Композиции перорально распадающихся таблеток, содержащие кортикостероиды, для лечения эозинофильного эзофагита - RU2678695C2
Код документа: RU2678695C2
Чертежи

Описание
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
В данной заявке заявляется приоритет согласно кодексу США, раздел 35, § 119(e) по предварительной заявке на патент США №61/874450, зарегистрированной 6 сентября 2013 г., описание которой включено в данный документ в полном объеме для любых целей посредством ссылки.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Изобретение относится к вводимым перорально композициям, содержащим низкие дозы местно действующих кортикостероидов, которые эффективны для лечения патологических состояний, связанных с воспалением пищевода.
УРОВЕНЬ ТЕХНИКИ
Заболевания, связанные с воспалением пищевода, такие как эозинофильный эзофагит (ЭоЭ), которые характеризуются высокими уровнями эозинофилов в пищеводе, а также очаговой базальной гиперплазией, все чаще диагностируются у детей и взрослых. Много аспектов этого заболевания остаются невыясненными, в том числе этиология, естественный ход развития и оптимальное лечение. ЭоЭ поражает все возрастные группы, но наиболее часто людей в возрасте от 20 до 50 лет. Симптомы ЭоЭ часто похожи на симптомы гастроэзофагеальной рефлюксной болезни (ГЭРБ) и включают рвоту, дисфагию, боль и задержку пищи. Обычно встречающаяся неправильная постановка диагноза ГЭРБ вместо ЭоЭ часто приводит к задержке лечения пациентов с ЭоЭ. На данный момент не существует утвержденных противовоспалительных лекарственных препаратов, вводимых местно, для лечения патологических состояний, связанных с воспалением верхнего отдела желудочно-кишечного тракта, в частности воспалительных патологических состояний пищевода, т.е. ЭоЭ. Для этого заболевания характерны боль, затрудненное глотание и предрасположенность пациентов к задержке пищи и другим осложнениям. Хотя системное лечение такими кортикостероидами, как преднизолон, является эффективным, они обладают значительным побочным действием, таким как подавление гипоталамо-гипофизарно-надпочечниковой системы (HPA), что отображается в уровнях кортизола в слюне, общее подавление иммунной функции, и, в частности у детей, неприятные побочные эффекты при долговременном системном применении включают задержку роста, что может привести к уменьшению роста взрослого человека.
В противоположность этому, способы лечения ЭоЭ включают введение дважды в день стероидных лекарственных препаратов с помощью дозирующего ингалятора (ДИ) на заднюю часть горла так, чтобы они практически не вдыхались, и указание для пациента держать рот закрытым при выдохе и глотании, и прополоскать рот непосредственно после введения, и не глотать пищу или воду в течение двух часов после введения. Полоскание рекомендуют, так как остаточные количества лекарственного вещества во рту и горле могут привести к инфекционному кандидозу, а глотание противопоказано, так как оно может смыть лекарственный препарат с пищевода. В другом исследовании 50% пациентов, которых лечили флутиказона пропионатом (ФП), продемонстрировали гистологическую ремиссию по сравнению с 9% пациентов, которые получали плацебо (Р=0,047). ФП уменьшал уровень эозинофилов в слизистой оболочке пищевода с более выраженным эффектом для индивидов, не имеющих аллергии. Однако, эта терапия особенно сложна для детей младшего возраста и людей с задержкой в развитии, которые вряд ли смогут эффективно применять данный способ «выдоха и глотания».
В другом рандомизированном двойном слепом плацебо-контролируемом исследовании, выполненном для оценки эффективности вязкого раствора 1 мг будезонида (содержимое контейнера массой 0,5 мг растворяют вместе с содержимым пяти пакетов сукралозы по 1 г в 10-15 мл жидкости), который вводят перорально дважды вдень, по сравнению с плацебо у подростков и взрослых пациентов с активной формой ЭоЭ в течение 15 дней, активность заболевания до лечения и после лечения оценивали клинически, эндоскопически и гистологически. Первичной конечной точкой являлось уменьшение среднего числа эозинофилов в эпителии пищевода (количество в поле зрения под большим увеличением - количество эозинофилов в пищеводе). 15-дневный курс лечения будезонидом хорошо переносится и является высокоэффективным для получения гистологической и клинической ремиссии у подростков и взрослых пациентов с активной формой ЭоЭ. После 15-дневного курса лечения значительно уменьшалось количество эозинофилов в эпителии пищевода пациентов, получающих будезонид (от 68,2 до 5,5 эозинофилов в поле зрения под большим увеличением; P<0,0001); но не в группе, получающей плацебо (от 62,3 до 56,5 эозинофилов в поле зрения под большим увеличением; P=0,48). Показатели дисфагии значительно уменьшались среди пациентов, получающих будезонид, по сравнению с пациентами, получающими плацебо (5,61 по сравнению с 2,22; P<0,0001). На основании эндоскопического исследования установлено, что у пациентов, получающих будезонид, уменьшены белые экссудаты и красные бороздки. Эта лекарственная форма не была утверждена Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) для коммерческого применения, и пероральное введение будет беспорядочно и вероятно приведет к несогласующимся результатам.
Когда необходимы твердые пероральные лекарственные формы с содержанием лекарственного вещества ≤5% по массе, лекарственное вещество или тонко измельчают и смешивают с по меньшей мере одним вспомогательным носителем, или гранулируют в грануляторе с кипящим слоем или с большим усилием сдвига обычно путем распыления раствора лекарственного вещества, чтобы достичь однородности/гомогенности смеси, а следовательно однородности состава готовых единиц дозирования согласно нормативным требованиям (FDA's Draft Guidance for Industry "Powder blend and finished dosage units - stratified in-process dosage unit sampling and assessment" October 2003). Многие тонкоизмельченные лекарственные вещества имеют тенденцию сегрегировать и образовывать более крупные частицы в смеси, чтобы уменьшить свою поверхностную энергию, возникающие сегрегация и агломерация могут быть причиной возникновения проблем неоднородности/негомогенности смеси, которые необходимо решать в первую очередь. Сегрегацию и агломерацию в смесях, содержащих низкие дозы очень плохо растворимых в воде лекарственных веществ, необходимо устранять не только в процессе смешивания порошка, но и до получения готовых лекарственных форм, капсул или таблеток, чтобы получить и поддерживать требуемую однородность/гомогенность смеси и/или устранить большие различия при растворении. Сегрегация частиц лекарственного вещества, особенно в смесях для прямого прессования, зависит от оборудования и материала. Таким образом, очень сложной является задача получения приемлемой гомогенности смеси для прямого прессования, содержащей низкую дозу лекарственного вещества и пригодные фармацевтически приемлемые вспомогательные вещества (т.е. при содержании лекарственного вещества в смеси <5% по массе), и поддержания гомогенности смеси до получения готовых лекарственных форм (например, таблеток или капсул) (McGinity J. W. et al. Dissolution and uniformity properties of ordered mixes of micronized griseofulvin and a directly compressible excipient. Drug Development and Industrial Pharmacy 1985; 11(4): 891-900; Yalkowsky S.H. and Bolton S. Particle size and content uniformity, Pharmaceutical Research. 1990; 7(9): 962-966; Ahmad H. and Shah N. Formulation of low dose medicines - Theory and Practice, Amer. Pharm. Rev. 2000; 3 (3): 1-5; Mahmoudi Z.N. et al. The influence of filler in blend uniformity of micronized drugs. Contributed poster, AAPS Annual Meeting (USA) 2010; Mahmoudi Z.N. et al. Effect of drug particle size on blend segregation and content uniformity. Contributed poster, AAPS Annual Meeting (USA) 2011).
В WO 2011041509 описано получение фармацевтической композиции, вводимой перорально, содержащей местно действующий кортикостероид в количестве менее 20 мг. Хотя нет конкретной информации о том, как достичь приемлемой однородности смеси, в демонстрационном примере о смеси для прессования, содержащей только 4% по массе флутиказона пропионата, которая будет обуславливать достижение приемлемой однородности состава ПРТ, то обстоятельство, что флутиказон гранулируется вместе с пригодными вспомогательными веществами для гранулирования, такими как быстро диспергирующиеся микрогранулы, содержащие маннит и кросповидон, свидетельствует о том, что грануляция флутиказона выполнена с целью достижения приемлемой однородности состава готовых таблеток. Однако тонкоизмельченные частицы местно действующих кортикостероидов могут присутствовать в агломерированных гранулах, и таким образом могут не быть легкодоступными для воспаленной ткани, пораженной ЭоЭ, при пероральном введении для быстрого вызывания ремиссии ЭоЭ.
Следовательно, существует потребность в композициях, содержащих низкие дозы кортикостероидов, которые имеют приемлемую однородность/гомогенность смеси, при перемешивании местно действующего кортикостероида с пригодными фармацевтически приемлемыми вспомогательными веществами, такими как носитель (т.е. при содержании лекарственного вещества в смеси <5%, особенно <3% по массе) и сохраняют гомогенность смеси до получения готовых дозированных лекарственных форм (например, таблеток или капсул), которые не только проявляют высокую однородность состава, но и пригодны для перорального введения пациентам, оказывая при этом местное (а не системное) лечение воспаления верхних отделов желудочно-кишечного тракта, в частности эозинофильного эзофагита (ЭоЭ), так как тонкоизмельченные частицы местно действующих кортикостероидов, в значительной степени адсорбированные на поверхности носителя, способны вызывать быструю ремиссию ЭоЭ.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к твердым пероральным фармацевтическим композициям, содержащим низкую дозу местно действующего кортикостероида и по меньшей мере один фармацевтически приемлемый носитель для адсорбции лекарственного вещества, причем количество лекарственного вещества составляет менее чем около 5% (масса лекарственного вещества/масса композиции), в частности - менее 3% по массе, а композиция не обладает значительной системной глюкокортикоидной или минералокортикоидной активностью после перорального введения людям. Смесь кортикостероида с носителем обладает высокой однородностью/ гомогенностью лекарственного вещества в смеси, что преобразуется в однородность состава готовых таблеток.
Композиция изобретения, которая может быть разработана в виде перорально распадающейся таблетки (далее именуемой ПРТ), которая распадается в течение 30 секунд при тестировании с использованием испытания на распадаемость согласно статье <701> фармакопеи США и/или распадается в течение 60 секунд при помещении в ротовую полость человека.
Настоящее изобретение также относится к способу изготовления низкодозированных фармацевтических композиций, включающему адсорбцию местно действующего кортикостероида, необязательно тонкоизмельченного, на по меньшей мере одном фармацевтически приемлемом носителе, таком как силикатированная микрокристаллическая целлюлоза, путем смешивания в течение достаточно длительного времени и прохождения через десегрегирующую/измельчающую мельницу по меньшей мере один раз до перемешивания с другими фармацевтически приемлемыми вспомогательными веществами, а затем изготовление из полученной смеси пригодных дозированных лекарственных форм, например, таблеток.
Настоящее изобретение описывает способ получения фармацевтических композиций, содержащих низкие дозы очень плохо растворимых в воде тонкоизмельченных лекарственных веществ и по меньшей мере один фармацевтически приемлемый носитель, которые необходимо перемешивать в течение достаточно длительного времени с использованием подходящего смесителя и мельницы тонкого помола, что позволяет устранить сегрегацию-агломерацию частиц лекарственного вещества в смеси не только в процессе перемешивания порошка, но также до получения готовых лекарственных форм, капсул или таблеток, чтобы достичь и поддерживать желаемую высокую однородность/гомогенность смеси и/или достичь высокой однородности состава готовых единиц дозирования, а также устранить большие различия при растворении.
Композиции настоящего изобретения пригодны для лечения различных патологических состояний, в том числе воспалительных патологических состояний желудочно-кишечного тракта. Таким образом, в настоящем изобретении также предложен способ лечения воспалительных патологических состояний верхних отделов желудочно-кишечного тракта у индивида, в частности пищевода (эозинофильный эзофагит), посредством местного введения с минимальным системным всасыванием и сопутствующим побочным действием, связанным с кортикостероидами. Способ включает введение индивиду фармацевтической композиции настоящего изобретения, содержащей частицы, необязательно тонкоизмельченные, местно действующего кортикостироида, адсорбированные на силикатированной микрокристаллической целлюлозе, для лечения эозинофильного эзофагита. В альтернативном варианте композиции настоящего изобретения могут содержать водорастворимое или набухающее в воде фармацевтически приемлемое вспомогательное вещество, такое как биогель или биоадгезивный полимер, которые будут усиливать биоадгезию кортикостероида к воспаленной слизистой оболочке пищевода.
ПОДРОБНОЕ ОПИСАНИЕ ФИГУРЫ
Фиг. 1 иллюстрирует места отбора проб в смесителе для получения представительных образцов согласно проекту руководства FDA (FDA's Draft Guidance for Industry "Powder blend and finished dosage units - stratified in-process dosage unit sampling and assessment" October 2003).
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Изобретение относится к твердой фармацевтической композиции, которая содержит кортикостероид в количестве менее чем около 5% (масса лекарственного вещества/масса композиции) и по меньшей мере один фармацевтически приемлемый носитель для адсорбции лекарственного вещества, причем композиция не обладает значительной системной глюкокортикоидной или минералокортикоидной активностью, а твердая фармацевтическая композиция распадается в течение 30 секунд при тестировании с использованием испытания на распадаемость согласно статье <701> фармакопеи США. Композиция распадается в течение около 60 секунд или менее при контакте со слюной в ротовой полости субъекта или пациента, которому необходима такая композиция.
Твердая фармацевтическая композиция настоящего изобретения обеспечивает воспаленные ткани верхних отделов желудочно-кишечного тракта, в частности воспаленные ткани пищевода, терапевтически эффективным количеством местного кортикостероида.
Как выше, так и далее по всему описанию изобретения, следующие термины, если не указано иное, имеют следующие значения.
Термин «лекарственное вещество», «активный» или «активный фармацевтический ингредиент», при использовании в данном документе, включает фармацевтически приемлемый и местно действующий кортикостероид, фармацевтически приемлемые соли, сложные эфиры, сольваты (в том числе гидраты), полиморфные модификации, стереоизомеры и/или пролекарства и их смеси. Термин «соль» относится к продукту, который образуется по реакции пригодной неорганической или органической кислоты с лекарственным веществом в форме «свободного основания». Пригодные кислоты включают те, которые обладают достаточной кислотностью для образования стабильной соли, например, кислоты с низкой токсичностью, образующие соли, утвержденные для употребления людьми и животными. Неограничивающими примерами кислот, которые можно использовать для получения солей перорально активного лекарственного вещества, включают неорганические кислоты, например: HCl, Н3РО4, H2SO4. Неограничивающие примеры органических кислот включают алкилсульфоновые кислоты и пропионовую кислоту.
Термины «перорально распадающаяся таблетка», «перорально диспергирующаяся таблетка» или «ПРТ» относятся к твердой лекарственной форме настоящего изобретения, которая быстро распадается в ротовой полости пациента после приема без разжевывания. Скорость перорального распадения может варьироваться, но она значительно выше, чем скорость перорального распадения традиционных твердых лекарственных форм или жевательных твердых лекарственных форм (т.е. таблеток или капсул), которые предполагается проглатывать непосредственно после приема.
Термин «около», при использовании в данном документе по отношению к численной величине, включает «точно». Например, «около 30 секунд» включает точно 30 секунд, а также величины, близкие к 30 секундам (например, 25 секунд, 29 секунд, 31 секунда, 35 секунд и т.д.). Когда термин «около» используется по отношению к интервалу значений, термин «около» относится как к минимальному, так и к максимальному значению интервала (например «около 1-50 мкм» означает «от около 1 мкм до около 50 мкм»).
Термин «тесно связанный», при использовании в данном документе для описания пространственного расположения двух или более компонентов композиции, относится к компонентам, которые тщательно перемешаны, как, например, в смесях, покрытиях и матрицах.
Если не указано иное, все процентные содержания и соотношения рассчитаны по массе. Если не указано иное, все процентные содержания и соотношения рассчитаны, исходя из общей массы композиции.
Термин «отсутствие значительной системной глюкокортикоидной или минералокортикоидной активности», при использовании в данном документе, относится к композициям на основе кортикостероидов, которые не обладают общим действием на организм посредством всасывания в кровоток, но обладает местным действием посредством местного контакта с пораженной тканью. Кортикостероиды, которые обладают высокой системной глюкокортикоидной активностью при пероральном введении, включают, например, гидрокортизон, преднизон, преднизолон, метилпреднизолон, дексаметазон, бетаметазон и т.д., или минералокортикоидной активностью (например, альдостерон). Кортикостероиды, которые, как правило, обладают системной глюкокортикоидной или минералокортикоидной активностью при пероральном введении, также могут использоваться в разбавленных композициях настоящего изобретения, при этом системное поглощение кортикостероида уменьшено или подавлено.
Пригодные местно действующие кортикостероиды, которые можно включать в фармацевтическую композицию настоящего изобретения, охватывают будезонид, флутиказон, флунизолид, циклесонид, мометазон, беклометазон, тиксокортол и соли или сложные эфиры или их смеси.
В конкретном варианте реализации изобретения композиция настоящего изобретения содержит флутиказон. В других вариантах реализации изобретения композиция настоящего изобретения содержит будезонид. В некоторых других вариантах реализации изобретения композиция настоящего изобретения содержит циклесонид.
В одном варианте реализации изобретения кортикостероид может быть в форме кристаллов со средним размером частиц около 100 мкм или менее, около 75 мкм или менее, около 50 мкм или менее, более конкретно - около 25 мкм или менее или около 15 мкм или менее. В конкретном варианте реализации изобретения кортикостероид измельчали так, чтобы получить частицы со средним размером менее около 10 мкм, менее около 8 мкм или меньше, менее около 6 мкм или, в частности менее около 4 мкм. В альтернативном варианте такие кристаллы могут иметь средний размер в субмикронном диапазоне (например, средний размер частиц<около 1 мкм), т.е. могут быть в форме наночастиц (например, средний размер частиц находится в интервале около 1-100 нм).
В другом варианте реализации изобретения кортикостероид может присутствовать в аморфной форме, например, быть связанным со стабилизирующим агентом, который ограничивает рекристаллизацию лекарственного вещества, например, с поливинилпирролидоном (ПВП), гидроксипропилметилцеллюлозой (ГПМЦ), гидроксипропилцеллюлозой, гидроксиэтилцеллюлозой, Soluplus®, Kollidon® VA64, лаурилсульфатом натрия, поверхностно-активными веществами ряда твинов, полимером Eudragit® ЕРО и их смесями.
Количество кортикостероида в фармацевтической композиции в соответствии с данным изобретением выбирают таким образом, чтобы получить максимальную терапевтическую пользу от местного применения, при этом максимально уменьшить побочное действие от системного всасывания. В случае твердых фармацевтических композиций настоящего изобретения количество кортикостероида в композиции составляет менее чем около 5% масс, (масса лекарственного вещества/масса композиции). В одном варианте реализации изобретения количество кортикостероида в фармацевтической композиции составляет менее чем около 4%. В другом варианте реализации изобретения оно составляет менее чем около 3%. Еще в одном варианте реализации изобретения оно составляет менее чем около 2%, менее чем около 1,5%, менее чем около 1%, менее чем около 0,5% по массе или меньше. В одном варианте реализации изобретения количество кортикостероида в фармацевтической композиции составляет между около 0,50 мг и около 18 мг. Еще в одном варианте реализации изобретения количество кортикостероида в фармацевтической композиции составляет между около 0,75 мг и около 12 мг. Еще в одном варианте реализации изобретения количество кортикостероида в фармацевтической композиции составляет между около 1,5 мг и около 9 мг. В других вариантах реализации изобретения количество кортикостероида составляет около 0,01 мг, около 0,05 мг, около 0,1 мг, около 0,15 мг, около 0,1 мг, около 0,2 мг, около 0,25 мг, около 0,3 мг, около 0,35 мг, около 0,4 мг, около 0,45 мг, около 0,5 мг, около 0,6 мг, около 0,7 мг, около 0,75 мг, около 0,8 мг, около 1 мг, около 1,5 мг, около 2 мг, около 3 мг, около 4 мг, около 4,5 мг, около 5 мг, около 6 мг, около 7 мг, около 8 мг, около 9 мг, около 10 мг, около 12 мг, около 18 мг, включая все возможные интервалы и подинтервалы между этими значениями.
В варианте реализации изобретения быстро распадающаяся композиция изобретения может содержать фармацевтически приемлемые вспомогательные вещества, которые набухают, растворяются или другим способом облегчают распадение композиции ПРТ с образованием однородной вязкой суспензии, содержащей тонкоизмельченные частицы кортикостероида, которая покрывает воспаленную слизистую пищевода для лечения эозинофильного эзофагита. В некоторых вариантах реализации настоящего изобретения общая масса лекарственной формы находится в интервале от 300 до 900 мг, чтобы включить максимально возможное количество быстро диспергирующихся микрогранул, содержащих по меньшей мере один альдит в сочетании с по меньшей мере одним разрыхлителем, для максимального покрытия поверхности, пораженной эозинофильным эзофагитом, тонкоизмельченным кортикостероидом. В другом варианте реализации изобретения быстро диспергирующиеся микрогранулы содержат по меньшей мере один разрыхлитель в сочетании с альдитом и/или сахаридом. Количество альдита и/или сахарида в быстро диспергирующихся гранулах изменяется в интервале около 99%-90% или около 95%-90% от общей массы гранул, содержащих разрыхлитель, включая все интервалы и подинтервалы между этими значениями. В одном варианте реализации изобретения средний размер частиц альдита и/или сахарида составляет около 30 мкм или менее, например: около 1-30 мкм, около 5-30 мкм, около 5-25 мкм, около 5-20 мкм, около 5-15 мкм, около 5-10 мкм, около 10-30 мкм, около 10-25 мкм, около 10-20 мкм, около 10-15 мкм, около 15-30 мкм, около 15-25 мкм, около 15-20 мкм, около 20-30 мкм, около 20-25 мкм или около 25-30 мкм.
В одном варианте реализации изобретения лекарственная форма обладает общей массой 300 мг и содержит около 0,05 мг (0,16%), около 0,75 мг (0,25% масс.), около 1,5 мг (0,5% масс.), около 3 мг (1% масс.), около 4,5 мг (1,5%), около 6 мг (2% масс.), около 9 мг (3% масс.), около 12 мг (4% масс.), около 16 мг (5%) кортикостероида.
В другом варианте реализации изобретения лекарственная форма обладает общей массой 600 мг и содержит около 0,75 мг (0,125% масс.), около 1,5 мг (0,25% масс.), около 3 мг (0,5% масс.), около 4,5 мг (0,75%), около 6 мг (0,1 % масс.), около 9 мг (1,5% масс.), около 12 мг (2% масс.), около 18 мг (3% масс.) кортикостероида. В одном варианте реализации изобретения местно действующий кортикостероид представляет собой флутиказона пропионат, количество которого в фармацевтической композиции изменяется от около 0,05 до около 15 мг при содержании лекарственного вещества от около 0,16% до 5% от массы композиции.
В другом варианте реализации изобретения количество флутиказона пропионата в композиции изменяется от около 0,75 до около 4,5 мг при содержании лекарственного вещества от около 0,25% до 1,5% от массы композиции.
В другом варианте реализации изобретения количество флутиказона пропионата в композиции изменяется от около 0,05 до около 18 мг при содержании лекарственного вещества от около 0,125% до 5% от массы композиции.
Фармацевтически приемлемый носитель, используемый в смесях настоящего изобретения, пригоден для адсорбции лекарственного вещества, он должен обладать свойствами превосходного носителя для сухих смесей, обеспечивая сыпучесть смеси и ее способность к обработке и предотвращая сегрегацию. Он может способствовать равномерному распределению кортикостероида. Его выбирают из группы, состоящей из микрокристаллической целлюлозы, силикатированной микрокристаллической целлюлозы, прежелатинизированного крахмала, кукурузного крахмала, коллоидного диоксида кремния, аморфного силиката алюминия магния (коммерчески доступен под названиями VEEGUM™ или NEUSILIN™). Предпочтительно используют силикатированную микрокристаллическую целлюлозу, которая состоит из тесно связанных частиц микрокристаллической целлюлозы и коллоидного диоксида кремния (PROSOLV® СМКЦ: МКЦ 98% и КДК 2%). Использование этого ингредиента в композиции изобретения улучшает сыпучесть и характеристики смешивания смеси на основе кортикостероида; улучшает однородность/гомогенность смеси и физическую устойчивость составов при хранении до заключительного изготовления готовых лекарственных форм, таких как таблетки или капсулы, т.е. также достигается устранение или уменьшение потенциального расслоения и сегрегации микрочастиц кортикостероида. Присутствие такого носителя в смеси с активным ингредиентом также обеспечивает воспроизводимость приготовления композиции изобретения (в частности для применяемой технологии прямого таблетирования). В одном варианте реализации настоящего изобретения описана смесь, содержащая низкую дозу кортикостероида и носитель, которая демонстрирует высокую однородность смеси, низкую способность к сегрегации и превосходную сыпучесть. Эта смесь, в частности, пригодна для приготовления быстро распадающейся разбавленной композиции на основе кортикостероида. В одном варианте реализации изобретения смесь содержит флутиказона пропионат, адсорбированный на силикатированной микрокристаллической целлюлозе, и быстро диспергирующиеся микрогранулы.
Скорость распадения композиций настоящего изобретения в ротовой полости индивида может составлять около 60 секунд или менее, около 50 секунд или менее, около 40 секунд или менее, около 30 секунд или менее, около 20 секунд или менее или около 10 секунд или менее.
Скорость распадения твердых фармацевтических композиций настоящего изобретения, определенная с использованием испытания на распадаемость согласно статье <701> фармакопеи США, составляет около 60 секунд или менее, около 45 секунд или менее, около 30 секунд или менее, около 20 секунд или менее или около 10 секунд или менее.
В дополнение к кортикостероиду и носителю смесь для композиций или пероральных лекарственных форм настоящего изобретения может содержать дополнительные фармацевтически приемлемые ингредиенты, которые набухают, растворяются или другим способом способствуют распадению. Такие ингредиенты могут включать разрыхлитель, альдит, сахарид или их смесь, водорастворимое полимерное связующее, полимер, образующий биогель, или биоадгезивный полимер, которые могут удерживать частицы кортикостероида, закрепленными на воспаленной ткани пищевода, дольше, чем в их отсутствие.
В одном варианте реализации в изобретении предложена твердая фармацевтическая композиция, содержащая кортикостероид и фармацевтически приемлемый полимер, образующий биогель, который обеспечивает более длительное удерживание кортикостероида на воспаленных тканях пищевода. Ингредиент, именуемый в данном документе «полимер, образующий биогель» или «биоадгезивный полимер», представляет собой агент, который способствует прикреплению кортикостероида к биологическим поверхностям, особенно к воспаленной слизистой, посредством гелеобразования в физиологических условиях желудочно-кишечного тракта, например, при контакте с физиологическими жидкостями и/или при физиологических температурах, и включает, но не ограничиваясь ими, представленные ниже полимеры, образующие биогель.
Полимер, образующий биогель, может представлять собой термочувствительный полимер. Пригодные термочувствительные полимеры включают полиакриламиды, такие как поли(N-изопропилакриламид), а также сополимеры поли(эфир-сложный эфир), такие как поли(этиленгликоль-(DL-молочная кислота-со-гликолевая кислота)-этиленгликоль). Такие термочувствительные полимеры могут частично или полностью покрывать воспаленные ткани пищевода, при этом частица(-ы) кортикостероида находится вблизи или в тесном контакте с воспаленными тканями, таким образом улучшая локальное соприкосновение кортикостероида с воспаленными тканями.
В одном варианте реализации изобретения композиция настоящего изобретения включает биоадгезивный агент, такой как липид или полимер. Примерами таких липидов являются глицерофосфолипиды, такие как фосфатидилхолин, и диацилглицеролы, такие как глицерилдиолеат. Примеры биоадгезивных полимеров включают хитозан, сложные полиортоэфиры и сополимеры, тройные сополимеры и их смеси.
В другом варианте реализации изобретения твердые фармацевтические композиции настоящего изобретения включают адгезивный агент. Пригодные адгезивные агенты включают комплекс сахарозы с сульфатом алюминия, хитозан и его производные, такие как триметилхитозан, поливинилпирролидон, метилцеллюлозу, гидроксипропилцеллюлозу, поперечно сшитые сополимеры полиакриловой кислоты, поливинилпирролидон, сополимер винилпирролидона-поливинилацетата (например, Kollidon® VA 64 производства BASF), Soluplus®, поли(этиленгликоль 6000-винилкапролактам-винилацетат) (13:57:30) (сополимер производства BASF), поливиниловый спирт, полиэтиленоксид, полиамид, альгиновую кислота и ее соли, каррагенан, ксантановую камедь, аммонийметакрилатные сополимеры, полимеры CARBOPOL, мальтодекстрины, пектины, сукралозу и их комбинации.
В некоторых вариантах реализации твердых фармацевтических композиций настоящего изобретения, кортикостероид и адгезивный агент тесно связаны друг с другом. В одном таком варианте реализации твердая фармацевтическая композиция содержит кортикостероид, который окружен адгезивным агентом или инкапсулирован в него. В другом таком варианте реализации твердая фармацевтическая композиция содержит кортикостероид, расположенный на поверхности адгезивного агента. В других вариантах реализации твердая фармацевтическая композиция содержит кортикостероид, смешанный или гранулированный с адгезивным агентом.
В некоторых вариантах реализации настоящего изобретения твердая фармацевтическая композиция включает любую твердую лекарственную форму, которая быстро распадается во рту с образованием суспензии порошкообразного кортикостероида, который, как предполагается, покрывает или прилипает к воспаленной слизистой пищевода при проглатывании.
В одном варианте реализации изобретения композиция настоящего изобретения находится в форме ПРТ. ПРТ содержит лекарственное вещество в количестве менее чем около 5% (масса лекарственного вещества/масса композиции) и фармацевтически приемлемый носитель, причем композиция не обладает значительной системной глюкокортикоидной или минералокортикоидной активностью после перорального введения людям. Частицы лекарственного вещества (например, кортикостероида, необязательно покрытые или необязательно соединенные с адгезивным агентом, как описано в данном документе) объединены с быстро диспергирующимися микрогранулами. Быстро диспергирующиеся микрогранулы содержат альдит, сахарид или их смесь и разрыхлитель или разрыхлитель в сочетании с фармацевтически приемлемыми добавками с многофункциональной активностью (например, прежелатинизированным крахмалом, гидроксипропилцеллюлозой и тому подобным).
Неограничивающий список разрыхлителей, пригодных для быстро диспергирующихся микрогранул, включает кросповидон (поперечно сшитый ПВП), гликолят крахмала натрия, поперечно сшитую натрий-карбоксиметилцеллюлозу, силикат кальция и гидроксипропилцеллюлозу с низкой степенью замещения.
Количество разрыхлителя в ПРТ, как правило, находится в интервале от около 1% до около 10% по массе.
Альдиты представляют собой гидрогенизированные формы углеводов, в которых карбонильная группа (т.е. альдегидная или кето-группа) была восстановлена до первичной или вторичной гидроксильной группы. Неограничивающие примеры пригодных альдитов для быстро диспергирующихся гранул фармацевтических композиций настоящего изобретения включают, например, арабит, изомальт, эритрит, грицерин, лактит, маннит, сорбит, ксилит, мальтит и их смеси. Термин «сахарид» является синонимом термина «сахара» и включает моносахариды, такие как глюкоза, фруктоза, лактоза и рибоза, и дисахариды, такие как сахароза, лактоза, мальтоза, трегалоза и целлобиоза. В одном варианте реализации изобретения неограничивающие примеры сахаридов, пригодных для использования в композициях настоящего изобретения, включают, например, лактозу, сахарозу, мальтозу и их смеси. В другом варианте реализации изобретения быстро диспергирующиеся гранулы содержат по меньшей мере один разрыхлитель в сочетании с альдитом. В другом варианте реализации изобретения быстро диспергирующиеся гранулы содержат по меньшей мере один разрыхлитель в сочетании с сахаридом. Еще в одном варианте реализации изобретения гранулы, содержащие разрыхлитель, состоят из по меньшей мере одного разрыхлителя в сочетании с альдитом и сахаридом.
Количество альдита и/или сахарида в быстро диспергирующихся гранулах изменяется в пределах около 99%-90% или около 95%-90% от общей массы гранул, содержащих разрыхлитель, включая все интервалы и подинтервалы между этими значениями.
Количество альдита и/или сахарида в ПРТ изменяется от около 30% до около 70% по массе.
В одном варианте реализации изобретения средний размер частиц альдита и/или сахарида составляет 30 мкм или менее, например: около 1-30 мкм, около 5-30 мкм, около 5-25 мкм, около 5-20 мкм, около 5-15 мкм, около 5-10 мкм, около 10-30 мкм, около 10-25 мкм, около 10-20 мкм, около 10-15 мкм, около 15-30 мкм, около 15-25 мкм, около 15-20 мкм, около 20-30 мкм, около 20-25 мкм или около 25-30 мкм.
Отношение разрыхлителя к альдиту, сахариду или их смеси в быстро диспергирующихся микрогранулах изменяется от около 90/10 до около 99/01, например: около 90/10, около 91/9, около 92/8, около 93/7, около 94/6, около 95/5, около 96/4, около 97/3, около 98/2, около 99/1, включая все значения, интервалы и подинтервалы между этими значениями.
Частицы кортикостероида, как правило, адсорбируются на носителе. Процесс получения включает многократное перемешивание кортикостероида и носителя, чтобы смесь адсорбировалась на носителе. Кортикостероид, как правило, тонко измельчен (средний размер частиц менее 10 мкм) по следующим причинам. Во первых, предполагается, что готовая лекарственная форма (такая как ПРТ) будет быстро распадаться при контакте со слюной в ротовой полости. Для достижения этого лекарственная форма (ПРТ) предпочтительно должна содержать как минимум 100 мг быстро диспергирующихся микрогранул, вне зависимости от дозы кортикостероида (например, 0,1 мг, 1 мг, 10 мг или 20 мг). Во вторых, для достижения однородности/гомогенности смеси и однородности состава готовых лекарственных форм, гомогенное распределение может быть достигнуто путем включения тонкоизмельченных частиц лекарственного вещества в силикатированную микрокристаллическую целлюлозу отдельно или в сочетании с быстро диспергирующимися микрогранулами путем по меньшей мере однократного смешивания или размалывания, как описано в примерах различных вариантов реализации настоящего изобретения. Первый вариант включения лекарственного вещества в силикатированную микрокристаллическую целлюлозу будет в значительной степени препятствовать сегрегации микрочастиц кортикостероида при временном хранении до заключительного изготовления готовых лекарственных форм, капсул или таблеток, демонстрирующих высокую однородность состава и/или небольшие различия при растворении.
Быстро диспергирующиеся гранулы или гранулят могут быть получены, как описано в патентах США 2005/0232988 или 2003/0215500, путем грануляции разрыхлителя с альдитом и/или сахаридом со средним размером частиц не более чем около 30 мкм. Грануляцию можно провести, например, в грануляторе с большим усилием сдвига с примерно 20-25% воды в качестве гранулирующей жидкости с использованием, если необходимо, влажного помола и высушивания, чтобы получить быстро диспергирующиеся микрогранулы со средним размером частиц не более чем около 300 мкм (например, около 175-300 мкм). В альтернативном варианте быстро диспергирующиеся микрогранулы можно получить, как описано в патенте США 13/310632, путем грануляции альдита, сахарида или их смеси и разрыхлителя в сочетании с фармацевтически приемлемой добавкой с многофункциональной активностью (например, с крахмалом, гидроксипропилцеллюлозой и тому подобным) на низком уровне 0,5-3% по массе в грануляторе с кипящим слоем.
Быстро диспергирующиеся микрогранулы, присутствующие в ПРТ, способствуют быстрому распадению таблетки, когда ее помещают в ротовую полость, создавая однородную суспензию, содержащую частицы кортикостероида. Желательно включать достаточное количество быстро диспергирующихся микрогранул, чтобы тщательно покрыть слизистую пищевода. Это создает проблему однородности состава для таких низко дозированных ПРТ (например, ПРТ массой 300 мг, содержащие 12 мг или менее кортикостероида). Как правило, эту проблему решают путем грануляции, которая включает распыление разбавленного раствора кортикостероида на слой порошка вспомогательных веществ. Частицы лекарственного вещества встраиваются в гранулы, а, следовательно, могут быть недоступными для воспаленной слизистой, что приводит к плохой эффективности. Неожиданно было замечено, что возможно не только достичь желаемой однородности состава, но также увеличить вероятность того, что частицы кортикостероида будут доступными для воспаленной слизистой, путем адсорбции тонкоизмельченных частиц местно действующего кортикостероида на фармацевтически приемлемом носителе (таком как силикатированная микрокристаллическая целлюлоза) до смешивания с быстро диспергирующимеся микрогранулами и другими вспомогательными веществами и прессования ПРТ.
Лекарственная форма, описанная в данном документе, может также содержать фармацевтически приемлемые вспомогательные вещества, как правило, используемые в составах для распадающихся таблеток, такие как наполнители, разбавители, вещества, способствующие скольжению, разрыхлители, связующие вещества и смазывающие вещества.
Примеры пригодных наполнителей, разбавителей и/или связующих веществ включают лактозу (например, лактозу, высушенную распылением, такую как FAST-FLO®), микрокристаллическую целлюлозу (различные марки Avicel®, CEOLUS®), гидроксипропилцеллюлозу, L-гидроксипропилцеллюлозу (с малой степенью замещения), низкомолекулярную гидроксипропилметилцеллюлозу (ГПМЦ) (например, Methocel™ E, F и K производства Dow Chemical, MethloloseE SH производства Shin-Etsu, Ltd), гидроксиэтилцеллюлозу, натрий-карбоксиметилцеллюлозу, карбоксиметилгидроксиэтилцеллюлозу и другие производные целлюлозы, сахарозу, агарозу, сорбит, маннит, декстрины, мальтодекстрины, крахмалы или модифицированные крахмалы (в том числе картофельный крахмал, кукурузный крахмал и рисовый крахмал), фосфат кальция (например, основной фосфат кальция, гидроортофосфат кальция, дикальцийфосфат гидрат), сульфат кальция, карбонат кальция, альгинат натрия и коллаген. Предпочтительный наполнитель для композиции изобретения представляет собой маннит, такой так высушенный распылением маннит.
Примеры пригодных разрыхлителей включают кросповидон (поперечно сшитый ПВП), гликолят крахмала натрия, поперечно сшитую натрий-карбоксиметилцеллюлозу, силикат кальция и гидроксипропилцеллюлозу с низкой степенью замещения. Предпочтительный разрыхлитель для композиции изобретения представляет собой кросповидон.
Конкретные примеры веществ, способствующих скольжению, и смазывающих веществ включают стеариновую кислоту, стеарат магния, стеарат кальция или стеараты других металлов, тальк, глицерилбегенат, коллоидный диоксид кремния, кукурузный крахмал и необязательно стеарат магния или стеарилфумарат натрия (смазывающее вещество, которое смешивается внутригранулярно или используется внешне для смазывания кристаллических или неровных поверхностей). Предпочтительное вещество, способствующее скольжению, для композиции изобретения представляет собой коллоидный диоксид кремния, а предпочтительное смазывающее вещество представляет собой стеарилфумарат натрия.
Твердые фармацевтические композиции настоящего изобретения могут включать помимо ПРТ другие лекарственные формы: капсулу, пленку или другую твердую лекарственную форму, которая быстро распадается во рту с образованием суспензии или дисперсии кортикостероида, и которую легко можно проглотить, чтобы покрыть поверхность слизистой, пораженную эозинофильным эзофагитом.
Например, капсулы могут включать высушенные или лиофилизированные композиции, такие как перорально распадающиеся или растворяющиеся лекарственные формы, полученные с использованием технологии лиофилизации Zydis® (например, как описано в патенте США №6316027), содержащие кортикостероид в качестве активного фармацевтического ингредиента. Пленочные лекарственные формы могут включать съедобные пленки, как, например, представленные в патенте США №6596298 или патенте США №6740332, содержащие кортикостероид в качестве активного фармацевтического ингредиента. В одном варианте реализации настоящего изобретения твердые композиции содержат лиофилизированную матрицу, причем лиофилизированная матрица содержит кортикостероид, носитель и вспомогательное вещество. Пригодные вспомогательные вещества включают маннит, ксилит, мальтол, мальтит, лактозу, сахарозу, мальтозу и их комбинации.
Местное введение кортикостероида в ротовую полость индивидов связано с кандидозом. Хотя изобретение разработано так, чтобы как можно меньше способствовать возникновению такой инфекции, однако, в другом варианте реализации изобретения фармацевтическая композиция может содержать противогрибковый агент. Пригодные противогрибковые агенты включают, но не ограничиваясь ими, ингибиторы митоза, производные пиримидина, полиены, бензимидазолы, имидазолы, полиены, триазолы, тиазолы, аллиламины, эхинокандины и другие «неклассифицированные» противогрибковые агенты, известные в данной области техники, которые не попадают в какую-либо из вышеприведенных категорий (например, толнафлат и циклопирокс). Например, пригодные противогрибковые агенты, которые могут входить в состав твердых фармацевтических композиций настоящего изобретения, включают абафунгин, аморолфин, анидулафунгин, бифоназол, бутенафин, бутоконазол, кандицин, каспофунгин, циклопирокс, клотримазол, эконазол, фентиконазол, филипин, флуконазол, флуцитозин, гризеофульвин, изавуконазол, изоконазол, итраконазол, кетоконазол, микафунгин, миконазол, миконазола нитрат, нафтифин, натамицин, нистатин, оксиконазол, посаконазол, прамиконазол, равуконазол, римоцидин, сертаконазол, сулконазол, тербафин, терконазол, тиоконазол, толнафтат, ундециленовая кислота и вориконазол.
В другом варианте реализации изобретения фармацевтические композиции настоящего изобретения включают противовирусный агент. Противовирусные агенты, которые могут входить в состав твердых фармацевтических композиций настоящего изобретения, включают интерфероны, нуклеозидные и нуклеотидные ингибиторы обратной транскриптазы, ненуклеозидные ингибиторы обратной транскриптазы, ингибиторы протеазы, ингибиторы интегразы, ингибиторы слияния, ингибиторы мейоза, производные гуанозина, производные пиридина, производные пиримидина и другие «неклассифицированные» противовирусные лекарственные препараты, известные в данной области техники, которые не попадают в какую-либо из вышеприведенных групп (например, фоскарнет и милтефозин). Например, пригодные противовирусные агенты, которые могут входить в состав твердых фармацевтических композиций настоящего изобретения, включают абакавир, ацикловир, адефовир, амантадин, амдоксовир, ампренавир, аплавирок, априцитабин, арбидол, атазанавир, бевиримат, BMS-488043, боцепревир, бривудин, цидофовир, DCM205, докозанол, делавирдин, диданозин, дарунавир, эфавиренз, элвитегравир, элвуцитабин, эмтрицитабин, энфувиртид, эпигаллокатехина галлат, этравирин, фамцикловир, фосампренавир, ганцикловир, глобоиднан А, гриффитсин, ибализумаб, идоксуридин, индинавир, ламивудин, лопинавир, ловирид, маравирок, нелфинавир, невирапин, озельтамивир, пегилированный интерферон альфа-2а, пегилированный интерферон альфа-2b, пенцикловир, перамивир, плериксафор, PRO 140, рацивир, ралтегравир, ритонавир, рибавирин, римантадин, рилпивирин, саквинавир, стампидин, ставудин, тенофовир, типранавир, TNX-355, трифлуридин, тромантадин, валацикловир, валганцикловир, викривирок, видарабин, вирамидин, вивекон, залцитабин, занамивир и зидовудин.
Таблеточные лекарственные формы, включая лекарственные формы ПРТ, содержащие низкую дозировку местно действующего кортикостероида и фармацевтически приемлемый носитель, причем количество лекарственного вещества составляет менее чем около 5% (масса лекарственного вещества/масса композиции), не обладают значительной системной глюкокортикоидной или минералокортикоидной активностью после перорального введения людям, распадаются за менее чем около 30 с (методика фармакопеи США) и обладают низкой истираемостью, что дает достаточную устойчивость при обработке, транспортировке и/или упаковке в блистеры с выдавливаемым защитным слоем. Истираемость составляет менее чем около 1%, например: менее чем около 0,9%, менее чем около 0,8%, менее чем около 0,7%, менее чем около 0,6%, менее чем около 0,5%, менее чем около 0,4%, менее чем около 0,3% и т.д., включая все интервалы и подинтервалы между этими значениями.
Для получения смесей кортикостероида с пригодным носителем, характеризующихся гомогенностью, т.е. приемлемой однородностью, а также пригодной для изготовления таблеток однородностью состава можно применять различные способы. Смесь вышеупомянутых ингредиентов можно получить как с помощью сухого смешивания, так и с помощью грануляции.
Настоящее изобретение дополнительно описывает способ изготовления пероральной композиции, такой как смесь для прессования с содержанием лекарственного вещества от около 0,5% до около 3% по массе, причем сложность представляет достижение и поддержание приемлемого уровня однородности смеси до превращения смеси для прессования в готовые дозированные лекарственные форма (например, перорально распадающиеся таблетки), демонстрирующие приемлемую однородность состава. Способ состоит из следующих стадий:
1) приготовление быстро диспергирующихся микрогранул или гранулята;
2) приготовление промежуточной смеси 1, включающее загрузку в V-образный смеситель четвертой части силикатированной микрокристаллической целлюлозы (СМКЦ, фармацевтический приемлемый носитель), тонкоизмельченного кортикостероида, коллоидного диоксида кремния (вещество, способствующее скольжению) и еще четвертой части СМКЦ и перемешивание содержимого в течение 10 минут;
3) приготовление промежуточной смеси 2, включающее загрузку в гранулятор с большим усилием сдвига сыпучего наполнителя (такого как маннит, высушенный распылением), промежуточной смеси 1, оставшегося количества СМКЦ, разрыхлителя (такого как кросповидон) и подсластителя (порошок сукралозы) и перемешивание содержимого в течение 10 минут при скорости мешалки 300±50 об/мин и скорости ножа 1500±50 об/мин;
4) приготовление целевой смеси для прессования, включающее загрузку в V-образный смеситель половины быстро диспергирующихся гранул, полученных на стадии 1, смазывающего вещества (такого как стеарилфумарат натрия), промежуточной смеси 2, полученной на стадии 3, и оставшейся половины быстро диспергирующихся гранул, полученных на стадии 1, и перемешивание в течение 30 минут, отбор пробы и дополнительное перемешивание в течение 10±1 минут, чтобы получить приемлемую однородность/гомогенность смеси в соответствии с нормативными требованиями;
5) приготовление перорально распадающихся таблеток, включающее смесь для прессования, полученную на стадии 4, которая демонстрирует приемлемую однородность состава в соответствии с нормативными требованиями.
В одном варианте реализации изобретения способ изготовления перорально распадающихся таблеток заключается в многократном сухом смешивании и размалывании. Способ включает следующие стадии:
1) приготовление быстро диспергирующихся микрогранул со средним размером частиц не более чем около 400 мкм путем грануляции одного или более альдитов и/или сахаридов, при этом средний диаметр частиц каждого составляет не более чем около 30 мкм, с разрыхлителем (таким как кросповидон) в присутствии воды или смеси спирт-вода, а затем высушивания гранулята (сушилка с кипящим слоем или традиционная печь);
2) приготовление размолотой промежуточной смеси 1 путем перемешивания фармацевтически приемлемого носителя (такого как силикатированная микрокристаллическая целлюлоза), тонкоизмельченного кортикостероида и вещества, способствующего скольжению (такого как коллоидный диоксид кремния), в V-образном смесителе в течение 10 минут при 25±1 об/мин, а затем размалывания на мельнице тонкого помола, оснащенной ситом 024R (30 меш) при примерно 2400±100 об/мин;
3) приготовление размолотой промежуточной смеси 2 путем смешивания половины сыпучего маннита, промежуточной смеси 1, полученной на стадии 2, разрыхлителя (кросповидон) и подсластителя (порошок сукралозы) в V-образном смесителе в течение 10 минут при 25±1 об/мин, а затем размалывания на мельнице тонкого помола, оснащенной ситом 024R при скорости примерно 2400±100 об/мин, и промывания мельницы оставшейся половиной сыпучего маннита;
4) приготовление смеси для прессования путем смешивания быстро диспергирующихся гранул, полученных на стадии 1, смазывающего вещества (такого как стеарилфумарат натрия), размолотой промежуточной смеси 2, полученной на стадии 3, и сыпучего маннита для промывки в течение в общей сложности 40 минут;
5) приготовление таблеток путем прессования смеси, полученной на стадии 4.
Способ многократного сухого смешивания и помола является предпочтительным для приготовления композиций изобретения.
В способе изобретения различные стадии и порядок добавления индивидуальных компонентов являются важными для достижения приемлемой однородности/гомогенности смеси для прессования, а также приемлемой однородности состава готовых единиц дозирования в соответствии с нормативными требованиями. Таблетки, полученные с помощью вышеупомянутого способа, характеризуются внешним видом, временем распадения, твердостью и истираемостью, подходящими для того, чтобы ПРТ выдерживали транспортировку в контейнерах для насыпных грузов, промышленную упаковку в блистеры или флаконы и транспортировку первично/вторично упакованной продукции для продажи и конечное применение согласно цели изобретения. Кроме того, изготовленные и затем упакованные в блистеры таблетки являются высокостабильными в условиях испытаний стабильности «методом ускоренного старения» и в условиях долгосрочных испытаний в соответствии с требованиями Международной конференции по гармонизации (ICH).
Твердые фармацевтические композиции настоящего изобретения пригодны для перорального введения местно действующего кортикостероида для лечения воспаления тканей верхних отделов желудочно-кишечного тракта, например, пищевода. Для лечения патологических состояний, связанных с воспалением желудочно-кишечного тракта, желательным является использование местно действующего кортикостероида, так как при этом возникает меньше побочных эффектов по сравнению кортикостироидом, имеющим высокую системную активность.
Воспалительные патологические состояния желудочно-кишечного тракта, которые можно лечить согласно настоящему изобретению, включают воспаление пищевода, воспаление голосовой щели, воспаление надгортанника, воспаление миндалин, воспаление ротовой части глотки, эозинофильный эзофагит, гастроэзофагеальную рефлюксную болезнь (ГЭРБ), неэрозивную рефлюксную болезнь (НЭРБ), эрозивный эзофагит, пищевод Барретта, эозинофильный гастроэнтерит, гиперэозинофильный синдром, коррозийный (щелочной) химический эзофагит, эзофагит, вызванный облучением, эзофагит, вызванный химиотерапией, временный эзофагит, вызванный лекарственными препаратами (также известный как медикаментозный эзофагит), постоянный эзофагит, вызванный лекарственными препаратами, болезнь Крона пищевода и псевдомембранозный эзофагит.
В одном конкретном варианте реализации изобретения фармацевтические композиции настоящего изобретения пригодны для лечения воспалительных патологических состояний верхних отделов желудочно-кишечного тракта, в частности эозинофильного эзофагита.
Таким образом, настоящее изобретение включает фармацевтическую композицию, используемую в качестве лекарственных препаратов при лечении воспалительных патологических состояний желудочно-кишечного тракта.
Изобретение также включает способ введения твердой фармацевтической композиции настоящего изобретения пациенту, который в этом нуждается. В одном варианте реализации настоящее изобретение включает способ лечения эозинофильного эзофагита, включающий введение фармацевтической композиции настоящего изобретения пациенту, который в этом нуждается. При введении твердой фармацевтической композиции настоящего изобретения индивиду, композиция распадается в ротовой полости пациента. В другом варианте реализации настоящее изобретение включает способ лечения гастроэзофагеальной рефлюксной болезни (ГЭРБ), неэрозивной рефлюксной болезни (НЭРБ) или эрозивного эзофагита, включающий введение фармацевтической композиции настоящего изобретения индивиду, который в этом нуждается. В другом варианте реализации настоящее изобретение включает способ лечения пищевой аллергии с установленным аллергеном, например «аллергического СРК» и «аллергических реакций кишечника».
Из вышеприведенного описания и экспериментальной части видно, что настоящее изобретение обладает несколькими важными преимуществами. В описанном изобретении предложены низкодозированные пероральные композиции, содержащие кортикостероид и фармацевтический носитель, которые характеризуются высокой однородностью состава и стабильностью композиции и лекарственных форм, причем микрочастицы кортикостероида в основном присутствуют возле или вблизи поверхности носителя, и поэтому вероятно будут иметь подходящее расположение для местного лечения воспаления желудочно-кишечного тракта, в частности ЭоЭ, после распадения ПРТ, содержащей кортикостероид, в ротовой полости пациента и проглатывания полученной вязкой суспензии.
ЭКСПЕРИМЕНТЫ
МЕТОДИКИ
Объемную/насыпную плотность определяли согласно методике 1 статьи <616> фармакопеи США.
Распределение частиц по размерам (РЧР) определяли на образцах массой 5-10 мг с использованием звукового просеивателя ATM.
Сыпучесть определяли с использованием прибора для определения сыпучести Sotax, используя около 110 г вещества и применяя стандартные шесть предварительных/вибрационных режимов; свойство сыпучести выражали в виде индекса сыпучести (α'/αст) и индекса Kappa.
Содержание воды определяли, используя титрование по Карлу Фишеру, или потери при высушивании определяли согласно методике 1а статьи <921> фармакопеи США.
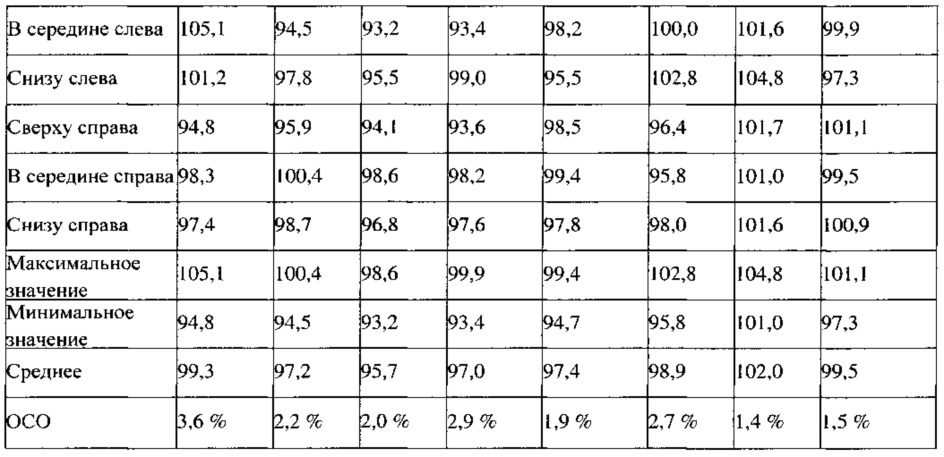
Определение однородности смеси. Определение однородности смеси проводили путем отбора шести произвольных проб с использованием пробоотборника из различных мест в целевой смеси для прямого прессования, содержащейся в V-образном смесителе, как показано на фиг. 1. Отобранные образцы анализировали на содержание в них лекарственного вещества с использованием проявляющей стабильность методики ВЭЖХ.
Однородность состава ПРТ. Перорально распадающиеся таблетки произвольно отбирали в начале, середине и конце каждого цикла прессования, для 10 таблеток определяли однородность состава, используя проявляющую стабильность методику ВЭЖХ.
Испытание на распадаемостъ выполняли в соответствии с методикой <701> фармакопеи США. Истираемость определяли в соответствии с методикой <1216> фармакопеи США.
ПРИМЕРЫ
Пример 1. Быстро диспергирующиеся микрогранулы
Быстро диспергирующиеся микрогранулы получают согласно процедуре, описанной в опубликованной заявке на патент США №2003/0215500, изданной 20 ноября 2003 г., содержание которой включено в данный документ в полном объеме для любых целей посредством ссылки. Более конкретно, D-маннит (152 кг) со средним размером частиц примерно 20 мкм или менее (PEARLITOL® 25 производства Roquette, Франция) смешивают с 8 кг поперечно сшитого повидона (Crospovidone® XL-10 производства ISP) в грануляторе с большим усилием сдвига (GMX 600 производства Vector), гранулируют с очищенной водой (примерно 32 кг), размалывают с использованием влажного помола на мельнице Comil производства Quadro и в конце высушивают на лотках, чтобы получить микрогранулы, которые характеризуются потерями при высушивании менее чем около 1,0%. В альтернативном варианте гранулы после влажного помола сушат в сушилке с кипящим слоем, чтобы получить микрогранулы, которые характеризуются потерями при высушивании менее чем около 1,0% по массе. Высушенные гранулы просеивают, и материал с превышенным размером снова размалывают, чтобы получить быстро диспергирующиеся микрогранулы со средним размером частиц в интервале примерно 175-300 мкм.
D-маннит со средним размером частиц <20 мкм (93 части) и кросповидон (5 частей) гранулируют путем распыления раствора крахмала (2 части крахмала (Starch 1500®) производства Colorcon) в грануляторе с кипящим слоем с распылителем, расположенным сверху, и высушивают так, чтобы потери при высушивании составляли <1,0%. Высушенные гранулы просеивают через сито 20 меш, а гранулы с превышенным размером снова размалывают и просеивают, если необходимо получить дополнительное количество быстро диспергирующихся гранул.
Пример 2. Приготовление смеси для ПРТ, содержащих 1,5 мг флутиказона, с использованием гранулятора с большим усилием сдвига; партия 1
Вначале получают промежуточную смесь 1 путем смешивания четвертой части силикатированной микрокристаллической целлюлозы (СМКЦ, коммерчески доступна под названием PROSOLV® HD90), тонкоизмельченного флутиказона пропионата, коллоидного оксида кремния и еще четвертой части СМКЦ в V-образном смесителе вместимостью 1 кварта в течение 10±1 минут (см. в Таблице 1 массы отдельных компонентов в смесях для прессования ПРТ, 1,5 и 3 мг). Получают вторую промежуточную смесь (промежуточная смесь 2): в емкость для гранулирования вместимостью 10 л гранулятора с большим усилием сдвига PMA 1 загружают маннит, высушенный распылением (PARTECK® М200), промежуточную смесь 1, оставшуюся часть СМКЦ, кросповидон и порошок сукралозы. Перемешивание выполняют в течение 10±1 минут при скорости мешалки 300±50 об/мин и скорости ножа 1500±50 об/мин, чтобы получить промежуточную смесь 2. Половину быстро диспергирующихся микрогранул, стеарилфумарат натрия, промежуточную смесь 2 и оставшуюся часть быстро диспергирующихся микрогранул перемешивают в V-образном смесителе вместимостью 8 кварт, через 30 и 40 минут отбирают образцы для проверки однородности смеси. Для целевой смеси определяли объемную/насыпную плотность, распределение частиц по размерам (РЧР), сыпучесть и содержание влаги.

Пример 3. Приготовление смесей для ПРТ, содержащих 1,5 и 3 мг флутиказона, путем многократного смешивания и размалывания; партии 2, 3, 5
Промежуточную смесь 1 (см. в Таблице 1 массы отдельных компонентов в смесях для прессования ПРТ, 1,5 и 3 мг) получают, последовательно загружая в V-образный смеситель вместимостью 2 кварты половину СМКЦ, тонкоизмельченный флутиказона пропионат, коллоидный диоксид кремния и оставшуюся половину СМКЦ и перемешивая в течение 10±1 минут. Промежуточную смесь 1 пропускают через устройство QUADRO Comil, оснащенное ситом 024R (30 меш) при примерно 2400±100 об/мин. Промежуточную смесь 2 получают следующим образом: половину высушенного распылением маннита, размолотую промежуточную смесь 1, кросповидон и порошок сукралозы смешивают в V-образном блендере вместимостью 4 кварты в течение 10±1 минут и размалывают через сито 024R. Устройство Comil промывают, пропуская оставшуюся половину высушенного распылением маннита через сито 024R. Половину быстро диспергирующихся микрогранул, стеарилфумарат натрия, размолотую промежуточную смесь 1, маннит для промывки и оставшуюся часть быстро диспергирующихся микрогранул смешивают, через 30 и 40 минут отбирают образцы для проверки однородности смеси. Для целевых смесей определяют объемную/насыпную плотность, распределение частиц по размерам (РЧР), сыпучесть и содержание влаги.
Пример 4. Результаты определения однородности смесей, полученных в примерах 2 и 3
Результаты испытаний смесей для прессования, полученных в примерах 2 и 3, представлены в Таблице 2. Смеси для прессования ПРТ 1,5 мг (партия 1) и ПРТ 1,5 мг (партия 2), которые были получены с использованием двух различных комбинаций оборудования: V-образный смеситель-гранулятор с большим усилием сдвига и V-образный смеситель-мельница тонкого помола, демонстрируют подобные физические свойства смеси (порошка), такие как объемная и насыпная плотности, распределения частиц по размерам, свойства сыпучести и величины однородности смеси, за исключением того, что партия, которую перемешивали 30 минут, демонстрирует немного более высокое ОСО, %.



Пример 5. Прессование ПРТ из примеров 2 и 3
Смеси для прессования, полученные в примерах 2 и 3, прессуют, используя роторный таблеточный пресс Manesty Beta Press, оснащенный 8 комплектами штампа (пуансон и матрица) с круглой ровной плоской радиусной кромкой (9,5 мм). Средняя масса таблетки составляет около 300 мг. Для циклов прессования прилагаемая сила основного прессования поддерживается на уровне 5-6 кН, при этом силу предварительного прессования устанавливают равной 2±0,2 кН (за исключение случаев, где указано другое). Во время прессования используют контрольно-измерительное оборудование для таблеточных прессов производства SMI для измерения скорости прессования и силы прессования. В процессе таблетирования периодически отбирают таблетки для визуального осмотра «внешнего вида», определяют массу, толщину, твердость и истираемость. Дополнительные таблетки также служат «составной пробой» для аналитического контроля. Подробно параметры прессования и свойства таблеток приведены в Таблице 4. Для таблеток, а именно круглых таблеток, проводят количественный анализ, определяют содержание действующего вещества, однородность состава, растворимость (высвобождается более 90% в течение 45 минут для всех партий); истираемость для них составляет не более 0,4%, твердость - около 4 килопонд, время распадения - менее 30 с.
Пример 6. Проверочные смеси для прессования и партии ПРТ (ПРТ 1,5 мг: партия 3, 3 мг: партия 5)
В случае смесей для прессования ПРТ, содержащих 1,5 и 3,0 мг флутиказона, различные партии смесей получают путем двухкратного смешивания и размалывания (повторяющийся процесс смешивания в V-образном смесителе в сочетании с помолом на мельнице Comil) (см. композиции в Таблице 1, результаты определения физических свойств/однородности смеси и однородности смеси в зависимости от места пробоотбора в Таблицах 2 и 3, соответственно). Хотя величины объемной и насыпной плотности, распределения частиц по размерам, величины однородности смеси для поверочных смесей для прессования подобны соответствующим характеристикам партии смеси для прессования ПРТ с дозировкой 1,5 мг (партия 2), вычисленные свойства сыпучести как для ПРТ с дозировкой 1,5 мг (партия 3), так и для ПРТ с дозировкой 3 мг (партия 5) не являются очень хорошими. Смеси для прессования обеих партий прессуют с использованием одного и того же оборудования Beta Press, оснащенного одним и тем же набором инструментов, при сравнимых параметрах прессования. В ходе прессования обеих смесей наблюдается некоторые дефекты поверхности и/или прилипание к пуансонам. Подробно параметры прессования и свойства таблеток приведены в Таблице 4.
Пример 7. Получение смесей для ПРТ, содержащих 1,5 и 3 мг флутиказона, путем многократного смешивания и помола, содержание смазывающего вещества - 1,5% по массе, партии 4 и 6
Для получения партий смесей для прессования, содержащих флутиказон, вначале готовят промежуточную смесь 1 и промежуточную смесь 2, как описано в примере 3, затем перемешивают компоненты целевой смеси без включения смазывающего вещества в течение 35 минут и дополнительно перемешивают в течение 5 минут после добавления смазывающего вещества (стеарилфумарат натрия, 1,5% по массе). Для целевой смеси определяют объемную и насыпную плотность, распределение частиц по размерам (РЧР), сыпучесть, содержание влаги, однородность смеси и однородность смеси в зависимости от места пробоотбора для обеих партий. Результаты представлены в Таблицах 2 и 3.
Пример 8. Прессование ПРТ из примера 7
Обе партии прессуют с использованием одного и того же роторного таблеточного пресса, одного и того же комплекта инструментов и при одинаковых условиях прессования, как описано выше.

Пример 9. Исследование стабильности
Партии №2 (1,5 мг) и №5 (3 мг) ПРТ, содержащих флутиказон, упаковывают во флаконы из ПВП вместимостью 30 см (30 таблеток/флакон) с вискозным жгутом и пакетом с влагопоглотителем массой 0,5 г (Sorb-it, 1/2 г пакет). Все ПРТ стабильны в условиях испытаний методом «ускоренного старения» (40°С /относительная влажность 75%) в течение 6 месяцев, а также в условиях долгосрочных испытаний (25°С / относительная влажность 60%) в течение 9 месяцев, что показано в Таблицах 5 и 6. Физические свойства, такие как внешний вид, твердость, истираемость и время распадения, для всех условий испытания стабильности также сопоставимы с исходными величинами для ПРТ, содержащих 1,5 и 3 мг флутиказона.




Пример 10. Приготовление материалов для клинических исследований, партии С1, С2
Партии для клинических исследований (ПРТ, содержащие флутиказона пропионат, 1,5 и 3 мг с 1,5% по массе стеарилфумарата натрия) получают с помощью процесса многократного смешивания - помола - смешивания с последующим прессованием, как описано в примерах 7 и 8. Результаты испытаний в ходе технологического процесса и результаты анализа партий для клинических исследований представлены в Таблицах 7 и 8, соответственно. Партии смесей для прессования демонстрируют физические (порошковые) свойства, подобные свойствам партий, на которых показана техническая применимость, за исключением того, что в партиях для клинических исследований больше более мелких частиц, которые проходят через сито 100 меш (размер 67-76% частиц <150 мкм), по сравнению с 47-57% более мелких частиц в партиях, на которых показана техническая применимость. Однако это не влияло значительно на характеристики таблетирования партий для клинических исследований: в партии С-2 (3 мг) наблюдаются немного более высокие значения ОСО (%) для однородности состава и превосходные результаты для однородности смеси.




Пример 11. Данные о стабильности партий для клинических исследований, ПРТ, содержащие флутиказон, 1,5 и 3 мг
Партии ПРТ, содержащих 1,5 мг и 3 мг флутиказона, упаковывают во флаконы из ПВП вместимостью 30 см (30 таблеток/флакон) с вискозным жгутом и пакетом с влагопоглотителем массой 0,5 г (Sorb-it, 1/2 г пакет). Все ПРТ стабильны в условиях испытаний методом «ускоренного старения» (40°С / относительная влажность 75%) в течение 9 месяцев, а также в условиях долгосрочных испытаний (25°С / относительная влажность 60%) в течение 24 месяцев, что показано в Таблице 9. Физические свойства, такие как внешний вид, твердость, истираемость и время распадения для всех условий испытания стабильности также сравнимы с исходными значениями для ПРТ, содержащих 1,5 и 3 мг флутиказона.


Пример 12. Клинические исследования
Исследование для проверки концепции проводят для партий предназначенных для клинических исследований ПРТ, содержащих флутиказон, с дозировками 1,5 мг и 3 мг с участием пациентов в возрасте от 12 лет до 55 лет с диагнозом ЭоЭ.
Используемые дозы составляют 1,5 мг два раза в день и 3,0 мг один раз в день. Исследование также включало группу пациентов, получающих плацебо. Каждая группа включала 8 субъектов. Данные анализа эффективности демонстрируют положительный сигнал, при этом наивысший отклик проявляется гистологически в уменьшении максимального количества эозинофильных клеток, наблюдаемых в поле зрения микроскопа под большим увеличением (отличительная черта заболевания и индикатор отклика на лечение). Для обеих групп (1,5 мг и 3 мг), получающих лечение, очевидна большая эффективность, определенная гистологически, по сравнению с группой, получающей плацебо. Доля субъектов, для которых наблюдается по меньшей мере 30% уменьшение общей тяжести симптомов ЭоЭ, что определено путем опроса пациентов, также больше для двух типов ПРТ, содержащих флутиказон, по сравнению с плацебо. Также наблюдаются эндоскопические улучшения с изменениями бороздок и кровеносных сосудов, что демонстрирует значительное отличие ПРТ, содержащих флутиказон, от плацебо и указывает на противовоспалительный эффект препаратов.
В общем, содержащие флутиказон ПРТ по данному изобретению демонстрируют улучшения гистологии, ослабление общих симптомов и общей эндоскопической активности.
Пример 13. Приготовление смесей для ПРТ, содержащих 0,75, 4,5 и 6 мг флутиказона; партии 7-9
Для получения партий смесей для прессования, содержащих 0,25% по массе флутиказона, вначале готовят промежуточную смесь 1 и промежуточную смесь 2, как описано в примере 3. Промежуточную смесь 1 (см. в Таблице 10 массы отдельных компонентов в смесях для прессования ПРТ, 0,75 мг, 4,5 мг и 6 мг) получают, последовательно загружая в V-образный смеситель вместимостью 2 кварты половину СМКЦ, тонкоизмельченный флутиказона пропионат, коллоидный диоксид кремния и оставшуюся половину СМКЦ и перемешивая при 25 об/мин в течение 10±1 минут. Промежуточную смесь 1 пропускают через устройство QUADRO Comil, оснащенное ситом 024R (30 меш) при примерно 2400±100 об/мин. Промежуточную смесь 2 получают следующим образом: половину высушенного распылением маннита, размолотую промежуточную смесь 1, кросповидон и порошок сукралозы смешивают в V-образном смесителе вместимостью 32 кварты при 25 об/мин в течение 10±1 минут и размалывают через сито 024R. Мельницу Comil промывают, пропуская оставшуюся часть высушенного распылением маннита через сито 024R. Половину быстро диспергирующихся микрогранул, размолотую промежуточную смесь 1, маннит для промывки и оставшуюся часть быстро диспергирующихся микрогранул смешивают в V-образном смесителе вместимостью 32 кварты при 25 об/мин без включения смазывающего вещества в течение 35 минут и дополнительно в течение 5 минут после добавления смазывающего вещества (стеарилфумарат натрия, 1,5% по массе).
Пример 14. Приготовление смесей для ПРТ, содержащих 1,5 мг флутиказона, объем партии: 30 кг; партия 10
Процесс изготовления партии смеси для прессования с более низким содержанием флутиказона 0,5% по массе осуществлен в полупромышленном масштабе (30 кг). Вначале процесс по существу включает приготовление промежуточной смеси 1 и промежуточной смеси 2, как описано в примере 3. Промежуточную смесь 1 (см. в Таблице 10 массы индивидуальных компонентов в смесях для прессования ПРТ, 1,5 мг) готовят, последовательно загружая V-образный смеситель вместимостью 32 кварты половину СМКЦ, тонкоизмельченный флутиказона пропионат, коллоидный диоксид кремния и оставшуюся часть СМКЦ и перемешивая при 25 об/мин в течение 10±1 минут. Промежуточную смесь 1 пропускают через устройство QUADRO Comil, оснащенное ситом 024R (30 меш) при примерно 2400±100 об/мин. Промежуточную смесь 2 получают следующим образом: половину высушенного распылением маннита, размолотую промежуточную смесь 1, кросповидон и порошок сукралозы смешивают в смесителе Galley с контейнером вместимостью 113 л при 12 об/мин в течение 20±1 минут и размалывают через сито 024R. Мельницу Comil промывают, пропуская оставшуюся часть высушенного распылением маннита через сито 024R. Половину быстро диспергирующихся микрогранул, размолотую промежуточную смесь 1, маннит для промывки и оставшуюся часть быстро диспергирующихся микрогранул смешивают при 12 об/мин в течение не более 40 минут.
В ходе технологического процесса отбирают пробы целевых смесей, полученных в примерах 13 и 14, которые анализируют в соответствии с требованиями фармакопеи США, а также проводят аналитический контроль объемной и насыпной плотности, распределения частиц по размерам, сыпучести, однородности смеси и содержания влаги. Впервые получена партия смеси для прямого прессования ПРТ с содержанием лекарственного вещества 0,25% по массе с использованием процедуры, установленной для смеси с содержанием лекарственного вещества 0,5% по массе. В процессе изготовления не обнаружено никаких технических ограничений. Результаты, представленные в Таблице 11, демонстрируют соответствующие требованиям физические свойства. Хотя данные об однородности смеси указывают на однородное распределение активного ингредиента, для партии 7 с наименьшим заявленным содержанием лекарственного вещества экспериментально определено заниженное содержание этого вещества, возможно вследствие потери активного ингредиента в процессе смешивания/помола. В случае полупромышленного прессования в процессе смешивания до введения смазывающего вещества, образцы смесей, отобранные через 20, 30 и 40 минут, демонстрируют приемлемые величины однородности смеси, как показано ниже.

Пример 15. Прессование ПРТ из примеров 13 и 14
Партии ПРТ с дозировками 0,75 мг, 4,5 мг и 6 мг прессуют, используя один и тот же роторный таблеточный пресс (Beta Press), оснащенный одним и тем же подающим фидером и одним и тем же комплектом инструментов (инструменты размера В, круглая плоская радиусная кромка, 9,5 мм) и при одних и тех же условиях прессования, как уже описано для ПРТ с дозировками 1,5 мг или 3 мг. Масса, время распадения, истираемость, твердость и толщина прессованных таблеток с дозировками 0,75 мг, 4,5 мг и 6 мг отвечают требованиям спецификации. Физические свойства таблеток подобны и сопоставимы со свойствами таблеток с другими дозировками. Результаты определения содержания действующего вещества и однородности состава для ПРТ с дозировкой 0,75 мг указывают на низкое содержание действующего вещества, подтверждающее низкую однородность смеси, что описано выше.
ПРТ с дозировкой 1,5 мг прессуют с использованием промышленного таблеточного пресса Korsch XL 400, оснащенного комплектом инструментов размера D с круглой плоской радиусной кромкой (9,5 мм), работающий со скоростью 1015 таблеток в минуту. Масса, время распадения, истираемость, твердость и толщина прессованных таблеток отвечают всем требованиям спецификации. Физические свойства таблеток подобны и сопоставимы со свойствами таблеток с такими же дозировками или другими дозировками, изготовленных в малом масштабе. Результаты определения содержания действующего вещества и однородности состава подтверждают результаты определения однородности смеси, следовательно, ПРТ соответствуют всем требованиям спецификации для продукции. Увеличение масштаба производства демонстрирует, что текущее мелкомасштабное производство при содержании лекарственного вещества 0,5% по массе или выше возможно расширить. Сопоставимые/подобные результаты, полученные для близких дозировок, и согласованность результатов, полученных для различных размеров партий, по меньшей мере при содержании действующего вещества 0,5% по массе или выше, указывают на робастный, пригодный процесс прямого прессования для производства ПРТ с дозировками от 1,5 мг до 6 мг или с содержанием лекарственного вещества от 0,5% до 2% по массе. Такое минимальное содержание лекарственного вещества в прошлом было возможно получить только с помощью распылительной грануляции, распыляя раствор лекарственного вещества при грануляции всех компонентов таблетки кроме смазывающего вещества.





Изучена стабильность партии 7 (0,75 мг), партии 8 (4,5 мг) и партии 9 (6 мг) ПРТ, содержащих флутиказон; эти партии упаковывают во флаконы из ПВП вместимостью 30 см3 (30 таблеток/флакон) с вискозным жгутом и пакетом влагопоглотителя силикагеля массой 0,5 г (Sorb-it, 1/2 г пакет). Все ПРТ стабильны в условиях испытаний методом «ускоренного старения» (40°С / относительная влажность 75%) в течение 6 месяцев, а также в условиях долгосрочных испытаний (25°С / относительная влажность 60%) в течение 9 месяцев; все величины, определенные в конкретные моменты времени (Т=0, 1 месяц, 2 месяца, 3 месяца, 6 месяцев, 9 месяцев), соответствуют критериям приемлемости (распадаемость: не более 30 с, количественный анализ на содержание действующего вещества: не менее 90,0% и не более 110,0%, каждая примесь: не более 0,5%, общее содержание примесей: не более 1,5%. Физические свойства, такие как внешний вид, твердость, истираемость и время распадения, для всех условий испытания стабильности также сопоставимы с исходными величинами для соответствующих ПРТ, содержащих флутиказон.
Хотя изобретение описано по отношению к конкретным вариантам реализации, представленным в данном документе, понятно, что оно допускает дополнительные изменения, и предполагается, что данная заявка предусматривает любое применение изобретения или любые разновидности и адаптации, в общем, вытекающие из принципов изобретения и включающие такие отступления от настоящего описания, которые подпадают под известную или общепринятую практику в области техники, к которой принадлежит изобретение, что может быть применено к существенным признакам, вышеизложенным в данном документе, и что следует далее из прилагаемой формулы изобретения.
Реферат
Изобретение относится к фармацевтической промышленности, а именно к перорально распадающейся таблетке, содержащей кортикостероиды, для лечения воспалительных состояний верхней части желудочно-кишечного тракта. Перорально распадающаяся таблетка для перорального введения для лечения воспалительных состояний верхней части желудочно-кишечного тракта, содержащая: местно действующий кортикостероид или его фармацевтически приемлемую соль, сложный эфир или полиморфную модификацию, адсорбированный на фармацевтически приемлемом носителе; быстро диспергирующиеся микрогранулы. Способ лечения воспалительного патологического состояния желудочно-кишечного тракта. Способ получения перорально распадающейся таблетки. Предложенная перорально распадающаяся таблетка, содержащая кортикостероиды, для лечения воспалительного патологического состояния желудочно-кишечного тракта, полученная предложенным способом, уменьшает побочное действие от системного всасывания и улучшает терапевтическую эффективность кортикостероидов. 3 н. и 42 з.п. ф-лы, 1 ил., 12 табл., 15 пр.
Комментарии