Способ модификации перфторполиэфиров - RU2034000C1
Код документа: RU2034000C1
Описание
Изобретение относится к химии высокомолекулярных соединений, а именно к способам получения перфторполиэфиров, используемых в виде водных эмульсий при защите строительных материалов.
Известен способ модификации перфорполиэфиров путем их взаимодействия с металлгидридами (алюминия, лития) или боргидридом натрия в инертном растворителе при нагревании. Получаемые перфторполиэфиры имеют только одну функциональную блокирующую группу (СН2ОН группа).
Предлагаемый способ позволяет получать перфторполиэфиры, способные к взаимодействию с органическими и неорганическими субстратами, и с концевыми группами, содержащими один или два атома галогена, не являющегося фтором. Они могут быть использованы в качестве поверхностных модификаторов для полимерных и неорганических материалов в целях придания им свойств, характерных для фторированных продуктов; водо- и маслоотталкивающие свойства, низкий коэффициент трения, низкий показатель преломления и т.д.
Предлагаемый способ модификации перфторполиэфиров осуществляют путем их взаимодействия с металлгидридами в инертном растворителе при
нагревании с использованием в качестве исходного перфторполиэфира общей формулы
TO(

m/n=0,01-0,5;
R=Cl, Br, F, J;
R'=Cl, Br, F, J;
Т группа, содержащая 1-3 атома углерода и один или два атома хлора, брома и иода;
Х=F,CF3, или их сложные эфиры.
Дополнительно могут проводить взаимодействие с соединениями общей формулы R"X, где Х атом галогена,
R″=
-H;-CH2-



[R1-алкил или -СН2(СНО)nH]
-

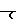
-CH


Дополнительно могут проводить взаимодействие с эпоксициклическими соединениями.
Фторполиэфиры, имеющие кислотную функциональную концевую группу, использующиеся в качестве исходных продуктов для получения соединений настоящего изобретения, могут быть получены путем фотоокисления перфторпропена и/или тетрафторэтилена в присутствии малых количеств тщательно галоидированного этилена, содержащего фтор и, по меньшей мере, один атом галогена, который не является фтором. Продукт фотоокисления затем подвергается термообработке в целях удаления содержащихся в нем пероксидных групп.
Способ иллюстрируется следующими примерами.
П р и м е р 1. 17 г смеси кислот формулы
ClC3F6O(C3F6O)n(CF2O)mCF2COOH где n является средним значением, равным 0,85, а m средним значением, равным 0,02, смешивают с 6 г Р2О5 и
полученную в результате смесь нагревают от 100 до 200оС в стеклянной колбе, совмещенной с колонкой Вигре (8х150 мм) и холодильником Либиха. Получают 12 г жидкости, подвергшейся перегонке
при 170-180оС. ИК-спектр характеризуется поглощением при 1805-1870 см-1, соответствующем карбоксильным группам, и отсутствием линий, относящихся к гидроксильным группам в области
от 3300 до 3600 см-1.
Таким образом, структура ангидрида формулы
[ClC3F6O(C3F6O)0,85(CF2O)0,
08CF2CO]2O соответствует этому соединению.
14 г той же кислоты подвергают взаимодействию с 10 см3тионилхлорида, нагревая реакционную смесь в
колбе с обратным холодильником в течение 8 ч в присутствии 0,1 г пиридина. По завершении реакции большую часть тионилхлорида отгоняют, полученный концентрат перегоняют при 105-120оС (760 мм
рт.ст. ) и получают 9 г продукта, который при ИК-анализе характеризуется значительным поглощением при 1805 см-1 и отсутствием поглощения при 3300-3600 см-1. На основании
процентного содержания гидролизованного хлора (водным NaOH 0,5 н.) установлено, что указанный продукт имеет формулу
СlC3F6O(C3F6O)0,85
(CF2O)0,02CF2COCl.
7 г полученного продукта смешивают с 1,5 г безводного бензола и добавляют в суспензию 3,5 г AlCl3 в 15 см3
СН2Сl2, затем охлаждают до 0оС и все время перемешивают. В течение реакции указанная смесь становится красной и однородной: после 4-часовой реакции смесь выливают в
ледяную воду, собирают отделенную органическую фазу, промывают в воде и бикарбонате, высушивают в присутствии Na2SO4, концентрируют в СН2Сl2 и перегоняют
при 195-198оС, получая при этом 3 г соединения, в ИК-спектре которого обнаружены линии поглощения 1720 см-1, соответствующие карбонилу, и 1500 и 1600 см-1,
соответствующие бензольному кольцу, следовательно, структура
ClC3F6О (C3F6O)0,85(CF2O)0,02CF2COC6H5 соответствует полученному продукту.
П р и м е р 2. 90 г смеси кислот формулы
ClC3F6O(C3F6O)n(CF2O)mCF2COOH, полученной по примеру 1, где n является средним значением, равным 1,27, а m=0,05, подвергают взаимодействию с 100 г этанола (99,9% ) в присутствии 2 г H2SO4 (96%) и 60 г бензола в колбе емкостью 250 см3 с ректификационной колонной (1х100), снабженной спиральной насадкой. При этом поддерживается температура, обеспечивающая
кипение смеси. В течение 3 ч дистиллят не отходит, а затем, при коэффициенте обратного потока около 20, через 6 ч, сверху собирают азеотропную смесь (вода, бензол, этанол 43 мл). При снижении
коэффициента обратного потока до 5, большую часть бензол-этаноловой азеотропной смеси (140 мл) перегоняют. Полученный в конечном счете продукт выливают в ледяную воду и немедленно отделяют и
высушивают в присутствии Na2SO4, перегоняют при 170-180оС, получают 82 г продукта. ИК-анализ указанного продукта не обнаруживают характерных для кислоты линий. Продукт
не может быть подвергнут титрованию раствором триэтиламина в метаноле в противоположность исходной смеси кислот.
При помощи ЯМР установлено присутствие эфирной группы
-

43 г полученного сложного этилового эфира растворяют в 150 см3простого этилового эфира, охлажденного до 0оС, и в полученный раствор в течение 2 ч добавляют газообразный аммиак. В течение указанного времени реакция завершается. После выпаривания
простого эфира оставшуюся жидкость ректифицируют при 248-260оС, получают продукт, который при ИК-анализе обнаруживает линии поглощения 1740 см-1 и 1610 см-1. Этот
продукт имеет следующую амидную структуру:
СlC3F6O(C3F6O)1,27(CF2O)0,05CF2CONH2
14
г вышеуказанного сложного этилового эфира растворяют в 10 см3простого этилового эфира и добавляют 2,4 см3 н-бутиламина. Эту реакционную смесь выдерживают в течение 1 ч, затем
раствор концентрируют, перегоняют и собирают при 220 и 230оС продукт, который подвергают элементному анализу (С=34,4% Cl= 7,9% F= 40,5% Н= 2%), ИК-анализу и анализу ЯМР19Г,
подтвердивших амидную структуру полученного соединения
ClC3F6O(C3F6O)1,27(CF2O)0,05CF2CONHC4
H9
П р и м е р 3. 9,4 г амида формулы
ClC3F6O(C3F6O)1,27(CF2O)0,05CF2CONH2 полученного в предыдущем примере смешивают в стеклянной колбе, снабженной холодильником, с 20 г Р2О5, и полученную смесь нагревают при 150-180оС в течение 2 ч.
Затем холодильник удаляют, а полученный жидкий продукт дважды перегоняют и отделяют 7 г кипящей фракции при 120-130оС.
Продукт, составляющий указанную фракцию, подвергают
ИК-анализу, при котором обнаруживается четкая линия поглощения при 2250 см-2, которая соответствует типичной CN-группе, а формула нитрила
ClC3F6O(C3
F6O)1,27(CF2O)0,05CF2CN соответствует полученному продукту.
П р и м е р 4. Образец (20 г) сложного фенилового эфира формулы
ClC3F6O(C3F6O)1,27(CF2O)0,05CF2COOC6H5 полученный из 10 см3 кислоты,
описанной в примере 2, путем реакции превращения в соответствующий ацилхлорид и последующей реакции с фенолом в присутствии пиридина и промыванием гидроспиртовым раствором подвергают взаимо- действию
с 3,7 г о-фенилендиамина при постепенном нагревании в течение 20 ч от 30 до 200оС. Продукт, полученный в виде твердого вещества зеленого цвета, имеет температуру плавления 65-70о
С и температуру кипения 230-260оС и в своем спектре обнаруживает четкие линии поглощения 1450, 1540, 1590 см-1 (соответствующие конденсированным кольцам и группировке -С=N- в
цикле), а также широкую линию поглощения в области 2700-3100 см-1, принадлежащую связям С-Н и N-Н.
Элементный анализ (С=32,5% Сl=6,4% H=0,1% F=54,9%), подтвердил, что
полученный продукт имеет следующую структуру:
ClC3F6O(C3F6O)1,27(CF2O)0,05=CF2C

П р и м е р 5. 10 г нитрила примера 3 подвергают взаимодействию в стеклянном сосуде при температуре -50оС с безводным аммиаком, взятом в избытке. Избыточное количество аммиака удаляют из продукта, структура которого соответствует амидину формулы
ClC3F6O(C3F6O)1,27(CF2O)0,05CF2C
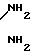
установленной на основе ИК-анализа, спектры которого показали линии поглощения в 1600 см-2, соответствующие иминовой группе, и в области 3400-3100 см-1, соответствующие N-H-связям.
5 г
полученного амидина постепенно нагревают до 300оС в течение 8 ч до тех пор, пока не закончится выделение аммиака, в результате получают высоковязкую жидкость, ИК-спектр которой обнаруживает
единственную четкую линию поглощения в 1550 см-1, соответствующую -С-N-группе триазинового кольца, в соответствии с чем структура полученного соединения идентифицирована с
полиоксахлорперфторалкилтриазином формулы
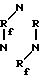
П р и м е р 6. 50 г сложного этилового эфира
СlC3F6O(C3 F6O)1,27(CF2O)0,05-CF2COOC2H5, полученного в соответствии с примером 2, добавляют в течение 2 ч в суспензию 4 г LiAlH4 в 250 см3безводного этилового эфира при температуре от комнатной до 25оС. После 3 ч реакции избыток LiAlH4 разлагают при помощи HCl (5%-ной, эфирную фазу отделяют, высушивают в присутствии Na2SO4, концентрируют и перегоняют при атмосферном давлении. Получают продукт, имеющий точку кипения от 150 до 170оС.
При ИК-анализе этого продукта не обнаружено линий, присущих карбонилу, но обнаружена широкая линия, присущая карбоксилам. Путем обработки взвешенным количеством ангидрида уксусной кислоты в присутствии пиридина и этилового эфира и через 12 ч уксусную кислоту подвергают анализу методом обратного титрования, в результате чего для указанного продукта получен эквивалентный вес 490, который соответствует гидроксилам.
ЯМР, Н анализ подтверждает следующую структуру продукта:
ClC3F6O(C3F6O)1,27(CF2
O)0,05CF2CH2OH.
П р и м е р 7. 55 г амида формулы
ClC3F6O(C3F6O)1,27(CF2
O)0,05CF2CONH2, полученного согласно примеру 3, добавляют в течение 2 ч в суспензию 4 г LiAlH4 в 300 см3 простого этилового эфира при комнатной
температуре.
После 2 часовой реакции при комнатной температуре и 3 2-часовой при 35оС избыток LiAlH4 разлагают при 0оС водным раствором тетрагидрофурана, затем добавляют 500 г Н2О и 50 см3 водного NaOH (40%).
Органический слой отделяют, сушат в присутствии Na2SO4 и перегоняют, получают фракцию (40 г) с температурой кипения 140-160оС. ИК-анализ не показал линий поглощения, присущих карбонильным группам.
Полученный продукт имеет структуру ClC3F6O(C3F6O)1,27(CF2O)1,27(CF2O)0,05CF2- -CH2NH2 аминовый эквивалентный вес, определенный путем титрования HClO4 0,1 н. в уксусной кислоте, составляет 495.
П р и м е р 8. Раствор 1,6 см3 акрилилхлорида в 20 см3 этилового
эфира добавляют в раствор 11 г амина формулы
ClC3F6O(C3F6O)1,98(CF2O)0,08CF2CH2NH2
, полученного по примеру 7 из амина соответствующей кислоты, и 2,8 см3триэтиламина в 100 см3 этилового эфира, охлажденного до 0оС, и перемешивают. Смесь поддерживают в
течение 3 ч при 0оС, после чего смесь отфильтровывают.
Полученный эфирный раствор добавляют в 0,1 г фентиазина

Полученный продукт имеет структуру,
соответствующую акриламиду формулы
ClC3F6O(C3F6O)1,98-(CF2O)0,08-CF2-
-CH2
HCO-CH=CH2.
Анализ, проведенный при помощи ЯМР (Н), также подтвердил указанную структуру соединения.
П р и м е р 9. Смесь кислот формулы
ТО(С2F4O)n(CF2O)mCF2COOH, полученную согласно известному способу [2] где Т= ClCF2 или ClCF2CF2, m/n=1,2 c
молярным отношением СООН/Т=1,1 и имеющую средний молекулярный вес, равный 900, подвергают этерификации в соответствии с методикой, описанной в примере 2.
После удаления спиртовой фазы
слой, содержащий сложные этиловые эфиры перфторированных кислот, очищают путем отгонки под вакуумом при 100оС в течение 2 ч, 150 г сложных эфиров перегоняют и получают продукты (195-210оС), ИК-спектр которых обнаруживают линию поглощения в 1790 см-1. Затем эти соединения восстанавливают до спиртов, содержащих одну функциональную группу и имеющих формулу
ТО(С2F4O)n)CF2O)mCF2CH2OH при помощи LiAlH4 способом, описанным в примере 6.
Полученный таким образом продукт имеет температуру кипения от 175 до 190оС.
П р и м е р 10. Смесь кислот формулы
TO(CF2C

Затем 20 г эфира растворяют в 100 см3 раствора в объеме 1/1 простого эфира и FC 113 (1,1,2-трихлоро-1,1,
2-трифторэтан), охлажденного до 0оС, после чего раствор обрабатывают газообразным аммиаком в течение 3 ч. После выпаривания растворителя получают остаток, состоящий из смеси амидов
формулы
TO(CF2C

П р и м е р 11. 10 г смеси амидов, полученной в примере 10, обрабатывают 20 г R2O5 в течение 3 ч при 150-180оС. Полученный жидкий
продукт выделяют при помощи перегонки и посредством ИК-анализа поглощение в 2250 см-1 и идентифицируют как смесь нитрилов формулы
TO(CF2C

П р и м е р 12. 50 г
сложного этилового эфира формулы ClC3F6O(C3F6O)1,27(CF2O)0,05CF2COOC2H5, полученного по
примеру 2, растворяют в 150 см3 смеси 1,1,24трихлоро-1,1,2-трифторэтан (FC 113) и метанола в отношении 1/1 по весу. Этот раствор добавляют в 15 г аминопропилтриэтоксисилана и нагревают в
колбе с обратным холодильником. После окончания реакции растворитель и избыток реагента выпаривают. В ИК-спектре получен- ного соединения не обнаружено характерной для эфира линии поглoщения в 1790
см-1, но обнаружена линия поглощения в 1740 см-1, соответствующая амидиновой группе. Анализ, проведенный при помощи ЯМР (ISF и Н), показал, что полученный продукт имеет следующую
структуру:
ClC3F6O(C3F6O)1,27(CF2O)0,05CF-

П р и м е р 13. 50 г смеси сложных метилэфиров, соответствующих формуле
BrC3F6O-(CF

Путем титрования ангидридом уксусной кислоты, как описано в примере 6, получен эквивалентный вес, равный 650, который соответствует гидроксилу формулы
BrC3F6O(CF

Реферат
Использование: водные эмульсии перфторполиэфиров могут быть использованы при защите строительных материалов. Сущность изобретения: способ модификации перфторполиэфиров путем их взаимодействия с металлгидридами в инертном растворителе при нагревании, в качестве перфторполиэфиров используют соединения общей формулы T-O-

Формула
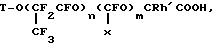
где n 1 15, m/n 0,01 0,5;
R Cl, Br, F, J;
R' Cl, br, F, J;
Т группа, содержащая 1-3 атома углерода и один или два атома хлора, брома, или йода;
X F, CF3,
или их сложные эфиры.
R"X,
где X галоген;
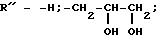

R1 алкил или

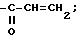
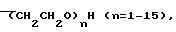
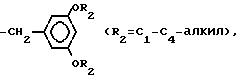


3. Способ по п.1, отличающийся тем, что дополнительно проводят взаимодействие с эпоксициклическими соединениями.
Комментарии