 2F1655, 2F1656 и 2F1657) и культивировали в соответствии с рекомендациями производителя в среде MSCGM (Cambrex). Клетки растили на стандартном культивированном тканью пластике при 37°C в атмосфере с 5% CO2.
Костный мозг гребня подвздошной кости человека получали из NDRI с согласия пациента. Костный мозг обрабатывали в соответствии со способом, описанном Ho, et al. (международная заявка № WO03/025149). Костный мозг смешивали с лизирующим буфером (155 мМ NH4Cl, 10 мМ KHCO3 и 0,1 мМ ЭДТА, pH 7,2) в соотношении 1 часть костного мозга к 20 частям лизирующего буфера. Суспензию клеток перемешивали на вортексе, инкубировали в течение 2 минут при температуре окружающей среды и центрифугировали в течение 10 минут при 500×g. Супернатант удаляли и клеточный осадок повторно растворяли в минимальной поддерживающей среде-альфа (Invitrogen) с добавлением 10% (об/об) эмбриональной бычьей сыворотки и 4 мМ глутамина. Клетки еще раз центрифугировали и клеточный осадок повторно растворяли в свежей среде. Количество жизнеспособных мононуклеарных клеток определяли тестом на вытеснение трипанового синего (Sigma, г. Сент-Луис, штат Миссури). Мононуклеарные клетки высевали в культивированные тканью пластиковые колбы с плотностью 5×104 клетки/см2. Клетки инкубировали при 37°C в атмосфере с 5% CO2 либо в условиях стандартной атмосферы O2, либо 5% O2. Клетки культивировали в течение 5 суток без замены среды. После 5 суток культивирования среду и незакрепившиеся клетки удалили. Закрепившиеся клетки поддерживали в культуре.
Выделение мРНК и анализ GENECHIP®. Активно растущие культуры клеток извлекали из колб с помощью скребка и переносили в холодный ФСБ. Клетки центрифугировали в течение 5 минут при 300×g. Супернатант удаляли, клетки повторно суспендировали в свежем ФСБ и центрифугировали еще раз. Супернатант удаляли и клеточный осадок немедленно замораживали и хранили при -80°C. Клеточную мРНК экстрагировали и транскрибировали в кДНК, которую затем транскрибировали в кРНК и метили биотином. Меченную биотином кРНК гибридизовали с олигонуклеотидным чипом GENECHIP® HG-U133A (Affymetrix, г. Санта-Клара, штат Калифорния). Гибридизацию и сбор данных проводили в соответствии с рекомендациями производителя. Анализ проводили с использованием программного обеспечения Significance Analysis of Microarrays (SAM) версии 1.21 (Stanford University; Tusher, V.G. et al., 2001 г., Proc. Natl. Acad. Sci. USA 98: 5116-5121).
Результаты
Проанализировали четырнадцать различных популяций клеток. Типы клеток, а также информация о пассажах, культуральном субстрате и культуральной среде перечислены в таблице 9-1.
Данные оценивали с использованием анализа главных компонентов, анализируя те 290 генов, которые дифференциально экспрессировались в клетках. Данный анализ позволяет провести относительное сравнение для поиска сходств между популяциями. В таблице 9-2 представлены евклидовы расстояния, рассчитанные для сравнения клеточных пар. Евклидовы расстояния были основаны на сравнении клеток на основе 290 генов, которые дифференциально экспрессировались в клетках разных типов. Евклидово расстояние обратно пропорционально степени сходства между экспрессией 290 генов (т.е. чем больше расстояние, тем меньше сходства).
В таблицах 9-3, 9-4 и 9-5 показана экспрессия генов, повышенная в клетках, полученных из плаценты, (таблица 9-3) повышенная в клетках, полученных из пуповины, (таблица 9-4) и пониженная в клетках, полученных из пуповины и плаценты (таблица 9-5). Колонка под названием «Идентификатор набора зондов» относится к идентификационному коду, присвоенному производителем наборам нескольких олигонуклеотидных зондов, расположенных в определенном месте чипа, которые гибридизуются с указанным геном (колонка «Название гена»), содержащим последовательность, которую можно найти в базе данных NCBI (GenBank) под указанным номером доступа (колонка «Номер доступа NCBI»).
В таблицах 9-6, 9-7 и 9-8 показана экспрессия генов, повышенная в фибробластах человека (таблица 9-6), клетках ICBM (таблица 9-7) и клетках MSC (таблица 9-8).
Обзор. Настоящее исследование провели для получения молекулярной характеризации клеток, полученных из пуповины и плаценты. Анализ включал клетки, полученные из трех различных пуповин и трех различных плацент. Исследование также включало две различные линии дермальных фибробластов, три линии мезенхимальных стволовых клеток и три линии клеток костного мозга гребня подвздошной кости. Экспрессируемые данными клетками мРНК анализировали с использованием олигонуклеотидного чипа, содержащего зонды на 22000 генов. Результаты показали, что в клетках данных пяти разных типов присутствует дифференциальная экспрессия 290 генов. Данные гены включают десять генов, экспрессия которых специфически повышается в клетках, полученных из плаценты, и семь генов, экспрессия которых специфически повышается в клетках, полученных из пуповины. Для пятидесяти четырех генов обнаружена специфически пониженная экспрессия в клетках плаценты и пуповины по сравнению с клетками других типов. Экспрессию выбранных генов подтвердили с использованием ПЦР (смотрите следующий пример). Данные результаты показывают, что клетки имеют вполне определенный профиль экспрессии генов, например, по сравнению с клетками костного мозга и фибробластами.
ПРИМЕР 10
Клеточные маркеры в клетках, полученных из ткани пуповины, и клетках, полученных из плаценты
В предыдущем примере проведена оценка сходств и различий клеток, полученных из плаценты человека и из пуповины человека, путем сравнения их профилей экспрессии генов с профилями клеток, полученных из других источников (с использованием олигонуклеотидного чипа). Были идентифицированы шесть «характерных» генов: ox-LDL R1 (рецептор 1 окисленных ЛПНП), интерлейкин-8, реннин, ретикулон, CXCL3 (лиганд хемокинового рецептора 3) и GCP-2 (гранулоцитарный хемотаксический белок-2). Данные «характерные» гены экспрессировались в клетках на относительно высоком уровне.
Описанные в данном примере процедуры провели для верификации данных анализа на микрочипах и выявления согласий/разногласий между экспрессией генов и белков, а также для создания серии надежных анализов для выявления уникальных идентификаторов клеток, полученных из плаценты и пуповины.
Способы и материалы
Клетки. Клетки, полученные из плаценты (три изолята, включая один преимущественно неонатальный изолят по результатам кариотипического анализа), клетки, полученные из пуповины (четыре изолята), и нормальные фибробласты кожи человека (NHDF; неонатальные и взрослые) выращивали в ростовой среде с добавлением пенициллина/стрептомицина в покрытой желатином колбе T75. Мезенхимальные стволовые клетки (MSC) выращивали в среде для выращивания мезенхимальных стволовых клеток Bulletkit (MSCGM; Cambrex, г. Уокерсвилл, штат Мэриленд).
Для выполнения протокола IL-8 клетки размораживали из жидкого азота и высевали в покрытые желатином колбы с плотностью 5000 клеток/см2, растили в течение 48 часов в ростовой среде и затем растили в течение еще 8 часов в 10 миллилитрах бессывороточной среды [среда DMEM с низким содержанием глюкозы (Gibco, г. Карлсбад, штат Калифорния), пенициллин/стрептомицин (Gibco, г. Карлсбад, штат Калифорния) и 0,1% (в/об) бычьего сывороточного альбумина (БСА; Sigma, г. Сент-Луис, штат Миссури)]. После данной обработки экстрагировали РНК и супернатанты центрифугировали при 150×g в течение 5 минут для удаления продуктов распада клеток. Затем супернатанты замораживали при температуре -80°C для проведения анализа ИФА.
Культивирование клеток для анализа ИФА. Клетки, полученные из плаценты и пуповины, а также фибробласты человека, полученные из неонатальной крайней плоти человека, культивировали в ростовой среде в покрытых желатином колбах T75. На пассаже 11 клетки замораживали в жидком азоте. Клетки размораживали и переносили в пробирки для центрифугирования объемом 15 миллилитров. После центрифугирования при 150×g в течение 5 минут супернатант удалили. Клетки повторно суспендировали в 4 миллилитрах культуральной среды и подсчитывали. Клетки выращивали в течение 24 часов в колбе с площадью поверхности 75 см2, содержащей 15 миллилитров ростовой среды, при посеве 375000 клеток/колбу. Среду на 8 часов заменили на бессывороточную среду. В конце инкубации бессывороточную среду собрали, центрифугировали при 14000×g в течение 5 минут и хранили при температуре -20°C.
Для оценки количества клеток в каждую колбу добавляли по 2 миллилитра трипсина/ЭДТА (Gibco, г. Карлсбад, штат Калифорния) на колбу. После открепления клеток от стенок колбы активность трипсина нейтрализовали добавлением 8 миллилитров ростовой среды. Клетки перенесли в пробирку для центрифугирования объемом 15 миллилитров и центрифугировали при 150×g в течение 5 минут. Супернатант удалили и в каждую пробирку добавили по 1 миллилитру ростовой среды для повторного суспендирования клеток. Количество клеток оценивали с помощью гемоцитометра.
Анализ ИФА. Количество IL-8, секретированного клетками в бессывороточную среду, анализировали с использованием наборов для анализа ИФА (R&D Systems, г. Миннеаполис, штат Миннесота). Все анализы проводили в соответствии с инструкциями, предоставленными производителем.
Выделение общей РНК. РНК экстрагировали из конфлюентных культур клеток, полученных из пуповины и плаценты, и фибробластов или, для оценки экспрессии IL-8, из клеток, обработанных, как описано выше. Клетки лизировали 350 микролитрами буферного раствора RLT, содержащего бета-меркаптоэтанол (Sigma, г. Сент-Луис, штат Миссури), в соответствии с инструкциями производителя (набор RNeasy Mini Kit; Qiagen, г. Валенсия, штат Калифорния). РНК экстрагировали в соответствии с инструкциями производителя (набор RNeasy Mini Kit; Qiagen, г. Валенсия, штат Калифорния) и обрабатывали ДНКазой (2,7 ед./образец) (Sigma, г. Сент-Луис, штат Миссури). РНК элюировали 50 микролитрами обработанной DEPC воды и хранили при температуре -80°C.
Обратная транскрипция. РНК также экстрагировали из плаценты и пуповины человека. Ткань (30 миллиграмм) суспендировали в 700 микролитрах буферного раствора RLT, содержащего 2-меркаптоэтанол. Образцы механически гомогенизировали и проводили экстракцию РНК в соответствии с рекомендациями производителя. РНК экстрагировали 50 микролитрами обработанной DEPC воды и хранили при температуре -80°C. Обратную транскрипцию РНК проводили с использованием случайных гексамеров, применяя реагенты для обратной транскрипции TaqMan (Applied Biosystems, г. Фостер Сити, штат Калифорния) при 25°C в течение 10 минут, 37°C в течение 60 минут и 95°C в течение 10 минут. Образцы хранили при -20°C.
Гены, идентифицированные кДНК-микрочипом как уникально регулируемые в клетках из послеродового материала (характерные гены - включая рецептор окисленных ЛПНП, интерлейкин-8, реннин и ретикулон), дополнительно исследовали с использованием ПЦР в реальном времени и стандартной ПЦР.
ПЦР в реальном времени. ПЦР проводили на образцах кДНК с использованием продуктов для экспрессии генов Assays-on-Demand™: рецептор окисленных ЛПНП (Hs00234028); реннин (Hs00166915); ретикулон (Hs00382515); CXCL3 (хемокиновый лиганд 3) (Hs00171061); GCP-2 (Hs00605742); IL-8 (Hs00174103); и GAPDH (Applied Biosystems, г. Фостер Сити, штат Калифорния) смешивали с кДНК и универсальным мастер-миксом для ПЦР TaqMan в соответствии с инструкциями производителя (Applied Biosystems, г. Фостер Сити, штат Калифорния), используя секвенатор 7000 с программным обеспечением ABI Prism 7000 SDS (Applied Biosystems, г. Фостер Сити, штат Калифорния). Использовали следующие условия термоциклирования: сначала 50°C в течение 2 мин. и 95°C в течение 10 мин., затем 40 циклов по 95°C в течение 15 с и 60°C в течение 1 мин. Данные ПЦР анализировали в соответствии с рекомендациями производителя (Бюллетень пользователя № 2 компании Applied Biosystems для секвенатора ABI Prism 7700 Sequence Detection System).
Стандартная ПЦР. Стандартную ПЦР проводили на секвенаторе ABI PRISM 7700 (Perkin Elmer Applied Biosystems, г. Бостон, штат Массачусетс, США) для подтверждения результатов ПЦР в реальном времени. ПЦР проводили с использованием 2 микролитров раствора кДНК, реакционного буферного раствора 1× AmpliTaq Gold universal mix PCR reaction buffer (Applied Biosystems, г. Фостер Сити, штат Калифорния) и первичного денатурирования при температуре 94°C в течение 5 минут. Амплификацию оптимизировали для каждого набора праймеров. Использовали следующие протоколы амплификации: для IL-8, CXCL3 (хемокинового лиганда 3) и ретикулона - 94°C в течение 15 секунд, затем 30 циклов по 55°C в течение 15 секунд и 72°C в течение 30 секунд; для реннина - 94°C в течение 15 секунд, затем 38 циклов по 53°C в течение 15 секунд и 72°C в течение 30 секунд; для рецептора окисленных ЛПНП и GAPDH - 94°C в течение 15 секунд, затем 33 цикла по 55°C в течение 15 секунд и 72°C в течение 30 секунд. Использованные для амплификации праймеры перечислены в таблице 1. Концентрация праймеров в конечной реакционной смеси ПЦР составляла 1 мкМ за исключением GAPDH, для которого она составляла 0,5 мкМ. Для GAPDH использовали те же праймеры, что и для ПЦР в реальном времени, за исключением того, что в конечную реакционную смесь ПЦР не добавляли прилагаемый производителем зонд TaqMan. Образцы прогнали на 2% (в/об) агарозном геле и окрасили бромидом этидия (Sigma, г. Сент-Луис, штат Миссури). Изображения получали с использованием пленки 667 Universal Twinpack (VWR International, г. Саут-Плэйнфилд, штат Нью-Джерси) и фокусной камеры Polaroid (VWR International, г. Саут-Плэйнфилд, штат Нью-Джерси).
Иммунофлуоресцентный анализ. Полученные клетки фиксировали в холодном 4% (в/об) параформальдегиде (Sigma-Aldrich, г. Сент-Луис, штат Миссури) на 10 минут при комнатной температуре. Использовали по одному изоляту для каждых из клеток, полученных из пуповины, и клеток, полученных из плаценты, пассажа 0 (P0) (непосредственно после выделения) и пассажа 11 (P11) (два изолята клеток, полученных из плаценты, два изолята клеток, полученных из пуповины) и для фибробластов (P11). Иммуноцитохимический анализ проводили с использованием антител к следующим эпитопам: виментин (1:500, Sigma, г. Сент-Луис, штат Миссури), дезмин (1:150; Sigma - выработанные на кролика; или 1:300; Chemicon, г. Темекула, штат Калифорния - выработанные на мышь), альфа-актин гладких мышц (SMA; 1:400; Sigma), цитокератин 18 (CK18; 1:400; Sigma), фактор фон Виллебранда (vWF; 1:200; Sigma) и CD34 (CD34 человека, класс III; 1:100; DAKOCytomation, г. Карпинтерия, штат Калифорния). Кроме того, для клеток из послеродового материала пассажа 11 проверили следующие маркеры: анти-человеческий GRO альфа - PE (1:100; Becton Dickinson, г. Франклин Лейкс, штат Нью-Джерси), анти-человеческий GCP-2 (1:100; Santa Cruz Biotech, г. Санта-Крус, штат Калифорния), рецептор 1 окисленных ЛПНП против человека (ox-LDL R1; 1:100; Santa Cruz Biotech) и анти-человеческий NOGA-A (1:100; Santa Cruz, Biotech).
Культуры промывали фосфатно-солевым буфером (ФСБ) и обрабатывали белковым блокирующим раствором, содержащим ФСБ, 4% (об/об) козьей сыворотки (Chemicon, г. Темекула, штат Калифорния) и 0,3% (об/об) Тритона (Triton X-100; Sigma, г. Сент-Луис, штат Миссури) в течение 30 минут для доступа к внутриклеточным антигенам. Когда рассматриваемый эпитоп размещался на клеточной поверхности (CD34, ox-LDL R1), Тритон X-100 исключали на всех стадиях процедуры, чтобы не допустить потери эпитопа. Кроме того, когда первичное антитело было выработано на козу (GCP-2, ox-LDL R1, NOGO-A), в течение всей процедуры вместо козьей сыворотки использовали 3% (об/об) ослиную сыворотку. Затем культуры в течение 1 часа обрабатывали при комнатной температуре разбавленными в блокирующем растворе первичными антителами. Растворы первичных антител удаляли и промывали культуры ФСБ перед добавлением растворов вторичных антител (1 час при комнатной температуре), содержащих блокирующий раствор вместе с конъюгатом козьего антимышиного IgG - Texas Red (1:250; Molecular Probes, г. Юджин, штат Орегон) и/или конъюгатом козьего анти-кроличьего IgG - Alexa 488 (1:250; Molecular Probes) или конъюгатом ослиного анти-козьего IgG - FITC (1:150, Santa Cruz Biotech). Затем культуры промывали и обрабатывали 10-микромолярным раствором DAPI (Molecular Probes) в течение 10 минут для визуализации клеточных ядер.
После иммунного окрашивания получали флуоресцентные изображения с использованием соответствующего фильтра для флуоресценции на инвертированном эпи-флуоресцентном микроскопе (Olympus, г. Мелвилл, штат Нью-Йорк). Во всех случаях положительное окрашивание соответствовало сигналу флуоресценции выше сигнала для контрольного окрашивания, при котором выполняли всю описанную выше процедуру за исключением обработки раствором первичных антител. Представительные изображения получали с использованием цветной цифровой видеокамеры и программного обеспечения ImagePro (Media Cybernetics, г. Карлсбад, штат Калифорния). Для образцов с тройным окрашиванием каждое изображение получали с использованием только одного эмиссионного фильтра. Затем получили послойные монтажи с использованием программного обеспечения Adobe Photoshop (Adobe, г. Сан-Хосе, штат Калифорния).
Подготовка клеток для анализа FACS. Закрепившиеся клетки в колбах промывали фосфатно-солевым буфером (ФСБ) (Gibco, г. Карлсбад, штат Калифорния) и отделяли с помощью трипсина/ЭДТА (Gibco, г. Карлсбад, штат Калифорния). Клетки собирали, центрифугировали и повторно суспендировали в 3% (об/об) растворе FBS в ФСБ при концентрации клеток 1×107 на миллилитр. Отбирали аликвоты по сто микролитров в конические пробирки. Клетки, окрашиваемые на внутриклеточные антигены, пермеабилизировали буферным раствором Perm/Wash (BD Pharmingen, г. Сан-Диего, штат Калифорния). Антитела добавляли к аликвотам согласно рекомендациям производителя и инкубировали клетки в темноте в течение 30 минут при 4°C. После инкубации клетки промывали ФСБ и центрифугировали для удаления избытка антител. Клетки, требующие вторичных антител, повторно суспендировали в 100 микролитрах 3% раствора FBS. Вторичные антитела добавляли к аликвотам согласно рекомендациям производителя и инкубировали клетки в темноте в течение 30 минут при 4°C. После инкубации клетки промывали ФСБ и центрифугировали для удаления избытка вторичных антител. Промытые клетки повторно суспендировали в 0,5 миллилитра ФСБ и анализировали методом проточной цитометрии. Применяли следующие антитела: рецептор 1 окисленных ЛПНП (sc-5813; Santa Cruz Biotech.), GROa (555042; BD Pharmingen, г. Бедфорд, штат Массачусетс), мышиный IgG1 каппа, (P-4685 и M-5284; Sigma), ослиный к козьему IgG (sc-3743; Santa Cruz Biotech.). Анализ методом проточный цитометрии проводили на анализаторе FACScalibur (Becton Dickinson, г. Сан-Хосе, штат Калифорния).
Результаты
Результаты ПЦР в реальном времени от выбранных «характерных» генов, проведенного на кДНК из клеток, полученных из плаценты человека, взрослых и неонатальных фибробластов и мезенхимальных стволовых клеток (MSC), показывают, что как рецептор окисленных ЛПНП, так и реннин в клетках, полученных из плаценты, экспрессировались на более высоком уровне по сравнению с другими клетками. Данные, полученные в результате ПЦР в реальном времени, проанализировали методом ΔΔCT и отразили на логарифмической шкале. Уровни экспрессии ретикулона и рецептора окисленных ЛПНП в клетках, полученных из пуповины, оказались выше по сравнению с другими клетками. Никакие существенные отличия в уровнях экспрессии CXCL3 (хемокинового лиганда 3) и GCP-2 между клетками, полученными из плаценты, и контролями не были обнаружены. Результаты ПЦР в реальном времени подтвердили стандартной ПЦР. Данные результаты дополнительно подтвердили секвенированием продуктов ПЦР. Никакие существенные отличия в уровнях экспрессии CXCL3 (хемокинового лиганда 3) между клетками, полученными из плаценты, и контролями при использовании перечисленных выше праймеров на CXCL3 стандартной ПЦР не были обнаружены.
Уровень продукции цитокина, IL-8, в послеродовом материале был повышен как в клетках, культивированных в ростовой среде, так и в клетках, культивированных в бессывороточной среде. Все данные ПЦР в реальном времени подтвердили стандартной ПЦР и секвенированием продуктов ПЦР.
При исследовании супернатантов от клеток, выращенных в бессывороточной среде, на наличие IL-8 самые высокие уровни обнаружили в средах, полученных от клеток пуповины и некоторых изолятов клеток плаценты (таблица 10-1). В среде, полученной от фибробластов кожи человека, IL-8 не обнаружено.
Клетки, полученные из плаценты, также исследовали на выработку рецептора окисленных ЛПНП, GCP-2 и GRO-альфа с использованием анализа FACS. Протестированные клетки оказались положительными на GCP-2. Рецептор окисленных ЛПНП и GRO обнаружить данным способом не удалось.
Клетки, полученные из плаценты, также тестировали на выработку выбранных белков с использованием иммуноцитохимического анализа. Немедленно после выделения (пассаж 0) клетки, полученные из плаценты человека, зафиксировали 4% раствором параформальдегида и обработали антителами на шесть белков: фактор фон Виллебранда, CD34, цитокератин 18, десмин, альфа-актин гладких мышц и виментин. Клетки положительно окрашивались как на альфа-актин гладких мышц, так и на виментин. Данная тенденция сохранялась вплоть до пассажа 11. Лишь небольшое количество клеток (<5%) на пассаже 0 окрашивалось положительно на цитокератин 18.
Клетки пассажа 0, полученные из пуповины человека, проверили на выработку выбранных белков с использованием иммуноцитохимического анализа. Немедленно после выделения (пассаж 0) клетки зафиксировали 4% раствором параформальдегида и обработали антителами на шесть белков: фактор фон Виллебранда, CD34, цитокератин 18, десмин, альфа-актин гладких мышц и виментин. Клетки, полученные из пуповины, оказались положительными на альфа-актин гладких мышц и виментин, причем данная тенденция окрашивания сохранялась вплоть до пассажа 11.
Обзор. Согласованность между уровнями экспрессии генов, измеренными с использованием микрочипа и ПЦР (как в реальном времени, так и стандартной), установлена для четырех генов: рецептор 1 окисленных ЛПНП, реннин, ретикулон и IL-8. Экспрессия данных генов дифференциально регулировалась на уровне мРНК в клетках, полученных из ткани пуповины, и клетках, полученных из плаценты, при этом IL-8 также дифференциально регулировался на уровне белка. Присутствие рецептора окисленных ЛПНП на уровне белка в клетках, полученных из плаценты, с использованием анализа FACS не было обнаружено. Дифференциальная экспрессия GCP-2 и CXCL3 (хемокинового лиганда 3) в клетках, полученных из плаценты, не была подтверждена на уровне мРНК, однако GCP-2 обнаружили на уровне белка с использованием анализа FACS. Хотя данный результат не отражен в данных, первоначально полученных в эксперименте с микрочипом, это может быть вызвано различиями в чувствительности данных методик.
Немедленно после выделения (пассаж 0) клетки, полученные из плаценты человека, положительно окрашивались как на альфа-актин гладких мышц, так и на виментин. Данную тенденцию также наблюдали для клеток пассажа 11. Данные результаты указывают, что экспрессия виментина и альфа-актина гладких мышц может сохраняться в клетках при пассировании, в ростовой среде и в условиях, используемых в данных процедурах. Клетки пассажа 0, полученные из пуповины человека, проверили на экспрессию альфа-актина гладких мышц и виментина, и они оказались положительными на оба белка. Данная тенденция по окраске сохранялась вплоть до пассажа 11.
ПРИМЕР 11
Иммунологическая оценка in vitro клеток, полученных из ткани пуповины, и клеток, полученных из плаценты
Проводили оценку in vitro иммунологических характеристик клеток UTC в попытке прогнозировать возможный иммунологический ответ на данные клетки при их трансплантации in vivo. Клетки UTC оценивали методом проточной цитометрии на присутствие в них HLA-DR, HLA-DP, HLA-DQ, CD80, CD86 и B7-H2. Данные белки экспрессируются антигенпредставляющими клетками (АПК) и необходимы для прямой стимуляции непримированных CD4+ Т-клеток (Abbas & Lichtman, Cellular and Molecular Immunology, 5-е изд. (2003 г.), г. Саундерс, штат Филадельфия, стр. 171). Клеточные линии также анализировали методом проточной цитометрии на экспрессию HLA-G (Abbas & Lichtman, 2003 г., supra), CD 178 (Coumans, et al., (1999 г.) Journal of Immunological Methods 224, 185-196) и PD-L2 (Abbas & Lichtman, 2003 г., supra; Brown, et. al. (2003 г.) The Journal of Immunology 170, 1257-1266). Считается, что экспрессия данных белков клетками, находящимися в плацентарных тканях, опосредует иммуно-привилегированный статус плацентарных тканей in utero. Для прогнозирования степени, в которой полученные из плаценты и пуповины клеточные линии вызывают иммунный ответ in vivo, клеточные линии тестировали в однонаправленной реакции смешанной культуры лимфоцитов (СКЛ-реакции).
Материалы и способы
Клеточная культура. Клетки культивировали до конфлюентности в ростовой среде, содержащей пенициллин/стрептомицин в колбах T75 (Corning, г. Корнинг, штат Нью-Йорк), покрытых 2% раствором желатина(Sigma, г. Сент-Луис, штат Миссури).
Окрашивание антителами. Клетки промывали фосфатно-солевым буфером (ФСБ) (Gibco, г. Карлсбад, штат Калифорния) и отделяли с помощью трипсина/ЭДТА (Gibco, г. Карлсбад, штат Калифорния). Клетки собирали, центрифугировали и повторно суспендировали в 3% (об/об) растворе FBS в ФСБ при концентрации клеток 1x107 на миллилитр. Антитело (таблица 11-1) добавляли к ста микролитрам клеточной суспензии в соответствии с инструкциями производителя и инкубировали в темноте в течение 30 минут при 4°C. После инкубации клетки промывали ФСБ и центрифугировали для удаления несвязавшихся антител. Клетки повторно суспендировали в пятистах микролитрах ФСБ и анализировали методом проточной цитометрии на анализаторе FACS Calibur (Becton Dickinson, г. Сан-Хосе, штат Калифорния).
Реакция смешанной культуры лимфоцитов. Криоконсервированные флаконы с клетками, полученными из пуповины, пассажа 10, меченными как клеточная линия A, и клетками, полученными из плаценты, пассажа 11, меченными как клеточная линия B, отправили на сухом льду в лабораторию CTBR (г. Сенвиль, провинция Квебек) для проведения реакции смешанной культуры лимфоцитов согласно стандартному протоколу CTBR SOP № CAC-031. Мононуклеарные клетки периферической крови (МКПК) собрали у множества добровольных доноров мужского и женского пола. Аллогенные МКПК стимулятора (донора), аутогенные МКПК и клетки полученных из пуповины и плаценты клеточных линий обработали митомицином C. Аутогенные и обработанные митомицином C стимуляторные клетки добавили к МКПК респондера (получателя) и культивировали в течение 4 суток. После инкубации в каждый образец добавили [3H]-тимидин и культивировали смесь в течение 18 часов. Затем клетки собрали, экстрагировали радиоактивно меченную ДНК и измерили потребление [3H]-тимидина с использованием сцинтилляционного счетчика.
Коэффициент стимулирования для клеток аллогенного донора (SIAD) рассчитали как сумму средней пролиферации клеток получателя и обработанных митомицином C клеток аллогенного донора, деленную на базовую пролиферацию клеток получателя. Коэффициент стимулирования для клеток UTC рассчитали как сумму средней пролиферации клеток получателя и обработанной митомицином C клеточной линии из послеродового материала, деленную на базовую пролиферацию клеток получателя.
Результаты
Реакция смешанной культуры лимфоцитов. Клетки, полученные из плаценты. Проанализировали семь человек, добровольных доноров крови, для выявления одного аллогенного донора, который бы показал устойчивый пролиферативный ответ в реакции смешанной культуры лимфоцитов с остальными шестью донорами крови. Данного донора выбрали в качестве аллогенного донора, положительного контроля. Остальных шесть доноров выбрали как получателей. Клетки аллогенного донора, положительного контроля, и клетки полученных из плаценты клеточных линий обработали митомицином C и культивировали в реакции смешанной культуры лимфоцитов с индивидуальными клетками каждого из шести аллогенных получателей. Реакции повторяли три раза, используя два планшета для культивирования клеток с тремя получателями на планшет (таблица 11-2). Средний коэффициент стимулирования для тестируемых клеток находился в диапазоне от 1,3 (планшет 2) до 3 (планшет 1), а для клеток аллогенного донора, положительного контроля, - в диапазоне от 46,25 (планшет 2) до 279 (планшет 1) (таблица 11-3).
Реакция смешанной культуры лимфоцитов. Клетки, полученные из пуповины. Проанализировали шесть человек, добровольных доноров крови, для выявления одного аллогенного донора, который бы показал устойчивый пролиферативный ответ в реакции смешанной культуры лимфоцитов с остальными пятью донорами крови. Данного донора выбрали в качестве аллогенного донора, положительного контроля. Остальных пять доноров выбрали как получателей. Клетки аллогенного донора, положительного контроля, и клетки полученных из пуповины клеточных линий обработали митомицином C и культивировали в реакции смешанной культуры лимфоцитов с индивидуальными клетками каждого из пяти аллогенных получателей. Реакции повторяли три раза, используя два планшета для культивирования клеток с тремя получателями на планшет (таблица 11-4). Средний коэффициент стимулирования для тестируемых клеток находился в диапазоне от 6,5 (планшет 1) до 9 (планшет 2), а для клеток аллогенного донора, положительного контроля, - диапазоне от 42,75 (планшет 1) до 70 (планшет 2) (таблица 11-5).
Маркеры антигенпредставляющих клеток. Клетки, полученные из плаценты. Гистограммы проанализированных методом проточной цитометрии клеток, полученных из плаценты, показали отрицательную экспрессию HLA-DR, DP, DQ, CD80, CD86 и B7-H2, на что указывают значения флуоресценции, согласующиеся с используемым в качестве контроля IgG, что указывает на отсутствие у клеток плацентарных клеточных линий на клеточной поверхности молекул, требуемых для прямой стимуляции CD4+ T-клеток.
Иммуномодулирующие маркеры. Клетки, полученные из плаценты. Гистограммы проанализированных методом проточной цитометрии клеток, полученных из плаценты, показали положительную экспрессию PD-L2, на что указывает повышение значения флуоресценции по сравнению с используемым в качестве контроля IgG, и отрицательную экспрессию CD178 и HLA-G, на что указывают значения флуоресценции, согласующиеся с используемым в качестве контроля IgG.
Маркеры антигенпредставляющих клеток. Клетки, полученные из пуповины. Гистограммы проанализированных методом проточной цитометрии клеток, полученных из пуповины, показали отрицательную экспрессию HLA-DR, DP, DQ, CD80, CD86 и B7-H2, на что указывают значения флуоресценции, согласующиеся с используемым в качестве контроля IgG, что указывает на отсутствие у клеток пуповинных линий на клеточной поверхности молекул, требуемых для прямой стимуляции CD4+ T-клеток.
Иммуномодулирующие клеточные маркеры. Клетки, полученные из пуповины. Гистограммы проанализированных методом проточной цитометрии клеток, полученных из пуповины, показали положительную экспрессию PD-L2, на что указывает повышение значения флуоресценции по сравнению с используемым в качестве контроля IgG, и отрицательную экспрессию CD178 и HLA-G, на что указывают значения флуоресценции, согласующиеся с используемым в качестве контроля IgG.
Обзор. В реакциях смешанной культуры лимфоцитов, проведенных с клетками полученных из плаценты клеточных линий, средний коэффициент стимулирования для тестируемых клеток находился в диапазоне от 1,3 до 3, а для клеток аллогенного донора, положительного контроля, - в диапазоне от 46,25 до 279. В реакциях смешанной культуры лимфоцитов, проведенных с клетками полученных из пуповины клеточных линий, средний коэффициент стимулирования для тестируемых клеток находился в диапазоне от 6,5 до 9, а для клеток аллогенного донора, положительного контроля, - в диапазоне от 42,75 до 70. По результатам измерения методом проточной цитометрии клетки полученных из плаценты и пуповины клеточных линий оказались отрицательными в отношении экспрессии стимуляторных белков HLA-DR, HLA-DP, HLA-DQ, CD80, CD86 и B7-H2. По результатам измерения методом проточной цитометрии клетки полученных из плаценты и пуповины клеточных линий оказались отрицательными в отношении экспрессии иммуномодуляторных белков HLA-G и CD178 и положительными в отношении экспрессии PD-L2. МКПК аллогенного донора содержат антигенпредставляющие клетки с экспрессией HLA-DR, DQ, CD8, CD86 и B7-H2, позволяя тем самым стимулировать непримированные CD4+ T-клетки. Отсутствие на клеточной поверхности полученных из плаценты и пуповины клеточных линий антиген-представляющих молекул, требуемых для прямой стимуляции непримированных CD4+ T-клеток, и присутствие иммуномодулирующего белка PD-L2 может объяснить низкий коэффициент стимулирования, показанный данными клетками в СКЛ-реакции по сравнению с аллогенными контролями.
ПРИМЕР 12
Секретирование трофических факторов клетками, полученными из ткани пуповины, и клетками, полученными из плаценты
Измеряли секретирование выбранных трофических факторов клетками, полученными из пуповины, и клетками, полученными из плаценты. Выбранные для детектирования факторы включали в себя: (1) факторы с известной ангиогенной активностью, такие как фактор роста гепатоцитов (HGF) (Rosen et al. (1997 г.) Ciba Found. Symp. 212: 215-26), моноцитарный хемотаксический фактор 1 (MCP-1) (Salcedo et al. (2000 г.) Blood 96; 34-40), интерлейкин-8 (IL-8) (Li et al. (2003 г.) J. Immunol. 170: 3369-76), фактор роста кератиноцитов (KGF), основной фактор роста фибробластов (bFGF), фактор роста эндотелия сосудов (VEGF) (Hughes et al. (2004 г.) Ann. Thorac. Surg. 77: 812-8), матриксная металлопротеиназа 1 (TIMP1), ангиопоэтин 2 (ANG2), тромбоцитарный фактор роста (PDGF-bb), тромбопоэтин (TPO), связывающийся с гепарином эпидермальный фактор роста (HB-EGF), стромальный фактор 1 альфа (SDF-1альфа); (2) факторы с известной нейротрофической/нейропротекторной активностью, такие как нейротрофический фактор головного мозга (BDNF) (Cheng et al. (2003 г.) Dev. Biol. 258; 319-33), интерлейкин-6 (IL-6), гранулоцитарный хемотаксический белок-2 (GCP-2), трансформирующий фактор роста бета2 (TGF-бета2); и (3) факторы с известной хемокиновой активностью, такие как макрофагальный белок воспаления 1альфа (MIP1a), макрофагальный белок воспаления 1бета (MIP1b), моноцитарный хемоатрактантный белок-1 (MCP-1), Rantes (хемокин, экспрессируемый и секретируемый нормальными T-клетками при активации), I309, регулируемый активацией и тимусом хемокин (TARC), эотаксин, хемокин макрофагов (MDC), IL-8.
Способы и материалы
Клеточная культура. Клетки, полученные из плаценты и пуповины, а также фибробласты человека, полученные из неонатальной крайней плоти человека, культивировали в ростовой среде с добавлением пенициллина/стрептомицина в покрытых желатином колбах T75. На пассаже 11 клетки криоконсервировали и замораживали в жидком азоте. После размораживания в клетки добавляли ростовую среду, переносили их в пробирку для центрифугирования объемом 15 миллилитров и центрифугировали при 150×g в течение 5 минут. Супернатант удаляли. Клеточный осадок повторно суспендировали в 4 миллилитрах ростовой среды и подсчитывали. Клетки высевали в количестве 375000 клеток на колбу с площадью поверхности 75 см2, содержащую 15 миллилитров ростовой среды, и культивировали в течение 24 часов. Среду заменили на бессывороточную среду (среда DMEM с низким содержанием глюкозы (Gibco), 0,1% (в/об) бычьего сывороточного альбумина (Sigma), пенициллин/стрептомицин (Gibco)) на 8 часов. В конце инкубации кондиционированную бессывороточную среду собрали центрифугированием при 14000×g в течение 5 минут и хранили при температуре -20°C. Для оценки количества клеток в каждой колбе клетки промыли ФСБ и отделили с использованием 2 миллилитров трипсина/ЭДТА. Активность трипсина ингибировали добавлением 8 миллилитров ростовой среды. Клетки центрифугировали при 150×g в течение 5 минут. Супернатант удалили и клетки повторно суспендировали в 1 миллилитре ростовой среды. Количество клеток оценивали с помощью гемоцитометра.
Анализ ИФА. Клетки выращивали при 37°C в присутствии 5% диоксида углерода и атмосферного кислорода. Клетки, полученные из плаценты (партия 101503), также выращивали в атмосфере с содержанием кислорода 5% или в среде с добавлением бета-меркаптоэтанола (BME). Количество MCP-1, IL-6, VEGF, SDF-1альфа, GCP-2, IL-8 и TGF-бета2, вырабатываемое каждым образцом клеток, измеряли методом анализа ИФА (R&D Systems, г. Миннеаполис, штат Миннесота). Все тесты проводили в соответствии с инструкциями производителя.
Мультиплексный анализ ИФА Searchlight™. Хемокины (MIP1a, MIP1b, MCP-1, Rantes, I309, TARC, эотаксин, MDC, IL8), BDNF и ангиогенные факторы (HGF, KGF, bFGF, VEGF, TIMP1, ANG2, PDGF-bb, TPO, HB-EGF) измеряли с использованием протеомных массивов SearchLight Proteome Arrays (Pierce Biotechnology Inc.). Протеомные массивы представляют собой мультиплексные иммуноферментные сэндвич-системы для количественного измерения от 2 до 16 белков на лунку. Массивы получают, нанося матрицу 2×2, 3×3 или 4×4 от 4 до 16 разных антител захвата в каждую лунку 96-луночного планшета. После проведения процедуры сэндвич-ИФА снимают изображение всего планшета для получения сигнала хемилюминесценции, генерируемого в каждой точке матрицы в каждой лунке планшета. Количество сигнала, генерируемого в каждой точке матрицы, пропорционально количеству целевого белка в исходном стандартном или анализируемом образце.
Результаты
Анализ ИФА. MCP-1 и IL-6 секретировались клетками, полученными из плаценты, клетками, полученными пуповины, и фибробластами кожи (таблица 12-1). SDF-1альфа секретировался клетками, полученными из плаценты и культивированными в атмосфере 5% O2, и фибробластами. GCP-2 и IL-8 секретировались клетками, полученными из пуповины, и клетками, полученными из плаценты, культивированными в присутствии BME или в атмосфере 5% O2. GCP-2 также секретировался фибробластами человека. TGF-бета2 не был определен методом анализа ИФА.
Мультиплексный анализ ИФА Searchlight™. TIMP1, TPO, KGF, HGF, FGF, HBEGF, BDNF, MIP1b, MCP1, RANTES, I309, TARC, MDC и IL-8 секретировались клетками, полученными из пуповины (таблицы 12-2 и 12-3). TIMP1, TPO, KGF, HGF, HBEGF, BDNF, MIP1a, MCP-1, RANTES, TARC, эотаксин и IL-8 секретировались клетками, полученными из плаценты (таблицы 12-2 и 12-3). Ang2, VEGF и PDGF-bb не были обнаружены.
Обзор. Клетки, полученные из пуповины, и клетки, полученные из плаценты, секретировали ряд трофических факторов. Некоторые из данных трофических факторов, такие как HGF, bFGF, MCP-1 и IL-8, играют важные роли в ангиогенезе. Другие трофические факторы, такие как BDNF и IL-6, играют важные роли в регенерации нервной ткани.
ПРИМЕР 13
Кратковременное нейронное дифференцирование клеток, полученных из ткани пуповины, и клеток, полученных из плаценты
Исследовали способность клеток, полученных из плаценты, и клеток, полученных из пуповины, к дифференцированию в клетки нейронной линии дифференцирования.
Материалы и способы
Выделение и размножение клеток, полученных из плаценты, и клеток, полученных из пуповины. Клетки из тканей плаценты и пуповины выделяли и размножали, как описано в примере 1.
Модифицированный протокол Вудбери-Блэка. (А) Данный анализ адаптирован с анализа, изначально проводимого для тестирования потенциала нейронного индуцирования стромальных клеток костного мозга (1). Клетки, полученные из пуповины (022803), P4, и клетки, полученные из плаценты (042203), P3, разморозили и культуры размножали в ростовых средах с плотностью посева 5000 клеток/см2 до достижения субконфлюентности (75%). Затем клетки трипсинизировали и высевали в количестве 6000 клеток на лунку предметного стекла Titretek II (VWR International, г. Бристоль, штат Коннектикут). В качестве контролей в тех же условиях высевали мезенхимальные стволовые клетки (P3; 1F2155; Cambrex, г. Уокерсвилл, штат Мэриленд), остеобласты (P5; CC2538; Cambrex), адипозные клетки (Artecel, US6555374 B1) (P6; донор 2) и неонатальные фибробласты кожи человека (P6; CC2509; Cambrex).
Все клетки сначала размножались в течение 4 суток в среде DMEM/F12 (Invitrogen, г. Карлсбад, штат Калифорния), содержащей 15% (об/об) эмбриональной бычьей сыворотки (FBS; Hyclone, г. Логан, штат Юта), основной фактор роста фибробластов (bFGF; 20 нанограмм/миллилитр; Peprotech, г. Роки Хилл, штат Нью-Джерси), эпидермальный фактор роста (EGF; 20 нанограмм/миллилитр; Peprotech) и пенициллин/стрептомицин (Invitrogen). Через четверо суток клетки промыли фосфатно-солевым буфером (ФСБ; Invitrogen) и затем культивировали в среде DMEM/F12+20% (об/об) FBS+пенициллин/стрептомицин в течение 24 часов. Через 24 часа клетки промыли ФСБ. Затем клетки культивировали в течение 1-6 часов в индуцирующей среде, представлявшей собой среду DMEM/F12 (бессывороточную), содержащую 200 мМ бутилированного гидроксианизола, 10 мкМ хлорида калия, 5 миллиграмм/миллилитр инсулина, 10 мкМ форсколина, 4 мкМ вальпроевой кислоты и 2 мкМ гидрокортизона (все производства Sigma, г. Сент-Луис, штат Миссури). Затем клетки зафиксировали в 100% ледяном метаноле и провели иммуноцитохимический анализ (смотрите способы, представленные ниже) для оценки уровня экспрессии белка нестина человека.
(B) Полученные из тканей клетки (пуповины (022803) P11; плаценты (042203) P11) и взрослые фибробласты кожи человека (1F1853, P11) разморозили и культуры размножали в ростовой среде с плотностью посева 5000 клеток/см2 до достижения субконфлюентности (75%). Затем клетки трипсинизировали и высевали с аналогичной плотностью, как в (A), но на (1) 24-луночные обработанные тканевой культурой планшеты (TCP, Falcon brand, VWR International), (2) лунки планшетов TCP, обработанных 2% (в/об) раствором желатина в течение 1 часа при комнатной температуре, или (3) лунки планшетов TCP, обработанных раствором 20 мкг/миллилитр мышиного ламинина (адсорбирован в течение по меньшей мере 2 часов при температуре 37°C; Invitrogen).
Точно так же, как в (А), сначала клетки размножали и среды заменяли в указанные ранее периоды времени. Один набор культур зафиксировали, как и ранее, через 5 суток и шесть часов, в данном случае с использованием ледяного раствора 4% (в/об) параформальдегида (Sigma) на 10 минут при комнатной температуре. Во втором наборе культур среду удалили и заменили на среду для размножения клеток-предшественников нейронов (NPE), представляющую собой среду Neurobasal-A (Invitrogen), содержащую B27 (добавка B27; Invitrogen), L-глутамин (4 мМ) и пенициллин/стрептомицин (Invitrogen). В среду NPE дополнительно добавили ретиноевую кислоту (RA; 1 мкМ; Sigma). Данную среду удалили через 4 суток, зафиксировали культуры ледяным раствором 4% (в/об) параформальдегида (Sigma) на 10 минут при комнатной температуре и провели окрашивание на уровни экспрессии белков нестин, GFAP и TuJ1 (смотрите таблицу 13-1).
Протокол двухстадийного дифференцирования. Клетки, полученные из пуповины (042203) P11, плаценты (022803) P11, взрослые фибробласты кожи человека (P11; 1F1853; Cambrex) разморозили и культуры размножали в ростовой среде с плотностью посева 5000 клеток/см2 до достижения субконфлюентности (75%). Затем клетки трипсинизировали и высевали с плотностью 2000 клеток/см2, но на 24-луночные планшеты, покрытые ламинином (BD Biosciences, г. Франклин Лейкс, штат Нью-Джерси) в присутствии сред NPE с добавлением bFGF (20 нанограмм/миллилитр; Peprotech, г. Роки Хилл, штат Нью-Джерси) и EGF (20 нанограмм/миллилитр; Peprotech) [целая композиция сред далее будет называться NPE+F+E]. Одновременно на 24-луночные планшеты, покрытые ламинином, в среды NPE+F+E также высевали выделенные из гиппокампа взрослые клетки-предшественники нейронов крысы (P4; (062603)). Все культуры поддерживали в таких условиях в течение 6 суток (за это время клетки подпитывали один раз), по истечении которых среды заменили на среды с условиями дифференцирования, перечисленными в таблице N1-2, и дополнительно культивировали клетки в течение 7 суток. Культуры фиксировали ледяным раствором 4% (в/об) параформальдегида (Sigma) на 10 минут при комнатной температуре и провели окрашивание на уровни экспрессии белков нестин крысы или человека, GFAP и TuJ1.
Протокол с использованием множества факторов роста. Клетки, полученные из пуповины (P11 (042203)), разморозили и культуры размножали в ростовой среде с плотностью посева 5000 клеток/см2 до достижения субконфлюентности (75%). Затем клетки трипсинизировали и высевали с плотностью 2000 клеток/см2 на 24-луночные покрытые ламинином планшеты (BD Biosciences) в присутствии среды NPE+F (20 нанограмм/миллилитр)+E (20 нанограмм/миллилитр). Кроме того, в некоторых лунках присутствовала среда NPE+F+E+2% FBS или 10% FBS. После четырех суток условий «предифференцирования» все среды удаляли и образцы переводили на среду NPE с добавлением sonic hedgehog (SHH; 200 нанограмм/миллилитр; Sigma, г. Сент-Луис, штат Миссури), FGF8 (100 нанограмм/миллилитр; Peprotech), BDNF (40 нанограмм/миллилитр; Sigma), GDNF (20 нанограмм/миллилитр; Sigma) и ретиноевой кислоты (1 мкМ; Sigma). Через семь суток после замены среды культуры зафиксировали ледяным раствором 4% (в/об) параформальдегида (Sigma) на 10 минут при комнатной температуре и провели окрашивание на уровни экспрессии нестина человека, GFAP, TuJ1, дезмина и альфа-актина гладких мышц.
Протокол совместного культивирования с клетками-предшественниками нейронов. Выделенные из гиппокампа взрослые клетки-предшественники нейронов крысы (062603) высевали в виде нейросфер или отдельных клеток (10000 клеток/лунку) на покрытые ламинином 24-луночные чашки (BD Biosciences) в среде NPE+F (20 нанограмм/миллилитр)+E (20 нанограмм/миллилитр).
Отдельно размораживали клетки, полученные из пуповины, (042203) P11 и клетки, полученные из плаценты клетки, (022803) P11, и культуры размножали в среде NPE+F (20 нанограмм/миллилитр)+E (20 нанограмм/миллилитр) при плотности посева 5000 клеток/см2 в течение 48 часов. Затем клетки трипсинизировали и высевали с плотностью 2500 клеток/лунку на существующие культуры клеток-предшественников нейронов. В данный момент имеющуюся среду заменяли на свежую среду. Через четверо суток культуры зафиксировали ледяным раствором 4% (в/об) параформальдегида (Sigma) на 10 минут при комнатной температуре и провели окрашивание на уровень экспрессии ядерного белка человека (hNuc; Chemicon) (таблица NU1-1, представленная выше) для идентификации клеток, полученных из ткани пуповины, и клеток, полученных из плаценты.
Иммуноцитохимический анализ. Иммуноцитохимический анализ проводили с использованием антител, перечисленных в таблице NU1-1. Культуры промывали фосфатно-солевым буфером (ФСБ) и обрабатывали белковым блокирующим раствором, содержащим ФСБ, 4% (об/об) козьей сыворотки (Chemicon, г. Темекула, штат Калифорния) и 0,3% (об/об) Тритона (Triton X-100; Sigma) в течение 30 минут для доступа к внутриклеточным антигенам. Затем культуры в течение 1 часа обрабатывали при комнатной температуре разбавленными в блокирующем растворе первичными антителами. Затем растворы первичных антител удаляли и культуры промывали ФСБ перед добавлением растворов вторичных антител (1 час при комнатной температуре), содержащих блокирующий раствор вместе с конъюгатом козьего антимышиного IgG - Texas Red (1:250; Molecular Probes, г. Юджин, штат Орегон) и конъюгатом козьего анти-кроличьего IgG - Alexa 488 (1:250; Molecular Probes). Затем культуры промывали и обрабатывали 10-микромолярным раствором DAPI (Molecular Probes) в течение 10 минут для визуализации клеточных ядер.
После иммунного окрашивания получали флуоресцентные изображения с использованием соответствующего фильтра для флуоресценции на инвертированном эпи-флуоресцентном микроскопе (Olympus, г. Мелвилл, штат Нью-Йорк). Во всех случаях положительное окрашивание соответствовало сигналу флуоресценции выше сигнала для контрольного окрашивания, при котором выполняли всю описанную выше процедуру за исключением обработки раствором первичных антител. Представительные изображения получали с использованием цветной цифровой видеокамеры и программного обеспечения ImagePro (Media Cybernetics, г. Карлсбад, штат Калифорния). Для образцов с тройным окрашиванием каждое изображение получали с использованием только одного эмиссионного фильтра. Затем получили послойные монтажи с использованием программного обеспечения Adobe Photoshop (Adobe, г. Сан-Хосе, штат Калифорния).
Результаты
Протокол Вудбери-Блэка. (Woodbury, D. et al. (2000 г.). J Neurosci. Research. 61(4): 364-70). (А) После инкубации в данной композиции нейронной индукции клетки всех типов трансформировались в клетки с морфологией биполярной клетки и протяженными отростками. Наблюдали также и другие небиполярные морфологии большего размера. Кроме того, индуцированные популяции клеток давали положительный результат при окрашивании на нестин, маркер мультипотентных нейронных стволовых клеток и клеток-предшественников нейронов.
(B) При повторе протокола на чашках из обработанного тканевой культурой пластика (TCP) экспрессию нестина наблюдали только в случае предварительно покрытых ламинином культуральных поверхностей. Для дальнейшей проверки того, смогут ли экспрессирующие нестин клетки затем дать зрелые нейроны, клетки, полученные из ткани пуповины, клетки, полученные из плаценты, и фибробласты обработали средой NPE+RA (1 мкМ), средой с композицией, которая, как известно, индуцирует дифференцирование нейронных стволовых клеток и клеток-предшественников нейронов в такие клетки (Jang, Y.K. et al. (2004 г.). J. Neurosci. Research. 75(4): 573-84; Jones-Villeneuve, E.M. et al. (1983 г.). Mol Cel Biol. 3(12): 2271-9; Mayer-Proschel, M. et al. (1997 г.). Neuron. 19(4): 773-85). Клетки окрашивали на TuJ1, маркер незрелых и зрелых нейронов, GFAP, маркер астроцитов, и нестин. TuJ1 не обнаружили ни при каких условиях, также не наблюдали клетки с нейронной морфологией, что указывает на отсутствие генерации нейронов в условиях кратковременной обработки. Более того, результаты иммуноцитохимического анализа показали, что клетки, полученные из пуповины и плаценты, более не экспрессировали нестин и GFAP.
Двухстадийное дифференцирование. Изоляты пуповины и плаценты (а также фибробласты человека и клетки-предшественники нейронов грызунов, использованные в качестве типов клеток для отрицательного и положительного контроля соответственно) высевали на покрытые ламинином (стимулятор нейронного дифференцирования) чашки и обрабатывали в 13 разных условиях роста (и двух контрольных условиях), которые, как известно, стимулируют дифференцирование клеток-предшественников нейронов в нейроны и астроциты. Кроме того, также добавили два условия для изучения влияния GDF5 и BMP7 на дифференцирование клеток. По существу следовали подходу двухстадийного дифференцирования, при котором клетки сначала помещали в условия для размножения клеток-предшественников нейронов на период 6 суток с последующим переводом в условия полного дифференцирования на 7 суток. Морфологически в ходе данной процедуры как клетки, полученные из пуповины, так и клетки, полученные из плаценты, продемонстрировали принципиальные изменения клеточной морфологии. Однако клетки с формой нейронов или астроцитов наблюдали только в контрольных условиях с клетками-предшественниками нейронов. Результаты иммуноцитохимического анализа, отрицательные на нестин человека, TuJ1 и GFAP, подтвердили выводы морфологических наблюдений.
Множество факторов роста. После недельной обработки различными агентами нейронного дифференцирования клетки окрашивали на маркеры, характерные для клеток-предшественников нейронов (нестин человека), нейроны (TuJ1) и астроциты (GFAP). Клетки, выращиваемые на первой стадии в бессывороточных средах, обладали морфологией, отличной от клеток, выращиваемых в содержащих сыворотку (2% или 10%) средах, что указывает на потенциальное нейронное дифференцирование. Более конкретно, после двухстадийной процедуры обработки клеток, полученных из пуповины, с помощью EGF и bFGF и затем SHH, FGF8, GDNF, BDNF и ретиноевой кислоты клетки демонстрировали длинные протяженные отростки, аналогичные морфологии культивированных астроцитов. При добавлении 2% FBS или 10% FBS в среду на первой стадии дифференцирования количество клеток увеличивалось, и клеточная морфология не изменялась по сравнению с контрольными культурами при высокой плотности. Потенциальное нейронное дифференцирование не было выявлено в ходе проведения иммуноцитохимического анализа на нестин человека, TuJ1 или GFAP.
Совместное культивирование клеток-предшественников нейронов и клеток UTC. Клетки, полученные из пуповины и плаценты, высевали на культуры клеток-предшественников нейронов крыс, посеянных двумя сутками ранее в условиях размножения нейронов (NPE+F+E). Хотя визуальное наблюдение высеянных клеток UTC и клеток, полученных из плаценты, подтвердило, что данные клетки были высеяны в качестве отдельных клеток, проведенное через 4 суток после высевания (6 суток от начала выращивания) специфическое окрашивание на ядерный белок человека (hNuc) показало, что они стремились собраться вместе и избегали контакта с клетками-предшественниками нейронов. Более того, когда клетки UTC закреплялись на поверхности, данные клетки распространялись и оказывались иннервированными дифференцированными нейронами, имевшими крысиное происхождение, что указывает на то, что клетки UTC могли подвергнуться дифференцированию в мышечные клетки. Данное наблюдение основано на результатах изучения морфологии клеток с использованием фазоконтрастной микроскопии. Другое наблюдение состояло в том, что, как правило, тела крупных клеток (более крупных, чем клетки-предшественники нейронов) обладали морфологиями, аналогичными морфологиям клеток-предшественников нейронов, причем тонкие отростки расходились по множеству направлений. Результаты окрашивания на HNuc (присутствующий в одной половине клеточного ядра) указывают на то, что в некоторых случаях данные клетки человека могли слиться с крысиными клетками-предшественниками и наследовать их фенотип. Контрольные лунки, содержащие только клетки-предшественники нейронов, имели меньшее общее количество клеток-предшественников и видимо дифференцированных клеток по сравнению с лунками, в которых происходило совместное культивирование, с клетками, полученными из плаценты или из пуповины, что дополнительно указывает на влияние как клеток, полученных из пуповины, так и клеток, полученных из плаценты, на дифференцирование и поведение клеток-предшественников нейронов, либо путем высвобождения хемокинов и цитокинов, либо через опосредованные контактами эффекты.
Обзор. Провели множество протоколов для определения краткосрочного потенциала клеток UTC и клеток, полученных из плаценты, к дифференцированию в клетки нейронной линии дифференцирования. Данные исследования включали в себя фазоконтрастную микроскопию морфологии в комбинации с иммуноцитохимическим окрашиванием на нестин, TuJ1 и GFAP, белки, ассоциируемые с мультипотентными нейронными стволовыми клетками и клетками-предшественниками нейронов, незрелыми и зрелыми нейронами и астроцитами соответственно. Получены данные, указывающие на прохождение нейронного дифференцирования в определенных случаях в данных протоколах краткосрочного исследования.
Несколько важных наблюдений были сделаны при совместном культивировании клеток UTC и клеток, полученных из плаценты, с клетками-предшественниками нейронов. Данный подход с использованием клеток UTC человека и клеток, полученных из плаценты, вместе с ксеногенным клеточным типом позволил провести абсолютное определение природы каждой клетки в данных культурах. Во-первых, в данных культурах наблюдали некоторые клетки с увеличенной клеточной цитоплазмой и выходящими из тела клетки нейритоподобными отростками, хотя только половина тела была мечена белком hNuc. Данные клетки могли представлять собой либо клетки UTC и клетки, полученные из плаценты, человека, которые дифференцировались в клетки нейронной линии дифференцирования, либо клетки UTC и клетки, полученные из плаценты, которые слились с клетками-предшественниками нейронов. Во-вторых, замечено, что клетки-предшественники нейронов выпускали нейриты к клеткам UTC и клеткам, полученным из плаценты, таким образом, который указывает на прошедшее дифференцирование клеток-предшественников в нейроны и иннервацию клеток UTC и клеток, полученных из плаценты. В-третьих, совместные культуры клеток-предшественников нейронов и клеток UTC и клеток, полученных из плаценты, имели больше клеток крысиного происхождения и большую степень дифференцирования, чем контрольные культуры, состоявшие только из клеток-предшественников нейронов, что дополнительно указывает на то, что высеянные клетки UTC и клетки, полученные из плаценты, обеспечивали растворимые факторы и/или контакт-зависимые механизмы, которые стимулировали выживание, пролиферацию и/или дифференцирование клеток-предшественников нейронов.
ПРИМЕР 14
Долговременное нейронное дифференцирование клеток, полученных из ткани пуповины, и клеток, полученных из плаценты
Оценивали способность клеток, полученных из плаценты, и клеток, полученных из пуповины, к долговременному дифференцированию в клетки нейронных линий дифференцирования.
Материалы и способы
Выделение и размножение клеток. Клетки выделяли и размножали как описано в предыдущих примерах.
Размораживание и посев клеток. Замороженные аликвоты клеток (пуповины (022803) P11; (042203) P11; (071003) P12; плаценты (101503) P7), предварительно выращенные в ростовой среде, размораживали и высевали с плотностью 5000 клеток/см2 в колбы T-75, покрытые ламинином (BD, г. Франклин Лейкс, штат Нью-Джерси), в среде Neurobasal-A (Invitrogen, г. Карлсбад, штат Калифорния), содержащей B27 (добавка B27, Invitrogen), L-глутамин (4 мМ) и пенициллин/стрептомицин (10 миллилитров), комбинация которых в настоящем документе будет называться средами для размножения клеток-предшественников нейронов (NPE). В среды NPE дополнительно добавляли bFGF (20 нанограмм/миллилитр, Peprotech, г. Роки Хилл, штат Нью-Джерси) и EGF (20 нанограмм/миллилитр, Peprotech, г. Роки Хилл, штат Нью-Джерси), такая комбинация в настоящем документе будет называться NPE+bFGF+EGF.
Посев клеток контроля. Кроме того, размораживали и высевали с той же плотностью посева на покрытые ламинином колбы T-75 в среде NPE+bFGF+EGF взрослые фибробласты кожи человека (P11, Cambrex, г. Уокерсвилл, штат Мэриленд) и мезенхимальные стволовые клетки (P5, Cambrex). В качестве дополнительного контроля фибробласты, клетки пуповины и плаценты выращивали в ростовой среде в течение периода времени, указанного для всех культур.
Размножение клеток. Среды во всех культурах заменяли на свежие среды раз в неделю и следили за размножением клеток. В общем, каждую культуру пассировали один раз за период продолжительностью один месяц из-за ограниченного роста в среде NPE+bFGF+EGF.
Иммуноцитохимический анализ. Через один месяц все культуры зафиксировали ледяным раствором 4% (в/об) параформальдегида (Sigma) на 10 минут при комнатной температуре. Иммуноцитохимический анализ проводили с использованием антител к TuJ1 (бета-III-тубулин; 1:500; Sigma, г. Сент-Луис, штат Миссури) и GFAP (глиофибриллярный кислый белок; 1:2000; DAKOCytomation, г. Карпинтерия, штат Калифорния). Вкратце, культуры промывали фосфатно-солевым буфером (ФСБ) и обрабатывали белковым блокирующим раствором, содержащим ФСБ, 4% (об/об) козьей сыворотки (Chemicon, г. Темекула, штат Калифорния) и 0,3% (об/об) Тритона (Triton X-100; Sigma) в течение 30 минут для доступа к внутриклеточным антигенам. Затем культуры в течение 1 часа обрабатывали при комнатной температуре разбавленными в блокирующем растворе первичными антителами. Затем растворы первичных антител удаляли и культуры промывали ФСБ перед добавлением растворов вторичных антител (1 час при комнатной температуре), содержащих блокирующий раствор вместе с конъюгатом козьего антимышиного IgG - Texas Red (1:250; Molecular Probes, г. Юджин, штат Орегон) и конъюгатом козьего анти-кроличьего IgG - Alexa 488 (1:250; Molecular Probes). Затем культуры промывали и обрабатывали 10-микромолярным раствором DAPI (Molecular Probes) в течение 10 минут для визуализации клеточных ядер.
После иммунного окрашивания получали флуоресцентные изображения с использованием соответствующего фильтра для флуоресценции на инвертированном эпи-флуоресцентном микроскопе (Olympus, г. Мелвилл, штат Нью-Йорк). Во всех случаях положительное окрашивание соответствовало сигналу флуоресценции выше сигнала для контрольного окрашивания, при котором выполняли всю описанную выше процедуру за исключением обработки раствором первичных антител. Представительные изображения получали с использованием цветной цифровой видеокамеры и программного обеспечения ImagePro (Media Cybernetics, г. Карлсбад, штат Калифорния). Для образцов с тройным окрашиванием каждое изображение получали с использованием только одного эмиссионного фильтра. Затем получили послойные монтажи с использованием программного обеспечения Adobe Photoshop (Adobe, г. Сан-Хосе, штат Калифорния).
Результаты
Среда NPE+bFGF+EGF замедляет пролиферацию клеток UTC и изменяет их морфологию. Непосредственно после посева лишь часть клеток закрепилась на поверхности колб для культивирования, покрытых ламинином. Это могло быть обусловлено гибелью клеток в результате проведенного процесса замораживания/размораживания или сменой условий роста. Клетки, которые закрепились на поверхности, изменили морфологию на отличную от морфологии клеток, наблюдаемых в ростовых средах.
При достижении конфлюентности культуры пассировали и наблюдали за их ростом. Для клеток, переживших пассирование, размножение было совершенно минимальным. На данной стадии в культурах клеток, полученных из пуповины, начали появляться очень маленькие клетки без морфологии распространения и с яркой окраской под фазоконтрастным микроскопом. За данными областями колбы следили дальше. Образовавшиеся малые клетки выпустили разветвляющиеся отростки с характерными утолщениями вдоль своей длины, что очень похоже на описанные ранее PSA-NCAM+ клетки-предшественники нейронов и TuJ1+ незрелые нейроны, полученные из головного и спинного мозга (Mayer-Proschel, M. et al. (1997 г.). Neuron. 19(4): 773-85; Yang, H. et al. (2000 г.). PNAS. 97(24): 13366-71). Со временем данные клетки становились более многочисленными, но, тем не менее, обнаруживались только в клонах.
Клоны клеток, полученных из пуповины, экспрессируют нейрональные белки. Культуры зафиксировали через месяц после размораживания/посева и провели окрашивание на нейрональный белок TuJ1 и на GFAP, присутствующий в астроцитах промежуточный микрофиламент. Хотя все контрольные культуры, выращенные в ростовой среде, фибробласты и клетки MSC человека, выращенные в среде NPE+bFGF+EGF, оказались типа TuJ1-/GFAP-, TuJ1 был обнаружен в клетках, полученных из пуповины и плаценты. Экспрессию наблюдали в клетках с нейроноподобной морфологией и без нее. Экспрессии GFAP не наблюдали ни в одной из культур. Процентное отношение экспрессирующих TuJ1 клеток с нейроноподобной морфологией составляло не более 1% от всей популяции клеток (n=3 тестированных изолята клеток, полученных из пуповины). Хотя количественное определение не проводилось, процентное отношение TuJ1+ клеток без нейрональной морфологии было выше в культурах клеток, полученных из пуповины, чем в культурах клеток, полученных из плаценты. Данные результаты оказались специфичными, поскольку согласованные по возрасту контроли в ростовой среде не экспрессировали TuJ1.
Обзор. Разработаны способы генерации дифференцированных нейронов (на основе экспрессии TuJ1 и нейрональной морфологии) из клеток, полученных из пуповины. Хотя экспрессию TuJ1 не исследовали ранее, чем через один месяц in vitro, ясно, что по меньшей мере малая популяция клеток, полученных из пуповины, может породить нейроны либо путем дифференцирования по умолчанию, либо путем долговременной индукции в результате обработки в течение месяца в минимальных средах с добавлением L-глутамина, основного bFGF и EGF.
ПРИМЕР 15
Трофические факторы для поддержания клеток-предшественников нейронов
Исследовали влияние клеток, полученных из пуповины, и клеток, полученных из плаценты, на выживаемость и дифференцирование взрослых нейронных стволовых клеток и клеток-предшественников нейронов через неконтакт-зависимые (трофические) механизмы.
Материалы и способы
Выделение взрослых нейронных стволовых клеток и клеток-предшественников нейронов. Взрослых крыс линии Fisher 344 умерщвляли удушением в атмосфере CO2 с последующим смещением шейных позвонков. Цельные мозги извлекали в интактном виде с помощью черепных кусачек и отсекали ткань гиппокампа, используя корональные надрезы за двигательным и соматосенсорным участками мозга (Paxinos, G. & Watson, C. 1997 г. The Rat Brain in Stereotaxic Coordinates). Ткань промывали в среде Neurobasal-A (Invitrogen, г. Карлсбад, штат Калифорния), содержащей B27 (B27, Invitrogen), L-глутамин (4 мМ) и пенициллин/стрептомицин (Invitrogen), комбинация которых в настоящем документе будет называться средой для размножения клеток-предшественников нейронов (NPE). В среду NPE дополнительно добавляли bFGF (20 нанограмм/миллилитр, Peprotech, г. Роки Хилл, штат Нью-Джерси) и EGF (20 нанограмм/миллилитр, Peprotech, г. Роки Хилл, штат Нью-Джерси), комбинация которых в настоящем документе будет называться NPE+bFGF+EGF.
После промывки удаляли покрывающие оболочки головного мозга и ткань измельчяли скальпелем. Измельченную ткань собирали и добавляли трипсин/ЭДТА (Invitrogen) в количестве 75% от общего объема. Также добавляли ДНКазу (100 микролитров на 8 миллилитров общего объема, Sigma, г. Сент-Луис, штат Миссури). Затем смесь среды/ткани последовательно пропускали по одному разу через иглу размером 18, иглу размером 20 и, наконец, иглу размером 25 (все иглы производства Becton Dickinson, г. Франклин Лейкс, штат Нью-Джерси). Смесь центрифугировали в течение 3 минут при 250 g. Супернатант удалили и добавили свежую среду NPE+bFGF+EGF для повторного суспендирования осадка. Полученную клеточную суспензию пропускали через клеточный фильтр с размером пор 40 микрометров (Becton Dickinson), высевали на покрытые ламинином колбы T-75 (Becton Dickinson) или 24-луночные планшеты с низкой кластеризацией (Becton Dickinson) и выращивали в средах NPE+bFGF+EGF до получения достаточного числа клеток для описанных исследований.
Посев клеток. Клетки, полученные из тканей (пуповина (022803) P12, (042103) P12, (071003) P12; плацента (042203) P12), выращенные ранее в ростовой среде, высевали с плотностью 5000 клеток/вставка трансвелл (размером под 24-луночный планшет) и выращивали в течение одной недели в ростовой среде во вставках для достижения конфлюентности.
Посев взрослых клеток-предшественников нейронов. Клетки-предшественники нейронов, выращенные в виде нейросфер или отдельных клеток, высевали на покрытые ламинином 24-луночные планшеты с приблизительной плотностью 2000 клеток/лунку в среде NPE+bFGF+EGF и выдерживали один день для стимуляции закрепления клеток. Через день добавили вставки трансвелл, содержащие клетки, полученные из тканей, в соответствии со следующей схемой:
(1) вставка трансвелл (клетки, полученные из пуповины, в ростовых средах, 200 микролитров) + клетки-предшественники нейронов (NPE+bFGF+EGF, 1 миллилитр)
(2) вставка трансвелл (клетки, полученные из плаценты, в ростовых средах, 200 микролитров) + клетки-предшественники нейронов (NPE+bFGF+EGF, 1 миллилитр)
(3) вставка трансвелл (взрослые фибробласты кожи человека [1F1853; Cambrex, г. Уокерсвилл, штат Мэриленд] P12 в ростовых средах, 200 микролитров) + клетки-предшественники нейронов (NPE+bFGF+EGF, 1 миллилитр)
(4) контроль: только клетки-предшественники нейронов (NPE+bFGF+EGF, 1 миллилитр)
(5) контроль: только клетки-предшественники нейронов (только NPE, 1 миллилитр)
Иммуноцитохимический анализ. После 7 суток совместного культивирования культуры всех условий зафиксировали в холодном 4% (в/об) параформальдегиде (Sigma-Aldrich, г. Сент-Луис, штат Миссури) на 10 минут при комнатной температуре. Иммуногистохимический анализ проводили с использованием антител, направленных на эпитопы, перечисленные в таблице 15-1. Вкратце, культуры промывали фосфатно-солевым буфером (ФСБ) и обрабатывали белковым блокирующим раствором, содержащим ФСБ, 4% (об/об) козьей сыворотки (Chemicon, г. Темекула, штат Калифорния) и 0,3% (об/об) Тритона (Triton X-100; Sigma) в течение 30 минут для доступа к внутриклеточным антигенам. Затем культуры в течение 1 часа обрабатывали при комнатной температуре разбавленными в блокирующем растворе первичными антителами. Затем растворы первичных антител удаляли и культуры промывали ФСБ перед добавлением растворов вторичных антител (1 час при комнатной температуре), содержащих блокирующий раствор вместе с конъюгатом козьего антимышиного IgG - Texas Red (1:250; Molecular Probes, г. Юджин, штат Орегон) и конъюгатом козьего анти-кроличьего IgG - Alexa 488 (1:250; Molecular Probes). Затем культуры промывали и обрабатывали 10-микромолярным раствором DAPI (Molecular Probes) в течение 10 минут для визуализации клеточных ядер.
После иммунного окрашивания получали флуоресцентные изображения с использованием соответствующего фильтра для флуоресценции на инвертированном эпи-флуоресцентном микроскопе (Olympus, г. Мелвилл, штат Нью-Йорк). Во всех случаях положительное окрашивание соответствовало сигналу флуоресценции выше сигнала для контрольного окрашивания, при котором выполняли всю описанную выше процедуру за исключением обработки раствором первичных антител. Представительные изображения получали с использованием цветной цифровой видеокамеры и программного обеспечения ImagePro (Media Cybernetics, г. Карлсбад, штат Калифорния). Для образцов с тройным окрашиванием каждое изображение получали с использованием только одного эмиссионного фильтра. Затем получили послойные монтажи с использованием программного обеспечения Adobe Photoshop (Adobe, г. Сан-Хосе, штат Калифорния).
Количественный анализ дифференцирования клеток-предшественников нейронов. Количественно исследовали дифференцирование клеток-предшественников нейронов из гиппокампа. Для каждого тестируемого условия подсчитывали минимум по 1000 клеток или при меньшем количестве анализировали общее количество клеток, наблюдаемых для данного условия. Процентное содержание клеток, положительных в отношении указанной окраски, определяли как частное от деления количества положительных клеток на общее количество клеток, определенное путем окрашивания DAPI (ядерного).
Масс-спектрометрический анализ и двумерный гель-электрофорез. Для идентификации уникальных факторов, секретируемых в результате совместного культивирования, образцы кондиционированных сред, отобранные перед фиксацией, замораживали до -80°C на ночь. Затем образцы обрабатывали на центрифугах для ультрафильтрования (порог отсечения по молекулярной массе 30 кДа). Ретентат пропустили через колонку для иммуноаффинной хроматографии (антитела к альбумину человека; IgY) (иммуноаффинная хроматография не выделила альбумин из образцов). Фильтрат проанализировали с помощью MALDI. Прошедшую фракцию пропустили через колонку для аффинной хроматографии Cibachron Blue. Образцы анализировали методом ДСН-ПААГ-электрофореза и двумерного гель-электрофореза.
Результаты
Совместное культивирование с клетками, полученными из ткани, стимулирует дифференцирование взрослых клеток-предшественников нейронов. После культивирования с клетками, полученными из пуповины или плаценты, совместно культивированные клетки-предшественники нейронов, полученные из гиппокампа взрослой крысы, показывали существенное дифференцирование по всем трем основным линиям дифференцирования в центральной нервной системе. Данный эффект ясно наблюдался после пяти суток совместного культивирования и проявлялся в появлении отростков сложной формы у множества клеток и потере характерного для делящихся клеток-предшественников фазоконтрастного изображения. С другой стороны, клетки-предшественники нейронов, выращиваемые отдельно в отсутствие bFGF и EGF, выглядели нездоровыми и имели ограниченную выживаемость.
После завершения процедуры культуры окрашивали на маркеры, характерные для недифференцированных стволовых клеток и клеток-предшественников (нестин), незрелых и зрелых нейронов (TuJ1), астроцитов (GFAP) и зрелых олигодендроцитов (MBP). Подтвердили дифференцирование по всем трем линиям дифференцирования, тогда как культуры в контрольных условиях не показали существенного дифференцирования, что проявилось в сохранении положительной окраски на нестин у большинства клеток. Хотя и клетки, полученные из пуповины, и клетки, полученные из плаценты, индуцировали клеточное дифференцирование, степень дифференцирования для всех трех линий дифференцирования при совместном культивировании с клетками, полученными из плаценты, оказалась меньше, чем при совместном культивировании с клетками, полученными из пуповины.
Количественно определили процентное содержание клеток-предшественников нейронов, дифференцировавшихся после совместного культивирования с клетками, полученными из пуповины (таблица 15-2). Клетки, полученные из пуповины, существенно увеличили количество зрелых олигодендроцитов (MBP) (24,0% по сравнению с 0% для каждого контрольного условия). Кроме того, совместное культивирование привело к увеличению количества GFAP+ астроцитов и TuJ1+ нейронов в культуре (47,2% и 8,7% соответственно). Данные результаты подтвердили окрашиванием на нестин, которое указывает на потерю статуса клетки-предшественника в результате совместного культивирования (13,4% по сравнению с 71,4% при контрольном условии 4).
Хотя оказалось, что взрослые фибробласты человека также влияли на дифференцирование, такие клетки были не в состоянии стимулировать дифференцирование зрелых олигодендроцитов, а также не могли генерировать ощутимые количества нейронов. Хотя количественное определение не проводилось, оказалось, что фибробласты улучшали выживаемость клеток-предшественников нейронов.
Идентификация уникальных соединений. Кондиционированные среды от совместных культур с клетками, полученными из пуповины, и клетками, полученными из плаценты, и соответствующие контроли (среды NPE±1,7% сыворотки, среды от совместных культур с фибробластами) проанализировали на обнаружение различий. Потенциально уникальные соединения идентифицировали и вырезали из соответствующих двумерных гелей.
Обзор. Совместное культивирование взрослых клеток-предшественников нейронов с клетками, полученными из пуповины, или клетками, полученными из плаценты, приводит к дифференцированию данных клеток. Представленные в данном примере результаты показывают, что дифференцирование взрослых клеток-предшественников нейронов оказывается особенно выраженным в результате совместного культивирования с клетками, полученными из пуповины. Более конкретно, при совместном культивировании с клетками, полученными из пуповины, был сгенерирован существенный процент зрелых олигодендроцитов. Из-за отсутствия контакта между клетками, полученными из пуповины, и клетками-предшественниками нейронов данный результат, скорее всего, является результатом секретирования растворимых факторов клетками, полученными из пуповины (трофический эффект).
Было сделано еще несколько наблюдений. Во-первых, в контрольном условии с удаленными EGF и bFGF количество клеток было очень низким. Большая часть клеток погибала, и в среднем на лунку оставалось приблизительно 100 клеток или менее. Во-вторых, следует ожидать очень низкой степени дифференцирования в контрольном условии с сохранением EGF и bFGF в среде на протяжении всего процесса выращивания, поскольку данная среда обычно представляет собой среду для размножения. Хотя было обнаружено, что приблизительно 70% клеток сохранили свой статус клетки-предшественника (нестин+), приблизительно 30% клеток оказались GFAP+ (характерно для астроцитов). Это может быть вызвано тем, что в процессе процедуры произошло столь существенное размножение клеток, что контакт между клетками-предшественниками индуцировал данное дифференцирование(Song, H. et al. 2002 г. Nature 417(6884): 39-44).
ПРИМЕР 16
Трансплантация клеток, полученных из пуповины и плаценты
Клетки, полученные из пуповины и плаценты, можно использовать в регенеративной терапии. Оценивали ткань, полученную трансплантированием клеток, полученных из ткани пуповины, и клеток, полученных из плаценты, мышам линии SCID с использованием биоразлагаемого материала. Оценивали следующие материалы: нетканый викрил, пеноматериал 35/65 PCL/PGA и самособирающийся пептидный гидрогель RAD16.
Способы и материалы
Клеточная культура. Клетки, полученные из плаценты и пуповины, выращивали в ростовой среде (среда DMEM с низким содержанием глюкозы (Gibco, г. Карлсбад, штат Калифорния), 15% (об/об) эмбриональной бычьей сыворотки (№ по кат. SH30070.03; Hyclone, г. Логан, штат Юта), 0,001% (об/об) бета-меркаптоэтанола (Sigma, г. Сент-Луис, штат Миссури), пенициллин/стрептомицин (Gibco)) в покрытых желатином колбах.
Приготовление образца. Один миллион жизнеспособных клеток высевали в 15 микролитрах ростовой среды на нетканые каркасы из викрила диаметром 5 мм и толщиной 2,25 мм (64,33 миллиграмма/см3; партия № 3547-47-1) или каркасы из пеноматериала 35/65 PCL/PGA диаметром 5 мм (партия № 3415-53). Клеткам дали закрепиться в течение двух часов и затем добавили еще ростовой среды, чтобы полностью покрыть каркасы. Клетки выращивали на каркасах в течение ночи. В среде также инкубировали каркасы без клеток.
Самособирающиеся пептиды RAD16 (3D Matrix, г. Кембридж, штат Массачусетс, получены в рамках соглашения о передаче материалов) получали в виде стерильного 1% (в/об) раствора в воде, который смешивали в соотношении 1:1 с суспензией 1×106 клеток в 10% (в/об) растворе сахарозы (Sigma, г. Сент-Луис, штат Миссури), 10 мМ HEPES в модифицированной по способу Дульбекко среде Игла (DMEM; Gibco) непосредственно перед использованием. Конечная концентрация клеток в гидрогеле RAD16 составляла 1×106 клеток/100 микролитров.
ТЕСТИРУЕМЫЕ МАТЕРИАЛЫ (N=4/Rx)
1. Нетканый викрил + 1×106 клеток, полученных из пуповины
2. Пеноматериал 35/65 PCL/PGA+1×106 клеток, полученных из пуповины
3. Самособирающийся пептид RAD16+1×106 клеток, полученных из пуповины
4. Нетканый викрил + 1×106 клеток, полученных из плаценты
5. Пеноматериал 35/65 PCL/PGA+1×106 клеток, полученных из плаценты
6. Самособирающийся пептид RAD16+1×106 клеток, полученных из плаценты
7. Пеноматериал 35/65 PCL/PGA
8. Нетканый викрил
Подготовка животных. С животными обращались в соответствии с действующими требованиями Акта о благополучии животных. Соответствия требованиям указанных выше публичных законов достигали за счет следования нормативным документам по обеспечению благосостояния животных (Раздел 9 Свода федеральных правил) и соответствия требованиям действующих стандартов, опубликованных в Руководстве по содержанию и использованию лабораторных животных, 7-е издание.
Мыши (Mus Musculus)/Fox Chase SCID/самцы (Harlan Sprague Dawley, Inc., г. Индианаполис, штат Индиана), возраст 5 недель. Все манипуляции с мышами линии SCID проводили в боксе. Мышей индивидуально взвешивали и анестезировали внутрибрюшинной инъекцией смеси 60 миллиграмм/кг KETASET (гидрохлорид кетамина, Aveco Co., Inc., г. Форт Додж, штат Айова) и 10 миллиграмм/кг ROMPUN (ксилазин, Mobay Corp., г. Шони, штат Канзас) в физиологическом растворе. После индуцирования анестезии всю спину животного от спинной шейной области до спинной пояснично-крестцовой области выбривали от шерсти с использованием электрической машинки для стрижки животных. Затем обритую область оттирали хлоргексидиндиацетатом, промывали спиртом, высушивали и красили водным раствором йодофора с 1% доступного йода. На глаза наносили глазную мазь для защиты ткани от высыхания в течение периода анестезии.
Методика подкожной имплантации. На спинке мыши выполняли четыре надреза кожи длиной приблизительной 1,0 см каждый. Два черепных надреза размещали в поперечном направлении в дорсальной боковой области груди, приблизительно на 5 мм в каудальном направлении к пальпируемому нижнему краю лопатки, один с левой и один с правой стороны от позвоночного столба. Другие два надреза выполняли в поперечном направлении в области ягодичных мышц на каудальном крестцово-поясничном уровне, приблизительно на 5 мм в каудальном направлении к пальпируемому гребню подвздошной кости, по надрезу с каждой стороны от срединной линии. Имплантаты размещали в данных местах случайным образом в соответствии с планом эксперимента. Кожу отделяли от находящейся под ней соединительной ткани для получения небольшого кармана и вводили (или инжектировали в случае RAD16) имплантат приблизительно на 1 см в каудальном направлении от надреза. Соответствующий тестируемый материал имплантировали в подкожное пространство. Надрез на коже закрывали металлическими скобками.
Содержание животных. На протяжении всего исследования мышей содержали отдельно в клетках-микроизоляторах, диапазон температуры составлял 17,8-26,1°C (64-79ºF), и относительная влажность составляла 30-70%, и поддерживали при цикле освещения приблизительно 12 часов света/12 часов темноты. Температуру и относительную влажность поддерживали в указанных диапазонах, насколько это было возможно. Диета состояла из облученного корма Pico Mouse Chow 5058 (Purina Co.) и воды ad libitum.
Мышей умерщвляли в предписанное им время ингаляцией диоксида углерода. Места подкожной имплантации с покрывающими их участками кожи вырезали и замораживали для гистологического исследования.
Гистологическое исследование. Вырезанный фрагмент кожи с имплантатом фиксировали 10% нейтральным буферным раствором формалина (Richard-Allan, г. Каламазу, штат Мичиган). Образцы с покрывающей и смежной тканью рассекали по центру, обрабатывали парафином и помещали в заливочный состав по поверхности надреза с использованием стандартных способов. Микротомом получали срезы толщиной пять микрон, которые окрашивали гематоксилином и эозином (Poly Scientific, г. Бэй Шор, штат Нью-Йорк) с использованием стандартных способов.
Результаты
Прорастание ткани в пеноматериалы (без клеток), имплантированные подкожно в мышей линии SCID, через 30 суток было минимальным. В отличие от этого для пеноматериалов, в которые были имплантированы клетки, полученные из пуповины, или клетки, полученные из плаценты, имело место обширное заполнение тканью. Для нетканых викриловых каркасов наблюдали некоторое прорастание ткани. Нетканые каркасы, засеянные клетками, полученными из пуповины, или клетками, полученными из плаценты, продемонстрировали повышенное отложение матрикса и зрелые кровеносные сосуды.
Обзор. Диски из синтетического рассасывающегося нетканого материала/пеноматериала (диаметром 5,0 мм и толщиной 1,0 мм) или гидрогель из самособирающегося пептида засевали клетками, полученными из пуповины, или клетками, полученными из плаценты, человека и имплантировали подкожно с двух сторон на участке позвоночника мышам линии SCID. Результаты показали, что клетки, полученные из послеродового материала, могут резко повысить формирование ткани хорошего качества в биоразлагаемых каркасах.
ПРИМЕР 17
Использование клеток, полученных из ткани пуповины, и клеток, полученных из плаценты, в восстановлении нервной ткани
Повреждения ганглиозных клеток сетчатки (RGC) широко используют в качестве моделей для различных стратегий восстановления в ЦНС взрослых млекопитающих. Результаты показали, что ретробульбарное сечение аксонов клеток RGC у взрослого грызуна приводит к незавершенному прорастанию (Zeng BY et al. J. Anat. 186: 495-508 (1995 г.)) и прогрессирующей гибели популяции родительских клеток (Villegas-Perez et al., J Neurosci. 8:265-80 (1988 г.)). Многочисленные исследования продемонстрировали стимулирующие эффекты различных экзогенных и эндогенных факторов на выживаемость аксотомированных клеток RGC и регенерацию их аксонов (Yip and So, Prog Retin Eye Res. 19: 559-75 (2000 г.); Fischer D. et al. Exp Neurol. 172: 257-72 (2001 г.)). Более того, другие исследования продемонстрировали, что клеточные трансплантаты можно использовать для стимуляции регенерации аксонов рассеченных нервных волокон (Li et al., 2003 г.; Ramon-CuetoA. et al., Neuron 25: 425-35 (2000 г.)). Таким образом, результаты данных и других исследований демонстрируют, что клеточную терапию можно применять для лечения неврологических расстройств, затрагивающих спинной мозг, периферические нервы, половые нервы, оптические нервы, а также других заболеваний/травм, сопровождающихся возможным повреждением нервных тканей.
Самособирающиеся пептиды (PuraMatrix™, патенты США №№ 5670483, 5955343, заявки на патент США/PCT №№ US2002/0160471, международная заявка № WO 02/062969) были разработаны для использования в качестве каркаса для закрепления клеток, для инкапсулирования клеток в трехмерных покрытиях, высевания клеток на двумерных покрытиях, либо в качестве микроносителей в суспензионных культурах. Объемное культивирование клеток требовало применения либо выделенных из животных материалов (экстракт саркомы мыши), с сопутствующими проблемами репродуктивности и клеточной сигнализации, либо значительно больших по размеру синтетических каркасов, которые не могли имитировать физические атрибуты нанометрового размера и химические свойства нативного ECM. Пептиды RAD16 (NH2-(RADA)3-COOH) и KLD (NH2-(KLDL)3-COOH) синтезируют в виде небольших (RAD16 имеет размер 5 нанометров) олигопептидных фрагментов, которые самособираются в нановолокна в масштабе, аналогичном in vivo внеклеточному матриксу (ECM) (3D Matrix, Inc г. Кембридж, штат Массачусетс). Самосборка инициируется одно- или двухвалентными катионами, присутствующими в культуральных средах или в физиологическом окружении. В протоколах, описанных в данном примере, RAD16 использовали в качестве микроносителя для имплантации клеток, полученных из ткани пуповины, и клеток, полученных из плаценты, в дефект оптического нерва. В данном примере результаты показали, что трансплантаты клеток, полученных из ткани пуповины, и клеток, полученных из плаценты, могут обеспечить эффективность в модели регенерации аксонов оптического нерва взрослой крысы.
Способы и материалы
Клетки. Культуры взрослых клеток, полученных из пуповины и плаценты, и клеток фибробластов человека (пассаж 10) размножали в течение 1 пассажа. Все клетки изначально высевали с плотностью 5000 клеток/см2 на покрытые желатином колбы T75 в ростовой среде с добавлением 100 единиц на миллилитр пенициллина, 100 микрограмм на миллилитр стрептомицина, 0,25 микрограмма на миллилитр амфотерицина B (Invitrogen, г. Карлсбад, штат Калифорния). На пассаже 11 клетки трипсинизировали и определяли их жизнеспособность окраской трипановым синим. Вкратце, 50 микролитров клеточной суспензии смешивали с 50 микролитрами 0,04% (в/об) трипанового синего (Sigma, г. Сент-Луис, штат Миссури) и оценивали количество жизнеспособных клеток с использованием гемоцитометра. Затем клетки трижды промывали в среде Лейбовица L-15, не содержащей добавок (Invitrogen, г. Карлсбад, штат Калифорния). Затем клетки суспендировали с концентрацией 200000 клеток в 25 микролитрах RAD16 (3DM Inc., г. Кембридж, штат Массачусетс), которые буферизовали и сделали изотоническим в соответствии с рекомендациями производителя. Чтобы поддерживать суспензию клеток/матрикса во влажном состоянии до использования, на нее сверху добавили сто микролитров среды Лейбовица L-15, не содержащей добавок. Данные культуры клетки/матрикса поддерживали в стандартных атмосферных условиях до проведения трансплантации. В месте проведения трансплантации избыточную среду удаляли.
Животные и хирургия. Использовали самок крыс линии Long Evans (вес тела 220-240 грамм). В условиях внутрибрюшинной трибромэтанольной анестезии (20 миллиграмм/100 грамм веса тела) обнажили оптический нерв и надрезали оболочку зрительного нерва интраорбитально на расстоянии приблизительно 2 миллиметра от оптического диска, нерв приподняли из оболочки для его полного перерезания маленькими ножницами (Li Y et al., 2003 г., J. of Neuro. 23(21): 7783-7788). Полноту пересечения проверили визуально по полному разделению проксимального и дистального обрезков нерва. Контрольная группа состояла из крыс с выполненным повреждением без трансплантатов. В группе крыс с трансплантатами животным вводили культивированные клетки из послеродового материала, высеянные в RAD16, которые устанавливали между проксимальным и дистальным обрезками нерва с использованием пары микрозажимов. В рассеченный оптический нерв имплантировали приблизительно 75000 клеток в RAD16. Клетку/матрикс распределили в надрез с использованием пары маленьких микрозажимов. Оболочку рассеченного оптического нерва закрыли черным монофиламентным нейлоном 10/0 (Ethicon, Inc., г. Эдинбург, Великобритания). Таким образом, зазор закрыли путем подтягивания надрезанных проксимального и дистального концов нерва друг к другу.
После проведения инъекции клеток животным с помощью инъекций ввели дексаметазон (2 миллиграмма/килограмм) в течение 10 суток после трансплантации. На протяжении всего исследования животных поддерживали на пероральном циклоспорине A (210 миллиграмм/литр питьевой воды; полученная концентрация в крови: 250-300 микрограмм/литр) (Bedford Labs, г. Бедфорд, штат Огайо), начиная с 2 суток перед трансплантацией и до окончания исследования. Корм и воду давали ad libitum. Животных умерщвляли через 30 или 60 суток после трансплантации.
Применение CTB. За трое суток до умерщвления животных, под анестезией, через склеру за хрусталиком касательно вставляли стеклянный микрокапилляр с рабочей частью длиной 30-50 миллиметров, и в стекловидное тело вводили аликвоты 4-5 микролитров 1% водного раствора ретроградной метки-холерогена B (CTB) (List Biologic, г. Кэмпбелл, штат Калифорния). Животных перфузировали фиксирующим раствором и оптические нервы собирали в тот же фиксирующий раствор на 1 час. Затем оптические нервы выдержали в сахарозе в течение ночи. Криостатные срезы толщиной двадцать микрометров инкубировали в 0,1-молярном растворе глицина в течение 30 минут и блокировали раствором ФСБ, содержащим 2,5% бычьего сывороточного альбумина (БСА) (Boeringer Mannheim, г. Мангейм, Германия) и 0,5% Triton X-100 (Sigma, г. Сент-Луис, штат Миссури), затем обработали раствором, содержащим козьи анти-CTB антитела (List Biologic, г. Кэмпбелл, штат Калифорния), разбавленные 1:4000 в ФСБ, содержащем 2% нормальной кроличьей сыворотки (NRS) (Invitrogen, г. Карлсбад, штат Калифорния), 2,5% БСА и 2% Triton X-100 (Sigma, г. Сент-Луис, штат Миссури) в ФСБ, и инкубировали в растворе биотинилированного кроличьего анти-козьего антитела IgG (Vector Laboratories, г. Берлингейм, штат Калифорния), разбавленного 1:200 в 2% растворе Triton-X100 в ФСБ в течение 2 часов при комнатной температуре. После этого провели окрашивание в 1:200 растворе стрептавидин-зеленый (Alexa Flour 438; Molecular Probes, г. Юджин, штат Орегон) в ФСБ в течение 2 часов при комнатной температуре. Окрашенные срезы затем промыли в ФСБ и докрасили пропидий-йодидом для конфокальной микроскопии.
Подготовка к гистологическому исследованию. Вкратце, через 5 суток после инъекции CTB крыс перфузировали 4% раствором параформальдегида. Крысам давали 4 кубических сантиметра уретана и затем перфузировали ФСБ (0,1 М) и впоследствии 4% раствором параформальдегида. Спинной мозг перерезали и удаляли кость из головы для получения доступа к бугорку. Затем бугорок извлекали и помещали в 4% раствор параформальдегида. Глаз извлекали путем максимально глубокого надреза вокруг внешнего периметра глаза. Следили за тем, чтобы не перерезать оптический нерв, проходящий с нижней стороны глаза. Глаз извлекали и перерезали мышцы, получая доступ к оптическому нерву, который затем помещали в 4% раствор параформальдегида.
Результаты
Только повреждение нерва. Через месяц после ретротубулярного рассечения оптического нерва в соединяющемся с сетчаткой сегменте оптического нерва идентифицировали ряд меченных CTB аксонов. Оказалось, что в ближайших 200 микрометрах от разреза аксоны выпустили ряд коллатеральных ветвей под прямыми углами к главной оси, прерывающихся в виде нейроматозного сгустка у поверхности разреза. В данном разрезе между проксимальным и дистальным обрезками нерва зазор прогрессивно заполнялся 2-3 миллиметровым сегментом васкуляризированной соединительной ткани; однако аксоны в данную заполненную область не перемещались. Таким образом, у животных, получивших только повреждение нерва, не наблюдали рост аксонов для достижения дистального обрезка нерва.
Трансплантация RAD16. После трансплантации RAD16 в надрез наблюдали видимое прорастание васкуляризированной соединительной ткани. Однако прорастания аксонов между проксимальным и дистальным обрезками нерва не наблюдали. Результаты показывают, что использование только RAD16 недостаточно для индуцирования регенерации аксонов в данной ситуации.
Трансплантация клеток, полученных из ткани пуповины, или клеток, полученных из плаценты. Трансплантация клеток в рассеченный оптический нерв стимулировала возобновление роста оптического нерва. Некоторое возобновление роста также наблюдали в условиях, в которых имплантировали клетки фибробластов, хотя оно было минимальным по сравнению с возобновлением роста, наблюдаемым с трансплантированными клетками, полученными из плаценты. Возобновление роста оптического нерва наблюдали у 4/5 животных, которым трансплантировали клетки, полученные из плаценты, у 3/6 животных, которым трансплантировали взрослые фибробласты кожи, и у 1/4 животных, которым трансплантировали клетки, полученные из пуповины. В ситуациях, когда наблюдали возобновление роста, метки CTB подтвердили регенерацию аксонов ганглиозных клеток сетчатки, для которых продемонстрировано проникновение через область трансплантата. Для определения уровня глиального рубцевания также проводили мечение GFAP. Экспрессия GFAP оказалась более интенсивной в проксимальном обрезке нерва, причем некоторое иммунное окрашивание наблюдалось и по повторно иннервированному трансплантату.
Обзор. Данные результаты показывают, что трансплантированные взрослые клетки, полученные из плаценты, и клетки, полученные из ткани пуповины, человека способны стимулировать и направлять регенерацию рассеченных аксонов ганглиозных клеток сетчатки.
ПРИМЕР 18
Использование клеток, полученных из пуповины, и клеток, полученных из плаценты, в восстановлении дофаминергической нервной ткани
Клетки, полученные из послеродового материала пуповины и плаценты, тестировали на способность давать функциональные улучшения у грызунов с поражением 6-гидроксидофамином (6-OHDA), используемых в качестве модели для лечения нейродегенеративного заболевания, такого как болезнь Паркинсона (Eisenhofer, G. et al. (2003 г.) FASEB J. 17: 1248-1255; Rios M. et al. (1999 г.) J. Neurosci. 1999:3519-26; Xu Y. et al. (1998 г.) J. Neurosci. Res. 54: 691-7).
Способы и материалы
Модель животных и разбиение на группы. Интрапаренхиматозное нейрохимическое поражение полосатого тела, отдела pars compacta черной субстанции или нигростриатного пути 6-гидроксидофамином (6-OHDA) широко используют как надежную модель грызунов для болезни Паркинсона. 6-OHDA разрушает дофаминергические нейроны, приводя к развитию болезни Паркинсона. Двухмесячных самок мышей линии Sprague-Dawley (275-300 г) для поражения 6-гидроксидофамином в медиальном пучке переднего мозга для индуцирования фенотипа болезни Паркинсона приобрели непосредственно в лаборатории Charles River Laboratories (г. Монреаль, Канада).
После получения животным дали одну неделю на адаптацию перед проведением трансплантации и давали доступ к корму ad libitum на протяжении всей продолжительности эксперимента за исключением периода голодания, необходимого для проведения описанного ниже теста обученного доставания пищи лапой. Крыс содержали по две в клетке, ежедневно проверяли на изменение веса и тестировали во время светлой фазы цикла 12ч:12ч света:темноты. Уход за животными и эксперименты проводили в соответствии с Канадским руководством по содержанию и использованию лабораторных животных, и все процедуры были одобрены Комитетом по содержанию и использованию лабораторных животных Университета Лаваля. Поведенческие нарушения, связанные с поражением 6-OHDA, оценивали через две с половиной недели после операции путем провокации апоморфином.
Животных разделили на четыре группы на основе результатов контроля количества поворотов. Трансплантацию проводили вслепую двумя участвующими в данном исследовании специалистами. Трем группам трансплантировали клетки различных типов (n=18 на клеточный тип; неизвестен исследовательской группе), и одна группа получила только несущую среду (среды для культивирования клеток) и выполняла роль контроля (n=6). Крыс умерщвляли через 4, 8 и 16 недель после трансплантации (n=6 на клеточные типы, и n=2 для контроля в каждый момент времени). Перед каждым умерщвлением крыс периодически оценивали с помощью трех поведенческих тестов: провокация апоморфином, тест обученного доставания пищи лапой и повороты головы.
Клеточные трансплантаты. Культуры взрослых клеток, полученных из пуповины, клеток, полученных из плаценты, и фибробластов (исходных) человека (пассаж 10) размножали в течение одного пассажа. Все клетки изначально высевали с плотностью 5000 клеток/см2 на покрытые желатином колбы T75 в ростовой среде. Для последующих пассирований все клетки обрабатывали следующим образом. После трипсинизации подсчитывали жизнеспособные клетки после окрашивания трипановым синим на жизнеспособность. Вкратце, 50 микролитров клеточной суспензии смешивали с 50 микролитрами трипанового синего (Sigma, г. Сент-Луис, штат Миссури) и количество жизнеспособных клеток оценивали с помощью гемоцитометра. Клетки трипсинизировали и трижды промывали в среде DMEM с низким содержанием глюкозы (Invitrogen, г. Карлсбад, штат Калифорния) (данная среда не содержит сыворотки и добавки). Культуры клеток, полученных из пуповины, клеток, полученных из плаценты, и фибробластов человека (пассаж 11) трипсинизировали и дважды промывали в среде Лейбовица L-15 (Invitrogen, г. Карлсбад, штат Калифорния). Клетки (2×105 клеток на инъекцию) повторно суспендировали в 2 микролитрах среды Лейбовица L-15 (Invitrogen, г. Карлсбад, штат Калифорния).
Оперирование животных. Все процедуры проводили в соответствии с протоколами, утвержденными Комитетом по содержанию и использованию лабораторных животных (Centre de Recherche du CHUL, Local RC-9800, 2705 Blvd Laurier, Ste-Foy, Квебек, Канада G1V 4G2). Трансплантацию проводили под кетамин/ксилазиновой (75/10 мг/кг интраперитонеально) анестезией на животных, установленных на раме для стереотаксической хирургии малых животных (модель 900, David Kopf Instruments, г. Тахунга, штат Калифорния). Каждую трансплантацию проводили путем инфузии клеток через скошенную (45°) иглу размером 26 из нержавеющей стали, установленную на микрошприце емкостью 5 мкл (Hamilton Company, г. Рино, штат Невада), установленном на инфузионном насосе с автоматическим блоком для микроинъекций (модель UMPII, David Kopf Instruments, г. Тахунга, штат Калифорния). Инфузию клеток (или культуральных сред) проводили в полосатое тело со скоростью 1,0 мкл/мин/место при средней концентрации 100000 клеток/мкл с общим объемом 2 мкл на животное в соответствии со следующими координатами (если концентрация оказывалась ниже, объем инъекции соответствующим образом корректировали, чтобы единообразно трансплантировать одно и то же количество клеток в каждое животное): A=0,5 мм перед теменем, L=3,0 мм в сторону от срединной линии, V=-4,7 мм (место 1) и -4,5 мм (место 2) вертикально ниже твердой мозговой оболочки, при резцовом упоре, установленном на -2,5 мм ниже интерауральной линии. После завершения инъекции клеток иглу оставляли на месте еще на 3 мин. для диффузии клеток перед выведением иглы. Крысам ввели 30 мг/кг циклоспорина A (25 мг/мл CsA, разбавленного в оливковом масле, Bedford Laboratories, г. Бедфорд, штат Огайо) за сутки до трансплантации и получали 15 мг/кг/сутки CsA в течение всего оставшегося периода эксперимента путем подкожной инъекции. Животные, выполнявшие функцию контролей, не получали CsA. Перед операцией все животные получили 10 мл лактата и 0,03 мг/кг бупренорфина в качестве предоперационной обработки и дважды в сутки (лактат один раз в сутки) в течение трех суток после операции.
Поведенческий тест на провоцируемое апоморфином вращение. Изначально с крысами проводили тест на провокацию за две с половиной недели до трансплантации и затем за 2 суток перед различными моментами умерщвления (4, 8 и 16 недель после трансплантации). Каждая крыса получала дозу 0,05 мг/кг апоморфина в виде интраперитонеальной инъекции, и ее сразу помещали в устройство для провокации апоморфином (сферическую чашу). Оснастку, состоящую из одного эластичного ремня, помещенного вокруг грудной клетки крысы сразу за локтями, закрепляли с помощью застежки на липучке на веревке длиной приблизительно 40,6 см (16 дюймов), соединенной с ротометром, подключенным к компьютеру, который фиксировал общее количество совершаемых крысой полных поворотов тела и направление вращения (Rotometer System, San Diego Instruments, г. Сан-Диего, штат Калифорния). Используемый для анализа конечный результат получали путем вычитания общего количества ипсилатеральных поворотов из общего количества контралатеральных поворотов. Статистический анализ проводили в модели повторных измерений ANOVA с помощью статистической программы SAS.
Повороты головы. Перед трансплантацией животных тестировали для установления базовой линии и затем проверяли каждые две недели после трансплантации. У каждой крысы оценивали положение головы относительно тела в течение 60 секунд каждые пять минут по три раза за тест. Общее количество отклонений положения головы (отклонение более 10º считали поворотом головы) фиксировали по отдельности для левой и правой сторон и для обеих сторон рассчитывали среднее за три минуты количество поворотов в минуту. Оценку проводили независимо от активности крысы, включая подъем на задние лапы и чистку. Для определения степени восстановления поведения рассчитывали разность между средним количеством левых и правых поворотов, как и в случае провокации апоморфином статистический анализ проводили в модели повторных измерений ANOVA.
Тест обученного доставания пищи лапой. Способность обученного доставания пищи передней лапой оценивали через 4, 8 и 16 недель после трансплантации в соответствии с ранее опубликованным протоколом (Moore et al., 2001 г., Exp Neurol. 2001 г., 172(2): 363-76). Устройство для тестирования представляет собой изготовленный из оргстекла контейнер с двумя отделениями. Основная камера (длина 300 мм × высота 115 мм × ширина 103 мм), в которую помещали крысу, имела еще один скользящий компонент с отверстиями для доступа воздуха. От данной камеры отходит более узкая часть (185 мм × 115 мм × 60 мм), по центру которой находится платформа шириной 22 мм, проходящая по всей длине на высоте 62 мм. С обеих сторон платформы находится желобок размером 19 мм, в котором размещается лестница с семью ступеньками. Крыса забирается на платформу и собирает гранулы корма массой 45 мг из маленьких лунок в каждой ступеньке. Платформа, на которую забирается крыса, достаточно узкая, чтобы не позволить животному разворачиваться и доставать корм из правого желобка левой лапой и наоборот. Чтобы гарантировать, что крыса не сможет просто выскрести гранулы корма наверх вдоль стенки платформы, верхняя часть платформы выступает на 5 мм с каждой стороны (Moore et al., 2001 г.). Данный вариант теста проводят в течение 12 суток, разделенных на четыре фазы: привыкание, тренировка, лишение пищи и тестирование. Привыкание: (1-3 сутки) крыс помещают в пустые боксы на 20 минут каждые сутки, после чего их достают из боксов и возвращают в свои домашние клетки. Тренировка: (4-5 сутки) крыс помещают в клетки для тестирования и на ступени 2-6 лестницы выкладывают по 6×45 мг гранул корма, всего по 30 гранул с каждой стороны для каждой стадии тестирования. Крыс оставляют в устройстве на 20 минут, после чего их возвращают в домашние клетки. Лишение пищи: (6-7 сутки) животных лишают пищи и позволяют есть только в течение четырех часов непосредственно после тестирования/тренировки каждые сутки (вода все время остается доступной). Тестирование: (8-12 сутки) каждую крысу тестируют по 20 минут в течение пяти суток. На ступеньки 2-6 помещают корм в соответствии с протоколом тренировки. После тестирования животных достают из клеток, возвращают в домашние клетки и позволяют свободно кормиться в течение четырех часов. Количество гранул корма, взятых и съеденных каждой крысой, подсчитывают и фиксируют по отдельности как для правой, так и для левой лап. Соотношения чисел усредняли по последним 5 суткам тестирования, получая средний результат для каждой крысы, и анализировали в модели повторных измерений ANOVA.
Гистологическое исследование. При умерщвлении животных глубоко анестезировали интраперитонеальной инъекцией пентобарбитала (60 мг/мл, [0,1 мл/100 г]) и перфузировали внутрисердечно 0,9% физиологическим раствором, содержащим 0,1% гепарина, и затем 4% раствором параформальдегида (PFA) в 0,1 М фосфатно-солевом буфере (ФСБ) с pH 7,4. После перфузирования головной мозг собирали и фиксировали в течение шести часов в 4% PFA, после чего помещали в 20% раствор сахарозы в ФСБ. Мозг нарезали на срезы толщиной 35 мкм на замораживающем микротоме (Leica Microsystems, г. Монреаль, Канада) и последовательно собирали и сохраняли в антифризе, затем извлекали и промывали в ФСБ для каждого эксперимента.
Для иммуногистологического анализа срезы трижды промывали в ФСБ (0,1 M pH 7,4) и предварительно инкубировали в растворе, содержащем 0,4% Triton X-100, 5% NGS в ФСБ в течение 60 минут. Затем срезы инкубировали в течение ночи при 4°C с первичными антителами в соответствии со следующими комбинациями: кроличий анти-Iba-1 (Wako Pure Chemicals Industries, г. Ричмонд, штат Вирджиния; 1:1000) и мышиный анти-ED1 (Serotech, г. Роли, штат Северная Каролина; 1:1000), кроличий анти-глиофибриллярный кислый белок (GFAP, DakoCytomation, г. Миссиссога, провинция Онтарио; 1:4000) и мышиные анти-человеческие митохондрии (Chemicon, г. Темекула, штат Калифорния; 1:500) или кроличий анти-GABA (Chemicon, г. Темекула, штат Калифорния; 1:200) (также в комбинации с анти-человеческими митохондриями), разбавленными в ФСБ с 0,4% Triton X-100. После промывки в ФСБ срезы инкубировали с вторичными антителами; Alexa Fluor® 488 козий анти-кроличий высокой очистки от кросс-реактивных антител (Molecular Probes, г. Юджин, штат Орегон; 1:200) и Rhodamine Red-X козий антимышиный высокой очистки от кросс-реактивных антител (Jackson Immunoresearch, г. Вест Гроув, штат Пенсильвания; 1:200) в ФСБ в течение двух с половиной часов при комнатной температуре. После промывания срезы инкубировали в ФСБ, содержащем 0,022% DAPI (Molecular Probes, г. Юджин, штат Орегон), промыли и установили на покрытые желатином предметные стекла, закрыли изготовленными в лаборатории средами для заключения ткани с DABCO (поливиниловый спирт, DABCO, Tris-HCl 1,0 M, pH 8,0, дистиллированная вода, глицерин) и герметизировали лаком для ногтей. Флуоресцентное окрашивание оценивали с помощью флуоресцентного микроскопа Nikon i90 с установленной монохромной камерой Hamamatsu 1394 ORCA-285 и анализировали с помощью программного обеспечения Simple PCI версии 5.3.0.1102 (Compix Inc Imaging Systems, штат Пенсильвания, США).
В случае двойного иммунофлуоресцентного окрашивания, когда оба первичных антитела были получены в том же организме-хозяине, срезы промывали 0,1 M ФСБ и предварительно инкубировали в 0,1 M ФСБ, содержащем 1% бычьего сывороточного альбумина (БСА) и 0,4% Triton X-100 (оба производства Sigma, г. Сент-Луис, штат Миссури). После часовой инкубации при комнатной температуре с первым первичным антителом, мышиным анти-виментином (Sigma, г. Сент-Луис, штат Миссури; 1:5000), антителом против изоформы бета-III тубулина (Chemicon, г. Темекула, штат Калифорния) или мышиным анти-нейрон-специфичным ядерным белком (NeuN, Chemicon, г. Темекула, штат Калифорния; 1:5000) срезы промывали и инкубировали в течение часа в растворе вторичного антитела, конъюгированного с FITC козьего антимышиного IgG (Santa Cruz Biotechnology, г. Санта-Крус, штат Калифорния; 1:400) в 0,1 M ФСБ, 1% БСА и 0,4% Triton X-100. После промывания в ФСБ срезы инкубировали в течение одного часа с 5% нормальной мышиной сывороткой (Jackson Immunoresearch, г. Вест Гроув, штат Пенсильвания) и затем еще раз промывали перед инкубацией с избытком содержащего фрагменты Fab антитела к видам организмов-хозяев первичных антител (20 мкг/мл, Jackson Immunoresearch, г. Вест Гроув, штат Пенсильвания) в течение одного часа с последующим промыванием ФСБ. Затем срезы инкубировали в течение одного часа при комнатной температуре со вторым первичным антителом, мышиными анти-человеческими митохондриями (Chemicon, г. Темекула, штат Калифорния; 1:500) в ФСБ, содержащем 1% БСА и 0,4% Triton X-100. После нескольких промываний ФСБ срезы, наконец, инкубировали с конъюгатом родамина с козьим антимышиным IgG (Santa Cruz Biotechnology, г. Санта-Крус, штат Калифорния; 1:400) в 0,1 M ФСБ, 1% БСА, 0,4% Triton X-100 в течение одного часа. Затем срезы промывали и инкубировали с DAPI в течение 7 минут (Molecular Probes, г. Юджин, штат Орегон), устанавливали на стекла и запечатывали, как описано выше.
Для тирозингидроксилазного (TH) иммунного окрашивания срезы трижды промывали в 0,1 M ФСБ, pH 7,4, и помещали в 3% раствор пероксида на 30 минут при комнатной температуре. Затем срезы промывали 0,1 M ФСБ и предварительно инкубировали в растворе, содержащем 0,1 M ФСБ, 0,1% Triton X-100 (Sigma, г. Сент-Луис, штат Миссури) и 5% нормальной козьей сыворотки (NGS, Wisent Inc., г. Сен-Жан-Батист-де-Рувиль, провинция Квебек, Канада), в течение 30 мин. при комнатной температуре. Срезы инкубировали в течение ночи при 4°C в анти-TH (Pel-Freez, г. Роджерс, штат Арканзас; 1:5000) в ФСБ, 0,1% Triton X-100 и 5% NGS. После ночной инкубации срезы промывали 0,1 M ФСБ и инкубировали в течение 1 часа при комнатной температуре в растворе ФСБ, содержащем 0,1% Triton X-100, 5% NGS и биотинилированные козьи анти-кроличьи антитела (Vector Laboratories, г. Берлингтон, провинция Онтарио; 1:1500). После трехкратного промывания в 0,1 M ФСБ срезы помещали в раствор авидин-биотинпероксидазного комплекса (ABC Elite kit, Vector Laboratories, г. Берлингтон, провинция Онтарио) на 1 час при комнатной температуре. Антитела проявляли путем помещения срезов в трис-буферный раствор, содержащий 0,05% 3,3’-диаминобензидинтетрагидрохлорида (DAB, Sigma, г. Сент-Луис, штат Миссури) и 0,1% 30%-ного раствора перекиси водорода при комнатной температуре. Реакцию останавливали промывкой в 0,05 M трис-буферном растворе и с последующими промываниями в ФСБ. Срезы помещали на покрытые желатином предметные стекла, высушивали на воздухе в течение ночи, дегидратировали нарастающей концентрацией этанола и заливали средами для заключения ткани с DPX (Electron Microscopy Science, г. Хатфилд, штат Пенсильвания).
Результаты
Контроль веса. Вес животных контролировали ежедневно. Как показано на фиг.1, животные демонстрировали медленный и постоянный прирост веса через 2 недели после трансплантации. Потеря веса, отмеченная на 3-4, 7 и 15 неделях после трансплантации, была просто связана с периодом голодания, необходимым для проведения теста на лестнице. Продолжение небольшой потери веса после первой провокации апоморфином, возможно, связано с временной потерей аппетита. Двоих животных для клеток типа 1, которых запланировано умертвить через 8 недель, поменяли на двух крыс, которых запланировано умертвить через 4 недели, из-за их прогрессирующей потери веса (крысы №№ 27, 49). Через 8 недель после трансплантации крысу № 60 для клеток типа 1 пришлось умертвить на одни сутки раньше запланированного срока по аналогичным причинам. Через 10 недель после трансплантации еще у n=2 животных для клеток типа 2 наблюдали диарею и существенную потерю веса. Данные животные получали ежедневные инъекции лактата, но были обнаружены мертвыми в своей клетке через 24 часа после принятия данных дополнительных мер. Еще у двух животных из группы 2 начали проявляться аналогичные проблемы со здоровьем, и они были умерщвлены раньше запланированного срока. Оставшиеся n=2 животных из группы 2 были умерщвлены в запланированное время (через 24 часа после последней перфузии животных из группы 2). Крысу № 40 из группы 3 также умертвили на 13 неделе экспериментального протокола из-за появившихся проблем со здоровьем, аналогичных наблюдавшимся у животных в группе 2. Крысу № 66 из группы 1 обнаружили мертвой в своей клетке через 15 недель. Наблюдавшиеся перед этим симптомы напоминали те, что имели место у нездоровых животных из групп 2 и 3. Всего животных из группы 2 (оставшиеся n=6) умертвили через 10 недель после трансплантации, животных из группы 1 и 3 умертвили через 16 недель после трансплантации, как и было запланировано (n=5 для каждой группы). Сокращения: BB: поведенческая базовая линия; TP: трансплантация.
Провокация апоморфином. Все четыре группы (клетки 1, 2, 3 типа и несущая среда) анализировали в модели повторных измерений ANOVA (переменная: количество поворотов). Анализ показал, что существенным оказался только фактор «время» (p=0,0048), указывая на эффект «времени» для всех групп. Множество сравнений показало существенное восстановление функции (уменьшение количества поворотов) во всех группах с момента времени 0 (базовая линия) к 4 неделям после трансплантации (фиг.2). Данное уменьшение сохранялось с течением времени после 4 недель. Последний момент времени, составляющий 16 недель после трансплантации, невозможно было проанализировать в модели повторных измерений ANOVA, поскольку животных из группы 2 умертвили до данного момента (фиг.2).
К анализу в модели повторных измерений ANOVA также добавляли группу из пяти животных, по отдельности пораженных 6-OHDA, но с которыми не проводили никаких хирургических вмешательств (трансплантации). При добавлении данной группы к расчету в модели повторных измерений ANOVA только взаимодействие группа/время имело тенденцию к повышению статистической значимости (p=0,0985). Возможно, более существенное количество животных могло бы усилить данную тенденцию до статистически значимых результатов. Принимая во внимание, что данная тенденция указывала на восстановление функции с течением времени, дополнительный анализ показал, что только клетки пуповины (клетки типа 1) давали существенные положительные эффекты с течением времени (p=0,0056) и что пораженные животные, с которыми не проводили внутричерепного хирургического вмешательства, продемонстрировали тенденцию к отсутствию восстановления функции с течением времени (данный результат не является статистически значимым при p=0,0655).
Повороты головы. Повороты головы отражают общее количество ипсилатеральных поворотов, совершаемых животными после трансплантации клеток. Во всех тестируемых моментах времени (2, 4, 6, 8, 10, 12, 14 и 16 недель после трансплантации) не обнаружено статистически значимых различий между животными с трансплантированными клетками и животными, получившими только несущую среду (фиг.3). Повороты головы являются чисто субъективными и определяют естественную тенденцию крыс поворачивать голову влево или вправо после повреждения. Все группы анализировали в модели повторных измерений ANOVA (переменная: разность между количеством поворотов головы влево и вправо). С увеличением прошедшего времени в данном исследовании наблюдали улучшение в плане отсутствия предпочтений в повороте головы, однако это проявлялось во всех группах (фиг.3).
Тест на лестнице. В данном тесте не обнаружили статистически значимых различий между животными с трансплантированными клетками и контрольными группами. В тесте на лестнице измеряли потребление корма за 20-минутные периоды тестирования, которое требовало точных перемещений для взятия корма со ступенек. Все группы анализировали в модели повторных измерений ANOVA (переменная: отношение количества съеденных и взятых гранул корма). С помощью данного теста определили, что со временем наблюдаются различия в пищевом поведении, однако статистически значимых различий между группами выявить не удалось (фиг.4).
Иммунное окрашивание. Окрашенные гематоксилином и эозином (H и E) срезы показали хорошее приживление клеточного трансплантата через 1 сутки после трансплантации. Трансплантированные клетки в местах трансплантации идентифицировали с помощью окрашивания антигеном к ядерному белку человека вплоть до 8 недель после трансплантации, хотя количество человеческих клеток уменьшалось со временем после трансплантации. В настоящее время нет данных, подтверждающих дифференцирование клеток, полученных из послеродового материала, в нейрональный фенотип после трансплантации in vivo в данной модельной системе.
Клеточные трансплантаты анализировали на присутствие микроглиального маркера Iba-1. Как показано на фиг.5а, Iba-1 обильно экспрессировался каждым клеточным типом, особенно в сравнении с контрольными группами, получавшими несущую среду. Обнаружили тенденцию к падению экспрессии маркера Iba-1 со временем (фиг.5а). Оценка результатов окрашивания на ED-1 у трансплантированных животных продемонстрировала, что после трансплантации наблюдается выраженный макрофаговый ответ, уровень окрашивания на ED-1 уменьшался со временем во всех группах кроме группы животных, которым трансплантировали фибробласты (фиг.5b). Результаты окрашивания на DAPI по существу оставались постоянными на протяжении исследования (фиг.5c).
Аналогичным образом определение уровней глиофибриллярного кислого белка (GFAP) в трансплантатах для определения количества реактивных астроцитов после трансплантации продемонстрировало, что реакционные астроциты первоначально идентифицировались после трансплантации или введения несущей среды, причем данный эффект со временем снижался (фиг.6а). В соответствии с другими наблюдениями, показывающими дифференцирование клеток в трансплантате, обнаружили обильную экспрессию виментина в клетках через четыре недели после трансплантации, но экспрессия постоянно падала в течение следующих двенадцати недель (фиг.6b).
Результат окрашивания на тирозингидроксилазу человека оказался отрицательным. Таким образом, ни клетки пуповины, ни клетки плаценты не дифференцировались в дофаминергические клетки в используемых условиях обработки.
Обзор
Результаты продемонстрировали, что имплантация клеток пуповины обеспечила улучшение функции со временем в модели 6-OHDA болезни Паркинсона по результатам оценки поведенческой восприимчивости к провокации апоморфином. Для определения влияния на активацию различных нейронных цепей и/или механизмов провели как тест на лестнице, так и тест на повороты головы. По полученным параметрам данных тестов никаких различий не выявлено, таким образом, в настоящее время механизм, связанный с положительной динамикой, наблюдавшейся на 4 и 8 неделе у животных с трансплантированными клетками пуповины, остается невыясненным.
Иммуногистохимическое окрашивание не показало никаких признаков дифференцирования клеток после их трансплантации. В данных исследованиях невозможно было выявить никакой нейрональной или, более конкретно, никакой дофаминергической дифференциации. Таким образом, невозможно было получить никаких доказательств дифференцирования клеток на месте трансплантации. Это дополнительно указывает на то, что улучшения в поведении, наблюдаемые при провокации апоморфином после трансплантации клеток пуповины, скорее всего, связаны с трофическим ответом и не являются результатам регенеративного потенциала клеток.
TH-иммуноположительных клеток не наблюдали ни для одного клеточного типа ни в один момент времени. Однако TH является не единственным клеточным способом выработки дофамина. Дофамин может вырабатываться независимо из тирозингидроксилазы по тирозиназному способу. Более того, в присутствии тирозиназы дофамин ковалентно модифицирует и инактивирует тирозингидроксилазу. Кроме того, трансплантированные клетки могут обеспечить переработку DOPA (поступающей с пищей аминокислоты) в плазме после приема пищи в DOPA.
ПРИМЕР 19
Наборы для мультиплексного анализа цитокинов RayBio® и BD Powerblot™
Для анализа экспрессии 120 белков в клетках, полученных из послеродового материала, и лизатах использовали набор антител для мультиплексного анализа цитокинов человека RayBio® Human Cytokine Antibody Array C Series 1000. Данный анализ позволил получить характеризацию клеток UTC и идентифицировал спектр экспрессии ключевых трофических факторов для данных клеток.
Материалы и способы
Выращивание и сбор клеток. Клетки, полученные из пуповины, высевали с плотностью 5000 клеток на см2 в покрытые желатином колбы с ростовыми средами и размножали в течение 3-4 суток (целевая плотность для сбора 25000 клеток на см2). Клетки трипсинизировали, собирали и центрифугировали при 300×g в течение 5 минут. Трипсин/среды отсасывали и клетки трижды промывали фосфатно-солевым буфером (ФСБ).
Промывание и разливание клеток. После промывания клетки повторно суспендировали в количестве 107 клеток/мл в ФСБ и разливали в виде аликвот объемом 1 мл в покрытые силиконом стерильные микроцентрифужные пробирки объемом 1,5 мл. Клетки центрифугировали при 300×g в течение 5 минут и отсасывали ФСБ. Клетки либо лизировали и анализировали с использованием набора антител, либо лизировали и лиофилизировали для анализа.
Подготовка лиофилизированных образцов. Три партии клеток (партии клеток UTC L040405, L052505, L050505) подготовили к последующей лиофилизации путем погружения в жидкий азот (LN2) на 60 секунд. Затем пробирки извлекли из жидкого азота и немедленно погрузили в водяную баню с температурой 37°C на 60 секунд или до размораживания (максимальное время инкубации 3 минуты). Данный процесс повторили еще дважды. Замороженные и затем размороженные образцы центрифугировали в течение 10 минут при 13000×g и 4°C и поместили на лед. Супернатант из каждой пробирки удалили. Для определения общего содержания белков лизат разводили в ФСБ и разведенный раствор анализировали методом Бредфорда.
Для лиофилизации множество стерильных криопробирок объемом 1,5 мл, меченных как содержащие лизат, помещали в автоклавированный и охлажденный термоблок. В криопробирки помещали аликвоты супернатанта от лизатов с определенной общей концентрацией белков. Термоблок, содержащий незакрытые криопробирки, в асептических условиях помещали в автоклавированный неиспользованный конверт для автоклавирования. Конверт загружали в лиофилизатор.
Тестируемые материалы с лизатами загружали в закрываемую лотковую сушилку Dura-Stop MP Stoppering Tray Dryer производства компании FTS Systems и лиофилизировали с помощью следующей программы подъема температуры. Все стадии сушки проводили при скорости подъема температуры 2,5°C/минуту при вакууме 100 мТорр.
Подготовка клеточных гранул. Замороженные клеточные гранулы (партии 063004B, 022803, 050604B, 072804, 120304, 071404C, 090304) лизировали с использованием смеси 1:1 буферного раствора RIPA (50 мМ Tris HCl, pH 8, 150 мМ NaCl, 1% NP-40, 0,5% дезоксихолата натрия и 0,1% ДСН) и буферного раствора для лизиса клеток, поставляемого в наборе для мультиплексного анализа цитокинов RayBio® серии 1000.1 (Raybiotech Inc. г. Норкросс, штат Джорджия). Для достижения полного лизиса клеток использовали стеклянные гранулы (Sigma, г. Сент-Луис, штат Миссури). Концентрацию белков измеряли с использованием набора для мультиплексного определения белков BCA (Pierce Biotechnology, Inc., г. Рокфорд, штат Иллинойс).
Мультиплексный анализ с набором RayBio®. Наборы для мультиплексного анализа RayBio® VI и VII, которые составляют набор для мультиплексного анализа серии 1000.1, обрабатывали в течение ночи с равными количествами белка из каждого образца. Остальной протокол проводили в соответствии с рекомендациями производителя. Для определения рассматриваемых белков качественно анализировали пятна на мембране. Для количественного сравнения разных образцов данные пятна можно было проанализировать с использованием денситометрии с подтверждением изменений в экспрессии с использованием анализа ИФА.
Результаты
Всего анализировали десять различных популяций клеток UTC. Качественно идентифицировали сорок восемь белков, перечисленных в таблице 19-1. Некоторые белки экспрессировались в относительно высоких концентрациях во всех тестированных образцах, тогда как экспрессия других наблюдалась только в определенных образцах.
Обзор
Мультиплексный анализ с помощью набора RayBio® подтвердил экспрессию белков, ранее идентифицированных с использованием мультиплексного анализа генов и/или ИФА. Идентифицированы различные трофические факторы, благоприятные для лечения конкретных заболеваний. Например, в клетках UTC идентифицированы FGF, TGF-b и GCSF, и данные факторы роста ранее были связаны с улучшениями в животных моделях острого инсульта и восстановления после инсульта. Кроме того, в клетках UTC идентифицированы BDNF, BMP-4, BMP-6 и TGF-b1, которые положительно ассоциированы с болезнью Паркинсона. Все представленные данные проанализированы на качественном уровне, количественный анализ экспрессии рассматриваемых белков планируется провести в ближайшее время.
ПРИМЕР 20
Экспрессия теломеразы в клетках, полученных из пуповины
Функции теломеразы состоят в синтезе теломерных повторов, которые защищают целостность хромосом и продлевают репликативно-активную жизнь клеток (Liu, K, et al., PNAS, 1999 г.; 96: 5147-5152). Теломераза состоит из двух компонентов - теломеразного РНК-шаблона (TERC) и теломеразной обратной транскриптазы (hTERT). Регулирование теломеразы определяется транскрипцией hTERT, но не TERC. Таким образом, полимеразная цепная реакция (ПЦР) в реальном времени для мРНК hTERT является общепринятым способом определения теломеразной активности клеток.
Выделение клеток. Для определения выработки теломеразы в клетках, полученных из ткани пуповины человека, провели эксперименты по ПЦР в реальном времени. Клетки, полученные из ткани пуповины человека, подготовили в соответствии с описанными выше примерами. По существу пуповины, полученные в Национальном центре по обмену материалами для исследования заболеваний (NDRI, г. Филадельфия, штат Пенсильвания) после нормально прошедших родов, промывали для удаления крови и продуктов распада клеток и механически диссоциировали. Затем ткань инкубировали с расщепляющими ферментами, включая коллагеназу, диспазу и гиалуронидазу, в культуральной среде при 37°C. Клетки, полученные из ткани пуповины человека, культивировали в соответствии со способами, описанными в приведенных выше примерах. Мезенхимальные стволовые клетки и нормальные фибробласты дермы кожи (cc-2509 партия № 9F0844) получили от компании Cambrex, г. Уолкерсвилл, штат Мэриленд. Плюрипотентные клетки nTera-2 клеточной линии эмбриональной карциномы (тератомы) яичка человека (NTERA-2 cl.Dl) (смотрите Plaia et al., Stem Cells, 2006 г.; 24(3): 531-546) приобрели в культуральной коллекции ATCC (г. Манассас, штат Вирджиния) и культивировали в соответствии с описанными выше способами.
Выделение общей РНК. РНК экстрагировали из клеток с использованием набора RNeasy® (Qiagen, г. Валенсия, штат Калифорния). РНК элюировали 50 микролитрами обработанной DEPC воды и хранили при температуре -80°C. Обратную транскрипцию РНК проводили с использованием случайных гексамеров, применяя реагенты для обратной транскрипции TaqMan® (Applied Biosystems, г. Фостер Сити, штат Калифорния) при 25°C в течение 10 минут, при 37°C в течение 60 минут и при 95°C в течение 10 минут. Образцы хранили при -20°C.
ПЦР в реальном времени. ПЦР проводили на образцах кДНК с использованием продуктов для экспрессии генов Assays-on-Demand™ компании Applied Biosystems (также известные как наборы для определения экспрессии генов TaqMan® Gene Expression Assays) в соответствии с инструкциями производителя (Applied Biosystems). Данный доступный в продаже набор широко используют для анализа клеток человека на теломеразу. Вкратце, hTERT (ген теломеразы человека) (HsOO162669) и GAPDH человека (внутренний контроль) смешивали с кДНК и универсальным мастер-миксом для ПЦР TaqMan®, при этом использовали секвенатор 7000 с программным обеспечением ABI Prism 7000 SDS (Applied Biosystems). Использовали следующие условия термоциклирования: сначала 50°C в течение 2 минут и 95°C в течение 10 минут, затем 40 циклов по 95°C в течение 15 секунд и 60°C в течение 1 минуты. Данные ПЦР анализировали в соответствии с рекомендациями производителя.
Клетки, полученные из ткани пуповины человека (номер доступа ATCC PTA-6067), фибробласты и мезенхимальные стволовые клетки человека анализировали на hTERT и 18S РНК. Как показано в таблице 20-1, hTERT и, следовательно, теломеразу, не обнаружили в клетках, полученных из ткани пуповины человека.
Анализировали клетки, полученные из ткани пуповины человека (изолят 022803, номер доступа ATCC PTA-6067), и клетки nTera-2, и результаты показали отсутствие экспрессии теломеразы в двух партиях клеток, полученных из ткани пуповины человека, тогда как клеточная линия тератомы экспрессировала теломеразу на высоком уровне (таблица 20-2).
Следовательно, можно сделать вывод, что клетки, полученные из ткани пуповины человека, которые составляют предмет настоящего изобретения, не экспрессируют теломеразу.
ПРИМЕР 21
Использование клеток, полученных из ткани пуповины, для лечения АБС
В настоящем исследовании оценивали модифицирующие течение заболевания эффекты клеток, полученных из ткани пуповины человека (клеток hUTC), либо в начале, либо через неделю после начала заболевания для моделирования клинической парадигмы, в рамках которой пациентам с симптомами АБС могут вводиться клетки hUTC. В частности, целью настоящего исследования являлось определение эффективности клеток, полученных из ткани пуповины человека (клеток hUTC), в крысиной модели SOD1 G93A амиотрофического бокового склероза (АБС). Оценивали эффект введения клеток hUTC на выживаемость и локомоторную функцию животных после подоболочечной инъекции клеток hUTC.
Основанием для локального введения клеток hUTC с использованием подоболочечной инъекции служило стремление доставить клетки непосредственно в место зарождения заболевания. Кроме того, при данном способе введения обходится гематоэнцефалический барьер, что обеспечивает быстрый доступ тестируемого препарата к потенциальным реакционным участкам спинного мозга(Ochs G. et al. (1999 г.) J Pain Symptom Manage. 18: 229-32). Функциональные оценки локомоторной активности проводили раз в неделю по BBB-шкале (Basso-Beattie-Bresnahan) (Basso DM. et al. (1996 г.) Exp. Neurol. 139: 244-256) и теста «наклонная плоскость» (Rivilin AS, Tator CH. (1977 г.) J Neurosurg 47(4): 577; Lindberg RL et al. (1999 г.) J. Clin Invest. 103(8): 1127-1134).
В настоящее время модели грызунов SOD1 G93A являются единственным типом животной модели для АБС. Они представляют собой эффективный, экономически приемлемый способ скрининга новых терапевтических подходов для данного заболевания, что продемонстрировано другими исследователями при оценке эффективности различных агентов/соединений при лечении АБС (Karussis et al. (2010 г.) Arch Neurol. 67(10): 1187-94; Xu et al. (2009 г.) J Comp Neurol. 514(4): 297-309; Xu et al. (2006 г.) Transplantation. 82(7): 865-75; Garbuzova-Davis S. et al. (2003 г.) J. Hematother Stem Cell Res. 12(3): 255-70; Galán et al. (2010 г.) Neurologia. 25(8): 467-469; Kim et al. (2010 г.) Neurosci Lett. 468(3): 190-4; Gros-Louis et al. (2010 г.) J Neurochem. 113(5): 1188-99; Israelson A et al. (2010 г.) Neuron. 26; 67(4): 575-87).
Способыиматериалы
Самцов крыс линии SOD1 G93A, предоставленных Dr. David S. Howland, Университет штата Вашингтон, г. Сиэтл, спаривали с самками крыс линии N (SD) компании Taconic (г. Джермантаун, штат Нью-Йорк). Крыс спаривали в течение недели в парах один самец: одна самка. Помет забирали и генотипировали в возрасте 21 сутки, выявляя положительно трансгенных детенышей для использования в исследовании. Колонию размножали путем обратных скрещиваний детенышей-самцов одного помета с исходными самками для снижения фенотипической вариации. Все процедуры по уходу за животными и хирургическому вмешательству в настоящем исследовании проводили в соответствии с протоколами, утвержденными Комитетом по содержанию и использованию лабораторных животных медицинских учреждений Университета Джона Хопкинса.
Клетки hUTC (выделенные из ткани пуповины человека) размораживали из сохраненного замороженного образца, как описано в патенте США № 7510873, и разводили в фосфатно-солевом буфере (ФСБ). Клетки центрифугировали при 1500×g в течение 5 минут, затем повторно суспендировали в ФСБ и подсчитывали. Концентрацию клеток доводили до 20 миллионов клеток на миллилитр и вводили подоболочечно каждой крысе по 50 микролитров данной суспензии.
В исследовании использовали животных, положительных по гену супероксиддисмутазы 1 (SOD1). Данных животных взвешивали дважды в неделю для определения момента, когда вес тела снижался в течение двух последовательных измерений, что считали началом заболевания. Это является чувствительным и очень объективным показателем для определения начала заболевания в моделях АБС грызунов (Xu et al. (2006 г.) Transplantation. 82(7): 865-75). Крыс с зафиксированным началом заболевания случайно распределяли попарно в группы, получающие клетки или несущую среду.
Исследование проводили на двух группах исследования (таблица 21-1). Каждая группа исследования состояла из двух групп. Группа 1 группы исследования 1 получала одну подоболочечную инъекцию фосфатно-солевого буфера (ФСБ, контрольная группа с несущей средой) в момент начала заболевания. Группа 2 группы исследования 1 получала 1 миллион клеток hUTC приблизительно в момент начала заболевания. Клетки вводили подоболочечной инъекцией в ФСБ. Группа 3 группы исследования 2 получала одну подоболочечную инъекцию ФСБ (контрольная группа с несущей средой) через 1 неделю после приблизительного момента начала заболевания. Группа 4 группы исследования 2 получала 1 миллион рекомбинантных клеток hUTC через 1 неделю после приблизительного момента начала заболевания. Клетки вводили подоболочечной инъекцией в ФСБ.
Для сведения к минимуму повреждения и максимального ускорения восстановления животного использовали способ чрескожной инъекции для доставки клеток подоболочечно в спинномозговую жидкость поясничной цистерны у крыс SOD1. Животных устанавливали на устройстве для стереотаксической хирургии компании Kopf и анестезировали с использованием газовой анестезии (изофлюран:кислород:оксид азота = 1:33:66). Поясничную область выбривали и под нижнюю часть живота животного подкладывали цилиндрический поддерживающий элемент для растяжения межпозвоночных промежутков в поясничном отделе позвоночника. Клетки вводили с использованием микрошприцев объемом 200-500 мкл с иглой размером 25 длиной 2,54 см (1 дюйм). Иглу с микрошприцем, содержащим 50 микролитров клеточной суспензии, медленно вставляли в подпаутинное пространство в межпозвоночных промежутках L4-5 или L6-S1. При наблюдении физических признаков раздражения заднего отдела спинного мозга (т.е. отдергивания хвоста или задней лапы) клетки медленно вливали в подпаутинное пространство в течение 1 минуты. Шприц оставляли на месте в течение 2 минут и затем извлекали иглу. Точку вхождения иглы зажимали на 1 минуту смоченным йодом ватным тампоном, после чего животное снимали с устройства и укладывали на подогреваемую подстилку для восстановления. После пробуждения животное возвращали в свою клетку.
Прогрессирование заболевания и потенциальную терапевтическую эффективность контролировали с помощью двух функциональных параметров: (1) локомоторная активность (измеряемая раз в неделю); и (2) вес тела (измеряемый дважды в неделю). Использованные локомоторные тесты включали в себя тест по BBB-шкале (Basso-Beattie-Bresnahan) (Basso et al. (1995 г.) J Neurotrauma 1995 г.; 12(1): 1; Basso et al. (1996 г.) Exp. Neurol. 139: 244-256) и тест «наклонная плоскость» (Rivlin AS, Tator CH. (1977 г.) J Neurosurg 47(4): 577).
Для получения результатов теста по BBB-шкале животных успокаивали и адаптировали к тесту «открытое поле». Когда животное начинало постоянно передвигаться по открытому полю, двое экспертов, не знающих о том, как обрабатывали животное, проводили 4-минутное тестирование с использованием BBB-шкалы локомоторной активности, которая представляет собой 21-шаговую шкалу с высокой степенью воспроизводимости как для одного наблюдателя, так и между разными наблюдателями. Тест «открытое поле» записывали на видео. По данной записи двое экспертов количественно определяли количество движений задней лапой за 1 минуту; данный период времени составляли из фрагментов эпизодов на протяжении проведенного 4-минутного тестирования, в течение которых каждая задняя лапа полностью находилась в поле зрения камеры. Установленный временной лимит в 1 минуту обеспечил одинаковую продолжительность наблюдения за правой и левой задними лапами каждого животного, поскольку обе задние лапы не всегда одновременно полностью находятся в поле зрения камеры. В случае расхождения между результатами экспертов отснятый материал повторно просматривали на замедленной скорости. Отсутствие наблюдаемого движения задней лапой оценивали как 0, исходные баллы по шкале оценок соответствовали выделенным движениям суставов. Выставляемые баллы повышали по мере увеличения количества задействованных в движении суставов и/или увеличения интенсивности движений. По мере усиления локомоторной активности выставляли баллы за помещение лапы на подошву, удерживание веса и координацию движений передней и задней лапами. Наивысшие баллы выставляли за отделение большого пальца, устойчивость туловища и удерживание хвоста.
Тест «наклонная плоскость» представляет собой другой поведенческий тест, в котором оценивают способность животного сохранять положение на волнистой резиновой поверхности; данная поверхность последовательно поднимается с шагом 5º. Максимальный угол, при котором животное может удержать свой вес в течение 5 секунд, определяется как максимальный угол. В данном тесте проверяется сенсорная обратная связь, координация и мышечная сила, требуемая для перемещения.
Конечную стадию заболевания определяли как паралич настолько тяжелой степени, что животное не могло подняться в течение 20 секунд после укладывания на бок. На данной стадии результаты как теста по BBB-шкале, так и теста «наклонная плоскость» равны нулю, что представляет собой конечную точку, часто используемую для мутантных по SOD1 крыс и согласующуюся с требованиями Комитета по содержанию и использованию лабораторных животных Университета Джона Хопкинса.
Результаты тестов по BBB-шкале и «наклонная плоскость» анализировали методом дисперсного анализа с учетом повторных измерений (ANOVA) с последующим апостериорным анализом по критерию наименьшей значимой разности Фишера для оценки различий между группами, получавшими клетки и несущую среду. Протекание заболевания в зависимости от проведенной обработки анализировали путем сравнения возраста в момент начала заболевания и возраста гибели между двумя группами (с использованием критерия Стьюдента), а также с использованием метода анализа выживаемости Каплана-Мейера с последующей оценкой по логранговому критерию.
Гистологический анализ
Состояние двигательных нейронов животного-хозяина оценивали путем окрашивания моторных нейронов поясничного утолщения (L4-5) крезиловым фиолетовым (данные не показаны). Количество двигательных нейронов значительно уменьшилось, и многие нейроны имели дегенеративные профили как в группе, получавшей стволовые клетки, так и в контрольной группе. Это поведение согласуется с тем, что все ткани спинного мозга отбирали на конечной стадии заболевания. В паренхиме спинного мозга при иммуноцитохимическом окрашивании с использованием специфического антитела к ядерному белку человека не обнаружили выживших стволовых клеток. Вентральные корешки из межпозвоночного промежутка L4-L5 для обеих групп исследовали путем окрашивания толуидиновым синим. Существенную и аналогичную дегенерацию и демиелинизацию аксонов наблюдали как в группе, получавшей стволовые клетки, так и в контрольной группе, что согласуется с отбором данных препаратов на конечной стадии заболевания.
Результаты
Полученные кривые Каплана-Мейера показывают повышение выживаемости животных в группе, получавшей клетки, по сравнению с животными контрольной группы (n=10 для групп 1 и 2, n=8 для групп 3 и 4) в ходе обработки, изначально проведенной либо в начале заболевания (фиг.7А, группа исследования 1), либо через неделю после начала заболевания (фиг.7B, группа исследования 2), за исключением одного животного в группе 1, которое прожило дольше всех остальных животных в данном исследовании (фиг.7А). Хотя в обеих группах исследования среднее время начала заболевания не различалось среди животных, получавших несущую среду, и животных, получавших инъекцию клеток hUTC (фиг.8), средняя продолжительность жизни увеличилась после подоболочечного введения клеток hUTC, особенно в группе исследования 2, в которой продолжительность жизни животных увеличилась на 15,75 суток (2,25 недели, P<0,035) по сравнению с группой, получившей несущую среду, несмотря на аналогичный возраст начала заболевания.
Как показано на фиг.9, введение клеток hUTC в начале заболевания (фиг.9А и 9B) или через 1 неделю после начала заболевания (фиг.9C и 9D) ассоциировалось с повышение результатов в тесте по BBB-шкале и тесте «наклонная плоскость», что указывает на более медленное прогрессирование мышечной слабости у животных с трансплантированными клетками hUTC по сравнению с животными, которые получали только несущую среду. Данные результаты предполагают, что клетки hUTC могут быть способны сохранять локомоторную активность в данной модели.
Таким образом, данные, представленные в данном примере, приведенные ниже в таблице 21-2, показывают, что подоболочечная инъекция клеток hUTC может сохранять функцию двигательных нейронов и увеличивать продолжительность жизни в крысиной модели АБС.
Определения для таблицы 21-2
Начало заболевания: момент, в который обнаружено снижение веса тела при двух последовательных взвешиваниях. Это является чувствительным и очень объективным показателем для определения начала заболевания в моделях АБС грызунов (Xu L. et al. (2006 г.)).
Определение конечной стадии заболевания: стадия, на которой паралич вступает в настолько тяжелую стадию, что животное не может подняться в течение 20 секунд после укладывания на бок (конечная точка, часто используемая для мутантных по SOD1 крыс) и требовалась эвтаназия животного (Israelson at al, 2010 г.).
Продолжительность заболевания: период времени от начала заболевания до конечной стадии заболевания.
Продолжительность жизни: период времени от рождения до эвтаназии.
ПРИМЕР 22
Применение клеток, полученных из ткани пуповины, для профилактики АБС
Оценивали модифицирующие течение заболевания эффекты клеток hUTC в качестве профилактического действия до начала заболевания (возраст 10 или 12 недель). Клетки вводили внутривенной инъекцией в хвостовую вену, что является клинически предпочтительным способом введения. Функциональную оценку локомоторной активности проводили раз в неделю с использованием теста по BBB-шкале (Basso-Beattie-Bresnahan) и теста «наклонная плоскость».
Цель и объем настоящего исследования
Цель данного исследования состояла в изучении профилактической роли клеток, полученных из пуповины, способных к самообновлению и размножению, на клинических аспектах АБС у трансгенных крыс в том виде, который проявляется у крыс линии SOD1 G93A, следуя основным методологическим направлениям, опубликованным в работе Koliatsos с соавторами (Yan et al. (2006 г.) Stem Cells 24: 1976-1985; Yan et al. (2007 г.) PLoS Med 4: e39; Xu et al. (2006 г.) Transplantation 82: 865-875). Выбрали две группы исследования: (1) 10-недельную группу исследования, которая получала клетки, полученные из пуповины, или несущую среду в возрасте 10 недель; и (2) 12-недельную группу исследования, которая получала клетки или несущую среду в возрасте 12 недель. В каждой группе исследования группа А (в большинстве случаев закодированная как животные с нечетными номерами) получала клетки, полученные из пуповины, и группа B (в большинстве случаев закодированная как животные с четными номерами) получала несущую среду. Иммунную защиту от реакций трансплантата против организма-хозяина, описанную в предыдущих исследованиях лаборатории Koliatsos Lab (Yan et al. (2006 г.); Xu et al. (2006 г.)) не проводили. Приведенный ниже анализ явно учитывает сдвиг фенотипа колонии от очень вариабельного до менее вариабельного и, в конце концов, до оптимизированной популяции по отношению к началу заболевания и степени его тяжести.
Размножение животных и оптимизация фенотипа - наблюдение по началу заболевания и продолжительности жизни
Колонию крыс SOD1 размножали путем обратных скрещиваний детенышей-самцов одного помета с исходными самками для снижения фенотипической вариации. В качестве стандарта принимали, что время начала заболевания участвовавшего в размножении самца должно находиться в диапазоне 90-120 суток. В противном случае наблюдали, что фенотип потомков становился «нестабильным», т.е. время начала заболевания могло сдвигаться вплоть до возраста 180 суток и далее. Исходные животные, которых использовали для получения животных, применимых в настоящем исследовании, не попадали в данный период времени от 90 до 120 суток, что оказалось проблемой, вызвавшей существенный фенотипический дрейф в их потомстве. Для уменьшения влияния фенотипического дрейфа каждому животному, отнесенному к группе А, изначально подбирали в пару животное того же пола из того же помета и относили его к группе B. Среднее время начала заболевания для групп А и B в 10-недельной группе исследования составляло 153,15±12,71 и 154,0±13,75 суток, и продолжительность жизни для групп А и B составила 202,0±10,32 и 202,75±17,31 суток соответственно. Среднее время начала заболевания для групп А и B в 12-недельной группе исследования составляло 149,0±27,58 и 146,5±28,53 суток; продолжительность жизни для групп А и B составила 180,5±30,25 и 174,88±35,87 суток соответственно. Хотя как среднее время начала заболевания, так и продолжительность жизни в группе A были больше, чем в группе B, данные различия не были статистически значимыми. Поскольку последние две пары в 12-недельной группе исследования были составлены из генотипически близких крыс SOD1, т.е. потомков крыс SOD1, у которых время начала заболевания находилось в диапазоне 90-120 суток, данных крыс анализировали отдельно от остальных крыс той же группы исследования. Среднее время начала заболевания для данных двух пар в группах А и B составляло 114,5±0,5 и 111,0±0,61 суток. Такое различие не было статистически значимым. Продолжительность жизни для особей из данных двух пар, отнесенных в группу А, составила 133,5±0,03, т.е. на 10 суток больше, чем продолжительность жизни для особей из группы B (123,0±0,41 суток).
Характеристика кривых выживаемости
Проанализированные по логранковому критерию кривые Каплана-Мейера не показывали существенных различий между группами А и B в экспериментах с 10- и 12-недельными группами исследования (фиг.10 и фиг.11). Однако в эксперименте с 12-недельной группой исследования наблюдалась тенденция к повышению выживаемости в группе А; медианы распределений по продолжительности выживания для групп А и B составили 194 и 185 суток соответственно (смотрите фиг.11). Хотя между двумя последними парами в эксперименте с 12-недельной группой исследования не наблюдали существенного различия, анализ кривых выживаемости показывает многообещающую тенденцию к потенциально более продолжительной выживаемости в группе А (фиг.12).
Поведенческие тесты на животных из двух групп исследования - результаты тестов «наклонная плоскость» и по BBB-шкале. Последовательные данные двух тестов в модели повторных измерений ANOVA (RM ANOVA) анализировали двумя способами. В первом способе собирали данные, начиная за одну неделю до момента появления признаков заболевания у первого животного, и проводили сравнительный анализ (левые верхние панели на фиг.13-18). Во втором способе собирали данные, начиная с момента начала заболевания (левые нижние панели на фиг.13-18). При проведении данных анализов также вводили «компенсирующую» поправку для устранения фактора отклонения кривых, вызванного очень большим расхождением кривых выживаемости (из-за генотипической вариации) (правые панели на фиг.13-18). При введении данной компенсации брали самый низкий результат поведенческого теста для животного в районе времени его смерти и вводили вместе с результатами для оставшихся в живых животных из той же группы в последующих моментах времени (альтернативный способ может состоять в отбрасывании данной точки информации из анализируемого в дальнейшем массива данных и проведении анализа на меньшем количестве животных). Результаты анализа в модели повторных измерений ANOVA для данных по тесту «наклонная плоскость» и тесту по BBB-шкале не выявили существенных различий между двумя группами в экспериментах с 10- и 12-недельными группами исследования, несмотря на введенные поправки и корректировки для начала заболевания и выживаемости (фиг.13-15, 17). Однако при анализе последних двух пар в эксперименте с 12-недельной группой исследования по причинам, описанных выше в разделе про размножение животных, для поведенческих результатов получили вариации, аналогичные полученным при исследовании выживаемости (смотрите выше; фиг.16, 18). Хотя статистически существенные различия между результатами поведенческих тестов для данных двух пар не были обнаружены, существует тенденция к более высоким результатам для группы А по сравнению с группой B.
Гистологический анализ
Всех крыс SOD1, получавших и не получавших инъекции стволовых клеток, умертвили, когда их заболевание доходило до конечной стадии (результат теста по BBB-шкале <3). После перфузирования 4% раствором PFA головной мозг и другие органы животного собирали, криоконсервировали и замораживали для дальнейшей обработки. Чтобы проверить наличие остатков стволовых клеток человека на срезах из соответствующих органов/тканей, проводили иммуноцитохимический анализ с использованием специфического антитела к ядерному белку человека (HNu). Данные ткани включали в себя поясничный отдел спинного мозга (как соответствующая терапевтическая мишень) и легкое (как орган-контроль с предполагаемой высокой концентрацией введенных стволовых клеток). Окрашивание на HNu не показало никакой выживаемости трансплантированных клеток в данных органах у животных на конечной стадии заболевания.
Наблюдения на срезах спинного мозга. Крысы линии G93A SOD1 повторяют многие основные симптомы АБС человека. Дегенерация двигательных нейронов в спинном мозге крыс SOD1 развивается от двигательных нейронов поясничного отдела, которые иннервируют мышцы нижних конечностей, к двигательным нейронам шейного и грудного отделов, иннервирующим дыхательные мышцы. Животные умирают от истощения и остановки дыхания. Гистологическая цель данного исследования была ограниченной в силу отсутствия соответствующих тканей от животных на промежуточных стадиях заболевания и существенного фенотипического разброса. Доступные для исследования ткани спинного мозга были взяты у животных, умерщвленных на конечной стадии заболевания, когда большинство двигательных нейронов уже дегенерировало, даже у животных с предполагаемым защитным эффектом от инъекции стволовых клеток. Следовательно, единственная ценность гистологического анализа состояла в фиксации глубокой нейропатологии спинного мозга животных, получавших живые или мертвые клетки. Обработали пять таких спинных мозгов. Из каждого мозга изучили по шесть пар срезов, равномерно покрывающих с шагом 300 мкм участок L4-6 поясничного отдела спинного мозга. Один срез из каждой пары окрашивали крезиловым фиолетовым, другой срез окрашивали гематоксилином и эозином. Три образца были взяты у животных с нечетными номерами, и два образца - у животных с четными номерами.
Окрашенные срезы имели нормальную крупномасштабную структуру спинного мозга с четко различимыми всеми основными сегментами спинного мозга (фиг.19А и фиг.19B). Опухоли не были обнаружены (фиг.19А и фиг.19B). Как предсказывалось на основе фенотипа двигательных нейронов животных линии SOD1 G93A, как у животных с нечетными номерами, так и у животных с четными номерами существовала выраженная потеря двигательных нейронов. При подсчете количества нейронов из двух групп на основе количества ядрышек, присутствующих в базофильных профилях брюшного рога при увеличении 100X, не было отмечено существенных тенденций в количестве клеток двигательных нейронов между двумя группами (животные с нечетными номерами, 423; животные с четными номерами, 418). Для коррекции возможных различий в диаметре ядрышек между двумя группами использовали коррекцию Аберкромби на ошибку рассечения клеток (Ni = ni.t/t+d, где ni = результат подсчета, t = толщина среза, и d = средний диаметр профиля).
Оценка результатов теста по BBB-шкале для животных, получавших клетки, показала лучшую тенденцию по сравнению с необработанными животными. Следовательно, результаты настоящего исследования предполагают, что введение клеток ближе к началу заболевания или после него может иметь большую терапевтическую эффективность.
ПРИМЕР 23
Трофические факторы для дифференцирования клеток-предшественников нейронов
Нейронные стволовые клетки в центральной нервной системе (ЦНС) могут самообновляться и являются мультипотентными, поскольку они способны дифференцироваться в нейроны, олигодендроциты и астроциты. Данные клетки могут активироваться после инсульта и инициировать нейрогенез. Однако эффективность данного дифференцирования очень низкая, а вновь сгенерированные нейроны большей частью имеют непродолжительный период жизни, возможно, из-за отсутствия формирования каких-либо функциональных синапсов. Экзогенная доставка клеточных продуктов может улучшить нейрогенез и формирование новых синапсов и тем самым решить данную проблему, и, таким образом, может улучшить восстановление функции в условиях травмы или дегенеративного состояния. Последние исследования предполагают, что клеточные технологии могут оказаться эффективной терапией для множества неврологических состояний. Для понимания механизма(ов) данных технологий определили эффект клеток hUTC на дифференцирование взрослых нейронных стволовых клеток.
Материалы и способы
Клеточная культура
Клетки, полученные из ткани пуповины человека, (hUTC) размораживали (партия № 22042008, на пассаже P3 или P4), высевали на колбы T75 и/или T225 и культивировали в ростовой среде Хейфлика (среда DMEM с низким содержанием глюкозы (Gibco, номер по каталогу 11885-084), 15% об/об эмбриональной бычьей сыворотки (FBS, Hyclone, номер по каталогу SH30070.03IR), 4 мМ глютамакса (Gibco, номер по каталогу 3505-061) и 1% пенициллина/стрептомицина (Gibco, номер по каталогу 15070063)) в течение ночи или в течение 48 часов до образования конфлюентного слоя. После формирования клетками конфлюентного слоя клетки трижды промыли фосфатно-солевым буфером (ФСБ, Invitrogen, номер по каталогу 25200-056) и инкубировали в бессывороточной среде DMEM в течение 8 или 24 часов, как указано. Клетки отделяли от поверхности с помощью TrypLE™ (Gibco, номер по каталогу 12604-021) и подсчитывали с помощью инструмента Guava® (Guava Technologies).
Взрослые нейронные стволовые клетки из гиппокампа крысы (партия № R0706F0009, Millipore, номер по каталогу SCR022) размораживали (P3 или P4), высевали на покрытые поли-L-орнитином (Sigma, номер по каталогу P3655) и ламинином (Sigma, номер по каталогу 2020) колбы T75 (Corning, номер по каталогу 430641) и культивировали в среде DMEM/F12 (Millipore, номер по каталогу DF-042-B), содержащей добавку B27 (Invitrogen, номер по каталогу 17504044), 1% пенициллина/стрептомицина и FGF-2 (Millipore, номер по каталогу GF003), в соответствии с инструкциями производителя. При достижении степени конфлюентности 80% клетки отделяли от поверхности с использованием Accutase® (Millipore, номер по каталогу SCR005) и подсчитывали с использованием инструмента Guava®. Для анализа на дифференцирование клетки высевали с плотностью 20000 клеток/см2 на 6-луночные планшеты с покрытием (Millipore, номер по каталогу PICL06P05).
Анализ с совместным культивированием
Для анализа с совместным культивированием использовали 6-луночную систему трансвелл (Corning, номер по каталогу 3450). Взрослые нейронные стволовые клетки из гиппокампа крысы высевали либо отдельно, либо вместе с клетками hUTC с плотностью 20000 клеток/см2 на дно каждой лунки. Культуры инкубировали в среде DMEM/F12 с добавкой B27 и без FGF; через сутки половину среды заменяли на свежую.
Экстрагирование РНК/белка
РНК и белок подготовили с использованием набора для экстрагирования РНК/белка AllPrep RNA/Protein Kit (Qiagen, номер по каталогу 80404) в соответствии с инструкциями производителя. Объем лизирующего буферного раствора для клеток, выращенных в 6-луночных планшетах, выбрали соответствующим образом.
Синтез кДНК и удаление геномной ДНК
Использовали процедуру выделения мРНК и последующего получения кДНК с применением набора для обратной транскрипции QuantiTect® Reverse Transcription Kit (Qiagen, номер по каталогу 205313). Перед синтезом кДНК провели удаление геномной ДНК в соответствии с инструкциями производителя. Синтез кДНК проводили с 0,5 микрограмма общей РНК, выделенной из взрослых нейронных стволовых клеток гиппокампа крысы, с помощью набора для обратной транскрипции QuantiTect® Reverse Transcription Kit в полном объеме 20 микролитров в соответствии с инструкциями производителя.
Количественная ПЦР TaqMan
ПЦР проводили на секвенаторе реального времени ABI 7000 Real time PCR System в оптических 96-луночных реакционных планшетах (Applied Biosystems, номер по каталогу 4306737) в конечном объеме 20 микролитров. Транскрипты грызунов определяли в реакционной смеси, содержащей 10 микролитров 2x универсального мастер-микса для ПЦР TaqMan (Applied Biosystems, номер по каталогу 4364338), 1 микролитр - реагент для анализа экспрессии генов 20X TaqMan (Applied Biosystems, номер по каталогу 4331182); GFAP (идентификатор анализа: Rn 00566603_m1), нестин (Rn00364394_R1), основной миелиновый белок (идентификатор анализа: Rn00566745_m1), Sox2 (rn00584808_m1), изоформа бета-III тубулина (идентификатор анализа: Rn 00594933_m1) и GAPDH (идентификатор анализа: 99999916_s1). Подготовили 1 микролитр матричной ДНК и 8 микролитров не содержащей РНКазы воды (Sigma, номер по каталогу W4502). Амплификации проводили, начиная со стадии активации UNG при 50°C в течение 2 минут с последующей 10-минутной стадией денатурации матрицы при 95°C. Затем провели 40 циклов денатурации при 95°C в течение 15 секунд и комбинированного отжига/удлинения праймера при 60°C в течение 1 минуты.
Белковый иммуноблоттинг
Цельноклеточные лизаты (20 микрограмм) денатурировали кипячением в буферном растворе Лэммли (Boston Bioproducts, номер по каталогу BP-111R) в течение 3 минут. Затем образцы расщепили с помощью ДСН-ПААГ-электрофореза на 10% Tris-глициновом геле (Invitrogen, номер по каталогу: EC6078BOX) в Tris-глицин-ДСН подвижном буферном растворе (Boston Bioproducts, номер по каталогу BP-150) при токе 40 мА в течение 1 часа. Белки перенесли на 0,45-микронную мембрану из ПВДФ (Invitrogen, номер по каталогу LC2005) с использованием полусухой электрофоретической ячейки Trans-Blot® Cell (Bio-Rad) в гибридизационном буферном растворе (25 мМ Tris-основания; 192 мМ глицина; 10% (об/об) метанол). Перенос проводили при напряжении 25 В в течение 45 минут. Мембрану блокировали 3% (в/об) раствором БСА в ФСБТ (ФСБ+0,1% (об/об) Твин 20) при комнатной температуре в течение 1 часа. Блоты зондировали первичными антителами в течение 1 часа при комнатной температуре. Затем мембраны трижды промыли ФСБТ с последующим инкубированием в течение 1 часа с конъюгированными с пероксидазой хрена вторичными антителами в соотношении 1:5000 в ФСБ при комнатной температуре. Сигналы обнаруживали с использованием субстрата продолжительного действия SuperSignal® West Dura (Pierce, номер по каталогу 34076). Применяли следующие антитела: антитело против изоформы бета-III тубулина (1:250, Millipore, номер по каталогу MAB 1637); анти-GFAP (1:2000, DAKO, номер по каталогу Z0334); анти-Sox (1:1000, Abcam, номер по каталогу ab59776); анти-MBP (1:500, Millipore, номер по каталогу MAB 382); анти-GAPDH конъюгат с пероксидазой (1:5000, Sigma, номер по каталогу G9295); козий антимышиный IgG конъюгат с пероксидазой (R&D Systems, номер по каталогу HAF007); и козий анти-кроличий IgG конъюгат с пероксидазой (Sigma, номер по каталогу A0545).
Проточная цитометрия
После анализа с совместным культивированием, взрослые нейронные стволовые клетки из гиппокампа крысы отделили от поверхности обработкой Accutase® и на короткое время суспендировали в ФСБ, содержащем 1% параформальдегида. Затем клетки повторно суспендировали и инкубировали с блокирующим буферным раствором (ФСБ с добавлением 0,3% Triton X-100 (Sigma, номер по каталогу T9284)) и 0,3% козьей сывороткой (Millipore, номер по каталогу S26) в течение 15-30 минут и затем с первичными антителами (1:200, кроме GFAP при 1:2000) в блокирующем буферном растворе в течение 1 часа при комнатной температуре. Затем клетки трижды промывали буферным раствором (ФСБ с добавлением 0,3% Triton-X100), после чего инкубировали с соответствующими конъюгированными с флуорофором вторичными антителами (1:400), такими как, например, конъюгированный с R-фикоэритрином козий анти-кроличий IgG (Molecular Probes, номер по каталогу P-2771MP) и конъюгированный с Alexa Fluor® 488 козий антимышиный IgG (Molecular Probes, номер по каталогу A-11029), в блокирующем буферном растворе в течение 45 минут. Затем окрашенные клетки трижды промыли в промывочном буферном растворе, суспендировали в ФСБ и анализировали методом проточной цитометрии (FACScalibur™).
Иммуноцитохимический анализ
Высеянные на 24-луночном планшете взрослые нейронные стволовые клетки из гиппокампа крысы на 5 сутки после анализа с совместным культивированием промывали холодным ФСБ и фиксировали 3% раствором параформальдегида в течение 20 минут при 4°C. Зафиксированные клетки дважды промыли ФСБ, затем пермеабилизовали в блокирующем буферном растворе (комнатная температура, 15 минут) и инкубировали при комнатной температуре в течение 1 часа с первичными антителами (1:200, кроме GFAP при 1:2000). Окрашенные клетки трижды промывали в промывочном буферном растворе и инкубировали с соответствующими конъюгированными с флуорофором вторичными антителами. После последнего промывания (пять раз в промывочном буферном растворе) окрашенные клетки анализировали на конфокальном флуоресцентном микроскопе (Olympus). Все изображения получали с использованием объектива 20X.
Результаты
Клетки hUTC индуцируют дифференцирование нейронных стволовых клеток
Для исследования влияния клеток hUTC на дифференцирование нейронных стволовых клеток провели эксперимент с совместным культивированием с добавлением или без добавления FGF-2. Без добавления FGF-2 клетки имели тенденцию к слабому дифференцированию. Чтобы проанализировать способность нейронных стволовых клеток к дифференцированию с клетками UTC или без них после 3 суток, провели количественное определение нейрональных и ненейрональных маркеров методом ПЦР в реальном времени, вестерн-блоттинга и проточной цитометрии. Фенотип клеток идентифицировали путем измерения экспрессии ключевых маркерных генов: SOX для клеток-предшественников; глиофибриллярный кислый белок (GFAP) для астроцитов; бета-III-тубулин для нейронов, а также основные миелиновые белки (MBP) для олигодендроцитов. Результаты показывают, что клетки hUTC индуцировали увеличение транскриптов GFAP; причем увеличение было еще более выраженным в присутствии экзогенного FGF-2. Экспрессия изоформы бета-III тубулина в присутствии клеток hUTC и FGF-2 возрастала втрое. Уровень белка GFAP также увеличивался. Результаты проточной цитометрии показали наличие: (1) явно выраженной популяции клеток с экспрессией GFAP; и (2) ярко выраженной, но небольшой популяции клеток, положительных на бета-III-тубулин. Аналогичную экспрессию белков наблюдали в отсутствие и в присутствии FGF-2 в среде. Те же эксперименты провели с использованием нейронных стволовых клеток, которые были химически повреждены обработкой азидом натрия и дезоксиглюкозой. При обработке ишемических нейронных стволовых клеток клетками hUTC наблюдали аналогичную тенденцию в транскрипции и экспрессии белков за исключением 100-200-кратного возрастания количества транскриптов GFAP в присутствии FGF-2.
Количественное определение нейрональных и ненейрональных транскриптов методом ПЦР в реальном времени
Взрослые нейронные стволовые клетки из гиппокампа крысы опосредованно (таблица 23-1) или непосредственно (таблица 23-2) совместно культивировали с клетками hUTC. Уровень транскриптов выразили как кратность увеличения по сравнению с контрольными клетками (только нейронные стволовые клетки). В таблицах представлены результаты, полученные для двух независимых наборов экспериментов. Данные приведены в виде среднего ± ст. откл.
Обзор. Секретируемые клетками hUTC факторы стимулировали дифференцирование нейронных стволовых клеток в глиальные и нейрональные клетки in vitro.
ПРИМЕР 24
Трофические факторы для дифференцирования клеток-предшественников нейронов с использованием клеток PC12 в качестве модели
Клетки PC12 являются удобной модельной системой для нейронального дифференцирования. Данные клетки бесконечно пролиферируют в культуре, но могут дифференцироваться в нейроноподобные клетки при обработке конкретными факторами, включая фактор роста нервов (NGF), основной фактор роста фибробластов (bFGF), эпидермальный фактор роста (EGF) и, возможно, интерлейкин-6 (IL-6), гранулоцитарный колониестимулирующий фактор (GCSF) и фактор ингибирования лейкемии (LIF). Предыдущие исследования показали, что клетки hUTC секретируют нейротрофические факторы in vitro. Цель настоящего исследования заключалась в выяснении, могут ли растворимые факторы из клеток hUTC стимулировать дифференцирование клеток PC12.
Материалы и способы
Клеточная культура и кондиционированная среда
Четыре миллиона клеток, полученных из ткани пуповины человека, (hUTC) размораживали (партия № 1027, пассаж 2 или 4), высевали на колбы T75 и культивировали в ростовой среде Хейфлика (среда DMEM с низким содержанием глюкозы (Gibco, номер по каталогу 11885-084), 15% об/об эмбриональной бычьей сыворотки (FBS, Hyclone, номер по каталогу SH30070.03IR), 4 мМ глютамакса (Gibco, номер по каталогу 3505-061) и 1% пенициллина/стрептомицина (Gibco, номер по каталогу 1070063)) в течение ночи. На следующие сутки клетки трижды промыли фосфатно-солевым буфером (ФСБ, Invitrogen, номер по каталогу 25200-056) и инкубировали в среде RPMI-1640 (RPMI, ATCC, номер по каталогу 30-2001), 0,5% термоинактивированной лошадиной сыворотке (Gibco, номер по каталогу 26050088) и 1% пенициллина/стрептомицина в течение 48 часов. Среду отделяли и центрифугировали при 2500 об/мин в течение 5 минут.
Клетки Neuroscreen-1, субклон клеток PC12 (партия № NS1081219, Cellomics, номер по каталогу R04-0001-C1) размораживали (P2 или P6), высевали на покрытые коллагеном I колбы (Nunc, номер по каталогу 132707) и культивировали в среде RPMI с добавлением 10% термоинактивированной лошадиной сыворотки, 5% FBS (ATCC, номер по каталогу 30-2020) и 1% пенициллина/стрептомицина в соответствии с инструкциями производителя. При достижении степени конфлюентности 70% клетки отделяли от поверхности с использованием TrypLE™ (Gibco, номер по каталогу 12604-021) и подсчитывали с использованием инструмента Guava®. Для анализа на дифференцирование клетки высевали с плотностью от 20 до 30000 клеток/см2 в покрытые коллагеном 6-луночные планшеты (Millipore, номер по каталогу PICL06P05).
Обработка факторами роста и кондиционированной средой
Клетки Neuroscreen-1 трижды промыли ФСБ и оставляли в фазе покоя в течение 18-24 часов в среде RPMI с добавлением 0,5% лошадиной сыворотки перед обработкой 10 нг/мл фактора роста нервов бета человека (NGF, Millipore, номер по каталогу GF028) или кондиционированной средой. Клетки культивировали в течение 4 суток.
Анализ вырастания нейритов
Иммуноцитохимический анализ вырастания нейритов проводили с использованием набора Neurite Outgrowth kit в соответствии с инструкциями производителя (Thermo Fisher Scientific). После обработки NGF или кондиционированной клетками hUTC средой в указанный момент времени клетки фиксировали на 20 минут с использованием 4% раствора параформальдегида в ФСБ при 37°C. Тела и отростки клеток метили первичным антителом против бета-III-тубулина и затем обработали вторичным антителом, конъюгированным Alexa Fluor® 488. Чтобы пометить клеточные ядра, в фиксирующий раствор добавили краситель Hoechst 33258. Затем планшеты поместили на высокопроизводительную платформу для получения изображений Cellomics ArrayScan® VTI для автоматического получения и анализа изображений. Данная система основана на инвертированном эпифлуоресцентном микроскопе, который автоматически фокусируется и сканирует поля в отдельных лунках с помощью моторизованной подвижки. Флуоресцентные изображения получали с использованием многополосного фильтра на эмиссию и согласованных фильтров на возбуждение для синего канала (ядра) и зеленого канала (тело и отростки клеток) и снимали с использованием ПЗС-камеры высокого разрешения. Полученные изображения анализировали с помощью программного обеспечения ArrayScan® с использованием биоприложения для профилирования нейронов Neuronal Profiling (Thermo Fisher Scientific) для измерения различных морфологических параметров, включая общую длину нейритов на клетку, количество нейритов на клетку, количество точек ветвления и площадь тела клетки для каждой пригодной клетки на изображении. Клетку определяли как дифференцировавшуюся, когда общая длина ее нейритов составляла >5 микрометров, что равно удвоенной общей длине нейритов у клеток в отсутствие NGF. Используя объектив 10×, получали достаточное количество полей зрения для анализа по меньшей мере 500 клеток на лунку.
Результаты
Для проверки того, индуцируют ли клетки hUTC нейрогенез в клетках Neuroscreen-1, использовали кондиционированную среду (от культивированных клеток hUTC) для культивирования клеток PC12 в фазе покоя в течение 4 суток. В качестве положительного контроля клетки Neuroscreen-1 обрабатывали 10 нг/мл NGF. Эффект обработки NGF или кондиционированной средой наблюдали с использованием микроскопа. В отсутствие NGF или кондиционированной клетками UTC среды клетки имели относительно небольшой размер и округлую форму, и очень небольшое количество видимых нейритов. Обработка NGF или кондиционированной средой в течение 2-4 суток приводила к некоторому увеличению тел клеток с расширением сети нейритов. После обработки NGF или кондиционированной средой имело место постепенное увеличение длины нейритов. Более 40% клеток оказались дифференцированными на четвертые сутки после обработки кондиционированной средой. Для количественной характеризации дифференцирования клеток Neuroscreen-1 клетки метили антителами против изоформы бета-III тубулина; получали изображения и анализировали их с использованием биоприложения для профилирования нейронов Neuronal Profiling BioApplication. В клетках PC12, обработанных как NGF, так и кондиционированной клетками hUTC средой, наблюдали увеличение длины нейритов, количества нейритов и количества точек ветвления на клетки. Таким образом, кондиционированная клетками hUTC среда достаточна для индуцирования дифференцирования клеток PC12.
Клетки Neuroscreen-1, индуцированные обработкой NGF и средой, кондиционированной клетками hUTC в течение 48 часов (таблица 24-1).
Клетки Neuroscreen-1 выращивали в течение 4 суток на 12-луночных планшетах в среде RPMI с добавлением 0,5% лошадиной сыворотки (отрицательный контроль), 20 нг/мл NGF (положительный контроль) или среде, кондиционированной в течение 48 часов. Через 4 суток клетки фиксировали и окрашивали, как описано в разделе «Материалы и способы». В таблицах представлены результаты, полученные для двух независимых наборов экспериментов. Данные приведены в виде среднего ± ст. откл.
Обзор. Результаты указывают, что секретируемые клетками hUTC растворимые факторы индуцируют нейрогенез в клетках PC12, что показывают морфологические изменения клеток.
ПРИМЕР 25
Возможный протокол множественного введения клеток hUTCпри амиотрофическом боковом склерозе
Результаты исследования предполагают, что клетки hUTCs могут быть эффективны в крысиной модели SOD1 G93A амиотрофического бокового склероза (АБС) при однократном введении. Таким образом, дополнительные исследования могут продемонстрировать, что повторное введение клеток hUTC может повысить эффективность в данной модели заболевания.
Крысы SOD1 G93A могут быть обработаны клетками hUTC в момент начала заболевания или незадолго до или после начала введением клеток hUTC непосредственно в подоболочечное пространство или заднюю мозжечково-мозговую цистерну. Инъекции клеток можно проводить раз в неделю или чаще или реже. Для сведения к минимуму повреждения и максимального ускорения восстановления животного можно использовать способ чрескожной инъекции для доставки продукта клеток hUTC интратекально в спинномозговую жидкость поясничной цистерны у крыс SOD1. Животных устанавливают на устройстве для стереотаксической хирургии компании Kopf и анестезируют с использованием газовой анестезии (изофлюран:кислород:оксид азота = 1:33:66). Поясничную область выбривают и под нижнюю часть живота животного подкладывают цилиндрический поддерживающий элемент для растяжения межпозвоночных промежутков в поясничном отделе позвоночника. Клетки вводят с помощью микрошприцев объемом 200-500 мкл с иглой размером 25 длиной 2,54 см (1 дюйм). Иглу с пустым микрошприцем медленно вставляют в подпаутинное пространство в межпозвоночных промежутках L4-5 или L6-S1. При наблюдении физических признаков раздражения заднего отдела спинного мозга, т.е. отдергивания хвоста или задней лапы, клетки медленно вливают в подпаутинное пространство в течение 1 минуты. Шприц оставляют на месте в течение 2 минут и затем извлекают. Точку вхождения иглы зажимают на 1 минуту смоченным йодом ватным тампоном, затем животное снимают с устройства и приводят в сознание на подогреваемой подстилке. После пробуждения животное возвращают в свою клетку.
Животные могут быть обработаны клетками hUTC с различными уровнями дозировок при введении постоянного количества общего вводимого объема. Животные могут быть обработаны клетками hUTC с интервалами приблизительно в две недели, и оценивать их состояние можно вплоть до конечной стадии заболевания.
Прогрессирование заболевания и потенциальную терапевтическую эффективность контролируют с помощью двух выходных параметров: (1) функциональная локомоторная активность (измеряемая раз в неделю); и (2) вес тела (измеряемый дважды в неделю). Локомоторные тесты включают в себя тест по BBB-шкале (Basso-Beattie-Bresnahan) (Basso et al. (1995 г.) J. Neurotrauma, 1995 г.; 12(1): 1; Basso et al. (1996 г.) Exp. Neurol. 139: 244-256) и тест «наклонная плоскость» (Rivlin AS, Tator CH. (1977 г.) J Neurosurg 47(4): 577).
Для получения результатов теста по BBB-шкале животных успокаивают и адаптируют к тесту «открытое поле». Когда животное начинает постоянно передвигаться по открытому полю, двое экспертов, не знающих о том, как обрабатывали животное, проводят 4-минутное тестирование с использованием BBB-шкалы локомоторной активности, которая представляет собой 21-шаговую шкалу с высокой степенью воспроизводимости как для одного наблюдателя, так и между разными наблюдателями. Тест «открытое поле» записывают на видео. По данной записи двое экспертов количественно определяли количество движений задней лапой за 1 минуту; данный период времени составляют из фрагментов эпизодов на протяжении проведенного 4-минутного тестирования, в течение которых каждая задняя лапа полностью находилась в поле зрения камеры. Установленный временной лимит в 1 минуту обеспечивает одинаковую продолжительность наблюдения за правой и левой задними лапами каждого животного, поскольку обе задние лапы не всегда одновременно полностью находятся в поле зрения камеры. В случае расхождения между результатами экспертов отснятый материал повторно просматривают на замедленной скорости. Отсутствие наблюдаемого движения задней лапой оценивают как 0, исходные баллы по шкале оценок соответствуют выделенным движениям суставов. Выставляемые баллы повышают по мере увеличения количества задействованных в движении суставов и/или увеличения интенсивности движений. По мере усиления локомоторной активности выставляют баллы за помещение лапы на подошву, удерживание веса и координацию движений передней и задней лапами. Наивысшие баллы выставляют за отделение большого пальца, устойчивость туловища и удерживание хвоста.
Тест «наклонная плоскость» представляет собой другой поведенческий тест, в котором оценивают способность животного сохранять положение на волнистой резиновой поверхности; данная поверхность последовательно поднимается с шагом 5º. Максимальный угол, при котором животное может удержать свой вес в течение 5 секунд, определяется как максимальный угол. В данном тесте проверяется сенсорная обратная связь, координация и мышечная сила, требуемая для перемещения.
Конечная стадия заболевания определяется как паралич настолько тяжелой степени, что животное не может подняться в течение 20 секунд после укладывания на бок, что является конечной точкой, часто используемой для мутантных по SOD1 крыс. На данной стадии результаты как теста по BBB-шкале, так и теста «наклонная плоскость» равны нулю.
Настоящее изобретение не ограничивается описанными и представленными выше вариантами осуществления. Существует возможность вариаций и модификаций в рамках объема прилагаемой формулы изобретения.
Реферат
Изобретение относится к области биотехнологии, конкретно к способам лечения амиотрофического бокового склероза, что может быть использовано в медицине. Пациенту вводят клетки, полученные из ткани пуповины, способные к самообновлению и размножению, обладающие потенциалом к дифференцированию в клетку нейронного фенотипа. Изобретение позволяет сохранить функцию двигательных нейронов у пациента с амиотрофическим боковым склерозом. 5 н. и 24 з.п. ф-лы, 19 ил., 25 табл., 25 пр.
Формула
Документы, цитированные в отчёте о поиске
Трансплантация человеческих нервных клеток для лечения нейродегенеративных состояний
Комментарии