Твердые фармацевтические композиции, содержащие ингибитор интегразы - RU2602865C2
Код документа: RU2602865C2
Чертежи

Описание
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке испрашивается приоритет временной заявки на патент США № 61/254869, поданной 26 октября 2009 г, полное описание которой включено в описание путем ссылки.
ОБЛАСТЬ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение направлено на твердые фармацевтические композиции для перорального введения, в частности таблетки, которые содержат ралтегравир в форме фармацевтически приемлемой соли.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Соединение N-(4-фторбензил)-5-гидрокси-1-метил-2-(1-метил-1-{[(5-метил-1,3,4-оксадиазол-2-ил)карбонил]амино}этил)-6-оксо-1,6-дигидропиримидин-4-карбоксамид (далее именуемое «ралтегравиром») представляет собой мощный ингибитор интегразы ВИЧ. Ралтегравир имеет следующую структуру:
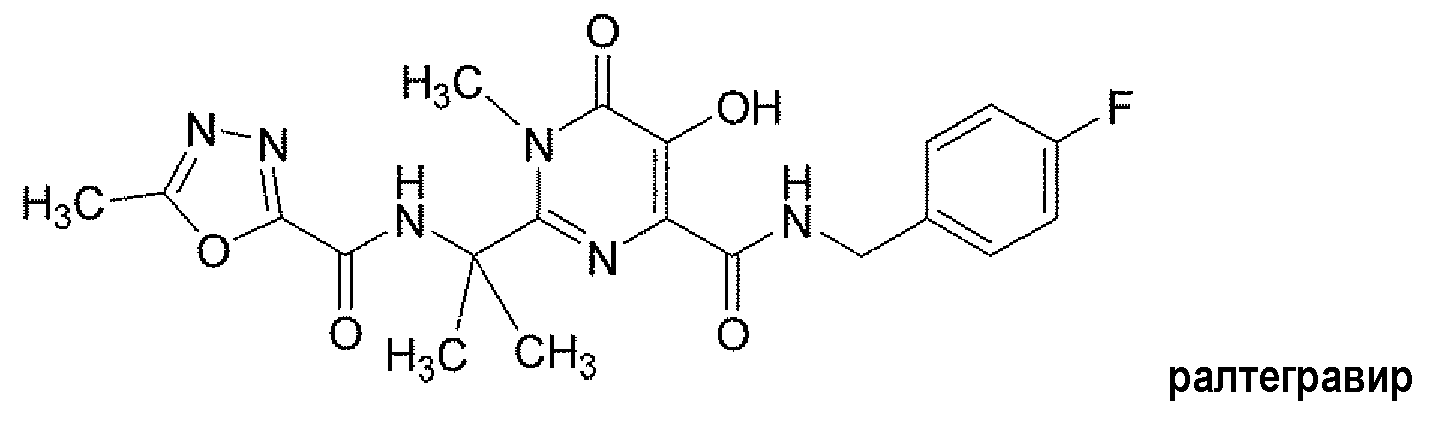
Ралтегравир, раскрытый в патенте США № 7169780, представляет собой активный фармацевтический ингредиент (API) в таблетках Изентресс®. Эти таблетки содержат 400 мг ралтегравира в форме калиевой соли и утверждены FDA (Управлением по контролю за качеством пищевых продуктов и лекарственных средств США) в комбинации с другими антиретровирусными средствами для лечения инфекции вирусом иммунодефицита человека (ВИЧ-1) у взрослых пациентов. Изентресс® является первым лекарственным продуктом в данном классе и важным средством в арсенале лекарственных препаратов, имеющихся для лечения ВИЧ-инфекции. Полезным дополнением к Изентресс® была бы содержащая ралтегравир таблетка, которая меньше по массе и объему и характеризуется обеспечением улучшенного фармакокинетического профиля.
Следующие ссылки представляют интерес в качестве документов предшествующего уровня техники:
В документе US 2006/0122205 A1 раскрыта кристаллическая калиевая соль ралтегравира.
В документе US 2007/0292504 A1 раскрыты фармацевтические препаративные формы для перорального введения в твердых лекарственных формах, которые содержат основную соль ралтегравира и композицию, регулирующую скорость высвобождения. В примере 3 описано получение посредством способа сухого гранулирования прессованных таблеток, содержащих калиевую соль ралтегравира (400 мг свободного фенола), микрокристаллическую целлюлозу, высушенную распылением водную лактозу, безводный двухосновный фосфат кальция, HPMC (гидроксипропилметилцеллюлозу) K4M, полоксамер 407, стеарилфумарат натрия и стеарат магния.
В документе US 2008/0118559 A1 раскрыты фармацевтические препаративные формы для перорального введения в твердых лекарственных формах, которые содержат соль щелочного металла ралтегравира и антинуклеирующий агент. В примере 3 описано получение посредством способа сухого гранулирования прессованных таблеток, содержащих калиевую соль ралтегравира (100 мг и 25 масс.% на основании свободного фенола), микрокристаллическую целлюлозу, моногидрат лактозы, кроскармеллозу натрия, HPMC 2910 (6 сП) и стеарат магния. В примере 6 описано получение посредством сухого гранулирования прессованных таблеток с пленочным покрытием красителем опадрай белым, содержащих калиевую соль ралтегравира (400 мг и 50% масс. на основании свободного фенола), микрокристаллическую целлюлозу, моногидрат лактозы, кроскармеллозу натрия, HPMC 2910 (6 сП) и стеарат магния.
В документе WO 2009/002823 A2 раскрыты прессованные таблетки, включающие ралтегравир и гранулы, содержащие атазанавира сульфат и внутригранулярный смазывающий агент, где гранулы имеют внутреннюю секцию и наружную поверхность, и по меньшей мере часть внутригранулярного смазывающего агента присутствует во внутренней секции гранул. Прессованные таблетки пригодны для лечения ВИЧ-инфекции.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение направлено на прессованные таблетки для перорального введения, которые содержат ралтегравир в качестве активного фармацевтического ингредиента в форме фармацевтически приемлемой соли. Более конкретно, настоящее изобретение включает прессованную таблетку, которая содержит:
(A) внутригранулярный компонент, содержащий:
(i) эффективное количество соли щелочного металла ралтегравира,
(ii) необязательно, первый суперразрыхлитель и
(iii) связывающий агент; и
(B) внегранулярный компонент, содержащий:
(i) второй суперразрыхлитель,
(ii) наполнитель и
(iii) смазывающий агент,
при условии, что в состав таблетки не входит атазанавир или его фармацевтически приемлемая соль.
Понятно, что прессованные таблетки могут включать один или более ингредиентов в дополнение к тем, которые конкретно указаны выше в пунктах (A) и (B), за исключением того, что в состав таблетки не входит атазанавир или фармацевтически приемлемая соль атазанавира. Используемый в настоящем описании термин «в состав таблетки не входит» в отношении определенного вещества (например, атазанавира или его фармацевтически приемлемой соли) означает, что прессованная таблетка по изобретению не содержит данное вещество. Прессованная таблетка может включать один или более дополнительных ингредиентов в Компоненте A или в Компоненте B или в каждом из Компонентов A и B. Прессованная таблетка может включать один или более ингредиентов в одном или более дополнительных компонентах. Дополнительные ингредиенты могут быть выбраны из API (кроме атазанавира и его фармацевтически приемлемых солей), эксципиентов, носителей и тому подобное.
Один вариант осуществления настоящего изобретения (альтернативно именуемый в настоящем описании Вариантом осуществления E1) представляет собой прессованную таблетку, как определено выше, где первый суперразрыхлитель присутствует во внутригранулярном компоненте A, т.е. присутствие первого суперразрыхлителя является обязательным. Соответственно, Вариант осуществления E1 представляет собой прессованную таблетку, которая содержит:
(A) внутригранулярный компонент, содержащий (i) эффективное количество соли щелочного металла ралтегравира, (ii) первый суперразрыхлитель и (iii) связывающий агент; и
(B) внегранулярный компонент, содержащий (i) второй суперразрыхлитель, (ii) наполнитель и (iii) смазывающий агент,
при условии, что таблетка не содержит атазанавир или его фармацевтически приемлемую соль.
Прессованные таблетки по настоящему изобретению могут обеспечить улучшенный фармакокинетический профиль по сравнению с содержащими полоксамер таблетками ралтегравира, такими как таблетки, описанные в заявке на патент США US 2007/0292504, который охватывает таблетки Изентресс®. Более конкретно, было обнаружено, что содержащие ралтегравир прессованные таблетки по настоящему изобретению обеспечивают значительное увеличение абсорбции лекарственного препарата (т.е. значительно более высокую AUC (площадь под кривой «концентрация-время»)) при значимо сниженной вариабельности абсорбции в отношении таблеток Изентресс® (см. пример 3 ниже). Состав, используемый в таблетках по настоящему изобретению, может обеспечить возможность получения таблеток с большей лекарственной загрузкой и меньшим размером, чем возможно для таблеток Изентресс®, что, таким образом, делает состав более привлекательным для использования в комбинациях с фиксированной дозой с другими API.
Настоящее изобретение также включает способы получения прессованных таблеток. Настоящее изобретение дополнительно включает применение прессованных таблеток для ингибирования интегразы ВИЧ, для лечения или профилактики ВИЧ-инфекции или для лечения, задержки начала или профилактики СПИДа.
Различные варианты осуществления, аспекты и признаки настоящего изобретения или описаны ниже, или будут очевидны из последующего описания, примеров и прилагаемой формулы изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг. 1 представляет собой график, показывающий отдельные и средние величины AUC ралтегравира для групп лечения дозой A и дозой B фармакокинетического исследования, описанного в примере 3.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Прессованные таблетки по настоящему изобретению включают внутригранулярный компонент и внегранулярный компонент, где внутригранулярный компонент содержит эффективное количество ралтегравира в форме фармацевтически приемлемой соли.
Как описано ниже, прессованные таблетки получают, используя способ, который включает гранулирование так, чтобы определенные ингредиенты комбинировать перед образованием гранул, а другие ингредиенты добавлять после гранулирования. Термин «внутригранулярный компонент» относится к ингредиентам прессованной таблетки, которые включают перед стадией гранулирования, а «внегранулярный компонент» относится к ингредиентам, которые включают после гранулирования.
Термин «фармацевтически приемлемая соль» относится к соли, которая обладает эффективностью материнского соединения и которая не является биологически или иным образом нежелательной (например, является ни токсичной, ни иным образом вредной для ее реципиента). Подходящие соли ралтегравира включают основные соли, т.е. соли, образованные в результате реакции лекарственного соединения с основанием. Соль ралтегравира представляет собой соль щелочного металла, такую как натриевая или калиевая соль, а обычнее калиевую соль. Соли щелочных металлов соединений могут быть образованы путем обработки соединения, растворенного в подходящем растворителе, водным раствором гидроксида щелочного металла (например, NaOH или KOH).
Термин «эффективное количество», используемый в настоящем описании, означает то количество API (например, ралтегравира), которое в ткани, системе, у животного или человека вызывает биологическую или лекарственную реакцию, искомую исследователем, ветеринаром, врачом или другим клиницистом. Эффективное количество может представлять собой «терапевтически эффективное количество» для облегчения симптомов подвергаемого лечению заболевания или состояния. Эффективное количество может также представлять собой «профилактически эффективное количество» для профилактики симптомов предотвращаемого заболевания или состояния. Когда лекарственное соединение ингибирует действие фермента (например, интегразы ВИЧ), данный термин также относится к количеству активного соединения, достаточному для того, чтобы ингибировать фермент и, тем самым, вызвать искомую реакцию (т.е. «эффективное ингибирующее количество»).
Внутригранулярный компонент включает, в дополнение к соли ралтегравира, связывающий агент и, необязательно, суперразрыхлитель. Термин «суперразрыхлитель» относится к веществу или смеси веществ, которые используются в таблетке для содействия ее разрушению или разрыхлению после введения. Внутригранулярный суперразрыхлитель может представлять собой кроскармеллозу натрия, кросповидон или натриевый гликолят крахмала и обычно представляет собой кроскармеллозу натрия или натриевый гликолят крахмала, а обычнее кроскармеллозу натрия. Суперразрыхлитель, используемый во внутригранулярном компоненте прессованной таблетки, может необязательно представлять собой комбинацию двух или более суперразрыхлителей, такую как комбинация кроскармеллозы натрия и натриевого гликолята крахмала. Суперразрыхлитель в комбинации может быть добавлен отдельно или в виде смеси для смешивания с другими ингредиентами внутригранулярного компонента.
Термин «связывающий агент» относится к веществу или смеси веществ, которые обеспечивают или улучшают когезионную способность гранул и могут также содействовать когезионной способности прессованных таблеток. Связывающий агент, например, обеспечивает то, что таблетка остается интактной после прессования. Подходящие связывающие агенты включают такие вещества, как желатин, гуаровая камедь, гидрированное растительное масло и различные целлюлозы. В одном аспекте изобретения связывающие агенты представляют собой связывающие агенты с низкой вязкостью. Термин «низкая вязкость» относится к связывающему агенту, который обеспечивает получение водного раствора 2 масс.% (т.е. масса полимера/масса воды), имеющего вязкость в диапазоне от примерно 2 до примерно 100 сантипуаз (сП) при 20°C (1 сП = 1 мПа·с). Связывающие агенты с низкой вязкостью, подходящие для использования в прессованных таблетках по изобретению, обычно обеспечивают получение раствора 2 масс.%, имеющего вязкость в диапазоне от примерно 2 до примерно 50 сП (например, от примерно 3 до примерно 20 сП) при 20°C. Подходящие связывающие агенты включают имеющие низкую вязкость растворимые в воде полимеры, такие как гидроксиалкилцеллюлозы, алкилцеллюлозы и поливинилпирролидоны. Имеющий низкую вязкость связывающий агент представляет собой обычно гидроксипропилметилцеллюлозу (HPMC), гидроксиэтилцеллюлозу (HEC), гидроксипропилцеллюлозу (HPC), поливинилпирролидон (PVP) или метилцеллюлозу. Имеющий низкую вязкость связывающий агент обычнее представляет собой HPMC, HPC или PVP. В одном аспекте изобретения имеющий низкую вязкость связывающий агент представляет собой HPMC, имеющую содержание гидроксипропила от примерно 7 до примерно 12 масс.%, содержание метокси от примерно 28 до примерно 30 масс.% и вязкость для 2% масс./масс. водных растворов от примерно 3 до примерно 20 сП. В другом аспекте связывающий агент представляет собой HPMC, которая представляет собой заместитель типа 2208, 2906 или 2910 стандарта Фармакопеи США, такой как HPMC 2910 (6 сП), который выпускается компанией Shin-Etsu Chemical Co в виде продукта с названием PHARMACOAT.
Связывающий агент может представлять собой комбинацию двух или более связывающих агентов. Например, связывающий агент может представлять собой комбинацию имеющих низкую вязкость растворимых в воде полимеров (например, двух или более полимеров HPMC), где полимерная смесь образует 2 масс.% раствор со средней вязкостью в диапазоне низкой вязкости. Средняя вязкость полимерной смеси обычно отличается от вязкости каждого компонентного полимера. Связывающие агенты в комбинации могут быть добавлены отдельно или в виде смеси для смешивания с другими ингредиентами во внутригранулярном компоненте.
Внегранулярный компонент включает суперразрыхлитель, наполнитель и смазывающий агент. Внегранулярный суперразрыхлитель (альтернативно именуемый в настоящем описании «второй суперразрыхлитель») может быть таким же или отличным от внутригранулярного суперразрыхлителя (альтернативно именуемого в настоящем описании «первым суперразрыхлителем»). Внегранулярный суперразрыхлитель может представлять собой кроскармеллозу натрия, кросповидон или натриевый гликолят крахмала, и обычно представляет собой кроскармеллозу натрия или натриевый гликолят крахмала, а обычнее представляет собой кроскармеллозу натрия. В одном аспекте изобретения внутригранулярный компонент прессованной таблетки не содержит суперразрыхлитель, а внегранулярный компонент включает суперразрыхлитель, такой как кроскармеллоза натрия. Прессованная таблетка предпочтительно содержит как внутригранулярный, так и внегранулярный суперразрыхлитель. Считается, что прессованные таблетки, содержащие как внутригранулярные, так и внегранулярные суперразрыхлители, являются более надежными, т.е. таблетки имеют лучшую воспроизводимость с точки зрения их характеристик уплотнения и растворения, чем аналогичные таблетки, содержащие только внегранулярный суперразрыхлитель. Таким образом, в одном аспекте изобретения таблетка содержит как внутригранулярный, так и внегранулярный суперразрыхлитель, и оба суперразрыхлителя представляют собой кроскармеллозу натрия или они оба представляют собой комбинацию кроскармеллозы натрия и натриевого гликолята крахмала. В другом аспекте таблетка содержит внутригранулярный и внегранулярный суперразрыхлители, и они оба представляют собой кроскармеллозу натрия.
Наполнитель (также именуемый в данной области «разбавителем») представляет собой вещество, используемое для придания объема таблетке. Подходящие наполнители включают безводный двухосновный фосфат кальция, двухосновный дигидрофосфат кальция, трехосновный фосфат кальция, сульфат кальция, карбоксиметилцеллюлозу кальция, микрокристаллическую целлюлозу, порошковую целлюлозу, глюкозу, фруктозу, лактозу, манит, декстрин, декстрозу, декстраты, каолин, лактит, карбонат магния, оксид магния, мальтит, мальтодекстрин, мальтозу, крахмал, сахарозу, трегалозу, тальк и их комбинации. В одном аспекте изобретения наполнитель представляет собой лактозу, микрокристаллическую целлюлозу, манит, безводный двухосновный фосфат кальция или двухосновный дигидрофосфат кальция. В другом аспекте изобретения наполнителем не является восстанавливающий сахар, т.е. в данном аспекте связывающий агент не представляет собой глюкозу, фруктозу, лактозу, мальтозу, декстрозу или тому подобное. В одном признаке данного аспекта наполнитель представляет собой микрокристаллическую целлюлозу, манит, безводный двухосновный фосфат кальция, двухосновный дигидрофосфат кальция или их комбинацию. В другом признаке данного аспекта наполнитель представляет собой микрокристаллическую целлюлозу, безводный двухосновный фосфат кальция, двухосновный дигидрофосфат кальция или их комбинацию.
В еще одном аспекте изобретения наполнитель представляет собой микрокристаллическую целлюлозу. Примером подходящей микрокристаллической целлюлозы является та, которая может характеризоваться как имеющая номинальный размер частиц примерно 100 мкм, содержание влаги от примерно 3% до примерно 5% и свободную насыпную плотность от примерно 0,28 до примерно 0,33 г/см3. Микрокристаллическая целлюлоза, имеющая указанные выше характеристики, представляет собой, например, AVICEL PH-102. Другие подходящие микрокристаллические целлюлозы представляют собой те, которые имеют следующие характеристики:
Соответственно, подходящие формы микрокристаллической целлюлозы для использования в прессованных таблетках по изобретению включают, но не ограничиваются ими, материалы, продаваемые под торговыми названиями AVICEL PH-101, AVICEL PH-102, AVICEL PH-103 и AVICEL PH-105 (все выпускаемые компанией FMC Corporation), и их комбинации. Так, например, микрокристаллическая целлюлоза, используемая в таблетке, может представлять собой AVICEL PH-102 или AVICEL PH-105 или их комбинацию.
В еще одном аспекте изобретения наполнитель представляет собой комбинацию микрокристаллической целлюлозы и безводного двухосновного фосфата кальция. Примером подходящей комбинации является порошок, содержащий примерно 75% микрокристаллической целлюлозы и примерно 25% безводного двухосновного фосфата кальция, где порошок получают диспергированием во влажном состоянии и распылительной сушкой целлюлозы и фосфата. Такой продукт выпускается компанией FMC Corporation под торговым названием AVICEL DG.
Роль смазывающего агента заключается в улучшении потока гранул, полученных на стадии гранулирования, перед их прессованием и/или в предотвращении адгезии прессованной таблетки к прессовочному оборудованию. Подходящие смазывающие агенты включают стеарат кальция, глицерилмоностеарат, глицерилпальмитостеарат, гидрированное касторовое масло, гидрированное растительное масло, легкое минеральное масло, стеарат магния, минеральное масло, полиэтиленгликоль, стеариновую кислоту, тальк, стеарат цинка и стеарилфумарат натрия. В одном аспекте изобретения смазывающий агент представляет собой стеарат магния, стеариновую кислоту, стеарилфумарат натрия или комбинацию двух или более из них. В другом аспекте смазывающий агент представляет собой стеарат магния. Если используют комбинацию смазывающих агентов, то смазывающие агенты могут быть добавлены отдельно или в виде смеси гранул.
Прессованная таблетка по изобретению не содержит атазанавир или его фармацевтически приемлемую соль, такую как атазанавир сульфат. Атазанавира сульфат имеет структуру:

Дополнительное описание атазанавира, атазанавира сульфата и способов их получения и применения можно найти, например, в патенте США № 6087383, заявке на патент США US 2005/0256202 A1 и заявке на Международный патент WO 2009/002823 A2. Атазанавир коммерчески доступен в виде прописываемого лекарственного средства от Bristol-Myers Squibb Company под торговым названием Reyataz® (атазанавир сульфат) в форме капсул по 100, 150, 200 и 300 мг.
Если из контекста ясно или прямо указано иное, то массовый процент ралтегравира в прессованной таблетке выражен в пересчете на свободный фенол, даже если он используется в форме соли. Величины массовых процентов ингредиентов таблетки (например, первого и второго суперразрыхлителей, связывающего агента, наполнителя, смазывающего агента и т.д.) основаны на общей массе таблетки.
Второй вариант осуществления настоящего изобретения (Вариант осуществления E2) представляет собой прессованную таблетку, как первоначально определено выше (т.е., как определено в Кратком изложении сущности изобретения), где:
(A)(i) соль щелочного металла ралтегравира используется на основе свободного фенола в количестве по меньшей мере примерно 30 масс.%;
(A)(ii) первый суперразрыхлитель используется в количестве в диапазоне от нуля до примерно 12 масс.%;
(A)(iii) связывающий агент используется в количестве в диапазоне от примерно 0,5 масс.% до примерно 7 масс.%;
(B)(i) второй суперразрыхлитель используется в количестве в диапазоне от примерно 3 масс.% до примерно 20 масс.%;
(B)(ii) наполнитель используется в количестве в диапазоне от примерно 10 масс.% до примерно 40 масс.%; и
(B)(iii) смазывающий агент используется в количестве в диапазоне от примерно 0,5 масс.% до примерно 2,5 масс.%;
где общее количество суперразрыхлителя находится в диапазоне от примерно 6 масс.% до примерно 20 масс.%; и
где массовый процент каждого ингредиента в прессованной таблетке основан на общей массе прессованной таблетки.
Третий вариант осуществления настоящего изобретения (Вариант осуществления E3) представляет собой прессованную таблетку, как определено в Варианте осуществления E1, где:
(A)(i) соль щелочного металла ралтегравира используется на основе свободного фенола в количестве по меньшей мере примерно 30 масс.%;
(A)(ii) первый суперразрыхлитель используется в количестве в диапазоне от примерно 3 масс.% до примерно 12 масс.%;
(A)(iii) связывающий агент (например, связывающий агент с низкой вязкостью) используется в количестве в диапазоне от примерно 0,5 масс.% до примерно 7 масс.%;
(B)(i) второй суперразрыхлитель используется в количестве в диапазоне от примерно 3 масс.% до примерно 15 масс.%;
(B)(ii) наполнитель используется в количестве в диапазоне от примерно 10 масс.% до примерно 40 масс.%; и
(B)(iii) смазывающий агент используется в количестве в диапазоне от примерно 0,5 масс.% до примерно 2,5 масс.%;
где общее количество суперразрыхлителя находится в диапазоне от примерно 6 масс.% до примерно 20 масс.%; и
где массовый процент каждого ингредиента в прессованной таблетке основан на общей массе прессованной таблетки.
Четвертый вариант осуществления настоящего изобретения (Вариант осуществления E4) представляет собой прессованную таблетку, как определено выше или как определено в Вариантах осуществления E1-E3, где:
(A)(ii) первый суперразрыхлитель выбран из группы, состоящей из кроскармеллозы натрия, натриевого гликолята крахмала, кросповидона и их комбинаций;
(A)(iii) связывающий агент имеет вязкость в диапазоне от примерно 2 до примерно 100 сантипуаз (сП) при 20°C и выбран из группы, состоящей из HPMC, HPC, PVP и их комбинаций;
(B)(i) второй суперразрыхлитель выбран из группы, состоящей из кроскармеллозы натрия, натриевого гликолята крахмала, кросповидона и их комбинаций;
(B)(ii) наполнитель выбран из группы, состоящей из микрокристаллической целлюлозы, маннита, лактозы, фосфата Ca и их комбинаций; и
(B)(iii) смазывающий агент выбран из группы, состоящей из стеарата Mg, стеариновой кислоты, стеарилфумарата натрия и их комбинаций.
В первом аспекте Варианта осуществления E4
(A)(ii) первый суперразрыхлитель выбран из группы, состоящей из кроскармеллозы натрия, натриевого гликолята крахмала и кросповидона;
(A)(iii) связывающий агент имеет вязкость в диапазоне от примерно 2 до примерно 100 сантипуаз (сП) при 20°C и выбран из группы, состоящей из HPMC, HPC и PVP;
(B)(i) второй суперразрыхлитель выбран из группы, состоящей из кроскармеллозы натрия, натриевого гликолята крахмала и кросповидона;
(B)(ii) наполнитель выбран из группы, состоящей из микрокристаллической целлюлозы, маннита, лактозы, фосфата Ca и их комбинаций; и
(B)(iii) смазывающий агент выбран из группы, состоящей из стеарата Mg, стеариновой кислоты, стеарилфумарата натрия и их комбинаций. В одном признаке первого аспекта наполнитель выбран из группы, состоящей из микрокристаллической целлюлозы, фосфата Ca и их комбинаций. В другом признаке первого аспекта Варианта осуществления E4 наполнитель представляет собой микрокристаллическую целлюлозу. В еще одном признаке первого аспекта Варианта осуществления E4 наполнитель представляет собой комбинацию микрокристаллической целлюлозы и двухосновного фосфата Ca (например, безводного двухосновного фосфата Ca).
Во втором аспекте Варианта осуществления E4 наполнитель выбран из группы, состоящей из микрокристаллической целлюлозы, фосфата Ca и их комбинаций. В третьем аспекте Варианта осуществления E4 наполнитель представляет собой микрокристаллическую целлюлозу. В четвертом аспекте Варианта осуществления E4 наполнитель представляет собой комбинацию микрокристаллической целлюлозы и двухосновного фосфата Ca (например, безводного двухосновного фосфата Ca).
Другой вариант осуществления настоящего изобретения (Вариант осуществления E5) представляет собой прессованную таблетку, как первоначально определено выше или определено в любом из предыдущих вариантов осуществления, где:
(A)(i) соль щелочного металла ралтегравира представляет собой соль Na или K, используемую в количестве в диапазоне от примерно 50 масс.% до примерно 65 масс.%;
(A)(ii) первый суперразрыхлитель используется в количестве в диапазоне от примерно 5 масс.% до примерно 10 масс.%;
(A)(iii) связывающий агент используется в количестве в диапазоне от примерно 2 масс.% до примерно 6 масс.%;
(B)(i) второй суперразрыхлитель используется в количестве в диапазоне от примерно 6 масс.% до примерно 12 масс.%;
(B)(ii) наполнитель используется в количестве в диапазоне от примерно 6 масс.% до примерно 25 масс.%; и
(B)(iii) смазывающий агент используется в количестве в диапазоне от примерно 1 масс.% до примерно 2,5 масс.%;
где общее количество суперразрыхлителя находится в диапазоне от примерно 10 масс.% до примерно 18 масс.%.
Другой вариант осуществления настоящего изобретения (Вариант осуществления E6) идентичен Варианту осуществления E5, за исключением того, что наполнитель - (B)(ii) - используется в количестве в диапазоне от примерно 10 масс.% до примерно 25 масс.%.
В одном аспекте Варианта осуществления E5 и Варианта осуществления E6 первый суперразрыхлитель и второй суперразрыхлитель представляют собой одинаковое вещество или одинаковую комбинацию веществ.
Другой вариант осуществления настоящего изобретения (Вариант осуществления E7) представляет собой прессованную таблетку, как первоначально определено выше или как определено в любом из Вариантов осуществления E1-E6, где:
(A)(ii) первый суперразрыхлитель представляет собой внутригранулярную кроскармеллозу Na;
(A)(iii) связывающий агент представляет собой HPMC;
(B)(i) второй суперразлыхлитель представляет собой внегранулярную кроскармеллозу Na;
(B)(ii) наполнитель представляет собой микрокристаллическую целлюлозу или комбинацию микрокристаллической целлюлозы и двухосновного фосфата Ca (например, безводный двухосновный фосфат Ca); и
(B)(iii) смазывающий агент представляет собой стеарат Mg.
Другой вариант осуществления настоящего изобретения (Вариант осуществления E8) идентичен Варианту осуществления E7, за исключением того, что наполнитель представляет собой микрокристаллическую целлюлозу (т.е. наполнитель не представляет собой комбинацию микрокристаллической целлюлозы и двухосновного фосфата Ca).
Другой вариант осуществления настоящего изобретения (Вариант осуществления E9) представляет собой прессованную таблетку, как определено в Варианте осуществления E8, где:
(A)(i) соль щелочного металла ралтегравира представляет собой соль Na или K, используемую в количестве в диапазоне от примерно 55 масс.% до примерно 60 масс.%;
(Α)(ii) внутригранулярная кроскармеллоза Na используется в количестве в диапазоне от примерно 5 масс.% до примерно 7 масс.%;
(A)(iii) HPMC используется в количестве в диапазоне от примерно 3 масс.% до примерно 5 масс.%;
(B)(i) внегранулярная кроскармеллоза Na используется в количестве в диапазоне от примерно 8 масс.% до примерно 10 масс.%;
(B)(ii) микрокристаллическая целлюлоза используется в количестве в диапазоне от примерно 16 масс.% до примерно 18 масс.%; и
(B)(iii) стеарат магния используется в количестве в диапазоне от примерно 1 масс.% до примерно 2 масс.%;
где общее количество кроскармеллозы натрия находится в диапазоне от примерно 13 масс.% до примерно 17 масс.%.
Другой вариант осуществления настоящего изобретения (Вариант осуществления E10) представляет собой прессованную таблетку, как определено в Варианте осуществления E7, где:
(A)(i) натриевая или калиевая соль ралтегравира используется в количестве в диапазоне от примерно 55 масс.% до примерно 65 масс.% на основании свободного фенола;
(A)(ii) внутригранулярная кроскармеллоза Na используется в количестве в диапазоне от примерно 5 масс.% до примерно 8 масс.%;
(A)(iii) HPMC используется в количестве в диапазоне от примерно 3 масс.% до примерно 5 масс.%;
(B)(i) внегранулярная кроскармеллоза Na используется в количестве в диапазоне от примерно 8 масс.% до примерно 10 масс.%;
(B)(ii) наполнитель представляет собой комбинацию микрокристаллической целлюлозы и двухосновного фосфата Ca и используется в количестве в диапазоне от примерно 7 масс.% до примерно 10 масс.%; и
(B)(iii) стеарат магния используется в количестве в диапазоне от примерно 1 масс.% до примерно 2 масс.%;
где общее количество кроскармеллозы натрия находится в диапазоне от примерно 13 масс.% до примерно 17 масс.%.
Другой вариант осуществления настоящего изобретения (Вариант осуществления E11) представляет собой прессованную таблетку, как первоначально определено выше или как определено в любом из Вариантов осуществления E1-E10, где соль щелочного металла ралтегравира используется на основании свободного фенола в количестве в диапазоне от примерно 200 мг до примерно 600 мг на стандартную дозу.
Другой вариант осуществления настоящего изобретения (Вариант осуществления E12) представляет собой прессованную таблетку, как первоначально определено выше или как определено в любом из Вариантов осуществления E1-E11, где соль щелочного металла представляет собой калиевую соль ралтегравира.
Другой вариант осуществления настоящего изобретения (Вариант осуществления E13) представляет собой прессованную таблетку, как определено в Варианте осуществления E7, где таблетка имеет следующий состав:
В одном аспекте настоящего варианта осуществления количество ралтегравира калия в стандартной лекарственной форме в виде таблетки составляет 434,4 мг (400 мг в пересчете на свободный фенол).
Другой вариант осуществления настоящего изобретения (Вариант осуществления E14) представляет собой прессованную таблетку, как определено в Варианте осуществления E7, где таблетка имеет следующий состав:
В одном аспекте настоящего варианта осуществления количество ралтегравира калия в стандартной лекарственной форме в виде таблетки составляет 434,4 мг (400 мг в пересчете на свободный фенол).
Другой вариант осуществления настоящего изобретения (Вариант осуществления E15) представляет собой прессованную таблетку, как первоначально определено выше или как определено в любом из Вариантов осуществления E1-E14, где соль щелочного металла ралтегравира представляет собой калиевую соль ралтегравира, которая представляет собой Форму 1 кристаллической калиевой соли ралтегравира. Форма 1 кристаллической соли представляет собой кристаллическую соль, описанную и охарактеризованную в примере 2 в заявке на патент США US 2006/0122205 A1. Форма 1 соли ралтегравира характеризуется типом рентгеновской порошковой дифракции, полученной с использованием Kα излучения меди (т.е. источник излучения представляет собой комбинацию излучения Cu Kα1 и Kα2), который включает 2Θ величины (т.е. отражения при 2Θ величинах) при градусах 5,9, 20,0 и 20,6. В одном аспекте данного варианта осуществления Форма 1 кристаллической калиевой соли ралтегравира характеризуется типом рентгеновской порошковой дифракции, полученной при использовании Kα излучения меди, которая включает 2Θ величины при градусах 5,9, 12,5, 20,0, 20,6 и 25,6. Репрезентативный тип XRPD для Формы 1 представлен на фиг. 1 заявки на патент США US 2006/0122205 A1.
Другой вариант осуществления настоящего изобретения (Вариант осуществления E16) представляет собой прессованную таблетку, как первоначально определено выше или как определено в любом из Вариантов осуществления E1-E15, где в состав таблетки не входят восстанавливающие сахара, т.е. восстанавливающий сахар не содержится в таблетке. Восстанавливающие сахара представляют собой сахара, которые действуют в качестве восстанавливающих агентов и легко восстанавливают щелочные растворы солей меди. Сахар, который вызывает появление кирпично-красного цвета при тестировании реактивом Бенедикта или раствором Фелинга, является восстанавливающим сахаром. Цвет в тестирующем растворе появляется вследствие восстановления ионов Cu(II) в оксид меди(I) сахаром. Восстанавливающие сахара включают глюкозу, фруктозу, лактозу, арабинозу и мальтозу.
Композиции, содержащие восстанавливающий сахар и амин, восприимчивы к реакции конденсации Мэйларда, которая может привести к образованию продуктов распада коричневого цвета. Таблетки по изобретению, в состав которых не входят восстанавливающие сахара, поэтому более совместимы с аминосодержащими веществами, которые могут присутствовать в таблетке. Применение таблеток, не содержащих восстанавливающие сахара, особенно привлекательно, когда таблетка включает второй активный фармацевтический ингредиент, имеющий одну или более аминогрупп (например, монолитная таблетка, содержащая комбинацию фиксированной дозы ралтегравира и аминосодержащего противовирусного средства против ВИЧ).
Другой вариант осуществления настоящего изобретения (Вариант осуществления E17) представляет собой прессованную таблетку, как первоначально определено выше или как определено в любом из Вариантов осуществления E1-E15, где в состав таблетки не входит полоксамер, т.е. таблетка не содержит полоксамер. Полоксамеры представляют собой блок-сополимеры этиленоксида и пропиленоксида. Сополимеры обычно имеют среднюю молекулярную массу в диапазоне от примерно 1000 до примерно 20000 и содержание оксиэтилена от примерно 40 до примерно 90 масс.%. Полоксамеры могут использоваться в фармацевтических препаративных формах в качестве, например, солюбилизирующих агентов, эмульгирующих агентов или смачивающих агентов. Репрезентативные полоксамеры включают полоксамер 188, полоксамер 237, полоксамер 338 и полоксамер 407. В определенных составах таблетки высокий уровень полоксамера может неблагоприятно воздействовать на сжатие и может привести к прилипанию материала таблетки к стенке головки экструдера во время образования таблетки прессованием. Высокий уровень полоксамера может также ингибировать абсорбцию определенных активных ингредиентов. Изентресс® содержит относительно высокий уровень полоксамера, и таблетки характеризуются наличием относительно медленного высвобождения ралтегравира после введения. Считается, что введение другого противовирусного средства против ВИЧ в такую препаративную форму для обеспечения комбинации фиксированной дозы с ралтегравиром может неблагоприятно воздействовать на абсорбцию противовирусного средства.
Другой вариант осуществления настоящего изобретения (Вариант осуществления E18) представляет собой прессованную таблетку, как первоначально определено выше или как определено в любом из Вариантов осуществления E1-E15, где в состав таблетки не входят полоксамеры и восстанавливающие сахара, т.е. таблетка не содержит ни полоксамер, ни восстанавливающий сахар.
Другой вариант осуществления настоящего изобретения (Вариант осуществления E19) представляет собой прессованную таблетку, как первоначально определено выше или как определено в любом из Вариантов осуществления E1-E18, где время распада прессованной таблетки составляет менее чем примерно 15 минут. В одном аспекте данного варианта осуществления время распада находится в диапазоне от примерно 5 минут до примерно 12 минут. Время распада определяется так, как описано в примере 2.
Прессованная таблетка, первоначально описанная выше и описанная в каждом из предыдущих аспектов и вариантов осуществления, может быть получена посредством влажного гранулирования, при котором общий размер частиц подходящей препаративной формы увеличивается за счет постоянной агрегации более мелких частиц. Влажное гранулирование включает смачивание тщательно смешанной смеси сухих внутригранулярных ингредиентов (например, соли ралтегравира, первого суперразрыхлителя и связывающего агента) достаточным количеством растворителя (например, водой или водой со спиртовым сорастворителем) для увлажнения сухой смеси так, чтобы частицы в смеси прилипли друг к другу с образованием более крупных частиц, и затем просеивание, измельчение или иное манипулирование размером частиц. После образования полученный влажный гранулят можно затем сушить и подвергать размолу в частицы подходящего размера (т.е. гранулы), гранулы смешивают со смазывающим агентом и, необязательно, с другими внегранулярными ингредиентами (например, вторым суперразрыхлителем и наполнителем) и смазанные гранулы прессуют в таблетки.
Прессованные таблетки могут быть покрыты сахаром для маскировки любого неприятного вкуса или покрыты пленкой (например, полимерным покрытием) для защиты таблетки от атмосферного распада. Покрытие также не должно неблагоприятно воздействовать на высвобождение лекарственного средства после перорального введения. Подходящей суспензией для пленочного покрытия является опадрай II (39K) (доступный от Colorcon, West Point, PA), который представляет собой полимер на основе гидроксипропилметилцеллюлозы (HPMC) с триацетином, лактозой и диоксидом титана. Пленки можно наносить напылением суспензии на таблетки и затем сушкой. Подходящие технологии пленочного покрытия описаны в руководствах Remington's Pharmaceutical Sciences. 18th edition, edited by A. R. Gennaro, 1990, Mack Publishing Co., pp. 1665-1675, и Remington - The Science and Practice of Pharmacy, 21 st edition, 2005, Chapter 46.
Технология и оборудование, подходящие для получения прессованных таблеток по настоящему изобретению, описаны в руководствах Remington's Pharmaceutical Sciences, 18th edition, edited by A. R. Gennaro, 1990, Chapter 89 и Remington - The Science and Practice of Pharmacy, 21 st edition, 2005, Chapter 45.
Настоящее изобретение включает способ (альтернативно именуемый в настоящем описании «Способом P1») получения прессованной таблетки, содержащей эффективное количество соли щелочного металла ралтегравира, необязательно, первый суперразрыхлитель, связывающий агент (например, связывающий агент с низкой вязкостью), второй суперразрыхлитель, наполнитель и смазывающий агент, где способ включает:
(A) сухое смешивание соли ралтегравира, первого суперразрыхлителя (необязательного) и связывающего агента с получением сухой смеси;
(B) влажное гранулирование сухой смеси и затем, необязательно, размол или просеивание смеси после влажного гранулирования;
(C) сушка смеси после влажного гранулирования на стадии B с получением высушенных гранул;
(D) помол и просеивание высушенных гранул стадии C;
(E) смешивание молотых просеянных гранул, полученных на стадии D, со вторым суперразрыхлителем, наполнителем и смазывающим агентом с получением смазанной гранулярной смеси; и
(F) прессование смазанной гранулярной смеси стадии E с получением таблетки; при условии, что в способе не используют и полученная таблетка не содержит атазанавир или его фармацевтически приемлемую соль.
Смешивание проводят на стадии A в течение времени, достаточного для получения относительно однородной смеси ингредиентов. Смешивание можно выполнять в любом подходящем оборудовании для смешивания, таком как гранулятор с высоким усилием сдвига, V-образный смеситель или бункерный смеситель. Влажное гранулирование стадии B можно проводить добавлением гранулирующей жидкости (обычно воды) в миксер, содержащий смешанные ингредиенты, и смешиванием влажных ингредиентов. Влажный гранулят может быть затем подвергнут помолу или просеиванию в отдельной операции (например, продавливанием влажного гранулята через сетчатый фильтр подходящего размера). Альтернативно, некоторые смесители оборудованы ножом фальцаппарата ударного типа, который работает независимо от смешивающих лопаток, исключая необходимость в отдельной операции помола/просеивания. Сушка на стадии C может быть проведена любым удобным образом, например путем лоточной сушки или сушки в псевдоожиженном слое при температуре в диапазоне от примерно 40°C до примерно 90°C. Гранулят обычно сушат до LOD (уровня выявления) примерно 0,5-3%. Помол и просеивание стадии D проводится для достижения подходящего размера частиц, например частиц со средним диаметром в диапазоне примерно от 50 до 1200 мкм. Смешивание на стадии E проводят в течение времени, достаточного для получения однородной смеси гранул с внегранулярными ингредиентами. В одном аспекте способа P1 стадия E включает (e-i) смешивание молотых, просеянных гранул, полученных на стадии D, со вторым суперразрыхлителем и наполнителем и затем (e-ii) добавление смазывающего агента к смеси, полученной на подстадии e(i), с получением смазанной гранулярной смеси. Затем гранулярную смесь прессуют в таблетку на стадии F, используя стандартный таблеточный пресс, такой как роторный пресс, с получением таблеток, например, круглой или овальной формы. Если прямо не указано иное (как на стадии C), то стадии способа P1 проводят в условиях окружающей среды, т.е. при температуре, равной или близкой примерно 25°C.
Гранулярные смеси, полученные в соответствии со способом P1, могут иметь благоприятные свойства текучести и сжатия. Например, гранулярные смеси, содержащие калиевую соль ралтегравира, полученную как описано в примере 1 ниже, имеют превосходные свойства текучести и сниженную тенденцию прилипать или сцепляться с прессовочным инструментом по сравнению с аналогичными сухими грануляционными смесями, описанными в заявке на патент США US 2007/0292504 A1 (см. пример 3 в патенте США '504 и ссылочный пример 1 ниже в настоящем описании) и в заявке на патент США US 2008/0118559 Al (см. пример 6 в патенте США '559).
Варианты осуществления способа P1 включают описанный выше способ, включающий один или более из следующих пунктов (i)-(xiv):
(i-a) соль щелочного металла ралтегравира представляет собой натриевую соль или калиевую соль соединения I;
(i-b) соль щелочного металла ралтегравира представляет собой калиевую соль ралтегравира; или
(i-c) соль щелочного металла ралтегравира представляет собой форму 1 кристаллической калиевой соли ралтегравира;
(ii-a) соль щелочного металла ралтегравира используется в количестве по меньшей мере примерно 30 масс.% на основании свободного фенола;
(ii-b) соль щелочного металла ралтегравира представляет собой соль Na или K, используемую в количестве в диапазоне от примерно 50 масс.% до примерно 65 масс.% на основании свободного фенола;
(ii-c) соль щелочного металла ралтегравира (например, калиевая соль ралтегравира) используется в количестве в диапазоне от примерно 55 масс.% до примерно 60 масс.% на основании свободного фенола; или
(ii-d) соль щелочного металла ралтегравира (например, калиевая соль ралтегравира) используется в количестве в диапазоне от примерно 55 масс.% до примерно 65 масс.% на основании свободного фенола;
(iii-a) первый суперразрыхлитель выбран из группы, состоящей из кроскармеллозы натрия, натриевого гликолята крахмала, кросповидона и их комбинаций;
(iii-b) первый суперразрыхлитель выбран из группы, состоящей из кроскармеллозы натрия, натриевого гликолята крахмала и кросповидона; или
(iii-c) первый суперразрыхлитель представляет собой кроскармеллозу натрия;
(iv-a) первый суперразрыхлитель используется в количестве в диапазоне от нуля масс.% до примерно 12 масс.%;
(iv-b) первый суперразрыхлитель используется в количестве в диапазоне от примерно 3 масс.% до примерно 12 масс.%;
(iv-c) первый суперразрыхлитель используется в количестве в диапазоне от примерно 5 масс.% до примерно 10 масс.%; или
(iv-d) первый суперразрыхлитель (например, кроскармеллоза Na) используется в количестве в диапазоне от примерно 5 масс.% до примерно 7 масс.%;
(v-a) связывающий агент имеет вязкость в диапазоне от примерно 2 до примерно 100 сантипуаз (сП) при 20°C и выбран из группы, состоящей из HPMC, HPC и PVP; или
(v-b) связывающий агент представляет собой HPMC;
(vi-a) связывающий агент используется в количестве в диапазоне от примерно 0,5 масс.% до примерно 7 масс.%; или
(vi-b) связывающий агент используется в количестве в диапазоне от примерно 2 масс.% до примерно 6 масс.%; или
(vi-c) связывающий агент (например, HPMC) используется в количестве в диапазоне от примерно 3 масс.% до примерно 5 масс.%;
(vii-a) второй суперразрыхлитель является необязательно таким же, как первый суперразрыхлитель, и выбран из группы, состоящей из кроскармеллозы натрия, натриевого гликолята крахмала, кросповидона и их комбинаций;
(vii-b) второй суперразрыхлитель является необязательно таким же, как первый суперразрыхлитель, и выбран из группы, состоящей из кроскармеллозы натрия, натриевого гликолята крахмала и кросповидона; или
(vii-c) второй суперразрыхлитель является необязательно таким же, как первый суперразрыхлитель, и представляет собой кроскармеллозу натрия;
(viii-a) второй суперразрыхлитель используется в количестве в диапазоне от примерно 3 масс.% до примерно 20 масс.% (в подпункте viii-a первый суперразрыхлитель используется в количестве, указанном в пункте iv-a, второй суперразрыхлитель используется в количестве, как указано в данном пункте, и общее количество суперразрыхлителя находится в диапазоне от примерно 6 масс.% до примерно 20 масс.%);
(viii-b) второй суперразрыхлитель используется в количестве в диапазоне от примерно 3 масс.% до примерно 15 масс.% (в подпункте viii-b первый суперразрыхлитель используется в количестве, указанном в пункте iv-b, второй суперразрыхлитель используется в количестве, как указано в данном пункте, и общее количество суперразрыхлителя находится в диапазоне от примерно 6 масс.% до примерно 20 масс.%);
(viii-c) второй суперразрыхлитель используется в количестве в диапазоне от примерно 6 масс.% до примерно 12 масс.%; (в подпункте viii-c первый суперразрыхлитель используется в количестве, указанном в пункте iv-c, второй суперразрыхлитель используется в количестве, как указано в данном пункте, и общее количество суперразрыхлителя находится в диапазоне от примерно 10 масс.% до примерно 18 масс.%); или
(viii-d) второй суперразрыхлитель (например, кроскармеллоза Na) используется в количестве в диапазоне от примерно 8 масс.% до примерно 10 масс.% (в подпункте viii-dпервый суперразрыхлитель используется в количестве, указанном в пункте iv-d, второй суперразрыхлитель используется в количестве, как указано в данном пункте, и общее количество суперразрыхлителя находится в диапазоне от примерно 13 масс.% до примерно 17 масс.%);
(ix-a) наполнитель выбран из группы, состоящей из микрокристаллической целлюлозы, маннита, лактозы, фосфата Ca и их комбинаций;
(ix-b) наполнитель представляет собой микрокристаллическую целлюлозу (например, AVICEL PH-102 или тому подобное); или
(ix-c) наполнитель представляет собой комбинацию микрокристаллической целлюлозы и двухосновного фосфата Ca (например, AVICEL DG или тому подобное);
(x-a) наполнитель используется в количестве в диапазоне от примерно 10 масс.% до примерно 40 масс.%; или
(x-b) наполнитель используется в количестве в диапазоне от примерно 10 масс.% до примерно 25 масс.%;
(x-c) наполнитель (например, микрокристаллическая целлюлоза) используется в количестве в диапазоне от примерно 16 масс.% до примерно 18 масс.%; или
(x-d) наполнитель (например, комбинация микрокристаллической целлюлозы и двухосновного фосфата Ca) используется в количестве в диапазоне от примерно 7 масс.% до примерно 10 масс.%;
(xi-a) смазывающий агент выбран из группы, состоящей из стеарата Mg, стеариновой кислоты, стеарилфумарата натрия и их комбинаций; или
(xi-b) смазывающий агент содержит стеарат магния;
(xii-a) смазывающий агент используется в количестве в диапазоне от примерно 0,5 масс.% до примерно 2,5 масс.%; или
(xii-b) смазывающий агент используется в количестве в диапазоне от примерно 1 масс.% до примерно 2,5 масс.%; или
(xii-c) смазывающий агент (например, стеарат магния) используется в количестве в диапазоне от примерно 1 масс.% до примерно 2 масс.%;
(xiii-a) способ дополнительно включает: (F) покрытие прессованной таблетки; или
(xiii-b) способ дополнительно включает: (F) покрытие прессованной таблетки суспензией пленочного покрытия (например, Опадрай II HP) с получением покрытой таблетки, в которой покрытие составляет от примерно 2 до примерно 4% массы прессованной таблетки; и
(xiv-a) соль щелочного металла ралтегравира (например, калиевая соль ралтегравира) используется в количестве на таблетку в диапазоне от примерно 200 мг до примерно 600 мг на основании свободного фенола; или
(xiv-b) соль щелочного металла ралтегравира (например, калиевая соль ралтегравира) используется в количестве на таблетку примерно 200 мг, 300 мг, 400 мг, 500 мг или 600 мг на основании свободного фенола.
Понятно, что каждое включение каждого из предыдущих пунктов (i)-(xiv) в способ P1, как первоначально описано, составляет один вариант осуществления способа P1. Понятно также, что каждое включение двух или более из пунктов (i)-(xiv) в способ P1, как первоначально описано, составляет один вариант осуществления способа P1. Любая комбинация пунктов (i)-(xiv) находится в пределах объема способа P1, пока такая комбинация по сути неприемлема или иным образом привела бы к неосуществимому способу.
Другой вариант осуществления способа P1 представляет собой способ P1, как первоначально описано выше, где идентичность и количество каждого из используемых в способе ингредиентов являются такими, как указано для прессованной таблетки, описанной выше в варианте осуществления E13.
Другой вариант осуществления способа P1 представляет собой способ P1, как первоначально описано выше, где идентичность и количество каждого из используемых в способе ингредиентов являются такими, как указано для прессованной таблетки, описанной выше в варианте осуществления E14.
Настоящее изобретение также включает прессованную таблетку, полученную способом P1, как первоначально изложено выше или изложено в любом из предыдущих вариантов осуществления способа P1.
Прессованные таблетки по настоящему изобретению применимы при ингибировании интегразы ВИЧ, лечении или профилактике инфекции ВИЧ и лечении, профилактике или задержке начала последующих патологических состояний, таких как СПИД. Лечение СПИДа, профилактика СПИДа, задержка начала СПИДа, лечение ВИЧ-инфекции или профилактика ВИЧ-инфекции определяются как включающие, без ограничения, лечение или профилактику широкого диапазона состояний ВИЧ-инфекции: СПИД, ARC (СПИД-связанный комплекс), как симптоматический, так и бессимптомный, и действительное или потенциальное воздействие ВИЧ. Например, таблетки по настоящему изобретению применимы при лечении или профилактике инфекции ВИЧ после подозреваемого прошлого воздействия ВИЧ в результате таких процедур, как гемотрансфузия, обменное переливание биологических жидкостей, укусы, случайный укол иглой или контакт с кровью пациента во время хирургического вмешательства.
Настоящее изобретение включает способ ингибирования интегразы ВИЧ (например, интегразы ВИЧ-1) у нуждающегося в этом пациента, который включает введение индивиду прессованной таблетки, как первоначально определено выше в «Кратком изложении сущности изобретения». Изобретение также включает способ лечения или профилактики ВИЧ-инфекции (например, инфекции ВИЧ-1) или лечения, профилактики или задержки начала СПИД (например, СПИД, вызванного ВИЧ-1) у нуждающегося в этом индивида, который включает введение индивиду прессованной таблетки по изобретению, как первоначально определено выше. В этих способах прессованная таблетка по настоящему изобретению может быть необязательно использована в комбинации с одним или более средств против ВИЧ, выбранных из анти-ВИЧ противовирусных препаратов, противоинфекционных средств и иммуномодуляторов. Варианты осуществления этих способов включают описанные выше способы, где прессованная таблетка представляет собой таблетку, как изложено в любом из предыдущих вариантов ее осуществления (например, таблетки, описанные в вариантах осуществления E1-E19, и прессованные таблетки, полученные способом P1).
Термин «индивид» (используемый в настоящем описании взаимозаменяемо с термином «пациент») относится к животному, предпочтительно млекопитающему, наиболее предпочтительно к человеку, который был объектом лечения, наблюдения или эксперимента.
Когда таблетку по настоящему изобретению используют или вводят в комбинации с другим средством (например, средством против ВИЧ), то таблетка и средство могут быть введены отдельно или вместе, и при введении отдельно таблетка и средство могут быть введены одновременно или в разное время (например, поочередно).
Настоящее изобретение также включает прессованную таблетку для перорального введения, которая представляет собой прессованную таблетку, как первоначально определено и описано в «Кратком изложении сущности изобретения» (i) для применения в, (ii) для применения в качестве лекарственного средства по поводу, или (iii) для применения при получении или изготовлении лекарственного средства для: (a) лечения (например, человека), (b) медицины, (c) ингибирования интегразы ВИЧ, (d) лечения или профилактики инфекции ВИЧ или (e) лечения, профилактики или задержки начала или прогрессирования СПИД. Варианты осуществления указанных видов применения включают виды применения, как описано выше, где прессованная таблетка по изобретению, как первоначально определено, заменяется ее описанными выше вариантами осуществления (которые включают, наряду с другими, прессованные таблетки, описанные в вариантах осуществления E1-E19, и прессованные таблетки, полученные способом P1). При этих видах применения прессованные таблетки по настоящему изобретению могут быть необязательно использованы в комбинации с одним или более средствами против ВИЧ, выбранными из анти-ВИЧ средств (отличных от атазанавира и его фармацевтически приемлемых солей), противоинфекционных средств и иммуномодуляторов.
«Средство против ВИЧ» представляет собой любое средство, которое непосредственно или опосредованно эффективно при ингибировании интегразы ВИЧ или другого фермента, требуемого для репликации или инфекции ВИЧ, лечения или профилактики ВИЧ-инфекции и/или лечения, профилактики или задержки начала или прогрессирования СПИД. Понятно, что средство против ВИЧ эффективно при лечении, предотвращении или задержке начала или прогрессирования ВИЧ-инфекции или СПИД и/или заболеваний или состояний, возникающих в их результате, или связанных с ними. Например, прессованные таблетки по настоящему изобретению могут быть эффективно введены или в периоды перед воздействием, и/или после воздействия, в комбинации с эффективными количествами одного или более противовирусных препаратов против ВИЧ, иммуномодуляторов, противоинфекционных средств или вакцин, используемых для лечения ВИЧ-инфекции или СПИД, таких как те, которые представлены в таблице 1 WO 01/38332 или в таблице WO 02/30930, за исключением атазанавира и его фармацевтически приемлемых солей. Подходящие противовирусные препараты против ВИЧ для применения в комбинации с соединениями по настоящему изобретению включают, например, те, которые перечислены ниже в таблице A:
Понятно, что объем комбинаций прессованной таблетки по настоящему изобретению со средствами против ВИЧ не ограничивается противовирусными препаратами против ВИЧ, перечисленными в таблице A и/или перечисленными в приведенных выше таблицах в WO 01/38332 и WO 02/30930, но включает в принципе любую комбинацию с любой фармацевтической композицией, которая пригодна для лечения или профилактики ВИЧ-инфекции или СПИД, исключая композиции, содержащие атазанавир или его фармацевтически приемлемую соль. Противовирусные препараты против ВИЧ и другие средства обычно используются в этих комбинациях в их обычных диапазонах дозировки и схемах, известных в данной области, включая, например, дозировки, описанные в изданиях справочника Physicians' Desk Reference, Thomson PDR, Thomson PDR, 57th edition (2003), the 58th edition (2004), the 59th edition (2005) и в его последующих изданиях.
Кроме того, понятно, что способы применения и способы лечения, изложенные в настоящем описании, исключают введение прессованных таблеток и комбинаций фиксированных доз (описанных ниже) по изобретению с атазанавиром или его фармацевтически приемлемой солью.
Прессованные таблетки по изобретению могут соответственно содержать от примерно 50 мг до примерно 800 мг ралтегравира на таблетку и обычно содержат от примерно 100 мг до примерно 700 мг на таблетку, а обычнее содержат от примерно 200 мг до примерно 600 мг на таблетку. Определенный уровень дозы и частота введения могут варьироваться от пациента к пациенту, например, в зависимости от возраста, массы тела, общего состояния здоровья, пола и рациона питания. Соответствующий уровень дозы ралтегравира, подходящий для конкретного пациента, средний специалист в данной области может определить без ненужного экспериментирования. Считается, что прессованные таблетки по изобретению, содержащие от 200 до 600 мг ралтегравира, вводимые перорально взрослым людям один или два раза в день, могут быть эффективны при лечении ВИЧ-инфекции.
Настоящее изобретение также включает твердую комбинацию с фиксированной дозой (альтернативно именуемой в настоящем описании комбинацией «FDC») для перорального введения, которая включает первую часть, содержащую эффективное количество соли щелочного металла ралтегравира, где первая часть содержит внутригранулярный компонент и внегранулярный компонент, используемые в прессованной таблетке, как первоначально описано в «Кратком изложении сущности изобретения» или как описано в любом из его аспектов или вариантов осуществления (например, в вариантах осуществления E1-E19); и вторую часть, которая включает состав, содержащий эффективное количество другого средства против ВИЧ, при условии, что комбинация с фиксированной дозой не содержит атазанавир или его фармацевтически приемлемую соль. Средство против ВИЧ во второй части может представлять собой любое средство против ВИЧ, как определено и описано выше, за исключением атазанавира или его фармацевтически приемлемой соли. В одном варианте осуществления (варианте осуществления FDC-E1) средство против ВИЧ во второй части представляет собой ингибитор прикрепления ВИЧ к рецептору, ингибитор CCR5 (хемокиновых рецепторов 5-го типа), ингибитор CXCR4 (хемокиновых рецепторов 4-го типа), ингибитор слияния вируса ВИЧ с клеткой, ингибитор интегразы ВИЧ, нуклеозидный ингибитор обратной транскриптазы ВИЧ, не нуклеозидный ингибитор обратной транскриптазы ВИЧ или ингибитор протеазы ВИЧ (кроме атазанавира или его фармацевтически приемлемой соли). В другом варианте осуществления (варианте осуществления FDC-E2) средство против ВИЧ во второй части выбрано из группы, состоящей из средств, перечисленных выше в таблице A.
Другой вариант осуществления комбинации с фиксированной дозой (вариант осуществления FDC-E3) представляет собой комбинацию, как первоначально описано или как описано в любом из вариантов осуществления FDC-E1 и FDC-E2, где комбинация представляет собой двухслойную прессованную таблетку, в которой первая часть находится в одном слое, а вторая часть находится во втором слое. Двухслойные таблетки могут быть получены прессованием вместе первой части и второй части. Двухслойные таблетки могут быть альтернативно получены введением первой или второй части в таблеточный пресс; прессованием этой части с образованием первого слоя таблетки; введением другой из первой и второй частей в таблеточный пресс; и прессованием как первой, так и второй частей с получением двухслойной таблетки.
Другой вариант осуществления комбинации с фиксированной дозой (вариант осуществления FDC-E4) представляет собой комбинацию, как первоначально описано или как описано в любом из вариантов осуществления FDC-E1 и FDC-E2, где комбинация представляет собой монолитную прессованную таблетку, где первая часть и вторая часть находятся в одном и том же слое. Монолитные таблетки могут быть получены смешиванием первой и второй частей вместе и прессованием смеси в таблеточном прессе.
Другой вариант осуществления комбинации с фиксированной дозой (вариант осуществления FDC-E5) представляет собой комбинацию, как первоначально описано или как описано в любом из предшествующих вариантов осуществления (FDC-E1)-(FDC-E4), где комбинация покрыта сахаром и/или пленочным покрытием описанным выше способом.
Аббревиатуры, используемые в настоящем описании, включают следующие: API = активный фармацевтический ингредиент; APCI = химическая ионизация при атмосферном давлении (масс-спектроскопия); сП = сантипуаз; CPCG-3 = гранулятор-3 Глатта для нанесения порошкового покрытия; EDTA = этилендиаминтетрауксусная кислота; EG = внегранулярная; г = грамм(ы); HEC = гидроксиэтилцеллюлоза; HPC = гидроксипропилцеллюлоза; HPMC = гидроксипропилметилцеллюлоза; ВЭЖХ = высокоэффективная жидкостная хроматография; IG = внутригранулярная; ЖЖ/МС = жидкостная хроматография/масс-спектрометрия; LOD = потеря при сушке; MRM = мониторинг множественных реакций; PK = фармакокинетика; PVP = поливинилпирролидон; SD = стандартное отклонение.
Следующие примеры служат только для иллюстрации изобретения и его практического осуществления. Примеры не следует рассматривать как ограничения объема или сущности изобретения. Ралтегравир может быть получен, как описано в примере 1 заявки на патент США US 2006/0122205 A1. Форма 1 кристаллической монокалиевой соли ралтегравира может быть получена, как описано в примере 2 заявки на патент США US 2006/0122205 A1.
ССЫЛОЧНЫЙ ПРИМЕР 1
Таблетки Изентресс®
Таблетки Изентресс® получают, используя процедуру сухого гранулирования, описанную в Примере 3 в заявке на патент США US 2007/0292504, после которой сердцевинную таблетку покрывают пленочным покрытием Опадрай II, причем таблетки Изентресс® имеют следующий состав:
ПРИМЕР 1
Получение прессованных таблеток, содержащих калиевую соль ралтегравира и внутригранулярную и внегранулярную кроскармеллозу Na (Таблетка Пр.l)
Прессованные таблетки, содержащие 400 мг ралтегравира на основании свободного фенола, получали сначала приготовлением смеси (примерно 4 кг) калиевой соли ралтегравира, HPMC и внутригранулярной части кроскармеллозы натрия в грануляторе с высоким усилием сдвига Fielder 10/25L при скорости вращения импеллера 500 об/мин и скорости вращения ножа фальцаппарата ударного типа 1800 об/мин в течение 0,5 минут, затем добавлением воды в соответствии с Фармакопеей США (40 масс.%; примерно 1,6 кг) в гранулятор и гранулированием при 250 об/мин в течение 5 минут при скорости распыления 320 г/мин. Затем гранулированный материал сушили в грануляторе в псевдоожиженном слое GPG-3 при температуре воздуха на впуске 80°C в течение 20-30 мин, где скорость воздушного потока исходно составляла 200 футов3/минуту (=5,66 м3/мин) и постепенно снижалась в течение периода сушки до конечной скорости потока 100 футов3/минуту (=1,42 м3/мин) с получением высушенного гранулята с LOD примерно 1 масс.%. Затем высушенный гранулят подвергали помолу и просеиванию с использованием мельницы Quadro 197 Comil с квадратным стержнем, работающей при 2000 об/мин, снабженной решетчатым ситом, имеющим отверстие 1,27 мм (т.е. сетчатым фильтром № 50) с получением гранул, которые смешивали с микрокристаллической целлюлозой и внегранулярной частью кроскармеллозы натрия в мешалке с V-образным корпусом емкостью 8 кварт (~8,8 л) при скорости вращения 25 об/мин в течение 5 минут. Стеарат магния (предварительно просеянный с использованием сетчатого фильтра размером № 40 меш) затем добавляли в мешалку и смесь перемешивали в течение еще 5 минут при скорости импеллера 25 об/мин. Смазанные гранулы зетам прессовали в таблетки по 700 мг с использованием роторного таблеточного пресса с простой инструментальной оснасткой овальной формы при силе сжатия, необходимой для достижения твердости таблетки примерно 15 килопонд (т.е. 147 Ньютон), по данным измерения с использованием тестера твердости Key модели HT-300. Затем сердцевинные таблетки покрывали Опадрай II в устройстве для пленочного покрытия Vector (чан емкостью 3,75 л), получая таблетки с пленочным покрытием с прибавкой массы приблизительно 3% относительно сердцевинной таблетки.
ПРИМЕР 2
Получение прессованных таблеток, содержащих ралтегравир калия и внутригранулярную и внегранулярную кроскармеллозу Na (Таблетка Пр.2)
Получение. Прессованные таблетки, содержащие 400 мг ралтегравира (лекарственная загрузка = 64,5%) и имеющие ингредиенты, показанные в таблице выше, получали в соответствии с процедурой, описанной в примере 1. Эти таблетки содержали те же ингредиенты, что и таблетки, полученные в примере 1, за исключением того, что AVICEL PH-102 (17,1 масс.%) в качестве внегранулярного наполнителя заменили на AVICEL DG (7,8 масс.%).
Дезинтеграция. Величины времени дезинтеграции таблеток Пр.1 и Пр.2 были получены в соответствии со способом Фармакопеи США <701> с использованием разрушающего устройства Vankel VK100 (Varian, Inc.), в корзину которого помещали одну таблетку и корзину погружали в 0,01 N водную HCl (деионизированная вода) при 37°C. Время до исчезновения таблетки после погружения представляет собой время дезинтеграции. Величины времени дезинтеграции (средняя величина двух замеров для каждого типа таблетки) таблеток Пр.1 и Пр.2 были почти одинаковыми, т.е. примерно 10 минут для таблетки Пр.1 и примерно 9 минут для таблетки Пр.2.
ПРИМЕР 3
Фармакокинетическое исследование ралтегравира, совместно введенного с атазанавиром, у здоровых мужчин и женщин
Открытое состоящее из 5 периодов рандомизированное, перекрестное исследование фармакокинетики однократных пероральных доз препаративных форм, содержащих калиевую соль ралтегравира и атазанавира сульфата, проводили у здоровых мужчин и женщин при введении, проводимом после приема легкой пищи. В первые два из пяти периодов каждый пациент получал последовательно одну дозу:
(A) таблетку ралтегравира 400 мг, полученную, по существу, способом, описанным в ссылочном примере 1 (т.е. Изентресс®), вводили совместно с реятазом® (300 мг), и
(B) таблетку ралтегравира 400 мг, полученную, по существу, способом, описанным в примере 1 (т.е. «Таблетку Пр.1»), вводили совместно с реятазом® (300 мг).
Образцы крови брали перед введением и через 0,5, 1, 1,5, 2, 3, 4, 6, 8, 12, 16, 24, 36 и 48 часов после введения. Имелся по меньшей мере 5-дневный период выведения препарата из организма между каждой из доз в получавших лечение группах A, B, C, D и E, начиная от введения дозы предыдущего периода. Безопасность пациента контролировали до и после каждого введения путем клинической оценки побочных явлений и проверки других параметров безопасности, включая лабораторные тесты безопасности крови и мочи, показатели жизненно важных функций, физикальное обследование и электрокардиограммы.
Подготовка и анализ образцов: Для анализа ралтегравира экстрагировали образцы плазмы, используя 96-луночную жидкостно-жидкостную экстракцию. Экстракты плазмы инжектировали на колонку ВЭЖХ Ace C18(50×3,0 мм, 3 мкм, титановые детали (rits) и анализировали в изократических условиях при подвижной фазе, состоящей из 42,5/57,5 (об./об.%) 0,1 мМ EDTA в 0,1% муравьиной кислоте/метаноле, при скорости потока 0,5 мл/минуту. Экстракты образцов ионизировали с использованием интерфейса APCI и контролировали MRM в режиме положительной ионизации. Динамический диапазон анализа ЖХ/МС/МС составил 2-1000 нг/мл в расчете на 200 мкл аликвоту человеческой плазмы.
PK расчеты: Площадь под кривой зависимости «концентрация-время» в плазме до последней выявляемой концентрации (AUC0-last) рассчитывали, используя некомпартментальную модель и способ расчета линейного подъема/логарифмического спуска с помощью программного обеспечения WinNonLin в версии v5.0.1. Величины AUC экстраполировали на бесконечность в соответствии со следующим уравнением: AUC0-∞=AUC0-last + Clast/β, где Clast представляет последнюю выявляемую концентрацию и β представляет наклон кривой снижения конечной фазы. Наблюдаемая максимальная концентрация в плазме (Cmax), время Cmax (Tmax) и концентрацию в плазме через 12 ч после введения (C12ч)определяли проверкой.
Фармакокинетические результаты исследования для доз A и B ралтегравира следующие:
1. Фармакокинетические величины представлены для ралтегравира. Величины Tmax представляют медиану (мин-макс); величины T1/2представляют среднююгармоническую ± псевдостандартное отклонение (SD); и величины всех других параметров представляют арифметическую среднюю ± SD.
2. Число пациентов n при каждом протоколе лечения составляло 20.
Фиг. 1 представляет собой диаграмму, показывающую отдельные и средние величины AUC для групп лечения дозой A и B.
По сравнению с таблетками Изентресса® (доза A) таблетки Пр.1 (доза B) привели к повышенному воздействию (т.е. более высоким величинам AUC) и заметно сниженной вариабельности AUC, Cmax и Cl2ч. Более высокие величины AUC указывают на потенциальную преимущественную эффективность таблеток Пр.1, и они могут обеспечить возможность такой же эффективности при более низкой дозе ралтегравира в таблетках с использованием состава примера 1. Сниженная вариабельность может также являться преимуществом, ведущим к более постоянным уровням ралтегравира в плазме.
Хотя в предшествующем описании речь идет о принципах настоящего изобретения с примерами, приведенными с целью иллюстрации, практическое осуществление изобретения охватывает все обычные изменения, адаптации и/или модификации, которые входят в объем следующей формулы изобретения. Все публикации, патенты и патентные заявки, приведенные в настоящем описании, полностью включены в описание путем ссылки.
Реферат
Описаны прессованные таблетки для перорального введения, содержащие ралтегравир в форме фармацевтически приемлемой соли. Таблетки содержат: (A) внутригранулярный компонент, содержащий (i) эффективное количество калиевой соли ралтегравира, (ii) первый суперразрыхлитель и (iii) связывающий агент; и (B) внегранулярный компонент, содержащий (i) второй суперразрыхлитель, (ii) наполнитель и (iii) смазывающий агент. Суперразрыхлитель представляет собой кроскармеллозу натрия, наполнитель представляет собой микрокристаллическую целлюлозу или комбинацию микрокристаллической целлюлозы и двухосновного фосфата кальция, смазывающий агент представляет собой стеарат магния. В состав таблетки не входит атазанавир или его фармацевтически приемлемая соль. Прессованные таблетки по изобретению характеризуются высокой нагрузкой по лекарственному веществу при приемлемом размере таблетки. 3 н. и 8 з.п. ф-лы, 1 ил., 3 пр.
Формула
(A) внутригранулярный компонент, содержащий:
(i) эффективное количество калиевой соли ралтегравира,
(ii) первый суперразрыхлитель, и
(iii) связывающий агент; и
(B) внегранулярный компонент, содержащий:
(i) второй суперразрыхлитель,
(ii) наполнитель, и
(iii) смазывающий агент,
при условии, что в состав таблетки не входит атазанавир или его фармацевтически приемлемая соль,
где:
(A)(i) калиевая соль ралтегравира используется в количестве в диапазоне от 50 мас. % до 65 мас. % на основании свободного фенола;
(A)(ii) первый суперразрыхлитель используется в количестве в диапазоне от 5 мас. % до 10 мас. %;
(A)(iii) связывающий агент используется в количестве в диапазоне от 2 мас.% до 6 мас.%;
(B)(i) второй суперразрыхлитель используется в количестве в диапазоне от 6 мас.% до 12 мас.%;
(В)(ii) наполнитель используется в количестве в диапазоне от 6 мас.% до 25 мас.%; и
(В)(iii) смазывающий агент используется в количестве в диапазоне от 1 мас.% до 2,5 мас.%;
где общее количество суперразрыхлителя находится в диапазоне от 10 мас.% до 18 мас.%,
где:
первый суперразрыхлитель представляет собой внутригранулярную кроскармеллозу Na;
связывающий агент представляет собой НРМС;
второй суперразлыхлитель представляет собой внегранулярную кроскармеллозу Na;
наполнитель представляет собой микрокристаллическую целлюлозу или комбинацию микрокристаллической целлюлозы и двухосновного фосфата Са; и
смазывающий агент представляет собой стеарат Mg.
(A) внутригранулярный компонент, содержащий:
(i) эффективное количество калиевой соли ралтегравира,
(ii) первый суперразрыхлитель, и
(iii) связывающий агент; и
(B) внегранулярный компонент, содержащий:
(i) второй суперразрыхлитель,
(ii) наполнитель, и
(iii) смазывающий агент,
при условии, что в состав таблетки не входит атазанавир или его фармацевтически приемлемая соль,
где:
(A)(i) калиевая соль ралтегравира используется в количестве в диапазоне от 55 мас.% до 60 мас.% на основании свободного фенола;
(A)(ii) первый суперразрыхлитель представляет собой внутригранулярную кроскармеллозу натрия, используемую в количестве в диапазоне от 5 мас.% до 7 мас.%;
(A)(iii) связывающий агент представляет собой НРМС, которая используется в количестве в диапазоне от 3 мас.% до 5 мас.%;
(B)(i) второй суперразрыхлитель представляет собой внегранулярную кроскармеллозу натрия, используемую в количестве в диапазоне от 8 мас.% до 10 мас.%;
(В)(ii) наполнитель представляет собой микрокристаллическую целлюлозу, используемую в количестве в диапазоне от 16 мас.% до 18 мас.%; и
(В)(iii) смазывающий агент представляет собой стеарат магния, используемый в количестве в диапазоне от 1 мас.% до 2 мас.%;
где общее количество кроскармеллозы натрия находится в диапазоне от 13 мас.% до 17 мас.%.
(A) внутригранулярный компонент, содержащий:
(i) эффективное количество калиевой соли ралтегравира,
(ii) первый суперразрыхлитель, и
(iii) связывающий агент; и
(B) внегранулярный компонент, содержащий:
(i) второй суперразрыхлитель,
(ii) наполнитель, и
(iii) смазывающий агент,
при условии, что в состав таблетки не входит атазанавир или его фармацевтически приемлемая соль,
где:
(A)(i) калиевая соль ралтегравира используется в количестве в диапазоне от 55 мас.% до 65 мас.% на основании свободного фенола;
(A)(ii) первый суперразрыхлитель представляет собой внутригранулярную кроскармеллозу натрия, используемую в количестве в диапазоне от 5 мас.% до 8 мас.%;
(A)(iii) связывающий агент представляет собой НРМС, которая используется в количестве в диапазоне от 3 мас.% до 5 мас.%;
(B)(i) второй суперразрыхлитель представляет собой внегранулярную кроскармеллозу натрия, используемую в количестве в диапазоне от 8 мас.% до 10 мас.%;
(В)(ii) наполнитель представляет собой комбинацию микрокристаллической целлюлозы и двухосновного фосфата кальция и используется в количестве в диапазоне от 7 мас.% до 10 мас.%; и
(В)(iii) смазывающий агент представляет собой стеарат магния, используемый в количестве в диапазоне от 1 мас.% до 2 мас.%,
где общее количество кроскармеллозы натрия находится в диапазоне от 13 мас.% до 17 мас.%.
Комментарии