Семейство арил, гетероарил, о-арил и о-гетероарил карбасахаров - RU2603769C2
Код документа: RU2603769C2
Чертежи




Описание
Данное изобретение относится к семейству фторированных арил, гетероарил, О-арил и О-гетероарил соединений гликозида, способу их получения, а также к применению их в фармацевтической или косметической отраслях, в частности для лечения или предотвращения диабета и ожирения, и в качестве депигментриующего или осветляющего вещества.
Сахара и их производные составляют один из наиболее распространенных классов соединений в природе. Благодаря своему химическому строению они проявляют разные физико-химические свойства и могут играть ключевую роль в большом числе биологических процессов.
В последние годы растет интерес к открытию новых гликозидов, обладающих полезными свойствами с точки зрения улучшенной эффективности, селективности и стабильности.
Среди этих соединений обнаружены, в частности, арил гликозиды или фенол гликозиды, применяемые в области косметики или в лечении или предотвращении заболеваний, таких как диабет, ожирение, рак, воспалительные заболевания, аутоиммунные заболевания, инфекции, тромбообразование, и в ряде других терапевтических областях. Исходя из их биологических свойств и их строения, многочисленные исследовательские группы проявляют интерес к этим соединениям.
Флоризин, в частности, можно привести в качестве молекулы, известной своей активностью ингибировать натрий-зависимые котранспортеры глюкозы (Journal of Clinical Investigation, vol. 79, р. 1510, (1987); там же vol. 80, р. 1037 (1987); там же vol. 87, р. 561 (1991); J. of Med. Chem., vol. 42, р. 5311 (1999); British Jpurnal of Pharmacology, vol. 132, р. 578, (2001)).
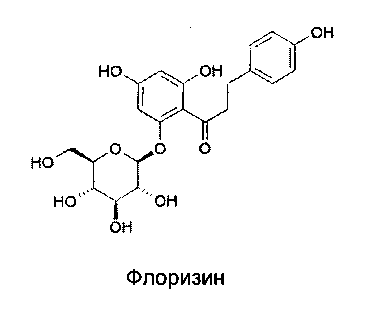
Ингибиторы натрий-зависимых котранспортеров глюкозы (SGLT), обнаруженные в частности в кишечнике и почках, вероятно пригодны для лечения диабета, и особенно диабета II типа, но также для гипергликемии, гиперинсулинемии, ожирения, гипертриглицеридемии, синдрома X (также известного как метаболический синдром, J. of Clin. Endocrinol. Metabol., 82, 727-734 (1997)), связанных с диабетом осложнений или еще атеросклероза. В действительности известно, что гипергликемия участвует в появлении и развитии диабета и приводит к снижению секреции инсулина и уменьшению чувствительности к инсулину, что приводит к повышению уровня глюкозы, таким образом, обостряя диабет. А значит, лечение гипергликемии можно рассматривать как средство лечения диабета.
В таком случае один из способов лечения гипергликемии должен способствовать выделению избытка глюкозы непосредственно в мочу, например при ингибировании натрий-зависимого котранспортера глюкозы в проксимальных канальцах почек, эффект этого состоит в ингибировании повторной абсорбции глюкозы и тем самым способствовании выделения ее в мочу, что приводит, таким образом, к снижению уровня сахара в крови.
В настоящее время существует большое количество лекарств, которые можно использовать для лечения диабета, такие как бигуаниды, сульфонилмочевины, вещества, улучшающие резистентность к инсулину и ингибиторы α-глюкозидаз. Однако эти соединения обладают многочисленными побочными эффектами, таким образом, повышая потребность в новых лекарствах.
Следовательно, согласно этому изобретению предложены новые соединения, которые полезны, в частности, для лечения или предотвращения диабета и ожирения.
Эти соединения представляют собой CF2-аналоги арил, гетероарил, О-арил, О-гетероарил гликозидов, где внутрициклический гликозидный кислород заменен на атом углерода, несущий два атома фтора. Эти соединения будут обладать отличительным признаком существующих стабильных аналогов О-арил и О-гетероарил гликозидов при столкновении с процессами ферментативного расщепления, в частности посредством ферментов типа глюкозидазы. Кроме того, двуфтористый углерод является хорошим имитатором атома кислорода.
Стабильные аналоги арил-гликозида, где есть аномерный кислород, который заменен на атом углерода, несущий два атома фтора, описаны в заявке на патент WO 2009/121939.
Синтез О-арил гликозидов, где внутрициклический или аномерный кислород заменен на атом углерода, несущий два атома фтора, описан в заявке на патент WO 2005/044256. А именно, описан синтез следующего соединения:

Об О-арил и арил аналогах, где эндоциклический кислород заменен на атом углерода, несущий два атома галогена, также сообщается в \Л/0 2009/076550, но не в качестве примера.
Таким образом, изобретатели разработали новые синтетические подходы, обеспечивающие доступ к новым арил, гетероарил, О-арил и О-гетероарил соединениям, полезным в качестве ингибиторов SGLT, в частности для лечения или предотвращения диабета и ожирения, и полезным в качестве ингибиторов тирозиназы, а именно для косметического применения и особенно в качестве депигментирующих или осветляющих веществ, и также в качестве антиоксидантов.
Следовательно, настоящее изобретение относится к соединению, имеющему следующую формулу (I):
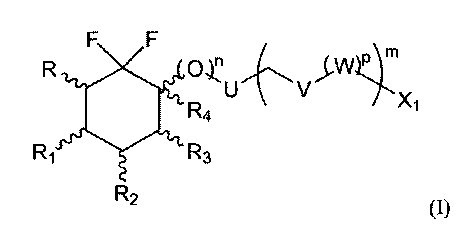
или его фармацевтически или косметически приемлемой соли, таутомеру, стереоизомеру или смеси стереоизомеров в любом соотношении, в частности смеси энантиомеров, и особенно рацемической смеси,
где
- n, m и р представляют собой независимо друг от друга 0 или 1,
- R представляет собой атом водорода или фтора или группу СН3, CH2F, СН2ОН, CH2OSiRaRbRc, CH2OR11, CH2OCOR11, CH2OCO2R11, CH2OCONR12R13, СН2ОР(O)(OR14)2 или CH2OSO3R14,
- R1 и R2 представляют собой независимо друг от друга атом фтора или группу ОН, OSiRdReRf, OR15, OCOR15, OCO2R15 или OCONR16R17,
- R3 представляет собой атом водорода или фтора или группу ОН, OSiRgRhRi, OR18, OCOR18, OCO2R18, OCONR19R20, NR19R20 или NR19COR18,
- R4 представляет собой атом водорода, когда n=1, и R4 представляет собой атом водорода, атом галогена или группу ОН, OSiRjRkRl, OR21, OCOR21, OCO2R21 или OCONR22R23, когда n=0,
или R и R1 вместе с атомами углерода, несущими их, образуют циклический ацеталь, имеющий следующую формулу:
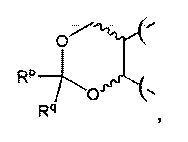
и/или (R1 и R2), (R2 и R3) и/или (R3 и R4) вместе с атомами углерода, несущими их, образуют циклический ацеталь, имеющий следующую формулу:
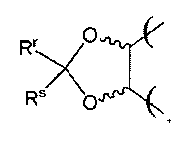
- Х1 представляет собой атом водорода, атом галогена, группу CN, ОН, SO2, SiRmRnRo, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24, CO2R24, NR25R26, NR25COR24, CONR25R26, SR24, SO2R24, CSR24 или OSO3R24, и
- U, V и W представляют собой независимо друг от друга кольцо фенила, пиразолила, N-(С1-С6)алкил-пиразолила или тиенила,
указанное кольцо возможно замещено одним или более заместителями, выбранными из группы, состоящей из атома галогена, групп CN, ОН, SO2, SiRmRnRo, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24, CO2R24, NR25R26, NR25COR24, CONR25R26, SR24, SO2R24, CSR24 и OSO3R24,
с:
- R11, R15, R18, R21 и R24, представляющими собой независимо друг от друга группу (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, гетероциклоалкил с 5-7 кольцевыми членами, арил, арил-(С1-С6)-алкил или (СгС6)-алкил-арил, эта группа возможно замещена одной или более группами, выбранными из атома галогена, групп ОН, СООН и СНО,
- R12, R13, R16, R17, R19, R20, R22, R23, R25 и R26, представляющими собой независимо друг от друга атом водорода или группу (С1-С6)-алкил или арил-(C1-С6)-алкил,
- R14, представляющим собой атом водорода или группу (С1-С6)-алкил,
- Ra до Ro, представляющими собой независимо друг от друга группу (С1-С6)-алкил, арил или арил-(С1-С6)-алкил, и
- Rp до Rs, представляющими собой независимо друг от друга атом водорода, группу (С1-С6)-алкил, арил или группу арил-(С1-С6)-алкил.
В данном изобретении подразумевается, что «фармацевтически или косметически приемлемый» означает, что это полезно в получении фармацевтической или косметической композиции, которая в целом безопасна, нетоксична и ни биологически, ни иным образом нежелательна, и которая приемлема для применения в ветеринарии и фармацевтического применения человеком, а также для косметического применения.
В данном изобретении подразумевается, что «фармацевтически или косметически приемлемые соли» соединения обозначают соли, которые фармацевтически или косметически приемлемы, как определено здесь, и которые обладают требуемой фармакологической активностью исходного соединения. Такие соли включают:
(1) гидраты и сольваты, такие как сольват (S)-пропиленгликоля,
(2) кислотно-аддитивные соли, образованные с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота или подобные; или образованные с органическими кислотами, такими как уксусная кислота, бензолсульфоновая кислота, бензойная кислота, камфорсульфоновая кислота, лимонная кислота, этансульфоновая кислота, фумаровая кислота, глюкогептоновая кислота, глюконовая кислота, глутаминовая кислота, гликолевая кислота, гидроксинафтойная кислота, 2-гидроксиэтансульфоновая кислота, молочная кислота, малеиновая кислота, яблочная кислота, миндальная кислота, метансульфоновая кислота, муконовая кислота, 2-нафталинсульфоновая кислота, пропионовая кислота, салициловая кислота, янтарная кислота, дибензоил-L-винная кислота, винная кислота, п-толуолсульфоновая кислота, триметилуксусная кислота, трифторуксусная кислота и подобные; и
(3) соли, образованные, когда кислотный протон, присутствующий в исходном соединении, либо замещен ионом металла, например ионом щелочного металла (например Na+, К+ или Li+), ионом щелочноземельного металла (подобного Са2+ или Mg2+) или ионом алюминия; либо координирует с органическим или неорганическим основанием. Приемлемые органические основания включают диэтаноламин, этаноламин, N-метилглюкамин, триэтаноламин, трометамин и подобные. Приемлемые неорганические основания включают гидроксид алюминия, гидроксид кальция, гидроксид калия, карбонат натрия и гидроксид натрия.
В данном изобретении подразумевается, что «таутомер» означает изомер, полученный в ходе прототропии, т.е. перемещения атома водорода и изменения расположения двойной связи. Разные таутомеры соединения, как правило, взаимообратимы и находятся в равновесии в растворе в разных соотношениях, которые зависят от используемого растворителя, от температуры или от рН.
В данном изобретении «стереоизомеры» означают изомеры, имеющие одинаковую молекулярную формулу и последовательность связанных атомов, но которые различаются трехмерным расположением этих атомов в пространстве. Они обозначают, таким образом, Е/Z изомеры, диастереоизомеры и энантиомеры. Е/Z изомеры являются соединениями, имеющими двойную связь, заместители, находящиеся на этой двойной связи, не располагаются с одной стороны двойной связи. Стереоизомеры, которые не являются зеркальным отображением друг друга, таким образом, обозначают как «диастереоизомеры», и стереоизомеры, которые являются неналагающимися зеркальными изображениями, обозначают как «энантиомеры».
А именно, сахарная группировка соединений по изобретению может принадлежать к D или L семейству, и предпочтительно к D семейству.
Атом углерода, связанный с четырьмя разными заместителями, называется «хиральным центром».
Эквимолярная смесь двух энантиомеров называется рацемической смесью.
В значении этого изобретения подразумевается, что «галоген» означает атом фтора, брома, хлора или йода.
В значении этого изобретения подразумевается, что группа «(С1-С6)-алкил» означает насыщенную, линейную или разветвленную углеводородную цепь, состоящую из 1-6 атомов углерода, в частности группы метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, н-гексил.
В значении этого изобретения подразумевается, что группа «(С2-С6)-алкенил» означает линейную или разветвленную углеводородную цепь, включающую по меньшей мере одну двойную связь и состоящую из 2 - 6 атомов углерода, например такую как группа этенил (винил) или пропенил.
В значении этого изобретения подразумевается, что группа «(С2-С6)-алкинил» означает линейную или разветвленную углеводородную цепь, включающую по меньшей мере одну тройную связь и состоящую из 2-6 атомов углерода, например такую как группа этинил или пропинил.
В значении этого изобретения подразумевается, что группа «(С3-С7)-циклоалкил» означает насыщенное углеводородное кольцо, состоящее из 3-7, предпочтительно 5-7, атомов углерода, в частности группу циклогексил, циклопентил или циклогептил.
В значении этого изобретения подразумевается, что группа «гетероциклоалкил с 5-7 кольцевыми членами» означает насыщенное углеводородное кольцо, имеющее 5-7 членов и содержащее один или более, предпочтительно один или два, гетероатомов вместо атомов углерода, например такие как атомы серы, азота или кислорода, например такая как группа тетрагидрофуранил, пиперидинил, пирролидинил, тетрагидропиранил, 1,3-диоксоланил.
В значении этого изобретения подразумевается, что группа «арил» означает углеводородную ароматическую группу, предпочтительно состоящую из 5 - 10 атомов углерода и включающую одно или более конденсированный колец, например такая как группа фенил или нафтил. Арил предпочтительно является фенилом.
В значении этого изобретения подразумевается, что группа «арил-(С1-С6)-алкил» означает любую арильную группу, как определено выше, которая связана с молекулой посредством (С1-С6)-алкильной группы, как определено выше. В частности группа, такая как эта, может представлять собой бензильную группу.
В значении этого изобретения подразумевается, что группа «(С1-С6)-алкил-арил» означает (С1-С6)-алкильную группу, как определено выше, которая связана с молекулой посредством арильной группы, как определено выше. В частности группа, такая как эта, может представлять собой метилфенильную группу.
В значении этого изобретения группа «N-(С1-С6)алкил-пиразолил» является группой следующей формулы, где X представляет собой группу (С1-С6)алкил, как определено выше:

эта группа связана с остатком молекулы через два атома углерода пиразолильной группировки.
Соединения по изобретению предпочтительно основаны на следующих формулах (Ia), (Ib) и (Ic), и в частности (Ia) и (Ic):
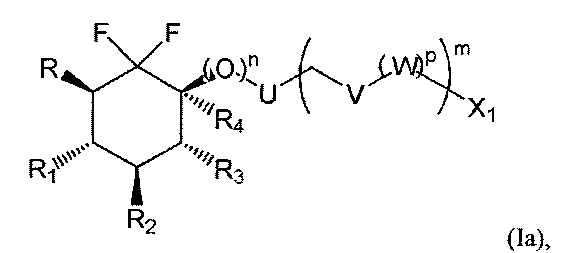
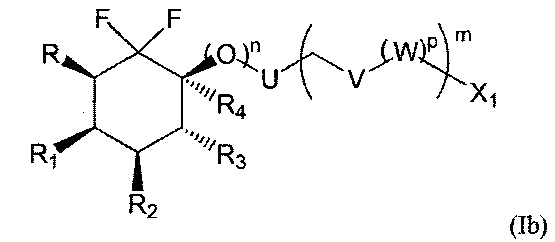

с R, R1, R2, R3, R4, X1, U, V, W, n, m и р такими, как определено выше.
Предпочтительно R1 и R2 представляют собой независимо друг от друга атом фтора или группу ОН, OSiRdReRf, OR15, OCOR15, OCO2R15 или OCONR16R17, и R3 представляет собой атом фтора или группу ОН, OSiRgRhRi, OR18, OCOR18, OCO2R18 или OCONR19R20.
Более предпочтительно R1 и R2 представляют собой независимо друг от друга группу ОН, OR15 или OCOR15, и R3 представляет собой группу ОН, OR18 или OCOR18.
Даже более предпочтительно R1, R2 и R3 могут быть выбраны независимо друг от друга из групп ОН, -O-(С1-С6)-алкил, -О-арил, -O-(С1-С6)-алкил-арил и -ОСО-(С1-С6)-алкил.
В частности R1, R2 и R3 могут быть выбраны независимо друг от друга из групп ОН, OSiMe3 и бензилокси (OBn), и предпочтительно из ОН и OBn.
Согласно особому воплощению R1, R2 и R3 являются одинаковыми.
Согласно другому особому воплощению R1, R2 и R3 являются одинаковыми, и каждый представляет собой группу ОН и R представляет собой группу СН2ОН.
Р предпочтительно представляет собой атом водорода или группу СН3, СН2ОН, CH2OR11, CH2OSiRaRbRc, CH2OCOR11, СН2ОР(O)(ОН)2 или CH2OSO3H, и в частности атом водорода или группу СН3, СН2ОН, CH2OR11, CH2OCOR11, СН2ОР(O)(ОН)2 или CH2OSO3H,
с Ra, Rb, Pc и R11 такими, как определено выше, и с CH2OR11, предпочтительно представляющим собой группу -CH2O-(С1-С6)-алкил, -CH2O-арил и -CH2O-(С1-С6)-алкил-арил, и CH2OCOR11, более предпочтительно представляющим собой группу -СН2ОСО-(С1-С6)-алкил.
Даже более предпочтительно Р представляет собой группу СН2ОН, CH2OSiRaRbRc, CH2OR11 или CH2OCOR11, и более предпочтительно группу СН2ОН, CH2OR11 или CH2OCOR11, с Ra, Rb, Rc и R11 такими, как определено выше.
Еще более предпочтительно R представляет собой группу СН2ОН, -CH2O-(С1-С6)-алкил, -CH2O-арил, -CH2O-(С1-С6)-алкил-арил и -СН2ОСО-(С1-С6)-алкил.
В частности R может представлять собой группу СН2ОН, CH2OSiMe3 или CH2OBn, и предпочтительно группу СН2ОН или CH2OBn.
Таким же образом R4 может предпочтительно представлять собой атом водорода или галогена или группу ОН или OR24, и в частности атом водорода или группу ОН или OR24 с R24 таким, как определено выше.
Еще более предпочтительно R4 может представлять собой атом водорода или галогена или группу ОН, -O-(С1-С6)-алкил, -О-арил и -O-(С1-С6)-алкил-арил, и в частности атом водорода или группу ОН, -O-(С1-С6)-алкил, -О-арил и -O-(С1-С6)-алкил-арил.
В частности R4 может представлять собой атом водорода или галогена (такой как Br, Cl, F) или группу ОН, и предпочтительно атом водорода или группу ОН, и особенно атом водорода.
Предпочтительно R4=Н, когда n=1, и R4=Н или ОН, когда n=0.
Предпочтительно X1 выбран из группы, состоящей из атома водорода, атома галогена, групп ОН, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24, CO2R24, NR25R26, NR25COR24 и CONR25R26; более предпочтительно из группы, состоящей из атома водорода, атома галогена, групп ОН, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24 и CO2R24; даже более предпочтительно из группы, состоящей из атома водорода, атома галогена, групп ОН, (С1-С6)-алкил и OR24.
Предпочтительно U, V и W представляют собой независимо друг от друга кольцо фенила, пиразолила, N-(С1-С6)алкил-пиразолила или тиенила, указанное кольцо возможно замещено одним или более заместителями, выбранными из группы, состоящей из атома галогена, групп ОН, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24, CO2R24, NR25R26, NR25COR24 и CONR25R26; более предпочтительно из группы, состоящей из атома галогена, групп ОН, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24 и CO2R24; даже более предпочтительно из группы, состоящей из атома галогена, групп ОН, (С1-С6)-алкил и OR24.
(1) В первом воплощении n представляет собой 1.
В первой подгруппе этого воплощения m=0 и U представляет собой возможно замещенный фенил. Соединения по изобретению, таким образом, могут быть представлены следующей формулой (I-1), и особенно следующими формулами (I-1а), (I-1b) и (I-1с), и в частности (I-1а) и (I-1с):
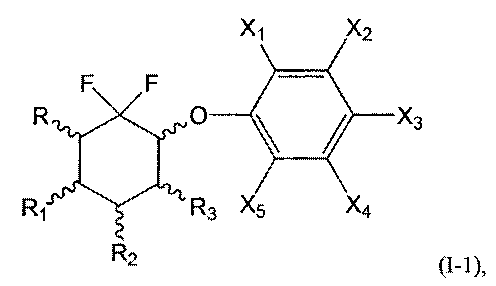


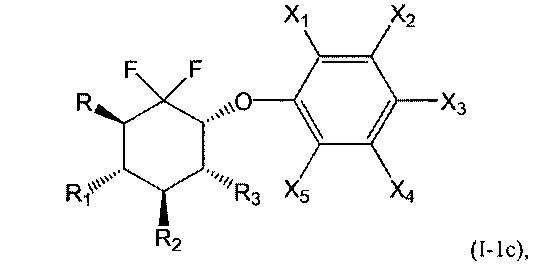
или их фармацевтически или косметически приемлемая соль, таутомер, стереоизомер или смесь стереоизомеров в любом соотношении, в частности смесь энантиомеров, и особенно рацемическая смесь,
где
- R, R1, R2 и R3 являются такими, как определено выше, и
- Х1, Х2, Х3, Х4 и Х5 представляют собой независимо друг от друга атом водорода, атом галогена, группу CN, ОН, SO2, SiRmRnRo, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24, CO2R24, NR25R26, NR25COR24, CONR25R26, SP24, SO2R24, CSR24 или OSO3R24; предпочтительно выбраны из группы, состоящей из атома водорода, атома галогена, групп ОН, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24, CO2R24, NR25R26, NR25COR24 и CONR25R26; более предпочтительно выбраны из группы, состоящей из атома водорода, атома галогена, групп ОН, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24 и CO2R24; даже более предпочтительно выбраны из группы, состоящей из атома водорода, атома галогена, групп ОН, (С1-С6)-алкил и OR24.
Примеры этой первой подгруппы включают, но не ограничиваются этим:

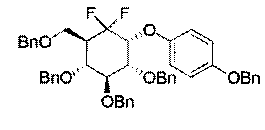
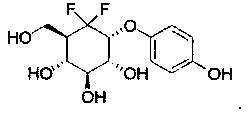
Во второй подгруппе этого воплощения m=1, р=0 и U и V представляют собой независимо друг от друга возможно замещенный фенил. Соединения по изобретению, таким образом, могут быть представлены следующей формулой (I-2), и особенно следующими формулами (I-2а) и (I-2b), и в частности (I-2а):



или их фармацевтически или косметически приемлемая соль, таутомер, стереоизомер или смесь стереоизомеров в любом соотношении, в частности смесь энантиомеров, и особенно рацемическая смесь,
где
- R, R1, R2 и R3 являются такими, как определено выше, и
- X1, Х2, Х3, Х4, Х5, Х6, Х7, Х8 и Х9 представляют собой независимо друг от друга атом водорода, атом галогена, группу CN, ОН, SO2, SiRmRnRo, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24, CO2R24, NR25R26, NR25COR24, CONR25R26, SR24, SO2R24, CSR24 или OSO3R24; предпочтительно выбраны из группы, состоящей из атома водорода, атома галогена, групп ОН, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24, CO2R24, NR25R26, NR25COR24 и CONR25R26; более предпочтительно выбраны из группы, состоящей из атома водорода, атома галогена, групп ОН, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24 и CO2R24; даже более предпочтительно выбраны из группы, состоящей из атома водорода, атома галогена, групп ОН, (С1-С6)-алкил и OR24.
Примеры этой второй подгруппы включают, но не ограничиваются этим:


В третьей подгруппе этого воплощения m=1, р=0, U представляет собой группу пиразолил или N-(С1-С6)алкил-пиразолил и V представляет собой возможно замещенный фенил. Соединения по изобретению, таким образом, могут быть представлены следующей формулой (I-3), и особенно следующими формулами (I-3а) и (I-3b), и в частности (I-3а):



или их фармацевтически или косметически приемлемая соль, таутомер, стереоизомер или смесь стереоизомеров в любом соотношении, в частности смесь энантиомеров, и особенно рацемическая смесь,
где
- R, R1, R2 и R3 являются такими, как определено выше,
- X1, Х2, Х3, Х4, Х5 и Х6 представляют собой независимо друг от друга атом водорода, атом галогена, группу CN, ОН, SO2, SiRmRnRo, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24, CO2R24, NR25R26, NR25COR24, CONR25R26, SR24, SO2R24, CSR24 или OSO3R24; предпочтительно выбраны из группы, состоящей из атома водорода, атома галогена, групп ОН, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24, CO2R24, NR25R26, NR25COR24 и CONR25R26; более предпочтительно выбраны из группы, состоящей из атома водорода, атома галогена, групп ОН, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24 и CO2R24; даже более предпочтительно выбраны из группы, состоящей из атома водорода, атома галогена, групп ОН, (С1-С6)-алкил и OR24, и
- X представляет собой атом водорода или группу (С1-С6)-алкил.
Примеры этой третьей подгруппы включают, но не ограничиваются этим:
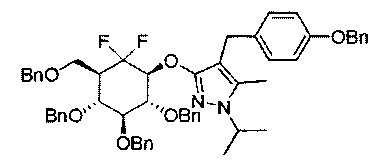

(2) Во втором воплощении n представляет собой 0.
В первой подгруппе этого воплощения m=1, р=0 и U и V независимо представляют собой возможно замещенный фенил. Соединения по изобретению, таким образом, могут быть представлены следующей формулой (I-4), и особенно следующими формулами (I-4а) и (I-4b), и в частности (I-4а):



или их фармацевтически или косметически приемлемая соль, таутомер, стереоизомер или смесь стереоизомеров в любом соотношении, в частности смесь энантиомеров, и особенно рацемическая смесь,
где
- R, R1, R2, R3 и R4 являются такими, как определено выше, и
- Х1, Х2, Х3, Х4, Х5, Х6, Х7, Х8 и Х9 представляют собой независимо друг от друга атом водорода, атом галогена, группу CN, ОН, SO2, SiRmRnRo, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24, CO2R24, NR25R26, NR25COR24, CONR25R26, SR24, SO2R24, CSR24 или OSO3R24; предпочтительно выбраны из группы, состоящей из атома водорода, атома галогена, групп ОН, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24, CO2R24, NR25R26, NR25COR24 и CONR25R26; более предпочтительно выбраны из группы, состоящей из атома водорода, атома галогена, групп ОН, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24 и CO2R24; даже более предпочтительно выбраны из группы, состоящей из атома водорода, атома галогена, групп ОН, (С1-С6)-алкил и OR24.
Примеры этой первой подгруппы включают, но не ограничиваются этим:

Во второй подгруппе этого воплощения m=1, р=1, U и W независимо представляют собой возможно замещенный фенил, и V представляет собой возможно замещенный тиенил. Соединения по изобретению, таким образом, могут быть представлены следующей формулой (I-5), и особенно следующими формулами (I-5а) и (I-5b), и в частности (I-5а):



или их фармацевтически или косметически приемлемая соль, таутомер, стереоизомер или смесь стереоизомеров в любом соотношении, в частности смесь энантиомеров, и особенно рацемическая смесь,
где
- R, R1, R2, R3 и R4 являются такими, как определено выше, и
- Х1, Х2, Х3, Х4, Х5, Х6, Х7, Х8, Х9, Х10 и Х11 представляют собой независимо друг от друга атом водорода, атом галогена, группу CN, ОН, SO2, SiRmRnRo, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24, CO2R24, NR25R26, NR25COR24, CONR25R26, SR24, SO2R24, CSR24 или OSO3R24; предпочтительно выбраны из группы, состоящей из атома водорода, атома галогена, групп ОН, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24, CO2R24, NR25R26, NR25COR24 и CONR25R26; более предпочтительно выбраны из группы, состоящей из атома водорода, атома галогена, групп ОН, (С1-С6)-алкил, (С2-С6)-алкенил, (С2-С6)-алкинил, (С3-С7)-циклоалкил, OR24, COR24, OCOR24 и CO2R24; даже более предпочтительно выбраны из группы, состоящей из атома водорода, атома галогена, групп ОН, (С1-С6)-алкил и OR24.
Примеры этой второй подгруппы включают, но не ограничиваются этим:
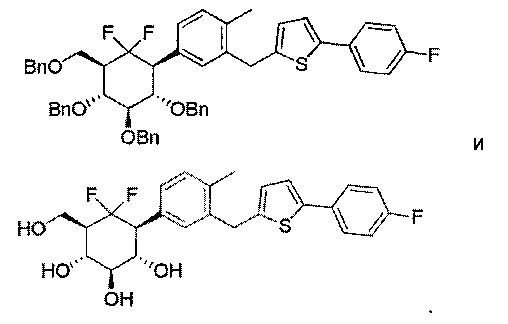
Таким образом, соединения по изобретению могут быть выбраны из следующих соединений:


Другим предметом данного изобретения является соединение, как определено выше, для применения в качестве лекарства, в частности в качестве ингибитора натрий-зависимого котранспортера глюкозы, такого как SGLT1, SGLT2 и SGLT3.
В значении данного изобретения подразумевается, что «ингибитор натрий-зависимого котранспортера глюкозы» означает соединение, способное ингибировать частично или полностью натрий-зависимый котранспортер глюкозы.
А именно, соединения по изобретению можно использовать для лечения или предотвращения диабета, и особенно диабета II типа, связанных с диабетом осложнений, таких как воспаление артерий нижних конечностей, инфаркт миокарда, почечная недостаточность, нейропатия или слепота, гипергликемии, гиперинсулинемии, ожирения, гипертриглицеридемии, синдрома X и артериосклероза. Соединения по изобретению применяются в частности для лечения или предотвращения диабета.
Подобным образом соединения по изобретению можно использовать в качестве противоракового, антибактериального, противовирусного, антитромботического или противовоспалительного средства.
Также изобретение относится к соединению по изобретению для применения его в лечении или предотвращении диабета, и особенно диабета II типа, связанных с диабетом осложнений, таких как воспаление артерий нижних конечностей, инфаркт миокарда, почечная недостаточность, нейропатия или слепота, гипергликемии, гиперинсулинемии, ожирения, гипертриглицеридемии, синдрома X и артериосклероза, а также для применения его в качестве противоракового, антибактериального, противовирусного, антитромботического или противовоспалительного средства, и в частности в лечении или предотвращении диабета.
Также изобретение относится к применению соединения по изобретению для изготовления лекарства, предназначенного для лечения или предотвращения диабета, и особенно диабета II типа, связанных с диабетом осложнений, таких как воспаление артерий нижних конечностей, инфаркт миокарда, почечная недостаточность, нейропатия или слепота, гипергликемии, гиперинсулинемии, ожирения, гипертриглицеридемии, синдрома X и артериосклероза, а также для изготовления противоракового, антибактериального, противовирусного, антитромботического или противовоспалительного средства, и в частности для лечения или предотвращения диабета.
Также изобретение относится к способу лечения или предотвращения диабета, и особенно диабета II типа, связанных с диабетом осложнений, таких как воспаление артерий нижних конечностей, инфаркт миокарда, почечная недостаточность, нейропатия или слепота, гипергликемии, гиперинсулинемии, ожирения, гипертриглицеридемии, синдрома X и артериосклероза, а также противоракового, антибактериального, противовирусного, антитромботического или противовоспалительного лечения, и в частности лечения или предотвращения диабета, согласно которому вводят эффективное количество по меньшей мере одного соединения по изобретению пациенту при необходимости этого.
Силилированные соединения по настоящему изобретению, а также соединения с R=CH2OBn, R1=OBn, R2=OBn и/или R3=OBn, не являются предпочтительными для их применения в качестве лекарства.
А именно, соединения, пригодные в качестве лекарства, и особенно в лечении или предотвращении диабета, являются соединениями формулы (Ia) или (Ib), и в частности (Ia); особенно соединения формулы (I-2)-(I-5), такие как (I-2а)-(I-5а) и (I-2b)-(I-5b), и в частности (I-2а)-(I-5а).
Другим предметом данного изобретения является косметическое применение соединения по изобретению, как определено выше, для осветления, отбеливания, депигментации кожи, удаления пятен с кожи, особенно старческих пятен и веснушек, или предотвращения пигментации кожи, или в качестве антиоксиданта, в частности посредством местного применения.
Таким образом, настоящее изобретение относится к способу осветления, отбеливания, депигментации кожи, удаления пятен с кожи, особенно старческих пятен и веснушек, или предотвращения пигментации кожи, согласно которому местно применяют по меньшей мере одно соединение по изобретению.
Силилированные соединения по настоящему изобретению, а также соединения с R=CH2OBn, R1=OBn, R2=OBn и/или R3=OBn, не являются предпочтительными для их косметического применения.
А именно, соединения, пригодные в косметической отрасли, в частности в качестве депигментирующих или осветляющих средств, являются соединениями формулы (Ia), (Ib) или (Ic), и в частности (Ic); особенно соединения формулы (I-1), такие как (I-1а), (I-1b) и (I-1с), а именно, (I-1с).
В частности соединения с депигментирующей активностью представляют собой ингибиторы тирозиназы. Они в частности являются соединениями следующей формулы:




Другим предметом данного изобретения является фармацевтическая или косметическая композиция, включающая по меньшей мере одно соединение по изобретению, как определено выше, и по меньшей мере один фармацевтически или косметически приемлемый наполнитель.
Соединения по изобретению могут быть введены перорально, под язык, парентерально, под кожу, внутримышечно, внутривенно, трансдермально, локально или ректально.
В фармацевтических составах по данному изобретению для перорального, подъязычного, парентерального, подкожного, внутримышечного, внутривенного, трансдермального, локального или ректального введения активный ингредиент может быть введен в стандартных формах введения, смешанных с общепринятыми фармацевтическими носителями, животным или людям. Подходящие стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, порошки, гранулы и пероральные растворы или суспензии, подъязычные или буккальные формы введения, парентеральные, подкожные, внутримышечные, внутривенные, интраназальные или интраокулярные формы введения и ректальные формы введения.
Когда твердую композицию получают в форме таблеток, главный активный ингредиент смешивают с фармацевтическим наполнителем, таким как желатин, крахмал, лактоза, стеарат магния, тальк, гуммиарабик или подобным. Таблетки можно покрыть сахарозой или другими подходящими веществами, или еще обработать таким образом, чтобы они обладали длительной или замедленной активностью и непрерывно высвобождали заранее определенное количество активного компонента.
Препарат желатиновых капсул получают, смешивая активный ингредиент с разбавителем и выливая полученную смесь в мягкие или твердые капсулы.
Препарат в форме сиропа или эликсира может содержать активный ингредиент вместе с подсластителем, антисептическим средством, а также ароматизатором и подходящим красителем.
Порошки или гранулы, диспергируемые в воде, могут содержать активный ингредиент, смешанный с диспергирующими агентами, смачивающими веществами или суспендирующими веществами, а также с корректорами вкуса или подсластителями.
Для ректального введения используют суппозитории, которые получают со связующими веществами, плавящимися при температуре прямой кишки, например какао-маслом или полиэтиленгликолями.
Для парентерального, интраназального или интраокулярного ведения используют водные суспензии, изотонические солевые растворы или стерильные и инъекционные растворы, которые содержат фармакологически совместимые диспергирующие агенты и/или смачивающие вещества.
Активный компонент также может быть приготовлен в виде микрокапсул возможно с одним или более дополнительными носителями.
Соединения по изобретению можно применять в дозах между 0,01 мг и 1000 мг в день, принимаемых в виде однократной дозы один раз в день или вводимых несколькими дозами в течение дня, например два раза в день равными дозами. Вводимая суточная доза предпочтительно находится между 0,1 мг и 100 мг, даже более предпочтительно между 2,5 мг и 50 мг. Может быть необходимо использовать дозы, превышающие эти пределы, что квалифицированные специалисты в данной области техники должны иметь в виду.
В одном особом воплощении изобретения фармацевтическая или косметическая композиция также может быть приготовлена для местного введения. Ее можно вводить в формах, хорошо известных для этого вида введения, т.е. в частности в виде лосьонов, пенок, гелей, дисперсий, спреев, шампуней, сывороток, масок, молочков для тела или кремов, например с эксципиентами, способствующими в частности прониканию через кожу, с тем, чтобы улучшить свойства и доступность активного компонента. Кроме композиции по изобретению эти композиции обычно также содержат физиологически приемлемую среду, которая, как правило, содержит воду или растворитель, например спирты, эфиры или гликоли. Также они могут содержать поверхностно-активные вещества, консерванты, стабилизаторы, эмульгаторы, загустители, другие активные компоненты, оказывающие дополнительное или возможно совместное действие, следовые элементы, эфирные масла, отдушки, красители, коллаген, химические или неорганические фильтры, гидратирующие агенты или термальные воды.
В одном особом воплощении фармацевтическая композиция по изобретению может включать по меньшей мере один другой активный компонент в дополнение к соединению по изобретению.
Примерами активных компонентов, которые можно привести, являются противодиабетические средства, такие как соединения типа сульфонилмочевины, которые представляют собой гипогликемические сульфамиды, которые увеличивают секрецию инсулина, подобные например хлорпропамиду, толбутамиду, толазамиду, глипизиду, гликлазиду, глибенкламиду, гликуидону и глимепириду, бигуаниды, которые уменьшают печеночный гликонеогенез и резистентность к инсулину, подобные метформину, тиазолидиндионы (также называемые глитазонами), которые увеличивают чувствительность к инсулину, подобные росиглитазону, пиоглитазону и циглитазону, ингибиторы альфа-глюкозидаз, которые замедляют всасывание в кишечнике углеводов, подобные акарбозе, миглитолу и воглибозе, меглитиниды (также называемые глитиниды), которые увеличивают поджелудочную секрецию инсулина, подобные репаглиниду и натеглиниду, миметики инкретина, подобные эксенатиду, или ингибиторы дипептидилпептидазы-4 (DPP4 от dipeptidylpeptidase-4, ДПП-4), подобные ситаглиптину, вилдаглиптину и инсулину, или антилипидные агенты, такие как статины, которые снижают холестерин, ингибируя фермент ГМГ-КоА редуктаза, подобные аторвастатину и церивастатину, фибраты, подобные безафибрату, гемфиброзилу и фенофибрату или эзетимибу.
Настоящее изобретение также относится к способам получения соединения по изобретению.
Таким образом, настоящее изобретение относится к способу получения соединения формулы (I) по изобретению, в котором R4=Н, согласно которому фторируют соединение следующей формулы (II):

где R, R1, R2, R3, Х1, U, V, W, n, m и р являются такими, как определено выше.
Фторирование проводят в присутствии фторирующего реагента, такого как DAST (трифторид диэтиламиносеры).
При необходимости можно осуществлять дополнительные стадии защиты, удаления защитных групп, замещения и т.п., эти стадии хорошо известны квалифицированному специалисту в данной области техники.
Полученное соединение формулы (I) можно выделить, отделяя от реакционной среды способами, хорошо известными квалифицированному специалисту в данной области техники, такими как экстракция, выпаривание растворителя или осаждение, или кристаллизация (с последующей фильтрацией).
Также соединение при необходимости можно очистить способами, хорошо известными квалифицированному специалисту в данной области техники, такими как перекристаллизация, перегонка, хроматография на колонке с силикагелем или высокоэффективная жидкостная хроматография (ВЭЖХ).
Соединение формулы (II) можно получить в ходе окисления соединения следующей формулы (III):

где R, R1, R2, R3, Х1, U, V, W, n, m и р являются такими, как определено выше.
Окисление проводят в присутствии окислителя согласно методикам, хорошо известным квалифицированному специалисту в данной области техники. Окислитель может представлять собой, например периодинан Десса - Мартина, ПХХ (хлорхромат пиридиния) и т.п.
Когда n=1, то способ получения соединения формулы (III) может включать следующие последовательные стадии, согласно которым:
(а1) связывают соединение следующей формулы (IV):

где R, R1, R2 и R3 являются такими, как определено выше,
с соединением следующей формулы (V):

где Х1, U, V, W, m и р являются такими, как определено выше,
что дает соединение следующей формулы (VI):

где R, R1, R2, R3, Х1, U, V, W, m и р являются такими, как определено выше, и (b1) осуществляют реакцию гидроборирования-окисления соединения формулы (VI), полученного на предыдущей стадии (а1), что дает соединение формулы (III) с n=1.
Стадию (а1) можно проводить в условиях реакции Мицунобу, хорошо известной квалифицированному специалисту в данной области техники, особенно используя ДЭАД (диэтилазодикарбоксилат), ДИАД (диизопропилазодикарбоксилат) или АДДП (азодикарбонилдипиперидин) в качестве связующего реагента и PPh3 или Р(nBu)3 в качестве фосфина.
Стадию (b1) можно проводить в условиях, хорошо известных квалифицированному специалисту в данной области техники, особенно в ходе реакции с бораном, таким как ВН3, и в частности ВН3. ТГФ или ВН3.Me2S, в растворителе, таком как ТГФ, с последующим добавлением пероксида водорода в присутствии основания, такого как гидроксид натрия.
Когда n=0, то способ получения соединения формулы (III) может включать следующие последовательные стадии, согласно которым:
(а2) связывают соединение следующей формулы (VII):

где R, R1, R2 и R3 являются такими, как определено выше,
с соединением следующей формулы (VIII):
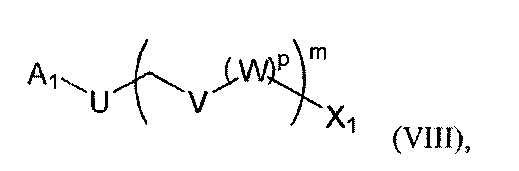
где Х1, U, V, W, m и р являются такими, как определено выше, и А1 представляет собой -Li или - Mg-Hal, Hal является атомом галогена, что дает соединение следующей формулы (IX):

где R, R1, R2, R3, Х1, U, V, m и р являются такими, как определено выше, (b2) восстанавливают соединение формулы (IX), полученное на предыдущей стадии (а2), что дает соединение следующей формулы (X):
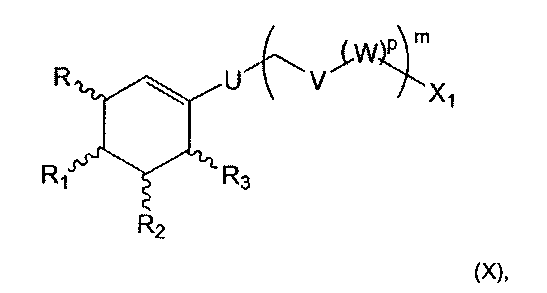
где R, R1, R2, R3, Х1, U, V, W, m и р являются такими, как определено выше, и (с2) осуществляют реакцию гидроборирования-окисления соединения формулы (X), полученного на предыдущей стадии (b2), что дает соединение формулы (III) с n=0.
Стадию (а2) можно проводить путем реакции соединения формулы (VIII), полученного из галогенированного производного в ходе реакции с магнием с образованием реактива Гриньяра или в ходе замены галогена, используя основание лития, такое как н-бутиллитий, с образованием соответствующего литированного соединения, с соединением формулы (VII) в растворителе, таком как ТГФ.
Подобное соединение формулы (VII) получают в условиях, хорошо известных квалифицированному специалисту в данной области техники, и особенно согласно способу, описанному в ЕР 0240175 или Carbohydrate Research 2010, 345, 1056-1060.
Соединение формулы (VIII) можно получить из галогенированного производного в ходе реакции с магнием с образованием реактива Гриньяра или в ходе замены галогена, используя основание лития, такое как н-бутиллитий, с образованием соответствующего литированного соединения.
Стадию (b2) можно проводить в присутствии восстановителя, такого как Et3SiH и кислоты Льюиса, такой как ВН3·Et2O.
Стадия (с2) соответствует предыдущей стадии (b1).
Способ получения соединений по изобретению с R4=Н более подробно будет описан ниже и в следующем экспериментальном разделе.
Схема А: Путь синтеза соединений первого воплощения (где n=1)

(а) На первой стадии циклогексенон Т1 подвергают восстановлению при стандартных условиях, таких как NaBH4, NaBH4/CeCl3, LiAlH4 или L-селектрид.
(b) Затем проводят реакцию сочетания Мицунобу между соединением Т2 и спиртом Т3 при стандартных условиях, используя ДЭАД, ДИАД или АДДП в качестве связующего реагента и PPh3 или Р(nBu)3 в качестве фосфина.
(с) Гидроборирование соединения Т4 при использовании ВН3.ТГФ или ВН3.Me2S дает соединение Т5.
(d) Спиртовую группировку соединения Т5 окисляют до кетона согласно обычным методикам, включая ПХХ, периодинан Десса - Мартина, что дает соединение Т6.
(е) Соединение Т6 можно фторировать, используя фторирующий реагент, такой как DAST, что дает дифторкарбасахар Т7. На последней стадии защитные группы можно удалить согласно обычным методикам, описанным в Protective groups (Protective groups in organic synthesis, Т.М. Greene). В частности:

(а) На первой стадии циклогексенон Т8 подвергают региоселективному восстановлению с участием три-втор-бутилборгидрида лития, как описано в Can. J Chem 2004, 82, 1361-1364.
(b) Затем проводят реакцию сочетания Мицунобу между соединением Т9 и спиртом Т3 при стандартных условиях, используя ДЭАД, ДИАД или АДДП в качестве связующего реагента и PPh3 или Р(nBu)3 в качестве фосфина.
(с) Гидроборирование соединения Т10 при использовании ВН3.ТГФ или ВН3.Me2S дает соединение Т11.
(d) Спиртовую группировку соединения Т11 окисляют до кетона согласно обычным методикам, включая ПХХ, периодинан Десса - Мартина, что дает соединение Т12.
(е) Соединение Т12 можно фторировать, используя фторирующий реагент, такой как DAST, что дает дифторкарбасахар Т13. На последней стадии защитные группы можно удалить согласно обычным методикам, описанным в Protective groups in organic synthesis, Т.W. Greene.
Циклогексенон Т8 получают согласно ЕР 0240175 или Cumpstey, I. Carbohydrate Research 2010, 345, 1056-1060, используя синтез до группы глюкозы из имеющейся в продаже 2,3,4,6-О-бензил-D-глюкопиранозы.
Соединение Т3 либо имеется в продаже (первая подгруппа), либо его синтезируют согласно:

(вторая подгруппа)
или

(R5 и R6 представляют собой группу (С1-С6)алкил) (третья подгруппа)
Схема В: Путь синтеза соединений второго воплощения (где n=0 и R4=Н)
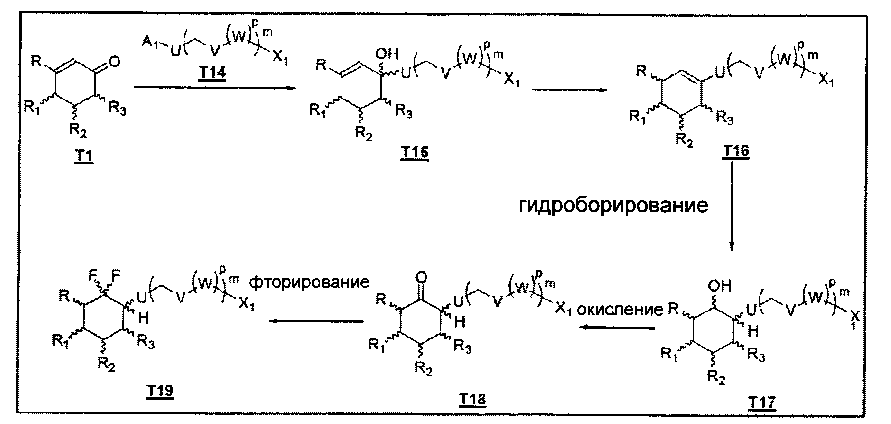
(а) На первой стадии реактив Гриньяра или литированное соединение Т14, полученное из соответствующего галогенированного соединения согласно обычной методике, добавляют к циклогексенону Т1.
(b) На следующей стадии синтеза соединение Т15 обрабатывают восстановителем, таким как Et3SiH в присутствии кислоты Льюиса, такой как BF3.Et2O, получая соединение Т16.
(с) Гидроборирование соединения Т16 при использовании ВН3.ТГФ или ВН3.Me2S дает соединение Т17.
(d) Спиртовую группировку соединения Т17 окисляют до кетона согласно обычным методикам, включая ПХХ, периодинан Десса - Мартина, что дает соединение Т18.
(е) Соединение Т18 можно фторировать, используя фторирующий реагент, такой как DAST, что дает дифторкарбасахар Т19. На последней стадии защитные группы можно удалить согласно обычным методикам, описанным в Protective groups in organic synthesis, Т.W. Creene.
И в частности:
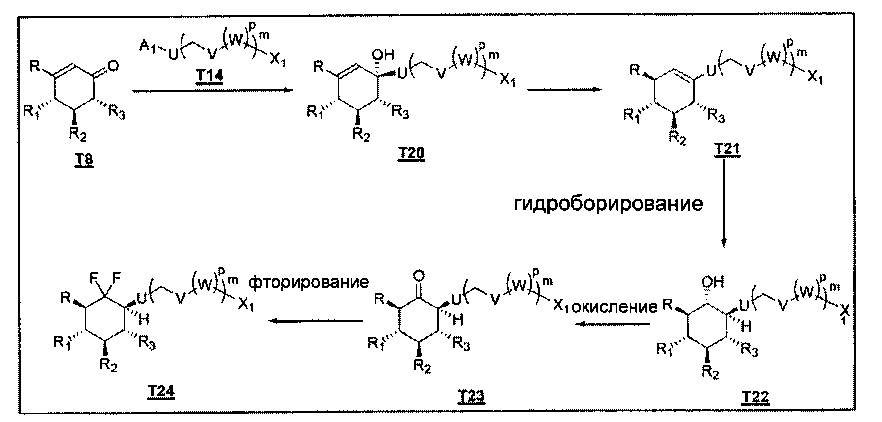
(а) На первой стадии реактив Гриньяра или литированное соединение Т14, полученное из соответствующего галогенированного соединения согласно обычной методике, добавляют к циклогексенону Т8.
(b) На следующей стадии синтеза соединение Т20 обрабатывают восстановителем, таким как Et3SiH в присутствии кислоты Льюиса, такой как BF3.Et2O, получая соединение Т21.
(с) Гидроборирование соединения Т21 при использовании ВН3.ТГФ или ВН3.Me2S дает соединение Т22.
(d) Спиртовую группировку соединения Т22 окисляют до кетона согласно обычным методикам, включая ПХХ, периодинан Десса - Мартина, что дает соединение Т23.
(е) Соединение Т23 можно фторировать, используя фторирующий реагент, такой как DAST, что дает дифторкарбасахар Т24. На последней стадии защитные группы можно удалить согласно обычным методикам, описанным в Protective groups in organic synthesis, Т.W. Creene.
Галогенированное соединение, дающее соединение Т14, можно синтезировать согласно следующей схеме:

Также настоящее изобретение относится к способу получения соединения формулы (I) по изобретению, где n=0 и R4≠Н, согласно которому связывают соединение формулы (VIII), как определено выше, и соединение следующей формулы (XI)
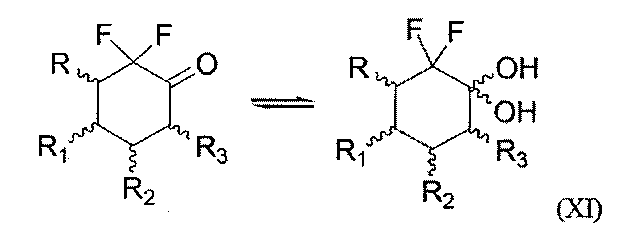
где R, R1, R2 и R3 являются такими, как определено выше,
что дает соединение формулы (I), где n=0 и R4=ОН,
с последующим возможным замещением группы ОН, что дает соединение формулы (I), где n=0 и R4= галоген, OSiRjRkRl, OR21, OCOR21, OCO2R21 или OCONR22R23.
Эти стадии связывания и замещения можно проводить в условиях, хорошо известных квалифицированному специалисту в данной области техники.
При необходимости можно осуществлять дополнительные стадии защиты, удаления защитных групп, замещения и т.п., эти стадии хорошо известны квалифицированному специалисту в данной области техники.
Полученное соединение формулы (I) можно выделить, отделяя от реакционной среды способами, хорошо известными квалифицированному специалисту в данной области техники, такими как экстракция, выпаривание растворителя или осаждение, или кристаллизация (с последующей фильтрацией).
Также соединение при необходимости можно очистить способами, хорошо известными квалифицированному специалисту в данной области техники, такими как перекристаллизация, перегонка, хроматография на колонке с силикагелем или высокоэффективная жидкостная хроматография (ВЭЖХ).
Способ получения соединения формулы (XI) может включать следующие последовательные стадии, согласно которым:
(а3) проводят реакцию гидроборирования-окисления соединения формулы (XII)

где R, R1, R2 и R3 являются такими, как определено выше, и R7=SiRa1Rb1Rc1 или СН2ОСН3 (метоксиметил - МОМ), где каждый Ra1, Rb1 и Rc1 независимо представляет собой группу (С1-С6)-алкил, арил или арил-(С1-С6)-алкил, что дает соединение следующей формулы (XIII)

где R, R1, R2 и R3 являются такими, как определено выше, и R7=SiRa1Rb1Rc1 или СН2ОСН3 (метоксиметил - МОМ),
(b3) окисляют соединение формулы (XIII), полученное на предшествующей стадии (а3), что дает соединение следующей формулы (XIV)

где R, R1, R2 и R3 являются такими, как определено выше, и R7=SiRa1Rb1Rc1 или СН2ОСН3 (метоксиметил - МОМ),
(с3) когда R7=SiRa1Rb1Rc1, то удаляют защитные группы у соединения формулы (XIV), полученного на предшествующей стадии (b3), что дает соединение формулы (XIV) с R7=Н,
(d3) когда R7=Н, то защищают соединение формулы (XIV) с R7=Н, полученное на предшествующей стадии (с3), что дает соединение формулы (XIV) с R7=COR8, где R8 представляет собой группу (С1-С6)-алкил, арил или арил-(С1-С6)-алкил,
(е3) фторируют соединение формулы (XIV) с R7=COR8 или СН2ОСН3, полученное на предшествующей стадии (d3) или (b3), что дает соединение следующей формулы (XV)
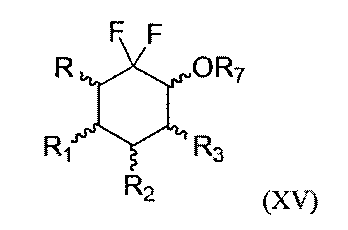
где R, R1, R2 и R3 являются такими, как определено выше, и R7=COR8 или СН2ОСН3,
(f3) удаляют защитные группы у соединения формулы (XV) с R7=COR8 или СН2ОСН3, полученного на предшествующей стадии (е3), что дает соединение формулы (XV) с R7=Н, и
(g3) окисляют соединение формулы (XV) с R7=Н, полученное на предшествующей стадии (f3), что дает соединение формулы (XI).
Стадия (а3) соответствует предыдущей стадии (b1). Соединение формулы (XII) можно получить из соединения формулы (IV) в ходе стадии защиты, хорошо известной квалифицированному специалисту в данной области техники.
Стадии (b3) и (g3) можно проводить в присутствии окислителя, такого как периодинан Десса - Мартина, ПХХ (хлорхромат пиридиния) и т.п.
Стадии (с3) и (d3) возможны и требуются только, когда R7=SiRa1Rb1Rc1 в исходном веществе формулы (XII).
Стадии (с3), (d3) и (f3) можно проводить в условиях, хорошо известных квалифицированному специалисту в данной области техники.
Стадию (е3) можно проводить в присутствии фторирующего реагента, такого как DAST (трифторид диэтиламиносеры).
Также настоящее изобретение относится к способу получения соединения формулы (I) по изобретению, где R4=Н, включающему следующие стадии, согласно которым:
(а4) бромируют соединение формулы (I) с R4=ОН, что дает соединение формулы (I) с R4=Br, и
(b4) восстанавливают соединение формулы (I) с R4=Br, полученное на предшествующей стадии (а4), что дает соединение формулы (I) с R4=Н.
Стадию (а4) можно проводить в присутствии бромирующего реагента, такого как SOBr2. Реакцию предпочтительно также проводят в присутствии основания, такого как пиридин. Исходное вещество можно получить согласно способу, описанному выше для получения соединений формулы (I) с R4≠Н.
Стадию (b4) можно проводить в присутствии гидрида, такого как Bu3SnH.
Способ получения соединений по изобретению с n=0 и R4=ОН или Н будет подробнее описан далее и в следующем экспериментальном разделе.
Схема С: Синтез соединений с n=0 и R4=ОН или Н

(а) На первой стадии циклогексенон Т1 подвергают восстановлению при стандартных условиях, таких как NaBH4, NaBH4/CeCl3, LiAlH4 или L-селектрид.
(b) Затем спирт Т2 защищают с образованием силилового эфира согласно хорошо известным методикам, описанным в Protective groups in organic synthesis, Т.W. Creene., что дает соединение Т25.
(с) Гидроборирование соединения Т25 при использовании ВН3.Me2S или ВН3.ТГФ дает соединение Т26.
(d) Соединение Т26 затем окисляют до соответствующего кетона Т27 согласно обычным методикам при участии ПХХ, периодинана Десса - Мартина и т.п.
(е) Когда Т27 содержит R7, который представляет собой силилированную защитную группу, то эту силилированную защитную группу соединения Т27 затем удаляют при кислотных условиях, используя обычные методики, описанные в Protective groups in organic synthesis, Т.W. Creene, что дает спирт Т28.
(f) Этот спирт Т28 защищают до эфира согласно хорошо известным методикам, описанным в Protective groups in organic synthesis, Т.W. Creene, что дает соединение Т29.
(g) Соединение Т27 (когда R7=МОМ) или Т29 фторируют, используя фторсодержащий реагент, такой как DAST, что дает фторированное соединение Т30.
(h) Эфирную или сложноэфирную защитную группу (OR7) соединения Т30 удаляют при обычных условиях, описанных в Protective groups in organic synthesis, Т.W. Creene, что дает спирт Т31.
(i) Затем этот спирт Т31 окисляют, используя периодинан Десса - Мартина, что дает соединение Т32.
(j) Реактив Гриньяра или литированное соединение Т14, полученное из соответствующего галогенированного соединения согласно обычной методике, добавляют к соединению Т32, что дает Т33.
(k) Соединение Т33 бромируют согласно обычным методикам, включая применение SOBr2 с последующим добавлением пиридина, что дает соединение Т34.
(l) Соединение Т34 затем восстанавливают в присутствии гидрида, такого как Bu3SnH.
(m) На последней стадии защитные группы можно удалить согласно обычным методикам, описанным в Protective groups in organic synthesis, Т.W. Creene.
Следует отметить, что стадии (k) и (l) проводят только для получения соединения формулы (I) с R4=Н.
И в частности:

(а) На первой стадии циклогексенон Т8 подвергают селективному восстановлению при участии NaBH4/CeCl3 в ТГФ и МеОН.
(b) Затем спирт Т36 защищают, используя имидазол и TBDMSCl, что дает соединение Т37 с R7=TBDMS; или диметоксиметан и P2O5, что дает соединение Т37 с R7=СН2ОСН3.
(с) Гидроборирование соединения Т37 при использовании ВН3.Me2S дает соединение Т38.
(d) Соединение Т38 затем окисляют до соответствующего кетона Т39 согласно обычным методикам при участии ПХХ, периодинана Десса - Мартина и т.п.
(е) Когда R7=TBDMS, то эту силилированную защитную группу соединения Т39 затем удаляют при кислотных условиях, таких как 12 н. HCl в метаноле и дихлорметане, что дает спирт Т40.
(f) Этот спирт Т40 защищают до ацетата, используя Ac2O, пиридин и каталитическое количество ДМАП (диметиламинопиридина), что дает соединение Т41.
(g) Соединение Т41 или соединение Т39 с R7=СН2ОСН3 фторируют, используя DAST в дихлорметане, что дает фторсодержащее соединение Т42.
(h) Когда R7=Ас, то ацетатную защитную группу соединения Т42 удаляют, используя метилат натрия в метаноле, что дает спирт Т43.
Когда R7=СН2ОСН3, то защитную группу МОМ Т42 удаляют, используя ТФК в дихлорметане, что дает спирт Т43.
(i) Затем этот спирт Т43 окисляют, используя периодинан Десса - Мартина, что дает соединение Т44.
(j) Реактив Гриньяра или литированное соединение Т14, полученное из соответствующего галогенированного соединения согласно обычной методике, добавляют к соединению Т44, что дает соединение Т45.
(k) Соединение Т45 бромируют с SOBr2 в дихлорметане с последующим добавлением пиридина, что дает соединение Т46.
(l) Соединение Т46 затем восстанавливают в присутствии Bu3SnH в толуоле, что дает соединение Т47.
(m) На последней стадии защитные группы можно удалить согласно обычным методикам, описанным в Protective groups in organic synthesis, Т.W. Creene. Следует отметить, что стадии (k) и (l) проводят только для получения соединения формулы (I) с R4=Н.
Также настоящее изобретение относится к способу получения соединения формулы (I) по изобретению, где R4=Н и n=1, согласно которому осуществляют реакцию связывания между соединением следующей формулы (XVI):
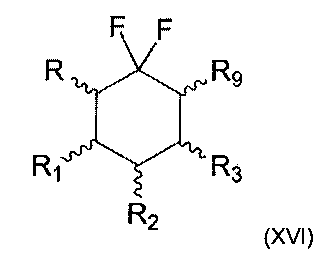
где R, R1, R2 и R3 являются такими, как определено выше, и R9 представляет собой уходящую группу, и соединением формулы (V), как определено выше.
Термин «уходящая группа», как используется в настоящем изобретении, относится к химической группе, которую можно легко заменить нуклеофилом в ходе реакции нуклеофильного замещения, нуклеофил в данном случае представляет собой спирт, т.е. молекулу, несущую группу ОН. Подобной уходящей группой может быть в частности атом галогена или сульфонат. Сульфонат в частности представляет собой группу -OSO2-R10, где R10 представляет собой группу (С1-С6)-алкил, арил, арил-(С1-С6)-алкил или (С1-С6)-алкил-арил. Сульфонат может находиться в мезилате (СН3-S(O2)O-), трифлате (CF3-S(O)2O-) или тозилате (п-Ме-С6Н4-S(O)2O).
Эту реакцию можно проводить в условиях, хорошо известных квалифицированному специалисту в данной области техники, особенно в присутствии основания, такого как NaH, K2CO3, или MeONa.
При необходимости можно проводить дополнительные стадии защиты, удаления защитных групп, замещения и т.п., эти стадии хорошо известны квалифицированному специалисту в данной области техники.
Полученное соединение формулы (I) можно выделить в ходе отделения от реакционной среды способами, хорошо известными квалифицированному специалисту в данной области техники, такими как экстракция, выпаривание растворителя или осаждение, или кристаллизация (с последующей фильтрацией).
Соединение также можно при необходимости очистить способами, хорошо известными квалифицированному специалисту в данной области техники, такими как перекристаллизация, перегонка, хроматография на колонке с силикагелем или высокоэффективная жидкостная хроматография (ВЭЖХ).
Соединение формулы (XVI) можно получить из соединения формулы (XV), где R7=Н, согласно методикам, хорошо известным квалифицированному специалисту в данной области техники. Например, когда уходящая группа представляет собой атом галогена, то реакцию можно проводить в присутствии галогенирующего реагента. Когда уходящая группа представляет собой сульфонат, то реакцию можно проводить в присутствии соответствующей сульфоновой кислоты и основания, такого как пиридин.
Способ получения соединений по изобретению с n=1 и R4=Н будет более подробно описан далее и в следующем экспериментальном разделе.

(а) На первой стадии спиртовую группу Т31 превращают в уходящую группу, такую как галоген или группа мезил, тозил или трифторметансульфонил, согласно методикам, хорошо известным квалифицированному специалисту в данной области техники.
(b) Затем Т48 замещают алкоголятом, полученным из Т3 при использовании основания, такого как NaH, K2CO3 или MeONa, что дает Т7.
(с) На последней стадии защитные группы можно удалить согласно обычным процедурам, описанным в Protective groups in organic synthesis, Т.W. Creene.
И в частности:

(а) На первой стадии спиртовую группу Т43 превращают в соответствующую трифторметансульфонильную группу в присутствии ангидрида трифторметансульфоновой кислоты и пиридина, что дает соединение Т49.
(b) Затем Т49 замещают алкоголятом, полученным из ТЗ при использовании NaH, что дает Т50. Реакцию проводят в диметилформамиде.
(с) На последней стадии защитные группы можно удалить согласно обычным процедурам, описанным в Protective groups in organic synthesis, Т.W. Creene.
Лучше понять изобретение можно после прочтения следующих примеров и фигур, эти примеры служат только для иллюстрации изобретения.
ФИГУРЫ
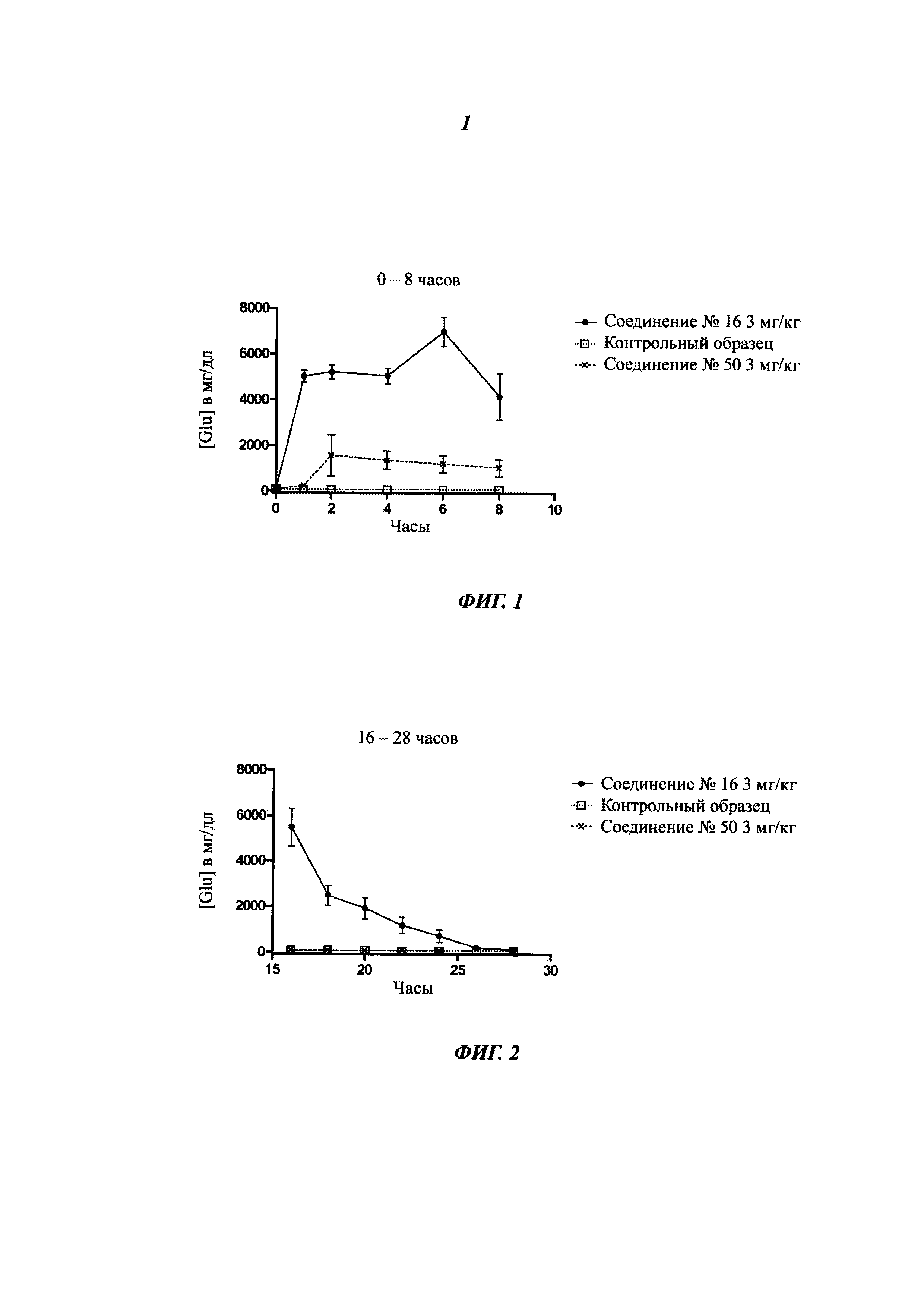
Фигура 1 представляет собой выведение глюкозы с мочой для соединения 16 и для соединения 50 между 0 и 8 часами после перорального введения (3 мг/кг по).
Фигура 2 представляет собой выведение глюкозы с мочой для соединения 16 и для соединения 50 между 16 и 28 часами после перорального введения (3 мг/кг по).
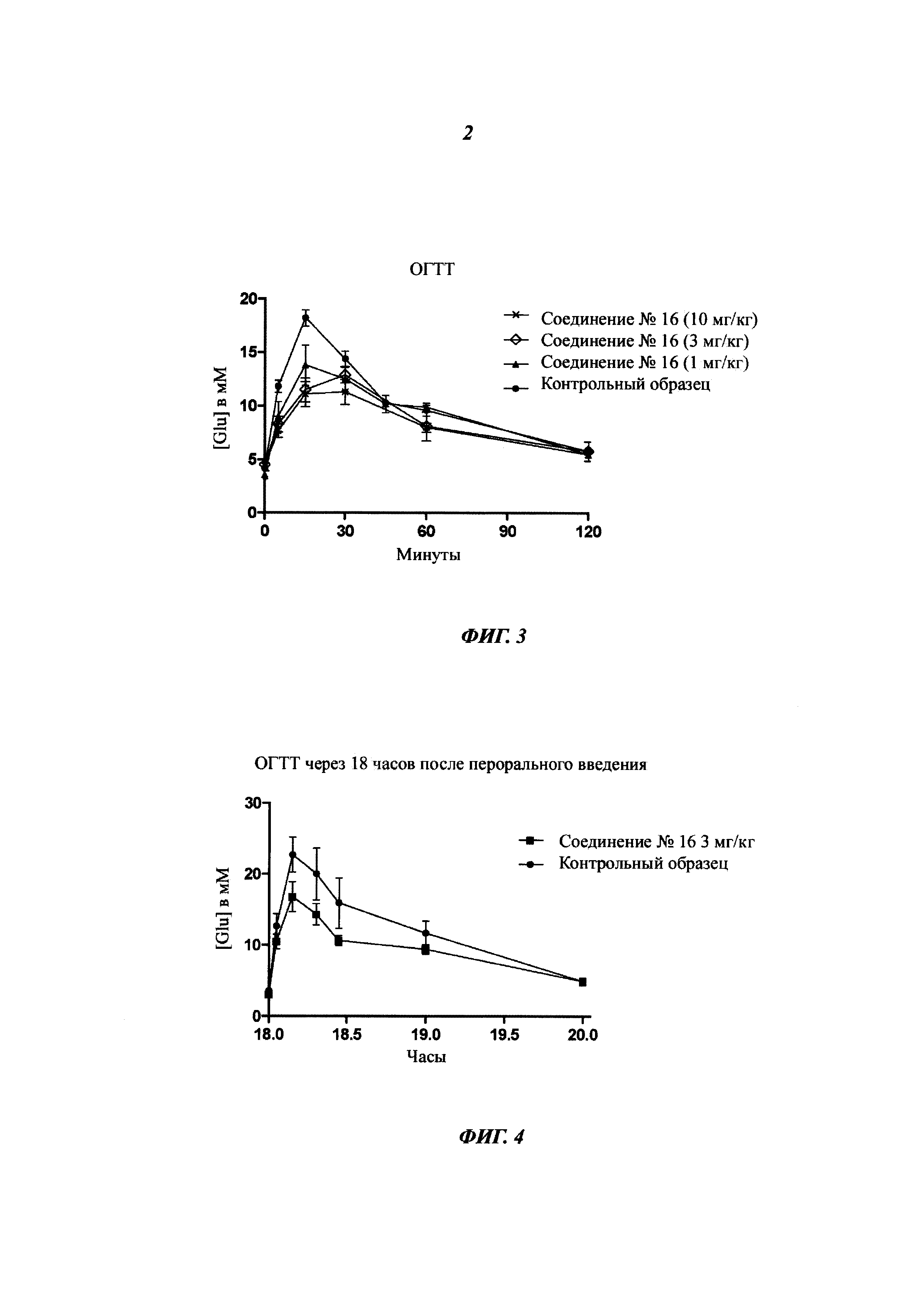
Фигура 3 представляет собой пероральный тест на толерантность к глюкозе для соединения 16 при 1, 3 и 10 мг/кг по.
Фигура 4 представляет собой пероральный тест на толерантность к глюкозе для соединения 16 через 18 часов после перорального введения соединения 16 (3 мг/кг по).
Фигура 5 представляет собой выведение глюкозы с мочой для соединения 16 и для соединения 50 между 16 и 28 часами после перорального введения (3 мг/кг по).
Фигура 6 представляет собой выведение глюкозы с мочой для соединения 16 и для соединения 9 из WO 2009/1076550 между 16 и 28 часами после перорального введения (3 мг/кг по).
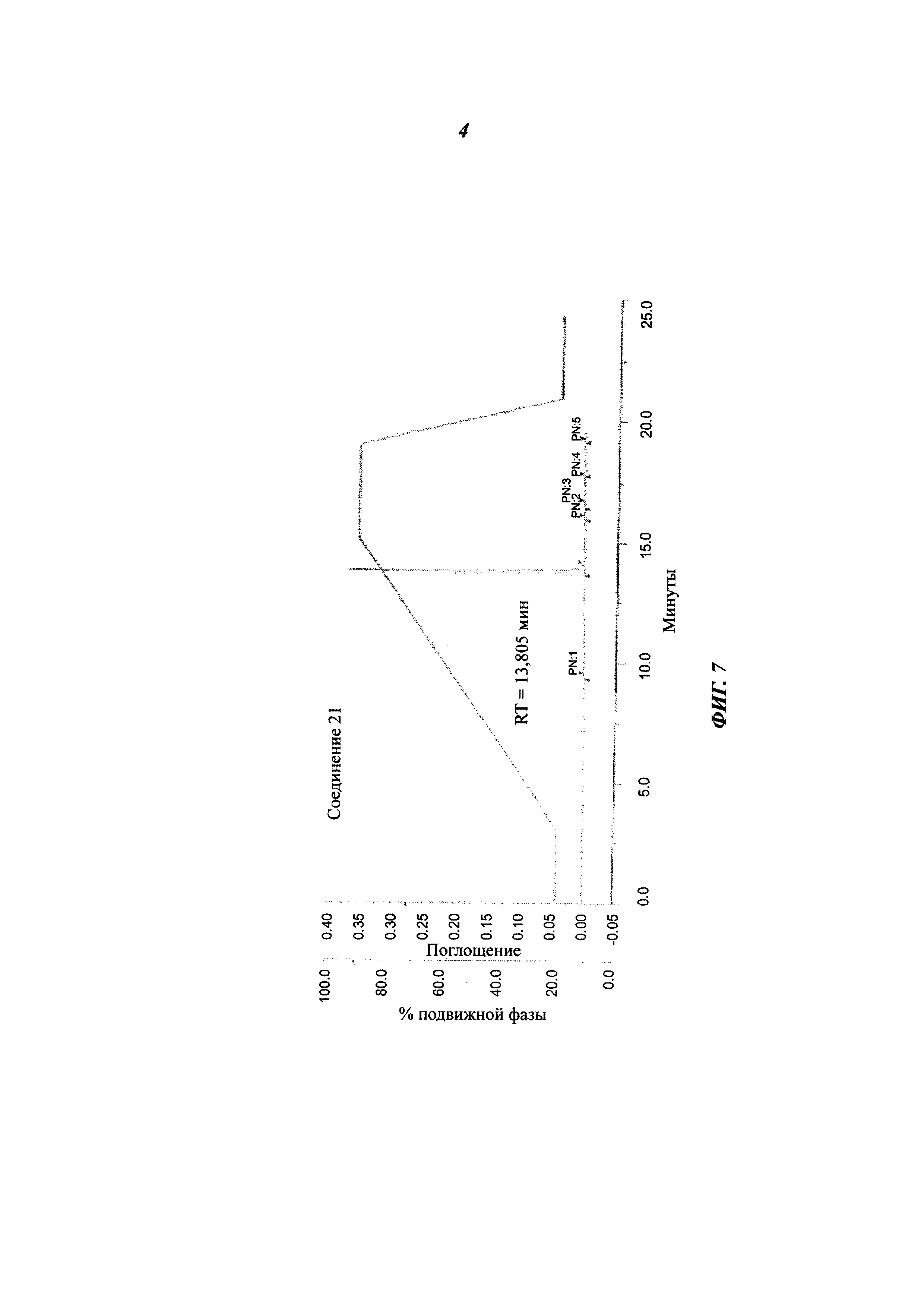
Фигура 7 представляет собой спектр ВЭЖХ соединения 21.
Фигура 8 представляет собой спектр ВЭЖХ соединения 26.
Фигура 9 представляет собой спектр ВЭЖХ соединения 26 после 4 часов инкубации при 37°С в присутствии β-глюкозидазы.
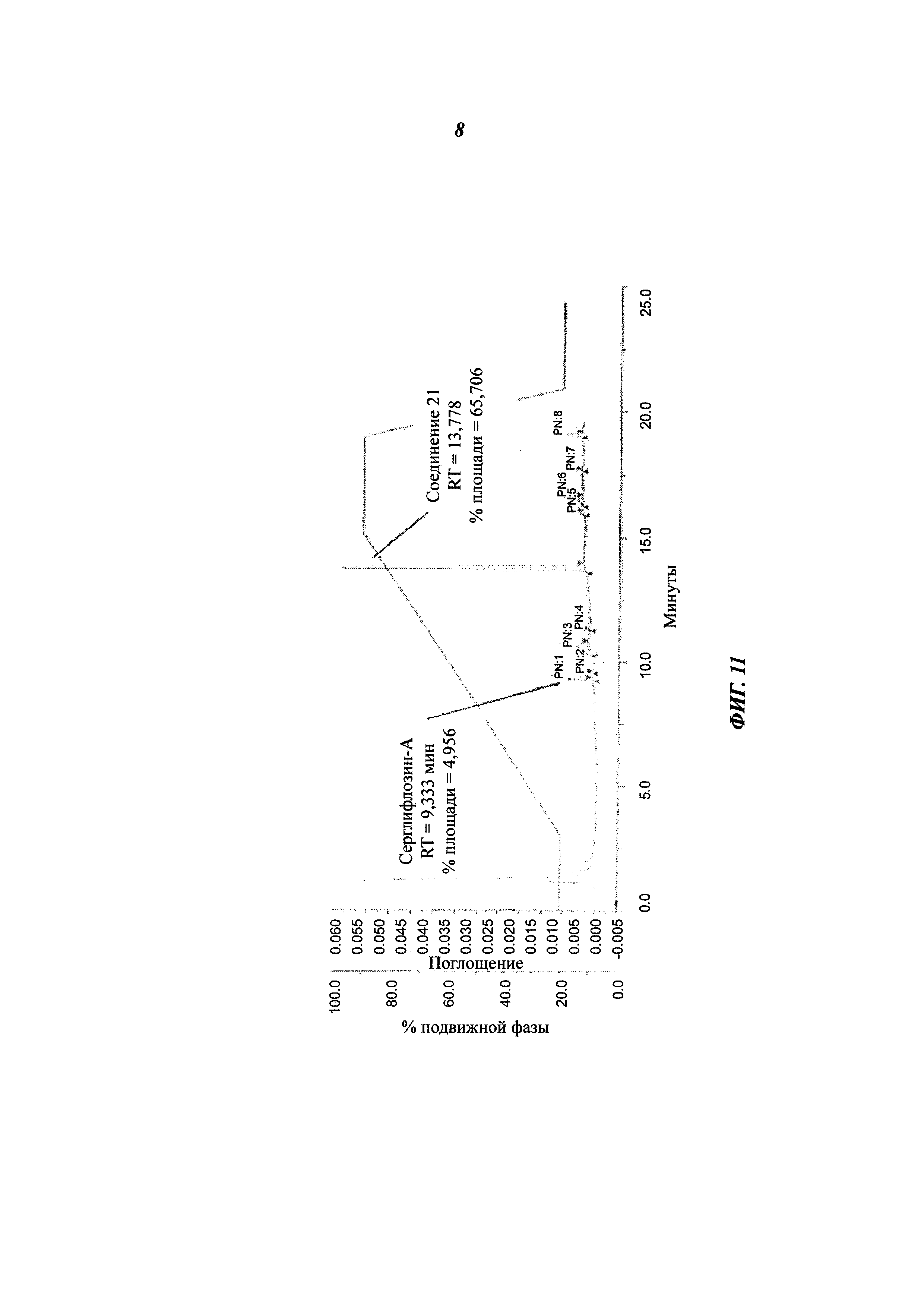
Фигура 10 представляет собой спектр ВЭЖХ серглифлозин-А.
Фигура 11 представляет собой спектр ВЭЖХ серглифлозин-А после 4 часов инкубации при 37°С в присутствии β-глюкозидазы.
ПРИМЕРЫ
1. Получение соединений по изобретению
Встречающиеся аббревиатуры определены ниже:
Ас - ацетил
АДДП - азодикарбонилдипиперидин
Bn - бензил
кат. - каталитический
DAST - трифторид диэтиламиносеры
ДХМ - дихлорметан
ди - диастереомерный избыток
ДМАП - 4-диметиламинопиридин
ДМФА - диметилформамид
ДМСО - диметилсульфоксид
экв. - эквивалент
ИЭР - ионизация электрораспылением
г - грамм
Гц - герц
мг - миллиграмм
МГц - мегагерц
мин - минута
мл - миллилитр
ммоль- миллимоль
мМ - миллимолярный
мкмоль - микромоль
нмоль - наномоль
ЯМР - ядерно-магнитный резонанс
по -перорально
ПЭГ - полиэтиленгликоль
QS - достаточное количество
Rf - скорость потока
к.т.- комнатная температура
ТФУА - трифторуксусный ангидрид
ТГФ - тетрагидрофуран
ТСХ - тонкослойная хроматография
TMS - триметилсилил
TBDMS - трет-бутилдиметилсилил
Характеристики устройств, используемых для проведения анализов всех соединений, описанных в этой заявке, указаны здесь ниже:
Спектры19F ЯМР регистрируют на спектрометре BRUKER DPX 300. Используемый внутренний стандарт представляет собой фтортрихлорметан CFCl3. Химические сдвиги (δ) выражены в миллионных долях (ppm от parts per million), и константы взаимодействия (J) - в герцах (Гц).
Были использованы следующие аббревиатуры:
s для синглета, bs для уширенного синглета, d для дублета, t для триплета, gdt для квартета, m для мультиплета или массива, dd для двойного дублета и т.п.
Масс-спектры получали на спектрофотометре Waters LCT Premier ХЕ, соединенном с ЖХ Waters Acquity.
Спектры ГХ-МС получали на Micromass Autospec 8 кВ, оборудованном ГХ НР 6890, капиллярной колонкой WCOT, НР 5MS, 30 м, Dl: 0,25 мм, при 50°С (0,5 мин), от 50 до 280°С при 10°С/мин, и 280°С для 5 мин, с ЭИ: 70 эВ.
Автоматизированную колоночную хроматографию осуществляли на оборудовании Biotage SP4, используя картриджи Biotage® SNAP. Проверку обеспечивали с помощью тонкослойной хроматографии (ТСХ) с пластинами Kieselgel 60F-254-0,25-мм. Отношение расстояния продвижения соединения на данной пластине к расстоянию продвижения элюента называется коэффициентом удерживания (Rf от retardation factor).
Получение примерных соединений по изобретению будет описано далее в целях иллюстрации, а не ограничения.
Синтез соединения 1
С34Н3406 М=538,63 г/моль
Масса: (ИЭР+): 561,2 (М+Na)

Уксусный ангидрид (420 мл) добавляли в инертной атмосфере в круглодонную колбу, содержащую 2,3,4,6-тетра-О-бензил-D-глюкопиранозу (100 г, 185 ммоль) в ДМСО (640 мл). Смесь перемешивали в течение ночи при комнатной температуре перед охлаждением до 0°С. Большой объем воды добавляли и перемешивание останавливали, чтобы реакционная смесь отстоялась в течение 3 часов (неочищенный лактон находился на дне колбы). Надосадочную жидкость удаляли, и неочищенную смесь разбавляли Et2O и промывали 3 раза водой, нейтрализовали насыщенным водным раствором NaHCO3 и промывали снова дважды водой. Органический слой потом сушили над сульфатом магния, фильтровали и концентрировали. Неочищенную смесь очищали с помощью хроматографии на силикагеле (циклогексан / этилацетат 8:2; И=0,61), что давало требуемый лактон 1 в виде бесцветного масла с выходом 80%.
Синтез соединения 2
С37Н43О9Р М=662,71 г/моль
Масса: (ИЭР+): 685,33 [М+Na]+; 1346,80 [2М+Na]+

В инертной атмосфере н-бутиллитий (1,6 М раствор в гексанах, 168 мл, 0,27 моль, 2,9 экв.) добавляли по каплям к раствору диметил метил-фосфоната (42 мл, 0,39 моль, 4,2 экв.) в ТГФ (390 мл), охлажденному до -78°С. Смесь перемешивали в течение 30 минут при этой температуре перед тем, как по каплям добавляли раствор лактона 1 (50 г, 93 ммоль, 1 экв.) в тетрагидрофуране (230 мл) при этой температуре. Смесь перемешивали в течение 30 минут перед нагреванием до 0°С, перемешивая.
Реакционную смесь выливали в ледяную смесь 10% насыщенного водного раствора хлорида аммония (100 мл) и этилацетата (300 мл). Органический слой отделяли, промывали водой, сушили над сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении, что давало в количественном отношении 3,4,5,7-тетра-O-бензил-1-деокси-1-(диметоксифосфорил)-D-глюко-2-гептилопиранозу 2 (63 г) в виде бледно-желтого масла, которое давало со временем белые кристаллы.
Синтез соединений 3а/b
С37Н45О9Р М=664,72 г/моль
Масса: (ИЭР+): 665,13 (М+Н); 687,27 (М+Na); 696,73 (М+МеОН)
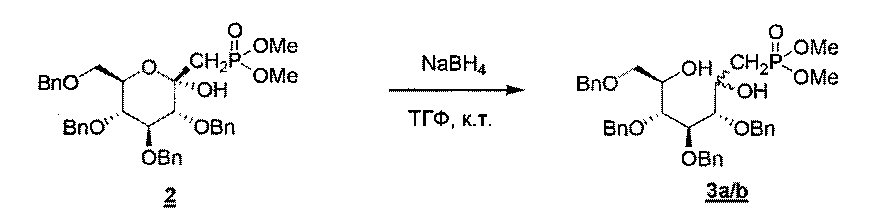
К раствору 2 (69,5 г, 105 ммоль, 1 экв.) в тетрагидрофуране (600 мл) добавляли частями боргидрид натрия (7,44 г, 210 ммоль, 2 экв.). Смесь перемешивали в течение ночи при комнатной температуре перед тем, как концентрировали при пониженном давлении. Остаток разделяли между этилацетатом и водой, и органический слой промывали водой, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенное соединение 3 (смесь диастереомеров а и b, 70,5 г, 100%) использовали на следующей стадии без дополнительной очистки.
Синтез соединения 4
С37Н41О9Р М=660,69 г/моль
Масса: (ИЭР+): 661,00 (М+Н); 683,20 (М+Na); 1343,0 (2М+Na)+

Раствор трифторуксусного ангидрида (27,1 мл, 0,19 моль, 4 экв.) в дихлорметане (130 мл), охлажденный до 0°С, добавляли по каплям в инертной атмосфере к раствору диметилсульфоксида (20,8 мл, 0,29 моль, 6 экв.) в дихлорметане (260 мл), полученному при температуре окружающей среды, перед охлаждением до -75°С. Смесь перемешивали в течение 45 минут при -75°С перед добавлением раствора 3 (32,43 г, 48,8 ммоль, 1 экв.) в дихлорметане (260 мл), охлажденного до -75°С. Смесь перемешивали в течение 1,5 часов при этой температуре. Триэтиламин (54,2 мл, 0,39 ммоль, 8 экв.) добавляли по каплям к реакционной смеси, которую затем нагревали до 0°С при перемешивании. Добавляли 2 н. водный раствор соляной кислоты к реакционной смеси. Органический слой отделяли, промывали насыщенным раствором гидрокарбоната натрия, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенное соединение 4 (36,3 г, 100%), полученное в виде желтоватого масла, использовали на следующей стадии без дополнительной очистки.
Синтез соединений 5а/b
С35Н40О6 М=556,69 г/моль
Масса: (ИЭР+): 557,20 (М+Н); 1135,07 (2М+Na)

2,3,4,6-тетра-О-бензил-D-глюкопиранозу (50 г, 92,7 ммоль, 1 экв.) растворяли в ТГФ (645 мл) и охлаждали до 0°С. Метилмагнийбромид (185 мл 1,4 М раствора в ТГФ/толуоле, 259,4 ммоль, 2,8 экв.) добавляли по каплям в инертной атмосфере и реакционную смесь перемешивали в течение 10 минут при 0°С и 3 часов 30 минут при 50°С. ТСХ (циклогексан-этилацетат, 7:3) показала полное превращение исходного вещества в два продукта (Rfa=0,17 и Rfb=0,25). Реакционную смесь выливали в насыщенный водный раствор хлорида аммония и экстрагировали этилацетатом. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали, что давало в количественном отношении требуемый неочищенный диол 5 (в виде смеси диастереомеров а и b) в виде желтого масла. Это соединение использовали на следующей стадии без дополнительной очистки.
Синтез соединения 6
С36Н36О6 М=552,66 г/моль
Масса: (ИЭР+): 575,40 (М+Na); 575,40 (М+К); 1127,07 (2М+Na); 1142,93 (2М+К)+.
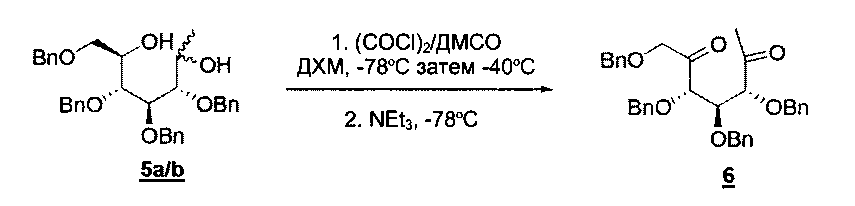
Раствор диметилсульфоксида (14 мл, 0,20 моль, 9 экв.) в дихлорметане (50 мл) добавляли по каплям к раствору оксалилхлорида (12,5 мл, 0,13 моль, 6 экв.) в дихлорметане (50 мл), охлажденному до -78°С, в инертной атмосфере. Смесь перемешивали при -78°С в течение 30 минут перед тем, как добавляли по каплям раствор диола 5 (12,2 г, 21,9 ммоль, 1 экв.) в дихлорметане (50 мл). Через 45 минут появлялся осадок и реакционную смесь нагревали до -40°С и перемешивали в течение дополнительных 30 минут. Затем смесь повторно охлаждали до -78°С и триэтиламин (55 мл, 0,39 моль, 18 экв.) добавляли по каплям. Через 15 минут охлаждающую ванну удаляли, и реакционная смесь достигала комнатной температуры. Образовывалось большое количество осадка. Через дополнительных 2 часа добавляли толуол (400 мл) и осадок удаляли в ходе фильтрации. Остаток промывали толуолом и фильтрат концентрировали и очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 97:3 до 70:30), что давало дикетон 6 (9,92 г, выход 76%) в виде оранжевого масла.
Синтез соединения 7
С35Н36О6 М=552,66 г/моль
Масса: (ИЭР+): 570,27 (М+H2O); 575,33 (М+Na)

L-пролин (7,35 г, 63,8 ммоль, 1 экв.) добавляли к раствору дикетона 6 (35,2 г, 63,7 ммоль, 1 экв.) в ДМСО (561 мл). Смесь перемешивали при 50°С на воздухе в течение 8 часов перед тем, как выливали в смесь воды и солевого раствора (2:1), экстрагировали этилацетатом, сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенную смесь очищали с помощью хроматографии на силикагеле (циклогексан / этилацетат 97:3 до 35:35), что давало соединение 7 (13,0 г, 37%) в виде оранжевого масла.
Синтез соединения 8
С35Н34О5 М=534,64 г/моль
Масса: (ИЭР+): 535,00 (М+Н); 552,00 (М+H2O); 785,87; 1086,67 (2М+H2O)

Методика А:
К раствору 4 (10,5 г, 15,89 ммоль, 1 экв.) в толуоле (400 мл) добавляли 18-краун-6 (168 мг, 0,64 ммоль, 0,04 экв.) и карбонат калия (6,69 г, 48,5 ммоль, 3,05 экв.). Смесь перемешивали в течение ночи при комнатной температуре, и затем оставшееся нерастворимое вещество отфильтровывали и промывали толуолом. Фильтрат и промывные воды объединяли, промывали 2 н. водным раствором соляной кислоты, затем насыщенным водным раствором гидрокарбоната натрия, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 98:2 до 80:20), что давало циклогексенон 8 (4,07 г; выход 48%) в виде желтоватого масла.
Методика В:
Раствор 7 (3,27 г, 5,92 ммоль, 1 экв.) в пиридине (14 мл) охлаждали до 0°С, затем POCl3 (2,75 мл, 29,6 ммоль, 5 экв.) добавляли по каплям. Смесь перемешивали при этой температуре в течение 10 минут перед тем, как удаляли охлаждающую ванну. Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем повторно охлаждали до 0°С. POCl3 (2,75 мл, 29,6 ммоль, 5 экв.) добавляли снова, пытаясь завершить реакцию. Смесь перемешивали в течение дополнительных 20 часов при комнатной температуре перед разбавлением Et2O (20 мл) и выливали в дробленый лед. Добавляли 1 М водный раствор HCl (100 мл) и смесь экстрагировали Et2O (200 мл и 100 мл). Объединенные органические экстракты промывали солевым раствором (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали, затем очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 98:2 до 80:20), что давало соединение 8 (1,46 г, выход 46%) в виде оранжевого масла.
Синтез соединения 9
C15H12BrClO2М=339,61 г/моль
Масса: (ГХ-МС): 338-340
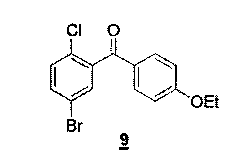
Синтез этого продукта описан в J. Meg. Chem. 2008, 51, 1145-1149.
Синтез соединения 10
C15H14BrClO М=325,63 г/моль
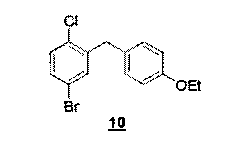
Синтез этого продукта описан в J. Med. Chem. 2008, 51, 1145-1149.
Синтез соединения 11
C50H49ClO6 М=781,37 г/моль
Масса: (ИЭР+): 798,20 (М+H2O)

В инертной атмосфере порошок Mg (265 мг, 10,9 ммоль, 2,4 экв.) помещали в трехгорлую колбу, затем добавляли 1/3 часть раствора 4-бром-1-хлор-2-(4-этилбензил)бензола (2,95 г, 9,1 ммоль; 2 экв.) в безводном ТГФ (25 мл) и 1,2-дибромэтан (10 мол. % Mg; 85 мг; 0,45 ммоль). Смесь нагревали с обратным холодильником. После начала реакции (экзотермической и расходующей Mg) остальной раствор 2-(4-этилбензил)-4-бром-1-хлорбензола в безводном ТГФ добавляли по каплям. Затем смесь взаимодействовала в течение еще одного часа при осторожном нагревании с обратным холодильником до израсходования большей части Mg.
Вышеприведенный реактив Гриньяра добавляли по каплям к раствору циклогексенона 8 (2,42 г, 4,53 ммоль, 1 экв.) в безводном ТГФ (25 мл) в инертной атмосфере при комнатной температуре (приблизительно 25°С), потом проводили реакцию в течение 3 часов. Добавляли насыщенный водный раствор хлорида аммония к смеси, чтобы погасить реакцию. Смесь экстрагировали Е120, промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 100:0 до 80:20), что давало заданное соединение 11 в виде желтого масла (3,01 г, 86%).
Синтез соединения 12
C50H49ClO5 М=765,37 г/моль
Масса: (ИЭР+): 782,13 (М+H2O)

Триэтилсилан (0,210 мл, 1,30 ммоль, 3 экв.) и эфират трехфтористого бора (48% BF3, 0,110 мл, 0,866 ммоль, 2 экв.) последовательно добавляли к раствору спирта Ц (338 мг, 0,433 ммоль, 1 экв.) в дихлорметане (5 мл) в инертной атмосфере при -20°С. После перемешивания в течение 2,5 часов добавляли насыщенный водный раствор хлорида натрия, чтобы погасить реакцию. Смесь экстрагировали CH2Cl2 (10 мл × 3) и органический слой промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 9,8:0,2 до 8:2), что давало заданное соединение 12 в виде белого порошка (278 мг, 0,363 ммоль, 84%).
Синтез соединения 13
C50H51ClO6 М=783,39 г/моль
Масса: (ИЭР+): 800 (М+H2O); 1581 (2М+H2O)

В инертной атмосфере комплекс боран-диметилсульфид (2 М в ТГФ, 16,7 мл, 33 ммоль, 10,5 экв.) добавляли к раствору 12 (2,41 г; 3,15 ммоль, 1 экв.) в безводном ТГФ (100 мл), охлажденному до 0°С. Реакционную смесь потом нагревали с обратным холодильником в течение 1 часа, охлаждали до 0°С и осторожно обрабатывали гидроксидом натрия (3 М в H2O, 10,5 мл, 31,5 ммоль, 10 экв.), затем пероксидом водорода (30% в H2O, 3,2 мл, 31,5 ммоль, 10 экв.) при комнатной температуре (выше 30°С). Смесь взаимодействовала в течение ночи при комнатной температуре (~25°С) перед добавлением насыщенного водного раствора хлорида аммония, чтобы погасить реакцию. Смесь экстрагировали этилацетатом, и органический слой промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 97:3 до 73:27), что давало требуемое соединение 13 (1,05 г; 43%) в виде желтоватого масла.
Синтез соединения 14
C50H49ClO6 М=781,37 г/моль
Масса: (ИЭР+): 798 (М+H2O); 1471; 1579 (2М+H2O)

Периодинан Десса - Мартина (81 мг; 1,91 ммоль; 1,5 экв.) добавляли частями к раствору спирта 13 (1,0 г; 1,28 ммоль, 1 экв.) в безводном дихлорметане (20 мл) при 0°С. Затем реакционную смесь перемешивали в течение ночи при комнатной температуре, потом гасили 1 н. водным раствором гидроксида натрия. Органический слой отделяли, и водный слой экстрагировали дихлорметаном. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 98:2 до 82:18), что давало заданный кетон 14 (783 мг, выход 79%) в виде бесцветного масла.
Синтез соединения 15
C50H49ClF2O6М=803,37 г/моль
19F ЯМР (CDCl3, 282m5 МГц): -100,3 (d, J=254 Гц, 1F, CFF); -113,3 (td, J1=254 Гц, J2=29 Гц, 1F, CFF).
Масса: (ИЭР+): 820,00 (М+H2O)
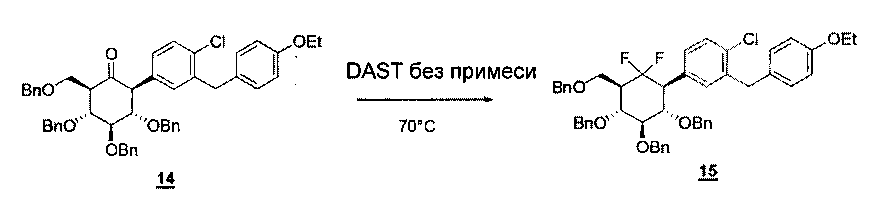
Раствор кетона 14 (421 мг, 0,539 ммоль, 1 экв.) в DAST (2 мл, 16,3 ммоль, 30 экв.) перемешивали в инертной атмосфере при 70°С в течение 12 часов. Потом смесь охлаждали до комнатной температуры и добавляли дихлорметан. Раствор выливали в смесь воды, льда и твердого NaHCO3. Продолжали перемешивать в течение 30 минут до достижения комнатной температуры. Водный слой экстрагировали дихлорметаном и органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 98:2 до 80:20), что давало требуемое соединение 15 в виде желтоватого масла (182 мг, выход 42%).
Синтез соединения 16
C22H25ClF2O5 М=442,88 г/моль
19F=ЯМР (MeOD, 282,5 МГц): -96,7 (d, J=254 Гц, 1F, CFF); -112,2 (td, J1=254 Гц, J2=28 Гц, 1F, CFF).
Масса: (ИЭР+): 465,3 (М+Na)
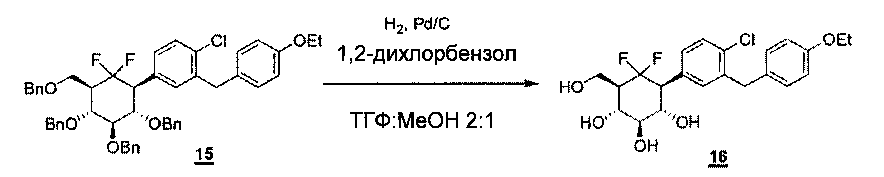
о-Дихлорбензол (0,320 мл, 2,82 моль, 10 экв.), затем 10% Pd/С (0,342 г, 0,32 моль, 1,1 экв.) добавляли к раствору 15 (228 мг, 0,28 ммоль, 1 экв.) в смеси ТГФ и МеОН (2:1, об./об., 160 мл). Реакционную смесь помещали в атмосферу водорода и перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь фильтровали и концентрировали перед очищением с помощью хроматографии на силикагеле (дихлорметан/метанол 100:1 до 90:10), что давало соединение 16 (105 мг, выход 83%).
Синтез соединения 17
С35Н36О5 М=536,66 г/моль
Масса: (ИЭР+): 554,13 (М+H2O); 1095 (2М+Na)
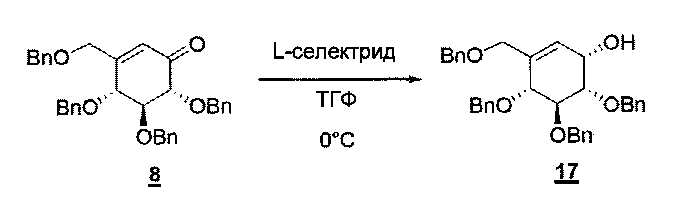
1 М раствор L-селектрида в ТГФ (0,84 мл, 0,84 ммоль, 1,5 экв.) добавляли по каплям к перемешиваемому и охлажденному (0°С) раствору циклогексенона 8 (0,300 г, 0,56 ммоль, 1 экв.) в ТГФ (14 мл) в инертной атмосфере. Смесь перемешивали в течение 18 часов, постепенно нагревая ее до комнатной температуры. Затем добавляли насыщенный водный раствор хлорида аммония и полученную в результате смесь перемешивали в течение дополнительных 15 минут. Добавляли воду, и водный раствор потом экстрагировали этилацетатом, и объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали, что давало в количественном отношении требуемое соединение 17 (350 мг) в виде желтого масла.
Синтез соединения 18
С14Н12О3 М=228,24 г/моль
Масса: (ГХ-МС): 228 (М)

Методика А.
2-Гидроксибензойную кислоту (13,8 г, 0,1 моль, 1 экв.) и анизол (10,9 мл, 0,1 моль, 1 экв.) добавляли к смеси графита (9,6 г, 0,8 моль, 8 экв.) и метансульфоновой кислоты (25 мл, 0,4 моль, 4 экв.), нагретой до 80°С. Реакционную смесь перемешивали при этой температуре в течение 12 часов перед охлаждением до комнатной температуры. Потом смесь экстрагировали дважды хлороформом, и объединенные органические слои промывали насыщенным водным раствором NaHCO3, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 70:30), что давало соединение 18 (4 г, выход 17%) в виде оранжевого масла.
Методика В.
BBr3.ДМСО (10,8 г, 34,42 ммоль, 1,1 экв.) добавляли частями к раствору 20 (7,58 г, 31,29 ммоль, 1 экв.) в дихлорметане (150 мл), охлажденному до 0°С. Реакционную смесь перемешивали при 0°С в течение 3 часов, потом выливали в смесь воды и льда. После 10 минут перемешивания слои разделяли и водный слой экстрагировали этилацетатом. Объединенные органические слои промывали водой и солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали, что давало соединение 18 (6,78 г) в виде пурпурного масла.
Синтез соединения 19
С16Н16О3 М=244,29 г/моль
Масса: (ИЭР+): 227,1 (М+Н-H2O)

Раствор 4-метоксифенилмагнийбромида (0,5 М в ТГФ, 300 мл, 0,150 моль, 1,1 экв.) добавляли по каплям в инертной атмосфере к раствору 2-метоксибензальдегида (18,75 г, 0,137 моль, 1 экв.) в ТГФ (188 мл), охлажденному до 0°С. Полученную в результате смесь перемешивали при комнатной температуре в течение ночи, затем выливали в насыщенный водный раствор NH4Cl. Водный слой экстрагировали этилацетатом, и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали, что давало соединение 19 (37,5 г) в виде коричневого масла.
Синтез соединения 20
C15H14O3 М=242,27 г/моль
Масса: (ГХ-МС): 51; 64; 77; 92; 107; 121; 128; 135; 139; 181; 197; 211; 225; 242

Пиридиния хлорхромат (34,3 г, 159 ммоль, 2 экв.) добавляли к раствору спирта 19 (19,4 г, 79,4 ммоль, 1 экв.) в дихлорметане (210 мл), содержащему молекулярные сита. Реакционную смесь перемешивали в течение ночи при комнатной температуре, фильтровали, чтобы удалить остатки ПХХ и молекулярных сит и концентрировали. Неочищенный остаток очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 100:0 до 85:15), что давало кетон 20 (12,6 г, выход 38%) в виде желтоватого твердого вещества.
Синтез соединения 21
C14H14O2 М=214,26 г/моль
Масса: (ГХ-МС): 214

Методика А.
10% Pd/С добавляли к раствору 18 (1,5 г, 6,6 ммоль, 1 экв.) в этаноле. Раствор перемешивали в атмосфере водорода при 30 бар до завершения реакции. Частицы палладия удаляли фильтрацией и раствор концентрировали, что давало соединение 21 (1,32 г, выход 93%) в виде белого порошка.
Методика В.
Раствор 18 (8,1 г, 35,5 ммоль, 1 экв.) в ацетонитриле (130 мл) в инертной атмосфере охлаждали до 0°С. ТМ5С1 (20,7 мл, 163,3 ммоль, 4,6 экв.), затем NaBH3CN (10,5 г, 1667 ммоль, 4,7 экв.) медленно добавляли (экзотермическая реакция). Полученную в результате желтую суспензию перемешивали при комнатной температуре в течение 2 часов, потом выливали в воду. Дихлорметан затем добавляли, и органический слой отделяли, промывали солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали. Неочищенный остаток очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 100:0 до 83:17), что давало заданное соединение 21 (выход 80%) в виде желтоватого твердого вещества.
Синтез соединения 22
C49H48O6 М=732,90 г/моль
Масса: (ИЭР+): 755,4 (М+Na); 771,4 (М+К)

К раствору 17 (50 мг, 0,093 ммоль, 1 экв.) в толуоле (0,30 мл), охлажденному до 0°С, в инертной атмосфере последовательно добавляли 21 (30 мг, 0,140 ммоль, 1,5 экв.), трибутилфосфин (0,35 мл, 0,140 ммоль, 1,5 экв.) и 1,1′-(азодикарбонил)дипиперидин (35 мг, 0,140 ммоль, 1,5 экв.). Реакционную смесь перемешивали при 0°С в течение 30 минут. Появлялся густой осадок, и смесь растворяли с дихлорметаном и концентрировали при пониженном давлении, что давало белый остаток, который очищали с помощью хроматографии на силикагеле (циклогексан / этилацетат 100:0 до 80:20), что давало заданное соединение 22 (63 мг, выход 93%) в виде бесцветного масла.
Синтез соединения 23
С49Н50О7 М=750,92 г/моль
Масса: (ИЭР+): 773,8 (М+Na); 789,7 (М+К)

К охлажденному раствору (0°С) 22 (62 мг, 0,085 ммоль, 1 экв.) в безводном ТГФ (0,837 мл) добавляли ВН3.Me2S (2 М раствор в ТГФ, 0,169 мл, 0,338 ммоль, 4 экв.). Полученный в результате раствор перемешивали в течение ночи при комнатной температуре, потом снова охлаждали до 0°С. Затем последовательно добавляли воду (0,107 мл, 23,6 ммоль, 70 экв.), пероксид водорода (30% водный раствор, 0,258 мл, 10,1 ммоль, 30 экв.) и гидроксид натрия (2 М водный раствор, 0,338 мл, 2,7 ммоль, 8 экв.) и смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли воду и этилацетат и органический слой промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Затем неочищенное соединение очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 100:0 до 75:25), что давало спирт 23 (34 мг, выход 53%) в виде белого твердого вещества.
Синтез соединения 24
С49Н48О7 М=748,90 г/моль
Масса: (ИЭР+): 771,7 (М+Na); 787,7 (М+К)

Периодинан Десса - Мартина (29 мг, 0,068 ммоль, 1,5 экв.) добавляли к раствору спирта 23 (34 мг, 0,045 ммоль, 1 экв.) в дихлорметане (0,680 мл), охлажденному до 0°С. Полученную в результате смесь перемешивали при комнатной температуре в течение 3 часов, затем раствор гидроксида натрия (1 н. водный раствор) и дихлорметана добавляли к смеси. Органический слой отделяли, сушили над сульфатом натрия, фильтровали и концентрировали, что давало требуемый кетон 24 (36 мг, выход 70%) в виде белого твердого вещества.
Синтез соединения 25
C49H48F2O6 М=770,90 г/моль
19F=ЯМР (CDCl3, 282,5 МГц): -109,3 (d, J=252 Гц, 1F, CFF); -120,3 (ddd, J1=252 Гц, J2=30 Гц, J3=19 Гц, 1F, CFF).
Масса: (ИЭР+): 773,4 (М-HF); 793,5 (М+Na)

DAST (0,72 мл, 4,96 ммоль, 20 экв.) добавляли к раствору кетона 24 (183 мг, 0,244 ммоль, 1 экв.) в дихлорметане (0,720 мл) в инертной атмосфере и реакционную смесь перемешивали в течение ночи при комнатной температуре. Раствор охлаждали до комнатной температуры, затем выливали в воду. Дихлорметан добавляли, и органический слой промывали насыщенным водным раствором NaHCO3, сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 100:0 до 90:10), затем с помощью препаративной ВЭЖХ (Kromasil С18, ацетонитрил/вода 89:11), что давало соединение 25 с выходом 32% в виде белого твердого вещества.
Синтез соединения 26
C21H24F2O6 М=410,41 г/моль
19F ЯМР (MeOD, 282,5 МГц): -109,6 (d, J=251 Гц, 1F, CFF); -122,4 (666, J1=251 Гц, J2=28 Гц, J3=20 Гц, 1F, CFF).
Масса: (ИЭР-): 445,2 (М+Cl)
Соединение 25 (48 мг, 0,06 ммоль, 1 экв.) растворяли в смеси ТГФ (6,3 мл) и метанола (6,3 мл). Добавляли 10% Pd/С (48 мг, 0,04 ммоль, 0,7 экв.), затем 2 капли 12 н. водного раствора соляной кислоты. Потом смесь перемешивали в течение 1 часа в атмосфере водорода при комнатной температуре, затем фильтровали и концентрировали. Неочищенную смесь очищали с помощью хроматографии на силикагеле (дихлорметан/метанол 100:0 до 90:10), что давало заданное соединение 26 (42 мг, выход 67%) в виде белого твердого вещества.
Синтез соединения 27
С48Н46О6 М=718,88 г/моль
Масса: (ИЭР+): 741,8 (М+Na), 757,7 (М+К)

Раствор 17 (30 мг, 0,056 ммоль, 1 экв.) в толуоле (0,180 мл) охлаждали до 0°С в инертной атмосфере и 4-(бензилокси)фенол (17 мг, 0,085 ммоль, 1,5 экв.), трибутилфосфин (0,42 мл, 0,168 ммоль, 3 экв.) и 1,1′-(азодикарбонил)дипиперидин (42 мг, 0,167 ммоль, 3 экв.) последовательно добавляли. Реакционную смесь перемешивали при 0°С в течение 30 минут. Реакционную смесь разбавляли дихлорметаном и концентрировали при пониженном давлении, что давало белый остаток, который очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 100:0 до 80:20), что давало соединение 27 (30 мг, выход 75%) в виде бесцветного масла.
Синтез соединения 28
С48Н48О7 М=736,88 г/моль
Масса: (ИЭР+): 759,8 (М+Na), 775,7 (М+К)

ВН3.Me2S (3,48 мл, 6,96 ммоль, 2 экв.) добавляли в инертной атмосфере к раствору 27 (1,00 г, 1,39 ммоль, 1 экв.) в безводном тетрагидрофуране (15 мл), охлажденному до 0°С. Эту смесь перемешивали в течение ночи при комнатной температуре. Потом добавляли воду (1,75 мл, 97,4 ммоль, 70 экв.) при 0°С, затем 30% водный раствор H2O2 (4,73 мл, 41,7 ммоль, 30 экв.) и 1 М водный раствор гидроксида натрия (11,1 мл, 11,1 ммоль, 8 экв.). Полученную в результате смесь перемешивали при комнатной температуре в течение 3 часов. Затем добавляли большое количество воды, потом экстрагировали EtOAc. Органический слой промывали солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Неочищенную смесь очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 95:5 до 60:40), что давало 28 (791 мг, выход 78%) в виде желтоватого масла.
Синтез соединения 29
С48Н46О7 М=734,87 г/моль
Масса: (ИЭР+): 757,8 (М+Na), 773,7 (М+К)

Периодинан Десса - Мартина (17 мг, 0,041 ммоль, 1,5 экв.) добавляли к раствору спирта 28 (682 мг, 1,61 ммоль, 1 экв.) в безводном дихлорметане (20 мл) при комнатной температуре. Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем разбавляли дихлорметаном и гасили 1 М водным раствором гидроксида натрия. После экстракции дихлорметаном органический слой сушили над MgSO4, фильтровали и концентрировали, что давало неочищенный кетон 29 (730 мг, выход 91%) в виде желтоватого масла.
Синтез соединения 30
С48Н46О7 М=756,87 г/моль
19F ЯМР (, 282,5 МГц): -120,6 (ddd, 1F, J1=251 Гц, J2=28 Гц, J3=20 Гц, CFF); -108,7 (d, 1F, J=251 Гц, CFF)
Масса: (ИЭР+): 779,3 [М+Na]+; 795,3 [М+К]+

Раствор кетона 29 (15 мг, 2,04 мкмоль, 1 экв.) в DAST (0,130 мл, 0,276 ммоль, 130 экв.) перемешивали в течение ночи в инертной атмосфере при 70°С. Затем неочищенную смесь разбавляли дихлорметаном и осторожно гасили H2O. Органический слой промывали насыщенным водным раствором NaHCO3, сушили над MgSO4, фильтровали и концентрировали. Неочищенную смесь очищали с помощью препаративной ТСХ (циклогексан/этилацетат 85:15), что давало соединение 30 в виде желтоватого масла.
Синтез соединения 31
C13H16F2O6М=306,26 г/моль
19F ЯМР (MeOD, 282,5 МГц): -109,2 (d, J=253 Гц, 1F, CFF); -123,0 (ddd, J1=253 Гц, J2=29 Гц, J3=20 Гц, 1F, CFF).
Масса: (ИЭР-): 341,0 [М+Cl]-
Соединение 30 (191 мг, 0,252 ммоль, 1 экв.) растворяли в ТГФ - этаноле (4:1, об./об., 120 мл) в инертной атмосфере. Добавляли 10% Pd/С (191 мг, 0,17 ммоль, 0,7 экв.) и 9 капель 12 н. водного раствора соляной кислота к смеси, которую дегазировали 5 раз с Н2. Полученную в результате черную суспензию перемешивали в атмосфере Н2 при комнатной температуре в течение 45 минут. Реакционную смесь фильтровали и фильтрат концентрировали. Неочищенный продукт очищали с помощью хроматографии на силикагеле (дихлорметан/метанол 100:0 до 90:10), что давало заданное соединение 31 с выходом 73% в виде бесцветного масла.
Синтез соединения 32
C14H12O2 М=212,24 г/моль
Масса: (Cl+): 213 (М+Н)

4-Гидроксибензальдегид (4 г, 32,8 ммоль, 1 экв.) и карбонат калия (4,75 г, 34,4 ммоль, 1,05 экв.) растворяли в безводном ДМФА (30 мл). Бензилбромид (4,1 мл, 34,4 ммоль, 1,05 экв.) медленно добавляли. Полученную в результате смесь перемешивали в течение ночи в инертной атмосфере при комнатной температуре. Ледяную воду добавляли к реакционной смеси, чтобы погасить реакцию, и затем ее разбавляли большим количеством воды. Смесь фильтровали, и остаток промывали водой и растворяли в этилацетате. Органический слой промывали солевым раствором, сушили над MgSO4, фильтровали и концентрировали, что давало в количественном отношении неочищенный альдегид 32 в виде желтоватого масла, которое медленно кристаллизовалось со временем.
Синтез соединения 33
C14H12O2 М=214,26 г/моль
Масса: (ГХ-МС): 91; 197; 214 (М).

Раствор альдегида 32 (6,5 г, 30,6 ммоль, 1 экв.) в безводном тетрагидрофуране (25 мл) добавляли по каплям к суспензии NaBH4 (1,51 г, 39,8 ммоль, 1,3 экв.) в безводном тетрагидрофуране (25 мл). Полученную в результате смесь перемешивали в течение 72 часов в инертной атмосфере при комнатной температуре, затем гасили ледяной водой, разбавляли диэтиловым эфиром, подкисляли водным раствором 4 н. HCl и экстрагировали диэтиловым эфиром. Органический слой промывали насыщенным водным раствором NaHCO3, сушили над MgSO4, фильтровали и концентрировали, что давало неочищенный спирт 33 (выход 97%) в виде белого аморфного твердого вещества.
Синтез соединения 34
C14H13BrO М=277,16 г/моль
Масса: (Cl+): 107; 197, 277 (М+Н)
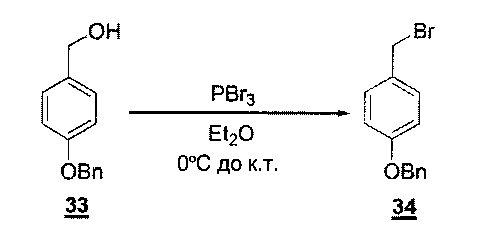
К ледяной суспензии неочищенного спирта 33 (6 г, 28,0 ммоль, 2,4 экв.) в диэтиловом эфире (50 мл) добавляли PBr3 (1,1 мл, 11,67 ммоль, 1 экв.) с такой скоростью, чтобы температура не превышала 8°С. Полученную в результате смесь перемешивали 2 часа в инертной атмосфере при комнатной температуре. Реакционную смесь затем охлаждали в ледяной ванне, гасили ледяной водой и разбавляли диэтиловым эфиром и этилацетатом. Органический слой промывали водным насыщенным раствором NaHCO3, сушили над MgSO4, фильтровали и концентрировали, что давало неочищенное соединение 34 (выход 99%) в виде белого аморфного твердого вещества.
Синтез соединения 35
С20Н22О4 М=326,39 г/моль
Масса: (ИЭР+): 349,1 (М+Na); 365,1 (М+К)

К 95% суспензии NaH (0,61 г, 25,26 ммоль, 1 экв.) в безводном ТГФ (30 мл) в инертной атмосфере добавляли раствор этилацетоацетата (3,5 мл, 27,79 ммоль, 1,1 экв.) в безводном ТГФ (10 мл). Полученную в результате смесь перемешивали 30 минут при комнатной температуре, затем добавляли по каплям раствор 34 (7 г, 25,26 ммоль, 1 экв.) в ТГФ (13 мл). Потом смесь перемешивали в течение ночи при 70°С и охлаждали до комнатной температуры перед тем, как концентрировали. Остаток переносили в Et2O (60 мл), промывали H2O и солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Полученную в результате неочищенную смесь очищали на колонке с силикагелем (99/1 до 85/15 циклогексан/этилацетат), что давало соединение 35 (выход 77%) в виде желтоватого масла.
Синтез соединения 36
C18H18N2O2 М=294,35 г/моль
Масса: (ИЭР+): 317,1 (М+Na); 333,1 (М+К)

К раствору 35 (6,5 г, 19,91 ммоль, 1 экв.) в этаноле (50 мл) добавляли 55% гидрат гидразина (1,25 мл, 22,10 ммоль, 1,1 экв.) при комнатной температуре. Полученную в результате смесь нагревали с обратным холодильником в течение 3 часов при комнатной температуре. Затем реакционную смесь охлаждали в ледяной ванне и фильтровали. Осадок промывали холодным этанолом, что давало соединение 36 (выход 77%) в виде белого твердого вещества.
Синтез соединения 37
C53H52N2O6М=812,99 г/моль
Масса: (ИЭР+): 813,5 (М+Н); 835 (М+Na); 851,4 (М+К).

Соединение 36 (328 мг, 1,11 ммоль, 1,5 экв.) добавляли к раствору 17 (400 мг, 0,75 ммоль, 1 экв.) в безводном ТГФ (6,4 мл) в инертной атмосфере, затем три-н-бутилфосфин (198 мг, 0,98 ммоль, 1,3 экв.) и азодикарбонилдипиперидин (376 мг, 1,49 ммоль, 2,0 экв.). Полученную в результате желтую суспензию перемешивали при 30°С в течение ночи. Растворитель удаляли и неочищенную смесь очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 100:0 до 60:40), что давало соединение 37 (262 мг, выход 43%) в виде желтоватого масла.
Синтез соединения 38
C56H58N2O6М=855,07 г/моль
Масса (ИЭР+): 854,43 (М+Na); 893,5 (М+К).

Карбонат цезия (4,1 г, 12,5 ммоль, 15 экв.), затем изопропилиодид (0,99 г, 5,83 ммоль, 7 экв.) добавляли к раствору 37 (0,68 г, 0,83 ммоль, 1 экв.) в ДМФА в инертной атмосфере. Полученную в результате суспензию перемешивали при комнатной температуре в течение 3 часов. Смесь разбавляли этилацетатом и водой. Органический слой промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенное желтое масло очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 100:0 до 77:23), что давало требуемое соединение 38 (549 мг, выход 77%) в виде желтоватого масла.
Синтез соединения 39
C56H60N2O7М=873,08 г/моль
Масса (ИЭР+): 873,6 (М+Н); 895,6 (М+Na); 911,5 (М+К)
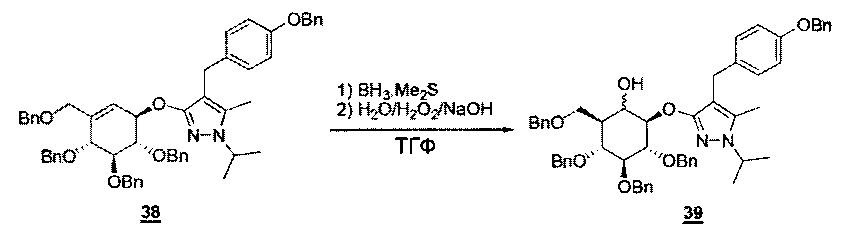
Раствор 9-BBN (0,5 М в ТГФ, 0,585 мл, 0,29 ммоль, 10 экв.) добавляли к раствору 38 (25 мг, 0,03 ммоль, 1 экв.) в безводном ТГФ в инертной атмосфере. Бесцветный раствор нагревали с обратным холодильником в течение ночи, затем охлаждали до 0°С. Последовательно добавляли воду (0,047 мл), водный раствор пероксида водорода (30% масс/масс, 0,100 мл) и 2 н. водный раствор гидроксида натрия (0,117 мл). Полученную в результате белую суспензию перемешивали в течение дополнительных 3 часов. Затем смесь разбавляли этилацетатом и выливали в воду. Потом органическую фазу сушили над сульфатом магния, фильтровали и концентрировали, что давало желтоватое масло. Очистка с помощью хроматографии на силикагеле (циклогексан/этилацетат 100:0 до 80:20) давала спирт 39 (2 мг, выход 8%).
Синтез соединения 40
C56H58N2O7 М=871,07 г/моль
Масса (ИЭР+): 871,6 (М+Н); 893,6 (М+Na); 909,5 (М+К)

Периодинан Десса - Мартина (9 мг, 0,021 ммоль, 1,5 экв.) добавляли к раствору 39 (12 мг, 0,014 ммоль, 1 экв.) в безводном дихлорметане в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем разбавляли дихлорметаном и 1 н. водным гидроксидом натрия. Потом водным слой экстрагировали дихлорметаном, и полученный в результате органический слой сушили над сульфатом натрия, фильтровали и концентрировали. Затем неочищенное желтое масло очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 100:0 до 72:28), что давало кетон 40 (8 мг, выход 67%) в виде желтоватого масла.
Синтез соединения 41
C56H58F2N2O6 М=893,07 г/моль
Масса (ИЭР+): 893,4 (М+Н); 911,5 (М+H2O)+

DAST (0,05 мл, 0,410 ммоль, 45 экв.) добавляли к раствору 40 (8 мг, 0,009 ммоль, 1 экв.) в безводном дихлорметане (0,05 мл) в инертной атмосфере. Реакционную смесь перемешивали при комнатной температуре в течение ночи и 3 часа при 35°С. Реакционную смесь доводили до комнатной температуры, затем разбавляли дихлорметаном и выливали в воду. Потом органический слой промывали насыщенным водным раствором NaHCO3, сушили над сульфатом магния, фильтровали и концентрировали, что давало неочищенное соединение 41 в виде оранжевого остатка.
Синтез соединения 42
C10H7FS М=178,23 г/моль
19F ЯМР (CDCl3, 282,5 МГц): -109,8 (m, 1F, Ar-F).
Масса (ГХ-МС): 133 (41%); 178 (100%)

В только что дегазированную смесь EtOH (69 мл) и H2O (9 мл) добавляли Pd2dba3 (534 мг, 0,58 ммоль, 0,025 экв.), РСу3 (660 мг, 2,35 ммоль, 0,1 экв.), 2-тиофен-бороновую кислоту (3,00 г, 23,4 ммоль, 1 экв.), K2CO3 (6,48 г, 46,9 ммоль, 2 экв.) и 4-бромфторбензол (5,17 мл, 47,0 ммоль, 2 экв.). Полученную в результате смесь перемешивали в течение ночи при 90°С и затем доводили до комнатной температуры. MgSO4 добавляли, чтобы погасить воду, и смесь фильтровали через прокладку из целита, используя этилацетат.Фильтрат концентрировали и очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 100:0 до 95:5), что давало соединение 42 (3,84 г, выход 92%) в виде белого твердого вещества.
Синтез соединения 43
C18H12BrFOS М=375,25 г/моль
19F ЯМР (CDCl3, 282,5 МГц): -111,3 (m, 1F, Ar-F).
Масса (ГХ-МС): 375,0 (97%); 376,0 (28%); 377,0 (100%); 416,0 (23%); 418,0 (23%)

5-Бром-2-метилбензойную кислоту (725 мг, 3,37 ммоль, 1 экв.) суспендировали в безводном дихлорметане (9,7 мл). Затем оксалилхлорид (0,32 мл, 3,74 ммоль, 1,1 экв.) и N,N-диметилформамид (0,013 мл, 0,17 ммоль, 0,05 экв.) добавляли при комнатной температуре, и смесь перемешивали в течение 6 часов. Потом растворитель выпаривали, что давало 5-бром-2-метилбензоилхлорид в виде желтого масла. Этот неочищенный продукт растворяли в безводном дихлорметане (19,3 мл), AlCl3 (49,5 мг, 3,71 ммоль, 1,1 экв.) и 42 (600 мг, 3,37 ммоль, 1 экв.) затем добавляли при 0°С (внутренняя температура). Полученную в результате смесь перемешивали при этой температуре в течение 30 минут и затем при комнатной температуре в течение ночи. Реакционную смесь выливали в лед и воду, органический слой отделяли и водный слой экстрагировали дихлорметаном. Органические слои собирали, сушили над MgSO4, фильтровали и концентрировали. Остаток обрабатывали н-гексаном с образованием осадка, который собирали в ходе фильтрации, промывали н-гексаном и сушили, что давало соединение 43 (выход 69%) в виде желтоватых кристаллов.
Синтез соединения 44
C18H14BrFS М=361,27 г/моль
19F ЯМР (CDCl3, 282.5 МГц): -115,0 (m, 1F, Ar-F).
Масса (ИЭР+): 133 (29%); 177 (49%); 182 (55%); 184 (70%); 191 (72%); 281 (39%); 360 (95%); 362 (100%)

Et3SiH (0,99 мл, 6,18 ммоль, 2,9 экв.) добавляли при комнатной температуре к раствору кетона 43 (800 мг, 2,13 ммоль, 1 экв.) в безводном дихлорметане -ацетонитриле (1:1, об./об., 16 мл). Полученную в результате смесь охлаждали до 0°С и медленно добавляли BF3.Et2O (0,75 мл, 5,97 ммоль, 2,8 экв.). Реакционную смесь затем перемешивали при комнатной температуре в течение 3 часов. Медленно добавляли насыщенный водный раствор NaHCO3 при 0°С. Водный слой экстрагировали дихлорметаном и полученный в результате органический слой сушили над MgSO4, фильтровали и концентрировали. Неочищенную смесь затем перекристаллизовывали с МеОН, что давало соединение 44 (выход 70%) в виде желтоватых кристаллов.
Синтез соединения 45
C53H49FO5S М=817,02 г/моль
19F ЯМР (CDCl3, 282,5 МГц): -115,2 (m, 1F, Ar-F)
Масса (ИЭР+): 839,5 [М+Na]+; 855,4 [М+К]+

н-Бутиллитий (1,4 М в гексанах, 0,30 мл, 0,412 ммоль, 1,1 экв.) медленно добавляли к охлажденному раствору (-70°С) 44 (149 мг, 0,412 ммоль, 1,1 экв.) в безводном ТГФ - толуоле (1:1, об./об., 4,8 мл) в инертной атмосфере. Полученный в результате темно-синий раствор перемешивали в течение 5 минут при этой температуре, затем медленно добавляли охлажденный раствор (-70°С) циклогексенона 8. Реакционную смесь перемешивали в течение15 минут при -70°С, потом выливали в воду. Органический слой затем сушили над сульфатом натрия, фильтровали и концентрировали, что давало неочищенное соединение 45 (300 мг, выход 98%) в виде желтого масла, которое использовали на следующей стадии без дополнительной очистки.
Синтез соединения 46
C53H49FO4S М=801,02 г/моль
19F ЯМР (CDCl3, 282.5 МГц): -115,3 (m, 1F, Ar-F)
Масса (ИЭР+): 823,5 [М+Na]+; 839,4 [М+К]+

Et3SiH (0,025 мл, 0,157 ммоль, 3 экв.) и BF3.Et2O (0,013 мл, 0,105 ммоль, 2 экв.) последовательно добавляли к охлажденному раствору (-20°С) 45 (43 мг, 0,052 ммоль, 1 экв.) в безводном дихлорметане (0,55 мл) в инертной атмосфере. Полученный в результате раствор перемешивали при -20°С в течение 1 часа 45 минут, разбавляли дихлорметаном и выливали в солевой раствор. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали, что давало зеленое масло, которое затем очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 100:0 до 82:18), что давало соединение 46 (27 мг, выход 64%) в виде зеленого масла.
Синтез соединения 47
C53H51FO5S М=819,03 г/моль
19F ЯМР (CDCl3, 282,5 МГц): -115,3 (m, 1F, Ar-F)
Масса (ИЭР+): 841,4 [М+Na]+; 857,4 [М+К]+

ВН3.Me2S (2 М в ТГФ, 0,065 мл, 0,130 ммоль, 4 экв.) добавляли к охлажденному раствору (0°С) 46 (26 мг, 0,032 ммоль, 1 экв.) в безводном ТГФ (0,335 мл) в инертной атмосфере. Полученный в результате раствор перемешивали при комнатной температуре в течение ночи перед охлаждением до 0°С. Затем осторожно добавляли воду (0,041 мл, 2,27 ммоль, 70 экв.), потом пероксид водорода (30% масс/об, 0,11 мл, 0,97 ммоль, 30 экв.) и 2 н. водный гидроксид натрия (0,13 мл, 0,26 ммоль, 8 экв.). Белую суспензию перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь потом разбавляли этилацетатом и выливали в воду. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали, что давало бесцветный остаток, который затем очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 100:0 до 77:23), что давало спирт 47 (7 мг, выход 26%) в виде желтоватого остатка.
Синтез соединения 48
C53H49FO5S М=817,02 г/моль
19F ЯМР (CDCl3, 282,5 МГц): -115,4 (m, 1F, Ar-F)
Масса (ИЭР+): 839,4 [М+Na]+; 855,4 [М+К]+
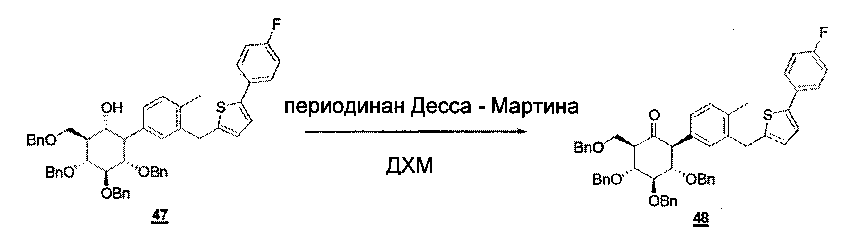
Периодинан Десса - Мартина (5 мг, 0,013 ммоль, 1,5 экв.) добавляли к раствору спирта 47 (7 мг, 0,009 ммоль, 1 экв.) в дихлорметане (0,150 мл). Полученную в результате смесь перемешивали при комнатной температуре в течение 1 часа 30 минут, затем выливали в 1 н. водный гидроксид натрия. Органический слой отделяли, и водный слой экстрагировали дихлорметаном. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Затем неочищенный остаток очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 100:0 до 80:20), что давало кетон 48 (6 мг, выход 86%) в виде желтоватого остатка.
Синтез соединения 49
C53H49F3O4S М=839,01 г/моль
19F ЯМР (CDCl3, 282,5 МГц): -115,3 (m, 1F, Ar-F); 113,75 (dt, J1=254 Гц, J2=29 Гц, 1F, CFF); -100,4 (d, J=254 Гц, 1F, CFF).
Масса (ИЭР+): 861,3 [М+Na]+; 877,4 [М+К]+

Кетон 48 (316 мг, 0,39 ммоль, 1 экв.) растворяли в DAST (1,4 мл, 11,4 ммоль, 30 экв.) и реакционную смесь перемешивали в течение ночи в инертной атмосфере при 70°С. Дихлорметан добавляли при комнатной температуре и реакционную смесь выливали в воду. Водную фазу экстрагировали дихлорметаном и органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 100:0 до 78:12), затем с помощью препаративной ВЭЖХ (Kromasil С18, МеОН/H2O 95:5), что давало 49 (84 мг, выход 26%) в виде бесцветного масла.
Синтез соединения 50
C25H25F3O4S М=478,52 г/моль
19F ЯМР (CDCl3, 282,5 МГц): -100,2 (d, J=254 Гц, 1F, CFF); -116,2 (dt, J1=254 Гц, J2=28 Гц, 1F, CFF); -117,6 (m, 1F, Ar-F).
Масса (ИЭР+): 501,2 [М+Na]+
Масса (ИЭР+): 513,2 [М+Cl]-

Соединение 49 (48 мг, 0,057 ммоль, 1 экв.) растворяли в ТГФ-МеОН (1:1, об./об., 4,2 мл) в инертной атмосфере. 10% Pd/С (96 мг, 0,02 ммоль, 0,35 экв.) и 5 капель 12 н. водной соляной кислоты добавляли к смеси, которую дегазировали 5 раз с Н2. Полученную в результате черную суспензию перемешивали в атмосфере Н2 при комнатной температуре в течение 72 часов. Реакционную смесь фильтровали через прокладку из Целита 545 и фильтрат концентрировали. Неочищенный продукт очищали с помощью хроматографии на силикагеле (дихлорметан/метанол 100:0 до 91:9), затем с помощью препаративной ВЭЖХ (5-амид С18, MeCN/H2O 38:62), что давало соединение 50 с выходом 27% в виде белого твердого вещества.
Синтез соединения 51
С35Н36О5 М=536,66 г/моль
Масса: (ИЭР+): 554,13 [М+H2O]+
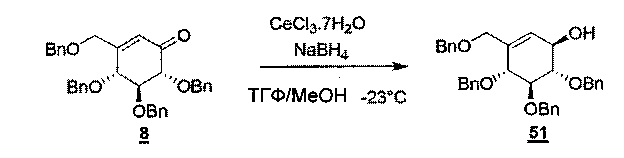
В инертной атмосфере гептагидрат хлорида церия (167 мг; 0,449 ммоль; 1,2 экв.) добавляли к раствору циклогексенона 8 (200 мг; 0,374 ммоль; 1 экв.) в МеОН-ТГФ (3:1, об./об., 5 мл), охлажденному до -23°С. Реакционную смесь перемешивали в течение 30 минут при этой температуре и добавляли боргидрид натрия (21 мг; 0,561 ммоль; 1,5 экв.). После дополнительных 45 минут добавляли насыщенный водный раствор хлорида аммония (15 мл) и хлорида натрия (15 мл). Водный слой экстрагировали этилацетатом, и объединенные экстракты сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/циклогексан 3/97 до 35/65), что давало спирт 51 (137 мг, выход 68%) в виде белого твердого вещества.
Синтез соединения 52
C41H50O5Si М=650,92 г/моль
Масса (ИЭР+): 673,5 [М+Na]+; 689,3 [М+К]+

К раствору 51 (3,80 г; 7,09 ммоль; 1 экв.) в безводном диметилформамиде (25 мл) в инертной атмосфере добавляли имидазол (1,45 г; 21,3 ммоль; 3 экв.). Реакционную смесь перемешивали в течение 30 минут при комнатной температуре перед добавлением трет-бутилдиметилсилилхлорида (1,70 г; 11,3 ммоль; 1,6 экв.). Смесь нагревали при 40°С в течение 12 часов, затем гасили водой и экстрагировали этилацетатом. Органические слои объединяли, промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали, что давало соединение 52 (4,57 г, выход 99%) в виде желтого масла. Это соединение использовали на следующей стадии без дополнительной очистки.
Синтез соединения 53
C41H52O6Si М=668,93 г/моль
Масса (ИЭР+): 691,4 [М+Na]+; 707,4 [М+К]+

К раствору 52 (4 г; 6,15 ммоль; 1 экв.) в безводном ТГФ (60 мл) в инертной атмосфере добавляли комплекс боран-диметилсульфид (12,3 мл; 2 М в ТГФ; 24,6 ммоль; 4 экв.) при 0°С. Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем последовательно добавляли воду (7,8 мл; 0,43 моль; 70 экв.), 30% пероксид водорода в воде (21,0 мл; 0,19 моль; 30 экв.) и 3 М водный гидроксид натрия (16,4 мл; 49,2 ммоль; 8 экв.) при 0°С. Смесь перемешивали в течение 2 часов при комнатной температуре, затем гасили насыщенным водным раствором хлорида аммония (300 мл). Водным слой экстрагировали этилацетатом, и объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат), что давало спирт 53 (754 мг, выход 63%) в виде желтого масла. (Неочищенное соединение 53 также можно использовать на следующей стадии без дополнительной очистки).
Синтез соединения 54
C41H50O6Si М=666,92 г/моль
Масса (ИЭР+): 689,5 [М+Na]+; 705,4 [М+К]+

К раствору 53 (1,51 г; 2,26 ммоль; 1 экв.) в безводном дихлорметане (23 мл) в инертной атмосфере добавляли периодинан Десса - Мартина (1,44 г; 3,39 ммоль; 1,5 экв.) при 0°С. Смесь перемешивали в течение ночи при комнатной температуре перед добавлением 1 М водного раствора гидроксида натрия (50 мл). Водный слой экстрагировали дихлорметаном (2×100 мл) и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/циклогексан 1/99 до 11/89), что давало кетон 54 (1,13 г, выход 75%) в виде желтого масла. Альтернативно кетон 54 можно получить с выходом 55% в ходе 3 стадий из 51 только с одной очисткой на этой последней стадии.
Синтез соединения 55
С35Н36О6М=552,66 г/моль
Масса (ИЭР+): 575,3 [М+Na]+; 591,3 [М+К]+

К раствору 54 (560 мг; 0,84 ммоль) в дихлорметане (4 мл) добавляли раствор 12 н. HCl в метаноле (2% об./об., 4 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Затем добавляли воду, потом насыщенный водный раствор гидрокарбоната натрия до нейтрализации. Смесь экстрагировали дихлорметаном, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток растирали в этаноле и фильтровали, что давало соединение 55 (337 мг, выход 73%) в виде белого твердого вещества.
Синтез соединения 56
С37Н38О7 М=594,69 г/моль
Масса (ИЭР+): 617,6 [М+Na]+; 633,6 [М+К]+

К раствору 55 (1,27 г; 2,30 ммоль; 1 экв.) в безводном дихлорметане (3 мл) в инертной атмосфере последовательно добавляли при 0°С пиридин (0,93 мл; 11,5 ммоль; 5 экв.), 4-диметиламинопиридин (60 мг; 0,46 ммоль; 0,2 экв.) и уксусный ангидрид (0,44 мл; 4,60 ммоль; 2 экв.). Смесь перемешивали при этой температуре в течение 45 минут. Затем добавляли воду, потом 1 н. водный раствор соляной кислоты. Водный слой экстрагировали дихлорметаном и объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали, что давало количественно кетон 56 (1,39 г) в виде светло-желтого масла. Неочищенное соединение 56 использовали на следующей стадии без дополнительной очистки.
Синтез соединения 57
C37H38F2O6 М=616,69 г/моль
19F ЯМР (CDCl3, 282,5 МГц): -110,0 (d, J=250 Гц, 1F, CFF); -119,4 (ddd, J1=249 Гц, J2=21 Гц, J3=29 Гц, 1F, CFF).
Масса (ИЭР+): 603,4 [М-HF+Li]+; 619,3 [М-HF+Li]+; 623,3 [М+Li]+; 639,3 [М+Na]+; 655,3 [М+К]+

К раствору 56 (1,30 г; 2,19 ммоль; 1 экв.) в безводном дихлорметане (5,2 мл) в инертной атмосфере добавляли трифторид диэтиламиносеры (5,2 мл; 42,4 ммоль; 19 экв.). Реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Затем раствор разбавляли дихлорметаном и добавляли твердый гидрокарбонат натрия. Смесь перемешивали в течение дополнительных 30 минут при 0°С, затем добавляли воду по каплям. Водный слой экстрагировали дихлорметаном, и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (EtOAc/циклогексан 2/98 до 12/88), что давало соединение 57 (471 мг, выход 35%) в виде светло-желтого масла.
Синтез соединения 58
С37Н40О6 М=580,71 г/моль
Масса (ИЭР+): 603,3 (М+Na)+; 619,3 (М+К)+
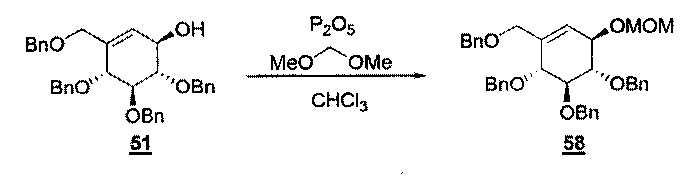
В инертной атмосфере неочищенное соединение 51 (53,7 г) растворяли в смеси безводного хлороформа (500 мл) и диметоксиметана (292 мл, 3,3 моль, 33 экв.). Добавляли P2O5 (73,9 г, 521 ммоль, 5,2 экв.). Реакционную смесь механически перемешивали в течение 1 часа при комнатной температуре. Затем смесь фильтровали через прокладку из Целит® 545 (элюирование дихлорметаном) и промывали насыщенным водным раствором NaHCO3 (700 мл). Затем добавляли воду (1 л) и смесь экстрагировали дихлорметаном (2×300 мл), промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали, что давало 58 (57,7 г) в виде коричневого масла, которое медленно кристаллизовалось. 58 использовали на следующей стадии без дополнительной очистки.
Синтез соединения 59
С37Н42О7 М=598,73 г/моль
Масса (ИЭР+): 621,3 (М+Na)+; 637,3 (М+К)+

В инертной атмосфере комплекс боран-диметилсульфид (2 М в ТГФ, 199 мл, 397 ммоль, 4 экв.) добавляли к раствору 58 (57,7 г) в безводном ТГФ (497 мл), охлажденному до 0°С. Реакционную смесь затем перемешивали в течение ночи при комнатной температуре перед охлаждением до 0°С и осторожно обрабатывали водой (125 мл, 6,96 моль, 70 экв.), затем пероксидом водорода (30% масс/об в H2O, 338 мл, 2,98 моль, 30 экв.) и гидроксидом натрия (2 М в H2O, 397 мл, 0,79 моль, 8 экв.). Смесь взаимодействовала в течение 2 часов при комнатной температуре (~25°С), затем добавляли насыщенный водный раствор хлорида аммония (700 мл) и воду (300 мл), чтобы погасить реакцию. Смесь экстрагировали этилацетатом (3×500 мл) и объединенные органические слои промывали водой (600 мл) и солевым раствором (600 мл), сушили над Na2SO4, фильтровали и концентрировали, что давало неочищенное соединение 59 (59,5 г) в виде желтого масла. 59 использовали на следующей стадии без дополнительной очистки.
Синтез соединения 60
С37Н40О7 М=596,71 г/моль
Масса (ИЭР+): 619,3 (М+Na)+; 635,3 (М+К)+

Периодинан Десса - Мартина (84,3 г; 199 ммоль; 2 экв.) добавляли частями к раствору неочищенного соединения 59 (59,5 г) в безводном дихлорметане (1 л) при 0°С. Затем реакционную смесь перемешивали в течение 18 часов при комнатной температуре, потом добавляли гидроксид натрия (1 н. в H2O, 1 л) и воду (500 мл). Потом водным слой экстрагировали дихлорметаном (2×400 мл) и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 98:2 до 86:14, об./об. на 750 г картридже Biotage SNAP), что давало заданный кетон 60 (32 г, выход 48% после 4 стадий) в виде желтого твердого вещества.
Синтез соединения 61
C37H40F2O6М=618,71 г/моль
19F ЯМР (CDCl3, 282,5 МГц): -108,5 (d, J=252 Гц, 1F, CFF); -121,0 (ddd, J1=252 Гц, J2=30 Гц, J3=20 Гц, 1F, CFF).
Масса (ИЭР+): 641,3 (М+Na)+; 657,3 (М+К)+
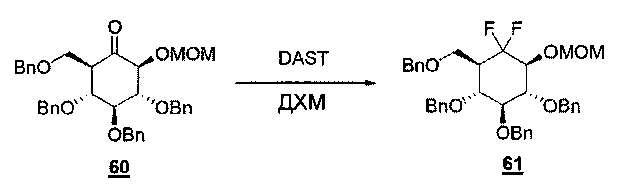
DAST (125 мл, 1,02 моль, 19 экв.) медленно добавляли к охлажденному раствору (0°С) 60 (32 г, 53,6 ммоль, 1 экв.) в безводном дихлорметане (145 мл). Реакционную смесь затем доводили до комнатной температуры и перемешивали в течение ночи. Дихлорметан (400 мл) потом добавляли, и смесь медленно выливали в смесь льда (1 л), дихлорметана (300 мл) и NaHCO3 (400 г). Смесь энергично перемешивали в течение 15 минут. Добавляли воду (500 мл) и водный слой экстрагировали дихлорметаном (2×300 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали, что давало неочищенное соединение 61 (32,6 г) в виде желтоватого масла. 61 использовали на следующей стадии без дополнительной очистки.
Синтез соединения 62
C35H36F2O5 М=574,65 г/моль
19F ЯМР (CDCl3, 282,5 МГц): -110,7 (d, J=249 Гц, 1F, CFF); -123,7 (ddd, J1=248 Гц, J2=29 Гц, J3=19 Гц, 1F, CFF).
Масса (ИЭР+): 577,5 [М-HF+Na]+; 592,5 [М+H2O]+; 597,5 [М+Na]+; 613,5 [М+К]+

А. К раствору 57 (70 мг; 0,114 ммоль; 1 экв.) в безводном метаноле в инертной атмосфере добавляли метилат натрия (8 мг; 0,142 ммоль; 1,25 экв.). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Затем добавляли воду, потом 1 н. водный раствор соляной кислоты, который добавляли до рН=6. Смесь экстрагировали этилацетатом, промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали, что давало спирт 62 (65 мг) в виде светло-оранжевого твердого вещества с количественным выходом.
В. Трифторуксусную кислоту (98,0 мл, 1,32 моль, 25 экв.) добавляли к раствору 61 (32,6 г) в безводном дихлорметане (260 мл) в инертной атмосфере. Реакционную смесь перемешивали в течение ночи при комнатной температуре. Смесь охлаждали до 0°С и добавляли воду (500 мл). Слои разделяли, и органический слой промывали водой (500 мл). Объединенные водные слои объединяли и экстрагировали дихлорметаном (2×100 мл). Объединенные органические слои промывали насыщ. NaHCO3 (250 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенную смесь очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 98:2 до 82:18 об./об. на 750 г картридже Biotage SNAP), что давало 62 (13,6 г, 30% после 2 стадий) в виде белого твердого вещества.
Синтез соединения 63
C35H36F2O6/C35H34F2O5 М=590,65 г/моль / 572,64 г/моль
19F ЯМР (CDCl3, 282,5 МГц):
Гидратная форма: -117,3 (dd, J1=257 Гц, J2=30 Гц, 1F, CFF); -125,6 (d, J1=258 Гц, 1F, CFF).
Кетонная форма: -112,1 (ddd, J1=260 Гц, J2=32 Гц, J3=6 Гц, 1F, CFF); -119,4 (dd, J1=260 Гц, J2=4 Гц, 1F, CFF).
Масса (ИЭР+): 608,4 [М+H2O]+; 613,5 [М+Na]+; 619,5 [М+К]+
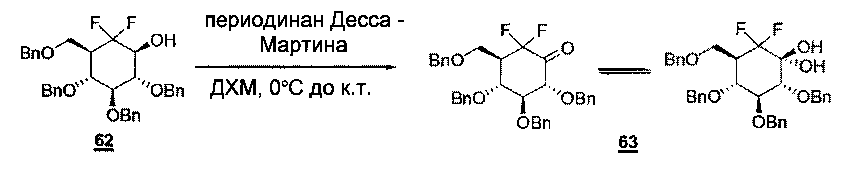
К раствору 62 (200 мг; 0,35 ммоль; 1 экв.) в безводном дихлорметане в инертной атмосфере добавляли периодинан Десса - Мартина (295 мг; 0,70 ммоль; 2 экв.). Реакционную смесь перемешивали в течение 3 часов при комнатной температуре перед добавлением 1 н. водного раствора гидроксида натрия (10 мл). Водный слой экстрагировали дихлорметаном и сушили над сульфатом натрия, фильтровали и концентрировали, что давало кетон 63 (158 мг, выход 77%) в виде светло-оранжевого твердого вещества, которое быстро переходило в гидратную форму до достижения равновесия.
Синтез соединения 64
C50H49ClF2O6М=819,37 г/моль
19F ЯМР (CDCl3, 282,5 МГц): -112,3 (dd, J1=266 Гц, J2=27 Гц, 1F, CFF); -113,7 (dd, J1=266 Гц, J2=6 Гц, 1F, CFF).
Масса (ИЭР+): 836,7 [М+H2O]+; 841,8 [М+Na]+; 857,7 [М+К]+

В инертной атмосфере в сосуд Шленка, содержащий стружки магния (50 мг, 2,04 ммоль, 1,2 экв.), добавляли 2 мл (из 5 мл) раствора 10 (552 мг, 1,70 ммоль, 1 экв.) и 1,2-дибромэтан (15 мкл, 0,17 ммоль, 0,1 экв.) в безводном ТГФ (5 мл). Смесь нагревали при 75°С в течение 5 минут, чтобы вызвать реакцию, и затем добавляли по каплям оставшиеся 3 мл раствора 10 и 1,2-дибромэтан при комнатной температуре. Этот раствор потом перемешивали при 75°С в течение 1 часа.
2,4 мл этого раствора Гриньяра, предварительно охлажденного до комнатной температуры, потом добавляли к раствору 63 (158 мг, 0,27 ммоль) в безводном ТГФ (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем добавляли насыщенный водный раствор хлорида аммония. Водный слой экстрагировали диэтиловым эфиром, и объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (циклогексан/этилацетат 100:0 до 77:23), что давало соединение 64 (152 мг) в виде смеси двух диастереомеров с выходом 69%. Эти диастереомеры можно разделить с помощью полупрепаративной ВЭЖХ.
Синтез соединения 65
C22H25ClF2O6 М=458,88 г/моль
19F ЯМР (MeOD, 282,5 МГц): -114,0 (dd, J1=262 Гц, J2=7 Гц, 1F, CFF); -115,4 (dd, J1=262 Гц, J2=26 Гц, 1F, CFF).
Масса (ИЭР+): 481,3 [М+Na]+; 497,3 [М+К]+

о-Дихлорбензол (53 мкл, 0,47 ммоль, 10 экв.), потом 10% Pd/С (56,0 мг, 53,3 мкмоль, 1,1 экв.) добавляли к раствору 64 (38,0 мг, 46,4 мкмоль, 1 экв.) в смеси ТГФ и МеОН (2:1, об./об., 26 мл). Реакционную смесь помещали в атмосферу водорода и перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь фильтровали и концентрировали, затем очищали с помощью хроматографии на силикагеле, что давало заданное соединение 65.
Синтез соединения 66
C50H48BrClF2O5 М=882,27 г/моль
19F ЯМР (CDCl3, 282,5 МГц): Основной аномер: -97,8 (dd, J1=246 Гц, J2=30 Гц, CFF); -102,6 (d, J=246 Гц, CFF).
Масса (ИЭР+): 4881,2 (М+Н)+; 898,3 (М+H2O)+.

SOBr2 (85 мкл, 1,10 ммоль, 15 экв.) добавляли при -40°С к раствору 65 (60 мг, 0,07 ммоль, 1 экв.) в безводном дихлорметане (0,73 мл) в инертной атмосфере. Смесь перемешивали, постепенно увеличивая температуру до 0°С, в течение 5 часов. Затем добавляли пиридин (89 мкл, 1,10 ммоль, 15 экв.) и раствор перемешивали в течение дополнительного 1 часа при 0°С. Добавляли 1 М водный раствор HCl и раствор доводили до комнатной температуры. Органический слой собирали, и водный слой экстрагировали дихлорметаном. Затем объединенный органический слой сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенную смесь очищали с помощью хроматографии на силикагеле (10 г Biotage SNAP, циклогексан/этилацетат 100:0 до 92/8), что давало 66 (15 мг, 23%) в виде бесцветного масла. Собранная фракция содержит один основной изомер.
Синтез соединения 15
C50H49ClF2O6 М=803,37 г/моль
19F ЯМР (CDCl3, 282,5 МГц): -100,3 (d, J=254 Гц, 1F, CFF); -113,3 (td, J1=254 Гц, J2=29 Гц, 1F, CFF).
Масса: (ИЭР+): 820,00 (М+H2O)

Bu3SnH (7 мкл, 25,5 ммоль, 1,5 экв.) добавляли к раствору 66 (15 мг, 17,0 ммоль) в безводном толуоле (170 мкл) при комнатной температуре. Затем смесь нагревали и перемешивали при 110°С в течение 3 часов. Потом одну дополнительную порцию Bu3SnH (7 мкл, 25,5 ммоль, 1,5 экв.) добавляли, и смесь перемешивали при 110°С в течение дополнительных 3 часов. Эту стадию повторяли еще раз, пока с помощью ТСХ не отмечали отсутствие реакции. Смесь концентрировали и очищали с помощью препаративной ТСХ (циклогексан/этилацетат 90:10, об./об.), что давало 15 (2 мг, 17%, β-аномер и 4 мг содержит α-аномер).
Синтез соединения 67
C36H35F5O7S М=706,72 г/моль
19F ЯМР (CDCl3, 282,5 МГц): -74,0 (d, J=12 Гц, CF3); -108,2 (dq, J1=252 Гц, J2=12 Гц, CFF); -119,5 (ddd, J1=253 Гц, J2=31 Гц, J3=18 Гц, CFF).
Масса (ИЭР+): 724,3 (М+H2O+); 729,2 (М+Na)+; 745,2 (М+К)+

Трифторметансульфоновый ангидрид (9,5 мл, 57,4 ммоль, 3 экв.) и пиридин (4,6 мл, 57,4 ммоль, 3 экв.) добавляли к охлажденному раствору (0°С) 62 (11,0 г, 19,1 ммоль, 1 экв.) в безводном дихлорметане (190 мл) в инертной атмосфере. Раствор нагревали до комнатной температуры и перемешивали в течение ночи. Затем добавляли воду (400 мл) к охлажденной смеси (0°С), которую потом экстрагировали дихлорметаном (2×150 мл), сушили над сульфатом натрия, фильтровали и концентрировали, что давало неочищенное соединение 67 (13,6 г) в виде коричневого твердого вещества. 67 использовали на следующей стадии без дополнительной очистки.
Синтез соединения 68
C48H46F2O6 М=756,87 г/моль
19F ЯМР (CDCl3, 282,5 МГц): -107,9 (brd, J1=256 Гц, CFF); -110,8 (ddd, J1=257 Гц, J2=30 Гц, J3=3 Гц, CFF).
Масса (ИЭР+): 779,4 (М+Na)+; 795,3 (М+К)+

Гидрид натрия (95%, 1,38 г, 57,3 ммоль, 3 экв.) добавляли к охлажденному (0°С) раствору 4-(бензилокси)фенола (13,4 г, 66,9 ммоль, 3,5 экв.) в безводном ДМФА (95 мл). Реакционную смесь перемешивали 1 час при этой температуре, затем добавляли раствор 67 (11,0 г) в безводном ДМФА (95 мл). Реакционную смесь перемешивали при 50°С в течение ночи, затем снова охлаждали до 0°С.Потом добавляли воду (250 мл), затем 1 н. водный раствор гидроксида натрия (600 мл). Смесь экстрагировали диэтиловым эфиром (300 мл, потом 2×150 мл) и объединенные органические слои промывали водой (2×600 мл) и солевым раствором (600 мл), затем сушили над сульфатом натрия, фильтровали и концентрировали, что давало неочищенное соединение 68 (13,5 г) в виде пурпурного масла. 68 использовали на следующей стадии без дополнительной очистки.
Синтез соединения 69
C13H16F2O6М=306,26 г/моль
19F ЯМР (D2O, 282,5 МГц): -107,6 (brd, J=262 Гц, 1F, CFF); -111,6 (brdd, J1=262 Гц, J2=31 Гц, 1F, CFF).
Масса (ИЭР-): 285,1 (М-Н-НРГ; 305,1 (М-Н)-; 341,1 (М+Cl)-; 351,1 (М+HCO2)-

Неочищенное соединение 68 (13,5 г) растворяли в смеси этанола /12 н. HCl 4% (об./об., 117 мл), тетрагидрофуране (63 мл). Палладий на активированном угле (10%, 3,8 г, 0,2 экв.) затем суспендировали в растворе, и реакционную смесь помещали в атмосферу водорода и перемешивали в течение 3 дней при комнатной температуре. Реакционную смесь фильтровали и концентрировали, затем очищали с помощью хроматографии на силикагеле (дихлорметан / метанол 100:0 до 85:15, об./об., на 340 г картридже Biotage SNAP), что давало соединение 69 (4,92 г, 90%), которое сушили сублимацией в виде белого твердого вещества.
2. Биологическая активность
а) Исследование облегчения выведения глюкозы.
В качестве подопытных животных использовали самок мышей CD1 (CDM или Charles River). Исследуемое соединение растворяли в наполнителе (5% N-метилпирролидона, 20% ПЭГ 400, 75% 20 мМ буфера Na4Р2O7, об./об./об.) при концентрации 1 мг/мл. Затем измеряли массу тела мышей и мышей распределяли по группам случайным образом, исследуемый образец перорально вводили в дозе 1 мг/кг, 3 мг/кг и 10 мг/кг. В качестве контроля перорально вводили только наполнитель (5% N-метилпирролидона, 20% ПЭГ 400, 75% 20 мМ буфера Na4P2O7, об./об./об.). Пероральное введение осуществляли с помощью желудочного зонда для мышей и 1 мл шприца. Минимальное число единиц в одной группе составляло 3, но могло достигать 12 для некоторых групп. Сбор мочи осуществляли вручную, слегка массируя брюшной отдел, чтобы собрать мочу (3 мкл) посредством градуированной пипетки. Мочу собирали через 1, 2, 4, 6, 8 и затем 16, 18, 20, 22, 24, 26 и 28 часов. Концентрацию глюкозы в моче измеряли, используя глюкозный набор WAKO, как указано ниже: 3 мкл мочи помещали в 96-луночный микропланшет для спектрометрического считывания. Аликвоту мочи разбавляли 350 мкл рабочего раствора WAKO. Для концентраций глюкозы, которые могут находиться за пределами глюкозного набора WAKO, аликвоту (35 мкл) последнего раствора помещали в другой 96-луночный микропланшет и дополнительно разбавляли (10 ×) 315 мкл рабочего раствора WAKO. Затем считывали поглощение 96-луночных планшетов при 505 нм, используя флуориметр/фотометр поглощения для планшетов BioTek SynergyMX, и вычисляли концентрацию глюкозы. Концентрации глюкозы для контрольных и исследуемых образцов в разные моменты времени усредняли, используя Excel 2007, и строили график, используя GraphPad Prism 5.
Полученные результаты для 16 и 50 показаны на фигурах 1 и 2. Из них следует, что 16 (3 мг/кг) вызывал продолжительную глюкозурию (вплоть до 26 часов, Фигура 2).
b) Анализы сравнения длительности действия соединений по изобретению с одним из соединений предшествующего уровня техники при изучении облегчения выведения глюкозы
Анализы проводили, как описано для а).
- Соединение 16 по изобретению сравнивали с дапаглифлозином, чтобы подчеркнуть улучшение длительности действия, т.е. большую длительность глюкозурии, соединения, когда внутрициклический атом кислорода глюкозной группировки заменен на группировку CF2.

Этот анализ проводили при дозе 3 мг/кг.
Полученные результаты представлены на Фигуре 5. Из них следует, что 16 (3 мг/кг) вызывал глюкозурию, которая продолжалась свыше 24 часов по сравнению с дапаглифлозином.
- Соединение 16 по изобретению сравнивали с соединением 9 из WO 2009/1076550, чтобы подчеркнуть улучшение длительности действия соединения, когда имитатор глюкозы, несущий группировку СН-ОН вместо внутрициклического атома кислорода, заменен на имитатор глюкозы, несущий CF2 вместо группировки СН-ОН.

Этот анализ проводили при дозе 3 мг/кг.
Полученные результаты представлены на Фигуре 6. Из них следует, что 16 (3 мг/кг) вызывал более длительную глюкозурию (вплоть до 24 часов), что не могло быть обнаружено в течение этого времени для соединения 9 из WO 2009/1076550.
с) Исследование облегчения в уменьшающихся колебаниях глюкозы в крови после сахарной нагрузки.
В качестве подопытных животных использовали самок мышей CD1 (CDM или Charles River), не получавших корма в течение 18 часов. Исследуемое соединение растворяли в наполнителе (5% N-метилпирролидона, 20% ПЭГ 400, 75% 20 мМ буфера Na4P2O7, об./об./об.) при концентрации 1 мг/мл. Затем измеряли массу тела мышей и мышей распределяли по группам случайным образом, исследуемый образец перорально вводили в дозе 1 мг/кг, 3 мг/кг и 10 мг/кг.В качестве контроля перорально вводили только наполнитель (5% N-метилпирролидона, 20% ПЭГ 400, 75% 20 мМ буфера Na4P2O7, об./об./об.). Через 15 минут после этого перорального введения всем мышам перорально вводили 20% раствор глюкозы в деионизованной воде. Пероральное введение осуществляли с помощью желудочного зонда для мышей и 1 мл шприца. Минимальное число единиц в одной группе составляло 3, но могло достигать 5 для некоторых групп. Сбор крови осуществляли через подкожную вену. Кровь собирали через 5, 10, 30, 45, 60 и 120 минут после сахарной нагрузки. Один эксперимент состоял во введении исследуемого образца за 18 часов до сахарной нагрузки, т.е. 18 часов после перорального введения исследуемого образца. Концентрацию глюкозы в крови измеряли, используя систему контроля уровня глюкозы в крови OneTouch® Ultra от Johnson and Johnson. Концентрации глюкозы для контрольных и исследуемых образцов в разные моменты времени усредняли, используя Excel 2007, и строили график, используя GraphPad Prism 5.
Полученные результаты для 16 показаны на фигурах 3 и 4.
Из них следует, что 16 уменьшал уровни глюкозы в крови в зависимости от дозы у обычных мышей после сахарной нагрузки (Фигура 3). Кроме того, вводимый перорально за 18 часов до сахарной нагрузки 16 (3 мг/кг) еще уменьшал колебания глюкозы в крови после сахарной нагрузки (Фигура 4).
d) Анализ оценки и сравнения устойчивости по отношению к глюкозидазе соединения 26 по изобретению с соединением предшествующего уровня техники (серглифлозин-А).
Анализ устойчивости к ферменту осуществляли с соединением 26 по изобретению и соединением А, используемым в качестве эталонного соединения, чтобы проверять эффективность β-глюкозидазы. Также оценивали устойчивость серглифлозин-А, чтобы сравнить улучшение метаболической устойчивости, полученной при замене внутрициклического атома кислорода группировки глюкозы на группировку CF2.

Все соединения обрабатывали β-глюкозидазой. Устойчивость соединения 26 и серглифлозин-А оценивали в ходе анализа ВЭЖХ после инкубации с β-глюкозидазой.
Использовали систему ВЭЖХ Gilson, оборудованную ручной системой ввода проб (V=20 мкл), детектором на диодной матрице (DAD172), настроенным на длину волны 230 нм, и 150 мм × 4,6 мм, 5 мкм HICHROM Kromasil 100-5С18 обращенно-фазовой колонкой. Использовали линейный бинарный градиент ВЭЖХ, как указано ниже: растворителем А была вода и растворителем В был ацетонитрил. После ввода 20 мкл образца растворитель В поддерживали на уровне 20% в течение 3 минут, увеличивали от 20% до 90% в течение 17 минут, поддерживали на уровне 90% в течение 4 минут; в конце растворитель В снижали обратно к 20% за 5,5 минут и поддерживали на уровне 20% в течение 3,5 минут.
Методика основана на J. Agric. Food Chem. 2005, 53, 4918-4924.
100 мкл раствора соединения 26 при 4,5*10-4 моль/л в ацетонитриле добавляли к раствору, содержащему 800 мкл фосфатного буфера (73173 Fluka, рН 7) в присутствии β-глюкозидазы от Almonds (10 Ед, 100 мкл 5,6 мг/мл раствора в фосфатном буфере (G4511sigma 18,7 Ед на мг)), и выдерживали 4 часа при 37°С.
100 мкл раствора серглифлозин-А при 4,5*10-4 моль/л в ацетонитриле обрабатывали в присутствии β-глюкозидазы таким же способом.
Параллельно 100 мкл раствора п-нитрофенил-β-глюкозида (соединение А) при 4,5*10-4 моль/л в фосфатном буфере (73173 Fluka, рН 7) добавляли к раствору, содержащему 700 мкл фосфатного буфера и 100 мкл ацетонитрила в присутствии β-глюкозидазы от Almonds (10 Ед, 100 мкл 5,6 мг/мл раствора в фосфатном буфере (G4511sigma 18,7 Ед на мг)), и выдерживали 4 часа при 37°С. В ходе процесса в присутствии β-глюкозидазы наблюдалось желтое окрашивание, что указывает на разложение соединения А.
Известно, что серглифлозин-А, как указывается в некоторых публикациях (Discov. Med. 2011, (58):255-263; Nature Reviews Drug Discovery 2010, 9, 551-559), подвергается расщеплению под действием β-глюкозидазы.
ВЭЖХ соединения 21 (Фигура 7), соединения 26 (Фигура 8) и серглифлозин-А (Фигура 10) осуществляли, чтобы проверить в экспериментах образование соединения 21 (агликоновая часть), что означает разрушение исходного вещества.

Выполняли ВЭЖХ соединения 26 в присутствии β-глюкозидазы (Фигура 9) и тем самым подчеркивали, что разрушения не происходит, поскольку образование соединения 21 не наблюдается.
Выполняли ВЭЖХ серглифлозин-А в присутствии β-глюкозидазы (Фигура 11) и тем самым подчеркивали, что разрушение происходит, поскольку наблюдается образование соединения 21. Чтобы оценить процент разрушения, выполняли калибровку по соединению 21, которая дала следующие результаты:

По данным строили график (% площади к концентрации) и полученную линейную регрессию описывали уравнением у=53629х и R2=0,994.
На фигуре 11 спектр ВЭЖХ серглифлозин-А в присутствии β-глюкозидазы указывает на то, что разрушение сопровождается образованием соединения 21 (% площади = 416).
Из предыдущего уравнения определяли, что концентрация соединения 21 составляет 7,76*10-3 г/л, что соответствует 3,6*10-8 моль.
Это составляет 80% разрушения серглифлозин-А после 4 часов инкубации при 37°С с β-глюкозидазой, тогда как разрушения соединения 26 не происходит при этих же условиях.
е) Анализ ингибирования реакции тирозин-тирозиназа
Ингибирование тирозиназы, т.е. ингибирование гидроксилирования тирозина до ДОФА, измеряли с помощью спектрофотометрии видимой части спектра, а именно, измеряя поглощение при 477 нм, показывающее количество меланина, вырабатываемого in vitro из субстратата тирозина под действием тирозиназы.
Чтобы убедиться, что измеренное поглощение пропорционально ферментативной активности в пределах изучаемых концентраций, готовили пять стандартных растворов, как указано ниже.
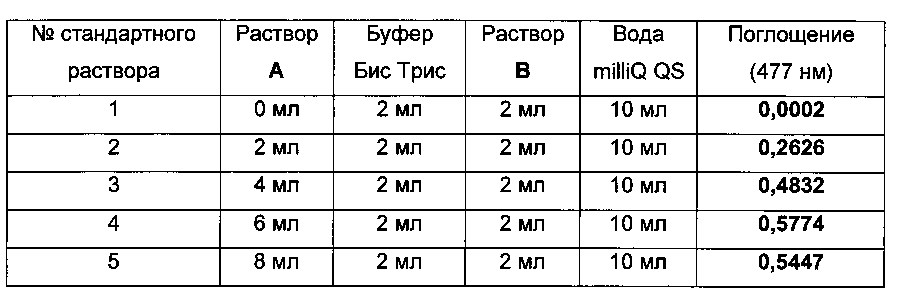
Поглощение измеряли на спектрометре УФ и видимой областей спектра Lambda 12 от Perkin Elmer.
Раствор А (1,000 Ед/мл маточный раствор тирозиназы) получали, растворяя 40 мг 1,250 Ед/мг грибной тирозиназы в 1 мл 100 мМ буфера Бис Трис рН 6,5, и доводили до 50 мл с водой milliQ.
Буфер Бис Трис (100 мМ буфер Бис Трис рН 6,5) получали, растворяя 2,09 г Бис Трис в воде milliQ, и доводили до 100 мл.
Раствор В (маточный раствор тирозина) получали, растворяя 100 мг тирозина в воде milliQ, и доводили до 100 мл.
Стандартные растворы инкубировали в течение 2 часов при 37°С, затем быстро охлаждали до 4°С. Поглощение растворов №2-5 измеряли при 477 нм относительно холостого раствора, не содержащего тирозиназу (раствор №1). Данные представляли графически (поглощение к концентрации тирозиназы) и полученную прямую линию в диапазоне поглощения от 0 до 0,5 описывали уравнением у=0,2415х-0,2343 и R2=0,9975.
Получали следующие исследуемые растворы и их поглощение измеряли при 477 нм:

С поглощением 0,1292 соединение 31 (раствор О) показывает ингибирование тирозиназы относительно гидрохинона (раствор Е).
f) Анализ оценки и сравнения ИК50 соединения 31 по изобретению с соединением предшествующего уровня техники (β-арбутином).
Протокол проведения опыта является таким же, как в анализе е.
Раствор А (1,000 Ед/мл маточный раствор тирозиназы) получали, растворяя все 50 кЕд грибной тирозиназы в 1 мл 100 мМ буфера Бис Трис рН 6,5, и доводили до 50 мл с водой milliQ.
Буфер Бис Трис (100 мМ буфер Бис Трис рН 6,5) получали, растворяя 2,09 г Бис Трис в воде milliQ, и доводили до 100 мл. рН доводили до 6,5, используя соляную кислоту.
Раствор В (маточный раствор тирозина) получали, растворяя 20 мг тирозина в воде milliQ, и доводили до 20 мл.

Основной раствор соединения 31 получали следующим образом: 10 мг соединения 31 растворяли в буфере Бис Трис до 1 мл.
Основной раствор β-арбутина получали следующим образом: 20 мг соединения β-арбутин растворяли в буфере Бис Трис до 1 мл.
Растворы инкубировали в течение 1 часа 30 минут при 37°С, затем быстро охлаждали до 4°С. По 100 мкл каждого раствора помещали в 96-луночный 5 планшет. Поглощение разных растворов измеряли при 477 нм (молекулярные устройства: Spectra Мах 340РС).
Разные растворы готовили, как описано в разных таблицах ниже, и их поглощение регистрировали. Поглощение раствора сравнения (без ингибитора) принимали за 100% ферментативной активности, что позволяло определить 10 процент ферментативной активности разных растворов.


По данным строили график (% активности к концентрации ингибиторов) и 15 линейную регрессию кривой использовали, чтобы рассчитать ИК50 обоих соединений. Полученные результаты представлены в таблице ниже:

Результаты ясно показывают, что соединение 31 является лучшим ингибитором тирозиназы, чем β-арбутин.
Реферат
Настоящее изобретение относится к соединению и его фармацевтически или косметически приемлемым солям, применимым в качестве ингибитора натрий-зависимого котранспортера глюкозы, антиоксиданта и для депигментации кожи в медицине и косметологии, следующей формулы (I):а также к способам его получения и композициям на его основе, где n, m и р представляют собой независимо друг от друга 0 или 1, R представляет собой СНОН или CHOR, Rи Rпредставляют собой ОН или OR, Rпредставляет собой ОН или OR, Rпредставляет собой атом водорода, когда n=1, или атом водорода, атом галогена или группу ОН, когда n=0; Xпредставляет собой атом водорода, атом галогена, группу ОН, (С-С)-алкил или OR; U, V и W представляют собой фенил, пиразолил, N-(С-С)алкил-пиразолил или тиенил, необязательно замещенные одним или более заместителями, выбранными из атома галогена, ОН, (С-С)-алкила и OR; R, Rи Rпредставляют собой арил-(С-С)-алкил и Rпредставляет собой (С-С)-алкил или арил-(С-С)-алкил. В предложенных способах фторируют соединение формулы (II) или связывают соединения формул (VIII) и (XI),причем в продукте связывания формулы (I) необязательно замещают группу ОН в положении Rгалогеном, или бромируют соединение формулы (I) с последующим восстановлением, или связывают соединение формулы (XVI) с соединением формулы (V), где R, R, R, R, Х, U, V, W, n, m и р указаны выше, Апредставляет собой -Li или -Mg-Hal, Hal представляет собой атом галогена, Rпредставляет собой уходящую группу. Предложено новое вещество, пригодное для отбеливания и депигментации кожи, в качестве антиоксиданта, для ингибирования натрий-зависимого котранспортера глюкозы, такого как SGLT1, SGLT2 и SGLT3, и заболеваний, где полезно такое ингибирование, в частности при лечении или предотвращении диабета и связанных с диабетом осложнений. 9 н. и 12 з.п.
Формула

или его фармацевтически или косметически приемлемая соль, где
- n, m и р представляют собой независимо друг от друга 0 или 1,
- R представляет собой СН2ОН или CH2OR11, R1 и R2 представляют собой независимо друг от друга группу ОН или OR15, R3 представляет собой группу ОН или OR18, R4 представляет собой атом водорода, когда n=1, и R4представляет собой атом водорода, атом галогена или группу ОН, когда n=0,
- X1 представляет собой атом водорода, атом галогена, группу ОН, (С1-С6)-алкил или OR24, и
- U, V и W представляют собой независимо друг от друга кольцо фенила, пиразолила, N-(С1-С6)алкил-пиразолила или тиенила,
указанное кольцо, возможно, замещено одним или более заместителями, выбранными из группы, состоящей из атома галогена, групп ОН, (С1-С6)-алкил и OR24,
- R11, R15и R18представляют собой независимо друг от друга группу арил-(С1-С6)-алкил и
- R24 представляет собой группу (С1-С6)-алкил или арил-(С1-С6)-алкил.
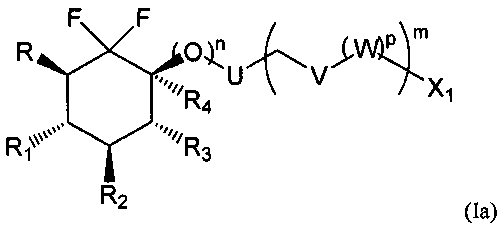


где R, R1, R2, R3, R4, X1, U, V, W, n, m и p являются такими, как определено в п. 1.


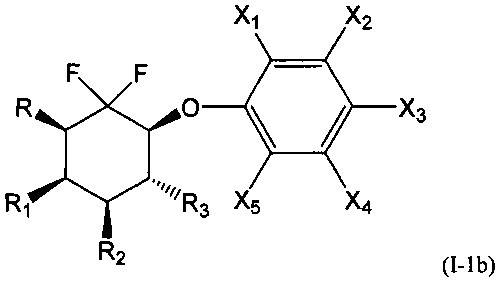
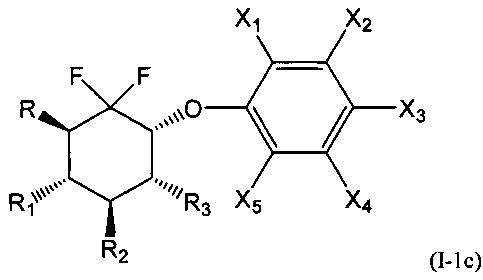
или его фармацевтически или косметически приемлемая соль, где
- R, R1, R2 и R3 являются такими, как определено в п. 1, и
- Х1, Х2, Х3, X4 и Х5 представляют собой независимо друг от друга атом водорода, атом галогена, группу ОН, (С1-С6)-алкил или OR24.
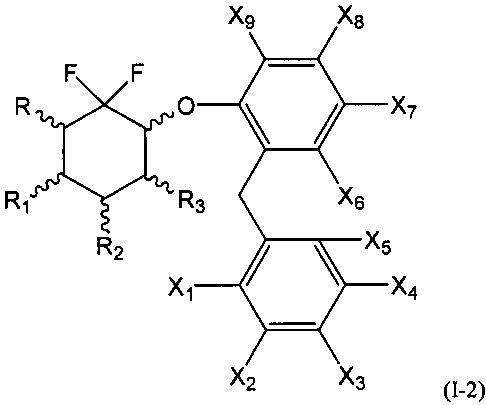
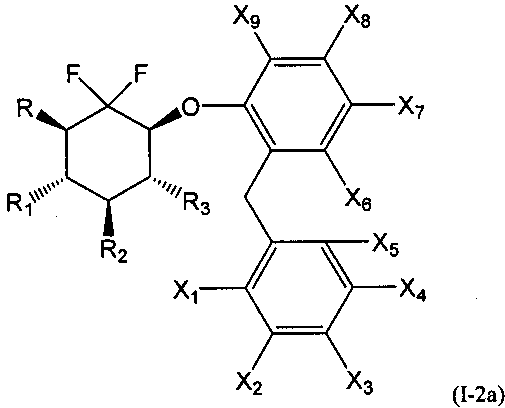

или его фармацевтически или косметически приемлемая соль,
где
- R, R1, R2 и R3 являются такими, как определено в п. 1, и
- X1, Х2, Х3, Х4, Х5, Х6, Х7, Х8 и Х9 представляют собой независимо друг от друга атом водорода, атом галогена, группу ОН, (С1-С6)-алкил или OR24.



или его фармацевтически или косметически приемлемая соль, где
- R, R1, R2 и R3 являются такими, как определено в п. 1,
- X1, Х2, Х3, Х4, Х5 и Х6 представляют собой независимо друг от друга атом водорода, атом галогена, группу ОН, (С1-С6)-алкил или OR24, и
- X представляет собой атом водорода или группу (С1-С6)-алкил.
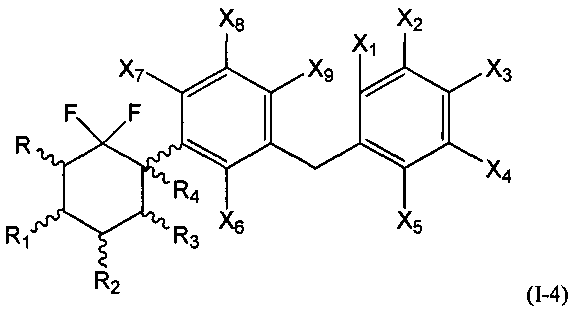
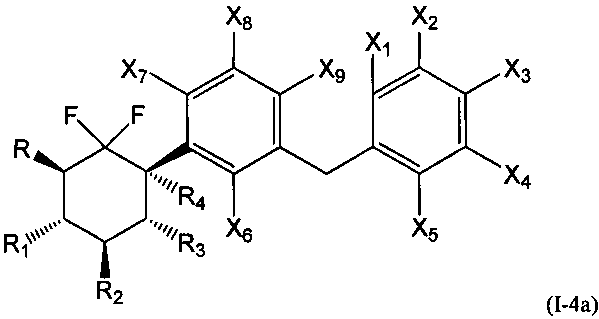

или его фармацевтически или косметически приемлемая соль, где
- R, R1, R2, R3 и R4 являются такими, как определено в п. 1, и
- X1, Х2, Х3, Х4, Х5, Х6, Х7, Х8 и Х9 представляют собой независимо друг от друга атом водорода, атом галогена, группу ОН, (С1-С6)-алкил или OR24.
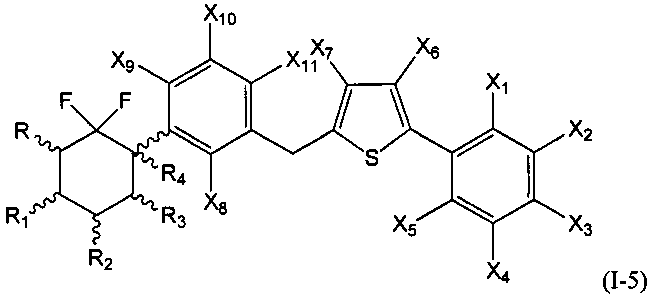


или его фармацевтически или косметически приемлемая соль, где
- R, R1, R2, R3 и R4 являются такими, как определено в п. 1, и
- X1, Х2, Х3, Х4, Х5, Х6, Х7, Х8, Х9, Х10 и Х11 представляют собой независимо друг от друга атом водорода, атом галогена, группу ОН, (С1-С6)-алкил или OR24.



где R, R1, R2, R3, Х1, U, V, W, n, m и р являются такими, как определено в п. 1.

где Х1, U, V, W, m и р являются такими, как определено в п. 1, и А1 представляет собой -Li или -Mg-Hal, Hal представляет собой атом галогена, и соединение следующей формулы (XI)

где R, R1, R2 и R3 являются такими, как определено в п. 1, что дает соединение формулы (I) по п. 1, где n=0 и R4=ОН.

где X1, U, V, W, m и р являются такими, как определено в п. 1, и А1 представляет собой -Li или -Mg-Hal, Hal представляет собой атом галогена, и соединение следующей формулы (XI)
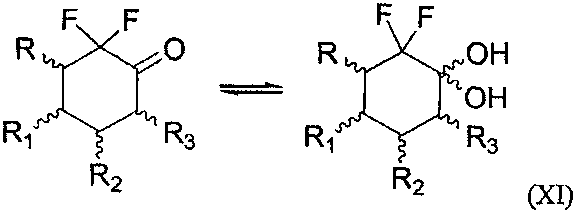
где R, R1, R2 и R3 являются такими, как определено в п. 1,
что дает соединение формулы (I) по п. 1, где n=0 и R4=ОН,
с последующим замещением группы ОН, что дает соединение формулы (I) по п. 1,
где n=0 и R4=галоген.
(а4) бромируют соединение формулы (I) по п. 1 с R4=ОН, что дает соединение формулы (I) по п. 1 с R4=Br, и
(b4) восстанавливают соединение формулы (I) по п. 1 с R4=Br, полученное на предшествующей стадии (а4), что дает соединение формулы (I) по п. 1 с R4=Н.
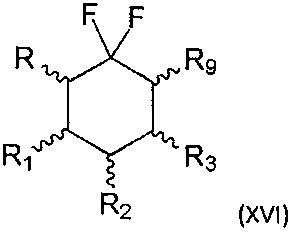
где R, R1, R2 и R3 являются такими, как определено в п. 1, и R9 представляет собой уходящую группу, с соединением следующей формулы (V):

где X1, U, V, W, m и р являются такими, как определено в п. 1.
Комментарии