Дендритная макромолекула, способ ее получения - RU2109764C1
Код документа: RU2109764C1
Описание
Изобретение относится к дендритной макромолекуле, состоящей из ядра и ответвлений, идущих от ядра.
Дендритные макромолекулы представляют собой трехмерные высокоупорядоченные олигомерные и полимерные молекулы с очень четкой химической структурой. Такие молекулы являются известными и описаны, например, в работе Д.А. Томалиа и др. в Angew. Chem. Int. Ed. Engl. 29(1990), стр. 138-175. В данной работе описан ряд различных дендритных молекул, например дендримеры полиамидоамина (РАМАМ).
Многочисленные и разнообразные дендритные макромолекулы находят широкое применение в самых различных областях.
Публикации упоминают некоторые из видов применения таких молекул (например, в электронике, для калибровки сит, в качестве катализаторов (и носителей катализаторов), в качестве мембран и покрытий селективного действия, но можно применять такие молекулы и в качестве модификаторов ударной прочности или агентов сшивания в некоторых пластмассах.
Недостаток вышеуказанных дендритных молекул состоит в том, что они очень подвержены деградации в результате реакций гидролиза, а дендримеры РАМАМ, в частности, разлагаются при повышении температуры, так как эти макромолекулы в значительной мере теряют стабильность при повышенных температурах.
Задачей изобретения является создание дендритной макромолекулы, крайне мало подверженной деградации в результате реакций гидролиза и обладающей хорошей термостабильностью.
Дендритная макромолекула по изобретению отличается тем, что ответвления получают из винилцианидных групп. Дендритная макромолекула обладает очень высокой термостабильностью и очень мало подвержена деградации в результате реакций гидролиза. Кроме того, предлагаемая макромолекула обладает очень компактной структурой.
Дендритные макромолекулы, известные также как дендримеры или звездчатые дендриты, представляют собой трехмерные высокоупорядоченные молекулы полимеров и олигомеров, обладающих очень четкой химической структурой. Эти макромолекулы образуются с помощью чередующихся реакций, начиная с ядра или инициатора-ядра. Обычно реакции, происходящие в процессе синтеза, являются практически завершенными и селективными реакциями, а это значит, что в ходе синтеза практически не происходит никаких нежелательных побочных реакций и получаемая дендритная макромолекула обладает строго заданной химической структурой.
На чертеже показана двухмерная проекция дендритной макромолекулы.
Молекулы, которые могут быть использованы в качестве ядер, содержат по меньшей мере одну функциональную группу. В рамках изобретения функциональная группа - это группа, которая может вступать в реакцию с винилцианидной группой, возможно в присутствии катализатора. В числе групп, которые при благоприятных условиях могут вступать в реакцию с винилцианидной группой, например гидроксильные группы, первичные и вторичные амины, тиольные группы, углеродные соединения с электроотрицательными заместителями, такие, как эфирные группы, амидные группы, кетонные группы, альдегидные группы, остатки карбоновых кислот и их солей. Предпочтительно ядро содержит гидроксильную группу, первичный и/или вторичный амин.
В зависимости от природы функциональной группы она может вступать в реакцию с одним или несколькими винилцианидными. группами. Если функциональная группа может вступать в реакцию с F-винилцианидными группами, то эта функциональная группа обладает функциональностью F. Гидроксильная группа может вступать в реакцию с винилцианидной группой и таким образом обладает функциональностью F I. Группа первичного амина может вступать в реакцию с двумя винилцианидными группами и, таким образом, обладает функциональностью F2. Функциональность F имеет значение 1, 2 или 3.
Молекула является подходящим ядром в том случае, если она содержит по меньшей мере одну функциональную группу C. Такая молекула предпочтительно должна содержать 1 - 10 функциональных групп C. Подходящее ядро можно выбрать, например, из группы, в которую входят аммиак, вода, метанол, полиметилендиамины, диэтилентриамин, диэтилентетрамин, тетраэтиленпентамин, полиэтиленимин с линейной и разветвленной цепью, метиламин, гидроксиэтиламин, октадециламин, полиаминоалкиларены, такие, как 1,3,5-трис(аминометил)бензол, трис(аминоалкил)амины, такие как трис(аминоэтил)-амин, гетероциклические амины, такие, как имидазолины и пиперидины, гидроксиэтиламиноэтиламин, меркаптоиламин, морфолин, пиперазин, пентаэритрит, сорбит, маннит, дулейтол, инозит, полиалкиленполиолы, такие, как полиэтиленгликоль и полипропиленгликоль, гликоли, такие, как этиленгликоль, 1,2-димеркартоэтан, полиалкиленполимеркаптаны, фосфин, эпсилон-амино-капроновая кислота, глицин, тиофенилы, фенолы, меламин и его производные, такие, как меламинтрис (гексаметилендиамин). В способе по изобретению предпочтительно использовать ядро, выбираемое из группы, в которую входят полиметилендиамины, гликоли и трис(1,3,5-аминометил)бензол. Полиметилендиамины, являющиеся более предпочтительными соединениями для использования в качестве ядер, включают гексаметилендиамин, этилендиамин и 1,4-диаминобутан (DAB). Лучше всего в качестве ядра использовать 1,4-диаминобутан.
При желании в качестве ядра дендритной макромолекулы можно использовать сополимер, содержащий вышеуказанные функциональные группы. Примерами таких сополимеров являются сополимер стирола и имида малеиновой кислоты, сополимер стирола и акрилонитрила, полиэтиленимина и таких полимеров, как полипропиленоксид, полистирол и этилен-пропилендиен, в которые вводят одну или более из вышеуказанных функциональных групп, такую, как группы NH2.
Форма выбранного ядра в значительной степени определяет форму макромолекулы. Если в качестве ядра используют небольшую молекулу, то получают макромолекулу сферической формы. Если в качестве ядра используют полимер, то полученная дендритная макромолекула будет иметь более вытянутую форму.
От ядра отходит ряд ответвлений, получаемых от винилцианидных групп. Если проводимые для этого реакции завершены, то общее число ответвлений в нужном поколении N можно вычислить следующим способом. Если G представляет собой число функциональных групп, содержащихся в молекуле ядра, а F представляет собой функциональность каждой отдельной функциональной группы, то число участков взаимодействия R на молекуле ядра равно сумме функциональностей F всех функциональных групп G. Максимальное число ответвлений N-ого поколения можно представить как число участков взаимодействия R, умноженное на 2N-1. Если же проведенные реакции остались незавершенными, тогда число ответвлений будет меньше, т.е. от R до (R*2N-1). Обычно дендритная макромолекула содержит 1-10 поколений ответвлений, предпочтительно 2-10, в частности 3-9.
Молекулярная масса дендритной макромолекулы по изобретению составляет 100-1000000, предпочтительно 700-100000, в частности 1600-100000.
Винилцианидные группы, подходящие для целей
изобретения, содержат двойную связь и группу, притягивающую электроны, которая непосредственно связана с этой двойной связью; эти группы можно выбрать из группы соединений формулы 1

где
R1 - -H или -CH3;
A- -C=N;
В качестве винилцианидных групп хорошо подходят акрилонитрил и метакрилонитрил (MAC N).
Дендритная макромолекула включает ядро, как описано выше, и ответвления. Ответвления дендритной
макромолекулы включают по меньшей мере четыре группы формулы 2

где
R1 - ядро или группа предыдущего поколения;
R2=-H или -CH3;
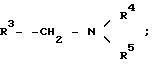

R4 - H или группа следующего поколения;
R5 - H или группа следующего поколения, где группы R4 и R5 в каждой из групп

могут иметь одинаковые или различные значения.
Ответвления обычно содержат менее 1000, предпочтительно менее 250 групп формулы 2. Предпочтительно ответвления содержат более 6, в частности более 10 групп формулы 2. При желании ответвления дендритной макромолекулы могут содержать различные группы формулы 2.
Изобретение относится также к способу получения дендритной макромолекулы по изобретению.
В работе Angew. Chem.Int.Ed.Engl. 29 (1990), стр. 138-175 описаны два способа синтеза, используемые для получения дендритных макромолекул. В ходе одного из этих способов синтеза используется так называемый "способ защищенных групп", когда состав дендритных макромолекул, например дендримеров полиэтиленимина (PEI), очень точно контролируют посредством целенаправленного использования защищенных групп, что предотвращает нежелательные побочные реакции и появление дефектов в структуре дендритной макромолекулы. В ходе синтеза по второму способу, который называется "способ избыточных реактивов", с помощью которого получают, например, дендримеры полиамидоамина (PAMA), используемые реактивы применяются в большом избытке, что статистически сводит к минимуму опасность нежелательных реакций и дефектов.
Вышеописанный "способ защищенных групп" основан на присутствии защищенных азиридиновых колец. Азиридиновые кольца открывают с помощью первичных аминов, после чего с помощью сильной кислоты производят депротекцию. Сложные процедуры выделения конечного продукта, низкий выход этого продукта в результате синтеза и использование дорогих реактивов делают этот способ получения дендритных макромолекул непригодным для широкого применения.
Вышеуказанный "способ избыточных реактивов" включает этап завершенной конденсации по Михаэлю групп первичных аминов до метилметакрилата, после чего проводят амидирование с использованием этилендиамина. Однако синтез получаемых таким способом дендримеров полиамидамина требует большого избытка реактивов для предотвращения нежелательных побочных реакций. Этот недостаток данного способа описан также в работе Д.А.Томалоа в "Angew.Chem.Int. Ed.Engl. 29 (1990), стр. 148. Большая часть избыточного количества реактивов удаляется выпариванием, например в "Rotavapor", после чего остатки реактивов удаляют из вязкого продукта реакции на этапе осаждения. Однако промежуточный продукт различных этапов синтеза должен быть совершенно чистым, а значит этап осаждения необходимо повторять несколько раз. Эти осложнения делают данный способ синтеза дендритных макромолекул также непригодным для широкого применения.
Недостатки каждого из вышеупомянутых способов настолько серьезным, что использование их на коммерческой основе представляет сложную проблему, о чес также говорится в работе Дж. Альпера в "Science" 251 (март 1991), стр. 1562-1564: "Основным препятствием на пути использования таких макромолекул является то, что необходимо разработать способы их синтеза, пригодные для применения в широком масштабе".
Изобретение описывает способ получения дендритных макромолекул, в котором отсутствуют вышеуказанные недостатки.
Способ получения дендритных макромолекул по изобретению отличается тем, что включает этапы от а) до с):
a) практически каждую функциональную группу ядра заставляют вступать в реакцию с
винилцианидной группой;
в) практически каждую нитриловую группу восстанавливают до аминогруппы;
с) практически каждую аминогруппу заставляют вступать в реакцию с винилцианидными
группами; причем этапы в) и с) проводят поочередно в количестве (H-1) раз до получения макромолекулы требуемого поколения N. Значение N обычно варьируется от 1 до 10; предпочтительно N имеет значение
от 3 до 10. Возможно остановить процесс получения макромолекулы после этапа в). Это позволяет получать дендритную макромолекулу поколения 1 1/2, 2 1/2 или выше. В рамках изобретения слово
"практически" означает по меньшей мере 80%. Предпочтительно, чтобы это значение составляло по меньшей мере 90%, еще лучше - по меньшей мере 95%, лучше всего - не менее 99%.
Способ синтеза для получения дендритной макромолекулы по изобретению не обладает вышеуказанными недостатками, а значит этот способ пригоден для широкомасштабного применения. Более того, согласно способу по изобретению нет необходимости каждый раз выделять и очищать продукт реакции, получаемый на каждом промежуточном этапе. Таким образом, дендритные макромолекулы по изобретению могут быть получены простым способом и в широком масштабе.
На этапе а) в способе по изобретению практически каждую функциональную группу ядра с функциональностью F заставляют вступать в реакцию с F-винилцианидными группами. Соответствующая реакция, например конденсация по Михаэлю, группы первичного амина до винилцианидной группы обычно происходит в растворе. Раствор, используемый для этой цели, обычно выбирают таким образом, чтобы не оказывать отрицательного воздействия на ход самой реакции и не вызывать появления нежелательных побочных реакций. Таким образом, очень важно, чтобы выбранный раствор не взаимодействовал с функциональными группами ядра в данных условиях реакции. Нужные растворители можно выбирать, например, из группы, в которую входят вода, тетрагидрофуран, различные спирты, такие, как метанол, этанол, изопропанол и т.п., различные простые эфиры и смеси этих растворителей. Окончательный выбор растворителя будет зависеть главным образом от природы функциональных групп ядра. Предпочтительными растворителями являются вода, метанол и их смеси.
Если нужно, чтобы каждый активный участок R ядра вступил в реакцию с винилцианидной группой в ходе данного этапа, то соотношение реактивов, которое можно описать как отношение числа винилцианидных групп к числу активных участков, должно быть не менее 1. Обычно это соотношение составляет от 1 до 5, предпочтительно от 1 до 3, если это соотношение менее 1, тогда не каждый активный участок R вступит в реакцию с винилцианидной группой.
Температура в ходе этапа а) обычно 0-100oC, предпочтительно 5-70oC.
При желании можно добавить в реакционную смесь катализатор (на данном этапе), что ускорит протекание реакции между функциональными группами и винилцианидными звеньями. В числе подходящих катализаторов слабые кислоты, например уксусная кислота. Обычно катализатор добавляют в реакционную смесь в количестве 0-5 мол.% относительно к числу активных участков R.
На экране b) способа по изобретению практически каждую введенную винилцианидную группу восстанавливают до аминогруппы, предпочтительно до первичной аминогруппы. Если введенная винилцианидная группа представляет собой акрилонитрил, то образуется группа пропиламина (PA). Соответствующая реакция восстановления обычно происходит в растворе. Растворитель, обычно используемый для этой цели, выбирают из группы, в которую входят диамины (например, алкандиамины, такие, как этилендиамин), вода, аммиак, различные спирты, такие, как метанол, этанол, изопропанол и т.п., различные простые эфиры такие, как тетрагидрофуран и диоксан, а также смеси этих растворителей. Предпочтительными являются вода, метанол, этилдиамин, 1,3-аминопропан или смеси этих растворителей.
Реакцию восстановления можно осуществить, например, путем взаимодействия введенной винилцианидной группы с газообразным H2. Если нужно осуществить полностью завершенную реакцию, то молярное отношение H2/нитрил должно быть не менее 2. Если молярное отношение меньше 2, то реакция полностью не завершится. Обычно этап восстановления проводят в присутствии подходящего катализатора. Применяют восстанавливающие катализаторы, предпочтительно гетерогенный восстанавливающий катализатор.
Катализатор, используемый в способе по изобретению, представляет собой катализатор, содержащий металл группы VIII периодической системы, как показано на обложке Краткого справочника по химии и физике, 58-е издание, "Си-Ар-Си-Пресс", 1977-1978. Известно, что, металлы VIII группы активизируют гидрирование нитрилов. См. например EP-A-0077911. Хорошо подходят для этой цели никель, кобальт, платина, палладий и родий. Для того, чтобы катализатор обладал достаточной активностью, металл должен иметь поверхность контакта. Металл может быть использован в чистом виде или вместе с соответствующим носителем.
В качестве катализатора в способе по изобретению лучше всего подходит никель Ранея или кобальт Ранея. См. US-A-1628190, где описаны эти катализаторы Ранея и их получение.
Никель Ранея включает никель и алюминий, причем последний может быть в виде металлического алюминия, оксидов алюминия или гидроксидов алюминия. К никелю Ранея можно добавить небольшие количества других металлов, таких, как железо и/или хром, в чистом или связанном виде, с целью повысить активность катализатора и его селективность в процессе гидрирования определенных групп соединений. Известно, что никель Ранея в сочетании с железом и/или хромом очень хорошо подходит для восстановления нитрильных групп; см., например, работу С. Р. Монтгомери "CATALYSIS OF ORGANIC REACTIONS", 5,стр. 383-409 (1981).
Кобальт Ранея также содержит алюминий и может быть снабжен промоторами. Известно, например, что кобальт Ранея с промотором хромом очень хорошо подходит для гидрирования нитрилов.
Перед использованием никеля или кобальта Ранея эти катализаторы необходимо предварительно обработать щелочным раствором, например, KOH или NaOH, что позволит повысить селективность реакции восстановления. Количество гидроксида, используемого для этой цели, зависит от количества катализатора. Обычно используют 0,01 - 0,2 г гидроксида на 1 г катализатора (сухого веса). Предпочтительно использовать 0,03-0,18 г гидроксида на 1 г катализатора, лучше всего 0,05 - 0,15 г гидроксида на 1 г катализатора. Предварительную обработку проводят путем растворения нужного количества гидроксида в минимальном количестве подходящего растворителя, например в воде, после чего полученный раствор добавляют в катализатор, который предварительно промывают водой. Полученную смесь интенсивно перемешивают.
Концентрация катализатора по отношению к суммарной массе реакционной смеси обычно составляет от 1 - 35 мас.%, предпочтительно 5 - 20 мас.%, лучше всего 6 -12 мас%.
Реакцию восстановления (этап b) можно проводить, например, в герметичном реакторе в атмосфере H2. Давление водорода в этом герметичном реакторе обычно составляет 1 - 500 бар, предпочтительно 10 - 200 бар, лучше всего - 10 - 100 бар. Температура проведения реакции не играет большой роли, обычно реакцию проводят при 0 - 200oC, предпочтительно 10 - 100o C.
На этапе c) способа по изобретению практически каждую функциональную группу заставляют взаимодействовать с винилцианидной группой (реакция конденсации по Михаэлю). Если функциональная группа представляет собой группу первичного амина, то эта группа может взаимодействовать с двумя винилцианидными группами. Условия проведения реакции на этом этапе можно подобрать так, чтобы они совпадали с условиями проведения реакции на этапе a).
По проведению этапов a) - c) получают дендритную макромолекулу второго поколения (N = 2). Дендритную макромолекулу последующих поколений можно получить путем поочередного повторения этапов b) и c). Если этапы b) и c) поочередно повторяются N-е число раз, то получают дендритную макромолекулу (N + 1)-ого поколения. При желании продукт реакции можно изолировать после этапа b), что позволяет получить дендритную макромолекулу поколения 1,5, 2,5 и т.д. Полученный продукт реакции можно изолировать после любого выбранного этапа реакции.
Полученную дендритную макромолекулу можно при желании полностью или частично модифицировать любыми функциональными группами. Это можно осуществить, например, путем полного или частичного взаимодействия аминогрупп или нитрильных групп (при желании в присутствии подходящего катализатора) с соответствующими реактивами. В числе примеров таких реактивов - минеральные соли, такие, как HCl, ненасыщенные алифатические эфиры и амиды, такие, как акриловый эфир, метакриловый эфир, кротиловый эфир и акриламид, галогенангидриды, такие, как кислый хлорид, акрилоилхлорид, алкилгалогениды, такие как этилбромацетат и аллилбромид, арилгалогениды, такие, как бензилхлорид, гидроксиэтилметакрилат, тозилгалогениды, такие, как тозилхлорид, ангидриды, такие, как ангидрид малеиновой кислоты, ангидрид фталиевой кислоты, дикарбоновые кислоты, такие, как терефталиевая кислота и адипиновая кислота, циклические радикалы, такие, как этиленоксид и эпихлоргидрид, (a) циклические альдегиды, такие, как формальдегид, этанол и гексанал, p-формилфенилуксусная кислота, а также 1,4,5,8-нафталинтетраацетальальдегид.
Полученные дендритные макромолекулы имеют концевые группы, которые полностью или частично модифицированы функциональными группами. Эти функциональные группы, например, выбирают из группы, в которую входят амин, нитрил, гидроксид, эфир, кислота, соль, амид, имид, тозилат и тиол. При необходимости может быть использована смесь нескольких различных функциональных групп.
Частично благодаря хорошей термостабильности и очень малой подверженности гидролизу дендритные молекулы по изобретению могут быть смешаны с термопластичным полимером или полимерной композицией.
Термопластичный полимер может быть из группы, в которую входят полиолефины, такие, как полиэтилен и полипропилен, полиэфиры, такие, как полиалкилентерефталаты (такие, как полиэтилентерефталат и полибутилентерефталат) и поликарбонаты, полиамиды, такие, как найлон 6, найлон 4, 6, найлон 8, найлон 6, 10 и т.п., полистирол, полиоксиметилен, сополимеры акрилонитрила-бутадиенастирола, сополимеры стирола-акрилонитрила, сополимеры стирола-имида малеиновой кислоты, полисульфокислота, полиимиды, сополимеры стирола-ангидрида малеиновой кислоты, поли(метилметакрилат), поли(виниловый спирт) или полимерные композиции, включающие несколько из этих полимеров. Однако этот список ни в коем случае не следует считать исчерпывающим.
При желании в смесь дендритных макромолекул по изобретению и термопластичного полимера или полимерной композиции могут быть введены добавки. В числе подобных добавок модификаторы ударной прочности, стабилизаторы, антиокислители, смазочные вещества, наполнители, противовоспламеняющиеся вещества, красители, пигменты, армирующие волокна и волокнистые проводники.
Нижеследующие примеры иллюстрируют изобретение, но не ограничивают его рамки.
Пример 1. 1200 мл метанола и 150 г (1,7 моль) 1,4-диаминобутана (DAB, субстрат) помещают в двухлитровую трехгорлую колбу, оснащенную мешалкой, холодильником, термометром и капельной воронкой. После того как смесь охладилась до 10oC, в течение 2 ч по капле добавляют раствор 400 г (7,6 ммоль) акрилонитрила (ASN) в 100 мл метанола. После этого полученную реакционную смесь нагревают 16 ч (температура 40oC).
После того как смесь охладилась практически до комнатной температуры, метанол и избыток акрилонитрила выпаривают под пониженным давлением. Полученный остаток растворяют в метаноле при 50oC, после чего, проведя кристаллизацию и изоляцию, получают искомый продукт, т.е. искомый тетранитрил, в чистом виде в виде белых игольчатых кристаллов; температура плавления продукта составляет 52,8oC. Выход - 92%.
Анализ выделенного продукта с применением1H и13C ЯМР- спектроскопии и масс-спектрометрии показал, что полученный продукт представляет собой DAB (ACN).
13C ЯМР (50 МГц, D2O):119 ммд, CN; 53,1 ммд, NCH2(CH2)3; 48,4 ммд,
NCH2CH2CN; 24,9 ммд, NCH2CH2CN; 16,9 ммд CH2CN.
1H ЯМР (200 МГц, CDCl3): 2,85
ммд, t, 2H, NCH2CH2CN;
2,55 ммд, m, 1H, NCH2(CH2)3; 2,48 ммд, t, 2H, CH2CN;
1,55 ммд, m, 1H, CH2CH2N.
Пример 2. 8,0 г никеля Ранея ("BLW 112 WR "фирмы "Дегусса"; состав, указанный поставщиком, мас.%: N 85; 2,0 Fe 2,0; Cr 2,5; Al 9,7) предварительно обработанные 0,8 г KOH, растворяли в 10 мл деминерализованной воды. Затем катализатор "промывали" три раза, используя 50 мл этилендиамина (EDA). Температура в процессе этой предварительной обработки составляла 20oC.
Затем катализатор и 100 мл EDA помещали в автоклав объемом 160 мл. Автоклав закрывали, продували несколько раз H2, затем нагревали до 40oC при давлении 60 атм. H2, а содержимое реактора постоянно перемешивали.
Затем 4 г DAB (ACN)4, растворенного в 10 г EDA, помещали в автоклав (не снижая давления) в так называемом аппорционном сосуде, который продували H2 несколько раз и давление в котором довели до 70 атм. Реакция восстановления проходила под давлением 70 атм. Восстановление заняло 120 мин. Анализ выделенного продукта, проведенный с применением13C ЯМР-спектроскопии, показал, что полученный продукт является 1,4-диаминобутан-n,n'-тетра-1-пропиламином, DAB(PA)4 .
1C ЯМР (50 МГц, D2O): 53,4 ммд, NCH2CH2CH2CH2 (2x); 51,1 ммд, NCH2CH2CH2HH2 (4x); 39,5 ммд, CH2NH2 (4x); 28,8 ммд, CH2CH2NH2 (4x); 23,9 ммд, NCH2CH2CH2CH2N (2x).
Пример 3. Повторяли процедуру по примеру 1, только вместо 1,4-диаминобутана в качестве субстрата использовали 5,0 г DAB(PA)4.
Анализ выделенного продукта, проведенный с применением13C ЯМР-спектроскопии, показал, что полученный продукт является DAB(PA)4(ACN)8. Выход - 91%.
13C ЯМР (50 МГц, CDCl3): 118,9 ммд, CN (8x); 53,9 ммд NCH2CH2CH2CH2 (2x); 51,5 и 51,4 ммд, NCH2CH2CH2N (8x); 49,6 ммд, NCH2CH2CN (8x); 25,0 и 24,9 ммд, NCH2CH2CH2CH2 и NCH2CH2CH2N (6x); 16,9 ммд, CH2CN (8x).
Пример 4. Повторяли процедуру по примеру 2, только в данном примере 2,0 г DAB(PA)4(ACN)8 восстанавливали в течение 1200 мин до получения DAB(PA)4(PA)8, о чем свидетельствует анализ выделенного продукта, проведенный с использованием13C ЯМР-спектроскопии.
13C ЯМР (50 МГц, D2O): 53,6 ммд, N CH2CH2 CH2CH2 (2x); 51,7 ммд, NCH2CH2CH2N (8x); 51,2 ммд, NCH2CH2CH2NH2 (8x); 39,6 ммд, CH2NH2 (8x); 28,9 ммд, CH2CH2NH2 (8x); 24,1 ммд, NCH2CH2CH2CH2N (2x); 22,3 ммд, NCH2CH2CH2N (4x).
Пример 5. Повторяли процедуру по примеру 3, только вместо DAB (PA)4 в качестве субстрата использовали 2,0 г DAB(PA)4 (PA)8.
Выделенный продукт подвергли анализу с применением13C ЯМР-спектроскопии, который показал, данный продукт является DAB(PA)4(PA)8 (ACN)16.
13C ЯМР (50 МГц, CDCl3): 119,0 ммд, CN (16x); 54 ммд, NCH2CH2CH2CH2 (2x); 52,2 ммд, NCH2 CH2CH2 (8x); 51,5 и 51,4 ммд, NCH2CH2CH2 (16x); 49,5 ммд, CH2CH2CN (16x); 25,0 и 24,9 ммд, NCH2CH2 CH2 и NCH2CH2N(10x); 24,3 ммд, NCH2CH2CH2N (4x); 16,9 ммд, CH2CN (16x).
Пример 6. Повторяли процедуру по примеру 4, только в данном примере 2,0 г DAB(PA)4(PA)8 (ACN)16 восстанавливали при 40oC в течение 4200 мин до получения DAB(PA)4(PA)8 (PA)16, о чем свидетельствует анализ результатов, полученных с помощью13C ЯМР-спектроскопии.
13C ЯМР (50 Мгц, D2O):53,6 ммд, NCH2 CH2CH2CH2 (2x); 51,7 ммд, NCH2CH2CH2N (24x); 51,2 ммд, NCH2CH2CH2NH2 (16x); 39,6 ммд, CH2NH2 (16x); 28,9 ммд, CH2CH2NH2 (16x); 24,1 ммд, NCH2CH2CH2CH2N (2x); 22,3 ммд, NCH2 CH2CH2N (12x).
Пример 7. Повторяли процедуру по примеру 5, только вместо DAB (PA)4(PA)8 в качестве субстрата использовали DAB(PA)4 (PA)8(PA)16.
Провели анализ выделенного продукта с помощью13C ЯМР-спектроскопии, который показал, что полученный продукт является DAB(PA)4 (PA)8(PA)16(ACN)32.
13C ЯМР (50 МГц, CD Cl3): 119,0 ммд, CN (32x); 54,2 ммд NCH2CH2CH2CH2 (2x); 52,2 ммд, NCH2CH2CH2 (24x); 51,4 ммд, NCH2CH2CH2 (32x); 49,4 ммд, NCH2CH2CH (32x); 24,9 ммд, NCH2CH2CH2CH2 и NCH2CH2CH2N (18x); 24,4 ммд, NCH2CH2CH2N (12x); 16,8 ммд, CH2CN (32x).
Пример 8. Повторяли процедуру по примеру 6, только в данном случае 2,0 г DAB(PA)4(PA)8(PA)16 (ACN)32 восстанавливали в течение 4200 мин при 60oC до получения DAB(PA)4(PA)8(PA)16(PA)32, о чем свидетельствует анализ результатов, полученный с помощью13C ЯМР-спектроскопии.
13C ЯМР (50 МГц, D2O): 51,7 ммд, NCH2CH2CH2N (56x); 51,2 ммд, NCH2CH2CH2NH2 (32x); 39,6 ммд, CH2NH2 (32x); 28,8 ммд, CH2CH2NH2 (32x); 22,3 ммд, NCH2CH2CH2N (28x).
Пример 9. Повторяли процедуру по примеру 7, только вместо DAB (PA)4(PA)8(PA)16 использовали в качестве субстрата DAB (PA)4(PA)8(PA)16 (PA)32.
Провели анализ полученного продукта с применением13C ЯМР-спектроскопии, который показал, что данный продукт является DAB(PA)4 (PA)8 (PA)16(PA)32 (ACN)64.
13C ЯМР (50 МГц, CDCl3): 119,0 ммд; CN (64x); 54,2 ммд, NCH2CH2CH2CH2 (2x); 52,2 ммд, NCH2CH2CH2 (56x); 51,4 ммд, NCH2CH2CH2 (64x); 49,5 ммд, NCH2CH2CH2 (64x); 25, 0 ммд, NCH2CH2CH2CH2 и NCH2CH2 CH2 N (34x); 24,2 ммд, NCH2CH2CH2N (28x); 16,9 ммд, CH2CH (64x).
Пример 10. Повторяли процедуру по примеру VIII, только в данном случае 2,0 г DAB(PA)4(PA)8(PA)16(PA)32 (ACN)64 восстанавливали в течение 4200 мин при 80oC до получения DAB(PA)4(PA)8(PA)16(PA)32 (PA)64, о чем свидетельствует анализ результатов, полученный с использованием13C ЯМР спектроскопии.
13C ЯМР (50 МГц, D20): 51,7 ммд, NCH2CH2CH2N (120x); 51,2 ммд, NCH2 CH2CH2NH2 (64x); 39,6 ммд, CH2NH2 (64x); 28,8 ммд, CH2CH2NH2 (64x); 22,3 ммд, NCH2CH2 CH2N (60x).
Пример 11. 20 г акрилонитрила растворяли в 10 мл метанола. Затем при 10oC в этот раствор добавляли по капле раствор 5,0 г этаноламина (ETAM) в метаноле. Затем реакционную смесь нагревали 16 ч (температура = 40oC). После выпаривания растворителя и промывки остатка эфиром анализ полученного продукта с использованием1H и13C ЯМР-спектроскопии показал, что данный продукт представляет собой динитрилэтанол (ETAM (ACN)2).
13C ЯМР (50 МГц, CDCl3): 119,0 ммд, C; 59, 5 ммд, CH2OH; 55,5 ммд, CH2CH2OH; 49,7 ммд, NCH2CH2CH; 17,4 ммд, CH2CN.
1H ЯМР (200 МГц, CDCl3): 3,66 ммд, t, 1H, CH2OH; 2,91 ммд, t, 2H, CH2CH2N; 2,72 ммд, 1H, t, NCH2CH2OH; 2,53 ммд, 2H,t, CH2CN.
Пример 12. Повторяли процедуру по примеру 2, только в качестве основы использовали 2,0 г ЕТАМ (ACN)2, растворенного в метаноле. Через 60 мин при 40oC в метаноле завершалось селективное восстановление, анализ полученного продукта с применением13C ЯМР-спектроскопии показал, что получено искомое соединение - ЕТАМ (PA)2.
13C ЯМР (50 МГц, D2O): 59,1 ммд, CH2OH; 55,0 ммд, NCH2CH2OH; 51,8 ммд, NCH2CH2CH2NH2 (2x); 39,5 ммд, CH2NH2 (2x); 28,9 ммд, CH2CH2NH2 (2x).
Пример 13. При 5oC 10 г акрилонитрила (189 ммоль) добавляли по капле в 0,5 г анионообменного соединения ("Lewatit MP 500 MBR, обработанного 3% NaOH, затем промытого водой до нейтрального значения pH) и 2,0 г полиэтиленгликоля ("PEG", Mn = 455, 4,4 ммоль). Полученную смесь перемешивали 12 ч при 20oC. Полученный продукт отфильтровывали и промывали дихлорметаном. Дихлорметан и избыток акрилонитрила выпаривали, полученный продукт промывали диэтиловым эфиром (трижды). Анализ полученного продукта с помощью13C ЯМР-спектроскопии показал, что выделенное масло представляет собой PEG(ACN)2.
13C ЯМР (50 МГц, CDCl3): 70,9 ммд, OCHoC-CH2О; 65,9 ммд, OCH2-CH2CN; 18,8 ммд, CH2CH; 118,2 ммд, CN.
Пример 14, Повторяли процедуру по примеру 11, только в качестве субстрата использовали 2,0 г PEG(ACN)2, а в качестве растворителя - метанол. Через 300 мин при 37oC селективное восстановление завершалось, анализ полученного продукта с применением13C ЯМР-спектроскопии показал, что получено искомое соединение - PEG(PA)2.
13C ЯМР (50 МГц, D2O): 70,0 ммд, OCH2-CH2O; 69,3 ммд, OCH2CH2CH2NH2; 38,2 ммд, CH2NH2; 32,0 ммд, CH2CH2NH2.
Пример 15. 1,0 г эпсилон-аминокапроновой кислоты (эпсилон-АС, 8,0 ммоль) растворяли в 10 мл воды и депротонировали 0,5 эквивалентами K2CO3. Затем при 0oC добавляли акрилонитрил в избыточном количестве (4 молярных эквивалента). Затем смесь нагревали 12 ч (при 40oC). Анализ результатов с применением13C ЯМР-спектроскопии показал, что бесцветное масло, полученное после выпаривания растворителей и избытка акрилонитрила, представляет собой эпсилон-AC(ACN)2.
13C ЯМР (50 МГц, CDCl3): 184,0 ммд, CO, 121,4 ммд, CN; 53,0 МИД, NCH2CH2CH2CH2; 48,8 ммд, NCH2CH2CH; 38,1 ммд, CH2CO; 27,0 ммд, NCH2 CH2CH2; 26,2/26,1 ммд, CH2CH2CH2CH2CO; 15,6 ммд, CH2CN.
Пример 16. Повторяли процедуру по примеру 11, только в данном случае 2,0 г эпсилон-АС (ACN)2 растворяли в воде и использовали в качестве субстрата. Спустя 120 мин при 40oC селективное восстановление завершалось, анализ полученного продукта с применением13C ЯМР-спектроскопии показал, что полученный продукт представляет собой искомое соединение - эпсилон - AC(PA)2.
13C ЯМР (50 МГц, CDCl3); 182,6 ммд, CO; 53,9 ммд, NCH2CH2CH2CH2; 51,6 ммд, NCH2CH2CH2NH2 (2x); 40,0 ммд, CH2NH2 (2x); 38,8 ммд, CH2CO; 29,5 ммд, CH2CH2NH2 (2x); 27,8 ммд, NCH2CH2; 26,5 ммд/25,8 ммд, NCH2 CH2CH2CH2CH2. Пример 17. Повторяли процедуру по примеру 11, только в данном случае использовали н-бутанол в качестве соединения для промывки катализатора, и в качестве растворителя субстрата и растворителя, применяемого в процессе реакции. После 180 мин реакции при 40oC селективное восстановление завершалось до получения искомого соединения - DAB(PA)4.
13C ЯМР (50 МГц, D2O); 53,4 ммд, NCH2CH2CH2CH2 (2x); 51,1 ммд, NCH2CH2
CH2NH2 (4x); 39,5 ммд, CH2NH2 (4x); 28,8 ммд, CH2CH2NH2 (4x); 23,9 ммд, NCH2CH2CH2CH2N (2x);
Пример 18. Повторяли процедуру по примеру 11, только в данном случае катализатор промывали тетрагидрофураном (THF). Затем 2,0 г DAB(ACN)4 растворяли в
тетрагидрофуране, и тетрагидрофуран также использовали в качестве растворителя в процессе реакции. Давление H2 составляло 30 атмосфер, температура - 80oC. Через 120 мин
селективное восстановление завершалось до получения искомого соединения - DAB(PA)4.
13C ЯМР (50 МГц, D2O): 53,4 ммд, NCH2CH2CH2CH2 (2x); 51,1 ммд, NCH2CH2CH2NH2 (4x); 39,5 ммд, CH2NH2 (4x); 28,8 ммд, CH2CH2NH2 (4x); 23,9 ммд, NCH2CH2CH2CH2N (2x).
Пример 19. Повторяли процедуру по примеру 18, только в данном случае реакцию проводили при 40oC. Спустя 240 мин селективное восстановление завершалось до образования искомого соединения - DAB(PA)4.
13C ЯМР (50 МГц, D2O); 53,4 ммд, NCH2CH2CH2CH2 (2x); 51,1 ммд, NCH2CH2CH2NH2 (4x); 39,5 ммд, CH2NH2 (4x); 28,8 ммд, CH2
CH2NH2 (4x); 23,9 ммд, NCH2CH2CH2CH2N (2x);
Пример 20. 8,0 г никеля Ранея ("BLM 112WR" фирмы "Дегусса"; состав,
мас. %: Ni 85; Fe 2,0; Cr 2,5; Al 9,7) предварительно обрабатывали KOH так же, как в примере 11. После этой обработки катализатор промывали 50 мл деминерализованной воды. Затем катализатор помещали в
автоклав вместе с 100 мл деминерализованной воды, после чего автоклав продували H2 и нагревали до 60oC. Затем 4,0 г DAB(ACN)4 растворяли в 5,0 мл метанола и помещали в
автоклав. Через 90 мин при давлении H2 в 70 атмосфер селективное восстановление завершалось до образования DAB(PA)4.
13C ЯМР (50 МГц, D2O); 54,4 ммд, NCH2CH2CH2CH2 (2x); 51,1 ммд, NCH2CH2CH2NH2 (4x); 39,5 ммд, CH2NH2 (4x); 28,8 ммд, CH2CH2NH2 (4x); 23,9 ммд, NCH2CH2CH2N (2x).
Пример 21. Повторяли процедуру по примеру 20, только в качестве катализатора использовали кобальт Ранея (тип "Grace 2724R", активизированный G). Через 15 мин селективное восстановление завершалось до образования искомого соединения - DAB(PA)4 .
13C ЯМР (50 МГц, D2O); 53,4 ммд, NCH2CH2CH2CH2 (2x); 51,1 ммд, NCH2CH2CH2NH2 (4x); 39,5 ммд, CH2NH2 (4x); 28,8 ммд, CH2CH2NH2 (4x); 23,9 ммд, NCH2CH2CH2CH2N (2x);
Пример 22. 10 г меламин (1,3,5-трисгексаметиленамина) (23,6 ммоль MEL(HMA)3) растворяли в 150 мл метанола. Полученный раствор добавляли в 15 г акрилонитрила (283 ммоль) при 0oC.
Полученную смесь перемешивали 1 ч при 20oC, а затем 12 ч при 45oC. Растворитель и избыток акрилонитрила удаляли при пониженном давлении с применением "ROTAVAPOR" при 40oC. Анализ продукта с применением13C ЯМР-спектроскопии показал, что продукт, полученный после охлаждения в диэтиловом эфире и выделения, представляющий собой вязкое масло красного
цвета, является чистым MEL(HMA)3(ACN)6.
13C ЯМР (50 МГц, CDCl3); 165,8 ммд, NCN; 118,7 ммд, 53,4 ммд, NCH2CH2CH2CH2 (3x); 49,6 ммд, NCH2CH2CN (6x); 40,5 ммд, NHCH2 (3x); 29,7 ммд, NHCH2CH2 (3x); 27,3 ммд, 26,8 ммд, 26,7 ммд, NCH2 CH2CH2CH2CH2CH2NH (9x); 17,0 ммд, CH2CN (6x).
Пример 23. Повторяли процедуру по примеру 20, только в данном случае 2,3 г MEL(HMA)3(ACN)6 растворяли в субстрате. Реакцию восстановления проводили при 60oC.13C и1H ЯМР-спектры показали, что селективное восстановление до получения искомого соединения - меламин (HMA)3(PA)6 завершилось через 1020 мин.
13C ЯМР (50 МГц, D2O): 165,7 ммд, NCN (3x);
53,7 ммд,
NCH2CH2CH2CH2 (3x); 51,3 ммд, NCH2CH2HH2 (6x); 40,8 ммд, NHCH2 (3x);
39,7 ммд,
CH2NH2 (6x); 29,8 ммд, NHCH2CH2 (3x); 29,1 ммд,
CH2CH2NH2 (6x); 27,6 ммд, 26,9 ммд, 25,6 ммд, NCH2
CH2CH2CH2CH2CH2NH (9x).
Пример 24. 25 г "Teffamine D - 2000R" (модифицированный полипропиленоксид, Mw = 2000, фирмы "Техасо Кемикал Компани") растворяли в 50 мл метанола. Полученный раствор добавляли в 6,0 г акрилонитрила при 0oC. Полученную смесь перемешивали 1 ч при 20oC, а затем 12 ч при 40oC. Затем полученный продукт растворяли в смеси 100 мл пентана и 5,0 мл диэтилового эфира. Результаты13C ЯМР-спектроскопического анализа показали, что полученный после выделения продукт представляет собой Teff (ACN)4 (бесцветная жидкость, выход - 94%).
13C ЯМР (50 МГц, CDCl3): 118,7 ммд, CN; 75,1-75,7 ммд, OCH2; 73,0-73,6 ммд, NCH; 52,2-52,5 ммд; NCH2CH2CN; 17,2-17,5 ммд, CCH3; 19,1 ммд, CH2CH.
Пример 25. 8,0 г никеля Ранея ("BLM112 WR" фирмы "Дегусса", состав, мас. %: Ni 85; Fe 2,0; Cr 2,5; Al 9,7) предварительно обрабатывали 0,8 г KOH, растворенного в 10 мл деминерализованной воды. После выпадения катализатора в осадок водный слой декантировали, затем добавляли 50 мл этилендиамина при перемешивании. После этого промытый таким образом катализатор отфильтровывали и добавляли в 100 мл этилендиамина в автоклаве на 160 мл. Автоклав закрывали и несколько раз продували H2. Затем в автоклав подавали H2 под давлением 70 бар при 38oC, причем содержимое автоклава интенсивно перемешивали.
Затем в автоклав помещали 2,0 г Teff (ACN)4, растворенного в 10 г этилендиамина. Восстановление полностью завершилось спустя 3 ч13C ЯМР-спектроскопия показала, что полученный продукт является Teff(PA)4.
13C ЯМР (50 Мгц, D2O): 74,8-75,9 ммд, OCH2; 72,4-73,3 ммд, NCH; 53, 0-52,7 ммд, NCH2CH2NH2; 39,1 ммд, CH2NH2; 32,3 ммд, CH2CH2NH2; 16,5-17,3 ммд, CCH3.
Пример 26. 900 мл воды и 75 г (0,85 моль) 1,4-диаминобутана (субстрат) помещали в трехгорлую колбу объемом 2 л, оснащенную мешалкой, холодильником, термометром и капельной воронкой. После этого смесь охлаждали до 10oC, затем в нее по капле в течение трех часов добавляли раствор 200 г (3,8 моль) акрилонитрила. Полученную реакционную смесь нагревали в течение 9 ч (температура - 65oC).
После того, как смесь охладилась до комнатной температуры, воду и избыток акрилонитрила подвергали азеотропному выпариванию. Полученный остаток, содержащий DAB-диаминобутан DAB(ACN)4 и воду, восстанавливали с помощью кобальта Ранея, который не подвергали предварительной обработке, через 1 ч реакцию останавливали, после чего получали искомое соединение в виде бесцветного масла. Анализ этого продукта с применением13C ЯМР-спектроскопии показал, что он является чистым DAB(PA)4. Выход - 98%.
13C ЯМР (50 МГц, D2O); 53,4 ммд, NCH2CH2CH2CH2 (2x); 51,1 ммд, NCH2CH2CH2NH2 (4x); 39,5 ммд, CH2NH2 (4x); 28,8 ммд, CH2CH2NH2 (4x); 23,9 ммд, NCH2CH2CH2CH2N (2x).
Пример 27. Термостабильность дендритных молекул, полученных в примерах 1-8, измеряли с применением термографического анализа (TGA). При этом приблизительно 2,5 мг испытуемого продукта нагревали с помощью "Perkin Elmer" (7 серия) в неоновой атмосфере с температуры 30oC до 900oC со скоростью возрастания температуры 20oC/мин. Ниже даны температуры, при которых разрушение продукта достигало максимума.
Результаты анализов TGA, продуктов, полученных в примерах 1-7
Продукт - Температура (oC)
DAB(ACN)4 - 330,1
DAB(PA)4 - 330,0
DAB(PA)4(ACN)8 - 331,8
DAB(PA)4(PA)8 - 378,0
DAB(PA)4(PA)8(ACN)16 - 332,0
DAB(PA)4(PA)8(PA)16 - 424,0
DAB(PA)4(PA)8(PA)16(ACN)32 - 331,5
Пример 28. 900 мл воды и 75 г (0,85
моль) диаминобутана помещали в 2- литровую трехгорлую колбу, оснащенную мешалкой, холодильником, термометром и капельной воронкой. После того, как смесь охлаждали до 10oC, в нее по капле
добавляли раствор 200 г акрилонитрила в 50 мл метанола с такой скоростью, чтобы температура реакционной смеси не поднималась выше 15oC. После того, как все требуемое количество раствора
добавляли, смесь выдерживали при комнатной температуре в течение двух часов, после чего нагревали до 65oC в течение 9 ч. Затем реакционную смесь охлаждали до комнатной температуры и
выделяли полученный продукт.
Результаты1H и13C ЯМР-спектроскопии и масс-пектрометрии показали, что изолированный продукт является DAB(ACN)4.
13C ЯМР (50 МГц, D2O): 119 ммд, CN; 53,1 ммд, NCH2(CH2)3; 49,4 ммд, NCH2CH2CN; 24,9 ммд, NCH2CH2CN; 16,9 ммд, CH2CN.
1H ЯМР (200 МГц, CDCl3): 2,85 ммд, t, 2H, NCH2CH2CN; 2,55 ммд, 1H, NCH2(CH2 )3; 2,48 ммд,t,2H, CH2CN; 1,55 ммд, m, 1H, CH2CH2N.
Пример 29. 30 мл воды и 5,0 г (58 ммоль) диаминобутана помещали в трехгорлую колбу объемом 250 мл, оснащенную мешалкой, холодильником, термометром и капельной воронкой. После того, как смесь охладилась до 10oC, в нее по капле добавляли раствор 15 г (280 моль акрилонитрила с такой скоростью, чтобы температура не поднималась выше 15oC. После того, как весь раствор был добавлен, смесь выдерживали при комнатной температуре в течение двух часов, после чего реакционную смесь нагревали до 45oC в течение 16 ч.
После охлаждения реакционной смеси до комнатной температуры воду и избыток акрилонитрила выпаривали. 2,5 г полученного продукта (DAB(ACN)4) растворяли в 4 мл метанола. Полученный раствор помещали в автоклав на 160 мл вместе с 8,0 г кобальта Ранея (типа "Grace 2724R", активированный Cr). Затем автоклав закрывали, несколько раз продували H2 и нагревали до 80oC при давлении H2 в 30 атмосфер, при этом содержимое реактора перемешивали. Такие условия поддерживали в течение часа.
После удаления катализатора фильтрацией и выпаривания воды 2,0 г остатка (DAB(PA)4) растворяли в 20 мл воды, в которую по капле добавляли раствор 5,4 г акрилонитрила при 10oC. Смесь выдерживали при комнатной температуре в течение двух часов, после чего нагревали в течение 16 ч до 40oC. После охлаждения воду и избыток акрилонитрила выпаривали при пониженном давлении. Полученный бесцветный остаток (чистый DAB(PA)4(ACN)8) восстанавливали так же, как DAB(ACN)4, как описано в настоящем примере. Селективное восстановление полностью завершалось через 90 мин.
Образовавшийся DAB(PA)4(PA)8 растворяли в 30 мл воды. Затем по капле добавляли 5,0 г акрилонитрила при 10oC. Затем реакционную смесь выдерживали при комнатной температуре в течение двух часов, после чего ее нагревали до 40oC в течение 16 ч. После охлаждения смеси воду и избыток акрилонитрила выпаривали при пониженном давлении, после чего в течение двух часов бесцветный осадок, т.е. DAB(PA)4(PA)8(ACN)16 селективно восстанавливали до DAB(PA)4(PA)8(PA)16 способом, описанным в настоящем примере в связи с восстановлением DAB(ACN)4.
Пример 30. Этилакрилат (EAC, 6,3 г; 63 ммоль) растворяли в 20 мл метанола. Полученный раствор охлаждали на ледяной бане и при перемешивании добавляли 0,5 г DAB(PA)4. Полученную смесь перемешивали при комнатной температуре 20 ч, после чего продукт, светло-желтую жидкость, выделяли.13C ЯМР-спектроскопия показала, что этот продукт является чистым DAB(PA)4(EAC)8.
13C ЯМР (50 МГц, CDCl3): 172,5 ммд, CO (8x); 60,2 ммд, COOCH2 (8x); 54,1 ммд, NCH2CH2CH2CH2 (2x); 51,9 ммд, NCH2CH2CH2N (8x); 49,1 ммд, NCH2CH2CO (8x); 32,6 ммд, CH2CO (8x); 25,0 ммд, NCH2CH2CH2CH2 (2x); 24,7 ммд, NCH2CH2CH2N (4x); 14,2 ммд, CH3 (8x).
Пример 31. DAB(PA)4(EAC)8 (0,5 г, 0,45 ммоль) растворяли в 3,0 мл метанола. Полученный раствор охлаждали до 0oC с помощью ледяной бани, при этом по капле добавляли этаноламин (EA) в избыточном количестве. После этого продукт изолировали.13C ЯМР-спектрометрия показала, что полученный продукт, желтое масло, является чистым DAB(PA)4(EA)8.
13C ЯМР (50 МГц, D2O): 175,6 ммд, CONH (8x); 60,3 ммд, CH2OH (8x); 53,2 ммд, CH2CH2CH2CH2 (2x); 51,5 ммд и 51,2 ммд, и NCH2CH2CH2N (8x); 49,1 ммд, NCH2CH2CO (8x); 41,8 ммд, CONHCH2 (8x); 32,9 ммд, CH2CO (8x); 24,0 ммд, NCH2CH2CH2CH2 (2x).
Приведенные примеры показывают, что возможно синтезировать различные поколения дендритных макромолекул по изобретению. Синтезированные макромолекулы по изобретению не подвергаются деградации путем реакций гидролиза. Синтез можно проводить с использованием различных растворителей, различных катализаторов и при различных условиях проведения реакций. Было показано, что можно осуществлять различные этапы реакций без выделения (промежуточного) продукта после каждого этапа, а это означает, что весь процесс значительно упрощается. Наиболее удаленное поколение дендритных макромолекул по изобретению можно модифицировать несколькими функциональными группами. И, наконец, примеры показывают, что дендритные макромолекулы по изобретению обладают очень хорошей термостабильностью.
Реферат
Изобретение относится к дендритной макромолекуле, включающей ядро и ответвления, идущие от ядра, отличающейся тем, что ответвления получают из винилцианидных групп. Изобретение относится также к способу получения предлагаемой дендритной макромолекулы. Дендритные макромолекулы по изобретению не подвержены деградации в ходе реакций гидролиза и обладают высокой устойчивостью к высоким температурам. Способ получения хорошо подходит для использования его в широких масштабах, в ходе синтеза получаемые промежуточные соединения не требуется выделять. 19 з.п.ф-лы, 1 ил.

Формула

где R1 - ядро или группа предыдущего поколения;
R2 - H или -СН3;
R3 -

R4 - Н или группа следующего поколения;
R5 - Н или группа следующего поколения, где R4 и R5 в каждой группе

13.11.92 - по пп.1 - 10 и 13;
12.11.92 - по пп.11, 12, 14 и 15.
Комментарии