Полимерная композиция и способ ее получения - RU2059670C1
Код документа: RU2059670C1
Чертежи
Описание
Изобретение относится к композиции кристаллических пропиленовых полимеров, предназначенным для изготовления термогерметизируемой пленки, и к способу получения указанных композиций.
Использование кристаллических сополимеров пропилена и олефинов в основном, этилена и/или 1-бутена, или их смеси с другими олефиновыми полимерами, в качестве материалов, обладающих герметизирующей способностью, хорошо известно специалистам.
Указанные кристаллические сополимеры получают путем полимеризации пропилена с небольшими количествами олефиновых сомономеров в присутствии катализаторов координации.
Полученный в результате полимер имеет статистическое распределение сомономеров, а точка его плавления ниже точки плавления кристаллических гомополимеров пропилена. Однако введение сомономеров ведет к частичной деградации кристаллической структуры вместе с образованием относительно больших количеств полимерной фракции, которая является растворимой в холодном ксилоле (при 25оС). Следовательно, механические свойства полимера ухудшаются, и даже если указанный полимер используется для получения многослойной пленки, например, путем совместной экструзии с полипропиленом, то могут возникнуть проблемы несовместимости с полипропиленовым слоем, в результате чего не может быть получена достаточная прочность запаивания. Более того, присутствие больших количеств растворимых в ксилоле фракций приводит к тому, что полученный полимер становится легко подверженным действию органических веществ, что делает его непригодным для хранения пищевых продуктов.
Указанные выше недостатки трудно устранить при использовании смесей указанных кристаллических сополимеров пропилена с другими полимерами, поскольку герметизирующие свойства, очевидно, связаны с природой и относительных количеств кристаллических фракций и растворимых в ксилоле фракций, и, вероятно, с их распределением в полимерном материале. Более того, изготовление смеси требует дорогостоящих обработок, как в отношении времени, так и в отношении энергетических затрат (например, гранулирование), которые необходимы для получения гомогенной дисперсии компонентов. Отсюда следует, что дальнейшие разработки должны быть направлены на способ получения полимеров, которые могли бы быть использованы непосредственно для изготовления герметизируемых пленок, имеющих низкую начальную температуру запаивания и низкий уровень растворимости (т.е. высокий уровень кристалличности).
Новые пропиленовые полимерные композиции, которые были получены посредством конкретных способов полимеризации, отвечают упомянутым выше требованиям. В частности, кристаллические полимерные композиции из пропиленов изобретения включают в себя, мас.
А) 30-65% предпочтительно 35-65% более предпочтительно 45-65% сополимера пропилена и С4-С8-α-олефина содержащего от 98 до 80% предпочтительно от 95-85%
пропилена;
В) 35-70% предпочтительно 35-65% а более предпочтительно 35-55% сополимера пропилена и этилена, и необязательно от 2-10% предпочтительно 3-6% С4-С8-α
-олефина. причем если С4-С8-α-олефин отсутствует, то указанный сополимер содержит 2-10% этилена, а предпочтительно 7-9% а при наличии С4-С8-α
-олефина, указанный сополимер содержит 0,5-5% а предпочтительно 1-3% этилена.
Указанный С4-С8-α-олефин выбирают предпочтительно из 1-бутена; 1-пентена; 1-гексена; 4-метил-1-пентена; и 1-октена. Особенно предпочтительным является 1-бутен.
Предпочтительно, если в сополимере указанной композиции (вариант В) С4-С8 -α-олефин отсутствует.
Более того, что вышеуказанные композиции имеют следующие свойства:
точка плавления около 125-140оС; начальная температура запаивания
(определенная ниже) составляет 100-110оС; фракция, растворимая в ксилоле при 25оС составляет менее 20% а предпочтительно менее 15% а наиболее предпочтительно менее 10 мас.
фракция, растворимая в н-гексане при 50оС, составляет менее 5,5 мас.
термин "начальная температура запаивания", или 1,Т означает минимальную температуру сваривания, при которой сварной шов многослойной пленки, имеющей, по крайней мере, один слой полипропилена и один слой композиции настоящего изобретения, не разрывается при приложении к указанной пленке нагрузки 200 г. Более подробно композиции данного изобретения иллюстрируются в примерах, приведенных ниже.
Композиции данного изобретения могут быть получены путем последовательной полимеризации мономеров в присутствии стереоспецифического катализатора Циглера-Натта на носителе, представляющем собой дигалид магния в активной форме. Указанный катализатор содержит в качестве главного элемента твердый каталитический компонент, включающий в себя титановое соединение, имеющее, по крайней мере, одну связь титан-галоген, и электронодонорное соединение, оба из которых нанесены на галид магния в активной форме.
Катализаторы, используемые в способе данного изобретения характеризуются тем, что они обладают способностью к продуцированию полипропилена, имеющего индекс стереорегулярности более 90% а предпочтительно более 95% Катализаторы, имеющие вышеуказанные характеристики, хорошо известны.
Твердые каталитические компоненты, используемые при изготовлении указанных катализаторов, содержат в качестве электронодонорных соединений соединения, выбранные из простых эфиров, кетонов, лактонов; соединений, содержащих N, P, и/или S-атомы; и сложных эфиров моно- и дикарбоновых кислот.
Из перечисленных соединений наиболее подходящими являются сложные эфиры фталевой кислоты, такие, как диизобутил, диоктил- и дифенилфталат и бензилбутилфталат; сложные эфиры малоновой кислоты, такие, как диизобутил- и диэтилалонат; алкил и арилпивалаты; алкил-, циклоалкил- и арилмалеаты; алкил- и арилкарбонаты, такие, как диизобутилкарбонат, этилфенилкарбонат, и дифенилкарбонат; сложные эфиры янтарной кислоты, такие, как моно- и диэтилсукцинат.
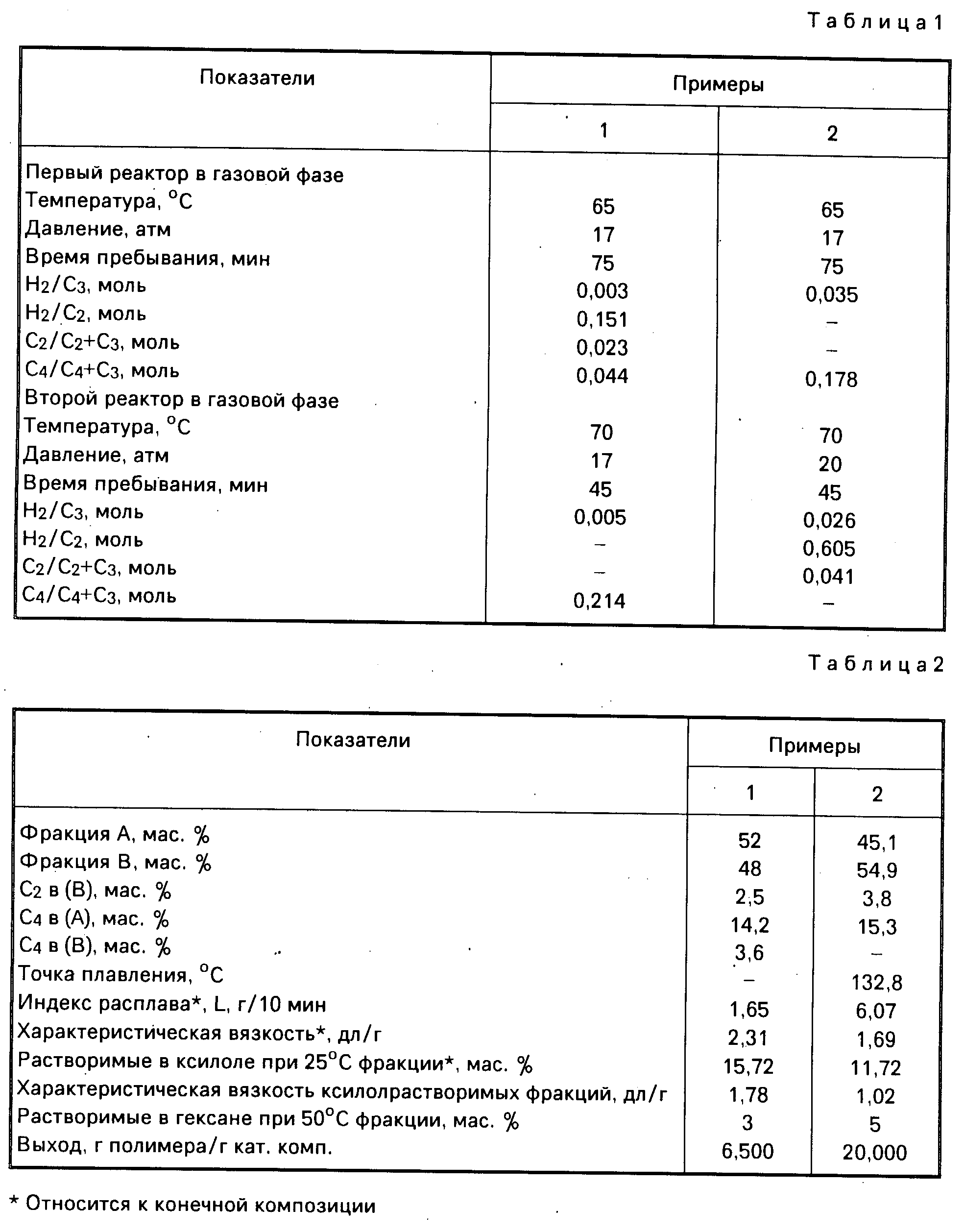
Другими наиболее подходящими донорами электронов являются 1,
3-диэфиры формулы
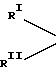

RIII и RIV являются одинаковыми или различными, и представляют собой алкилы с 1-4 атомами углерода.
Наиболее характерными из указанных соединений являются: 2-метил-2-изопропил-1,3-диметоксипропан; 2,2-диизобутил-1,3-диметоксипропан; и 2-изопропил-2- циклопентил-1,3-диметоксипропан.
Упомянутые выше каталитические компоненты могут быть получены различными способами.
Например, галид магния в виде безводного соединения, содержащего менее 1% воды, титановое соединение и электронодонорное соединение могут быть измельчены или размолоты вместе в условиях, при которых галид магния является активированным; после чего измельченный продукт один или два раза обрабатывают избыточным количеством TiCl4 при температуре в пределах 80-135оС, и несколько раз промывают углеводородом (например, таким, как гексан) до тех пор, пока в промывочной жидкости не обнаружат ионы хлора.
Согласно другому способу, безводный галид магния предварительно активируют стандартными методами, а затем подвергают взаимодействию с избытком TiCl4, содержащимся в растворе электронодонорного соединения. Температура процесса также составляет 80-135оС. Необязательно, обработку четыреххлористым титаном (TiCl4) повторяют, после чего твердый продукт промывают гексаном или другим углеводородным растворителем для удаления следовых количеств непрореагировавшего TiCl4.
Еще один способ заключается в том, что аддукт MgCl2•nROH в виде сфероидных частиц, где n равно 1-3, а ROH является этанолом, бутанолом или изобутанолом, подвергают реакции с избытком TiCl4, содержащимся в растворе электронодонорного соединения. Температура реакции составляет, в основном, 80-120оС. Затем твердое вещество выделяют, и еще раз подвергают реакции с TiCl4, после чего его выделяют и промывают углеводородом до тех пор, пока в промывочной жидкости не обнаружится присутствие ионов хлора.
Согласно другому способу, алкоголяты магния и хлоралкоголяты (хлоралкоголяты получают известным способом) подвергают реакции с избыточным TiCl4, содержащимся в растворе электронодонорного соединения, в условиях, аналогичных описанным выше.
В твердом компоненте катализатора, титановое соединение, (из расчета на Тi), в основном присутствует в количестве 0,5-10 мас. количество электронодонорного соединения, которое остается фиксированным на твердом носителе (внутренний донор) составляет, в основном, 5-20 мас. по отношению к дигалиду магния.
Титановыми соединениями, используемыми для получения компонентов катализатора, являются галиды и галогеналкоголяты. Предпочтительным соединением является тетрахлорид титана.
Удовлетворительные результаты могут быть также получены и с использованием тригалидов титана, в частности TiCl3HR, TiCl3ARA, а также галогеналкоголятов, таких, как TiCl3OR, где R является фенильным радикалом.
Описанные выше реакции приводят к образованию галида магния в активной форме. Помимо только что упомянутых имеются и другие известные специалистам реакции, которые приводят к образованию галида магния в активной форме, и которые в качестве исходных используют другие магниевые соединения, не являющиеся галидами, например такие, как карбоксилаты магния.
Присутствие активной формы галида магния в твердых компонентах катализатора может быть обнаружено с помощью рентгеновской спектроскопии, а именно, в том случае, максимальное интенсивное отражение, которое имеет место в спектре неактивированного галида магния (с поверхностной площадью менее 3 м3/г), исчезает, а вместо него поясняется гало с максимумом интенсивности, сдвинутым относительно положения максимума интенсивности неактивированного галида магния; либо, в этом случае, полуширина максимума интенсивности отражения, по меньшей мере, на 30% выше, чем максимальная интенсивность спектра неактивированного галида магния. Наиболее активная форма является формой, при которой рентгеновский спектр показывает гало.
Из галидов магния хлорид является наиболее предпочтительным соединением. В случае наиболее активной формы хлорида магния, рентгеновский спектр показывает гало вместо линии отражения, которая появляется на расстоянии 2,56

А1 алкиловыми соединениями, которые могут быть использованы в качестве сокатализаторов, являются
А1-триалкилы, такие, как А1-триэтил, А1-триизобутил, А1-три-н-бутил, и линейные или циклические А1-алкиловые соединения, содержащие два или более атомов А1, связанных между собой посредством О или N
или посредством SO4 и SO3-групп. Примерами указанных соединений являются
(C2H5)2Al-O-Al(C2H5)2
(C2H5)2Al-
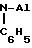
(C2H5)2Al-SO2-Al(C2H5)2
CH
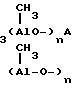
Могут быть также использованы соединения AlR2ORI, где RI является арилом, замещенным в одном или более положениях, а R является алкилом, имеющим 1-6 атомов углерода, и соединения AlR2H, где R имеет значение, указанное выше.
А1-алкиловое соединение используется в количествах, дающих отношение A1/Ti в пределах от 1 до 1000.
Электронодонорными соединениями, которые могут быть использованы в качестве внешних доноров (добавленных к А1-алкиловому соединению) являются сложные эфиры ароматических кислот, такие, как алкилбензоаты, а в частности, кремниевые соединения, содержащие, по крайней мере, одну связь Si-OR (R углеводородный радикал), 2,2,6,6-тетраметилпиперидин, и 2, 6-диизопропилпиперидин.
Примерами кремниевых соединений являются (трет-бутил)2Si(OCH3)2, (циклогексил)2Si(OCH3)3 и
(фенил)2Si(OCH3)2. 1,3-диэфиры, имеющие указанную
выше формулу, также с успехом могут быть использованы. Если внутренним донором является один из указанных
диэфиров, то внешние доноры могут отсутствовать.
Полимеризацию осуществляют, по крайней мере, в две стадии, а именно отдельные и последовательные стадии получения фракций (А) и (В), при этом каждую стадию проводят в присутствии пoлимера и катализатора, используемого в предыдущей стадии. Например, в одной стадии может быть получена фракция (В), а в последующей стадии может быть получена фракция (А). Порядок получения фракций (А) и (В) не является критическим, однако предпочтительно, сначала получать (А), а затем (В).
Полимеризация может быть осуществлена непрерывным или прерывным способом в соответствии со стандартной технологией, в жидкой фазе, в присутствии или в отсутствие инертного разбавителя; или в газовой фазе, или с использованием смешанной жидкой и газовой фазы. Предпочтительно проводить полимеризацию в газовой фазе.
Реакционное время и температуры указанных двух стадий не являются критическими; однако предпочтительно, если температура составляет 20-100оС. Регулирование мол. м. осуществляют с помощью стандартных регуляторов, таких, как водород.
Катализаторы могут быть предварительно подвергнуты взаимодействию с небольшими количествами олефинов (предпочтительная полимеризация). Предварительная полимеризация способствует улучшению эксплуатационных свойств катализаторов и морфологии полимеров.
Предварительную полимеризацию осуществляют путем выдерживания катализатора в суспензии в углеводородном растворителе (например, гексане, гептане и т.п.), и полимеризации при температуре от комнатной до 60оС в течение времени, достаточного для продуцирования полимера, составляющего 0,5-3-кратную массу от твердого компонента катализатора. Эта реакция может быть проведена также в жидком пропилене при указанных выше температурных условиях, в результате чего может быть получено до 1000 г полимера на 1 г каталитического компонента.
Поскольку фракции (А) и (В) получают непосредственно при полимеризации, то композиции данного изобретения имеют форму неэкструдированных частиц. Фракции (А) и (В) в указанных частицах смешаны оптимальным образом, так, что композиции настоящего изобретения могут быть использованы непосредственно для получения термосвариваемых пленок, не прибегая при этом к предварительной обработке, такой, как гранулирование.
Предпочтительные композиции имеют форму полимеризованных сферических или сфероидальных частиц диаметром 0,5-4,5 мм, а более предпочтительно, если указанные частицы имеют узкий спектр распределения, то есть, по крайней мере, 90% из них имеют диаметр 0,5-3,5 мм. Частицы указанного типа могут быть получены, например, с использованием известных катализаторов.
Получение твердого компонента катализатора.
В присутствии инертного газа и при комнатной температуре, 48 г безводного хлорида магния, 77 г безводного этилового спирта и 830 мл керосина загружали в 2-литровый автоклав, снабженный смесителем турбинного типа и трубопроводом для пропитки.
Содержимое автоклава нагревали до 120оС, размешивая при этом, и образовавшийся продукт присоединения между МgCl и спиртом расплавляли и подмешивали к диспергирующему агенту. Давление азота внутри автоклава поддерживали равным 15 атм. Трубопровод для пропитки нагревали при 120оС снаружи с помощью нагревательной рубашки, внутренний диаметр которой составлял 1 мм, а ее длина от одного конца до другого составляла 3 м.
Затем смесь протекала по трубопроводу со скоростью около 7 м/с. У выхода из трубопровода, дисперсию, взбалтывая, собирали в 5-литровую колбу, содержащую 2,5 л керосина и охлаждаемую снаружи с помощью рубашки при начальной температуре -40оС. Конечная температура эмульсии составляла 0оС. Твердые сферические частицы, состоящие из дисперсной фазы эмульсии, выделяли путем осаждения и фильтрации, промывали гептаном и осушали.
Все операции выполняли в атмосфере инертного газа.
Таким образом, было получено 130 г МgCl2•C2 H5OH в виде твердых сферических частиц с максимальным диаметром менее 50 мкм. После осушки указанного твердого продукта в вакууме в течение 2 ч, его масса составляла 105 г.
Полученный твердый продукт нагревали в потоке азота до температуры около 60оС для частичного удаления спирта из аддукта, в результате чего получали аддукт МgCl2•2,1C2H5OH.
Используя полученный таким образом аддукт, изготавливали каталитический компонент.
В 1-литровую стеклянную колбу, снабженную конденсатором, механической мешалкой и термометром, вводили, размешивая при 0оС в атмосфере азота, 625 мл ТiCl4, после чего добавляли 25 г МgCl2•2,1C2H2 OH-аддукта.
Содержимое колбы нагревали в течение 1 ч до 100оС. Когда температура достигала 40оС, вводили 9 ммоль диизобутилфталата. Указанную температуру поддерживали при 100оС в течение 2 ч, после чего продукт осаждали, а жидкость сифонировали. Затем добавляли 550 мл ТiCl4, и температуру доводили до 120оС в течение 1 ч. И, наконец, твердое содержимое колбы осаждали, а жидкость сифонировали; твердый остаток 6 раз промывали 200 см3 безводного гексана при 60оС и 3 раза при комнатной температуре.
П р и м е р 1 и 2. Осуществляли две стадии полимеризации с использованием компонента катализатора, полученного в соответствии с приведенным выше описанием.
Основные рабочие условия.
Полимеризацию осуществляли непрерывным способом в ряде реакторов, снабженных устройствами для переноса продукта из одного реактора в следующий, в присутствии азота.
Полимеризацию проводили в газовой фазе, водород и мономеры непрерывно анализировали и подавали для поддержания нужных постоянных концентраций.
В приведенных ниже примерах смесь триэтилалюминиевого (ТЕАl) активатора и дициклогексилдиметоксисилонового донора электронов, взятых в таких количествах, чтобы массовоe отношение ТЕАl/силан составляло 6,4, подвергали взаимодействию, с твердым компонентом катализатора, которое протекало в реакторе при -5оС в течение примерно 15 мин, так, чтобы массовое отношение ТЕА1/Ti составляло 65.
Затем катализатор переносили в другой реактор, содержащий избыток жидкого пропилена, и осуществляли полимеризацию в течение 3 мин при 20оС.
В первой фазе, форполимер переносили в другой реактор в целях полимеризации мономеров в газовой фазе, в результате чего образовывались фракция В (в примере 1) и фракция А (в примере 2).
Во второй фазе продукт, полученный в реакторе после удаления всех непрореагировавших мономеров, непосредственно подавали в другой реактор в газовой фазе в целях полимеризации мономеров, в результате чего получали другую из указанных двух фракций.
В конце второй фазы полимеризации полимер выгружали в промывочный аппарат, где непрореагировавшие мономеры и летучие вещества удаляли путем паровой обработки при 105оС и атмосферном давлении в течение 10 минут, после чего полимер осушали стандартными способами.
Исходные материалы и рабочие условия представлены в табл. 1; результаты испытаний на полимеризацию в отношении фракций (А) и (В), а также свойства конечных композиций представлены в табл. 2.
Для получения данных, представленных в табл. 2, использовали следующие аналитические методы. Содержание этилена (С2) определяли путем ИК-спектроскопии. Содержание 1-бутена (С4) определяли путем ИК-спектроскопии. Точку плавления определяли путем дифференциальной сканирующей калориметрии (ДСК).
Фракцию, растворимую в ксилоле, определяли путем солюбилизации образца материала в ксилоле при 125оС с последующим охлаждением раствора до комнатной температуры. Растворимую и нерастворимую фракции разделяли фильтрацией. Фракцию, растворимую в гексане, определяли следующим образом. Пленку продукта толщиной 100 мкм подвергали экстракции гексаном в автоклаве в течение 2 ч при 50оС. После чего гексан выпаривали и определяли сухой остаток. Индекс расплава определяли в соответствии с АSTMD 1238, условие L. Характеристическую вязкость определяли в тетрагидронафталине при 135оС.
Начальную температуру запаивания (SIT) композиций примеров 1 и 2 определяли следующими методами.
Получение пленки.
С помощью экструзии композиций примеров 1 и 2, проводимой при 200оС, получали различные пленки толщиной 50 мкм.
Каждую пленку, полученную таким образом, накладывали на полипропиленовую пленку, полученную из полипропилена с показателем стереорегулярности 97 (в кипящем н-гептане) и индексом расплава 4,5 г/10 мин. Толщина полипропиленовой пленки составляла 560 мкм.
Наложенные друг на друга пленки закрепляли в рамном прессе при 200оС с нагрузкой 9000 кг, выдерживаемой в течение 5 мин.
Соединенные пленки растягивали до их 6-кратной длины в обоих направлениях с помощью вытяжного устройства, изготовленного ТМ 1 0 G, в результате чего получали пленки толщиной 20 мкм. От каждой из упомянутых пленок получали образец размером 5х10 см.
Определение S I. T.
Испытание проводили путем приложения к термоспаянным образцам нагрузки 200 г.
В каждом испытании два вышеуказанных образца накладывали друг на друга, причем так, чтобы термоспаянные слои, изготовленные из композиций примеров 1 и 2, находились друг против друга. Эти наложенные друг на друга слои спаивали вдоль их 5-сантиметровой стороны с помощью лабораторного аппарата для сварки модель 12-12 AS (Sentinel combination Laboratory sealer).
Врем сварки составляло 5 ч, давление 1,2 атм. а ширина сварного шва составляла 2,5 см. Температуру сварки для каждого испытуемого образца повышали на 2оС.
Затем спаянные образцы разрезали на куски размером 2,5х10 см, а неспаянные концы прикрепляли к динамометру.
Как было указано выше, минимальной температурой сварки (обозначенной S. I. T. ) считают температуру, при которой сварной шов не разрывается при приложении к нему нагрузкой 200 г.
Значения S. I. T. для композиций образцов 1 и 2 составляли 100 и 125оС, соответственно.
Реферат
Использование: в промышленности пластмасс. Сущность изобретения: композиция включает, %: каучукообразный сополимер пропилена 45,1 - 52,0; статистический сополимер пропилена 48,0 - 54,9. В качестве первого компонента - сополимер 84,7 - 85,8% пропилена с 14,2 - 15,3% бутена-1, а в качестве второго - сополимер 96,2% пропилена, 2,5% этилена и 3,6% бутена-1. Композиция содержит фракцию, растворимую в ксилоле, в количестве 11,72 - 15,72% и фракцию, растворимую в н-гексане при 50oС, в количестве 3 - 5%. Композицию получают сополимеризацией мономеров в двух последовательно расположенных зонах. 2 с. и 3 з. п. ф-лы, 2 табл.
Формула
Статистический сополимер пропилена 48,0 54,9
2. Композиция по п. 1, отличающаяся тем, что содержит фракцию, растворимую в ксилоле при 25oС, в количестве 11,72 15,72 мас. на 100 мас. композиции.
Комментарии